

Abstract

Nicotinamide adenine dinucleotide kinase (NADK) controls the intracellular NADPH content and provides reducing power for the synthesis of macromolecules and anti-ROS. Moreover, NADK is considered to be a synthetic lethal gene for KRAS mutations. To discover NADK-targeted probes, a high-throughput screening assay was established and optimized with a Z factor of 0.71. The natural product (−)-epigallocatechin gallate (EGCG) was found to be a noncompetitive inhibitor of NADK with Ki = 3.28 ± 0.32 μΜ. The direct binding of EGCG to NADK was determined by several biophysical methods, including NMR spectroscopy, surface plasmon resonance (SPR) assay, and hydrogen–deuterium exchange mass spectrometry (HDX-MS). The SPR assay showed a Kd of 1.78 ± 1.15 μΜ. The HDX-MS experiment showed that EGCG was bound at the non-substrate-binding sites of NADK. Besides, binding mode prediction and derivative activity analysis revealed a potential structure–activity relationship between EGCG and NADK. Furthermore, EGCG can specifically inhibit the proliferation of KRAS-mutated lung cancer cell lines without affecting KRAS wild-type lung cancer cell lines.
Keywords: NAD kinase inhibitor, (−)-epigallocatechin gallate, hydrogen−deuterium exchange mass spectrometry, KRAS
Nicotinamide adenine dinucleotide (NAD+/NADH) and nicotinamide adenine dinucleotide phosphate (NADP+/NADPH) are two key cofactors in intracellular redox reactions,1 of which NAD+/NADH is called coenzyme I and NADP+/NADPH is called coenzyme II. NAD+/NADH are the key cofactors in cellular catabolism and energy synthesis. NADP+/NADPH is primarily involved in anabolism and provides reducing power for intracellular anti-ROS reactions. NADP+ is a key cofactor required by the pentose phosphate pathway (PPP), through which NADP+ is reduced to NADPH. The PPP produces NADPH and ribose 5-phosphate (R5P) and is essential for cell survival and proliferation. Therefore, the PPP has been shown to be a potential therapeutic target for cancer treatment.2
Rapidly proliferating tumor cells require a large amount of NADPH for the biosynthesis and neutralization of high reactive oxygen species (ROS).3 Therefore, inhibiting NADPH production may represent a novel direction of cancer treatment. NAD kinase (NADK) phosphorylates NAD+ to NADP+,4 and NADP+ is rapidly converted to NADPH by various and abundant intracellular reductases. Therefore, NADK is critical for controlling the intracellular NADP+/NADPH pool.5 It has been reported that NADK overexpression promotes cellular NADPH content and reduces ROS-induced DNA damage and cell death, whereas knockdown of NADK reduces cellular NADPH content and increases ROS-induced DNA damage and cell death.6,7
KRAS mutations are the most frequently mutated oncogenes in tumors such as pancreatic (90%), colorectal (30–40%), and lung (15–20%) tumors.8 Over the past few decades, KRAS structure, biochemistry, and biology have been intensively studied for “anti-KRAS” therapy. Sotorasib, a covalent small-molecule inhibitor of KRAS-G12C developed by Amgen, was approved by the U.S. Food and Drug Administration (FDA) for the treatment of nonsmall cell lung cancer with KRAS-G12C mutation.9,10 Mirati Therapeutics also announced its KRAS-G12D-selective inhibitor MRTX1133.11 However, targeting non-KRAS-G12C/D still seems elusive, and inhibitors targeting KRAS-G12C have developed drug resistance after clinical use.12,13 Attempts to find genes, signaling pathways, or metabolic processes that have a synthetic lethal relationship with KRAS mutations have shown great potential for drug development.14,15 Using the CRISPR-Cas9 tool, Yau et al.16 determined many metabolic enzymes with a synthetic lethal relationship to KRAS mutations in an in vivo colorectal cancer system, including NADK. In HCT116 cells with normal KRAS, knockout of NADK or addition of NADK inhibitor did not affect the tumorigenic growth of tumor cells, but in HCT116 with KRAS-activating mutation, knockout of NADK or addition of an NADK inhibitor significantly reduced the tumor growth. Thus, NADK is considered a potential synthetic lethal target in KRAS-activated tumors.
For the design of inhibitors of NADK, it is generally believed that binding to the ATP-binding domain may not be a good choice because the ATP-binding domain of NADK is solvent-exposed and does not interact closely with the enzyme.17 Several NADK inhibitors have been reported, all of which are NAD analogues, such as benzamide adenine dinucleotide (BAD) (Ki = 90 μM), di-5′-thioadenosine (DTA) (Ki = 45 μM), 8Br-DTA (Ki = 6 μM), and Di-8Br-DTA (Ki = 6 μM), which showed weak inhibitory activity against human NADK.18 However, BAD was found to cause skeletal muscle loss and hepatotoxicity in animal toxicity research. Thionicotinamide (TN), a prodrug of the NADK inhibitor, is transformed to NADPS and acts as an analogue of NAD with an IC50 of approximately 10 μM in the CEM-CCRF, MOLT-4, RL, and C85 cell lines (Figure 1).17,19
Figure 1.

