

Abstract
Background
Glypican 1 (GPC1) is a heparan sulphate proteoglycan cell membrane protein. It is implicated in driving cancers of the breast, brain, pancreas, and prostate; however, its role in esophagogastric cancer (EGAC) remains unexplored. The aim of the study was to investigate and elucidate the molecular mechanistic of GPC1 in human EGAC.
Methods
Thirty tissue and 120 microarray sections of EGAC were evaluated with Anti-GPC1 immunohistochemistry. Loss and gain of GPC1 function were performed using lentivirus transfection in EGAC cell lines. Mechanistically, AKT/GSK/β-catenin pathway was evaluated using AKT inhibitor MK-2206 and Wnt/β-catenin stimulant LiCl.
Results
GPC1 overexpression was found in 102 cases (68%). Overexpression of GPC1 correlated with lymph node metastasis, poor differentiation and decreased overall survival. Lentivirus mediated GPC1 knockdown resulted in decreased cell proliferation, migration, invasion, and colony formation. Knockdown caused G0/G1 cell cycle arrest, increased apoptosis, and reduced epithelial mesenchymal transition (EMT). GPC1 mediated its effects by activation of AKT/GSK/β-catenin pathway.
Conclusions
This is the first descriptive study to decipher the role of GPC1 in EGAC. Our results suggest that GPC1 regulates cell proliferation and growth and may serve as an attractive oncotarget in EGAC.
Keywords: Glypican 1 (GPC1), esophagogastric adenocarcinoma (EGAC), lentivirus, AKT/GSK/β-catenin signaling, proliferation, apoptosis, epithelial mesenchymal transition (EMT)
Introduction
Esophagogastric adenocarcinoma (EGAC) is a deadly disease with increasing incidence in the Western world (1). EGAC is common in Europe and North America accounting for 46% of global EGAC cases (2). Overall prognosis and survival for EGAC patients remain poor (3). Despite advancements in multimodal therapies, EGAC is difficult to treat due to advanced disease at the time of diagnosis necessitating research into molecular mechanisms which control tumor behavior to improve its detection and treatment options (4). Glypican-1 (GPC1) is a membrane bound heparan sulfate proteoglycans (HSPG) (5). Physiologic expression of GPC1 regulates cell-cell and cell-matrix signaling as well as growth factor response. Due to their HS chains, glypicans can sequester growth factors, chemokines, cytokines, and other signaling molecules, and can act as co-receptors with other cell membrane receptors, activating pathways such as fibroblast growth factor (FGF), vascular endothelial growth factor (VEGF), and Wnt (6-9). However, abnormal expression of GPC1 has been shown to be associated with tumorigenesis, chemoresistance, angiogenesis, and metastasis (10,11). GPC1 overexpression has also been linked to poor prognosis in pancreatic ductal adenocarcinoma, hepatocellular carcinoma, and glioblastoma, and has shown to affect the tumor microenvironment in prostate cancer by increasing tumor migration markers and growth (12-14). In esophageal squamous cell carcinoma (ESCC) patients, GPC1 overexpression was seen to correlate with poor overall survival (15). However, there are no studies in the literature regarding the role of GPC1 in esophagogastric cancer. The aim of the present study was to elucidate the biologic role of GPC1 in EGAC human tissues and cell lines. The findings of this study may provide insight and form basis for expanding new therapeutic options in EGAC patients. We present the following article in accordance with the MDAR reporting checklist (available at https://jgo.amegroups.com/article/view/10.21037/jgo-22-240/rc).
Methods
Human EGAC tissue and microarray
Thirty EGAC and adjacent normal tissue specimens were collected from patients at University of Colorado Anschutz Medical Center. In addition, EGAC tissue microarray (Figure S1) comprising of 120 EGAC and 25 adjacent normal tissue were purchased from US Biomax Inc. (Rockville, MD, USA). The experimental work with human tissue samples was conducted in accordance with the Declaration of Helsinki (as revised in 2013). The ethics committee at the University of Colorado (IRB No. 19-1319) approved the study. Each patient consented for the study.
Chemical reagents
MK-2206 was purchased from Merck & Co., Inc., (Whitehouse Station, NJ, USA) and lithium chloride (LiCl) was purchased from Millipore Sigma (St Louis, MO, USA). Dimethyl sulfoxide was used to reconstitute MK-2206 and LiCl.
Cell lines and culture
HEK-293T cells and human esophageal epithelial cells HET-1A were purchased from the American Type Culture Collection (Manassas, VA, USA). The EGAC cell lines OE33, FLO1 and OE19 were purchased from Millipore Sigma (St Louis, MO, USA). HET-1A cells were grown in complete Bronchial Epithelial Growth Media (Lonza, Basel, Switzerland). OE33 and OE19 cells were grown in Roswell Park Memorial Institute Medium (RPMI 1640; Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS), 0.5% penicillin/streptomycin, and gentamicin/amphotericin (1:500 dilution). FLO1 and HEK 293Ta cells were grown in Dulbecco’s Modified Eagle Medium (DMEM, Gibco, Grand Island, NY, USA) supplemented with 10% FBS, 0.5% penicillin/streptomycin, and gentamicin/amphotericin (1:500 dilution). All cells were incubated in a humidified atmosphere with 95% air and 5% carbon dioxide at 37 ℃. Serum-reduced media consisted of growth medium with 2–5% FBS, 0.5% penicillin/streptomycin, and gentamicin/amphotericin (1:500 dilution).
Construction of GPC1 knockdown and overexpression clones
Primers and restriction enzymes were purchased from Integrated DNA Technology and NEB Bio labs. For GPC1 knockdown, three unique LVshRNA clones (LVshGPC1#α, LVshGPC1#β, and LVshGPC-1#γ) were designed based on the human GPC1 gene (GenBank accession number: NM https://www.ncbi.nlm.nih.gov/gene/2817). A scrambled shRNA was designed and synthesized for negative control (NC). The primer sequences for the shRNA clones (Table S1). The primer oligonucleotides were annealed using the following parameters: 80 ℃ for 2 min, 65 ℃ for 10 min, 37 ℃ for 10 min and 25 ℃ for 5 min. The PCR product was gel extracted, purified and cloned into the psi-LVRH1GH vector (Genecopeia, MA, USA) using easy cloning kit (New England Bio labs, MA, USA). GPC1 overexpression plasmid, which will be referred to as LV-GPC1 hereon, was constructed by cloning the coding sequence of GPC1 gene into pEZ-Lv201(Genecopeia, MA, USA) at Bam HI/Eco RI site on the vector using easy cloning kit (New England Bio labs, MA, USA). The vectors carried an extended green fluorescent protein (e-GFP) reporter gene driven by SV-40 (Figure S2). The ligated products were transformed into competent DH5α cells (Zymo Research, Irvine, CA, USA) using a heat shock method. The transformed cells were grown on an LB-agar plates containing ampicillin at 37 ℃ and 5% CO2 overnight. The overnight culture was used for plasmid extraction using commercially available ZymoPURE Plasmid Miniprep kit (Zymo Research, Irvine, CA) per manufacturer’s instructions. GPC1 knockdown and GPC1 overexpressed clones were verified by restriction digest analysis on a 1.3% agarose gel (Figure S2).
Lentivirus production
Human embryonal kidney (HEK293T) cells were grown in DMEM supplemented with 10% FBS. Cells were transfected with a mixture of lentiplasmid and packaging plasmids using Endofectin Lenti transfection agent according to the manufacturer’s instructions (Genecopeia, MA, USA). Culture media containing shed lentivirus was harvested at 48 hours and 72 hours, centrifuged at 5,000 ×g and filtered through 0.2 µm PES filter. Titration of lentivirus yield was performed using real time qPCR assay.
Quantitative real-time PCR (qRT-PCR)
qRT-PCR was performed in 96-well plates using the iQ SYBR Green Supermix (Bio-Rad, Hercules, CA, USA) according to manufacturer’s instructions. qRT-PCR was carried out using a Roche Light Cycler 96. Total RNA was extracted from cells using the TRIzol reagent (Thermo Fisher Scientific, Waltham, MA). Next, 1 µg of RNA was reverse-transcribed into cDNA using the All-in-One First-Strand cDNA Synthesis Supermix kit (Applied Biological Materials, Richmond, BC, Canada) per manufacturer’s instructions. Relative expression of target genes was calculated with the 2−ΔΔCt method based on Ct values, using β-actin as the internal control. Primer sequences are listed in Table S2.
