

Abstract
Cord blood (CB)-derived natural killer (NK) cells that are genetically engineered to express a chimeric antigen receptor (CAR) are an attractive off-the-shelf therapy for the treatment of cancer, demonstrating a robust safety profile in vivo. For poor prognosis brain tumors such as glioblastoma multiforme (GBM), novel therapies are urgently needed. Although CAR-T cells demonstrate efficacy in preclinical GBM models, an off-the-shelf product may exhibit unwanted side effects like graft-versus-host disease. Hence, we developed an off-the-shelf CAR-NK cell approach using a B7H3 CAR and showed that CAR-transduced NK cells have robust cytolytic activity against GBM cells in vitro. However, transforming growth factor (TGF)-β within the tumor microenvironment has devastating effects on the cytolytic activity of both unmodified and CAR-transduced NK cells. To overcome this potent immune suppression, we demonstrated that co-transducing NK cells with a B7H3 CAR and a TGF-β dominant negative receptor (DNR) preserves cytolytic function in the presence of exogenous TGF-β. This study demonstrates that a novel DNR and CAR co-expression strategy may be a promising therapeutic for recalcitrant CNS tumors like GBM.
Keywords: cord blood, NK cells, CAR-NK cells, B7H3 CAR, GBM, TGF-β
Graphical abstract
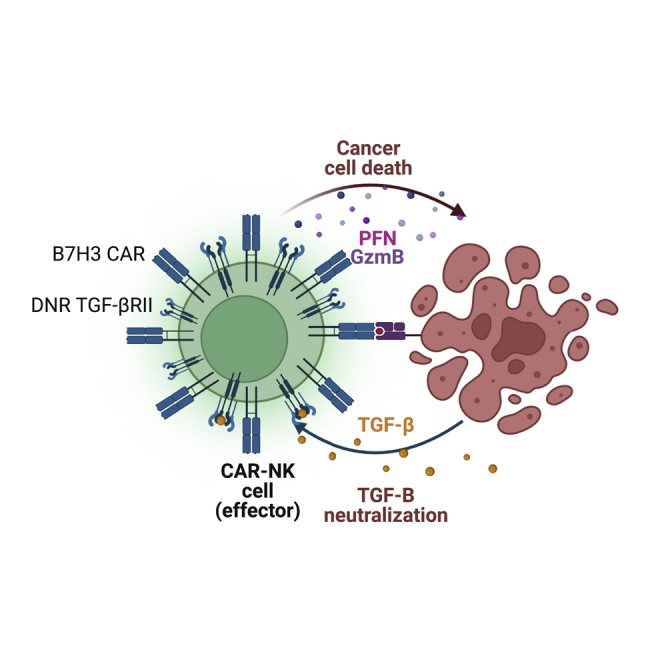
B7H3 CAR-NK cells show potent antitumor activity against GBM tumor cells in vitro. However, TGF-β secreted by tumor cells negatively affects CAR-NK cell cytolytic function. We can overcome this limitation by co-expressing B7H3 CAR-NK cells with a dominant negative TGF-β receptor.
Introduction
Glioblastoma multiforme (GBM) is one of the most lethal primary brain cancers, with an overall survival rate of approximately 15 months after standard treatment including surgery, radiotherapy, and chemotherapy.1,2 Hence, there is an urgent need to develop novel therapies. While immune-based therapies have been successful for the treatment of some hematologic malignancies, their use against brain tumors have so far been relatively limited. Tumor antigen heterogeneity,3 limited numbers of infiltrating lymphocytes at the tumor site, and the failure of checkpoint inhibitors to cross the blood-brain barrier4 are major obstacles to effective immunotherapies for the treatment of GBM.
It is postulated that altered immunity in patients with GBM (either because of intrinsic disease-related factors or because of the immune suppressive nature of the treatment, which includes high-dose steroids)5,6 contributes to the poor prognosis of these tumors and their relative resistance to vaccine and checkpoint inhibitors. Adoptive transfer of healthy donor immune cells as an off-the-shelf adoptive cell therapy may therefore offer an attractive alternative. Natural killer (NK) cells, one of the arms of the innate immune system, can elicit anti-tumor activity without the need for prior sensitization. Allogeneic NK cells can induce target cell killing in a major histocompatibility complex (MHC)-independent manner; and recognize recipient tumors as non-self, while sparing healthy tissues because of the absence of activating receptor ligand expression. Unlike T cells, NK cells do not usually induce graft-versus-host disease (GVHD), even in an allogeneic “off-the-shelf” setting, and KIR mismatching with HLA ligands on cancer cells can elicit a graft-versus-tumor response.7,8 Further, several clinal trials have highlighted the robust safety profile of allogeneic NK cells.9,10,11
In this study, we have used cord blood (CB) as the donor source for allogeneic NK cell product generation. CB offers a unique advantage of immediate availability from established CB banks and widespread scalability as an off-the-shelf product.12,13 Peripheral blood,14 human embryonic stem cells, induced pluripotent stem cells, and artificial NK cell lines are also available alternative donor sources for NK cell product manufacture.15,16
There is evidence that GBM can downregulate NK cell-activating receptor ligands such as ULBP2 by shedding, a common strategy used by human cancers.17 To overcome tumor immune evasion, NK cells can be engineered to recognize additional tumor targets (e.g., B7H3). B7H3 is a member of the B7-family with two isoforms in humans (2Ig-B7H3 and 4Ig-B7H3). It is highly expressed in more than 70% of GBM biopsy samples18,19 and undetectable in the normal brain, making it an ideal target for immune therapies.20,21,22
The GBM tumor microenvironment is highly immune suppressive.23 Tumors cells and tumor-associated macrophages are known to secrete transforming growth factor-beta (TGF-β)24,25,26 and TGF-β has documented immune dampening sequelae on NK cell effector functions.12,27,28,29,30,31 However, the effect of TGF-β on chimeric antigen receptor (CAR)-modified NK cells has not been previously investigated. Therefore, we posited that it was important to study the impact of TGF-β on the cytolytic function of CAR-NK cells in vitro.
In this report, we demonstrated that otherwise potent B7H3 CAR NK cells have decreased effector function against GBM in the presence of exogenous TGF-β, thereby providing a compelling rationale to nullify TGF-β signaling in B7H3 CAR-NK cells using a dominant negative TGF-β receptor (DNR). We, therefore, determined whether we could co-express the DNR and the B7H3 CAR on CB-derived NK cells to simultaneously enhance the specificity of the NK cell product and overcome the immune suppressive effects of TGF-β. In this report, we demonstrate that, by co-expressing the DNR on B7H3 CAR-NK cells, their cytolytic activity was maintained against glioma tumor cells, even after exposure to TGF-β. Our study provides compelling evidence that this novel DNR/CAR-engineered NK cell therapy holds the potential to treat patients with glioblastomas and other TGF-β-rich pediatric and adult solid tumors.
Results
B7H3 is homogenously expressed on GBM cell lines
GBM cell lines U87 MG and A172 and primary GBM cell lines derived from pediatric patients (FHTC GBM 511 and GBM 110) used in this study were stained for the expression of B7H3. All cell lines stained positive for B7H3 (>97% positivity within each cancer cell line). Representative flow data are shown in Figure 1A. In addition to evaluating B7H3 expression, we also evaluated the expression of the stress ligands MICA/MICB (Figure 1B) and PVR (ligand for DNAM1, Figure 1C) on these cancer cells. Both adult GBM cell lines (U87 MG and A172) showed expression of MICA/MICB (>70%) and PVR (>80%). Expression of these stress ligands by cancer cells offers an advantage for NK cell therapies, since NK cells innately express receptors that recognize these ligands.
Figure 1.

GBM tumor cells express NK cell receptors ligands
(A) Flow expression of B7H3 expression in adult and primary pediatric GBM cell lines. (B and C) NK cells receptors ligand present in GBM cells lines. (B) MICA/MICB (ligand for NKG2D) flow expression in GBM cell lines. (C) PVR (ligand for DNAM1) flow expression in GBM cell lines. Unstained cells (Tumor cells) were used as a negative control. Tumor cells + AB are stained with respective antibodies.
CB NK cells are stably transduced with a retroviral vector expressing a B7H3/CD28z CAR with preservation of activation marker and chemokine receptor expression
NK cells isolated from CB units were stimulated with K562-expressing membrane-bound IL-15 (mbIL-15) and 4-1BB ligand (clone 4),31 supplemented with IL-2 and IL-21 for 14 days (untransduced [UT] control) or transduced on day 4 with a retroviral vector to express a B7H3/CD28z CAR20,21 and cultured for an additional 10 days (see materials and methods) (Figure 2A). The mean CAR-NK transduction efficiency on day 12 of culture was 93.51% (range, 72.50%–98.00%; n = 9) (Figures 2B and 2C). Figure 2D summarizes the fold-expansion data including the numbers of B7H3 CAR transduced NK cells expanded from nine different CB units. At 6 days after transduction (10 days of culture), the mean NK cell expansion was 203-fold (range, 89–500).
Figure 2.

