

Abstract
Background and purpose:
Ghrelin is known as a hunger hormone and plays a pivotal role in appetite, food intake, energy balance, glucose metabolism, and insulin secretion, making it a potential target for the treatment of obesity and type 2 diabetes. The essential maturation step of ghrelin to activate the GHS-R1a is the octanoylation of the Ser3, which is catalyzed by the ghrelin O-acyltransferase enzyme (GOAT) enzyme. Therefore, the inhibition of GOAT may be useful for treating ghrelin-related diseases.
Experimental approach:
To discover the novel inhibitors against GOAT enzyme by a fast and accurate computational method, here, we tried to develop the homology model of GOAT. Subsequently, the generated model was stabilized by molecular dynamics simulation. The consecutive process of docking, pharmacophore mapping, and large-scale virtual screening were performed to find the potential hit compounds.
Findings / Results:
The homology model of the GOAT enzyme was generated and the quality of 3D structures was increased to the highest level of > 99.8% of residue in allowed regions. The model was inserted into the lipid bilayer and was stabilized by molecular dynamics simulation in 200 ns. The sequential process of pharmacophore-based virtual screening led to the introduction of three compounds including ethaverine, kaempferitrin, and reglitazar as optimal candidates for GOAT inhibition.
Conclusion and implications:
The results of this study may provide a starting point for further investigation for drug design in the case of GOAT inhibitors and help pave the way for clinical targeting of obesity and type 2 diabetes.
Keywords: Ghrelin O-acyltransferase enzyme, Molecular dynamics simulation, Obesity, Type 2 Diabetes, Virtual screening
INTRODUCTION
Ghrelin is a 28 amino acid peptide produced and secreted from the stomach to activate the growth hormone secretagogue receptor 1a (GHS-R1a) and growth hormone secretion (1). Beyond its primary mentioned role, ghrelin is known as a hunger hormone that plays a role in appetite, food intake, energy balance, glucose metabolism, and insulin secretion (2). Thereby ghrelin may be identified as a potential target for treating disorders such as obesity, anorexia nervosa, Prader-Willi syndrome, and type 2 diabetes. In addition, ghrelin has been proposed to be involved in some neurological processes including memory, learning, depression, and addiction. Besides, ghrelin has been linked to a neuroprotective role in Alzheimer's and Parkinson's diseases (3).
Ghrelin is expressed as a 117 amino-acids precursor preproghrelin and needs a series of modifications and cleavage processing before reaching the mature form of ghrelin to activate the GHS-R1a.
The preproghrelin undergoes cleavage by signal peptidase to produce the proghrelin with 94-amino acid. Subsequently, the proghrelin specifically octanoylated at the position Ser3. Then the acyl proghrelin is modified by the prohormone convertase and produces the mature acyl ghrelin containing 28 amino-acid (4). A unique and essential maturation step of ghrelin is the octanoylation of the Ser3 residue which is catalyzed by the ghrelin O-acyltransferase enzyme (GOAT). GOAT is a 45 kDa integral membrane of the membrane-bound O-acyltransferase (MBOAT) enzyme superfamily (5). The other known members of the MBOAT superfamily are Hedgehog acyltransferase and Porcupine (Porcn) (6). The enzymatic function of GOAT required the highly conserved residue Asp307 and His338 of the MBOAT family (7). Till now, ghrelin is the only substrate for GOAT, and due to the unique activity of GOAT in the maturation of the ghrelin, there is increasing interest in the development of GOAT inhibitors for therapeutic benefits (8). However, the hydrophobic nature of the integral membrane proteins has led to difficulty in their purification and structural determination by X-ray or cryogenic electron microscopy (cryo-EM) (9); hence the crystallographic structure of the GOAT enzyme has not been reported yet. The crystal structure of a bacterial MBOAT D-alanyl transferase DltB has been recently reported (10). Therefore, the lack of crystallographic structure of the GOAT enzyme has created a barrier to recognizing the accurate structure and also the drug discovery process.
In this study, in the lack of crystal structure of GOAT and also the absence of potent GOAT inhibitors in the clinic, we tried to use the computer aid drug design method to solve the homology model of GOAT and also find potential inhibitors by pharmacophore mapping and virtual screening procedures. The results of this study will expand the understanding of the possible structure of GOAT which may provide a starting point for further investigation in GOAT inhibitor design and help pave the way for clinical targeting for its related diseases. The flowchart of the applied procedures is presented in Fig. 1.
Fig. 1.
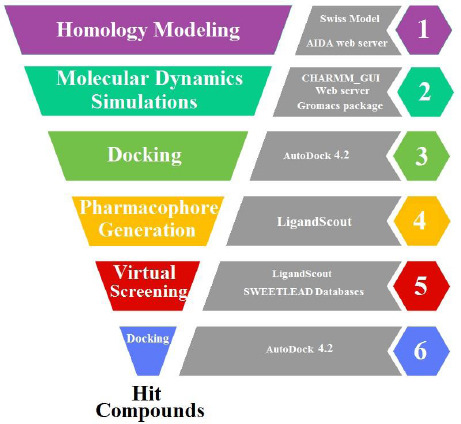
Flowchart illustrating the process to find hit compounds.
MATERIALS AND METHODS
Homology modeling
The amino acid sequence of the ghrelin O-acyltransferase was retrieved from the Uniprot web server with ID: Q96T53. The Swiss Model through the ExPAsy suite was used to sequence alignment and modeling of the GOAT (11). The server predicted the crystal structure of the D-alanyl transfer protein DltB the crystal form III with PDB ID: 6BUI-A, and coverage of the residue of 39 to 405 (365 out of 435) as the best template for GOAT. The homology model prediction and assembly of the rest of the 70 missing residues were done by the AIDA server (12), which uses ab-initio knowledge-based to find the best domain arrangements. The complete generated model was submitted to loop refinement by GalaxyRefine (13) and YASARA energy minimization (14). The Ramachandran plot was obtained from the MolProbity server for the assessment of the local stereochemistry quality (15). The model was further analyzed by the secondary structure server of PDBsum (16). The potential tunnel around the key residues of GOAT was calculated by Caver 3.0.1 software. The parameters were assigned as a maximum probe radius of 9 Å, shell depth of 4 Å, and clustering threshold of 3.5 Å (17).
Molecular dynamics simulation
Molecular dynamics simulation was performed to relax and refine the homology model in the membrane bilayer environment. The coordination of the homology model was fixed by -princ command in the GROMACS package (18). CHARMM-GUI web server (19) using PPM server to predict the orientation of the homology model with respect to a membrane bilayer and embedded the generated model into the bilayer membrane which containing the ratio of 1:1 of dipalmitoylphosphatidylcholine and dioleoyl phosphatidylcholine in an explicit solvent with 150 mM NaCl. GROMACS package Version 2020.1 was carried out on a high-performance Linux cluster for 200 ns. Finally, root means square deviation (RMSD) was analyzed from the final trajectory file and the final output in PDB format was used for the rest of the study.
