

Abstract
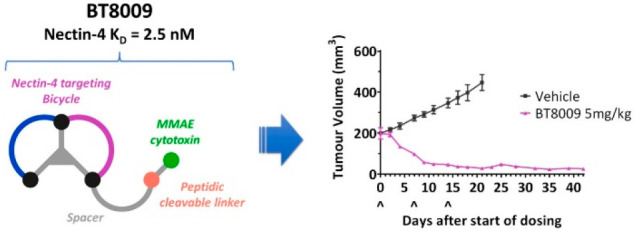
Bicycle toxin conjugates (BTCs) are a promising new class of molecules for targeted delivery of toxin payloads into tumors. Herein we describe the discovery of BT8009, a Nectin-4 targeting BTC currently under clinical evaluation. Nectin-4 is overexpressed in multiple tumor types and is a clinically validated target for selective delivery of cytotoxic payloads. A Nectin-4 targeting bicyclic peptide was identified by phage display, which showed highly selective binding for Nectin-4 but suffered from low plasma stability and poor physicochemical properties. Multiparameter chemical optimization involving introduction of non-natural amino acids resulted in a lead Bicycle that demonstrated high affinity for Nectin-4, good stability in biological matrices, and a much-improved physicochemical profile. The optimized Bicycle was conjugated to the cytotoxin Monomethyl auristatin E via a cleavable linker to give the targeted drug conjugate BT8009, which demonstrates potent anticancer activity in in vivo rodent models.
Introduction
Bicycle toxin conjugates (BTCs) are a new class of anticancer agents that allow efficient and targeted delivery of toxin payloads into tumors.1−3 They consist of a highly constrained, tumor-targeting synthetic bicyclic peptide that is conjugated to a cytotoxic payload via a cleavable linker, which allows payload release in the tumor microenvironment.1,4
BTCs aim to address the shortcomings of antibody drug conjugates (ADCs) in several ways. First, the small size of BTCs (∼4 kDa) compared to large biologic entities such as monoclonal antibody (mAb)-based conjugates (∼150 kDa) allows rapid distribution to tissues and extensive tumor penetration, which enables rapid delivery of payload into the tumor.1,5 Second, the peptidic nature of BTCs results in a short duration of systemic exposure (∼1 h) and liver-sparing renal elimination. These properties limit the body’s exposure to payload and should therefore minimize damage to normal tissue. 1
Two previously reported BTCs, namely, BT1718 and BT5528 (Figure 1), which target MT1-MMP and EphA2, respectively, show potent anticancer activity in preclinical studies1,3 and are currently being assessed in the clinic,6 with preliminary clinical pharmacokinetic (PK) results for BT1718 demonstrating concentrations of payload in tumors are similar to those observed in preclinical rodent models.2 We have now sought to employ BTCs against additional cancer targets, including Nectin-4.
Figure 1.

Structures of previously reported BTCs BT1718 and BT5528.
Nectin-4 is a cell adhesion molecule from the Nectin and Nectin-like family. It is a clinically validated tumor target and has been reported to be highly expressed in a wide range of solid tumors, including bladder, esophageal, pancreatic, and lung, but with limited distribution in healthy tissues.7−11 An antibody drug conjugate (PADCEV, enfortumab vedotin) that targets Nectin-4 was recently approved for the treatment of bladder cancer following the generation of positive data in a number of clinical studies.12
Herein we describe the discovery via phage display and subsequent chemical optimization of a Nectin-4 binding Bicycle and its incorporation into BT8009, a BTC that is currently under clinical evaluation. A detailed report of the pharmacologic properties of BT8009 has recently been described.13 In this, BT8009 shows potent efficacy in multiple tumor models, including patient-derived xenografts, across a variety of tumor indications and is well-tolerated in preclinical safety studies. In several models it demonstrated superior or equivalent efficacy to an analogue of the ADC PADCEV.13
Results and Discussion
Bicycle Phage Screening against Nectin-4
Bicycles are peptides that are constrained by a small molecular scaffold via three cysteine residues to form a bicyclic structure. The high level of constraint induced by the scaffold holds the Bicycle in the preferred bioactive conformation for optimal binding with a target protein, allowing high-affinity interactions to be achieved, while the large binding footprint of Bicycles often results in exquisite selectivity for the target.14 Hit Bicycles may be identified by phage display, and the large amount of screening data generated through this process informs on structure–activity relationships.15 Due to the synthetic nature of Bicycles, chemical optimization can be employed to introduce drug-like properties and enable their use as therapeutics. The phage screening process also self-selects for molecules that are amenable to conjugation to other molecular payloads such as radionuclides, cytotoxins, or immune cell engaging Bicycles.4,5,16
In order to identify a Nectin-4 binding peptide suitable for use in a BTC, naiv̈e Bicycle phage libraries with completely randomized residues between the three Bicycle-forming cysteine amino acids were screened against the soluble extracellular domain of Nectin-4. Each library presented Bicycles with different loop sizes when cyclized using the scaffold 1,3,5-triacryoyl-1,3,5-triazinane (TATA). Three distinct families of Bicycles, grouped by their conserved binding motifs, were identified as active binders. Fluorescein-labeled analogues of representative sequences from all three Bicycle families were synthesized by solid-phase peptide synthesis (SPPS), and their affinities were measured using fluorescence polarization (FP). These are summarized in Table 1.
Table 1. Sequences and Binding Affinities of Fluorescein-Labeled Analogues Bicycle Families Identified through Phage Display.
| family | Bicycle number | screening stage | sequence | affinity Kd (nM) |
|---|---|---|---|---|
| 1 | 1 | Naïve | ACPFGCHTDWSWPIWCA(Sar6)K(Fl) | 508 |
| 2 | Affinity maturation | ACPFGCMKNWSWPIWCTNK(Sar6)K(Fl) | 20.0 | |
| 2 | 3 | Naïve | ACWPLDSYWCARICA(Sar6)K(Fl) | 208 |
| 4 | Affinity maturation | QKWCAPLSDYWCNRICA(Sar6)K(Fl) | 65.5 | |
| 3 | 5 | Naïve | ACVTTSYDCFLHLLGCA(Sar6)K(Fl) | 888 |
| 6 | Affinity maturation | ACVTTSYDCWVTLGHCSRF(Sar6)K(Fl) | 242 |
Motif residues are highlighted in bold. All sequences cyclized using TATA scaffold. Affinities were measured using fluorescence polarization direct binding. The Geomean of Kd values displayed are all generated from n ≥ 4. Full assay details, including the number of replicates, errors, and competing tracer molecule are in the Supporting Information. (Sar6) = 6 × Sarcosine, (Fl) = 5/6-carboxyfluorescein.