CB NK cells are genetically modified to express the B7H3 CAR and maintain their expression of activating receptors
(A) Generation of CAR NK cells. (B and C) CB NK cells were transduced with a retroviral vector encoding for the B7H3 CAR. CD56+ NK cells (blue circular cells) selected from CB were rested overnight in recombinant human IL-15 (15 ng/mL) in Cell Genix Stem Cell Growth Medium (with 10% FBS). The next day (day 0), NK cells were stimulated with irradiated (100 Gy) clone 4 K562 cells (2:1 feeder cell:NK ratio) and IL-2 (200 IU/mL) and IL-21 (25 ng/mL). Activated NK cells were transduced with retroviral supernatants (red virus particles) on day +4 in retronectin-coated plates. Three days later (day +7), NK cells were stimulated again with irradiated clone 4 K562 cells. On day +12, CAR-transduced NK cells (blue cells with spikes) were collected for use. Media change with IL-2 (200 IU/mL) and IL-21 (25 ng/mL) was done every 2–3 days throughout the NK cells culture duration. Illustration created using Biorender.com. (B) Representative flow analysis of B7H3 CAR expression on CB NK cells. (C) Transduction efficiency of CB NK cells (n = 9). (D) Mean fold expansion of B7H3 CAR-NK cell products (n = 9). (E) The NK cell phenotypes after 10 days of ex vivo expansion of UT CB NK (black bar) cells versus B7H3 CAR (Red bar) expressing CB NK cells are shown (n = 8). Bar shows mean value. Individual point represents individual donor and error bars represent standard deviation.
Expanded B7H3 CAR-transduced NK cells and UT NK cells also showed similar activation and exhaustion marker profiles (Figure 2E) as evidenced by staining for natural activating receptors (NKG2D [B7H3 CAR-NK: 64.83% ± 21.63%; UT NK 65.99% ± 27.24%; p > 0.99], NKp46 [B7H3 CAR-NK: 94.06 ± 5.46%; UT NK 95.74% ± 3.6%; p > 0.99], and NKp30 [B7H3 CAR-NK: 89.32% ± 7.7%; UT NK 90.12% ± 3.81%; p > 0.99], and CD16 [B7H3 CAR-NK: 73.92% ± 10.32%; UT NK 67.45% ± 12.38%; p = 0.98]). As shown in Figure 2E, no significant difference in the expression of these receptors between B7H3 CAR-NK and UT NK was observed (p = 0.69). Similarly, there was no difference in the expression of other NK cell activation markers such as CD69 (B7H3 CAR-NK: 72.15% ± 23.79%; UT NK 68.91% ± 21.92%; p > 0.99) and CD25 (B7H3 CAR-NK: 85.64% ± 11.85%; UT NK 88.76% ± 14.38%; p > 0.99). We also verified that B7H3 CAR-NK cells showed no significant difference in expression of the exhaustion markers TIM3 (B7H3 CAR-NK: 96.60% ± 3.40%; UT NK 97.50% ± 2.78%; p > 0.99) and PD1 compared with UT NK cells (B7H3 CAR-NK: 23.91% ± 13.06% vs. UT NK 13.48% ± 8.74%; p = 0.71) (Figure 2E).
Finally, CB-derived NK cells expressed a wide range of chemokine receptors, indicating that they can potentially migrate to various tissues, further supporting their use as an off-the-shelf product for multiple cancers (Table 1). The major chemokine receptor-ligand pairs documented to modulate the trafficking of NK cells include CCR1-CCL3/CCL5, CCR5-CCL3/CCL5, CCR7-CCL19/CCL21, CXCR3-CXCL9/CXCL10, and CXCR4-CXCL12 (also known as stromal derived factor-1 alpha).32,33,34,35 We observed that CB-derived NK cells showed a diverse chemokine receptor repertoire and transduction with the B7H3 CAR did not alter chemokine receptor expression (n = 6) (Table 1). Specifically, CXCR3 (B7H3 CAR-NK, 99.32% ± 0.240%; UT NK, 98.87% ± 0.388%; p = 0.03), CXCR4 (B7H3 CAR-NK, 99.45% ± 0.48%; UT NK, 96.95% ± 3.10%; p = 0.07) and CCR10 (B7H3 CAR-NK, 99.10% ± 1.58%; UT NK, 97.47% ± 3.48%; p = 0.32) were expressed on transduced and non-transduced CB NK cells. In contrast, other chemokine receptors including CCR3, CCR4, CCR5, CCR7, CCR8, CCRL1, CXCR5, and CXCR7 had moderate donor-specific variation (Table 1). Finally, other chemokine receptors including CXCR1, CXCR2, CXCR6, CCR1, CCR2, and CCR9 had minimal expression on transduced and non-transduced CB-derived NK cells (Table 1).
Table 1.
CB NK cells genetically modified to express the B7H3 CAR maintain chemokine receptor expression
| B7H3 CAR-NK |
UT-NK |
|||||
|---|---|---|---|---|---|---|
| Mean | Median | SD | Mean | Median | SD | |
| CD181 (CXCR1) | 22.47 | 10.19 | 24.11 | 18.91 | 4.7 | 24.56 |
| CD182 (CXCR2) | 7.30 | 5.705 | 3.89 | 4.73 | 4.8 | 2.23 |
| CD183 (CXCR3) | 99.32 | 99.25 | 0.24 | 98.87 | 98.9 | 0.39 |
| CD184 (CXCR4) | 99.45 | 99.65 | 0.48 | 96.95 | 98.5 | 3.09 |
| CD186(CXCR6) | 20.52 | 18.15 | 8.41 | 23.73 | 20.7 | 12.00 |
| CD191 (CCR1) | 15.24 | 10.33 | 12.88 | 12.29 | 7.6 | 11.57 |
| CD192 (CCR2) | 12.93 | 8.68 | 9.08 | 9.65 | 8.6 | 3.81 |
| CD193(CCR3) | 76.53 | 84.3 | 19.65 | 52.73 | 42.9 | 29.12 |
| CD194 (CCR4) | 79.15 | 93.35 | 24.23 | 74.03 | 83.4 | 25.86 |
| CD195 (CCR5) | 86.63 | 91.8 | 11.24 | 80.57 | 84.3 | 17.99 |
| CD196 (CCR6) | 34.05 | 18.45 | 38.29 | 27.77 | 10.3 | 34.29 |
| CD197 (CCR7) | 83.32 | 97.95 | 24.26 | 75.57 | 95.9 | 32.56 |
| CD198 (CCR8) | 62.98 | 63.95 | 27.12 | 59.00 | 46.9 | 26.70 |
| CD199 (CCR9) | 23.75 | 26 | 10.19 | 13.56 | 12.6 | 5.17 |
| CCR10 | 99.10 | 99.75 | 1.58 | 97.47 | 99.6 | 3.48 |
| CCX-CCR (CCRL1) | 72.62 | 63.8 | 17.13 | 64.63 | 53.4 | 24.08 |
| CXCR5 | 79.87 | 89.65 | 17.50 | 62.20 | 72.6 | 25.54 |
| CXCR7 | 62.73 | 64.1 | 27.65 | 59.98 | 50.7 | 24.99 |
| XCR1 | 19.83 | 17.1 | 17.36 | 19.66 | 24.6 | 14.08 |
Data shown as percentage (%) mean, median, and standard deviation (S.D) of receptors expression (n = 6).
B7H3 CAR-transduced CB NK cells have enhanced cytolytic activity against B7H3+ cancer cells in vitro
We next tested whether B7H3 CAR-expressing NK cells showed enhanced cytotoxicity against B7H3-expressing targets compared with UT NK cells (Figures 3 and S1A–S1C). Expanded B7H3 CAR-NK cells and UT NK cells were co-cultured with glioblastoma cell lines U87 MG (Figure 3A) and A172 (Figures 3B, S1A, and S1B) (n = 7), or primary pediatric cell lines GBM-110 (Figure 3C) and GBM-511 (Figures 3D and S1C) (n = 4), at different effector to target ratios (E:T) and their target lysis was quantified using Celigo Imaging Cytometer. Across all E:T ratios, CAR-expressing CB NK cells showed increased killing of B7H3+ target cells compared with UT NK cells for both adult and pediatric GBM cell lines (Figures 3A and 3D). We observed that while there was donor-specific variation in the baseline killing efficiency of UT NK cells (Figure S1), there was a significant increase in the cytolytic activity of B7H3 CAR-NK cells against the same target cells (p < 0.05). Pediatric GBM cell lines seem to be more sensitive to killing by B7H3 CAR-NK cells in comparison with adult GBM lines (Figure S1).
Figure 3.

B7H3 CAR-NK cells show potent cytolytic activity against multiple GBM cell lines in vitro
(A–D) Cytotoxic activity summary of B7H3 CAR-NK (red) versus UT-NK cells (black), as measured by a 4-h cytotoxicity assay, against adult GBM cell lines (A) U87 MG (n = 7), and A172 cells (n = 7) and primary pediatric GBM lines (C) GBM-110 (n = 4) and (D) GBM-511 (n = 4). Representative data from the same donor are shown. (E) B7H3 CAR-NK and UT-NK showed enhanced IFN-y secretion in response to the U87 MG cell line. The red bar represents NK cells cocultured with U87 MG cells and the black bar represents NK cell cultured alone conditions (n = 3). Individual points represent the mean of replicates, and the error bars represent standard deviation. (∗p < 0.05, all comparisons). The p values were generated by the Holm-Sidak multiple comparison test after one-way analysis of variance. For more donors, see Figure S1.
