

Abstract
Purpose:
To evaluate AZD4635, an adenosine A2A receptor antagonist, as monotherapy or in combination with durvalumab in patients with advanced solid tumors.
Patients and Methods:
In phase Ia (dose escalation), patients had relapsed/refractory solid tumors; in phase Ib (dose expansion), patients had checkpoint inhibitor–naïve metastatic castration-resistant prostate cancer (mCRPC) or colorectal carcinoma, non–small cell lung cancer with prior anti–PD-1/PD-L1 exposure, or other solid tumors (checkpoint-naïve or prior anti–PD-1/PD-L1 exposure). Patients received AZD4635 monotherapy (75–200 mg once daily or 125 mg twice daily) or in combination with durvalumab (AZD4635 75 or 100 mg once daily). The primary objective was safety; secondary objectives included antitumor activity and pharmacokinetics; exploratory objectives included evaluation of an adenosine gene signature in patients with mCRPC.
Results:
As of September 8, 2020, 250 patients were treated (AZD4635, n = 161; AZD4635+durvalumab, n = 89). In phase Ia, DLTs were observed with monotherapy (125 mg twice daily; n = 2) and with combination treatment (75 mg; n = 1) in patients receiving nanosuspension. The most common treatment-related adverse events included nausea, fatigue, vomiting, decreased appetite, dizziness, and diarrhea. The RP2D of the AZD4635 capsule formulation was 75 mg once daily, as monotherapy or in combination with durvalumab. The pharmacokinetic profile was dose-proportional, and exposure was adequate to cover target with 100 mg nanosuspension or 75 mg capsule once daily. In patients with mCRPC receiving monotherapy or combination treatment, tumor responses (2/39 and 6/37, respectively) and prostate-specific antigen responses (3/60 and 10/45, respectively) were observed. High versus low blood-based adenosine signature was associated with median progression-free survival of 21 weeks versus 8.7 weeks.
Conclusions:
AZD4635 monotherapy or combination therapy was well tolerated. Objective responses support additional phase II combination studies in patients with mCRPC.
Translational Relevance.
Extracellular adenosine exerts immunosuppressive effects on the tumor microenvironment. Adenosine A2A receptor (A2AR) blockade improves antigen presentation, T-cell activation, regulatory T-cell function, and NK-cell activity, which may lead to a more robust antitumor immune response. AZD4635, an oral A2AR inhibitor, was well tolerated both as a monotherapy and in combination with the anti–PD-L1 antibody durvalumab in patients with relapsed/refractory solid tumors. Objective tumor and prostate-specific antigen responses were observed in patients with metastatic castration-resistant prostate cancer (mCRPC), which is generally resistant to immune checkpoint blockade. A 14 gene blood-based adenosine signature was associated with differential PFS and may facilitate selection of patients to optimize treatment strategies for those with advanced mCRPC. The findings support ongoing phase II studies of AZD4635 combination therapy in patients with mCRPC.
Introduction
Purinergic signaling regulates the immune response by balancing extracellular levels of adenosine triphosphate (ATP) and adenosine to exert pro- and anti-inflammatory signaling effects, respectively (1, 2). Extracellular levels of adenosine can accumulate within the tumor microenvironment (TME) at levels 10- to 20-fold higher than in normal tissue and can reach micromolar concentrations, particularly within the hypoxic tumor core (1, 3). The presence of adenosine within the tumor microenvironment affects both adaptive and innate immunity by suppressing T-cell and natural killer cell function (1, 4), providing a mechanism for resistance and immune escape. Extracellular adenosine is generated by ectoenzymes within the TME. CD39 (ectonucleoside triphosphate-1) converts ATP to adenosine monophosphate (AMP), and CD73 (ecto-5′-nucleotidase) then converts AMP to free adenosine (2). CD73 expression, thought to be a primary driver of elevated intratumoral adenosine, is increased in multiple tumor types, and is associated with poor prognosis in patients with several solid tumors and hematologic malignancies (5). Prostatic acid phosphatase (PAP; ACP3) also metabolizes extracellular AMP to free adenosine, independent of CD39 and CD73 (2, 6), and PAP mRNA levels are higher in prostate tumors compared with CD73 in other cancer types (cBioPortal data available ACP3 and NT5E), suggesting that prostate cancers may be particularly reliant on adenosine. Adenosine exerts its suppressive effects on immune cells primarily through the adenosine A2A receptor (A2AR; ref. 5), although the A2B receptor may also play a role in some tumor types (reviewed in ref. 7).
AZD4635 is an orally bioavailable A2AR antagonist that inhibits adenosine binding to A2AR in a dose-dependent manner and has immunomodulatory and antineoplastic activity (8, 9). Preclinical studies in murine and human in vitro models, and in vivo in mouse models with MC38-OVA tumors, demonstrated that AZD4635 increased dendritic cell activation, antigen presentation, and cytotoxic T-cell infiltration and activity (8, 10). Modulating the TME with AZD4635 may facilitate an improved antitumor immune response, especially when used in combination with immune checkpoint inhibitors (ICI; ref. 8). One such ICI is durvalumab, a selective, high-affinity, engineered, human, immunoglobulin G1 monoclonal antibody, that inhibits the binding of programmed death-ligand 1 (PD-L1) to programmed cell death protein-1 (PD-1) and CD80, facilitating T-cell activation and tumor cell elimination (11).
The identification of predictive biomarkers may facilitate optimal patient-specific treatment strategies for adenosine pathway modulation. Prior studies identified a 14-gene tissue-level expression signature (PPARG, CYBB, COL3A1, FOXP3, LAG3, APP, CD81, GP1, PTGS2, CASP1, FOS, MAPK1, MAPK3, and CREB1) that reflects adenosine signaling activity via A2AR within a variety of solid tumors (e.g., breast, cholangiocarcinoma, colon, myxofibrosarcoma, ovary, pancreas, sarcoma; ref. 12). This signature was previously postulated as a biomarker to identify patients with tumors having significant adenosine drive, and for whom targeted adenosine pathway therapy may prove beneficial (12). High adenosine signaling in baseline tumor samples was associated with shorter overall survival (OS) and progression-free survival (PFS). Those patients who responded to immune checkpoint inhibitor therapy (classified by best overall response) tended to have lower baseline tumor-adenosine-signaling levels compared with those patients who had disease progression following anti–PD-1 therapy (12).
This first-in-human, phase Ia/Ib study evaluated the safety, tolerability, pharmacokinetics (PK), and preliminary efficacy of orally administered AZD4635 in patients with relapsed, refractory solid tumors. The results presented here focus on the dose escalation phase for patients with relapsed or refractory solid tumors, and the dose expansion cohorts of patients with immune-oncology therapy (IO)–naïve metastatic castration-resistant prostate cancer (mCRPC) or microsatellite stable colorectal cancer (MSS-CRC), post-IO non–small cell lung cancer (NSCLC), or other solid tumors (either IO-naïve or post-IO) who were treated with AZD4635 monotherapy or AZD4635 plus durvalumab.
Patients and Methods
Study design
This phase I, open-label, multicenter study enrolled patients with relapsed or refractory solid tumors at 18 sites in the United States. While the trial evaluated multiple therapy combinations in different tumor types, the results presented here focus on the initial dose-finding phase Ia for patients with relapsed or refractory solid tumors, and the expansion phase Ib in patients with IO-naïve mCRPC and colorectal cancer, IO-refractory NSCLC, as well as other solid tumors (IO-naïve or IO-refractory). Patients were assigned to receive either AZD4635 monotherapy or AZD4635 plus durvalumab in the phase Ia/dose-finding phase using a nanosuspension formulation. In phase Ib, expansions included patients with mCRPC and NSCLC who were randomly assigned to receive AZD4635 monotherapy or AZD4635 plus durvalumab. Patients with MSS-CRC were enrolled as 2 separate sequential cohorts treated with AZD4635 monotherapy and AZD4635 plus durvalumab. In addition, patients with other relapsed, refractory solid tumors were enrolled to receive AZD4635 monotherapy in either IO-naïve or IO-refractory cohorts.
Patients were enrolled into 5 cohorts via a Bayesian logistic regression model–based dose escalation design (13) and treated with AZD4635 monotherapy in 3 cohorts (doses of: 125 mg twice daily, 75 mg once daily, or 100 mg once daily), and in 2 cohorts with AZD4635 (doses of: 75 mg once daily or 100 mg once daily) plus durvalumab. For subsequent patients after completing enrollment to phase Ib expansions, a capsule formulation was developed for better tolerability, dose consistency, and overall ease of AZD4635 administration. To evaluate the safety and PK of the capsule formulation, patients were enrolled and treated with AZD4635 capsule monotherapy (75 mg once daily, 150 mg once daily, 200 mg once daily). Patients in the durvalumab combination cohorts received 2 weeks of AZD4635 as monotherapy prior to the first dose of durvalumab, which was given at the FDA-approved dose of 1,500 mg once every 4 weeks. AZD4635 treatment was delayed and reduced for any grade ≥3 and/or clinically significant nonhematologic toxicity event. If such an adverse event (AE) did not resolve to ≤grade 2 within 14 days, AZD4635 treatment was discontinued.
Patients
Patients were ≥18 years of age with relapsed, refractory solid tumors and an Eastern Cooperative Oncology Group (ECOG) performance status of 0–1. Patients in the phase Ia dose escalation combination cohorts had histologic or cytologic confirmation of a solid, malignant tumor (excluding CNS tumors) that was refractory to standard therapies or for which no standard therapies existed.
