

Abstract
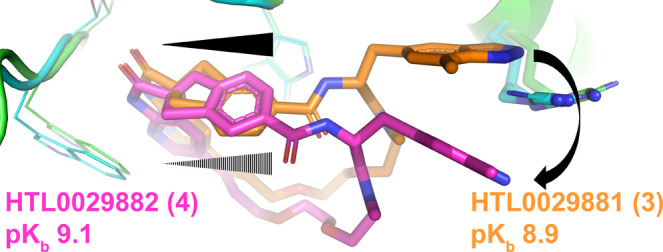
The diastereomeric macrocyclic calcitonin gene-related peptide (CGRP) antagonists HTL0029881 (3) and HTL0029882 (4), in which the stereochemistry of a spiro center is reversed, surprisingly demonstrate comparable potency. X-ray crystallographic characterization demonstrates that 3 binds to the CGRP receptor in a precedented manner but that 4 binds in an unprecedented, unexpected, and radically different manner. The observation of this phenomenon is noteworthy and may open novel avenues for CGRP receptor antagonist design.
Keywords: GPCR, CGRP, CGRPR, Macrocycle, Macrocyclic, Antagonist
Blockade of the function of the 37 amino acid calcitonin gene-related peptide (CGRP) at its cognate CGRP receptor (CGRPR) is a clinically validated mechanism for migraine treatment.1,2 Several small-molecule CGRP antagonists3−5 are now marketed as oral medicines, and antibodies targeting the CGRP receptor or the CGRP peptide itself are likewise marketed drugs.6
The quest to discover small-molecule CGRP antagonist drugs has an extensive history, the associated medicinal chemistry of which has been thoroughly reviewed.7,8 The approval of oral agents ubrogepant,3 rimegepant,4,5 and atogepant,9 and the regulatory submission of the nonoral agent zavegepant10 (Figure 1) represent the realization of decades of pharmaceutical industry effort toward overcoming the considerable challenges of identifying small-molecule drugs for this target.
Figure 1.
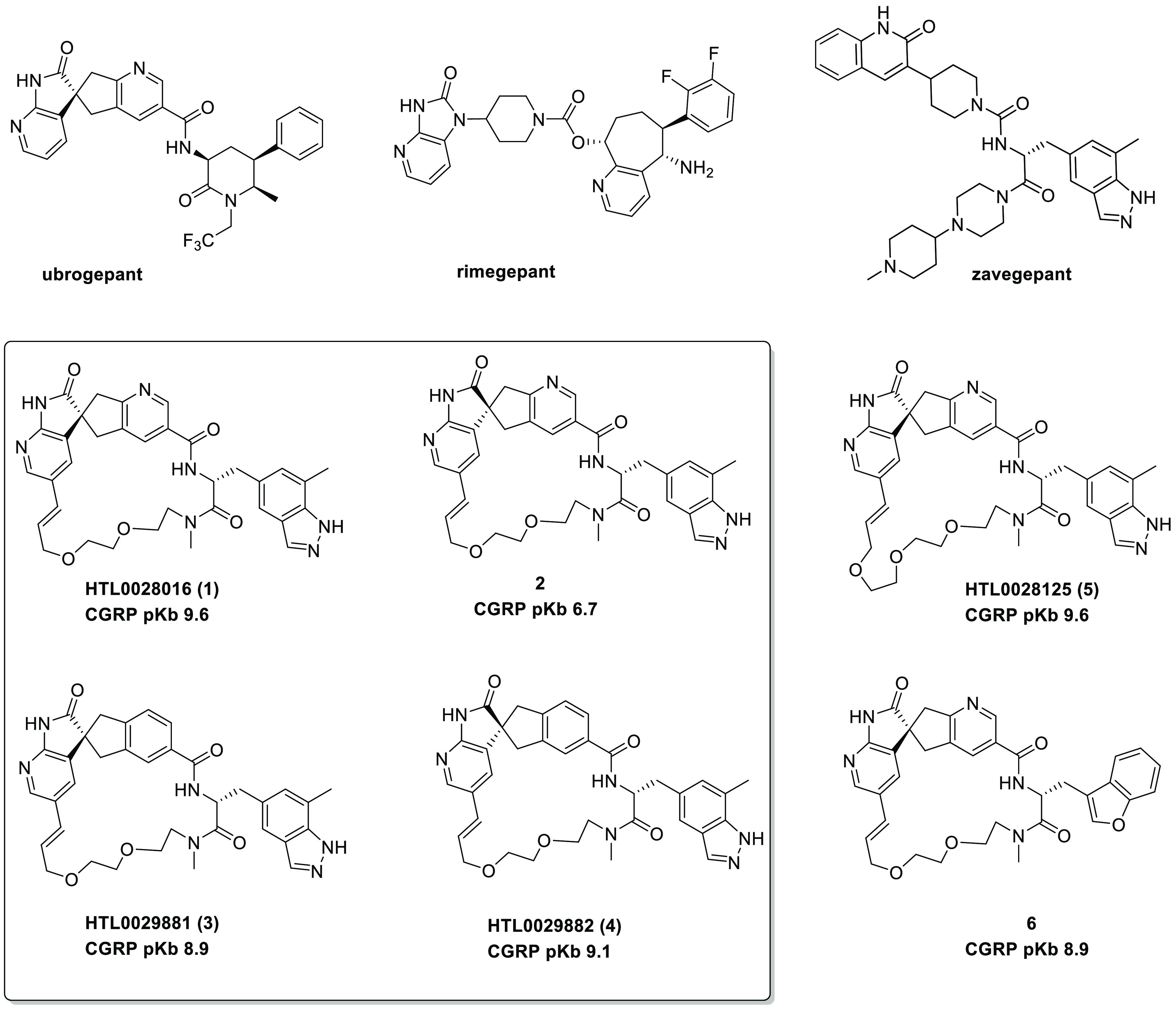
Ubrogepant,3 rimegepant,4,5 zavegepant,10 and macrocyclic CGRP antagonists.11
We recently reported11 the identification of novel macrocyclic CGRP antagonists, exemplified by HTL0028016 (1), and characterized their binding to the CGRP receptor by X-ray crystallography. Compound 1 is a highly potent CGRP antagonist (pKb 9.6), and its corresponding diasteroisomer (2) in which the spiro center stereochemistry is reversed is ca. three orders of magnitude less potent, which we ascribed to the stereospecificity of the ligand–receptor interaction.11 In addition, we also reported11 that the analogue of 1 in which the nicotinamide nitrogen is replaced with a CH in the corresponding spiroindene (HTL0029881, 3) exhibits a modest comparative reduction in potency (pKb 8.9)