CB NK cells also showed innate cytotoxicity, independent of CAR expression, as demonstrated by the modest killing of targets by UT-NK cells (Figure S1) via recognition of stress ligands expressed by cancer cells. We also measured cytokine release by B7H3 CAR-NK cells in the coculture supernatant harvested 24 h after cancer cell exposure. Both B7H3 CAR-transduced (282.28 pg/mL ±95.02; p = 0.03) and UT-NK cells (162.22 ± 45.91 pg/mL; p = 0.04) showed significantly increased interferon (IFN)-γ secretion in response to U87 MG cells compared with non-stimulated control conditions/NK cells alone conditions (B7H3 CAR-NK, 43.90 ± 18.73 pg/mL; UT NK, 17.04 ± 5.74 pg/mL) (Figure 3E).
In addition, a 72-h cytotoxicity assay revealed that B7H3 CAR-NK cells generated a significantly higher target lysis at the low E:T ratio (1:1 and 1:5) in comparison with UT-NK cells suggesting the capability of this product to elicit multiple/serial killing (p < 0.05) (Figure S2). Potent killing efficiency at the low E:T ratio also demonstrated the potency of CAR NK cell products in high cancer cell load conditions.
B7H3 CAR-NK cells are sensitive to TGF-β-induced immune suppression by GBM cell lines
To test whether TGF-β is secreted by the GBM tumor microenvironment (TME), cytokine levels were measured in supernatants harvested from U87-MG cells cultured at different levels of confluencies. We demonstrated that U87 MG cells do secrete different amounts of TGF-β1, depending on cell confluence (Table 2). Despite showing the anti-tumor activity of B7H3 CAR-NK cells against multiple GBM cell lines, we wanted to evaluate whether this potent anti-tumor response would be impaired in a TGF-β-rich immunosuppressive setting. As shown in Figure 4A, the cytolytic activity of B7H3 CAR-NK cells was significantly decreased after exposure to exogenous TGF-β (p < 0.0001). Specifically, B7H3 CAR-NK cell-mediated lysis of U87 MG target cells decreased from 89.73 ± 2.44% to 61.75 ± 3.42% in the presence of TGF-β at an E:T of 20:1 (p < 0.001) (Figure 4B) To elucidate the mechanism that could account for this shift in CAR NK cell effector function,25 we showed a decrease in the expression of NK cell activating receptors including CD16 and NKG2D in the presence of exogenous TGF-β, as shown in Figures 4C–4F.12,30,36 Specifically, the median fluorescence intensity (MFI) of NKG2D decreased from 16,422.00 ± 6,104.50 to 5,564.00 ± 1,712.16 for B7H3 CAR-NK cells after exposure to TGF-β (n = 4) (Figures 4C and 4E). Similarly, in the presence of TGF-β, CD16 receptor MFI decreased from 32,820.00 ± 3,587.57 to 12,535.00 ± 3,669.77 for B7H3 CAR-NK cells (n = 4) (Figures 4D and 4F). In contrast, there was no downregulation of CAR expression on NK cells after exposure to exogenous TGF-β, suggesting that expression of the genetically engineered CAR is resistant to TGF-β-mediated downregulation (Figure S3).
Table 2.
TGF- β1 level in U87 cell MG cells at different levels of confluency
| Confluency | Mean TGF-β 1 conc (pg/mL) | SD |
|---|---|---|
| 90%–100% | 316.7528 | 13.86 |
| 70%–80% | 427.7014 | 82.10 |
| 60% | 759.0877 | 137.52 |
| 50% | 243.4502 | 82.64 |
| Serum-free media | 0 |
Data shown as mean and standard deviation (S.D) of receptors expression (n = 2).
Figure 4.

TGF-β-mediated immune suppression in CAR-NK cells
Exogenous TGF-β decreases the cytolytic activity of B7H3 CAR-NK cells by downregulating NK cells' endogenous receptors. (A) Percentage of U87 MG lysis by TGF-β-treated (dashed lines) and untreated or control (solid lines) by UT-NK (black line) and B7H3 CAR-NK cells (red line). Representative experiment example out of n = 3 lines generated. (B) The mean specific U87 MG cells lysis at 20:1 E:T ratio. Red bar represents the NK cell’s target lysis in presence of TGF-β and the black bar represents the NK cell’s lysis in absence of TGF-β. The individual point represents the individual donor and error bars represent the standard deviation. (C and D) Representative surface expression NKG2D (C) and of CD16 (D) on UT-NK and B7H3 CAR-NK cells after culture in the presence (red histogram) or absence (blue histogram) of TGF-β (5 ng/mL). (E and F) Mean MFI of CD16 (F), NKG2D (E) for UT-NK and B7H3 CAR-NK cells after culture in the presence or absence of TGF-β (5 ng/mL) (n = 4). Red bar represents the NK cell’s mean MFI in presence of TGF-β and the Black bar represents the NK cell’s mean MFI in absence of TGF-β. The individual point represents the individual donor and error bars represent the standard deviation. (∗p < 0.05, all comparisons). (B) The p values were generated by the Holm-Sidak multiple comparison test after two-way analysis of variance. (E and F) The p values were generated by the Mann-Whitney test.
DNR abrogates TGF-β signaling in NK cells
We next hypothesized that blocking TGF-β signaling in B7H3 CAR-NK would rescue their effector function in the presence of TGF-β. Hence, we co-transduced B7H3 CAR-NK cells with a dominant-negative TGF-β receptor.12,37 We have previously shown that gene engineering T cells and NK cells with a dominant-negative TGF-β receptor effectively abrogates TGF-β signaling, with a consequent lack of receptor SMAD (SMAD2 and SMAD3) proteins phosphorylation required for the production of immune-suppressive genes (Figure 5A).12,27,30,38
Figure 5.

DNR abrogates TGF-β signaling in NK cells
(A) Schematic depicting the effects of TGF-β binding to the receptor complex: UT NK cells express the wild-type TGF-βRII, which, when engaged with soluble TGF-β in the TME, initiates a signaling cascade that culminates in impaired NK cell phenotype and cytotoxicity. NK cells transduced with the DNR variant TGF-β receptors alter the intracellular signaling and allow for maintained or enhanced NK cell phenotype and cytotoxicity in the setting of tumor-associated TGF-β. Illustration created using Biorender.com. (B) CB NK cells can be genetically modified to co-express the B7H3 CAR and DNRII. (C) Representative flow analysis for B7H3 CAR and TGF-β RII expression on NK cells. (Right) Results for the transduction efficiency of CB-NK cells (n = 6). (D and E) Inhibition of SMAD2/3 phosphorylation by TGF-β in CB-NK cells expressing DNRII. (D) Representative flow analysis showing the pSMAD2/3 expression in UT-NK and transduced NK cells after exposure to exogenous TGF-β (red histogram) in comparison to non-treated NK cells (blue histogram). (E) Mean upregulation (%) of the pSMAD2/3 in NK cell products (n = 3) after culture in the presence (red bar) or absence (black bar) of TGF-β (2 ng/mL). The individual point represents the individual donor and error bars represent standard deviation. (∗p < 0.05, all comparisons.) The p values were generated by the Holm-Sidak multiple comparison test following two-way analysis of variance.
As shown in Figures 5B and 5C, we were able to successfully transduce NK cells to express both the B7H3 CAR and the DNR with a mean transduction efficiency of 72.21 ± 23.05% (n = 6) gated on double-positive cells. To investigate whether co-transducing B7H3 CAR-NK cell with the DNR abrogated TGF-β-mediated signaling, we cocultured UT, B7H3 CAR-transduced, DNR-transduced, and DNR.B7H3 CAR co-transduced NK cells with exogenous TGF-β (2 ng/mL). Cells were harvested 30 mins after TGF-β exposure and phospho flow analysis was performed to characterize and quantify intracellular protein expression. As shown in Figures 5D and 5E, phospho flow analysis demonstrated rapid phosphorylation of SMAD2/3 when UT NK cells (UT + TGF-β 94.83% ± 4.03% vs. UT 1.96% ± 0.44%; p < 0.0001; n = 4) and B7H3 CAR-NK cells (B7H3+TGF-β 95.70% ± 1.76% vs. B7H3 3.30% ± 1.38%; p < 0.0001; n = 4) were exposed to TGF-β. In contrast, after TGF-β exposure, phosphorylation of SMAD2/3 was not observed in NK cells transduced with the DNR alone (DNR + TGF-β 13.43% ± 1.89% vs. DNR 8.71% ± 0.77%; p = 0.19; n = 3) or in NK cells co-transduced with the DNR.B7H3 CAR (DNR.B7H3+TGF-β 11.41% ± 5.43% vs. DNR.B7H3 4.90% ± 2.48%; n = 4; p = 0.042) (Figures 5D and 5E). These results confirmed that while SMAD2/3 phosphorylation is upregulated in UT-NK and B7H3, CAR-NK cells in the presence of TGF-β the DNR was able to inhibit SMAD2/3 phosphorylation in both DNR NK cells and DNR.B7H3 CAR-NK cells.