Family 2 showed the highest binding affinity for Nectin-4 following naïve phage screens (Kd = 208 nM), whereas families 1 and 3 gave Bicycles with affinities of 508 and 888 nM, respectively. Affinity maturation was carried out on all families, which consists of additional rounds of phage panning where motif residues are fixed while other residues are randomized, in order to find the optimal sequence for binding. In addition, small linear peptide chains (3–4 amino acids) were incorporated outside the Bicycle loops on either the N-terminus or C-terminus in an attempt to gain additional interactions with the target. Family 1 demonstrated a potency increase of approximately 20-fold to 20 nM upon affinity maturation screening. Families 2 and 3, which have less well-defined motifs, failed to demonstrate a significant improvement in affinity, and based on these data, Family 1 was selected for further investigation.
Hit Bicycle Structure and Binding Profile
The structure of the Family 1 hit Bicycle is shown in Figure 2. The Bicycle has an asymmetric structure with loops containing three residues and nine residues, along with a C-terminal three amino acid extension. The sequence within the loops is highly hydrophobic in nature, with multiple aromatic rings and many nonpolar amino acids. Two lysine residues, one in loop two and the other in the C-terminal extension, are the only charged residues present.
Figure 2.

Structure of hit Nectin-4 binding Bicycle 7 and corresponding fluorescein labeled analogue 2. The three cysteines used for cyclization are highlighted in orange; TATA scaffold highlighted in purple.
A fluorescein-labeled analogue of the hit Bicycle (2) was screened by FP against the extracellular domain (ECD) of Nectin-4 from mouse, rat, and nonhuman primate (cynomolgus monkey) and against the ECD of closely related homologues of Nectin-4. Bicycle 2 demonstrated very good species cross reactivity as well as exquisite selectivity for Nectin-4 (Table 2).
Table 2. Species Cross Reactivity and Selectivity Binding Data for Fluorescein-Labeled Bicycle 2 As Determined by FP Direct Binding.
| species cross reactivity | selectivity | |||||
|---|---|---|---|---|---|---|
| protein | human Nectin-4 ECD | mouse Nectin-4 ECD | rat Nectin-4 ECD | NHP (Cyno) Nectin-4 ECD | Nectin 1–3, 5 ECD | Nectin-like 1–5 ECD |
| Kd (nM) | 19.4 | 42.9 | 91.8 | 49.2 | >2000 | >2000 |
Affinities were measured using fluorescence polarization competition. The Geomean of Ki values displayed are all generated from n ≥ 2. Full assay details, including the number of replicates, errors, and competing tracer molecule are in the Supporting Information.
N-Terminal Capping and Alanine Scan
The phage screening process produces peptides that are flanked at each terminus by an alanine residue. In order to stabilize the peptide to exopeptidases,17 the N-terminal alanine residue from 7 was removed, and the N-terminus was acetylated, which led to a slight improvement in binding affinity (8, Table 3). A modest drop in binding affinity was observed when the C-terminal extension was removed (9); however, by removing the terminal alanine residues and performing N-terminal acetylation on the core Bicycle sequence, it was found that this loss in affinity could be recovered (10).
Table 3. Affinities of Initial Bicycles and Profiles in Physical Chemistry and Plasma Stability Assaysa.
| in vitro
plasma stability (t1/2, h) |
|||||
|---|---|---|---|---|---|
| Bicycle number | sequence† | Ki (nM) | solubility phosphate buffer pH 7.4 (mg/mL) | mouse | human |
| 7 | ACPFGCMKNWSWPIWCTNK | 27.3 | nt | nt | nt |
| 8 | Ac-CPFGCMKNWSWPIWCTNK | 13.5 | 4.4 | 2.2 | >24 |
| 9 | ACPFGCMKNWSWPIWCA | 57.6 | nt | nt | nt |
| 10 | Ac-CPFGCMKNWSWPIWC | 17.6 | 0.16 | 1.9 | >24 |
Core Bicycle sequence highlighted in bold. nt = not tested. Affinities were measured using fluorescence polarization competition. The Geomean of Ki values displayed are all generated from n ≥ 2. Full assay details, including the number of replicates, errors, and competing tracer molecule are in the Supporting Information.
In the absence of structural information, replacing each noncysteine amino acid in the Bicycle structure sequentially with alanine (alanine scanning) provides information on the contribution of each amino acid side chain to the binding affinity. Replacement of amino acids that are involved in key interactions either with the protein or in intramolecular interactions to stabilize the Bicycle secondary structure will result in a large loss of affinity. These residues can then be targeted for further medicinal chemistry optimization through replacement with non-natural amino acids, with the aim to enhance those key interactions and increase binding affinity. Conversely, positions that do not show a significant reduction in binding affinity upon replacement with alanine are not contributing greatly to binding. Therefore, less conservative mutations are often well-tolerated, and therefore these positions can be used to optimize other properties of the Bicycle. For example, solubility may be increased through introduction of hydrophilic or charged amino acids in these positions. d-Alanine scanning in parallel reveals positions where incorporation of d-amino acids is tolerated. This is useful, as introduction of d-amino acids into a peptide sequence is a well-known strategy for stabilizing against proteolytic degradation.18,19
Table S7 shows the results of the alanine scan performed on 7. These data demonstrate that approximately half of the amino acids appear essential for binding, namely, phenylalanine 2, methionine 4, tryptophan 7, serine 8, proline 10, and tryptophan 12. Conversely, several residues do not appear to contribute greatly to binding, and replacement with alanine has little or no effect on affinity, namely, lysine 5, asparagine 6, isoleucine 11, and the entire linear C-terminal extension. The results of the d-alanine scan also proved to be very valuable, in particular, the observation that replacing glycine 3 with d-alanine was very well-tolerated.
The fact that the three amino acids within the C-terminal extension could be equally well replaced by l- or d-alanine reinforced the earlier observation that they are not contributing greatly to binding affinity. Bicycle 10, which lacks the C-terminal extension but shows comparable binding affinity to that of hit compound 7, was therefore chosen as the basis for chemical optimization.
Although this Bicycle family demonstrates high affinity and a high degree of selectivity for Nectin-4, further enhancement of affinity was desired to allow optimal targeting. In addition, further profiling of 10 revealed poor physicochemical properties, as evidenced by low thermodynamic solubility of 0.16 mg/mL in phosphate buffer at pH 7.4 (Table 3). This can be attributed to the lack of polar residues in the sequence and the high number of aromatic amino acids. Removal of the C-terminal extension also contributed to the poor solubility, as early hit 8 was much more soluble in the same buffer (4.4 mg/mL). Bicycles with highly hydrophobic sequences have previously been linked with poor tolerability when incorporated into BTCs;4 therefore, decreasing the overall hydrophobicity of this series was required. 10 was also found to be poorly stable in mouse plasma (t1/2 = 1.9 h), and as our primary in vivo efficacy readout was to be carried out in mice, stabilization of the peptide was also necessary. Therefore, chemical optimization was required to improve the affinity, solubility, and stability of the lead compound before incorporation into a BTC.
