

Abstract
The presence of lipopolysaccharide (LPS) in gram-negative bacterial repeats-in-toxin (RTX) toxin preparations, as well as the harsh conditions required to remove it, suggests that LPS may complex with RTX toxins. Concentrated culture supernatant (CCS) preparations of the RTX toxin Pasteurella haemolytica leukotoxin (LKT) contained LKT and LPS as the most prominent components, with LKT and LPS constituting ≈30 and 50% of the density of the silver-stained fraction on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), respectively. CCS LKT contained 3.69 ± 0.46 mg of LPS per mg of protein, which was estimated to indicate an LPS/LKT molar ratio of ≈60:1. Subjection of the CCS LKT to preparative SDS-PAGE resulted in separation of LPS from LKT as detected by silver-stained analytical SDS-PAGE; however, the LKT fraction (SDS-PAGE LKT) contained significant endotoxin activity as detected by the Limulus amebocyte lysate assay. Subjection of the SDS-PAGE LKT to a second preparative SDS-PAGE run resulted in a reduction of the LPS/LKT molar ratio to 1:20. The target cell specificity of LKT for bovine leukocytic cells was retained by the SDS-PAGE LKT, and isolated LPS at comparable concentrations to that in CCS LKT exhibited no leukolytic activity. Addition of isolated LPS back to SDS-PAGE LKT resulted in reconstitution of an LPS-LKT complex. Immediately following reconstitution of the LPS-LKT complex, there was minimal change in leukolytic activity of the complex, but following 9.5 h at temperatures from −135 to 37°C, the LPS-LKT complex exhibited increased leukolytic activity and thermal stability compared to SDS-PAGE LKT. Therefore, it appears that LPS complexes with LKT, resulting in enhanced and stabilized leukolytic activity.
Pasteurella haemolytica, the causative agent of shipping-fever pneumonia in cattle, produces a leukotoxin (LKT) which is a member of the “repeats-in-toxin” (RTX) family of gram-negative bacterial pore-forming exotoxins (8, 31). The RTX family comprises a medically important group of toxins composed of two leukocyte-specific toxins, P. haemolytica LKT and Actinobacillus actinomycetemcomitans leukotoxin, as well as several hemolysins, e.g., Escherichia coli alpha-hemolysin, with broad host cell susceptibility (3, 28, 30). The RTX family members share several features including mechanisms of activation, secretion, and intoxication. The genes encoding RTX toxins exhibit sequence homologies, and RTX toxins have similar structural and functional elements including putative N-terminal hydrophobic membrane-spanning domains, posttranslational acylation modification sites, Ca2+ binding tandem nanopeptide repeats domains, and C-terminal secretion signal domains (31). Because preparations of RTX toxins also contain substantial amounts of lipopolysaccharides (LPS), the possible role of LPS in RTX toxin structure and function has been questioned (9).
LPS is a major component of gram-negative bacterial outer membranes. The LPS molecule has amphiphilic properties and consists of a hydrophobic fatty-acyl-containing lipid A; a highly charged and hydrophilic core containing 2-keto-3-deoxyoctosonic acid (KDO) substituted with phosphate and ethanolamine; and a polar, noncharged, hydrophilic repeating polysaccharide containing an O-specific chain (20). LPS readily interacts with numerous biomolecules including phospholipids and membrane and serum proteins (22). Some of these interactions are nonspecific and probably involve nonsaturable binding, whereas LPS binds stoichiometrically to certain proteins, suggesting a specific binding process (27, 34).
The relationship of LPS to RTX toxins has been controversial. The LPS content of most RTX toxin preparations have not been reported, but for those which have, the results have varied. One preparation of purified E. coli alpha-hemolysin was reported to be free of LPS based on low or nondetectable levels of KDO, lipid, and endotoxin activity, but a more highly purified preparation from the same laboratory was later reported to contain substantial amounts of the lipid A fatty acid 3-hydroxytetradecanoic acid (4, 5). The detection of significant levels of LPS in purified E. coli alpha-hemolysin lead Bohach and Snyder to conclude that LPS may be a component of the native aggregated form of alpha-hemolysin (4).
Evidence for a functional role of LPS in RTX toxins has also been found recently. An anti-P. haemolytica LKT monoclonal antibody (MAb) which blocked LKT-induced cytolysis of bovine monocytes was not able to neutralize monokine release from these cells, suggesting that LPS in the LKT preparation was responsible for the monokine response (29). It has also been observed that LKT in cooperation with LPS may cause the release of inflammatory mediators from cells which are not susceptible to LKT-induced cytolysis (25). Czuprynski and Welch have reviewed the evidence suggesting that the RTX toxins and LPS may act cooperatively and have questioned the possible functional and structural relationship of LPS and RTX toxins (9). LPS and RTX toxins are further related by participation of a transcriptional elongation factor, RfaH, which is required for maximal production of E. coli alpha-hemolysin (2, 19). However, RfaH is required for both LPS and alpha-hemolysin synthesis, rather than mediating LPS induction of alpha-hemolysin synthesis.
We have experienced difficulty in attempts to remove LPS from preparations of P. haemolytica LKT. We observed that our preparations of partially purified P. haemolytica LKT consisted primarily of a 102-kDa LKT band and a ≈10-kDa band compatible with LPS on silver-stained sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels (6, 12). Yoo et al. determined that LPS could be removed from LKT by denaturation in SDS and electrophoresis (35). However, dissociating agents such as 3 M guanidine, 8 M urea, or 0.25% Tween 20 diminished but did not completely resolve LPS from LKT (11). Therefore, we questioned whether LPS is a contaminant of LKT preparations or a component of an LKT complex. We tested the hypothesis that LPS forms a structural or functional complex with LKT by comparing the stability, target cell specificity, and leukolytic activity of LPS-free LKT with those of a reconstituted LPS-LKT complex preparation.
MATERIALS AND METHODS
Preparation of P. haemolytica CCS LKT.