The phase Ib dose expansion included 8 cohorts. Patients in the prostate cancer cohorts (2 cohorts of up to 40 patients each) had histologically or cytologically confirmed IO-naïve mCRPC. Patients were considered prostate-specific antigen (PSA) evaluable if baseline PSA was ≥1 ng/mL. Patients with mCRPC receiving androgen deprivation therapy with gonadotropin-releasing hormone analogues continued this treatment during the study. Two cohorts of up to 15 patients each with NSCLC (post-IO) were enrolled to receive AZD4635 monotherapy or AZD4635 plus durvalumab. Patients with NSCLC were to have received 1 line of previous therapy with an anti–PD-1/PD-L1 mAb therapy, either alone or in combination, and had either experienced disease progression or responded and then stopped responding. Two cohorts of up to 20 patients each with colorectal cancer (IO-naïve) were enrolled to receive AZD4635 monotherapy or AZD4635 plus durvalumab. Patients with colorectal cancer must have previously received at least 1 prior chemotherapy regimen and experienced subsequent disease progression. Two cohorts of up to 20 patients each with either IO-naïve or post-IO solid tumors were enrolled to receive AZD4635 monotherapy. Finally, patients in the post-IO group had previously received at least 1 line (and not more than 2 lines) of therapy with an anti–PD-1/PD-L1 mAb therapy, either alone or in combination, and had either experienced disease progression or responded and then stopped responding. Patients in the IO-naïve group had received and had disease progression on standard-of-care therapies. A subset of patients with mCRPC, colorectal cancer, or other solid tumors who had ≥1 tumor lesion amenable to biopsy underwent a tumor biopsy during screening and, unless clinically contraindicated, again after 2 weeks of monotherapy treatment.
Patients were excluded if they had received systemic anticancer chemotherapy, small-molecule, biologic, or hormonal agents (except androgen deprivation therapy) within 21 days (or 5 half-lives, whichever was shorter) prior to the first dose of study drug; prior therapy with AZD4635, or any other A2AR antagonist; active or prior documented evidence of autoimmune or inflammatory disorders within 3 years prior to the start of treatment (with the exception of vitiligo, alopecia, hypothyroidism, which was stable on hormone replacement, psoriasis, or eczema that did not require systemic treatment), prior grade ≥3, serious, or life-threatening immune-mediated reactions following previous anti–PD-1 or other immune-oncology therapies. Additional exclusion criteria included treatment with nitrosourea or mitomycin C within 6 weeks of the first dose of study drug; patients could not take prescription or nonprescription medication, or other products known to be sensitive substrates for breast cancer resistance protein/ATP-binding cassette subfamily G member 2 (BCRP/ABCG2) or organic anion transporter 1 (OAT1), or potent inhibitors or inducers of cytochrome P450 family 1 subfamily A member 2 (CYP1A2), which could not be discontinued 2 weeks prior to day 1 of dosing and withheld throughout the study until 2 weeks after the last dose of AZD4635; concomitant medications with another A1R antagonist that would increase the risk of seizure (e.g., theophylline, aminophylline); ongoing treatment with coumadin; major surgery, as defined by the Investigator or Medical Monitor (excluding placement of vascular access) within 4 weeks prior to the first dose of study treatment; radiotherapy with a wide field within 4 weeks, or radiotherapy with a limited field of radiation for palliation within 2 weeks, of the first dose of study treatment; any unresolved toxicities (except alopecia) of grade >1, per common terminology criteria for adverse events (CTCAE), from prior therapy at the time of study commencement were to be discussed with the medical monitor; history or presence of another primary invasive malignancy, except malignancy treated with curative intent with no known active disease ≥2 years before the first dose of study drug, and of low potential risk recurrence; adequately treated nonmelanoma skin cancer, or lentigo maligna without evidence of disease; adequately treated carcinoma in situ without evidence of disease; localized noninvasive primary under surveillance; inadequate bone marrow reserve or organ function (i.e., absolute neutrophil count <1.5 × 109/L; platelet count <100 × 109/L; hemoglobin <9.0 g/dL; alanine aminotransferase (ALT) >2.5 times the upper limit of normal (ULN) if no demonstrable liver metastases, or >5 times ULN if liver metastases are present; aspartate aminotransferase (AST) >2.5 times ULN if no demonstrable liver metastases, or >5 times ULN if liver metastases are present; total bilirubin >1.5 times ULN; creatinine >1.5 times ULN concurrent with creatinine clearance <50 mL/min, as measured or calculated by the Cockcroft and Gault equation; confirmation of creatinine clearance was only required when creatinine was >1.5 times ULN; organ transplantation that required immunosuppressive therapy.
The study was conducted in accordance with the ethical principles of the Declaration of Helsinki and was consistent with the International Conference on Harmonisation/Good Clinical Practice guidelines, and applicable regulatory requirements. The study protocol was approved by an Institutional Review Board or independent ethics committee at each study site prior to study initiation. Prior to participation in the study, all patients provided written informed consent. The study was registered with ClinicalTrials.gov as NCT02740985.
Study endpoints
The primary objective was the safety and tolerability of AZD4635 monotherapy (nanosuspension or capsule) or AZD4635 plus durvalumab. Secondary objectives were the preliminary antitumor activity and PK of AZD4635 monotherapy or AZD4635 plus durvalumab. Endpoints included objective response rate (ORR); PFS; PSA response; and single-dose and multiple-dose PK parameters of AZD4635. Tumor–response assessments were performed by the investigator and were based on Response Evaluation Criteria in Solid Tumors (RECIST) version (v) 1.1 (14). Exploratory pharmacodynamic objectives included the evaluation, by gene expression analyses, of molecular responses in the tumor and in peripheral whole blood that were associated with improved immune response.
Dose-limiting toxicity
In phase Ia (dose escalation), a dose-limiting toxicity (DLT) was defined as any toxicity not attributable to the disease or disease-related processes, occurring prior to the end of cycle 1 (day 21) for the monotherapy group, and before the end of cycle 2 for the combination therapy group. The following were considered as DLTs: neutropenia grade ≥4 present for >7 days, or febrile neutropenia; grade 4 thrombocytopenia, or grade ≥3 associated with bleeding; blood pressure ≥180/110 mm Hg or grade 4 hypertension; grade ≥3 nonhematologic toxicity, including infection (e.g., febrile neutropenia) attributed to AZD4635 administration; increased QTc interval; convulsions, seizures, or stroke; nausea, vomiting, or diarrhea that did not resolve to grade ≤1 within 7 days; increased hepatic transaminases (≥5 to ≤8 × the ULN) that did not resolve to grade 2 within 5 days following onset; transaminase elevation >8 × ULN or total bilirubin >5 × ULN, regardless of duration; any other toxicity that was greater than baseline, was considered to be clinically significant, and/or unacceptable, did not respond to supportive care, resulted in a disruption to the dosing schedule of >14 days, or was judged to be a DLT by the Investigator, Medical Monitor, or the Safety Review Committee. DLT exclusions included alopecia (any grade), lymphopenia (any grade), and isolated laboratory changes (any grade) without clinical sequelae or clinical significance.
All patients with mCRPC had PSA levels assessed at the beginning of each treatment cycle, at the end of study treatment, and at disease progression.
Pharmacokinetics
In phase Ia and Ib, venous blood samples were taken predose and 0.5, 1, 2, 4, 6, 8, and 24 hours postdose after a single dose on day 1 and after multiple doses on day 15 to determine the plasma concentrations of AZD4635 and active metabolite SSP-005174 (with similar potency as AZD4635). In addition, in phase Ia dose escalation cohorts, PK sampling was extended up to 96 hours after a single dose of AZD4635 during the lead-in (cycle 0) to characterize terminal half-life. Concentrations of AZD4635 and its metabolites were determined using a validated bioanalytical method by LabCorp (Covance Laboratories).
Primary PK variables, including area under the plasma concentration (AUClast); maximum plasma drug concentration (Cmax); time to maximum plasma concentration (Tmax); and terminal half-life (t1/2,) were determined using noncompartmental analysis with Phoenix WinNonlin version 6.4 (Certara), using standard methods.
Adenosine signaling signature scoring
Retrospective analysis of PFS in patients with mCRPC, based upon blood adenosine signaling levels, was performed using a 14-gene expression signature score obtained from peripheral blood at baseline. Ninety-five patients with mCRPC and treated with either AZD4635 monotherapy (n = 52) or AZD4635 plus durvalumab combination (n = 43) from the dose escalation and dose expansion cohorts, and who had baseline peripheral blood gene expression data, were scored on the previously published adenosine signaling signature (originally developed in tumor tissues by Sidders and colleagues; ref. 12). The blood-based adenosine signaling score reported here was calculated as the median of normalized, batch-corrected log2 gene expression values across the same 14 genes from the original intratumoral adenosine signature (12): PPARG, CYBB, COL3A1, FOXP3, LAG3, APP, CD81, GPI, PTGS2, CASP1, FOS, MAPK1, MAPK3, and CREB1. Then, the median signature score across patients was used as the cutoff for assigning patients to groups with high versus low levels of blood-based adenosine signaling.
RNA extraction and NanoString gene expression
Clinical patient whole-blood samples collected in PAXgene Blood RNA tubes (PreAnalytiX GmbH) were processed for total RNA using the PAXgene Blood RNA Kit (PreAnalytiX GmbH) per the manufacturer's recommended protocol. RNA concentration and quality were assessed by RNA ScreenTape assays using the TapeStation 2200 System (Agilent). NanoString gene expression assays were performed, and raw count data were processed as described previously (12). Briefly, 25 to 100 ng RNA was hybridized with the 770-gene, off-the-shelf, PCIP panel (NanoString). Background subtraction and normalization of the raw data were performed in nSolver 4.0 software (NanoString) and then the normalized data were log2 transformed. All data used in downstream analyses passed QC using nSolver default parameters.
Statistical analyses
Descriptive statistics were used to summarize patient characteristics, efficacy, PK, and safety data. PFS was analyzed using Kaplan–Meier methodology (15). PFS was defined as the time interval from the first dose of AZD4635 until the date of objective disease progression or death (by any cause in the absence of progression) regardless of whether the patient withdrew from study treatment or received another anticancer therapy prior to progression. A Cox proportional hazards model was used to calculate hazard ratios. NanoString data were corrected for differences in processing and sampling times using ComBat from the sva R package (16) from BioConductor (17). Gene signatures representing a variety of immune cell types and states were collected from the literature (18–24) and scored using either GSVA (25) for pre- versus posttreatment pharmacodynamic analysis or the median for baseline levels of the adenosine signature (12). Differential expression of genes and signatures were determined using repeated measures ANOVA (26) and P values were corrected for false discovery (27) within the R programming language (28). Volcano plot was generated in GraphPad Prism 9 for Windows (GraphPad Software).
