

Abstract

Efforts to combine advantages of fragment-based drug design (FBDD) and dynamic combinatorial chemistry (DCC) for the development of selective α-glucosidase inhibitors were described. Starting from 5 rationally designed fragments, two iterative dynamic combinatorial libraries (DCLs) comprising 29 acylhydrazone products were generated and screened using α-glucosidase and α-amylase as the templates. The optimal ligand identified showed substantial α-glucosidase inhibition with high selectivity over α-amylase as well as low cytotoxicity. Furthermore, inhibition type and detailed ligand/enzyme binding interactions were elucidated by the binding kinetic study and docking simulation, respectively.
Keywords: Dynamic combinatorial chemistry, α-Glucosidase, α-Amylase, Fragment-based drug design, Protein inhibition, Binding mechanism
Over the past two decades, dynamic combinatorial chemistry (DCC) has been established as a powerful tool to discover biologically active lead compounds in medicinal chemistry.1,2 Based on covalent or noncovalent reversible reactions between building blocks with complementary functional groups, spontaneous generation of dynamic combinatorial library (DCL) can be accomplished under thermodynamic control.3−6 Due to the adaptive nature of DCL, its equilibrium can be altered by the addition of external stimuli, thereby changing the composition of the dynamic system (Figure 1a).7−10 Enzyme protein is most commonly used in the screening of active components as external template.11 DCL driven by this type of stimulus achieves amplification of constituent(s) with higher binding affinity at the expense of other less favored combinations, resulting in a thermodynamically controlled evolutive process on molecular level.12,13
Figure 1.
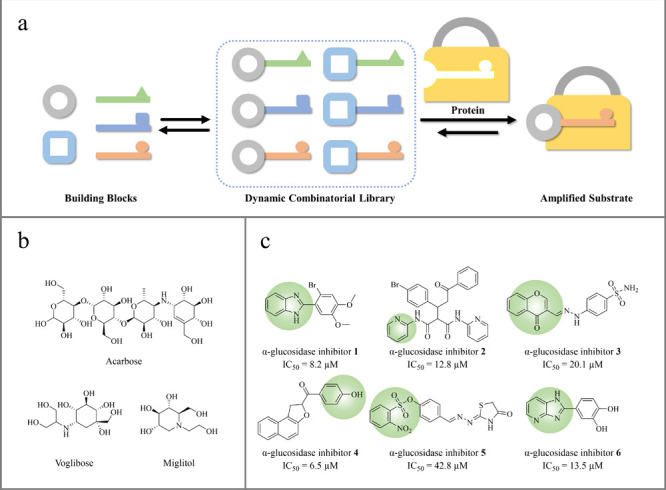
(a) Schematic diagram of dynamic combinatorial chemistry; (b) structure of some commercial α-glucosidase inhibitors; (c) α-glucosidase inhibitors reported in recent years.
Fragment-based drug design (FBDD) has gained wide application in the field of drug discovery in recent years due to its success in obtaining bioactive substances, and a variety of clinical drugs have been developed using this strategy.14 FBDD starts from low-affinity binding fragments, which eventually grows into highly active compounds through fragment modification.15 Compared with traditional high-throughput screening (HTS), FBDD has the advantages of target specificity and cost efficiency.16,17 Nevertheless, it still involves tedious synthesis and biological verification, same as the other conventional methods. This inherent disadvantage of FBDD can be effectively overcome by combining with DCC to yield a one-pot substance generation and affinity screening protocol, significantly accelerating the drug R&D process.12
Diabetes mellitus (DM) is a metabolic disease featured by elevated level of glucose in the blood due to insufficient insulin production, and it usually leads to severe complications in the liver, heart, brain, and kidneys.18 α-Amylase (EC 3.2.1.1), which is secreted primarily by the human pancreas, is the primary enzyme responsible for converting polysaccharides to oligosaccharides.19 The resulting oligosaccharides are further hydrolyzed into absorbable α-d-glucose by α-glucosidase (EC 3.2.1.20) located at the intestine’s brush boundary, leading to an increase in blood sugar level (Figure 2).20−22 Thus, inhibition on either of these enzymes can be a solution to blood sugar regulation. Indeed, acarbose, miglitol and voglibose are clinically prescribed α-amylase and α-glucosidase inhibitors for the treatment of T2DM.23,24 However, serious side effects such as diarrhea, gastrointestinal distress, and abdominal distension are often reported during the application of those medicines.25,26 Several enzymatic studies have documented that α-amylase inhibition is one of the leading causes for gastrointestinal side effects, as it can also facilitate the transfer of undigested polysaccharides to the intestine.27 Under such circumstances, selective inhibition of α-glucosidase can reduce the production of glucose on the one hand and circumvent the possible intestinal side effects of α-amylase inhibition on the other.28 Therefore, the development of efficient and selective α-glucosidase inhibitors is in great demand.
