

Abstract
BACKGROUND:
Ibrutinib is a Bruton tyrosine kinase inhibitor with remarkable efficacy against B-cell cancers. Ibrutinib also increases the risk of atrial fibrillation (AF), which remains poorly understood.
METHODS:
We performed electrophysiology studies on mice treated with ibrutinib to assess inducibility of AF. Chemoproteomic analysis of cardiac lysates identified candidate ibrutinib targets, which were further evaluated in genetic mouse models and additional pharmacological experiments. The pharmacovigilance database, VigiBase, was queried to determine whether drug inhibition of an identified candidate kinase was associated with increased reporting of AF.
RESULTS:
We demonstrate that treatment of mice with ibrutinib for 4 weeks results in inducible AF, left atrial enlargement, myocardial fibrosis, and inflammation. This effect was reproduced in mice lacking Bruton tyrosine kinase, but not in mice treated with 4 weeks of acalabrutinib, a more specific Bruton tyrosine kinase inhibitor, demonstrating that AF is an off-target side effect. Chemoproteomic profiling identified a short list of candidate kinases that was narrowed by additional experimentation leaving CSK (C-terminal Src kinase) as the strongest candidate for ibrutinib-induced AF. Cardiac-specific Csk knockout in mice led to increased AF, left atrial enlargement, fibrosis, and inflammation, phenocopying ibrutinib treatment. Disproportionality analyses in VigiBase confirmed increased reporting of AF associated with kinase inhibitors blocking Csk versus non-Csk inhibitors, with a reporting odds ratio of 8.0 (95% CI, 7.3–8.7; P<0.0001).
CONCLUSIONS:
These data identify Csk inhibition as the mechanism through which ibrutinib leads to AF.
REGISTRATION:
URL: https://ww.clinicaltrials.gov; Unique identifier: NCT03530215.
Keywords: atrial fibrillation; BTK protein, human; CSK tyrosine-protein kinase; electrophysiology; ibrutinib; protein kinase inhibitors
Ibrutinib received U.S. Food and Drug Administration breakthrough designation and was approved for the treatment of relapsed mantle cell lymphoma in 2013 and for chronic lymphocytic leukemia (CLL) in early 20141,2. Ibrutinib recently demonstrated efficacy in fron-tline setting in patients with CLL.3,4 However, treatment with ibrutinib is associated with an increased risk of atrial fibrillation (AF),5–13 with a 10-fold increased rate of AF in the ibrutinib arm in 1 study compared with the control arm.1 Longer term studies show AF rates as high as 16% in patients treated with ibrutinib for a median follow-up of 28 months.12 Because none of the trials involving ibrutinib prospectively screened for AF, most of the reported patients had AF requiring hospitalization or other interventions, suggesting that the true incidence of ibrutinib-associated AF may be significantly higher. AF is a common reason for discontinuing ibrutinib therapy,14 can result in significant morbidity, and is poorly understood. The management of ibrutinib-associated AF is challenging because of the difficulties in balancing the benefits of anticoagulation to mitigate the risk of stroke with an ibrutinib-related bleeding diathesis.15,16 Thus, there is a clear and pressing need to improve management of these patients, requiring a better understanding of the pathophysiology of AF induced by ibrutinib.17
METHODS
Detailed methods are provided in the Data Supplement. The data that support the findings of this study are available from the corresponding author on reasonable request.
Mice
All animal studies were approved by the Institutional Animal Care and Use Committee at Massachusetts General Hospital and were in compliance with relevant ethical regulations. All experiments were performed on 3- to 4-month-old and sex-matched C57BL/6J, CBA/CaJ, CBA/CaHN-BTKxid/J (BTKxid, a specific mutation abrogates Bruton tyrosine kinase (BTK) kinase activity in this strain), B6.129S7-FYNm1SorJ (FYN−/−, Fyn knockout mice), and Cskfl/fl mice18 which were bred with αMHCMerCreMer mice19 to generate cardiomyocyte-specific knockout of Csk, αMHCMerCreMerCskfl/fl mice.
In Vivo Interventions
Mice were treated with ibrutinib (Selleckchem) via intraperitoneal (IP) injection on a daily dose of 25 mg·kg−1·d−1.20–23 Acalabrutinib, saracatinib, or BIX 02188 (Selleckchem) were administered at a daily dose of 25 mg·kg−1·d−1 via IP injection in C57BL/6J mice for 28 days. Mice injected with vehicle in parallel were used as controls. Three-month-old αMHCMerCreMerCskfl/fl mice were treated with tamoxifen (Sigma-Aldrich) to induce cardiomyocyte-specific Csk knockout. Littermate-matched Cskfl/fl mice treated with tamoxifen were used as controls and characterization experiments were performed 4 weeks after the last dose of tamoxifen.
In Vivo Electrophysiology Study
Mice were anaesthetized by administering 5% isoflurane driven by an oxygen source and were subsequently maintained with 1% to 2% isoflurane in 95% O2. Surface ECG was recorded using subcutaneous needle electrodes connected to the Octal Bio amplifier and PowerLab station (AD Instruments) in a lead I configuration. An octapolar catheter (EPR-800; Millar Instruments) was inserted into the right jugular vein and positioned in the right atrium and ventricle. Programmed electric stimulation was performed using a standard protocol to measure function of the atrioventricular node and the conduction properties of atrial and ventricular tissues.24 Arrhythmias were induced by standard burst pacing protocol. AF was defined as a rapid irregular atrial rhythm with irregular R-R intervals lasting at least 1 second.25 Data were analyzed with LabChart8 Pro.
Blood Pressure
Blood pressure measurement was performed on conscious mice using volume pressure recording sensor technology with the CODA mouse tail-cuff system (Kent Scientific).
Echocardiography
Transthoracic echocardiography was performed on conscious mice using a E90 cardiac ultrasound system (GE Healthcare) with a L8–18i-D transducer at 100 frames/s. M-mode recordings were acquired in the short-axis view at the level of the papillary muscles. Left atrial (LA) diameter was measured from the parasternal long axis view.
MRI
MRI was performed on mice anaesthetized with 2% isoflurane in oxygen, using a 4.7 Tesla horizontal bore Pharmascan (Bruker) with a custom-built mouse cardiac coil (RAPID Biomedical) with respiratory gating (SA Instruments). Short axis images over the entire heart were sequentially acquired using a cine fast low angle shot sequence with intragate cardiac gating (Cine_FLASH_IG). Images were analyzed using Horos software to calculate the LA volume or the Segment software (Medviso) to calculate the left ventricular (LV) ejection fraction.
Plasma Cytokine Measurements
Plasma proinflammatory cytokine levels were determined using the mouse cytokine 9-plex ELISA kit (PBL Assay Science) and the Quansys Q-View imager (Quansys Biosciences).
Histology
Masson trichrome stain and hematoxylin and eosin stain were performed according to the manufacturer’s instructions (MilliporeSigma). Apoptotic cells were analyzed using the In Situ Cell Death Detection Kit, TMR red (MilliporeSigma). Cardiac macrophages were stained with CD68 (clone: FA-11, BioLegend) followed by a goat anti-rat IgG Alexa Fluor 488 antibody (A-11006, ThermoFisher Scientific). 4′,6-diamidino-2-phenylindole (ThermoFisher Scientific) was used for nuclear counterstaining. All images were obtained with NanoZoomer 2.0-RS (Hamamatsu) or confocal microscopy (Leica SP8). Cardiac fibrosis was analyzed using ImageJ (National Institutes of Health).
RNA Extraction and Quantitative Real-Time Polymerase Chain Reaction
Total RNA from LA or LV tissue was isolated using TRIzol reagent (Invitrogen) and the RNeasy Mini Kit (QIAGEN), and was reverse transcribed to cDNA using the iScript real-time polymerase chain reaction kit (Biorad). Quantitative real-time polymerase chain reaction was performed using the CFX384 Touch thermocycler (Biorad) and the iTaq Universal Probes Supermix (Biorad) with TaqMan probes (Thermo Fisher Scientific). Gene expression was normalized to Gapdh using the 2−ΔΔCT method and expressed as fold changes over control group.
Bulk RNA Sequencing
RNA sequencing and analysis was performed by MGH Sequencing Core. Libraries were constructed using the PolyA+ NEBNext Ultra Directional RNA Library Prep Kit and sequenced on an Illumina HiSeq 2500 in high-output mode. Differentially expressed genes were analyzed using the EdgeR method26 and were classified based on the cutoffs of 2-fold change in expression value and false discovery rates below 0.1. Gene set enrichment analysis was performed with gene set enrichment analysis tool (Broad Institute) on the hallmark set of pathways using the default false discovery rate cutoff of 0.25. Data were deposited to the Gene Expression Omnibus database under ID codes GSE139427.
Immunoblotting
Proteins extracted from murine LA and LV tissue were analyzed with standard Western blot protocols with the following primary antibodies: rabbit monoclonal anti–CSK (C-terminal Src kinase 1:1000, ab125005; Abcam); rabbit polyclonal anti-CaMKII-phospho-T287 (1:500, ab182647; Abcam); rabbit polyclonal anti-CaMKII (1:1000, A010; Badrilla); mouse monoclonal anti-GAPDH (1:2000, ab8245; Abcam); and horseradish peroxidase-conjugated goat anti-rabbit or donkey anti-mouse secondary antibodies (1:10 000; Jackson ImmunoResearch Laboratories). Proteins were detected using the ChemiDoc Touch Imaging System (Biorad) and were quantified with Quantity-One software (Bio-Rad).
Chemoproteomic Kinase Profiling
In situ chemoproteomic kinase profiling was performed by ActivX Biosciences using the KiNativ platform27 to quantitatively profile ibrutinib and acalabrutinib targets against native kinase in the mouse heart. Mouse heart lysate was incubated with ibrutinib or acalabrutinib at 10 μM for 15 minutes and were then labeled with a desthiobiotin ATP acylphosphate probe (ATP probe) at 20 μmol/L. Desthiobiotinylated peptides were analyzed by liquid chromatography–mass spectrometry/mass spectrometry using the mouse kinase-ATP probe master target list. Data were presented as % inhibition.
