

Abstract

Drug resistance to first-line antimalarials—including artemisinin—is increasing, resulting in a critical need for the discovery of new agents with novel mechanisms of action. In collaboration with the Walter and Eliza Hall Institute and with funding from the Wellcome Trust, a phenotypic screen of Merck’s aspartyl protease inhibitor library identified a series of plasmepsin X (PMX) hits that were more potent than chloroquine. Inspired by a PMX homology model, efforts to optimize the potency resulted in the discovery of leads that, in addition to potently inhibiting PMX, also inhibit another essential aspartic protease, plasmepsin IX (PMIX). Further potency and pharmacokinetic profile optimization efforts culminated in the discovery of WM382, a very potent dual PMIX/X inhibitor with robust in vivo efficacy at multiple stages of the malaria parasite life cycle and an excellent resistance profile.
Keywords: Malaria, plasmepsin X (PMX), plasmepsin IX (PMIX), WM382, dual inhibitor
Malaria is a life-threatening disease caused by Plasmodium parasites that is transmitted by infected female Anopheles mosquitoes. In 2020, there were an estimated 241 million cases of malaria worldwide and 627 000 malaria-related deaths.1 Approximately 95% of malaria cases and 96% of deaths occur in Africa, where Plasmodium falciparum (P. falciparum) is the predominant parasitic species. Current first-line treatments for P. falciparum malaria include artemisinin-based combination therapies (ACTs), in which an artemisinin is combined with a longer-half-life partner drug. Resistance to artemisinins first emerged in Cambodia in 2008, and resistance in the Mekong region is now widespread.2 There is more recent evidence that in Africa P. falciparum has developed genetic variants that confer resistance to artemisinin.3 While longer courses of therapy currently remain clinically effective against the artemisinin resistance observed to date, these data serve as a warning sign that potential treatment failure is on the horizon. The development of antimalarial agents with novel mechanisms of action is thus of paramount importance.
In the malaria parasite there is a family of 10 aspartic proteases called plasmepsins (PMI to PMX).4 In an effort to identify antimalarial agents with a novel mechanism of action, we initiated a phenotypic screen using red blood cells infected with P. falciparum and 1000 drug-like representative aspartic protease inhibitors from a 20 000-member library of Merck aspartic protease compounds. This study resulted in the identification of the hits (R,S)-WM4 and (R,S)-WM5, which displayed excellent in vitro potency (P. falciparum EC50 = 6.2 and 17 nM, respectively; Figure 1). Selection of in vitro resistance with WM4 and WM5 suggested that PMX is the target of these compounds.5 This was subsequently confirmed by a number of criteria, including a direct PMX protease biochemical FRET assay, in which the most potent diastereomers, (R)-WM4 and (R)-WM5, displayed IC50 values of 1.9 and 5.7 nM, respectively (Figure 1).5 Both inhibitors employ an iminopyrimidone ring, previously discovered in a β-secretase inhibitor program at Schering-Plough/Merck, that acts as “the aspartic warhead” binding to the two aspartic acid residues through three hydrogen bonds (Figure 2).6−8
Figure 1.

Structure and potencies of phenotypic screening hits.
Figure 2.

Aspartic warhead interactions of indinavir and WM4. While WM4 forms a network of three hydrogen bonds, indinavir’s central alcohol mimics an amide hydrolysis transition state.
After the identification of WM4 and WM5, potency and rodent pharmacokinetic (PK) optimization studies were initiated with the aim of identifying a compound suitable for in vivo efficacy studies of this novel mechanism of action. The structure–activity relationship (SAR) development of this class of inhibitors was guided by a PMX homology model. The model was built using the sequence of P. falciparum PMX and the crystal structure of renin (PDB ID 4S1G), a related human aspartic protease, as a template. Compounds were docked to the active site of PMX. The interactions predicted by the model are consistent with those seen in the recently solved PMX X-ray crystal structures.9
Evaluation of key interactions of the PMX active site in the aspartic warhead region of the model revealed not only that the iminopyrimidone functionality forms an efficient hydrogen-bonding network with the two aspartic acid residues at the PMX active site but also that the two alkyl groups at the 6-position fill two separate hydrophobic pockets (Figure 3A). With respect to alkyl substitution at the 6-position of the iminopyrimidone, diethyl and (R)-6-(cyclopropylmethyl)-6-phenyl result in the most potent warheads (compounds 6 and 7 in Table 1). Other geminal disubstitutions such as 6-isopropyl-6-methyl (compound 1) and 6,6-dimethyl (compound 5) are not as efficient at grasping hydrophobicity at the two distinct lipophilic cavities mentioned above. Monosubstitution at the 6-position, as in compound 3, results in lack of potency, and different ways to substitute the iminopyrimidone warhead, such as with a spirocycle (compound 2) or a fused bicycle (compound 4) were not well-tolerated due to a lack of shape complementarity at the PMX active site in this region of the molecule. A six-membered iminopyrimidone (compound 7) is preferable to the five-membered-ring iminohydantoin analogue (compound 8) to efficiently form the hydrogen-bonding network required for potency.