Concentrated culture supernatants (CCS) from a wild-type P. haemolytica biotype A, serotype 1 strain, Ok 1 (23), were prepared by inoculating 1.0 liter of RPMI medium containing 2.2 g of NaHCO3 per liter to an optical density at 600 nm (OD600) = 0.25 with the organism prepared by growth overnight on 5% bovine blood agar and then in brain-heart infusion medium to late logarithmic phase. The cultures in RPMI were grown at 37°C with stirring at 70 oscillations/min for approximately 2.5 h to an OD600 of 0.9 to 1.0. All subsequent steps were conducted at 4°C. The bacteria were removed by centrifugation at 13,000 × g for 30 min, and the culture supernatant was concentrated and partially purified by fractional precipitation (0 to 60% saturation by addition of 361 g of solid ammonium sulfate per liter). Following collection of the precipitate by centrifugation at 13,000 × g for 30 min, it was resuspended at ≈0.5 mg of protein/ml in 10 ml of 50 mM sodium phosphate–100 mM sodium chloride buffer (pH 7.0) (phosphate-buffered saline [PBS]), dialyzed against the same buffer, and stored at −135°C. Samples for analytical SDS-PAGE were mixed 1:1 (vol/vol) with SDS-PAGE sample buffer (Sigma Chemical Co., St. Louis, Mo.), incubated for 3 min in a boiling-water bath, and run on either 10 or 15% gels. The gels were silver stained (Daiichi Silver Stain II; Integrated Separation Systems, Natick, Mass.) (12) or transferred (Hoefer TE semi-dry transfer unit; Pharmacia Biotech, Inc., Piscataway, N.J.) to nitrocellulose membranes and immunoblotted with a murine anti-LKT MAb, C6 (6). Protein concentrations were measured by a bicinchoninic acid method (Micro BCA protein assay; Pierce Chemical Co., Rockford, Ill.).
Isolation of LPS from CCS LKT and whole P. haemolytica cells.
A modified phenol-water procedure was used to isolate LPS (32). To isolate LPS from whole cells, the wet P. haemolytica pellet from a preparation of the CCS LKT was resuspended in 10 ml of acetone, collected by centrifugation, and dried. The dried bacterial pellet (1 g) in glass centrifuge tubes was resuspended in 10 ml of 10 mM Tris–1 mM EDTA (pH 8.0) buffer saturated with phenol (Tris-phenol) and vortexed for 30 s, and 10 ml of distilled water was added. For isolation of LPS from CCS LKT, 10 ml of CCS LKT was mixed with the same Tris-phenol buffer as described above. The bacterial pellet or CCS LKT mixtures were incubated at 68°C for 10 min, vortexed three times, and placed on ice. Following centrifugation at 7,000 × g for 30 min at 4°C, the aqueous phase was transferred to glass centrifuge tubes. For the whole-cell LPS preparations, 0.1 mg of RNase (Boehringer Mannheim Corp., Indianapolis, Ind.) was added and the mixture was incubated at room temperature for 30 min. The aqueous phase from each sample was mixed with 5 ml of Tris-phenol buffer and incubated at 68°C for 10 min. The mixture was vortexed three times during the incubation, and after centrifugation at 7,000 × g for 30 min at 4°C, the aqueous phase was transferred to another set of centrifuge tubes. Chloroform (0.5 volume) was added and mixed, and after centrifugation as above, the chloroform phase was removed and discarded and the isolated LPS in the aqueous phase was dialyzed into 12.5 mM Tris–96 mM glycine–196 mM KCl (pH 8.5) buffer and stored at −135°C. The protein and nucleic acid content of the isolated LPS was <0.001 mg of protein and <0.01 mg of nucleic acid per mg of 2-keto-3-deoxyoctosonic acid (KDO) as measured by the Coomassie method and OD260 measurement, respectively. For the Coomassie method, 100-μl aliquots of sample were mixed with 1.0 ml of Coomassie blue reagent (0.01% Coomassie brilliant blue G250, 4.75% ethanol, 8.5% phorphoric acid) (10).
KDO assay.
KDO concentrations were determined by a colorimetric microassay (17), in which 45-μl aliquots of samples were mixed with 5 μl of 2 N H2SO4 and boiled for 30 min in sealed microcentrifuge tubes. Following boiling, the tubes were centrifuged at low speed to return all liquid to the bottom of the tubes, and 25 μl of periodate reagent (0.04 M NaIO4 in 0.125 N H2SO4) was added. The mixture was incubated at room temperature for 20 min, and then 25 μl of arsenite reagent (2% NaAsO2 in 0.5 N HCl) was added with mixing. A transient brown color developed, and after it disappeared, 50 μl of 0.6% thiobarbituric acid (Sigma Chemical Co.) was added, the reaction mixture was vortexed and incubated in a boiling-water bath for 15 min, 150 μl of dimethyl sulfoxide was added immediately to each sample, and the samples were vortexed. After the assay tubes had cooled, the OD550 was measured. Buffer served as the blank, and a 0.1- to 6.0-μg-of-KDO standard range (Sigma Chemical Co.) was used to quantify the KDO present in unknowns.
Endotoxin activity assay.
Endotoxin activity was quantified as endotoxin units (EU) by a kinetic chromogenic Limulus amebocyte lysate (LAL) assay (Kinetic-QCL LAL; BioWhittaker, Inc., Walkersville, Md.). Assays were conducted in duplicate on 100-μl serial dilutions of samples with LAL reagent water and low-endotoxin pipette tips (USA Scientific Plastics, Inc., Ocala, Fla.) in flat-bottom 96-well low-endotoxin plates (Costar, Cambridge, Mass.). Samples in plates were warmed to 37°C in an incubator for 10 min before the addition of 100 μl of reagent to the wells. The plates were immediately placed in a thermally controlled microplate reader (ThermoMax; Molecular Devices, Palo Alto, Calif.), and the OD405 was read for 15 min at 37°C. The endotoxin activity was determined with a standard program for quantification of EU (SoftMax; Molecular Devices).
LPS dry-weight determination.
LPS isolated from four preparations of CCS LKT (4.55 mg of protein) as described in a previous section was extensively dialyzed against water, and the dialysate was lyophilized. The lyophilized LPS was weighed on a semi-micro balance (model B6; Mettler Instrument Corp., Princeton, N.J.) to the nearest 0.1 mg. The amount of residual NaCl in the lyophilized LPS was determined by ion-specific electrode measurement, and the dry weight was corrected for the contribution of NaCl. The KDO content and endotoxin activity of the isolated, lyophilized LPS was also determined, such that the dry weight of LPS could be calculated from either the KDO content or EU value of various LKT preparations.
Separation of LPS from LKT by preparative SDS-PAGE.