Co-transducing NK cells with the DNR and B7H3 CAR rescues B7H3 CAR-NK cell cytolytic activity in the presence of TGF-β
We next investigated whether DNR-expressing B7H3 CAR-NK cells could maintain their enhanced cytolytic activity against B7H3-positive targets after exposure to TGF-β. UT-NK and B7H3-CAR, DNR and DNR.B7H3-CAR-expressing NK cells were cultured for 5 days in the presence or absence of TGF-β (5 ng/mL). After 5 days of treatment with TGF- β, the cytotoxic activity was evaluated using the Celigo Imaging Cytometer system (Figures 6A and 6B). After exposure to TGF-β, the ability of the UT-NK cell to kill the U87 cell line was significantly decreased from 69.67% ± 4.62% to 41.17% ± 3.97% at an E:T ratio of 20:1 (p = 0002; n = 3) (Figures 6A and 6B). Further, B7H3 CAR-NK cell target lysis decreased from 89.73% ± 2.44% to 61.75% ± 3.42% in the presence of TGF-β (p = 0.002) (Figures 6A and 6B) at an E:T ratio of 20:1. In contrast, DNR-transduced NK cells cultured in the presence of TGF-β maintained their cytolytic capability (p = 0.86) and DNR.B7H3 CAR-NK cells also retained their enhanced cytolytic activity against B7H3+ U87 MG cells (p = 0.99) (Figures 6A and 6B) at an E:T ratio of 20:1.
Figure 6.

Co-transducing NK cells with DNR and B7H3-CAR rescues B7H3-CAR NK cell cytolytic activity in the presence of TGF-β
(A) Representative experiment out of n = 5 NK cell products generated, show the percentage of U87 MG target lysis by NK cells cultured in the presence (red lines) and absence (black lines) of TGF-β. The individual point represents a mean of the replicates. The error bars represent the standard deviation. (B) The mean target cells lysis in (n = 5 in each group) presence (red bar) or absence (black bar) of TGF-β (5 ng/mL). (C and D) Representative surface expression of NKG2D (C) and CD16 (D) for UT-NK versus transduced NK cell products after culture in the presence (red histogram) or absence (blue histogram) of TGF-β. (E and F) Mean percent MFI decrease in NKG2D (E) and CD16 (F) receptor expression in transduced versus UT-NK cell products after culture in the presence or absence of TGF-β (n = 4). See materials and methods for percent MFI calculation. The individual point represents each donor and error bars represent standard deviation (∗p < 0.05, all comparisons). The p values were generated by the Holm-Sidak multiple comparison test following two-way analysis of variance.
We also investigated whether NK cell activating receptor expression was preserved in NK cells co-transduced with the DNR and B7H3 CAR in the presence of exogenous TGF-β. Our results demonstrate that the DNR was able to mitigate the downregulation of CD16 and NKG2D receptors in DNR NK cells and DNR.B7H3 CAR-NK cells (Figure 6) in the presence of TGF-β. This is in contrast to NKG2D (Figures 6C and 6E) and CD16 (Figures 6D and 6F) receptor downregulation observed in UT NK and B7H3 CAR-NK cells in the presence of TGF-β (n = 4; CD16, DNR.B7H3 CAR-NK vs. B7H3 CAR-NK [p < 0.0001]; NKG2D, DNR.B7H3 CAR-NK vs. B7H3 CAR-NK [p = 0.005]; CD16, DNR NK vs. UT-NK [p ≤ 0.0001]; NKG2D-DNR NK vs. UT NK [p < 0.0001]).
We also evaluated whether the DNR.B7H3 CAR-NK cell product would maintain cytolytic activity in the presence of conditioned media collected from the U87 MG cells line compared with NK cell products not expressing the DNR. We observed that B7H3 CAR-NK cells elicited decreased target cell lysis (61.81% ± 8.32%) after 24 h co-culture in comparison with DNR.B7H3 CAR-NK cells (75.21% ± 17.50%) at a 1:1 E:T ratio. Both untreated B7H3 CAR-NK cells (95.33% ± 5.70%) and DNR.B7H3 CAR-NK cells (99.39% ± 0.35%) (used as controls) showed equivalent target lysis at the 1:1 E:T ratio (Figure S4).
We then investigated whether the potent cytolytic activity of the DNR CAR-NK cell product persisted after repetitive cancer cell and TGF-β restimulation. We cocultured TGF-β-treated and untreated NK cells with U87 MG cells for 72 h at E:T ratios of 5:1 and 1:1. After 72 h, target lysis was recorded and NK cells were restimulated with targets with or without TGF-β (Figure 7A). Repetitive restimulation with TGF-β at a concentration of 5 ng/mL was performed to saturate the system (and was intentionally above the clinically reported concentration of active TGF-β detected in GBM patients, 0.60 ± 2.30 ng/mL).39 Conditions (transduced versus UT NK cells; with or without TGF-β) with a target lysis of less than 30% were removed from the next round of stimulation, because we assumed that these cells had already lost their effector functions.
Figure 7.

DNR and B7H3 co-transduced NK cells show persistent cancer cell lysis after restimulation
Representative example of the cytotoxic activity of UT-NK (black), B7H3 CAR-NK (red), DNR NK (green), and DNR.B7H3 CAR-NK cells (purple) from the same donor, as measured by a luciferase cytotoxicity assay after repetitive tumor and TGF-β restimulation (n = 2). (A) Schematic illustrating the experiment design (created using Biorender.com). (B) Target lysis by NK cells at 1:1 E:T ratio after each tumor with or without TGF-β restimulation. (C) Target lysis by NK cells at the 5:1 E:T ratio after each tumor with or without TGF-β restimulation. Individual points represent the mean of the replicates, and the error bars represent the standard deviation (∗p < 0.05, all comparisons). (B) The p values were generated using the Kruskal-Wallis test. (C) The p values were generated using the Holm-Sidak multiple comparison test after two-way analysis of variance.
At the 1:1 E:T ratio, we observed that all transduced NK cells (DNR.B7H3-100.53% ± 0.17%, DNR-98.51% ± 1.83%, and B7H3 CAR-94.20% ± 1.01%) showed robust killing in comparison with UT-NK (61.23% ± 1.91%; p < 0.05), only after the first stimulation with U87 MG cells (Figure 7B). However, DNR.B7H3 CAR-NK cells also showed significantly higher target lysis compared with the B7H3 CAR-NK cells (p < 0.05) (Figure 7B). Moreover, after the second stimulation, DNR.B7H3 CAR-NK cells (91.93% ± 1.86%) maintained their significantly higher target killing efficiency in comparison with the B7H3 CAR-NK (33.75% ± 20.47%; p < 0.05) and UT-NK (5.69% ± 2.90%; p < 0.05) cells (Figure 7B). DNR.B7H3 CAR-NK cell products also showed higher target lysis in comparison with DNR NK cell products (73.337% ± 9.49%). When the NK cell products were TGF-β treated, there was lower target lysis elicited by B7H3 CAR-NK cells after both the first and second stimulations, in comparison to DNR.B7H3 CAR-NK cells and DNR NK cells (p < 0.05).
In experiments conducted using U87 MG cell line restimulations at the 5:1 E:T ratio, we observed persistent and robust target lysis by DNR.B7H3 CAR-NK products even after the fourth stimulation, while other conditions showed a gradual loss in cytolytic function (Figure 7C). Specifically, DNR NK, B7H3 CAR-NK cells, and UT NK cells showed significantly less target lysis compared with DNR.B7H3 CAR-NK cells after the fourth stimulation (p < 0.05). Further, in conditions using cancer cells plus TGF-β restimulation at the 5:1 E:T ratio, we observed a complete loss of effector function in both the UT and B7H3 CAR-NK cell products after restimulation. In contrast, DNR.B7H3-CAR-NK and DNR NK cells showed significant resistance to TGF-β-mediated blunting of effector function even after the fourth restimulation compared with B7H3 CAR-NK cells and UT-NK cells (p < 0.05) (Figure 7C). In summary, these results further confirm that the DNR abrogates the negative signaling of TGF-β in DNR.B7H3 CAR-NK cell products and B7H3 CAR NK cells are resistant to TGF-β-mediated immune suppression only when engineered to co-express a DNR receptor (n = 6) (Figure 6).
Discussion
B7H3 CAR-T cells have shown potent antitumor activity against GBM in preclinical models.20,21,22 However, autologous cell therapy products can be complex and time consuming to manufacture, especially when generated from highly immune-suppressed patients. Conventional αβT cells have limitations in the off-the-shelf setting because allogeneic αβT cell products can pose a serious risk of GVHD40 and additional engineering to knockout the TCR to manage GVHD may be required.41,42,43 NK cells and γδT cells are an attractive alternative for CAR-based immunotherapies, especially in an allogeneic off-the-shelf setting.44,45 NK cells have an innate ability to detect and eliminate malignant cells and may be safer in the off-the-shelf setting because of the low rates of GVHD observed with adoptively transferred allogeneic NK cells in numerous clinical settings.46 Like NK cells, γδT cells also elicit MHC-independent anti-tumor responses.47 Similar to NK cells, they express the NKG2D receptor to recognize malignant cells and they mediate cytotoxicity via the granzyme-perforin axis and antibody (Ab)-dependent cellular cytotoxicity via FcγRIII (CD16) expression.48 However, unlike NK cells, expansion of γδT cells is still limited and insufficient numbers for clinical use have been reported, which is a major hurdle for their broad applicability as an off-the-shelf adoptive cell therapy.47,49
In this study, we demonstrate that NK cells naturally express receptors for the stress ligands present on GBM adult and pediatric cell lines and exhibit cytolytic ability against GBM cells in vitro. Additionally, we show that CB-derived NK cells can be gene engineered to express a B7H3 CAR. Here, we demonstrated that these CAR-engineered NK cells are highly cytolytic against GBM cell lines and primary GBM cells in vitro. However, we also observed that B7H3 CAR-NK cells are susceptible to the loss of effector function mediated by TGF-β within the TME and this susceptibility can be rescued by co-transduction with a DNR.