Figure 3.

Model of compound 35 in the active site of PMX: (A) aspartic warhead and benzylic substituent; (B) “right-hand” amine.
Table 1. Aspartic Warhead SAR.
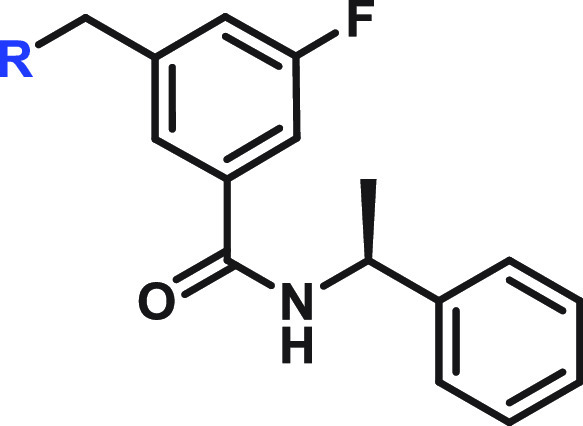

Single enantiomer. Both R and S were tested.
Phenyl, heteroaryl, and n-alkyl groups at the benzylic position with an R configuration (assumed from the known absolute configuration of compounds 10 and 11; Table 2) can dramatically increase the potency, since these groups stabilize the bioactive conformation as well as fill a narrow hydrophobic cavity in the active site (Figure 3A). The S configuration results in a significant loss of potency, as represented by compound 11. Therefore (R)-phenyl and presumably (R)-pyridin-3-yl and (R)-n-propyl benzylic substitution (compounds 10, 12, and 14, respectively) provide the highest potency in this region. Addition of cycloalkyl groups such as cyclopropyl (compound 13) results in a significant loss of potency, while the introduction of polarity at the benzylic position in the form of an ether (compound 16) or a tertiary amine such as a pyrrolidine (compound 17) retains a high level of potency. However, polarity is not well-tolerated at all positions in this area. This is exemplified by the incorporation of a hydrogen-bond donor via an ethanol functionality in compound 15, which results in a loss of potency.
Table 2. Benzylic Substituent SAR.
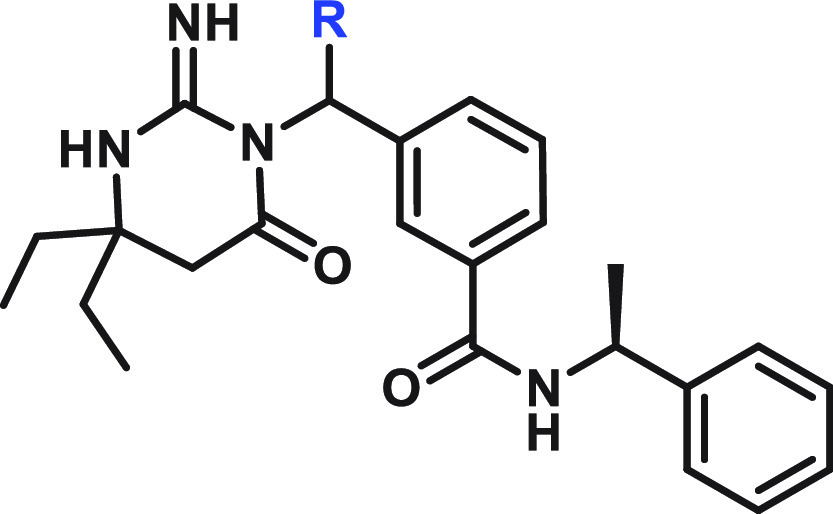

Single enantiomer. The absolute configuration of the carbon attached to R was not determined.
