

Abstract

Tyrosine kinase 2 (TYK2) mediates the interleukin-23 (IL-23), IL-12, and type I interferon (IFN)-driven signal responses that are critical in autoimmune diseases. Here, a series of novel derivatives with an N-(methyl-d3)pyridazine-3-carboxamide skeleton that bind to the TYK2 pseudokinase domain were designed, synthesized, and evaluated. Among them, compound 30 demonstrated more excellent inhibitory potency against STAT3 phosphorylation than the positive control deucravacitinib. In addition to JAK isoform selectivity, compound 30 exhibited good in vivo and in vitro pharmacokinetic properties. Furthermore, compound 30 was orally highly effective in both IL-23-driven acanthosis and anti-CD40-induced colitis models. Together, these findings support compound 30 as a promising candidate for therapeutic applications in autoimmune diseases.
Keywords: TYK2, pseudokinase, inhibitor, autoimmune diseases
Autoimmune diseases are characterized by dysregulated cytokine signaling. Targeting cytokines or their receptors has proved to be effective in multiple autoimmune diseases. Many pathogenic cytokines transmit signals through the JAK-STAT pathway.1−5 As a member of the JAK family, tyrosine kinase 2 (TYK2) regulates downstream signal pathways of the interleukin-23 (IL-23), IL-12, and type I and III interferon (IFN) receptors,6−9 which are critical in the pathobiology of psoriasis, systemic lupus erythematosus (SLE), and inflammatory bowel disease (IBD).10−13 The human IL-12/23 monoclonal antibody ustekinumab has been approved by the U.S. Food and Drug Administration for the treatment of psoriasis,14 ulcerative colitis,15 and Crohn’s disease.16 As a result, TYK2 is a promising target for the development of orally active small molecules for autoimmune diseases.17−19
According to previous studies and available data, there is currently no commercially available inhibitor for clinical use.20−25 Some of the TYK2 inhibitors are summarized in Figure 1. Deucravacitinib is currently in the registration stage for the treatment of psoriasis. Unlike previous JAK inhibitors, deucravacitinib selectively binds to the TYK2 pseudokinase (JH2) domain and inhibits its signaling pathway by an allosteric mechanism.26−28 Based on this mechanism, deucravacitinib achieves higher selectivity for TYK2 than JAK 1–3, avoiding undesirable side effects resulting from the high homology of the adenosine triphosphate (ATP) active site (JH1) within the JAK family.13,29 Besides, BMS-986202 with a six-membered pyrimidine group is also a TYK2 JH2 inhibitor and in a phase II clinical trial.30 While brepocitinib31,32 and PF-0682664733 are also under development, they both bind to the TYK2 JH1 domain.
Figure 1.

Representative structures of TYK2 inhibitors.
Herein, to discover structurally diverse TYK2 inhibitors that bind to the JH2 domain, 28 new compounds have been designed, synthesized, and tested with various biological assays. Most compounds have moderate to good potencies in the TYK2 JH2 binding affinity assay and the inhibition of STAT3 phosphorylation activity assay. Among them, compound 30 demonstrated excellent inhibitory potency against STAT3 phosphorylation. To better understand the selectivity of 30, binding affinity and cellular function assays for other members of JAK family were performed. In addition, compound 30 had reasonable pharmacokinetic (PK) exposure in mice. Finally, 30 was orally effective in both IL-23-driven acanthosis (psoriasis-like) and anti-CD40-induced colitis models.
Based on the crystal structure of TYK2 JH2 in complex with deucravacitinib (PDB ID 6NZP),13,34,35 we found that the N-(methyl-d3)pyridazine-3-carboxamide skeleton of the ligand could form key hydrogen-bonding interactions with Val690 and Glu688 in the hinge region (Figure 2). In this way, the deuteromethyl group of the C3 amide could be anchored toward the adjacent Ala671, which is critical for maintaining the high selectivity. Thus, we preserved the N-(methyl-d3)pyridazine-3-carboxamide skeleton and tried to introduce other substituents. Although the triazole nitrogen atom N2 engages in a direct hydrogen bond with Arg738, we found that there was still an unoccupied pocket below the P loop, which was particularly attractive since it might improve the potency upon the introduction of other groups. In order to facilitate the study of the structure–activity relationship (SAR) of this pocket, we replaced the terminal triazole group of deucravacitinib with an amide. Subsequently, the introduction of different substituents was carried out to investigate the effect of the electronic properties and steric hindrance on the activity. In this case, the target compounds 5–20, 23–30, and 32 were generated. Previous work by BMS showed that pyridylamine could be substituted for the C6 pendent cyclopropylamide with retention of activity using both in silico and traditional techniques.13 In addition, we found that the cyclopropylamide reached out into the solvent based on the crystal structural information. Hence, we designed the target compounds 21, 22, and 31 substituted with pyridylamine groups. Here we describe the synthesis and SAR of compounds with an N-(methyl-d3)pyridazine-3-carboxamide skeleton.