Figure 2.

Effects of α-amylase and α-glucosidase on polysaccharide hydrolysis.
In this study, we report the development of efficient and selective α-glucosidase inhibitors using fragment-based DCC. Beginning with several rationally designed fragments, two iteratively evolved DCLs containing a total of 29 compounds were generated and screened in the presence of α-glucosidase, leading to the identification of a highly selective and potent α-glucosidase inhibitor. This in situ one-pot ligand synthesis and bioactivity screening methodology significantly outperforms the other conventional methods in term of overall efficiency, making it a valuable reference for the discovery of other protein inhibitors.
Inspired by the structures of previously reported α-glucosidase inhibitors 1–6 (Figure 1c), molecules 1–5, all of which followed the “rule of three” (MW < 300, hydrogen bond acceptors ≤3, log P ≤ 3) for appropriate initial fragments, were designed as starting points for fragment growth.29−34 By docking with the α-glucosidase crystal structure, it was revealed that all fragments could deeply localize to the enzyme’s active site with binding energy less than −5 kcal/mol. More importantly, the target zone highlighted in Figure 3b offered as an ideal space for fragment growth to enhance the ligand binding affinity through numbers of extra binding interactions. Acylhydrazone formation has been proved as a useful reversible reaction to be applied in DCC-related medicinal chemistry due to its controllable reaction kinetic property and product stability under acidic and physiological conditions. Therefore, we envisaged using acylhydrazone-based DCC to link extra components to fragments 1–5, aiming to obtain optimized ligands with higher binding affinities (Figure 3a).
Figure 3.

(a) Fragment growing design concept based on DCC; (b) overlay of the crystal structures of fragments 1 (orange), 2 (magenta), 3 (gray), 4 (green), and 5 (cyan) bound to α-glucosidase.
We designed and synthesized three acylhydrazides C1–C3 (Scheme 1) based on the structures of known glucosidase inhibitors, intending to form additional hydrophobic and π–π stacking interactions with residues in the target region. Meanwhile, initial fragments-derived aromatic aldehydes E1–E5 were used as complementary components to construct the first DCL. Three acylhydrazides and five aldehydes were mixed in PBS buffer (pH 6.25) in the presence of aniline to generate DCL-1, leading to the formation of 15 acylhydrazone species (Scheme 1). Equilibrium of DCL-1 was confirmed in 16 h using HPLC chromatography with all acylhydrazones detected. By adding α-glucosidase as the template, amplification of compounds C1E3, C2E3, C3E3, and C3E5 was observed in comparison to the blank reaction (Figure 4a,b). In addition to that, α-amylase was spontaneously used as template to evaluate its influence on DCL-1. To our delight, no significant difference between two equilibria was observed with/without α-amylase, as displayed in Figure 4c.
Scheme 1. Generation of DCL-1.

Figure 4.

HPLC chromatographic analysis of acylhydrazones in DCL-1: (a) at equilibrium; (b) equilibrium with α-glucosidase; (c) equilibrium with α-amylase; (d) amplified compounds.