The DLT analysis set included all patients who completed >75% of the specified study drug doses during the first 3 weeks of continuous dosing (AZD4635 monotherapy), or first 6 weeks (first 2 cycles) for the combination therapy (AZD4635 plus durvalumab); or all patients who experienced a DLT during the DLT evaluation period. The PK analysis set included patients for whom an adequate PK profile was obtained. The tumor response analysis set included patients with a baseline tumor assessment and measurable disease at baseline. The evaluable for efficacy analysis set included all patients with a baseline tumor assessment. The PSA evaluable set included all patients with PSA levels ≥1 ng/mL. All patients who received ≥1 dose of study medication were included in the safety analysis set. For the safety and other nonefficacy analyses, patients were classified according to the treatment/dose schedule they actually received. For all efficacy analyses, patients were classified according to the planned dose. All data summaries and listings were produced using SAS version 9.4 (SAS Institute, Cary, NC).
Data availability statement
Data underlying the findings described in this article may be obtained in accordance with AstraZeneca's data sharing policy described at: https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure.
Results
Patient demographics and clinical characteristics
As of September 8, 2020, 250 patients with solid tumors had been treated; 161 patients received AZD4635 monotherapy, and 89 patients received AZD4635 plus durvalumab therapy in the cohorts reported in this article (Fig. 1). Demographics and baseline disease characteristics for patients are described in Table 1; Supplementary Tables S1 and S2, and were generally balanced between monotherapy and durvalumab combination cohorts in the phase Ib expansion cohorts including mCRPC. Patients had previously received, and progressed on, standard-of-care therapies. The number of prior regimens for each of the disease cohorts is summarized in Table 1; Supplementary Table S2.
Figure 1.

Patient flow diagram. aDurvalumab was administered at 1,500 mg, once every 4 weeks. Combo, combination therapy; DCO, data cutoff; Durva, durvalumab; IO, immune-oncology therapy; Mono, monotherapy; n, number; QD, once per day; R/R, relapsed/refractory. Patient numbers are based on actual treatment received.
Table 1.
Patient demographics and clinical characteristics for patients with mCRPCa.
| AZD4635 Monotherapy | AZD4635 + Durva combo | |
|---|---|---|
| Characteristic | (n = 65) | (n = 43) |
| Age in years, mean (range) | 71.2 (40–88) | 70.1 (52–82) |
| ECOG, PS, n (%) | ||
| 0 | 25 (38.5) | 15 (34.9) |
| 1 | 40 (65.1) | 27 (62.8) |
| 2 | 0 | 1 (2.3)b |
| Site of disease, n (%) | ||
| Bone | 62 (95.4) | 35 (81.4) |
| Distant lymph nodes | 25 (38.5) | 28 (65.1) |
| Local or regional lymph nodes | 21 (32.3) | 21 (48.8) |
| Lung | 10 (15.4) | 8 (18.6) |
| Liver | 8 (12.3) | 10 (23.3) |
| Otherc: | 20 (30.8) | 12 (27.9) |
| Bladder | 2 (3.1) | 1 (2.3) |
| Pelvis | 0 | 2 (4.7) |
| Peritoneum | 1 (1.5) | 1 (2.3) |
| Number of prior regimens, n (%) | ||
| 1 | 0 | 4 (9.3) |
| 2 | 5 (7.7) | 1 (2.3) |
| 3 | 9 (13.8) | 7 (16.3) |
| ≥4 | 51 (78.5) | 31 (72.1) |
| Prior regimens, median (range) | 5.0 (2–10) | 5.0 (1–10) |
| Prior chemotherapy, n (%) | 42 (64.6) | 26 (60.5) |
| Prior hormonal therapy, n (%) | 65 (100) | 41 (95.3) |
| Prior NHAs, n (%) | 60 (92.3) | 37 (86.0) |
| Prior sipuleucel-T, n (%) | 24 (36.9) | 19 (44.2) |
| Prior radiation | 1 (1.5) | 0 |
Abbreviations: Combo, combination; Durva, durvalumab; ECOG PS, Eastern Cooperative Oncology Group performance status; n, number; NHA, new hormonal agent.
aData presented are from phases Ia and Ib.
bOne patient's ECOG PS changed from 1 at screening to 2 at the date of first dose.
cOther sites of disease listed are those with ≥2 patients across both cohorts.
Safety
In phase Ia, there were 2 patients out of 3 with DLTs in the monotherapy group treated with 125 mg twice daily of the AZD4635 nanosuspension [grade 3 nausea (n = 1) and grade 2 upper abdominal pain (n = 1)]. Based on these events and an observed t1/2 of AZD4635 that was longer than had been predicted based on preclinical studies (29, 30), the dose of AZD4635 was reduced to 75 mg once daily. No patients treated with AZD4635 75 mg or 100 mg once daily nanosuspension monotherapy experienced a DLT. In the phase Ia combination therapy group among 16 evaluable patients, one patient with mCRPC who was treated with 75 mg AZD4635 once daily nanosuspension plus durvalumab experienced 2 DLTs (grade 2 fatigue, grade 2 nausea). The nanosuspension formulation was used for the expansion cohorts at 100 mg once daily for both AZD4635 monotherapy and AZD4635 plus durvalumab. As the capsule formulation became available, AZD4635 doses of up to 200 mg were explored and considered tolerable with no DLTs at any of the doses tested for the capsule formulation. However, only 3 patients in the expansion part transitioned to the capsule prior to the data cut-off date.
The most common treatment-related AEs occurring in >10% of the total patient population who received monotherapy or the total patient population who received combination therapy are presented in Supplementary Table S3. These included nausea, fatigue, vomiting, decreased appetite, dizziness, and diarrhea. Treatment-related AEs of grade 3 or greater severity were observed in 12.9% (19/147) patients treated with AZD4635 monotherapy and in 21.8% (17/78) patients treated with AZD4635 plus durvalumab (Table 2). Immune-related AEs (IRAE) in patients with mCRPC, colorectal cancer, NSCLC, or other solid tumors treated with either AZD4635 monotherapy or AZD4635 plus durvalumab are presented in Supplementary Table S4. IRAEs observed in the AZD4635 plus durvalumab group were similar in classification and magnitude to those previously reported for durvalumab monotherapy (31). In the current study, immune-mediated, treatment-related AEs of grade 3 or greater severity observed with AZD4635 + durvalumab included dermatitis psoriasiform, type I diabetes mellitus (each n = 2), obliterative bronchiolitis, rash, and myositis (each n = 1; Supplementary Table S4).
Table 2.
Causally related adverse events of grade ≥3 severity in patients treated with AZD4635 monotherapy or AZD4635 plus durvalumab combination therapy.
| mCRPC | CRC | NSCLC | Otherb,c | Otherb,d | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| IO-naïvea | IO-naïvea | Post-IOb | IO-naïve | Post-IO | ||||||
| AZD4635 Mono | AZD4635 + Durva | AZD4635 Mono | AZD4635 + Durva | AZD4635 Mono | AZD4635 + Durva | AZD4635 Mono | AZD4635 Mono | AZD4635 Mono Total | AZD4635 + Durva Total | |
| Patients with adverse event, n (%) | (n = 65) | (n = 43) | (n = 25) | (n = 22) | (n = 17c) | (n = 13) | (n = 19) | (n = 21) | (n = 147) | (n = 78) |
| ≥Grade 3 causally related AE | 9 (13.8) | 12 (27.9) | 3 (12.0) | 3 (13.6) | 1 (5.9) | 2 (15.4) | 5 (26.3) | 1 (4.8) | 19 (12.9) | 17 (21.8) |
| Abdominal pain | 1 (1.5) | 0 | 0 | 1 (4.5) | 0 | 0 | 1 (5.3) | 0 | 2 (1.4) | 1 (1.3) |
| Diarrhea | 0 | 0 | 0 | 0 | 0 | 1 (7.7) | 1 (5.3) | 0 | 1 (0.7) | 1 (1.3) |
| Nausea | 1 (1.5) | 2 (4.7) | 0 | 0 | 0 | 0 | 0 | 0 | 1 (0.7) | 2 (2.6) |
| Vomiting | 1 (1.5) | 0 | 1 (4.0) | 0 | 0 | 0 | 0 | 0 | 2 (1.4) | 0 |
| Fatigue | 1 (1.5) | 0 | 1 (4.0) | 1 (4.5) | 1 (5.9) | 0 | 2 (10.5) | 1 (4.8) | 6 (4.1) | 1 (1.3) |
| Hyperglycemia | 0 | 1 (2.3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (1.3) |
| Hypophosphatemia | 0 | 0 | 0 | 1 (4.5) | 0 | 0 | 0 | 0 | 0 | 1 (1.3) |
| Type I diabetes mellitus | 0 | 2 (4.7) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 (2.6) |
| Anemia | 3 (4.6) | 0 | 0 | 0 | 0 | 0 | 0 | 1 (4.8) | 4 (2.7) | 0 |
| Insomnia | 1 (1.5) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (0.7) | 0 |
| Dizziness | 0 | 1 (2.3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (1.3) |
| Headache | 1 (1.5) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (0.7) | 0 |
| Hypertension | 0 | 1 (2.3) | 0 | 0 | 0 | 0 | 2 (10.5) | 0 | 2 (1.4) | 1 (1.3) |
| Dyspnea | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (4.8) | 1 (0.7) | 0 |
| Obliterative bronchiolitis | 0 | 1 (2.3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (1.3) |
| Dermatitis psoriasiform | 0 | 2 (4.7) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 (2.6) |
| Rash | 0 | 1 (2.3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (1.3) |
| Arthralgia | 1 (1.5) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (0.7) | 0 |
| Myositis | 0 | 0 | 0 | 0 | 0 | 1 (7.7) | 0 | 0 | 0 | 1 (1.3) |
| ALT increased | 0 | 0 | 0 | 1 (4.5) | 0 | 0 | 0 | 0 | 0 | 1 (1.3) |
| Blood creatinine increased | 0 | 1 (2.3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (1.3) |
| CRP increased | 1 (1.5) | 1 (2.3) | 0 | 0 | 0 | 0 | 0 | 0 | 1 (0.7) | 1 (1.3) |
| Lipase increased | 0 | 1 (2.3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (1.3) |
| Lymphocyte count decreased | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (4.8) | 1 (0.7) | 0 |
| Neutrophil count decreased | 0 | 1 (2.3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (1.3) |
| Sudden death | 0 | 0 | 1 (4.0) | 0 | 0 | 0 | 0 | 0 | 1 (0.7) | 0 |
Note: Two patients with NSCLC post-IO who were assigned to cohort AZD4635 100 mg once daily + Durva received AZD4635 monotherapy and were therefore counted in the AZD4635 100 mg once daily monotherapy cohort for the safety analysis.