Reported inhibitors of NADK. Abbreviations: BAD, benzamide adenine dinucleotide; DTA, di-5′-thioadenosine; TN, thionicotinamide.
NADK has been proved to be a potential target in cancer, but existing inhibitors are all NAD analogues with poor selectivity. There is an urgent need to identify potent and selective NADK inhibitors. In the present work, by developing and optimizing a high-throughput screening assay for NADK, we found that the natural product (−)-epigallocatechin gallate (EGCG) is a noncompetitive inhibitor of NADK, and we showed that EGCG directly binds to NADK through various biophysical experiments. Docking modeling found that EGCG inhibited NADK with a potential structure–activity relationship. At the same time, EGCG specifically inhibited the proliferation of KRAS mutant A459, H23, and H358 cells but not KRAS wild-type PC-9 cells.
NADK physiologically catalyzes the phosphorylation of NAD+ to NADP+ utilizing ATP as a phosphate donor. NADH and NADPH have an absorption peak at 340 nm in addition to the peak at 260 nm compared with NAD(P). Based on this characteristic, we developed a high-throughput screening (HTS) assay by coupling G6PD with NADK to convert the generated NADP+ to NADPH and then measured the light absorption at 340 nm (Figure 2A).20
Figure 2.

Development and optimization of the high-throughput screening assay of NADK inhibitors. (A) Schematic diagram of the principle of the high-throughput screening assay. Abbreviations: G6PD, glucose-6-phosphate dehydrogenase; NADK, NAD kinase. (B) Parameters for evaluating the quality of the high-throughput screening assay. (C) Flowchart of high-throughput screening.
According to the Michaelis–Menten equation, it must be ensured that within a certain time range, the enzymatic reaction is a first-order reaction in which V is equal to V0. To determine the appropriate reaction conditions, we used different amounts of protein. With a protein amount of 0.5 to 1 μg (100 to 200 nM) in the reaction mixture, the enzyme time course was linear over the first 10 min (Figure S1B). To further determine the substrate concentration used in the screening, we determined the Km values for the two substrates NAD and ATP to be 1.6 and 6.5 mM, respectively (Figure S2). Thus, the final substrate concentrations used were 2 and 4 mM, respectively. The Z factor of the HTS assay was calculated to be 0.71,21 which indicates good quality (Figure 2B).
Many clinical drug candidates are derived from natural products and their derivatives, which provide novel chemical backbones.22 Therefore, we used the established HTS assay to screen an in-house compound library containing 3000 natural products to discover novel NADK inhibitors. The workflow for identifying hits is depicted in Figure 2C. The absorbance at 340 nm was monitored for 15 min at 37 °C in continuous kinetic detection mode. The compounds with percentage of inhibition greater than 70% compared with the DMSO control were chosen for secondary screening. To rule out the effect of inhibitors on G6PD, we performed another enzyme assay: with glucose-6-phosphate (G6P) and NADP+ as substrates, G6PD catalyzes the production of 6-phospho-d-glucono-1,5-lactone and NADPH. We evaluated the IC50 values of the potential compounds in a dose-dependent manner.
We identified several active compounds from more than 3000 compounds with structural similarities. (−)-Gallocatechin and (−)-epigallocatechin exhibited IC50 values greater than 100 μM, and the IC50 value for EGCG was 1.30 ± 1.06 μM (Figure 3A,B). EGCG was chosen for further study since it has the strongest inhibitory effect. The EGCG–NADK interaction was verified using ligand-based NMR spectroscopy. Carr–Purcell–Meiboom–Gill (CPMG) and saturation transferred difference (STD) spectroscopy indicated that EGCG binds specifically to NADK (Figure 3C). A surface plasmon resonance (SPR)-based binding assay was carried out to determine the binding affinity of EGCG with NADK, and the results showed a Kd of 1.78 ± 1.15 μΜ (Figure 3D).
Figure 3.

Discovery and identification of EGCG as an inhibitor targeting NADK. (A) Chemical structures of several candidate compounds that were screened from an in-house compound library. (B) Determination of the IC50 of EGCG using the enzymatic activity assay. (C) Carr–Purcell–Meiboom–Gill data from the incubation of 200 μΜ EGCG with 10 μM NADK. (D) Determination of the affinity of EGCG and NADK by the SPR assay. (E) EGCG is a noncompetitive inhibitor of NADK with Ki = 3.28 ± 0.32 μΜ. Data were fitted using GraphPad Prism software in noncompetitive mode. The IC50, Kd, and Ki values are shown as mean ± SD.
We aimed to study the mechanism of action of EGCG to inhibit NADK. A competitive experiment showed that EGCG does not compete with the substrates NAD and ATP to exert an inhibitory effect with Ki = 3.28 ± 0.32 μΜ (Figure 3E). This suggests that EGCG does not bind to the substrate pocket of NADK but instead regulates the activity of NADK in an unknown mode.