Figure 2.

Design and modification strategies for the target compounds and X-ray crystal structure of TYK2 JH2 complexed with deucravacitinib (green) (PDB ID 6NZP).
As shown in Scheme 1, compounds 5–27 were first prepared starting from commercially available methyl 4,6-dichloropyridazine-3-carboxylate (33), which was hydrolyzed and then amidated to afford intermediate 35 in high yield. The C4 chloride of 35 was selectively displaced by 3-amino-2-methoxybenzoic acid to give key acid 36. Under palladium-catalyzed conditions, 36 reacted with cyclopropanecarboxamide to give intermediate 37, which was condensed with various amines using EDCI to provide the desired amides 5–19 and 23–27. Alternatively, treatment of intermediate 37 with ammonium chloride easily gave amide 40, which was further converted to the expected compound 20. In addition, pyridines 41 were prepared by the coupling reaction of acid 36 with the corresponding amines. The condensation reactions of 41 with various amines gave compounds 21 and 22 in moderate yields.
Scheme 1. Synthetic Route to the Desired Compounds 5–27.

Reagents and conditions: (a) LiBr, N,N-diisopropylethylamine, CH3CN, H2O, rt, 10 h, 98%; (b) CD3NH2 hydrochloride salt, T3P, N,N-diisopropylethylamine, rt, 10 h, 94%; (c) 3-amino-2-methoxybenzoic acid hydrochloride salt, (CH3COO)2Zn, i-PrOH, H2O, 85 °C, 10 h, 74%; (d) cyclopropanecarboxamide, Pd2(dba)3, Xantphos, Cs2CO3, 1,4-dioxane, 120 °C, 10 h, 53%; (e) ArNH2 or 39a–h, EDCI, pyridine, 55 °C, 2 h, 53–70%; (f) ammonium chloride, EDCI, HOBt, Et3N, DMF, rt, 12 h, 80%; (g) 1-((4-bromophenyl)sulfonyl)pyrrolidine, Pd2(dba)3, Xantphos, Cs2CO3, 1,4-dioxane, 108 °C, 4 h, 61%; (h) pyridin-2-amine or 5-fluoropyridin-2-amine, Pd2(dba)3, Xantphos, Cs2CO3, 1,4-dioxane, 120 °C, 10 h, 63–83%; (i) (4-aminophenyl)(pyrrolidin-1-yl)methanone, EDCI, pyridine, 55 °C, 37–52%; (j) R–NH2, EDCI, HOBt, Et3N, DMF, rt, 2 h.
Compounds 28–32 were synthesized according to the route shown in Scheme 2. Nucleophilic substitution of commercially available 1-fluoro-4-nitrobenzene (42) with various amines in the presence of K2CO3 gave intermediates 43a–c, which were reduced and further condensed with intermediate 37 or 41a to obtain target compounds 28–31. Fluorobenzene 46 was obtained from the starting material 3-amino-5-fluoro-2-methoxybenzoic acid by a synthetic procedure similar to that for 37 and then condensed with intermediate 44c to give target compound 32.
Scheme 2. Synthetic Route to the Desired Compounds 28–32.

Reagents and conditions: (a) R–NH2, K2CO3, DMF, 105 °C, 2 h, 86–87%; (b) H2, Pd/C, CH3OH, rt, 12 h, 93–94%; (c) 37 or 41a, EDCI, pyridine, rt, 12 h, 49–61%; (d) 3-amino-5-fluoro-2-methoxybenzoic acid, (CH3COO)2Zn, i-PrOH, H2O, 85 °C, 10 h, 70%; (e) cyclopropanecarboxamide, Pd2(dba)3, Xantphos, Cs2CO3, 1,4-dioxane, 120 °C, 10 h, 70%; (f) 44c, EDCI, pyridine, rt, 12 h, 22%.
All of the targeted compounds were evaluated for their abilities to bind to the TYK2 JH2 domain by thermal shift analysis (TSA)36 and the inhibition activities of IFNα-stimulated STAT3 phosphorylation dependent on TYK2 in Jurkat cells. High-affinity binding to the TYK2 JH2 domain resulted in an increase in the melting temperature (Tm). Deucravacitinib was used as a positive control and exhibited high affinity, with Tm values of 62.26 and 56.72 °C at 10 and 1 μM, respectively, as well as excellent inhibition activity against STAT3 phosphorylation with an IC50 value of 3.2 nM.