To verify the inhibitory activity of the amplified hits, acylhydrazones C1E3, C2E3, C3E3, and C3E5 were synthesized for biological evaluation. Among them, compound C3E5 showed moderate α-glucosidase inhibitory activity (IC50 = 32.53 μM, Table 1). As comparison, although components with sulfonate groups (C1E3, C2E3, C3E3) were clearly amplified in DCL-1 in the presence of α-glucosidase, only compound C3E3 (IC50 = 167.98 μM) displayed very low inhibitory activity. A possible explanation was that those ligands might bind to other parts of enzyme instead of the catalytic pocket. In accordance with the α-amylase templated DCL outcome, no α-amylase inhibitory activity (IC50 > 200 μM) was observed for all four ligands, allowing us to continue developing more potent and selective α-glucosidase inhibitors based on acylhydrazone C3E5. Besides, cytotoxicity of compound C3E5 was assessed using the standard CCK-8 method on the HepG2 cell line.35,36 The toxicity of compound C3E5 was found to be insignificant (CC50 > 200 μM), indicating its good biocompatibility.
Table 1. in Vitro Biological Activity of Amplified Hits and Acarbose.
| IC50 (μM) |
|||||
|---|---|---|---|---|---|
| entry | compound | α-glucosidase | α-amylase | CC50 (μM) | amplification folda |
| 1 | C1E3 | >200 | >200 | 1.59 | |
| 2 | C2E3 | >200 | >200 | 2.10 | |
| 3 | C3E3 | 167.98 ± 5.59 | >200 | 4.86 | |
| 4 | C3E5 | 32.53 ± 2.38 | >200 | >200 | 1.95 |
| 5 | C4E5 | 6.49 ± 0.53 | 128.10 ± 4.62 | >200 | 2.68 |
| 6 | C4E6 | 22.60 ± 1.12 | 151.46 ± 5.69 | >200 | 2.13 |
| 7 | C4E8 | 2.52 ± 0.21 | >200 | >200 | 2.32 |
| 8 | acarbose | 8.06 ± 0.33 | 5.32 ± 0.21 | ||
Calculated as (%P/%B), where %P and %B were the relative peak areas of the compounds in the HPLC spectra of the protein-templated reaction and the blank reaction, respectively.
Subsequently, an evolved DCL-2 was established using acylhydrzone C3E5 as the model compound. Four chromone aldehydes and two heterocyclic acylhydrazides were mixed together with building blocks C3 and E5, leading to the formation of 15 optimized acylhydrazones (Scheme 2). Upon addition of α-glucosidase, compounds C4E5, C4E6, and C4E8 were clearly amplified (Figure 5b). Same as DCL-1, no significant amplification was observed when α-amylase was used as the template, as expected (Figure 5c,d).
Scheme 2. Generation of DCL-2.

Figure 5.

HPLC chromatographic analysis of acylhydrazones in DCL-2: (a) at equilibrium; (b) equilibrium with α-glucosidase; (c) equilibrium with α-amylase; (d) amplified compounds.
The same in vitro biological assay was applied to evaluate the amplified ligands from DCL-2. As shown in Table 1, all hits exhibited stronger α-glucosidase inhibitory activity than compound C3E5. Among them, the activities of compounds C4E5 (IC50 = 6.49 μM) and C4E8 (IC50 = 2.52 μM) were higher than that of the standard acarbose (IC50 = 8.06 μM). Excitingly, the most active inhibitor C4E8 exhibited a nearly 80-fold enhancement in activity over the initial fragment 5 (IC50 = 187.12 μM), undoubtedly confirming the success of the evolved DCL-2. From Table 1 it could be seen that acarbose had similar inhibitory effects on both enzymes (α-glucosidase: IC50 = 8.06 μM; α-amylase: IC50 = 5.32 μM), while ligand C4E8 displayed excellent selectivity on α-glucosidase against α-amylase (compound C4E8 did not show obvious inhibition on amylase at up to 200 μM concentration). Also, cytotoxicity of the new hits was also shown to be negligible at concentrations up to 200 μM (Table 1).