Proteomics on Human Heart Tissue Biopsies
Cardiac tissue biopsies (LA, right atrial, LV) were obtained from 7 individuals undergoing mitral valve surgery. Proteins extracted from the biopsies were digested, fractionated and analyzed by online reversed-phase liquid chromatography coupled to a Q-Exactive Plus quadrupole Orbitrap tandem mass spectrometer as described previously.28,29 Raw mass spectrometry data were processed using the MaxQuant software and proteins were identified with the built-in Andromeda search engine using a database containing all reviewed human SwissProt protein entries. A total of 6588 proteins were measured of which 5941 were quantified (Linscheid N, Poulsen PC, Lundby A, unpublished data). Raw intensities were normalized by quantile-based normalization. CSK was identified by mass spectrometry/mass spectrometry identification in all 12 biopsies (6 LA and 6 LV). CSK quantitation was based on 20 distinct peptides, (19 of which were unique to CSK) that in total covered 54% of the CSK amino acid sequence. Intensities were compared using a paired 2-sided Student t test.
Cellular Electrophysiology
Action potentials (APs) and sodium currents (INa) were recorded using whole-cell patch-clamp techniques in isolated mouse LA cardiomyocytes at room temperature with an Axopatch 200B amplifier and digitized at 10 kHz with a Digidata 1440A A/D converter. Data were recorded with Clampex 10.3 and analyzed with Clampfit 10.3 (Molecular Devices Inc).
Data Analysis and Statistics
Statistical analyses were conducted with GraphPad Prism 8.4.3 software. Data are presented as mean±SEM. Data were tested for normality using the Shapiro–Wilk normality test. Statistical significance was assessed by the 2-tailed Student’s t test for normally distributed data, or by the 2-sided Mann–Whitney test if data did not pass the normality test. For repeated measures, paired Student t test was performed. For multiple comparisons, 1-way ANOVA followed by a Sidak posttest was performed. AF inducibilities were assessed by Fisher exact test. P values of 0.05 or less were considered significant.
VigiBase Data Analysis and Statistics
A disproportionality analysis was performed based on adverse drug reactions reported within VigiBase, the World Health Organization deduplicated database of more than 19.5 million individual case safety reports, from more than 130 countries.30 This study was approved by the Vanderbilt University Medical Center institutional review board (No. 181337) and was registered with the national clinical trials database (NCT03530215). The supplied data in VigiBase come from a variety of sources. The likelihood of a causal relationship is not the same in all reports. The information does not represent the opinion of the World Health Organization. A case/noncase analysis (disproportionality analysis) was performed to study whether drug-induced AF events were differentially reported with any of the 16 kinase inhibitors studied compared with AF events reported in the entire database. A collection of CSK IC50 inhibition levels for all U.S. Food and Drug Administration–approved anticancer kinase inhibitors from the literature and public databases identified CSK IC50 values for 16 kinases. Cmax levels for each drug were obtained from the literature. Association between magnitude of reporting for AF versus full database (information component) and CSK inhibition at Cmax for each kinase inhibitor (Cmax/IC50) was assessed by Spearman correlation test.
RESULTS
Ibrutinib Treatment Increases Susceptibility to Atrial Arrhythmias in Mice
To investigate the mechanism by which ibrutinib leads to AF, we first sought to develop a small animal model of ibrutinib-mediated AF. In contrast to other reports,31 we found that short duration exposure to ibrutinib did not result in increased inducible AF (Figure IA and IB in the Data Supplement). Given that AF may take weeks or even months to occur in humans treated with ibrutinib,5 we treated mice with daily injections of 25 mg/kg for 4 weeks (Figure 1A). This dose has been commonly used in mice to study effects of ibrutinib on tumor suppression20,21,23 and its cardiotoxicity.22 After 4 weeks of ibrutinib treatment, treated mice demonstrated more inducible atrial fibrillation than vehicle injected mice (Figure 1B and 1C), accompanied by prolonged atrial effective refractory periods (Figure IC in the Data Supplement) without significant changes in the action potential duration (Figure ID and IE in the Data Supplement). Ibrutinib treatment also led to atrial fibrosis, evident on histological imaging (Figure 1D and 1E) as well as by increased expression of fibrosis markers fibronectin, and collagens 1α1 and 3α1 (Figure 1F). Echocardiography and cardiac MRI to assess cardiac structure (Figure 1G) showed preserved left ventricular ejection fraction (Figure 1I), but significant atrial dilation (Figure 1H; Table I in the Data Supplement) in ibrutinib-treated mice compared with vehicle injected controls. Circulating tumor necrosis factor α (TNFα) and interleukin-6 (IL-6) were both elevated in ibrutinib-treated mice compared with vehicle injected controls (Figure 1J and 1K) demonstrating increased inflammation in ibrutinib-treated mice. Finally, RNA sequencing of atrial tissue showed 40 differentially expressed genes at a false discovery rate of 0.1 (Figure 1L; Table II in the Data Supplement), with enrichment of 18 gene clusters/pathways including epithelial to mesenchymal transition, IL-6/Jak/Stat, and interferon α and γ and inflammatory responses (Figure 1M; Figure II in the Data Supplement).
Figure 1. Long-term ibrutinib treatment predisposes mice to AF.
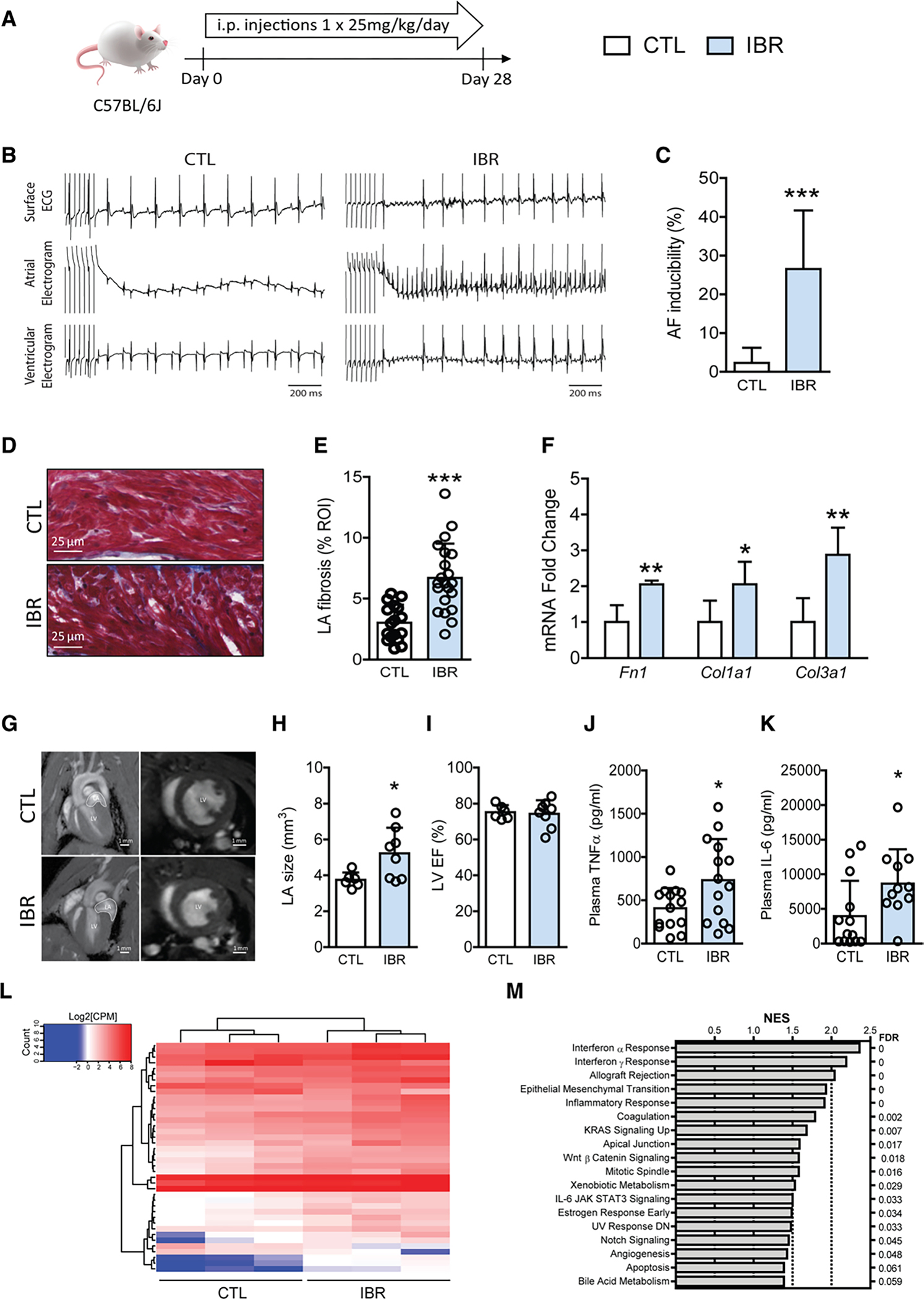
Evaluation of ibrutinib effects on C57BL/6J wild-type mice. A, Experimental outline of ibrutinib administration in C57BL/6J mice. Mice received intraperitoneal injection of ibrutinib (IBR) or vehicle (CTL) at 25 mg·kg−1·d−1 for 4 weeks (28 days), followed by in vivo electrophysiology studies. B, Representative surface lead II ECG, intracardiac atrial and ventricular electrogram recordings showing AF after atrial burst pacing in ibrutinib-treated mice (IBR), but not in CTL. C, AF inducibility in CTL (n = 9) and IBR (n = 8). Data are mean±SEM percentage of successfully induced atrial arrhythmia episodes during invasive EP studies. ***P<0.0001, Fisher exact test. D, Histological images of Masson trichrome stain of LA fibrosis in CTL and IBR. E, Quantification of tissue fibrosis levels (%) in LA sections from CTL and IBR mice. Data are mean±SEM, n = 17 LA regions of interest from 5 CTL mice, n=22 LA regions of interest from 5 IBR mice. ***P<0.0001, unpaired Student t test. F, Mean±SEM of Fn1, Col1a1, and Col3a1 mRNA levels in LA tissues from CTL and IBR mice. n=5 mice per group. *P<0.05, **P<0.01, unpaired Student t test. G, Representative cardiac magnetic resonance images of 4-chamber views (left) and ventricular short axis views (right) from CTL and IBR mice. LA is highlighted in the 4-chamber view. H and I, Quantification of LA sizes (H) and left ventricular ejection fraction (I) measured by MRI after 4 weeks of ibrutinib treatment. Data are mean±SEM, n=6 mice in CTL and n=8 mice in IBR. *P<0.05, unpaired Student t test. J and K, Quantification of plasma levels of TNFα (J) and IL-6 (K) after 4 weeks of ibrutinib treatment. Data are mean±SEM, n=13 mice in CTL and n=11 mice in IBR. *P<0.05, unpaired Student t test (J) or Mann–Whitney test (K). L, Heatmap of expression levels of 40 differentially expressed genes from RNAseq data of 3 CTL and 3 IBR LA tissues. M, Gene set enrichment analysis shows 18 differentially regulated gene categories/pathways in LA after 4 weeks of ibrutinib treatment. Barplot of gene set enrichment analysis NES with false discovery rate on the right. AF indicates atrial fibrillation; CPM, counts per million; ECG, electrocardiography; LA, left atrial; and NES, normalized enrichment score.