Herein, we report that in contrast to the diastereoisomer pair 1 and 2, the reversed spiro chemistry indene HTL0029882 (4) exhibits a potency comparable to that of its nonreversed counterpart compound 3. X-ray crystallography demonstrated that 4 had a different binding mode relative to 3, where the indazolylmethyl moiety was involved in radically different interactions. This finding of comparable potencies for the two diastereoisomers 3 and 4 demonstrates the remarkable plasticity of the CGRP receptor protein and previously unreported high ligand inducibility.
The macrocycles 3 and 4 were prepared using homochiral spiro intermediates 7 and 8, respectively (Figure 2), according to the previously reported route11 (described in detail toward the end of this manuscript). Intermediates 7 and 8 were separated by chiral SFC chromatography, and their absolute stereochemistries were unknown at the initial point of final compound synthesis. Both enantiomers were progressed separately through the synthetic procedure to afford the final compounds, with the expectation that only one enantiomer would afford a potent macrocycle; this macrocycle would thus define the absolute stereochemistries of the spiro intermediates 7 and 8. However, the final products of the two spiro intermediates 7 and 8 both proved to be potent macrocyclic CGRP antagonists (pKb 8.9 and 9.1 respectively)
Figure 2.

Homochiral intermediates of 3 and 4.
This surprising finding prompted a thorough analytical reinterrogation of the final compounds, all of which confirmed they were indeed different diastereoisomers, and that the observed CGRP pharmacology could be unambiguously ascribed to the assigned structures of the two compounds (Figures S2 and S3).
Compounds 3 and 4 were then ultimately cocrystallized with the CGRP receptor ectodomain, which provided an explanation for the comparable activities of the two isomers. The receptor–ligand complexes were characterized by X-ray crystallography, which revealed two diastereomeric compounds that differed in their absolute spiro stereochemistry and exhibited two distinct receptor binding modes (Figure 3). Macrocycle 3 demonstrated a HTL0028016 (1) like spiro stereochemistry (hereafter referred to as the “classical” stereochemistry, also shared with ubrogepant), and 4 demonstrated the reverse spiro stereochemistry (hereafter referred to as the “nonclassical” stereochemistry) of compound 2. These structural characterizations allowed the absolute stereochemistries of intermediates 7 and 8 to be assigned.
Figure 3.

