

Abstract

Schistosomiasis is a neglected tropical disease caused by parasitic flatworms. Current treatment relies on just one partially effective drug, praziquantel (PZQ). Schistosoma mansoni Venus Kinase Receptors 1 and 2 (SmVKR1 and SmVKR2) are important for parasite growth and egg production, and are potential targets for combating schistosomiasis. VKRs consist of an extracellular Venus Flytrap Module (VFTM) linked via a transmembrane helix to a kinase domain. Here, we initiated a drug discovery effort to inhibit the activity of the SmVKR2 kinase domain (SmVKR2KD) by screening the GSK published kinase inhibitor set 2 (PKIS2). We identified several inhibitors, of which four were able to inhibit its enzymatic activity and induced phenotypic changes in ex vivoS. mansoni. Our crystal structure of the SmVKR2KD displays an active-like state that sheds light on the activation process of VKRs. Our data provide a basis for the further exploration of SmVKR2 as a possible drug target.
Keywords: kinase domain, drug discovery, inhibitor, Schistosoma, schistosomiasis, crystal structure, docking, inhibition of autophosphorylation
Hundreds of millions of people worldwide suffer from the parasitic disease known as schistosomiasis, which is caused by a trematode blood fluke of the genus Schistosoma.1,2 The three most medically important species are Schistosoma hematobium, Schistosoma mansoni, and Schistosoma japonicum. S. hematobium is the most common species with a presence in 54 countries, particularly in Africa.3S. mansoni is endemic in sub-Saharan Africa, Brazil, the Caribbean islands, Puerto Rico, Suriname, and Venezuela.3 Finally, S. japonicum is endemic in parts of the People’s Republic of China and the Philippines.3 The eggs of the parasite induce an inflammatory response that then leads to tissue fibrosis and portal vein hypertension or occlusion (intestinal schistosomiasis caused by S. mansoni and S. japonicum) or hydronephrosis and squamous bladder cancer (urinary schistosomiasis caused by S. hematobium).4,1,5 The greatest infection intensities are among children and adolescents, and, if left untreated, this painful and debilitating disease impairs academic performance and undermines social and economic development.6−8 Of note, female genital schistosomiasis has been linked to an increased risk of HIV infections9,10 and is now a major focus of World Health Organization (WHO) awareness campaigns.11
The current strategy to treat and control schistosomiasis focuses on decreasing morbidity through periodic treatment with the drug, praziquantel (PZQ), which is an acylated quinoline-pyrazine derivative.12,13 The WHO estimated that 236.6 million people required treatment for schistosomiasis in 2019 (https://www.who.int/news-room/fact-sheets/detail/schistosomiasis). PZQ acts on a calcium-permeable ion channel that is a member of the transient receptor potential melastatin channel subfamily.14,15 The drug causes rapid paralysis of the adult schistosome and damage to the worm’s surface (tegument).12 Although stable clinical resistance to the drug has yet to be reported, concern remains regarding the reliance on just one drug to treat whole populations of people. Further, the drug has a number of pharmaceutical and pharmacological drawbacks that encourage the search for new drugs.13,16
In recent years, the discovery of Venus Kinase Receptors (VKRs) in S. mansoni has offered new directions for schistosomiasis research drug discovery.17,18 VKR proteins are composed of a unique extracellular domain similar to class C G-protein coupled receptors that adopt a Venus Flytrap Module (VFTM).18,17 The VFTM of VKRs is connected to an intracellular tyrosine kinase domain via a single transmembrane helix. VKR proteins form homo- and heterodimers, a key requirement for VKR activation.19 VKR proteins are important in schistosome growth and egg production.17 Two VKR proteins, SmVKR1 and SmVKR2, have been cloned and characterized in S. mansoni, with l-arginine and calcium ions as the respective putative ligands.20 SmVKR1 activates the c-Jun N-terminal kinase (JNK) signal transduction pathway as determined by yeast two-hybrid screening.17 The JNK pathway is involved in oogenesis and the resumption of meiosis in Caenorhabditis elegans(21) and Drosophila melanogaster.22
VKRs are unique to invertebrates, making them attractive candidates for small molecule inhibition. To date, no VKR structure has been elucidated. The intracellular kinase domain of VKRs shares 41% sequence identity with that of insulin receptors (IRs).23 A known IR inhibitor, tyrphostin AG1024, inhibited both SmVKR1 and SmVKR2 and caused concentration-dependent apoptosis and cessation of egg production in schistosomes.24 The dual action of AG1024 on IR and VKR kinase domains is due to their conserved sequences and, possibly, structures.24
Here, we focused on small molecule discovery for the SmVKR2 kinase domain (SmVKR2KD). Specifically, we screened the GlaxoSmithKline (GSK) published kinase inhibitor set 2 (PKIS2; 645 molecules)25 against SmVKR2KD recombinantly expressed in Sf9 insect cells and identified several low micromolar inhibitors. These were then screened against ex vivo adult S. mansoni for phenotypic changes; one inhibitor was markedly bioactive and a further three less so. The inhibitors inhibit the autophosphorylation activity of SmVKR2KD. We also determined the crystal structure of the SmVKR2KD in complex with ADP, and based on the conformation of conserved kinase motifs, the SmVKR2KD is in an active-like dimer state. The structure was used for in silico docking to predict the binding pose of the bioactive compounds.