Hydrogen–deuterium exchange mass spectrometry (HDX-MS) is a novel technology that is widely used to study the dynamic changes, folding, and interactions of protein structures. Changes in the rate of hydrogen–deuterium exchange represent a binding event of the receptor upon ligand binding. Therefore, we studied the interaction of EGCG with NADK using HDX-MS. NADK has 446 amino acids, divided into 95 amino acids at the N-terminus as the regulatory domain and the C-terminal kinase domain, which has complete activity. We purified the kinase domain Δ95 of NADK and tested EGCG for Δ95 with IC50 = 0.96 ± 1.23 μΜ (Figure S6). For this reason, the protein used for HDX-MS was NADK-Δ95. HDX-MS revealed that peptides 410VRDPVSDWF418 (region I), 155GAVKKKFCTF164 (region II), and 345ITPICPHSLSFRPIVVPAGVE365 (region III) showed increased protection against solvent exchange upon EGCG binding (Figure 4A,B). Specifically, regions I, II, and III demonstrated 21.8%, 12.9%, and 21.1% decreases in deuterium uptake upon binding of NADK to EGCG, respectively (Figure 4A,B). The HDX-MS data suggest that these regions are involved in the binding event of EGCG.
Figure 4.

HDX-MS analysis of EGCG-bound NADK. (A) Differential HDX consolidation view mapped to the NADK crystal structure (PDB ID 3PFN). Percentages of deuterium differences are color-coded according to the HDX perturbation key in Figure S6. Black indicates regions that have no sequence coverage and include proline residues that have no amide hydrogen exchange activity; gray indicates no statistically significant changes between the compared states. (B) Deuterium uptake plots in the presence or absence of EGCG at the indicated time points for regions I, II, and III.
The complete structure of the NADK protein has not yet been resolved, and a modified human NADK truncated protein (PDB ID 3PFN) contains residues 68–421 in the PDB database.23 We mapped the peptide identified by HDX-MS to this crystal structure. Consistent with the previous results, none of these regions are substrate binding sites for NADK (Figure 4A).
Molecular docking was performed to explore the binding mode of EGCG with NADK. In the tetrameric structure, region I and region III are spatially close, suggesting that EGCG may bind to these two regions simultaneously. Besides, we tried to dock EGCG to region II, but the docking score was poor, and the pocket formed by region II is too small to accommodate EGCG. In the predicted complex structure, EGCG forms several hydrogen-bonding interactions with R411(A), D412(A), G363(B), and E365(B) to maintain high affinity with NADK (Figure 5). The three compounds obtained by screening have a strong structure similarity. The IC50 value of (−)-gallocatechin is 255.10 ± 1.68 μΜ, and that of (−)-epigallocatechin is 182.80 ± 1.22 μM. We docked the two compounds at the same site. (−)-Gallocatechin forms hydrogen-bonding interactions with D412(A) and G363(B), while (−)-epigallocatechin forms three hydrogen-bonding interactions with R411(A), D412(A), and D304(B), indicating that the pyrogallol structure may be the core group that exerts inhibitory activity. The bonds at C2 and C3 of (−)-epigallocatechin are in the same orientation as in EGCG, and the two bonds of (−)-gallocatechin point in opposite directions, indicating that when the two bonds point in the same direction, the steric hindrance of the compound entering the pocket may be lower, allowing the compound to bind more easily to the protein. As shown in the predicted model, (−)-epigallocatechin and EGCG are closer than (−)-gallocatechin. EGCG is synthesized by esterification of gallic acid with the hydroxyl group at C3. According to the docking model, the hydroxyl on the benzene ring of the gallic acid part forms two hydrogen-bonding interactions with E365(B) of NADK, which may be the reason why its inhibitory activity is greatly improved compared to (−)-epigallocatechin.
Figure 5.

Docking model predicting the interaction of EGCG and NADK. EGCG was docked at the site located between region I and region III on NADK tetramer using Schrödinger software. The right panel is an enlarged view of the binding mode, and the critical residues are shown as sticks. The yellow dotted lines represent hydrogen-bonding interactions. Single letters represent amino acids. Numbers indicate the positions of residues in full-length NADK.
We assessed the effect of EGCG on cell proliferation in lung cancer cell lines. Surprisingly, EGCG hardly affected the proliferation of KRAS wild-type PC-9 cells compared to the DMSO control group. When the concentration of EGCG reached 60 μM, the number of cells decreased significantly at the p < 0.01 level at the fourth day. A549 cells harboring the KRAS G12S mutation showed significantly inhibited proliferation after treatment with 30 μM EGCG. When the treatment concentration reached 50 μM, the growth of A549 cells was completely inhibited. For H358 and H23 cells carrying the KRAS G12C mutation, the growth and proliferation of these two cell lines were completely inhibited when the dose reached 30 μM (Figure 6). Overall, the inhibitory effect of EGCG on KRAS wild-type PC-9 cells was weak, and the inhibitory effect on A549 cells of KRAS G12S was shown at 30 μM, while the inhibition was strongest on H358 and H23 cells of KRAS-G12C, indicating that EGCG has a certain selectivity for lung cancer cell lines with different KRAS mutation types.