Structures of CGRPR ectodomain complexes bound to (A) HTL0029882 (4, magenta) 8AX6 and (B) HTL0029881 (3) (orange) 8AX5 as determined by X-ray crystallography. Cartoon representations of the binding site with interacting residues, where ligands are shown in stick representation with nitrogen and oxygen atoms colored blue and red, respectively. Water molecules are displayed as spheres and colored as ligands. CLR is colored green or light green, and RAMP1 is colored blue or teal. 2Fo–Fc electron density maps for the ligands are shown as a gray mesh and displayed at 1σ. (C and D) Physicochemical properties of the cavity characterized with GRID. CLR and RAMP1 residues and the cartoon representations are colored as described above. The GRID C3 surface used the pocket surface in terms of how close a ligand carbon atom can reside (1.0 kcal mol–1, gray mesh) and C1 lipophilic hotspots (−2.8 kcal mol–1, yellow transparent solid) are shown.
We previously reported11 that CGRP macrocycle antagonists occupy the binding site of CGRPR ectodomain formed by the N-termini of calcitonin-like receptor (CLR) and receptor activity-modifying protein 1 (RAMP112,13). Consistent with the cocrystal structures of CGRPR bound to macrocycle antagonist HTL0028125 (5) (PDB ID 7P0F) and benzofuran (6) (PDB ID 7P0I),103 binds in a shallow and solvent-exposed binding pocket with an extended conformation (Figure 3B). The druggability of the the apo binding side was assessed using GRID molecular interaction fields,14,15 and the results highlighted the lipophilic character of the binding site groove and the right-hand-side cavity formed at the interface between the CLR and RAMP1 domains. The binding mode of 3 replicates the extended conformation of cocrystallized compound 5, conserving key protein–ligand interactions.
The spiroazaindolone-indene fragment of 3 is anchored to Thr122 CLR backbone, while the 7-methyl-1H-indazol-5-yl group (a group that is also common to zavegepant10 and HTL2256216) is hydrogen bonded to Asp71 in the RAMP1 side chain. Other direct hydrogen bond interactions of 5 that are conserved in 3 are those between the carbonyl of the N-methylated amide and Trp72 CLR and between the benzamide nitrogen and the Gly71 CLR backbone (Figure 3D). As for the crystallographic structures of CGRPR-5 and CGRPR-6,11 the nine-atom linking chain of 3 is solvent-exposed above the bulky Trp72 CLR side chain at the front of the binding pocket.
As observed from comparing cocrystallized CGRPR structures with different ligands10,16 the binding pocket shows flexibility and plasticity. The various subpockets adjust to complement the bound ligand, particularly around the lipophilic cavity at the CLR–RAMP1 interface formed by the Arg38, Ile41, and Met42 residues of CLR, and the Asp71, Trp74, and Trp84 residues of RAMP1. Arg38 of CLR and Trp74 and Asp71 of RAMP1 showed high flexibility in other ligand-bound CGRPR structures, contributing to the shape and physicochemical properties of the right-hand-side cavity occupied by the indazolyl groups of 5 and 3 and the benzofuran group of compound 6.11 The Arg119 residue of the flexible side chain of CLR, which forms favorable interactions with the nicotinamide pyridyl nitrogen of 5,11 moves slightly away from the corresponding and the nonoptimal benzamide carbon of 3, consistent with its lower potency compared to 1.
The great extent of the ligand specific induced fit propensity of the right-hand-side cavity was revealed by the CGRPR-4 structure (Figure 3A). Despite its opposite spiro-stereochemistry, the spiroazaindolone-indene fragment of 4 is anchored to Thr122 on the CLR backbone and embedded in a cavity formed by Arg119, Trp121, Tyr124, and Trp72 of CLR in the same manner as 3, and the olefin of the linker chain overlaps between the two cocrystallized ligands (Figure 4). Moreover, the overall CLR-RAMP1 binding domain structure is highly conserved, with a backbone RMSD between the two different structures of just 0.5 Å. However, side chains in the cavity at the CLR–RAMP1 interface undergo significant ligand-induced fit rearrangements in order to optimize the ligand–protein contacts. Beside the direct interactions with Thr122 of CLR, the other hydrogen bond interactions of 4 are water-mediated. Water molecules bridge the N-methylated amide carbonyl ligand and Trp72 of CLR in addition to the benzamide nitrogen and the Gly71 CLR backbone. Furthermore, in order to accommodate the benzamide amide group of 4, Trp74 of RAMP1 flips the side chain. This opens a channel between the Trp74 of and Arg119 side chains of RAMP1 and CLR, respectively, that is filled with water molecules, with one water bridging the benzamide oxygen and the flipped Trp74 side chain of RAMP1. The high resolution of the crystallographic structure (1.9 Å) allows the appreciation of the tight water network that fills the newly formed cavities and coordinates and mediates protein–ligand interactions.
Figure 4.