Although both SmVKR1 and SmVKR2 are important for schistosome growth and egg production,17 we targeted SmVKR2 in our drug discovery approach as its expression is higher than that of SmVKR1.20 Because the kinase domain of the VKRs is linked to an extracellular domain by a transmembrane helix, several constructs varying in length were generated for expression in Sf9 insect cells. Specifically, 12 constructs with N- and C-terminal truncations were designed based on the sequence alignment of VKR1 and VKR2 from S. hematobium and S. mansoni using the Phyre2 and Pfam servers to try to maintain conserved kinase motifs (Figure S1). Small scale expression and purification experiments identified a SmVKR2KD construct (residues 967–1308) suitable for further studies based on milligram expression levels as judged by Western blot. The protein displayed a monodisperse profile by size exclusion chromatography (Figure S2). After purification, the protein did not appear to be post-translationally modified or autophosphorylated as revealed by mass spectrometry analysis (Figure S2).
We employed surface plasmon resonance (SPR) to screen the PKIS2 library against the SmVKR2KD. The PKIS2 library comprises 645 small molecule inhibitors representing 86 diverse chemotypes.25 Twelve showed the strongest binding to the SmVKR2KD with affinities between 0.57 and 170 μM (Table 1 and Figure S3). The identified inhibitors display a similar backbone consisting of either a 1H-pyrrolo[2,3-b]pyridine linked to benzene or a pyrimidine linked to benzene or pyrazolo[1,5-b]pyridazine (Table 1). GSK1520489 and GSK986310 had the highest affinities of 0.44 μM and 1.96 μM, respectively. Both compounds contain a 2,4-diaminopyrimidine as the putative hinge-binding moiety that likely drives binding.
Table 1. Small Molecule Inhibitors of the SmVKR2KDa.

Compounds are grouped by common compound substructure; red, 2,4-diaminopyrimidine; green, 7-azaindole; blue, 3-aminoindazole. Compounds in black do not have a common substructure.
The 12 compounds identified from the SPR screen were tested against ex vivo adult S. mansoni worms to investigate whether they induce phenotypic alterations in the parasite. Compound effects at 10 μM were assessed at 2, 5, and 24 h, and activity was partially quantified using an observation-based severity scoring system that is designed to holistically assess the many different responses to chemical insult of which the schistosome is capable.26,27 For those compounds eliciting phenotypic changes, WormAssay was also employed as an additional readout to measure average worm motility per well.28,29 Four compounds, GW696155, GSK986310, GSK1520489, and SB-710363, produced a variety of effects in the worm (Figure 1; Table 2; Table S1). GW696155 generated the strongest responses. By 2 h, the worms had decreased motility and lost their ability to adhere to the floor of the well (severity score of 2; Table 2). By 24 h, additional responses included worm degeneration and damage to the surface tegument in the form of blebs or bubbles (severity score of 4). These time-dependent observations were consistent with a decrease in average worm motility over time as measured by WormAssay (Figure 1). SB-710363 and GSK986310 had a milder effect in decreasing worm movement (severity score of 1), again registered by WormAssay. GSK1520489 caused a mild uncoordinated motility in the worm as assessed visually without a long-term decrease in average motility as measured by WormAssay. Although, SPR identified 12 compounds against the SmVKR2KD (Table 1), the absence of phenotypic activity for eight of these compounds (Table S2) could be due to their poor uptake by the parasite. It is also possible that the phenotypic changes noted for the four active compounds are only partially related or unrelated to engagement of the SmVKR2 target.
Figure 1.

Changes in the average motility of adult S. mansoni as a function of time, as measured by WormAssay. Significance was determined by Student’s t-test (two-tailed): *P < 0.05, **p < 0.005, ***P < 0.0005. Data were derived from three to five biological assays with 10 μM compound.