Preparation of total cell extract
Cells were plated on 24-well plates at a density of 0.05×106 cells per well and cultured in full strength media until 80% confluent. After 72 h of transfection, cells were washed with ice cold phosphate-buffered saline (PBS) and then lysed with ice cold RIPA buffer (Sigma Aldrich, St. Louis, MO, USA). Protein concentration was determined using BCA assay (DC assay, Bio-Rad, Hercules, CA) using bovine serum albumin to generate a standard curve.
Isolation of nuclear and cytoplasmic extract
Nuclear and cytoplasmic fractions were prepared using an NE-PER Nuclear Cytoplasmic Extraction Reagent kit (Pierce, Rockford, IL, USA) according to the manufacturer’s instruction.
Western blotting
Total cell lysates and nuclear or cytoplasmic proteins were isolated as described above. Equal amounts of the protein concentrations (20 µg) were separated using sodium dodecyl sulfate-polyacrylamide gel (Bio-Rad 4–20%) electrophoresis and transferred to nitrocellulose membranes. Membranes were blocked in TBS-Tween 20 with 5% nonfat milk for 1 hour. Antibodies and dilutions are listed in Table S3. Primary antibodies were diluted in 5% BSA and incubated at 4 ℃ overnight. Secondary antibodies were diluted in TBS-Tween 20 with 5% nonfat milk. Protein quantification with densitometry analysis was performed using Image Lab Software (Bio-Rad Laboratories, Inc., 2017).
Flow cytometry cell cycle analysis and cell growth
For cell cycle analysis, cells were seeded in 12-well flat-bottom plates at a density of 200,000 cells per well for 24 hours in antibiotic free media. After 48 hours of transfection, cells harvested for cell cycle status using Propidium Iodide Flow Cytometry Kit (ab139418; Abcam, Cambridge, UK) per manufacturer’s instructions. Samples were analyzed by flow cytometry using a BD FACS Calibur flow cytometer (BD Biosciences, San Diego, CA, USA). Cell proliferation was determined by CCK-8 assay according to the manufacturer’s instructions. The 96 well plates for cell proliferation were read on BioTek microplate reader at 450 nm at 48 hours and 72 hours after transfection.
Cell apoptosis
Cells were seeded into 24 well plates at 200,000 cells per well and allowed to adhere for 24 hours. Cells were then transfected with lentivirus particles. At 48 hours, cells were prepared for flow cytometry using Tonbo Bioscience PE Annexin V Apoptosis Kit (Tonbo Bioscience, San Diego, CA, USA) per manufacturer’s instructions. Samples were analyzed using a BD FACS Canto II. Data was analyzed using Flowjo v.10.8 software.
Wound healing scratch assay
Transfected FLO1 and OE19 cells were seeded into 12-well plates. After reaching 90% confluence the monolayer was scratched with a 200 µL sterile pipette tip. Cells were cultured in serum free media for an additional 48 hours. The scratch size was monitored with Nikon Ti2 microscope. The remaining wound area was measured using ImageJ software (nih.gov, Bethesda, MD, USA) and normalized to time 0 h wounds.
Transwell migration and invasion assay
Equal number of cells were seeded in 8 µm polycarbonate inserts (Corning Costar) combined with 24-well culture plates for migration (uncoated) and invasion (Matrigel-coated) assays. After 24 h cells were fixed with 4% paraformaldehyde and stained for 30 min in a 0.1% crystal violet solution in PBS. The number of cells on the underside of each insert was determined using light microscopy (Nikon Ti2 microscope). Five randomly selected fields were counted per insert.
Spheroid formation assay
For spheroid formation assay, 50,000 cells were seeded in U bottom plates and transfected with either LVshRNA#α or overexpressing GPC1 lentivirus (LV-GPC1). A thin layer of Matrigel matrix (diluted with serum free media in a 1:3 ratio) was added onto the spheroids to assess their invasiveness. Bright field and florescence photographs were taken on day 0, 3 and 7. The diameter of spheroid was measured with NIS image software on Nikon Ti2 microscope.
Immunohistochemistry
Five µm tissue sections were prepared from paraffin embedded blocks. Tissue sections were deparaffinized with xylene and rehydrated with graded ethanol. Anti-GPC-1 antibody staining was performed using Rapid IHC kit (Bio Vision) according to the manufacturer’s protocol. Immunostaining was scored as: 0, no staining; 1, normal staining; 2, strong staining. The ‘density’ of staining (termed the positivity score) was as follows: 1, indicates less than 50% positivity; 2, indicates more than 50% positivity. The final IHC score was determined by multiplying the intensity score by the positivity score, with a maximum positive score of 4. These data were referred to as the GPC-1 H score to categorize them into low and high expression groups.
TUNEL analysis
Apoptotic DNA fragmentation was detected using double fluorescence with CF-594 labeling terminal UTP-nick end labeling kit (Biotium, Fremont, California, USA) and 4',6-diamidino-2-phenylindole (DAPI; Thermofisher, Waltham, MA, USA). Cells were imaged under a fluorescent microscope (Nikon, TE2000).
Public data sets
Survival analysis was downloaded from Kaplan Meier plotter database (https://kmplot.com/analysis/). Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) data was downloaded from DAVID (https://david.ncifcrf.gov/gene2gene.jsp).
Statistical analysis
Experiments were performed three times. Clinicopathological variables were compared using chi square test. Data represents mean ± standard deviation (SD) and comparisons made using Student’s t-test. Data with multiple comparisons were analyzed by ANOVA followed by Fisher’s least significant difference post hoc test. Chi-square tests or Fisher’s exact tests were used to assess correlation of GPC1 with clinical pathological characteristics and Student’s t-test was used for comparisons between two groups. All statistical analysis was performed using Graph Pad prism software. A P value <0.05 was considered significant.
Results
GPC1 is upregulated in human EGAC
We first investigated expression of GPC1 in EGAC tissues using immunohistochemistry in paraffin embedded samples and tissue microarray. Clinical and pathology data of patients are summarized in Table 1. GPC1 was highly expressed in 102/150 (68%) tissue samples (Figure 1A, Figure S1), with IHC score of 3.2±2.2 compared to 0.78±0.84 in adjacent benign tissue (Figure 1B). Furthermore, expression of GPC1 was higher in poorly differentiated compared to well differentiated tumors (Figure 1C) and in those with positive lymph node metastasis (Figure 1D). Univariate and multivariate analysis showed positive correlation of GPC1 expression with T stage and N stage and overall survival (Table 2). Five-year survival data was available for 102 patients. Kaplan Meier survival curve showed that patients with high GPC1 expression had a shorter overall survival (P=0.0024), however there was no statistical association of GPC1 with relapse free survival (Figure 1E). We also queried the Kaplan-Meier Plot database and affy. ID GPC1(202756_s_at) which has a total of 875 patients with EGAC adenocarcinoma. The number of patients with high and low expression groups was 608 and 267 respectively. Kaplan Meier analysis showed that patients with high GPC1 expression had lower OS compared to low GPC1 expression patients (Figure 1F).
Table 1. Clinical and pathological characteristics.
| Characteristics | Cases (n=150) | GPC1 expression | P value | |
|---|---|---|---|---|
| Low | High | |||
| Age, years | 0.922 | |||
| <60 | 76 | 22 | 54 | |
| >60 | 74 | 26 | 48 | |
| Gender | 0.129 | |||
| Male | 110 | 35 | 75 | |
| Female | 40 | 13 | 27 | |
| T stage | <0.050 | |||
| T1 | 10 | 7 | 3 | |
| T2 | 52 | 16 | 36 | |
| T3 | 82 | 23 | 59 | |
| T4 | 6 | 2 | 4 | |
| Lymph nodes | <0.005 | |||
| Yes | 110 | 22 | 88 | |
| No | 40 | 25 | 15 | |
| Differentiation | <0.050 | |||
| Well | 44 | 21 | 23 | |
| Moderate | 77 | 22 | 55 | |
| Poor | 29 | 5 | 24 | |
GPC1, glypican 1.
Figure 1.