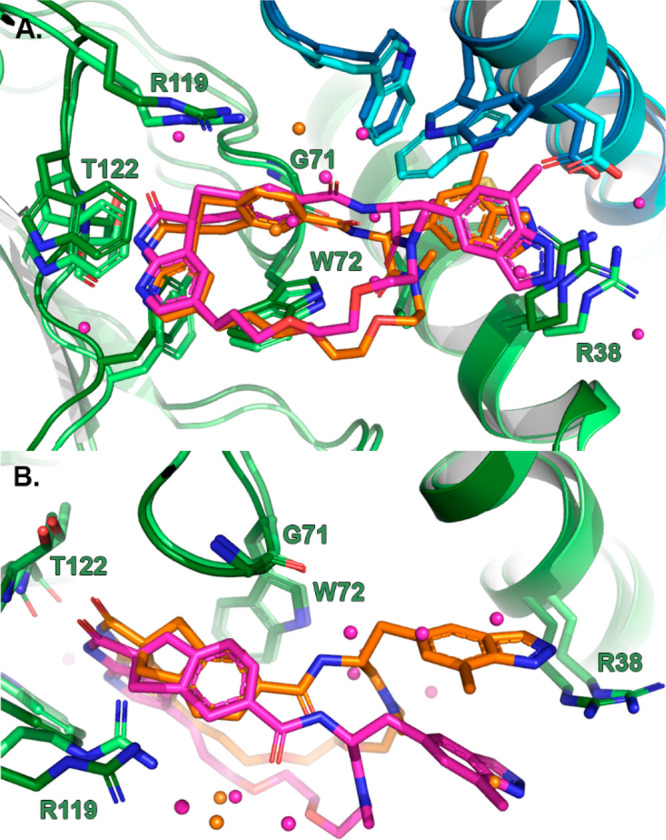
Superposition of the binding cavities of 3 (orange) and 4 (magenta) viewed (A) as previously shown and (B) from “overhead”. Protein and waters are colored as described in Figure 3.
The new orientation of Trp74 of RAMP1 in the presence of 4 and the unexpectedly large movement of the Arg38 side chain of CLR form a new groove for the indazolyl group, which has a new induced-fit lipophilic hotspot formed by the Trp74 and Asp71 side chains of RAMP1 and Arg38 of CLR to accommodate the indazole methyl group (Figure 2C). The Arg38 side chain of CLR shows high flexibility, with interpretations of protein electron density maps leading to two different side chain orientations or rotamers. In both rotomers, the Arg38 side chain of CLR engages with Asp71 of RAMP1 via salt-bridge and hydrogen bond interactions. Furthermore, Arg38 of CLR exhibits a strong π-cation stacking with the indazolyl moiety. Coordinating with Arg38 CLR side chain, a well-connected water network fills the differently shaped cavity at the CLR–RAMP1 interface occupied by 3 indazolyl moiety (Figure 2D).
If the nonclassical stereochemistry macrocycle 2 were to adopt a binding mode similar to that of 4, its nicotinamide nitrogen would then be positioned at the bottom of the pocket groove and buried in an unfavorable lipophilic and hydrophobic environment, thus precluding the adoption of the nonclassical binding mode. This would explain the lower potency of 2 relative to both 4 and its classical stereochemistry counterpart 1
The structurally characterized classical and nonclassical binding modes of 3 and 4 were further substantiated by the methylation of their respective indazol-1-yl nitrogen atoms, which formed compounds 9 and 10, respectively (Figure 5).
Figure 5.

Indazole N1-methylated analogues of macrocyclic CGRP antagonists 3 and 4.
In the classical binding mode of 3, the indazole NH engages in a key hydrogen bonded interaction with Asp71 of RAMP1 (Figure 2 and 3), suggesting that the methylation of the indozole nitrogen should be detrimental to potency. In contrast, in the nonclassical binding mode of 4, the indazolyl NH appears not to be involved in a hydrogen bonding interaction with the CGRP receptor, suggesting that 4 could accommodate N-methylation. The above conjecture was experimentally confirmed with compound 9 (the classical stereochemistry and binding mode) displaying a ca. 30-fold decrease in potency with respect to its NH counterpart, consistent with a perturbation of the interaction with Asp 71, whereas the N-methyl analogue 10 (nonclassical) actually demonstrated a potency comparable to that of its parent 4. Indeed, the structure of CGRPR bound 10 subsequently revealed that both the protein rearrangements induced and the binding mode were the same as those of 4, with the indazolyl N-methyl adding favorable van der Waals ligand–protein contacts with the Arg38 and Trp74 side chains of CLR and RAMP, respectively (Figure S1).