Recently it has been shown that long-term ibrutinib treatment enhances CaMKII signaling and dysregulates calcium handling in the murine atrial cardiomyocytes.22 We replicated the finding that ibrutinib treatment significantly increased the ratio of phosphorylated CaMKII to total CaMKII levels in LA tissues compared with vehicle treatment (Figure III in the Data Supplement), indicating elevated CaMKII activity associated with long-term ibrutinib administration.
Ibrutinib Increases Susceptibility to Atrial Arrhythmias in Mice Lack of BTK
Ibrutinib not only inhibits BTK, but also potently inhibits multiple other kinases.32 We sought to determine whether ibrutinib’s proarrhythmic side effect is because of BTK inhibition, or rather, an off-target effect. BTKxid mice, which harbor a mutation in the BTK gene resulting in loss of BTK activity and X-linked immune deficiency,33,34 were treated with either ibrutinib or vehicle control according to the same 4-week protocol (Figure 2A), and subjected to electrophysiology studies for AF inducibility. If BTK is the relevant target, we might expect that even vehicle treated BTKxid mice would have inducible AF, but BTKxid mice did not demonstrate AF inducibility (Figure 2B). On the other hand, ibrutinib treatment of BTKxid mice resulted in a significant increase in AF inducibility compared with vehicle-treated mice, which, accompanied by LA enlargement (Figure 2C) and fibrosis (Figure 2D and 2E strongly suggests non-BTK targets play a role in ibrutinib-induced AF.
Figure 2. Ibrutinib causes atrial fibrillation through off-target effects.
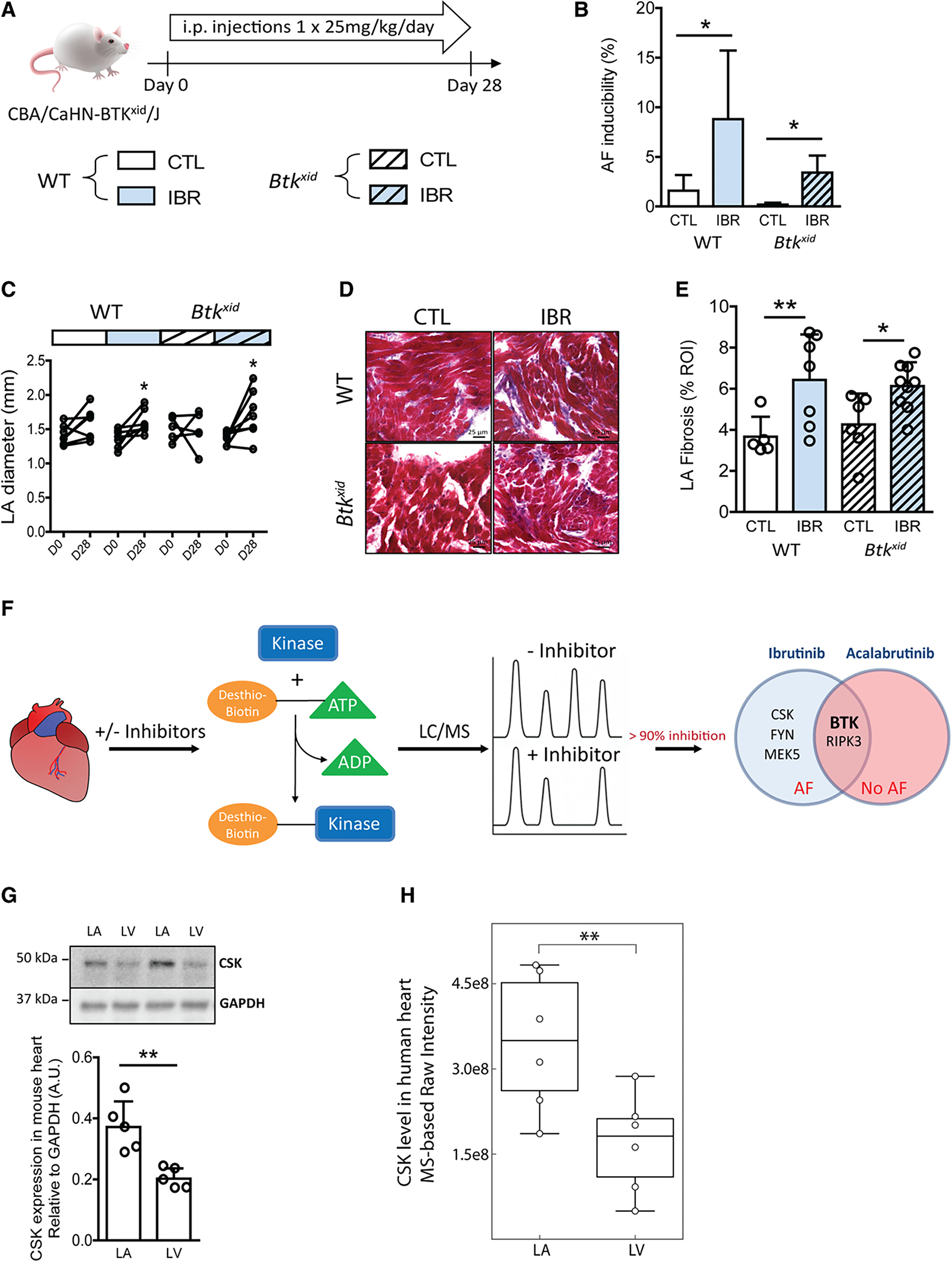
Evaluation of ibrutinib effects on mice lacking BTK. A, Experimental outline of ibrutinib administration in BTK mutant CBA/CaHN-BTKxid/J mice lacking active BTK (Btkxid) and strain-matched wild-type CBA/CaJ (WT). Mice received intraperitoneal injection of ibrutinib (IBR) or vehicle (CTL) at 25 mg·kg−1·d−1 for 4 weeks (28 days), followed by in vivo electrophysiology studies. B, AF inducibility in WT and Btkxid after 4-week ibrutinib or vehicle treatment. Data are mean±SEM percentage of successfully induced atrial arrhythmia episodes during invasive EP studies in WT CTL, n=5, WT IBR, n=7, Btkxid CTL, n=6, and Btkxid IBR n=8 mice. *P<0.05, Fisher exact test. C, Data are LA diameters measured by echocardiography before (D0) and after 4 weeks (D28) of ibrutinib (IBR) or vehicle (CTL) injections in WT and Btkxid mice (n=7 mice in WT CTL; n=7 in WT IBR; n=5 in Btkxid CTL; n=8 in Btkxid IBR). *P<0.05, D0 vs D28, paired Student t test. D, Representative sections of Masson trichrome stain of LA fibrosis in CTL and IBR of WT and Btkxid mice. E, Mean±SEM LA fibrosis levels (%) in LA sections of WT CTL (n=5 regions of interest from 2 mice), WT IBR (n=7 from 3 mice), Btkxid CTL (n=6 from 3 mice), and Btkxid IBR (n=9 from 4 mice). *P<0.05, ** P<0.01, 1-way ANOVA with Sidak posttest. F, Workflow of chemoproteomic kinase profiling of ibrutinib and acalabrutinib targets in mouse heart. Five kinases and 2 kinases detected to be inhibited by >90% were identified as ibrutinib or acalabrutinib targets in the heart. A Venn diagram shows the overlap of 2 kinases inhibited by both ibrutinib and acalabrutinib. G, Mean±SEM CSK protein levels normalized to GAPDH levels in paired LA and LV tissues from adult wild-type C57BL/6J mice (n=5 mice; bottom). **P<0.01, paired Student t test. CSK protein is detected at ≈50 kDa on Western blot (top). H, The expression level of CSK in human heart was evaluated from MS-based protein intensity measurements performed on heart biopsies collected from LA and LV from 6 living humans. Normalized raw intensities measured for CSK are shown. Individual measurements from LA or LV are depicted dots. The relative protein expression levels of CSK in atrial and ventricular biopsies were compared by a paired Student t test. P=0.009. The box in the box-and-whisker plot shows the quartiles of the dataset while the whiskers represent the CI (95%) by bootstrapping. AF indicates atrial fibrillation; BTK, Bruton tyrosine kinase; CSK, c-Src terminal kinase; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; i.p., intraperitoneal; LA, left atria; LC, liquid chromatography; LV, left ventricle; and MS, mass spectrometry.
Ibrutinib Cardiac Kinase Targets Associated With AF
To identify candidate targets for ibrutinib-induced AF, we performed chemoproteomic profiling of ibrutinib in cardiac tissue. In these experiments, cardiac lysates were incubated with a biotinylated acylphosphate ATP derivative that irreversibly transfers biotin to conserved lysine residues in the ATP-binding pocket of protein kinases and other ATP binding proteins.35,36 Isolation of biotin tagged proteins in the presence and absence of ibrutinib allowed identification of ibrutinib targets in the murine cardiac tissue lysates (Figure 2F). We identified 5 kinases that were inhibited by >90% with 10 uM ibrutinib: BTK, FYN, MEK5, CSK, and RIPK3 (Figure IVA in the Data Supplement). We then studied the second generation BTK inhibitor, acalabrutinib, which to date has not been associated with AF in CLL patients.37 We first confirmed that a 4-week exposure of mice to daily acalabrutinib injections did not cause AF—further supporting a non-BTK, off-target mechanism for AF induction (Figure VA and VE in the Data Supplement). We then repeated the chemoproteomic experiments using acalabrutinib in place of ibrutinib, identifying BTK and RIPK3 as specific cardiac acalabrutinib targets (Figure IVB in the Data Supplement). Comparison of ibrutinib and acalabrutinib targets allowed us to narrow the candidate kinases associated with AF susceptibility to FYN, MEK5, and CSK (Figure 2F).
