

Abstract
Objective
To assess the efficacy and tolerability of tocilizumab in a multicenter, randomized, double‐blind, placebo‐controlled trial in refractory adult patients with dermatomyositis (DM) and polymyositis (PM).
Methods
Thirty‐six subjects with probable or definite DM/PM were enrolled in a 6‐month phase 2B clinical trial and randomized 1:1 to receive tocilizumab (8 mg/kg intravenously) or placebo every 4 weeks for 24 weeks. Eligible subjects had either a DM rash, a myositis‐associated autoantibody or an adjudicated PM diagnosis. Active disease was defined by at least three of six abnormal core set measures (CSMs), including a manual muscle testing (MMT)‐8 score of less than 136/150. If the MMT‐8 score was greater than 136, then a cutaneous score of 3 or more (10 cm visual analogue scale) was required along with three additional abnormal CSMs indicating disease activity. The primary endpoint compared the Total Improvement Score (TIS) between both arms from week 4 to 24. Secondary outcomes included time to meeting minimal TIS improvement, changes in CSMs, time to worsening, steroid‐sparing effect, proportion of subjects meeting more stringent improvement criteria, and safety outcomes.
Results
There was no significant difference (P = 0.86) in the TIS over 24 weeks between tocilizumab and placebo arms. The secondary endpoints of time to improvement (minimal, moderate, or major), time to worsening, CSM changes, safety outcomes, and steroid‐sparing effect were also not significantly different between arms.
Conclusion
Tocilizumab was safe and well tolerated but did not meet the primary or secondary efficacy outcomes in refractory DM and PM in this 24‐week phase 2B study.
INTRODUCTION
The idiopathic inflammatory myopathies (IIMs) are rare, heterogeneous diseases characterized by muscle weakness and systemic involvement with substantial morbidity and mortality. The treatment of IIMs is unsatisfactory, and currently there are now only two Food and Drug Administration (FDA)‐approved medications, Acthar gel and intravenous immunoglobulin (IVIg). The rarity of IIMs has led to a paucity of controlled prospective clinical trials. Other than a large multicenter trial assessing the efficacy of rituximab (1) and several other recently completed or ongoing trials studying intravenous gamma globulin, abatacept, an immunoproteasome inhibitor, and lenabasum, most trials involve single referral centers reporting on small numbers of subjects. Furthermore, our ability to accurately assess the effects of therapeutic interventions was previously limited by unreliable and insensitive outcome measures. However, through the original efforts of the International Myositis Assessment and Clinical Studies (IMACS) Group validating core set measures (CSMs), and a recently revised definition of improvement (DOI), we have made considerable progress in the design of myositis clinical trials (2). Therefore, we adopted this recently proposed DOI for our study in order to more effectively assess the effects of therapeutic intervention.
Based on several studies supporting the efficacy of tocilizumab (anti‐interleukin 6 receptor [IL6R]) in rheumatoid arthritis (RA) and systemic onset juvenile idiopathic arthritis, the use of this agent in other autoimmune disorders has also been proposed (3, 4). Interleukin 6 (IL‐6) plays a critical role in both adaptive and innate immune responses. It is produced by T cells, B cells, monocytes, and endothelial cells; binds to its receptor (IL‐6R); and triggers several intracellular pathways leading to the release of inflammatory mediators and stimulation of the immune system. Inhibition of IL‐6 in RA decreases acute phase reactants and other indicators of chronic inflammation (5). Although a previously published mouse model reported IL‐6 as a mediator of muscle inflammation (6), another mouse model noted that the pathology of C protein‐induced myositis (CIM) mimics that seen in human polymyositis (PM) (7). In mice treated with tocilizumab, CIM was ameliorated (8). Supporting these preclinical studies, a small number of patients with refractory PM responded favorably to treatment with tocilizumab (9). Furthermore, serum IL‐6 levels in adult and juvenile dermatomyositis (DM) paralleled disease activity (10), and IL‐6 levels correlated with both the type I interferon gene and chemokine signatures. These observations have suggested a coordinated dysregulation of IL‐6 production and type I interferon signaling that potentially contributes to disease pathogenesis in DM. Based on these human and animal studies and using the aforementioned new myositis outcome measures, we examined the efficacy and safety of tocilizumab as a therapeutic advance for PM and DM in a placebo‐controlled, randomized clinical trial.