To gain further insight into the inhibition mechanism of the most active compound C4E8, kinetic characterization was performed using a slightly modified method that had been previously described. As depicted in Figure 6a, the Lineweaver–Burk plot obtained by linear fitting intersected in the second quadrant, indicating that compound C4E8 had a mixed type inhibition on α-glucosidase, and the Michaelis constant (Km) of α-glucosidase was determined to be 0.93 mM, which was in agreement with other earlier studies.37,38 The Dixon plot (Figure 6b) revealed that the competitive inhibition constant (Ki) and the noncompetitive inhibition constant (αKi) of compound C4E8 were 1.98 μM and 2.75 μM, respectively, resulting in conclusion that this inhibitor would preferentially bind to free enzyme than the enzyme–substrate complex.
Figure 6.

(a) Lineweaver–Burk plot of 1/[S] vs 1/V at various inhibitor concentrations; (b) Dixon plot of [I] vs 1/V at different substrate concentrations.
Finally, molecular docking was used to disclose the possible enzyme–ligand binding interactions. According to the docked conformation of compound C4E8 in Figure 7a, the ligand was well accommodated in the center of the enzyme active pocket. Thermodynamically beneficial binding interactions, including hydrogen bonding, π–π stacking, and hydrophobic interactions were formed between α-glucosidase and compound C4E8, yielding binding energy of −10.8 kcal/mol (Figure 7b,c). Specifically, the indole moiety formed two strong face-to-face π–π stacking interactions with Phe 177 residue and a set of edge-to-face π–π stacking interaction with His 239 residue. Moreover, the carbonyl group of compound C4E8 established a hydrogen bonding with Arg 312 residue, and a similar hydrogen bonding was also established between its imino group and Phe 157 residue. It was worth noting that the methyl group of the chromone moiety also formed a hydrophobic interaction with Phe 177 residue, a key factor that might be attributed to the enhanced inhibitory activity of compound C4E8 compared to compound C3E5. Stacked docking simulation highlighted the varied binding mechanisms between compound C4E8 and fragment 5 (Figure 7d). While the aldehyde-derived moieties of two compounds had very similar docking poses, the indole moiety of compound C4E8 extended into the target region of the enzyme active site, as we initially anticipated, and was consequently stabilized by numerous residues, resulting in dramatic enhancement in inhibitory efficiency.
Figure 7.
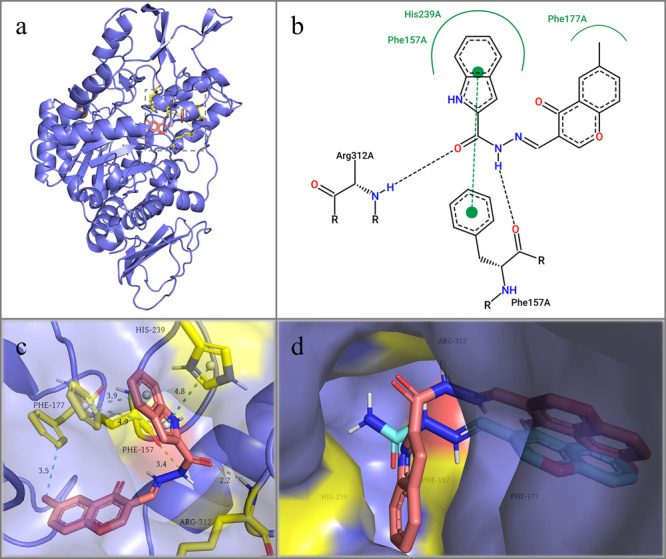
(a) 3D representation of the docking state of compound C4E8 against α-glucosidase, with the dashed box representing the binding site of compound C4E8; (b) 2D display of the docking status; (c) 3D depiction of the docking status of compound C4E8 against α-glucosidase. Hydrogen bonding, π–π stacking, and hydrophobic interactions were indicated by dashed yellow, green, and blue lines, respectively; (d) Comparison of binding poses in enzyme crystal structure between compound 5 (cyan) and compound C4E8 (red).