To evaluate the 3 remaining kinases, we performed additional experiments in mice. Treatment of mice with the FYN inhibitor saracatinib38 for 4 weeks did not result in increased AF (Figure VB and VE in the Data Supplement). Further, the global Fyn knockout mouse Fyn−/−39 had no increase in AF either (Figure VC and VE in the Data Supplement). Because Fyn has been reported to phosphorylate and modify the activity of the cardiac sodium channel,40 we recorded the sodium currents from atrial cardiomyocytes isolated from ibrutinib or control treated mice. We observed no difference in the activation and inactivation voltage dependence between these groups (Figure IF–IH in the Data Supplement). In aggregate, these data argue against FYN as the AF relevant target of ibrutinib. Similarly, treatment of mice with MEK5 inhibitor BIX-0218841 for 4 weeks did not lead to an increase in AF inducibility (Figure VD and VE in the Data Supplement).
Cardiac Knockout of CSK Predisposes the Heart to Atrial Arrhythmia
We turned our focus to CSK. CSK is a nonreceptor protein tyrosine kinase that phosphorylates residues on the c-terminal end of Src kinase family members, rendering them inactive.42,43 CSK is expressed in the adult heart at higher levels in the atrium than in the ventricle in both mouse (Figure 2G) and humans (Figure 2H), which may suggest a greater reliance on CSK to keep Src family kinases quiescent in the atrium and could account for the observation of more atrial than ventricular arrhythmias in ibrutinib-treated patients. To obtain a cardiac specific knockout of Csk, we generated αMHCmercremerCSKfl/fl mice where CSK can be temporally deleted in cardiomyocytes by treatment with tamoxifen (αMHC Csk−/−).18,19 To mimic the ibrutinib treatment protocol, mice were studied 28 days after a five day tamoxifen treatment (Figure 3A and 3B). The αMHC Csk−/− mice demonstrated increased AF (Figure 3C) compared with controls (tamoxifen treated Cskfl/fl mice), with slowed AV node conduction properties but without changes in atrial effective refractory period or PR interval (Figure VIA–VIE and Table III in the Data Supplement). Histological studies revealed marked cardiac fibrosis (Figure 3D and 3E, and Figure VIG and VIH in the Data Supplement), as well as a cellular infiltrate in the atria of αMHC Csk−/− mice (Figure VII in the Data Supplement). The cells were positive for staining with CD68, suggesting that the infiltrate consists of monocyte/macrophage lineage cells (Figure 3F and 3G). MRI studies showed preserved ventricular ejection fraction and increased LA size (Figure 3H and 3I; Figure VIJ and VIK in the Data Supplement). Circulating TNFα and IL-6 levels were both higher in the αMHC Csk−/− mice compared with controls (Figure 3J and 3K). TUNEL staining showed increased apoptotic cells in the LA tissue of αMHC Csk−/− mice (Figure VIL and VIM in the Data Supplement). RNA sequencing of atrial tissue from αMHC Csk−/− versus control mice demonstrated 896 differentially expressed genes at an false discovery rate of 0.1 (Figure 3L; Table IV in the Data Supplement) and 18 enriched gene clusters/pathways including interferon α and γ, epithelial to mesenchymal transition, inflammatory response, and IL6/JAK/STAT (Figure 3M; Figure VIN in the Data Supplement). Of the 18 enriched pathways, 15 are in common with the ibrutinib-treated mice (Figure 3N). Taken together, these data suggest that CSK loss in the myocardium recapitulates cardiac findings in the ibrutinib-AF model and suggests that CSK is the likely kinase target for ibrutinib-mediated AF.
Figure 3. Cardiac-specific knockout of CSK causes AF and increases LA interstitial fibrosis, LA size, CD68+ macrophages, and plasma TNFα and IL-6 levels.
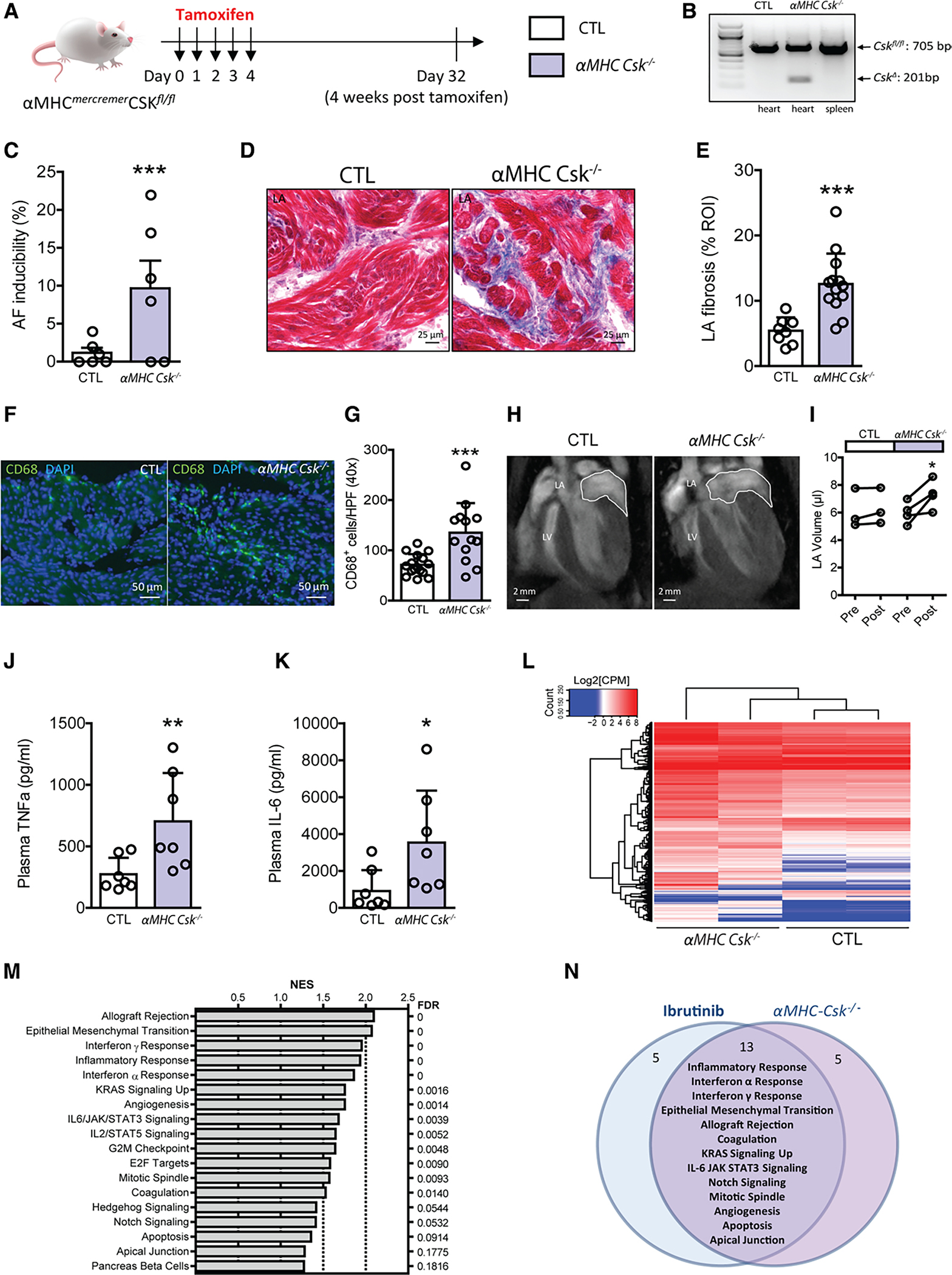
A, Experimental outline. Cardiac specific deletion of Csk was induced in αMHCmercremer Cskfl/fl mice after 5 daily doses of tamoxifen injection (αMHC Csk−/−). Littermate Cskfl/fl mice receiving the 5-dose tamoxifen treatment were used as control (CTL). In vivo electrophysiology studies were performed on day 32, 4 weeks after the last dose of tamoxifen. B, Polymerase chain reaction analyses of Cskfl/fl (CTL) and αMHCmercremerCskfl/fl (αMHC Csk−/−) heart and spleen tissues 4 weeks posttamoxifen show cardiac specific deletion of Csk allele (CskΔ) in αMHC Csk−/− mice. C, Atrial fibrillation inducibility in CTL and αMHC Csk−/− mice 4 weeks after tamoxifen treatment. Data are mean±SEM percentage of successfully induced atrial arrhythmia episodes during invasive EP studies. n=6 mice per group, ***P<0.0001, Fisher exact test. D, Images of Masson trichrome stain of LA fibrosis in CTL and αMHC Csk−/− mice. E, Mean±SEM LA fibrosis levels (%) in LA sections of CTL (n=8 ROIs from 3 mice) and αMHC Csk−/− (n=13 from 3 mice) mice. ***P<0.0001, unpaired Student t test. F, Immunofluorescence images of LA sections stained for CD68 (green) and DAPI(blue) in CTL and αMHC Csk−/− mice. G, Quantification of CD68+ cells in LA sections of CTL (n=16 HPFs from 3 mice) and αMHC Csk−/− (n=13 from 3 mice). Data are mean±SEM; ***P<0.0001, unpaired Student t test. H, Representative cardiac magnetic resonance images of CTL and αMHC Csk−/− mice. I, Data are LA volumes obtained by cardiac MRI on CTL and αMHC Csk−/− mice before and 4 weeks posttamoxifen treatment (n=3 mice for CTL and n=4 mice for αMHC Csk−/−). Data are mean±SEM, *P<0.05, paired Student t test. J and K, Quantification of plasma TNFα (J) and IL-6 (K) levels in CTL (n=7) and αMHC Csk−/− (n=7) mice. Data are mean±SEM, *P<0.05, ** P<0.01, nonparametric Mann–Whitney test. L, Heatmap of expression levels of 896 differentially expressed genes from RNAseq data of 2 CTL and 2 αMHC Csk−/− LA tissues. M, Gene set enrichment analysis shows 18 differentially regulated categories/pathways in LA after cardiac Csk knockout. Barplot of gene set enrichment analysis NES with false discovery rate on the right. N, A Venn diagram shows the overlap of 13 differentially regulated gene categories/pathways between ibrutinib-treated mice and cardiac Csk knockout mice. AF indicates atrial fibrillation; CPM, counts per million; CSK, C-terminal Src kinase; DAPI, 4′,6-diamidino-2-phenylindole; HPF, high power fields under 40× objective; LA, left atria; NES, normalized enrichment score; and ROI, region of interest.