Both UT and CAR-expressing NK cells showed increased secretion of IFN-γ in response to U87 MG cells, confirming that NK cells have an innate ability to recognize cancer cell stress ligands without the need for prior sensitization. This could represent an advantage for NK cells over conventional αβT cells for CAR-based immunotherapies since the innate ability of NK cells to recognize and kill tumor cells remains intact, even in the setting of tumor immune evasion with loss of the tumor antigen targeted by the CAR.50
UT-NK cells are, however, sensitive to the immune-suppressive, TGF-β rich TME.12,27,29,30,36 To our knowledge, this study is the first to demonstrate that CAR-transduced NK cells are not immune to TGF-β-mediated immune suppression. We showed that the cytolytic activity of B7H3 CAR-transduced NK cells was significantly decreased in the presence of exogenous TGF-β against the U87 GBM cell line. However, this decrease in effector function was not related to a downregulation of CAR expression since B7H3 CAR-NK cells retained their CAR expression after exposure to exogenous TGF-β. This observation indicates that the CAR moiety is more stably expressed on the NK cell surface and, therefore, less susceptible to downregulation and recycling, unlike endogenous receptors (e.g., NKG2D, DNAM1, and CD16). The stable expression of the CAR moiety is often attributed to the hinge and transmembrane domains.51 The use of the CD8α transmembrane domain in our construct may be a factor contributing to the transduction efficiencies. This would need further investigation. Further, it would be interesting to study the effect of TGF-β on CARs with different transmembrane domains and hinge regions. These observations could educate us about the ideal CAR design for TGF-β-rich solid tumors and will be explored in future studies.
In contrast with the stable CAR expression, we observed a significant downregulation of NK cell-activating receptors expressed by B7H3 CAR-NK cells in the presence of TGF-β. Therefore, we posit that the decreased cytolytic function of B7H3 CAR-NK cells is linked to the downregulation of activating NK cells receptors, suggesting that the innate cytolytic activity of NK cells also contributes to the anti-tumor potency of CAR-engineered NK cell products. This observation is potentially useful when designing next generation engineered NK cell therapies.
Our group has previously shown that a mutant TGF-β receptor 2 (DNR) can effectively protect CB-derived NK cells against the detrimental effects of TGF-β.12,27,30 Building on this experience, we genetically engineered NK cells to co-express a B7H3 CAR and a DNR to overcome the immune suppressive effects of TGF-β-mediated signaling. We demonstrated that expression of the DNR on B7H3 CAR-NK cells abrogated the TGF-β signaling cascade. This cascade, initiated by phosphorylation of Smad2/3, ultimately decreases surface receptor expression with consequent impairment of the antitumor effector functions of our CAR NK cells. In this study, we show that DNR-expressing B7H3 CAR-NK cells are resistant to the inhibitory effects of TGF-β. Since DNR.B7H3 CAR-NK cells preserved their NK cell-activating receptor expression in the presence of exogenous TGF-β, these unique cell products could also retain their enhanced cytolytic activity against TGF-β -secreting GBM cells in vitro.
When administering adoptive immunotherapy, several cell injection or delivery routes (e.g., intravenous injection, focal injection via the feeding vessels of the tumor, and direct intratumoral injection) may be selected. Intravenous injection is generally preferred for the delivery of adoptive cell therapies, including NK cells, since previous studies have shown that intravenously injected NK cells can migrate into tumor tissues. However, only a low number of NK cells localized to the tumor sites even at high NK cell doses. Furthermore, Holladay et al. have shown that intravenously injected NK cells failed to exert antitumor activity against established intracerebral gliomas.52,53 Moreover, intratumoral/intracavitary T cell therapies have successfully achieved tumor regression in some cases.54,55,56,57,58 It is therefore posited that direct injection into a tumor cavity could lead to direct cytotoxicity and this will be explored in our future work.
In summary, we have shown that TGF-β-rich GBM cells suppress CAR-NK cell effector functions in vitro, which can be overcome by co-expressing a dominant-negative receptor for TGF-β. We have successfully demonstrated the feasibility of CAR and DNR co-expression in NK cells, and these products have enhanced potency against GBM cells. We assert that utilizing a strategy to co-express a TGF-βRII DNR and a CAR on NK cells may be a promising therapeutic advance for the treatment of GBM and other TGF-β-secreting solid tumors.
Materials and methods
Experimental model and subject details
Cell lines and cell culture
U87 MG cells and A172 cells were purchased from ATCC. U87 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM):F12 media (1:1) (ThermoFisher Scientific) with 10% FBS and 1% Glutamax. A172 cells were cultured in DMEM high glucose media (ThermoFisher Scientific) with 10% fetal bovine serum (FBS) and 1% Glutamax. GBM-110 and GBM 511 FHTC primary pediatric cancer cell lines were purchased from Fred Hutchinson Cancer Research Center and grown in predefined serum-free neural stem cell medium, supplemented with epidermal growth factor and fibroblast growth factor, on laminin (ThermoFisher Scientific) coated plates. Tissue culture plates were coated with 1 mL laminin working solution prepared in phosphate-buffered saline (PBS) (10 μg/mL) and incubated at 37°C for a minimum of 1 h. Plates were washed once with PBS before seeding cells. Membrane-bound interleukin 15 (IL15) and 41BB ligand-expressing K562 cells were kindly provided by Dr. Dario Campana (St. Jude Children’s Research Hospital, Memphis, TN) and cultured in RPMI 1640 media (ThermoFisher Scientific) with 10% FBS and 1% Glutamax. We purchased 293T cells from ATCC and were grown in DMEM high glucose media with 10% FBS and 1% Glutamax. Samples were obtained under the guidelines of the Institutional Review Board at CNH - Pro0004033 and CR00000266.
Plasmid construction and retrovirus production
Generation of the retroviral vector encoding the truncated human type II TGF-β receptor (DNRII)12,38 and B7H3 CAR20,21 has been previously described (Figures S6A and S6B). Briefly, for the DNRII retrovirus vector, a cassette encoding the DNRII transgene from Dr. Joan Massagué’s group was cloned into an SFG retroviral vector (Figures S6B and S6C).59 Transient viral supernatant generated using the Phoenix-eco cell line was used to transduce (using multiple rounds of transduction) the packaging cell line PG13. PG13 retrovirus supernatant encoding the DNRII transgene was harvested and stored at −80°C until needed. B7H3.CAR.SFG retrovirus was kindly provided by Dr. Barbara Savoldo.20,21
Transduction and expansion of NK cells
We used a B7H3 CAR derived from the B7H3 376.96 mAb (Figure S6A).60,61 Du et al. have previously shown that this B7H3 CAR recognizes tumor cells expressing either the 2Ig-B7H3 or 4Ig-B7H3 isoform of human B7H3.21 CB units for research were provided by the INOVA CB Bank (IRB Pro00003869) or were purchased from STEMCELL Technologies and Organa Bio. CB MNCs were isolated by density-gradient centrifugation (Lymphoprep, STEMCELL Technologies). Isolated NK cells were expanded from CB MNCs in Stem Cell Growth Media (CellGenix GMP SCGM) supplemented with human recombinant IL-2 (200 IU/mL, R&D System) and IL-21 (25 ng/mL, R&D System) as previously described.62 NK cells were transduced with B7H3 CAR20 or with the DNRII retrovirus as previously described.30,38 For co-transduction, NK cells were transduced with B7H3 CAR and DNR retrovirus supernatants by sequential spinoculation (Figure S6D).
Transduction of cancer cell lines
U87 MG and A172 GBM cell lines were transduced with pCDH-EF1a-eFFly-mCherry lentivirus plasmid. pCDH-EF1a-eFFly-mCherry was a gift from Irmela Jeremias (Addgene plasmid # 104833; http://n2t.net/addgene:104833; RRID:Addgene_104833).63 Briefly, 2 × 106 293T cells were seeded in T75 flask and transfected with the plasmid mixture of the lentiviral vector, the pCDH-EF1a-eFFly-mCherry along with RTR2, VSVG, pCAG KGP1 (kindly provided by Dr. Allistair Abraham and St Jude Children’s Research Hospital) using the lipofectamine 3000 (ThermoFisher Scientific) according to the manufacturer’s instruction. Supernatant containing the lentivirus was collected at 72 h after transfection and filtered with 0.45-μm filters. Virus culture media (VCM) was frozen and stored at −80°C. One day before transduction, 50,000–75,000 cells were seeded in a 24-well plate. The next day, cell culture media was replaced with 2 mL prepared lentivirus VCM per well with 10 μg/mL polybrene (Millipore Sigma). Expression was determined 72 h after transduction by flow cytometry. mCherry-expressing cells were sorted to ensure 100% purity. The multiplicity of infection of pCDH-EF1a-eFFly-mCherry Lentivirus was not determined for this study but is being evaluated for future studies. Further, while it is unclear whether the transduction efficiency (77.5%) will pose a potential risk, future in vivo studies will evaluate this question in greater detail.