Exploration of SAR through Single Amino Acid Substitutions
In the first round of chemical optimization, single non-natural amino acid replacements were introduced into the sequence of 10 with the aim of improving the affinity, solubility, or stability of the peptide (Table 4).18
Table 4. Affinities of Compound 10 Analogues Containing Non-Natural Amino Acids Determined by FP Competitiona.
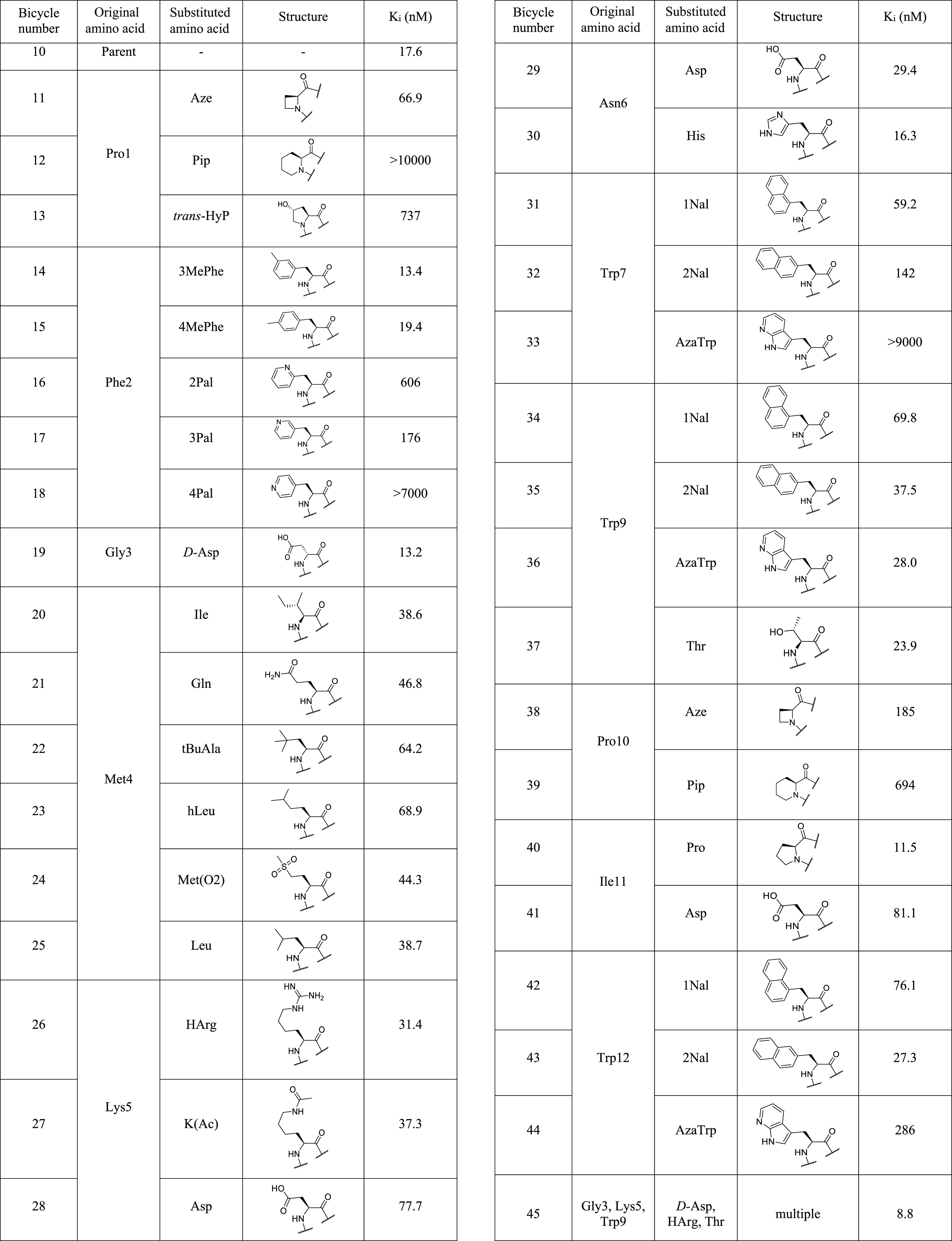
Affinities were measured using fluorescence polarization competition. The Geomean of Ki values displayed are all generated from n ≥ 2. Full assay details, including the number of replicates, errors, and competing tracer molecule are in the Supporting Information. Amino acid sequence of parent peptide 10 Ac-CPFGCMKNWSWPIWC–CONH2 (TATA scaffold).
Both proline 1 and proline 10 were replaced with azetidine-2-carboxylic acid (Aze) and pipecolinic acid (Pip) in order to explore expansion and contraction of the five-membered ring; however, in both positions these substitutions led to a substantial loss in affinity. Pro1 was also replaced with hydrophilic analogue trans-hydoxyproline (trans-HyP); however, a large drop in binding affinity was observed, and this substitution was not considered further.
Phenylalanine 2 was targeted for substitution next; 14 and 15 were prepared to explore the effect of introducing a methyl group in the 3 and 4 positions of the aromatic ring. These analogues maintained the affinity of the parent peptide (13.4 and 19.4 nM, respectively), suggesting that there may be room for larger substituents around the aromatic ring. 2-, 3-, and 4-Pyridylalanine analogues were also prepared to investigate the effect of introducing polarity to the otherwise hydrophobic aromatic residue; however, this was not well-tolerated and led to a substantial decrease in affinity in all cases, with the 4-pyridylalanine analogue (18) being the least well-tolerated with no detectable binding affinity.
The d-alanine scan showed that a slight improvement in affinity was achieved through replacement of glycine 3 with d-alanine. This prompted us to assess d-aspartic acid (d-Asp) in this position (19), which was well-tolerated with a Ki of 13.2 nM and, importantly, allowed introduction of polarity and charge into the sequence.
Methionine residues are often associated with both chemical and metabolic instability, though the extent of this is dependent on peptide sequence. The alanine scan data suggested that the methionine 4 side chain contributed greatly to binding; therefore, only structurally related substitutions were investigated. Replacements were reasonably well-tolerated, with analogues incorporating isoleucine, glutamine, leucine, and methionine-dl-sulfoxide (Met(O)) in this position showing affinities within threefold of parent, whereas replacement with tert-butylalanine (tBuAla) and homoleucine (hLeu) caused a reduction in affinity to 64.2 and 68.9 nM, respectively.
Lysine 5 was targeted for replacement, as removal of the primary amine present in the side chain would allow downstream in-solution functionalization of the Bicycle via acylation of the N-terminus (i.e., for toxin conjugation) without the need for incorporation of orthogonal protecting groups. The alanine scan results suggested that the lysine side chain is not essential for binding, and this was evidenced by the fact that both homoarginine (hArg) and N-ε-acetyl-lysine (Lys(Ac)) were well-tolerated in this position (31.4 and 37.3 nM, respectively). Homoarginine was preferred, as it retained the positive charge of lysine, which may aid with solubility. Replacement with negatively charged aspartic acid led to reduced affinity (28, 77.7 nM).