Preparative SDS-PAGE was performed on the ammonium sulfate-precipitated CCS LKT, which was resuspended in 1.0 ml of PBS and dialyzed against the same buffer. CCS LKT was mixed with an equal volume of SDS-PAGE sample buffer, heated in boiling water for 3 min, and then cooled on ice. Insoluble material was removed by centrifugation at 5,000 × g for 10 min at 4°C, and the clear supernatant was loaded onto the electrophoresis column of a preparative SDS-PAGE apparatus (model 491 prep cell; Bio-Rad Laboratories, Hercules, Calif.) with a 1-cm 3.75% stacking gel and an 11-cm 7% resolving gel. The electrophoresis was performed at constant current of 35 mA at 4°C for 15 h. Collection of fractions was begun when the dye front had migrated to approximately 3 cm from the bottom of the gel column. The flow rate of elution buffer was 0.235 ml/min, the OD280 was recorded (Pharmacia-LKB control and optical unit UV-1; Pharmacia Biotech, Inc.), and 4.75-ml fractions were collected (GradiFrac; Pharmacia Biotech, Inc.). SDS was removed from fractions by precipitation by adding 0.34 ml of 3 M KCl to fractions (the final concentration of KCl was 0.2 M) (26). Following a 10-min incubation at room temperature, the precipitated potassium dodecyl sulfate was collected by centrifuged at 700 × g for 15 min at 4°C and the supernatant was transferred to new tubes and stored at −20°C. The protein concentration in fractions was measured on 100-μl aliquots of fractions by the Coomassie blue method described above, the KDO content was measured on 45-μl aliquots of fractions, leukotoxin activity was measured on serially diluted 25-μl aliquots of fractions, and analytical SDS-PAGE was conducted on various fractions. The fractions containing LKT were pooled and concentrated with a Centriprep 30 concentrator (Amicon, Inc., Beverly, Mass.). The leukolytic activity of the pooled SDS-PAGE LKT was quantified as described below. To determine whether additional endotoxic activity present in the SDS-PAGE LKT could be removed, an aliquot of SDS-PAGE LKT was mixed with an equal volume of SDS-PAGE sample buffer and subjected to a second run of preparative SDS-PAGE as described above.
Assay of leukolytic activity and quantitation of LKT activity.
BL3 (CRL8037), HL60 (CCL240), and Raji (CCL86) cells were obtained from and cultured as described by the American Type Culture Collection (Rockville, Md.). Aliquots of CCS LKT, preparative SDS-PAGE fractions, or LKT samples treated in various ways were assayed for leukolytic activity by serially diluting these aliquots in wells of round-bottom 96-well microplates containing 100 μl of RPMI 1640 (pH 7.2) (Sigma Chemical Co.). A BL3 cell suspension (100 μl; 4 × 106 cells/ml) was added to each well, and the plate was incubated at 37°C for 120 min. The exposure was ended by centrifugation at 700 × g for 5 min. Leukolysis was determined by measuring the leakage of the large cytoplasmic enzyme lactate dehydrogenase (LDH) into the supernatant. LDH was assayed by transfer of 100 μl of incubation supernatant to wells of a flat-bottom 96-well plates, the plates were warmed to 37°C, 100 μl of LDH assay reagent at 37°C was added (LDL-50 [Sigma Chemical Co.] rehydrated by addition of 25 ml of H2O), and the LDH activity was measured in a thermally controlled kinetic microtiter plate reader (ThermoMax) at 340 nm for 2 min at 37°C. Data was reported as 10−3 OD/min. Maximal LDH leakage was determined by exposing cells to 0.1% Triton X-100, and buffer in the place of sample aliquot was used as the background LDH leakage. The specific leakage was determined as follows:
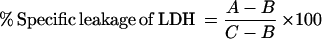 |
where A is the LDH leakage induced by LKT, B is the leakage in PBS, and C is the total LDH leakage induced by 0.1% Triton X-100. One toxic unit (TU) is defined as the reciprocol of the sample dilution causing 50% LDH leakage from 4 × 105 BL3 cells in 200 μl at 37°C for 2 h. The samples were tested in triplicate. For testing the target cell specificity, Raji and HL60 cells were tested for LKT- or LPS-induced LDH leakage as described above for BL3 cells.
Reconstitution of a putative LPS-LKT complex.
LPS (370 μg) isolated from whole cells in 100 μl of 12.5 mM Tris–96 mM glycine–196 mM KCl (pH 8.3) buffer (buffer A) and SDS-PAGE LKT (10 μg of protein in 100 μl of the same buffer) were mixed and incubated for 5 h at 4°C. This mixture was loaded onto an anion-exchange column (Mono Q HR 5/5; Pharmacia Biotech, Inc.) equilibrated in buffer A on a fast protein liquid chromatography system (Pharmacia Biotech, Inc.). The sample was eluted with the application buffer A and an elution buffer B (0.1 N acetic acid, 2 M NaCl) with a 1-h gradient composed of 100% buffer A for 0 to 10 min, 0 to 20% buffer B for 10 to 30 min, 20 to 50% buffer B for 30 to 48 min, and 100% buffer B for 48 to 60 min at a flow rate of 0.5 ml/min. LPS treated identically to the reconstituted LPS-LKT complex was subjected to chromatography as described above and used as a control to locate the LPS elution peak. The OD280 and conductivity were measured, and 1-ml fractions were collected. Fractions containing peaks were precipitated by addition of an equal volume of 72% trichloroacetic acid at 4°C incubated for 30 min, the precipitate was collected by centrifugation at 10,000 × g for 10 min at 4°C, the supernatant was discarded, and the pellet was resuspended in 80 μl of SDS-PAGE sample buffer. LPS and LKT were detected in the precipitated fractions by analytical SDS-PAGE with silver staining and immunoblotting with anti-LKT MAb as described in a previous section.
Immunoprecipitation of the putative LPS-LKT complex.
Immunoprecipitation was conducted on 50 μl of the reconstituted LPS-LKT complex prepared as described above. The mixture was initially centrifuged at 13,000 × g for 30 min at 4°C (all subsequent steps were conducted at 4°C) to remove any insoluble material, and then 50 μl of 1:20-diluted anti-LKT MAb C6 (4 μg) was added and the mixture was incubated for 2 h. This was followed by the addition of 7 μg of goat anti-murine immunoglobulin G (IgG) antisera (Sigma Chemical Co.) in a volume of 10 μl and incubation for 15 h. The immunoprecipitate was collected by centrifugation at 13,000 × g for 30 min and washed twice with 200 μl of PBS containing 0.03% Tween 20. The washed pellet was dissolved in 60 μl of SDS-PAGE sample buffer and subjected to analytical SDS-PAGE.