Mixture of two diastereomers.
Compounds 18–22 (Table 3) exemplify the importance of a primary amide (which forms a hydrogen bond with a carbonyl oxygen atom of the protein backbone) and a phenyl group at the right spatial location as key features in the pharmacophore of these inhibitors. Simple ethylamine compound 19, which lacks the key phenyl group, and secondary amide compound 20 are inactive. Compound 21 shows that a two-carbon linker between the amide nitrogen atom and the phenyl group is likewise not well-tolerated. The introduction of a nitrogen atom in the phenyl group (compound 22) results in a loss of potency versus reference compound 18. Interestingly, fused bicyclic amines result in the most potent “right-hand” amines because of the superior ability of this functionality to lock the bioactive conformation versus (S)-phenylethanamine compound 18. A high level of potency is achieved with 5,6- and 6,6-fused bicycles (compounds 23 and 24), and potency is retained when a heteroatom is introduced into the saturated ring, such as in isochromane compound 25 and tetrahydroquinoline compound 26. The most potent “right-hand” amine found in this study is a chromane group (compound 27). While the phenyl group of the chromane efficiently fills a hydrophobic cavity in the PMX active site, its oxygen atom may also form a favorable hydrogen bond with an asparagine residue (Figure 3B).
Table 3. “Right-Hand” Amine SAR.


Mixtures of two diastereomers.
The central benzamide group acts as a connector, efficiently positioning the key pharmacophore functionalities of these inhibitors, the aspartic warhead and the “right-hand” amine, at the correct spatial locations in the PMX active site. The preferred central group is phenyl (compound 28; Table 4). While heteroaryls such as pyridine regioisomers 30–32 are well-tolerated, pyridine regioisomer 29, pyrimidine compound 33, and pyridazine compound 34 display significantly reduced potency.
Table 4. Central Ring SAR.

Introduction of the most potent moieties shown in Tables 1–4 (diethylaminopyrimidone, n-propyl at the benzylic position, a phenyl central core, and a chromane amide) in the same molecule resulted in compound 35 (Figure 4), a very potent plasmepsin inhibitor that displays subnanomolar potency (EC50 = 0.4 nM) inP. falciparumin vitro growth assays and IC50 values of 0.06 and 4.5 nM in the biochemical PMX and PMIX assays, respectively.
Figure 4.

Lead compound 35 displays moderately good cell membrane permeability (Papp = 16 × 10–6 cm/s) and subnanomolar potency in inhibiting P. falciparum growth in vitro. The next step was to identify a compound that retains a high level of potency and displays adequate mouse PK to evaluate the potential of this novel mechanism of action in an in vivo malaria model. PK studies on compound 35 showed short mean residence times (MRTs), high clearance in rats and mice, and low oral bioavailability in rats (see Table 6). One possible reason for the poor rodent PK parameters of compound 35 is its relatively high lipophilicity (clogP = 5.8 and logD7.4 = 2.9). Efforts then focused on inventing compounds that retain a high level of potency but display superior rodent PK parameters. Attempts to reduce the lipophilicity and block one or more potential metabolic soft spots10 did not improve the rodent PK. Modeling indicated that these inhibitors adopt a U-shaped conformation in the PMX active site (Figure 3). This bioactive conformation inspired a number of constrained design proposals such as attaching the 2-position of the central ring to the substituent of the benzylic group through a six-membered ring (Figure 5). This strategy resulted in a very potent series of central bicyclic core PMX inhibitors (Table 5), exemplified by compound 36. In addition to highly potent cell-based activity (EC50 = 2.5 nM), compound 36 displayed significantly improved rat clearance (CLp = 79 mL min–1 kg–1 and CLu = 518 mL min–1 kg–1) and MRT (2.7 h), although its oral bioavailability in mouse was low (Table 6).
Table 6. Physicochemical Properties and Rodent PK Profiles.