GPC1 is overexpressed in human EGAC. (A) Representative immunohistochemical staining of GPC1 in normal paratumor tissue (I) and human EGAC tissues (II, III, IV) showing variable intensity of GPC1 staining. Tissue sections were stained with anti GPC1 antibody followed by counterstaining with hematoxylin and eosin and visualized under light microscope with 40× magnification. (B) GPC1 IHC score in normal paratumor tissue and tumor tissue. Data represent mean histological score (IHC score) ± SD, ***, P<0.001. (C) GPC1 expression according to the degree of differentiation of tumor (HE and immunohistochemical staining, 20× magnification). (D) GPC1 expression in tumor tissue with lymph node metastasis (immunohistochemical staining, 20× magnification). (E) KM plot based on level of GPC1 expression in tumor tissue. (F) KM survival plot in KM plotter database estimating survival in 875 patients with EGAC. Red line: patients with high GPC1 (n=704); black line: low expression GPC1 patients (n=171). High GPC1 patient group was associated with decreased overall survival (P=2.2e−12, 706 log-rank test). GPC1, glypican 1; EGAC, esophagogastric cancer; IHC, immunohistochemistry; SD, standard deviations; HE, hematoxylin and eosin; KM, Kaplan-Meier.
Table 2. Univariate and multivariate analysis of prognostic variables in EGAC.
| Variable | Univariate analysis | Multivariate analysis | |||||
|---|---|---|---|---|---|---|---|
| P value | HR | 95% CI | P value | HR | 95% CI | ||
| GPC1 expression (low/high) | 0.005* | 1.491 | 1.126–2.046 | 0.024* | 1.392 | 1.041–2.011 | |
| TNM stage (I + II/III + IV) | <0.001 | 2.394 | 1.811–3.345 | 0.185 | 1.482 | 0.835–2.256 | |
| T stage (T1 + T2/T3 + T4) | <0.005* | 2.129 | 1.243–3.554 | 0.04* | 1.68 | 1.002–2.650 | |
| N stage (N0/N1 + N2 + N3) | <0.001* | 2.509 | 1.873–3.270 | 0.001* | 1.909 | 1.290–2.799 | |
| Age (<60/>60) | 0.646 | 1.045 | 0.786–1.478 | – | – | – | |
| Sex (male/female) | 0.716 | 1.066 | 0.810–1.430 | – | – | – | |
*, P<0.05. EGAC, esophagogastric cancer; HR, hazard ratio; CI, confidence interval; GPC1, glypican 1. TNM, tumour, node and metastasis.
High expression of GPC1 in EGAC cell lines
We analyzed GPC1 expression in normal human epithelial cells HET-1A and human EGAC cell lines OE19, FLO1 and OE33. Consistent with the human EGAC tissue results we found that GPC1 expression was significantly high in all three EGAC cell lines compared to HET-1A cells (Figure 2A,2B, Figure S3). Among the cancer cell lines, FLO1 showed the highest GPC1 expression and OE19 the lowest GPC1 expression and were selected for GPC1 knockdown and overexpression experiments respectively. Confocal microscopy showed cytoplasmic and nuclear envelope localization of GPC1 protein with co-localization with cytoplasmic vesicles (Figure 2C). The findings of increased GPC1 expression in EGAC tissues as well as cell lines provided us rationale to further explore the role of GPC1 in EGAC.
Figure 2.

GPC1 is highly expressed in esophagogastric cell lines. (A) Real time qPCR mRNA expression of GPC1 in HET-1A and EGAC cell lines (OE19, FLO1 and OE33). (B) Western blot and densitometry analysis of GPC1 protein in HET-1A and EGAC cell lines (OE19, FLO1 and OE33). Data represent mean ± SD. n=3. *, P<0.05, **, P<0.005, ***, P<0.001 vs. HET-1A. (C) Confocal images showing GPC-1 protein (green) predominantly in cytoplasm. Co-localization of GPC1 (orange-yellow staining) with cytoplasmic organelles. No expression was detected in nucleus or nuclear membrane. Green marks GPC-1, red marks β tubulin and blue marks DAPI-stained DNA. Scale bar: 100 µm. GPC1, glypican 1; ns, not significant; DAPI, 4',6-diamidino-2-phenylindole; FITC, fluorescein isothiocyanate; qPCR, quantitative polymerase chain reaction; EGAC, esophagogastric cancer; HET-1A, normal human epithelial cell line; SD, standard deviations.
Validation of efficient knockdown and overexpression of GPC1 with Lentivirus
We next evaluated the functional role of GPC1 in EGAC cells by knocking down its expression in FLO1 cells as well as overexpressing GPC1 in OE19 cells using lentivirus transfection as described in material and methods section. Transfection efficiency of LVshGPC1#α and negative control (NC) in FLO1 cells and LV-GPC1 and empty vector (EV) in OE19 cells was observed under florescence microscope by visualizing green GFP expression (Figure 3A,3B) 72 h after transfection. As shown in Figure 3C, transfection efficiencies >80% was achieved. Functional knockdown and overexpression of GPC1 was confirmed by qRT-PCR and Western blotting. LVshGPC1#α resulted in 95% knockdown of GPC1mRNA and 76% reduction of GPC1 protein relative to NC. Similarly, LV-GPC1 produced a 110% and 85% overexpression of GPC1 at mRNA and protein level respectively (Figure 3D).
Figure 3.

Validation of efficient knockdown and overexpression of GPC1 with lentivirus. (A) Fluorescent microscopy images of GPC1 knockdown FLO1 cells and negative control. (B) Fluorescent microscopy images of GPC1 overexpressed OE19 cells and empty vector. (C) Bar chart showing transfection efficiencies for lentiviral plasmids in FLO1 and OE19 cells. Western blot and densitometry analysis of GPC1 after knockdown of FLO1 cells with LVshGPC1#α, LVshGPC1#β, and LVshGPC-1#γ in FLO1 cells and GPC1 overexpression with LV-GPC1 in OE19 cells. β-actin loading control. Fold change of GPC1 expression was normalized to the expression of control cells. Scale bar: 50 µm. Data represent mean ± SD. n=3. **, P<0.005, ***, P<0.001, ****, P<0.0001 compared with control. LVshGPC1#α, LVshGPC1#β, and LVshGPC-1#γ, GPC1 knockdown plasmids; NC, negative scrambled control; DAPI, 4',6-diamidino-2-phenylindole-stained DNA; e-GFP, enhanced green fluorescent protein; EV, empty vector control; LV-GPC1, overexpressing GPC1 lentivirus; GPC1, glypican 1; SD, standard deviations.
GPC1 regulates cell morphology
There is mounting evidence that remodeling of actin filaments promote determine cell motility, migration and proliferation (16,17). Disruption of actin cytoskeleton is associated with decreased migration, increased apoptosis, and features of interest for anticancer therapy. Therefore, we examined the effects of GPC1 on cytoskeleton and morphology. FLO1 cells are spindle shaped and fusiform in appearance. They have well defined F-actin filament network arranged parallel to the long axis of cell. GPC1 knockdown in FLO1 cells altered their morphology and they appeared larger and polygonal often with binuclear or multinuclear appearance (Figure 4A). GPC1 knockdown resulted in attenuation of F-actin staining and disruption of actin fibers into depolymerized aggregates (Figure 4B). Over expression of GPC1 in OE19 cells resulted thick actin stress fibers in a whorled pattern mimicking lamellipodia (Figure 4C). These results indicate that GPC1 has a critical role in maintaining cytoskeletal integrity and may confer enhanced migratory properties to cancer cells.
Figure 4.

GPC1 regulates cell morphology. (A) Cell morphology of parental FLO1, GPC1 knockdown FLO1 cells was examined with immunofluorescence staining. Compared to spindle shaped fusiform structure of parental FLO1 cells the knockdown cells became more rounded, polygonal with enlarged binuclear and multinuclear morphology (white circle). (B) Reduced F-actin staining and its depolymerization into aggregates (white arrows) in GPC1 knockdown cells compared to negative control. (C) LV-GPC1 cells showed thick coarse actin bundles of stress fibers (white arrow) compared to empty vector control. Red indicates F-actin, green indicates e-GFP, blue indicates DAPI-stained DNA. Scale bar: 100 µm. LVshGPC1#α, LVshGPC1#β, and LVshGPC-1#γ, GPC1 knockdown plasmids; e-GFP, enhanced green fluorescent protein; DAPI, 4',6-diamidino-2-phenylindole-stained DNA; NC, negative scrambled control; EV, empty vector control; LV-GPC1, overexpressing GPC1 lentivirus; GPC1, glypican 1.