PATIENTS AND METHODS
This study was conducted at seven sites (University of Pittsburgh School of Medicine Division of Rheumatology and Clinical Immunology, Pittsburgh, Pennsylvania; Wallace Rheumatic Studies Center, Beverly Hills, California; Vanderbilt University Medical Center Division of Rheumatology, Nashville, Tennessee; University of Kansas Medical Center Department of Neurology, Kansas City, Kansas; Mayo Clinic Division of Rheumatology, Rochester, Minnesota; Medical College of Wisconsin Division of Rheumatology, Milwaukee, Wisconsin; and Hofstra Northwell School of Medicine Division of Rheumatology, New York, New York) with the University of Pittsburgh as the coordinating center. The institutional review boards at each location approved the protocol. Written informed consent was obtained from each subject. Eligible patients included adults over the age of 18 with a diagnosis of definite or probable DM or PM by the criteria of Bohan and Peter (11). Subjects had the classic rash(es) of DM (heliotrope, Gottron sign, or Gottron papules), possessed one of the myositis‐associated autoantibodies (antisynthetase, anti‐signal recognition particle [SRP], anti‐Mi‐2, anti‐PM‐Scl, anti transcription intermediary factor 1‐γ [TIF1‐γ] or anti‐melanoma differentiation‐associated protein 5 [MDA5]), or had the diagnosis of PM agreed on by a three‐member adjudication committee consisting of a rheumatologist, neurologist, and neuromuscular pathologist. Refractory disease was defined as having failed (or demonstrating intolerance to) an adequate course of glucocorticoids or having failed glucocorticoids and at least one other immunosuppressive (IS) or immunomodulatory agent (eg, methotrexate, azathioprine, cyclosporine, tacrolimus, mycophenolate mofetil, cyclophosphamide, intravenous immunoglobulin [IVIg], an anti‐tumor necrosis factor [anti‐TNF] agent, and rituximab). An adequate trial of glucocorticoids generally included prednisone (or an equivalent glucocorticoid) at a dose of at least 60 mg/day for 1 month, whereas an adequate trial of an IS agent was at least 3 months at a dose known to be effective for rheumatological diseases. Enrolled subjects had “active” disease defined by at least three of six abnormal myositis CSMs (12), including a manual muscle testing (MMT)‐8 score of less than 136/150 (Supplementary Table A). However, if the MMT was greater than 136/150, subjects had to have a cutaneous visual analogue scale (VAS) score of 3.0 cm or more on a 10 cm scale on the Myositis Disease Activity Assessment Tool (MDAAT) in addition to at least three other abnormal myositis CSMs (13): 1) patient global VAS score of 2.0 cm or more, 2) physician global VAS score of 2.0 cm or more, 3) health assessment questionnaire (HAQ) disability index of 0.25 or more, 4) elevation of at least one muscle enzyme (creatine kinase [CK], aspartate aminotransferase, alanine aminotransferase, aldolase, or lactate dehydrogenase) 1.3 or more times the upper limit of normal and 5) global extramuscular disease activity (a composite of constitutional, cutaneous, skeletal, gastrointestinal, pulmonary, and cardiac activity) VAS score of 1.0 cm or more on the MDAAT. All VAS scales were 10 cm and anchored at the ends and midpoint. Physicians were allowed to review the previous visits' extramuscular and physician global scores. To minimize bias, the same clinical investigator was directed to complete all of the clinical assessments longitudinally on study subjects.
Concomitant IS medications permitted included methotrexate, azathioprine, cyclosporine, mycophenolate mofetil, and tacrolimus. IVIg was allowed as an immunomodulatory agent. If an IS agent was discontinued prior to screening, then a previously specified washout (1) was done. If the IS agent was continued as a concomitant IS medication, then the patient should have been on this IS medication for 3 months or more and the dose had to be stable for 4 weeks prior to screening and remain unchanged in the trial until the DOI was met during the treatment phase of the study. Prednisone (or an equivalent glucocorticoid) was allowed in addition to concomitant IS medication and the dose had to be stable for 4 weeks before screening. Tapering of prednisone (or an equivalent glucocorticoid) was allowed (generally 20%‐25% of the existing dose every 4 weeks) only after the subject met the criteria for improvement or if safety issues related to continued glucocorticoid use supervened. A prednisone dose of less than 1 mg/kg/day of prednisone at study entry was encouraged. If in the clinical site investigator's opinion there was disease worsening during the trial, then the smallest reasonable prednisone dose increase was instituted. No concomitant biologic agents (rituximab, anti‐TNF agents, and abatacept), cyclophosphamide, or tofacitinib were allowed. Investigators assessed adverse events as being secondary to either study drug or concomitant IS therapy.