As shown in Table 1, our SAR analysis started with the R1 group and showed that compounds bearing different aromatic rings, such as benzene (5), pyridine (6), and thiophene (7), showed similar binding affinities. Among them, derivative 5 displayed better inhibition activity in the cell assay, so we kept the benzene ring for further study. Interestingly, when hydrophobic groups were introduced on the terminal amide, compounds 8–11 were obtained and displayed significantly improved affinities. Among them, compounds 10 and 11 with tertiary amines substituted with hydrophobic aliphatic groups were superior to deucravacitinib in regard to both binding affinities and inhibition activities. We speculated that the hydrophobic aliphatic substituents interacted with the nearby residues in the hydrophobic P pocket, which increased their binding affinities. Docking of compound 11 with TYK2 JH2 (PDB ID 6NZP) was generated and verified the speculation (Figure 3A). However, one-atom extension of the hydrophobic group of 11 to give compound 12 dramatically decreased the binding affinity, suggesting that a sterically hindered substituent might not be accepted in the TYK2 JH2 domain. Unfortunately, further evaluation of stability in vitro showed that only 24% and 12% of compound 11 remained after 60 min of incubation in human (H) and mouse (M) liver microsomes (LM), respectively. The stability could be slightly increased by deuterating the hydrogen atoms of the pyrrolidine ring at C2′ and C5′ of compound 13 without affecting the inhibition activity. When we introduced a bulky bridged ring, an electron-withdrawing group, or a heteroatom on the pyrrolidine ring to obtain compounds 14, 15, and 16, no better metabolic stability was achieved.
Table 1. SAR of Modifications at R1 and R2.



All data represents an average of n ≥ 3 test occasions.
LM%rem refers to liver microsomal stability as measured by% remaining.
The nitrogen atom is directly attached to the pyridazine ring.
The reference drug deucravacitinib.
Figure 3.
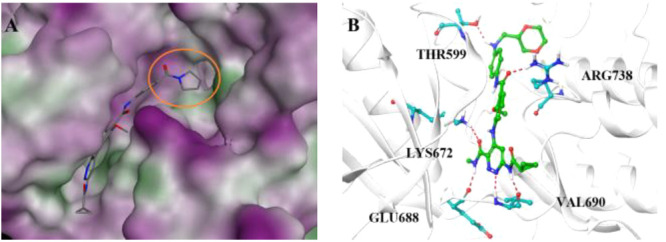
(A) Binding mode of 11 in the hydrophobic pocket of the TYK2 JH2 domain. The hydrophobic pocket is depicted as an orange circle. (B) Binding mode of 30 with TYK2 JH2. The hydrogen bonds formed are shown as red dashed lines.
To further investigate the SAR at the R1 position, compounds 17–19 were synthesized by changing the benzene ring into pyridine (17), fluorobenzene (18), or cyclohexane (19). However, these derivatives did not show improved microsome stabilities and inhibition activities. Replacement of the R2 cyclopropanecarboxamide of 11 with pyridine (21) or fluoropyridine (22) maintained or slightly enhanced the stability, but these compounds were not stable enough to be used as oral drugs.
Further modification of the R1 group with bicyclic groups in place of the benzene ring led to the preparation of analogues 23–27. Although compounds 23–25 exhibited significantly increased stabilities, poor binding affinities and inhibition activities were obtained. An attempt to improve the binding affinity by introducing a hydrophobic cyclopropyl group (26) was also unsuccessful. To our delight, changing the position of amide bond substitution to give compound 27 appeared to slightly improve the potency compared with 23. Subsequently, opening the ring and replacing the amide bond with an amino group afforded 28, the affinity of which was further improved, consistent with previous findings. Excitingly, a significant improvement in the metabolic stability in HLM was observed (30), possibly due to avoidance of amide bond cleavage. In contrast to 21, reduced stability of compound 31 bearing a pyridine moiety at R2 was detected in comparison with 30. Further modification at the 5-position of the benzoic acid nucleus afforded compound 32, which also led to some loss of stability while retaining the potent inhibition. At the end, the SAR found 30 as a potent and stable TYK2 inhibitor.
To clarify the interactions between TYK2 JH2 and 30, a molecular docking study was performed. As shown in Figure 3B, the N-(methyl-d3)pyridazine-3-carboxamide skeleton maintained hydrogen-bonding interactions to the hinge region, whereas the benzenecarboxamide carbonyl group could form a hydrogen-bonding interaction with Arg738. Additionally, a hydrophobic interaction of the alkyl group with nearby residues was formed, which might be responsible for the improvement in the inhibitory potency compared to the unsubstituted control deucravacitinib. The excellent binding affinity of compound 30 (Kd < 0.017 nM) by TYK2 JH2 kinase assays also further verified the docking result (Figure S4).
Considering the safety risk of chronic administration of pan-JAK inhibitors,37,38 binding affinity and cellular function assays for other members of JAK family were performed to determine the selectivity. As shown in Table 2, 30 exhibited poor binding affinities in the TYK2, JAK1, JAK2, and JAK3 JH1 domain assays (IC50 >1000 nM). In a JAK2-dependent EPO-stimulated STAT3 phosphorylation assay in platelet cells, 30 displayed weak potency (IC50 = 3496 nM), indicating ∼1800-fold selectivity for TYK2 over JAK2 signaling. In the IL-2 induced p-STAT5 mediated by JAK1/3 and IL-6 mediated p-STAT3 assays, 30 showed moderate potencies and indicated good selectivities over JAK1/3.