Figure 6.
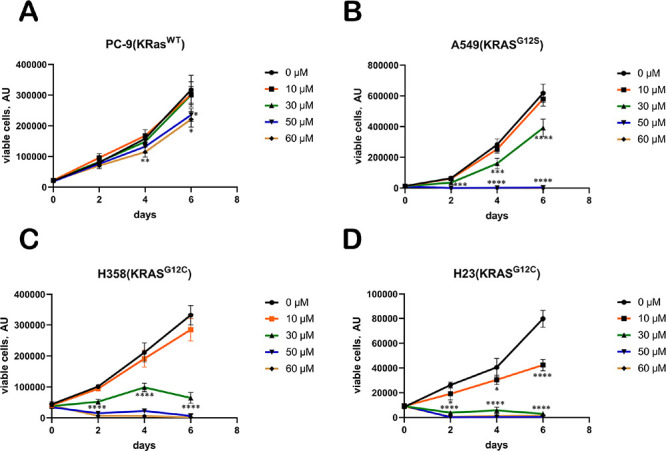
Growth curves of several lung cancer cell lines treated with EGCG at various concentrations (n = 3 replicates): (A) PC-9 cells (KRAS WT); (B) A549 cells (KRAS G12S); (C) H358 cells (KRAS G12C); (D) H23 cells (KRAS G12C). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. Data analysis was performed using the two-tailed unpaired Student’s t test.
NADK controls the cellular NADP+/NADPH content and provides important reducing power for cellular anabolism and resistance to ROS.5,24 The rapid growth of tumor cells requires a large amount of NADP+/NADPH to meet the synthesis requirements of nucleotides, fats, and other macromolecules and to neutralize ROS toxicity.17,24,25 KRAS is the most commonly mutated oncogene in tumors and is considered an untargetable protein for tumors.26 In general, mutant KRAS fosters tumor growth by shifting cancer cell metabolism toward anabolic pathways27 and inducing ROS production. This may render the NADK–NADP+/NADPH pathway vulnerable to KRAS mutant tumor cells. Studies have shown that NADK and KRAS-activating mutations have a synthetic lethal relationship in the process of tumor transformation growth.16,28 The development of small-molecule inhibitors targeting NADK may specifically kill tumor cells with KRAS-activating mutations.29
Although there have been preliminary reports of small-molecule inhibitors of NADK, such as BAD and TN, these compounds all inhibit NADK activity through NAD+ analogues.18,30,31 There are many proteins that bind NAD+ in cells, and it is expected that these compounds can also interfere with other NAD+ binding proteins, which means that these compounds do not achieve selective inhibition of NADK. Although certain cell and mouse experiments have shown that these compounds can effectively inhibit tumor cell growth, there is insufficient evidence that these functions are directly achieved by inhibiting NADK enzymatic activity.18,30,31
We obtained a highly active NADK inhibitor compound EGCG through high-throughput screening, and this compound is not a competitive inhibitor of NAD+ or ATP, indicating that EGCG does not bind to the NAD+ or ATP binding pocket of NADK. In addition, the chemical structure of EGCG is significantly different from that of NAD+ or ATP. This suggests that there are other targetable pockets in NADK. HDX-MS results revealed that region I may be such a pocket that regulates NADK activity in an allosteric manner. The changes observed experimentally in regions II and III may be the protein conformational changes caused by binding of EGCG to NADK. As a natural product, EGCG is widely present in plants such as tea, and the compound backbone is considered safe and worthy of further development.32 However, the water solubility, stability, and in vivo utilization of EGCG are relatively low, and the related modified compounds need to be further studied. The most typical attack energy of EGCG is its antioxidant activity, which plays an important role in anti-inflammatory, anti-cardiovascular disease, and antitumor activities.33 EGCG is a polyhydroxyl structure, which can directly neutralize ROS. EGCG can directly chelate a variety of metal ions, including iron ions, thereby inhibiting membrane lipid oxidation and possibly inhibiting ferroptosis.34 EGCG can directly or indirectly activate NRF2-mediated antioxidant pathways.35 In addition, EGCG can directly or indirectly inhibit the expression of pro-oxidative enzymes such as cyclooxygenase-2 (COX-2) and inducible nitric oxide synthase (iNOS) and inhibit the production of ROS.36
Nevertheless, some studies have found that EGCG has a pro-ROS function. The antioxidant and pro-oxidative activities of EGCG appear to be related to the pH of its environment. For example, EGCG exhibits pro-oxidative activity under low pH (2–4) conditions and exhibits antioxidant activity under high pH (5–7) conditions. EGCG can promote the production of cellular ROS through a certain mechanism. In addition, EGCG promotes the reduction of Fe3+ to Fe2+, which in turn further oxidizes H2O2 to more reactive ROS, such as hydroxyl radicals, through the Fenton reaction.37
There are some reports that EGCG can directly bind to intracellular proteins and exert inhibitory functions. EGCG can bind to G3BP protein (Kd = 0.4 μM) to inhibit its binding to the RAS-GAP protein and cGAS, thereby inhibiting the activation of RAS-cGAS downstream signaling.38,39 EGCG binds to chaperones Pin1 and HSP90 and inhibits their molecular chaperone function.40−42 Many cell membrane receptors are also direct targets of EGCG, including fibronectin receptor 67LR (Kd = 40 nM)43 and TGFβRII,44 which inhibit tumor cell metastasis and migration. We have now found that EGCG is an inhibitor of NADK (Kd = 1.78 ± 1.15 μM), further enriching the complex functions of EGCG in cells and providing a new mechanism by which EGCG regulates cellular ROS. The structural modification of compounds with EGCG as the backbone will help to obtain more efficient NADK inhibitors and provide new therapeutic drugs for tumor treatment. Moreover, the development of inhibitors targeting NADK is also a new strategy for the treatment of KRAS mutant tumors.