Table 2. Phenotypic Changes of S. mansoni As a Function of Time Expressed As Descriptors and Severity Scoresa.

Descriptor terms: degenerating (deg); uncoordinated (unc); slow (S); on sides, inability of worms to adhere to well floor with either the oral or ventral sucker; teg bleb, damage to the surface tegument. Representative data from three to five biological singleton assays with 10 μM compound.
To understand whether the bioactive compounds can inhibit the SmVKR2KD, we measured their IC50 values in the presence of 10 μM ATP; SB-710363 was not included in the measurements due to its weak ex vivo activity. In the absence of a known substrate for SmVKR2KD, we measured the ability of the compounds to inhibit the autophosphorylation capability of SmVKR2KD. GSK1520489 and GSK986310 inhibited activity with IC50 values of 6.47 μM and 5.69 μM, respectively, whereas GW696155 was less potent with a value of 20.15 μM, which could be attributed to its low aqueous solubility (a theoretical logP of 2.76; Figure 2). All three compounds inhibit the autophosphorylation activity of SmVKR2KD and likely display competitive inhibition as the PKIS2 compounds bind to the ATP site of kinases.25
Figure 2.

Compounds that are active against the parasite inhibit the autophosphorylation activity of SmVKR2KD. Concentration–response curves were developed for (A) GSK1520489, (B) GSK986310, and (C) GW696155. The inhibition measured for GW696155 is less than that of the other compounds, possibly due to its low solubility in the assay buffers. Assays were performed in triplicate, and error bars portray the standard deviations around the mean.
To support our drug discovery approach, we determined for the first time a crystal structure of the SmVKR2KD at 3.0 Å resolution in the presence of ADP. SmVKR2 adopts a canonical bilobal kinase fold with an ADP molecule bound in the cleft between the two lobes (Figure 3). The N- and C-terminal lobes are formed mainly by β-sheets and α-helices, respectively, and both lobes are connected by a hinge. A novel feature of the SmVKR2KD is the presence of an extended helix at the N-terminal lobe, which we termed α0. This helix probably extends toward the membrane as part of the transmembrane helix that links the kinase domain with the VFTM module. The SmVKR2KD was crystallized as a dimer with the interface being stabilized by interactions between the N-lobe from one protomer and the C-lobe of the opposite protomer, related by a 2-fold symmetry. Although the crystals were grown in the presence of ATP-γ-S, the electron density maps only corresponded to the ADP moiety and not the thiophosphate group. Despite ATP-γ-S being a nonhydrolyzable ATP analogue, it can be slowly hydrolyzed at a rate 0.5% of that of ATP.30 As the crystals took over one month to grow, we believe that the ATP-γ-S was slowly hydrolyzed during crystallization and that we captured the posthydrolysis state of the kinase. In the electron density maps, we also observe weak density near the ADP and the catalytic D1118 that could correspond to the cleaved thiophosphate group (Figure S4): the distance between the ADP and this density is too far to correspond to a magnesium cation.
Figure 3.
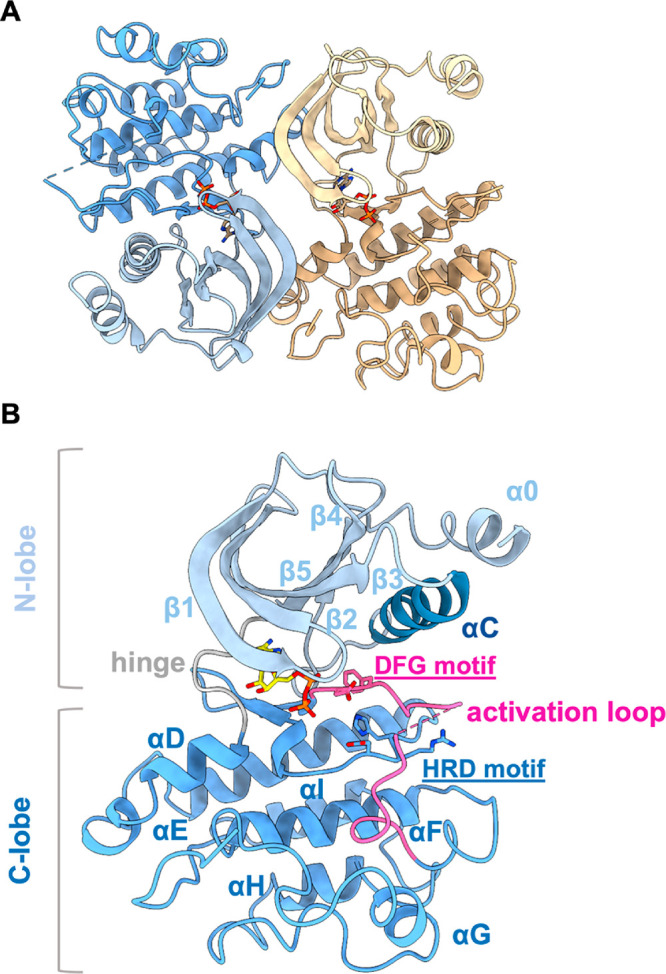
Crystal structure of the SmVKR2KD. (A) The SmVKR2KD adopts a canonical kinase domain fold. In the presence of ADP, from ATP-γ-S hydrolysis, the structure has adopted a dimeric architecture stabilized by interactions between the N- and C-terminal lobes of the opposite monomers. Each monomer is shown as a cartoon with the N- and C-terminal lobes in light and dark colors, respectively. ADP is shown as sticks. (B) The SmVKR2KD has adopted an active-like conformation based on the orientation of key motifs, including the αC-in and DFG-in conformations. Key motifs have been labeled. Same color scheme as panel A.