Flow cytometry
NK cell phenotype and chemokine receptor expression were assessed by flow cytometry using antibodies specific for human CD3, CD16, CD25, CD56, CD57, CD69, NKp30, NKG2D, B7H3, PD-1, and TIM3, and PD-1, CCX-CKR (CCRL1), CD195 (CCR5), CD198 (CCR8), CXCR7, CD184 (CXCR4), CD185 (CXCR5), CD193 (CCR3), CD197 (CCR7), CD183 (CXCR3), CD192 (CCR2), CD199 (CCR9), XCR1, CD181 (CXCR1), CD191 (CCR1), CD196 (CCR6), CCR10, CD182 (CXCR2), CD186 (CXCR6), CD194 (CCR4) (from BD bioscience, ThermoFisher Scientific, and Biolegend) conjugated with BV421, BV510, BV605, BV650, FITC, AF488, PerCP-cy5.5, PE, PE-cy7, APC, and APC-cy7 fluorochromes. B7H3 ligand and NK cell receptors on human GBM cell lines were assessed by antibodies specific for B7H3, MICA/MICB, PVR and B7H6 (Biolegend and ThermoFisher Scientific). Antibody clones and manufacturer’s details can be found in Table S1. Expression of B7H3 CAR was detected using two step staining. Briefly, CAR NK cells were stained with recombinant human B7H3 Fc Chimera Protein purchased from R&D and then stained with secondary F(ab')₂ Fragment Goat Anti-Human IgG (H + L) purchased from Jackson Immunoresearch to detect the B7-H3 protein. To distinguish between live and dead populations, samples were stained with Fixable Viability Dyes (ThermoFisher Scientific). After that, samples were fixed using BD Cytofix/Cytoperm Fixation/Permeabilization Kit (BD Biosciences). Expression of DNRII was determined by quantifying the increase in TGFBRII expression on transduced NK cells in comparison with UT NK cells. Samples were acquired with CytoFLEX, Research Flow Cytometry (Beckman Coulter). For each sample a minimum of 10,000 events were acquired and data were analyzed using FlowJo 10.
Short-term cytotoxicity assay
The Celigo Imaging Cytometer based cytolytic assay was performed to determine the cytolytic activity of transduced NK cells at 4 h, as previously described.64,65 Briefly, B7H3+ targets were labeled with Calcein AM dye (Invitrogen). After staining, targets were cocultured with transduced and UT control NK cells in a flat-bottom microplate at E:T ratios of 10:1, 5:1, and 1:1. Live cells were detected by imaging plates after 4 h of incubation using the Celigo Imaging Cytometer system. “Target lysis” was calculated according to the manufacturer’s recommendations as follows:
Signaling assessment of NK cells after exposure to TGF-β
TGF-β was purchased from PeproTech, and stock solution was prepared at a concentration of 50 ng/mL and stored at −80°C until needed. Before using TGF-β with NK cells, stock solution was diluted 1/10 in HCl (4 mmol/L) for a final concentration of 5 ng/mL and incubated for 30 min at room temperature as previously described.12 NK cells were serum starved overnight and were then treated with 2 ng/mL TGF-β for 30 min. To distinguish between live and dead populations, samples were stained with Fixable Viability Dyes (ThermoFisher Scientific). Cells were then fixed by adding fixation buffer (BioLegend 420801) and permeabilized with True Phos perm (BioLegend 425401) for 1 h. Subsequently, cells were stained with antibodies to SMAD2 (pS465/pS467)/SMAD3 (pS423/pS425), CD3, CD56, and TGFBRII. Samples were then acquired using the CytoFLEX, Research Flow Cytometer (Beckman Coulter). For each sample, a minimum of 10,000 gated events in live cell populations were acquired and data were analyzed using FlowJo 10.
Phenotypic and functional assessment of NK cells after exposure to TGF-β
NK cells were cultured with 5 ng/mL TGF-β (activated with 4 mmol/L HCl) added on days 1 and 3. After 5 days of treatment, NK cells were examined by flow cytometry and cytotoxicity assays. The cytolytic ability of the NK cell products after exposure to TGF-β was determined using the Celigo Imaging Cytometer cytotoxicity assay as described previously.66,67 The %MFI decrease for NKG2D and CD16 NK cells receptors is calculated as follows:
Repeat stimulation of B7H3.DNR CAR-NK
We plate 5 × 103 effector cells at a 1:1 E:T ratio with firefly luciferase expressing U87 MG cells, in the presence and absence of exogenous TGF-β. Three days later, antitumor activity was determined by a luciferase-based assay as previously described.66,67 Fresh U87 MG cells and TGF-β were added to the restimulated CAR-NK cells after the luciferase reading.
TGF-β1 ELISA of U87 cells conditioned media
To measure TGF-β1 concentrations secreted by the GBM cell line, U87 MG cells were allowed to grow to different levels of confluency. Before collection, culture media was changed to serum free media. Supernatants were collected after 24 h and were frozen at −80°C for batch analysis using the TGF-β1 ELISA kit from R&D Systems. The kit was run according to the manufacturer’s protocol and the TGF-β1 concentration determined using the provided standards.
Quantification and statistical analysis
Measurements were summarized as mean ± standard deviation as noted in the figure legends. Experimental sample numbers are indicated in the figure legends. Normality was determined using the Shapiro-Wilk test. For normally distributed data, analysis of variance or Welch’s t-test (unequal variances t-test) were used to compare quantitative differences (mean ± standard deviation) between groups. The Holm-Sidak t-test was used for multiple comparison; p values were two-sided; A p value of less than 0.05 was considered a significant difference. Whenever the data were not normally distributed, means were compared using the Kruskal-Wallis test (for multiple comparison) or the Mann-Whitney test with error correction (between groups). The indicated statistical tests were performed using GraphPad Prism software.
Data availability statement
The data that support the findings of this study are available upon request from the corresponding author, C.B.
Acknowledgments
Supported by the NIH/NCI Moonshot grant (5 U01 CA239258-02). We thank Dr. Allistair Abraham and St. Jude’s Children’s Research Hospital for providing the RTR2, VSVG, pCAG, and KGP1 plasmids; Dr. Dario Campana and St. Jude’s Children’s Research Hospital for providing the mbIL-15- and 4-1BBL-expressing K562 cell line; and Dr. Gianpietro Dotti and Dr. Barbara Savoldo for providing the B7H3/CD28z CAR virus culture media used in the study.
Author contributions
Conceptualization, K.C., C.M.B., and C.R.Y.C.; Methodology, K.C., B.S., C.L., E.Y, C.M.B., and C.R.Y.C; Investigation, K.C., A.G., H.L., D.K.S., and E.D.; Writing – Original Draft, K.C., C.M.B., and C.R.Y.C.; Writing – Review & Editing, all authors; Resources, E.G., R.J., M.H., and B.S. Supervision, C.M.B. and C.R.Y.C.
Declaration of interests
C.M.B. is a scientific co-founder and scientific advisory board member for Catamaran Bio and Mana Therapeutics. C.M.B also serves on the Board of Directors of Cabaletta Bio and has stock in Neximmune and Repertoire Immune Medicine. C.R.Y.C. is a consultant for Catamaran Bio and is a scientific co-founder and scientific advisory board member of Mana Therapeutics. C.R.Y.C also has sponsored research agreements with Catamaran Bio. E.Y. is a scientific advisory board member for Catamaran Bio. B.S. has sponsored research agreements with Bluebird Bio, Cell Medica, Bellicum Pharmaceutical and Tessa Therapeutics. C.R.Y.C, E.Y., B.S., and C.M.B have intellectual property related to developing NK cell therapies. All other authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2022.10.010.
Contributor Information
Conrad Russell Y. Cruz, Email: ccruz@childrensnational.org.
Catherine M. Bollard, Email: cbollard@childrensnational.org.