GPC1 promotes EGAC cell proliferation in vitro
We further investigated the effect of GPC1 on proliferation using standard colorimetric cell counting technique (CCK8) and colony forming assays. Cell viability was measured at 24, 48 and 72 h at OD 450 nm. As shown in Figure 5A, GPC1 overexpressing OE19 cells demonstrated significantly higher viability compared to empty vector. Conversely, GPC1 knockdown suppressed proliferation of FLO1 cells. Anchorage dependent and soft agar colony forming capacity of GPC1 knockdown FLO1 and GPC1 overexpressing OE19 cells were also tested. As represented in Figure 5B, GPC1 promoted colony formation of OE19 cells while GPC1 knockdown FLO1 cells formed significantly less colonies compared to controls. Anchorage independent colonies of GPC1 overexpressing OE19 cells were larger as well as more in number compared to empty vector controls (Figure 5C). To assess the effect of GPC1 in relation to mimicking a native tumor environment instead of a monolayer assay we studied the spheroid formation capacities of GPC1 knockdown and overexpressing cells grown in matrigel. GPC1 overexpressed OE19 cells formed larger diameter spheroids compared to GPC1 knockdown FlO1 cells (Figure 5D). Taken together, the in vitro assay results suggest that GPC1 stimulates cell proliferation and colony formation.
Figure 5.

GPC1 promotes EGAC cell proliferation in vitro (A) CCK-8 assay determination of proliferation of cells after GPC1 knockdown (left) and GPC1 overexpression in FLO1 and OE19 cells respectively. (B) Representative images of anchorage dependent colony formation. FLO1 and OE19 cells were seeded in 6 well plates and treated with either negative control, GPC1 knockdown, empty vector or GPC1 overexpression plasmid and grown for 14 days. Colonies were stained with 0.1% crystal violet and visualized under microscope at 40× magnification. Bar chart represents mean ± SD values for number of colonies in indicated groups (n=3, ***, P<0.001). (C) Representative image (left) and bar chart (right) of soft agar colony formation after transfection of cells with LVshGPC1#α and LV-GPC1 grown in soft agar for 14 days, stained with 0.5% crystal violet and visualized under microscope at 40× magnification (**, P<0.01; ***, P<0.001). (D) Representative images and bar chart of sphere formation capacity of GPC1 knockdown and overexpressed cells in matrigel at 100× magnification. Bar chart represents mean ± SD values for number of colonies in indicated groups. n=3, ***, P<0.001. OD, optical density; NC, negative scrambled control; LVshGPC1#α, GPC1 knockdown plasmid; EV, empty vector control; LV-GPC1, overexpressing GPC1 lentivirus; GPC1, glypican 1; EGAC, esophagogastric cancer; SD, standard deviations.
GPC1 regulates cell cycle
Having shown that GPC1 promotes cell proliferation and survival, we next investigated effect of GPC1 on cell cycle phase distribution and proteins by flow cytometry. Flow cytometric analysis of GPC1 knockdown FLO1 cells showed higher percentage of cell counts in G0/G1 phase and a reduced percentage of cell counts in S phase compared to negative control. Conversely, GPC1 overexpressed OE19 cells showed significantly higher percentage of cell counts in S phase and lower percentage of cell counts in G0/G1 phase compared to empty vector (Figure 6A). The data suggests that GPC1 increased cell proliferation by enhancing transition of cells from G0/G1 to S phase committing them to cell division and proliferation. Further evidence of GPC1 role in cell cycle phase regulation was sought by analyzing expression of cell cycle specific proteins which control transition of cells from G1 to S phase. Western blot analysis showed that GPC1 overexpression increased cyclin D1, cyclin A, cyclin E1, CDK4 and CDK6 while GPC1 knockdown decreased cyclin D1, cyclin E1, CDK4 and CDK6 expression (Figure 6B). These results demonstrate that GPC1 affects key cell cycle regulators which promote entry of cells from G1 to S phase.
Figure 6.

GPC1 regulates cell cycle progression. Propidium iodide staining, and flow cytometry was performed after GPC1 knockdown and overexpression to assess distribution of cell cycle phases. (A) Cell cycle was arrested in G0/G1 phase after knockdown of GPC1 in FLO1 cells. GPC1 overexpression with LV-GPC1 in OE19 cells showed an increase in S phase. (B) Western blot analysis after knockdown and overexpression of GPC1 showing protein levels of key cyclins and cyclin dependent kinases. Beta actin was used as loading control. Representative images and quantification of cell phases are shown. Data are means from three experiments ± SD; ordinary one ANOVA with multiple comparisons, ****, P<0.0001. NC, negative scrambled control; LVshGPC1#α, GPC1 knockdown plasmid; PI-A, Propidium Iodide-Annexin; EV, empty vector control; LV-GPC1, overexpressing GPC1 lentivirus; GPC1, glypican 1; SD, standard deviations.
GPC1 knockdown induced apoptosis
Annexin/PI flow cytometry was performed to assess effect of GPC1 on apoptosis. The percentage of apoptotic cells in GPC1 knockdown FLO1 cells were higher (14.22%±0.26%) compared to control (2.22%±0.64%). GPC1 overexpression in OE19 did not have any significant effect on apoptosis (Figure 7A). TUNEL staining showed an increased number of apoptotic cells in GPC1 knockdown FLO1 cells compared to controls (Figure 7B). Western blot analysis of GPC1 knockdown FLO1 cells showed increased levels of active cleaved caspase and Bax whereas expression of Bcl-1 was reduced (Figure 7C).
Figure 7.

GPC1 knockdown induced apoptosis. (A) After knockdown of GPC1 in FLO1 cells and overexpressing GPC1 in OE19 cells, they were double-stained with annexin FITC/PI and analyzed by flow cytometry. The percentage of apoptotic cells increased in GPC1 knockdown cells compared to controls. There was no difference in apoptosis after overexpression of GPC1 compared to control empty vector. Representative images, and a bar chart for quantification of the percentage of apoptosis (B) GPC1 knockdown FLO1 cells were visualized under a fluorescence microscope for TUNEL staining using a TUNEL/TRITC staining kit. Blue denotes DAPI, red denotes TRITC and pink denotes merged condensed chromatin within the nucleus. Scale bar: 50 µm. (C) Western blot assay of GPC1 knockdown FLO1 cell lysate showing protein levels of procaspase, cleaved caspase 3, Bax, and Bcl2. Beta actin was used as a loading control. Data represents n=3, mean ± SD; ordinary one ANOVA with multiple comparisons, **, P<0.01, ***, P<0.001. NC, negative scrambled control; LVshGPC1#α, GPC1 knockdown plasmid; EV, empty vector control; LV-GPC1, overexpressing GPC1 lentivirus; ns, not significant; DAPI, 4',6-diamidino-2-phenylindole-stained DNA; TRITC, tetramethylrhodamine; GPC1, glypican 1; FITC/PI, fluorescein isothiocyanate/Propidium Iodide; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling; SD, standard deviations.
GPC1 promotes cell migration, invasion, and epithelial mesenchymal transformation
To determine whether GPC1 promoted cell migration and invasion, wound healing and invasion assays were performed. We observed that GPC1 overexpressed OE19 cells showed increased closure of the scratch compared to GPC1 knockdown FLO1 cells (Figure 8A). In Boyden’s chamber assay, GPC1 overexpressed OE19 cells showed increased migration and invasion (with matrigel) of cells when compared to controls. In contrast number of migrating and invading GPC1 knockdown FLO1 cells were significantly reduced (Figure 8B,8C). There are a number of studies which have shown synergy between epithelial mesenchymal transition (EMT) signaling and metastasis (18,19). After GPC1 knockdown in FLO1 cells there was an increased expression of epithelial markers (E- cadherin and ZO-1) and reduced expression of mesenchymal markers (vimentin, SLUG, and ZEB1) (Figure 8D,8E). GPC1 overexpressing OE19 cells showed a reciprocal effect on EMT markers. Taken together, our data indicates that GPC1 promotes migration and invasion in EGAC cells via activating EMT signaling.
Figure 8.