Subjects were excluded for the following reasons: 1) a global muscle damage score of greater than 5 on a 10 cm VAS scale using the Muscle Damage Index (14), 2) cancer‐associated myositis (ie, diagnosis of myositis within 2 years of cancer diagnosis), or 3) previous treatment with tocilizumab.
Design overview
This was a phase 2B double‐blind, placebo‐controlled trial randomized 1:1 for active drug (tocilizumab, a recombinant humanized antihuman IL‐6 receptor monoclonal antibody of the IgG1ĸ subclass) placebo for 6 months with a targeted enrollment of 40 subjects. The myositis subset, PM or DM, was treated as a stratification variable and a “balanced coin” approach was employed to control treatment assignment within enrollment sites. Patients received intravenous tocilizumab (8 mg/kg; maximum dose 800 mg) or placebo at baseline (week 0) and every 4 weeks thereafter (weeks 4, 8, 12, 16, and 20). All six CSMs were assessed at the screening visit and every 4 weeks thereafter (weeks 0, 4, 8, 12, 16, 20, and 24). A post‐trial 6‐month nondrug observational follow‐up was conducted with two additional visits, 3 and 6 months after the closing visit, to assess the durability of response to study drug and safety.
Study definitions/statistical considerations
The sample size of 40 subjects was justified based on this trial being a proof‐of‐concept pilot study allowing us to determine an estimate of the effect size of the study drug and placebo and to assess feasibility and safety in a pilot trial. The primary response criterion for this trial was the achievement of a Total Improvement Score (TIS) of 20 or more (minimal improvement) at any time point during the 24‐week period. The TIS, derived from the 2016 American College of Rheumatology/European Alliance of Associations for Rheumatology myositis response criteria and established by international consensus, is a composite score calculated by adding the weighted improvement scores of all six CSMs (2) (Supplementary Table B). The primary outcome measure of the trial was the average TIS at weeks 4, 8, 12, 16, 20, and 24 during the 6‐month treatment period. The primary outcome in the placebo and treatment groups were compared using a generalize estimating equation (GEE) model because of potential missing data and repeated measures over time. In the GEE model, we included treatment group, myositis subset (PM or DM), and visit time points, and both unadjusted and adjusted model runs were performed. Each of the baseline CSM was investigated to determine 1) whether they were predictive of the follow‐up TIS and 2) whether adjustment for them in our model would affect our finding that outcome was not related to treatment.
Additional secondary endpoints analyzed between both arms include 1) comparison of the time to meeting the primary response (Kaplan‐Meier curves and log rank tests), 2) comparison of the individual CSM in subjects over time (repeated measures analyses using GEE model that included treatment group, myositis subset [PM or DM], and visit time points), 3) comparison of the average steroid dose change from baseline to the 6‐month follow‐up in the patients randomized to tocilizumab versus placebo (Wilcoxon test), 4) comparison of the proportion of subjects having TISs of 20 or more (minimal), 40 or more (moderate) and 60 or more (major) (Supplementary Table B), and 5) the difference in adverse events and serious adverse events (χ2). The durability of the treatment response was assessed by 6) comparing the proportion (χ2) and time to meeting the DOI on two consecutive visits (Kaplan‐Meier curves and log rank tests) and 7) determining the time to disease worsening between treatment and placebo groups in those subjects who first met the DOI (Kaplan‐Meier curve and log rank test). Finally, the previously published IMACS DOI for adult myositis (15) (three of six CSMs improved by 20% or more, with no more than two CSMs worsening by 25% or more [worsening measure cannot be the MMT]) was assessed by 8) comparing the proportion of subjects meeting the previous IMACS DOI in the treatment and placebo arms (Kaplan‐Meier curve and log rank test).