Abbreviations: ALT, alanine aminotransferase; CRC, colorectal carcinoma; CRP, C-reactive protein; Durva, durvalumab; IO, immunotherapy; Mono, monotherapy.
aData are from phase Ia and Ib.
bData are from phase Ib only.
cPrimary tumor site includes n = 3 each breast and uterus; n = 2 each pancreas, soft tissue, ovary, stomach; and n = 1 each bone, esophagus, vulva, anus, salivary gland.
dPrimary tumor site includes n = 6 prostate/(m)CRPC; n = 2 each melanoma, kidney, liver, urinary bladder; and n = 1 each pancreas, soft tissue, bone, esophagus, stomach, head/neck, vulva.
Three patients with mCRPC in the AZD4635 monotherapy group discontinued treatment due to AEs considered possibly related to treatment (fatigue, n = 2; diarrhea, n = 1; nausea, n = 1; vomiting, n = 1). One patient with mCRPC in the AZD4635 plus durvalumab group discontinued durvalumab treatment due to an AE (encephalopathy, n = 1), which was considered to be disease related. There was one patient with colorectal cancer treated with AZD4635 monotherapy who had sudden death 15 days after the last dose of AZD4635, which was considered treatment-related by the investigator; additional details were unavailable.
Pharmacokinetics
The primary PK profile and parameters of AZD4635 and its metabolites following single dose and multiple doses (75 and 100 mg once daily of the nanosuspension; and 75, 150, and 200 mg once daily of the capsule) are presented in Supplementary Fig. S1 and Supplementary Tables S5 and S6, respectively.
AZD4635 appears rapidly in the plasma following a single oral administration—Tmax of 0.5 to 2.0 hours (range, 0.2–6.0 hours). Following Tmax, AZD4635 levels then declined in a biphasic manner, with a median t1/2 of 7.92 to 14.5 hours after administration of a single dose. After a single dose of AZD4635 at either 75 or 100 mg as a nanosuspension, the mean Cmax values were 656.00 and 653.80 ng/mL, and the mean AUClast values were 4,062.50 ng*h/mL and 4,292.84 ng*h/mL, respectively. After a single oral dose of the AZD4635 capsule formulation at either 75, 150, or 200 mg, the mean Cmax values were 428.83, 1,054.43, and 942.57 ng/mL, and the mean AUClast values were 3,328.33 ng*h/mL, 9,292.86 ng*h/mL, and 7,168.57 ng*h/mL, respectively.
Interindividual variabilities in Cmax and AUC values for AZD4635 were high, with overlapping exposures for the 75 and 100 mg nanosuspension, as well as for the 150 and 200 mg capsule formulation. There was a minimal accumulation in exposure (Cmax and AUC) following multiple dosing. Based on unbound AUC, the active metabolite SSP-005174 exposure was about 35% that of AZD4635, and it appeared in the systemic circulation with a median Tmax of 1 hour and was eliminated with a mean t1/2 of approximately 9 hours (reported PK for the 100 mg once daily nanosuspension). Steady-state free AZD4635 plasma concentrations were above the in vitro 50% inhibitory concentration (IC50; 10 nmol/L at 1 μmol/L adenosine) over the dosing interval for AZD4635 doses ≥75 mg once daily (both nanosuspension and capsule; Supplementary Fig. S1). For the 125 mg nanosuspension twice daily dose, steady-state AZD4635 plasma concentration was only available for 1 patient (data not shown). A clinical relative-bioavailability study in healthy volunteers indicated that AZD4635 Cmax and AUC values from the capsule formulation were approximately 27% and 10% higher compared with the nanosuspension (32). Subsequently, the RP2D of the AZD4635 capsule formulation was determined to be 75 mg once daily, either as monotherapy or in combination with durvalumab.
Pharmacodynamics
The pharmacodynamic profile of AZD4635 in peripheral blood (PB) was evaluated in 56 patients with mCRPC across phase Ia and phase Ib cohorts. PB gene expression data were generated for each patient at baseline and after monotherapy AZD4635 treatment (2–4 weeks). Gene set variation analysis (GSVA) was used to evaluate changes in immune-related gene expression signatures relative to baseline. The analysis included 144 gene expression signatures, all with at least 60% gene coverage. After correcting for multiple testing, 6 signatures were significantly modulated [q < 0.05, false discovery rate (FDR)] with signatures for natural killer T cells (NKT) and plasmacytoid dendritic cells (pDC) decreasing and signatures for tertiary lymphoid structure associated chemokines, T helper 17 cells (Th17), immunosuppressive tumor-associated macrophage (TAM), and innate lymphoid cells group 3 (ILC3) increasing, relative to baseline (Supplementary Fig. S2; Supplementary Table S7). These data indicate that AZD4635 treatment modulates gene expression signatures associated with both innate and adaptive immune responses in peripheral blood. Furthermore, we show that the IFNγ and the T helper 1 cell (Th1) signatures, both of which were previously reported to have increased expression in tumor biopsies post-AZD4635 treatment (4, 12), trend toward increased expression in the peripheral blood following AZD4635 treatment (Supplementary Table S7).
Efficacy
The best percent changes from baseline in target lesion sizes for all patients enrolled in phase Ia (with measurable disease at baseline) are shown in Fig. 2A. Three patients with prostate cancer in phase Ia had objective responses: 1 complete response (CR) and 2 partial responses (PR). In addition, 3 patients [with bladder cancer, chondrosarcoma, and head and neck tumors (each, n = 1)] who received combination treatment had long-term stable disease (>1 to >3.5 years).
Figure 2.

A–C, Best percentage change from baseline in phase Ia patients with measurable disease at baseline (A); best percentage change from baseline in patients with mCRPC and measurable disease at baseline (B); and PSA responses relative to baseline over time in patients with IO-naïve mCRPC treated with AZD4635 + durvalumab (C). Durva, durvalumab; IO, immune-oncology therapy; Mono, AZD4635 monotherapy; RECIST v1.1, Response Evaluation Criteria In Solid Tumors version 1.1. Best objective response was based on the investigator-assessed RECIST v1.1 response at each tumor assessment. Target lesion sum of diameters are scaled up if ≤1/3 of measurements are missing. If >1/3 of measurements are missing, then the percentage change is not calculated. * and Δ indicate that the patient is still on treatment. 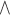 Percentage change from baseline in tumor lesion size exceeds + 100%; waterfall plot was truncated for the patient at this point. PSA response is defined as ≥50% reduction from baseline. Spider plot is truncated for some patients where PSA relative to baseline exceeds 5.
Percentage change from baseline in tumor lesion size exceeds + 100%; waterfall plot was truncated for the patient at this point. PSA response is defined as ≥50% reduction from baseline. Spider plot is truncated for some patients where PSA relative to baseline exceeds 5.
In the phase Ib expansion cohorts, objective responses were observed in patients with mCRPC. No objective responses were observed in patients with NSCLC or colorectal cancer. The best responses observed in colorectal cancer and NSCLC patients treated with monotherapy or combination with durvalumab were stable disease after a minimum of 2 dosing cycles. These were seen in both IO-naïve and IO-treated patients. In patients with NSCLC, 5 (33.3%) patients who received AZD4635 monotherapy, and 4 (26.7%) patients who received AZD4635 plus durvalumab, had a best response of SD with disease control at 22 weeks (3 scans) seen in 1 (6.7%) patient treated with monotherapy and 3 (20%) patients treated with AZD4635 plus durvalumab (Supplementary Table S8). In patients with colorectal cancer, 9 (45.0%) who received AZD4635 monotherapy, and 9 (47.4%) patients who received AZD4635 plus durvalumab, had a best response of stable disease (SD) after a minimum of 2 cycles of therapy, according to RECIST v1.1, with disease control at 22 weeks observed in 5 patients (20%) treated with AZD4635 plus durvalumab. In patients with other solid tumors who received AZD4635 monotherapy, 4 (21.1%) patients who were IO-naïve and 6 (28.6%) patients who had previously received IO achieved SD at after a minimum of 2 cycles of therapy, according to RECIST v1.1.
For patients with mCRPC in both phase Ia and phase Ib, the median (minimum, maximum) treatment duration of AZD4635 was 1.45 (0.07, 21.39) months for monotherapy and 3.22 (0.56, 36.14) months for combination therapy. The median (minimum, maximum) number of cycles of durvalumab was 3.0 (1.0, 35.0). The time on treatment and best objective response for each patient is shown for IO naïve patients with mCRPC receiving AZD4635 monotherapy or AZD4635 plus durvalumab in Fig. 3A and B, respectively.
Figure 3.

Time from first dose to end of treatment for IO-naïve patients with mCRPC for monotherapy cohort (A), and combination cohort (B) (safety analysis set). CR, complete response; IO, immune-oncology therapy; PR, partial response.