Docking and pharmacophore model generation
GOAT has several reported inhibitors of various types of peptidomimetic, small molecules, and steroid-based with well-understood efficacy. Therefore, to predict the possible binding site of reported inhibitors and the homology model of the GOAT, ten inhibitors with the highest inhibitory effect were selected from different derivatives for molecular docking (20). The preparation of the receptor and ligands and analyzing the result were done by AutoDockTools 1.5.7. Polar hydrogen was added to the receptor, non-polar hydrogen was merged, and the Kollman charges were assigned. MarvinSketche version 15.2.2 was used for drawing the ligands 2D structure, and Chem3D ultra version 8.0 minimized and converted the 2D files to 3D structures. 50 × 50 × 50 Å (x, y, and z) grid box was centered on the key residues with 0.375 nm grid point spacing in each dimension. The grid maps of each atom type were prepared by AutoGrid4.2. Docking parameters were set as previously published method (21,22) and Lamarckian genetic algorithm in run job equals to 50. Subsequently, all ten protein-ligand complexes from docking results were used for pharmacophore generating by the LigandScout (Ver. 3.1) software (23) to find the functional groups from different pharmacophore models.
Pharmacophore-based virtual screening
Generated pharmacophore maps were used as a filter for virtual screening of the SWEETLEAD database containing 9127 compounds of approved drugs, herbal isolates, and regulated chemical compounds in SDF format to find the potential hit compounds. It should be noted that this is the free and highly accurate database in the case of a 2D format of the chemical structure and also contain the precise information for each compound, which is not seen in other databases such as Maybridge and NCI (24). A large number of compounds and the variety of structural features in this database could ensure the achievement of probable hit compounds. Finally, the obtained hit compounds from pharmacophore-based virtual screening were docked over the GOAT enzyme, and the result was sorted based on the docking binding energy. The visualization of the result was performed by PyMol version 1.1eval.
RESULT
Homology modeling generation
The 3D structure of GOAT is not determined by X-ray crystallography or nuclear magnetic resonance (NMR), so the computational method was used to predict the possible 3D homology model of GOAT. The FASTA sequence of GOAT with 435 residues and ID: Q96T53 was retrieved from the UniProt database. The Swiss-Model web server (11) was utilized for 3D structure prediction. The top matches’ template for 3D structure prediction to the GOAT sequences was the electron microscopy structure of human Hedgehog acyltransferase with PDB ID: 7MHY and sequence identity of 16.08% and coverage of the residue of 37 to 416. The second template was the crystal structure of the first MBOAT reported structure D-alanyl transfer protein DltB form III with PDB ID: 6BUI-A with a sequence identity of 12.62% and coverage of the residue of 39 to 405.
Both templates were used for homology modeling generation, and the generated models were compared to each other. Then the quality of both models was analyzed by Ramachandran plot. Interestingly the homology model generated by the template of PDB ID: 6BUI has slight superiority in geometry with 90.7% (331 residues out of 365) residues in favored (98%) regions, 97.8% (357 residues out of 365) residues in allowed (> 99.8%) regions, and 2.1% (8 residues out of 365) residues in outliers, while the model generated by the template of PDB ID: 7MHY presented the 91.0% (344 residues out of 378) residues in favored (98%) region, 97.6% (369 residues out of 378) residues in allowed (> 99.8%) regions, and 2.3% (9 residues out of 378) residues in outliers. The superimposition of both generated homology models presented a slight difference in alpha-helices positions and the loops as shown in Fig. 2.
Fig. 2.
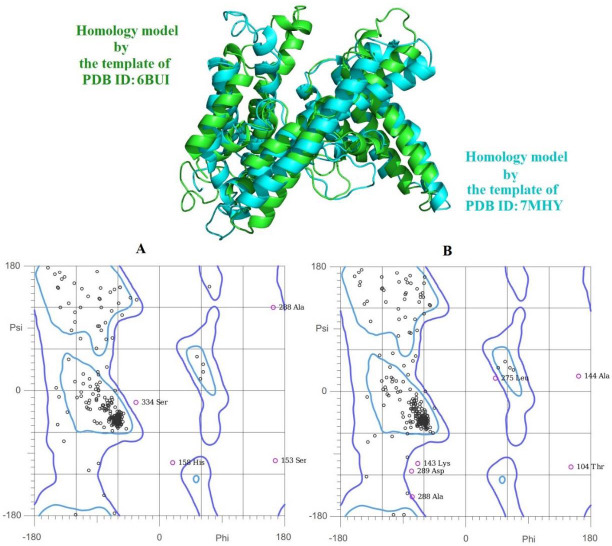
Superimpose position of two generated homology models by the template of PDB ID: 6BUI in green and the template of PDB ID: 7MHY in cyan. (A) and (B) are the Ramachandran plots of the two different homology models, respectively.
The DltB contains eleven transmembranes and has belonged to the superfamily of MBOAT, the same as the GOAT. Interestingly despite the low similarity in total sequence alignment of GOAT and DltB, there is a high similarity between the conserved residues in both enzymes (25). Moreover, the structural determination of the DltB was done by the X-ray method and not the electron microscopy method presented in 7MHY (26), therefore the generated homology model by the template of the DltB with PDB ID: 6BUI was selected for further evaluation. The 70 missing residues in the first model were generated and assembled by the knowledge-based ab-initio method of the AIDA server (12). In this step, besides completing the length of the sequence, the geometry of the model was also improved and reached 94.5% (409 residues out of 433) residues in favored (98%) regions and 1.1% (5 residues out of 433) residues in outliers. In the last attempt to increase the quality of 3D structures and stabilize the confirmation of the homology model, the loop refinement and energy minimization were performed, which increased the geometry quality to the highest desirable level of 95.6% (414 residues out of 433) residues in favored (98%) regions, 99.5% (431 residues out of 433) residues in allowed (> 99.8%) regions, and 0.4% (2 out of 433) residuesin outliers (Fig. 3).
Fig. 3.
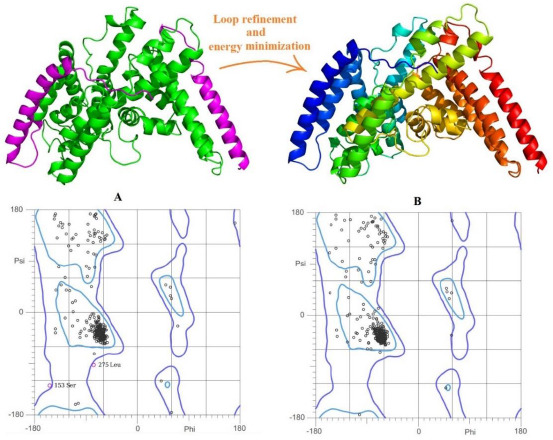
Homology modeling and the Ramachandran plots. (A) Generated homology model by the template of PDB ID: 6BUI in green the 3D structure of 70 missing residues was predicted and added by the AIDA web server in magenta color and its Ramachandran plots and (B) the final homology model after the loop refinement and energy minimization and related Ramachandran plots.