CSK Inhibitors Are Associated With Increased Risk of AF in Patients
We next asked whether other kinase inhibitors approved for cancer treatment which inhibit CSK also increase the risk of AF. We performed a disproportionality analysis44 using the international pharmacovigilance database, VigiBase.30 We selected U.S. Food and Drug Administration–approved anticancer kinase inhibitors whose activity against CSK was available in the literature or in public databases, identifying 5 CSK inhibitors (Cmax>IC50) and 11 weak/noninhibitors (Cmax<IC50; Figure VII and Table V in the Data Supplement). Disproportionality analysis in 19.5 million individual case safety reports in VigiBase were first evaluated by the information component, a Bayesian estimator, comparing AF reporting rate with each of these 16 kinase inhibitors versus all other drugs in the entire database. Information component reporting for AF with each of these kinase inhibitors was found to be associated with their Cmax/IC50 values (r=0.72; P=0.003; Figure 4A) demonstrating that the reporting rate of AF as a side effect correlates with the level of CSK inhibition. Then, we used the reporting odds ratio, a frequentist estimator, to compare AF reporting rate with CSK inhibitors (n=5) versus CSK noninhibitors (n=11). In these analyses, the numerator is the number of AF adverse drug reactions reported and the denominator is the total number of adverse drug reactions reported with a drug or a group of drugs. Reporting rate of AF with CSK inhibitors (2157 of 79 987 [2.7%]) versus noninhibitors (626 of 180 858 [0.35%]) resulted in a reporting odds ratio of 8.0 (95% CI, 7.3–8.7; P<0.0001; Figure 4B). The sensitivity analysis after removing ibrutinib from the CSK inhibitor group revealed that the association between CSK inhibitors and AF (425 of 50 986 [0.84%]) versus CSK noninhibitors (649 of 182 691 [0.35%]) remained significant, with a reporting odds ratio of 2.4 (95% CI, 2.1–2.7; P<0.0001). These data suggest that off-target inhibition of CSK increases the risk of AF even for drugs other than ibrutinib.
Figure 4. CSK inhibition is highly associated with atrial fibrillation in individual case safety reports gathered in the international pharmacovigilance database, VigiBase.
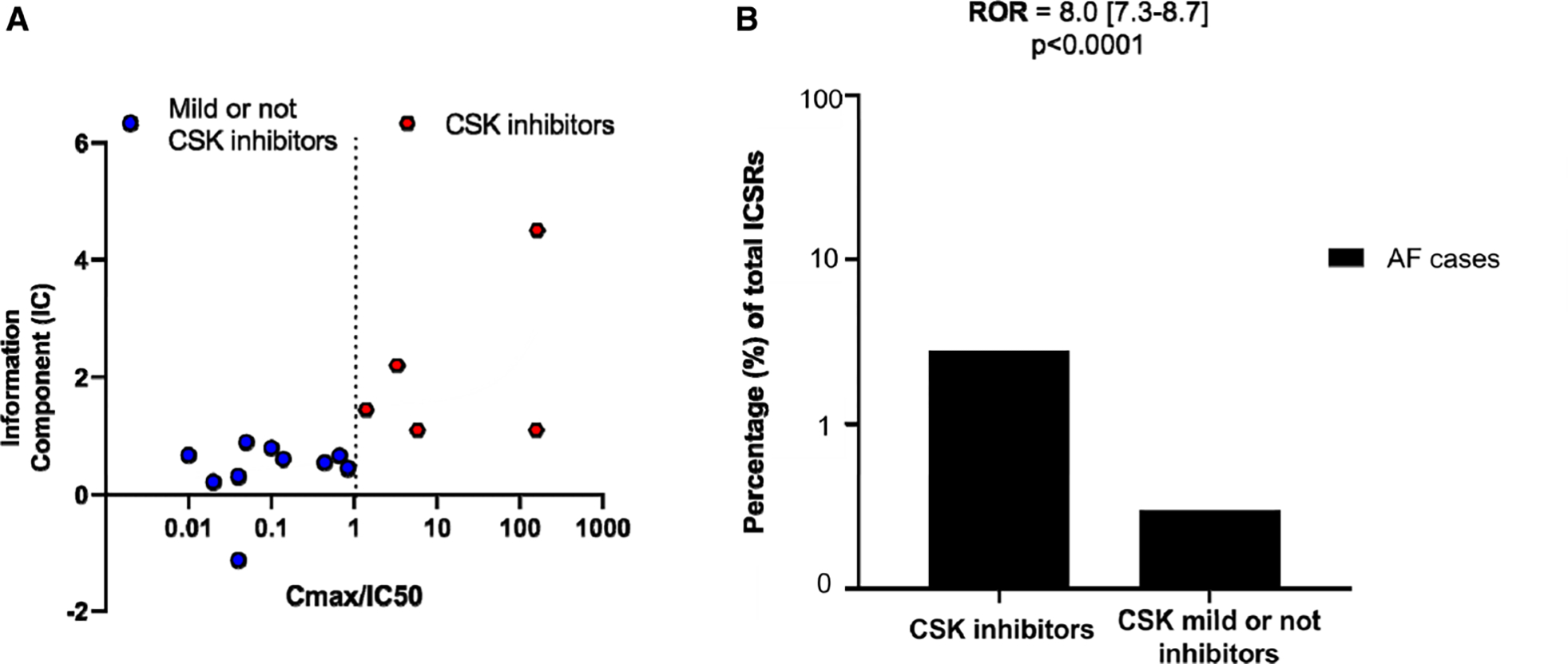
A, Data are Cmax/CSK IC50 against atrial fibrillation information component (a Bayesian disproportionality estimator) for 16 kinase inhibitors that are reported to inhibit CSK with various potency (IC50s range from 2.2 nmol/L to 66 μmol/L; Table V in the Data Supplement) out of all kinase inhibitors approved by U.S. Food and Drug Administration as anticancer drugs. CSK inhibitors are defined as Cmax/CSK IC50>1, and mild or not CSK inhibitors have Cmax/IC50<1. B, ROR for AF cases over total number of ICSRs on CSK inhibitors compared with the non-CSK inhibitors (mild or not CSK inhibitors). AF indicates atrial fibrillation; CSK, C-terminal Src kinase; ICSR, individual case safety reports; and ROR, reporting odds ratio.
DISCUSSION
Although ibrutinib has demonstrated impressive efficacy in the treatment of CLL and other B-cell lymphomas, its use can be limited by AF. We used a combination of approaches ranging from murine models to human studies to determine that ibrutinib-mediated AF is attributable to off-target inhibition of CSK. Consistent with these findings, there are no reports of increased AF in BTK-deficient patients suffering from Bruton X-linked agammaglobulinemia.
CSK is an endogenous inhibitor, or brake, on Src family tyrosine kinases (SFKs).45 C-terminal phosphorylation by CSK inactivates SFKs, and conversely, CSK inhibition leads to increased SFK activity. SFKs are involved in many cellular functions: cell proliferation, differentiation, survival, adhesion, migration, programmed death, inflammation, and others.45 There is a fine balance between CSK phosphorylation and SFK activation and altering this balance can lead to deleterious effects. Germline Csk knockout in mice is embryonically lethal with homozygous knockout embryos exhibiting defects in the neural tube, increased activity of SFKs, and death on days E9.5 to E10.5.43,46 Conditional systemic or cell-type specific knock out of Csk shows increased SFK activity and increased inflammation.18,43,47,48 The role of CSK in the heart is unclear. We found CSK is present in the heart and is enriched in the atria compared with ventricle. Furthermore, deleting Csk in adult mouse heart results in cardiac fibrosis, atrial enlargement, increased atrial macrophages, increased circulating cytokines, apoptosis, and atrial arrhythmias. Differential gene expression in Csk knockout mice demonstrated that 15 of the 18 dysregulated pathways are in common with ibrutinib-treated mice, including several inflammatory pathways. Ibrutinib potently inhibits CSK in the heart. Pharmacokinetic studies of ibrutinib show that CSK is tonically inhibited by ibrutinib as plateau concentrations of ibrutinib continuously exceed its IC50 for CSK.49 It is possible that long-term administration of ibrutinib suppresses CSK, which leads to increased inflammation and fibrosis predisposing the heart to AF.
Limited studies have started to investigate the arrhythmogenic mechanisms contributed to ibrutinib-associated AF. In a mouse model of 4-week ibrutinib administration, Jiang et al observed higher AF inducibility, increased LA mass and fibrosis, and abnormal calcium handling related to increased CaMKII activity.22 They concluded that structural remodeling and Ca2+ handling disorders in the atrium are likely the underlying mechanisms for ibrutinib-induced AF. Our results support structural remodeling of the left atrium accounting for the proarrhythmic effects of ibrutinib. Furthermore, our observation that short-term ibrutinib treatment did not increase inducible AF and long-term ibrutinib did not alter AP and INa in LA cardiomyocytes suggests that electric remodeling is not the underlying mechanism for ibrutinib-associated AF. We also observed increased CaMKII phosphorylation in the LA after long-term ibrutinib treatment. CaMKII is a multifunctional protein in the cardiomyocytes acting as a link between Ca2+ dysregulation and AF.50 Activation of CaMKII can trigger inflammatory gene expression and activation of the nucleotide oligomerization domain–like receptor pyrin domain-containing protein 3 inflammasome in the cardiomyocytes in response to stress signals.51 Increased CaMKII activity in long-term ibrutinib exposure likely contributes to increased inflammation and may increase phosphorylation of downstream Ca2+ handling proteins.22,50 However, further work will be required to determine the links between CSK and CaMKII.
To determine whether CSK inhibition may increase risk of AF in other settings, we performed a pharmacoepidemiologic study of approved anticancer kinase inhibitors and their associated AF cases using the VigiBase drug side effects database. We found that drugs that inhibit CSK are more highly associated with AF adverse drug reactions than those that do not. This points to CSK as a general AF target that should be avoided during drug development. Interestingly, there are more selective BTK inhibitors, including acalabrutinib, that do not inhibit CSK. While the data with acalabrutinib are preliminary, it is thought to have less risk of AF compared with ibrutinib. CSK inhibition is not thought to be necessary for ibrutinib’s efficacy. While early results from acalabrutinib studies have demonstrated efficacy in the same range as ibrutinib, we await the results of direct head to head comparison between these 2 drugs, which is underway (https://www.clinicaltrials.gov; Unique identifier: NCT02477696).37,52
In recent years, ibrutinib has been moved to earlier and front-line therapy for a number of different hematologic malignancies particularly CLL; however, this has been accompanied by considerable challenges for the clinician. In particular, the presence of AF in ibrutinib-treated patients is accompanied by an increased risk of bleeding making the introduction of anticoagulation challenging.53 Therefore, there is a pressing need to better delineate mechanisms for ibrutinib-associated AF. Our data also has implications for other BTK inhibitors that are being tested clinically. Although the data are still unclear with respect to AF risk associated with these new agents, the inhibition of CSK by these other kinase inhibitors may be determinative in any AF signals seen. Our data suggests a previously unappreciated role for CSK in AF and may have implications for AF more broadly. Cardiooncology has emerged as a clinical need because of the explosion of cancer therapies and adverse effects on the cardiovascular system. Because of the targeted nature of novel oncology drugs, cardiovascular sequelae (initially observed in patients) may provide new insights into cardiovascular biology.54
We acknowledge several limitations in our studies. We observed a less severe cardiac phenotype in ibrutinib-treated mice compared with cardiac deletion of Csk. This may be explained by the fact that ibrutinib not only inhibits CSK, activating Src family kinase members, but also less potently inhibits several of the Src family kinases themselves, possibly blunting the effects of CSK inhibition. The pharmacological agents we used have off-target effects, as well. For instance, saracatinib inhibits FYN, but also SRC and ABL.38 Whenever possible we also analyzed genetic knockout models, which are more specific. We acknowledge that murine AF is different in many ways from human AF and thus may not model many aspects of the human arrhythmia.
