

Abstract
Peripherally active tetrapeptides as selective κ opioid receptor (KOR) agonists have been prepared in good overall yields and high purity following solid-phase peptide synthesis via Fmoc protection strategy. Structural modifications at the first and second position of the lead compound FF(d-Nle)R-NH2 (FE200041) were contemplated with aromatic side chains containing d-amino acids, such as (d)-pF-Phe, (d)-mF-Phe, (d)-oF-Phe, which led to highly selective and efficacious KOR agonists endowed with strong antinociceptive activity in vivo following intravenous (i.v.) and subcutaneous (s.c.) administration in the tail flick and formalin tests. These results suggest potential clinical applications in the treatment of neuropathic and inflammatory pain.
Keywords: κ opioid receptors, Selectivity, Nociception, Peptides, Tail flick, G protein stimulation
Opioid analgesics currently employed in therapy act primarily on μ opioid receptors (MOR). However, they can induce undesirable side effects, including euphoria/dysphoria, addiction, constipation, and urinary retention.1 Unlike μ and δ receptor agonists (MOR/DOR agonists),2,3 κ opioid receptor agonists (KOR agonists) do not cause constipation and urinary retention, thus, they can be considered in the treatment of postoperative, inflammatory/visceral pain, burn/neuropathic pain, and rheumatoid arthritis.4 In the case of dental surgery, pentazocine and butorphanol are KOR agonists able to induce a consistent analgesia in women (with less potency in men), which suggests possible benefits in some types of patients.5 Unfortunately KOR agonists have been found to cause severe central side effects, generically described as “dysphoric behaviors,” that have limited further clinical development.5,6 In order to reduce central side effects in favor of an improvement in peripheral beneficial activity, a common strategy is to prevent their penetration into the central nervous system (CNS).7 Actually, some KOR agonists, as well as peripheral KOR agonists such as ICI 204448, GR 94839, and EMD61753 (asimadoline), produced analgesia and decreased inflammation in rheumatoid arthritis rat models following local administration (e.g., terfenadine, astemizole, and mequitazine) (Figure 1).8,9 However, when asimadoline was tested on patients undergoing knee surgery, they reported a strong increase in pain sensation.10 Compounds of this type have been discontinued in clinical trials because of low bioavailability, poor efficacy, and the emergence of central side effects at therapeutic doses. So far, peptide-based KOR agonists including E-2078 and SK-9709 have also been developed as analgesics; both of them are centrally active dynorphin peptide fragments (Figure 1).11 Recently, Dooley described the development of a KOR agonist with high affinity (Ki = 1.2 nM) and selectivity (μ/κ and δ/κ ratios >3000) by scanning a combinatorial library of tetrapeptides.12
Figure 1.

Structures of the most representative KOR agonists.
This tetrapeptide called FE200041 [sequence: FF(d-Nle)R-NH2] consists entirely of d-amino acids. It is a highly selective KOR agonist able to inhibit cAMP formation stimulated by forskolin (Figure 1).13 FE200041 is an extremely potent antinociceptive agent without side effects on the CNS at doses higher than those necessary to obtain the analgesic effect. It is capable of producing a peripheral analgesic effect in the hindpaw ipsilateral in rats and significantly inhibiting acetic acid writing and formalin-induced flinching,13 which paves the way for the development of peripherally acting FE200041 peptide analogues. A classical structure–activity relationship (SAR) study was difficult to delineate;14 however, the structural modifications applied on a tetrapeptide combinatorial library indicate that d-amino acids are mandatory in the sequence.12 In fact, the inclusion of l-Trp in the third