Supplemental information
References
- 1.Stupp R., Mason W.P., van den Bent M.J., Weller M., Fisher B., Taphoorn M.J.B., Belanger K., Brandes A.A., Marosi C., Bogdahn U., et al. European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups. National Cancer Institute of Canada Clinical Trials Group Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 2.Hanif F., Muzaffar K., Perveen K., Malhi S.M., Simjee S.U. Glioblastoma multiforme: a review of its epidemiology and pathogenesis through clinical presentation and treatment. Asian Pac. J. Cancer Prev. 2017;18:3–9. doi: 10.22034/APJCP.2017.18.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alexander B.M., Cloughesy T.F. Adult glioblastoma. J. Clin. Oncol. 2017;35:2402–2409. doi: 10.1200/jco.2017.73.0119. [DOI] [PubMed] [Google Scholar]
- 4.Filley A.C., Henriquez M., Dey M. Recurrent glioma clinical trial, CheckMate-143: the game is not over yet. Oncotarget. 2017;8:91779–91794. doi: 10.18632/oncotarget.21586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aldea M., Orillard E., Mansi L., Marabelle A., Scotte F., Lambotte O., Michot J.-M. How to manage patients with corticosteroids in oncology in the era of immunotherapy? Eur. J. Cancer. 2020;141:239–251. doi: 10.1016/j.ejca.2020.09.032. [DOI] [PubMed] [Google Scholar]
- 6.Cain D.W., Cidlowski J.A. Immune regulation by glucocorticoids. Nat. Rev. Immunol. 2017;17:233–247. doi: 10.1038/nri.2017.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heipertz E.L., Zynda E.R., Stav-Noraas T.E., Hungler A.D., Boucher S.E., Kaur N., Vemuri M.C. Current perspectives on "Off-The-Shelf" allogeneic NK and CAR-NK cell therapies. Front. Immunol. 2021;12:732135. doi: 10.3389/fimmu.2021.732135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ruggeri L., Mancusi A., Capanni M., Martelli M.F., Velardi A. Exploitation of alloreactive NK cells in adoptive immunotherapy of cancer. Curr. Opin. Immunol. 2005;17:211–217. doi: 10.1016/j.coi.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 9.Rubnitz J.E., Inaba H., Ribeiro R.C., Pounds S., Rooney B., Bell T., Pui C.H., Leung W. NKAML: a pilot study to determine the safety and feasibility of haploidentical natural killer cell transplantation in childhood acute myeloid leukemia. J. Clin. Oncol. 2010;28:955–959. doi: 10.1200/jco.2009.24.4590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ruggeri L., Capanni M., Urbani E., Perruccio K., Shlomchik W.D., Tosti A., Posati S., Rogaia D., Frassoni F., Aversa F., et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science. 2002;295:2097–2100. doi: 10.1126/science.1068440. [DOI] [PubMed] [Google Scholar]
- 11.Shenouda M.M., Gillgrass A., Nham T., Hogg R., Lee A.J., Chew M.V., Shafaei M., Aarts C., Lee D.A., Hassell J., et al. Ex vivo expanded natural killer cells from breast cancer patients and healthy donors are highly cytotoxic against breast cancer cell lines and patient-derived tumours. Breast Cancer Res. 2017;19:76. doi: 10.1186/s13058-017-0867-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yvon E.S., Burga R., Powell A., Cruz C.R., Fernandes R., Barese C., Nguyen T., Abdel-Baki M.S., Bollard C.M. Cord blood natural killer cells expressing a dominant negative TGF-β receptor: implications for adoptive immunotherapy for glioblastoma. Cytotherapy. 2017;19:408–418. doi: 10.1016/j.jcyt.2016.12.005. [DOI] [PubMed] [Google Scholar]
- 13.Liu E., Tong Y., Dotti G., Shaim H., Savoldo B., Mukherjee M., Orange J., Wan X., Lu X., Reynolds A., et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia. 2018;32:520–531. doi: 10.1038/leu.2017.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu E., Marin D., Banerjee P., Macapinlac H.A., Thompson P., Basar R., Nassif Kerbauy L., Overman B., Thall P., Kaplan M., et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N. Engl. J. Med. 2020;382:545–553. doi: 10.1056/NEJMoa1910607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Woll P.S., Grzywacz B., Tian X., Marcus R.K., Knorr D.A., Verneris M.R., Kaufman D.S. Human embryonic stem cells differentiate into a homogeneous population of natural killer cells with potent in vivo antitumor activity. Blood. 2009;113:6094–6101. doi: 10.1182/blood-2008-06-165225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chouaib S., Pittari G., Nanbakhsh A., El Ayoubi H., Amsellem S., Bourhis J.-H., Spanholtz J. Improving the outcome of leukemia by natural killer cell-based immunotherapeutic strategies. Front. Immunol. 2014;5:95. doi: 10.3389/fimmu.2014.00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zingoni A., Vulpis E., Loconte L., Santoni A. NKG2D ligand shedding in response to stress: role of ADAM10. Front. Immunol. 2020;11:447. doi: 10.3389/fimmu.2020.00447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Picarda E., Ohaegbulam K.C., Zang X. Molecular pathways: targeting B7-H3 (CD276) for human cancer immunotherapy. Clin. Cancer Res. 2016;22:3425–3431. doi: 10.1158/1078-0432.ccr-15-2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang C., Zhang Z., Li F., Shen Z., Qiao Y., Li L., Liu S., Song M., Zhao X., Ren F., et al. Large-scale analysis reveals the specific clinical and immune features of B7-H3 in glioma. Oncoimmunology. 2018;7:e1461304. doi: 10.1080/2162402x.2018.1461304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nehama D., Di Ianni N., Musio S., Du H., Patané M., Pollo B., Finocchiaro G., Park J.J.H., Dunn D.E., Edwards D.S., et al. B7-H3-redirected chimeric antigen receptor T cells target glioblastoma and neurospheres. EBioMedicine. 2019;47:33–43. doi: 10.1016/j.ebiom.2019.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Du H., Hirabayashi K., Ahn S., Kren N.P., Montgomery S.A., Wang X., Tiruthani K., Mirlekar B., Michaud D., Greene K., et al. Antitumor responses in the absence of toxicity in solid tumors by targeting B7-H3 via chimeric antigen receptor T cells. Cancer Cell. 2019;35:221–237.e8. doi: 10.1016/j.ccell.2019.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Majzner R.G., Theruvath J.L., Nellan A., Heitzeneder S., Cui Y., Mount C.W., Rietberg S.P., Linde M.H., Xu P., Rota C., et al. CAR T cells targeting B7-H3, a pan-cancer antigen, demonstrate potent preclinical activity against pediatric solid tumors and brain tumors. Clin. Cancer Res. 2019;25:2560–2574. doi: 10.1158/1078-0432.ccr-18-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DeCordova S., Shastri A., Tsolaki A.G., Yasmin H., Klein L., Singh S.K., Kishore U. Molecular heterogeneity and immunosuppressive microenvironment in glioblastoma. Front. Immunol. 2020;11:1402. doi: 10.3389/fimmu.2020.01402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andersen R.S., Anand A., Harwood D.S.L., Kristensen B.W. Tumor-associated microglia and macrophages in the glioblastoma microenvironment and their implications for therapy. Cancers. 2021;13:4255. doi: 10.3390/cancers13174255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nduom E.K., Weller M., Heimberger A.B. Immunosuppressive mechanisms in glioblastoma. Neuro Oncol. 2015;17:vii9–vii14. doi: 10.1093/neuonc/nov151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frei K., Gramatzki D., Tritschler I., Schroeder J.J., Espinoza L., Rushing E.J., Weller M. Transforming growth factor-β pathway activity in glioblastoma. Oncotarget. 2015;6:5963–5977. doi: 10.18632/oncotarget.3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burga R.A., Yvon E., Chorvinsky E., Fernandes R., Cruz C.R., Bollard C. Engineering the TGFβ receptor to enhance the therapeutic potential of natural killer cells as an immunotherapy for neuroblastoma. Clin. Cancer Res. 2019;25:4400–4412. doi: 10.1158/1078-0432.ccr-18-3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang L., Pang Y., Moses H.L. TGF-β and immune cells: an important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010;31:220–227. doi: 10.1016/j.it.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Viel S., Marçais A., Guimaraes F.S.-F., Loftus R., Rabilloud J., Grau M., Degouve S., Djebali S., Sanlaville A., Charrier E., et al. TGF-β inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci. Signal. 2016;9:ra19. doi: 10.1126/scisignal.aad1884. [DOI] [PubMed] [Google Scholar]
- 30.Powell A.B., Yadavilli S., Saunders D., Van Pelt S., Chorvinsky E., Burga R.A., Albihani S., Hanley P.J., Xu Z., Pei Y., et al. Medulloblastoma rendered susceptible to NK-cell attack by TGFβ neutralization. J. Transl. Med. 2019;17:321. doi: 10.1186/s12967-019-2055-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Imai C., Iwamoto S., Campana D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood. 2005;106:376–383. doi: 10.1182/blood-2004-12-4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grégoire C., Chasson L., Luci C., Tomasello E., Geissmann F., Vivier E., Walzer T. The trafficking of natural killer cells. Immunol. Rev. 2007;220:169–182. doi: 10.1111/j.1600-065X.2007.00563.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bernardini G., Antonangeli F., Bonanni V., Santoni A. Dysregulation of chemokine/chemokine receptor axes and NK cell tissue localization during diseases. Front. Immunol. 2016;7:402. doi: 10.3389/fimmu.2016.00402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bernardini G., Sciumè G., Santoni A. Differential chemotactic receptor requirements for NK cell subset trafficking into bone marrow. Front. Immunol. 2013;4:12. doi: 10.3389/fimmu.2013.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Levy E.R., Clara J.A., Reger R.N., Allan D.S.J., Childs R.W. RNA-seq analysis reveals CCR5 as a key target for CRISPR gene editing to regulate in vivo NK cell trafficking. Cancers. 2021;13:872. doi: 10.3390/cancers13040872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wilson E.B., El-Jawhari J.J., Neilson A.L., Hall G.D., Melcher A.A., Meade J.L., Cook G.P. Human tumour immune evasion via TGF-β blocks NK cell activation but not survival allowing therapeutic restoration of anti-tumour activity. PLoS One. 2011;6:e22842. doi: 10.1371/journal.pone.0022842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Foster A.E., Dotti G., Lu A., Khalil M., Brenner M.K., Heslop H.E., Rooney C.M., Bollard C.M. Antitumor activity of EBV-specific T lymphocytes transduced with a dominant negative TGF-β receptor. J. Immunother. 2008;31:500–505. doi: 10.1097/CJI.0b013e318177092b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bollard C.M., Rössig C., Calonge M.J., Huls M.H., Wagner H.-J., Massague J., Brenner M.K., Heslop H.E., Rooney C.M. Adapting a transforming growth factor β–related tumor protection strategy to enhance antitumor immunity. Blood. 2002;99:3179–3187. doi: 10.1182/blood.V99.9.3179. [DOI] [PubMed] [Google Scholar]