The difference in the binding modes of the diastereoisomers 3 and 4 is perhaps best appreciated from a “overhead” view of the superpositioned structures (Figure 4B), where the radically different positions of the macrocyclic PEG chain and the indazole substituents are clearly evident. As described above, despite the reversal of the spiro stereochemistry, the spiroazaindolone-indene fragments of the two isomers 3 and 4 bind to the receptor in the same manner; it is the position of the carboxamide substituent that is different and leads to the alternative binding mode. Indeed, 4 can be conceived of as having the classical 3 spiro configuration but with the position of the amide attachment transposed by one atom (Figure 6). Interestingly, in this context, Wood et. al. made a similar conjecture17 to rationalize the activities of enantiomers of spirohydantoin CGRPR antagonists of chemotype 11 , and used this thinking in subsequent design hypotheses.
Figure 6.

Contrasting relationships of classical and nonclassical stereochemistries and binding modes (the asterisk (*) represents the macrocycle chain attachment points), and the spiro-hydantoin CGRP antagonist17 (11).
Compounds 3, 4, 9, and 10 were prepared according to the previously described procedure,11 as shown in Scheme 1; indeed, the synthesis and characterization of 3 itself was previously reported.11
Scheme 1. Synthesis of Macrocyclic CGRP Antagonists Described Herein.
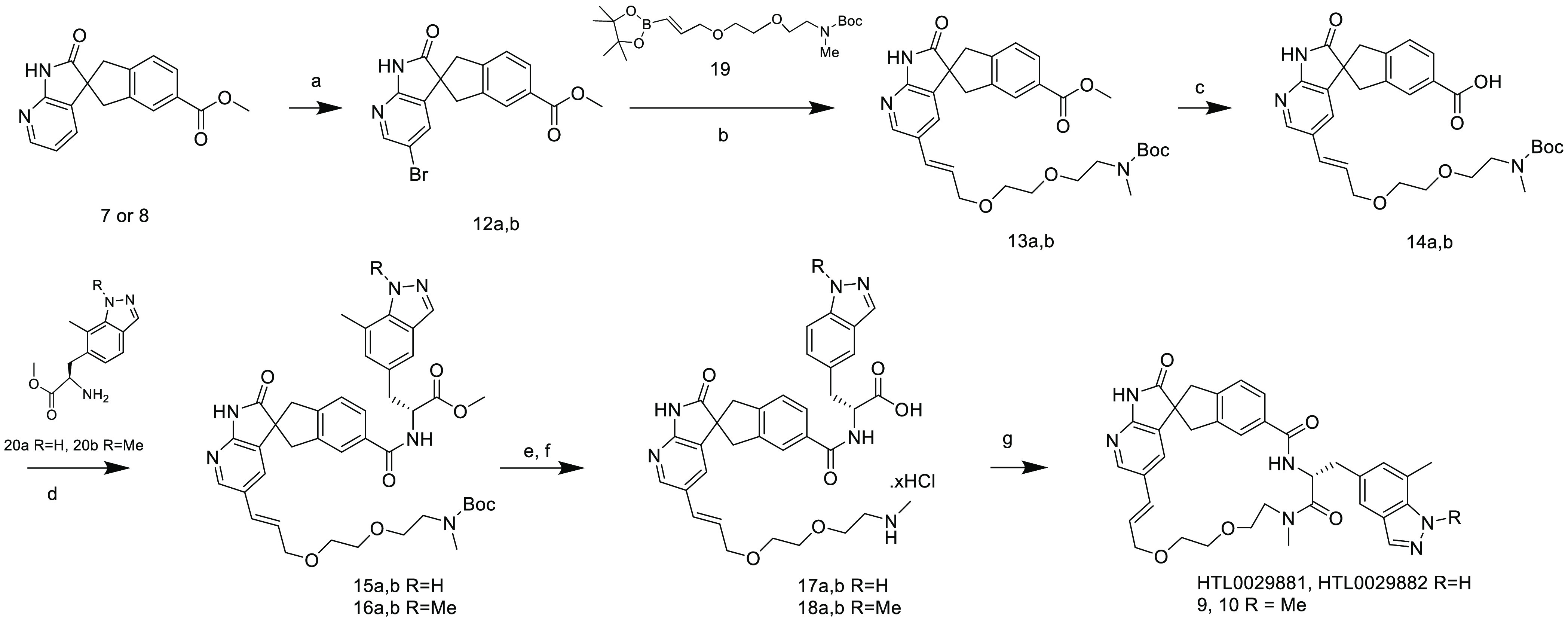
Reagents and Conditions are as follows: (a) N-bromosuccinimide, DMF, 0 °C to RT (92–93%); (b) K3PO4, XPhos Pd G2, 1,4-dioxane/water (4:1), 100 °C (44–49%); (c) 1 M LiOH (aq), THF, RT (88–91%); (d) HATU, DIPEA, DMF, 0 °C to RT (76–100%); (e) 1 M LiOH (aq), THF, RT (97–100%); (f) 4 M HCl/1,4-dioxan, RT; and (g) HATU or PyAOP, DIPEA, DMF, RT (7.5–29%).