All three tryptophan residues 7, 9, and 12 were individually replaced with 1-naphthylalanine (1Nal) and 2-naphthylalanine (2Nal). Although some compounds maintained affinity (e.g., 43, 27.3 nM), none led to an improvement in binding and were not considered further due to the resulting increase in hydrophobicity. The 7-azaindole (AzaTrp) analogue of tryptophan was also tested in each of the three positions but led to a decrease in affinity in all cases.
Examination of the phage screening data revealed that, while a tryptophan in position 9 was the most abundant residue observed, there were also a number of hits in which a threonine was present at this position. Compound 37, which incorporated threonine at position 9, retained potency of the parent compound (23.9 nM) and was highlighted as a modification to include in future analogues due to the much-reduced lipophilicity of threonine compared to tryptophan. The strategy that led to this beneficial replacement highlights the power of the Bicycle screening platform, which provides a large volume of screening data that can be tapped into during the optimization process.
This approach was also used to introduce aspartic acid and histidine into position 6 as replacements of asparagine, both of which were well-tolerated (29.4 and 16.3 nM, respectively). In addition, proline and aspartic acid were assessed in position 11 as isoleucine replacements. Proline was very well-tolerated, with compound 40 exhibiting a Ki of 11.5 nM.
From the first round of SAR screening, d-Asp3, hArg5, and Thr9 were identified as substitutions that maintained good binding affinity and were considered beneficial for stability and/or physicochemical optimization. These were combined to give 45, which binds to Nectin-4 with a Ki of 8.8 nM. Although the sequence of 45 is overall more polar than the parent peptide, as evidenced by a lower CHIlogP (a chromatographic measure of hydrophilicity),20 only a modest improvement in thermodynamic solubility was observed (Table 6). This could be attributed to the peptide having an overall neutral charge; therefore, introduction of an additional negative or positive charge could be advantageous. These initial substitutions did, however, lead to improved stability of the compound, as evidenced by the increased in vitro half-life in mouse plasma of more than 24 h, compared with 1.9 h seen for the parent compound 10. Bicycle 45 was progressed as a lead compound and used as the base sequence for the next round of optimization, where affinity and solubility enhancements were the key objectives.
Table 6. Profiles of Hit Bicycle 10 Versus Partially Optimized Lead 45 and Final Optimized Bicycle 58.
| in vitro
plasma stability (t 1/2, h) |
|||||
|---|---|---|---|---|---|
| Bicycle number | affinity Ki (nM) | solubility phosphate buffer pH 7.4 (mg/mL) | mouse | human | CHIlogP |
| 10 | 17.6 | 0.16 | 1.9 | >24 | 4.07 |
| 45 | 8.9 | 0.50 | >24 | >24 | 2.12 |
| 58 | 2.8 | 4.6 | >24 | >24 | 1.50 |
Optimization of Affinity and Solubility
The results of the second round of single amino acid substitutions based on 45 as the parent sequence are summarized in Table 5. Here, more focused substitutions were introduced based on learnings from the previous round of optimization.
Table 5. Affinities of Bicycle 45 Analogues Containing Non-Natural Amino Acidsa.
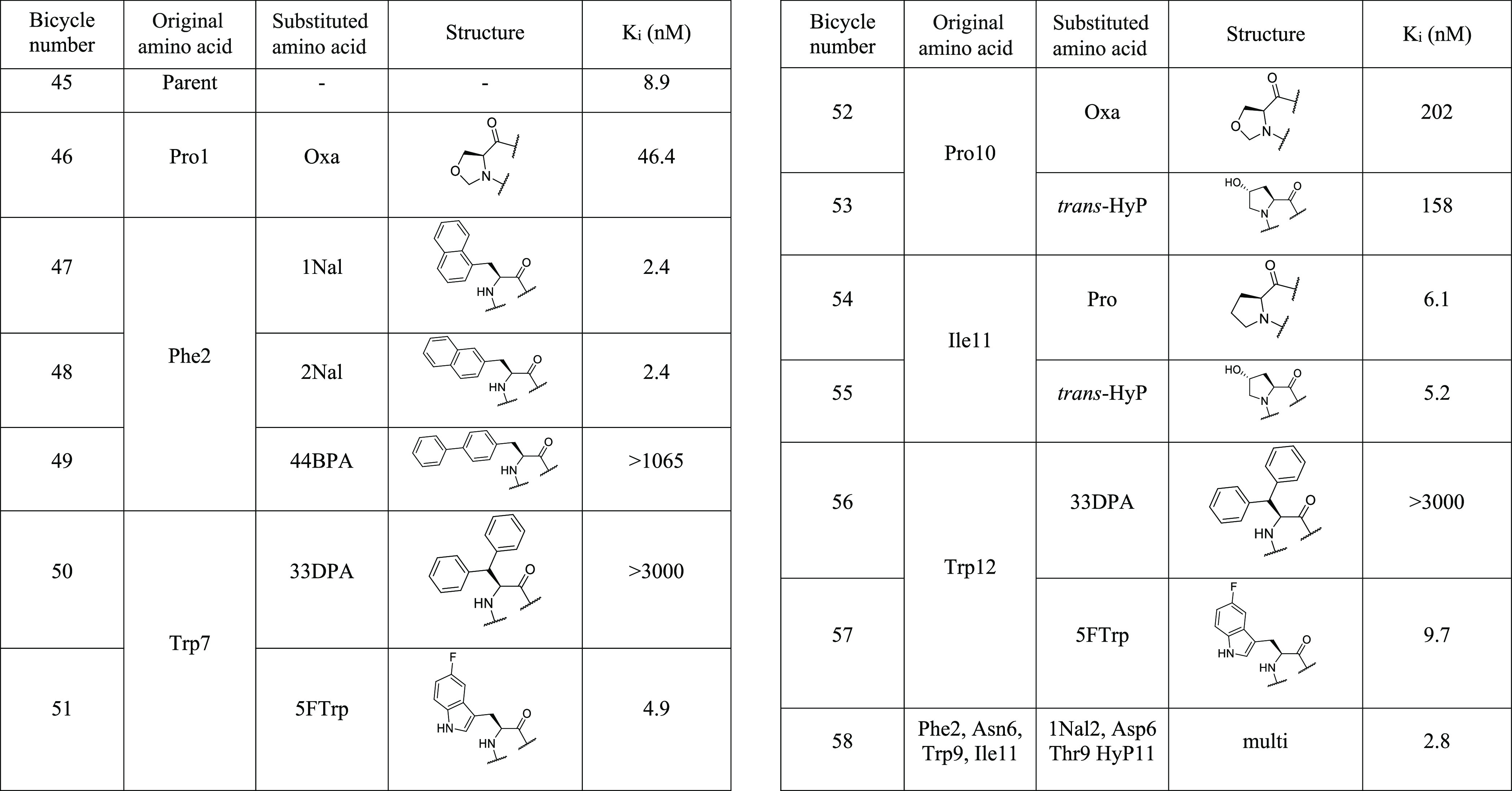
Affinities were measured using fluorescence polarization competition. The Geomean of Ki values displayed are all generated from n ≥ 2. Full assay details, including the number of replicates, errors, and competing tracer molecule, are in the Supporting Information. Amino acid sequence of parent peptide 45 Ac-CPF[d-Asp]CM[HArg]NWSTPIWC–CONH2 (TATA scaffold).