GPC1 promotes cell migration, invasion, and epithelial mesenchymal transformation. (A) Light microscopy images (magnification 20×) of scratch assay taken at 0 and 48 h after transfection. The bar chart showing mean ± SEM of scratch width (relative to that at 0 h; n=3, ANOVA, *, P<0.05, **, P<0.01). (B) Light microscopy images (magnification 20×) of Transwell assay in either uncoated (for migration) or Matrigel coated (for invasion) polycarbonate 8 µm chambers. Migrated cells at the bottom of inserts were stained with 0.1% crystal violet. (C) Bar chart represents data from n=3, mean ± SD of number of migrated and invaded cells in 5 random fields (Student’s t-test, **, P<0.01, ***, P<0.001). (D) Western blot analysis of EMT proteins E-cadherin, N-cadherin, ZEB1, vimentin, in GPC1 knockdown FLO1 cells and GPC1 overexpressed OE19 cells. (E) Densitometry analysis of EMT proteins in GPC1knockdown FLO1 cells and GPC1 overexpressed OE19 cells versus respective controls. Data represent mean ± SD, n=3. **, P<0.01, ***, P<0.001. GPC1, glypican 1; NC, negative scrambled control; LVshGPC1#α, GPC1 knockdown lentivirus; EV, empty vector control; LV-GPC1, overexpressing GPC1 lentivirus; SEM, standard error of mean; ANOVA, analysis of variance; SD, standard deviations.
KEEG and GO analysis of GPC1
To explore the mechanistic of GPC-1 biological actions, functional and pathway enrichment analysis was performed using DAVID. Biologic processes were enriched in proteoglycan catabolic and fibroblast signaling regulation pathways (Figure 9A). Molecular functions were enriched in “protein and DNA binding” (Figure 9B). Cellular components analysis showed GPC1 was enriched in Golgi apparatus. Interestingly pathway analysis showed association of HSPGs with “Wnt”, “AKT” and “ERK” (Figure 9C).
Figure 9.

Gene ontology and KEEG analysis of GPC1: (A) biologic processes; (B) molecular function; (C) KEEG pathway analysis of HSPG proteins. FDR, false discovery rate; HSPGS, heparan sulfate proteoglycans; KEEG, Kyoto Encyclopedia of Genes and Genomes; GPC1, glypican 1.
GPC1 increases β-catenin expression and activates Wnt signaling via AKT/GSK-3β pathway
The β-catenin pathway has been shown to be activated in various cancers (20,21). Based on KEEG and GO data which we obtained we explored whether GPC1 activates β-catenin pathway in EGAC. Results in Figure 10A show that knockdown of GPC1 decreased the expression of active phospho-AKT, phospho-β-catenin and phospho-GSK-3β. In contrast, over expression of GPC1 correspondingly increased active phosphorylated AKT, β-catenin and GSK-3β. Total AKT and total GSK-3β remained unchanged in both groups. In the AKT/GSK-3β pathway, AKT is upstream of GSK-3β and β-catenin (22). AKT can directly phosphorylate GSK-3β to inhibit its activity, which in its inactive state increases phosphorylation of β-catenin. Phospho-β-catenin becomes stabilized for nuclear translocation and activates nuclear transcription factors notably TCF/LEF/cyclin D1 stimulating cell growth and proliferation (23-25). To further characterize the distribution of β-catenin, cytoplasmic and nuclear separation of protein lysates from GPC1 overexpressed OE19 and GPC1 knockdown FLO1 cells was performed. Western blot assay showed that GPC1 overexpressed OE19 cells showed increased β-catenin in the nucleus compared to controls, whereas GPC1 knockdown resulted in reduced β-catenin in the nucleus when compared to negative scrambled control (Figure 10B). There was also increased expression of LEF and TCF1/7 in GPC1 overexpression cells (Figure 10B) and reverse effects with GPC1 knockdown. Additionally, immunofluorescence staining revealed that GPC1 knockdown FLO1 cells showed less nuclear staining and enhanced membrane staining of β-catenin staining compared to negative controls, while GPC1 overexpression resulted in increased nuclear staining and reduced membrane staining of β-catenin compared to empty vector control (Figure 10C). Bioinformatics data also showed significant positive correlation between GPC1 and β-catenin, c-MYC, cyclin D1 and LEF/TCF factors indicating cooperation of β-catenin pathway in GPC1 signaling (Figure 10D). Collectively these results indicate that GPC1 is upstream of and activates AKT/GSK-3β/β-catenin pathway.
Figure 10.

GPC1 activates AKT/GSK-3β/β‐catenin pathway. (A) Western blot analysis of key proteins of AKT/GSK-3β/β‐catenin pathway. (B) Western blot assay examining the nuclear and cytoplasmic expression of catenin (left) and nuclear expression of β‐catenin downstream target transcription factors (LEF and TC-F1) in GPC1 knockdown FLO1 and GPC1 overexpressed OE19 cells. (C) Dual immunofluorescence staining for β‐catenin (red) and nucleus with DAPI (blue) was performed in GPC1 knockdown FLO1 and GPC1 overexpressed OE19 cells. Compared to controls, knockdown of GPC1 showed increased membrane staining and reduced nuclear staining (pink) of β‐catenin, while GPC1 overexpression resulted in reduced membrane staining and increased nuclear accumulation (pink) in OE19 cells. Scale bar: 50 µm. (D) The expression of GPC1 correlated with β‐catenin, c-myc, TCF4, cyclin D1, and LEF1 levels which were analyzed in the GEPIA database. Lamin B and β-actin were used for loading controls nuclear and cytoplasmic fractions respectively. NC, negative scrambled control; LVshGPC1#α, GPC1 knockdown plasmid; EV, empty vector control; LV-GPC1, overexpressing GPC1 lentivirus; DAPI, 4',6-diamidino-2-phenylindole-stained DNA; TRITC, tetramethylrhodamine; TPM, transcripts per million; GPC1, glypican 1.
MK-2206 inhibited AKT signaling in GPC1 overexpressed cells
AKT is regarded as a key prosurvival and proliferation factor (26,27). GSK3-β and β-catenin are important downstream substrates for phosphorylaytion by AKT (28). We studied the effect of AKT inhibition with MK-2206 on GPC1 regulation of AKT/GSK-3β/β-catenin pathway. For this experiment, we transfected OE19 cells with LV-GPC1 in presence of MK-2206 (1 µM) for 24 hours and examined the effects on proteins of AKT/GSK-3β/β-catenin pathway. As shown in Figure 11, Western blot assay showed MK-2206 significantly attenuated the levels of active phospho-AKT, phospho-GSK3b, and phospho-β-catenin induced by overexpression of GPC1 in OE19 cells. These results indicate that MK2206 is effective in decreasing activation of AKT and its downstream pathway in EGAC cells and may be a beneficial combinatorial strategy with standard chemotherapy in esophagogastric cancer treatment.
Figure 11.
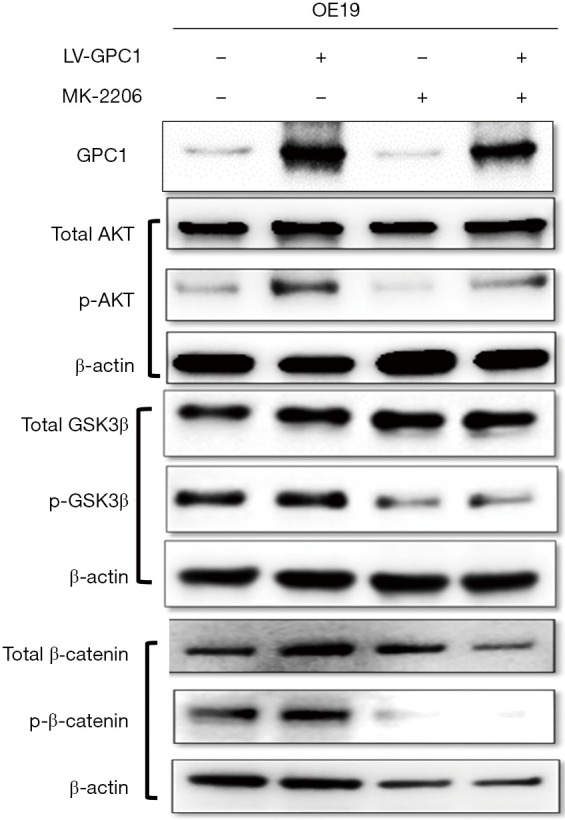
MK-2206 inhibited phosphorylation of AKT downstream targets in GPC1 overexpressed cells. OE19 cells were transfected with GPC1 overexpression plasmid or EV and treated with 1 µM of MK-2206 for 24 h. Western blot was performed and probed for active p-AKT, p-GSK3-β and p-β-catenin. MK2206 attenuated increased levels of p-AKT, p-GSK3-β, and p-β-catenin induced by LV-GPC1. LV-GPC1, overexpressing GPC1 lentivirus; GPC1, glypican 1.