In the tumor response–evaluable population for patients with mCRPC (n = 76), the ORR in the AZD4635-monotherapy group was 5.1% [95% confidence interval (CI), 0.6–17.3; 2 confirmed PR among 39 patients] versus 16.2% (95% CI, 6.2–32.0; 2 confirmed PR; 4 confirmed CR among 37 patients) for those in the AZD4635-plus-durvalumab group. The best changes from baseline in target lesion sizes are shown in Fig. 2B. CR was observed in 2 of 37 (5.4%) patients who received AZD4635 plus durvalumab. The average baseline alkaline phosphatase was 108 IU/L in responders versus 138 IU/L in nonresponders. All mCPRC responders had lymph node involvement and some also had bone or visceral involvement (e.g., lung and liver). Absolute target lesion diameter among responders ranged from 18 to 102 mm (Supplementary Table S9). Because of the small numbers of patients, it is difficult to draw any conclusions regarding the correlation between treatment response and these disease characteristics.
Thirty-two patients in the mCRPC cohort had SD as their best response after a minimum of 2 treatment cycles: 14 (35.9%) in the AZD4635-monotherapy group, and 18 (48.6%) in the AZD4635-plus-durvalumab group. Duration of response ranged from 2.3 to 34.0 months; 9 patients were on study at the data cut-off date (September 8, 2020): AZD4635-monotherapy cohort: 5 of 63 (8.1%) patients; AZD4635-plus-durvalumab cohort: 4 of 45 (8.9%) patients. The disease control rate (DCR; defined as the percentage of patients who have achieved best objective response of CR, PR, or SD for at least 22 weeks) was 18.0% in the AZD4635-monotherapy group and 33.3% in the AZD4635-plus-durvalumab group (Supplementary Table S8).
Evaluation of the PSA response over time demonstrated that 3 of 60 (5.0%; 95% CI, 1.0–13.9) patients in the AZD4635-monotherapy group and 10 of 45 (22.2%; 95% CI, 11.2−37.1) patients in the AZD4635-plus-durvalumab group, had a PSA50 response (i.e., a decline of at least 50% in PSA levels from baseline; Fig. 2C). Confirmed PSA responses were observed in 2 of 60 (3.3%; 95% CI, 0.41−11.53) patients in the AZD4635-monotherapy group and 7 of 45 (15.6%; 95% CI, 6.49−29.46) patients in the AZD4635-plus-durvalumab group (Supplementary Table S10). In the AZD4635-plus-durvalumab group, 8 of the 10 patients with PSA responses received prior sipuleucel-T, whereas none of the PSA responders in the AZD4635-monotherapy group received prior sipuleucel-T.
Median PFS was 10.1 weeks (95% CI, 6.3–15.3) and 14.9 weeks (95% CI, 8.7–29.3) for patients with mCRPC receiving AZD4635 monotherapy or AZD4635 plus durvalumab, respectively (Fig. 4A). Analysis of median PFS by peripheral whole-blood adenosine-signaling signature is shown in Fig. 4B. Patients whose blood adenosine-signaling signature was greater than the median value (termed “high”; using a similar method to that in ref. 12) had a numerically longer median PFS of 21 weeks (95% CI, 14.1–34.4) compared with patients whose blood adenosine-signaling signature was lower than the median (termed “low”) with a median PFS of 8.7 weeks (95% CI, 6.3–14.7). The PFS probability at 24 weeks was 48.9% (95% CI, 31.5–64.2) in patients with a high blood-adenosine signature versus 20.6% (95% CI, 9.4–34.8) in patients with a low blood-adenosine signature.
Figure 4.

Progression-free survival for patients with mCRPC by treatment (A), and by adenosine level (B) (efficacy analysis set). CI, confidence interval; combo, AZD4635 plus durvalumab combination therapy; Durva, durvalumab; HR, hazard ratio; IO, immune-oncology therapy; Mono, AZD4635 monotherapy. Patients not known to have died or progressed are censored at their last evaluable investigator overall response assessment. The plots are truncated at week 75. Two patients in the adenosine high group and the adenosine low group remained progression free after week 75.
Discussion
This first-in-human phase Ia/b study evaluated the safety, tolerability, PK, and preliminary clinical activity of the oral A2AR antagonist, AZD4635, as monotherapy and in combination with the anti–PD-L1 monoclonal antibody, durvalumab. The results presented here focus on initial dose-finding results in patients with relapsed/refractory solid tumors and the expansion phase in patients with IO-naïve mCRPC or colorectal cancer, post-IO NSCLC, or other solid tumors (IO-naïve or post-IO).
AZD4635 was well tolerated both as a monotherapy and in combination with durvalumab in all patients, with nausea and fatigue being among the most frequently reported AEs related to study treatment. Notably, the incidence of treatment-related AEs of grade 3 or greater severity increased from 12.9% with AZD4635 monotherapy to 21.8% with the addition of durvalumab. The increased incidence was observed across a range of immune and nonimmune mediated AEs. The percentage of grade 3−4 treatment-related events observed with durvalumab monotherapy in the PACIFIC study was 11.8% (33), so there may be an additive effect when combining the two immunotherapies. For the initial dose-finding and expansion phases, AZD4635 was evaluated initially as a nanosuspension and later switched to a capsule formulation, developed for dose consistency and improved ease of administration. The relative bioavailability, PK, and tolerability profiles of the AZD4635 75 mg once daily capsule formulation were comparable with those of the 100 mg once daily nanosuspension formulation, supporting the switch to the capsule formulation for the latter part of the study and for future clinical trials with AZD4635, although only a small number of the patients reported here were treated with capsules. Overall, AZD4635 exhibited a linear and dose-proportional PK profile, with steady-state trough concentrations above IC50 at doses ≥75 mg once daily. In addition, the t1/2 of approximately 14 hours (range, 11–24 hours) supported the once-daily dosing regimen. Based on receptor occupancy data from the brains of nonhuman primates following positron emission tomography imaging and PK/pharmacodynamic modeling, AZD4635 doses above 75 mg once daily are predicted to provide about 90% A2AR occupancy in the tumor microenvironment (assuming similarity of occupancy in tumor versus brain at steady state; ref. 29). AZD4635 is primarily metabolized by CYP1A2, based on a clinical drug–drug interaction study with the strong CYP1A2 inhibitor fluvoxamine (34). Variability among individuals in expression and activity of CYP1A2 is high, and various factors contribute to differences in the pharmacokinetics of CYP1A2 substrates including sex, race, smoking history, and genetic polymorphisms (34). In the current study, high interindividual variability in AZD4635 pharmacokinetics was observed, with overlapping exposures at 75 and 100 mg nanosuspension, as well as at 150 and 200 mg capsule formulation.
Efficacy data demonstrated that 8 heavily pretreated (i.e., ≥4 prior therapies) patients with mCRPC achieved a confirmed objective response: 2 patients achieved a PR with AZD4635 monotherapy, while 2 patients achieved a CR and 4 patients achieved a PR with AZD4635 plus durvalumab (duration of response ranged from 2.3 to 34.0 months). Other ICI therapies (either as monotherapy or in combination) have been evaluated in patients with mCRPC: phase III clinical trials evaluating the anti–CTLA-4 antibody, ipilimumab, or the anti–PD-L1 antibody, atezolizumab, failed to demonstrate an OS benefit in patients with mCRPC (35–37). In a phase II study of pembrolizumab in patients with mCRPC, the ORR was 5% (9/199 patients; ref. 38). In a phase II study of nivolumab in combination with ipilimumab in patients with mCRPC, the ORRs were 25% (8/32) and 10% (3/30), respectively, for patients with mCRPC who were chemotherapy-naïve and chemotherapy-exposed (39). AZD4635 plus durvalumab resulted in a longer PFS in patients with mCRPC compared with AZD4635 monotherapy (median, 14.9; 95% CI, 8.7–29.3 vs. 10.1; 95% CI, 6.3–15.3 weeks, respectively), with a PFS probability at 24 weeks of 41.8% (95% CI, 25.9–56.9) vs. 24.2% (95% CI 12.1–38.7), respectively. Patients with mCRPC treated with approved NHAs or cabazitaxel in this late line showed a median PFS ranging from 1.4 to 4.4 months (40, 41), which suggests that the treatment is generally consistent with previously observed PFS. Furthermore, 22.2% of patients in the AZD4635-plus-durvalumab group had a PSA50 response compared with 5.0% of patients in the AZD4635-monotherapy group. In patients with colorectal cancer, although no objective tumor responses were observed, 6 patients in the AZD4635-plus-durvalumab group remained on study for ≥6 months. In contrast, patients with NSCLC who had previously progressed after an anti–PD-1/anti–PD-L1–containing therapy had limited evidence of clinical benefit with AZD4635 monotherapy or AZD4635 plus durvalumab.
Targeting the tumor microenvironment to reduce adenosine levels or to inhibit adenosine signaling, via blockade of A2AR, has been explored as a promising approach to improve antitumor immune responses (4, 8). In addition to AZD4635, other agents in clinical trials targeting A2AR include ciforadenant (NCT03454451 and NCT02655822), AB928 (NCT04381832 and NCT03629756), NIR178 (NCT03549000 and NCT03207867), and EOS100850 (NCT03873883). Like AZD4635, these other adenosine-receptor inhibitors were generally well tolerated in phase I and II studies (42). The most common treatment-related AEs for ciforadenant were fatigue, nausea, and pruritus (42, 43). Grade ≥3 treatment-related AEs for NIR178 monotherapy included nausea, increased AST, ALT, and lipase levels, and pneumonitis at the highest doses (480 and 640 mg; refs. 42, 44), while no grade ≥3 treatment-related AEs occurred in patients who received EOS100850 up to doses of 160 mg twice daily (42, 45). Taken together, these findings suggest an overall favorable safety profile for this class of agents.