In conclusion, we successfully developed a highly potent and selective α-glucosidase inhibitor by exploring fragment-based dynamic combinatorial chemistry. Starting from rationally designed fragments 1–5, two successively evolved DCLs containing a total of 29 acylhydrazone substrates were generated and screened using α-glucosidase as template. The best ligand C4E8 identified from other candidates was not only more than three times more active than the reference acarbose but also showed low cytotoxicity. In addition, control experiments were also performed using α-amylase as template, to which the acylhydrazone substrates did not show clear response. Possible binding mechanism of ligand C4E8 against α-glucosidase was further investigated in the kinetic studies and molecular docking. Overall, compound C4E8 exhibited the potential as an excellent lead compound for the development of commercial T2DM medicine.
Acknowledgments
This work was supported by the Natural Science Foundation of Jiangsu Province (BK20180853), China Postdoctoral Science Foundation (2019 M651717). and Six Talent Peaks Project in Jiangsu Province (SWYY-162).
Glossary
Abbreviations
- FBDD
fragment-based drug design
- DCC
dynamic combinatorial chemistry
- DCL
dynamic combinatorial library
- HTS
high-throughput screening
- DM
diabetes mellitus.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.2c00405.
Experimental procedures, characterization data, and biological data (PDF)
Author Contributions
Yao Wu: conceptualization, project administration, visualization, writing–original draft. Changming Liu: conceptualization, project administration. Lei Hu: conceptualization, project administration, writing–original draft.
The authors declare no competing financial interest.
Supplementary Material
References
- Huc I.; Lehn J.-M. Virtual combinatorial libraries: Dynamic generation of molecular and supramolecular diversity by self-assembly. PNAS 1997, 94 (6), 2106–2110. 10.1073/pnas.94.6.2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J.; Zhang S.; Zhao S.; Hu L. Identification and synthesis of an efficient multivalent E. coli heat labile toxin inhibitor - A dynamic combinatorial chemistry approach. Bioorg. Med. Chem. 2020, 28 (9), 115436. 10.1016/j.bmc.2020.115436. [DOI] [PubMed] [Google Scholar]
- Ramström O.; Lohmann S.; Bunyapaiboonsri T.; Lehn J. M. Dynamic combinatorial carbohydrate libraries: probing the binding site of the concanavalin A lectin. Chem. 2004, 10 (7), 1711–5. 10.1002/chem.200305551. [DOI] [PubMed] [Google Scholar]
- Hu L.; Ramström O. Silver-catalyzed dynamic systemic resolution of alpha-iminonitriles in a 1,3-dipolar cycloaddition process. Chem. Commun. 2014, 50 (29), 3792–4. 10.1039/C4CC00944D. [DOI] [PubMed] [Google Scholar]
- Soubhye J.; Gelbcke M.; Van Antwerpen P.; Dufrasne F.; Boufadi M. Y.; Neve J.; Furtmuller P. G.; Obinger C.; Zouaoui Boudjeltia K.; Meyer F. From Dynamic Combinatorial Chemistry to in Vivo Evaluation of Reversible and Irreversible Myeloperoxidase Inhibitors. ACS. Med. Chem. Lett. 2017, 8 (2), 206–210. 10.1021/acsmedchemlett.6b00417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S.; Wu Y.; Hu L. Identification and synthesis of selective cholesterol esterase inhibitor using dynamic combinatorial chemistry. Bioorg. Chem. 2022, 119, 105520. 10.1016/j.bioorg.2021.105520. [DOI] [PubMed] [Google Scholar]