| Physicochemical Properties | |||
|---|---|---|---|
| 35 | 36 | WM382 | |
| LDH P. falciparum EC50 (nM) | 0.4 | 2.5 | 0.4 |
| FRET P. falciparum IC50 PMX/PMIX (nM) | 0.06/4.5 | 0.07/2.2 | 0.03/1.4 |
| LLE PMX/PMIX | 7.4/5.5 | 8.1/6.4 | 8.0/6.3 |
| clogP/TPSA | 5.8/95 | 3.6/104 | 4.6/104 |
| logD7.4/kin. sol. (μM) | 2.9/0.2 | 2.1/0.2 | 2.5/0.1 |
| MDCK (A:B) (10–6cm/s) | 16.3 | 8.8 | 11.6 |
| Rodent PK Profiles | ||||||
|---|---|---|---|---|---|---|
|
35 |
36 |
WM382 |
||||
| rat | mouse | rat | mouse | rat | mouse | |
| MRT (h) | 1.1 | 1.1 | 2.7 | 1.3 | 3.7 | 2.5 |
| CLP/CLu (mL min–1 kg–1) | 103/5,648 | 36/3,148 | 79/518 | 68/1,278 | 42/1,022 | 42/774 |
| Vd/Vd,u (L/kg) | 6.7/340 | 2.3/204 | 12/80 | 5.4/102 | 9.4/222 | 6.6/120 |
| F (%) | 6 | 14 | 27 | 4 | 8 | 38 |
| PPB (%) | 98.0 | 98.9 | 84.8 | 94.7 | 95.2 | 94.5 |
Figure 5.

Strategy that resulted in potent PMX/IX dual inhibitors with improved rodent PK.
Table 5. Bicyclic Core Inhibitor Optimization.


Mixture of two diastereomers.
Percent inhibition at 10 nM.
In silico evaluation of compound 36 metabolism revealed that the “right-hand” aminochromane is the main metabolic soft spot, specifically the carbon atom at the position α to chromane oxygen. With this information in hand, attempts to further improve the rodent PK were initiated (Table 5). Although lactone compound 37 is inactive, thiochromane compound 38, benzoxepine compound 39, and dihydrobenzofuran compound 40 are all potent dual PMIX and PMX inhibitors. The introduction of a gem-dimethyl functionality at the position α to the “right-hand” amine chromane oxygen resulted in WM382, a compound that displayed a potency enhancement versus compound 36P. falciparumEC50, FRET PMX IC50, and FRET PMIX IC50 = 0.4, 0.03, and 1.4 nM, respectively, for WM382 versus 2.5, 0.07, and 2.2 nM, respectively, for compound 36; Table 6) that is likely due to an increase in lipophilicity and permeability. In addition, a mouse MRT of 2.5 h and a significantly improved oral bioavailability of 38% indicated that WM382 is an appropriate compound for in vivo evaluation of this novel mechanism of action.5
WM382 was evaluated in theP. berghei and P. falciparum mouse models of blood stage infection.11 In both models, WM382 administration resulted in no detectable parasitemia after 4 days of 3 mpk BID dosing and cured mice at 10 and 30 mpk BID dosing with a rate of parasite clearance equivalent to that of chloroquine.5WM382 displays a remarkable parasiticidal efficacy after 4 days of dosing, with a free drug concentration at trough that is less than its in vitro EC50. Additionally, WM382 has shown in vitro and in vivo efficacy at the mosquito and liver stages of the parasite life cycle.5 The high level of efficacy of WM382 in a broad spectrum of in vitro and in vivo studies is likely due to its potent inhibition of both PMIX and PMX enzymes across the life cycle. The first indication of the dual activity of WM382 was suggested during cross-resistance studies, in which a low level of cross-resistance was observed with 3D7-WM5.1- and 3D7-WM4.1-resistant parasite lines. 3D7-WM5.1- and 3D7-WM4.1-resistant parasites were selected against the more PMX-selective inhibitors WM5 and WM4, respectively.5 Computational evaluation of amino acid residues at the active sites of the 10 different P. falciparum plasmepsins (Figure 6)12−16 indicated the possibility that in addition to PMX, PMIX could be a second target of WM382. Although PMIX and PMX have very distinct electrostatic surfaces9 and are expressed at different subcellular locations in the malaria parasite,17 their active sites are very similar (Figure 6). The high potency of WM382 against PMIX was confirmed after the development of a PMIX protease biochemical FRET assay. Inhibition of both PMIX and PMX blocks processing of several parasite proteins that are essential for parasitic functions including erythrocyte invasion, egress, blood-stage parasite development, parasite transmission to mosquitoes, and exoerythrocytic merozoite infectivity.5
Figure 6.