Although these agents have generally been well tolerated, blockade of adenosine-receptor signaling has shown variable clinical efficacy with different agents. Efficacy of ciforadenant was observed in clinical trials in patients with renal cell carcinoma (RCC) and mCRPC. In a pretreated population of patients with RCC, Fong and colleagues reported that 3% of patients had a PR with ciforadenant monotherapy versus 11% of patients treated with ciforadenant plus atezolizumab (43). Harshman and colleagues reported that 5 of 33 patients with mCRPC and treated with ciforadenant, with or without atezolizumab, had tumor regression, and 1 patient in the combination cohort had a PR (46). In phase I trials, EOS100850 had a best response of SD in 6 patients (45). Furthermore, the dual A2AR/A2B receptor antagonist, AB928 in combination with carboplatin, pemetrexed, and pembrolizumab resulted in a PR in 3 of 6 evaluable patients with advanced NSCLC (47), while NIR178 monotherapy, when given to 17 evaluable patients with advanced NSCLC, resulted in 1 CR and 1 PR (both patients were IO-naïve; ref. 44). Our findings with AZD4635 compare somewhat favorably to those from other adenosine receptor antagonists.
Given the variable response to targeting A2AR, predictive biomarkers to identify patients who may benefit most from treatment are needed. Using a blood-based adenosine-signaling gene expression signature originally derived from tumor tissue by Sidders and colleagues (12), we observed that patients with mCRPC with a high blood-adenosine signature score had a numerically longer PFS compared with patients with a low blood-adenosine signature score (medians of 21.0 vs. 8.7 weeks, respectively), and a PFS probability at 24 weeks of 48.9% versus 20.6%, respectively. The observed PFS in patients with a high peripheral blood-adenosine signature score compares favorably with available therapies in later-line treatment of patients with mCRPC [e.g., cabazitaxel, with a median PFS of 3.9 (95% CI, 3.5–4.6) months; ref. 48]. These preliminary findings suggest that adenosine signaling in peripheral blood may be a potential predictive biomarker for patients with mCRPC; however, additional studies are needed to confirm these observations.
Importantly, gene expression in this study was measured from peripheral blood rather than tumor tissue. We applied this method based on the knowledge that A2AR is predominantly expressed in not only tumor-infiltrating but also peripherally circulating immune cells and that a blood-based assay, due to relative ease of collection and availability of patient blood samples, would potentially be more scalable than a tumor-based assay. As the genes chosen for this signature were confined to those present in the commercially available 770-gene PanCancer Immune Profiling (PCIP) gene-expression panel (NanoString), use of a broader panel of transcripts may potentially enhance sensitivity (12). In addition, as gene signature changes were evaluated in peripheral blood alone and not linked to changes in the tumor tissue collected in this study, no definitive conclusions can be drawn. To our knowledge, the data reported herein represent the first application of the 14-gene panel developed by Sidders and colleagues (12) to categorize levels of adenosine signaling in patients’ peripheral blood. Further refinement of the signature and the identification of an optimal cut-off value may permit the prospective selection of patients. Consequently, evaluation of additional biomarkers of response is ongoing.
Finally, in the publication from Sidders and colleagues (12) describing characterization of the intratumoral adenosine signature via the same 14-gene-expression panel, 7 patients from the current study treated with AZD4635 nanosuspension (75 mg, n = 6; 100 mg, n = 1) underwent paired tumor biopsies prior to treatment, following 2 weeks of monotherapy and had data available for analysis. RNA analysis using the PCIP NanoString panel confirmed that in 5 of 7 patients, intratumoral adenosine signaling decreased; 4 of this group also had concordant increases in gene-expression signatures of cytolytic activity and IFNG signaling (12). This suggests that at the RP2D, effects on markers of adenosine signaling and other immune pathways are impacted (12).
In conclusion, AZD4635, either as a monotherapy or in combination with durvalumab, was well tolerated with no major safety concerns, and was associated with preliminary evidence of clinical activity in patients with mCRPC (30). Although higher doses of AZD4635 were tolerated (i.e., 150 and 200 mg once daily), the PK and safety profiles of AZD4635 support once-daily dosing at the RP2D of 75 mg once daily for the capsule formulation and 100 mg once daily for the nanosuspension. Patient selection using a 14-gene-expression signature to measure baseline peripheral blood-adenosine signaling levels may facilitate optimal patient-specific treatment strategies for adenosine-pathway modulation. Refinement and validation of this signature is ongoing.
These findings support phase II studies of AZD4635 combination therapy in patients with mCRPC. Several chemotherapies lead to increased extracellular adenosine and, thus combining AZD4635 with chemotherapy may lead to greater immune activation, reduced tumor growth, and improved patient outcomes (49, 50). A phase II study to assess the safety and efficacy of AZD4635 plus durvalumab and AZD4635 plus oleclumab in patients with prostate cancer (NCT04089553; ref. 51) and a phase II nonrandomized study to assess the efficacy and safety of AZD4635 plus durvalumab ± cabazitaxel in patients with progressing mCRPC (NCT04495179) are ongoing (52).
Supplementary Material
Acknowledgments
The authors would like to thank all patients and their families, and the study site staff at each study center for their participation. The authors would also like to thank Lara McGrath at AstraZeneca, for bioinformatics assistance; Lindsey Jung, Sudhakar Yelchuri, Jun Tian, Jack Field, and Grace Lu for biostatistical analysis; Ganesh Mugundu for PK analysis and design; Bolan Linghu for bioinformatic data analysis; and Melinda Merchant for assistance with study design and data interpretation. Medical writing and editorial support, conducted in accordance with Good Publication Practice 3 (GPP3) and the International Committee of Medical Journal Editors (ICMJE) guidelines, were provided by Eli Berdougo, PhD, of Oxford PharmaGenesis Inc., Newtown, PA, and funded by AstraZeneca, Gaithersburg, MD.
This study was sponsored by AstraZeneca.
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Authors' Disclosures
E.A. Lim reports other support from Pfizer outside the submitted work. J.C. Bendell reports other support from Gilead, Genetech/Roche, BMS, Five Prime, Lilly, Merck, MedImmune, Celgene, EMD Serono, Taiho, Macrogenics, GSK, Novartis, OncoMed, LEAP, TG Therapeutics, AstraZeneca, BI, Bayer, Daiichi Sankyo, Incyte, Apexigen, Koltan, Forty Seven, AbbVie, SynDevRex, Array, Onyx, Sanofi, Takeda, Celldex, Agios, Eisai, Nektar, ARMO, Cytomx, Ipsen, Merrimack, Tarveda, Boston Biomedical, Tyrogenex, Oncogenex, Marshall Edwards, Pieris, Mersana, Blueprint, Calithera, Evelo, MERUS, Jacobio, FORMA, Novocare, Arrys, Tracon, Effector, Sierra, Innate, Arch Oncology, Prelude Oncology, Vyriad, Harpoon, ADC, Unum Therapeutics, Angem, Pfizer, Imclone, Millennium, Rgenix, Bellicum, Acerta Pharma, Arcus Bio, Seattle Genetics, Gossamer Bio, Shattuck Labs, TempestTx, Synthorx Inc, Revolution Medicines, Inc., Bicycle Therapeutics, Relay Therapeutics, Scholar Rock, Zymeworks, NGM Biopharma, Beigene, CALGB, Stemcentrx, Cyteir Therapeutics, Foundation Bio, Innate Pharma, Morphotex, OncXerna, NuMab, AtlasMedx, IGM Biosciences, Mabspace, Treadwell Therapeutics, REPARE Therapeutics, Hutchinson MediPharma, Regeneron, PureTech Health, NeoImmune Tech, Molecilar Partners, Innate, Phoenix Bio, Torque, Tizona, Janssen, Tolero, Amgen, Moderna Therapeutics, Agios, Continuum Clinical, Fusion Therapeutics, and Samsung Bioepios during the