position leads to a mixture of compounds with very poor activity in guinea pig brain homogenates in vitro.12,13 The literature highlights the key role exerted by d-Nle3 in the receptor binding pocket’s recognition; in fact, the replacement of this residue with l-Trp causes a drastic drop in activity.15 Analogues with a high binding affinity for KOR possess d-Arg4, which promotes the anchoring of the molecule in the receptor.16d-Arg4 represents a pharmacophoric residue that allows hydrogen bond formation and ionic interactions with Glu209 in the binding pocket, which is fundamental for KOR selectivity.15 Aromatic d-amino acids such as Phe1,2, as well as aliphatic side-chain-containing residues, e.g., Nle2, are well tolerated even if a generalization to similar amino acids is not already proved.17 Thus, in order to clarify the SAR of this peptide with peripheral analgesic activity, 12 novel C-terminal amide tetrapeptide analogues of the lead compound FE200041 were designed and synthesized as selective KOR agonists. The Phe1 and Phe2 residues in the FE200041 sequence have been replaced one by one with diverse aromatic side chains containing d-amino acids, namely, o-(F)-phenylalanine, m-(F)-phenylalanine, p-(F)-phenylalanine, tyrosine, tryptophan, and 1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid, in order to investigate their influence on the binding and selectivity of opioid receptors (Figure 2).18 The aim of this work is to study the influence of diverse hydrophobic/aromatic side-chain-containing amino acids in the first and second position of the lead compound while retaining the key role of d-Arg4 and d-Nle3. These C-terminal amide-containing peptides have been used for in vitro determination of receptor binding affinity toward the three opioid receptors/G-protein-coupled stimulation and in vivo to evaluate their effective peripheral analgesic activity. Full d-amino acids containing peptides may possess improved in vivo efficacy, considering the long-lasting effect of FE200041 after intravenous (i.v.) administration in the formalin-induced paw flinch test.13
Figure 2.

Amino acid sequences of the lead compound and novel tetrapeptides.
The novel tetrapeptides as C-terminal amides were obtained in good overall yields and high purity after RP-HPLC purification [for details see the Supporting Information (SI)];19 LRMS and 1H NMR were applied for structural identification.20 The final products as TFA salts were used for in vitro biological assays (Table 1).21−23
Table 1. Displacement of [3H]HS665 by HS665, FF(d-Nle)R-NH2 (FE200041), and 1–12 Analogues in Brain Membranes of Rat and Guinea Piga.
| ligand | ligand
affinity |
efficacy | potency | ||
|---|---|---|---|---|---|
| Ki + SEM (nM) | Emax ± SEM (%) | LogEC50 ± SEM | |||
| MORb | DORb | KORc | |||
| HS665 | n.d.d | n.d.d | 1.91 ± 0.91 | n.d.d | n.d.d |
| U-69 | n.d.d | n.d.d | n.d.d | 237.8 ± 14.5 | –5.84 ± 0.19 |
| FE200041 | N.B.e | N.B.e | 2.33 ± 0.52 | 127.7 ± 2.3 | –7.58 ± 0.31 |
| 1 | N.B.e | N.B.e | N.B.e | n.d.d | n.d.d |
| 2 | N.B.e | N.B.e | 1.28 ± 0.21 | 124.0 ± 2.9 | –7.65 ± 0.29 |
| 3 | N.B.e | N.B.e | 4.35 ± 0.62 | 210.9 ± 8.0 | –5.87 ± 0.12 |
| 4 | N.B.e | N.B.e | N.B.e | n.d.d | n.d.d |
| 5 | N.B.e | N.B.e | 3.07 ± 0.87 | 122.0 ± 2.9 | –8.29 ± 0.55 |
| 6 | N.B.e | N.B.e | 1.11 ± 0.04 | 117.4 ± 2.3 | –5.02 ± 0.72 |
| 7 | N.B.e | N.B.e | 0.92 ± 0.09 | 156.7 ± 7.9 | –6.02 ± 0.27 |
| 8 | N.B.e | N.B.e | 3.00 ± 0.52 | 135.6 ± 6.3 | –5.73 ± 0.25 |
| 9 | N.B.e | N.B.e | N.B.e | n.d.d | n.d.d |
| 10 | N.B.e | N.B.e | N.B.e | n.d.d | n.d.d |
| 11 | N.B.e | N.B.e | N.B.e | n.d.d | n.d.d |
| 12 | N.B.e | N.B.e | 3.63 ± 1.19 | 97.6 ± 1.4 | –8.10 ± 0.93 |
The stimulation efficacy (Emax) and potency (LogEC50) of the G protein by U-69, LEAD, and 1–12 ligands in [35S]GTPγS binding assays were evaluated in guinea pig brain membrane homogenates.
Rat brain membrane.