Enantiomeric spiro esters (7 and 8) were brominated with N-bromo succinimide, and the resultant bromides (12a and 12b) were coupled with the boronylallyloxy species (19) to afford intermediates 13a and 13b, respectively. Hydrolysis of intermediates 13a and 13b with lithium hydroxide and subsequent coupling with amino acid esters (20a10 and 20b) using HATU afforded the protected macrocycle precursors 15 and 16, respectively. Hydrolysis with lithium hydroxide and Boc removal with HCl afforded the macrocycle precursors 17 and 18, and macrolactamisation with HATU or PyAOP, afforded the final compounds.
Functional CGRPR antagonist potencies, i.e., pKb values, of the compounds were determined according to previously reported methods.11,16
Purified MBP-RAMP1 ECD-CLR ECD fusion protein samples bound to 3, 4, and 10 were prepared according to previously reported methods.11,16
Data and refinement statistics are listed in the Supporting Information (pp 2 and 3). Structure coordinates were deposited in the Protein Data Bank with IDs 8AX5, 8AX6, and 8AX7 for HTL0029881 (3), HTL0029882 (4), and HTL0031448 (10), respectively.
In conclusion, the diastereomeric macrocyclic CGRP antagonists 3 and 4 demonstrate a remarkable phenomenon in which reversal of the stereochemistry of a spiro center in the core of the molecule induces a radical change in the ligand binding mode.
Compound 3 binds to the CGRP receptor in the structurally precedented “classical” binding mode of HTL00285125 (5), whereas compound 4 effects a profound reorganization of the RAMP–CLR interface to engage in the unprecedented nonclassical binding mode (Figures 3 and 4). This phenomenon is facilitated by the pyridine in 1 and 2, where this phenomenon does not occur, being changed to a phenyl group in 3 and 4. In the pyridine of compound 2, a repulsive interaction between the pyridine nitrogen and the protein restricts access to the nonclassical binding mode, whereas the phenyl ring of 4 facilitates access to this mode. The associated plasticity of the CGRP binding site is notable, with the receptor in its role as host showing generous hospitality by accommodating the needs of its different guests.
The phenomenon of different enantiomers or diastereoisomers of a molecule exhibiting comparable potencies while binding to their protein target in radically different modes is precedented18,19 and has been characterized structurally for other classes of protein targets (see notable examples for CK1 Inhibitors20 and PPARγ agonists21). However, this phenomenon has not been previously observed or structurally characterized for a GPCR protein target and is as such noteworthy. Furthermore, the identification and characterization of the nonclassical binding mode may open novel avenues for CGRP receptor antagonist design.
Glossary
Abbreviations
- CLR
calcitonin receptor-like receptor
- ECD
extracellular domain
- HATU
1-[bis(dimethylamino)methylene]1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate
- MBP
maltose-binding protein
- PyAOP
7-azabenzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate
- RAMP
receptor activity-modifying protein
- DIPEA
N,N-diisopropylethylamine
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.2c00400.
Crystallographic data and refinement statistics, structure of the compound 10–CGRPR ectodomain complex, synthetic experimental procedure, and purity and surety of compounds 3 and 4 (PDF)
Author Present Address
∇ Isomorphic Laboratories, London EC4V 6JA, U.K
Author Contributions
The manuscript describes practical and/or design and strategic contributions of all authors.
None.
The authors declare no competing financial interest.