Amino acid residues within the active sites of the 10 different plasmepsins. Residue numbers shown are in reference to PMX. Those highlighted in yellow are residues that are different in PMIX and PMX. Protein sequences were aligned with Clustal Omega,12 and active-site pockets are labeled using the nomenclature of Schecter and Berger,13 similar to a prior plasmepsin modeling study.14 See Genbank and PlasmoDB15 for P. falciparum 3D7 strain protein database accessions.16
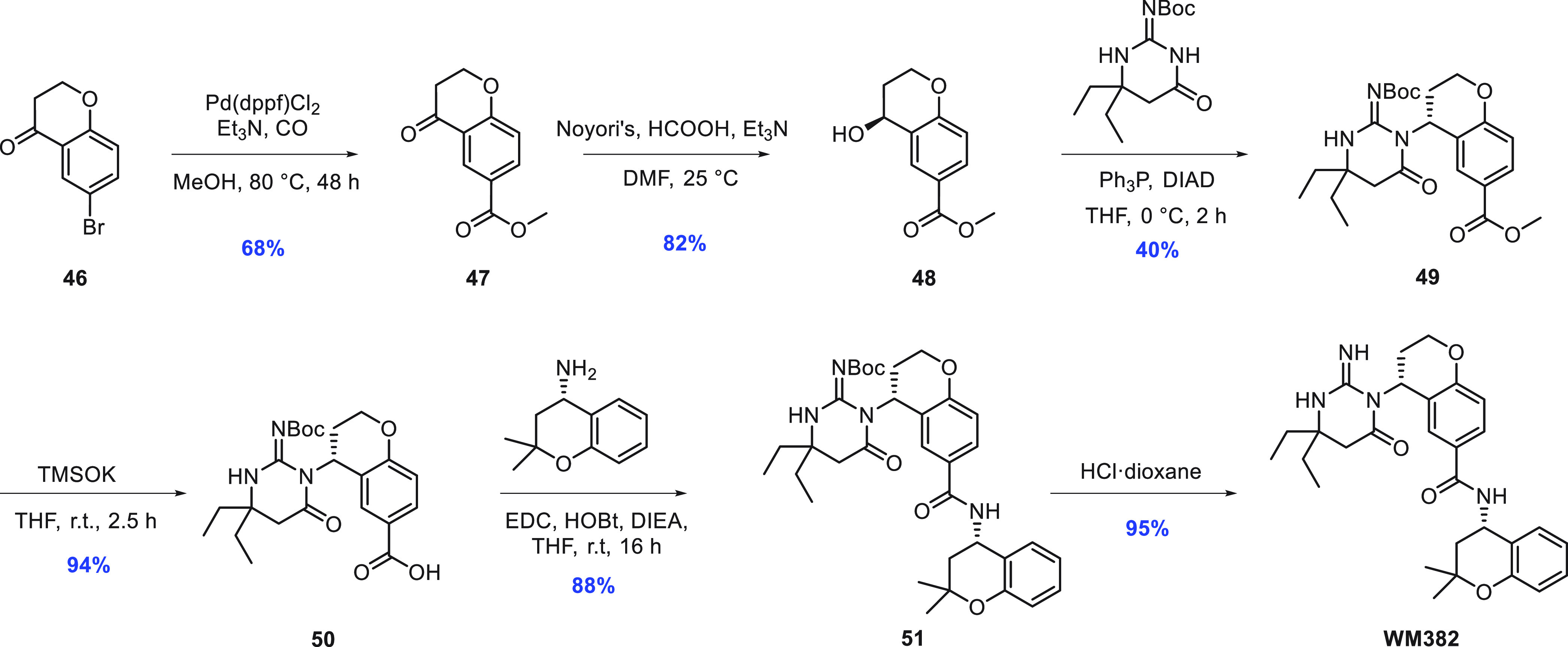
The synthesis of WM382 (Scheme 1) is a straightforward convergent synthesis using three different fragments with an overall yield of 17%. This synthesis allows the possibility of low cost of goods, which is of paramount importance for the development of any antimalarial agent. After the formation of keto ester 47 from keto bromide 46 by carbonylation in the presence of methanol, chiral alcohol 48 was obtained by asymmetric hydrogenation of the ketone using Noyori’s reduction protocol.18 The aspartic warhead was introduced through a Mitsunobu reaction, and the “right-hand” amine was finally added after ester hydrolysis followed by amide coupling and removal of the iminopyrimidone Boc protecting group.