PDBsum server (16) analysis of the secondary structure consists of 63.4% (276 out of 435 residues) of the structure in helix form, 3.9% in 3-10 helix form, and 32.6% in other (or coil) form without any percentage of residues in beta-strands and represented in Fig. 4. All members of the MBOAT superfamily contain a tunnel in which the key amino acids are located, and probably this tunnel is the place of the enzyme's catalytic activity. The homology model was further analyzed by CAVER 3.0.1. (17) to predict the possible tunnels. The Caver computed the internal tunnel approximately 17 Å long, from the endoplasmic reticulum lumen to the cytoplasmic face (Fig. 4C). The three helixes, which contain amino acids from Ile299 to His338 and consist of key residues Asp307 and His338, are involved in forming this tunnel and may play an effective role in the catalytic activity of the enzyme.
Fig. 4.
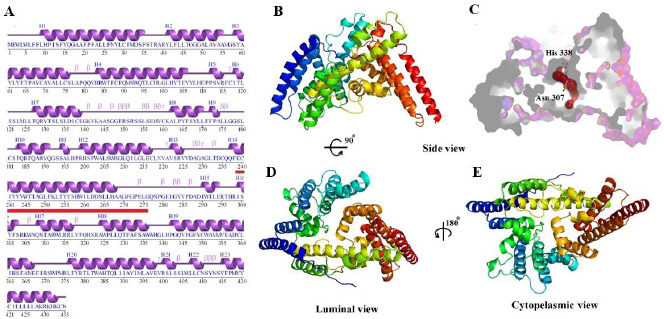
Sequence analyzing and the display of the homology model in luminal and cytoplasmic view. (A) Secondary structure analysis of ghrelin O-acyltransferase enzyme by the PDBsum server, the red line indicates the amino acids involved in the tunnel; (B) generated homology model of the enzyme from the side view; (C) the surface display of the homology model and predicted inside the tunnel; (D) and (E) are the luminal and cytoplasmic faces of the enzyme, respectively.
Molecular dynamics simulations studies
A generated homology model was further optimized by the molecular dynamics simulation and embedded in the membrane bilayer environment with the aid of the CHARMM_GUI server (19) for 200 ns to ensure the stability of the model in hydrophobic conditions. The stability of the homology model was examined by backbone RMSD. The backbone RMSD plot shows that the model reached an equilibrium state and remained stable after 50 ns of simulations with an average RMSD value of 0.42 nm. Thus, the length of the time allotted for the simulation was sufficient. The optimal structure of the molecular dynamics simulations was further used for the rest of the study (Fig. 5).
Fig. 5.
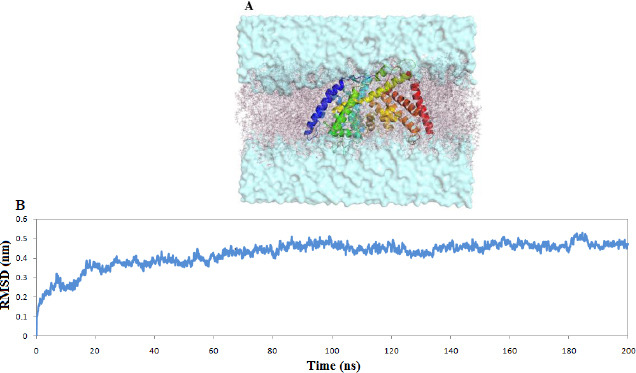
(A) The homology model of ghrelin O-acyltransferase enzyme embedded in lipid bilayer environment and (B) the backbone RMSD plots.
Docking and pharmacophore model generation
The GOAT's unique function and its effect on ghrelin activation provided the potential opportunity to design and develop new inhibitors for the management of diabetes and obesity. In recent years, many inhibitors such as peptidomimetics, small molecules, and terpenoids have been reported as GOAT inhibitors (20). Due to the concern about the bio-stability of peptide inhibitors, the design of small-molecule inhibitors was considered.
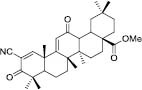
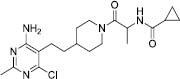
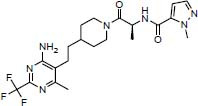
To investigate the possible interaction between existing inhibitors and the homology model of the GOAT, some derivatives with a high biological inhibitory effect were selected from each category of inhibitors and were docked into the active site of the GOAT enzyme which is around the key residues Asp307 and His338. The chemical structures of the inhibitors and docking binding energy and also the inhibitory concentration are listed in Table 1.
Table 1.
Reported ghrelin O-acyltransferase enzyme inhibitor and the result of docking and pharmacophore screening.
| No. | Structure | Reported IC50 (nM) | Binding energy (kcal/mol) | No. of retrieved hit compounds |
|---|---|---|---|---|
| 1 |
|
6000 | -8.47 | 934 |
| 2 |
|
192 | -7.02 | 124 |
| 3 |
|
69 | -7.77 | 564 |
| 4 |
|
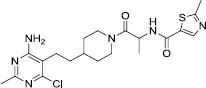
96.4 | -7.92 | 8 |
| 5 |
|
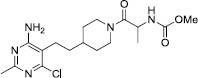
149 | -7.06 | 1855 |
| 6 |
|
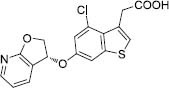
50 | -7.99 | 4 |
| 7 |
|
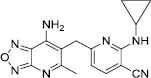
0.02 | -8.96 | 1267 |
| 8 |
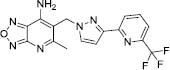
|
0.028 | -8.98 | 0 |
| 9 |
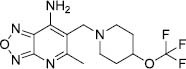
|
0.56 | -5.75 | 42 |
| 10 |
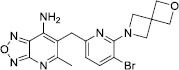
|
0.02 | -9.06 | 2170 |
Subsequently, the protein-ligand complexes of the docking results were considered for pharmacophore mapping features of hydrogen bond donors (HBD) and acceptors (HBA), aromatic rings (AR), negative and positive ionizable (NI and PI) areas and hydrophobic areas (HY) to find the functional groups from different complexes to be able to cover all the main possible interactions and features between the inhibitors and GOAT enzyme. The superimpose position of all inhibitors in the active site of GOAT is shown in [Figure 6A]. Terpene inhibitors, known as antiangiogenic and antitumor agents, were discovered as GOAT inhibitors by screening the NCI library. Compound 1 was the most potent derivative with methyl ester substituent from this category (27). Docking results indicated that compound 1 stabilized in the active site by forming the hydrogen bond with Tyr255 and hydrophobic interactions with Leu180, Trp259, and Trp337. So, the pharmacophore map contains the three hydrophobic areas and one hydrogen bond acceptor feature (Fig. 6B).