Based on the conformation and orientation of the hydrophobic spine, the αC-in DFG-in conformation,31 and the E1014 and D1118 pointing toward the ATP-binding site, the SmVKR2KD has adopted an active-like conformation. The hydrophobic spine comprises the catalytic and regulatory spines. The complete catalytic spine or C-spine forms upon ATP binding (in our ADP-bound structure, the adenine ring of ADP brings the two lobes together) and consists of hydrophobic residues from both lobes, including A995 from the VAVK motif, and L1107 from the β7-strand, which sandwiches the ADP adenine ring. The regulatory spine or R-spine consists of L1029 from the αC β4 loop, M1018 from the αC helix, F1119 from the DFG motif, and H1098 from the HRD motif. Another feature of active kinases is the phosphorylation and conformation of the activation loop that is tightly associated with the C-lobe. In the SmVKR2KD, the loop is confined within the C-lobe reminiscent of active kinases, whereas in inactive kinases, it extends toward the N-lobe.31 Although the entire activation loop could be traced, we decided to partially model it as the density between residues 1124 and 1141 is too weak/disordered to confidently add side chains. The interaction of K997 and E1014, although weak at a distance of 3.8 Å, resembles the salt-bridge seen in active kinases that anchor the αC helix to the β3 strand.31 Overall, based on apparent similarities between the key structural features of the SmVKR2KD and active kinases, we propose that the SmVKR2KD structure represents an active-like state. Further, because the structure is in the presence of ADP, it most likely resembles the posthydrolysis state, and even though αC appears to be in the in-conformation, it shows a small degree of displacement toward the out-conformation relative to fully active kinases.31,32
Our efforts to capture the SmVKR2KD in complex with the identified inhibitors yielded weakly diffracting crystals that were not suitable for further analysis. In an attempt to identify the binding pose of the inhibitors, we performed in silico docking using AutoDock Vina.33 As the PKIS2 compounds are ATP-competitive kinase inhibitors,25 we focused the search area within the ATP binding site. Although the resolution is limited to 3.0 Å and the density for water molecules or the magnesium cation was not observed, docking can still provide insights into the possible binding pose and interaction of the inhibitors within the ATP binding site. To evaluate our setup, we docked ATP and ADP and compared the latter to our crystal structure: the pose of the docked ADP displayed minor deviations from the crystal structure but within the acceptable limitations of the low-resolution structure (Figure S5; Table 3). The four compounds GSK1520489, GSK986310, GW696155, and SB-710363 that showed antischistosomal activity, of which the first three were also tested for and found to inhibit the enzymatic activity of the SmVKR2KD were selected for docking, and their in silico affinities and binding poses measured (Table 3 and Figure 4, respectively). GSK1520489 is coordinated by interactions between its sulfone and amide groups with the side and main chains of D1051, respectively, and the amine (between the pyrimidine and sulfone group) within the main chain of R1104. The N-methylbenzamide of GSK1520489 is positioned within the hydrophobic spine of the ATP binding site and mimics the binding of the adenine ring of ADP. Binding of GSK986310 is mediated between the compound’s cyclohexylamine and the backbone carboxylate of R1104, and its amide and the side chain of either D1051 or S1054. The inhibitor’s meta-tolyl substituent is placed within the hydrophobic spine. GW696155, which displayed the greatest ex vivo activity, displays binding pose interactions between the pyrazolo[1,5-b]pyridazine ring and the side chain of K997 and the pyrimidine ring with the side chain of D1051. The piperazine ring is pointing toward the hydrophobic spine. Binding by SB-710363 is stabilized by interactions between its phenol ring and the main chain of M1047, located at the hinge, and by its cyclopropane carboxamide and the side chain of D1118 in the DFG motif.