conduct of the study, as well as other support from Gilead, BMS, Five Prime, Genentech/Roche, Lilly, Merck, Celgene, MedImmune, Taiho, Macrogenics, GSK, EMD Serono, Novartis, Oncomed, LEAP, TG Therapeutics, AstraZeneca, BI, Bayer, Daiichi Sankyo, Incyte, Koltan, FortySeven, AbbVie, Array, Apexigen, Onyx, Sanofi, Takeda, Eisai, Celldex, Agios, Nektar, Cytomx, ARMO, Ipsen, Merrimack, Boston Biomedical, Tarveda, Oncogenex, Tyrogenex, Pieris, Mersana, Blueprint, Marshall Edwards, Evolo, FORMA, Merus, Calithera, Jacobio, NovoCare, Arrys, Effector, Tracon, Sierra, Innate, Arch Oncology, Unum Therapeutics, Prelude Oncology, Harpoon, ADC, Amgen, Pfizer, Vyriad, Imclone, Acerta Pharma, Millennium, Bellicum, Rgenix, Arcus Bio, Seattle Genetics, Gossamer Bio, Shattuck Labs, TempestTx, Synthorx Inc, Revolution Medicines, Inc., Zymeworks, Relay Therapeutics, Bicycle Therapeutics, Scholar Rock, NGM Biopharma, Beigene, CALGB, Cyteir Therapeutics, Stemcentrx, Foundtion Bio, Innate Pharma, Morphotex, NuMab, AtlasMedx, OncXerna, IGM Biosciences, Mabspace, Treadwell Therapeutics, REPARE Therapeutics, Hutchinson MediPharma, Regeneron, PureTech Health, NeoImmune Tech, Molecular Partners, Innate, Phoenix Bio, Torque, Tizona, Janssen, Tolero, Amgen, Moderna Therapeutics, Agios, Moderna Therapeutics, Fusion Therapeutics, and Samsung Bioepios outside the submitted work. G.S. Falchook reports grants from AstraZeneca during the conduct of the study, a patent for copyright with Wolters Kluwer with royalties paid, and royalties (self) from Wolters Kluwer (2014–present). G.S. Falchook also reports the following relationships: advisory role (to institution) with Fujifilm (2018), Silicon (2020, 2021), Navire (2021), Turning Point (2021), Predicine (2021), Inspirna (2021), and Regeneron (2021); advisory role (self) with EMD Serono (2010, 2011); speakers honorarium for CME from Total Health Conferencing (2019) and Rocky Mountain Oncology Society (2020); travel (self, for work and/or research related to institution) from Bristol-Myers Squibb (2015), EMD Serono (2011, 2012, 2013), Fujifilm (2018), Millennium (2013), and Sarah Cannon Research Institute (employer, at least once yearly); and research funding (to institution, for any trial for which G.S. Falchook has been the PI [ever] or subinvestigator [minimum last 4 years]) from 3-V Biosciences, Abbisko, AbbVie, ABL Bio ADC Therapeutics, Accutar, Aileron, American Society of Clinical Oncology, Amgen, ARMO/Eli Lilly, Artios, AstraZeneca, BeiGene, Bioatla, Bioinvent, Biothera, Bicycle, Black Diamond, Boehringer Ingelheim, Celldex, Celgene, Ciclomed, Curegenix, Curis, Cyteir, Daiichi, DelMar, eFFECTOR, Eli Lilly, EMD Serono, Epizyme, Erasca, Exelixis, Freenome, Fujifilm, Genmab, GlaxoSmithKline, Hutchison MediPharma, IGM Biosciences, Ignyta, ImmunoGen/MacroGenics, Incyte, Jacobio, Jounce, Jubilant, Kolltan, Loxo/Bayer, MedImmune, Millennium, Merck, miRNA Therapeutics, Molecular Templates, National Institutes of Health, Navire, NiKang, Novartis, OncoMed, Oncorus, Oncothyreon, Poseida, Precision Oncology, Prelude, PureTech, Pyramid, RasCal, Regeneron, Relay, Rgenix, Ribon, Samumed, Sapience, Seagen, Silicon, Simcha, Sirnaomics, Strategia, Syndax, Synthorx/Sanofi, Taiho, Takeda, Tarveda, Teneobio, Tesaro, Tocagen, Turning Point, UT MD Anderson Cancer Center, Vegenics, and Xencor. T.M. Bauer reports grants and personal fees from AstraZeneca during the conduct of the study, and grants and personal fees from Pfizer, Lilly, Bayer, and BMS outside the submitted work. C.G. Drake reports being an employee at Janssen Research and Development. D.J. George reports personal fees from Advanced Accelerator Applications, American Association for Cancer Research, Eisai, IdeoOncology, Medscape Education, Merck Sharp & Dohme, Michael J Hennessey Assoc, Millennium Medical Publishing, Myovant Sciences, NCI GU Steering Committee, Propella Therapeutics, RevHealth, LLC, Seattle Genetics, UroGPO, WebMD, and Xcures; grants and personal fees from Astellas, AstraZeneca, Janssen Pharma, and Pfizer; grants, personal fees, and nonfinancial support from AVEO Pharmaceuticals, Bayer H/C Pharma, Exelixis, and Sanofi; and grants from BMS, Calithera, and Novartis outside the submitted work. J.L. Karlix reports other support from AstraZeneca during the conduct of the study. S.V. Ulahannan reports grants from Array, Incyte, Bayer, Syros, Eisai, AstraZeneca, AbbVie, Inc., Adlai Nortye, ArQule, Inc., Atreca, Boehringer Ingelheim, Bristol-Myers Squibb, Celgene Corporation, Ciclomed LLC, Erasca, Evelo Biosciences, Inc., Exelexis, G1 Therapeutic, Inc., GlaxoSmithKline GSK, IGM Biosciences, Isofol, Klus Pharma, Inc., Macrogenics, Merck Co, Inc., Mersana Therapeutics, OncoMed Pharmaceuticals, Pfizer, Regeneron, Inc., Revolution Medicines, Inc., Synermore Biologics Co., Takeda, Tarveda Therapeutics, Tesaro, Tempest, and Vigeo Therapeutics, Inc. during the conduct of the study, grants from Array, Incyte, Bayer, Syros, Eisai, AstraZeneca, AbbVie, Inc., Adlai Nortye, ArQule, Inc., Atreca, Boehringer Ingelheim, Bristol-Myers Squibb, Celgene Corporation, Ciclomed LLC, Erasca, Evelo Biosciences, Inc., Exelexis, G1 Therapeutic, Inc., GlaxoSmithKline GSK, IGM Biosciences, Isofol, Klus Pharma, Inc., Macrogenics, Merck Co, Inc., Mersana Therapeutics, OncoMed Pharmaceuticals, Pfizer, Regeneron, Inc., Revolution Medicines, Inc., Synermore Biologics Co., Takeda, Tarveda Therapeutics, Tesaro, Tempest, and Vigeo Therapeutics, Inc. outside the submitted work. K.F. Sachsenmeier reports other support from AstraZeneca outside the submitted work, as well as a patent for 17/049,739, 2021 pending. D.L. Russell reports personal fees and other support from AstraZeneca during the conduct of the study and personal fees from AstraZeneca outside the submitted work. G. Moorthy reports other support from AstraZeneca during the conduct of the study and other support from AstraZeneca outside the submitted work. B. Sidders reports other support from AstraZeneca Ltd during the conduct of the study, other support from AstraZeneca Ltd outside the submitted work, as well as a patent for WO2021105232A1 issued. E.A. Pilling reports personal fees and other support from AstraZeneca outside the submitted work. H. Chen reports personal fees from AstraZeneca outside the submitted work. M.M. Hattersley reports personal fees from AstraZeneca outside the submitted work. M. Das reports other support from AstraZeneca outside the submitted work. R. Kumar reports other support from AstraZeneca outside the submitted work. G.P. Pouliot reports personal fees from AstraZeneca outside the submitted work. M.R. Patel reports other support from AstraZeneca outside the submitted work. No disclosures were reported by the other authors.
Authors' Contributions
E.A. Lim: Data curation, writing–review and editing. J.C. Bendell: Conceptualization, data curation, writing–review and editing. G.S. Falchook: Data curation, writing–review and editing. T.M. Bauer: Data curation, writing–review and editing. C.G. Drake: Data curation, writing–review and editing. J.H. Choe: Data curation, writing–review and editing. D.J. George: Writing–review and editing. J.L. Karlix: Conceptualization, writing–review and editing. S. Ulahannan: Data curation, formal analysis, writing–review and editing. K.F. Sachsenmeier: Conceptualization, data curation, formal analysis, methodology, writing–review and editing. D.L. Russell: Data curation, writing–review and editing. G. Moorthy: Data curation, formal analysis, methodology, writing–review and editing. B.S. Sidders: Formal analysis, methodology, writing–review and editing. E.A. Pilling: Formal analysis, methodology, writing–review and editing. H. Chen: Formal analysis, writing–review and editing. M.M. Hattersley: Formal analysis, writing–review and editing. M. Das: Formal analysis, writing–review and editing. R. Kumar: Writing–review and editing. G.P. Pouliot: Conceptualization, formal analysis, methodology, writing–review and editing. M.R. Patel: Data curation, formal analysis, writing–review and editing.