Scheme 1. Synthesis of WM382.
As shown by us previously,5WM4 is a more selective PMX inhibitor, whereas WM382 is a potent dual PMX/IX inhibitor. To date, the selection of resistance to WM382 has not been possible, presumably because of its potent dual inhibitory profile. In contrast, in vitro resistance to PMX-selective WM4 inP. falciparum was selected by slow, incremental increases in drug concentration (up to 8 × EC50) over a 6 month period.5 Genome sequencing of the resistant parasites did not identify any target enzyme point mutations. However, copy number analysis revealed amplification events on chromosome 8, a region including the gene encoding the aspartic protease PMX (PlasmoDB: PF3D7_0808200). The breakpoints of each gene amplification were established, and the copy number of PMX ranged from 6 to 10, with protein expression also increased in the resistant lines. These data suggested that PMX is the direct target and/or is involved in the mechanism of resistance for WM4. Similar experiments have not successfully generated resistance to WM382, most likely because this would require amplification of (or mutation in) both the pmix and pmx genes, which are on different chromosomes. Attempts to select for resistance with WM382 have not yielded resistant parasites, consistent with WM382 exerting its action through more than one target. “Risk to resistance” selections were performed for WM4 and WM382 and compared with atovaquone as a control.13 Parasites resistant to atovaquone were readily selected as expected, but no parasite recrudescence was observed for up to 90 days for inocula ranging from 106 to 109 when WM4 or WM382 was used. Therefore, a minimum inoculum for resistance (MIR)19,20 value could not be calculated. It is likely that the very high barrier to the selection of resistance (i.e., MIR > 9; Table 7) for WM382 results from the dual targeting of the two essential plasmepsin enzymes PMX and PMIX. This stands in stark contrast to selective (single-protein-targeting) inhibitors reported in the literature and highlights the value of our approach, which included phenotypic screening followed by target identification and structure-based drug design.
Table 7. Resistance Profiles for WM4 and WM382.
| compound | molecular target | resistance gene | EC90 fold pressure | MIR |
|---|---|---|---|---|
| WM4 | PMX | pmx (Pf3D7_0808200) | 80 nM | >9 |
| WM382 | PMX and PMIX | N/A | 1.5 nM | >9 |
In conclusion, WM382 was invented utilizing a structure-guided medicinal chemistry effort starting from phenotypic screening of a targeted aspartic protease library that yielded hits WM4 and WM5. Optimization of the potency and rodent pharmacokinetics resulted in the tool compound WM382, a very potent PMX/IX dual inhibitor with excellent antimalarial potency in vitro and in vivo against blood, mosquito, and liver stages. This series of compounds shows great promise as next-generation antimalarial agents.
Acknowledgments
This work was made possible through support of The Wellcome Trust [109662/Z/15/Z, 2027/Z/16/Z]. We thank Tanweer Khan, Andrew Stamford, Brian McKittrick, and Jared Cumming for their ideas and support of the initial screening campaign. We thank Michael J. Kelly III, Philippe G. Nantermet, and Jacqueline B. Fine for project and manuscript preparation support. We also thank Benjamin D. Sherry for the scale-up of WM382.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.2c00355.
Experimental procedures and NMR spectra of key compounds (PDF)
Author Contributions
⊥ M.d.l.R. and P.F. contributed equally
The authors declare no competing financial interest.
Author Status
The following authors are employees of Merck & Co.: M.d.L.R., Z.G., L.Z., N.M., C.W.B., M.V., J.R., D.B.O., and J.A.M.
Supplementary Material
References
- World Malaria Report 2021; World Health Organization, 2021.