Table 3. Binding Affinities for the Docked Nucleotides and Inhibitors As Calculated by Autodock Vina.
| compound name | affinity (kcal/mol) |
|---|---|
| ATP | –6.6 |
| ATPγS | –7.6 |
| ADP | –5.9 |
| GSK1520489 | –7.9 |
| GSK986310 | –7.7 |
| GW696155 | –8.0 |
| SB-710363 | –7.6 |
Figure 4.

In silico docking poses of the four antischistosomal inhibitors in the SmVKR2KD active site. The docked inhibitors display polar and hydrophobic interactions within the ATP binding site. The SmVKR2 kinase domain is shown as a gray cartoon and the interacting side chains and inhibitors as sticks. The carbon atoms of SmVKR2 and the inhibitor are colored in gray and light blue, respectively, and nitrogen, oxygen, and sulfur are colored dark blue, red, and yellow, respectively. Polar contacts are shown as black dashed lines.
Because treatment and control of schistosomiasis rely on just one partially effective drug, there is a need to identify alternative therapies. As a response to this insecure situation, we examined inhibitors of VKR as starting points for a new chemotherapy and, for the first time, identify inhibitors of the SmVKR2 receptor by targeting its kinase domain. Using the well characterized and freely available PKIS2 small molecule library from GSK,25 we identified a set of 12 compounds that displayed low micromolar SPR binding in vitro (Table 1), four of which, GSK1520489, GSK986310, GW696155, and SB-710363, were also active against ex vivoS. mansoni (Table 2). GSK1520489, GSK986310, and GW696155 inhibited the autophosphorylation activity of SmVKR2KD, and for the first two compounds, the IC50 values generated were similar to those derived from the SPR experiments.
Although the four compounds do not display significant chemical similarity based on the Tanimoto coefficient (despite the presence of either a pyrimidine or a pyrazolo[1,5-b]pyridazine group; Table S3), they can, nonetheless, efficiently bind and compete with ATP in the binding site. The compound with the strongest ex vivo antischistosomal activity was GW696155, which structurally resembles ATP, even though the Tanimoto coefficient is only 0.1. From our in silico docking studies, the pyrazolo[1,5-b]pyridazine and pyrimidine moieties of GW696155 are placed in a similar pose to the adenine and ribose moieties of ATP, respectively. GW696155 has been identified as an inhibitor of several human and parasite kinases25,34 and provides us with a useful starting point to explore analogues for improved potency and bioactivity against the schistosome parasite. The strong ex vivo activity of GW696155 could be attributed to its high membrane permeability but also to its nonselective nature25,34 by inhibiting other S. mansoni kinases.
Finally, our structural studies provide possible insights into how VKRs are activated. It has been suggested that VKRs need to dimerize upon ligand binding by the extracellular VFTM module that could then lead to dimerization of the kinase domain, autophosphorylation, and activation.19 The SmVKR2KD structure is in an active-like conformation due to the orientation of key motifs that could resemble the ATP bound state after the VFTM module has dimerized.19 This is consistent with the recent structures of the full-length IR receptor.35 In our structure, we have also resolved a new feature for tyrosine kinases, namely, helix a0 (Figure 3). In the full length VKR, this helix would extend toward the membrane to form part of the transmembrane helix that links the kinase domain with the VFTM module.
In conclusion, we have identified initial lead inhibitors against the SmVKR2KD that could pave the way to more potent inhibitors against the VKR2 receptor. Further, our resolved structure of the SmVKR2KD will aid drug discovery efforts using in silico methods.
Acknowledgments
We would like to thank Diamond Light Source synchrotron for beam time allocation and access. We would also like to thank Dr. David Drewry, Structural Genomics Consortium UNC, for kindly providing us with compounds for the enzymatic assays. We thank Dr. Andrew Quigley, Membrane Protein Lab, for access to the CLARIOstar plate reader. The Structural Genomics Consortium is a registered charity (number 1097737) which receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada, Genentech, Innovative Medicines Initiative (EU/EFPIA), Janssen, Merck KGaA Darmstadt Germany, MSD, Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, São Paulo Research Foundation-FAPESP, Takeda, and The Wellcome Trust. This work was funded by a Medical Research Council grant to K.B. (MR/N019113/1). C.R.C. acknowledges R21AI156554, which supported, in part, the maintenance of the S. mansoni life-cycle. Golden Syrian hamsters infected with the NMRI isolate S. mansoni were supplied by the NIAID Schistosomiasis Resource Center of the Biomedical Research Institute (Rockville, MD), NIH-NIAID Contract HHSN272201700014I.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.2c00248.