Coremoval of LPS and LKT from LKT preparations by batchwise treatment with polymyxin B-agarose.
Polymyxin B-agarose (1 ml; Sigma Chemical Co.) was washed twice with and resuspended in 0.8 ml of low-endotoxin water (Gibco BRL Products, Gaithersburg, Md.). Equal volumes (0.8 ml) of CCS LKT, SDS-PAGE LKT, or isolated LPS were mixed with washed polymyxin B-agarose at 4°C, and the polymyxin B-agarose was collected by centrifugation. A 200-μl aliquot of the supernatant was removed for endotoxin activity leukolytic, or protein assays (conducted as described above), and the remaining 600 μl was mixed with another 0.8 ml of washed polymyxin B-agarose; this above process was repeated two more times.
Comparison of the leukolytic activity and thermal stability of the reconstituted LPS-LKT complex with SDS-PAGE LKT.
The reconstituted LPS-LKT complex was prepared by mixing 0.2 ml containing 30 μg of LPS with 0.2 ml of SDS-PAGE LKT (2.8 μg of LKT), and the leukolytic activity on BL3 cells was measured immediately as described above. To assess the leukolytic activity and thermal stability of the LPS-LKT complex compared to the SDS-PAGE LKT following incubation at various temperatures, four sets of 15-μl aliquots of SDS-PAGE LKT (15 μg of protein/ml) were each mixed with either 15 μl of isolated CCS LPS (3.7 μg of LPS/ml) or buffer, and then one pair each of reconstituted LPS-LKT complex and SDS-PAGE LKT was incubated at −135, 4, 21, or 37°C for 570 min. Following incubation, the leukolytic activity with BL3 cells was determined by measuring LDH leakage (see above).
RESULTS
LPS content and type in CCS LKT preparations.
On silver-stained analytical SDS-PAGE, LKT and LPS were the most prominent components of CCS LKT preparations from P. haemolytica (Fig. 1A). Although the total protein and KDO content of CCS LKT varied by ±50% among several consecutive preparations, LKT and LPS consistently composed about 30 and 50%, respectively, of the total silver stain density of these CCS LKT preparations (data not shown). LPS isolated from the CCS LKT by phenol extraction consisted of two silver-stained bands with estimated molecular masses of 10 and 17 kDa on analytical SDS-PAGE (Fig. 1B, lane 1). Likewise, LPS isolated from whole P. haemolytica cells was composed of two bands with similar molecular masses and banding patterns on silver-stained SDS-PAGE (lane 2). The banding pattern reported herein is similar to that previously reported for LPS from P. haemolytica biotype A, serotype 1 (18). Therefore, it appears that the LPS in CCS LKT is similar, if not identical, to the LPS associated with the outer membrane. LPS isolated from CCS LKT (n = 4) contained 0.54% ± 0.06% (wt/wt) KDO and had a ratio of 720 × 103 ± 22 × 103 EU/mg of LPS. Using an LPS monomer mass of 10 kDa and assuming that LKT constitutes ≈60% of the CCS LKT protein (11), the molar ratio of LPS to LKT in CCS LKT would be estimated to be about 60 LPS monomers to 1 LKT monomer.
FIG. 1.

CCS LKT contains a similar rough-type LPS banding pattern to that of LPS extracted from whole P. haemolytica cells. CCS LKT (2 μg of protein) (A) and phenol-extracted LPS from CCS LKT (18.5 μg of LPS) in lane 1 or from whole P. haemolytica cells (18.5 μg of LPS) in lane 2 (B) were subjected to analytical SDS-PAGE on 15% gels and silver stained. Lane M contains molecular mass markers. LKT and LPS had been previously shown to run at 97 to 102 and 6 to 14 kDa, respectively (6, 11).
Separation of LPS from LKT.
The LPS and LKT from CCS LKT were separated by preparative SDS-PAGE, as previously reported by Yoo et al. (35). LPS eluted as two peaks in the early fractions, corresponding to its low molecular mass (Fig. 2A). A prominent protein peak eluted in the middle fractions of the run was coincident with the leukolytic activity peak following removal of free dodecyl sulfate by potassium precipitation. This peak was determined to contain ≈20% of the protein and ≈60% of the leukolytic activity applied to the preparative SDS-PAGE gel.
FIG. 2.

Separation of LPS from LKT by preparative SDS-PAGE. (A) CCS LKT (0.8 ml containing 2 mg of protein) was subjected to preparative SDS-PAGE, 4.75-ml fractions were collected, free SDS was removed, and the fractions were assayed for protein (solid line), KDO (dotted line), and leukolytic activity (dashed line) as described in Materials and Methods. (B and C) Fractions (100 μl) from the preparative SDS-PAGE (B) and from the pooled and concentrated LKT peak (1.0 μg of protein) (C) were subjected to analytical SDS-PAGE on 15 and 10% gels (B and C, respectively) and silver stained.
Analytical SDS-PAGE analysis of the fractions from preparative SDS-PAGE showed that the KDO-containing fractions were composed of the 10- and 17-kDa LPS bands and a 6-kDa band not seen in the LPS isolated from CCS LKT or whole P. haemolytica cells (Fig. 2B, lanes 6 and 7). The origin of the 6-kDa band is uncertain, but it may be associated with the residual bromophenol dye front from the preparative SDS-PAGE. The LKT activity peak was composed of the 102-kDa LKT band and a faint 6-kDa band (lane 21). Only the 102-kDa LKT band was observed for the pooled and concentrated LKT peak from preparative SDS-PAGE (Fig. 2C, lane c). Although no LPS band was detected by analytical SDS-PAGE, significant endotoxic activity was detected by the LAL assay in SDS-PAGE LKT (Table 1). Subjection of the SDS-PAGE LKT to a second run on preparative SDS-PAGE resulted in further reduction in the EU level, such that the calculated molar ratio of LPS to LKT was reduced to 1:20.
TABLE 1.