- Imwong M.; Suwannasin K.; Kunasol C.; Sutawong K.; Mayxay M.; Rekol H.; Smithuis F. M; Hlaing T. M.; Tun K. M; van der Pluijm R. W; Tripura R.; Miotto O.; Menard D.; Dhorda M.; Day N. P J; White N. J; Dondorp A. M The spread of artemisin-resistant Plasmodium falciparum in the Greater Mekong subregion: a molecular epidemiology observational study. Lancet Infect. Dis. 2017, 17 (5), 491–497. 10.1016/S1473-3099(17)30048-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehn B. M. Drug-resistant malaria detected in Africa will require monitoring. JAMA 2021, 325 (23), 2335. 10.1001/jama.2021.9133. [DOI] [PubMed] [Google Scholar]
- Nasamu A. S.; Polino A. J.; Istvan E. S.; Goldberg D. E. Malaria parasite plasmepsins: More than just plain old degradative pepsins. J. Biol. Chem. 2020, 295 (25), 8425–8441. 10.1074/jbc.REV120.009309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favuzza P.; de Lera Ruiz M.; Thompson J. K.; Triglia T.; Ngo A.; Steel R. W. J.; Vavrek M.; Christensen J.; Healer J.; Boyce C.; Guo Z.; Hu M.; Khan T.; Murgolo N.; Zhao L.; Penington J.; Reaksudsan K.; Jarman K.; Dietrich M. H.; Richardson L.; Guo K.-Y.; Lopaticki S.; Tham W.-H.; Rottmann M.; Papenfuss T.; Robbins J. A.; Boddey J. A.; Sleebs B. E.; Sabroux H. J.; McCauley J. A.; Olsen D. B.; Cowman A. F. Dual plasmepsin-targeting antimalarial agents disrupt multiple stages of the malaria parasite life cycle. Cell Host Microbe 2020, 27 (4), 642–658. 10.1016/j.chom.2020.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamford A. W.; Scott J. D.; Li S. W.; Babu S.; Tadesse D.; Hunter R.; Wu Y.; Misiaszek J.; Cumming J. N.; Gilbert E. J.; Huang C.; McKittrick B. A.; Hong L.; Guo T.; Zhu Z.; Strickland C.; Orth P.; Voigt J. H.; Kennedy M. E.; Chen X.; Kuvelkar R.; Hodgson R.; Hyde L. A.; Cox K.; Favreau L.; Parker E. M.; Greenlee W. J. Discovery of an Orally Available, Brain Penetrant BACE1 Inhibitor That Affords Robust CNS Aβ Reduction. ACS Med. Chem. Lett. 2012, 3 (11), 897–902. 10.1021/ml3001165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cumming J. N.; Smith E. M.; Wang L.; Misiaszek J.; Durkin J.; Pan J.; Iserloh U.; Wu Y.; Zhu Z.; Strickland C.; Voigt J.; Chen X.; Kennedy M. E.; Kuvelkar R.; Hyde L. A.; Cox K.; Favreau L.; Czarniecki M. F.; Greenlee W. J.; McKittrick B. A.; Parker E. M.; Stamford A. W. Structure based design of iminohydantoin BACE1 inhibitors: identification of an orally available, centrally active BACE1 inhibitor. Bioorg. Med. Chem. Lett. 2012, 22 (7), 2444–2449. 10.1016/j.bmcl.2012.02.013. [DOI] [PubMed] [Google Scholar]
- Scott J. D.; Li S. W.; Brunskill A. P. J.; Chen X.; Cox K.; Cumming J. N.; Forman M.; Gilbert E. J.; Hodgson R. A.; Hyde L. A.; Jiang Q.; Iserloh U.; Kazakevich I.; Kuvelkar R.; Mei H.; Meredith J.; Misiaszek J.; Orth P.; Rossiter L. M.; Slater M.; Stone J.; Strickland C. O.; Voigt J. H.; Wang G.; Wang H.; Wu Y.; Greenlee W. J.; Parker E. M.; Kennedy M. E.; Stamford A. W. Discovery of the 3-Imino-1,2,4-thiadiazinane 1,1-Dioxide Derivative Verubecestat (MK-8931)—A β-site amyloid precursor protein cleaving enzyme 1 inhibitor for the treatment of Alzheimer’s disease. J. Med. Chem. 2016, 59 (23), 10435–10450. 10.1021/acs.jmedchem.6b00307. [DOI] [PubMed] [Google Scholar]
- Hodder A.; Christensen J.; Scally S.; Triglia T.; Ngo A.; Birkinshaw R.; Bailey B.; Favuzza P.; Dietrich M.; Tham W.-H.; Czabotar P.; Lowes K.; Guo Z.; Murgolo N.; de Lera Ruiz M.; McCauley J.; Sleebs B.; Olsen D.; Cowman A. Basis for drug selectivity of plasmepsin IX and X inhibition for Plasmodium falciparum and vivax. Structure 2022, 30 (7), 947–961. 10.1016/j.str.2022.03.018. [DOI] [PubMed] [Google Scholar]
- Quant-qual hepatocyte MetID evaluation pointed to the bicyclic “right-hand” amine as a metabolic soft spot (to M+16).