Disease worsening (16) was defined as: 1) physician global worsening of 2 cm or more on a 10 cm VAS and/or worsening of the MMT by 20% or more, 2) global extramuscular organ disease activity (on the MDAAT) worsening by 2 cm or more on a 10 cm VAS, or 3) decline in any three of six CSMs by 30% or more. Subjects meeting these criteria during the treatment phase of the trial were considered treatment failures if they did not first meet criteria for DOI. Treatment failures were also defined as those subjects failing to achieve at least minimal improvement (Supplementary Table B) during the 24‐week treatment period. All analyses were intention to treat, in which patients remained in their randomized group irrespective of their actual treatment, rescue therapy, or nonprotocol treatment during the trial.
RESULTS
Thirty‐six subjects (23 DM/13 PM) were randomized (18 in each arm) and analyzed using intention to treat (Figure 1). The trial began enrolling on March 11, 2015, with the first patient visit on April 22, 2015, and the last patient visit on April 23, 2019. The plan was to enroll 40 subjects with refractory PM or DM, but an interim analysis revealed that the accrual and follow‐up of four additional patients was unlikely to change the primary outcome, so enrollment was stopped at 36 subjects. All but four subjects (two tocilizumab/two placebo) completed the 24‐week treatment phase due to two subjects missing the final study evaluation (week 24), one subject having the study drug interrupted because of an adverse event and lack of final study evaluation (week 24), and one subject withdrawing from the study because of an unrelated adverse event at week 12 and not returning for any additional follow‐up visits. Overall, 89% of subjects completed the 24‐week study visits. Two additional subjects stopped the study drug early because of disease worsening but completed all the study assessments, such that 83% completed 24 weeks of assessments. In the treatment arm, one subject had their azathioprine increased (100 to 125 mg/day), IVIg added (2 g/kg,) and mycophenolate mofetil (2 g/day) later added after the IVIg was stopped (poor venous access). In the placebo arm, one subject stopped the study drug at week 12 but continued to be followed with CSM until week 20. Prednisone was increased from 30 to 40 mg daily, and tacrolimus was increased from 1 to 2 mg/day. The baseline demographic characteristics and CSMs between both groups were similar as was their previous IS treatment at trial initiation (Table 1). Most patients (n = 14) were enrolled at the coordinating center (University of Pittsburgh), followed by Cedars Sinai (n = 8) and Northwell (n = 6), with the additional 8 subjects enrolled from the other four sites (1‐3 patients).
Figure 1.

Participant flow diagram. Participant flow diagram of subjects enrolled in tocilizumab in myositis trial. AE, adverse event.
Table 1.
Baseline demographic and clinical characteristics and core set measures by treatment group
| Characteristic | Tocilizumab | Placebo | P value |
|---|---|---|---|
| (n = 18) | (n = 18) | ||
| Caucasian, n (%) | 12 (67) | 14 (78) | 0.71 a |
| Age (y), mean (range) | 54.3 (31.3‐83.4) | 50.4 (21.5‐70.7) | 0.41 b |
| Female, n (%) | 15 (83) | 13 (72) | 0.69 a |
| IIM subset, n (%) | 0.30 | ||
| PM | 8 (44) | 5 (28) | |
| DM | 10 (56) | 13 (72) | |