Table 2. Selectivity Profiles of 30 in Binding and Cellular Assays.
| assay | IC50 (nM) |
|---|---|
| JAK1 JH1 | >1000 |
| JAK2 JH1 | >1000 |
| JAK3 JH1 | >1000 |
| TYK2 JH1 | >1000 |
| EPO | 3496 |
| IL-2 | 41 |
| IL-6 | 33 |
To further explore the druggability, compound 30 was evaluated for its PK properties in mice after intragastric (20 mg/kg) and intravenous (1 mg/kg) administrations. As shown in Table 3, 30 showed a high area under the curve (AUC0–∞ = 13476 ng h/mL) when dosed orally. The half-life (t1/2) and the oral bioavailability (F) of 30 were 1.18 h and 253.29%, respectively, indicating that 30 might serve as a potential candidate for further study. We speculated that the bioavailability greater than 100% might be due to the rapid clearance of intravenous injection.39
Table 3. Mouse PK Profile of Compound 30.
| admin. | dose (mg/kg) | Tmax (h) | C0 (ng h) | Cmax (ng h) | AUC(0–t) (ng h/mL) | AUC(0–∞) (ng h/mL) | Cl (L h–1 kg–1) | t1/2 (h) | MRT (0–t) (h) | F (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| iga | 20 | 0.25 | – | 4075 | 13379 | 13476 | – | 1.18 | 2.65 | 253.29 |
| ivb | 1 | – | 1075 | – | 264 | 264 | 3.78 | 0.20 | 0.26 | – |
Intragastric administration (oral gavage).
Intravenous injection.
Inhibition of hERG channels by drugs for autoimmune diseases is detrimental for the development of new drugs.40 Herein, compound 30 displayed 9.8% inhibition at 10 μM in the patch-clamp assay, which was not statistically significant.
The IL-23-dependent colitis model, which is one of the most commonly used mouse IBD models,41 was employed to evaluate the in vivo efficacy of compound 30. As shown in Figure 4A, it was observed that the mice in the model group had continuous weight loss during the treatment. Compound 30 showed a trend in weight loss relief nearly comparable to that of deucravacitinib, administered at an oral dose of 30 mg/kg BID after treatment for 6 days. At the end of the treatment period, we measured the colon weight per unit length, which indirectly reflects the degree of inflammation in the colon. Compared to vehicle-treated normal mice, anti-CD40 monoclonal antibody significantly increased the weight of colon and thickened it at the same time. Mice treated with compound 30 exhibited a decreasing trend in colonic weight, and no significant difference was observed relative to deucravacitinib.
Figure 4.

(A) Record of the initial body weight from day 1 to day 6. *, P < 0.05 and **, P < 0.01 vs control group; #, P < 0.05 and ##, P < 0.01 vs model group. (B) Weights of colons per unit length in each group. ***, P < 0.001 vs control group; ###, P < 0.001 vs model group.
We also constructed an acanthosis skin inflammation model,42 which was similar to the underlying mechanism of psoriasis, to evaluate the role of compound 30 in ameliorating epidermal hyperplasia. In this model, 30 significantly protected mice from IL-23-induced acanthosis, characterized by erythema, desquamation, and thickening (Figure 5A), and achieved results comparable to those with deucravacitinib at a dose of 20 mg/kg BID for 5 days (Figure 5B). Moreover, no significant weight losses were observed (Figure 5C).
Figure 5.

(A) At day 6, mice from each group were photographed to observe differences. (B) Mean dorsal skin thickness changes over time. **, P < 0.01 and ***, P < 0.001 vs vehicle group; #, P < 0.05 and ##, P < 0.01 vs model group. (C) Relative body weight changes over time. #, P < 0.05 vs model group.
In conclusion, to discover structurally diverse TYK2 inhibitors that bind to the JH2 domain, a series of novel derivatives with an N-(methyl-d3)pyridazine-3-carboxamide skeleton were designed, synthesized, and evaluated. Among them, compound 30 demonstrated better inhibition activity against STAT3 phosphorylation compared with the positive control deucravacitinib. Furthermore, 30 had good microsome stabilities and in vivo pharmacokinetic properties. Finally, compound 30 was orally highly efficacious in both IL-23-driven acanthosis and anti-CD40-induced colitis models. On the basis of these positive results, 30 might be a promising candidate for therapeutic applications in autoimmune diseases.
Acknowledgments
The authors thank Xueqin Ma for the in vitro DMPK experiments, Miao Tang for the TYK2 in vitro experiments, Na Li for the in vivo PK experiments, and Xin Li and Huibin Du for the NMR spectral data.
Glossary
Abbreviations
- TYK2
tyrosine kinase 2
- IL-23
interleukin-23
- JAK
Janus kinase
- STAT
signal transducer and activator of transcription
- IFN
interferon
- IBD
inflammatory bowel disease
- SLE
systemic lupus erythematosus
- JH2
pseudokinase
- ATP
adenosine triphosphate
- PK
pharmacokinetics
- SAR
structure–activity relationship
- T3P
propylphosphonic anhydride
- i-PrOH
isopropyl alcohol
- EDCI
1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide
- HOBt
1-hydroxybenzotriazole
- Et3N
triethylamine
- DMF
N,N-dimethylformamide
- Pd(dba)3
tris(dibenzylideneacetone)dipalladium
- TSA
thermal shift analysis
- Tm
melting temperature
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.2c00334.
