

Abstract

Enterovirus D68 (EV-D68) is a nonpolio enterovirus that is mainly transmitted through respiratory routes and poses a potential threat for large-scale spread. EV-D68 infections mostly cause moderate to severe respiratory diseases in children and potentially induce neurological diseases. However, there are no specific antiviral drugs or vaccines against EV-D68. Herein, through virtual screening and rational design, a series of novel quinoline analogues as anti-EV-D68 agents targeting VP1 were identified. Particularly, 19 exhibited potent antiviral activity with an EC50 value ranging from 0.05 to 0.10 μM against various EV-D68 strains and showed inhibition of viral replication verified by Western blot, immunofluorescence, and plaque formation assay. Mechanistic studies indicated that the anti-EV-D68 agents work mainly by interacting with VP1. The acceptable bioavailability of 23.9% in rats and significant metabolic stability in human liver microsome (Clint = 10.8 mL/min/kg, t1/2 = 148 min) indicated that compound 19 with a novel scaffold was worth further investigation.
Introduction
Enterovirus D68 (EV-D68) is a nonpolio enterovirus (NPEV) that belongs to the enterovirus D group of the Picornaviridae family. EV-D68 infections mostly cause moderate respiratory diseases but can also result in severe bronchiolitis or pneumonia and can occasionally lead to death, especially among children.1,2 Unlike other enteroviruses, EV-D68 is mainly transmitted through the respiratory route and primarily binds to sialic acid receptors in the upper respiratory tract,3−5 and this transmission route poses a potential threat for large-scale spread. In 2014, the largest outbreak of severe respiratory diseases that were associated with EV-D68 occurred in the United States, with 1153 confirmed infections and potentially millions of milder cases that were untested.6−9 During the same period, many countries in Europe and Asia successively reported that the EV-D68 epidemic was present.10,11 In addition to causing serious respiratory diseases, EV-D68 was found to be associated with a temporal polio-like neurological disorder known as acute flaccid myelitis (AFM).8,12−14 The discovery indicated that EV-D68 can potentially induce neurological diseases.11,12,15 Although being infected with EV-D68 can cause serious illness, there are currently no specific antiviral drugs or vaccines and the only treatments are supportive treaments.7 Therefore, developing novel structures and specific therapeutic drugs to better treat EV-D68 is urgent.
EV-D68, with a positive-sense single-stranded RNA genome, consists of a single open reading frame (ORF) and untranslated regions (UTR) on both sides16 and encodes a polyprotein that can be hydrolyzed to structural proteins and nonstructural proteins.1 EV-D68 is a nonenveloped virus and is surrounded by viral capsid proteins.14 The capsid shell is a structural protein that constitutes 60 protomers, and these protomers contain the surface proteins VP1, VP2, and VP3 and the internal VP4.7 The VP1 canyon floor is formed primarily by GH loop14 and usually harbors a small, hydrophobic pocket, which is ordinarily filled with a lipid moiety or pocket factor, and this pocket is involved in regulating particle stability and is important for cell receptor binding.7 One of the effective strategies to inhibit enterovirus infections is using capsid binders to target VP1. Only a few EV-D68 VP1 inhibitors have been reported thus far, and the typical representatives are pleconaril (1, EC50 = 0.43 ± 0.02 μM) and pirodavir (2, EC50 = 1.61 ± 0.30 μM) (Figure 1).14 Inhibitors occupy the hydrophobic pocket and displace the pocket factor, thereby increasing particle stability and preventing receptor binding or genome release.7,14,17,18 However, pleconaril, once as an antirhinoviral inhibitor, was not licensed primarily because of the clinical risk.14 Therefore, it is essential to discovery novel scaffold antivirals against EV-D68.
Figure 1.

Structures of the reported EV-D68 VP1 inhibitors.
In this paper, we screened a series of novel scaffold quinoline analogues that showed potent anti-EV-D68 activity, and through structural optimization and antiviral mechanism exploration, the oxadiazole-substituted quinoline scaffold was finally determined. Several compounds with good in vitro antiviral potency were obtained, as guided by structure–activity relationship (SAR) exploration and VP1 protein structural biology. Furthermore, these compounds also exhibited broad-spectrum antiviral potency against various EV-D68 strains. Preliminary mechanistic studies showed that 19 is a VP1 inhibitor against EV-D68 with acceptable pharmacokinetic properties. Hence, these newly discovered quinoline derivatives, especially 19, have the potential to be developed as anti-EV-D68 lead compounds.
Results and Discussion
Chemistry
Compounds 10–18 and 19–60 were prepared according to the general synthetic method depicted in Schemes 1 and 2, respectively. They were tested and verified by 1H NMR, 13C NMR, and HRMS, and all target compounds were purified to >95%, as determined by HPLC.
Scheme 1. Synthesis of Compounds 10–18.

Reagents and conditions: (a) Tin(II) chloride dihydrate, EtOH, reflux, 5 h; (b) R1-B (OH)2, PdCl2 (dppf), K2CO3, DME/H2O, 120 °C, 40 min, microwave.
Scheme 2. Synthesis of Compounds 19–60.

Reagents and conditions: (a) Tin(II) chloride dihydrate, EtOH, reflux, 5 h; (b) H2N-OH, EtOH, reflux, 5 h; (c) (R2-CO)2O, pyridine, reflux, 3 h; (d) R3-B (OH)2, PdCl2 (dppf), K2CO3, DME/H2O, 120 °C, 40 min, microwave.
Compound 5 was synthesized according to a method previously reported.21 In the presence of ethanol, the nitro group in starting material 3 was reduced using stannous chloride dihydrate as the reductant, and then cyclization product compound 5 was obtained after the reflux reaction with material 4. Fortunately, product 5 could be prepared directly with a yield of 61% through a one-pot method, which avoids the postprocessing and purification normally necessary for the reduced product of raw material 3. The Suzuki coupling reaction of intermediate 5 with different boric acid derivatives in the presence of PdCl2(dppf) and potassium carbonate provided compounds 10–18 (Scheme 1).
The preparation of intermediate 7 was similar to that of 5. Intermediate 7 was obtained through the Friedlander reaction, and raw material 6 was used to replace 4. Treatment of 7 with hydroxylamine (50 wt % in water) under reflux conditions in ethanol afforded intermediate 8. Subsequently, 9a–g were obtained by cyclization with the appropriate acid anhydrides. Similar to Scheme 1, compounds 19–60 were prepared correspondingly from intermediates 9a–g through Suzuki coupling with different boric acid derivatives in the presence of PdCl2 (dppf) and potassium carbonate (Scheme 2). 19·HCl and 21·HCl were obtained by salification in hydrogen chloride solution (Scheme 3).
Scheme 3. Synthesis of Compounds 19·HCl and 21·HCl.

Reagents and conditions: (a) HCl in ethyl acetate, ethyl acetate, rt.
Identification of Two Screening Hits
Although VP1 has been demonstrated to be an effective target against EV-D68,14,19,20 no specific treatment is available, and in view of the clinical risk of pleconaril,14 we were committed to the development of novel scaffold antivirals against EV-D68. Therefore, the combination of structure-based virtual screening and in vitro evaluation was adopted to identify novel hit compounds for VP1 inhibitors through the screening protocol summarized in Figure 2. More than 20 000 compounds from our in-house antiviral compound library were docked into the VP1 binding site, which was generated from the cocrystal structure of pleconaril (PDB: 4WM7). After being sorted by docking scores, compounds with scores better than −9 were collected. The docking poses were then superimposed, and nine compounds that fitted the VP1 hydrophobic pocket and with spatial structure similar to pleconaril were finally selected (Figure S1). Subsequently, screening hits 10 and 11, which exhibited anti-EV-D68 activity with an EC50 value greater than 10 μM concentrations, were identified by CPE assay (Table 1), and the other substandard compounds are shown in Table S1.
Figure 2.

Process of screening hit compounds.
Table 1. Antiviral Evaluation of the Screening Hits against EV-D68a in RD Cellsb.

EV-D68 strain, STL-2014-12.
EC50, concentration that effectively inhibited the cytopathic effect by 50% in Rhabdomyosarcoma cell lines (RD cells).
Compound Design and SAR Study
Although 10 and 11 acted as novel scaffold compounds against EV-D68, antiviral potencies did not achieve the expectation. So, further structural modification and SARs need to be performed to obtain better antiviral agents. We found that two screening hits with the same scaffold showed a 5-fold difference in antiviral potency (Table 1), which indicated that the 6-position substituent of quinoline core played a crucial role in SARs. Therefore, the main structure optimization strategy in the early stage was to explore the A substituent (Figure 3). Thus, series 1 compounds with different aromatic ring substituents at the 6-position of the quinoline core were synthesized and tested to discover a suitable structure for the VP1 pocket. The anti-EV-D68 activities of series 1 compounds are shown in Table 2, and the reported VP1 inhibitor pleconaril (1) was also tested as positive control.
Figure 3.

(A) SAR exploration regions of A and B. (B) The docking pose comparison of 10 (green), 11 (yellow), and pleconaril (blue, PDB: 4WM7) in the VP1 cavity.
Table 2. Antiviral Evaluation of Series 1 against EV-D68a in RD Cellsb.


EV-D68 strain: STL-2014-12.
EC50: concentration that effectively inhibited the cytopathic effect by 50% in Rhabdomyosarcoma cell lines (RD cells).
CC50: concentration that inhibited the normal uninfected cell viability by 50%, or the maximum soluble concentration which started from 200 μM in serial 3-fold dilutions.
Selectivity index (SI) calculated as CC50/EC50 ratio.
NA: no activity.
The SARs of series 1 compounds revealed that the species and position of the R1 substituent were essential for the anti-EV-D68 activities. When R1 was introduced into electron-donating groups, compounds 10–16 exhibited micromolar antiviral activity with EC50 values in the range of 0.47–6.73 μM, while electron-withdrawing groups such as amide (compound 18) led to inactivation. On one hand, starting from hit 10, alkane side chains of different lengths were expanded. It was found that the length of the para-substituted alkane side chain had little effect on the activity (compounds 10, 13, and 14). On the other hand, for optimization of hit 11, the antiviral activity of the amino group in the para position was significantly better than that in the ortho and meta positions (compounds 11, 15, and 16). Furthermore, substitution of the pyrazole resulted in a complete loss of activity (compound 17). In general, among R1 substituents, the para position was better than ortho and meta positions, and an amino group was better than an alkyl chain. Although compound 15 exhibited increased anti-EV-D68 potency compared with previous screening hits and similar activity to pleconaril through SARs of R1 in series 1, it was still unsatisfactory.
The ethyl carboxylate group, as a flexible group, could not fit firmly into hydrophobic residues that were deep inside the VP1 pocket of EV-D68. So, after R1 groups (A, Figure 3A) were modified, we turned our attention to the B substituent (Figure 3A). When analyzing the superimposed docking poses, we found that ethyl carboxylate group was in a position structurally equivalent to that of the trifluoromethyl-substituted oxadiazole moiety of pleconaril (B, Figure 3B). It has also been reported that, pleconaril (1) has antiviral activity better than that of pirodavir (2) due to the better fit of the substituted oxadiazole group rather than the ester to the VP1 pocket.14 Therefore, to further enhance the compound’s antiviral activity, trifluoromethyl-substituted oxadiazole was spliced into the 3-position of the quinoline scaffold in place of ethyl carboxylate as the next structural optimization strategy (Figure 4). In addition to trifluoromethyl, various other substituents, such as methyl, ethyl, n-propyl, isopropyl, n-butyl, and tert-butyl substituents of the oxadiazole moiety, were also explored for comprehensive analysis of SARs. Therefore, series 2 compounds (Table 3) were synthesized by fixing the p-aminophenyl group as the 6-position substituent of the quinoline scaffold, which was from the best performing compound 15 in series 1.
Figure 4.

Structural optimization strategy. Comparison of the docking modes of 15 (green) and pleconaril (blue, PDB: 4WM7). The results indicated that ethyl carboxylate- and trifluoromethyl-substituted oxadiazole were at structurally equivalent positions in the VP1 pocket (shown in the red box).
Table 3. Antiviral Evaluation of Series 2 against EV-D68a in RD Cellsb.

| compound | R2 | EC50 (μM)b | CC50 (μM)c | SId |
|---|---|---|---|---|
| 19 | CF3 | 0.05 ± 0.01 | >2.5 | >50 |
| 20 | CH3 | 0.28 ± 0.03 | >2.5 | >9 |
| 21 | C2H5 | 0.10 ± 0.007 | >2.5 | >25 |
| 22 | n-C3H7 | 0.41 ± 0.03 | >0.8 | >2 |
| 23 | i-C3H7 | 0.40 ± 0.37 | >0.8 | >2 |
| 24 | n-C4H9 | 0.71 ± 0.05 | >2.5 | >3 |
| 25 | t-C4H9 | 4.22 ± 0.12 | >7.4 | >2 |
EV-D68 strain: STL-2014-12.
EC50: concentration that effectively inhibited the cytopathic effect by 50% in Rhabdomyosarcoma cell lines (RD cells).
CC50: concentration that inhibited the normal uninfected cell viability by 50%, or the maximum soluble concentration which started from 200 μM in serial 3-fold dilutions.
Selectivity index (SI) calculated as CC50/EC50 ratio.
According to the in vitro anti-EV-D68 activities, most series 2 compounds exhibited significant advantages over series 1. Compound 19 was obtained with a nearly 10-fold increase in antiviral potency compared to compound 15. This verified that the ester group at the 3-position of the quinoline scaffold was in a position equivalent to that of substituted oxadiazole group in the VP1 protein cavity and also indicated that the structure optimization strategy in Figure 4 was effective. According to the results, the effect of R2 on antiviral activity was critical, and the priority of R2 was CF3 > C2H5 > CH3 > other groups. Due to scaffold changes after structural optimization, it became necessary to revalidate and enrich the SARs of the quinoline 6-position substituent. Therefore, series 3 compounds (Table 4) were obtained for the chemical expansion of R3, in which the R2 groups were based on the preferred trifluoromethyl, methyl, and ethyl groups.
Table 4. Antiviral Evaluation of Series 3 against EV-D68a in RD Cellsb.


EV-D68 strain: STL-2014-12.
EC50: concentration that effectively inhibited the cytopathic effect by 50% in Rhabdomyosarcoma cell lines (RD cells).
CC50: concentration that inhibited the normal uninfected cell viability by 50%, or the maximum soluble concentration which started from 200 μM in serial 3-fold dilutions.
Selectivity index (SI) calculated as CC50/EC50 ratio.
NA: no activity.
These results indicated that even if the ester moiety was changed, most of the SARs of R1 (in series 1) still apply to the R3 group (in series 3). When R3 was switched to different substituents in the para position, the length of the carbon chain had little effect on antiviral activity (compounds 27, 28, 38–41, and 48–51), but for the steric or cyclic structure, the activity decreased or even disappeared (compounds 30–32). For the amino substituent, the para position was still superior to ortho and meta positions (compounds 21, 46, and 47). When the R3 substituent was replaced with aminopyridine similar to p-aminobenzene, the antiviral activity was not improved (compounds 19–21, 26, 37, and 45). Furthermore, other para substituents were also explored, such as halogen, alkoxy, and cyano, where all the antiviral activities were reduced or completely disappeared (compounds 34, 35, 43, 44, 53–55, 59, and 60). In addition, for the structural expansion with the p-aminobenzene group, the antiviral activities of the compounds were also not enhanced (compounds 33, 36, and 56–58).
According to the preliminary in vitro antiviral activities of compounds 10–60, 19 and 21 exhibited potent antiviral activities against EV-D68 with an EC50 value of 0.05 μM and 0.10 μM, correspondingly, which warranted further exploration of the antiviral potency and pharmacokinetic properties.
Broad-Spectrum Antiviral Potency
To further study the broad-spectrum anti-EV-D68 activities of quinoline analogues, multiple EV-D68 strains (STL, KY, MO, and Fermon) were evaluated to determine the inhibitory activity of compounds 19·HCl and 21·HCl, which are the hydrochloride salt forms prepared from compounds 19 and 21, respectively, to improve the water solubility. As shown in Table 5, compounds 19·HCl and 21·HCl both exhibited excellent broad-spectrum antiviral potency against various EV-D68 strains, which were better than that of pleconaril.
Table 5. Antiviral Evaluation of the Selected Quinoline Analogues against Various EV-D68 Strainsa in RD cellsb.
| EV-D68
(STL)a |
EV-D68
(KY)a |
EVD-68
(MO)a |
EV-D68
(Fermon)a |
|||||
|---|---|---|---|---|---|---|---|---|
| compound | EC50(μM)b | SIc | EC50(μM)b | SIc | EC50(μM)b | SIc | EC50(μM)b | SIc |
| 19·HCl | 0.10 ± 0.004 | >111 | 0.09 ± 0.003 | >123 | 0.09 ± 0.0001 | >123 | 0.09 ± 0.007 | >123 |
| 21·HCl | 0.08 ± 0.01 | >139 | 0.07 ± 0.01 | >159 | 0.07 ± 0.01 | >159 | 0.06 ± 0.02 | >185 |
| pleconaril | 0.40 ± 0.16 | >250 | 0.24 ± 0.004 | >417 | 0.64 ± 0.06 | >156 | 0.30 ± 0.02 | >333 |
EV-D68 strain: STL (STL-2014-12), KY (US/KY/14-18953), MO (US/MO/14-18947) and Fermon (prototype strain Fermon).
EC50: concentration that effectively inhibited the cytopathic effect by 50% in Rhabdomyosarcoma cell lines (RD cells).
Selectivity index (SI) calculated as CC50/EC50 ratio.
Inhibition against EV-D68
Through modification and optimization of the compound structure, 19·HCl and 21·HCl, possessing the best antiviral activity, were selected by CPE assay. But it was also necessary to determine the inhibition of EV-D68 viral replication through different verification methods. The Western blot (Figure 5A) and immunofluorescence assays (Figure 5B) were performed, and the results showed that 19·HCl and 21·HCl significantly inhibited the expression level of viral VP1 in a dose-dependent manner. In addition, to further confirm the efficacy of 19·HCl and 21·HCl on virus proliferation, a plaque formation assay was conducted (Figure 5C). At a concentration of 1 μM, compound 19·HCl and 21·HCl significantly inhibited the replication of EV-D68 virus particles, superior to that of pleconaril.
Figure 5.

Inhibition of viral replication in EV-D68-infected cells. Replication of EV-D68 virions was characterized by (A) Western blot, (B) immunofluorescence assay, and (C) plaque assay.
Pharmacokinetic Study
Subsequently, we performed a pharmacokinetic evaluation of compounds 19 and 21·HCl in SD rats. Through intravenous injection of 1 mg/kg and oral administration of 5 mg/kg, plasma samples were prepared and then analyzed by LC-MS. As shown in Table 6 and Figure S2, the metabolic properties of 19 were significantly superior to those of 21·HCl, which might benefit from the improved metabolic stability imparted by the trifluoromethyl group. Compound 19 exhibited moderate half-life values in both iv and oral administrations (t1/2 = 1.92 and 1.21 h, respectively), rapid distribution at an oral dose of 5 mg/kg (tmax = 1 ± 0 h), and acceptable bioavailability (F = 23.9%). But the clearance (Cl = 64 ± 13.1 mL/min/kg) result indicated that the metabolic stability of 19 in rats was not ideal. Therefore, further metabolic properties needed to be determined.
Table 6. Pharmacokinetic Properties in Rats.
|
19 |
21·HCl |
|||
|---|---|---|---|---|
| PK parametersa | ivb | poc | ivd | poe |
| C0 (ng/mL) | 637 ± 227 | 1370 ± 166 | ||
| t1/2 (h) | 1.92 ± 0.225 | 1.21 ± 0.175 | 0.309 ± 0.131 | 1.06 ± 0.25 |
| Cmax (ng/mL) | 108 ± 48.5 | 36.1 ± 7.3 | ||
| tmax (h) | 1 ± 0 | 0.417 ± 0.144 | ||
| CL (mL/min/kg) | 64 ± 13.1 | 106 ± 10 | ||
| AUC0-t (h·ng/mL) | 253 ± 52.6 | 307 ± 115 | 157 ± 15 | 49.5 ± 10.9 |
| AUC0-inf (h·ng/mL) | 268 ± 58.5 | 320 ± 114 | 158 ± 15 | 53.8 ± 12.5 |
| F (%) | 23.9 | 6.8 | ||
The values are expressed as the mean ± SD.
iv (intravenous injection), dosed at 1 mg/kg (n = 3) with formulation in 5% DMSO/5% Solutol HS-15/90% water.
po (gastrointestinal route), dosed at 5 mg/kg (n = 3) with formulation in 5% DMSO/5% Solutol HS-15/90% water.
iv (intravenous injection), dosed at 1 mg/kg (n = 3) with formulation in 5% DMSO/95% HP-β-CD in water.
po (gastrointestinal route), dosed at 5 mg/kg (n = 3) with formulation in 5% DMSO/95% HP-β-CD in water.
In view of the clearance, three species of liver microsomes, including rat, dog, and human, were used to study the metabolic stability of compound 19. The trend of substrate remaining at different time points in the test systems was illustrated in Figure 6 and Table S2. The stability of compound 19 exhibited a species difference in rat liver microsomes (Clint = 61.2 mL/min/kg, t1/2 = 41 min), dog liver microsomes (Clint = 115.5 mL/min/kg, t1/2 = 17.3 min), and human liver microsomes (Clint = 10.8 mL/min/kg, t1/2 = 148 min), which indicated significant metabolic stability in human liver microsomes. As inferred from the results, despite the unsatisfactory pharmacokinetic stability in rats, compound 19 might exhibit better pharmacokinetic properties in humans and had the potential to be further developed.
Figure 6.
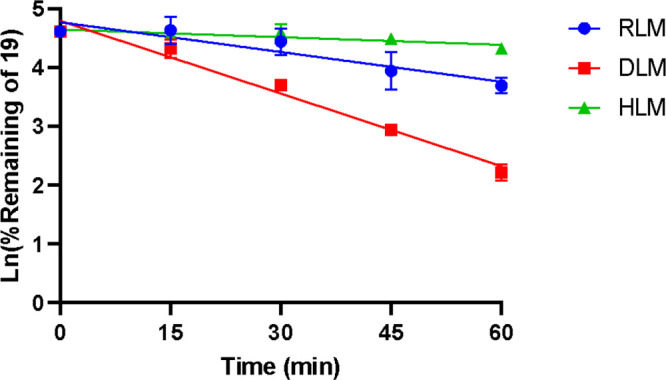
Compound 19 in rat liver microsomes (RLM), dog liver microsomes (DLM), and human liver microsomes (HLM).
Mechanistic Studies
To investigate the antiviral mechanism of compound 19, a time of drug addition experiment was conducted to simulate the virus proliferation cycle as previously described.19 The viral load was detected after compound 19 was added at different stages in the virus proliferation cycle. Pleconaril was used as a control compound as the known VP1 inhibitor. As shown in Figure 7A, the antiviral activity disappeared when compound 19 was added after 1 hpi (phases II and IV). However, the significant antiviral activity during the addition of 19 at 0–1 h (phase I) and the entire proliferation cycle (0–14 h, phase III) indicated that 19 inhibited virus replication only at the early stages, involving the attachment or uncoating of the virus. Furthermore, pleconaril, as a reported VP1 inhibitor, also showed the same experimental results as those for compound 19.
Figure 7.

(A) Time durations of drug addition. Compound 19 (2 μM) and pleconaril (2 μM) were added to RD cells before or after infection with 0.01 MOI EV-D68 at the corresponding time. Viral load was detected by qRT-PCR. Values are presented as mean ± SD, ****P < 0.0001, compared with the control group. (B) Compound 19 protected the EV-D68 virion from heat inactivation. 19 (12.5 μM) or pleconaril (12.5 μM) or DMSO was incubated with EV-D68 virions and heated to the appointed temperatures. Virus titers were quantified by TCID50 assay. The values are presented as mean ± SD. (C) Thermal stabilization by compound 19. The green line represents the control EV-D68 (1.5 μg) that was incubated with SYTO9 dye to detect RNA release. The red and blue lines represent EV-D68 virions incubated with 10 μg/mL compound 19 or pleconaril, respectively.
Considering that the VP1 inhibitor can improve the stability of virus virions and protect them from thermal inactivation, the antiviral mechanism of compound 19 was also verified by thermoprotection assay (Figure 7B), according to a previously reported method.19,22 The results showed that the presence of compound 19 or pleconaril could significantly reduce the thermal inactivation of virus at 55 °C. In contrast, the control group without drug was completely inactivated. Moreover, 19 maintained certain virus activity, even at 57 °C, which was better than that of pleconaril. These results suggested that VP1 may be the proposed target of compound 19. In addition, to further assess viral stability and the dynamics of uncoating,23,24 a particle stability thermal release assay (PaSTRy) was used as described previously.25,26 As a reported VP1 inhibitor, pleconaril was also used as a positive control. As shown in Figure 7C, the temperature at which pure virus released its RNA (TR) was approximately 52 °C. When 1.5 μg of purified virus was incubated with compound 19, TR increased to 55–56 °C, which indicated that compound 19 indeed acted on the capsid of EV-D68, resulting in an improved virus particle stability. However, at the same concentration, pleconaril showed almost no protection against viral particles, which was similar to DMSO and indicated that pleconaril exhibited weaker activity against EV-D68 compared to compound 19.
Molecular Docking
As shown in Figure 8, we used the ligand docking module to dock the preferred compound 19 into the EV-D68 VP1 pocket (PDB: 4WM7). The cocrystal structure of pleconaril indicated that the electron density of the ligand could be well filled in the hydrophobic pocket of VP1, and the original pocket factor was replaced14 (Figure 8A). As for compound 19, the docking result also exhibited a binding mode similar to that of pleconaril, which filled the cavity of VP1 (Figure 8B). By overlapping the docking poses of compound 19 and pleconaril, it could be seen that the spatial structures in the VP1 cavity were basically the same (Figure 8C). Although the scaffold structure of compound 19 was completely different from that of pleconaril, the common trifluoromethyl-substituted oxadiazole groups could both fill the hydrophobic pocket deep in the VP1. The docking results also further verified that the structural optimization strategy of introducing an oxadiazole group was rational. Most likely due to the open region of the protein cavity, the length of the carbon chain substituent at the 6-position of the quinoline scaffold was not a key factor for fitting the cavity.
Figure 8.

(A) The binding mode of pleconaril in the VP1 pocket of EV-D68 (PDB: 4WM7). (B) The docking pose for compound 19 in the same hydrophobic VP1 pocket. (C) The superimposition of docking poses of pleconaril (blue) and compound 19 (purple) in the EV-D68 VP1 pocket.
Conclusions
In general, through structure-based virtual screening, rational drug design, and structure–activity relationship studies, a series of novel scaffold quinoline analogues as anti-EV-D68 agents were identified, and the best compound, 19, was obtained with broad-spectrum antiviral activity against multiple EV-D68 strains and was superior to pleconaril. Among these analogues, the 1,2,4-oxadiazole substituent was essential for quinoline scaffold binding to the hydrophobic pocket of EV-D68 VP1 and enhanced antiviral activity. The in vitro antiviral potency confirmed that 19 could inhibit the expression of viral proteins and replication. Moreover, the drug addition experiment, thermoprotection, and PaSTRy assay verified that these analogues exerted inhibition against EV-D68 mainly by interacting with VP1 of the capsid protein. The acceptable bioavailability in rats and significant metabolic stability in human liver microsomes indicate that the novel scaffold structure of compound 19 may justify further exploration.
Experimental Section
Chemistry
Chemical reagents and solvents were purchased from commercial sources without further purification. 1H, 13C, and 19F NMR spectra were recorded on a Bruker Avance NEO (600 MHz) spectrometer and were determined in CDCl3, CD3OD-d4, DMSO-d6, or acetone-d6 with tetramethylsilane (TMS) functioning as the internal reference. HRMS was obtained using an Agilent TOF G6230A LC/MS with an ESI source. Purity of the compounds was analyzed by HPLC (Agilent Technologies 1260 Infinity) using CH3CN/H2O (gradient elution of 10:90 to 95:5) as the mobile phase with a flow rate of 1 mL/min on a C18 column under the condition of UV 254 nm. All compounds exhibited greater than 95% purity.
Ethyl 6-Bromoquinoline-3-carboxylate (5)
Tin(II) chloride dihydrate (9.03 g, 40.0 mmol) was added to a solution of 5-bromo-2-nitrobenzaldehyde 3 (2.30 g, 10.0 mmol) and ethanol (25 mL) followed by ethyl 3,3-diethoxypropanoate 4 (4.76 g, 25.0 mmol). The mixture was heated to reflux for 5 h. After the reaction was completed, the mixture was concentrated. The residue was dissolved in ethyl acetate and quenched with saturated aqueous NaHCO3. The resulting mixture was filtered and extracted with ethyl acetate. The combined organic layers were washed with saturated aqueous NaCl, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The crude compound was purified via flash chromatography with n-hexane/ethyl acetate to afford 5 (1.7 g, 61%) as a yellow solid. 1H NMR (600 MHz, DMSO-d6) δ 9.32 (t, J = 1.8 Hz, 1H), 8.98 (d, J = 2.2 Hz, 1H), 8.52 (t, J = 1.9 Hz, 1H), 8.03 (q, J = 1.6 Hz, 2H), 4.42 (q, J = 7.1 Hz, 2H), 1.38 (t, J = 7.1 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 164.52, 149.93, 147.83, 137.75, 135.16, 131.54, 131.04, 127.95, 123.64, 120.65, 61.50, 14.23. HRMS (ESI) calcd for C12H10BrNO2 [M + H]+ 279.9973, found 279.9968.
6-Bromoquinoline-3-carbonitrile (7)
Tin(II) chloride dihydrate (26.49 g, 117.39 mmol) was added to a solution of 5-bromo-2-nitrobenzaldehyde 3 (9.0 g, 39.13 mmol) and ethanol (250 mL) and was stirred for 10 min. Then ethyl 3,3-diethoxypropanoate 4 (14.0 g, 97.83 mmol) was added and heated to reflux for 5 h. After the reaction was completed, the resulting precipitated solid was filtered and rinsed well with ethanol. Product 7 (7.72 g, 85%) was obtained without further purification as a yellow solid powder. 1H NMR (600 MHz, DMSO-d6) δ 9.20 (d, J = 2.3 Hz, 1H), 9.02 (q, J = 2.0 Hz, 1H), 8.38 (q, J = 2.2 Hz, 1H), 8.11–8.03 (m, 2H). 13C NMR (151 MHz, DMSO-d6) δ 150.67, 146.73, 141.74, 135.96, 131.29, 130.75, 127.19, 121.42, 117.17, 106.83. HRMS (ESI) calcd for C10H5BrN2 [M + H]+ 232.9714, found 232.9704.
6-Bromo-N-hydroxyquinoline-3-carboximidamide (8)
6-Bromoquinoline-3-carbonitrile 7 (3.08 g, 13.3 mmol) and 50% aqueous solution of hydroxylamine (1.05 g, 16.0 mmol) in ethanol (30 mL) were heated to reflux for 4 h. After the reaction was completed, the reaction mixture and its white precipitate formation was filtered and rinsed well with ethyl acetate to afford 8 (3.2 g, 91%) as a white solid without further purification. 1H NMR (600 MHz, DMSO-d6) δ 10.08 (s, 1H), 9.23 (d, J = 2.1 Hz, 1H), 8.55 (d, J = 2.2 Hz, 1H), 8.25 (d, J = 2.2 Hz, 1H), 7.96 (d, J = 8.9 Hz, 1H), 7.88 (dd, J = 8.9, 2.2 Hz, 1H), 6.12 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 148.96, 148.80, 146.08, 132.91, 131.16, 131.00, 130.31, 128.52, 127.22, 120.09. HRMS (ESI) calcd for C10H8BrN3O [M + H]+ 265.9929, found 265.9920.
General Method A for the Preparation of 9a–g
6-Bromo-N-hydroxyquinoline-3-carboximidamide 8 (1 equiv) and acid anhydride (1.2 equiv) were added to pyridine (20 mL), and the reaction mixture was refluxed for 3 h and then cooled to room temperature. The reaction mixture was poured into water, adjusted to pH 4 by the addition of 1.0 N HCl, and then extracted with ethyl acetate. The combined organic layers were dried over anhydrous Na2SO4 and evaporated. The residue was purified via flash chromatography with n-hexane/ethyl acetate to give the corresponding intermediates.
3-(6-Bromoquinolin-3-yl)-5-(trifluoromethyl)-1,2,4-oxadiazole (9a)
Compound 9a was obtained by general method A as a yellow solid with a yield of 71%. 1H NMR (600 MHz, chloroform-d) δ 9.57 (d, J = 2.1 Hz, 1H), 8.84–8.80 (m, 1H), 8.12 (d, J = 2.2 Hz, 1H), 8.06 (d, J = 8.9 Hz, 1H), 7.91 (dd, J = 8.9, 2.2 Hz, 1H). 13C NMR (151 MHz, chloroform-d) δ 167.21, 166.38 (q, J = 44.8 Hz), 148.22, 148.10, 135.21, 134.99, 131.38, 130.64, 128.20, 122.01, 119.20, 115.88 (q, J = 273.7 Hz). 19F NMR (565 MHz, chloroform-d) δ −65.19. HRMS (ESI) calcd for C12H5BrF3N3O [M + H]+ 343.9646, found 343.9642.
3-(6-Bromoquinolin-3-yl)-5-methyl-1,2,4-oxadiazole (9b)
Compound 9b was obtained by general method A as a white solid with a yield of 57%. 1H NMR (600 MHz, chloroform-d) δ 9.54 (d, J = 2.1 Hz, 1H), 8.73 (d, J = 2.1 Hz, 1H), 8.06 (d, J = 2.1 Hz, 2H), 8.03 (d, J = 8.9 Hz, 1H), 7.85 (dd, J = 9.1, 2.2 Hz, 1H), 2.71 (s, 3H). 13C NMR (151 MHz, chloroform-d) δ 177.19, 166.29, 148.69, 147.56, 134.44, 134.07, 131.19, 130.46, 128.46, 121.52, 121.05, 12.44. HRMS (ESI) calcd for C12H8BrN3O [M + H]+ 289.9929, found 289.9921.
3-(6-Bromoquinolin-3-yl)-5-ethyl-1,2,4-oxadiazole (9c)
Compound 9c was obtained by general method A as a yellow solid with a yield of 52%. 1H NMR (600 MHz, chloroform-d) δ 9.55 (d, J = 2.1 Hz, 1H), 8.75 (d, J = 2.0 Hz, 1H), 8.07 (d, J = 2.2 Hz, 1H), 8.03 (d, J = 8.9 Hz, 1H), 7.85 (dd, J = 8.9, 2.2 Hz, 1H), 3.03 (q, J = 7.6 Hz, 2H), 1.49 (t, J = 7.6 Hz, 3H). 13C NMR (151 MHz, chloroform-d) δ 181.40, 166.16, 148.74, 147.52, 134.40, 134.12, 131.18, 130.48, 128.48, 121.49, 121.21, 20.37, 10.81. HRMS (ESI) calcd for C13H10BrN3O [M + H]+ 304.0085, found 304.0079.
3-(6-Bromoquinolin-3-yl)-5-propyl-1,2,4-oxadiazole (9d)
Compound 9d was obtained by general method A as a yellow solid with a yield of 50.2%. 1H NMR (600 MHz, chloroform-d) δ 9.57 (d, J = 2.1 Hz, 1H), 8.77 (d, J = 2.5 Hz, 1H), 8.11–8.03 (m, 2H), 7.86 (dd, J = 8.9, 2.2 Hz, 1H), 2.99 (t, J = 7.5 Hz, 2H), 1.96 (h, J = 7.4 Hz, 2H), 1.10 (t, J = 7.4 Hz, 3H). 13C NMR (151 MHz, chloroform-d) δ 180.61, 166.14, 148.70, 147.43, 134.49, 134.24, 131.13, 130.50, 128.53, 121.57, 121.26, 28.49, 20.20, 13.66. HRMS (ESI) calcd for C14H12BrN3O [M + H]+ 318.0242, found 318.0233.
3-(6-Bromoquinolin-3-yl)-5-isopropyl-1,2,4-oxadiazole (9e)
Compound 9e was obtained by general method A as a yellow solid with a yield of 60.1%. 1H NMR (600 MHz, chloroform-d) δ 9.55 (d, J = 2.1 Hz, 1H), 8.76 (d, J = 2.0 Hz, 1H), 8.08 (d, J = 2.2 Hz, 1H), 8.03 (d, J = 9.0 Hz, 1H), 7.85 (dd, J = 9.0, 2.2 Hz, 1H), 3.34 (m, J = 7.0 Hz, 1H), 1.50 (d, J = 7.0 Hz, 6H). 13C NMR (151 MHz, chloroform-d) δ 184.61, 166.05, 148.79, 147.51, 134.35, 134.13, 131.19, 130.47, 128.49, 121.46, 121.30, 27.61, 20.20. HRMS (ESI) calcd for C14H12BrN3O [M + H]+ 318.0242, found 318.0236.
3-(6-Bromoquinolin-3-yl)-5-butyl-1,2,4-oxadiazole (9f)
Compound 9f was obtained by general method A as a yellow solid with a yield of 56.2%. 1H NMR (600 MHz, chloroform-d) δ 9.56 (d, J = 2.2 Hz, 1H), 8.75 (d, J = 2.1 Hz, 1H), 8.07 (d, J = 2.2 Hz, 2H), 8.03 (d, J = 8.9 Hz, 1H), 7.85 (dd, J = 9.0, 2.2 Hz, 1H), 3.01 (t, J = 7.6 Hz, 2H), 1.90 (p, J = 7.5 Hz, 2H), 1.50 (h, J = 7.4 Hz, 2H), 1.00 (t, J = 7.4 Hz, 3H). 13C NMR (151 MHz, chloroform-d) δ 180.73, 166.13, 148.77, 147.54, 134.35, 134.06, 131.19, 130.45, 128.46, 121.45, 121.19, 28.62, 26.34, 22.18, 13.56. HRMS (ESI) calcd for C15H14BrN3O [M + H]+ 332.0398, found 332.0393.
3-(6-Bromoquinolin-3-yl)-5-(tert-butyl)-1,2,4-oxadiazole (9g)
Compound 9g was obtained by general method A as a yellow solid with a yield of 40%. 1H NMR (600 MHz, chloroform-d) δ 9.55 (d, J = 2.1 Hz, 1H), 8.76 (d, J = 2.0 Hz, 1H), 8.08 (d, J = 2.2 Hz, 1H), 8.03 (d, J = 8.9 Hz, 1H), 7.84 (dd, J = 9.0, 2.2 Hz, 1H), 1.53 (s, 9H). 13C NMR (151 MHz, chloroform-d) δ 186.95, 165.98, 148.84, 147.50, 134.29, 134.13, 131.20, 130.47, 128.49, 121.42, 121.39, 33.77, 28.42. HRMS (ESI) calcd for C15H14BrN3O [M + H]+ 332.0398, found 332.0390.
General Method B for the Preparation of Compounds 10–18
Ethyl 6-bromoquinoline-3-carboxylate 5 (1.0 equiv), boric acid derivative (1.3 equiv), PdCl2 (dppf) (0.05 equiv), and K2CO3 (2 equiv) were dissolved in DME (5 mL) and water (1 mL). The mixture was reacted by microwave irradiation at 120 °C for 40 min under nitrogen. The solvents were removed, and the residue was purified via flash chromatography with n-hexane/ethyl acetate to give the corresponding products.
Ethyl 6-(4-Propylphenyl)quinoline-3-carboxylate (10)
Compound 10 was obtained by general method B as a white solid with a yield of 70%. 1H NMR (600 MHz, DMSO-d6) δ 9.28 (d, J = 2.2 Hz, 1H), 9.02 (dd, J = 2.2, 0.8 Hz, 1H), 8.48 (d, J = 2.1 Hz, 1H), 8.23 (dd, J = 8.8, 2.1 Hz, 1H), 8.15 (dd, J = 8.8, 0.9 Hz, 1H), 7.78–7.73 (m, 2H), 7.37–7.32 (m, 2H), 4.42 (q, J = 7.1 Hz, 2H), 2.61 (dd, J = 8.3, 6.9 Hz, 2H), 1.68–1.59 (m, 2H), 1.39 (t, J = 7.1 Hz, 3H), 0.92 (t, J = 7.3 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 164.83, 149.23, 148.53, 142.37, 139.03, 138.69, 136.24, 131.22, 129.37, 129.22, 127.05, 126.92, 126.46, 123.16, 61.34, 36.94, 24.08, 14.24, 13.74. HRMS (ESI) calcd for C21H21NO2 [M + H]+ 320.1651, found 320.1645.
Ethyl 6-(3-Aminophenyl)quinoline-3-carboxylate (11)
Compound 11 was obtained by general method B as a yellow solid with a yield of 41.3%. 1H NMR (600 MHz, DMSO-d6) δ 9.29 (d, J = 2.2 Hz, 1H), 9.05 (d, J = 2.1 Hz, 1H), 8.40 (d, J = 2.0 Hz, 1H), 8.18–8.10 (m, 2H), 7.24 (t, J = 7.8 Hz, 1H), 7.10 (t, J = 2.0 Hz, 2H), 7.07 (dt, J = 7.7, 1.2 Hz, 1H), 6.77–6.72 (m, 1H), 4.42 (q, J = 7.1 Hz, 2H), 1.39 (t, J = 7.1 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 164.85, 149.24, 148.59, 147.46, 139.83, 139.76, 138.77, 131.33, 129.86, 129.32, 126.88, 126.55, 123.20, 116.31, 114.95, 113.63, 61.35, 14.25. HRMS (ESI) calcd for C18H16N2O2 [M + H]+ 293.1290, found 293.1287.
Ethyl 6-(4-Isopropylphenyl)quinoline-3-carboxylate (12)
Compound 12 was obtained by general method B as a white solid with a yield of 83%. 1H NMR (600 MHz, DMSO-d6) δ 9.27 (d, J = 2.2 Hz, 1H), 8.99 (d, J = 2.4 Hz, 1H), 8.45 (d, J = 2.2 Hz, 1H), 8.19 (dd, J = 8.7, 2.2 Hz, 1H), 8.13 (d, J = 8.7 Hz, 1H), 7.74 (d, J = 7.9 Hz, 2H), 7.38 (d, J = 7.9 Hz, 2H), 4.41 (q, J = 7.1 Hz, 2H), 2.94 (m, J = 6.9 Hz, 1H), 1.38 (t, J = 7.1 Hz, 3H), 1.24 (d, J = 7.0 Hz, 6H). 13C NMR (151 MHz, DMSO-d6) δ 164.83, 149.24, 148.58, 139.07, 138.69, 136.41, 131.27, 129.38, 127.18, 126.92, 126.49, 123.16, 61.35, 33.23, 23.88, 14.25. HRMS (ESI) calcd for C21H21NO2 [M + H]+ 320.1651, found 320.1646.
Ethyl 6-(4-Butylphenyl)quinoline-3-carboxylate (13)
Compound 13 was obtained by general method B as a white solid with a yield of 30%. 1H NMR (600 MHz, DMSO-d6) δ 9.29 (d, J = 2.1 Hz, 1H), 9.03 (d, J = 2.1 Hz, 1H), 8.49 (d, J = 2.1 Hz, 1H), 8.23 (dd, J = 8.7, 2.2 Hz, 1H), 8.15 (d, J = 8.8 Hz, 1H), 7.78–7.73 (m, 2H), 7.37–7.32 (m, 2H), 4.42 (q, J = 7.1 Hz, 2H), 2.64 (t, J = 7.7 Hz, 2H), 1.64–1.55 (m, 2H), 1.39 (t, J = 7.1 Hz, 3H), 1.33 (h, J = 7.4 Hz, 2H), 0.91 (t, J = 7.4 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 164.83, 149.23, 148.53, 142.58, 139.03, 138.69, 136.20, 131.22, 129.37, 129.17, 127.07, 126.93, 126.45, 123.17, 61.34, 34.52, 33.11, 21.85, 14.25, 13.86. HRMS (ESI) calcd for C22H23NO2 [M + H]+ 334.1807, found 334.1803.
Ethyl 6-Phenylquinoline-3-carboxylate (14)
Compound 14 was obtained by general method B as a white solid with a yield of 20.0%. 1H NMR (600 MHz, DMSO-d6) δ 9.30 (d, J = 2.1 Hz, 1H), 9.04 (d, J = 2.1 Hz, 1H), 8.52 (d, J = 2.1 Hz, 1H), 8.25 (dd, J = 8.7, 2.2 Hz, 1H), 8.17 (d, J = 8.8 Hz, 1H), 7.88–7.83 (m, 2H), 7.55 (t, J = 7.7 Hz, 2H), 7.48–7.42 (m, 1H), 4.42 (q, J = 7.1 Hz, 2H), 1.39 (t, J = 7.1 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 164.81, 149.40, 148.63, 139.08, 138.88, 138.78, 131.32, 129.44, 129.24, 128.25, 127.23, 126.93, 126.89, 123.21, 61.36, 14.25. HRMS (ESI) calcd for C18H15NO2 [M + H]+ 278.1181, found 278.1176.
Ethyl 6-(4-Aminophenyl)quinoline-3-carboxylate (15)
Compound 15 was obtained by general method B as a yellow solid with a yield of 76%. 1H NMR (600 MHz, DMSO-d6) δ 9.21 (d, J = 2.1 Hz, 1H), 8.94 (d, J = 2.1 Hz, 1H), 8.31 (d, J = 2.2 Hz, 1H), 8.15 (dd, J = 8.8, 2.2 Hz, 1H), 8.06 (d, J = 8.7 Hz, 1H), 7.60–7.54 (m, 2H), 6.73–6.69 (m, 2H), 5.40 (s, 2H), 4.40 (q, J = 7.1 Hz, 2H), 1.38 (t, J = 7.1 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 164.93, 149.26, 148.37, 148.00, 139.64, 138.26, 130.69, 129.09, 127.83, 127.13, 125.67, 124.08, 123.00, 114.37, 61.26, 14.24. HRMS (ESI) calcd for C18H16N2O2 [M + H]+ 293.1290, found 293.1288.
Ethyl 6-(2-Aminophenyl)quinoline-3-carboxylate (16)
Compound 16 was obtained by general method B as a yellow solid with a yield of 67%. 1H NMR (600 MHz, DMSO-d6) δ 9.30 (d, J = 2.2 Hz, 1H), 9.00 (d, J = 2.0 Hz, 1H), 8.23 (d, J = 2.0 Hz, 1H), 8.14 (d, J = 8.7 Hz, 1H), 7.97 (dd, J = 8.7, 2.0 Hz, 1H), 7.15–7.08 (m, 2H), 6.83 (dd, J = 7.9, 1.0 Hz, 1H), 6.70 (td, J = 7.4, 1.2 Hz, 1H), 5.05 (s, 2H), 4.42 (q, J = 7.1 Hz, 2H), 1.39 (t, J = 7.1 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 164.90, 149.14, 148.31, 145.43, 139.09, 138.60, 133.68, 130.49, 129.10, 128.89, 128.84, 126.89, 124.48, 122.93, 116.97, 115.67, 61.33, 14.26. HRMS (ESI) calcd for C18H16N2O2 [M + H]+ 293.1290, found 293.1287.
Ethyl 6-(1H-Pyrazol-4-yl)quinoline-3-carboxylate (17)
Compound 17 was obtained by general method B as a black solid with a yield of 9.4%. 1H NMR (600 MHz, DMSO-d6) δ 13.12 (s, 1H), 9.22 (t, J = 1.6 Hz, 1H), 8.88 (d, J = 1.9 Hz, 1H), 8.45–8.37 (m, 2H), 8.21 (dt, J = 8.7, 1.6 Hz, 1H), 8.12 (s, 1H), 8.07 (d, J = 8.7 Hz, 1H), 4.41 (q, J = 7.1 Hz, 2H), 1.39 (t, J = 7.1 Hz, 3H).13C NMR (151 MHz, DMSO-d6) δ 164.84, 148.39, 148.06, 137.86, 136.84, 132.32, 130.67, 129.26, 127.13, 126.53, 123.71, 123.11, 120.26, 61.29, 14.22. HRMS (ESI) calcd for C15H13N3O2 [M + H]+ 268.1086, found 268.1081.
Ethyl 6-(4-Carbamoylphenyl)quinoline-3-carboxylate (18)
Compound 18 was obtained by general method B as a white solid with a yield of 51%. 1H NMR (600 MHz, DMSO-d6) δ 9.32 (d, J = 2.1 Hz, 1H), 9.07 (d, J = 2.1 Hz, 1H), 8.67–8.57 (m, 1H), 8.36–8.29 (m, 1H), 8.19 (d, J = 8.8 Hz, 1H), 8.09 (s, 1H), 8.05 (d, J = 8.0 Hz, 2H), 7.96 (d, J = 7.9 Hz, 2H), 7.45 (s, 1H), 4.43 (q, J = 7.1 Hz, 2H), 1.40 (t, J = 7.1 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 167.48, 164.78, 149.69, 148.83, 141.40, 138.91, 138.12, 133.79, 131.25, 129.54, 128.42, 127.47, 127.02, 126.88, 123.31, 61.40, 14.25. HRMS (ESI) calcd for C19H16N2O3 [M + H]+ 321.1239, found 321.1234.
General Method C for the Preparation of Compounds 19–50
A corresponding compound of 9a–g (1.0 equiv), boric acid derivative (1.3 equiv), PdCl2 (dppf) (0.05 equiv), and K2CO3 (2 equiv) were dissolved in DME (5 mL) and water (1 mL). The mixture was reacted by microwave irradiation at 120 °C for 40 min under nitrogen. The solvents were removed, and the residue was purified via flash chromatography with n-hexane/ethyl acetate to give the corresponding products.
4-(3-(5-(Trifluoromethyl)-1,2,4-oxadiazol-3-yl)quinolin-6-yl)aniline (19)
Compound 19 was obtained by general method C as a yellow solid with a yield of 58%. 1H NMR (600 MHz, DMSO-d6) δ 9.35 (d, J = 2.2 Hz, 1H), 9.07 (d, J = 2.1 Hz, 1H), 8.37 (d, J = 2.1 Hz, 1H), 8.17 (dd, J = 8.8, 2.2 Hz, 1H), 8.09 (d, J = 8.8 Hz, 1H), 7.60–7+.55 (m, 2H), 6.74–6.69 (m, 2H), 5.42 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 167.13, 165.28 (q, J = 44.5, 44.1 Hz), 149.33, 147.63, 146.39, 139.98, 135.96, 130.53, 129.24, 127.85, 127.47, 125.58, 123.78, 118.25, 115.87 (q, J = 273.0 Hz), 114.36. 19F NMR (565 MHz, DMSO) δ −64.65. HRMS (ESI) calcd for C18H11F3N4O [M + H]+ 357.0963, found 357.0958.
4-(3-(5-Methyl-1,2,4-oxadiazol-3-yl)quinolin-6-yl)aniline (20)
Compound 20 was obtained by general method C as a yellow solid with a yield of 29%. 1H NMR (600 MHz, DMSO-d6) δ 9.31 (d, J = 2.0 Hz, 1H), 8.95 (d, J = 2.0 Hz, 1H), 8.30 (d, J = 2.0 Hz, 1H), 8.12 (dd, J = 8.8, 2.1 Hz, 1H), 8.07 (d, J = 8.8 Hz, 1H), 7.60–7.55 (m, 2H), 6.74–6.69 (m, 2H), 5.41 (s, 2H), 2.72 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 177.91, 166.19, 149.24, 147.32, 146.74, 139.72, 134.91, 129.91, 129.20, 127.84, 127.62, 125.76, 123.62, 119.92, 114.36, 12.16. HRMS (ESI) calcd for C18H14N4O [M + H]+ 303.1246, found 303.1241.
4-(3-(5-Ethyl-1,2,4-oxadiazol-3-yl)quinolin-6-yl)aniline (21)
Compound 21 was obtained by general method C as a yellow solid with a yield of 48%. 1H NMR (600 MHz, DMSO-d6) δ 9.32 (d, J = 2.0 Hz, 1H), 8.96 (d, J = 2.0 Hz, 1H), 8.33 (d, J = 2.0 Hz, 1H), 8.13 (dd, J = 8.8, 2.0 Hz, 1H), 8.07 (d, J = 8.7 Hz, 1H), 7.58 (d, J = 8.1 Hz, 2H), 6.71 (d, J = 8.2 Hz, 2H), 5.40 (s, 2H), 3.07 (q, J = 7.6 Hz, 2H), 1.39 (t, J = 7.5 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 181.67, 166.08, 149.24, 147.32, 146.74, 139.72, 134.93, 129.90, 129.19, 127.83, 127.63, 125.77, 123.66, 119.97, 114.36, 19.73, 10.53. HRMS (ESI) calcd for C19H16N4O [M + H]+ 317.1402, found 317.1396.
4-(3-(5-Propyl-1,2,4-oxadiazol-3-yl)quinolin-6-yl)aniline (22)
Compound 22 was obtained by general method C as a yellow solid with a yield of 48.1%. 1H NMR (600 MHz, DMSO-d6) δ 9.31 (d, J = 2.2 Hz, 1H), 8.96 (d, J = 2.3 Hz, 1H), 8.32 (d, J = 2.2 Hz, 1H), 8.13 (dd, J = 8.8, 2.1 Hz, 1H), 8.07 (d, J = 8.7 Hz, 1H), 7.58 (d, J = 8.2 Hz, 2H), 6.71 (d, J = 8.2 Hz, 2H), 5.40 (s, 2H), 3.03 (t, J = 7.4 Hz, 2H), 1.85 (h, J = 7.4 Hz, 2H), 1.02 (t, J = 7.4 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 180. 70, 166.08, 149.24, 147.32, 146.74, 139.71, 134.94, 129.89, 129.19, 127.83, 127.62, 125.77, 123.65, 119.95, 114.35, 27.69, 19.60, 13.48. HRMS (ESI) calcd for C20H18N4O [M + H]+ 331.1559, found 331.1553.
4-(3-(5-Isopropyl-1,2,4-oxadiazol-3-yl)quinolin-6-yl)aniline (23)
Compound 23 was obtained by general method C as a yellow solid with a yield of 36.6%. 1H NMR (600 MHz, DMSO-d6) δ 9.32 (d, J = 2.1 Hz, 1H), 8.97 (d, J = 2.0 Hz, 1H), 8.34 (d, J = 2.4 Hz, 1H), 8.13 (dd, J = 8.8, 2.1 Hz, 1H), 8.07 (d, J = 8.8 Hz, 1H), 7.61–7.56 (m, 2H), 6.74–6.68 (m, 2H), 5.40 (s, 2H), 3.41 (m, J = 7.0 Hz, 1H), 1.43 (d, J = 7.0 Hz, 6H). 13C NMR (151 MHz, DMSO-d6) δ 184.54, 166.03, 149.24, 147.32, 146.74, 139.72, 134.97, 129.89, 129.19, 127.83, 127.64, 125.78, 123.69, 119.97, 114.36, 26.95, 19.92. HRMS (ESI) calcd for C20H18N4O [M + H]+ 331.1559, found 331.1553.
4-(3-(5-Butyl-1,2,4-oxadiazol-3-yl)quinolin-6-yl)aniline (24)
Compound 24 was obtained by general method C as a yellow solid with a yield of 20.3%. 1H NMR (600 MHz, DMSO-d6) δ 9.32 (d, J = 2.1 Hz, 1H), 8.96 (d, J = 2.0 Hz, 1H), 8.33 (d, J = 2.0 Hz, 1H), 8.13 (dd, J = 8.8, 2.1 Hz, 1H), 8.07 (d, J = 8.8 Hz, 1H), 7.61–7.55 (m, 2H), 6.74–6.68 (m, 2H), 5.40 (s, 2H), 3.05 (t, J = 7.5 Hz, 2H), 1.85–1.77 (m, 2H), 1.47–1.38 (m, 2H), 0.94 (t, J = 7.3 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 180.85, 166.09, 149.25, 147.32, 146.74, 139.72, 134.96, 129.90, 129.19, 127.83, 127.63, 125.76, 123.66, 119.95, 114.35, 28.09, 25.57, 21.64, 13.56. HRMS (ESI) calcd for C21H20N4O [M + H]+ 345.1715, found 345.1711.
4-(3-(5-(tert-Butyl)-1,2,4-oxadiazol-3-yl)quinolin-6-yl)aniline (25)
Compound 25 was obtained by general method C as a yellow solid with a yield of 48.4%. 1H NMR (600 MHz, DMSO-d6) δ 9.32 (d, J = 2.2 Hz, 1H), 8.96 (d, J = 2.1 Hz, 1H), 8.35 (d, J = 2.3 Hz, 1H), 8.13 (dd, J = 8.7, 2.1 Hz, 1H), 8.07 (d, J = 8.8 Hz, 1H), 7.61–7.54 (m, 2H), 6.74–6.67 (m, 2H), 5.40 (s, 2H), 1.49 (s, 9H). 13C NMR (151 MHz, DMSO-d6) δ 186.68, 166.01, 149.24, 147.33, 146.74, 139.72, 134.99, 129.88, 129.19, 127.83, 127.63, 125.78, 123.71, 119.97, 114.36, 33.49, 28.05. HRMS (ESI) calcd for C21H20N4O [M + H]+ 345.1715, found 345.1710.
5-(3-(5-(Trifluoromethyl)-1,2,4-oxadiazol-3-yl)quinolin-6-yl)pyridin-2-amine (26)
Compound 26 was obtained by general method C as a yellow solid with a yield of 12%. 1H NMR (600 MHz, DMSO-d6) δ 9.39 (t, J = 2.3 Hz, 1H), 9.11 (q, J = 2.4 Hz, 1H), 8.49–8.43 (m, 2H), 8.20 (dt, J = 8.8, 2.2 Hz, 1H), 8.13 (dd, J = 8.8, 2.4 Hz, 1H), 7.91 (dt, J = 8.7, 2.0 Hz, 1H), 6.60 (d, J = 8.6 Hz, 1H), 6.27 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 167.08, 165.32 (q, J = 43.7 Hz), 159.77, 147.80, 146.75, 146.59, 137.53, 136.06, 135.68, 130.25, 129.48, 127.43, 124.10, 122.33, 118.39, 115.87 (q, J = 273.4 Hz), 108.21. 19F NMR (565 MHz, DMSO) δ −64.64. HRMS (ESI) calcd for C17H10F3N5O [M + H]+ 358.0916, found 358.0910.
3-(6-(4-Butylphenyl)quinolin-3-yl)-5-(trifluoromethyl)-1,2,4-oxadiazole (27)
Compound 27 was obtained by general method C as a white solid with a yield of 35%. 1H NMR (600 MHz, chloroform-d) δ 9.55 (d, J = 2.0 Hz, 1H), 8.96 (d, J = 2.0 Hz, 1H), 8.25 (d, J = 9.2 Hz, 1H), 8.11 (m, J = 4.8, 2.1 Hz, 2H), 7.67–7.62 (m, 2H), 7.34 (d, J = 7.8 Hz, 2H), 2.72–2.68 (m, 2H), 1.71–1.64 (m, 2H), 1.46–1.38 (m, 2H), 0.97 (t, J = 7.4 Hz, 3H). 13C NMR (151 MHz, chloroform-d) δ 167.53, 166.24 (q, J = 44.7 Hz), 148.61, 147.48, 143.27, 140.78, 136.84, 136.36, 131.62, 129.88, 129.23, 127.42, 127.28, 125.82, 118.66, 115.94 (q, J = 274.4 Hz), 35.33, 33.60, 22.41, 13.97. 19F NMR (565 MHz, CDCl3) δ −65.20. HRMS (ESI) calcd for C22H18F3N3O [M + H]+ 398.1480, found 398.1475.
3-(6-(p-Tolyl)quinolin-3-yl)-5-(trifluoromethyl)-1,2,4-oxadiazole (28)
Compound 28 was obtained by general method C as a white solid with a yield of 25%. 1H NMR (600 MHz, chloroform-d) δ 9.55 (d, J = 2.1 Hz, 1H), 8.95 (d, J = 2.0 Hz, 1H), 8.24 (d, J = 9.2 Hz, 1H), 8.10 (dt, J = 5.2, 2.6 Hz, 2H), 7.65–7.60 (m, 2H), 7.34 (d, J = 7.8 Hz, 2H), 2.44 (s, 3H). 13C NMR (151 MHz, chloroform-d) δ 167.54, 166.23 (q, J = 44.7 Hz), 148.69, 147.55, 140.71, 138.22, 136.68, 136.29, 131.54, 129.95, 129.86, 127.40, 127.28, 125.80, 118.66, 115.94 (q, J = 273.7 Hz), 21.17. 19F NMR (565 MHz, CDCl3) δ −65.20. HRMS (ESI) calcd for C19H12F3N3O [M + H]+ 356.1011, found 356.1005.
3-(6-Phenylquinolin-3-yl)-5-(trifluoromethyl)-1,2,4-oxadiazole (29)
Compound 29 was obtained by general method C as a white solid with a yield of 53.6%. 1H NMR (600 MHz, chloroform-d) δ 9.57 (d, J = 2.2 Hz, 1H), 8.97 (d, J = 2.0 Hz, 1H), 8.27 (d, J = 8.6 Hz, 1H), 8.15–8.09 (m, 2H), 7.75–7.71 (m, 2H), 7.53 (t, J = 7.6 Hz, 2H), 7.45 (t, J = 7.4 Hz, 1H). 13C NMR (151 MHz, chloroform-d) δ 167.49, 166.26 (q, J = 44.7 Hz), 148.71, 147.68, 140.83, 139.59, 136.41, 131.64, 129.99, 129.15, 128.23, 127.47, 127.38, 126.23, 118.73, 115.94 (q, J = 273.3 Hz). 19F NMR (565 MHz, chloroform-d) δ −65.20. HRMS (ESI) calcd for C18H10F3N3O [M + H]+ 342.0854, found 342.0849.
3-(6-(4-Isopropylphenyl)quinolin-3-yl)-5-(trifluoromethyl)-1,2,4-oxadiazole (30)
Compound 30 was obtained by general method C as a white solid with a yield of 45%. 1H NMR (600 MHz, chloroform-d) δ 9.55 (d, J = 2.1 Hz, 1H), 8.95 (d, J = 2.0 Hz, 1H), 8.24 (d, J = 9.2 Hz, 1H), 8.11 (m, J = 4.6, 2.1 Hz, 2H), 7.69–7.64 (m, 2H), 7.42–7.37 (m, 2H), 3.01 (h, J = 6.9 Hz, 1H), 1.33 (d, J = 6.9 Hz, 6H). 13C NMR (151 MHz, acetone-d6) δ 168.41, 166.58 (q, J = 43.7 Hz), 149.75, 149.60, 148.10, 140.98, 137.74, 136.86, 131.80, 130.67, 128.28, 128.05, 127.91, 126.85, 119.64, 116.99 (d, J = 272.5 Hz), 34.45, 24.12. 19F NMR (565 MHz, CDCl3) δ −65.19. HRMS (ESI) calcd for C21H16F3N3O [M + H]+ 384.1324, found 384.1319.
3-(6-(4-(tert-Butyl)phenyl)quinolin-3-yl)-5-(trifluoromethyl)-1,2,4-oxadiazole (31)
Compound 31 was obtained by general method C as a white solid with a yield of 44%. 1H NMR (600 MHz, chloroform-d) δ 9.56 (d, J = 2.1 Hz, 1H), 8.97 (d, J = 2.1 Hz, 1H), 8.26 (d, J = 9.3 Hz, 1H), 8.15–8.10 (m, 2H), 7.71–7.65 (m, 2H), 7.59–7.53 (m, 2H), 1.40 (s, 9H). 13C NMR (151 MHz, chloroform-d) δ 167.52, 166.24 (q, J = 44.7 Hz), 151.47, 148.63, 147.49, 14 0.65, 136.61, 136.38, 131.61, 129.89, 127.42, 127.11, 126.13, 125.85, 118.66, 115.94 (q, J = 273.7 Hz), 34.67, 31.32. 19F NMR (565 MHz, CDCl3) δ −65.20. HRMS (ESI) calcd for C22H18F3N3O [M + H]+ 398.1480, found 398.1475.
3-(6-([1,1′-Biphenyl]-4-yl)quinolin-3-yl)-5-(trifluoromethyl)-1,2,4-oxadiazole (32)
Compound 32 was obtained by general method C as a white solid with a yield of 29%. 1H NMR (600 MHz, chloroform-d) δ 9.58 (d, J = 2.1 Hz, 1H), 8.99 (d, J = 2.0 Hz, 1H), 8.29 (d, J = 8.5 Hz, 1H), 8.20–8.15 (m, 2H), 7.83–7.80 (m, 2H), 7.76 (d, J = 8.3 Hz, 2H), 7.69–7.66 (m, 2H), 7.49 (t, J = 7.7 Hz, 2H), 7.43–7.37 (m, 1H). 13C NMR (151 MHz, chloroform-d) δ 167.49, 166.26 (q, J = 44.8 Hz), 148.76, 147.70, 141.12, 140.31, 140.29, 138.38, 136.39, 131.47, 130.07, 128.91, 127.84, 127.81, 127.65, 127.43, 127.08, 126.06, 118.78, 115.94 (q, J = 273.3 Hz). 19F NMR (565 MHz, CDCl3) δ −65.18. HRMS (ESI) calcd for C24H14F3N3O [M + H]+ 418.1167, found 418.1162.
4-(4-(3-(5-Ttrifluoromethyl)-1,2,4-oxadiazol-3-yl)quinolin-6-yl)phenyl)morpholine (33)
Compound 33 was obtained by general method C as a yellow solid with a yield of 16.7%. 1H NMR (600 MHz, DMSO-d6) δ 9.41 (d, J = 2.2 Hz, 1H), 9.15 (d, J = 2.1 Hz, 1H), 8.50 (d, J = 2.1 Hz, 1H), 8.25 (dd, J = 8.8, 2.2 Hz, 1H), 8.15 (d, J = 8.7 Hz, 1H), 7.78 (d, J = 8.9 Hz, 2H), 7.11 (d, J = 8.9 Hz, 2H), 3.84–3.69 (m, 4H), 3.26–3.18 (m, 4H). 13C NMR (151 MHz, DMSO-d6) δ 165.33, 163.55 (q, J = 43.6 Hz), 149.31, 146.13, 145.07, 137.47, 134.46, 128.95, 127.65, 127.08, 126.00, 125.65, 123.10, 116.62, 114.10 (q, J = 273.4 Hz), 113.56, 64.34, 46.23. 19F NMR (565 MHz, DMSO-d6) δ −64.64. HRMS (ESI) calcd for C22H17F3N4O2 [M + H]+ 427.1382, found 427.1376.
3-(6-(4-Fluorophenyl)quinolin-3-yl)-5-(trifluoromethyl)-1,2,4-oxadiazole (34)
Compound 34 was obtained by general method C as a white solid with a yield of 17.5%. 1H NMR (600 MHz, DMSO-d6) δ 9.43 (d, J = 2.2 Hz, 1H), 9.15 (d, J = 2.0 Hz, 1H), 8.54 (d, J = 2.1 Hz, 1H), 8.23 (dd, J = 8.7, 2.2 Hz, 1H), 8.18 (d, J = 8.7 Hz, 1H), 7.92–7.88 (m, 2H), 7.41–7.36 (m, 2H). 13C NMR (151 MHz, DMSO-d6) δ 167.02, 165.34 (q, J = 44.5, 44.0 Hz), 162.42 (d, J = 245.4 Hz), 148.18, 147.49, 138.36, 136.46, 135.30 (d, J = 3.4 Hz), 131.13–131.00 (m), 129.60, 129.30 (d, J = 8.1 Hz), 127.18, 126.58, 118.52, 116.12 (d, J = 21.3 Hz), 115.86 (q, J = 273.2 Hz). 19F NMR (565 MHz, DMSO-d6) δ −64.66, −114.30. HRMS (ESI) calcd for C18H9F4N3O [M + H]+ 360.0760, found 360.0755.
3-(6-(4-Chlorophenyl)quinolin-3-yl)-5-(trifluoromethyl)-1,2,4-oxadiazole (35)
Compound 35 was obtained by general method C as a white solid with a yield of 9.52%. 1H NMR (600 MHz, DMSO-d6) δ 9.45 (d, J = 2.2 Hz, 1H), 9.18 (d, J = 2.1 Hz, 1H), 8.59 (d, J = 2.2 Hz, 1H), 8.25 (dd, J = 8.8, 2.2 Hz, 1H), 8.20 (d, J = 8.8 Hz, 1H), 7.91–7.87 (m, 2H), 7.64–7.60 (m, 2H). 13C NMR (151 MHz, DMSO-d6) δ 166.30, 165.33–163.76 (m), 147.63, 146.96, 137.35, 136.93, 135.84, 132.60, 130.23, 129.00, 128.54, 128.29, 126.48, 126.10, 117.88, 115.17 (d, J = 273.4 Hz). 19F NMR (565 MHz, DMSO-d6) δ −64.65. HRMS (ESI) calcd for C18H9ClF3N3O [M + H]+ 376.0464, found 376.0459.
N,N-Dimethyl-4-(3-(5-(trifluoromethyl)-1,2,4-oxadiazol-3-yl)quinolin-6-yl)aniline (36)
Compound 36 was obtained by general method C as a yellow solid with a yield of 13.6%. 1H NMR (600 MHz, DMSO-d6) δ 9.39 (d, J = 2.1 Hz, 1H), 9.14 (d, J = 2.3 Hz, 1H), 8.46 (d, J = 2.2 Hz, 1H), 8.24 (dd, J = 8.8, 2.1 Hz, 1H), 8.14 (d, J = 8.8 Hz, 1H), 7.78–7.72 (m, 2H), 6.91–6.85 (m, 2H), 2.99 (s, 6H). 13C NMR (151 MHz, DMSO-d6) δ 166.95, 165.13 (q, J = 43.8 Hz), 150.26, 147.56, 146.37, 139.40, 135.91, 130.41, 129.16, 127.54, 127.33, 125.59, 123.95, 118.16, 115.70 (q, J = 273.0, 272.6 Hz), 112.56, 29.82. 19F NMR (565 MHz, DMSO-d6) δ −64.63. HRMS (ESI) calcd for C20H15F3N4O [M + H]+ 385.1276, found 385.1271.
5-(3-(5-Methyl-1,2,4-oxadiazol-3-yl) quinolin-6-yl)pyridin-2-amine (37)
Compound 37 was obtained by general method C as a yellow solid with a yield of 32%. 1H NMR (600 MHz, DMSO-d6) δ 9.33 (d, J = 2.2 Hz, 1H), 8.96 (d, J = 2.4 Hz, 1H), 8.46 (d, J = 2.5 Hz, 1H), 8.36 (d, J = 2.1 Hz, 1H), 8.14 (dd, J = 8.8, 2.1 Hz, 1H), 8.09 (d, J = 8.7 Hz, 1H), 7.90 (dd, J = 8.6, 2.6 Hz, 1H), 6.60 (d, J = 8.6 Hz, 1H), 6.25 (s, 2H), 2.73 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 177.94, 166.14, 159.70, 147.47, 147.07, 146.52, 137.24, 135.70, 134.96, 129.61, 129.42, 127.56, 123.94, 122.48, 120.02, 108.20, 12.16. HRMS (ESI) calcd for C17H13N5O [M + H]+ 304.1198, found 304.1191.
3-(6-(4-Butylphenyl)quinolin-3-yl)-5-methyl-1,2,4-oxadiazole (38)
Compound 38 was obtained by general method C as a white solid with a yield of 17%. 1H NMR (600 MHz, chloroform-d) δ 9.53 (d, J = 2.1 Hz, 1H), 8.88 (d, J = 1.9 Hz, 1H), 8.22 (d, J = 8.6 Hz, 1H), 8.09–8.03 (m, 2H), 7.67–7.61 (m, 2H), 7.35–7.31 (m, 2H), 2.72 (s, 3H), 2.71–2.67 (m, 2H), 1.70–1.63 (m, 2H), 1.41 (dt, J = 14.8, 7.4 Hz, 2H), 0.96 (t, J = 7.4 Hz, 3H). 13C NMR (151 MHz, chloroform-d) δ 177.03, 166.67, 148.24, 148.07, 143.04, 140.33, 137.12, 135.38, 130.84, 129.78, 129.16, 127.63, 127.29, 125.73, 120.50, 35.32, 33.60, 22.40, 13.97, 12.45. HRMS (ESI) calcd for C22H21N3O [M + H]+ 344.1763, found 344.1757.
5-Methyl-3-(6-(4-propylphenyl)quinolin-3-yl)-1,2,4-oxadiazole (39)
Compound 39 was obtained by general method C as a white solid with a yield of 11%. 1H NMR (600 MHz, chloroform-d) δ 9.53 (d, J = 2.1 Hz, 1H), 8.88 (d, J = 2.2 Hz, 1H), 8.23 (d, J = 8.6 Hz, 1H), 8.10–8.04 (m, 2H), 7.64 (d, J = 8.0 Hz, 2H), 7.33 (d, J = 7.9 Hz, 2H), 2.72 (s, 3H), 2.67 (t, J = 7.6 Hz, 2H), 1.71 (h, J = 7.4 Hz, 2H), 1.00 (t, J = 7.3 Hz, 3H). 13C NMR (151 MHz, chloroform-d) δ 177.04, 166.64, 148.13, 148.00, 142.83, 140.37, 137.14, 135.45, 130.89, 129.71, 129.21, 127.64, 127.28, 125.73, 120.52, 37.70, 24.53, 13.87, 12.45. HRMS (ESI) calcd for C21H19N3O [M + H]+ 330.1606, found 330.1601.
3-(6-(4-Ethylphenyl)quinolin-3-yl)-5-methyl-1,2,4-oxadiazole (40)
Compound 40 was obtained by general method C as a white solid with a yield of 10%. 1H NMR (600 MHz, chloroform-d) δ 9.54 (d, J = 2.0 Hz, 1H), 8.88 (d, J = 2.0 Hz, 1H), 8.22 (d, J = 8.6 Hz, 1H), 8.09–8.03 (m, 2H), 7.68–7.63 (m, 2H), 7.35 (d, J = 7.9 Hz, 2H), 2.77–2.70 (m, 5H), 1.31 (t, J = 7.6 Hz, 3H). 13C NMR (151 MHz, chloroform-d) δ 177.04, 166.67, 148.26, 148.09, 144.36, 140.32, 137.20, 135.38, 130.84, 129.79, 128.62, 127.63, 127.38, 125.76, 120.51, 28.56, 15.56, 12.45. HRMS (ESI) calcd for C20H17N3O [M + H]+ 316.1450, found 316.1442.
5-Methyl-3-(6-(p-tolyl)quinolin-3-yl)-1,2,4-oxadiazole (41)
Compound 41 was obtained by general method C as a white solid with a yield of 44%. 1H NMR (600 MHz, chloroform-d) δ 9.53 (d, J = 2.0 Hz, 1H), 8.87 (d, J = 2.0 Hz, 1H), 8.21 (d, J = 8.5 Hz, 1H), 8.08–8.02 (m, 2H), 7.64–7.59 (m, 2H), 7.32 (d, J = 7.8 Hz, 2H), 2.72 (s, 3H), 2.43 (s, 3H). 13C NMR (151 MHz, acetone-d6) δ 178.51, 167.35, 149.23, 148.53, 140.62, 138.61, 137.54, 135.71, 131.02, 130.60, 130.50, 128.47, 127.91, 126.60, 121.49, 20.98, 12.15. HRMS (ESI) calcd for C19H15N3O [M + H]+ 302.1293, found 302.1290.
5-Methyl-3-(6-phenylquinolin-3-yl)-1,2,4-oxadiazole (42)
Compound 42 was obtained by general method C as a white solid with a yield of 44%. 1H NMR (600 MHz, chloroform-d) δ 9.54 (d, J = 2.1 Hz, 1H), 8.88 (d, J = 2.4 Hz, 1H), 8.24 (d, J = 8.7 Hz, 1H), 8.10–8.03 (m, 2H), 7.74–7.69 (m, 2H), 7.52 (t, J = 7.7 Hz, 2H), 7.46–7.40 (m, 1H), 2.72 (s, 3H). 13C NMR (151 MHz, chloroform-d) δ 177.05, 166.61, 148.27, 148.21, 140.36, 139.83, 135.44, 130.86, 129.84, 129.06, 128.04, 127.58, 127.45, 126.11, 120.57, 12.44. HRMS (ESI) calcd for C18H13N3O [M + H]+ 288.1137, found 288.1131.
4-(3-(5-Methyl-1,2,4-oxadiazol-3-yl)quinolin-6-yl)phenol (43)
Compound 43 was obtained by general method C as a white solid with a yield of 24%. 1H NMR (600 MHz, DMSO-d6) δ 9.72 (s, 1H), 9.35 (t, J = 1.5 Hz, 1H), 8.99 (d, J = 2.1 Hz, 1H), 8.37 (d, J = 2.0 Hz, 1H), 8.17–8.08 (m, 2H), 7.69 (d, J = 8.4 Hz, 2H), 6.96–6.90 (m, 2H), 2.73 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 177.95, 166.15, 157.88, 147.55, 147.22, 139.23, 135.13, 130.24, 129.59, 129.36, 128.41, 127.49, 124.92, 120.03, 116.05, 12.16. HRMS (ESI) calcd for C18H13N3O2 [M + H]+ 304.1086, found 304.1081.
3-(6-(4-Methoxyphenyl)quinolin-3-yl)-5-methyl-1,2,4-oxadiazole (44)
Compound 44 was obtained by general method C as a white solid with a yield of 17%. 1H NMR (600 MHz, DMSO-d6) δ 9.38 (d, J = 2.1 Hz, 1H), 9.04 (d, J = 2.0 Hz, 1H), 8.45 (d, J = 2.0 Hz, 1H), 8.20 (dd, J = 8.7, 2.2 Hz, 1H), 8.15 (d, J = 8.7 Hz, 1H), 7.85–7.79 (m, 2H), 7.14–7.08 (m, 2H), 3.84 (s, 3H), 2.74 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 178.00, 166.14, 159.56, 147.68, 147.42, 138.86, 135.25, 131.21, 130.33, 129.44, 128.41, 127.48, 125.40, 120.09, 114.70, 55.36, 12.17. HRMS (ESI) calcd for C19H15N3O2 [M + H]+ 318.1243, found 318.1237.
5-(3-(5-Ethyl-1,2,4-oxadiazol-3-yl)quinolin-6-yl)pyridin-2-amine (45)
Compound 45 was obtained by general method C as a yellow solid with a yield of 41.6%. 1H NMR (600 MHz, DMSO-d6) δ 9.33 (d, J = 2.2 Hz, 1H), 8.96 (d, J = 2.0 Hz, 1H), 8.47 (d, J = 2.5 Hz, 1H), 8.38 (d, J = 2.0 Hz, 1H), 8.14 (dd, J = 8.8, 2.1 Hz, 1H), 8.09 (d, J = 8.8 Hz, 1H), 7.90 (dd, J = 8.7, 2.6 Hz, 1H), 6.60 (d, J = 8.6 Hz, 1H), 6.24 (s, 2H), 3.07 (q, J = 7.5 Hz, 2H), 1.38 (t, J = 7.5 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 181.68, 166.01, 159.70, 147.46, 147.05, 146.55, 137.23, 135.66, 134.97, 129.56, 129.41, 127.56, 123.95, 122.49, 120.05, 108.17, 19.72, 10.51. HRMS (ESI) calcd for C18H15N5O [M + H]+ 318.1355, found 318.1348.
3-(3-(5-Ethyl-1,2,4-oxadiazol-3-yl)quinolin-6-yl)aniline (46)
Compound 46 was obtained by general method C as a yellow solid with a yield of 33.5%. 1H NMR (600 MHz, DMSO-d6) δ 9.39 (d, J = 2.2 Hz, 1H), 9.05 (d, J = 2.1 Hz, 1H), 8.38 (d, J = 2.1 Hz, 1H), 8.15 (d, J = 8.7 Hz, 1H), 8.09 (dd, J = 8.7, 2.1 Hz, 1H), 7.18 (t, J = 7.7 Hz, 1H), 7.01 (t, J = 2.0 Hz, 1H), 6.96 (dt, J = 7.7, 1.2 Hz, 1H), 6.67–6.62 (m, 1H), 5.25 (s, 2H), 3.08 (q, J = 7.6 Hz, 2H), 1.39 (t, J = 7.6 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 181.73, 166.02, 149.39, 147.88, 147.56, 140.21, 139.78, 135.36, 130.57, 129.72, 129.35, 127.37, 125.99, 120.11, 114.88, 113.86, 112.54, 19.73, 10.53. HRMS (ESI) calcd for C19H16N4O [M + H]+ 317.1402, found 317.1396.
2-(3-(5-Ethyl-1,2,4-oxadiazol-3-yl)quinolin-6-yl)aniline (47)
Compound 47 was obtained by general method C as a yellow solid with a yield of 34.5%. 1H NMR (600 MHz, DMSO-d6) δ 9.41 (d, J = 2.2 Hz, 1H), 9.02 (d, J = 2.1 Hz, 1H), 8.23 (d, J = 2.0 Hz, 1H), 8.14 (d, J = 8.6 Hz, 1H), 7.94 (dd, J = 8.6, 2.0 Hz, 1H), 7.15–7.08 (m, 2H), 6.82 (dd, J = 8.1, 1.2 Hz, 1H), 6.73–6.67 (m, 1H), 5.01 (s, 2H), 3.08 (q, J = 7.6 Hz, 2H), 1.39 (t, J = 7.6 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 181.75, 166.05, 147.63, 147.51, 145.53, 139.20, 135.27, 132.87, 130.47, 129.19, 128.86, 128.40, 127.40, 124.53, 119.91, 116.86, 115.58, 19.73, 10.54. HRMS (ESI) calcd for C19H16N4O [M + H]+ 317.1402, found 317.1397.
3-(6-(4-Butylphenyl)quinolin-3-yl)-5-ethyl-1,2,4-oxadiazole (48)
Compound 48 was obtained by general method C as a white solid with a yield of 38.2%. 1H NMR (600 MHz, chloroform-d) δ 9.55 (d, J = 2.0 Hz, 1H), 8.90 (d, J = 2.0 Hz, 1H), 8.23 (d, J = 8.7 Hz, 1H), 8.11–8.04 (m, 2H), 7.66–7.62 (m, 2H), 7.33 (d, J = 8.1 Hz, 2H), 3.04 (q, J = 7.6 Hz, 2H), 2.72–2.66 (m, 2H), 1.70–1.63 (m, 2H), 1.51 (t, J = 7.6 Hz, 2H), 1.41 (h, J = 7.4 Hz, 2H), 0.96 (t, J = 7.4 Hz, 3H). 13C NMR (151 MHz, acetone-d6) δ 182.41, 167.18, 149.16, 148.50, 143.62, 140.63, 137.75, 135.74, 131.03, 130.55, 129.89, 128.48, 127.93, 126.64, 121.55, 35.70, 34.30, 22.87, 20.56, 14.07, 10.80. HRMS (ESI) calcd for C23H23N3O [M + H]+ 358.1919, found 358.1914.
5-Ethyl-3-(6-(4-propylphenyl)quinolin-3-yl)-1,2,4-oxadiazole (49)
Compound 49 was obtained by general method C as a white solid with a yield of 44.2%. 1H NMR (600 MHz, chloroform-d) δ 9.55 (d, J = 2.2 Hz, 1H), 8.90 (d, J = 2.2 Hz, 1H), 8.23 (d, J = 8.7 Hz, 1H), 8.11–8.04 (m, 2H), 7.67–7.62 (m, 2H), 7.35–7.31 (m, 2H), 3.04 (q, J = 7.6 Hz, 2H), 2.70–2.64 (m, 2H), 1.76–1.66 (m, 2H), 1.51 (t, J = 7.6 Hz, 3H), 1.00 (t, J = 7.4 Hz, 3H). 13C NMR (151 MHz, acetone-d6) δ 182.42, 167.19, 149.21, 148.53, 143.40, 140.63, 137.81, 135.72, 131.02, 130.59, 129.94, 128.48, 127.92, 126.65, 121.56, 38.09, 25.16, 20.55, 13.94, 10.80. HRMS (ESI) calcd for C22H21N3O [M + H]+ 344.1763, found 344.1758.
5-Ethyl-3-(6-(4-ethylphenyl)quinolin-3-yl)-1,2,4-oxadiazole (50)
Compound 50 was obtained by general method C as a white solid with a yield of 48.3%. 1H NMR (600 MHz, chloroform-d) δ 9.55 (d, J = 2.1 Hz, 1H), 8.90 (d, J = 2.4 Hz, 1H), 8.23 (d, J = 8.7 Hz, 1H), 8.11–8.03 (m, 2H), 7.65 (d, J = 8.0 Hz, 2H), 7.35 (d, J = 7.9 Hz, 2H), 3.04 (q, J = 7.6 Hz, 2H), 2.74 (q, J = 7.6 Hz, 2H), 1.51 (t, J = 7.6 Hz, 2H), 1.31 (t, J = 7.6 Hz, 3H). 13C NMR (151 MHz, acetone-d6) δ 182.41, 167.18, 149.18, 148.51, 145.02, 140.63, 137.78, 135.73, 131.03, 130.56, 129.33, 128.47, 128.01, 126.64, 121.54, 28.95, 20.55, 15.88, 10.80. HRMS (ESI) calcd for C21H19N3O [M + H]+ 330.1606, found 330.1601.
5-Ethyl-3-(6-(p-tolyl)quinolin-3-yl)-1,2,4-oxadiazole (51)
Compound 51 was obtained by general method C as a white solid with a yield of 38.5%. 1H NMR (600 MHz, chloroform-d) δ 9.54 (d, J = 2.1 Hz, 1H), 8.89 (d, J = 1.9 Hz, 1H), 8.22 (d, J = 8.7 Hz, 1H), 8.09–8.02 (m, 2H), 7.64–7.60 (m, 2H), 7.32 (d, J = 7.8 Hz, 2H), 3.04 (q, J = 7.6 Hz, 2H), 2.43 (s, 3H), 1.50 (t, J = 7.7 Hz, 3H). 13C NMR (151 MHz, methanol-d4) δ 183.28, 167.51, 148.78, 148.72, 141.82, 139.37, 137.83, 137.15, 132.07, 130.84, 129.80, 129.23, 128.30, 126.88, 122.21, 21.22, 21.06, 11.02. HRMS (ESI) calcd for C20H17N3O [M + H]+ 316.1450, found 316.1444.
5-Ethyl-3-(6-phenylquinolin-3-yl)-1,2,4-oxadiazole (52)
Compound 52 was obtained by general method C as a white solid with a yield of 35.2%. 1H NMR (600 MHz, chloroform-d) δ 9.56 (d, J = 2.1 Hz, 1H), 8.91 (d, J = 2.0 Hz, 1H), 8.24 (d, J = 8.7 Hz, 1H), 8.12–8.04 (m, 2H), 7.75–7.70 (m, 2H), 7.52 (t, J = 7.7 Hz, 2H), 7.46–7.40 (m, 1H), 3.04 (q, J = 7.6 Hz, 2H), 1.51 (t, J = 7.6 Hz, 3H). 13C NMR (151 MHz, acetone-d6) δ 182.40, 167.15, 149.29, 148.70, 140.62, 140.44, 135.76, 131.05, 130.65, 129.83, 128.76, 128.41, 128.06, 127.06, 121.58, 20.55, 10.79. HRMS (ESI) calcd for C19H15N3O [M + H]+ 302.1293, found 302.1289.
4-(3-(5-Ethyl-1,2,4-oxadiazol-3-yl)quinolin-6-yl)phenol (53)
Compound 53 was obtained by general method C as a white solid with a yield of 23.4%. 1H NMR (600 MHz, DMSO-d6) δ 9.72 (s, 1H), 9.36 (d, J = 2.1 Hz, 1H), 9.00 (d, J = 2.2 Hz, 1H), 8.40 (d, J = 2.2 Hz, 1H), 8.17–8.09 (m, 2H), 7.73–7.65 (m, 2H), 6.96–6.90 (m, 2H), 3.07 (q, J = 7.5 Hz, 2H), 1.39 (t, J = 7.6 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 181.71, 166.02, 157.87, 147.55, 147.22, 139.21, 135.15, 130.21, 129.59, 129.34, 128.40, 127.50, 124.94, 120.07, 116.04, 19.73, 10.53. HRMS (ESI) calcd for C19H15N3O2 [M + H]+ 318.1243, found 318.1237.
5-Ethyl-3-(6-(4-methoxyphenyl)quinolin-3-yl)-1,2,4-oxadiazole (54)
Compound 54 was obtained by general method C as a white solid with a yield of 61.8%. 1H NMR (600 MHz, DMSO-d6) δ 9.37 (d, J = 2.0 Hz, 1H), 9.02 (d, J = 2.0 Hz, 1H), 8.45 (d, J = 2.1 Hz, 1H), 8.18 (dd, J = 8.7, 2.0 Hz, 1H), 8.13 (d, J = 8.8 Hz, 1H), 7.81 (d, J = 8.6 Hz, 2H), 7.13–7.08 (m, 2H), 3.83 (s, 3H), 3.07 (q, J = 7.5 Hz, 2H), 1.39 (t, J = 7.6 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 181.71, 166.00, 159.54, 147.66, 147.39, 138.80, 135.23, 131.19, 130.25, 129.41, 128.37, 127.47, 125.38, 120.10, 114.67, 55.34, 19.72, 10.51. HRMS (ESI) calcd for C20H17N3O2 [M + H]+ 332.1399, found 332.1394.
5-Ethyl-3-(6-(4-fluorophenyl)quinolin-3-yl)-1,2,4-oxadiazole (55)
Compound 55 was obtained by general method C as a white solid with a yield of 23.8%. 1H NMR (600 MHz, DMSO-d6) δ 9.39 (d, J = 2.1 Hz, 1H), 9.04–9.00 (m, 1H), 8.48 (d, J = 2.0 Hz, 1H), 8.20–8.12 (m, 2H), 7.92–7.86 (m, 2H), 7.41–7.34 (m, 2H), 3.07 (q, J = 7.5 Hz, 2H), 1.39 (t, J = 7.6 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 181.73, 165.94, 163.18, 161.55, 147.82 (d, J = 10.3 Hz), 138.07, 135.44 (d, J = 3.4 Hz), 135.37, 130.39, 129.54, 129.27 (d, J = 8.5 Hz), 127.33, 126.40, 120.20, 116.06 (d, J = 21.3 Hz), 19.72, 10.51. 19F NMR (565 MHz, DMSO-d6) δ −114.44. HRMS (ESI) calcd for C19H14FN3O [M + H]+ 320.1199, found 320.1194.
N-(4-(3-(5-Ethyl-1,2,4-oxadiazol-3-yl)quinolin-6-yl)phenyl)acetamide (56)
Compound 56 was obtained by general method C as a white solid with a yield of 21.7%. 1H NMR (600 MHz, DMSO-d6) δ10.12 (s, 1H), 9.39 (d, J = 2.1 Hz, 1H), 9.03 (d, J = 2.0 Hz, 1H), 8.48 (d, J = 2.0 Hz, 1H), 8.20 (dd, J = 8.8, 2.1 Hz, 1H), 8.15 (d, J = 8.7 Hz, 1H), 7.82 (d, J = 8.7 Hz, 2H), 7.76 (d, J = 8.6 Hz, 2H), 3.09 (q, J = 7.5 Hz, 2H), 2.10 (s, 3H), 1.40 (t, J = 7.6 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 181.56, 168.38, 165.82, 147.59, 147.33, 139.38, 138.55, 135.10, 133.11, 130.03, 129.28, 127.33, 126.26, 125.42, 119.97, 119.29, 24.00, 19.55, 10.34. HRMS (ESI) calcd for C21H18N4O2 [M + H]+ 359.1508, found 359.1503.
4-(4-(3-(5-Ethyl-1,2,4-oxadiazol-3-yl)quinolin-6-yl)phenyl)morpholine (57)
Compound 57 was obtained by general method C as a white solid with a yield of 24%. 1H NMR (600 MHz, DMSO-d6) δ9.37 (d, J = 2.1 Hz, 1H), 9.04 (d, J = 2.2 Hz, 1H), 8.46 (d, J = 2.1 Hz, 1H), 8.20 (dd, J = 8.8, 2.2 Hz, 1H), 8.13 (d, J = 8.7 Hz, 1H), 7.81–7.75 (m, 2H), 7.14–7.08 (m, 2H), 3.80–3.75 (m, 4H), 3.24–3.19 (m, 4H), 3.09 (q, J = 7.6 Hz, 2H), 1.40 (t, J = 7.6 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 181.56, 165.87, 150.84, 147.40, 146.98, 138.80, 134.99, 129.89, 129.18, 128.86, 127.57, 127.40, 124.55, 119.90, 115.15, 65.93, 47.85, 19.55, 10.35. HRMS (ESI) calcd for C23H22N4O2 [M + H]+ 387.1821, found 387.1816.
2-Chloro-4-(3-(5-ethyl-1,2,4-oxadiazol-3-yl)quinolin-6-yl)aniline (58)
Compound 58 was obtained by general method C as a white solid with a yield of 26.1%. 1H NMR (600 MHz, DMSO-d6) δ9.33 (d, J = 2.2 Hz, 1H), 8.97 (d, J = 2.0 Hz, 1H), 8.39 (d, J = 2.1 Hz, 1H), 8.14 (dd, J = 8.9, 2.1 Hz, 1H), 8.07 (d, J = 8.7 Hz, 1H), 7.75 (d, J = 2.2 Hz, 1H), 7.58 (dd, J = 8.4, 2.2 Hz, 1H), 6.94 (d, J = 8.4 Hz, 1H), 5.66 (s, 2H), 3.07 (q, J = 7.5 Hz, 2H), 1.39 (t, J = 7.6 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 181.68, 166.01, 147.50, 147.11, 144.98, 138.18, 135.07, 129.76, 129.31, 127.53, 127.49, 127.24, 126.52, 124.40, 120.06, 117.81, 115.83, 19.72, 10.51. HRMS (ESI) calcd for C19H15ClN4O [M + H]+ 351.1013, found 351.1007.
3-(6-(4-Chlorophenyl)quinolin-3-yl)-5-ethyl-1,2,4-oxadiazole (59)
Compound 59 was obtained by general method C as a white solid with a yield of 27.3%. 1H NMR (600 MHz, DMSO-d6) δ 9.42 (s, 1H), 9.07 (s, 1H), 8.56 (s, 1H), 8.20 (q, J = 9.3, 8.8 Hz, 2H), 7.89 (d, J = 8.0 Hz, 2H), 7.61 (d, J = 8.0 Hz, 2H), 3.08 (d, J = 8.9 Hz, 2H), 1.40 (d, J = 8.1 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 181.61, 165.76, 147.84, 147.80, 137.62, 135.31, 133.01, 130.11, 129.47, 129.03, 128.81, 127.18, 126.48, 120.10, 19.55, 10.34. HRMS (ESI) calcd for C19H14ClN3O [M + H]+ 336.0904, found 336.0898.
4-(3-(5-Ethyl-1,2,4-oxadiazol-3-yl)quinolin-6-yl)benzonitrile (60)
Compound 60 was obtained by general method C as a white solid with a yield of 27.9%. 1H NMR (600 MHz, DMSO-d6) δ 9.45 (d, J = 2.1 Hz, 1H), 9.10 (d, J = 2.0 Hz, 1H), 8.67 (d, J = 2.0 Hz, 1H), 8.28 (dd, J = 8.8, 2.2 Hz, 1H), 8.22 (d, J = 8.7 Hz, 1H), 8.11–8.06 (m, 2H), 8.05–8.00 (m, 2H), 3.09 (q, J = 7.5 Hz, 2H), 1.40 (t, J = 7.6 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 181.65, 165.71, 148.26, 148.15, 143.27, 137.03, 135.51, 132.95, 130.10, 129.63, 127.92, 127.53, 127.12, 120.23, 118.67, 110.59, 19.55, 10.34. HRMS (ESI) calcd for C20H14N4O [M + H]+ 327.1246, found 327.1239.
4-(3-(5-(Trifluoromethyl)-1,2,4-oxadiazol-3-yl)quinolin-6-yl)aniline Hydrochloride (19·HCl)
Compound 19 (0.1 g, 0.28 mmol) was stirred in ethyl acetate (10 mL) at room temperature, and 2 M HCl in ethyl acetate (0.28 mL, 0.56 mmol) was added. The mixture was stirred for 30 min, filtered, and then dried to give compound 19·HCl as a solid (0.08 g, 73%). 1H NMR (600 MHz, DMSO-d6) δ 9.47 (d, J = 2.2 Hz, 1H), 9.23 (d, J = 2.1 Hz, 1H), 8.62 (d, J = 2.0 Hz, 1H), 8.28 (dd, J = 8.8, 2.1 Hz, 1H), 8.23 (d, J = 8.8 Hz, 1H), 8.00–7.94 (m, 2H), 7.61–7.56 (m, 2H). 13C NMR (151 MHz, DMSO-d6) δ 166.73, 165.22 (q, J = 43.5 Hz), 147.52, 147.22, 138.27, 138.11, 136.89, 132.21, 131.17, 129.05, 128.33, 127.11, 126.78, 123.90, 118.48, 118.46–112.68 (m). 19F NMR (565 MHz, DMSO-d6) δ −64.64. HRMS (ESI) calcd for C18H12F3N4O+ [M + H]+ 357.0963, found 357.0959.
4-(3-(5-Ethyl-1,2,4-oxadiazol-3-yl)quinolin-6-yl)aniline Hydrochloride (21·HCl)
Compound 21·HCl was obtained with the same synthetic method as described of 19·HCl. 68% yield from compound 21. 1H NMR (600 MHz, DMSO-d6) δ 9.45 (d, J = 2.1 Hz, 1H), 9.16 (d, J = 2.1 Hz, 1H), 8.60 (d, J = 2.0 Hz, 1H), 8.29–8.21 (m, 2H), 8.00–7.95 (m, 2H), 7.60–7.54 (m, 2H), 3.08 (q, J = 7.6 Hz, 2H), 1.39 (t, J = 7.6 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 181.73, 165.51, 147.08, 146.36, 138.20, 138.02, 136.49, 132.31, 130.86, 128.32, 127.38, 126.62, 123.80, 120.23, 19.56, 10.33. HRMS (ESI) calcd for C19H17N4O+ [M + H]+ 317.1402, found 317.1396.
Structure-Based Virtual Screening
The cocrystal structure of pleconaril (4WM7) was used as the VP1 receptor for structure-based virtual screening. The Schrödinger suite (Schrödinger, New York, NY) was used for the docking work, and all other settings were default unless otherwise noted. Ligands from the antiviral library were prepared with the LigPrep module. The cocrystal structure was processed via Protein Preparation Wizard, and all waters were removed. The ligands were docked into Grid, which was generated from Receptor Grid Generation. Compounds with high scores were collected to manually identify and reject those in which the docking pose was unreasonable. Subsequently, the selected compounds were tested for in vitro evaluation.
Cell Lines and Viruses
Human rhabdomyosarcoma cell lines (RD) were purchased from the American Type Culture Collection (ATCC, CCL-136) and maintained in Dulbecco’s modified Eagle’s medium (DMEM, Gibco) that was supplemented with 10% fetal bovine serum (FBS, Gibco) and 100 U/mL penicillin/streptomycin at 37 °C in 5% CO2. The enterovirus D68 strains US/KY/14-18953 (GenBank accession no. KM851231), US/MO/14-18947 (GenBank accession no. KM851225), the clinical strain STL-2014-12 (GenBank accession no. KM881710),27 and the prototype strain Fermon (GenBank accession no. AY426531) were provided by Professor Tong Cheng from Xiamen University.
Antiviral Assay
The anti-EV-D68 activity in RD cells was determined by using the cytopathic effect (CPE) reduction assay based on ATP levels, which was positively correlated with cellular viability. The assay was performed as described previously.28 RD cells (1.5 × 104 per well) were inoculated in 96-well plates and cultured for 24 h. One hundred TCID50 virus and serial 3-fold dilutions of the compounds ranging from 0.01 to 200 μM were added to RD cells, and DMSO was added as a control. After culturing at 37 °C for 72 h, the antiviral activity of compounds was assessed by measurement of ATP levels using CellTiter-Glo Luminescent Cell Viability Assay (Promega). Raw data of the luminescence signal were measured by a SpectraMax M5 microplate reader (Molecular Devices) and normalized to the average values of uninfected and virus infected cells. The antiviral activity was expressed as EC50 value, which represented the concentration of a 50% increase in ATP levels (50% reduction of CPE) compared to virus and no-virus controls.
Cytotoxicity Assay
RD cells (1.5 × 104 per well) were inoculated in 96-well plates and cultured for 24 h. Then, serial 3-fold dilutions of the compounds ranging from 0.01 to 200 μM were added to the cells, and DMSO was added as a cell control. After culturing for 72 h, the cytotoxicity of test compounds was assessed by measurement of ATP levels using CellTiter-Glo Luminescent Cell Viability Assay (Promega) and was expressed as CC50 value of 50% cytotoxic concentration.
Western Blot Assay
Western blot assay was performed in RD cells. Briefly, RD cells seeded in six-well plates with 8.0 × 104 per well. After 24 h, the cells were inoculated with EV-D68 virus at an MOI of 0.01 in the presence of gradient-diluted compounds or DMSO for 1.5 h. After 12 hpi, cells were harvested and lysed by 140 μL of RIPA lysis buffer. For Western blot analysis, equal amounts of protein samples were separated on SDS-PAGE (10% polyacrylamide) and then were transferred onto PVDF membrane (Roche, Indianapolis, IN). Then the membrane was blockaded with 5% skimmed milk and incubated with EV-D68 primary antibodies (GeneTex, GTX132313) and tublin primary antibodies (abcam, ab7291). Then the membrane was incubated with the corresponding HRP-conjugated secondary antibody (abcam, ab6721, and ab6789). The proteins were detected with the ECL luminescence reagent (ThermoFisher Scientific, Waltham, MA).
Immunofluorescence Assay
RD cells cultured in 96-well black plates and then infected with EV-D68 virus at an MOI of 1 and corresponding compounds for 12 h. Then cells were fixed with 4% formaldehyde for 30 min and permeabilized with 0.1% Triton X-100 for another 30 min. After blocking with 5% bovine serum, cells were stained with rabbit anti-EV-D68 VP1 antibody (GeneTex: GTX132313) and antirabbit secondary antibody conjugated to Alexa-546 (ThermoFisher Scientific, A10040) with 1 μM Hoechst33342 (ThermoFisher Scientific) as a nucleus stain. Fluorescent images were visualized using a Nikon Eclipse TS2-LS microscope equipped with NIS-Elements 3.0 software (Tokyo, Japan).
Plaque Assay
Plaque assays were performed in RD cells. Briefly, cells were seeded in 12-well plates and grown overnight. The virus stocks and the test compounds were serially diluted, added to cells, and then incubated for 1.5 h. The supernatant was aspirated, and the cells were incubated in 2× DMEM media overlay containing 2% low melting point agarose (Promega, Madison, WI). Cells were fixed at 72 hpi with 4% paraformaldehyde at room temperature for 4 h and stained with 1% crystal violet dye solution prepared with 4% paraformaldehyde.
Pharmacokinetic Study In Vivo
The pharmacokinetic experiments were performed by WuXi Apptec Co., Ltd. (compound 19) and Pharmaron Beijing Co., Ltd. (compound 21·HCl), and all procedures performed on animals were approved by the Institutional Animal Care and Use Committee, Shanghai Site (IACUC-SH) (WuXi Corporate Committee for Animal Research Ethics (WX-CCARE)) and Institutional Animal Care and Use Committee (IACUC) of Pharmaron, respectively. Briefly, the pharmacokinetic parameters of compound 19 (male SD rats, 6–9 weeks old, Beijing Vital River Laboratory Animal Technology Co., Ltd.) and compound 21·HCl (male SD rats, 6–8 weeks old, SPF (Beijing) Biotechnology Co., Ltd.) were evaluated after a single dose of 1 mg/kg intravenous (iv) and 5 mg/kg oral (po) administration. Each blood collection (approximately 0.15 mL per time point) was performed from the jugular vein of each animal at 0.083 (only for iv), 0.25, 0.5, 1, 2, 4, 6, and 24 h after administration and stored in a tube containing heparin sodium. Blood samples were processed for plasma by centrifugation at approximately 4 °C and 4000 rpm for 10 min. Plasma was collected, transferred into prelabeled 96-well plates or polypropylene tubes, quickly frozen over dry ice, and kept at −60 °C or lower. The compound concentrations in plasma were determined via LC-MS/MS analysis.
Liver Microsome Stability Assay
The metabolic stability of testing compound was evaluated using rat, beagle dog, and human liver microsomes to predict intrinsic clearance. The reaction mixtures which contained a final concentration of 1 μM compound, 0.5 mg/mL liver microsome protein, and 1 mM NADPH were incubated in 96-well plates at 37 °C. At the time points of 0, 5, 15, 30, and 60 min, respectively, 25 μL of the mixture was added to 125 μL of quenching solution with internal standard (5 ng/mL, verapamil) and mixed to terminate the reaction. A parallel negative control group was set up (no NADPH). The mixture was sealed, vortexed, and centrifuged at 4 °C for 15 min. The samples were then subjected to LC-MS/MS analysis. The extent of metabolism was calculated, and t1/2 values and intrinsic clearance were determined.
Time of Drug Addition Assay
The procedures were described previously.30 Briefly, RD cells were seeded in 12-well plates and cultured for 24 h. The cells were infected with EV-D68 at 0 h (MOI = 0.01), and the virus was washed off after 1 hpi. Compounds 19 (2 μM) or pleconaril (2 μM) or DMSO were added at specified periods of time according to the schedule of drug addition. The supernatants were harvested at 14 hpi. Viral load was detected by qRT-PCR.
Quantitative Real-Time PCR
The total cellular RNA was harvested by using the RNeasy mini kit (QIAGEN, USA). Quantitative real-time PCR was performed with One Step PrimeScript RT-PCR Kit (Takara, Japan) according to the manufacturer’s instructions using specific primers and probes for EV68 virus (forward primer: TTGTGGCAGCTCATGGCAA; reverse primer: AACATGGCGGCTCTCTGGT; probe: ACTCCTCCAGGTGGACATGCCCGACA). Virus RNA copy numbers were calculated by the cycle threshold values obtained for each sample compared to the known copy number standard curve.
Thermoprotection Assay
The procedures were described previously.19 Briefly, compound 19 (12.5 μM) or pleconaril (12.5 μM) or DMSO was incubated with virus (2 × 106 PFU/ml) at 33 °C for 30 min. The mixture was heated to the appropriate temperatures in the range of 37–59 °C for 2 min and then rapidly cooled to 4 °C on a PCR machine. Viral titers of the harvested mixture were assessed by TCID50 assay.