| Prednisone dose (mg/day c ), mean (SD) | 8.3 (7.5) | 17.8 (20.5) | 0.07 b |
| Patients on prednisone, n (%) | 12 (67) | 15 (83) | 0.44 a |
| Patients taking nonglucocorticoid IS and/or immunomodulatory agents, n (%) | 15 (83) | 15 (83) | 1.00 a |
| Mtx | 4 (22) | 6 (33) | |
| Mtx + Aza | 1 (6) | 1 (6) | |
| Mtx + IVIg | 1 (6) | 1 (6) | |
| Mtx + MMF | 0 (0) | 1 (6) | |
| MMF | 6 (33) | 0 (0) | |
| Aza | 1 (6) | 0 (0) | |
| Aza + IVIg | 0 (0) | 1 (6) | |
| Tacrolimus | 1 (6) | 2 (11) | |
| IVIg | 1 (6) | 3 (17) | |
| Patients with positive myositis autoantibodies, n (%) | 8 (44) | 10 (56) | 0.51 |
| Antisynthetase | 0 (0) | 4 (22) | |
| Anti‐TIF1‐γ | 2 (11) | 1 (6) | |
| Anti‐Mi‐2 | 2 (11) | 2 (11) | |
| SRP d | 2 (11) | 0 (0) | |
| HMGCR e | 0 (0) | 1 (6) | |
| MJ/NXP‐2 | 1 (6) | 1 (6) | |
| MDA‐5 | 1 (6) | 0 (0) | |
| Ro60 | 0 (0) | 1 (6) | |
| MMT‐8 score, mean (SD) | 126.7 (15.3) | 132.8 (11.5) | 0.19 b |
| Global assessment by VAS (0‐10 cm scale), mean (SD) | |||
| Physician | 5.1 (1.7) | 4.7 (1.4) | 0.47 b |
| Patient | 6.1 (2.1) | 5.4 (2.4) | 0.37 b |
| HAQ disability index (range 0‐3), mean (SD) | 1.3 (0.7) | 1.3 (0.6) | 0.94 b |
| Muscle enzyme (xULN) f , median (IQR) | 9.1 (1.0‐9.8) | 1.5 (1.0‐2.4) | 0.41 g |
| Extramuscular score by VAS (0‐10 cm scale), mean (SD) | 3.3 (2.5) | 3.3 (1.6) | 0.95 b |
Abbreviations: Aza, azathioprine; DM, dermatomyositis; HAQ, health assessment questionnaire; HMGCR, 3‐hydroxy‐3‐methylglutaryl‐CoA reductase; IIM, idiopathic inflammatory myopathy; IQR, interquartile range; IS, immunosuppressive; IVIg, intravenous immunoglobulin; MDA, melanoma differentiation‐associated protein 5; MMF, mycophenolate mofetil; MMT, manual muscle testing; Mtx, methotrexate; NXP‐2, anti‐nuclear matrix protein‐2; PM, polymyositis; SRP, signal recognition particle; VAS, visual analog scale; xULN, times the upper limit of normal.
Fisher's exact test P value (χ2 tests were performed for all other categorical variables listed in the above table to generate the P values).
Two sample t test P value.
Prednisone dose (mg/day) in subjects taking any dose of prednisone.
Signal recognition particle.
3‐hydroxy‐3‐methyl glutaryl‐CoA reductase.
Upper limit normal.
Wilcoxon test P value.
Primary outcome
There was no significant difference (P = 0.86) in the TIS (primary outcome) over 24 weeks between tocilizumab‐ and placebo‐treated subjects in the entire cohort or by myositis subset (ie, PM vs. DM). The mean (SD) TIS while on treatment for patients randomized to placebo was 29.3 (16.8) compared with a mean of 26.4 (16.8) for patients randomized to tocilizumab. Figure 2 represents the average TIS at each time point by treatment groups. The differences in average scores between the follow‐up visits within the treatment phase of the trial was significant (P < 0.001) for all subjects, but this occurred whether randomized to tocilizumab or placebo. In the tocilizumab group, the score at the first follow‐up visit was 20.0 and the score at the last follow‐up visit (week 24) was 31.5. In the placebo group, the score at first follow‐up visit was 14.0 and the score at the last follow‐up visit was 36.7 (Figure 2). The primary outcome was also unaffected by any of the baseline values of the six CSMs.
Figure 2.
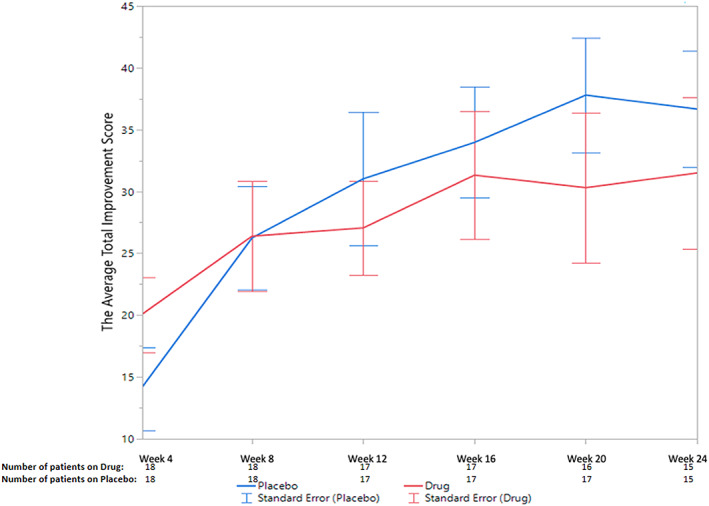
The average total improvement scores of the placebo and treatment groups at each of the six follow‐up visits.