Materials and Methods, SPR data analysis, electron density maps, table with Tanimoto coefficient analysis of the inhibitors, crystallographic data collection, and refinement statistics table (PDF)
Accession Codes
The coordinates and structure factors of the SmVKR2KD have been deposited into the Protein Data Bank with the PDB ID code 7ZVS.
Author Present Address
⧫ Roche Pharma Research and Early Development, Roche Innovation Center Basel, F. Hoffmann—La Roche AG, 4070 Basel, Switzerland
Author Present Address
¶ Department of Science and Technology—Advanced Science and Technology Institute, UP Technology Park Complex, C.P. Garcia Ave., Diliman, Quezon City, Philippines
Author Present Address
+ Department of Chemistry, Eastern Visayas State University, Tacloban City, Philippines
Author Contributions
■ These authors contributed equally
Author Contributions
K.B. and C.R.C. designed and managed the study. I.M., D.G., and R.O. cloned and expressed the SmVKR2KD . I.M. purified and crystallized the SmVKR2KD. I.M. and K.B. determined the crystal structure and refined the structure. W.Z. provided the PKIS2 screen and analyzed data. L.J.L., N.E.-S., and C.R.C. performed the phenotypic screening and analyzed the data. K.B. performed enzymatic inhibition assays. S.W.R. and I.N. performed SPR measurements and analysis. J.E., R.N., and K.B. performed in silico docking and analysis. K.B. and C.R.C. wrote the paper with help from all the authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Colley D. G.; Bustinduy A. L.; Secor W. E.; King C. H. Human schistosomiasis. Lancet 2014, 383 (9936), 2253–64. 10.1016/S0140-6736(13)61949-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross A. G.; Bartley P. B.; Sleigh A. C.; Olds G. R.; Li Y.; Williams G. M.; McManus D. P. Schistosomiasis. N Engl J. Med. 2002, 346 (16), 1212–20. 10.1056/NEJMra012396. [DOI] [PubMed] [Google Scholar]
- McManus D. P.; Dunne D. W.; Sacko M.; Utzinger J.; Vennervald B. J.; Zhou X. N. Schistosomiasis. Nat. Rev. Dis Primers 2018, 4 (1), 13. 10.1038/s41572-018-0013-8. [DOI] [PubMed] [Google Scholar]
- Gryseels B.; Polman K.; Clerinx J.; Kestens L. Human schistosomiasis. Lancet 2006, 368 (9541), 1106–18. 10.1016/S0140-6736(06)69440-3. [DOI] [PubMed] [Google Scholar]
- Colley D. G.; Secor W. E. Immunology of human schistosomiasis. Parasite Immunol 2014, 36 (8), 347–57. 10.1111/pim.12087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King C. H. Parasites and poverty: the case of schistosomiasis. Acta Trop 2010, 113 (2), 95–104. 10.1016/j.actatropica.2009.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utzinger J.; N’Goran E. K.; Caffrey C. R.; Keiser J. From innovation to application: social-ecological context, diagnostics, drugs and integrated control of schistosomiasis. Acta Trop 2011, 120, S121–S137. 10.1016/j.actatropica.2010.08.020. [DOI] [PubMed] [Google Scholar]
- Kang S.; Damania A.; Majid M. F.; Hotez P. J. Extending the global worm index and its links to human development and child education. PLoS Negl Trop Dis 2018, 12 (6), e0006322. 10.1371/journal.pntd.0006322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjetland E. F.; Hegertun I. E.; Baay M. F.; Onsrud M.; Ndhlovu P. D.; Taylor M. Genital schistosomiasis and its unacknowledged role on HIV transmission in the STD intervention studies. Int. J. STD AIDS 2014, 25 (10), 705–15. 10.1177/0956462414523743. [DOI] [PubMed] [Google Scholar]