Synthetic procedures and analytical data for compounds reported in this Letter and procedures for in vitro and in vivo assays (PDF)
Author Contributions
§ F.L. and B.W. contributed equally to this work. The manuscript was written through contributions of all authors. All of the authors approved the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- O’Sullivan L. A.; Liongue C.; Lewis R. S.; Stephenson S. E.; Ward A. C. Cytokine receptor signaling through the Jak-Stat-Socs pathway in disease. Mol. Immunol. 2007, 44, 2497–2506. 10.1016/j.molimm.2006.11.025. [DOI] [PubMed] [Google Scholar]
- Hoffman H. M.; Broderick L. JAK inhibitors in autoinflammation. J. Clin. Invest. 2018, 128, 2760–2762. 10.1172/JCI121526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X.; Li J.; Fu M.; Zhao X.; Wang W. The JAK/STAT signaling pathway: from bench to clinic. Signal Transduction Targeted Ther. 2021, 6, 402. 10.1038/s41392-021-00791-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Hu Y.; Song B.; Xiong Y.; Wang J.; Chen D. Targeting JAK/STAT signaling pathways in treatment of inflammatory bowel disease. Inflamm. Res. 2021, 70, 753–764. 10.1007/s00011-021-01482-x. [DOI] [PubMed] [Google Scholar]
- Villarino A. V.; Kanno Y.; O’Shea J. J. Mechanisms and Consequences of Jak-STAT Signaling in the Immune System. Nat. Immunol. 2017, 18, 374–384. 10.1038/ni.3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velazquez L.; Fellous M.; Stark G. R.; Pellegrini S. A protein tyrosine kinase in the interferon αβ signaling pathway. Cell 1992, 70, 313–322. 10.1016/0092-8674(92)90105-L. [DOI] [PubMed] [Google Scholar]
- Karaghiosoff M.; Steinborn R.; Kovarik P.; Kriegshäuser G.; Baccarini M.; Donabauer B.; Reichart U.; Kolbe T.; Bogdan C.; Leanderson T.; Levy D.; Decker T.; Müller M. Central role for type I interferons and Tyk2 in lipopolysaccharide-induced endotoxin shock. Nat. Immunol. 2003, 4, 471–477. 10.1038/ni910. [DOI] [PubMed] [Google Scholar]
- Tokumasa N.; Suto A.; Kagami S.; Furuta S.; Hirose K.; Watanabe N.; Saito Y.; Shimoda K.; Iwamoto I.; Nakajima H. Expression of Tyk2 in dendritic cells is required for IL-12, IL-23, and IFN-gamma production and the induction of Th1 cell differentiation. Blood 2007, 110, 553–560. 10.1182/blood-2006-11-059246. [DOI] [PubMed] [Google Scholar]
- Ghoreschi K.; Balato A.; Enerbäck C.; Sabat R. Therapeutics targeting the IL-23 and IL-17 pathway in psoriasis. Lancet 2021, 397, 754–766. 10.1016/S0140-6736(21)00184-7. [DOI] [PubMed] [Google Scholar]
- Clark J. D.; Flanagan M. E.; Telliez J.-B. Discovery and Development of Janus Kinase (JAK) Inhibitors for Inflammatory Diseases. J. Med. Chem. 2014, 57, 5023–5038. 10.1021/jm401490p. [DOI] [PubMed] [Google Scholar]
- Schwartz D. M.; Kanno R.; Villarino A.; Ward M.; Gadina M.; O’Shea J. J. JAK Inhibition as a Therapeutic Strategy for Immune and Inflammatory Diseases. Nat. Rev. Drug Discovery 2017, 16, 843–862. 10.1038/nrd.2017.201. [DOI] [PubMed] [Google Scholar]
- Schwartz D. M.; Bonelli M.; Gadina M.; O’Shea J. J. Type I/II Cytokines, JAKs, and New Strategies for Treating Autoimmune Diseases. Nat. Rev. Rheumatol. 2016, 12, 25–36. 10.1038/nrrheum.2015.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrobleski S. T.; Moslin R.; Lin S.; Zhang Y.; Spergel S.; Kempson J.; Tokarski J. S.; Strnad J.; Zupa-Fernandez A.; Cheng L.; Shuster D.; Gillooly K.; Yang X.; Heimrich E.; McIntyre K. W.; Chaudhry C.; Khan J.; Ruzanov M.; Tredup J.; Mulligan D.; Xie D.; Sun H.; Huang C.; D’Arienzo C.; Aranibar N.; Chiney M.; Chimalakonda A.; Pitts W. J.; Lombardo L.; Carter P. H.; Burke J. R.; Weinstein D. S. Highly Selective Inhibition of Tyrosine Kinase 2 (TYK2) for the Treatment of Autoimmune Diseases: Discovery of the Allosteric Inhibitor BMS-986165. J. Med. Chem. 2019, 62, 8973–8995. 10.1021/acs.jmedchem.9b00444. [DOI] [PubMed] [Google Scholar]
- FDA Approves Stelara (ustekinumab) to Treat Psoriasis. Drugs.com, September 25, 2009. https://www.drugs.com/newdrugs/fdaapproves-stelara-ustekinumab-psoriasis-1667.html (accessed 2020-11-08).