Supplementary Material
References
- Edvinsson L.; Haanes K. A.; Warfvinge K.; Krause D. N. CGRP as the target of new migraine therapies - successful translation from bench to clinic. Nat. Rev. Neuro. 2018, 14 (6), 338–350. 10.1038/s41582-018-0003-1. [DOI] [PubMed] [Google Scholar]
- Ha D. K.; Kim M. J.; Han N.; Kwak J. H.; Baek I. H. Comparative Efficacy of Oral Calcitonin-Gene-Related Peptide Antagonists for the Treatment of Acute Migraine: Updated Meta-analysis. Clinical drug investigation. 2021, 41 (2), 119–132. 10.1007/s40261-020-00997-1. [DOI] [PubMed] [Google Scholar]
- Moore E.; Fraley M. E.; Bell I. M.; Burgey C. S.; White R. B.; Li C.-C.; Regan C. P.; Danziger A.; Michener M. S.; Hostetler E.; Banerjee P.; Salvatore C. Characterization of Ubrogepant: A Potent and Selective Antagonist of the Human Calcitonin Gene–Related Peptide Receptor. J. Pharmacol. Exp. Ther. 2020, 373, 160–166. 10.1124/jpet.119.261065. [DOI] [PubMed] [Google Scholar]
- Luo G.; Chen L.; Conway C. M.; Denton R.; Keavy D.; Signor L.; Kostich W.; Lentz K. A.; Santone K. S.; Schartman R.; Browning M.; Tong G.; Houston J. G.; Dubowchik G. M.; Macor J. E. Discovery of (5S,6S,9R)-5-Amino-6-(2,3-difluorophenyl)-6,7,8,9-tetrahydro-5H-cyclohepta[b]pyridin-9-yl 4-(2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-1-yl)piperidine-1-carboxylate (BMS-927711): An Oral Calcitonin Gene-Related Peptide (CGRP) Antagonist in Clinical Trials for Treating Migraine. J. Med. Chem. 2012, 55, 10644–10651. 10.1021/jm3013147. [DOI] [PubMed] [Google Scholar]
- Croop R.; Lipton R. B.; Kudrow D.; Stock D. A.; Kamen L.; Conway C. M.; Stock E. G.; Coric V.; Goadsby P. J. Oral rimegepant for preventive treatment of migraine: a phase 2/3, randomised, double-blind, placebo-controlled trial. Lancet 2021, 397 (10268), 51–60. 10.1016/S0140-6736(20)32544-7. [DOI] [PubMed] [Google Scholar]
- Sacco S.; Bendtsen L.; Ashina M.; Reuter U.; Terwindt G.; Mitsikostas D.-D.; Martelletti P. European headache federation guideline on the use of monoclonal antibodies acting on the calcitonin gene related peptide or its receptor for migraine prevention. J. Headache Pain 2019, 20, 6. 10.1186/s10194-018-0955-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubowchik G. M.; Conway C. M.; Xin A. W. Blocking the CGRP Pathway for Acute and Preventive Treatment of Migraine: The Evolution of Success. J. Med. Chem. 2020, 63, 6600–6623. 10.1021/acs.jmedchem.9b01810. [DOI] [PubMed] [Google Scholar]
- Bell I. M. Calcitionin Gene-Related Peptide Receptor Antagonists: New Therapeutic Agents for Migraine. J. Med. Chem. 2014, 57, 7838–7858. 10.1021/jm500364u. [DOI] [PubMed] [Google Scholar]
- Deeks E. D. Atogepant: First Approval. Drugs 2022, 82, 65–70. 10.1007/s40265-021-01644-5. [DOI] [PubMed] [Google Scholar]
- Chaturvedula P. V.; Mercer S. E.; Pin S. S.; Thalody G.; Xu C.; Conway C. M.; Keavy D.; Signor L.; Cantor G. H.; Mathias N.; Moench P.; Denton R.; Macci R.; Schartman R.; Whiterock V.; Davis C.; Macor J. E.; Dubowchik G. M. Discovery of (R)-N-(3-(7-methyl-1H-indazol-5-yl)-1-(4-(1-methylpiperidin-4-yl)-1-oxopropan-2-yl)-4-(2-oxo-1,2-dihydroquinolin-3-yl)-piperidine-1-carboxamide (BMS-742413): A Potent Human CGRP Antagonist with Superior Safety Profile for the Treatment of Migraine Through Intranasal Delivery. Bioorg. Med. Chem. Lett. 2013, 23, 3157–3161. 10.1016/j.bmcl.2013.04.012. [DOI] [PubMed] [Google Scholar]
- Cansfield A. D.; Ator M. A.; Banerjee J.; Bestwick M.; Bortolato A.; Brown G. A.; Brown J.; Butkovic K.; Cansfield J. E.; Christopher J. A.; Congreve M.; Cseke G.; Deflorian F.; Dugan B.; Hunjadi M. P.; Hutinec A.; Inturi T. K.; Landek G.; Mason J.; O’Brien A.; Ott G. R.; Rupcic R.; Saxty G.; Southall S. M.; Zadravec R.; Watson S. P. Novel Macrocyclic Antagonists of the Calcitonin Gene-Related Peptide Receptor: Design, Realization, and Structural Characterization of Protein-Ligand Complexes. ACS Chem. Neurosci. 2022, 13, 751–765. 10.1021/acschemneuro.1c00696. [DOI] [PubMed] [Google Scholar]