Guinea pig brain membrane.
n.d., not determined.
N.B., no binding.
The novel analogues present similar equilibrium binding affinities (Ki value) as HS665 on KOR (Figure 1, see SI), with the exception of 9–11 and 1 and 4, which are completely inactive. For MOR and DOR, these peptides did not show specific binding (Table 1). In general, we can observe the following trend in Ki values for KOR: 3 > 12 > 5 > 8 > FF(d-Nle)R-NH2 > 2 > 6 > 7. It is worth noting that peptides 7, 2, and 6 exhibit a binding affinity value lower than those of the reference compound HS665 and FF(d-Nle)R-NH2 (Ki = 0.92, 1.28, and 1.11 nM, respectively, for 7, 2, and 6 compared with 1.91 and 2.33 nM for HS665 and the lead compound). Compounds endowed with a high affinity for KOR were tested in functional [35S]GTPγS binding assays in homogenates of guinea pig brain membranes (Figures S3 and S4, see SI).24−26 The analogues were measured at 10–10–10–5 M concentration, and the KOR-selective agonist U-69 was used as a reference compound. The lead compound, 2, 5, 6, and 8 produced a weak dose-dependent increase in comparison with U-69. Peptide 7 (Emax = 156.7.3%) showed higher efficacy than the lead compound FF(d-Nle)R-NH2 (Emax = 127.7%). Peptide 3 (Emax= 210.9%) almost reached the level of U-69-induced G protein activation (Emax = 237.8%), while compound 12 did not stimulate G protein (Table 1). The large orthosteric site of KOR is optimal for endogenous peptide binding.27 The presence of pF-Phe2 in 7 is responsible for a strong increase in binding affinity and efficacy for KOR with respect to the lead compound, considering that its structural isomer 1 is completely inactive. A significant binding ability is also preserved for tetrapeptides containing Tyr1 and mF-Phe1 (6 and 2, respectively). Martinez-Mayorga et al. reported a conformational search for the lead compound FF(d-Nle)R-NH2 within the KOR binding site.15 The conformers obtained present the canonical salt bridge with Asp138 and the positioning of a Phe side chain at the bottom of the pocket. Other favorable interactions involve H-bond formation with Lys227, Glu297, and Tyr312 and a strong H-bond and ionic interactions with Glu209, which promotes the KOR-subtype selectivity.15 This peptide allows the Trp287 side-chain rotation, which is recognized as a fundamental residue in GPCR activation.28 It is feasible to assume that the inclusion of a hydrophobic substituent, such as a fluorine atom, on an aromatic ring reinforces the hydrophobic interaction of our peptide with key residues in the binding pocket of KOR and is responsible for its selectivity.15,28
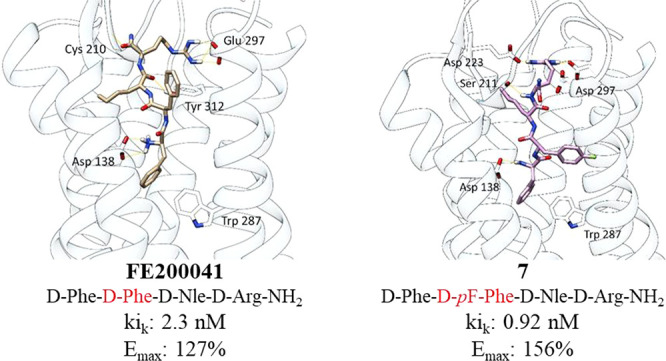
In order to compare the interactions of peptide 7 at the KOR receptors with those of the parent compound FF(d-Nle)R-NH2 and the crystallographic ligand found in the receptor–ligand complex 6B73,29−31 an in silico docking study was performed. Results show that the lead compound FF(d-Nle)R-NH2 docked at KOR is able to establish several polar interactions involving guanidinium groups and Glu297 residues, an H-bond between Cys210 and the C-terminal amide of 7, and a π–π interaction with Trp297 (Table 2).