LPS content of LKT preparations
| LKT preparation | LKT sp act (103 TU/mg of protein) | LPS content
|
||
|---|---|---|---|---|
| mg of LPS/mg of protein | μg of KDO/mg of protein | 103 EU/mg of protein | ||
| CCS LKTa | 3.88 ± 0.85 | 3.69 ± 0.46 | 22.6 ± 4.6 | 2,660 ± 330 |
| SDS-PAGE LKTb | ||||
| 1st run | 125 | 0.063 | 1.38 | 47 |
| 2nd run | 100 | 0.005 | <0.1 | 3.6 |
Values are means ± standard deviations (n = 4).
Values are for a single CCS preparation subjected to two successive preparative SDS-PAGE runs.
The SDS-PAGE LKT retained its target cell specificity for bovine leukocytes. The SDS-PAGE LKT was leukolytic for bovine BL3 lymphoma cells but not for human Raji lymphoma cells or HL60 myelomonocytic leukemic cells (Table 2). Isolated LPS used at a similar concentration to that observed in CCS LKT had no leukolytic activity (data not shown).
TABLE 2.
SDS-PAGE LKT retains bovine leukocyte target cell specificity
| LKT preparation | LKT sp act (103 TU/mg of protein)a in:
|
||
|---|---|---|---|
| BL3 cells | Raji cells | HL60 cells | |
| CCS LKT | 73 ± 10 | 0 ± 0 | 0 ± 0 |
| SDS-PAGE LKT | 314 ± 22 | 0 ± 0 | 0 ± 0 |
Values are means ± standard deviations (n = 3).
Coremoval of LPS and LKT by polymyxin B-agarose.
The natural occurrence of an LPS-LKT complex was suggested by the removal of both LPS and LKT from CCS LKT preparations by polymyxin B-agarose, which binds the lipid A portion of LPS (21). Batchwise treatment of CCS LKT with three successive rounds of polymyxin B-agarose resulted in complete removal of both EU and LKT leukolytic activity from CCS LKT (Table 3). EU and LKT activity were reduced at different rates by successive polymyxin B-agarose treatments, but both activities were completely removed by three treatments. Three rounds of polymyxin B-agarose treatment were also required to remove a similar amount of isolated LPS in the absence of added LKT (data not shown). As with CCS LKT, polymyxin B-agarose treatment of SDS-PAGE LKT which contained LPS and LKT at a ratio of 1:20 resulted in removal of 98% of the applied LKT activity and 93% of the applied protein.
TABLE 3.
Coremoval of LPS and LKT from CCS LKT by successive batchwise treatment with polymyxin B-agarose
| No. of polymyxin B-agarose treatments | LPS content (103 EU/ml) | LKT activity (103 TU/ml) |
|---|---|---|
| 0 | 26.3 | 148 |
| 1 | 25.0 | 45.0 |
| 2 | 10.6 | 4.13 |
| 3 | 0.0 | 0.0 |
Detection of reconstituted LPS-LKT complex.
Addition of isolated LPS back to SDS-PAGE LKT resulted in the formation of a reconstituted LPS-LKT complex, which was detected by anion-exchange chromatography. Chromatography of isolated LPS on a quaternary ammonium-type anion-exchange MonoQ HR 5/5 column developed with a nonlinear 60-min gradient of 30 ml of 0 to 0.1 N acetic acid and 0 to 2.0 M NaCl resulted in detection of a small breakthrough peak and a large peak eluting at 20.9 ml (Fig. 3A). Detection of LPS by analytical SDS-PAGE showed that the 20.9-ml peak contained LPS but that the breakthrough peak and the interpeak region did not (Fig. 3C, lanes 1 to 3). As expected, no LKT was detected as assessed by Western blotting (Fig. 3D, lanes 1 to 3). To test whether LPS could complex with LKT, 370 μg of LPS was mixed with 10 μg of LKT and incubated for 5 h at 4°C. Chromatography of this mixture on the MonoQ HR 5/5 column resulted in detection of the same breakthrough and 20.9-ml peaks as well as three additional peaks eluting in the interpeak region (Fig. 3B). As before, the 20.9-ml peak contained LPS and the breakthrough peak did not (Fig. 3C, lanes 4 and 8). Peaks eluting at 13.0 and 15.1 ml consisted of both LKT (Fig. 3D, lanes 6 and 7) and LPS (Fig. 3C, lanes 6 and 7). The slightly retained peak eluting at 3.0 ml did not contain either LPS or LKT (Fig. 3B and C, lane 5).
FIG. 3.

Detection of a reconstituted LPS-LKT complex by anion-exchange chromatography. Isolated LPS (A) (0.1 ml containing 370 μg of LPS) or reconstituted LPS-LKT complex (B) (0.2 ml containing 370 μg of LPS and 10 μg of protein) prepared as described in Materials and Methods was applied to a MonoQ HR5/5 column and developed with a nonlinear 30-ml acetic acid-NaCl gradient, and fractions were collected. The vertical axis is the OD280 measured in absorbance units (AU). (C and D) Fractions (1.0 ml) from various peaks and interpeak regions were pooled and analyzed for LPS and LKT by analytical SDS-PAGE (C) and for LKT by immunoblotting with an anti-LKT MAb C6 (D).
The detection of the reconstituted LPS-LKT complex was also examined by using immunoprecipitation by a murine anti-LKT MAb C6 and goat anti-murine IgG antisera. Following mixing of SDS-PAGE LKT and LPS as described above, immunoprecipitation of the mixture or CCS LKT preparation with anti-LKT MAb C6 followed by goat anti-murine IgG antisera resulted in detection of both the LKT and LPS bands in the washed precipitate on analytical SDS-PAGE (Fig. 4, lanes 5 and 2, respectively). The buffer control contained precipitated Igs but no LKT or LPS (Fig. 4, lane 1). Immunoprecipitation of isolated LPS in the absence of added LKT resulted in no detected immunoprecipitated LPS (lane 3). Likewise, immunoprecipitation of SDS-PAGE LKT in the absence of added isolated LPS resulted in detection of LKT but no immunoprecipitated LPS (lane 4).
FIG. 4.