- Ramström O.; Lehn J. M. Drug discovery by dynamic combinatorial libraries. Nat. Rev. Drug. Discovery 2002, 1 (1), 26–36. 10.1038/nrd704. [DOI] [PubMed] [Google Scholar]
- Xu J.; Zhao S.; Zhang S.; Pei J.; Li Y.; Zhang Y.; He X.; Hu L. Development of a multivalent acetylcholinesterase inhibitor via dynamic combinatorial chemistry. Int. J. Biol. Macromol. 2020, 150, 1184–1191. 10.1016/j.ijbiomac.2019.10.127. [DOI] [PubMed] [Google Scholar]
- Caraballo R.; Dong H.; Ribeiro J. P.; Jimenez-Barbero J.; Ramström O. Direct STD NMR identification of beta-galactosidase inhibitors from a virtual dynamic hemithioacetal system. Angew. Chem., Int. Ed.Engl. 2010, 49 (3), 589–93. 10.1002/anie.200903920. [DOI] [PubMed] [Google Scholar]
- Hu L.; Schaufelberger F.; Timmer B. J. J.; Flos M. A.; Ramström O., Constitutional Dynamic Chemistry. In Kirk-Othmer Encyclopedia of Chemical Technology, 2014; pp 1–25. [Google Scholar]
- Mondal M.; Hirsch A. K. Dynamic combinatorial chemistry: a tool to facilitate the identification of inhibitors for protein targets. Chem. Soc. Rev. 2015, 44 (8), 2455–88. 10.1039/C4CS00493K. [DOI] [PubMed] [Google Scholar]
- Wu Y.; Zhao S.; Liu C.; Hu L. Development of urease inhibitors by fragment-based dynamic combinatorial chemistry. ChemMedChem. 2022, 17 (19), e202200307 10.1002/cmdc.202200307. [DOI] [PubMed] [Google Scholar]
- Zhao S.; Xu J.; Zhang S.; Han M.; Wu Y.; Li Y.; Hu L. Multivalent butyrylcholinesterase inhibitor discovered by exploiting dynamic combinatorial chemistry. Bioorg. Chem. 2021, 108, 104656. 10.1016/j.bioorg.2021.104656. [DOI] [PubMed] [Google Scholar]
- Hajduk P. J.; Greer J. A decade of fragment-based drug design: strategic advances and lessons learned. Nat. Rev. Drug. Discovery 2007, 6 (3), 211–9. 10.1038/nrd2220. [DOI] [PubMed] [Google Scholar]
- Bancet A.; Raingeval C.; Lomberget T.; Le Borgne M.; Guichou J. F.; Krimm I. Fragment Linking Strategies for Structure-Based Drug Design. J. Med. Chem. 2020, 63 (20), 11420–11435. 10.1021/acs.jmedchem.0c00242. [DOI] [PubMed] [Google Scholar]
- Keseru G. M.; Erlanson D. A.; Ferenczy G. G.; Hann M. M.; Murray C. W.; Pickett S. D. Design Principles for Fragment Libraries: Maximizing the Value of Learnings from Pharma Fragment-Based Drug Discovery (FBDD) Programs for Use in Academia. J. Med. Chem. 2016, 59 (18), 8189–206. 10.1021/acs.jmedchem.6b00197. [DOI] [PubMed] [Google Scholar]
- Gong H.; Yuan Z.; Zhan L. High-throughput screening against ∼ 6.1 million structurally diverse, lead-like compounds to discover novel ROCK inhibitors for cerebral injury recovery. Mol. Divers. 2016, 20 (2), 537–549. 10.1007/s11030-015-9650-y. [DOI] [PubMed] [Google Scholar]
- Dhameja M.; Gupta P. Synthetic heterocyclic candidates as promising alpha-glucosidase inhibitors: An overview. Eur. J. Med. Chem. 2019, 176, 343–377. 10.1016/j.ejmech.2019.04.025. [DOI] [PubMed] [Google Scholar]
- Wu Y.; Zhao S.; Hu L. Identification of potent alpha-amylase inhibitors via dynamic combinatorial chemistry. Bioorg. Med. Chem. 2022, 55, 116609. 10.1016/j.bmc.2022.116609. [DOI] [PubMed] [Google Scholar]