Table 2. Interactions Found for the Best Docked Poses of FE200041 [FF(d-Nle)R-NH2], 7, and a Crystallographic Ligand on KOR.
| FE200041 | 7 | crystallographic ligand | |
|---|---|---|---|
| Asp 138 | 1 ionic interaction | 1 ionic interaction | 1 ionic interaction |
| Asp 223 | 1 H-bond | ||
| Trp 287 | π–π | π–π | |
| Cys 210 | 1 H-bond | ||
| Tyr 312 | π–π | ||
| Glu 297 | 1 H-bond and 1 ionic interaction | 2 H-bond and 1 ionic interaction | |
| Ser 211 | 1 H-bond |
Some of these interactions are in common with the tetrapeptide 7: it does not bind to Cys210 but shows additional interactions with Asp223 and Ser211. It is noteworthy that the NH3+ group of Phe1 of both compounds is able to establish an ionic interaction with the key residue Asp138, which is considered pivotal for their agonist activity and also fundamental for crystallographic ligand–receptor complex interaction.15,29 The high number of interactions and residues accounted for 7 suggest a good stabilization of the binding pose in the KOR binding pocket, thereby leading to a strong affinity and specificity for it (Figure 3).
Figure 3.

Best binding poses of FE200041 (A) and 7 (C) on KOR. Significant H-bond interactions with key amino acid residues are depicted in yellow dots (docking score values; FE200041, −11.156; 7, −11.223). Panels (B) and (D) represent the main interactions found for ligand–KOR complexes (FE200041 and 7, respectively).
The positioning of the fluorine substituent on the aromatic moiety is not discriminant to guarantee the agonist activity since all the peptides incorporating this structural modification are active on KOR; however, it sensibly influences their binding affinity value and efficacy/potency profile. Conversely, the position of the amino acid incorporating such modifications inside the peptide sequence is determinant for the binding affinity of such tetrapeptide isomers (e.g., 7 vs 9). Despite its high binding affinity value (Ki: 4.35 nM), compound 3 is able to stimulate the G-protein-coupled receptor expressed in guinea pig brain with an efficacy of 210.9%, which almost reaches that of the synthetic reference compound U-69.
Prompted by these findings, in vivo antinociceptive assays have been performed to test the capacity of the most interesting tetrapeptides, 2, 3, 7, and 8, to induce analgesic effects following diverse administration routes. The results obtained in the tail flick test after intracerebroventricular (i.c.v.) administration at the dose of 10 nmol/mouse are reported in Figure 4. Tetrapeptides 2, 3, 7, and 8 are able to induce a strong antinociceptive effect higher than those of the lead compound and the synthetic compound U50,488H. This could be due to a strong metabolic stability or better capacity in intracerebral diffusion.
Figure 4.

Effects induced by peptides 2, 3, 7, and 8 in the tail flick test. Compounds were administered i.c.v. at the dose of 10 nmol/mouse. Data are expressed as the area under the curve and statistically analyzed using one-way ANOVA followed by Tukey’s multiple comparisons test. *p < 0.05, ***p < 0.001, and ****p < 0.0001 vs V (vehicle-treated animals); °°p < 0.01, °°°p < 0.001, and °°°°p < 0.0001 vs U50,488H; #p < 0.05, ##p < 0.01, and ###p < 0.001 vs lead compound; N = 7.
The best tetrapeptides, 7 and 2, were selected to test their in vivo efficacy after intravenous (i.v.) administration in the same assay at the dose of 20 μmol/kg (Figure 5). Both of them were able to produce an intense antinociceptive effect, albeit lower than that of the reference compound U50,488H.
Figure 5.

Effects induced by peptides 7 and 2 in the tail flick test. Compounds were administered i.v. at the dose of 20 μmol/kg. Data are expressed as the area under the curve and statistically analyzed using one-way ANOVA followed by Tukey’s multiple comparisons test. * p < 0.05 and **p < 0.01 vs V (vehicle-treated animals); N = 8.
With the aim to explore the stability of the selected peptides through different administration routes, we performed the formalin test after subcutaneous administration (s.c.). The formalin flinch test allowed analysis of the effect of FE200041 on acute and tonic inflammatory pain in rat. Administration of the lead compound (1 mg/kg i.v.) produced a total inhibition of 2% formalin-induced flinching in phase I and over 80% in phase II.13,32 Our novel tetrapeptides were administered in the mouse paw (100 nmol/mouse) 15 min before formalin (Figure 6).