References
- 1. Sek K, Molck C, Stewart GD, Kats L, Darcy PK, Beavis PA. Targeting adenosine receptor signaling in cancer immunotherapy. Int J Mol Sci 2018;19:3837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vigano S, Alatzoglou D, Irving M, Menetrier-Caux C, Caux C, Romero P, et al. Targeting adenosine in cancer immunotherapy to enhance T-cell function. Front Immunol 2019;10:925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Blay J, White TD, Hoskin DW. The extracellular fluid of solid carcinomas contains immunosuppressive concentrations of adenosine. Cancer Res 1997;57:2602–5. [PubMed] [Google Scholar]
- 4. Bendell J, Bauer T, Patel M, Falchook G, Karlix JL, Lim E, et al. Evidence of immune activation on the first-in-human phase 1a dose escalation study of the adenosine 2a receptor antagonist AZD4635, in patients with advanced solid tumors. Cancer Res 2019;79: 13s (suppl. abstr.CT026). [Google Scholar]
- 5. Allard B, Allard D, Buisseret L, Stagg J. The adenosine pathway in immuno-oncology. Nat Rev Clin Oncol 2020;17:611–29. [DOI] [PubMed] [Google Scholar]
- 6. Kong HY, Byun J. Emerging roles of human prostatic Acid phosphatase. Biomol Ther 2013;21:10–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gao ZG, Jacobson KA. A2B adenosine receptor and cancer. Int J Mol Sci 2019;20:5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Borodovsky A, Barbon CM, Wang Y, Ye M, Prickett L, Chandra D, et al. Small molecule AZD4635 inhibitor of A2AR signaling rescues immune cell function including CD103(+) dendritic cells enhancing anti-tumor immunity. J Immunother Cancer 2020;8:e000417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Borodovsky A, Wang Y, Ye M, Deng N, Shaw JC, Sachsenmeier K, et al. Inhibition of A2AR by AZD4635 induces anti-tumor immunity alone and in combination with anti-PD-L1 in preclinical models. Cancer Res 2018;78:13s, (suppl. abstr 3751). [Google Scholar]
- 10. Barbon CM, Borodovsky A, Wang Y, Prickett L, Sachsenmeier K, Schuller A, et al. The A2AR antagonist AZD4635 prevents adenosine-mediated immunosuppression of CD103+ dendritic cells. Cancer Res 2019;79:13s (suppl. abstr LB-192). [Google Scholar]
- 11. Stewart R, Morrow M, Hammond SA, Mulgrew K, Marcus D, Poon E, et al. Identification and characterization of MEDI4736, an Antagonistic Anti-PD-L1 monoclonal antibody. Cancer Immunol Res 2015;3:1052–62. [DOI] [PubMed] [Google Scholar]
- 12. Sidders B, Zhang P, Goodwin K, O'Connor G, Russell DL, Borodovsky A, et al. Adenosine signaling is prognostic for cancer outcome and has predictive utility for immunotherapeutic response. Clin Cancer Res 2020;26:2176–87. [DOI] [PubMed] [Google Scholar]
- 13. Neuenschwander B, Branson M, Gsponer T. Critical aspects of the Bayesian approach to phase I cancer trials. Stat Med 2008;27:2420–39. [DOI] [PubMed] [Google Scholar]
- 14. Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009;45:228–47. [DOI] [PubMed] [Google Scholar]
- 15. Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Am Statist Assoc 1958;53:457–81. [Google Scholar]
- 16. Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012;28:882–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat Methods 2015;12:115–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Danaher P, Warren S, Dennis L, D'Amico L, White A, Disis ML, et al. Gene expression markers of tumor infiltrating leukocytes. J Immunother Cancer 2017;5:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015;160:48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Prat A, Navarro A, Pare L, Reguart N, Galvan P, Pascual T, et al. Immune-related gene expression profiling after PD-1 blockade in Non-small cell lung carcinoma, head and neck squamous cell carcinoma, and melanoma. Cancer Res 2017;77:3540–50. [DOI] [PubMed] [Google Scholar]
- 21. Crinier A, Milpied P, Escaliere B, Piperoglou C, Galluso J, Balsamo A, et al. High-dimensional Single-cell analysis identifies Organ-specific signatures and conserved NK cell subsets in humans and mice. Immunity 2018;49:971–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Becker M, De Bastiani MA, Parisi MM, Guma FT, Markoski MM, Castro MA, et al. Integrated transcriptomics establish macrophage polarization signatures and have potential applications for clinical health and disease. Sci Rep 2015;5:13351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Du X, Tang Y, Xu H, Lit L, Walker W, Ashwood P, et al. Genomic profiles for human peripheral blood T cells, B cells, natural killer cells, monocytes, and polymorphonuclear cells: comparisons to ischemic stroke, migraine, and Tourette syndrome. Genomics 2006;87:693–703. [DOI] [PubMed] [Google Scholar]
- 24. Bezman NA, Kim CC, Sun JC, Min-Oo G, Hendricks DW, Kamimura Y, et al. Molecular definition of the identity and activation of natural killer cells. Nat Immunol 2012;13:1000–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinf 2013;14:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Singh V, Rana RK, Singhal R. Analysis of repeated measurement data in the clinical trials. J Ayurveda Integr Med 2013;4:77–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Benjamini YH., Y.;. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol 1995;57:289–300. [Google Scholar]
- 28. R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing; 2021. Available from: https://www.R-project.org/. [Google Scholar]
- 29. Johnström P, Gutierrez PM, Varnäs K, Schou M, Takano A, Jones L, et al. Abstract 2641: AZD4635 A2A receptor occupancy in cynomolgus monkey using PET and its application to an oncology clinical development program. Cancer Res 2017;77:13s (suppl. abstr 2641). [Google Scholar]
- 30. Lim EA, Bauer TM, Patel MR, Falchook GS, Karlix JL, Choe JH, et al. A phase I, open-label, multicenter study to assess the safety, pharmacokinetics, and preliminary antitumor activity of AZD4635 both as monotherapy and in combination in patients with advanced solid malignancies: Results from prostate cancer patients (NCT02740985). 2020; Virtual Meeting. Journal of Clinical Oncology. p Abstr 5518. [Google Scholar]
- 31. Powles T, O'Donnell PH, Massard C, Arkenau HT, Friedlander TW, Hoimes CJ, et al. Efficacy and safety of durvalumab in locally advanced or metastatic urothelial carcinoma: updated results from a Phase 1/2 Open-label Study. JAMA Oncol 2017;3:e172411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Moorthy G, Pouliot GP, Graham L, Wilks C, Sarvotham T, Mitchell P, et al. Abstract CT168: Clinical pharmacology of AZD4635 (A2ARi): Integration of PK data from cancer patients (CP) and healthy volunteer (HV) clinical trials to provide dosing recommendations. Cancer Res 2020;80:16s (suppl. abstr CT168). [Google Scholar]
- 33. Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, et al. Durvalumab after chemoradiotherapy in Stage III Non-small-cell lung cancer. N Engl J Med 2017;377:1919–29. [DOI] [PubMed] [Google Scholar]
- 34. Gunes A, Dahl ML. Variation in CYP1A2 activity and its clinical implications: influence of environmental factors and genetic polymorphisms. Pharmacogenomics 2008;9:625–37. [DOI] [PubMed] [Google Scholar]
- 35. Kwon ED, Drake CG, Scher HI, Fizazi K, Bossi A, van den Eertwegh AJ, et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184–043): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol 2014;15:700–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Beer TM, Kwon ED, Drake CG, Fizazi K, Logothetis C, Gravis G, et al. Randomized, Double-blind, Phase III Trial of Ipilimumab versus placebo in asymptomatic or minimally symptomatic patients with metastatic chemotherapy-naive castration-resistant prostate cancer. J Clin Oncol 2017;35:40–7. [DOI] [PubMed] [Google Scholar]
- 37. Powles T, Yuen KC, Gillessen S, Kadel EE III, Rathkopf D, Matsubara N, et al. Atezolizumab with enzalutamide versus enzalutamide alone in metastatic castration-resistant prostate cancer: a randomized phase 3 trial. Nat Med 2022;28:144–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Antonarakis ES, Piulats JM, Gross-Goupil M, Goh J, Ojamaa K, Hoimes CJ, et al. Pembrolizumab for treatment-refractory metastatic castration-resistant prostate cancer: multicohort, Open-Label Phase II KEYNOTE-199 Study. J Clin Oncol 2020;38:395–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sharma P, Pachynski RK, Narayan V, Flechon A, Gravis G, Galsky MD, et al. Nivolumab plus ipilimumab for metastatic castration-resistant prostate cancer: preliminary analysis of patients in the CheckMate 650 Trial. Cancer Cell 2020;38:489–99. [DOI] [PubMed] [Google Scholar]
- 40. de Bono JS, Oudard S, Ozguroglu M, Hansen S, Machiels JP, Kocak I, et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet 2010;376:1147–54. [DOI] [PubMed] [Google Scholar]
- 41. de Wit R, de Bono J, Sternberg CN, Fizazi K, Tombal B, Wulfing C, et al. Cabazitaxel versus abiraterone or enzalutamide in metastatic prostate cancer. N Engl J Med 2019;381:2506–18. [DOI] [PubMed] [Google Scholar]
- 42. Willingham SB, Hotson AN, Miller RA. Targeting the A2AR in cancer; early lessons from the clinic. Curr Opin Pharmacol 2020;53:126–33. [DOI] [PubMed] [Google Scholar]
- 43. Fong L, Hotson A, Powderly JD, Sznol M, Heist RS, Choueiri TK, et al. Adenosine 2A receptor blockade as an immunotherapy for treatment-refractory renal cell cancer. Cancer Discov 2020;10:40–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chiappori A, Williams CC, Creelan BC, Tanvetyanon T, Gray JE, Haura EB, et al. Phase I/II study of the A2AR antagonist NIR178 (PBF-509), an oral immunotherapy, in patients (pts) with advanced NSCLC. J Clin Oncol 2018;36:15s, (suppl. abstr 9089). [Google Scholar]
- 45. Buisseret L, Rottey S, de Bono J, Mossakowski M, Delafontaine B, Manickavasagar T, et al. First in human study with EOS100850, a novel potent A2A antagonist, shows excellent tolerance and clinical benefit in immune resistant advanced cancers. Cancer Res 2020;80:16s (suppl. abstr CT152). [Google Scholar]
- 46. Harshman LC, Chu M, George S, Hughes BGM, Carthon BC, Fong L, et al. Adenosine receptor blockade with ciforadenant ± atezolizumab in advanced metastatic castration-resistant prostate cancer (mCRPC). J Clin Oncol 2020;38:6s, (suppl. abstr 129). [Google Scholar]
- 47. Spira AI, Conkling PR, Johnson ML, Gardner O, Gilbert HN, Scharville M, et al. ARC-4 study: Efficacy and safety of AB928 plus carboplatin, pemetrexed and a PD-1 antibody in participants with metastatic non-small cell lung cancer (mNSCLC). J Clin Oncol 2020;38(suppl. abstr e21659). [Google Scholar]
- 48. Rouyer M, Oudard S, Joly F, Fizazi K, Tubach F, Jove J, et al. Overall and progression-free survival with cabazitaxel in metastatic castration-resistant prostate cancer in routine clinical practice: the FUJI cohort. Br J Cancer 2019;121:1001–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Powderly J, Spira A, Gutierrez R, DiRenzo D, Udyavar A, Karakunnel JJ, et al. Phase 1 evaluation of AB928, a novel dual adenosine receptor antagonist, combined with chemotherapy or AB122 (anti-PD-1) in patients (pts) with advanced malignancies. Abstract 4854. Ann Oncol 2019;30:v475–532. [Google Scholar]
- 50. Walters MJ, Piovesan D, Tan J, DiRenzo D, Yin F, Miles D, et al. Abstract 5556: Combining adenosine receptor inhibition with AB928 and chemotherapy results in greater immune activation and tumor control. Cancer Res 2018;78:13s (suppl. abstr 5556). [Google Scholar]
- 51. Clinicaltrials.gov. An open-label, phase II study of AZD4635 in patients with prostate cancer. Available from: https://clinicaltrials.gov/ct2/show/NCT04089553?term=AZD4635&draw=2&rank=5. [February 9, 2021].
- 52. Clinicaltrials.gov. A study of AZD4635 with durvalumab and with cabazitaxel and durvalumab in patients with mCRPC. (AARDVARC). 2020. Available from: https://clinicaltrials.gov/ct2/show/NCT04495179?term=AZD4635&draw= 2&rank=2. [February 9, 2021].
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data underlying the findings described in this article may be obtained in accordance with AstraZeneca's data sharing policy described at: https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure.