

Abstract
Activating mutations in KRAS are the most frequent oncogenic alterations in cancer. The oncogenic hotspot position 12, located at the lip of the switch II pocket, offers a covalent attachment point for KRASG12C inhibitors. To date, KRASG12C inhibitors have been discovered by first covalently binding to the cysteine at position 12 and then optimizing pocket binding. We report on the discovery of the in vivo active KRASG12C inhibitor BI-0474 using a different approach, in which small molecules that bind reversibly to the switch II pocket were identified and then optimized for non-covalent binding using structure-based design. Finally, the Michael acceptor containing warhead was attached. Our approach offers not only an alternative approach to discovering KRASG12C inhibitors but also provides a starting point for the discovery of inhibitors against other oncogenic KRAS mutants.
Introduction
The KRAS oncoprotein is a GTPase acting as a key node in intracellular signaling pathways that are involved in cell growth and survival.1,2 In normal cells, KRAS functions as a molecular switch, alternating between an inactive GDP-bound and an active GTP-bound state.3 The transition between these states is modulated by guanine-nucleotide exchange factors such as SOS and GTP hydrolysis, which is catalyzed by GTPase-activating proteins (GAPs) to inactivate KRAS. GTP-bound KRAS allows binding of effector proteins to trigger downstream signaling pathways, including the RAF-MEK-ERK (MAPK) pathway.4−7 Activating mutations in KRAS are the most frequent oncogenic driver events in cancer.8 Oncogenic KRAS mutations have been shown to shift the equilibrium between the two KRAS states toward the active GTP-bound form. However, the major oncogenic versions of KRAS still undergo GAP-mediated GTP/GDP exchange and reactivation.9
Targeting KRAS has been of interest for the last 30 years. However, the surface of KRAS is thought to be devoid of druggable pockets except for the binding location for GTP and GDP, which have picomolar affinity for KRAS. Together, this has given rise to the assumption that KRAS is an undruggable target.
In 2012, progress was made when the workers at Genentech identified small molecules that bind to the switch I/II pocket of KRAS.11 Simultaneously, we conducted a fragment-based screen and also found small molecules that bind to this site, which were subsequently optimized using structure-based design.12−14
Another pocket on KRAS, the switch II pocket, was identified by Shokat and colleagues.15 The current clinical KRASG12C inhibitors16 bind to inactive GDP-bound KRAS in the switch II pocket and form a covalent bond with the mutant cysteine of KRASG12C. In doing so, the oncoprotein is trapped in its inactive state, preventing its reactivation by nucleotide exchange. Sotorasib has become the first approved KRASG12C drug, receiving accelerated approval by the US Food and Drug Administration in May 2021 for adult patients with advanced KRASG12C-mutant NSCLC who received at least one prior systemic therapy.
The reported KRASG12C inhibitors were discovered from hits or weakly binding analogues that contained an electrophilic warhead. The electrophilic nature of the compounds was retained throughout the entire optimization process until the final candidates were identified. Here, we describe a covalent KRASG12C inhibitor showing in vivo efficacy that was discovered using a different approach. In search of compounds that bound to a second site, we conducted a fragment-based screen in which a first-site ligand that bound to switch I/II was covalently linked to a KRAS mutant (S39C) to hold it in place.10 The hits that were found did not bind near to the first-site ligand as anticipated but instead bound reversibly to the switch II site. Starting from these fragments, we grew toward Cys12 and optimized reversible binding affinity. Finally, the resulting most promising binder was decorated with an acrylamide warhead to form a covalent bond to KRASG12C and boost its potency at the last stage of optimization.
Results and Discussion
In reported fragment screens on KRAS, non-covalent fragments preferentially bind to the switch I/II pocket. Upon binding to KRAS, a conformational change occurs, which opens both the switch I/II pocket and a nearby subpocket. To identify fragments which bind to this subpocket, we attempted a fragment-based screen in the presence of high concentrations of a first-site ligand. However, the compounds in the second site screen simply displaced the weakly bound first-site ligand. As previously reported,10 we solved this problem by covalently blocking the first site with a small-molecule ligand. To block the site in this way, we initially evaluated cysteine mutations of six residues adjacent to the switch I/II binding site. We then evaluated covalent modification of each of these KRAS mutants with various electrophiles. Using both NMR and X-ray crystallography, we verified that reaction of a KRASG12V,S39C mutant with (1H-benzo[d]imidazol-2-yl)methanethiol (BIT) produced a properly folded, covalently modified KRASG12V,S39C-BIT protein with the switch I/II binding site occupied (Figure 1).10 This modified protein was then used to conduct a HSQC-based screen of ∼13,000 fragments, revealing 20 fragment hits that displayed chemical shift perturbation patterns different than those observed with compounds that bind to switch I/II.10
Figure 1.

Schematic overview of the second site fragment screening approach: a KRASG12V construct with a S39C mutation was covalently modified with BIT to block the switch I/II pocket on KRAS (purple circle) and to stabilize the S II helix conformation (PDB 4PZZ).10 A HSQC NMR screen was performed with the blocked KRASG12V,S39C protein, and hits in the switch II pocket (green circle) were identified (exemplified with the fragment co-crystal structure with PDB code 7U8H).
Among these compounds was a cluster that included both aminocyanothiophenes and aminothiazoles (Table 1). NMR titration of the hits using the modified KRAS construct revealed that 1 bound to the modified protein with an affinity of 116 μM, around two-fold more tightly than the corresponding five-membered ring analogue 2. Aminothiazole 3 also showed reduced affinity compared to 1, and its five-membered ring analogue 4 displayed no shifts at all. Based upon these initial results from the screen, we chose to follow-up on fragment 1 with the goal of improving affinity and elucidating the binding mode.
Table 1. Aminocyanothiophene and Related Fragment Hits Measured on KRASG12V,S39C-BIT.

KD values are shown as mean ± SD and were determined, as described in the Experimental Section.
Using a combination of targeted commercial purchasing and discrete compound synthesis, we first sought to identify the key features of the hit that govern the binding to KRAS (Table 2). The amine and the cyano groups were shown to be important, based on the loss of affinity observed for both 5 and 6. Furthermore, elimination of the alkyl ring resulted in loss of binding (7), and aromatization of the ring system (8) also reduced affinity compared with the initial fragment hit. Interestingly, 9 a ring-opened alkyl variant of the aminocyanothiophene, bound with an affinity nearly equal to the hit, and a seven-membered ring analogue, 10, bound with affinity comparable to the five-membered ring compound 2.
Table 2. Compounds from Fragment Optimization Measured on KRASG12V,S39C-BIT.

KD values are shown as mean ± SD and were determined, as described in the Experimental Section.
The crystal structure of 1 in complex with GDP·KRASG12V,C118S,S39C-BIT (PDB code 7U8H) revealed the binding mode of the fragment (Figure 2). The compound was found to indeed bind to the switch II pocket similar to that occupied by previously reported covalent inhibitors of KRASG12C.17,18 The cyclohexyl ring of the fragment hit occupies the lipophilic base defined by Val9, Met72, Phe78, Val103, and Ile100. The amine engages in hydrogen bond interactions with the side chain of Asp69 and the backbone carbonyl of Glu63, with distances of 2.7 and 2.8 Å, respectively. Furthermore, the cyano moiety interacts with both the amide NH of Glu63 (distance 2.9 Å) as well as nearby bound water. This network of interactions readily explains the rigid requirements for both of these functional groups for best affinity.
Figure 2.

Crystal structure of 1 in complex with GDP·KRASG12V,C118S,S39C-BIT (PDB code 7U8H, 1.7 Å resolution; monomer B in yellow, color-coded by atom type, hydrogen bonds are shown as blue dotted lines). The gray protein surface illustrates the channel toward amino acid position 12 and is highlighted with a blue arrow indicating the distance between both ends.
Analysis of the crystal structure revealed that the 4-position of the cyclohexyl ring was oriented directly toward the 12-position residue of the protein (G12V), which was approximately 11 Å away. Furthermore, a mostly open channel lay between the hit and position 12 with only one major salt-bridge between Glu62 and His95 keeping the switch II pocket in a stabilized conformation.
To confirm our ability to access the channel, we prepared and evaluated derivatives of 1 with methyl groups in the 4-position of the six-membered ring. The single 4-methyl of compound 11 conferred a five-fold increase in affinity, and the gem-dimethyl of analogue 12 produced a further gain. However, as NMR-derived KD determination below 10 μM becomes inaccurate19 due to the high protein concentration, an exact value for this analogue could not be provided using KRASG12V,S39C-BIT. We also evaluated this analogue using 15N-labeled KRASG12V without the mutated Ser 39 and blocking benzimidazole (Table3). We obtained a slightly weaker affinity of 15 μM, confirming the ability of these fragments to bind to an unmodified oncogenic KRAS mutant.
Table 3. Compounds from Fragment Growing Measured on KRASG12V.

KD values are shown as mean ± SD and were determined, as described in the Experimental Section.
The crystal structure of 12 in complex with KRASG12C (PDB code 8AFC) revealed that the binding mode of the compounds is preserved when the switch I/II site is not occupied. Furthermore, the addition of the gem-dimethyl did not affect the binding mode of the core scaffold and maintained all important polar interactions (Figures 2 and 3).
Figure 3.
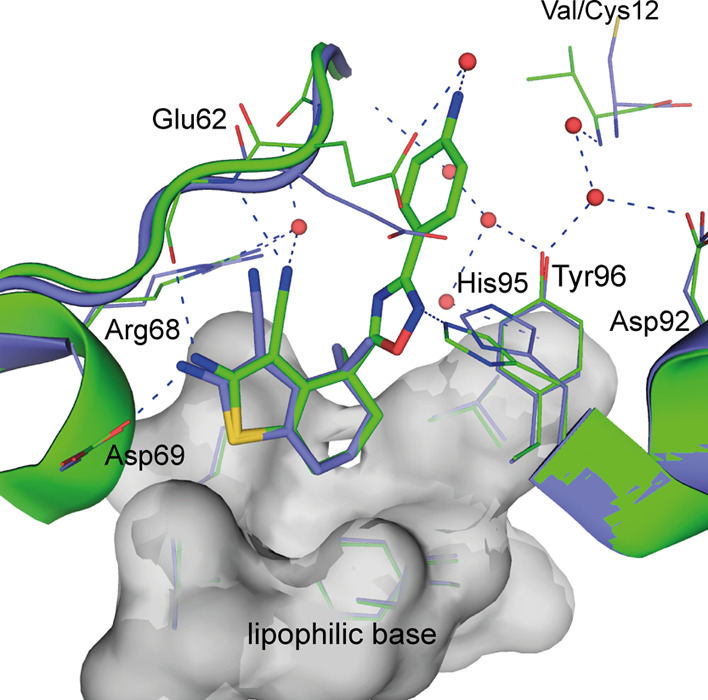
Comparison of the binding modes observed in the respective crystal structures. Crystal structure of 12 in complex with GDP·KRASG12C (PDB Code 8AFC, 2.4 Å resolution; blue, color-coded by atom type) and 20a in complex with GDP·KRASG12V,S39C-BIT (PDB code 8AFD, resolution 1.63 Å; monomer B in green, color-coded by atom type). The lipophilic base with Val9, Met72, Phe78, Ile100, and Val103 is shown as gray transparent surface (water molecules are shown as red spheres, hydrogen bonds as blue dotted lines).
Together, these data suggested that it would be feasible to design a covalent KRASG12C inhibitor from the non-covalent binders discovered in this study. In contrast, a previously described fragment (GNE-2897,2013) and also the switch II pocket binding core groups of two reported KRASG12C inhibitors (14,171518) did not display tight binding to KRAS without their covalent warheads. We hypothesized that the use of a KRAS binding core with native affinity to the protein should enable the discovery of a covalent inhibitor with greater selectivity against other cysteines and therefore lower toxicity as well as a higher second-order rate constant and improved KRASG12C inhibition, resulting in a stronger antiproliferative effect.
After the identification of a suitable exit vector, we turned our attention to growing the fragment toward Cys12. We hypothesized that growing the molecule into the channel leading to Cys12 should lead to a significant improvement in KRAS binding affinity. To deal with the aforementioned limitations of the KD determination below 10 μM, KRASG12V was used instead of KRASG12V,S39C-BIT for the NMR measurements during fragment growing toward amino acid position 12. As a first step, amides 16 and 17a/b (Table 3) were prepared, but a significant drop in affinity was observed. To rigidify the structure, the corresponding oxadiazoles, accessible from a common acid intermediate 18, which itself was in a similar KD range as the corresponding amides, were synthesized. The minimally decorated oxadiazole 19 still showed a slightly weaker affinity compared to the dimethyl analogue, but potency could be enhanced by further growing. Decorated aniline analogues 20a and 20b, synthesized from the (S)-isomer 18a of acid 18, showed a significantly improved KD, exceeding the binding affinity of the dimethyl analogue 12 and again reaching the limitation of accurate KD determination (Table 3). To get an estimate on the binding selectivity for the inactive versus active state of KRAS, the affinity of 20a and 20b to KRASG12D loaded with a non-hydrolyzable GTP analogue was determined as well. Although an exact KD determination was in this case not possible (KD > 2 mM, as shown in Table S6), a clear preference for the inactive GDP-bound state of KRAS was observed.
The comparison of the KRAS co-crystal structures of 12 and 20a provided an explanation for the initial potency drop that needed to be compensated by further growing: to expand the pocket toward Cys12, the salt bridge between Glu62 and His95 observed in the structure of 12 bound to KRAS is broken up by the aniline of 20a causing a reorientation of the Glu62 side chain. By installation of the oxadiazole moiety, affinity can be regained via an H-bond with His95 (Figure 3, PDB codes 8AFC and 8AFD).
After synthesizing the first single-digit micromolar reversible binders and reaching the HSQC assay limit at 10 μM, we turned our attention toward adding acrylamide warheads to analogues 20a/b. A mass spectrometry (MS)-based assay to detect covalent modification of KRASG12C and determine a rate constant readout by measuring the time course of Cys12 modification was used as an initial driving assay for optimization.21 To extend the dynamic range of our driving assay for more potent analogues, an AlphaScreen assay measuring disruption of the GDP-KRASG12C::SOS1 interaction22 was applied. Anilides 21a/b carrying the acrylamide warhead directly attached to the fragment showed measurable second-order rates in the MS-based assay but were not detectable in the AlphaScreen format. However, based on the co-crystal structure of 20a, we hypothesized that the introduction of a linker between the central phenyl core and the acrylamide should lead to a preferable positioning of the electrophile. Switching the exit vector from 21a to 21b and introducing a piperazine linker resulted in acrylamide 22 which did not lead to an improved second-order rate, but nonetheless showed an IC50 of ∼3 μM in biochemical assay.
Because of this AlphaScreen data and the lower intrinsic reactivity of piperazine acrylamides compared to aryl acrylamides,25,2622 was considered the more attractive analogue for further optimization, despite its lower MS-based activity compared to 21b.
Based on analysis of the co-crystal structures, we hypothesized that both His95 and Glu6227 could be addressed by appropriately occupying the subpocket formed by His95, Glu62, and Asp92. Changing the central phenyl ring to a pyridine improved the interaction with His95 and facilitated the introduction of a basic residue addressing the acidic environment. A (3S)-1,3-dimethylpiperazine substituent was found to be a suitable choice for this purpose. Together, this led to identification of BI-0474 which demonstrated around 500-fold improvement in both the second-order rate constant and the GDP-KRAS::SOS1 protein–protein interaction assay (Table 4) and showed a tight interaction network in the pocket (Figure 4, PDB code 8AFB). Intriguingly, the improvement in potency for BI-0474 compared to 22 seemed to be attributable mainly to an increase in the kinact despite the fact that no structural variation was made directly to the covalent warhead or the piperazine linker. Furthermore, BI-0474 showed potent antiproliferative activity of 26 nM on NCI-H358 cells carrying a G12C mutation (Table 4 and Figure S9). However, an antiproliferative effect on cells carrying a KRASG12D mutation (GP2D or LS513 cells) was only observed above 4 μM (data shown in Table S8). In comparison with AMG-510, BI-0474 already showed a slightly higher second-order rate and lower KI, comparable kinact and IC50 in the biochemical assay, and an only slightly weaker antiproliferative effect on NCI-H358 cells.
Table 4. Second-Order Rate Constants, Biochemical Activity, and Antiproliferative Activity of Acrylamide Analogues.

kinact/KI, kinact, KI, IC50, and EC50 values are shown as geometric mean ± SD (n ≥ 3) and were determined, as described in the Experimental Section.
6 nM previously reported.24
Figure 4.
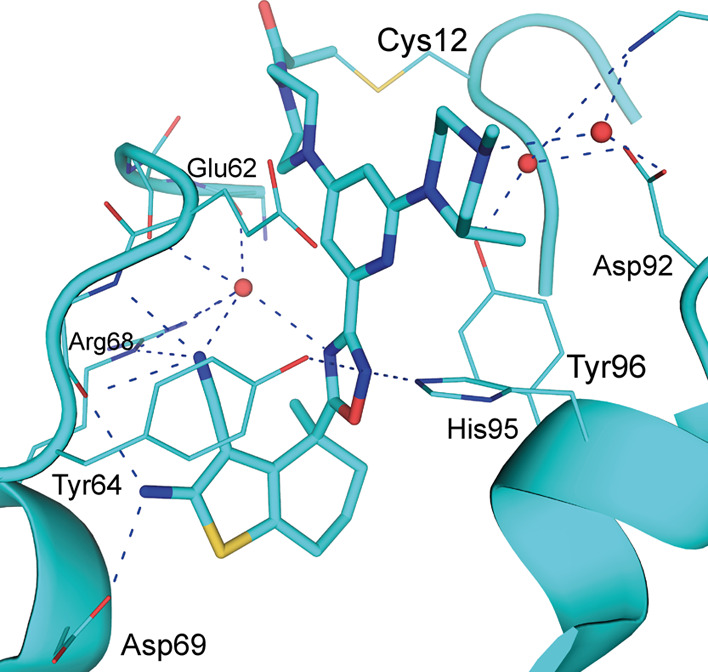
Structure of BI-0474 (23) in complex with GDP·KRASG12C (PDB Code 8AFB, 1.12 Å resolution; cyan, color-coded by atom type), BI-0474 is covalently bound to Cys12 of KRAS (water molecules are shown as red spheres, hydrogen bonds as blue dotted lines).
Intraperitoneal (ip) administration of BI-0474 at 40 mg/kg in NMRI nude mice resulted in plasma exposure expected to lead to TO > 60% after multiple dosing based on a mouse PK model and on a model prediction in the KRASG12C mutant NCI-H358 xenograft model (Figure 5a).21 Encouraged by these results, we assessed TO as well as pharmacodynamic (PD) biomarker modulation in the KRASG12C mutant NCI-H358 model after a daily treatment with 40 mg/kg ip on 3 consecutive days. As shown in Figure 5b, MS-based TO analysis showed a strong and treatment-induced reduction of unmodified KRASG12C protein on the third day of consecutive treatment (five-fold and nine-fold reduction compared to respective vehicle controls at 2 and 6 h post last dose). This was confirmed by the analysis of RAS-GTP levels in the tumor samples (G-LISA, Figure 5c). Importantly, and in agreement with the TO and RAS-GTP data, p-ERK levels were also strongly reduced (five-fold and six-fold at 2 and 6 h post last dose, Figure 5d). Finally, we also measured cleaved caspase 3 levels from the tumor lysates (Figure 5e) and, while 2 h post-dose induction of apoptosis was only detectable in one out of five treated samples, apoptosis induction was detectable in all five treated samples 6 h post-dose on day 3, indicating that KRASG12C inhibition leads to induction of programmed cell death in this xenograft model. Given these data, anti-tumor efficacy was also assessed in this KRASG12C mutant NCI-H358 model (Figure 5f). At this time in our KRAS-G12C program, bioavailability was not yet optimized; hence, we wanted to explore whether infrequent systemic administration of covalent KRASG12C inhibitors could be efficacious. BI-0474 was administered ip weekly or twice weekly on 2 consecutive days at 40 mg/kg and showed anti-tumor efficacy in both dose groups with 68% tumor growth inhibition (TGI) on day 19 post treatment start for the dose group receiving a weekly dose of 40 mg/kg ip and 98% TGI for the dose group receiving a weekly dose of 80 mg/kg ip (= 40 mg/kg ip on 2 consecutive days). These data confirm dose-dependent efficacy for BI-0474 in the NCI-H358 model. Compared to animals receiving the vehicle only, both dose groups showed some body weight loss, which is less pronounced for the low dose group compared to the high dose group.
Figure 5.

BI-0474 shows efficacy and PD biomarker modulation in an NCI-H358 cell line-derived non-small cell lung cancer xenograft model. (a) Top graph: PK model of BI-0474 40 mg/kg ip qdx3 including measured free plasma concentration of BI-0474 2, 6, and 24 h post last treatment (n = 5, mean value ± SD); bottom graph: target occupancy (TO) model of BI-0474 40 mg/kg ip qdx3 based on PK and MS-based second-order rate. (b–e) Treatment with BI-0474 leads to TO, reduction of RAS-GTP and pERK levels, as well as apoptosis induction in vivo. Five individual tumors per treatment group were analyzed, the individual values are plotted, as is the mean ± SD. (b) BI-0474 treatment increases TO as measured by reduction of unmodified KRASG12C protein measured by MS, (c) reduces RAS-GTP levels as measured by RAS G-LISA, (d) reduces pERK levels as measured by a pERK1/2 immunoassay (MSD), and (e) induces cleaved caspase-3 levels measured by a caspase-3 immunoassay (MSD). (f) BI-0474 shows efficacy in an NCI-H358 non-small cell lung cancer xenograft model. Left graph: mean tumor volume ± SD for vehicle (n = 8, black circles) compared to 40 mg/kg BI-0474 administered ip once a week (q7d) (n = 8, red squares) and 40 mg/kg BI-474 administered ip twice a week on 2 consecutive days (qdx2) (n = 8, blue triangles). Tumor volumes for each individual mouse measured on day 19 are included in the Supporting Information (Figure S10). Right graph: mean body weight changes.
Chemistry
Compounds in Tables 1 and 2 were commercial or accessible via single-step procedures from commercial starting materials (Scheme 1). 12 was synthesized via the Gewald reaction using dimethyl cyclohexanone. 14 was obtained by dimethylation of the corresponding naphthylamine. Suzuki coupling of 2-chloro-3-fluoropyridine and the suitable phenylboronic acid yielded 15.
Scheme 1. Synthesis of Fragment Analogues 12, 14, and 15.

Reagents and conditions: (a) S8, malono-nitrile, l-proline, DMF, BMIMCl, 12 h, 70 °C, 61%; (b) K2CO3, CH3I, DMF, 18 h, 50 °C, 35%; and (c) XPhos Pd G3, Cs2CO3, dioxane, 1 h, 80 °C, 48%.
As described in Scheme 2, amides 16 and 17a/b as well as oxadiazole 19 could be obtained via the corresponding racemic carboxylic acids 30 and 18: the Gewald reaction on 28 or its methylated analogue 31 gave aminocyanothiophenes 29 and 32.28 Saponification resulted in the desired acid building blocks 30 and 18 which could be converted to the corresponding amides 16 and 17a/b or to oxadiazole 19 via a two-step procedure by coupling the acid with N-hydroxyacetamidine followed by ring closure. Aryl-substituted oxadiazoles 20–22 were accessible by a common procedure (Scheme 3): reaction of a BOC-protected amino benzonitrile with hydroxylamine resulted in an amidoxime. Coupling with the enantiopure acid 18a, which was obtained by chiral SFC from the racemic material 18, followed by ring closure yielded the corresponding oxadiazole 37/38. After BOC-deprotection, the acrylamides were obtained by amidation using acryloyl chloride.29
Scheme 2. Synthesis of Amides and Oxadiazole 19.

Reagents and conditions: (a) S8, morpholine, malononitrile, EtOH, 1 h, 55 °C, 55%; (b) LiOH, dioxane/MeCN/water, 48 h 50 °C, 86%; (c) HATU, DIPEA, PhNH2 or CH3NH3Cl, DMF, 30 min., 0 °C–RT, 32%; (d) NaH, CH3I, DMF, 12 h, 0 °C–RT, 86%; (e) S8, l-proline, malononitrile, EtOH, 48 h, 70 °C, 26%; (f) KOH, EtOH/water, 12 h, 80 °C, 72%; (g) (1) triethylamine, HATU, N-hydroxyacetamidine, DMSO, 1 h, RT; (2) Bu4NOH, THF/water, 20 min., RT, 34%.
Scheme 3. Synthesis of Aryl-Substituted Oxadiazoles and Corresponding Acrylamides.

Reagents and conditions: (a) NH2OH·HCl, NaHCO3, EtOH, 16 h, 80 °C, 57–65%; (b) 18a, HATU, DIPEA, DMF, 1 h, 25 °C then 12 h, 80 °C, 32–45%; (c) TFA, DCM, 1 h, 0 °C–RT, 68–77%; and (d) acryloyl chloride, K2CO3, acetone/water, 5 h, 0 °C, 32–63%.
For the synthesis of BI-0474 (Scheme 4), a suitably substituted nitrile (42) was obtained from its dichloro analogue 40 by regioselective nucleophilic aromatic substitutions followed by a Pd-catalyzed amination to introduce the second piperazine substituent. The desired acrylamide was then synthesized, as described above via reaction of the nitrile with hydroxylamine, coupling of the resulting amidoxime 43 with the enantiopure acid 18a followed by ring closure to the oxadiazole 45. Deprotection and amidation resulted in the acrylamide BI-0474.
Scheme 4. Synthesis of BI-0474 (23).
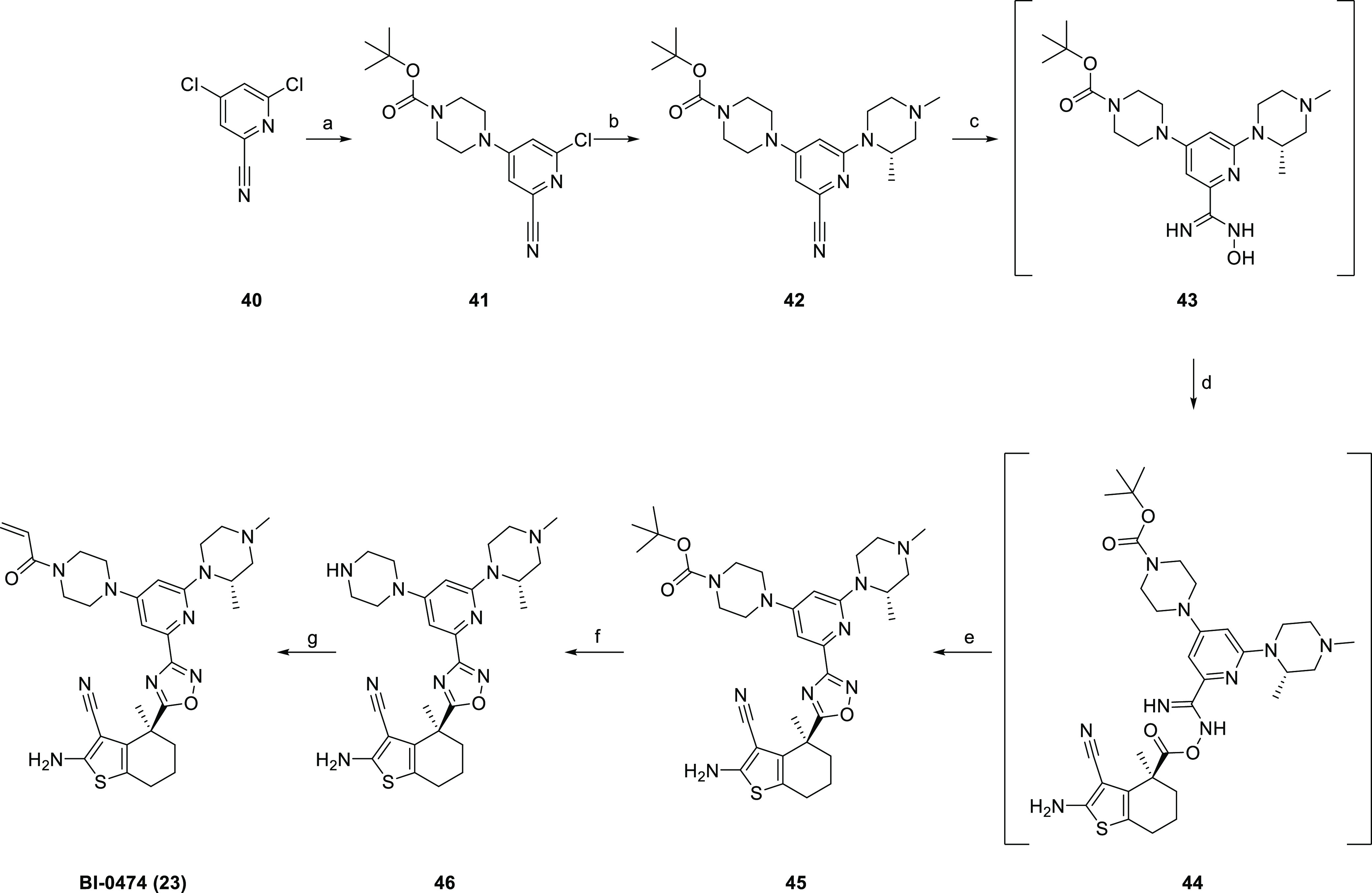
Reagents and conditions: (a) BOC-piperazine, DIPEA, DMSO, 1 h, 60 °C, 69%; (b) (S)-1,3-dimethylpiperazine, Pd2dba3, xantphos, Cs2CO3, dioxane, 16 h, 110 °C, 19%; (c) NH2OH·HCl, Na2CO3, EtOH, 1 h, 85 °C, 99%; (d) 18a, HATU, DIPEA, DMF, 16 h, 25 °C, 29%; (e) DBU, 16 h, 90 °C, 56%; (f) HCl, dioxane, 1 h, 40 °C, 67%; and (g) acryloyl chloride, K2CO3, acetone/water, 15 min, RT, 47%.
Conclusions
Here, we report the discovery of an in vivo active KRASG12C inhibitor derived from a reversibly bound fragment hit. We identified fragments binding to KRAS using a second-site screening approach with a covalently blocked switch I/II pocket. Using structure-based design, we then optimized the initial hits into compounds that bind with single digit μM affinity to KRAS. Further optimization of these compounds via installation of an acrylamide warhead led to single digit nanomolar IC50 values in a biochemical assay. This strategy produced BI-0474 which shows in vivo biomarker modulation and efficacy in an NCI-H358 model after intermittent treatment via ip administration. In contrast to known KRASG12C inhibitors such as sotorasib or adagrasib, which are dosed orally, BI-0474 has only shown efficacy using ip dosing. However, a more advanced orally available analogue from this series has recently entered phase I clinical trials (NCT04973163).24,30,31 Based on its non-covalent binding features, the chemical matter identified via our approach has not only led to the discovery of KRASG12C inhibitors but also molecules that are highly attractive for further use in the discovery of inhibitors against other KRAS mutants.
Experimental Section
Chemistry
General Procedures
Commercial starting materials were used without further purification. Solvents used for reactions were of commercial “dry” or “extra-dry” or “analytical” grade. All other solvents used were of reagent grade. Unless specifically mentioned, all reactions were carried out in standard commercially available glassware using standard synthetic chemistry methods. Air-sensitive and moisture-sensitive reactions were performed under an atmosphere of dry nitrogen or argon with dried glassware. If not explicitly mentioned otherwise, the purity of all tested compounds was ≥95%.
Preparative reversed-phase high-performance liquid chromatography (RP-HPLC) was carried out on Agilent or Gilson systems using columns from Waters (Sunfire C18 OBD, 5 or 10 μm, 20 mm × 50 mm, 30 mm × 50 mm, or 50 mm × 150 mm; X-Bridge C18 OBD, 5 or 10 μm, 20 mm × 50 mm, 30 mm × 50 mm, or 50 mm × 150 mm) or YMC (Triart C18, 5, or 10 μm, 20 mm × 50 mm, or 30 mm × 50 mm). Unless specifically mentioned, compounds were eluted with MeCN/water gradients using either acidic [0.2% HCOOH or trifluoroacetic acid (TFA)] or basic water (5 mL of 2 M NH4HCO3 + 2 mL of NH3 (32%) made up to 1 L with water).
NMR experiments were recorded on Bruker AVANCE HD 400, 500, and 600 MHz spectrometers equipped with a TCI cryoprobe. Samples were dissolved in 600 μL of DMSO-d6, and TMS was added as an internal standard. The temperature was set to 298 K. Processing of spectra was performed with Bruker Topspin 3.6 software. Spectra were analyzed with ACD/NMR Workbook 2019. Chemical shifts are reported in ppm on the δ scale. No zero filling was performed, and the spectra were manually integrated after automatic baseline correction. The 1D 1H spectra were acquired with 30° excitation pulses and an interpulse delay of 4.2 s with 64k data points and a 20 ppm sweep width. The 1D 13C spectra were acquired with broadband composite pulse decoupling (WALTZ16) and an interpulse delay of 3.3 s with 64k data points and a sweep width of 240 ppm. The 2D HSQC spectra were recorded on all samples to aid the interpretation of the data and to identify signals hidden underneath solvent peaks. The spectra were acquired with sweep widths obtained by automatic sweep width detection from the 1D reference spectra in the direct dimension with 1k data points and with 210 ppm and 256 data points in the indirect dimension.
All samples were analyzed on an Agilent 1200 series LC system coupled with an Agilent 6140 mass spectrometer. Purity was determined via UV detection with a bandwidth of 170 nm in the range from 230 to 400 nm. LC parameters were as follows: Waters XBridge C18 column; 2.5 μm particle size; 2.1 × 20 mm; run time, 2.1 min; flow, 1 mL/min; column temperature, 60 °C; 5 μL injections; solvent A (20 mM NH4HCO3/NH3, pH 9) and solvent B (MS-grade acetonitrile); start 10% B, gradient 10 to 95% B from 0.0 to 1.5 min, 95% B from 1.5 to 2.0 min, gradient 95 to 10% B from 2.0 to 2.1 min.
HRMS data were recorded using an LTQ Orbitrap XL (Thermo Scientific) coupled with a Triversa Nanomate Nanospray ion source (ADVION Bioscience Inc.). The mass calibration was performed using the Pierce LTQ Velos ESI positive ion calibration solution from Thermo Scientific (product no. 88323). The scan window was set to 100–500 amu with a maximum injection time of 500 ms and 1 microscan. Resolution of the Orbitrap was 60,000 with a mass accuracy of ≤5 ppm. The ion mode was set to positive with a capillary temperature of 200 °C and a voltage of 60 eV. The tube lens potential was set to 110 eV. The NanoESI voltage was 1.45 kV, and the N2 gas pressure was set to 0.45 psi. The total sample volume was 5 μL, and the acquisition time was 60 s, with 15 scans of averaging per spectrum. The sample dilution [10 mM dimethyl sulfoxide (DMSO) stock solution] was diluted 1:200 in 50% MeOH + 0.01% formic acid.
Synthesis of esters 29 and 32 has been previously described,28 and a detailed description of the methods used is provided in the Supporting Information.
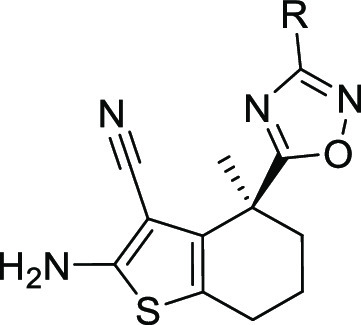
2-Amino-4,4-dimethyl-4,5,6,7-tetrahydro-1-benzothiophene-3-carbonitrile (12)
To a solution of 2,2-dimethylcyclohexanone (24, 1.50 g; 11.9 mmol) in dimethyl formamide (DMF) (15.0 mL) was added BMIMCl (0.65 g, 2.4 mmol), sulfur (1.14 g, 35.7 mmol), malononitrile (1.73 g, 26.2 mmol), and l-proline (1.37 g, 11.9 mmol), and the reaction mixture was allowed to stir at 70 °C for 12 h. The reaction mixture was extracted with EtOAc, washed with water, dried, and concentrated in vacuo to obtain the crude product. This was purified by flash column chromatography (EtOAc/hexane) to afford 1.50 g of 12 (61%).
1H NMR (DMSO-d6, 600 MHz): δ 6.86 (s, 2H), 2.37 (t, 2H, J = 6.1 Hz), 1.6–1.7 (m, 2H), 1.5–1.6 (m, 2H), 1.25 (s, 6H).
13C NMR (DMSO-d6, 151 MHz): δ 164.3, 139.2, 118.0, 116.8, 82.1, 39.4, 33.6, 28.7, 24.8, 20.3.
HRMS (m/z): [M + H]+ calcd for C11H14N2S, 207.09505; found, 207.09516.
8-Chloro-N,N-dimethylnaphthalen-1-amine (14)
8-Chloronaphthalen-1-amine (25, 50 mg, 0.27 mmol) and potassium carbonate (164 mg, 1.19 mmol) were suspended in DMF (0.5 mL) and treated with CH3I (170 mg, 0.594 mmol). The reaction mixture was heated to 50 °C and stirred overnight. Additional potassium carbonate (82 mg, 0.59 mmol) and CH3I (85 mg, 0.59 mmol) were added, and stirring was continued at 50 °C for additional 5 h until the reaction showed acceptable conversion. The reaction mixture was filtered, and the vial and remaining solids were washed with DMSO. The product was isolated from the filtrate via preparative RP HPLC (Agilent, Waters XBridge C18 30 × 50 mm 5 μm, 30–80% MeCN in water + 0.1% formic acid) to afford 20 mg (35%) of 14.
1H NMR (DMSO-d6, 600 MHz): δ 7.86 (dd, 1H, J = 0.9, 8.3 Hz), 7.6–7.6 (m, 1H), 7.55 (dd, 1H, J = 1.3, 7.3 Hz), 7.46 (t, 1H, J = 7.8 Hz), 7.41 (t, 1H, J = 7.8 Hz), 7.21 (dd, 1H, J = 1.1, 7.5 Hz), 2.72 (s, 6H).
13C NMR (DMSO-d6, 151 MHz): δ 150.5, 137.6, 129.4, 129.4, 128.8, 127.1, 126.2, 125.0, 123.4, 116.5, 45.7.
HRMS (m/z): [M + H]+ calcd for C12H12ClN, 206.07310; found, 206.07327.
3-Fluoro-2-(3-fluoropyridin-2-yl)phenol (15)
2-Chloro-3-fluoropyridine (26, 50 mg, 0.37 mmol) was dissolved in dioxane (1.0 mL) and heated to 80 °C. 2- fluoro-6-hydroxyphenylboronic acid (27, 122 mg, 0.745 mmol) was dissolved in dioxane (0.5 mL) and added, followed by cesium carbonate (2.0 M aqueous solution, 0.47 mL, 0.93 mmol) and XPhos Pd G3 (33.2 mg, 0.037 mmol), and the reaction mixture was stirred for 1.5 h at 80 °C. The mixture was diluted with water and brine, extracted with dichloromethane (DCM), and the combined extracts were dried, filtered, and concentrated. The crude product was purified via preparative RP HPLC (Agilent, Waters-Sunfire C18 30 × 50 mm 5 μm, 10–70% MeCN in water + 0.1% HCOOH), yielding 37 mg (48%) of 15 as an off-white solid.
1H NMR (DMSO-d6, 600 MHz): δ 10.09 (br s, 1H), 8.52 (td, 1H, J = 1.6, 4.6 Hz), 7.80 (ddd, 1H, J = 1.3, 8.4, 9.6 Hz), 7.52 (td, 1H, J = 4.4, 8.5 Hz), 7.31 (dt, 1H, J = 7.0, 8.3 Hz), 6.81 (d, 1H, J = 8.3 Hz), 6.7–6.8 (m, 1H).
13C NMR (DMSO-d6, 151 MHz): δ 160.9, 157.4, 157.8, 145.8, 140.7, 131.3, 125.4, 123.9, 112.1, 111.5, 106.1.
HRMS (m/z): [M + H]+ calcd for C11H7F2NO, 208.05685; found, 208.5674.
2-Amino-3-cyano-4,5,6,7-tetrahydro-1-benzothiophene-4-carboxylic Acid (30)
Ethyl 2-amino-3-cyano-4,5,6,7-tetrahydro-1-benzothiophene-4-carboxylate (29, 1.0 g, 4.00 mmol) was dissolved in dioxane/MeCN/water (1:1:5, 7 mL), lithium hydroxide (382 mg, 16.0 mmol) was added, and the mixture was stirred for 48 h at 50 °C. HCl (6 M in water) was added until pH 3, and the product was extracted with EtOAc. The combined organic layers were dried over MgSO4, filtered, and concentrated, yielding 760 mg (86%) of 30 as orange crystals which were used for the next step without purification.
2-Amino-3-cyano-N-phenyl-4,5,6,7-tetrahydro-1-benzothiophene-4-carboxamide (16)
2-Amino-3-cyano-4,5,6,7-tetrahydro-1-benzothiophene-4-carboxylic acid (30, 100 mg, 0.427 mmol) and O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HATU) (23.5 mg, 0.598 mmol) were dissolved in DMF (2.0 mL) and cooled to 0 °C. Diisopropyl ethyl amine (DIPEA) (166 mg, 1.28 mmol) and aniline (52 mg, 0.56 mmol) were added, and the mixture was stirred at 0 °C for 30 min and then allowed to reach RT. The reaction mixture was filtered, and the product was isolated via preparative RP HPLC (Agilent, Waters XBridge C18 30 × 50 mm 5 μm, 5–70% MeCN in water + 0.1% ammonium carbonate/NH3), yielding 40 mg (32%) of 16 as an off-white solid.
1H NMR (DMSO-d6, 600 MHz): δ 10.08 (s, 1H), 7.59 (dd, 2H, J = 0.9, 8.6 Hz), 7.3–7.3 (m, 2H), 7.0–7.1 (m, 1H), 6.93 (s, 2H), 3.59 (t, 1H, J = 6.0 Hz), 2.4–2.5 (m, 2H), 2.0–2.0 (m, 1H), 1.8–1.9 (m, 2H), 1.6–1.7 (m, 1H).
13C NMR (DMSO-d6, 151 MHz): δ 169.8, 161.0, 137.6, 127.6, 126.9, 121.5, 118.0, 117.7, 114.2, 81.5, 40.9, 25.9, 21.9, 19.1.
HRMS (m/z): [M + H]+ calcd for C16H15N3OS, 298.10086; found, 298.10105.
2-Amino-3-cyano-4-methyl-4,5,6,7-tetrahydro-1-benzothiophene-4-carboxylic Acid (18)
Ethyl 2-amino-3-cyano-4-methyl-4,5,6,7-tetrahydro-1-benzothiophene-4-carboxylate (32, 78 mg, 0.295 mmol) was dissolved in ethanol (1.5 mL), potassium hydroxide (370 μL, 4 M in water, 1.48 mmol) was added, and the mixture was stirred for 12 h at 80 °C. The mixture was diluted with water and EtOAc, KHSO4 (10% in water) was added to adjust to pH 4, and the mixture was extracted with EtOAc. The organic layers were combined and concentrated, and the crude product was purified via preparative RP HPLC (Gilson, 20–90% MeCN in water + 0.1% HCOOH) to afford 50 mg (72%) of 18 as an off-white solid.
1H NMR (DMSO-d6, 600 MHz): δ 12.50 (br s, 1H), 6.86 (s, 2H), 2.42 (t, 2H, J = 6.1 Hz), 1.9–2.0 (m, 1H), 1.73 (quin, 2H, J = 6.0 Hz), 1.6–1.7 (m, 1H), 1.42 (s, 3H).
13C NMR (DMSO-d6, 151 MHz): δ 177.2, 163.3, 134.0, 118.6, 117.0, 83.4, 44.4, 35.9, 24.3, 24.3, 20.2.
HRMS (m/z): [M + H]+ calcd for C11H12N2O2S, 237.06922; found, 237.06961.
Chiral SFC of enantiomers of 18 was performed on a Thar SFC Pre80 using a Chiralpak AD-H 5 μm, 21 mm × 250 mm column at a flow rate of 25 g/min at 35 °C and 120 bar with 75% CO2 + 25% MeOH (+0.5% isopropylamine) as the mobile phase to obtain 18a as the first eluting stereoisomer.
2-Amino-3-cyano-N,4-dimethyl-6,7-dihydro-5H-benzothiophene-4-carboxamide (17a)
The preparation was carried out as described for 16 using 2-amino-3-cyano-4-methyl-6,7-dihydro-5H-benzothiophene-4-carboxylic acid (18, 22 mg, 0.093 mmol) and methyl ammonium chloride (7.5 mg, 0.11 mmol) to afford 8 mg (32%) of 17a as an off-white solid.
1H NMR (DMSO-d6, 600 MHz): δ 7.11 (q, 1H, J = 4.2 Hz), 6.94 (s, 2H), 2.63 (d, 3H, J = 4.6 Hz), 2.48 (t, 2H, J = 6.2 Hz), 1.9–2.0 (m, 1H), 1.74 (quin, 2H, J = 6.0 Hz), 1.6–1.7 (m, 1H), 1.43 (s, 3H).
13C NMR (DMSO-d6, 151 MHz): δ 175.6, 163.8, 134.2, 119.7, 116.8, 83.0, 44.9, 36.7, 26.9, 24.5, 24.4, 20.2.
HRMS (m/z): [M + H]+ calcd for C12H15N3OS, 250.10086; found, 250.10099.
2-Amino-3-cyano-4-methyl-N-phenyl-6,7-dihydro-5H-benzothiophene-4-carboxamide (17b)
The preparation was carried out as described for 16 using 2-amino-3-cyano-4-methyl-6,7-dihydro-5H-benzothiophene-4-carboxylic acid (18, 100 mg, 0.423 mmol) and aniline (51 mg, 0.55 mmol) to afford 70 mg (29%) of 17b as an off-white solid.
1H NMR (DMSO-d6, 600 MHz): δ 8.93 (s, 1H), 7.5–7.6 (m, 2H), 7.2–7.3 (m, 2H), 7.0–7.1 (m, 1H), 6.90 (s, 2H), 2.5–2.6 (m, 1H), 2.4–2.5 (m, 1H), 2.04 (ddd, 1H, J = 3.2, 10.0, 13.0 Hz), 1.7–1.8 (m, 3H), 1.50 (s, 3H).
13C NMR (DMSO-d6, 151 MHz): δ 174.3, 163.6, 139.4, 133.9, 128.7, 124.0, 121.8, 119.9, 116.7, 82.9, 45.9, 35.9, 24.4, 24.2, 20.2.
HRMS (m/z): [M + H]+ calcd for C17H17N3OS, 312.11651; found, 312.11654.
2-Amino-4-methyl-4-(3-methyl-1,2,4-oxadiazol-5-yl)-4,5,6,7-tetrahydro-1-benzothiophene-3-carbonitrile (19)
2-Amino-3-cyano-4-methyl-6,7-dihydro-5H-benzothiophene-4-carboxylic acid (18, 120 mg, 0.498 mmol) and triethylamine (143 μL, 0.995 mmol) were dissolved in DMSO (1.0 mL). After addition of HATU (203 mg, 0.23 mmol) and stirring at RT for 15 min, N-hydroxyacetamidine (42 mg, 0.547 mmol) was added, and the mixture was stirred for 1 h at RT. The reaction mixture was filtered, and the product was isolated via preparative RP HPLC (Agilent, YMC Triart C18 19 × 50 mm 5 μm, MeCN in water + 0.1% formic acid), yielding a crude intermediate which was used directly for the next step. The intermediate was dissolved in tetrahydrofuran (THF) (1.0 mL), tetrabutylammonium hydroxide (40% in water, 170 μL, 0.26 mmol) was added, and the mixture was stirred for 20 min at RT. The reaction mixture was concentrated, diluted with DMSO, and the product was isolated via preparative RP HPLC (Agilent, YMC Triart C18 19 × 50 mm 5 μm, MeCN in water + 0.1% formic acid) to afford 46 mg (34%) of 19 as an off-white solid.
1H NMR (DMSO-d6, 600 MHz): δ 7.04 (s, 2H), 2.32 (s, 3H), 2.00 (ddd, 1H, J = 2.9, 9.5, 13.0 Hz), 1.8–1.9 (m, 2H), 1.7–1.8 (m, 1H), 1.72 (s, 3H).
13C NMR (DMSO-d6, 151 MHz): δ 183.6, 167.2, 164.1, 132.1, 119.9, 116.3, 82.1, 38.9, 37.4, 25.1, 24.1, 19.9, 11.7.
HRMS (m/z): [M + H]+ calcd for C13H14N4OS, 275.09611; found, 275.09648.
tert-Butyl N-{4-[(Z)-N′-Hydroxycarbamimidoyl]phenyl}carbamate (35a)
To a stirred solution of tert-butyl N-(4-cyanophenyl)carbamate (33a, 3.0 g, 13.8 mmol) in ethanol (30 mL) was added hydroxylamine HCl (1.9 g, 27.5 mmol) and sodium bicarbonate (2.3 g, 27.45 mmol) at RT, and the reaction mixture was stirred for 16 h at 80 °C. The mixture was cooled to RT, filtered, and the precipitate was washed with EtOAc. The filtrate was concentrated, and the crude product was purified by NP chromatography (10–20% EtOAc in PE) to yield 2.0 g of (57%) 35a as an off-white solid.
1H NMR (DMSO-d6, 400 MHz): δ 9.46 (s, 1H), 9.43 (s, 1H), 7.55 (d, 2H, J = 8.8 Hz), 7.43 (d, 2H, J = 8.8 Hz), 5.69 (s, 2H), 1.48 (s, 9H).
tert-Butyl N-(4-{5-[(4S)-2-Amino-3-cyano-4-methyl-4,5,6,7-tetrahydro-1-benzothiophen-4-yl]-1,2,4-oxadiazol-3-yl}phenyl)carbamate (37a)
To a stirred solution of (4S)-2-amino-3-cyano-4-methyl-4,5,6,7-tetrahydro-1-benzothiophene-4-carboxylic acid (18a, 329 mg 1.39 mmol) in dry DMF (7.0 mL) was added DIPEA (1.25 mL, 7.00 mmol) followed by HATU (581 mg, 1.53 mmol). The reaction mixture was stirred for 20 min at RT, then tert-butyl N-{4-[(Z)-N′-hydroxycarbamimidoyl]phenyl}carbamate (35a, 350 mg, 1.39 mmol) was added, and the reaction mixture was stirred at 25 °C for 1 h and then heated to 80 °C for 12 h. The reaction was quenched with water, stirred for 30 min, extracted with EtOAc, and concentrated. The crude product was purified by NP chromatography (0–30% EtOAc in PE) to yield 209 mg (41%) of 37a as an off-white solid.
1H NMR (DMSO-d6, 400 MHz): δ 9.71 (s, 1H), 7.87 (d, 2H, J = 8.8 Hz), 7.63 (d, 2H, J = 8.8 Hz), 7.06 (s, 2H), 2.53 (br d, 2H, J = 6.4 Hz), 2.0–2.1 (m, 1H), 1.8–2.0 (m, 3H), 1.78 (s, 3H), 1.49 (s, 9H).
(4S)-2-Amino-4-[3-(4-aminophenyl)-1,2,4-oxadiazol-5-yl]-4-methyl-4,5,6,7-tetrahydro-1-benzothiophene-3-carbonitrile (20a)
To a stirred solution of tert-butyl N-(4-{5-[(4S)-2-amino-3-cyano-4-methyl-4,5,6,7-tetrahydro-1-benzothiophen-4-yl]-1,2,4-oxadiazol-3-l}phenyl)carbamate (37a, 200 mg 0.44 mmol) in dry DCM (8.0 mL) was added 15% TFA in DCM (3.0 mL) at 0 °C, and the reaction mixture was stirred for 1 h. The reaction was quenched with saturated NaHCO3 solution, the layers were separated, and the aqueous phase was extracted with DCM. The combined organic layers were dried and concentrated, and the crude product was purified by preparative RP HPLC to afford 120 mg (77%) of 20a as an off-white solid.
1H NMR (DMSO-d6, 600 MHz): δ 7.6–7.7 (m, 2H), 7.04 (s, 2H), 6.6–6.7 (m, 2H), 5.74 (s, 2H), 2.5–2.6 (m, 2H), 2.0–2.1 (m, 1H), 1.8–1.9 (m, 3H), 1.76 (s, 3H).
13C NMR (DMSO-d6, 151 MHz): δ 183.3, 168.2, 164.0, 152.2, 132.3, 128.8, 119.9, 116.3, 114.0, 113.2, 82.2, 39.0, 37.4, 25.3, 24.1, 20.0.
HRMS (m/z): [M + H]+ calcd for C18H17N5OS, 352.12266; found, 352.12279.
N-(4-{5-[(4S)-2-Amino-3-cyano-4-methyl-4,5,6,7-tetrahydro-1-benzothiophen-4-yl]-1,2,4-oxadiazol-3-yl}phenyl)prop-2-enamide (21a)
To a stirred solution of acryloyl chloride (60 mg, 0.67 mmol) in acetone (3.3 mL) and water (1.1 mL), (4S)-2-amino-4-[3-(4-aminophenyl)-1,2,4-oxadiazol-5-yl]-4-methyl-4,5,6,7-tetrahydro-1-benzothiophene-3-carbonitrile (20a, 55 mg 0.16 mmol) in acetone and water (2:6, 0.5 mL) was slowly added at 0 °C. Then potassium carbonate (43 mg, 0.67 mmol) was added, and the mixture was stirred for 5 h at 0 °C. The mixture was concentrated, and the crude product was purified by preparative RP HPLC to yield 35 mg (55%) of 21a as an off-white solid.
1H NMR (DMSO-d6, 600 MHz): δ 10.45 (s, 1H), 7.9–8.0 (m, 2H), 7.8–7.9 (m, 2H), 7.07 (s, 2H), 6.47 (dd, 1H, J = 10.2, 17.0 Hz), 6.31 (dd, 1H, J = 1.8, 16.9 Hz), 5.8–5.9 (m, 1H), 2.5–2.6 (m, 2H), 2.1–2.1 (m, 1H), 1.94 (ddd, 1H, J = 2.7, 7.8, 13.3 Hz), 1.8–1.9 (m, 1H), 1.80 (s, 3H).
13C NMR (DMSO-d6, 151 MHz): δ 184.2, 167.7, 164.1, 163.9, 142.3, 132.1, 128.4, 128.1, 121.4, 120.0, 120.0, 116.3, 82.0, 39.1, 37.3, 25.2, 24.1, 20.0.
HRMS (m/z): [M + H]+ calcd for C21H19N5O2S, 406.13322; found, 406.13328.
tert-Butyl N-{3-[(Z)-N′-Hydroxycarbamimidoyl]phenyl}carbamate (35b)
The preparation was carried out as described for 35a using tert-butyl N-(3-cyanophenyl)carbamate (33b, 3.0 g, 13.8 mmol) to afford 2.1 g (58%) of 35b as an off-white solid.
1H NMR (DMSO-d6, 400 MHz): δ 9.58 (s, 1H), 9.36 (s, 1H), 7.82 (s, 1H), 7.4–7.5 (m, 1H), 7.2–7.3 (m, 2H), 5.68 (s, 2H), 1.47 (s, 9H).
tert-Butyl N-(3-{5-[(4S)-2-Amino-3-cyano-4-methyl-4,5,6,7-tetrahydro-1-benzothiophen-4-yl]-1,2,4-oxadiazol-3-yl}phenyl)carbamate (37b)
The preparation was carried out as described for 37a using tert-butyl N-{3-[(Z)-N′-hydroxycarbamimidoyl]phenyl}carbamate (35b, 250 mg, 0.995 mmol) to afford 200 mg (45%) of 37b as an off-white solid.
1H NMR (DMSO-d6, 400 MHz): δ 9.60 (s, 1H), 8.21 (s, 1H), 7.5–7.6 (m, 2H), 7.4–7.5 (m, 1H), 7.07 (s, 2H), 2.5–2.6 (m, 2H), 2.0–2.1 (m, 1H), 1.8–2.0 (m, 3H), 1.79 (s, 3H), 1.49 (s, 9H).
(4S)-2-Amino-4-[3-(3-aminophenyl)-1,2,4-oxadiazol-5-yl]-4-methyl-4,5,6,7-tetrahydro-1-benzothiophene-3-carbonitrile (20b)
The preparation was carried out as described for 20a using tert-butyl N-(3-{5-[(4S)-2-amino-3-cyano-4-methyl-4,5,6,7-tetrahydro-1-benzothiophen-4-yl]-1,2,4-oxadiazol-3-yl}phenyl)carbamate (37b, 200 mg, 0.443 mmol) to afford 120 mg (77%) of 20b as an off-white solid.
1H NMR (DMSO-d6, 600 MHz): δ 7.24 (t, 1H, J = 1.9 Hz), 7.1–7.2 (m, 1H), 7.1–7.1 (m, 1H), 7.06 (s, 2H), 6.7–6.8 (m, 1H), 5.42 (s, 2H), 2.55 (dt, 2H, J = 2.8, 6.0 Hz), 2.0–2.1 (m, 1H), 1.93 (ddd, 1H, J = 2.7, 7.8, 13.3 Hz), 1.8–1.9 (m, 2H), 1.78 (s, 3H).
13C NMR (DMSO-d6, 151 MHz): δ 184.0, 168.5, 164.1, 149.8, 132.2, 130.1, 127.2, 119.9, 117.1, 116.2, 114.7, 112.5, 82.0, 39.1, 37.4, 25.2, 24.1, 20.0.
HRMS (m/z): [M + H]+ calcd for C18H17N5OS, 352.12266; found, 352.12276.
N-(3-{5-[(4S)-2-Amino-3-cyano-4-methyl-4,5,6,7-tetrahydro-1-benzothiophen-4-yl]-1,2,4-oxadiazol-3-yl}pheyl) prop-2-enamide (21b)
The preparation was carried out as described for 21a using (4S)-2-amino-4-[3-(3-aminophenyl)-1,2,4-oxadiazol-5-yl]-4-methyl-4,5,6,7-tetrahydro-1-benzothiophene-3-carbonitrile (20b, 55 mg, 0.16 mmol) to afford 40 mg (63%) of 21b as an off-white solid.
1H NMR (DMSO-d6, 600 MHz): δ 10.42 (s, 1H), 8.30 (d, 1H, J = 1.5 Hz), 7.98 (dd, 1H, J = 0.8, 8.2 Hz), 7.71 (dd, 1H, J = 0.9, 7.7 Hz), 7.5–7.6 (m, 1H), 7.08 (s, 2H), 6.45 (dd, 1H, J = 10.1, 16.9 Hz), 6.3–6.3 (m, 1H), 5.80 (dd, 1H, J = 1.7, 10.2 Hz), 2.5–2.6 (m, 2H), 2.10 (ddd, 1H, J = 3.2, 9.9, 13.1 Hz), 1.9–2.0 (m, 1H), 1.8–1.9 (m, 2H), 1.80 (s, 3H).
13C NMR (DMSO-d6, 151 MHz): δ 184.5, 167.9, 164.1, 163.9, 140.3, 132.1, 132.1, 130.3, 127.9, 127.2, 122.5, 122.4, 120.0, 118.1, 116.2, 81.9, 39.2, 37.3, 25.1, 24.1, 19.9.
HRMS (m/z): [M + H]+ calcd for C21H19N5O2S, 406.13322; found, 406.13316.
tert-Butyl-4-{3-[(Z)-N′-hydroxycabamimidoyl]phenyl}piperazine-1-carboxylate (36)
The preparation was carried out as described for 35a using tert-butyl 4-(3-cyanophenyl)piperazine-1-carboxylate (34, 3.0 g, 12.1 mmol) to afford 2.2 g (65%) of 36 as an off-white solid.
1H NMR (DMSO-d6, 400 MHz): δ 9.54 (s, 1H), 7.2–7.2 (m, 2H), 7.1–7.2 (m, 1H), 6.96 (dd, 1H, J = 1.9, 8.0 Hz), 5.76 (br d, 2H, J = 1.6 Hz), 3.4–3.5 (m, 4H), 3.1–3.1 (m, 4H), 1.42 (s, 9H).
tert-Butyl 4-(3-{5-[(4S)-2-Amino-3-cyano-4-methyl-4,5,6,7-tetrahydro-1-benzothiophen-4-yl]-1,2,4-oxadiazol-3-yl}phenyl)piperazine-1-carboxylate (38)
The preparation was carried out as described for 37a using tert-butyl-4-{3-[(Z)-N′-hydroxycabamimidoyl]phenyl}piperazine-1-carboxylate (36, 350 mg, 1.21 mmol) to afford 200 mg (32%) of 38 as an off-white solid.
1H NMR (DMSO-d6, 400 MHz): δ 7.49 (s, 1H), 7.4–7.4 (m, 2H), 7.19 (td, 1H, J = 2.2, 7.4 Hz), 7.07 (s, 2H), 3.48 (br d, 4H, J = 5.0 Hz), 3.18 (br t, 4H, J = 5.4 Hz), 2.5–2.6 (m, 2H), 2.0–2.2 (m, 1H), 1.8–2.0 (m, 3H), 1.79 (s, 3H), 1.43 (s, 9H).
(4S)-2-Amino-4-methyl-4-{3-[3-(piperazin-1-yl)phenyl]-1,2,4-oxadiazol-5-yl}-4,5,6,7-tetrahydro-1-benzothiophene-3-carbonitrile (39)
The preparation was carried out as described for 20a using tert-butyl 4-(3-{5-[(4S)-2-amino-3-cyano-4-methyl-4,5,6,7-tetrahydro-1-benzothiophen-4-yl]-1,2,4-oxadiazol-3-yl}phenyl)piperazine-1-carboxylate (38, 200 mg, 0.384 mmol) to afford 110 mg (68%) of 39 as an off-white solid.
1H NMR (DMSO-d6, 600 MHz): δ 7.38 (d, 1H, J = 1.7 Hz), 7.3–7.3 (m, 2H), 7.1–7.1 (m, 1H), 7.00 (s, 2H), 3.0–3.1 (m, 4H), 2.8–2.8 (m, 4H), 2.5–2.5 (m, 2H), 2.03 (ddd, 1H, J = 2.9, 9.8, 13.2 Hz), 1.87 (ddd, 1H, J = 2.6, 7.8, 13.3 Hz), 1.7–1.8 (m, 2H), 1.72 (s, 3H).
13C NMR (DMSO-d6, 151 MHz): δ 184.2, 168.4, 164.1, 152.4, 132.0, 130.3, 127.4, 120.0, 118.9, 117.9, 116.2, 113.3, 82.0, 49.3, 45.8, 39.1, 37.2, 25.3, 24.1, 20.0.
HRMS (m/z): [M + H]+ calcd for C22H24N6OS, 421.18051; found, 421.18058.
(4S)-2-Amino-4-methyl-4-(3-{3-[4-(prop-2-enoyl)piperazin-1-yl]phenyl}-1,2,4-oxadiazol-5-yl)-4,5,6,7-tetrahydro-1-benzothiophene-3-carbonitrile (22)
The preparation was carried out as described for 21a using (4S)-2-amino-4-methyl-4-{3-[3-(piperazin-1-yl)phenyl]-1,2,4-oxadiazol-5-yl}-4,5,6,7-tetrahydro-1-benzothiophene-3-carbonitrile (39, 110 mg, 0.262 mmol) to afford 40 mg (32%) of 22 as an off-white solid.
1H NMR (DMSO-d6, 600 MHz): δ 7.5–7.5 (m, 1H), 7.4–7.5 (m, 1H), 7.4–7.4 (m, 1H), 7.2–7.2 (m, 1H), 7.07 (s, 2H), 6.86 (dd, 1H, J = 10.5, 16.7 Hz), 6.15 (dd, 1H, J = 2.4, 16.7 Hz), 5.7–5.8 (m, 1H), 3.73 (br d, 4H, J = 25.9 Hz), 3.24 (br s, 4H), 2.5–2.6 (m, 2H), 2.11 (ddd, 1H, J = 3.0, 9.9, 13.2 Hz), 1.94 (ddd, 1H, J = 2.7, 7.7, 13.3 Hz), 1.8–1.9 (m, 2H), 1.80 (s, 3H).
13C NMR (DMSO-d6, 151 MHz): δ 184.3, 168.3, 164.7, 164.1, 151.6, 132.0, 130.4, 128.6, 128.0, 127.5, 120.0, 119.4, 118.5, 116.2, 113.8, 82.0, 49.0, 48.5, 45.2, 41.7, 39.1, 37.2, 25.3, 24.1, 20.0.
HRMS (m/z): [M + H]+ calcd for C25H26N6O2S, 475.19107; found, 475.19107.
tert-Butyl 4-(2-Chloro-6-cyanopyridin-4-yl)piperazine-1-carboxylate (41)
To a stirred solution of 4,6-dichloropyridine-2-carbonitrile (40, 5.00 g, 28.9 mmol) in DMSO (50 mL) was added piperazine-1-carboxylic acid tert-butyl ester (5.92 g, 31.8 mmol). Then, DIPEA (11.2 g, 86.7 mmol) was added, and the reaction mixture was stirred at 60 °C for 1 h. The mixture was dissolved in EtOAc and washed with water. The organic phase was dried, filtered, and concentrated under reduced pressure. The crude product was purified via NP chromatography (90–100% EtOAc in hexane), yielding 6.44 g (69%) of 41 as a white solid.
1H NMR (DMSO-d6, 500 MHz): δ 7.55 (d, 1H, J = 1.6 Hz), 7.12 (s, 1H), 3.5–3.5 (m, 4H), 3.4–3.4 (m, 4H), 1.42 (s, 9H).
tert-Butyl 4-{2-Cyano-6-[(2S)-2,4-dimethylpiperazin-1-yl]pyridin-4-yl}piperazine-1-carboxylate (42)
tert-Butyl 4-(2-chloro-6-cyanopyridin-4-yl)piperazine-1-carboxylate (41, 1.00 g, 3.10 mmol), (S)-1,3-dimethylpiperazine (0.99 g, 8.7 mmol), tris(dibenzylideneacetone)dipalladium(0) (141.9 mg, 0.154 mmol), xantphos (184.8 mg, 0.31 mmol), cesium carbonate (2.02 g, 6.20 mmol), and dry dioxane (8.0 mL) were combined and stirred in a closed vessel under an argon atmosphere for 16 h at 110 °C. Brine was added to the mixture, and the product was extracted with DCM. The combined organic phases were dried, filtered, and concentrated under reduced pressure. The crude product was purified via preparative RP HPLC (Gilson, Waters XBridge C18 50 × 150 mm 10 μm, 30 to 98% MeCN in basic water), yielding 240 mg (19%) of 42 as an off-white solid.
1H NMR (DMSO-d6, 500 MHz): δ 2.76 (td, 1H, J = 2.8, 11.8 Hz), 2.6–2.7 (m, 2H), 2.5–2.6 (m, 2H), 2.11 (s, 3H), 1.77 (dt, 1H, J = 3.2, 11.0 Hz), 1.44 (t, 1H, J = 10.1 Hz), 0.90 (d, 3H, J = 6.3 Hz).
tert-Butyl 4-{2-[(2S)-2,4-Dimethylpiperazin-1-yl]-6-[(Z)-N′-hydroxycarbamimidoyl]pyridin-4-yl}piperazine-1-carboxylate (43)
tert-Butyl 4-{2-cyano-6-[(2S)-2,4-dimethylpiperazin-1-yl]pyridin-4-yl}piperazine-1-carboxylate (42, 200 mg, 0.499 mmol), hydroxylamine hydrochloride (90 mg, 1.3 mmol), and sodium carbonate (68.8 mg 0.649 mmol) were dissolved in dry EtOH (2.0 mL), and the mixture was stirred at 85 °C for 1 h. The mixture was diluted with water and extracted with 10% MeOH in DCM. The combined organic layers were washed with brine, dried, filtered, and concentrated under reduced pressure to afford 215 mg (99%) of 43 as a brown oil which was used for the next step without further purification.
tert-Butyl 4-{2-[(Z)-N′-[(Z)-(4S)-2-Amino-3-cyano-4-methyl-4,5,6,7-tetrahydro-1-benzothiophene-4-carbonyloxy]carbamimidoyl]-6-[(2S)-2,4-dimethylpiperazin-1-yl]pyridin-4-yl}piperazine-1-carboxylate (44)
(4S)-2-Amino-3-cyano-4-methyl-4,5,6,7-tetrahydro-1-benzothiophene-4-carboxylic acid (18a, 150 mg, 0.64 mmol) in DMF (1.5 mL) was treated with HATU (241 mg, 0.64 mmol) and DIPEA (331 μL, 1.90 mmol), and the mixture was stirred for 20 min at RT. Then, tert-butyl 4-{2-[(2S)-2,4-dimethylpiperazin-1-yl]-6-[(Z)-N′-hydroxycarbamimidoyl]pyridin-4-yl}piperazine-1-carboxylate (43, 215 mg, 0.495 mmol) was added, and the mixture was stirred for 16 h at RT. The reaction mixture was diluted with water and extracted with EtOAc. The combined organic layers were washed with brine, dried, filtered, and concentrated to afford 120 mg (29%) of 44 which was used directly for the next step.
tert-Butyl 4-(2-{5-[(4S)-2-Amino-3-cyano-4-methyl-4,5,6,7-tetrahydro-1-benzothiophen-4-yl]-1,2,4-oxadiazol-3-yl}-6-[(2S)-2,4-dimethylpiperazin-1-yl]pyridin-4-yl)piperazine-1-carboxylate (45)
A mixture of tert-butyl 4-{2-[(Z)-N′-[(Z)-(4S)-2-amino-3-cyano-4-methyl-4,5,6,7-tetrahydro-1-benzothiophene-4-carbonyloxy]carbamimi-doyl]-6-[(2S)-2,4-dimethylpiperazin-1-yl]pyridin-4-yl}piperazine-1-carboxylate (44, 120 mg, 0.184 mmol) and DBU (140 mg, 0.92 mmol) was stirred at 90 °C for 16 h. The reaction mixture was diluted with water, extracted with EtOAc, and the combined organic layers were washed with brine, dried, filtered, and concentrated. The crude product was purified via NP chromatography (MeOH/DCM) followed by preparative RP HPLC (Gilson, Waters XBridge C18 50 × 150 mm 5 μm, 40–98% MeCN in basic water), yielding 65 mg (56%) of 45 as a light brown solid.
1H NMR (DMSO-d6, 400 MHz): δ 7.08 (s, 2H), 6.89 (s, 1H), 6.18 (s, 1H), 4.5–4.6 (m, 1H), 4.08 (br d, 1H, J = 13.4 Hz), 3.45 (br s, 4H), 2.99 (dt, 1H, J = 3.0, 12.4 Hz), 2.83 (br d, 1H, J = 10.1 Hz), 2.7–2.7 (m, 1H), 2.19 (s, 3H), 2.0–2.1 (m, 2H), 1.8–2.0 (m, 4H), 1.78 (s, 3H), 1.42 (s, 9H), 1.13 (d, 3H, J = 6.6 Hz).
(4S)-2-Amino-4-(3-{6-[(2S)-2,4-dimethylpiperazin-1-yl]-4-(piperazin-1-yl)pyridin-2-yl}-1,2,4-oxadiazol-5-yl)-4-methyl-4,5,6,7-tetrahydro-1-benzothiophene-3-carbonitrile (46)
tert-Butyl 4-(2-{5-[(4S)-2-amino-3-cyano-4-methyl-4,5,6,7-tetrahydro-1-benzothiophen-4-yl]-1,2,4-oxadiazol-3-yl}-6-[(2S)-2,4-dimethylpiperazin-1-yl]pyridin-4-yl)piperazine-1-carboxylate (45, 140 mg, 0.208 mmol) was dissolved in 1,4-dioxane (5 mL), HCl (4 M in 1,4-dioxane, 0.52 mL, 2.08 mmol) was added, and the mixture was stirred for 1 h at 40 °C. The mixture was concentrated, two drops of triethylamine were added, and the product was isolated via preparative RP HPLC (Agilent, Waters XBridge C18 50 × 150 mm 5 μm, 10–70% MeCN in basic water), yielding 75 mg (67%) of 46 as an off-white solid.
1H NMR (DMSO-d6, 600 MHz): δ 7.07 (s, 2H), 6.88 (d, 1H, J = 1.8 Hz), 6.14 (d, 1H, J = 1.8 Hz), 4.54 (br s, 1H), 4.06 (br d, 1H, J = 12.5 Hz), 3.2–3.3 (m, 4H), 2.99 (dt, 1H, J = 3.1, 12.5 Hz), 2.84 (br s, 1H), 2.8–2.8 (m, 4H), 2.69 (br d, 1H, J = 10.8 Hz), 2.5–2.6 (m, 2H), 2.19 (s, 3H), 2.1–2.1 (m, 2H), 1.8–2.0 (m, 4H), 1.78 (s, 3H), 1.12 (d, 3H, J = 6.6 Hz).
13C NMR (DMSO-d6, 151 MHz): δ 183.9, 168.9, 164.1, 160.1, 158.0, 144.9, 132.1, 119.9, 116.2, 100.6, 91.7, 82.0, 60.4, 55.5, 47.7, 47.2, 46.6, 45.8, 39.6, 39.1, 37.3, 25.4, 24.1, 20.0, 14.3.
HRMS (m/z): [M + H]+ calcd for C27H35N9OS, 534.27580; found, 534.27579.
(4S)-2-Amino-4-(3-{6-[(2S)-2,4-dimethylpiperazin-1-yl]-4-[4-(prop-2-enoyl)piperazin-1-yl]pyridin-2-yl}-1,2,4-oxadiazol-5-yl)-4-methyl-4,5,6,7-tetrahydro-1-benzothiophene-3-carbonitrile (23)
Acryloyl chloride (1 M in acetone, 419 μL, 0.419 mmol) was added to a mixture of potassium carbonate (57.8 mg, 0.419 mmol), acetone (3.7 mL), and water (0.74 mL). After 10 min, (4S)-2-amino-4-(3-{6-[(2S)-2,4-dimethylpiperazin-1-yl]-4-(piperazin-1-yl)pyridin-2-yl}-1,2,4-oxadiazol-5-yl)-4-methyl-4,5,6,7-tetrahydro-1-benzothiophene-3-carbonitrile (46, 74.5 mg, 0.14 mmol) in acetone/water (5:1, 0.5 mL) was added, and the mixture was stirred for 15 min at RT. The mixture was concentrated, and the product was isolated via preparative RP HPLC (Agilent, Waters XBridge C18 30 × 50 mm 5 μm, 10–80% MeCN in basic water), yielding 39 mg (47%) of 23 as a white solid.
1H NMR (DMSO-d6, 600 MHz): δ 7.00 (s, 2H), 6.83 (d, 1H, J = 1.8 Hz), 6.76 (dd, 1H, J = 10.5, 16.8 Hz), 6.12 (d, 1H, J = 1.7 Hz), 6.08 (dd, 1H, J = 2.4, 16.7 Hz), 5.6–5.7 (m, 1H), 4.49 (br s, 1H), 4.01 (br d, 1H, J = 12.7 Hz), 3.62 (br d, 4H, J = 28.1 Hz), 3.32 (br s, 4H), 2.92 (dt, 1H, J = 3.1, 12.5 Hz), 2.76 (br d, 1H, J = 10.8 Hz), 2.63 (br d, 1H, J = 11.0 Hz), 2.5–2.5 (m, 2H), 2.12 (s, 3H), 2.0–2.1 (m, 2H), 1.7–1.9 (m, 4H), 1.71 (s, 3H), 1.06 (d, 3H, J = 6.6 Hz).
13C NMR (DMSO-d6, 151 MHz): δ 184.0, 168.9, 164.8, 164.1, 160.0, 157.3, 145.1, 132.1, 128.6, 128.0, 119.9, 116.2, 100.6, 92.1, 81.9, 60.4, 55.5, 47.2, 46.8, 46.6, 46.2, 44.9, 41.4, 39.6, 39.1, 37.3, 25.4, 24.1, 20.0, 14.3.
HRMS (m/z): [M + H]+ calcd for C30H37N9O2S, 588.28637; found, 588.28629.
Protein Purification
Proteins of KRASG12C, KRASG12V, KRASG12V-S39C, and KRASG12V-C118S-S39C-BIT (uniprot reference ID P01116) were cloned, expressed, and purified, as previously described.13 Protein batches of BIT-modified proteins (indicated by -BIT) were obtained by addition of 1H-benzimidazol-2-ylmethanethiol hydrochloride (BIT) to the unmodified protein at a concentration of 2 mg/mL in 50 mM Tris-HCL (pH 8.5), 100 mM NaCl, 2 mM GDP, and 5 mM. MgCl2. After overnight incubation at 4 °C, the protein was transferred to a HiPrep Desalting column 26/10, washed with the BIT free buffer, and concentrated to 25–40 mg/mL for further usage.
Crystallization and Structure Determination
Crystals of KRASG12V,C118S,S39C-BIT in complex with (1) were obtained by co-crystallization mixing 0.2 μL of protein solution [24.6 mg/mL protein in 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) pH 7.5, 150 mM NaCl, 5 mM MgCl2, 2 mM GDP, and 5 mM (1)] and 0.2 μL of reservoir solution (30% PEG3350, 0.1 M bicine pH 8.5, and 0.2 M ammonium sulfate) at 4 °C. Crystals of KRASG12C in complex with 12 were obtained by co-crystallization using 0.2 μL of protein solution [40 mg/mL in 20 mM HEPES pH 7.5, 150 mM NaCl, 3.2 mM (12), and 3.2% DMSO] and 0.2 μL of reservoir solution [100 mM NaAc buffer pH 5, 1.5 M (NH4)2SO4] at 4 °C. The structure of KRASG12V,S39C-BIT in complex with 20a was obtained using a soaking procedure. Crystals of KRASG12V,S39C-BIT were obtained using the hanging drop method by mixing 1 μL of protein solution (29 mg/mL in 50 mM Tris-HCL pH 8.5, 100 mM NaCl, 2 mM GDP, and 5 mM MgCl2) with 1 μL of reservoir solution (100 mM PCB buffer pH 5.9–6.2 and 23–26% PEG1500, 2% at 20 °C). Cubic crystals appeared within a week and were soaked overnight with 50 mM 20a. Crystals of KRASG12C in complex with 23 were obtained by co-crystallization using 0.2 μL of protein solution (40 mg/mL in 20 mM HEPES pH 7.5, 150 mM NaCl, 1 mM 23, and 1% DMSO) and 0.2 μL of reservoir solution (10% PEG8000, 10% PEG1000) at 4 °C. All crystals were flesh frozen with 10–25% ethylene glycol before measurements. Details of data collection, data processing, and refinement (using standard procedures described in the Supporting Information) and the respective statistics are shown in Tables S1–S4. Final models were analyzed with MolProbity revealing 97.1/2.70/0.2 (1), 96.5/3.5/0/0 (12), 96.2/3.8/0 (20a), and 96.3/3.1/0 (23) of the residues in Ramachandran favored, allowed, and disallowed regions, respectively. The refined 2Fo – Fc electron density maps are shown in Figure S1. Structures are deposited in the Protein Data Bank (PDB, http://www.rcsb.org) with the following accession numbers (1) 7U8H, (12) 8AFC, (20a) 8AFD, and (23) 8AFB.
KD Determination by NMR Spectroscopy
Dissociation constants of the fragments were determined, as described previously14 by titrating compounds from a 50 mM DMSO-d6 stock solution to a 70 μM solution of KRAS. 4–5 titration points were performed with individual NMR samples for every concentration and careful correction of the total DMSO-d6 content. A minimum of three resonances of the protein were monitored, and the SD was calculated from the KDs of the individually monitored resonances (Figures S6 and S7). KD values are indicated as <10 μM when the CSPs could no longer be reliably fitted because all CSPs were already close to saturation at the first titration point of 125 μM. This limitation is due to the relatively high protein concentration required in the NMR assay of 70 μM. KD values are indicated as >2000 μM if we observed dose-responsive CSPs but they could not be fitted at the highest concentration used of 1000 μM. KD values are indicated as >10,000 μM if we observed no CSPs at a ligand concentration of 500 μM.
kinact/KI Determination by Mass Spectrometry
Samples were prepared at a protein concentration of 1 μM in HBS-N buffer, pH 7.4, 1% DMSO. Compounds were added in concentrations between 0.78 and 100 μM, and incubation was performed at room temperature with incubation times between 30 s and 24 h. Samples were quenched by adding an equal volume of 4% formic acid, diluted with 0.1% formic acid to a final protein concentration of 0.2 μM, and immediately frozen until they were analyzed by liquid chromatography/MS (LC/MS).
LC/MS analysis was performed on a Waters Xevo XS instrument equipped with an M-Class LC system. A Waters BEH C4 column (50 mm, 5 μm, 300 Å) was used for desalting (T = 60 °C, flow rate: 40 μL/min). 5 μL of the sample was injected onto the column. Proteins were eluted with a 1.5 min gradient of 5–95% acetonitrile in 0.1% formic acid. The mass spectrometer was calibrated with direct infusion of sodium iodide solution (200 femtomoles/μL, Sigma-Aldrich) at a flow rate of 20 μL/min. A low-flow ESI probe was used, and instrument settings were as follows: 1 scan/s, mass range: 400–3500 m/z, sensitivity mode, capillary voltage 3 kV, sampling cone: 70 V, source offset: 80 V, source temperature: 120 °C, and desolvation temperature: 400 °C.
Deconvolution of mass spectra was performed in Unifi using MaxEnt (batch mode). Ratios between unmodified and modified proteins were extracted from the deconvoluted spectra in Unifi and fitted in GraphPad Prism (one-phase decay model). The obtained kobs values were used as input into the GraphPad Prism Michaelis–Menten model and kinact and KI values obtained. Mean values are reported with standard deviation (n = 3 independent experiments).
NCI-H358 Proliferation Assay
NCI-H358 (ATCC #CRL-5807) cells were seeded in flat bottom 96 well microtiter plates (white PerkinElmer CulturePlate 96, cat. no. 6005689) in 90 μL RPMI/10% fetal calf serum (FCS), 1% l-Glu, 1× NEAA, and 1× Na-Pyrovat at a density of 2000 cells/well. On day 2, 10 μL of dilutions of the test compounds covering a concentration range between app. 1 and 10,000 nM were added to the cells. Cells were incubated for 3 days in a humidified, CO2 controlled incubator at 37 °C. 100 μL of the cell titer glow reagent (CellTiter Glo Luminescent cat. no. G7571, Promega) was added to each well and incubated for additional 10 min at RT (with agitation). Luminescence was measured on a Wallac Victor using standard luminescence read out. IC50 values were calculated using standard Levenburg Marquard algorithms (GraphPad Prism).
GP2D and LS513 Proliferation Assay
Cells (GP2D or LS513 cells) were plated in 96-well round bottom plates (Costar 7007 ULA Round Bottom 96 well) in 180 μL of medium (Dulbecco’s modified Eagle’s medium for GP2D; RPMI for LS513) containing 2% FCS (1000 cells per well) and incubated overnight at 37 °C. The next day, 20 μL of appropriate serial compound dilutions in medium was added, spanning a concentration range of 10 μM down to sub nanomolar concentrations. Cells were incubated for 5 days at 37 °C in a CO2 incubator. After addition of 20 μL of the CTG reagent (Promega) and 10 to 30 min agitation, luminescence was measured. Data analysis (dose–response curves) was carried out using GraphPad Prism-based software.
Cell Line-Derived Efficacy and Biomarker Study in the NCI-H358 Model
To establish the non-small cell lung cancer cell line-derived NCI-H358 xenograft model, 7–8 weeks old female NOG mice (NOD.Cg-Prkdcscid Il2rgtm1Sug/JicTac) with a bodyweight of 20 g from Taconic, Denmark were engrafted subcutaneously with 5 million NCI-H358 cells (ATCC #CRL-5807) suspended in growth factor reduced, phenol red-free Matrigel (Corning). Mice were group-housed (eight mice per cage) under pathogen-free and controlled environmental conditions (21 ± 1.5 °C temperature, 55 ± 10% humidity, and a 12 h light–dark cycle) and handled according to the internal institutional, Austrian governmental and European Union guidelines (Austrian Animal Protection Laws, ETS-123). All animal studies were approved by the internal ethics and the local governmental committee. For the biomarker study, 40 mice were injected subcutaneously with tumor cells into the right flank, and 20 of these were randomized to vehicle versus treatment arm based on tumor size once the tumors reached a mean size of 180 mm3 with n = 5 mice per group and sampling timepoint (2, 6 h). Tumor-bearing mice were treated for 3 consecutive days with either vehicle or BI-0474 40 mg/kg, and tumors were explanted and frozen to analyze biomarker modulation 2 and 6 h hours post last dose on the third day. For the efficacy study, 50 mice were injected subcutaneously with tumor cells into the right flank, and 24 mice were randomized based on tumor size into three groups of n = 8 mice once the tumors reached a mean size of 160 mm3. In the efficacy study, a vehicle control arm was compared with two treatment arms, testing two dose schedules [BI-0474 40 mg/kg once a week (q7d) and BI-0474 40 mg/kg twice a week on 2 consecutive days (qdx2)]. Tumor size was measured with an electronic caliper three times per week, and the results were converted to tumor volume (mm3) by the formula: length × width2 × π/6. Body weight changes were assessed on a daily basis as a measure of tolerability. For statistical evaluation, a one-sided non-parametric Mann–Whitney–Wilcoxon U-test was used to compare treatment groups with the vehicle control. BI-0474 was formulated in 25% HP-γ-CD, and the vehicle control group was treated with 25% HP-γ-CD once a week in the efficacy study and daily for 3 consecutive days in the biomarker study. All dose groups were treated intraperitonally (ip) using an application volume of 10 mL/kg. The studies described were not blinded.
Cell Lysis and Protein Lysate Generation
Approximately 5 mm3 pieces/tumors were excised from the bulk tumors and placed in lysis buffer. The tumor pieces were ground carefully before centrifugation. The protein concentration was measured according to manufacturer’s recommendations (BRADFORD).
TO Measurements
KRASG12C TO was addressed by measuring the disappearance of the peptide mapping the Ras G12C locus upon drug binding in a targeted proteomics approach using Ras G12 wild-type- and G12C mutant-mapping peptides LVVVGAGGVGK and LVVVGAGCVGK and their stable isotope-labeled (SIL) variants as internal standards. Protein from tumor lysates was precipitated using the methanol-chloroform method. Air-dried pellets were reduced, alkylated, and digested with 5 mM tris(2-carboxyethyl) phosphine hydrochloride, 10 mM 2-chloroacetamide, 1% sodium deoxycholate, SIL peptides (K+8, C alkylated, SpikeTide TQL, JPT Technologies), and trypsin in 50 mM triethylammonium bicarbonate pH 8 at 37 °C. Digests were acidified with TFA and supernatants purified by solid-phase extraction (SepPak tC18, Waters). Eluted peptides were dried to completion, dissolved in 0.1% TFA, and injected into the LC/MS system. LC/MS analyses were performed on a nanoflow HPLC system (EASY-nLC II) interfaced to a triple quadrupole mass spectrometer (Quantiva) through a nano-ESI source (EASYspray). Nano-LC separation was performed with a gradient from 2 to 40% solvent B (80 : 20 acetonitrile: water, 0.1% formic acid) in solvent A (0.1% formic acid in water) in 120 min over a pre-column (Acclaim PepMap 100 C18, 5 μm particle size, 200 μm × 2 cm) connected in line with an analytical column (EASYspray C18, 2 μm particle size, 75 μm × 50 cm) in a vented column setup at 50 °C analytical column temperature. MS data were acquired by multiple reaction monitoring of Ras G12-mapping peptides with Q1 set to [M + 2H]2+ and Q3 to y7+ and y8+ for Ras G12 wild-type peptides and y7+, y8+, and y9+ for Ras G12C mutant peptides. All LC and MS instruments and LC columns were purchased from Thermo Fisher Scientific. Peak integration was performed with Skyline (University of Washington). Analytical process variance mapped by both SIL peptides was 10% CV across all samples; to account for additional inter-sample variance, endogenous/standard peak area ratios of the Ras G12C-mutant peptides were normalized to those of the Ras G12 wild-type peptide for each individual animal. Assuming no target engagement in the control animal groups, TO in the treated animals groups was then calculated as 1 – median(G12C/G12 ratio)treated animals/median(G12C/G12 ratio)control animals (Table S4).
RAS-GTP Measurements
The protein concentration was adjusted to 1 μg/μL right before the assay performance (G-LISA Ras Activation Assay Biochem Kit, Cytoskeletion Inc.). Measurements were taken according to the manufacturer’s recommendations. 25 μg of the sample was added per well of RAS GTP G-LISA plates. Absorbance was measured at 490 nm on a PerkinElmer Enspire Multilabel Reader.
Phospho/Total ERK1/2 Measurements
Measurements were performed according the manufacturers recommendations (Phospho/Total ERK1/2 Whole Cell Lysate Kit, MSD). 20 μg of samples was added per well of the MSD plate. The plate was analyzed on an MSD SECTOR S600 Imager. Results were saved as the text file and converted to an Excel file. Averages of all pERK and total ERK ratios (two measurements per sample) were calculated for all samples.
Cleaved Caspase-3 Measurements
Measurements were performed according to the manufacturer’s recommendations (Apoptosis Whole Cell Lysate Kit, MSD). 20 μg of samples were added per well of the MSD plate. The plate was analyzed on an MSD SECTOR S600 Imager.
Acknowledgments
In honor of his deep commitment to science, the authors dedicate this article to the memory of our friend and colleague Christian Smethurst, who, tragically and unexpectedly, passed away on July 13, 2022. Boehringer-Ingelheim is grateful for financial support by the Austrian Research Promotion Agency FFG (grant numbers: 854341, 861507, 867897, 874517, 883626, and 892584). The authors would like to thank Christian Salamon, Onur Kaya, Eva Strauss, Maximilian Scharnweber, Daniela Häring, Jale Karolyi-Oezguer, Michael Kulhanek, Jacqueline Guggenberger, Johannes Huber, David Covini, Gabriel Kiesenhofer, Wolfgang Hela, Barbara Müllauer, Bernhard Wolkerstorfer, Gabriela Siszler, Alexander Weiss-Puxbaum, Andreas Zöphel, and Georg Dahmann for their support in this project.
Glossary
Abbreviations
- BMIMCl
1-butyl-3-methyl-imidazolium-chloride
- DBU
1,8-diazabicyclo[5.4.0]undec-7-ene
- DCM
dichloromethane
- DIPEA
diisopropyl ethyl amine
- DMF
dimethyl formamide
- EtOAc
ethyl acetate
- HATU
O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate
- ip
intraperitoneal
- NP
normal phase
- PD
pharmacodynamic
- PE
petroleum ether
- RP
reversed phase
- RT
room temperature
- TGI
tumor growth inhibition
- TO
target occupancy
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c01120.
Synthetic procedures for intermediates previously described in the literature; crystallographic data collection and refinement statistics; electron density maps of ligands with the respective compounds from the crystal structures; raw data from in vivo TO measurements; HPLC traces for acrylamides used in biochemical and in vivo profiling; examples of HSQC-NMR spectra, titration curves, and experimental details on KD determinations; KD values for 20a and 20b measured on GDP·KRASG12V, GDP·KRASG12V,S39C-BIT, GDP·KRASG12D,S39C-BIT, and GppCp·KRASG12D; MS assay results of single assay runs and examples of hyperbolic fit curves for the determination of kinact and KI; GP2D and LS513 proliferation data for BI-0474 and AMG-510 and titration curves for NCI-H358 proliferation assay; single tumor volumes of individual animals from the NCI-H358 in vivo efficacy study (PDF)
Molecular formula strings (CSV)
Accession Codes
The atomic coordinates of the KRAS crystal data have been deposited in the PDB and will be released upon article publication (PDB IDs: 7U8H, 8AFB, 8AFC, 8AFD).
Author Present Address
∥ Abbvie, 381 Plantation Street, Worcester, MA 01605, USA
Author Present Address
⊥ Abbvie, 1 North Waukegan Road, North Chicago, IL 60064, USA.
Author Present Address
# Abbvie, 1 North Waukegan Road, North Chicago, IL 60064, USA.
Author Contributions
J.B. and A.G.W. contributed equally. J.B. supervised the BI medicinal chemistry team, designed compounds and synthetic strategies, and wrote the manuscript. A.G.W. supervised the VU medicinal chemistry team, designed and selected compounds, contributed to synthetic strategies, and wrote the manuscript. C.S. designed compounds and synthetic strategies and contributed to the manuscript. D.K. managed the BI/VU collaboration, performed crystallography, and contributed to the manuscript. J.B. performed crystallography and contributed to the manuscript. M.M. performed analytics, NMR spectroscopy, and contributed to the manuscript. G.G. performed analytics and NMR spectroscopy. J.P. performed crystallography and contributed to the manuscript. A.L. and J.R.A. designed compounds and synthetic strategies. Q.S. performed NMR spectroscopy and crystallography. M.G. supervised the BI biology team, performed in vitro assays, and contributed to the manuscript. D.R. performed in vivo studies and contributed to the manuscript. H.A. performed TO modeling and contributed to the manuscript. K.R. performed kinact/KI measurements and contributed to the manuscript. F.S. performed PD studies and contributed to the manuscript. T.G. performed AlphaScreen measurements and contributed to the manuscript. N.M. performed in vivo TO measurements and contributed to the manuscript. M.T. designed compounds and synthetic strategies and contributed to the manuscript. L.H., T.W., A.G., H.W., and A.M. contributed to the manuscript. O.K. supervised the BI medicinal chemistry team during hit finding and contributed to the finding strategy and compound design. D.B.M. was responsible for the medicinal chemistry strategy and reviewed the manuscript. S.W.F. was responsible for the finding and fragment optimization strategy and reviewed the manuscript.
Funding for the Vanderbilt portions of this work came from US National Institutes of Health (NIH) grants: 5DP1OD006933 (NIH Director’s Pioneer award, S.W.F.), P50A95103-12 (NCI SPORE in GI Cancer; R. J. Coffey), and 5RC2CA148375 to L. J. Marnett as well as The Lustgarten Foundation Award (S.W.F.). The authors would like to thank the Vanderbilt University Biomolecular NMR Facility, with assistance provided by Donald Stec. This facility receives support from an NIH SIG grant (1S-10RR025677-01) and Vanderbilt University matching funds. Finally, the authors would like to thank the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences for use of the Advanced Photon Source (contract: DE-AC02-06CH11357).
The authors declare the following competing financial interest(s): Joachim Brker, Christian Smethurst, Dirk Kessler, Jark Bttcher, Moriz Mayer, Gerhard Gmaschitz, Michael Gmachl, Dorothea Rudolph, Heribert Arnhof, Klaus Rumpel, Fabio Savarese, Thomas Gerstberger, Nikolai Mischerikow, Matthias Treu, Lorenz Herdeis, Tobias Wun-berg, Andreas Gollner, Andreas Mantoulidis, Oliver Krmer, and Darryl B. McConnell were full time employ-ees of Boehringer-Ingelheim at the time this study was performed. Alex G. Waterson, Jason Phan, Andrew Little, Jason R. Abbott, Qi Sun, and Stephen Fesik received additional funding from Boehringer-Ingelheim to support some of this work and may be eligible to receive future royalties as part of this agreement.
Author Status
§ This author is recently deceased.