Fig. 6.
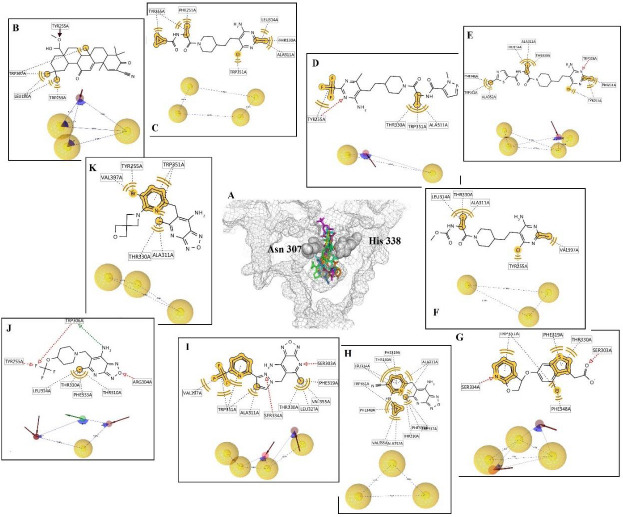
(A) The 3D display of the reported inhibitor in the active site of ghrelin O-acyltransferase enzyme. In the 2D display, the yellow line and sphere are related to hydrophobic interactions; the green and red dash indicate the hydrogen bond donor and acceptor, respectively. In the pharmacophore map, the yellow sphere means the hydrophobic area, and the green and red flashes mean the hydrogen bond donor and acceptor features, respectively. (B-K) compounds 1-10, respectively.
Pyrimidine-substituted compounds (2,3,4,5) were introduced as inhibitors of GOAT by the pharmaceutical company of Eli Lilly (28). Compound 2 was docked into the active site with the major hydrophobic interactions by Phe251, Tyr255, Ala311, Leu314, Thr330, and Trp351. Therefore, the hydrophobic areas are the dominant pharmacophore map [Figure 6C].
Compound 3 formed several hydrophobic interactions with Tyr255, Ala311, Thr330, and Trp351 and another interaction with Tyr255 through a hydrogen bond. The pharmacophore model contains two hydrophobic areas and one hydrogen bond acceptor (Fig. 6D).
Compound 4 interacts with the active site of GOAT through hydrophobic interactions with Phe251, Tyr255, Ala311, Leu314, Thr330, Phe348, Trp351, and Ala352. And also forms one hydrogen bond with Trp306. Four hydrophobic areas and one hydrogen bond acceptor are the features of the pharmacophore model (Fig. 6E).
Compound 5 trapped in the active site of GOAT through hydrophobic interactions with Tyr255, Ala311, Leu314, Thr330, and Val397. The pharmacophore map contains three hydrophobic areas (Fig. 6F).
Compound 6 was disclosed by the pharmaceutical company of GlaxoSmithKline as a potent inhibitor against GOAT with bicyclic pyridocyclopentyl ether scaffold (29). Compound 6 accommodated in the active site by forming the hydrophobic interactions with Phe319, Thr330, Phe348, and Trp351, and also the hydrogen bonds were observed with Ser303 and Ser334. Two hydrogen bond acceptors and three hydrophobic areas are the features of the pharmacophore map (Fig. 6G).
Compounds 7-10 with oxadiazolopyridine scaffold were revealed as a potent inhibitor of GOAT by Boehringer Ingelheim Company (30). Compound 7 interacted with the active site through multiple hydrophobic interactions with Thr310, Ala311, Leu314, Phe319, Thr330, Phe333, Trp337, Phe348, Trp351, Ala352, and Val355, which occurred due to the presence of cyclopropyl ring and methyl substitution. So, the pharmacophore map consists of three hydrophobic areas (Fig. 6H).
Compound 8 established many hydrophobic interactions with Ala311, Phe319, Leu327, Thr330, Trp351, and Val397. Same as compound 6, Ser303 and Ser334 are involved in forming hydrogen bonds. The pharmacophore model included four hydrophobic areas and two hydrogen bond acceptor features (Fig. 6I).
Compound 9 was involved in hydrophobic interactions with Thr310, Leu314, Thr330, and Phe333. Two hydrogen bonds were formed through Tyr255, Arg304, and two hydrogen bonds with Trp306. In particular, this compound presents the pharmacophore model with the hydrogen bond donor group and also two hydrogen bonds acceptor, and one hydrophobic area (Fig. 6J).
Compound 10 interacts with the active site with the lowest binding energy -9.06 kcal/mol by forming several hydrophobic interactions with Tyr255, Ala311, Thr330, Trp351, and Val397. The pharmacophore model contains three hydrophobic areas (Fig. 6K).
According to Fig. 6, the optimal pharmacophore features are the hydrophobic area and the hydrogen bond donor in some models. All ten pharmacophore models were individually used as filters for virtual screening over the SWEETLEAD database.
The acyl-CoA and ghrelin binding site on GOAT
The biological activities of ghrelin need the octanoylation of the Ser3, which is catalyzed by the GOAT enzyme (4). However, the exact binding locations of the acyl-CoA and ghrelin on the GOAT enzyme and the mechanism of its action have remained unclear. However, two approaches to catalytic activity seem to be possible. The first possible process is related to the two-step ping-pong mechanism, which is done by an acylated intermediate of the GOAT in the same way as seen in DHHC acyltransferases (31). The next GOAT mechanism is related to the design of the bisubstrate analog of the GO-CoA-Tat inhibitor, which was inspired by a ternary complex of acyl-CoA, ghrelin, and GOAT. The inhibitory effect of the ghrelin acylation by this inhibitor indicates the presence of two binding sites and the possibility of synchronous transmission of the acyl group (20). So, it's expected that both substrates should be placed close together on the GOAT till the transferring of the octanoyl group could occur. For this purpose, two consecutive docking procedures were performed. First, the structure of the octanoyl-CoA was docked over the GOAT enzyme by the AutoDock package, and then the complex of GOAT and octanoyl-CoA was used as a receptor for docking with ghrelin by the PatchDock server. The ghrelin PDB structure was obtained from the Beevers et al. group (32). The docking result indicated that octanoyl-CoA is located in the predicted tunnel close to the key residue with a docking binding energy of -2.55 kcal/mol. The octanoyl-CoA interacts with the GOAT by forming the three hydrogen bonds with Trp306, key residue Asp307, and Trp337. Several van der Waals interactions were formed, especially with key residue His338. His98 is also involved in pi-sulfur interaction. Besides the pi interaction with Val397 observed. The top docking result of the PatchDock proposed that ghrelin placed on the GOAT's surface near the octanoyl-CoA, probably the octanoyl-CoA and ghrelin get closer during the transition state, and the octanoylation occurs Fig. 7.