- 39.Schneider T., Sailer M., Ansorge S., Firsching R., Reinhold D. Increased concentrations of transforming growth factor β1 and β2 in the plasma of patients with glioblastoma. J. Neuro Oncol. 2006;79:61–65. doi: 10.1007/s11060-005-9116-7. [DOI] [PubMed] [Google Scholar]
- 40.Sanber K., Savani B., Jain T. Graft-versus-host disease risk after chimeric antigen receptor T-cell therapy: the diametric opposition of T cells. Br. J. Haematol. 2021;195:660–668. doi: 10.1111/bjh.17544. [DOI] [PubMed] [Google Scholar]
- 41.Torikai H., Reik A., Liu P.Q., Zhou Y., Zhang L., Maiti S., Huls H., Miller J.C., Kebriaei P., Rabinovich B., et al. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood. 2012;119:5697–5705. doi: 10.1182/blood-2012-01-405365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ren J., Liu X., Fang C., Jiang S., June C.H., Zhao Y. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin. Cancer Res. 2017;23:2255–2266. doi: 10.1158/1078-0432.Ccr-16-1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Osborn M.J., Webber B.R., Knipping F., Lonetree C.L., Tennis N., DeFeo A.P., McElroy A.N., Starker C.G., Lee C., Merkel S., et al. Evaluation of TCR gene editing achieved by TALENs, CRISPR/Cas9, and megaTAL nucleases. Mol. Ther. 2016;24:570–581. doi: 10.1038/mt.2015.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alnaggar M., Xu Y., Li J., He J., Chen J., Li M., Wu Q., Lin L., Liang Y., Wang X., et al. Allogenic Vγ9Vδ2 T cell as new potential immunotherapy drug for solid tumor: a case study for cholangiocarcinoma. J. Immunother. Cancer. 2019;7:36. doi: 10.1186/s40425-019-0501-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Basar R., Daher M., Rezvani K. Next-generation cell therapies: the emerging role of CAR-NK cells. Blood Adv. 2020;4:5868–5876. doi: 10.1182/bloodadvances.2020002547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Björklund A.T., Carlsten M., Sohlberg E., Liu L.L., Clancy T., Karimi M., Cooley S., Miller J.S., Klimkowska M., Schaffer M., et al. Complete remission with reduction of high-risk clones following haploidentical NK-cell therapy against MDS and AML. Clin. Cancer Res. 2018;24:1834–1844. doi: 10.1158/1078-0432.ccr-17-3196. [DOI] [PubMed] [Google Scholar]
- 47.Yazdanifar M., Barbarito G., Bertaina A., Airoldi I. γδ T cells: the ideal tool for cancer immunotherapy. Cells. 2020;9:1305. doi: 10.3390/cells9051305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Couzi L., Pitard V., Sicard X., Garrigue I., Hawchar O., Merville P., Moreau J.F., Déchanet-Merville J. Antibody-dependent anti-cytomegalovirus activity of human γδ T cells expressing CD16 (FcγRIIIa) Blood. 2012;119:1418–1427. doi: 10.1182/blood-2011-06-363655. [DOI] [PubMed] [Google Scholar]
- 49.Xiao L., Chen C., Li Z., Zhu S., Tay J.C., Zhang X., Zha S., Zeng J., Tan W.K., Liu X., et al. Large-scale expansion of Vγ9Vδ2 T cells with engineered K562 feeder cells in G-Rex vessels and their use as chimeric antigen receptor-modified effector cells. Cytotherapy. 2018;20:420–435. doi: 10.1016/j.jcyt.2017.12.014. [DOI] [PubMed] [Google Scholar]
- 50.Sekine T., Marin D., Cao K., Li L., Mehta P., Shaim H., Sobieski C., Jones R., Oran B., Hosing C., et al. Specific combinations of donor and recipient KIR-HLA genotypes predict for large differences in outcome after cord blood transplantation. Blood. 2016;128:297–312. doi: 10.1182/blood-2016-03-706317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fujiwara K., Tsunei A., Kusabuka H., Ogaki E., Tachibana M., Okada N. Hinge and transmembrane domains of chimeric antigen receptor regulate receptor expression and signaling threshold. Cells. 2020;9:1182. doi: 10.3390/cells9051182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ishikawa E., Tsuboi K., Saijo K., Harada H., Takano S., Nose T., Ohno T. Autologous natural killer cell therapy for human recurrent malignant glioma. Anticancer Res. 2004;24:1861–1871. [PubMed] [Google Scholar]
- 53.Holladay F.P., Heitz T., Wood G.W. Antitumor activity against established intracerebral gliomas exhibited by cytotoxic T lymphocytes, but not by lymphokine-activated killer cells. J. Neurosurg. 1992;77:757–762. doi: 10.3171/jns.1992.77.5.0757. [DOI] [PubMed] [Google Scholar]
- 54.Barba D., Saris S.C., Holder C., Rosenberg S.A., Oldfield E.H. Intratumoral LAK cell and interleukin-2 therapy of human gliomas. J. Neurosurg. 1989;70:175–182. doi: 10.3171/jns.1989.70.2.0175. [DOI] [PubMed] [Google Scholar]
- 55.Nakagawa K., Kamezaki T., Shibata Y., Tsunoda T., Meguro K., Nose T. Effect of lymphokine-activated killer cells with or without radiation therapy against malignant brain tumors. Neurol. Med. Chir. 1995;35:22–27. doi: 10.2176/nmc.35.22. [DOI] [PubMed] [Google Scholar]
- 56.Hayes R.L., Koslow M., Hiesiger E.M., Hymes K.B., Hochster H.S., Moore E.J., Pierz D.M., Chen D.K., Budzilovich G.N., Ransohoff J. Improved long term survival after intracavitary interleukin-2 and lymphokine-activated killer cells for adults with recurrent malignant glioma. Cancer. 1995;76:840–852. doi: 10.1002/1097-0142. [DOI] [PubMed] [Google Scholar]
- 57.Tsuboi K., Saijo K., Ishikawa E., Tsurushima H., Takano S., Morishita Y., Ohno T. Effects of local injection of ex vivo expanded autologous tumor-specific T lymphocytes in cases with recurrent malignant gliomas. Clin. Cancer Res. 2003;9:3294–3302. [PubMed] [Google Scholar]
- 58.Tsurushima H., Liu S.Q., Tuboi K., Matsumura A., Yoshii Y., Nose T., Saijo K., Ohno T. Reduction of end-stage malignant glioma by injection with autologous cytotoxic T lymphocytes. Jpn. J. Cancer Res. 1999;90:536–545. doi: 10.1111/j.1349-7006.1999.tb00781.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Massagué J. TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 2012;13:616–630. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fauci J.M., Sabbatino F., Wang Y., Londoño-Joshi A.I., Straughn J.M., Jr., Landen C.N., Ferrone S., Buchsbaum D.J. Monoclonal antibody-based immunotherapy of ovarian cancer: targeting ovarian cancer cells with the B7-H3-specific mAb 376.96. Gynecol. Oncol. 2014;132:203–210. doi: 10.1016/j.ygyno.2013.10.038. [DOI] [PubMed] [Google Scholar]
- 61.Kasten B.B., Arend R.C., Katre A.A., Kim H., Fan J., Ferrone S., Zinn K.R., Buchsbaum D.J. B7-H3-targeted (212)Pb radioimmunotherapy of ovarian cancer in preclinical models. Nucl. Med. Biol. 2017;47:23–30. doi: 10.1016/j.nucmedbio.2017.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kellner J.N., Cruz C.R., Bollard C.M., Yvon E.S. Gene modification of human natural killer cells using a retroviral vector. Methods Mol. Biol. 2016;1441:203–213. doi: 10.1007/978-1-4939-3684-7_17. [DOI] [PubMed] [Google Scholar]
- 63.Ebinger S., Özdemir E.Z., Ziegenhain C., Tiedt S., Castro Alves C., Grunert M., Dworzak M., Lutz C., Turati V.A., Enver T., et al. Characterization of rare, dormant, and therapy-resistant cells in acute lymphoblastic leukemia. Cancer Cell. 2016;30:849–862. doi: 10.1016/j.ccell.2016.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chan L., Somanchi S., Rosbach K., Lee D. High-throughput direct cell counting-based natural killer cell-mediated cytotoxicity assay using Celigo Imaging Cytometry (TUM2P.1027) J. Immunol. 2015;194:69.24. [Google Scholar]
- 65.Chan L.L.-Y., Rosbach K., Somanchi S.S., Lee D. Abstract 1303: quantification of natural killer cell-mediated cytotoxicity using Celigo imaging cytometry. Cancer Res. 2015;75:1303. doi: 10.1158/1538-7445.Am2015-1303. [DOI] [Google Scholar]
- 66.Brown C.E., Wright C.L., Naranjo A., Vishwanath R.P., Chang W.-C., Olivares S., Wagner J.R., Bruins L., Raubitschek A., Cooper L.J.N., Jensen M.C. Biophotonic cytotoxicity assay for high-throughput screening of cytolytic killing. J. Immunol. Methods. 2005;297:39–52. doi: 10.1016/j.jim.2004.11.021. [DOI] [PubMed] [Google Scholar]
- 67.Karimi M.A., Lee E., Bachmann M.H., Salicioni A.M., Behrens E.M., Kambayashi T., Baldwin C.L. Measuring cytotoxicity by bioluminescence imaging outperforms the standard chromium-51 release assay. PLoS One. 2014;9:e89357. doi: 10.1371/journal.pone.0089357. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available upon request from the corresponding author, C.B.