- Saddique F. A.; Ahmad M.; Ashfaq U. A.; Muddassar M.; Sultan S.; Zaki M. E. A. Identification of Cyclic Sulfonamides with an N-Arylacetamide Group as alpha-Glucosidase and alpha-Amylase Inhibitors: Biological Evaluation and Molecular Modeling. Pharmaceuticals 2022, 15, 106. 10.3390/ph15010106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges de Melo E.; da Silveira Gomes A.; Carvalho I. α- and β-Glucosidase inhibitors: chemical structure and biological activity. Tetrahedron 2006, 62 (44), 10277–10302. 10.1016/j.tet.2006.08.055. [DOI] [Google Scholar]
- Mohammadi-Khanaposhtani M.; Nikraftar A.; Asgari M. S.; Emadi M.; Mojtabavi S.; Faramarzi M. A.; Rastegar H.; Larijani B.; Mahdavi M. Synthesis, in vitro and in silico enzymatic inhibition assays, and toxicity evaluations of new 4,5-diphenylimidazole-N-phenylacetamide derivatives as potent α-glucosidase inhibitors. Med. Chem. Res. 2021, 30 (6), 1273–1283. 10.1007/s00044-021-02734-5. [DOI] [Google Scholar]
- Tundis R.; Loizzo M. R.; Menichini F. Natural Products as α-Amylase and α-Glucosidase Inhibitors and their Hypoglycaemic Potential in the Treatment of Diabetes: An Update. Mini. Rev. Med. Chem. 2010, 10 (4), 315–331. 10.2174/138955710791331007. [DOI] [PubMed] [Google Scholar]
- Shamim S.; Khan K. M.; Ullah N.; Chigurupati S.; Wadood A.; Ur Rehman A.; Ali M.; Salar U.; Alhowail A.; Taha M.; Perveen S. Synthesis and screening of (E)-3-(2-benzylidenehydrazinyl)-5,6-diphenyl-1,2,4-triazine analogs as novel dual inhibitors of alpha-amylase and alpha-glucosidase. Bioorg. Chem. 2020, 101, 103979. 10.1016/j.bioorg.2020.103979. [DOI] [PubMed] [Google Scholar]
- Hameed S.; Kanwal; Seraj F.; Rafique R.; Chigurupati S.; Wadood A.; Rehman A. U.; Venugopal V.; Salar U.; Taha M.; Khan K. M. Synthesis of benzotriazoles derivatives and their dual potential as alpha-amylase and alpha-glucosidase inhibitors in vitro: Structure-activity relationship, molecular docking, and kinetic studies. Eur. J. Med. Chem. 2019, 183, 111677. 10.1016/j.ejmech.2019.111677. [DOI] [PubMed] [Google Scholar]
- Nawaz M.; Taha M.; Qureshi F.; Ullah N.; Selvaraj M.; Shahzad S.; Chigurupati S.; Waheed A.; Almutairi F. A. Structural elucidation, molecular docking, alpha-amylase and alpha-glucosidase inhibition studies of 5-amino-nicotinic acid derivatives. BMC. Chem. 2020, 14, 43. 10.1186/s13065-020-00695-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y.; Zhang B.; Sun W.; Xing Y., Chapter 36 - Intervention of Prediabetes by Flavonoids From Oroxylum indicum. In Bioactive Food as Dietary Interventions for Diabetes (Second ed.), Watson R. R.; Preedy V. R., Eds. Academic Press: 2019; pp 559–575. [Google Scholar]
- Sepehri N.; Azizian H.; Ghadimi R.; Abedinifar F.; Mojtabavi S.; Faramarzi M. A.; Moghadamnia A. A.; Zabihi E.; Mohebbi G.; Larijani B.; Hamedifar H.; Mohammadi-Khanaposhtani M.; Mahdavi M. New 4,5-diphenylimidazole-acetamide-1,2,3-triazole hybrids as potent α-glucosidase inhibitors: synthesis, in vitro and in silico enzymatic and toxicity evaluations. Monatsh 2021, 152 (6), 679–693. 10.1007/s00706-021-02779-7. [DOI] [Google Scholar]