Figure 6.

Effects induced by tetrapeptides 2, 3, 7, 8, and U50,488H in the formalin test. Compounds were administered s.c. at the dose of 100 mmol/mouse. Data were analyzed using one-way ANOVA followed by Tukey’s multiple comparisons test. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 vs V (vehicle-treated animals); N = 8.
All the tested peptides seemed to retain their antinociceptive activity with an efficacy almost comparable with that of the reference compound in the early and late phase of the formalin test, but among them peptide 2 gave the best result. These data let us suppose a stronger metabolic resistance of tetrapeptide 2 compared with 7 against plasma degradation. In light of these results, we finally determined the plasma and brain concentrations of the lead compound and peptide 7 after bolus i.v. administration at 13.9 mg/kg dose in mouse (for further details see SI). The pharmacokinetic parameters of both compounds were measured in plasma (Table 3). The exposure of FP200041 and peptide 7 was confirmed in plasma, with compound 7 exhibiting an improved plasma half-life and AUC versus the lead compound. Of note, there was no quantifiable drug exposure in brain homogenates for either compound (Table S1). Ranges for the calibration curves were determined on the basis of the peak areas observed for the samples. The calibration curve was fit using a 1/×2 weighting.
Table 3. Pharmacokinetic Analysis of FP200041 and Peptide 7 Following a Single i.v. Bolus Dose (13.9 mg/kg) in Mouse Plasmaa.
| parameter | FP200041 | 7 |
|---|---|---|
| Cmax (nM) | 93.0 | 109 |
| Tmax (min) | 5.00 | 15.0 |
| t1/2 (min) | 12.7 | 20.6 |
| AUCinf (min·nM) | 2181 | 5054 |
Noncompartmental pharmacokinetic analysis was performed using Phoenix 64 Build 8.0.0.3176 (see SI).
The calibration range for both analytes was 10–1000 nM, with a correlation coefficient of >0.99 in both instances. Even if both peptides are not detectable in the brain, it is clear that the novel compound 7 possesses a half-life in plasma that is sensibly higher than that of the lead compound, which confirms a stronger metabolic stability.
Overall, these results substantiate the efficacy of compounds 2 and 7 as potent, systemically active KOR-selective agonists, which suggests that these peptides might be promising candidates for further pharmacological characterization.33−35 The novel compounds are efficacious as antinociceptive agents following peripheral administration on KOR. Among them, peptide 7 shows a favorable pharmacokinetic profile since it is not detectable in mouse brain. It is well stated that peripheral KOR agonists could act as potential broad-spectrum analgesics for chronic pain. Our peptides are highly specific for KOR without agonist/antagonist activity for MOR and DOR, which represents a promising starting point for further development. However, a lot of work is still needed to assess their toxicological profile and oral bioavailability.
Glossary
Abbreviations
- MOR
μ opioid receptor
- KOR
κ opioid receptor
- DOR
δ opioid receptor
- CNS
central nervous system
- BBB
blood-brain barrier
- cAMP
3′,5′-cyclic adenosine monophosphate
- SAR
structure–activity relationship
- RP-HPLC
reverse-phase high-performance liquid chromatography
- LRMS
low-resolution mass spectroscopy
- 1H-NMR
proton nuclear magnetic resonance
- TFA
trifluoroacetic acid
- GPCR
G-protein-coupled receptor
- i.c.v.
intracerebroventricular
- i.v.
intravenous
- s.c.
subcutaneous
- MPE
maximum possible effect
- Cmax
maximum serum concentration
- Tmax
time to maximum plasma concentration
- AUCinf
area under the curve extrapolated to infinity
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.2c00237.
Experimental procedures, analytical data, in vitro binding assays and G protein stimulation, in vivo procedures, pharmacokinetic assessment in mouse, RP-HPLC traces, and LRMS spectra of the novel tetrapeptides (PDF)
Author Contributions
A.S. and A.M. designed and synthesized the novel compounds; they wrote the manuscript with the help of all authors. A.D.V., G.S., and L.M. purified and characterized the novel tetrapeptides. E.S. and S.B. performed the in vitro binding experiments. S.P. and P.M. designed and performed the in vivo experiments. K.C., P.T., K.H., D.B., and J.M.S. performed the pharmacokinetic studies. D.M. revised the draft.