- Moschen A. R.; Tilg H.; Raine T. IL-12, IL-23 and IL-17 in IBD: Immunobiology and Therapeutic Targeting. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 185–196. 10.1038/s41575-018-0084-8. [DOI] [PubMed] [Google Scholar]
- Andreou N. P.; Legaki E.; Gazouli M. Inflammatory Bowel Disease Pathobiology: the Role of the Interferon Signature. Ann. Gastroenterol. 2020, 33, 125–133. 10.20524/aog.2020.0457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y.; Zhu Y.; Xia Y.; Peng H.; Yang X.-K.; Liu Y. Y.; Xu W. D.; Pan H. F.; Ye D. Q. Therapeutic Potential of Tyrosine Kinase 2 in Autoimmunity. Expert Opin. Ther. Targets 2014, 18, 571–580. 10.1517/14728222.2014.892925. [DOI] [PubMed] [Google Scholar]
- He X.; Chen X.; Zhang H.; Xie T.; Ye X. Y. Selective Tyk2 Inhibitors as Potential Therapeutic Agents: a Patent Review (2015–2018). Expert Opin. Ther. Pat. 2019, 29, 137–149. 10.1080/13543776.2019.1567713. [DOI] [PubMed] [Google Scholar]
- Weinstein D. W.; Moslin R. M. Advances in the Discovery and Development of Selective Tyrosine Kinase 2 (TYK2) Inhibitors. Med. Chem. Rev. 2018, 53, 177–200. 10.29200/acsmedchemrev-v53.ch10. [DOI] [Google Scholar]
- Liang J.; van Abbema A.; Balazs M.; Barrett K.; Berezhkovsky L.; Blair W.; Chang C.; Delarosa D.; DeVoss J.; Driscoll J.; Eigenbrot C.; Ghilardi N.; Gibbons P.; Halladay J.; Johnson A.; Kohli P. B.; Lai Y.; Liu Y.; Lyssikatos J.; Mantik P.; Menghrajani K.; Murray J.; Peng I.; Sambrone A.; Shia S.; Shin Y.; Smith J.; Sohn S.; Tsui V.; Ultsch M.; Wu L. C.; Xiao Y.; Yang W.; Young J.; Zhang B.; Zhu B. Y.; Magnuson S. Lead optimization of a 4-aminopyridine benzamide scaffold to identify potent, selective, and orally bioavailable TYK2 inhibitors. J. Med. Chem. 2013, 56, 4521–4536. 10.1021/jm400266t. [DOI] [PubMed] [Google Scholar]
- Moslin R.; Gardner D.; Santella J.; Zhang Y.; Duncia J. V.; Liu C.; Lin J.; Tokarski J. S.; Strnad J.; Pedicord D.; Chen J.; Blat Y.; Zupa-Fernandez A.; Cheng L.; Sun H.; Chaudhry C.; Huang C.; D’Arienzo C.; Sack J. S.; Muckelbauer J. K.; Chang C.; Tredup J.; Xie D.; Aranibar N.; Burke J. R.; Carter P. H.; Weinstein D. S. Identification of imidazo[1,2-b]pyridazine TYK2 pseudokinase ligands as potent and selective allosteric inhibitors of TYK2 signalling. MedChemComm 2017, 8, 700–712. 10.1039/C6MD00560H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez Lopez de Turiso F.; Guckian K. Selective TYK2 inhibitors as potential therapeutic agents: a patent review (2019–2021). Expert Opin. Ther. Pat. 2022, 32, 365–379. 10.1080/13543776.2022.2026927. [DOI] [PubMed] [Google Scholar]
- Akahane K.; Li Z.; Etchin J.; Berezovskaya A.; Gjini E.; Masse C. E.; Miao W.; Rocnik J.; Kapeller R.; Greenwood J. R.; Tiv H.; Sanda T.; Weinstock D. M.; Look A. T. Anti-leukaemic activity of the TYK2 selective inhibitor NDI-031301 in T-cell acute lymphoblastic leukaemia. Br. J. Haematol. 2017, 177, 271–282. 10.1111/bjh.14563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C.; Lin J.; Moslin R.; Tokarski J. S.; Muckelbauer J.; Chang C.; Tredup J.; Xie D.; Park H.; Li P.; Wu D. R.; Strnad J.; Zupa-Fernandez A.; Cheng L.; Chaudhry C.; Chen J.; Chen C.; Sun H.; Elzinga P.; D’arienzo C.; Gillooly K.; Taylor T. L.; McIntyre K. W.; Salter-Cid L.; Lombardo L. J.; Carter P. H.; Aranibar N.; Burke J. R.; Weinstein D. S. Identification of Imidazo[1,2-b]pyridazine Derivatives as Potent, Selective, and Orally Active Tyk2 JH2 Inhibitors. ACS Med. Chem. Lett. 2019, 10, 383–388. 