Fig. 7.
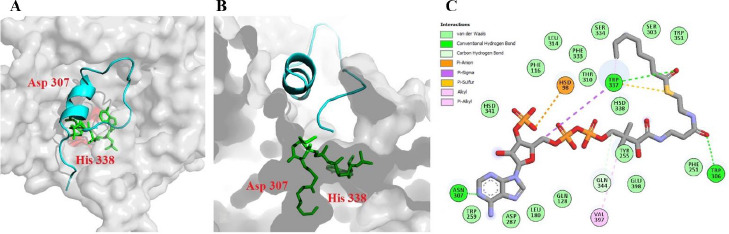
Docking result and the placement of octanoyl-CoA and ghrelin over the GOAT enzyme. (A) The surface shows octanoyl-CoA in green and ghrelin in cyan; (B) the cutaway mode of the placement of octanoyl-CoA in green and ghrelin in cyan; (C) the 2D interactions of octanoyl-CoA with GOAT enzyme. GOAT, Ghrelin O-acyltransferase enzyme.
Pharmacophore-based virtual screening
Virtual screening is a beneficial method to predict the potential interactions between the ligand and receptor and the efficient strategy to discover the hit compound in an express and economical way. Virtual screening based on the generated pharmacophore map was performed over the SWEETLEAD database consisting of 9127 compounds of approved drugs, herbal isolates, and regulated chemical compounds with the aid of LigandScout (Ver. 3.1) software to discover the potential hit compounds with the focus on pharmacophore features. The number of extracted hit compounds from each pharmacophore map was listed in Table 1. About 6968 compounds were found based on being fit with pharmacophore features. To ensure that the proposed final compounds have suitable binding energy and proper interactions with the active site of GOAT, all extracted hit compounds from pharmacophore-based virtual screening were docked into the active site of GOAT. The docking results were sorted based on the docking binding energy. The top five compounds were selected from each pharmacophore map, which leads to the retrieval of 27 hit compounds out of 9127 starting points. The virtual screening of the pharmacophore map of compound 8 retrieved zero hit compound, so there is no information on this map in [Table 2).
Table 2.
The structures and mechanism of actions of the top retrieved compounds from the final docking study.
| Pharmacophore map | Official name of hit compounds and mechanism of action | Structure | Pharmacophore fit score | Binding energy (kcal/mol) |
|---|---|---|---|---|
Compound 1
|
Indian approved Bisoctrizole UV-filter |
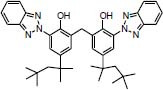
|
46.23 | -11.21 |
| Traditional herbal isolate Prosapogenin induces apoptosis in human cancer |
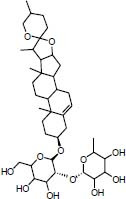
|
46.48 | -11.14 | |
| NPC approved Narasin antibacterial |
|
45.69 | -10.96 | |
Compound 2
|
Traditional herbal isolate Physoxanthin Pro-vitamin A |
|
46.18 | -10.61 |
| Traditional herbal isolate Withaphysalin anticancer |
|
45.97 | -10.44 | |
| Traditional herbal isolate Alpha-carotene-5 6-epoxide |
|
45.85 | -9.70 | |
Compound 3
|
Traditional herbal isolate Kaempferitrin Antidiabetic |
|
36.53 | -11.42 |
| Traditional herbal isolate 7-methoxy baicalein |
|
37.53 | -10.45 | |
| FDA approved Conivaptan inhibitor of antidiuretic hormone |
|
36.88 | -10.41 | |
Compound 4
|
FDA approved Montelukast Eukotriene receptor antagonist |
|
55.72 | -8.03 |
| China approved Penfluridol antipsychotic |
|
56.94 | -7.51 | |
| NPC approved Manidipine antihypertensive |
|
54.96 | -7.27 | |
Compound 5
|
FDA Approved Rifaximin antibiotic |
|
37.84 | -10.49 |
| FDA Approved Irinotecan antineoplastic |
|
37.97 | -10.45 | |
| Traditional herbal isolate Periplocin antitumor |
|
36.98 | -10.29 | |
Compound 6
|
FDA approved Halofantrine antimalarial |
|
55.50 | -7.490 |
| FDA approved Dexlansoprazole proton pump inhibitor |
|
56.06 | -6.79 | |
| NPC approved Ethaverine Peripheral vasodilators |
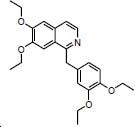
|
57.33 | -6.31 | |
Compound 7
|
Traditional herbal isolate Guggulsterol anti-inflammatory |
|
37.39 | -10.04 |
| Traditional herbal isolate Tigogenin induces apoptosis in rheumatoid arthritis |
|
35.70 | -9.90 | |
| Traditional herbal isolate Solanine pneumonia |
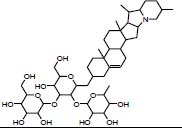
|
36.18 | -9.83 | |
Compound 9
|
Traditional herbal Isolate Darutoside antiandrogen |
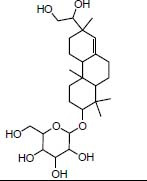
|
55.17 | -9.89 |
| NPC approved Reglitazar Anti-diabetic |
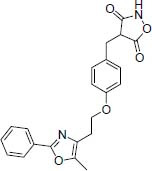
|
53.88 | -8.28 | |
| Traditional herbal isolate Kaempferol-3-Galactoside antimicrobial agent |
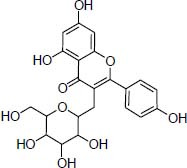
|
55.60 | -8.25 | |
Compound 10
|
FDA approved Ergocalciferol vitamin D analog |
|
35.91 | -10.51 |
| Traditional herbal isolate Methyl amentoflavone inhibitor of CYP3A4 |
|
35.32 | -10.48 | |
| Traditional herbal isolate Hemerocallin |
|
37.61 | -10.32 |
As seen in Table 2, the top three compounds based on the pharmacophore fit score are ethaverine, penfluridol, and dexlansoprazole with a value of 57.33, 56.94, and 56.06 on their relative pharmacophore map, respectively. Based on the docking binding energy, the top three compounds are kaempferitrin, bisoctriazole, and prosapogenin with binding energies of -11.42, -11.21, -11.14 kcal/mol, respectively.
Kaempferitrin, kaempferol-3-galactoside, and methyl amentoflavone contain flavonoid's scaffold.