As larger and smaller ring sizes were not tolerated as replacements for either proline residue, a more modest replacement was considered, and Oxazolidine-4-carboxylic acid (Oxa) was evaluated. This was intended to introduce hydrophilic character into this otherwise hydrophobic residue. This substitution led to a 5- and greater than 20-fold decrease in affinity versus parent in positions 1 and 10, respectively. Substitution with trans-HyP was also assessed in position 10 but was not well-tolerated (53, 158 nM). However, trans-HyP was also introduced in position 11, where replacement of Ile11 with Pro was tolerated in the last screening round. This resulted in a modest improvement in binding affinity (55, 5.2 nM), and this substitution was taken forward due to the increased polarity of trans-HyP versus Ile.
Phenylalanine 2 was targeted as a key residue for affinity enhancement. The alanine scan highlighted this residue as critical for binding, and methylation of the phenyl ring in positions 3 and 4 was well-tolerated in the first round of screening, suggesting that there was room available for growth around this side chain. Larger aromatic amino acids were therefore introduced, and both 1Nal and 2Nal led to a substantial improvement in affinity (2.4 nM in both cases). Incorporation of these highly hydrophobic groups would, however, need to be compensated for through the introduction of hydrophilic groups elsewhere in the sequence. A larger aromatic amino acid 4,4-biphenylalanine (44BPA) was also assessed in this position but resulted in a loss of binding affinity.
The bulky aromatic amino acid 3,3-diphenylalanine (33DPA) was tested as a replacement for Trp7 and Trp12, but it led to a loss of binding in both cases. 5-Fluorotryptophan (5FTrp) was also introduced into these positions and was very well-tolerated. Replacement of Trp7 with 5FTrp resulted in a modest increase in binding affinity (51, 4.9 nM); however, this slight improvement did not justify introduction of a potentially costly reagent, and it was therefore not taken forward.
Although replacement of Phe2 for 1Nal or 2Nal resulted in potent binders with affinities in the desired range, introduction of the hydrophobic naphthyl ring deviated from our design goal of improving the physicochemical properties. Therefore, additional well-tolerated solubility enhancing residues that had been identified over the previous rounds of screening were incorporated to give 58, which maintained high affinity for Nectin-4 and showed a great improvement in solubility (4.6 mg/mL, Table 6). Bicycle 58 also shows excellent stability in both human and mouse plasma ex vivo.
Affinities were measured using fluorescence polarization competition. The Geomean of Ki values displayed are all generated from n ≥ 6. Full assay details, including the number of replicates, errors, and competing tracer molecule are in the Supporting Information.
Bicycle Toxin Conjugate BT8009
Due to its well-balanced properties of solubility, stability, and affinity, Bicycle 58 was progressed to formatting as a BTC, using an architecture similar to BT5528 (structure shown in Figure 1).1 The structure of the Nectin-4 targeting BTC is shown in Figure 3 and includes a monomethyl auristatin E (MMAE) payload and a valine-citrulline dipeptide linker, which is cleavable by proteases found in the tumor microenvironment. A 10 unit polysarcosine spacer was introduced between the Bicycle and the linker payload construct to reduce potential steric clashes that could negatively impact on binding to the tumor target or access of enzymes to the cleavable linker.
Figure 3.

Structure of Bicycle Toxin Conjugate (BT8009).
The profile of the resulting BTC BT8009 is shown in Table 7. The conjugate retains the high affinity for Nectin-4 of the parent Bicycle (KD = 2.50 nM determined by surface plasmon resonance (SPR)) and, as reported elsewhere, binds to endogenous Nectin-4 on cells with an apparent Kd of 12.9 nM.13 Plasma protein binding is low in both human and mouse, and the conjugate possesses high stability in human plasma with lower stability in mouse plasma. The reduced stability in mouse plasma is likely due to cleavage of the linker by a mouse-specific enzyme (Ces1c), which is well-documented;21 however, the stability was still sufficient for in vivo profiling.
Table 7. Profile of Bicycle Toxin Conjugate BT8009a.
| plasma
protein binding (%) |
in vitro
plasma stability (t1/2, h) |
mouse
pharmacokinetics 3 mg/kg, IV (bolus) |
||||||
|---|---|---|---|---|---|---|---|---|
| human Nectin-4 KD (SPR) | mouse | human | solubility phosphate buffer pH 7.4(mg/mL) | mouse | human | T1/2 (h) | Clp (mL/min/kg) | Vss (L/kg) |
| 2.50 | 88.2 | 79.3 | 0.8 | 4.4 | >57.8 | 1.0 | 3.5 | 0.25 |
Affinity was measured using SPR. The Geomean of KD value displayed is generated from n = 13. Full assay details, including the number of replicates and errors, are in the Supporting Information.
Pharmacokinetic profiling in mouse demonstrated that BT8009 has low plasma clearance and a low volume of distribution resulting in a terminal half-life of ca. 1 h. This PK profile is consistent with the intended mode of action of BTCs, which involves rapid payload delivery into the tumor and rapid elimination from systemic circulation to spare healthy tissue from long-term toxin exposure.
When the solubility of BT8009 was tested in phosphate buffer at pH 7.4, it was found to be 0.8 mg/mL, much lower than that of the precursor peptide. This was expected due to conjugation of the relatively nonpolar MMAE toxin; however, exploration of additional formulations identified 25 mM histidine HCl, 10% sucrose, and 0.2 mg/mL tween at pH 8.5, which solubilized BT8009 to a concentration of 5.5 mg/mL. This was projected to be sufficient to permit intravenous dosing at the required levels in the clinic.
The efficacy of BT8009 was evaluated in a cell-derived xenograft (CDX) model using breast adenocarcinoma (MDA-MB-468) cells, which express Nectin-4 (Figure 4).22 When tested at a dose of 3 mg/kg once weekly, significant antitumor activity was observed. At a dose of 3 mg/kg twice weekly or 5 mg/kg once weekly, potent efficacy was achieved with almost complete regression of the tumor after 18 days. Importantly, following cessation of dosing after 18 days, animals from the 5 mg/kg once weekly dosing group were monitored up to day 42, and no tumor regrowth was observed. Consistent animal body weights throughout the study indicate that BT8009 appeared to be well-tolerated at all doses tested (see Supporting Information). In additional studies reported elsewhere, BT8009 has shown preclinical efficacy in a wide range of CDX and PDX tumor types with full tumor regression seen in small and large tumors, where efficacy broadly correlates with Nectin-4 expression. Additionally, when BT8009 was codosed with an excess of an MMAE-free analogue, efficacy was attenuated, and a BTC incorporating a nonbinding Bicycle analogue showed a demonstrably lower rate and degree of tumor regression.13
Figure 4.