- Angulo-Barturen I.; Jiménez-Diaz M. B.; Mulet T.; Rullas J.; Herreros E.; Ferrer S.; Jiménez E.; Mendoza A.; Regadera J.; Rosenthal P. J.; Bathurst I.; Pompliano D. L.; Gómez de las Heras F.; Gargallo-Viola D. A murine model of falciparum-malaria by in vivo selection of competent strains in non-myelodepleted mice engrafted with human erythrocytes. PLoS One 2008, 3, e2252. 10.1371/journal.pone.0002252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F.; Wilm A.; Dineen D.; Gibson T. J.; Karplus K.; Li W.; López R.; McWilliam H.; Remmert M.; Söding J.; Thompson J. D.; Higgins D. G. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7 (1), 539. 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schechter I.; Berger A. On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 1967, 27 (2), 157–162. 10.1016/S0006-291X(67)80055-X. [DOI] [PubMed] [Google Scholar]
- Bobrovs R.; Jaudzems K.; Jirgensons A. Exploiting structural dynamics to design open-flap inhibitors of malarial aspartic proteases. J. Med. Chem. 2019, 62 (20), 8931–8950. 10.1021/acs.jmedchem.9b00184. [DOI] [PubMed] [Google Scholar]
- Aurrecoechea C.; Brestelli J.; Brunk B. P.; Dommer J.; Fischer S.; Gajria B.; Gao X.; Gingle A.; Grant G.; Harb O. S.; Heiges M.; Innamorato F.; Iodice J.; Kissinger J. C.; Kraemer E.; Li W.; Miller J. A.; Nayak V.; Pennington C.; Pinney D. F.; Roos D. S.; Ross C.; Stoeckert C. J. Jr.; Treatman C.; Wang H. PlasmoDB: a functional genomic database for malaria parasites. Nucleic Acids Res. 2009, 37 (Database issue), D539–D543. 10.1093/nar/gkn814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PMI, XP_001348249.1 PF3D7_1407900; PMII, XP_001348250.1 PF3D7_1408000; PMIII, XP_001348251.1 PF3D7_1408100; PMIV, XP_001348248.1 PF3D7_1407800; PMV, XP_001349975.1 PF3D7_1323500; PMVI, XP_001351190.1 PF3D7_0311700; PMVII, XP_001347613.1 PF3D7_1033800; PMVIII, XP_001348799.2 PF3D7_1465700; PMIX, XP_001348455.1 PF3D7_1430200; PMX, XP_001349441.1 PF3D7_0808200.
- Nasamu A. S.; Glushakova S.; Russo I.; Vaupel B.; Oksman A.; Kim A. S.; Fremont D. H.; Tolia N.; Beck J. R.; Meyers M. J.; Niles J. C.; Zimmerberg J.; Goldberg D. E. Plasmepsins IX and X are essential and druggable mediators of malaria parasite egress and invasion. Science 2017, 358 (6362), 518–522. 10.1126/science.aan1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkuma T. Asymmetric hydrogenation of ketones: Tactics to achieve high reactivity, enantioselectivity, and wide scope. Proc. Jpn. Acad., Ser. B: Phys. Biol. Sci. 2010, 86 (3), 202–219. 10.2183/pjab.86.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding X. C.; Ubben D.; Wells T. N. C. A framework for assesing the risk of resistance for anti-malarials in development. Malar. J. 2012, 11 (1), 292. 10.1186/1475-2875-11-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffey M.; Blasco B.; Burrows J. N.; Wells T. N. C.; Fidock D. A.; Leroy D. Assessing risks of Plasmodium falciparum resistance to select next-generation antimalarials. Trends Parasitol. 2021, 37 (8), 709–721. 10.1016/j.pt.2021.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.