Secondary outcomes
Several different criteria were used to compare the time to improvement between placebo and tocilizumab‐treated subjects, including the time to first DOI (ie, minimal improvement) as well as time to first moderate or major improvement according to the TIS (Supplementary Table B). At some time point during the 24‐week trial, 30, 19, and 7 patients met the criteria for minimum, moderate, and major improvement, respectively. However, the time to minimal improvement was not significantly different in the two treatment groups (P = 0.77), nor were the times to moderate or major improvement (P = 0.61 and P = 0.64, respectively). A comparison on the proportion of patients having minimal improvement across the six follow‐up visits was similarly not significant (P = 0.51). Further, there was no significant difference in individual CSMs over time between the two treatment groups (Table 2). Two additional secondary endpoints were (a) the time to two consecutive visits at which minimal improvement was noted (26 subjects met this criterion) and (b) the time to meeting the previously published IMACS DOI (15) (25 subjects met this criterion). Comparison of the two treatment groups using these two metrics also did not result in significant differences (P = 0.98 and P = 0.97, respectively). Using the aforementioned criteria for disease worsening, two patients in the placebo group and four subjects in the treatment group satisfied this criterion, and there was no significant difference in the time to worsening between the two treatment groups (P = 0.55).
Table 2.
Change in core set measures from baseline to week 24
| Core set measures | Tocilizumab | Placebo | |||
|---|---|---|---|---|---|
| Baseline | Week 24 | Baseline | Week 24 | ||
| (average) | (average) | (average) | (average) | P value a | |
| Extramuscular global assessment (MDAAT) | 3.3 | 3.0 | 3.3 | 1.9 | 0.49 |
| Physician global disease activity | 5.1 | 3.9 | 4.7 | 2.3 | 0.19 |
| Muscle enzyme (xULN b ) | 9.1 | 3.5 | 2.5 | 2.8 | 0.22 |
| Patient global disease activity | 6.1 | 4.2 | 5.4 | 4.1 | 0.07 |
| MMT‐8 | 126.7 | 135.1 | 132.8 | 139.7 | 0.82 |
| HAQ DI | 1.3 | 1.0 | 1.3 | 1.0 | 0.60 |
Abbreviations: HAQ DI, Health Assessment Questionnaire Disability Index; MDAAT, Myositis Disease Activity Assessment Tool; MMT‐8, manual muscle testing.
GEE model P values, for comparing the overtime changes (4‐24 weeks) in each core set measure between the two treatment groups.
Times upper limit normal.
The number of subjects randomized to placebo having no improvement, minimal improvement, moderate improvement, and major improvement at their last visit (24 weeks or earlier) was 3 (16.7%), 7 (38.9%), 6 (33.3%), and 2 (11.1%). This compares to 8 (44.4%), 3 (16.7%), 4 (22.2%), and 3 (16.7%) in those randomized to tocilizumab (P = 0.22).
Thirty‐three subjects had information on glucocorticoid use at both baseline and the 6‐month follow‐up visit. Six subjects receiving tocilizumab and four receiving placebo were on no prednisone at study enrollment. The average change from baseline to week 24 in subjects randomized to tocilizumab was −1.0 mg/day whereas subjects randomized to placebo increased their daily prednisone dose by 7.2 mg/day. Due to the small sample sizes and the presence of outliers, the two groups were compared using the Wilcoxon test, and there was no significant change in the glucocorticoid dose (P = 0.40; Figure 3).
Figure 3.

Change in glucocorticoid dose between baseline and completion of the 24‐week treatment phase. TCZ, tocilizumab.