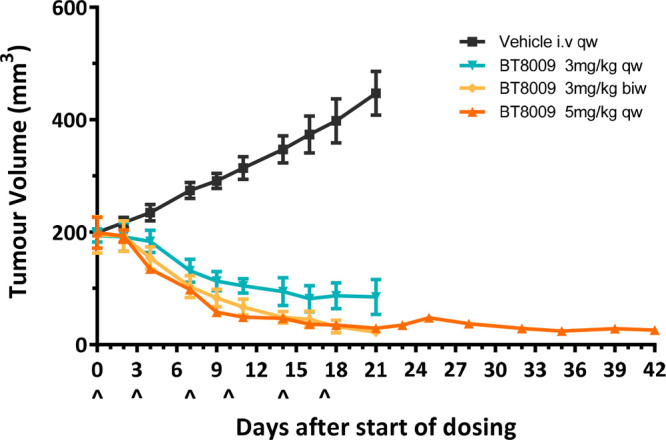
Efficacy of BT8009 in a breast adenocarcinoma (MDA-MB-468) CDX model.
Conclusions
We describe the first example of a low molecular weight and fully synthetic construct that selectively targets Nectin-4, a protein previously only targeted through large biological agents. Screening against Nectin-4 for bicyclic peptides by phage display produced three hit series with excellent affinity for the target protein. After an initial evaluation, a single series was chosen as the lead, which was shown to be highly selective for Nectin-4 over other members of the target family. Several iterative rounds of chemical optimization produced a bicyclic peptide with low nanomolar affinity for Nectin-4, which shows good stability when tested in in vitro matrix assays and has an excellent physicochemical profile. As reported elsewhere, in a selectivity screen against 5528 human plasma membrane proteins, the optimized Bicycle only shows interaction with Nectin-4, demonstrating its high degree of specificity for the target.13 It is noteworthy that the large body of data generated during the phage screening campaign played a pivotal role in the optimization process by identifying important substitutions that, in particular, proved vital in optimizing the physical chemistry properties.
The fully optimized bicyclic peptide was incorporated into a toxin conjugate construct, BT8009, which retained the affinity of the parent bicyclic peptide and demonstrated potent antitumor activity in a CDX mouse model and was well-tolerated at efficacious doses. BT8009 was selected as a candidate for development in 2018 and is currently undergoing clinical evaluation.23
This is the third reported example of high-affinity and selective tumor-targeting Bicycles being employed in toxin conjugates to give potent and well-tolerated anticancer agents,1,3,4 further demonstrating that this approach is generalizable and has the potential to generate a pipeline of such compounds for targeted cancer therapy. In addition, Nectin-4 targeting Bicycles have been employed in fully synthetic, Nectin-4 targeting immune cell agonists (TICAs), which induce tumor-localized CD137 agonism and show robust antitumor activity in preclinical in vivo studies.16,24,25 Tumor-targeting Bicycles can be utilized as delivery moieties for a variety of payloads and have the potential to generate a diverse range of oncology medicines.
Experimental Section
Synthetic schemes and analytical traces can be found in the Supporting Information. Compounds were isolated at greater than 95% purity, as assessed by high-performance liquid chromatography (HPLC) (apart from screening peptides, which were greater than 85% pure).
General Peptide Synthesis
Bicycle peptides were synthesized as previously described.4 Briefly, Bicycle peptides were synthesized on Rink amide resin using standard 9-fluorenylmethyloxycarbonyl (Fmoc) solid-phase peptide synthesis, either by manual coupling (for large scale) or using a Biotage SyroII automated peptide synthesizer (for small scale). Following trifluoroacetic acid based cleavage from the resin, peptides were precipitated with diethyl ether and dissolved in 50:50 acetonitrile/water. The crude peptides (at ∼1 mM concentration) were then cyclized with 1.3 equiv of TATA scaffold, using ammonium bicarbonate (100 mM) as a base. Completion of cyclization was determined by matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) or liquid chromatography-mass spectrometry (LC–MS). Once complete, the cyclization reaction was quenched using N-acetyl cysteine (10 equiv with respect to TATA), and the solutions were lyophilized. The residue was dissolved in an appropriate solvent and purified by reversed-phase (RP) HPLC. Peptide fractions of sufficient purity and the correct molecular weight (verified by either MALDI-TOF and HPLC or LC–MS) were pooled and lyophilized. Concentrations were determined by UV absorption using the extinction coefficient at 280 nm, which was based on Trp/Tyr content. Standard Fmoc amino acids, as well as nonproteinogenic Fmoc amino acids, were obtained from Sigma-Aldrich, Iris Biotech GmbH, Apollo Scientific, ChemImpex, and Fluorochem.
Val-Cit-PAB-MMAE was prepared as described in the literature.26
Glutarate-Val-Cit-PAB-MMAE
To a solution of Val-Cit-PAB-MMAE (1.00 g, 890 μmol, 1 equiv) in dimethylformamide (DMF) (2.00 mL) was added glutaric anhydride (203 mg, 1.78 mmol, 2 equiv) and then N,N-diisopropylethylamine (DIEA) (230 mg, 1.78 mmol, 2 equiv). The reaction mixture was stirred at 25 °C for 3 h. LC-MS showed that Val-Cit-PAB-MMAE had been consumed and that one main peak with the desired mass was detected. The mixture was filtered, and the filtrate was directly purified by preparative HPLC. Glutarate-Val-Cit-PAB-MMAE was obtained as a white solid (900 mg, 727 μmol, 82% yield). MS (electrospray ionization (ESI)) exact mass calcd: 1237.5, found: m/z 1238.7 ([M + H]+), 1260.6 ([M + Na]+), 619.6 ([M+2H]2+). Purity >95% by HPLC (see Supporting Information).
NHS-Glutarate-Val-Cit-PAB-MMAE
To a solution of Glutarate-Val-Cit-PAB-MMAE (8.5 g, 6.87 mmol, 1 equiv) and N-hydroxysuccinimide (3.95 g, 34.34 mmol, 5 equiv) in dimethylacetamide (DMA) (30 mL) and dichloromethane (DCM) (10 mL) was added 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI)·HCl (6.58 g, 34.34 mmol, 5 equiv) and 4-dimethylaminopyridine (DMAP) (83.91 mg, 686.86 umol, 0.1 equiv). The mixture was stirred at 20 °C for 16 h. LC-MS showed Glutarate-Val-Cit-PAB-MMAE was consumed, and one main peak with desired mass was detected. DCM was removed under reduced pressure, and the reaction was purified by preparative HPLC to give NHS-Glutarate-Val-Cit-PAB-MMAE, which was obtained as a white solid (7.0 g, 4.72 mmol, 68.65% yield). MS (ESI) exact mass calcd: 1334.6, found: m/z 1334.6 ([M + H]+), 667.9 ([M+2H]2+). Purity 90% by HPLC (see Supporting Information).