- Galappaththi-Arachchige H. N.; Zulu S. G.; Kleppa E.; Lillebo K.; Qvigstad E.; Ndhlovu P.; Vennervald B. J.; Gundersen S. G.; Kjetland E. F.; Taylor M. Reproductive health problems in rural South African young women: risk behaviour and risk factors. Reprod Health 2018, 15 (1), 138. 10.1186/s12978-018-0581-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engels D.; Hotez P. J.; Ducker C.; Gyapong M.; Bustinduy A. L.; Secor W. E.; Harrison W.; Theobald S.; Thomson R.; Gamba V.; Masong M. C.; Lammie P.; Govender K.; Mbabazi P. S.; Malecela M. N. Integration of prevention and control measures for female genital schistosomiasis, HIV and cervical cancer. Bull. World Health Organ 2020, 98 (9), 615–624. 10.2471/BLT.20.252270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews P. Praziquantel: mechanisms of anti-schistosomal activity. Pharmacol Ther 1985, 29 (1), 129–56. 10.1016/0163-7258(85)90020-8. [DOI] [PubMed] [Google Scholar]
- Caffrey C. R. Chemotherapy of schistosomiasis: present and future. Curr. Opin Chem. Biol. 2007, 11 (4), 433–9. 10.1016/j.cbpa.2007.05.031. [DOI] [PubMed] [Google Scholar]
- Gunaratne G. S.; Yahya N. A.; Dosa P. I.; Marchant J. S. Activation of host transient receptor potential (TRP) channels by praziquantel stereoisomers. PLoS Negl Trop Dis 2018, 12 (4), e0006420. 10.1371/journal.pntd.0006420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S. K.; Marchant J. S. The Journey to Discovering a Flatworm Target of Praziquantel: A Long TRP. Trends Parasitol 2020, 36 (2), 182–194. 10.1016/j.pt.2019.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caffrey C. R.; El-Sakkary N.; Mader P.; Krieg R.; Becker K.; Schlitzer M.; Drewry D. H.; Vennerstrom J. L.; Grevelding C. G.. Drug Discovery and Development for Schistosomiasis. In Neglected Tropical Diseases: Drug Discovery and Development; Swinney D.; Pollastri M. P., Eds.; Wiley-VCH: Weinheim, Germany, 2019; Vol. 77, pp 187–225. [Google Scholar]
- Vanderstraete M.; Gouignard N.; Cailliau K.; Morel M.; Hahnel S.; Leutner S.; Beckmann S.; Grevelding C. G.; Dissous C. Venus kinase receptors control reproduction in the platyhelminth parasite Schistosoma mansoni. PLoS Pathog 2014, 10 (5), e1004138. 10.1371/journal.ppat.1004138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicogne J.; Pin J. P.; Lardans V.; Capron M.; Noel C.; Dissous C. An unusual receptor tyrosine kinase of Schistosoma mansoni contains a Venus Flytrap module. Mol. Biochem. Parasitol. 2003, 126 (1), 51–62. 10.1016/S0166-6851(02)00249-9. [DOI] [PubMed] [Google Scholar]
- Ahier A.; Rondard P.; Gouignard N.; Khayath N.; Huang S.; Trolet J.; Donoghue D. J.; Gauthier M.; Pin J. P.; Dissous C. A new family of receptor tyrosine kinases with a venus flytrap binding domain in insects and other invertebrates activated by aminoacids. PLoS One 2009, 4 (5), e5651. 10.1371/journal.pone.0005651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouignard N.; Vanderstraete M.; Cailliau K.; Lescuyer A.; Browaeys E.; Dissous C. Schistosoma mansoni: structural and biochemical characterization of two distinct Venus Kinase Receptors. Exp Parasitol 2012, 132 (1), 32–9. 10.1016/j.exppara.2011.05.007. [DOI] [PubMed] [Google Scholar]
- Smith P.; Leung-Chiu W. M.; Montgomery R.; Orsborn A.; Kuznicki K.; Gressman-Coberly E.; Mutapcic L.; Bennett K. The GLH proteins, Caenorhabditis elegans P granule components, associate with CSN-5 and KGB-1, proteins necessary for fertility, and with ZYX-1, a predicted cytoskeletal protein. Dev. Biol. 2002, 251 (2), 333–47. 10.1006/dbio.2002.0832. [DOI] [PubMed] [Google Scholar]
- Sackton K. L.; Buehner N. A.; Wolfner M. F. Modulation of MAPK activities during egg activation in Drosophila. Fly (Austin) 2007, 1 (4), 222–7. 10.4161/fly.5200. [DOI] [PubMed] [Google Scholar]