A Data Safety Monitoring Committee monitored adverse events, and there were 14 events in eight tocilizumab‐treated subjects (44.4%) and 9 events in six placebo‐treated subjects (33.3%). The proportion of patients in the two groups having an adverse event was not different (Fisher exact P = 0.73), and the average number of adverse events during the follow‐up period in the two groups was compared assuming a Poisson distribution with no significant difference (P = 0.40). There was only one serious adverse event (3%) in the trial occurring in a tocilizumab‐treated patient experiencing bilateral pulmonary emboli. Infection was the most common adverse event, occurring in eight tocilizumab‐treated patients and seven placebo‐treated subjects. However, tocilizumab was generally well tolerated, and only one subject stopped treatment early because of disease worsening.
DISCUSSION
In this phase 2B randomized, placebo‐controlled trial, there was no significant difference in the TIS during the 24‐week treatment phase between myositis patients randomized to either placebo or tocilizumab (P = 0.86). This conclusion was unaffected by statistical adjustment for myositis subset (PM vs. DM), the time of the follow‐up visit, or any of the baseline values of the six CSMs. Three secondary outcomes compared the time to improvement in the treatment groups using three different definitions: 1) a TIS metric of minimal (≥20), moderate (≥40), or major (≥60) improvement; 2) a TIS meeting minimal improvement on two consecutive visits; and 3) the previously published IMACS criteria for improvement (15). As with the primary outcome measure, there were no significant differences in the time to improvement for any of these various definitions. Furthermore, when the two treatment groups were compared by classifying patients on their last follow‐up visit into the categories of no, minimal, moderate, and major improvement, the differences between myositis subjects randomized to tocilizumab or placebo were not statistically significant (P = 0.22). The proportion of patients having minimal improvement across the six follow‐up visits was also not significant. Only two subjects randomized to placebo and four randomized to tocilizumab worsened during the trial. There was no significant difference in glucocorticoid dosage (ie, steroid‐sparing effect) from trial initiation to conclusion between treatment groups, nor were the adverse events proportionally different.
This study used recently published criteria for determining the response to treatment in patients with PM and DM (2) and allowed the opportunity to compare these findings with a previously published DOI (15). The TIS is the metric being used in recent and most ongoing myositis clinical trials and was the endpoint used in the ProDERM (Progress in DERMatomyositis) study, which led to FDA approval of IVIg in DM in 2021. There was no difference in the data analysis and outcome of this study using the newer DOI supporting the more recently reported TIS as a reasonable metric for use in subsequent myositis clinical trials. Many current and active myositis clinical trials are indeed comparing these two definitions of improvement, which should lead to additional data regarding the relative utility of different metrics of improvement.
Several factors may have contributed to the lack of efficacy of tocilizumab in either PM or DM. Preliminary data and animal models have suggested that targeting IL‐6 might be efficacious in myositis as serum IL‐6 levels, particularly in DM, correlated with disease activity (1). Furthermore, IL‐6 levels correlated with overexpression of the type I interferon signature lending support for the potential effectiveness of anti‐IL‐6 therapy in myositis. Despite these biologically plausible considerations, tocilizumab may simply be a poor therapeutic choice for inflammatory myopathy. Nevertheless, analyzing pre‐ and post‐protein and gene expression profiles on myositis subjects in this trial and correlating such findings with the prospectively collected clinical data could potentially shed light on the contribution of selected cytokines and altered gene expression to disease activity in PM or DM. An observable outcome in many rheumatologic trials is a high placebo effect, and this was again observed in our trial in which subjects in the placebo arm improved in much the same fashion as tocilizumab‐treated patients. In fact, the differences in the TIS between follow‐up visits in all subjects was highly significant (P < 0.001), and in patients randomized to either tocilizumab or placebo, the pattern of scores showed a statistically significant improving trend in the TIS over the 24‐week trial period (P < 0.02 for both arms). In the design of future myositis clinical trials, overcoming a strong placebo effect may require enrollment of patients with more substantial myositis disease activity, such that the magnitude of the treatment effect in the active arm is more likely. Of course, this still requires choosing a therapeutic agent that effectively mitigates disease activity in patients with myositis and that is supported by preliminary data demonstrating immunopathogenic relevance. The CSMs in myositis continue to be potentially problematic. Certainly, both the CSM and the definitions of improvement have been validated and agreed on by myositis experts (13, 14, 15, 17), but these include subjective metrics such as the physician and patient global activity scores, the disability index of the Health Assessment Questionnaire, and the assessment of extramuscular disease activity using the MDAAT. Even the MMT‐8, though quantitative, may be subject to investigator variability and patient effort. Moreover, muscle enzymes may be more helpful as a measure of disease activity and treatment response in some myositis subsets (eg, necrotizing myopathy and PM), but they may correlate poorly with disease activity—particularly in patients with DM in which the CK is lower, even with active disease. As more validated, quantitative measures of myositis disease activity—including cutaneous metrics (18), muscle strength assessment using dynamometry, and functional measures of muscle strength—are available to myositis investigators, our ability to more accurately assess change in clinical trials will presumably be achieved. Finally, muscle damage must always be addressed in the enrollment of myositis subjects because excessive damage may certainly mitigate the treatment effect. Although we used agreed on metrics of muscle damage to exclude subjects, future clinical trials could consider more objective measures such as muscle magnetic resonance imaging to address this troublesome factor.