Bicycle Toxin Conjugate Precursor (59)
Sequence: [B-Ala][Sar10]CP[1Nal][dD]CM[HArg]DWSTP[HyP]WC[CONH2].
67 g of Rink Amide MBHA Resin (0.3 mmol/g, 20 mmol) was used to generate 5.06 g of 59 using the general methods outlined above (1.71 mmol, 8.6% yield). MS (ESI) exact mass calcd: 2954.4, found: m/z 1476.6 ([M+2H]2+), 984.7 ([M+3H]3+). Purity >95% by HPLC (see Supporting Information).
BT8009
To a solution of 59 (66.4 mg, 22.5 umol, 1 equiv) in DMA (4 mL) was added DIEA (8.7 mg, 67.4 umol, 11.7 uL, 3 equiv) and NHS-Glutarate-Val-Cit-PAB-MMAE (30.0 mg, 22.5 umol, 1 equiv). The mixture was stirred at 20 °C for 16 h. LC-MS showed that 59 was consumed completely, and one main peak with the desired m/z was detected. The reaction was diluted and then purified by preparative HPLC. Compound BT8009 (42.7 mg, 10.2 umol, 45% yield) was obtained as a white solid. MS (ESI): exact mass calcd, 4173.9; found, m/z 1391.3 ([M + 3H]3+), 1043.9 ([M + 4H]4+). Purity >99% by HPLC (see Supporting Information).
Acknowledgments
The authors thank K. McDonnell for critical review of this manuscript. We thank Wuxi AppTec Ltd. for support with chemistry, DMPK, and in vivo pharmacology studies.
Glossary
Abbreviations
- ADC
Antibody Drug Conjugate
- BTC
Bicycle Toxin Conjugate
- CDX
cell derived xenograft
- Ces1c
Carboxylesterase 1C
- DM1
Mertansine
- ECD
extracellular domain
- EphA2
Erythropoietin-producing hepatocellular receptor A2
- FP
Fluorescence polarization
- MMAE
Monomethyl auristatin E
- MT1-MMP
Membrane type 1 matrix metalloproteinase
- PK
Pharmacokinetic
- PDX
Patient derived xenograft
- SPR
Surface plasmon resonance
- TATA
1,3,5-Triacryloyl-1,3,5-triazinane
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c00065.
General procedures; compound analytics and synthetic schemes, sequences of representative Nectin-4 binding Bicycles from phage screens, body weights of CDX animals (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): GM, HS, LC, KvR, GIB, KD, AB, SW, PP, PJ, MR, and PB are/were shareholders or option holders in Bicycle Therapeutics Plc, the parent company of BicycleTx Ltd.
Supplementary Material
References
- Bennett G.; Brown A.; Mudd G.; Huxley P.; Van Rietschoten K.; Pavan S.; Chen L.; Watcham S.; Lahdenranta J.; Keen N. MMAE delivery using the bicycle toxin conjugate BT5528. Molecular Cancer Therapeutics 2020, 19 (7), 1385. 10.1158/1535-7163.MCT-19-1092. [DOI] [PubMed] [Google Scholar]
- Cook N.; Banerji U.; Evans J.; Biondo A.; Germetaki T.; Randhawa M.; Godfrey L.; Leslie S.; Jeffrey P.; Rigby M.; Bennett G.; Blakemore S.; Koehler M.; Niewiarowski A.; Pittman M.; Symeonides S. N. 464P - Pharmacokinetic (PK) assessment of BT1718: A phase I/II a study of BT1718, a first in class bicycle toxin conjugate (BTC), in patients (pts) with advanced solid tumours. Annals of Oncology 2019, 30, v174. 10.1093/annonc/mdz244.026. [DOI] [Google Scholar]
- Bennett G.; Lutz R.; Park P.; Harrison H.; Lee K. Abstract 1167: Development of BT1718, a novel bicycle drug conjugate for the treatment of lung cancer. Cancer Res. 2017, 77, 1167. 10.1158/1538-7445.AM2017-1167. [DOI] [Google Scholar]
- Mudd G. E.; Brown A.; Chen L.; van Rietschoten K.; Watcham S.; Teufel D. P.; Pavan S.; Lani R.; Huxley P.; Bennett G. S. Identification and optimization of EphA2-selective bicycles for the delivery of cytotoxic payloads. J. Med. Chem. 2020, 63 (8), 4107–4116. 10.1021/acs.jmedchem.9b02129. [DOI] [PubMed] [Google Scholar]
- Eder M.; Pavan S.; Bauder-Wüst U.; van Rietschoten K.; Baranski A.-C.; Harrison H.; Campbell S.; Stace C. L.; Walker E. H.; Chen L.; Bennett G.; Mudd G.; Schierbaum U.; Leotta K.; Haberkorn U.; Kopka K.; Teufel D. P. Bicyclic peptides as a new modality for imaging and targeting of proteins overexpressed by tumors. Cancer Res. 2019, 79 (4), 841. 10.1158/0008-5472.CAN-18-0238. [DOI] [PubMed] [Google Scholar]
- Bendell J. C.; Wang J. S.-Z.; Bashir B.; Richardson D. L.; Bennett G.; Campbell C.; Hennessy M. G.; Jeffrey P.; Kirui J.; Mahnke L.; Shapiro G. BT5528–100 phase I/II study of the safety, pharmacokinetics, and preliminary clinical activity of BT5528 in patients with advanced malignancies associated with EphA2 expression. Journal of Clinical Oncology 2020, 38, TPS3655–TPS3655. 10.1200/JCO.2020.38.15_suppl.TPS3655. [DOI] [Google Scholar]
- Deng H.; Shi H.; Chen L.; Zhou Y.; Jiang J. Over-expression of Nectin-4 promotes progression of esophageal cancer and correlates with poor prognosis of the patients. Cancer Cell International 2019, 19 (1), 106. 10.1186/s12935-019-0824-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiwada S.; Sho M.; Yasuda S.; Shimada K.; Yamato I.; Akahori T.; Kinoshita S.; Nagai M.; Konishi N.; Nakajima Y. Nectin-4 expression contributes to tumor proliferation, angiogenesis and patient prognosis in human pancreatic cancer. Journal of experimental & clinical cancer research: CR 2015, 34 (1), 30–30. 10.1186/s13046-015-0144-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reymond N.; Fabre S.; Lecocq E.; Adelaıde J.; Dubreuil P.; Lopez M. Nectin4/PRR4, a new afadin-associated member of the Nectin family that trans-interacts with Nectin1/PRR1 through V domain interaction *. J. Biol. Chem. 2001, 276 (46), 43205–43215. 10.1074/jbc.M103810200. [DOI] [PubMed] [Google Scholar]