- Vanderstraete M.; Gouignard N.; Ahier A.; Morel M.; Vicogne J.; Dissous C. The venus kinase receptor (VKR) family: structure and evolution. BMC Genomics 2013, 14, 361. 10.1186/1471-2164-14-361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderstraete M.; Gouignard N.; Cailliau K.; Morel M.; Lancelot J.; Bodart J. F.; Dissous C. Dual targeting of insulin and venus kinase Receptors of Schistosoma mansoni for novel anti-schistosome therapy. PLoS Negl Trop Dis 2013, 7 (5), e2226. 10.1371/journal.pntd.0002226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drewry D. H.; Wells C. I.; Andrews D. M.; Angell R.; Al-Ali H.; Axtman A. D.; Capuzzi S. J.; Elkins J. M.; Ettmayer P.; Frederiksen M.; Gileadi O.; Gray N.; Hooper A.; Knapp S.; Laufer S.; Luecking U.; Michaelides M.; Muller S.; Muratov E.; Denny R. A.; Saikatendu K. S.; Treiber D. K.; Zuercher W. J.; Willson T. M. Progress towards a public chemogenomic set for protein kinases and a call for contributions. PLoS One 2017, 12 (8), e0181585. 10.1371/journal.pone.0181585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long T.; Rojo-Arreola L.; Shi D.; El-Sakkary N.; Jarnagin K.; Rock F.; Meewan M.; Rascon A. A. Jr.; Lin L.; Cunningham K. A.; Lemieux G. A.; Podust L.; Abagyan R.; Ashrafi K.; McKerrow J. H.; Caffrey C. R. Phenotypic, chemical and functional characterization of cyclic nucleotide phosphodiesterase 4 (PDE4) as a potential anthelmintic drug target. PLoS Negl Trop Dis 2017, 11 (7), e0005680. 10.1371/journal.pntd.0005680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdulla M. H.; Ruelas D. S.; Wolff B.; Snedecor J.; Lim K. C.; Xu F.; Renslo A. R.; Williams J.; McKerrow J. H.; Caffrey C. R. Drug discovery for schistosomiasis: hit and lead compounds identified in a library of known drugs by medium-throughput phenotypic screening. PLoS Negl Trop Dis 2009, 3 (7), e478. 10.1371/journal.pntd.0000478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcellino C.; Gut J.; Lim K. C.; Singh R.; McKerrow J.; Sakanari J. WormAssay: a novel computer application for whole-plate motion-based screening of macroscopic parasites. PLoS Negl Trop Dis 2012, 6 (1), e1494. 10.1371/journal.pntd.0001494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibo-Verdugo B.; Wang S. C.; Almaliti J.; Ta A. P.; Jiang Z.; Wong D. A.; Lietz C. B.; Suzuki B. M.; El-Sakkary N.; Hook V.; Salvesen G. S.; Gerwick W. H.; Caffrey C. R.; O’Donoghue A. J. The Proteasome as a Drug Target in the Metazoan Pathogen, Schistosoma mansoni. ACS Infect Dis 2019, 5 (10), 1802–1812. 10.1021/acsinfecdis.9b00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuoka K.; Kawakita M.; Kaziro Y. Interaction of adenosine-5′-O-(3-thiotriphosphate) with Ca2+,Mg2+-adenosine triphosphatase of sarcoplasmic reticulum. J. Biochem 1982, 91 (5), 1629–37. 10.1093/oxfordjournals.jbchem.a133854. [DOI] [PubMed] [Google Scholar]
- Modi V.; Dunbrack R. L. Jr. Defining a new nomenclature for the structures of active and inactive kinases. Proc. Natl. Acad. Sci. U. S. A. 2019, 116 (14), 6818–6827. 10.1073/pnas.1814279116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathea S.; Salah E.; Tallant C.; Chatterjee D.; Berger B. T.; Konietzny R.; Muller S.; Kessler B. M.; Knapp S. Conformational plasticity of the ULK3 kinase domain. Biochem. J. 2021, 478 (14), 2811–2823. 10.1042/BCJ20210257. [DOI] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31 (2), 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cestari I.; Haas P.; Moretti N. S.; Schenkman S.; Stuart K. Chemogenetic Characterization of Inositol Phosphate Metabolic Pathway Reveals Druggable Enzymes for Targeting Kinetoplastid Parasites. Cell Chem. Biol. 2016, 23 (5), 608–617. 10.1016/j.chembiol.2016.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scapin G.; Dandey V. P.; Zhang Z.; Prosise W.; Hruza A.; Kelly T.; Mayhood T.; Strickland C.; Potter C. S.; Carragher B. Structure of the insulin receptor-insulin complex by single-particle cryo-EM analysis. Nature 2018, 556 (7699), 122–125. 10.1038/nature26153. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.