TCID50 Assay
The procedures were described previously.29 Briefly, RD cells were seeded in 96-well plates and cultured for 24 h. Then serial 10-fold dilutions of the test virus ranging from 100 to 10–8 were added to the cells. After 72 h of incubation, the cytopathic effect was observed. The 50% tissue culture infectious dose (TCID50) was calculated using the method of Reed and Muench.
Particle Stability Thermal Release Assay
The procedures were described previously.25,26 Briefly, compound 19 (10 μg/mL) or pleconaril (10 μg/mL) or DMSO was added to a 50 μL reaction containing 1.5 μg of EV-D68 and 5 μL of SYTO9. DMSO was used as 5%. The mixture was incubated at 33 °C for 15 min and then heating ramped from 25 to 99 °C. The fluorescence was recorded in triplicate at 1 °C intervals. The negative first derivative curved based on fluorescence values was plotted to determine the melting temperature.
Acknowledgments
This research was supported by grants from the National Key Research and Development Project (2021YFC2300704) (W.Z.) and in part from the National Science and Technology Major Projects for “Major New Drugs Innovation and Development” (2018ZX09711003) (W.Z.).
Glossary
Abbreviations
- AFM
acute flaccid myelitis
- CPE
cytopathic effect
- DME
1,2-dimethoxyethane
- ESI
electrospray ionization
- EV-D68
enterovirus D68
- hpi
hour post infection
- HRMS
high-resolution mass spectrometry
- LC/MS
liquid chromatography–mass spectrometry
- NPEV
nonpolio enterovirus
- ORF
open reading frame
- RD
rhabdomyosarcoma cell lines
- SAR
structure–activity relationship
- TMS
tetramethylsilane
- UTR
untranslated regions
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c01311.
Author Contributions
∥ X.L., Y.L., and S.F. equally contributed to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Sun J.; Hu X. Y.; Yu X. F. Current Understanding of Human Enterovirus D68. Viruses 2019, 11, 490. 10.3390/v11060490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassidy H.; Poelman R.; Knoester M.; Van Leer-Buter C. C.; Niesters H. G. M. Enterovirus D68 - The New Polio?. Front. Microbiol. 2018, 9, 2677. 10.3389/fmicb.2018.02677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberste M. S.; Maher K.; Schnurr D.; Flemister M. R.; Lovchik J. C.; Peters H.; Sessions W.; Kirk C.; Chatterjee N.; Fuller S.; Hanauer J. M.; Pallansch M. A. Enterovirus 68 is Associated with Respiratory Illness and Shares Biological Features with Both the Enteroviruses and the Rhinoviruses. J. Gen. Virol. 2004, 85, 2577–2584. 10.1099/vir.0.79925-0. [DOI] [PubMed] [Google Scholar]
- Imamura T.; Okamoto M.; Nakakita S.; Suzuki A.; Saito M.; Tamaki R.; Lupisan S.; Roy C. N.; Hiramatsu H.; Sugawara K. E.; Mizuta K.; Matsuzaki Y.; Suzuki Y.; Oshitani H. Antigenic and Receptor Binding Properties of Enterovirus 68. J. Virol. 2014, 88, 2374–2384. 10.1128/JVI.03070-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Sheng J.; Baggen J.; Meng G.; Xiao C.; Thibaut H. J.; Van Kuppeveld F. J.; Rossmann M. G. Sialic Acid-Dependent Cell Entry of Human Enterovirus D68. Nat. Commun. 2015, 6, 8865. 10.1038/ncomms9865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midgley C. M.; Watson J. T.; Nix W. A.; Curns A. T.; Rogers S. L.; Brown B. A.; Conover C.; Dominguez S. R.; Feikin D. R.; Gray S.; Hassan F.; Hoferka S.; Jackson M. A.; Johnson D.; Leshem E.; Miller L.; Nichols J. B.; Nyquist A. C.; Obringer E.; Patel A.; Patel M.; Rha B.; Schneider E.; Schuster J. E.; Selvarangan R.; Seward J. F.; Turabelidze G.; Oberste M. S.; Pallansch M. A.; Gerber S. I. Severe Respiratory Illness Associated with a Nationwide Outbreak of Enterovirus D68 in the USA (2014): A Descriptive Epidemiological Investigation. Lancet Respir. Med. 2015, 3, 879–887. 10.1016/S2213-2600(15)00335-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baggen J.; Thibaut H. J.; Strating J.; Van Kuppeveld F. J. M. The Life Cycle of Non-Polio Enteroviruses and How to Target It. Nat. Rev. Microbiol. 2018, 16, 368–381. 10.1038/s41579-018-0005-4. [DOI] [PubMed] [Google Scholar]
- Holm-Hansen C. C.; Midgley S. E.; Fischer T. K. Global Emergence of Enterovirus D68: A Systematic Review. Lancet Infect. Dis. 2016, 16, e64–e75. 10.1016/S1473-3099(15)00543-5. [DOI] [PubMed] [Google Scholar]
- Kreuter J. D.; Barnes A.; McCarthy J. E.; Schwartzman J. D.; Oberste M. S.; Rhodes C. H.; Modlin J. F.; Wright P. F. A Fatal Central Nervous System Enterovirus 68 Infection. Arch. Pathol. Lab. Med. 2011, 135, 793–796. 10.5858/2010-0174-CR.1. [DOI] [PubMed] [Google Scholar]
- Poelman R.; Schuffenecker I.; Van Leer-Buter C.; Josset L.; Niesters H. G.; Lina B. European Surveillance for Enterovirus D68 during the Emerging North-American Outbreak in 2014. J. Clin. Virol. 2015, 71, 1–9. 10.1016/j.jcv.2015.07.296. [DOI] [PubMed] [Google Scholar]
- Esposito S.; Bosis S.; Niesters H.; Principi N. Enterovirus D68 Infection. Viruses 2015, 7, 6043–6050. 10.3390/v7112925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messacar K.; Schreiner T. L.; Maloney J. A.; Wallace A.; Ludke J.; Oberste M. S.; Nix W. A.; Robinson C. C.; Glodé M. P.; Abzug M. J.; Dominguez S. R. A Cluster of Acute Flaccid Paralysis and Cranial Nerve Dysfunction Temporally Associated with an Outbreak of Enterovirus D68 in Children in Colorado, USA. Lancet 2015, 385, 1662–1671. 10.1016/S0140-6736(14)62457-0. [DOI] [PubMed] [Google Scholar]
- Messacar K.; Asturias E. J.; Hixon A. M.; Van Leer-Buter C.; Niesters H. G. M.; Tyler K. L.; Abzug M. J.; Dominguez S. R. Enterovirus D68 and Acute Flaccid Myelitis-Evaluating the Evidence for Causality. Lancet Infect. Dis. 2018, 18, e239–e247. 10.1016/S1473-3099(18)30094-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Sheng J.; Fokine A.; Meng G.; Shin W. H.; Long F.; Kuhn R. J.; Kihara D.; Rossmann M. G. Structure and Inhibition of EV-D68, a Virus that Causes Respiratory Illness in Children. Science 2015, 347, 71–74. 10.1126/science.1261962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hixon A. M.; Frost J.; Rudy M. J.; Messacar K.; Clarke P.; Tyler K. L. Understanding Enterovirus D68-Induced Neurologic Disease: A Basic Science Review. Viruses 2019, 11, 821. 10.3390/v11090821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opanda S. M.; Wamunyokoli F.; Khamadi S.; Coldren R.; Bulimo W. D. Genetic Diversity of Human Enterovirus 68 Strains Isolated in Kenya Using the Hypervariable 3′-End of VP1 Gene. PLoS One 2014, 9, e102866. 10.1371/journal.pone.0102866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egorova A.; Ekins S.; Schmidtke M.; Makarov V. Back to the Future: Advances in Development of Broad-Spectrum Capsid-Binding Inhibitors of Enteroviruses. Eur. J. Med. Chem. 2019, 178, 606–622. 10.1016/j.ejmech.2019.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossmann M. G.; He Y.; Kuhn R. J. Picornavirus-Receptor Interactions. Trends Microbiol. 2002, 10, 324–331. 10.1016/S0966-842X(02)02383-1. [DOI] [PubMed] [Google Scholar]
- Ma C.; Hu Y.; Zhang J.; Musharrafieh R.; Wang J. A Novel Capsid Binding Inhibitor Displays Potent Antiviral Activity against Enterovirus D68. ACS Infect. Dis. 2019, 5, 1952–1962. 10.1021/acsinfecdis.9b00284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnard D. L.; Hubbard V. D.; Smee D. F.; Sidwell R. W.; Watson K. G.; Tucker S. P.; Reece P. A. In Vitro Activity of Expanded-Spectrum Pyridazinyl Oxime Ethers Related to Pirodavir: Novel Capsid-Binding Inhibitors with Potent Antipicornavirus Activity. Antimicrob. Agents Chemother. 2004, 48, 1766–1772. 10.1128/AAC.48.5.1766-1772.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesan H.; Hocutt F. M.; Jones T. K.; Rabinowitz M. H. A One-Step Synthesis of 2,4-Unsubstituted Quinoline-3-Carboxylic Acid Esters from O-Nitrobenzaldehydes. J. Org. Chem. 2010, 75, 3488–3491. 10.1021/jo100392x. [DOI] [PubMed] [Google Scholar]
- Lacroix C.; Laconi S.; Angius F.; Coluccia A.; Silvestri R.; Pompei R.; Neyts J.; Leyssen P. In Vitro Characterisation of a Pleconaril/Pirodavir-Like Compound with Potent Activity against Rhinoviruses. Virol. J. 2015, 12, 106. 10.1186/s12985-015-0330-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J.; Wang X.; Hu Z.; Gao Q.; Sun Y.; Li X.; Porta C.; Walter T. S.; Gilbert R. J.; Zhao Y.; Axford D.; Williams M.; McAuley K.; Rowlands D. J.; Yin W.; Wang J.; Stuart D. I.; Rao Z.; Fry E. E. Picornavirus Uncoating Intermediate Captured in Atomic Detail. Nat. Commun. 2013, 4, 1929. 10.1038/ncomms2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Peng W.; Ren J.; Hu Z.; Xu J.; Lou Z.; Li X.; Yin W.; Shen X.; Porta C.; Walter T. S.; Evans G.; Axford D.; Owen R.; Rowlands D. J.; Wang J.; Stuart D. I.; Fry E. E.; Rao Z. A Sensor-Adaptor Mechanism for Enterovirus Uncoating from Structures of EV71. Nat. Struct. Mol. Biol. 2012, 19, 424–429. 10.1038/nsmb.2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter T. S.; Ren J.; Tuthill T. J.; Rowlands D. J.; Stuart D. I.; Fry E. E. A Plate-Based High-Throughput Assay for Virus Stability and Vaccine Formulation. J. Virol. Methods 2012, 185, 166–170. 10.1016/j.jviromet.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Colibus L.; Wang X.; Spyrou J. A. B.; Kelly J.; Ren J.; Grimes J.; Puerstinger G.; Stonehouse N.; Walter T. S.; Hu Z.; Wang J.; Li X.; Peng W.; Rowlands D.; Fry E. E.; Rao Z.; Stuart D. I. More-Powerful Virus Inhibitors from Structure-Based Analysis of HEV71 Capsid-Binding Molecules. Nat. Struct. Mol. Biol. 2014, 21, 282–288. 10.1038/nsmb.2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Q.; Zhu R.; Xu L.; He M.; Yan X.; Liu D.; Yin Z.; Wu Y.; Li Y.; Yang L.; Hou W.; Li S.; Li Z.; Chen Z.; Li Z.; Yu H.; Gu Y.; Zhang J.; Baker T. S.; Zhou Z. H.; Graham B. S.; Cheng T.; Li S.; Xia N. Atomic structures of enterovirus D68 in complex with two monoclonal antibodies define distinct mechanisms of viral neutralization. Nat. Microbiol. 2019, 4 (1), 124–133. 10.1038/s41564-018-0275-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao T.; Li Y.; Fan S.; Li W.; Wang S.; Li S.; Cao R.; Zhong W. Design, Synthesis and Pharmacological Evaluation of a Novel mTOR-targeted Anti-EV71 Agent. Eur. J. Med. Chem. 2019, 175, 172–186. 10.1016/j.ejmech.2019.04.048. [DOI] [PubMed] [Google Scholar]
- Zhao L.; Yan Y.; Dai Q.; Li X.; Xu K.; Zou G.; Yang K.; Li W.; Guo X.; Yang J.; Li Y.; Xia Q.; Cao R.; Zhong W. Development of Novel Anti-influenza Thiazolides with Relatively Broad-Spectrum Antiviral Potentials. Antimicrob. Agents Chemother. 2020, 64, e00222. 10.1128/AAC.00222-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Xu Y.; Yan Y.; Li W.; Zhao L.; Dai Q.; Li Y.; Li S.; Zhong J.; Cao R.; Zhong W. Small Molecule Inhibitor of ATPase Activity of HSP70 as a Broad-Spectrum Inhibitor against Flavivirus Infections. ACS Infect. Dis. 2020, 6, 832–843. 10.1021/acsinfecdis.9b00376. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.