Detection of a reconstituted LPS-LKT complex by coimmunoprecipitation with anti-LKT MAb C6. Isolated LPS, SDS-PAGE LKT, and reconstituted LPS-LKT complex were incubated with anti-LKT C6 murine IgG MAb followed by precipitation with goat anti-mouse antiserum, and the precipitate was washed and subjected to analytical SDS-PAGE and silver staining. Lanes: 1, anti-LKT C6 murine IgG MAb and goat anti-mouse antiserum only; 2, CCS LKT (74 μg of LPS and 13 μg of protein); 3, isolated LPS (185 μg); 4, SDS-PAGE LKT (5.0 μg of protein); 5, reconstituted LPS-LKT complex (185 μg of LPS and 5.0 μg of protein); 6, molecular mass markers; 7, isolated LPS (18.5 μg); 8, SDS-PAGE LKT (1.0 μg of protein), respectively, directly subjected to analytical SDS-PAGE without immunoprecipitation to serve as markers for LPS and LKT, respectively.
Leukolytic activity and stability of reconstituted LPS-LKT complex.
The effect of LPS complexation with LKT on leukolytic activity was tested immediately after complexation or following 9.5 h of incubation at various temperatures. When the leukolytic activity of SDS-PAGE LKT was compared to the reconstituted LPS-LKT complex immediately following mixing, the leukolytic activity was similar (Table 4). The reconstituted LPS-LKT complex had enhanced leukolytic activity at −135, 4, 21, and 37°C following 9.5 h of incubation, whereas SDS-LKT did not. The maximum enhancement was seen at 4°C. At 37°C, both the reconstituted LPS-LKT complex and SDS-PAGE LKT were partially inactivated, but the reconstituted LPS-LKT complex appeared more thermally stable than the SDS-PAGE LKT did.
TABLE 4.
Complexation of LPS with LKT enhances and stabilizes LKT activity
| Preparation | LKT sp act (103 TU/mg of protein)a
|
||||
|---|---|---|---|---|---|
| Immediately after mixing | After incubation for 9.5 h at:
|
||||
| −135°C | 4°C | 21°C | 37°C | ||
| LPS | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 |
| SDS-PAGE LKT | 113 ± 19 | 113 ± 20 | 147 ± 35 | 127 ± 54 | 7 ± 4 |
| Reconstituted LPS-LKT complex | 120 ± 78 | 163 ± 15 | 400 ± 100 | 153 ± 11 | 57 ± 15 |
Values are means ± standard deviations (n = 3).
DISCUSSION
Circumstantial findings suggest that LPS may play a structural and possibly a functional role in RTX toxins, but the exact nature of the relationship of LPS to RTX toxins is unclear. With respect to RTX toxin activity, it has been suspected that LPS in RTX toxin preparations may be responsible for some of the intoxication phenomena attributed to RTX toxins. Although RTX cytolytic activity appears to reside entirely with the RTX toxin proteins, particular sublytic activities attributed entirely to RTX toxins, such as cytokine release, may be caused by LPS rather than RTX toxins (29). Part of this uncertainty about the relationship of LPS to RTX toxins arises from whether LPS is a contaminant of RTX toxin preparations or whether it is a component of an LPS-RTX toxin structural complex (9).
Based on significant levels of the lipid A fatty acid 3-hydroxytetradecanoic acid in purified preparations of alpha-hemolysin, Bohach and Snyder proposed that E. coli alpha-hemolysin consists of an aggregated complex of alpha-hemolysin protein and LPS (4). Likewise, we came to believe that LPS might play a role in RTX toxin structure and function based on difficulty in removal of LPS from the RTX toxin P. haemolytica LKT (11). Based on these observations, we hypothesized that LPS is a structural component of LKT rather than a contaminant.
Distinguishing between LPS as a contaminant and LPS as a component of a complex is difficult. The notion of LPS as a contaminant assumes that LPS is unnaturally associated with LKT, but there are no specific tests for unnatural association. LPS and LKT are major components of culture supernatants (11), and both are observed in histologic sections of lung tissue from cattle naturally infected with P. haemolytica (33). However, the coincidental occurrence of LPS and LKT in both culture supernatants and infected lungs does not directly implicate a natural complexation of LPS with LKT. The natural occurrence of an LPS-LKT complex is supported by the coprecipitation of LPS and LKT by an anti-LKT MAb. Immunoprecipitation has been used previously to provide evidence for a natural LPS-protein complexation (15). In addition to coprecipitation of LPS and LKT by anti-LKT MAb, both LPS and LKT from CCS LKT were bound by the lipid A affinity resin polymyxin B agarose (21), suggesting that these components may be naturally complexed. The reconstitution of an LPS-LKT complex provides further support that an LPS-LKT complex may exist naturally.
The manner in which LPS interacts with particular proteins varies from crystalline lattice forms as typified by E. coli major outer membrane protein O-8, where there is a 1:1 molar ratio of LPS monomers to protein monomers (34), to proteins which bind to LPS aggregates or micelles, as typified by mammalian lipoprotein binding protein or phospholipid transfer protein, where the amount of LPS greatly exceeds that of protein (16, 36). The high LPS content of CCS LPS-LKT complex suggests that LKT is more like the later type in which LKT is bound to aggregated LPS or LPS micelles.
The functional role of LPS in the LPS-LKT complex was not extensively examined in this study. Isolated LPS at the concentrations found in CCS LKT was not cytolytic for an LKT-susceptible target cell line, and the SDS-PAGE LKT retained bovine leukocyte specific leukolytic activity. Stevens and Czupyrnski reported that LPS associated with LKT may be involved in certain sublytic effects attributed to LKT (29). In some sublytic RTX effects LPS may augment the LKT-elicited effect, whereas in others LPS may be the effector with LKT functioning as an LPS carrier. We did find that LPS may play a structural role in stabilizing LKT activity. Like other RTX toxins, LKT has unstable activity (7), although the mechanism of this instability is not completely understood. LKT loses activity when incubated at 37 to 57°C, and this inactivation does not involve proteolysis. The reconstituted LPS-LKT complex was more stable at low to moderate temperatures than the SDS-PAGE LKT was, suggesting that LPS complexation with LKT may stabilize LKT activity. The thermal instability of LKT may be associated with self-aggregation. LKT forms large aggregates with molecular masses of 1,000 to 8,000 kDa which are less active than LKT disaggregated to octomers or tetramers (7). It is possible that LPS reduces LKT aggregation, thereby stabilizing LKT activity.