This work was partially funded by MIUR-PRIN 2017 (2017PHRC8X_004) and NIH R01DA052340, provided to J.M.S. and K.H.
The authors declare no competing financial interest.
Supplementary Material
References
- Kiguchi N.; Ko M. C. Potential therapeutic targets for the treatment of opioid abuse and pain. Adv. Pharmacol. 2022, 93, 335–371. 10.1016/bs.apha.2021.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollica A.; Costante R.; Stefanucci A.; Pinnen F.; Lucente G.; Fidanza S.; Pieretti S. Antinociceptive profile of potent opioid peptide AM94, a fluorinated analogue of biphalin with non-hydrazine linker. J. Pept. Sci. 2013, 19, 233–239. 10.1002/psc.2465. [DOI] [PubMed] [Google Scholar]
- Feliciani F.; Pinnen F.; Stefanucci A.; Costante R.; Cacciatore I.; Lucente G.; Mollica A. Structure-activity relationships of biphalin analogs and their biological evaluation on opioid receptors. Mini Rev. Med. Chem. 2013, 13, 11–33. 10.2174/138955713804484776. [DOI] [PubMed] [Google Scholar]
- Beck T. C.; Hapstack M. A.; Beck K. R.; Dix T. A. Therapeutic potential of kappa opioid agonists. Pharmaceuticals 2019, 12, 95. 10.3390/ph12020095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machelska H.; Pfluger M.; Weber W.; Piranvisseh-Volk M.; Daubert J. D.; Dehaven R.; Stein C. Peripheral effects of the κ-opioid agonist EMD 61753 on pain and inflammation in rats and humans. J. Pharmacol Exp Ther. 1999, 290, 354–361. [PubMed] [Google Scholar]
- Spasov A. A.; Grechko O. Y.; Eliseeva N. V.; Litvinov R. A.; Shamshina D. D. Toxic effect of single treatment with kappa-opioid agonist, Ru-1205 compound, on the neurological status of wild type mice. J. Clin. Pharm. 2017, 3, 1014. [Google Scholar]
- Bagal S.; Bungay P. Restricting CNS penetration of drugs to minimise adverse events: role of drug transporters. Drug Discovery Today Technol. 2014, 12, 79–85. 10.1016/j.ddtec.2014.03.008. [DOI] [PubMed] [Google Scholar]
- Hindmarch I.; Easton J. C. A placebo-controlled assessment of mequitazine and astemizole in tests of psychomotor ability. Int. J. Clin. Pharm. Res. 1986, 6, 457–464. [PubMed] [Google Scholar]
- Wang X.; Gou X.; Yu X.; Bai D.; Tan B.; Cao P.; Qian M.; Zheng X.; Wang H.; Tang P.; Zhang C.; Ye F.; Ni J. Antinociceptive and antipruritic effects of HSK21542, a peripherally-restricted kappa opioid receptor agonist, in animal models of pain and itch. Frontiers in Pharmacol. 2021, 12, 773204. 10.3389/fphar.2021.773204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber A.; Bartoszyk G. D.; Bender H. B.; Gottschlich R.; Greiner H. E.; Harting J.; Mauler F.; Minck K.-O.; Murray R. D.; Simon M.; Seyfried C. A. A pharmacological profile of the novel, peripherally-selective kappa-opioid receptor agonist. Br. J. Pharmacol. 1994, 113, 1317–1327. 10.1111/j.1476-5381.1994.tb17142.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall S. M.; Lee Y. S.; Hruby V. J. Dynorphin A analogs for the treatment of chronic neuropathic pain. Future Med. Chem. 2016, 8, 165–177. 10.4155/fmc.15.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dooley C. T.; Ny P.; Bidlack J. M.; Houghten R. A. Selective ligands for the mu, delta, and kappa opioid receptors identified from a single mixture based tetrapeptide positional scanning combinatorial library. J. Biol. Chem. 1998, 273, 18848–18856. 10.1074/jbc.273.30.18848. [DOI] [PubMed] [Google Scholar]