Some hits contain the triterpenoid core, such as prosapogenin, withaphysalin, periplocin, guggulsterol, tigogenin, solanine, and β-sitosterol- β -d-glucopyranoside.
Overall, most compounds are traditional herbal isolates; while some compounds are from FDA-approved drugs like conivaptan, montelukast, rifaximin, irinotecan, and halofantrine.
Three compounds, ethaverine with the highest pharmacophore fit score (57.33), kaempferitrin with the highest binding energy (-11.43 kcal/mol), and reglitazar with previously known anti-diabetic properties were chosen for further investigation and visualization. As seen in Fig. 8, all three compounds placed in the cavity which contained the key amino acids Asp307 and His338 and were in the same place that octanoyl-CoA was placed, but with better docking binding energy therefore these three compounds will probably prevent the placement of the octanoyl-CoA and show their inhibitory effects (Fig. 8A).
Fig. 8.
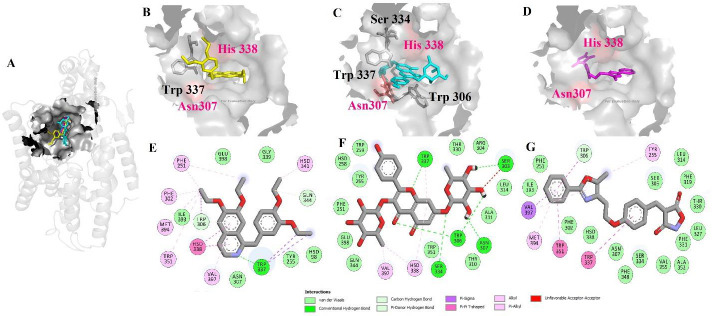
Docking results of (A) the superimpose position of three compounds ethaverine, kaempferitrin, and reglitazar over the ghrelin O-acyltransferase enzyme. 3D and 2D representation of (B and E) ethaverine, (C and F) kaempferitrin, and (D and G) reglitazar.
Ethaverine, the ethyl analog of papaverine, is a peripheral vasodilator. This agent relaxes all smooth muscles. It notes that anorexia is one of its adverse effects, which would be associated with inhibition of the GOAT enzyme (33). Ethaverine stabilized in the cavity by forming the hydrogen bond with Trp337. Interestingly His338 forms the pi interactions with the isoquinoline group and several pi interactions were seen with Phe251, Phe302, His341, Trp351, Met394, and Val397 as seen in Fig. 8B.
Kaempferitrin is herbal isolation from bauhinia forficate, which is used as an antidiabetic agent in Brazil. It is worth noting that kaempferitrin leads to a decrease in blood glucose and also stimulates glucose uptake as much as insulin. Thus, this compound is known as an insulin-mimetic remedy (34). Kaempferitrin was placed well in the cavity, forming five hydrogen bonds with Ser303, Trp306, Asp307, Ser334, and also Trp337; less pi interaction was seen in comparison with ethaverine (Fig. 8C).
Reglitazar is an isoxazolidine-3,5-dione derivative agent with a promising antidiabetic effect on non-insulin-dependent diabetes mellitus patients (35). Reglitazar stabilized in the cavity with the aid of pi interactions with Tyr 255, Trp337, Trp351, Met394, and Val397. Besides, no hydrogen bond was seen (Fig. 8D).
Generally, all 27 compounds could be regarded as potential GOAT inhibitors to treat obesity or type 2 diabetes, however, compounds ethaverine, kaempferitrin, and reglitazar were chosen for further investigation due to the highest binding energy of kaempferitrin (-11.42 kcal/mol) and highest pharmacophore fit score in ethaverine (57.33) and also the antidiabetic property of reglitazar. It seems that kaempferitrin and reglitazar, which are known as antidiabetic agents, probably have an inhibitory effect on GOAT receptor and can be used for further examination and structure optimization and may help the way to reach potential inhibitor of GOAT.
DISCUSSION
The Swiss-Model webserver was used to predict the homology of the GOAT. Two models were generated from the human Hedgehog acyltransferase top template and D-alanyl transfer protein DltB. Based on the Ramachandran plot's result and the similarity between the superfamily of the GOAT and DltB, the homology model by the template of the DltB was selected for the rest of the study. Then the knowledge-based ab-initio method of the AIDA server was utilized for generating the missing residues in the first model. Finally, loop refinement and energy minimizationwere performed for the sake of increasing the quality of the 3D structures of the homology model.
Interestingly, the possible tunnel inside the model was predicted by the CAVER 3.0.1, and the key residues Asp307 and His338 are found in this tunnel and may indicate the location of the enzyme's active site. As previously mentioned, the GOAT is a trans membranes protein, so to increase the compatibility of the generated model in the hydrophobic conditions of the membrane, the bilayer environment was created with the aid of CHARMM_GUI and generated homology model embedded in the membrane. Then 200 ns of molecular dynamics simulation were performed using the GROMACS package. The generated homology model of the GOAT reached the equilibrium state after 50 ns of simulations. Finally, the output of this step was used as the optimal model of GOAT for further investigation. Subsequently, a docking procedure was performed over the selected existing inhibitors of the GOAT and the active site of the generated homology model to investigate the posture of the interactions. Then to cover all important functional groups in the active site of the GOAT, multiple pharmacophore mapping was done with the aid of the complexes of docking results (36). The result indicated that the hydrophobic area feature was more frequent in pharmacophore maps. Then to speed up the filtering process in discovering the novel hit compounds for the GOAT enzyme, the generated pharmacophore map was used as a filter for virtual screening over the SWEETLEAD database (37). The pharmacophore-based screening helps to increase the accuracy in selecting the hit compound from the diverse structures. GOAT enzyme catalyzes the octanoylation of the Ser3 of the ghrelin with the aid of the acyl-CoA. So, to investigate the possible binding site of the acyl-CoA and ghrelin on the GOAT enzyme, a docking procedure was done. The docking result showed the strong hydrogen bond forming between octanoyl-CoA and the key residue Asp307, and also van der Waals interaction with residue His338. This placement probably contributes to the catalytic process of the enzyme and separates the octanoyl fraction. The ghrelin docking placement was also near the octanoyl-CoA. However, the exact mechanism of the action of the GOAT has remained unclear.
Inhibitors against GOAT enzyme that have been reported before have scaffolds such as terpenes, bicyclic pyridocyclopentyl ether, oxadiazolopyridine, and others(20), however, the result of the pharmacophore-based virtual screening from the 9127 compounds suggests that ethaverine with isoquinolines scaffold, kaempferitrin with flavonoid scaffold, and reglitazar oxazolidine-dione and oxazole moiety which have not been seen in previously reported inhibitors or have been used less, therefore, all three compounds with novel scaffold probably have a potential inhibitory effect on GOAT and may be used as starting point for structure optimization for the discovery of novel GOAT inhibitor.