Preparative SDS-PAGE reduced the LPS content of LKT to <0.05 LPS monomer per LKT monomer. Although the preparative SDS-PAGE LKT did not contain detectable LPS by silver-stained analytical SDS-PAGE or by a KDO assay, appreciable endotoxin activity was detected by a chromogenic LAL assay. The most straightforward explanation for this endotoxin activity is that the small amount of residual LPS in the SDS-PAGE LKT is the source of the endotoxin activity.
Another possible explanation for the residual endotoxin activity in the preparative SDS-PAGE LKT is that LKT itself can react with the LAL endotoxin assay. Some substances which cause false-positive reactions with the LAL reagent have been characterized, but others have not (13, 24). β-Glycans from fungal and plant cell walls and LAL-reactive cellulosic material from extracts of cuprophane dialyzers activate the LAL reagent via an alternative pathway. In one LKT preparation, reducing carbohydrate material was present in the CCS LKT preparation, and diminution of this reducing carbohydrate was associated with diminution of endotoxin activity (14). In the present study, although KDO, the major carbohydrate component of rough LPS, was markedly reduced, endotoxin activity, although reduced, was not eliminated. It seems unlikely that significant levels of β-glycans in LKT preparations are responsible for the endotoxin activity observed. In addition to β-glycans, proteolytic enzymes can cause a true “false-positive” reaction by direct degradation of the chromogenic substrate. Proteolytic activity has been detected in P. haemolytica culture supernatants, but this proteolytic activity can be partitioned from LKT (1). Therefore, the cause of the observed LAL endotoxin activity in SDS-PAGE LKT is low residual LPS associated with LKT.
ACKNOWLEDGMENTS
Jun Li was supported by a graduate research assistantship from Anthony W. Confer, Food Animal Research Endowed Chair, Department of Anatomy, Pathology, and Pharmacology, College of Veterinary Medicine, Oklahoma State University. This study was supported in part by the Oklahoma Agricultural Experiment Station through Section 1433 Animal Health Formula Funds and Hatch grant OKL02249 from the U.S. Department of Agriculture.
REFERENCES
- 1.Abdullah K M, Udoh E A, Shewen P E, Mellors A. A neutral glycoprotease of Pasteurella haemolytica A1 specifically cleaves O-sialoglycoproteins. Infect Immun. 1992;60:56–62. doi: 10.1128/iai.60.1.56-62.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bailey M J, Hughes C, Koronakis V. Increased distal gene transcription by the elongation factor RfaH, a specialized homologue of NusG. Mol Microbiol. 1996;22:729–737. doi: 10.1046/j.1365-2958.1996.d01-1726.x. [DOI] [PubMed] [Google Scholar]
- 3.Bhakdi S, Mackman N, Nicaud J M, Holland I B. Escherichia coli hemolysin may damage target cells by generating transmembrane pores. Infect Immun. 1986;52:63–69. doi: 10.1128/iai.52.1.63-69.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bohach G A, Snyder I S. Chemical and immunological analysis of the complex structure of Escherichia coli alpha hemolysin. J Bacteriol. 1985;164:1071–1080. doi: 10.1128/jb.164.3.1071-1080.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cavalieri S J, Snyder I S. Cytotoxic activity of partially purified Escherichia coli alpha hemolysin. J Med Microbiol. 1982;15:11–21. doi: 10.1099/00222615-15-1-11. [DOI] [PubMed] [Google Scholar]
- 6.Clinkenbeard K D, Clarke C R, Hague C, Clinkenbeard P A, Srikumaran S, Morton R J. Pasteurella haemolytica leukotoxin-induced synthesis of eicosanoids by bovine neutrophils in vitro. J Leukoc Biol. 1994;56:644–649. doi: 10.1002/jlb.56.5.644. [DOI] [PubMed] [Google Scholar]
- 7.Clinkenbeard K D, Clinkenbeard P A, Waurzyniak B J. Chaotropic agents cause disaggregation and enhanced activity of Pasteurella haemolytica leukotoxin. Vet Microbiol. 1995;45:201–209. doi: 10.1016/0378-1135(94)00131-f. [DOI] [PubMed] [Google Scholar]
- 8.Confer A W, Clinkenbeard K D, Murphy G M. Pathogenesis and virulence of Pasteurella haemolytica in cattle: an analysis of current knowledge and future approaches. In: Donachie W, Lainson F A, Hodgson J C, editors. Haemophilus, Actinobacillus, and Pasteurella. London, United Kingdom: Plenum Publishing Corp.; 1995. pp. 51–62. [Google Scholar]
- 9.Czuprynski C J, Welch R A. Biological effects of RTX toxins: the possible role of lipopolysaccharide. Trends Microbiol. 1995;3:480–483. doi: 10.1016/s0966-842x(00)89016-2. [DOI] [PubMed] [Google Scholar]
- 10.Daniels L. Chemical analysis. In: Gerhardt P, Murry R G E, Krieg N R, Wood W A, editors. Methods for general and molecular bacteriology. Washington, D.C: American Society for Microbiology; 1994. p. 543. [Google Scholar]
- 11.El Rassi Z, Clinkenbeard P A, Clinkenbeard K D. High-performance liquid chromatography of Pasteurella haemolytica leukotoxin using anion-exchange perfusion columns. J Chromatogr Ser A. 1998;808:167–176. doi: 10.1016/s0021-9673(98)00131-9. [DOI] [PubMed] [Google Scholar]
- 12.Fomsgaard A, Freudenberg M A, Galanos C. Modification of the silver staining technique to detect lipopolysaccharide in polyacrylamide gels. J Clin Microbiol. 1990;28:2627–2631. doi: 10.1128/jcm.28.12.2627-2631.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gagliardi N C, Nolan J P, Moreno Feind D, DeLissio M. A rapid sensitive monoclonal assay for lipid A in solution. J Immunol Methods. 1986;91:243–247. doi: 10.1016/0022-1759(86)90485-0. [DOI] [PubMed] [Google Scholar]