- Arshad T.; Khan K. M.; Rasool N.; Salar U.; Hussain S.; Asghar H.; Ashraf M.; Wadood A.; Riaz M.; Perveen S.; Taha M.; Ismail N. H. 5-Bromo-2-aryl benzimidazole derivatives as non-cytotoxic potential dual inhibitors of alpha-glucosidase and urease enzymes. Bioorg. Chem. 2017, 72, 21–31. 10.1016/j.bioorg.2017.03.007. [DOI] [PubMed] [Google Scholar]
- Barakat A.; Islam M. S.; Al-Majid A. M.; Soliman S. M.; Ghabbour H. A.; Yousuf S.; Choudhary M. I.; Ul-Haq Z. Synthesis, molecular structure, spectral analysis, and biological activity of new malonamide derivatives as α-glucosidase inhibitors. J. Mol. Struct. 2017, 1134, 253–264. 10.1016/j.molstruc.2016.12.093. [DOI] [Google Scholar]
- Wang G.; Chen M.; Wang J.; Peng Y.; Li L.; Xie Z.; Deng B.; Chen S.; Li W. Synthesis, biological evaluation and molecular docking studies of chromone hydrazone derivatives as alpha-glucosidase inhibitors. Bioorg. Med. Chem. Lett. 2017, 27 (13), 2957–2961. 10.1016/j.bmcl.2017.05.007. [DOI] [PubMed] [Google Scholar]
- Spasov A. A.; Babkov D. A.; Prokhorova T. Y.; Sturova E. A.; Muleeva D. R.; Demidov M. R.; Osipov D. V.; Osyanin V. A.; Klimochkin Y. N. Synthesis and biological evaluation of 2-acylbenzofuranes as novel alpha-glucosidase inhibitors with hypoglycemic activity. Chem. Biol. Drug. Des. 2017, 90 (6), 1184–1189. 10.1111/cbdd.13038. [DOI] [PubMed] [Google Scholar]
- Taha M.; Ismail N. H.; Imran S.; Ainaa I.; Selvaraj M.; baharudin M. s.; Ali M.; Khan K. M.; Uddin N. Synthesis of 2-phenyl-1H-imidazo[4,5-b]pyridine as type 2 diabetes inhibitors and molecular docking studies. Med. Chem. Res. 2017, 26 (5), 916–928. 10.1007/s00044-017-1806-0. [DOI] [Google Scholar]
- Kaur R.; Kumar R.; Dogra N.; Yadav A. K. Design, synthesis, biological evaluations and in silico studies of sulfonate ester derivatives of 2-(2-benzylidenehydrazono)thiazolidin-4-one as potential α-glucosidase inhibitors. J. Mol. Struct. 2022, 1247, 131266. 10.1016/j.molstruc.2021.131266. [DOI] [Google Scholar]
- Kawata K.; Osawa M.; Okabe S. In Vitro Toxicity of Silver Nanoparticles at Noncytotoxic Doses to HepG2 Human Hepatoma Cells. Environ. 2009, 43 (15), 6046–6051. 10.1021/es900754q. [DOI] [PubMed] [Google Scholar]
- Zhang H. Y.; Xu W. Q.; Wang Y. W.; Omari-Siaw E.; Wang Y.; Zheng Y. Y.; Cao X.; Tong S. S.; Yu J. N.; Xu X. M. Tumor targeted delivery of octreotide-periplogenin conjugate: Synthesis, in vitro and in vivo evaluation. Int. J. Pharm. 2016, 502 (1–2), 98–106. 10.1016/j.ijpharm.2016.02.024. [DOI] [PubMed] [Google Scholar]
- Li Y. S.; He M.; Zhou T. S.; Wang Q.; He L.; Wang S. J.; Hu B.; Wei B.; Wang H.; Cui Z. N. 2,5-Disubstituted furan derivatives containing 1,3,4-thiadiazole moiety as potent alpha-glucosidase and E. coli beta-glucuronidase inhibitors. Eur. J. Med. Chem. 2021, 216, 113322. 10.1016/j.ejmech.2021.113322. [DOI] [PubMed] [Google Scholar]
- Kawde A. N.; Taha M.; Alansari R. S.; Almandil N. B.; Anouar E. H.; Uddin N.; Rahim F.; Chigurupati S.; Nawaz M.; Hayat S.; Ibrahim M.; Elakurthy P. K.; Vijayan V.; Morsy M.; Ibrahim H.; Baig N.; Khan K. M. Exploring efficacy of indole-based dual inhibitors for alpha-glucosidase and alpha-amylase enzymes: In silico, biochemical and kinetic studies. Int. J. Biol. Macromol. 2020, 154, 217–232. 10.1016/j.ijbiomac.2020.03.090. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.