CONCLUSION
Ghrelin is a 28 amino-acid peptide known as a hunger hormone that plays a role in appetite, food intake, energy balance, glucose metabolism, and insulin secretion and is a potential target for the treatment of obesity and type 2 diabetes. Ghrelin requires a series of modifications before reaching the active form of 28 amino-acid lengths of ghrelin. An essential posttranslational maturation step of ghrelin is the octanoylation of the Ser3, which is catalyzed by the GOAT enzyme, so the inhibition of GOAT may be useful for the treatment of ghrelin-related disease. The hydrophobic nature of the GOAT enzyme has led to the difficulty in determining its structure by X-ray or cryo-electron microscopy. In this study, with the aid of the computational method, the homology model of GOAT was developed, and its stereochemical quality was assessed and improved. To stabilize the homology modeled in the lipid bilayer, the molecular dynamics simulation was carried out for 200 ns, and the stable structure was used as input for the rest of the study. The consecutive process of docking, pharmacophore mapping, virtual screening, and again docking led to finding the potential hit compound against GOAT enzyme. These compounds, ethaverine, kaempferitrin, and reglitazar were introduced as optimal options for GOAT inhibition. The results of this study could be optimized and may provide a starting point for further investigation for drug design in the case of GOAT inhibitors.
Conflict of interest statement
The authors declared no conflict of interest in this study.
Authors’ contribution
F.S. Hosseini participated in the investigation, methodology, software, visualization, and drafting of the manuscript; A. Ghassempour conceptualized the study; and M. Amanlou contributed to conceptualization, methodology, supervision, and project administration, funding acquisition, and writing, review, and editing of the article. The final version of the article was approved by all authors.
Acknowledgments
This study was financially supported by the Research Council of Tehran University of Medical Sciences and Health Services, Tehran, Iran (Grant No. 98-3-104-46109).
REFERENCES
- 1.Diano S, Farr SA, Benoit SC, McNay EC, da Silva I, Horvath B, et al. Ghrelin controls hippocampal spine synapse density and memory performance. Nat Neurosci. 2006;9(3):381–388. doi: 10.1038/nn1656. doi: 10.1038/nn1656. [DOI] [PubMed] [Google Scholar]
- 2.Lim CT, Kola B, Korbonits M. The ghrelin/GOAT/GHS-R system and energy metabolism. Rev Endocr Metab Disord. 2011;12(3):173–186. doi: 10.1007/s11154-011-9169-1. doi: 10.1007/s11154-011-9169-1. [DOI] [PubMed] [Google Scholar]
- 3.Gahete MD, Córdoba-Chacón J, Kineman RD, Luque RM, Castaño JP. Role of ghrelin system in neuroprotection and cognitive functions: implications in Alzheimer's disease. Peptides. 2011;32(11):2225–2228. doi: 10.1016/j.peptides.2011.09.019. doi: 10.1016/j.peptides.2011.09.019Get. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu X, Cao Y, Voodg K, Steiner DF. On the processing of proghrelin to ghrelin. J Biol Chem. 2006;281(50):38867–38870. doi: 10.1074/jbc.M607955200. doi: 10.1074/jbc.M607955200. [DOI] [PubMed] [Google Scholar]
- 5.Hofmann K. A superfamily of membrane-bound O-acyltransferases with implications for wnt signaling. Trends Biochem. Sci. 2000;25(3):111–112. doi: 10.1016/s0968-0004(99)01539-x. doi: 10.1016/s0968-0004(99)01539-x. [DOI] [PubMed] [Google Scholar]
- 6.Masumoto N, Lanyon-Hogg T, Rodgers UR, Konitsiotis AD, Magee AI, Tate EW. Membrane bound O-acyltransferases and their inhibitors. Biochem Soc Trans. 2015;43(2):246–252. doi: 10.1042/BST20150018. doi: 10.1042/BST20150018. [DOI] [PubMed] [Google Scholar]
- 7.Taylor MS, Ruch TR, Hsiao P-Y, Hwang Y, Zhang P, Dai L, et al. Architectural organization of the metabolic regulatory enzyme ghrelin O-acyltransferase. J Biol Chem. 2013;288(45):32211–32228. doi: 10.1074/jbc.M113.510313. doi: 10.1074/jbc.M113.510313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buglino JA, Resh MD. Identification of conserved regions and residues within hedgehog acyltransferase critical for palmitoylation of Sonic hedgehog. PLoS One. 2010;5(6):e11195, 1–10. doi: 10.1371/journal.pone.0011195. doi: 10.1371/journal.pone.0011195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taylor MS, Dempsey DR, Hwang Y, Chen Z, Chu N, Boeke JD, et al. Mechanistic analysis of ghrelin-O-acyltransferase using substrate analogs. Bioorg Chem. 2015;62:64–73. doi: 10.1016/j.bioorg.2015.07.003. doi: 10.1016/j.bioorg.2015.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma D, Wang Z, Merrikh CN, Lang KS, Lu P, Li X, et al. Crystal structure of a membrane-bound O-acyltransferase. Nature. 2018;562(7726):286–290. doi: 10.1038/s41586-018-0568-2. DOI: 10.1038/s41586-018-0568-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwede T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 2003;31(13):3381–3385. doi: 10.1093/nar/gkg520. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu D, Jaroszewski L, Li Z, Godzik A. AIDA: ab initio domain assembly server. Nucleic Acids Res. 2014;42(W1):W308–W313. doi: 10.1093/nar/gku369. doi: 10.1093/nar/gku369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heo L, Park H, Seok C. GalaxyRefine: protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013;41(W1):W384–W388. doi: 10.1093/nar/gkt458. doi: 10.1093/nar/gkt458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krieger E, Joo K, Lee J, Lee J, Raman S, Thompson J, et al. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: four approaches that performed well in CASP8. Proteins. 2009;77(Suppl9):114–122. doi: 10.1002/prot.22570. doi: 10.1002/prot.22570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hashemi S, Sharifi A, Zareei S, Mohamedi G, Biglar M, Amanlou M. Discovery of direct inhibitor of KRAS oncogenic protein by natural products: a combination of pharmacophore search, molecular docking, and molecular dynamic studies. Res Pharm Sci. 2020;15(3):226–240. doi: 10.4103/1735-5362.288425. doi: 10.4103/1735-5362.288425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laskowski RA. PDBsum: summaries and analyses of PDB structures. Nucleic Acids Res. 2001;29(1):221–222. doi: 10.1093/nar/29.1.221. doi: 10.1093/nar/29.1.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hosseini FS, Amanlou A, Amanlou M. Tankyrase inhibitor for cardiac tissue regeneration: an in-silico approach. Iran J Pharm Res. 2021;20(4):315–328. doi: 10.22037/ijpr.2021.115367.15339. doi: 10.22037/ijpr.2021.115367.15339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1:19–25. doi: 10.1016/j.softx.2015.06.001. [Google Scholar]