GPC1 knockdown abolished LiCl driven β-catenin stimulation
GSK-3β is regarded as key enzyme mediating Wnt/β catenin pathway (29). Inhibition of GSK-3β results in stabilization of β-catenin and its nuclear translocation ultimately promoting β-catenin-driven transcriptional activity (30). Pharmacologic inhibition of GSK3-β activity has recently gained interest in cancer therapy (30). LiCl is a classic inhibitor of GSK-3β and activates Wnt pathway. We examined the effect of LiCl in OE19 cells and sought to see if GPC1 knockdown could downregulate LiCl induced β-catenin stimulation. For this experiment, we transfected OE19 cells with either overexpressing GPC1 (LV-GPC1) or GPC1 knockdown (LVshGPC1#α) lentivirus in the presence of LiCl (40 mM) followed by probing for proteins of β-catenin pathway. As shown in Figure 12, LiCl and GPC1 overexpression lentivirus activated Wnt/β catenin and increased levels of phospho-GSK-3β, phospho-β-catenin, TCF1/7 and c-myc indicating that both LiCl and GPC1 activate β-catenin pathway. Remarkably, GPC1 knockdown in LiCl treated cells abolished the surge of p-GSK β, β-catenin, TCF and c-myc induced by LiCl. Collectively these results indicate that GPC1 inhibition effectively abrogates activated AKT/Wnt/β-catenin signaling.
Figure 12.
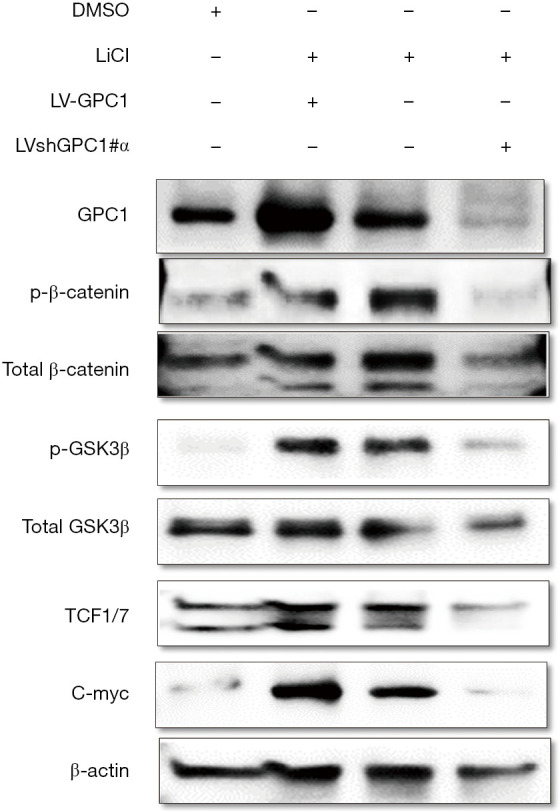
GPC1 knockdown abolished LiCl driven β-catenin stimulation. OE19 cells were transfected with either LV-GPC1 or LVshGPC1#α in the presence of LiCl (40 mM) for 48 h. Western blot analysis of active p-GSK3-β, active p-β-catenin, TCF1/7 and c-myc. DMSO, dimethylsulphoxide; LiCl, lithium chloride; LV-GPC1, overexpressing GPC1 lentivirus; LVshGPC1#α, GPC1 knockdown lentivirus; GPC1, glypican 1.
GPC1 promoted HET-1A proliferation, migration and invasion
We next asked if GPC1 could promote a normal epithelial cell to acquire a neoplastic phenotype? To answer this question, we over expressed GPC1 in OE19 cells and analyzed proliferation, migration, invasion capabilities as well as apoptosis and EMT proteins. Results showed that GPC1 significantly increased HET-1A proliferation at 48 and 72 h (Figure 13A). GPC1 overexpressed cells showed larger area of gap closure (Figure 13B), significantly greater number of cells migrated and invaded the membrane (Figure 13C). Western blot assay (Figure 13D) showed that GPC1 repressed apoptosis by downregulating PARP and caspase 3. Furthermore, GPC1 expressing HET-1A cells showed upregulation of Vimentin and downregulation of E-cadherin indicating GPC1 promotes EMT.
Figure 13.

Overexpression of GPC1 in normal HET-1A cells enhanced cell proliferation, suppressed apoptosis and enhanced migration and invasion. (A) HET-1A cells were transfected with LV-GPC1 and cell proliferation was measured by CCK-8 assay. (B) Light microscopy images (magnification 20×) of scratch assay from indicated groups at 0 and 48 h after transfection. The bar chart represents mean ± SD of scratch wound closure compared to 0 h width. n=3, ***, P<0.01. (C) Light microscopy images (magnification 20×) of Transwell assay in either uncoated (migration) or matrigel coated (invasion) polycarbonate 8 μm chambers. Migrated cells at the bottom of inserts were stained with 0.1% crystal violet. Bar chart showing the number of migrated cells. Data represents n=3, mean ± SD number of invaded cells in 5 objective fields, ***, P<0.001. (D) Western blot assay showing protein expression of pro-PARP, cleaved PARP, procaspase3, cleaved caspase3, E-cadherin and Vimentin in HET-1A and GPC1 overexpressed HET-1A cells. β-actin loading control. OD, optical density; LV-GPC1, overexpressing GPC1 lentivirus; EV, empty vector control; GPC1, glypican 1; SD, standard deviations; PARP, poly(ADP-ribose) polymerase 1.
Discussion
EGAC remains one of the deadliest cancers worldwide. Despite current progress in surgical resection combined with chemoradiotherapy and immunotherapy, it remains a leading cause of cancer related deaths (31). Often patients are diagnosed with advanced disease and are not eligible for curative therapy. Esophagogastric cancer often arises from Barrett’s esophagus, which is premalignant. Despite tremendous advances in understanding the genetic and epigenetic aberrations which drive cancer, efforts aimed at translating molecular candidates to clinical practice have proven to be challenging, underscoring the need to identify novel markers for early diagnosis and therapy. GPC1, a membrane anchored heparan sulphate molecule, has recently gained interest not only as a biomarker but also of therapeutic potential (32,33).
High levels of GPC1 have been reported in cancers of breast, esophagus, pancreas and brain (10-12). Studies have also attributed high GPC1 expression to development of chemoresistance and poor patient survival (34,35). However, role of GPC1 in esophagogastric cancer is relatively unexplored.
In this study we show that GPC1 was increased in human EGAC tissue and correlated with T/N/M classification, lymph node metastasis and poor overall survival of patients. Using bioinformatics analysis and cell line experiments we showed that expression of GPC1 was variable in human EGAC cell line but higher than normal epithelial cell line. GPC1 expression correlated with viability, migration, invasion and EMT, demonstrated by knockdown and overexpressing GPC1 in 2 EGAC cell lines. We also showed GPC1 regulates AKT/GSK-3β/β-catenin signaling. This study strengthens the possibility of GPC1 to be a potentially attractive biomarker for not only early diagnosis but also for patient prognosis. Nevertheless, future in vivo animal studies and clinical studies are required to validate the clinical utility of GPC1.