- Vanderah T. W.; Schteingart C. D.; Trojnar J.; Junien J. L.; Lai J.; Riviere P. J. FE200041 (d-Phe-d-Phe-d-Nle-d-Arg-NH2): A peripheral efficacious kappa opioid agonist with unprecedented selectivity. J. Pharmacol. Exp. Ther. 2004, 310, 326–333. 10.1124/jpet.104.065391. [DOI] [PubMed] [Google Scholar]
- Placzek M. S.; Schroeder F. A.; Che T.; Wey H. Y.; Neelamegam R.; Wang C.; Roth B. L.; Hooker J. M. Discrepancies in kappa opioid agonist binding revealed through PET imaging. ACS Chem. Neurosci. 2019, 10, 384–395. 10.1021/acschemneuro.8b00293. [DOI] [PubMed] [Google Scholar]
- Martinez-Mayorga K.; Byler K. G.; Yongye A. B.; Giulianotti M. A.; Dooley C. T.; Houghten R. A. Ligand/kappa-opioid receptor interactions: insights from the X-ray crystal structure. Eur. J. Med. Chem. 2013, 66, 114–121. 10.1016/j.ejmech.2013.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes F. M.; Shaner B. E.; Brower J. O.; Woods R. J.; Dix T. A. Development of a Peptide-derived orally-active kappa-opioid receptor agonist targeting peripheral pain. Open Med. Chem. J. 2013, 7, 16–22. 10.2174/1874104501307010016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Wan H.; Dong P.; Wang B.; Zhang L.; Hu Q.; Zhang T.; Feng J.; He F.; Bai C.; Zhang L.; Tao W. Discovery of SHR0687, a highly potent and peripheral nervous system-restricted KOR agonist. ACS Med. Chem. Lett. 2020, 11, 2151–2155. 10.1021/acsmedchemlett.0c00287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanucci A.; Pinnen F.; Feliciani F.; Cacciatore I.; Lucente G.; Mollica A. Conformationally constrained histidines in the design of peptidomimetics: strategies for the χ-space control. Int. J. Mol. Sci. 2011, 12, 2853–2890. 10.3390/ijms12052853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanucci A.; Iobbi V.; Della Valle A.; Scioli G.; Pieretti S.; Minosi P.; Mirzaie S.; Novellino E.; Mollica A. In Silico identification of tripeptides as lead compounds for the design of KOR ligands. Molecules 2021, 26, 4767. 10.3390/molecules26164767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollica A.; Costante R.; Novellino E.; Stefanucci A.; Pieretti S.; Zador F.; Samavati R.; Borsodi A.; Benyhe S.; Vetter I.; Lewis R. J. Design, synthesis and biological evaluation of two opioid agonist and Cav 2.2 blocker multitarget ligands. Chem. Biol. Drug Des. 2015, 86, 156–162. 10.1111/cbdd.12479. [DOI] [PubMed] [Google Scholar]
- Cheng Y.-C.; Prusoff W. H. Relationship between the Constant (Ki) and the Concentration of Inhibitor Which Causes 50% Inhibition (I50) of an Enzymatic Reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Guerrieri E.; Mallareddy J. R.; Tóth G.; Schmidhammer H.; Spetea M. Synthesis and pharmacological evaluation of [(3)H]HS665, a novel, highly selective radioligand for the kappa opioid receptor. ACS Chem. Neurosci. 2015, 6, 456–63. 10.1021/cn5002792. [DOI] [PubMed] [Google Scholar]
- Oktem H. A.; Moitra J.; Benyhe S.; Toth G.; Lajtha A.; Borsodi A. Opioid receptor labeling with the chloromethyl ketone derivative of [3H]Tyr-d-Ala-Gly-(Me)Phe-Gly-ol (DAMGO) II: Covalent labeling of mu opioid binding site by 3H-Tyr-d-Ala-Gly-(Me)Phe chloromethyl ketone. Life Sci. 1991, 48, 1763–1768. 10.1016/0024-3205(91)90214-V. [DOI] [PubMed] [Google Scholar]