In conclusion, our results demonstrate a previously unappreciated role for CSK and downstream targets in arrhythmogenesis and AF specifically. Further, our findings that kinase inhibitors with potent activity against CSK are significantly associated with AF, compared with those that do not, strongly suggest that CSK inhibition should be avoided during drug development. It remains to be seen whether perturbations in CSK and downstream targets contribute to AF seen in the general population.
Supplementary Material
Clinical Perspective.
What Is New?
We found that ibrutinib-induced atrial fibrillation in a murine model is accompanied by atrial fibrosis, remodeling, and increased inflammation.
We also found that the proarrhythmic side effect of ibrutinib, causing atrial fibrillation, is not a result of its on-target inhibition of Bruton tyrosine kinase, but instead is a result of off-target inhibition of C-terminal Src kinase.
What Are the Clinical Implications?
Second generation Bruton tyrosine kinase inhibitors with increased specificity that avoid C-terminal Src kinase inhibition may also avoid the proarrhythmic side effects of ibrutinib.
Caution should be exercised with drugs that potently inhibit C-terminal Src kinase as they confer increased risk of atrial fibrillation.
Acknowledgments
The authors thank Dr Federico Damilano for his help in murine cardiomyocyte isolation. Dr Xiao conceived the project, designed and performed experiments, analyzed and interpreted data, created the figures, and wrote the manuscript. Dr Salem designed and performed pharmacovigilance analysis, interpreted data, and wrote the related part of the manuscript. Drs Clauss, Hanley, Bapat, and Hulsmans performed experiments and analyzed data. Y. Iwamoto, G. Wojtkiewicz, and J. Tedeschi performed experiments. Drs Cetinbas and Schloss analyzed data. Dr Lebrun-Vignes helped to access the VigiBase database. Dr Lundby provided human proteomic data. Drs Lundby, Sadreyev, Moslehi, Nahrendorf, and Ellinor provided intellectual input and edited the manuscript. Dr Milan conceived the project, designed experiments, interpreted data, and wrote the manuscript.
Sources of Funding
Dr Xiao is supported by an American Heart Association Career Development Award (20CDA35260081). Dr Clauss is supported by a Marie Curie International Outgoing Fellowship within the 7th European Community Framework Program (PIOF-GA-2012–328352) and by the German Center for Cardiovascular Research (DZHK; 81X2600210, 81X2600204). Dr Bapat is supported by the NIH grant T32HL007604. Dr Hulsmans was supported by an American Heart Association Career Development Award (19CDA34490005). Dr Schloss is supported by the NIH grant T32HL076136 and Deutsche Forschungsgemeinschaft (SCHL 2221/1–1). Dr Moslehi was supported by a Vanderbilt-Ingram Cancer Center Young Ambassadors and the NIH (R56 HL141466 and R01 HL141466). Dr Nahrendorf is supported by the NIH grant HL139598. Dr Ellinor is supported by grants from the Fondation Leducq (14CVD01), the American Heart Association (18SFRN34110082), and the NIH (1RO1HL092577, R01HL128914, K24HL105780). Dr Milan is supported by NIH grant HL132905.
Footnotes
Disclosures
None.
The online-only Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/circulationaha.120.049210.
Contributor Information
Ling Xiao, Cardiovascular Research Center, Massachusetts General Hospital and Harvard Medical School, Boston, MA..
Joe-Elie Salem, Clinical Pharmacology, Sorbonne University, INSERM, APHP, UNICO-GRECO Cardio-oncology Program, Sorbonne University, ISERM, APHP, UNICO-GRECO Cardio-oncology Program, Hospital Pitié-Salpêtrière, Paris, France.; Clinical Investigation Center, Paris, France Vanderbilt University Medical Center, Cardio-Oncology Program, Division of Cardiovascular Medicine, Nashville, TN.
Sebastian Clauss, Cardiovascular Research Center, Massachusetts General Hospital and Harvard Medical School, Boston, MA.; Department of Medicine I, Klinikum Grosshadern, University of Munich, Germany
Alan Hanley, Cardiovascular Research Center, Massachusetts General Hospital and Harvard Medical School, Boston, MA..
Aneesh Bapat, Cardiovascular Research Center, Massachusetts General Hospital and Harvard Medical School, Boston, MA..
Maarten Hulsmans, Center for Systems Biology, Department of Radiology, Massachusetts General Hospital and Harvard Medical School, Boston, MA..
Yoshiko Iwamoto, Center for Systems Biology, Department of Radiology, Massachusetts General Hospital and Harvard Medical School, Boston, MA..
Gregory Wojtkiewicz, Center for Systems Biology, Department of Radiology, Massachusetts General Hospital and Harvard Medical School, Boston, MA..
Murat Cetinbas, Department of Molecular Biology, Massachusetts General Hospital and Harvard Medical School, Boston, MA.; Department of Genetics, Harvard Medical School, Boston, MA
Maximilian J. Schloss, Center for Systems Biology, Department of Radiology, Massachusetts General Hospital and Harvard Medical School, Boston, MA..
Justin Tedeschi, Cardiovascular Research Center, Massachusetts General Hospital and Harvard Medical School, Boston, MA..
Bénédicte Lebrun-Vignes, Clinical Pharmacology, Sorbonne University, INSERM, APHP, UNICO-GRECO Cardio-oncology Program, Sorbonne University, ISERM, APHP, UNICO-GRECO Cardio-oncology Program, Hospital Pitié-Salpêtrière, Paris, France.; Clinical Pharmacology and Regional Pharmacovigilance Center, Sorbonne University, ISERM, APHP, UNICO-GRECO Cardio-oncology Program, Hospital Pitié-Salpêtrière, Paris, France. Université Paris Est (UPEC), IRMB- EA 7379 EpiDermE (Epidemiology in Dermatology and Evaluation of Therapeutics), F-94010, Créteil, France
Alicia Lundby, Department of Biomedical Sciences, Faculty of Health and Medical Sciences and NNF Center for Protein Research, Københavns Universitet, Copenhagen, Denmark.
Ruslan I. Sadreyev, Department of Pathology, Massachusetts General Hospital and Harvard Medical School, Boston, MA..
Javid Moslehi, Vanderbilt University Medical Center, Cardio-Oncology Program, Division of Cardiovascular Medicine, Nashville, TN.
Matthias Nahrendorf, Cardiovascular Research Center, Massachusetts General Hospital and Harvard Medical School, Boston, MA.; Center for Systems Biology, Department of Radiology, Massachusetts General Hospital and Harvard Medical School, Boston, MA.