10.1021/acsmedchemlett.9b00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J.; Van Abbema A.; Balazs M.; Barrett K.; Berezhkovsky L.; Blair W. S.; Chang C.; Delarosa D.; DeVoss J.; Driscoll J.; Eigenbrot C.; Goodacre S.; Ghilardi N.; MacLeod C.; Johnson A.; Bir Kohli P.; Lai Y.; Lin Z.; Mantik P.; Menghrajani K.; Nguyen H.; Peng I.; Sambrone A.; Shia S.; Smith J.; Sohn S.; Tsui V.; Ultsch M.; Williams K.; Wu L. C.; Yang W.; Zhang B.; Magnuson S. Identification of an imidazopyridine scaffold to generate potent and selective TYK2 inhibitors that demonstrate activity in an in vivo psoriasis model. Bioorg. Med. Chem. Lett. 2017, 27, 4370–4376. 10.1016/j.bmcl.2017.08.022. [DOI] [PubMed] [Google Scholar]
- Papp K.; Gordon K.; Thaçi D.; Morita A.; Gooderham M.; Foley P.; Girgis I. G.; Kundu S.; Banerjee S. Phase 2 Trial of Selective Tyrosine Kinase 2 Inhibition in Psoriasis. N. Engl. J. Med. 2018, 379, 1313–1321. 10.1056/NEJMoa1806382. [DOI] [PubMed] [Google Scholar]
- Danese S.; Peyrin-Biroulet L. Selective Tyrosine Kinase 2 Inhibition for Treatment of Inflammatory Bowel Disease: New Hope on the Rise. Inflamm. Bowel Dis. 2021, 27, 2023–2030. 10.1093/ibd/izab135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min X.; Ungureanu D.; Maxwell S.; Hammarén H.; Thibault S.; Hillert E. K.; Ayres M.; Greenfield B.; Eksterowicz J.; Gabel C.; Walker N.; Silvennoinen O.; Wang Z. Structural and Functional Characterization of the JH2 Pseudokinase Domain of JAK Family Tyrosine Kinase 2 (TYK2). J. Biol. Chem. 2015, 290, 27261–27270. 10.1074/jbc.M115.672048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke J. R.; Cheng L.; Gillooly K. M.; Strnad J.; Zupa-Fernandez A.; Catlett I. M.; Zhang Y.; Heimrich E. M.; McIntyre K. W.; Cunningham M. D.; Carman J. A.; Zhou X.; Banas D.; Chaudhry C.; Li S.; D’Arienzo C.; Chimalakonda A.; Yang X.; Xie J. H.; Pang J.; Zhao Q.; Rose S. M.; Huang J.; Moslin R. M.; Wrobleski S. T.; Weinstein D. S.; Salter-Cid L. M. Autoimmune pathways in mice and humans are blocked by pharmacological stabilization of the TYK2 pseudokinase domain. Sci. Transl. Med. 2019, 11, eaaw1736 10.1126/scitranslmed.aaw1736. [DOI] [PubMed] [Google Scholar]
- Liu C.; Lin J.; Langevine C.; Smith D.; Li J.; Tokarski J. S.; Khan J.; Ruzanov M.; Strnad J.; Zupa-Fernandez A.; Cheng L.; Gillooly K. M.; Shuster D.; Zhang Y.; Thankappan A.; McIntyre K. W.; Chaudhry C.; Elzinga P. A.; Chiney M.; Chimalakonda A.; Lombardo L. J.; Macor J. E.; Carter P. H.; Burke J. R.; Weinstein D. S. Discovery of BMS-986202: A Clinical Tyk2 Inhibitor that Binds to Tyk2 JH2. J. Med. Chem. 2021, 64, 677–694. 10.1021/acs.jmedchem.0c01698. [DOI] [PubMed] [Google Scholar]
- Fensome A.; Ambler C. M.; Arnold E.; Banker M. E.; Brown M. F.; Chrencik J.; Clark J. D.; Dowty M. E.; Efremov I. V.; Flick A.; Gerstenberger B. S.; Gopalsamy A.; Hayward M. M.; Hegen M.; Hollingshead B. D.; Jussif J.; Knafels J. D.; Limburg D. C.; Lin D.; Lin T. H.; Pierce B. S.; Saiah E.; Sharma R.; Symanowicz P. T.; Telliez J.-B.; Trujillo J. I.; Vajdos F. F.; Vincent F.; Wan Z.-K.; Xing L.; Yang X.; Yang X.; Zhang L. Dual Inhibition of TYK2 and JAK1 for the Treatment of Autoimmune Diseases: Discovery of ((S)-2,2-Difluorocyclopropyl)((1R,5S)-3-(2-((1-methyl-1H-pyrazol-4-yl)-amino)-pyrimidin-4-yl)-3,8-diazabicyclo[3.2.1]octan-8 yl)methanone (PF-06700841). J. Med. Chem. 2018, 61, 8597–8612. 10.1021/acs.jmedchem.8b00917. [DOI] [PubMed] [Google Scholar]