- Hay D. L.; Garelja M. L.; Poyner D. R.; Walker C. S. Update on the pharmacology of calcitonin/CGRP family of peptides: IUPHAR Review 25. Br. J. Pharmacol. 2018, 175, 3–17. 10.1111/bph.14075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay D. L.; Pioszak A. A. Receptor Activity-Modifying Proteins (RAMPs): New Insights and Roles. Annual review of pharmacology and toxicology 2016, 56, 469–487. 10.1146/annurev-pharmtox-010715-103120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodford P. J. A computational procedure for determining energetically favourable binding sites on biologically important macromolecules. J. Med. Chem. 1985, 28, 849–857. 10.1021/jm00145a002. [DOI] [PubMed] [Google Scholar]
- Sciabola S.; Stanton R. V.; Mills J. E.; Flocco M. M.; Baroni M.; Cruciani G.; Perruccio F.; Mason J. S. High-throughput virtual screening of proteins using GRID molecular interaction fields. J. Chem. Inf. Model. 2010, 50, 155–169. 10.1021/ci9003317. [DOI] [PubMed] [Google Scholar]
- Bucknell S. J.; Ator M. A.; Brown A.; Brown J.; Cansfield A. D.; Cansfield J. E.; Christopher J. A.; Congreve M.; Cseke G.; Deflorian F.; Jones C. R.; Mason J. S.; O’Brien M. A.; Ott G. R.; Pickworth M.; Southall S. M. Structure-Based Drug Discovery of N-((R)-3-(7-Methyl-1H-indazol-5-yl)-1-oxo-1-(((S)-1-oxo-3-(piperidin-4-yl)-1-(4-(pyridin-4-yl)piperazin-1-yl)propan-2-yl)amino)propan-2-yl)-2’-oxo-1’,2’-dihydrospiro[piperidine-4,4’-pyrido[2,3-d][1,3]oxazine]-1-carboxamide (HTL22562): A Calcitonin Gene-Related Peptide Receptor Antagonist for Acute Treatment of Migraine. J. Med. Chem. 2020, 63, 7906–7920. 10.1021/acs.jmedchem.0c01003. [DOI] [PubMed] [Google Scholar]
- Wood M. R.; Schirripa K. M.; Kim J. J.; Bednar R. A.; Fay J. F.; Bruno J. G.; Moore E. L.; Mosser S. D.; Roller S.; Salvatore C. A.; Vacca J. P.; Selnick H. G. Novel CGRP receptor antagonists from central amide replacements causing a reversal of preferred chirality. Bioorganic & medicinal chemistry letters 2010, 20 (22), 6827–6830. 10.1016/j.bmcl.2010.08.105. [DOI] [PubMed] [Google Scholar]
- Klebe G.Differences in Binding of Stereoisomers to Protein Active Sites. In Supramolecular Structure and Function 8; Pifat-Mrzljak G., Eds.; Springer: Boston, MA, 2005; pp 31–52. 10.1007/0-306-48662-8_3 [DOI] [Google Scholar]
- Karlsson J.; Morgillo C. M.; Deplano A.; Smaldone G.; Pedone E.; Luque F. J.; Svensson M.; Novellino E.; Congiu C.; Onnis V.; Catalanotti B.; Fowler C. J. Interaction of the N-(3-Methylpyridin-2-yl)amide Derivatives of Flurbiprofen and Ibuprofen with FAAH: Enantiomeric Selectivity and Binding Mode. PLoS One 2015, 10 (11), e0142711 10.1371/journal.pone.0142711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luxenburger A.; Schmidt D.; Ianes C.; Pichlo C.; Krüger M.; von Drathen T.; Brunstein E.; Gainsford G. J.; Baumann U.; Knippschild U.; Peifer C. Design, Synthesis and Biological Evaluation of Isoxa-zole-Based CK1 Inhibitors Modified with Chiral Pyrrolidine Scaffolds. Molecules 2019, 24 (5), 873. 10.3390/molecules24050873. [DOI] [PMC free article] [PubMed] [Google Scholar]; PMID: 30832206. PMCID: PMC6429214.
- Jang J. Y.; Koh M.; Bae H.; An D. R.; Im H. N.; Kim H. S.; Yoon J. Y.; Yoon H. J.; Han B. W.; Park S. B.; Suh S. W. Structural basis for differential activities of enantiomeric PPARγ agonists: Binding of S35 to the alternate site. Biochim Biophys Acta Proteins Proteom 2017, 1865 (6), 674–681. 10.1016/j.bbapap.2017.03.008. [DOI] [PubMed] [Google Scholar]; PMID: 28342850.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.