Patrick T. Ellinor, Cardiovascular Research Center, Massachusetts General Hospital and Harvard Medical School, Boston, MA.; Cardiovascular Disease Initiative, Broad Institute of Harvard and MIT, Cambridge, MA
David J. Milan, Cardiovascular Research Center, Massachusetts General Hospital and Harvard Medical School, Boston, MA.; Leducq Foundation, Boston, MA
REFERENCES
- 1.Burger JA, Tedeschi A, Barr PM, Robak T, Owen C, Ghia P, Bairey O, Hillmen P, Bartlett NL, Li J, et al. ; RESONATE-2 Investigators. Ibrutinib as initial therapy for patients with chronic lymphocytic leukemia. N Engl J Med. 2015;373:2425–2437. doi: 10.1056/NEJMoa1509388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang ML, Rule S, Martin P, Goy A, Auer R, Kahl BS, Jurczak W, Advani RH, Romaguera JE, Williams ME, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2013;369:507–516. doi: 10.1056/NEJMoa1306220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Woyach JA, Ruppert AS, Heerema NA, Zhao W, Booth AM, Ding W, Bartlett NL, Brander DM, Barr PM, Rogers KA, et al. Ibrutinib regimens versus chemoimmunotherapy in older patients with untreated CLL. N Engl J Med. 2018;379:2517–2528. doi: 10.1056/NEJMoa1812836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shanafelt TD, Wang XV, Kay NE, Hanson CA, O’Brien S, Barrientos J, Jelinek DF, Braggio E, Leis JF, Zhang CC, et al. Ibrutinib-rituximab or chemoimmunotherapy for chronic lymphocytic leukemia. N Engl J Med. 2019;381:432–443. doi: 10.1056/NEJMoa1817073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown JR, Moslehi J, O’Brien S, Ghia P, Hillmen P, Cymbalista F, Shanafelt TD, Fraser G, Rule S, Kipps TJ, et al. Characterization of atrial fibrillation adverse events reported in ibrutinib randomized controlled registration trials. Haematologica. 2017;102:1796–1805. doi: 10.3324/haematol.2017.171041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gustine JN, Meid K, Dubeau TE, Treon SP, Castillo JJ. Atrial fibrillation associated with ibrutinib in Waldenström macroglobulinemia. Am J Hematol. 2016;91:E312–E313. doi: 10.1002/ajh.24366 [DOI] [PubMed] [Google Scholar]
- 7.Lee HJ, Chihara D, Wang M, Mouhayar E, Kim P. Ibrutinib-related atrial fibrillation in patients with mantle cell lymphoma. Leuk Lymphoma. 2016;57:2914–2916. doi: 10.3109/10428194.2016.1169408 [DOI] [PubMed] [Google Scholar]
- 8.Leong DP, Caron F, Hillis C, Duan A, Healey JS, Fraser G, Siegal D. The risk of atrial fibrillation with ibrutinib use: a systematic review and meta-analysis. Blood. 2016;128:138–140. doi: 10.1182/blood-2016-05-712828 [DOI] [PubMed] [Google Scholar]
- 9.McMullen JR, Boey EJ, Ooi JY, Seymour JF, Keating MJ, Tam CS. Ibrutinib increases the risk of atrial fibrillation, potentially through inhibition of cardiac PI3K-Akt signaling. Blood. 2014;124:3829–3830. doi: 10.1182/blood-2014-10-604272 [DOI] [PubMed] [Google Scholar]
- 10.Thompson PA, Lévy V, Tam CS, Al Nawakil C, Goudot FX, Quinquenel A, Ysebaert L, Michallet AS, Dilhuydy MS, Van Den Neste E, et al. Atrial fibrillation in CLL patients treated with ibrutinib. An international retrospective study. Br J Haematol. 2016;175:462–466. doi: 10.1111/bjh.14324 [DOI] [PubMed] [Google Scholar]
- 11.Thorp BC, Badoux X. Atrial fibrillation as a complication of ibrutinib therapy: clinical features and challenges of management. Leuk Lymphoma. 2018;59:311–320. doi: 10.1080/10428194.2017.1339874 [DOI] [PubMed] [Google Scholar]
- 12.Farooqui M, Valdez J, Soto S, Bray A, Tian X, Wiestner A. Atrial fibrillation in cll/sll patients on ibrutinib. Blood. 2015;126:2933. [Google Scholar]
- 13.Mato AR, Clasen S, Pickens P, Gashonia L, Rhodes J, Svoboda J, Hughes M, Nabhan C, Ali N, Schuster S, et al. Left atrial abnormality (LAA) as a predictor of ibrutinib-associated atrial fibrillation in patients with chronic lymphocytic leukemia. Cancer Biol Ther. 2018;19:1–2. doi: 10.1080/15384047.2017.1394554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mato AR, Hill BT, Lamanna N, Barr PM, Ujjani CS, Brander DM, Howlett C, Skarbnik AP, Cheson BD, Zent CS, et al. Optimal sequencing of ibrutinib, idelalisib, and venetoclax in chronic lymphocytic leukemia: results from a multicenter study of 683 patients. Ann Oncol. 2017;28:1050–1056. doi: 10.1093/annonc/mdx031 [DOI] [PubMed] [Google Scholar]
- 15.Caron F, Leong DP, Hillis C, Fraser G, Siegal D. Current understanding of bleeding with ibrutinib use: a systematic review and meta-analysis. Blood Adv. 2017;1:772–778. doi: 10.1182/bloodadvances.2016001883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown JR, Moslehi J, Ewer MS, O’Brien SM, Ghia P, Cymbalista F, Shanafelt TD, Fraser G, Rule S, Coutre SE, et al. Incidence of and risk factors for major haemorrhage in patients treated with ibrutinib: An integrated analysis. Br J Haematol. 2019;184:558–569. doi: 10.1111/bjh.15690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ganatra S, Sharma A, Shah S, Chaudhry GM, Martin DT, Neilan TG, Mahmood SS, Barac A, Groarke JD, Hayek SS, et al. Ibrutinib-associated atrial fibrillation. J Am Coll Cardiol. Clin Electrophysiol. 2018;4:1491–1500. doi: 10.1016/j.jacep.2018.06.004 [DOI] [PubMed] [Google Scholar]
- 18.Schmedt C, Saijo K, Niidome T, Kühn R, Aizawa S, Tarakhovsky A. Csk controls antigen receptor-mediated development and selection of T-lineage cells. Nature. 1998;394:901–904. doi: 10.1038/29802 [DOI] [PubMed] [Google Scholar]
- 19.Sohal DS, Nghiem M, Crackower MA, Witt SA, Kimball TR, Tymitz KM, Penninger JM, Molkentin JD. Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ Res. 2001;89:20–25. doi: 10.1161/hh1301.092687 [DOI] [PubMed] [Google Scholar]
- 20.Dubovsky JA, Flynn R, Du J, Harrington BK, Zhong Y, Kaffenberger B, Yang C, Towns WH, Lehman A, Johnson AJ, et al. Ibrutinib treatment ameliorates murine chronic graft-versus-host disease. J Clin Invest. 2014;124:4867–4876. doi: 10.1172/JCI75328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ponader S, Chen SS, Buggy JJ, Balakrishnan K, Gandhi V, Wierda WG, Keating MJ, O’Brien S, Chiorazzi N, Burger JA. The Bruton tyrosine kinase inhibitor PCI-32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo. Blood. 2012;119:1182–1189. doi: 10.1182/blood-2011-10-386417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang L, Li L, Ruan Y, Zuo S, Wu X, Zhao Q, Xing Y, Zhao X, Xia S, Bai R, et al. Ibrutinib promotes atrial fibrillation by inducing structural remodeling and calcium dysregulation in the atrium. Heart Rhythm. 2019;16:1374–1382. doi: 10.1016/j.hrthm.2019.04.008 [DOI] [PubMed] [Google Scholar]
- 23.Decker S, Zwick A, Khaja Saleem S, Kissel S, Rettig A, Aumann K, Dierks C. Optimized xenograft protocol for chronic lymphocytic leukemia results in high engraftment efficiency for all cll subgroups. Int J Mol Sci. 2019;20(24):6277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mahida S, Mills RW, Tucker NR, Simonson B, Macri V, Lemoine MD, Das S, Milan DJ, Ellinor PT. Overexpression of KCNN3 results in sudden cardiac death. Cardiovasc Res. 2014;101:326–334. doi: 10.1093/cvr/cvt269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yao C, Veleva T, Scott L Jr, Cao S, Li L, Chen G, Jeyabal P, Pan X, Alsina KM, et al. Enhanced cardiomyocyte NLRP3 inflammasome signaling promotes atrial fibrillation. Circulation. 2018;138:2227–2242. doi: 10.1161/CIRCULATIONAHA.118.035202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nomanbhoy TK, Sharma G, Brown H, Wu J, Aban A, Vogeti S, Alemayehu S, Sykes M, Rosenblum JS, Kozarich JW. Chemoproteomic evaluation of target engagement by the cyclin-dependent kinase 4 and 6 inhibitor palbociclib correlates with cancer cell response. Biochemistry. 2016;55:5434–5441. doi: 10.1021/acs.biochem.6b00629 [DOI] [PubMed] [Google Scholar]
- 28.Lundby A, Rossin EJ, Steffensen AB, Acha MR, Newton-Cheh C, Pfeufer A, Lynch SN, Olesen SP, Brunak S, Ellinor PT, et al. ; QT Interval International GWAS Consortium (QT-IGC). Annotation of loci from genome-wide association studies using tissue-specific quantitative interaction proteomics. Nat Methods. 2014;11:868–874. doi: 10.1038/nmeth.2997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lundby A, Andersen MN, Steffensen AB, Horn H, Kelstrup CD, Francavilla C, Jensen LJ, Schmitt N, Thomsen MB, Olsen JV. In vivo phosphoproteomics analysis reveals the cardiac targets of β-adrenergic receptor signaling. Sci Signal. 2013;6:rs11. doi: 10.1126/scisignal.2003506 [DOI] [PubMed] [Google Scholar]
- 30.Lindquist M. Vigibase, the who global icsr database system: basic facts. Drug Information Journal. 2008;42:409–419. [Google Scholar]
- 31.Tuomi JM, Xenocostas A, Jones DL. Increased susceptibility for atrial and ventricular cardiac arrhythmias in mice treated with a single high dose of ibrutinib. Can J Cardiol. 2018;34:337–341. doi: 10.1016/j.cjca.2017.12.001 [DOI] [PubMed] [Google Scholar]
- 32.Wu H, Wang W, Liu F, Weisberg EL, Tian B, Chen Y, Li B, Wang A, Wang B, Zhao Z, et al. Discovery of a potent, covalent BTK inhibitor for B-cell lymphoma. ACS Chem Biol. 2014;9:1086–1091. doi: 10.1021/cb4008524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berning AK, Eicher EM, Paul WE, Scher I. Mapping of the X-linked immune deficiency mutation (xid) of CBA/N mice. J Immunol. 1980;124:1875–1877. [PubMed] [Google Scholar]
- 34.Rawlings DJ, Saffran DC, Tsukada S, Largaespada DA, Grimaldi JC, Cohen L, Mohr RN, Bazan JF, Howard M, Copeland NG. Mutation of unique region of Bruton’s tyrosine kinase in immunodeficient XID mice. Science. 1993;261:358–361. doi: 10.1126/science.8332901 [DOI] [PubMed] [Google Scholar]
- 35.Cravatt BF, Wright AT, Kozarich JW. Activity-based protein profiling: from enzyme chemistry to proteomic chemistry. Annu Rev Biochem. 2008;77:383–414. doi: 10.1146/annurev.biochem.75.101304.124125 [DOI] [PubMed] [Google Scholar]
- 36.Ferguson FM, Gray NS. Kinase inhibitors: the road ahead. Nat Rev Drug Discov. 2018;17:353–377. doi: 10.1038/nrd.2018.21 [DOI] [PubMed] [Google Scholar]
- 37.Byrd JC, Harrington B, O’Brien S, Jones JA, Schuh A, Devereux S, Chaves J, Wierda WG, Awan FT, Brown JR, et al. Acalabrutinib (ACP-196) in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374:323–332. doi: 10.1056/NEJMoa1509981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kaufman AC, Salazar SV, Haas LT, Yang J, Kostylev MA, Jeng AT, Robinson SA, Gunther EC, van Dyck CH, Nygaard HB, et al. Fyn inhibition rescues established memory and synapse loss in Alzheimer mice. Ann Neurol. 2015;77:953–971. doi: 10.1002/ana.24394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stein PL, Lee HM, Rich S, Soriano P. pp59fyn mutant mice display differential signaling in thymocytes and peripheral T cells. Cell. 1992;70:741–750. doi: 10.1016/0092-8674(92)90308-y [DOI] [PubMed] [Google Scholar]