Acknowledgments
We are very grateful for the instrumental support and technical support of Shanghai National Protein Science Center (Shanghai Science Research Center Protein Expression and Purification System).
Glossary
Abbreviations
- EGCG
(−)-epigallocatechin gallate
- NADK
nicotinamide adenine dinucleotide kinase
- G6PD
glucose 6-phosphate dehydrogenase
- SPR
surface plasmon resonance
- HDX-MS
hydrogen–deuterium exchange mass spectrometry
- PPP
pentose phosphate pathway
- COX-2
cyclooxygenase-2
- iNOS
inducible nitric oxide synthase
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.2c00163.
Development and optimization of the high-throughput screening assay of NADK inhibitors (Figure S1); Km of the substrate of NADK (Figure S2); enzyme activity curve of EGCG against G6PD (Figure S3); STD data for incubation of 200 μΜ EGCG with 10 μM NADK (Figure S4); two independent SPR experiments of EGCG (Figure S5); determination of the IC50 of EGCG against truncated NADK-Δ95 using the enzymatic activity assay (Figure S6); schematic diagram of HDX rate changes of EGCG binding to Δ95 (Figure S7); docking models of (−)-gallocatechin and (−)-epigallocatechin to NADK (Figure S8) (PDF)
Author Contributions
# T.L. and W.S. contributed equally to this work. Q.L. and C.L. conceived and designed the study. T.L. performed most of the experiments. W.S. performed the HDX-MS experiments. Y.D. and N.Z. performed NMR experiments. T.L. and Q.L. wrote and edited the manuscript. Q.W. and B.Z. purified the protein. D.G. provided instrument support. D.D. and H.Z. provided help with the compound library. M.W. and B.H. proofread the manuscript. All of the authors read and approved the final manuscript.
This work was supported by the National Key R&D Program of China (2022YFC3400500 and 2021ZD0203900 to C.L.), the National Natural Science Foundation of China (32000915 to Q.L. and 81821005 and 91853205 to C.L.), and the project of the National Multidisciplinary Innovation Team of Traditional Chinese Medicine supported by the National Administration of Traditional Chinese Medicine (ZYYCXTD-202004 to C.L.), High-level new R&D institute of Guangdong Province (2019B090904008), High-level Innovative Research Institute Guangdong Province (2021B0909050003),Guangdong Basic and Applied Basic Research Foundation, Department of Science and Technology of Guangdong Province, Zhongshan Municipal Bureau of Science and Technology.
The authors declare no competing financial interest.
Supplementary Material
References
- Ying W. NAD+/NADH and NADP+/NADPH in Cellular Functions and Cell Death: Regulation and Biological Consequences. Antioxid. Redox Signaling 2008, 10 (2), 179–206. 10.1089/ars.2007.1672. [DOI] [PubMed] [Google Scholar]
- Ge T.; Yang J.; Zhou S.; Wang Y.; Li Y.; Tong X. The Role of the Pentose Phosphate Pathway in Diabetes and Cancer. Front. Endocrinol. 2020, 11, 365. 10.3389/fendo.2020.00365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden M. G.; Cantley L. C.; Thompson C. B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324 (5930), 1029–1033. 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner F.; Niere M.; Ludwig A.; Ziegler M. Structural and Functional Characterization of Human NAD Kinase. Biochem. Biophys. Res. Commun. 2001, 288 (1), 69–74. 10.1006/bbrc.2001.5735. [DOI] [PubMed] [Google Scholar]
- Pollak N.; Niere M.; Ziegler M. NAD Kinase Levels Control the NADPH Concentration in Human Cells. J. Biol. Chem. 2007, 282 (46), 33562–33571. 10.1074/jbc.M704442200. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Xu Y.; Lu W.; Ghergurovich J. M.; Guo L.; Blair I. A.; Rabinowitz J. D.; Yang X. Upregulation of Antioxidant Capacity and Nucleotide Precursor Availability Suffices for Oncogenic Transformation. Cell Metab. 2021, 33 (1), 94–109. 10.1016/j.cmet.2020.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray J. P.; Alavian K. N.; Jonas E. A.; Heart E. A. NAD Kinase Regulates the Size of the NADPH Pool and Insulin Secretion in Pancreatic β-Cells. Am. J. Physiol.: Endocrinol. Metab. 2012, 303 (2), E191–E199. 10.1152/ajpendo.00465.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh H.; Longo D. L.; Chabner B. A. Improving Prospects for Targeting RAS. JCO 2015, 33 (31), 3650–3659. 10.1200/JCO.2015.62.1052. [DOI] [PubMed] [Google Scholar]
- Lanman B. A.; Allen J. R.; Allen J. G.; Amegadzie A. K.; Ashton K. S.; Booker S. K.; Chen J. J.; Chen N.; Frohn M. J.; Goodman G.; Kopecky D. J.; Liu L.; Lopez P.; Low J. D.; Ma V.; Minatti A. E.; Nguyen T. T.; Nishimura N.; Pickrell A. J.; Reed A. B.; Shin Y.; Siegmund A. C.; Tamayo N. A.; Tegley C. M.; Walton M. C.; Wang H.-L.; Wurz R. P.; Xue M.; Yang K. C.; Achanta P.; Bartberger M. D.; Canon J.; Hollis L. S.; McCarter J. D.; Mohr C.; Rex K.; Saiki A. Y.; San Miguel T.; Volak L. P.; Wang K. H.; Whittington D. A.; Zech S. G.; Lipford J. R.; Cee V. J. Discovery of a Covalent Inhibitor of KRASG12C (AMG 510) for the Treatment of Solid Tumors. J. Med. Chem. 2020, 63 (1), 52–65. 10.1021/acs.jmedchem.9b01180. [DOI] [PubMed] [Google Scholar]
- Hong D. S.; Fakih M. G.; Strickler J. H.; Desai J.; Durm G. A.; Shapiro G. I.; Falchook G. S.; Price T. J.; Sacher A.; Denlinger C. S.; Bang Y.-J.; Dy G. K.; Krauss J. C.; Kuboki Y.; Kuo J. C.; Coveler A. L.; Park K.; Kim T. W.; Barlesi F.; Munster P. N.; Ramalingam S. S.; Burns T. F.; Meric-Bernstam F.; Henary H.; Ngang J.; Ngarmchamnanrith G.; Kim J.; Houk B. E.; Canon J.; Lipford J. R.; Friberg G.; Lito P.; Govindan R.; Li B. T. KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383 (13), 1207–1217. 10.1056/NEJMoa1917239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Allen S.; Blake J. F.; Bowcut V.; Briere D. M.; Calinisan A.; Dahlke J. R.; Fell J. B.; Fischer J. P.; Gunn R. J.; Hallin J.; Laguer J.; Lawson J. D.; Medwid J.; Newhouse B.; Nguyen P.; O’Leary J. M.; Olson P.; Pajk S.; Rahbaek L.; Rodriguez M.; Smith C. R.; Tang T. P.; Thomas N. C.; Vanderpool D.; Vigers G. P.; Christensen J. G.; Marx M. A. Identification of MRTX1133, a Noncovalent, Potent, and Selective KRASG12D Inhibitor. J. Med. Chem. 2022, 65 (4), 3123–3133. 10.1021/acs.jmedchem.1c01688. [DOI] [PubMed] [Google Scholar]
- Xue J. Y.; Zhao Y.; Aronowitz J.; Mai T. T.; Vides A.; Qeriqi B.; Kim D.; Li C.; de Stanchina E.; Mazutis L.; Risso D.; Lito P. Rapid Non-Uniform Adaptation to Conformation-Specific KRAS(G12C) Inhibition. Nature 2020, 577 (7790), 421–425. 10.1038/s41586-019-1884-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunnett-Kane V.; Nicola P.; Blackhall F.; Lindsay C. Mechanisms of Resistance to KRASG12C Inhibitors. Cancers 2021, 13 (1), 151 10.3390/cancers13010151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre A. J.; Hahn W. C. Synthetic Lethal Vulnerabilities in KRAS-Mutant Cancers. Cold Spring Harbor Perspect. Med. 2018, 8 (8), a031518. 10.1101/cshperspect.a031518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downward J. RAS Synthetic Lethal Screens Revisited: Still Seeking the Elusive Prize?. Clin. Cancer Res. 2015, 21 (8), 1802–1809. 10.1158/1078-0432.CCR-14-2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yau E. H.; Kummetha I. R.; Lichinchi G.; Tang R.; Zhang Y.; Rana T. M. Genome-Wide CRISPR Screen for Essential Cell Growth Mediators in Mutant KRAS Colorectal Cancers. Cancer Res. 2017, 77 (22), 6330–6339. 10.1158/0008-5472.CAN-17-2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedeschi P. M.; Bansal N.; Kerrigan J. E.; Abali E. E.; Scotto K. W.; Bertino J. R. NAD+ Kinase as a Therapeutic Target in Cancer. Clin. Cancer Res. 2016, 22 (21), 5189–5195. 10.1158/1078-0432.CCR-16-1129. [DOI] [PubMed] [Google Scholar]
- Petrelli R.; Sham Y. Y.; Chen L.; Felczak K.; Bennett E.; Wilson D.; Aldrich C.; Yu J. S.; Cappellacci L.; Franchetti P.; Grifantini M.; Mazzola F.; Di Stefano M.; Magni G.; Pankiewicz K. W. Selective Inhibition of Nicotinamide Adenine Dinucleotide Kinases by Dinucleoside Disulfide Mimics of Nicotinamide Adenine Dinucleotide Analogues. Bioorg. Med. Chem. 2009, 17 (15), 5656–5664. 10.1016/j.bmc.2009.06.013. [DOI] [PubMed] [Google Scholar]
- Tedeschi P. M.; Lin H.; Gounder M.; Kerrigan J. E.; Abali E. E.; Scotto K.; Bertino J. R. Suppression of Cytosolic NADPH Pool by Thionicotinamide Increases Oxidative Stress and Synergizes with Chemotherapy. Mol. Pharmacol. 2015, 88 (4), 720–727. 10.1124/mol.114.096727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoxhaj G.; Ben-Sahra I.; Lockwood S. E.; Timson R. C.; Byles V.; Henning G. T.; Gao P.; Selfors L. M.; Asara J. M.; Manning B. D. Direct Stimulation of NADP+ Synthesis through Akt-Mediated Phosphorylation of NAD Kinase. Science 2019, 363 (6431), 1088–1092. 10.1126/science.aau3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.-H.; Chung T. D. Y.; Oldenburg K. R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol Screening 1999, 4 (2), 67–73. 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- Barnes E. C.; Kumar R.; Davis R. A. The Use of Isolated Natural Products as Scaffolds for the Generation of Chemically Diverse Screening Libraries for Drug Discovery. Nat. Prod. Rep. 2016, 33 (3), 372–381. 10.1039/C5NP00121H. [DOI] [PubMed] [Google Scholar]
- Wang H.; et al. 3PFN: Crystal Structure of human NAD kinase. In RCSB Protein Data Bank, November 10, 2010. 10.2210/pdb3PFN/pdb. [DOI]
- Rather G. M.; Pramono A. A.; Szekely Z.; Bertino J. R.; Tedeschi P. M. In Cancer, All Roads Lead to NADPH. Pharmacol. Ther. 2021, 226, 107864. 10.1016/j.pharmthera.2021.107864. [DOI] [PubMed] [Google Scholar]
- Pramono A. A.; Rather G. M.; Herman H.; Lestari K.; Bertino J. R. NAD- and NADPH-Contributing Enzymes as Therapeutic Targets in Cancer: An Overview. Biomolecules 2020, 10 (3), 358. 10.3390/biom10030358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai X.; Jiang Y.; Tan C. Let-7 Sensitizes KRAS Mutant Tumor Cells to Chemotherapy. PLoS One 2015, 10 (5), e0126653 10.1371/journal.pone.0126653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pupo E.; Avanzato D.; Middonti E.; Bussolino F.; Lanzetti L. KRAS-Driven Metabolic Rewiring Reveals Novel Actionable Targets in Cancer. Front. Oncol. 2019, 9, 848. 10.3389/fonc.2019.00848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang Y. H.; Dogruluk T.; Tedeschi P. M.; Wardwell-Ozgo J.; Lu H.; Espitia M.; Nair N.; Minelli R.; Chong Z.; Chen F.; Chang Q. E.; Dennison J. B.; Dogruluk A.; Li M.; Ying H.; Bertino J. R.; Gingras M.-C.; Ittmann M.; Kerrigan J.; Chen K.; Creighton C. J.; Eterovic K.; Mills G. B.; Scott K. L. Functional Annotation of Rare Gene Aberration Drivers of Pancreatic Cancer. Nat. Commun. 2016, 7 (1), 10500. 10.1038/ncomms10500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schild T.; McReynolds M. R.; Shea C.; Low V.; Schaffer B. E.; Asara J. M.; Piskounova E.; Dephoure N.; Rabinowitz J. D.; Gomes A. P.; Blenis J. NADK is Activated by Oncogenic Signaling to Sustain Pancreatic Ductal Adenocarcinoma. Cell Rep. 2021, 35 (11), 109238. 10.1016/j.celrep.2021.109238. [DOI] [PubMed] [Google Scholar]
- Roussel B.; Johnson-Farley N.; Kerrigan J. E.; Scotto K. W.; Banerjee D.; Felczak K.; Pankiewicz K. W.; Gounder M.; Lin H.; Abali E. E.; Bertino J. R. A Second Target of Benzamide Riboside: Dihydrofolate Reductase. Cancer Biol. Ther. 2012, 13 (13), 1290–1298. 10.4161/cbt.21786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh Y.-C.; Tedeschi P.; Adebisi Lawal R.; Banerjee D.; Scotto K.; Kerrigan J. E.; Lee K.-C.; Johnson-Farley N.; Bertino J. R.; Abali E. E. Enhanced Degradation of Dihydrofolate Reductase through Inhibition of NAD Kinase by Nicotinamide Analogs. Mol. Pharmacol. 2013, 83 (2), 339–353. 10.1124/mol.112.080218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D.; Wan S. B.; Yang H.; Yuan J.; Chan T. H.; Dou Q. P. EGCG, Green Tea Polyphenols and Their Synthetic Analogs and Prodrugs for Human Cancer Prevention and Treatment. Adv. Clin. Chem. 2011, 53, 155–177. 10.1016/B978-0-12-385855-9.00007-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggarwal V.; Tuli H. S.; Tania M.; Srivastava S.; Ritzer E. E.; Pandey A.; Aggarwal D.; Barwal T. S.; Jain A.; Kaur G.; Sak K.; Varol M.; Bishayee A. Molecular Mechanisms of Action of Epigallocatechin Gallate in Cancer: Recent Trends and Advancement. Semin. Cancer Biol. 2022, 80, 256–275. 10.1016/j.semcancer.2020.05.011. [DOI] [PubMed] [Google Scholar]
- Xie L.-W.; Cai S.; Zhao T.-S.; Li M.; Tian Y. Green Tea Derivative (−)-Epigallocatechin-3-Gallate (EGCG) Confers Protection against Ionizing Radiation-Induced Intestinal Epithelial Cell Death Both in Vitro and in Vivo. Free Radical Biol. Med. 2020, 161, 175–186. 10.1016/j.freeradbiomed.2020.10.012. [DOI] [PubMed] [Google Scholar]
- Talebi M.; Talebi M.; Farkhondeh T.; Mishra G.; İlgün S.; Samarghandian S. New Insights into the Role of the Nrf2 Signaling Pathway in Green Tea Catechin Applications. Phytother. Res. 2021, 35 (6), 3078–3112. 10.1002/ptr.7033. [DOI] [PubMed] [Google Scholar]
- Yu Y.; Deng Y.; Lu B.; Liu Y.; Li J.; Bao J. Green Tea Catechins: A Fresh Flavor to Anticancer Therapy. Apoptosis 2014, 19 (1), 1–18. 10.1007/s10495-013-0908-5. [DOI] [PubMed] [Google Scholar]
- Lambert J. D.; Elias R. J. The Antioxidant and Pro-Oxidant Activities of Green Tea Polyphenols: A Role in Cancer Prevention. Arch. Biochem. Biophys. 2010, 501 (1), 65–72. 10.1016/j.abb.2010.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim J.-H.; Su Z.-Y.; Chae J.-I.; Kim D. J.; Zhu F.; Ma W.-Y.; Bode A. M.; Yang C. S.; Dong Z. Epigallocatechin Gallate Suppresses Lung Cancer Cell Growth through Ras–GTPase-Activating Protein SH3 Domain-Binding Protein 1. Cancer Prev. Res. 2010, 3 (5), 670–679. 10.1158/1940-6207.CAPR-09-0185. [DOI] [PubMed] [Google Scholar]
- Liu Z.-S.; Cai H.; Xue W.; Wang M.; Xia T.; Li W.-J.; Xing J.-Q.; Zhao M.; Huang Y.-J.; Chen S.; Wu S.-M.; Wang X.; Liu X.; Pang X.; Zhang Z.-Y.; Li T.; Dai J.; Dong F.; Xia Q.; Li A.-L.; Zhou T.; Liu Z.; Zhang X.-M.; Li T. G3BP1 Promotes DNA Binding and Activation of CGAS. Nat. Immunol 2019, 20 (1), 18–28. 10.1038/s41590-018-0262-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Z.; Henry E. C.; Gasiewicz T. A. (−)-Epigallocatechin-3-Gallate is a Novel Hsp90 Inhibitor. Biochemistry 2009, 48 (2), 336–345. 10.1021/bi801637q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandelwal A.; Hall J.; Blagg B. S. J. Synthesis and Structure Activity Relationships of EGCG Analogues, A Recently Identified Hsp90 Inhibitor. J. Org. Chem. 2013, 78 (16), 7859–7884. 10.1021/jo401027r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urusova D. V.; Shim J.-H.; Kim D. J.; Jung S. K.; Zykova T. A.; Carper A.; Bode A. M.; Dong Z. Epigallocatechin-Gallate Suppresses Tumorigenesis by Directly Targeting Pin1. Cancer Prev. Res. 2011, 4 (9), 1366–1377. 10.1158/1940-6207.CAPR-11-0301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana H.; Koga K.; Fujimura Y.; Yamada K. A Receptor for Green Tea Polyphenol EGCG. Nat. Struct Mol. Biol. 2004, 11 (4), 380–381. 10.1038/nsmb743. [DOI] [PubMed] [Google Scholar]
- Kim J.-H.; Kim W. Alleviation Effects of Rubus Coreanus Miquel Root Extract on Skin Symptoms and Inflammation in Chronic Atopic Dermatitis. Food Funct. 2022, 13 (5), 2823–2831. 10.1039/D1FO03580K. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Wang H.; et al. 3PFN: Crystal Structure of human NAD kinase. In RCSB Protein Data Bank, November 10, 2010. 10.2210/pdb3PFN/pdb. [DOI]