- 14.Gentry M J, Srikumaran S. Neutralizing monoclonal antibodies to Pasteurella haemolytica leukotoxin affinity-purify the toxin from crude culture supernatants. Microb Pathog. 1991;10:411–417. doi: 10.1016/0882-4010(91)90086-p. [DOI] [PubMed] [Google Scholar]
- 15.Gulig P A, Hansen E J. Coprecipitation of lipopolysaccharide and the 39,000-molecular-weight major outer membrane protein of Haemophilus influenzae type b by lipopolysaccharide-directed monoclonal antibodies. Infect Immun. 1985;49:819–827. doi: 10.1128/iai.49.3.819-827.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hailman E, Albers J J, Wolfbauer G, Tu A-Y, Wright S D. Neutralization and transfer of lipopolysaccharide by phospholipid transfer protein. J Biol Chem. 1996;271:12172–12178. doi: 10.1074/jbc.271.21.12172. [DOI] [PubMed] [Google Scholar]
- 17.Karkhanis A D, Zeltner J Y, Jackson J, Carlo D J. A new and improved microassay to determine 2-keto-3-deoxyoctonate in lipopolysaccharide of Gram-negative bacteria. Anal Biochem. 1978;85:595–601. doi: 10.1016/0003-2697(78)90260-9. [DOI] [PubMed] [Google Scholar]
- 18.Lacroix R P, Duncan R, Jenkins R P, Leitch R A, Perry J A, Richards J C. Structural and serological specificities of Pasteurella haemolytica lipopolysaccharides. Infect Immun. 1993;61:170–181. doi: 10.1128/iai.61.1.170-181.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leeds J A, Welch R A. RfaH enhances elongation of Escherichia coli hlyCABD mRNA. J Bacteriol. 1996;178:1850–1857. doi: 10.1128/jb.178.7.1850-1857.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lüderitz O, Freudenberg M A, Galanos C, Lehmann V, Rietschel E T, Shaw D H. Lipopolysaccharides of Gram-negative bacteria. Curr Top Membr Transp. 1982;17:79–151. [Google Scholar]
- 21.Mølvig J, Baek L. Removal of endotoxin from culture media by a polymyxin B sepharose column. Scand J Immunol. 1987;26:611–619. doi: 10.1111/j.1365-3083.1987.tb02296.x. [DOI] [PubMed] [Google Scholar]
- 22.Morrison D C. Nonspecific interactions of bacterial lipopolysaccharides with membranes and membrane components. In: Berry L J, editor. Cellular biology of endotoxin. Handbook of endotoxin. Vol. 3. Amsterdam, The Netherlands: Elsevier; 1985. pp. 25–55. [Google Scholar]
- 23.Panciera R J, Corstvet R E, Confer A W, Gresham C N. Bovine pneumonic pasteurellosis: effect of vaccination with live Pasteurella species. Am J Vet Res. 1984;45:2538–2542. [PubMed] [Google Scholar]
- 24.Roslansky P A. Reactivity of Limulus amebocyte lysate (LAL) to glycans. In: Novitsky T, editor. LAL update 8(2). Woods Hole, Mass: Associates of Cape Cod, Inc.; 1990. pp. 2–5. [Google Scholar]
- 25.Saban R, Broadstone R V, Haak-Frendscho M, Skoyen S, Fialkowski J, Maheswaran S K, Bjorling D E, Czuprynski C. Effects of Pasteurella haemolytica leukotoxin and lipopolysaccharide on histamine, prostanoid, and leukotriene release by bovine lung parenchyma in vitro. Am J Vet Res. 1997;58:1227–1230. [PubMed] [Google Scholar]
- 26.Sandri M, Rizzi C, Catani C, Carraro U. Selective removal of free dodecyl sulfate from 2-mercaptoethanol-SDS-solubilized proteins before KDS-protein precipitation. Anal Biochem. 1993;213:34–39. doi: 10.1006/abio.1993.1382. [DOI] [PubMed] [Google Scholar]
- 27.Schumann R R, Leong S R, Flaggs G W, Gray P W, Wright S D, Mathison J C, Tobias P S, Ulevitch R J. Structure and function of lipopolysaccharide binding proteins. Science. 1990;249:1429–1431. doi: 10.1126/science.2402637. [DOI] [PubMed] [Google Scholar]
- 28.Shewen P E, Wilkie B N. Cytotoxin of Pasteurella haemolytica acting on bovine leukocytes. Infect Immun. 1982;35:911–914. doi: 10.1128/iai.35.1.91-94.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stevens P, Czupyrnski C. Dissociation of cytolysis and monokine release by bovine mononuclear phagocytes incubated with Pasteurella haemolytica partially purified leukotoxin and lipopolysaccharide. Can J Vet Res. 1995;59:110–117. [PMC free article] [PubMed] [Google Scholar]
- 30.Tsai C C, McArthur W P, Baehni P C, Hammond B F, Taichman N S. Extraction and partial characterization of a leukotoxin from a plaque-derived gram-negative microorganism. Infect Immun. 1979;25:427–439. doi: 10.1128/iai.25.1.427-439.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Welch R A, Bauer M E, Kent A D, Ledds J A, Moayeri M, Regassa L B, Swenson D L. Battling against host phagocytes: the wherefore of the RTX family of toxins? Infect Agents Dis. 1996;4:254–272. [PubMed] [Google Scholar]
- 32.Westphal Q, Jann K. Bacterial lipopolysaccharides: extraction with phenol-water and further applications of the procedure. Methods Carbohydr Chem. 1965;5:83–89. [Google Scholar]
- 33.Whiteley L O, Maheswaran S K, Weiss D J, Ames T R. Immunohistochemical localization of Pasteurella haemolytica A1-derived endotoxin, leukotoxin, and capsular polysaccharide in experimental bovine pasteurella pneumonia. Vet Pathol. 1990;27:150–161. doi: 10.1177/030098589002700302. [DOI] [PubMed] [Google Scholar]
- 34.Yamada H, Mizushima S. Interaction between major outer membrane protein (O-8) and lipopolysaccharide in Escherichia coli K12. Eur J Biochem. 1980;103:209–218. doi: 10.1111/j.1432-1033.1980.tb04305.x. [DOI] [PubMed] [Google Scholar]
- 35.Yoo H S, Rajagopal B S, Maheswaran S K, Ames T R. Purified Pasteurella haemolytica leukotoxin induces expression of inflammatory cytokines from bovine alveolar macrophages. Microb Pathog. 1995;18:237–252. doi: 10.1016/s0882-4010(05)80001-4. [DOI] [PubMed] [Google Scholar]
- 36.Yu B, Wright S D. Catalytic properties of lipopolysaccharide (LPS) binding protein. Transfer of LPS to soluble CD14. J Biol Chem. 1996;271:4100–4105. doi: 10.1074/jbc.271.8.4100. [DOI] [PubMed] [Google Scholar]