- Takano A.; Ishikawa N.; Nishino R.; Masuda K.; Yasui W.; Inai K.; Nishimura H.; Ito H.; Nakayama H.; Miyagi Y.; Tsuchiya E.; Kohno N.; Nakamura Y.; Daigo Y. Identification of Nectin-4 oncoprotein as a diagnostic and therapeutic target for lung cancer. Cancer Res. 2009, 69 (16), 6694. 10.1158/0008-5472.CAN-09-0016. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Zhang J.; Shen Q.; Yin W.; Huang H.; Liu Y.; Ni Q. High expression of Nectin-4 is associated with unfavorable prognosis in gastric cancer. Oncology letters 2018, 15 (6), 8789–8795. 10.3892/ol.2018.8365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powles T.; Rosenberg J. E.; Sonpavde G. P.; Loriot Y.; Durán I.; Lee J.-L.; Matsubara N.; Vulsteke C.; Castellano D.; Wu C.; Campbell M.; Matsangou M.; Petrylak D. P. Enfortumab Vedotin in previously treated advanced urothelial carcinoma. New England Journal of Medicine 2021, 384 (12), 1125–1135. 10.1056/NEJMoa2035807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigby M.; Bennett G.; Chen L.; Mudd G. E.; Harrison H.; Beswick P. J.; van Rietschoten K.; Watcham S. M.; Scott H. S.; Brown A. N.; Park P. U.; Campbell C.; Haines E.; Lahdenranta J.; Skynner M. J.; Jeffrey P.; Keen N.; Lee K. BT8009; a Nectin-4 targeting Bicycle Toxin Conjugate for treatments of solid tuours. Mol. Cancer Ther 2022, 10.1158/1535-7163.MCT-21-0875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeriswyl V.; Rapley H.; Pollaro L.; Stace C.; Teufel D.; Walker E.; Chen S.; Winter G.; Tite J.; Heinis C. Bicyclic peptides with optimized ring size inhibit human plasma kallikrein and its orthologues while sparing paralogous proteases. ChemMedChem. 2012, 7 (7), 1173–1176. 10.1002/cmdc.201200071. [DOI] [PubMed] [Google Scholar]
- Heinis C.; Rutherford T.; Freund S.; Winter G. Phage-encoded combinatorial chemical libraries based on bicyclic peptides. Nat. Chem. Biol. 2009, 5 (7), 502–507. 10.1038/nchembio.184. [DOI] [PubMed] [Google Scholar]
- Upadhyaya P.; Lahdenranta J.; Hurov K.; Battula S.; Dods R.; Haines E.; Kleyman M.; Kristensson J.; Kublin J.; Lani R.; Ma J.; Mudd G.; Repash E.; Van Rietschoten K.; Stephen T.; You F.; Harrison H.; Chen L.; McDonnell K.; Brandish P.; Keen N. Anticancer immunity induced by a synthetic tumor-targeted CD137 agonist. J. Immunother Cancer 2021, 9, 1. 10.1136/jitc-2020-001762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teufel D. P.; Bennett G.; Harrison H.; van Rietschoten K.; Pavan S.; Stace C.; Le Floch F.; Van Bergen T.; Vermassen E.; Barbeaux P.; Hu T.-T.; Feyen J. H. M.; Vanhove M. Stable and Long-Lasting, Novel bicyclic peptide plasma kallikrein inhibitors for the treatment of diabetic macular edema. J. Med. Chem. 2018, 61 (7), 2823–2836. 10.1021/acs.jmedchem.7b01625. [DOI] [PubMed] [Google Scholar]
- Di L. Strategic approaches to optimizing peptide ADME properties. AAPS journal 2015, 17 (1), 134–143. 10.1208/s12248-014-9687-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werle M.; Bernkop-Schnürch A. Strategies to improve plasma half life time of peptide and protein drugs. Amino Acids 2006, 30 (4), 351–367. 10.1007/s00726-005-0289-3. [DOI] [PubMed] [Google Scholar]
- Valko K.; Ivanova-Berndt G.; Beswick P.; Kindey M.; Ko D. Application of biomimetic HPLC to estimate lipophilicity, protein and phospholipid binding of potential peptide therapeutics. ADMET and DMPK 2018, 6 (2), 162–175. 10.5599/admet.544. [DOI] [Google Scholar]
- Dorywalska M.; Dushin R.; Moine L.; Farias S. E.; Zhou D.; Navaratnam T.; Lui V.; Hasa-Moreno A.; Casas M. G.; Tran T.-T.; Delaria K.; Liu S.-H.; Foletti D.; O'Donnell C. J.; Pons J.; Shelton D. L.; Rajpal A.; Strop P. molecular basis of valine-citrulline-PABC linker instability in site-specific ADCs and its mitigation by linker design. Molecular Cancer Therapeutics 2016, 15 (5), 958. 10.1158/1535-7163.MCT-15-1004. [DOI] [PubMed] [Google Scholar]
- Noyce R. S.; Bondre D. G.; Ha M. N.; Lin L.-T.; Sisson G.; Tsao M.-S.; Richardson C. D. tumor cell marker PVRL4 (Nectin 4) is an epithelial cell receptor for measles virus. PLOS Pathogens 2011, 7 (8), e1002240. 10.1371/journal.ppat.1002240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKean M.; Falchook G. S.; Bendell J. C.; Bashir B.; Palmisiano N.; Watson R.; McKenna M.-A.; Campbell C.; Smethurst D. P.; Jeffrey P.; Kirui J.; Arkenau H.-T.; Fontana E. Association of combined phase I/II study of a novel bicyclic peptide and MMAE conjugate BT8009 in patients with advanced malignancies with Nectin-4 expression. Journal of Clinical Oncology 2021, 39, TPS2668–TPS2668. 10.1200/JCO.2021.39.15_suppl.TPS2668. [DOI] [Google Scholar]
- Hurov K.; Lahdenranta J.; Upadhyaya P.; Haines E.; Cohen H.; Repash E.; Kanakia D.; Ma J.; Kristensson J.; You F.; Campbell C.; Witty D.; Kelly M.; Blakemore S.; Jeffrey P.; McDonnell K.; Brandish P.; Keen N. BT7480, a novel fully synthetic Bicycle tumor-targeted immune cell agonist (Bicycle TICA) induces tumor localized CD137 agonism. Journal for ImmunoTherapy of Cancer 2021, 9 (11), e002883. 10.1136/jitc-2021-002883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhyaya P. Discovery and optimization of a synthetic class of Nectin-4-targeted CD137 agonists for immuno-oncology. J. Med. Chem. 2022, 65 (14), 9858–9872. 10.1021/acs.jmedchem.2c00505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker J. A.; Sorkin M. R.; Ledesma F.; Kabaria S. R.; Barfield R. M.; Rabuka D.; Alabi C. A. Hydrophilic sequence-defined cross-linkers for antibody–drug conjugates. Bioconjugate Chem. 2019, 30 (11), 2982–2988. 10.1021/acs.bioconjchem.9b00713. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.