- Banfield C.; Scaramozza M.; Zhang W.; Kieras E.; Page K. M.; Fensome A.; Vincent M.; Dowty M. E.; Goteti K.; Winkle P. J.; Peeva E. The Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of a TYK2/JAK1 Inhibitor (PF-06700841) in Healthy Subjects and Patients with Plaque Psoriasis. J. Clin. Pharmacol. 2018, 58, 434–447. 10.1002/jcph.1046. [DOI] [PubMed] [Google Scholar]
- Gerstenberger B. S.; Ambler C.; Arnold E. P.; Banker M.-E.; Brown M. F.; Clark J. D.; Dermenci A.; Dowty M. E.; Fensome A.; Fish S.; Hayward M. M.; Hegen M.; Hollingshead B. D.; Knafels J. D.; Lin D. W.; Lin T. H.; Owen D. R.; Saiah E.; Sharma R.; Vajdos F. F.; Xing L.; Yang X.; Yang X.; Wright S. W. Discovery of Tyrosine Kinase 2 (TYK2) Inhibitor (PF-06826647) for the Treatment of Autoimmune Diseases. J. Med. Chem. 2020, 63, 13561–13577. 10.1021/acs.jmedchem.0c00948. [DOI] [PubMed] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H.; Henrick K.; Nakamura H. Announcing the worldwide Protein Data Bank. Nat. Struct. Biol. 2003, 10, 980–980. 10.1038/nsb1203-980. [DOI] [PubMed] [Google Scholar]
- Tokarski J. S.; Zupa-Fernandez A.; Tredup J. A.; Pike K.; Chang C.; Xie D.; Cheng L.; Pedicord D.; Muckelbauer J.; Johnson S. R.; Wu S.; Edavettal S. C.; Hong Y.; Witmer M. R.; Elkin L. L.; Blat Y.; Pitts W. J.; Weinstein D. S.; Burke J. R. Tyrosine Kinase 2-mediated Signal Transduction in T Lymphocytes Is Blocked by Pharmacological Stabilization of Its Pseudokinase Domain. J. Biol. Chem. 2015, 290, 11061–11074. 10.1074/jbc.M114.619502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Jeldres T.; Tyler C. J.; Boyer J. D.; Karuppuchamy T.; Yarur A.; Giles D. A.; Yeasmin S.; Lundborg L.; Sandborn W. J.; Patel D. R.; Rivera-Nieves J. Targeting Cytokine Signaling and Lymphocyte Traffic via Small Molecules in Inflammatory Bowel Disease: JAK Inhibitors and S1PR Agonists. Front Pharmacol. 2019, 10, 212. 10.3389/fphar.2019.00212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danese S.; Argollo M.; Le Berre C.; Peyrin-Biroulet L. JAK selectivity for inflammatory bowel disease treatment: does it clinically matter?. Gut 2019, 68, 1893–1899. 10.1136/gutjnl-2019-318448. [DOI] [PubMed] [Google Scholar]
- Ward K. W.; Azzarano L. M.; Evans C. A.; Smith B. R. Apparent absolute oral bioavailability in excess of 100% for a vitronectin receptor antagonist (SB-265123) in rat. I., Investigation of potential experimental and mechanistic explanations. Xenobiotica 2004, 34, 353–366. 10.1080/0049825042000205540. [DOI] [PubMed] [Google Scholar]
- Kalyaanamoorthy S.; Barakat K. H. Development of Safe Drugs: The hERG Challenge. Med. Res. Rev. 2018, 38, 525–555. 10.1002/med.21445. [DOI] [PubMed] [Google Scholar]
- Uhlig H. H.; McKenzie B. S.; Hue S.; Thompson C.; Joyce-Shaikh B.; Stepankova R.; Robinson N.; Buonocore S.; Tlaskalova-Hogenova H.; Cua D. J.; Powrie F. Differential activity of IL-12 and IL-23 in mucosal and systemic innate immune pathology. Immunity. 2006, 25, 309–318. 10.1016/j.immuni.2006.05.017. [DOI] [PubMed] [Google Scholar]
- Zheng Y.; Danilenko D. M.; Valdez P.; Kasman I.; EasthamAnderson J.; Wu J.; Ouyang W. Interleukin-22, a TH17 Cytokine, Mediates IL-23-Induced Dermal Inflammation and Acanthosis. Nature 2007, 445, 648–651. 10.1038/nature05505. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.