We demonstrate that GPC1 regulated cytoskeletal structure as knockdown of GPC1 in FLO1 cells disrupted F-actin cytoskeleton, reduced cell viability, proliferation, migration, and invasion. Meanwhile, GPC1 overexpression in OE19 showed opposite effects. Along the same lines, colony formation was inhibited by GPC1 knockdown compared to GPC1 overexpressing cells. To understand the possible mechanism of GPC1 role in cell growth, flow cytometry analysis of GPC1 knockdown and overexpressing cells was performed. GPC1 knockdown cells showed G0/G1 phase arrest in contrast to GPC1 overexpressing cells which showed a larger proportion of cells in S phase. These results are in line other studies which show that GPC1 inactivates G1/S checkpoint and promotes S phase entry and DNA replication (36). Cell cycle progression requires transit through check points which is regulated by cyclins and cyclin dependent kinases (CDK’s) (37). Cyclin D1, cyclin E and cyclin A are master regulators of G1 and S phase of cell cycle (38). Cyclin D1 determines the length of G1 phase while cyclin E and cyclin A regulate G1 to S transition. Western blot analysis showed decreased levels of cyclin D, cyclin E1, cyclin A, cdk4 and cdk6 after GPC1 knockdown, whereas levels of these cyclins and kinases were increased after GPC1 overexpression suggesting that GPC1 enhances transit of cells from G1 to S phase for cell division and proliferation. To further clarify the role of GPC1 in invasion and migration, EMT of cells was studied. EMT contributes to invasiveness of cancers and is characteristic of aggressive malignant cancers (39). We show that GPC1 overexpression in OE19 cells induced EMT by upregulating N-cadherin, ZEB1, vimentin and SLUG and reduced expression of E-Cadherin. Opposite effects on EMT proteins were seen in GPC1 knockdown FLO1 cells. Moreover, tissue samples from EGAC patients showed that higher expression of GPC1 correlated with positive lymph node metastasis. The mechanisms by which GPC1 promotes cancer progression are not entirely clear. There is data showing that GPC1 is regulated by microRNA’s such as miR-96-5 and miR-149 in colon and pancreas cancer (40,41). These regulatory miRNA’s promote GPC1 activation by various mechanisms including promotor hypomethylation (42), KRAS and ecotropic viral integration site 1 (EVI1) (43) or GPC1 gene amplification (44). Activated GPC1 then promotes interaction between FGF and PI3/AKT pathway, which is essential for cell survival by inhibiting apoptosis (45). GPC1 is also known to stimulate Wnt/β-catenin signaling (46) and there is also strong evidence of cooperation and synergy between PI3/AKT and Wnt/β-catenin driving certain gastrointestinal cancers (47,48). Our results are in accordance with these studies, as we noted an increased active p-AKT, p-GSK-β and p-β-catenin and increased accumulation of nuclear β-catenin in GPC1 overexpressed cells, whereas their levels were markedly reduced in GPC1 knockdown FLO1 cells. Additionally, GPC1 stimulated β-catenin downstream transcription factors LEF and TCF1/7 which promote cell proliferation. Since AKT is the key component of PI3/AKT signaling which drives processes for cancer propagation, targeting AKT seems to be a highly attractive targeted treatment strategy. Inhibition of AKT with small molecules like MK-2206 has recently gained attention in treatment of aggressive cancers (49,50). MK-2206 blocked the activation of AKT/GSK-3β/β-catenin in GPC1 overexpressed OE19 cells. We also showed that knockdown of GPC1 in LiCl stimulated cells effectively abolished the LiCl driven β-catenin surge. Future research using combination inhibitors of AKT/GSK-3β/β-catenin to disrupt GPC1 downstream signaling to treat this devastating cancer may be extremely beneficial. Lastly, we show that GPC1 increased cell viability, migration/invasion and repressed apoptosis in normal HET-1A cells. Furthermore, these normal cells acquired EMT properties which suggest GPC1 may play an oncogenic role in progression of a normal cell to acquire malignant phenotype. Future in vivo tumor implantation of GPC1 expressed normal HET-1A cells are needed to validate the in vitro effects of our study.
There are potential limitations of our study. First, this was an in vitro cell study, therefore results shown here do not necessarily replicate the effects of human tumor microenvironment. Further in vivo experiments are required to validate the role of GPC1 in EGAC. Secondly, we tested only 2 cell lines and it is not known how other EGAC cell lines would respond to GPC1, MK-2206 or LiCl. Thirdly, as no previous studies were available in the literature describing the effect of GPC1 in EGAC our experiments and data gathered from them provide first evidence for future research. Nevertheless, findings of our study theorize that GPC1 is a novel protein which has an important role in progression of EGAC, and it might be an attractive novel therapeutic target for its early diagnosis and treatment.
Supplementary
The article’s supplementary files as
Acknowledgments
Funding: This work was supported by Academic Enrichment Fund, Department of Surgery (No. 2350453) and ACS IRG #16-184-56 from the American Cancer Society to the University of Colorado Cancer Center.
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. The experimental work with human tissue samples was conducted in accordance with the Declaration of Helsinki (as revised in 2013). The ethics committee at the University of Colorado (IRB No. 19-1319) approved the study. Each patient consented for the study.
Footnotes
Reporting Checklist: The authors have completed the MDAR reporting checklist. Available at https://jgo.amegroups.com/article/view/10.21037/jgo-22-240/rc
Data Sharing Statement: Available at https://jgo.amegroups.com/article/view/10.21037/jgo-22-240/dss
Peer Review File: Available at https://jgo.amegroups.com/article/view/10.21037/jgo-22-240/prf
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at https://jgo.amegroups.com/article/view/10.21037/jgo-22-240/coif). AP reports that he receives robotic intuitive foundation fellowship grant, which is awarded for training minimal invasive surgery fellows. Paul O Hara and Academic enrichment fund were awarded for this project. The other authors have no conflicts of interest to declare.
References
- 1.Sung H, Ferlay J, Siegel RL, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin 2021;71:209-49. 10.3322/caac.21660 [DOI] [PubMed] [Google Scholar]
- 2.Blot W, Tarone R. Esophageal Cancer. In: Thun MJ, Linet MS, Cerhan JR, et al. (editors). Schottenfeld and Fraumeni Cancer Epidemiology and Prevention. New York, NY: Oxford University Press, 2018. [Google Scholar]
- 3.Njei B, McCarty TR, Birk JW. Trends in esophageal cancer survival in United States adults from 1973 to 2009: A SEER database analysis. J Gastroenterol Hepatol 2016;31:1141-6. 10.1111/jgh.13289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lin D, Khan U, Goetze TO, et al. Gastroesophageal Junction Adenocarcinoma: Is There an Optimal Management? Am Soc Clin Oncol Educ Book 2019;39:e88-95. 10.1200/EDBK_236827 [DOI] [PubMed] [Google Scholar]
- 5.Sarrazin S, Lamanna WC, Esko JD. Heparan sulfate proteoglycans. Cold Spring Harb Perspect Biol 2011;3:a004952. 10.1101/cshperspect.a004952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kleeff J, Wildi S, Kumbasar A, et al. Stable transfection of a glypican-1 antisense construct decreases tumorigenicity in PANC-1 pancreatic carcinoma cells. Pancreas 1999;19:281-8. 10.1097/00006676-199910000-00009 [DOI] [PubMed] [Google Scholar]
- 7.Li J, Kleeff J, Kayed H, et al. Glypican-1 antisense transfection modulates TGF-beta-dependent signaling in Colo-357 pancreatic cancer cells. Biochem Biophys Res Commun 2004;320:1148-55. 10.1016/j.bbrc.2004.06.063 [DOI] [PubMed] [Google Scholar]
- 8.Gengrinovitch S, Berman B, David G, et al. Glypican-1 is a VEGF165 binding proteoglycan that acts as an extracellular chaperone for VEGF165. J Biol Chem 1999;274:10816-22. 10.1074/jbc.274.16.10816 [DOI] [PubMed] [Google Scholar]
- 9.Lund ME, Campbell DH, Walsh BJ. The Role of Glypican-1 in the Tumour Microenvironment. Adv Exp Med Biol 2020;1245:163-76. 10.1007/978-3-030-40146-7_8 [DOI] [PubMed] [Google Scholar]
- 10.Matsuda K, Maruyama H, Guo F, et al. Glypican-1 is overexpressed in human breast cancer and modulates the mitogenic effects of multiple heparin-binding growth factors in breast cancer cells. Cancer Res 2001;61:5562-9. [PubMed] [Google Scholar]