- 40.Ahern CA, Zhang JF, Wookalis MJ, Horn R. Modulation of the cardiac sodium channel NaV1.5 by Fyn, a Src family tyrosine kinase. Circ Res. 2005;96:991–998. doi: 10.1161/01.RES.0000166324.00524.dd [DOI] [PubMed] [Google Scholar]
- 41.Zhou Z, Rawnsley DR, Goddard LM, Pan W, Cao XJ, Jakus Z, Zheng H, Yang J, Arthur JS, Whitehead KJ, et al. The cerebral cavernous malformation pathway controls cardiac development via regulation of endocardial MEKK3 signaling and KLF expression. Dev Cell. 2015;32:168–180. doi: 10.1016/j.devcel.2014.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nada S, Okada M, MacAuley A, Cooper JA, Nakagawa H. Cloning of a complementary DNA for a protein-tyrosine kinase that specifically phosphorylates a negative regulatory site of p60c-src. Nature. 1991;351:69–72. doi: 10.1038/351069a0 [DOI] [PubMed] [Google Scholar]
- 43.Nada S, Yagi T, Takeda H, Tokunaga T, Nakagawa H, Ikawa Y, Okada M, Aizawa S. Constitutive activation of Src family kinases in mouse embryos that lack Csk. Cell. 1993;73:1125–1135. doi: 10.1016/0092-8674(93)90642-4 [DOI] [PubMed] [Google Scholar]
- 44.Salem JE, Manouchehri A, Moey M, Lebrun-Vignes B, Bastarache L, Pariente A, Gobert A, Spano JP, Balko JM, Bonaca MP, et al. Cardiovascular toxicities associated with immune checkpoint inhibitors: an observational, retrospective, pharmacovigilance study. Lancet Oncol. 2018;19:1579–1589. doi: 10.1016/S1470-2045(18)30608-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Okada M Regulation of the SRC family kinases by Csk. Int J Biol Sci. 2012;8:1385–1397. doi: 10.7150/ijbs.5141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Imamoto A, Soriano P. Disruption of the csk gene, encoding a negative regulator of Src family tyrosine kinases, leads to neural tube defects and embryonic lethality in mice. Cell. 1993;73:1117–1124. doi: 10.1016/0092-8674(93)90641-3 [DOI] [PubMed] [Google Scholar]
- 47.Berman-Booty LD, Eraslan R, Hanumegowda U, Cantor GH, Bounous DI, Janovitz EB, Jones BK, Buiakova O, Hayward M, Wee S. Systemic loss of C-terminal Src kinase expression elicits dpontaneous duppurative inflammation in conditional knockout mice. Vet Pathol. 2018;55:331–340. doi: 10.1177/0300985817747330 [DOI] [PubMed] [Google Scholar]
- 48.Thomas RM, Schmedt C, Novelli M, Choi BK, Skok J, Tarakhovsky A, Roes J. C-terminal SRC kinase controls acute inflammation and granulocyte adhesion. Immunity. 2004;20:181–191. doi: 10.1016/s1074-7613(04)00023-8 [DOI] [PubMed] [Google Scholar]
- 49.Scheers E, Leclercq L, de Jong J, Bode N, Bockx M, Laenen A, Cuyckens F, Skee D, Murphy J, Sukbuntherng J, et al. Absorption, metabolism, and excretion of oral ¹4C radiolabeled ibrutinib: an open-label, phase I, single-dose study in healthy men. Drug Metab Dispos. 2015;43:289–297. doi: 10.1124/dmd.114.060061 [DOI] [PubMed] [Google Scholar]
- 50.Heijman J, Voigt N, Wehrens XH, Dobrev D. Calcium dysregulation in atrial fibrillation: the role of CaMKII. Front Pharmacol. 2014;5:30. doi: 10.3389/fphar.2014.00030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Suetomi T, Willeford A, Brand CS, Cho Y, Ross RS, Miyamoto S, Brown JH. Inflammation and NLRP3 inflammasome activation initiated in response to pressure overload by Ca2+/calmodulin-dependent protein kinase II δ signaling in cardiomyocytes are essential for adverse cardiac remodeling. Circulation. 2018;138:2530–2544. doi: 10.1161/CIRCULATIONAHA.118.034621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang M, Rule S, Zinzani PL, Goy A, Casasnovas O, Smith SD, Damaj G, Doorduijn J, Lamy T, Morschhauser F, et al. Acalabrutinib in relapsed or refractory mantle cell lymphoma (ACE-LY-004): a single-arm, multicentre, phase 2 trial. Lancet. 2018;391:659–667. doi: 10.1016/S0140-6736(17)33108-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Salem JE, Manouchehri A, Bretagne M, Lebrun-Vignes B, Groarke JD, Johnson DB, Yang T, Reddy NM, Funck-Brentano C, Brown JR, et al. Cardiovascular toxicities associated with ibrutinib. J Am Coll Cardiol. 2019;74:1667–1678. doi: 10.1016/j.jacc.2019.07.056 [DOI] [PubMed] [Google Scholar]
- 54.Bellinger AM, Arteaga CL, Force T, Humphreys BD, Demetri GD, Druker BJ, Moslehi JJ. Cardio-oncology: how new targeted cancer therapies and precision medicine can inform cardiovascular discovery. Circulation. 2015;132:2248–2258. doi: 10.1161/CIRCULATIONAHA.115.010484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Anders S, Pyl PT, Huber W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bate A, Lindquist M, Edwards IR, Olsson S, Orre R, Lansner A, De Freitas RM. A Bayesian neural network method for adverse drug reaction signal generation. Eur J Clin Pharmacol. 1998;54:315–321. doi: 10.1007/s002280050466 [DOI] [PubMed] [Google Scholar]
- 58.Mertz P, Lebrun-Vignes B, Salem JE, Arnaud L. Characterizing drug-induced capillary leak syndromes using the World Health Organization VigiBase. J Allergy Clin Immunol. 2019;143:433–436. doi: 10.1016/j.jaci.2018.09.001 [DOI] [PubMed] [Google Scholar]
- 59.Norén GN, Hopstadius J, Bate A. Shrinkage observed-to-expected ratios for robust and transparent large-scale pattern discovery. Stat Methods Med Res. 2013;22:57–69. doi: 10.1177/0962280211403604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.De Bruin ML, Pettersson M, Meyboom RH, Hoes AW, Leufkens HG. Anti-HERG activity and the risk of drug-induced arrhythmias and sudden death. Eur Heart J. 2005;26:590–597. doi: 10.1093/eurheartj/ehi092 [DOI] [PubMed] [Google Scholar]
- 61.Grouthier V, Lebrun-Vignes B, Glazer AM, Touraine P, Funck-Brentano C, Pariente A, Courtillot C, Bachelot A, Roden DM, Moslehi JJ, et al. Increased long QT and torsade de pointes reporting on tamoxifen compared with aromatase inhibitors. Heart. 2018;104:1859–1863. doi: 10.1136/heartjnl-2017-312934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rothman KJ, Lanes S, Sacks ST. The reporting odds ratio and its advantages over the proportional reporting ratio. Pharmacoepidemiol Drug Saf. 2004;13:519–523. doi: 10.1002/pds.1001 [DOI] [PubMed] [Google Scholar]
- 63.Salem JE, Dureau P, Bachelot A, Germain M, Voiriot P, Lebourgeois B, Trégouët DA, Hulot JS, Funck-Brentano C. Association of oral contraceptives with drug-induced QT interval prolongation in healthy nonmenopausal women. JAMA Cardiol. 2018;3:877–882. doi: 10.1001/jamacardio.2018.2251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pfuma E, Bullock J, Orbach RC, Habtemariam B, Pan Y, Marathe A, Zhao P. FDA center for drug evaluation and research clinical pharmacology and biopharmaceutics review: ibrutinib. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/205552orig1s000clinpharmr.pdf. 2013. (Date accessed: June 1, 2019)
- 65.Agency EME. Tasigna European public assessment report scientific discussion. https://www.ema.europa.eu/en/documents/scientific-discussion/tasigna-epar-scientific-discussion_en.pdf. 2007. (Date accessed: June 1, 2019)
- 66.Christmann-Franck S, van Westen GJ, Papadatos G, Beltran Escudie F, Roberts A, Overington JP, Domine D. Unprecedently large-scale kinase inhibitor set enabling the accurate prediction of compound-kinase activities: a way toward selective promiscuity by design? J Chem Inf Model. 2016;56:1654–1675. doi: 10.1021/acs.jcim.6b00122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.FDA U. Highlights of prescribing information: Bosulif. https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/203341lbl.pdf. 2012. (Date accessed: June 1, 2019)
- 68.Wang X, Roy A, Hochhaus A, Kantarjian HM, Chen TT, Shah NP. Differential effects of dosing regimen on the safety and efficacy of dasatinib: retrospective exposure-response analysis of a Phase III study. Clin Pharmacol. 2013;5:85–97. doi: 10.2147/CPAA.S42796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.FDA U. Highlights of prescribing information: Zelboraf (vemurafenib). https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/202429s000lbl.pdf. 2011. (Date accessed: June 1, 2019)
- 70.Agency EME. Nexavar European public assessment report scientific discussion. https://www.ema.europa.eu/en/documents/scientific-discussion/nexavar-epar-scientific-discussion_en.pdf. 2006. (Date accessed: June 1, 2019)
- 71.Peng B, Lloyd P, Schran H. Clinical pharmacokinetics of imatinib. Clin Pharmacokinet. 2005;44:879–894. doi: 10.2165/00003088-200544090-00001 [DOI] [PubMed] [Google Scholar]
- 72.Schuck R, Yu J, Zhao H, Orbach RC, Zhao L, Wang Y. FDA center for drug evaluation and research clinical pharmacology and biopharmaceutics review: Iressa (gefitinib). https://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/206995orig1s000clinpharmr.pdf. 2014. (Date accessed: June 1, 2019)
- 73.Anastassiadis T, Deacon SW, Devarajan K, Ma H, Peterson JR. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat Biotechnol. 2011;29:1039–1045. doi: 10.1038/nbt.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Orbach RC, Zineh I. FDA center for drug evaluation and research clinical pharmacology and biopharmaceutics review: Crizotinib. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/206995orig1s000clinpharmr.pdf. 2011. (Date accessed: June 1, 2019)
- 75.FDA U. Highlights of prescribing information: Inlyta (axitinib). https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/202324lbl.pdf. 2012. (Date accessed: June 1, 2019)
- 76.Ling J, Fettner S, Lum BL, Riek M, Rakhit A. Effect of food on the pharmacokinetics of erlotinib, an orally active epidermal growth factor receptor tyrosine-kinase inhibitor, in healthy individuals. Anticancer Drugs. 2008;19:209–216. doi: 10.1097/CAD.0b013e3282f2d8e4 [DOI] [PubMed] [Google Scholar]
- 77.Agency EME. Sutent European Public Assessment report product information. https://www.ema.europa.eu/en/documents/product-information/sutent-epar-product-information_en.pdf. 2016. (Date accessed: June 1, 2019)
- 78.Moon YJ, Song P, Marathe A, Booth B, Garnett C. FDA center for drug evaluation and research clinical pharmacology and biopharmaceutics review: Vandetanib. https://www.accessdata.fda.gov/drugsatfda_docs/nd
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.