Supplementary Material
References
- Hofmann M. H.; Gerlach D.; Misale S.; Petronczki M.; Kraut N. Expanding the Reach of Precision Oncology by Drugging All KRAS Mutants. Cancer Discovery 2022, 12, 924–937. 10.1158/2159-8290.cd-21-1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simanshu D. K.; Nissley D. V.; McCormick F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. 10.1016/j.cell.2017.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colicelli J. Human RAS Superfamily Proteins and Related GTPases. Sci. Stke 2004, 2004, RE13. 10.1126/stke.2502004re13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman T. S.; Sondermann H.; Friedland G. D.; Kortemme T.; Bar-Sagi D.; Marqusee S.; Kuriyan J. A Ras-Induced Conformational Switch in the Ras Activator Son of Sevenless. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 16692–16697. 10.1073/pnas.0608127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigil D.; Cherfils J.; Rossman K. L.; Der C. J. Ras Superfamily GEFs and GAPs: Validated and Tractable Targets for Cancer Therapy?. Nat. Rev. Cancer 2010, 10, 842–857. 10.1038/nrc2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore A. R.; Rosenberg S. C.; McCormick F.; Malek S. RAS-Targeted Therapies: Is the Undruggable Drugged?. Nat. Rev. Drug Discovery 2020, 19, 533–552. 10.1038/s41573-020-0068-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boriack-Sjodin P. A.; Margarit S. M.; Bar-Sagi D.; Kuriyan J. The Structural Basis of the Activation of Ras by Sos. Nature 1998, 394, 337–343. 10.1038/28548. [DOI] [PubMed] [Google Scholar]
- Prior I. A.; Hood F. E.; Hartley J. L. The Frequency of Ras Mutations in Cancer. Cancer Res. 2020, 80, 2969–2974. 10.1158/0008-5472.can-19-3682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lito P.; Solomon M.; Li L.-S.; Hansen R.; Rosen N. Allele-Specific Inhibitors Inactivate Mutant KRAS G12C by a Trapping Mechanism. Science 2016, 351, 604–608. 10.1126/science.aad6204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q.; Phan J.; Friberg A. R.; Camper D. V.; Olejniczak E. T.; Fesik S. W. A Method for the Second-Site Screening of K-Ras in the Presence of a Covalently Attached First-Site Ligand. J. Biomol. NMR 2014, 60, 11–14. 10.1007/s10858-014-9849-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer T.; Garrenton L. S.; Oh A.; Pitts K.; Anderson D. J.; Skelton N. J.; Fauber B. P.; Pan B.; Malek S.; Stokoe D.; Ludlam M. J. C.; Bowman K. K.; Wu J.; Giannetti A. M.; Starovasnik M. A.; Mellman I.; Jackson P. K.; Rudolph J.; Wang W.; Fang G. Small-Molecule Ligands Bind to a Distinct Pocket in Ras and Inhibit SOS-Mediated Nucleotide Exchange Activity. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 5299–5304. 10.1073/pnas.1116510109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q.; Burke J. P.; Phan J.; Burns M. C.; Olejniczak E. T.; Waterson A. G.; Lee T.; Rossanese O. W.; Fesik S. W. Discovery of Small Molecules That Bind to K-Ras and Inhibit Sos-Mediated Activation. Angew. Chem., Int. Ed. 2012, 51, 6140–6143. 10.1002/anie.201201358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler D.; Bergner A.; Böttcher J.; Fischer G.; Döbel S.; Hinkel M.; Müllauer B.; Weiss-Puxbaum A.; McConnell D. B. Drugging All RAS Isoforms with One Pocket. Future Med. Chem. 2020, 12, 1911–1923. 10.4155/fmc-2020-0221. [DOI] [PubMed] [Google Scholar]
- Kessler D.; Gmachl M.; Mantoulidis A.; Martin L. J.; Zoephel A.; Mayer M.; Gollner A.; Covini D.; Fischer S.; Gerstberger T.; Gmaschitz T.; Goodwin C.; Greb P.; Häring D.; Hela W.; Hoffmann J.; Karolyi-Oezguer J.; Knesl P.; Kornigg S.; Koegl M.; Kousek R.; Lamarre L.; Moser F.; Munico-Martinez S.; Peinsipp C.; Phan J.; Rinnenthal J.; Sai J.; Salamon C.; Scherbantin Y.; Schipany K.; Schnitzer R.; Schrenk A.; Sharps B.; Siszler G.; Sun Q.; Waterson A.; Wolkerstorfer B.; Zeeb M.; Pearson M.; Fesik S. W.; McConnell D. B. Drugging an Undruggable Pocket on KRAS. Proc. Natl. Acad. Sci. U.S.A. 2019, 116, 15823–15829. 10.1073/pnas.1904529116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrem J. M.; Peters U.; Sos M. L.; Wells J. A.; Shokat K. M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan A. K.; Piazza G. A.; Keeton A. B.; Leite C. A. The Path to the Clinic: A Comprehensive Review on Direct KRASG12C Inhibitors. J. Exp. Clin. Cancer Res. 2022, 41, 27. 10.1186/s13046-021-02225-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fell J. B.; Fischer J. P.; Baer B. R.; Blake J. F.; Bouhana K.; Briere D. M.; Brown K. D.; Burgess L. E.; Burns A. C.; Burkard M. R.; Chiang H.; Chicarelli M. J.; Cook A. W.; Gaudino J. J.; Hallin J.; Hanson L.; Hartley D. P.; Hicken E. J.; Hingorani G. P.; Hinklin R. J.; Mejia M. J.; Olson P.; Otten J. N.; Rhodes S. P.; Rodriguez M. E.; Savechenkov P.; Smith D. J.; Sudhakar N.; Sullivan F. X.; Tang T. P.; Vigers G. P.; Wollenberg L.; Christensen J. G.; Marx M. A. Identification of the Clinical Development Candidate MRTX849, a Covalent KRASG12C Inhibitor for the Treatment of Cancer. J. Med. Chem. 2020, 63, 6679–6693. 10.1021/acs.jmedchem.9b02052. [DOI] [PubMed] [Google Scholar]
- Lanman B. A.; Allen J. R.; Allen J. G.; Amegadzie A. K.; Ashton K. S.; Booker S. K.; Chen J. J.; Chen N.; Frohn M. J.; Goodman G.; Kopecky D. J.; Liu L.; Lopez P.; Low J. D.; Ma V.; Minatti A. E.; Nguyen T. T.; Nishimura N.; Pickrell A. J.; Reed A. B.; Shin Y.; Siegmund A. C.; Tamayo N. A.; Tegley C. M.; Walton M. C.; Wang H.-L.; Wurz R. P.; Xue M.; Yang K. C.; Achanta P.; Bartberger M. D.; Canon J.; Hollis L. S.; McCarter J. D.; Mohr C.; Rex K.; Saiki A. Y.; San Miguel T. S.; Volak L. P.; Wang K. H.; Whittington D. A.; Zech S. G.; Lipford J. R.; Cee V. J. Discovery of a Covalent Inhibitor of KRASG12C (AMG 510) for the Treatment of Solid Tumors. J. Med. Chem. 2020, 63, 52–65. 10.1021/acs.jmedchem.9b01180. [DOI] [PubMed] [Google Scholar]
- Williamson M. P. Using Chemical Shift Perturbation to Characterise Ligand Binding. Prog. Nucl. Magn. Reson. Spectrosc. 2013, 73, 1–16. 10.1016/j.pnmrs.2013.02.001. [DOI] [PubMed] [Google Scholar]
- Davies C. W.; Oh A. J.; Mroue R.; Steffek M.; Bruning J. M.; Xiao Y.; Feng S.; Jayakar S.; Chan E.; Arumugam V.; Uribe S. C.; Drummond J.; Frommlet A.; Lu C.; Franke Y.; Merchant M.; Koeppen H.; Quinn J. G.; Malhotra S.; Do S.; Gazzard L.; Purkey H. E.; Rudolph J.; Mulvihill M. M.; Koerber J. T.; Wang W.; Evangelista M. Conformation-Locking Antibodies for the Discovery and Characterization of KRAS Inhibitors. Nat. Biotechnol. 2022, 40, 769–778. 10.1038/s41587-021-01126-9. [DOI] [PubMed] [Google Scholar]
- Patricelli M. P.; Janes M. R.; Li L.-S.; Hansen R.; Peters U.; Kessler L. V.; Chen Y.; Kucharski J. M.; Feng J.; Ely T.; Chen J. H.; Firdaus S. J.; Babbar A.; Ren P.; Liu Y. Selective Inhibition of Oncogenic KRAS Output with Small Molecules Targeting the Inactive State. Cancer Discovery 2016, 6, 316–329. 10.1158/2159-8290.cd-15-1105. [DOI] [PubMed] [Google Scholar]
- Hofmann M. H.; Gmachl M.; Ramharter J.; Savarese F.; Gerlach D.; Marszalek J. R.; Sanderson M. P.; Kessler D.; Trapani F.; Arnhof H.; Rumpel K.; Botesteanu D.-A.; Ettmayer P.; Gerstberger T.; Kofink C.; Wunberg T.; Zoephel A.; Fu S.-C.; Teh J. L.; Böttcher J.; Pototschnig N.; Schachinger F.; Schipany K.; Lieb S.; Vellano C. P.; O’Connell J. C.; Mendes R. L.; Moll J.; Petronczki M.; Heffernan T. P.; Pearson M.; McConnell D. B.; Kraut N. BI-3406, a Potent and Selective SOS1-KRAS Interaction Inhibitor, Is Effective in KRAS-Driven Cancers through Combined MEK Inhibition. Cancer Discovery 2021, 11, 142–157. 10.1158/2159-8290.cd-20-0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K. S.; Quinn J. G.; Saabye M. J.; Guerrero J. F. S.; Nonomiya J.; Lian Q.; Phung W.; Izrayelit Y.; Walters B. T.; Gustafson A.; Endres N. F.; Beresini M. H.; Mulvihill M. M. High-Throughput Kinetic Characterization of Irreversible Covalent Inhibitors of KRASG12C by Intact Protein MS and Targeted MRM. Anal. Chem. 2022, 94, 1230–1239. 10.1021/acs.analchem.1c04463. [DOI] [PubMed] [Google Scholar]
- Canon J.; Rex K.; Saiki A. Y.; Mohr C.; Cooke K.; Bagal D.; Gaida K.; Holt T.; Knutson C. G.; Koppada N.; Lanman B. A.; Werner J.; Rapaport A. S.; San Miguel T. S.; Ortiz R.; Osgood T.; Sun J.-R.; Zhu X.; McCarter J. D.; Volak L. P.; Houk B. E.; Fakih M. G.; O’Neil B. H.; Price T. J.; Falchook G. S.; Desai J.; Kuo J.; Govindan R.; Hong D. S.; Ouyang W.; Henary H.; Arvedson T.; Cee V. J.; Lipford J. R. The Clinical KRAS(G12C) Inhibitor AMG 510 Drives Anti-Tumour Immunity. Nature 2019, 575, 217–223. 10.1038/s41586-019-1694-1. [DOI] [PubMed] [Google Scholar]
- Lonsdale R.; Burgess J.; Colclough N.; Davies N. L.; Lenz E. M.; Orton A. L.; Ward R. A. Expanding the Armory: Predicting and Tuning Covalent Warhead Reactivity. J. Chem. Inf. Model. 2017, 57, 3124–3137. 10.1021/acs.jcim.7b00553. [DOI] [PubMed] [Google Scholar]
- Dahal U. P.; Gilbert A. M.; Obach R. S.; Flanagan M. E.; Chen J. M.; Garcia-Irizarry C.; Starr J. T.; Schuff B.; Uccello D. P.; Young J. A. Intrinsic reactivity profile of electrophilic moieties to guide covalent drug design: N-α-acetyl-l-lysine as an amine nucleophile. Medchemcomm 2016, 7, 864–872. 10.1039/c6md00017g. [DOI] [Google Scholar]
- Wang X.; Allen S.; Blake J. F.; Bowcut V.; Briere D. M.; Calinisan A.; Dahlke J. R.; Fell J. B.; Fischer J. P.; Gunn R. J.; Hallin J.; Laguer J.; Lawson J. D.; Medwid J.; Newhouse B.; Nguyen P.; O’Leary J. M.; Olson P.; Pajk S.; Rahbaek L.; Rodriguez M.; Smith C. R.; Tang T. P.; Thomas N. C.; Vanderpool D.; Vigers G. P.; Christensen J. G.; Marx M. A. Identification of MRTX1133, a Noncovalent, Potent, and Selective KRASG12D Inhibitor. J. Med. Chem. 2022, 65, 3123–3133. 10.1021/acs.jmedchem.1c01688. [DOI] [PubMed] [Google Scholar]
- Naumann B.; Böhm R.; Fülöp F.; Bernath G. Preparation of New Trifunctional Thiophene Derivatives. Pharmazie 1996, 51, 4–6. [Google Scholar]
- Reßing N.; Marquardt V.; Gertzen C. G. W.; Schöler A.; Schramm A.; Kurz T.; Gohlke H.; Aigner A.; Remke M.; Hansen F. K. Design, synthesis and biological evaluation of β-peptoid-capped HDAC inhibitors with anti-neuroblastoma and anti-glioblastoma activity. Medchemcomm 2018, 10, 1109–1115. 10.1039/c8md00454d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallin J.; Engstrom L. D.; Hargis L.; Calinisan A.; Aranda R.; Briere D. M.; Sudhakar N.; Bowcut V.; Baer B. R.; Ballard J. A.; Burkard M. R.; Fell J. B.; Fischer J. P.; Vigers G. P.; Xue Y.; Gatto S.; Fernandez-Banet J.; Pavlicek A.; Velastagui K.; Chao R. C.; Barton J.; Pierobon M.; Baldelli E.; Patricoin E. F.; Cassidy D. P.; Marx M. A.; Rybkin I. I.; Johnson M. L.; Ou S.-H. I.; Lito P.; Papadopoulos K. P.; Jänne P. A.; Olson P.; Christensen J. G. The KRASG12C Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discovery 2020, 10, 54–71. 10.1158/2159-8290.cd-19-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savarese F.; Gollner A.; Rudolph D.; Lipp J.; Popow J.; Hofmann M. H.; Arnhof H.; Rinnenthal J.; Trapani F.; Gmachl M.; Gerlach D.; Broeker J.; Ettmayer P.; Mantoulidis A.; Phan J.; Smethurst C. A.; Treu M.; Waterson A. G.; Lu H.; Machado A.; Daniele J.; Fesik S. W.; Vellano C. P.; Heffernan T. P.; Marszalek J. R.; McConnell D. B.; Petronczki M.; Kraut N.; Waizenegger I. C. Abstract 1271: In Vitro and in Vivo Characterization of BI 1823911 - a Novel KRASG12C Selective Small Molecule Inhibitor. Cancer Res. 2021, 81, 1271. 10.1158/1538-7445.am2021-1271. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.