- Sim L. J.; Selley D. E.; Childers S. R. In Vitro autoradiography of receptor-activated G proteins in rat brain by agonist-stimulated guanylyl 5′-[gamma-[35S]thio]-triphosphate binding. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 7242–7246. 10.1073/pnas.92.16.7242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szűcs E.; Büki A.; Kékesi G.; Horváth G.; Benyhe S. Mu-Opioid (MOP) receptor mediated G-protein signaling is impaired in specific brain regions in a rat model of schizophrenia. Neurosci. Lett. 2016, 619, 29–33. 10.1016/j.neulet.2016.02.060. [DOI] [PubMed] [Google Scholar]
- Traynor J. R.; Nahorski S. R. Modulation by mu-opioid agonists of guanosine-5′-O-(3-[35S]thio)triphosphate binding to membranes from human neuroblastoma SH-SY5Y cells. Mol. Pharmacol. 1995, 47, 848–854. [DOI] [PubMed] [Google Scholar]
- Zheng Z.; Huang X. P.; Mangano T. J.; Zou R.; Chen X.; Zaidi S. A.; Roth B. L.; Stevens R. C.; Katritch V. Structure-based discovery of new antagonist and biased agonist chemotypes for the kappa opioid receptor. J. Med. Chem. 2017, 60, 3070–3081. 10.1021/acs.jmedchem.7b00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanucci A.; Dimmito M. P.; Macedonio G.; Ciarlo L.; Pieretti S.; Novellino E.; Lei W.; Barlow D.; Houseknecht K. L.; Streicher J. M.; Mollica A. Potent, efficacious, and stable cyclic opioid peptides with long lasting antinociceptive effect after peripheral administration. J. Med. Chem. 2020, 63, 2673–2687. 10.1021/acs.jmedchem.9b01963. [DOI] [PubMed] [Google Scholar]
- Che T.; Majumdar S.; Zaidi S. A.; Ondachi P.; McCorvy J. D.; Wang S.; Mosier P. D.; Uprety R.; Vardy E.; Krumm B. E.; Han G. W.; Lee M. Y.; Pardon E.; Steyaert J.; Huang X. P.; Strachan R. T.; Tribo A. R.; Pasternak G. W.; Carroll F. I.; Stevens R. C.; Cherezov V.; Katritch V.; Wacker D.; Roth B. L. Structure of the nanobody-stabilized active state of the kappa opioid receptor. Cell 2018, 172, 55–67. 10.1016/j.cell.2017.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrödinger Release 2021-4: Protein Preparation Wizard; Epik, Schrödinger, LLC: New York, NY, 2021.
- Harder E.; Damm W.; Maple J.; Wu C.; Reboul M.; Xiang J. Y.; Wang L.; Lupyan D.; Dahlgren M. K.; Knight J. L.; Kaus J. W.; Cerutti D. S.; Krilov G.; Jorgensen W. L.; Abel R.; Friesner R. A. OPLS3: A force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput. 2016, 12, 281–296. 10.1021/acs.jctc.5b00864. [DOI] [PubMed] [Google Scholar]
- Mollica A.; Carotenuto A.; Novellino E.; Limatola A.; Costante R.; Pinnen F.; Stefanucci A.; Pieretti S.; Borsodi A.; Samavati R.; Zador F.; Benyhe S.; Davis P.; Porreca F.; Hruby V. J. Novel cyclic biphalin analogue with improved antinociceptive properties. ACS Med. Chem. Lett. 2014, 5, 1032–1036. 10.1021/ml500241n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck T. C.; Dix T. A. Targeting peripheral ϰ-opioid receptors for the non-addictive treatment of pain. Future Drug Discovery 2019, 1, 1–9. 10.4155/fdd-2019-0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck T. C.; Reichel C. M.; Helke K. L.; Bhadsavle S. S.; Dix T. A. Non-addictive orally-active kappa opioid agonists for the treatment of peripheral pain in rats. Eur. J. Pharmacol. 2019, 856, 172396. 10.1016/j.ejphar.2019.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minervini V.; Dahal S.; France C. P. Behavioral characterization of κ opioid receptor agonist spiradoline and cannabinoid receptor agonist CP55940 mixtures in rats. J. Pharmacol. Exp. Ther. 2017, 360, 280–287. 10.1124/jpet.116.235630. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.