- 19.Lee J, Cheng X, Swails JM, Yeom MS, Eastman PK, Lemkul JA, et al. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J Chem Theory Comput. 2016;12(1):405–413. doi: 10.1021/acs.jctc.5b00935. doi: 10.1021/acs.jctc.5b00935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moose JE, Leets KA, Mate NA, Chisholm JD, Hougland JL. An overview of ghrelin O-acyltransferase inhibitors: a literature and patent review for 2010-2019. Expert Opin Ther Pat. 2020;30(8):581–593. doi: 10.1080/13543776.2020.1776263. doi: 10.1080/13543776.2020.1776263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Asadi M, Mohammadi-Khanaposhtani M, Hosseini FS, Gholami M, Dehpour AR, Amanlou M. Design synthesis, and evaluation of novel racecadotril-tetrazole-amino acid derivatives as new potent analgesic agents. Res Pharm Sci. 2021;16(4):341–357. doi: 10.4103/1735-5362.319573. doi: 10.4103/1735-5362.319573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hosseini FS, Amanlou M. Anti-HCV and anti-malaria agent, potential candidates to repurpose for coronavirus infection: Virtual screening, molecular docking, and molecular dynamics simulation study. Life Sci. 2020:118205. doi: 10.1016/j.lfs.2020.118205. doi: 10.1016/j.lfs.2020.118205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wolber G, Langer T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J Chem Inf Model. 2005;45(1):160–169. doi: 10.1021/ci049885e. doi: 10.1021/ci049885e. [DOI] [PubMed] [Google Scholar]
- 24.Novick PA, Ortiz OF, Poelman J, Abdulhay AY, Pande VS. SWEETLEAD: an in silico database of approved drugs, regulated chemicals, and herbal isolates for computer-aided drug discovery. PLoS One. 2013;8(11):e79568. doi: 10.1371/journal.pone.0079568. doi: 10.1371/journal.pone.0079568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Campaña MB, Irudayanathan FJ, Davis TR, McGovern-Gooch KR, Loftus R, Ashkar M, et al. The ghrelin O-acyltransferase structure reveals a catalytic channel for transmembrane hormone acylation. J Biol Chem. 2019;294(39):14166–14174. doi: 10.1074/jbc.AC119.009749. doi: 10.1074/jbc.AC119.009749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shoemaker SC, Ando N. X-rays in the cryo-electron microscopy era: Structural biology's dynamic future. Biochemistry. 2018;57(3):277–285. doi: 10.1021/acs.biochem.7b01031. doi: 10.1021/acs.biochem.7b01031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McGovern-Gooch KR, Mahajani NS, Garagozzo A, Schramm AJ, Hannah LG, Sieburg MA, et al. Synthetic triterpenoid inhibition of human ghrelin O-acyltransferase: the involvement of a functionally required cysteine provides mechanistic insight into ghrelin acylation. Biochemistry. 2017;56(7):919–931. doi: 10.1021/acs.biochem.6b01008. doi: 10.1021/acs.biochem.6b01008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Galka CS, Hembre EJ, Honigschmidt NA, Keding SJ, Martinez-Grua MA, Plaza GR, et al. inventors; Preparation of n-acylamino acid derivatives as Preparation of n-acylamino acid derivatives as ghrelin O-acyl transferase inhibitors. USA. 2016. Pub. NO:WO/2016/168225 A1. International Application No:PCT/EP2019/027180.
- 29.Bandyopadhyay A, Cheung M, Eidam HS, Joshi H, Su D-S. inventors; Preparation of ghrelin O-acyltransferase inhibitors for treatment of metabolic disorders. UK. 2019. Pub. NO. WO/2019/149959 A1. International Application No:PCT/US2016/052770.
- 30.Godbout C, Trieselmann T, Vintonyak V. inventors; Preparation of oxadiazolopyridine derivatives for use as ghrelin O-acyl transferase (GOAT) inhibitors. Germany. 2018. NO. WO/2018/024653 A1. International Application No:PCT/EP2017/069274.
- 31.Jennings BC, Linder ME. DHHC protein S-acyltransferases use similar ping-pong kinetic mechanisms but display different acyl-CoA specificities. J Biol Chem. 2012;287(10):7236–7245. doi: 10.1074/jbc.M111.337246. DOI: 10.1074/jbc.M111.337246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beevers AJ, Kukol A. Conformational flexibility of the peptide hormone ghrelin in solution and lipid membrane bound: a molecular dynamics study. J Biomol Struct Dyn. 2006;23(4):357–364. doi: 10.1080/07391102.2006.10531231. doi: 10.1080/07391102.2006.10531231. [DOI] [PubMed] [Google Scholar]
- 33.Fleckenstein A, Grün G, Döring HJ, Haastert HP, Tritthart H. Ethaverin a coronary dilator with beta-receptor depressing and antiarrhythmia myocardial effects. Die Medizinische Welt. 1973;24(12):441–449. PMID: 4144783. [PubMed] [Google Scholar]
- 34.Jorge AP, Horst H, de Sousa E, Pizzolatti MG, Silva FR. Insulinomimetic effects of kaempferitrin on glycaemia and on 14C-glucose uptake in rat soleus muscle. Chem Biol Interact. 2004;149(2-3):89–96. doi: 10.1016/j.cbi.2004.07.001. doi: 10.1016/j.cbi.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 35.Shibata T, Matsui K, Nagao K, Shinkai H, Yonemori F, Wakitani K. Pharmacological profiles of a novel oral antidiabetic agent, JTT-501, an isoxazolidinedione derivative. Eur J Pharmacol. 1999;364(2-3):211–219. doi: 10.1016/s0014-2999(98)00832-2. doi: 10.1016/S0014-2999(98)00832-2. [DOI] [PubMed] [Google Scholar]
- 36.Razzaghi-Asl N, Mirzayi S, Mahnam K, Adhami V, Sepehri S. In silico screening and molecular dynamics simulations toward new human papillomavirus 16 type inhibitors. Res Pharm Sci. 2022;17(2):189–208. doi: 10.4103/1735-5362.335177. doi: 10.4103/1735-5362.335177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mojaddami A, Sakhteman A, Fereidoonnezhad M, Faghih Z, Najdian A, Khabnadideh S, et al. Binding mode of triazole derivatives as aromatase inhibitors based on docking, protein ligand interaction fingerprinting, and molecular dynamics simulation studies. Res Pharm Sci. 2017;12(1):21–30. doi: 10.4103/1735-5362.199043. doi: 10.4103/1735-5362.199043. [DOI] [PMC free article] [PubMed] [Google Scholar]