- 11.Aikawa T, Whipple CA, Lopez ME, et al. Glypican-1 modulates the angiogenic and metastatic potential of human and mouse cancer cells. J Clin Invest 2008;118:89-99. 10.1172/JCI32412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frampton AE, Prado MM, López-Jiménez E, et al. Glypican-1 is enriched in circulating-exosomes in pancreatic cancer and correlates with tumor burden. Oncotarget 2018;9:19006-13. 10.18632/oncotarget.24873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen G, Wu H, Zhang L, et al. High glypican-1 expression is a prognostic factor for predicting a poor clinical prognosis in patients with hepatocellular carcinoma. Oncol Lett 2020;20:197. 10.3892/ol.2020.12058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oh S, Yeom J, Cho HJ, et al. Integrated pharmaco-proteogenomics defines two subgroups in isocitrate dehydrogenase wild-type glioblastoma with prognostic and therapeutic opportunities. Nat Commun 2020;11:3288. 10.1038/s41467-020-17139-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li J, Chen Y, Zhan C, et al. Glypican-1 Promotes Tumorigenesis by Regulating the PTEN/Akt/β-Catenin Signaling Pathway in Esophageal Squamous Cell Carcinoma. Dig Dis Sci 2019;64:1493-502. 10.1007/s10620-019-5461-9 [DOI] [PubMed] [Google Scholar]
- 16.Svitkina T. The Actin Cytoskeleton and Actin-Based Motility. Cold Spring Harb Perspect Biol 2018;10:a018267. 10.1101/cshperspect.a018267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fife CM, McCarroll JA, Kavallaris M. Movers and shakers: cell cytoskeleton in cancer metastasis. Br J Pharmacol 2014;171:5507-23. 10.1111/bph.12704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vu T, Datta PK. Regulation of EMT in Colorectal Cancer: A Culprit in Metastasis. Cancers (Basel) 2017;9:171. 10.3390/cancers9120171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liao TT, Yang MH. Revisiting epithelial-mesenchymal transition in cancer metastasis: the connection between epithelial plasticity and stemness. Mol Oncol 2017;11:792-804. 10.1002/1878-0261.12096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Soutto M, Peng D, Katsha A, et al. Activation of β-catenin signalling by TFF1 loss promotes cell proliferation and gastric tumorigenesis. Gut 2015;64:1028-39. 10.1136/gutjnl-2014-307191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jung YS, Park JI. Wnt signaling in cancer: therapeutic targeting of Wnt signaling beyond β-catenin and the destruction complex. Exp Mol Med 2020;52:183-91. 10.1038/s12276-020-0380-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yun SH, Park JI. PGC-1α Regulates Cell Proliferation and Invasion via AKT/GSK-3β/β-catenin Pathway in Human Colorectal Cancer SW620 and SW480 Cells. Anticancer Res 2020;40:653-64. 10.21873/anticanres.13995 [DOI] [PubMed] [Google Scholar]
- 23.Shimizu T, Kagawa T, Inoue T, et al. Stabilized beta-catenin functions through TCF/LEF proteins and the Notch/RBP-Jkappa complex to promote proliferation and suppress differentiation of neural precursor cells. Mol Cell Biol 2008;28:7427-41. 10.1128/MCB.01962-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cross DA, Alessi DR, Cohen P, et al. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995;378:785-9. 10.1038/378785a0 [DOI] [PubMed] [Google Scholar]
- 25.Gayet J, Zhou XP, Duval A, et al. Extensive characterization of genetic alterations in a series of human colorectal cancer cell lines. Oncogene 2001;20:5025-32. 10.1038/sj.onc.1204611 [DOI] [PubMed] [Google Scholar]
- 26.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell 2007;129:1261-74. 10.1016/j.cell.2007.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Plas DR, Thompson CB. Akt-dependent transformation: there is more to growth than just surviving. Oncogene 2005;24:7435-42. 10.1038/sj.onc.1209097 [DOI] [PubMed] [Google Scholar]
- 28.Sharma M, Chuang WW, Sun Z. Phosphatidylinositol 3-Kinase/Akt Stimulates Androgen Pathway through GSK3β Inhibition and Nuclear β-Catenin Accumulation. J Biol Chem 2002;277:30935-41. 10.1074/jbc.M201919200 [DOI] [PubMed] [Google Scholar]
- 29.Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci 2003;116:1175-86. 10.1242/jcs.00384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sahin I, Eturi A, De Souza A, et al. Glycogen synthase kinase-3 beta inhibitors as novel cancer treatments and modulators of antitumor immune responses. Cancer Biol Ther 2019;20:1047-56. 10.1080/15384047.2019.1595283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Y. Epidemiology of esophageal cancer. World J Gastroenterol 2013;19:5598-606. 10.3748/wjg.v19.i34.5598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li N, Spetz MR, Ho M. The Role of Glypicans in Cancer Progression and Therapy. J Histochem Cytochem 2020;68:841-62. 10.1369/0022155420933709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ghosh S, Huda P, Fletcher N, et al. Clinical development of an anti-GPC-1 antibody for the treatment of cancer. Expert Opin Biol Ther 2022;22:603-13. 10.1080/14712598.2022.2033204 [DOI] [PubMed] [Google Scholar]
- 34.Yeh MC, Tse BWC, Fletcher NL, et al. Targeted beta therapy of prostate cancer with 177Lu-labelled Miltuximab® antibody against glypican-1 (GPC-1). EJNMMI Res 2020;10:46. 10.1186/s13550-020-00637-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou CY, Dong YP, Sun X, et al. High levels of serum glypican-1 indicate poor prognosis in pancreatic ductal adenocarcinoma. Cancer Med 2018;7:5525-33. 10.1002/cam4.1833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qiao D, Meyer K, Friedl A. Glypican-1 stimulates Skp2 autoinduction loop and G1/S transition in endothelial cells. J Biol Chem 2012;287:5898-909. 10.1074/jbc.M111.325282 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 37.Ding L, Cao J, Lin W, et al. The Roles of Cyclin-Dependent Kinases in Cell-Cycle Progression and Therapeutic Strategies in Human Breast Cancer. Int J Mol Sci 2020;21:1960. 10.3390/ijms21061960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bertoli C, Skotheim JM, de Bruin RA. Control of cell cycle transcription during G1 and S phases. Nat Rev Mol Cell Biol 2013;14:518-28. 10.1038/nrm3629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yuan XW, Wang DM, Hu Y, et al. Hepatocyte nuclear factor 6 suppresses the migration and invasive growth of lung cancer cells through p53 and the inhibition of epithelial-mesenchymal transition. J Biol Chem 2013;288:31206-16. 10.1074/jbc.M113.480285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li J, Li B, Ren C, et al. The clinical significance of circulating GPC1 positive exosomes and its regulative miRNAs in colon cancer patients. Oncotarget 2017;8:101189-202. 10.18632/oncotarget.20516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li C, Du X, Tai S, et al. GPC1 regulated by miR-96-5p, rather than miR-182-5p, in inhibition of pancreatic carcinoma cell proliferation. Int J Mol Sci 2014;15:6314-27. 10.3390/ijms15046314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu H, Niu F, Liu F, et al. Elevated glypican-1 expression is associated with an unfavorable prognosis in pancreatic ductal adenocarcinoma. Cancer Med 2017;6:1181-91. 10.1002/cam4.1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tanaka M, Ishikawa S, Ushiku T, et al. EVI1 modulates oncogenic role of GPC1 in pancreatic carcinogenesis. Oncotarget 2017;8:99552-66. 10.18632/oncotarget.20601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Witkiewicz AK, McMillan EA, Balaji U, et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun 2015;6:6744. 10.1038/ncomms7744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Javadinia SA, Shahidsales S, Fanipakdel A, et al. The Esophageal Cancer and the PI3K/AKT/mTOR Signaling Regulatory microRNAs: a Novel Marker for Prognosis, and a Possible Target for Immunotherapy. Curr Pharm Des 2018;24:4646-51. 10.2174/1381612825666190110143258 [DOI] [PubMed] [Google Scholar]
- 46.Lv J, Cao XF, Ji L, et al. Association of Wnt1/beta-catenin with clinical pathological characteristics and prognosis of esophageal squamous cell carcinoma. Genet Test Mol Biomarkers 2010;14:363-9. 10.1089/gtmb.2009.0173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang H, Deng G, Ai M, et al. Hsp90ab1 stabilizes LRP5 to promote epithelial-mesenchymal transition via activating of AKT and Wnt/β-catenin signaling pathways in gastric cancer progression. Oncogene 2019;38:1489-507. 10.1038/s41388-018-0532-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saif MW, Chu E. Biology of colorectal cancer. Cancer J 2010;16:196-201. 10.1097/PPO.0b013e3181e076af [DOI] [PubMed] [Google Scholar]
- 49.Cheng Y, Zhang Y, Zhang L, et al. MK-2206, a novel allosteric inhibitor of Akt, synergizes with gefitinib against malignant glioma via modulating both autophagy and apoptosis. Mol Cancer Ther 2012;11:154-64. 10.1158/1535-7163.MCT-11-0606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ji D, Zhang Z, Cheng L, et al. The combination of RAD001 and MK-2206 exerts synergistic cytotoxic effects against PTEN mutant gastric cancer cells: involvement of MAPK-dependent autophagic, but not apoptotic cell death pathway. PLoS One 2014;9:e85116. 10.1371/journal.pone.0085116 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The article’s supplementary files as