In summary, tocilizumab was well tolerated but was not more effective than placebo in this phase 2B trial of patients with PM and DM based on a newly reported measure of clinical response. Future clinical trials in myositis should continue to incorporate previously validated CSMs but should consider studying newly validated objective metrics of myositis disease activity.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Oddis had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Oddis, Rockette, Lacomis, Aggarwal.
Acquisition of data
Oddis, Koontz, Venturupalli, Moghadam‐Kia, Crofford, Dimachkie, Ernste, Gazeley, Marder, Aggarwal.
Analysis and interpretation of data
Oddis, Rockette, Zhu, Koontz, Ascherman, Aggarwal.
Supporting information
Disclosureform
APPENDIX A: MMT‐8
APPENDIX B: Final myositis response criteria for minimal, moderate, and major improvement in adult dermatomyositis/polymyositis (DM/PM) and combined adult DM/PM and juvenile DM clinical trials and studies
This trial was registered with ClinicalTrials.gov on October 29, 2014, before any subject was enrolled (Registration Number for this trial is NCT 02043548).
Author disclosures are available at https://onlinelibrary.wiley.com/action/downloadSupplement?doi=10.1002%2Facr2.11493&file=acr211493‐sup‐0001‐Disclosureform.pdf.
REFERENCES
- 1. Oddis CV, Reed AM, Aggarwal R, Rider LG, Ascherman DP, Levesque MC, et al. Rituximab in the treatment of refractory adult and juvenile dermatomyositis and adult polymyositis: a randomized, placebo‐phase trial. Arthritis Rheum 2013;65:314–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Aggarwal R, Rider LG, Ruperto N, Bayat N, Erman B, Feldman BM, et al. 2016 American College of Rheumatology/European League against rheumatism criteria for minimal, moderate, and major clinical response in adult dermatomyositis and polymyositis: an international myositis assessment and clinical studies group/paediatric rheumatology international trials organisation collaborative initiative. Ann Rheum Dis 2017;69:898–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kaly L, Rosner I. Tocilizumab ‐ a novel therapy for non‐organ‐specific autoimmune diseases. Best Pract Res Clin Rheumatol 2012;26:157–65. [DOI] [PubMed] [Google Scholar]
- 4. Schoels MM, van der Heijde D, Breedveld FC, Burmester GR, Dougados M, Emery P, et al. Blocking the effects of interleukin‐6 in rheumatoid arthritis and other inflammatory rheumatic diseases: systematic literature review and meta‐analysis informing a consensus statement. Ann Rheum Dis 2013;72:583–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Smolen JS, Schoels MM, Nishimoto N, Breedveld FC, Burmester GR, Dougados M, et al. Consensus statement on blocking the effects of interleukin‐6 and in particular by interleukin‐6 receptor inhibition in rheumatoid arthritis and other inflammatory conditions. Ann Rheum Dis 2012;72:482–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Scuderi F, Mannella F, Marino M, Provenzano C, Bartoccioni E. IL‐6‐deficient mice show impaired inflammatory response in a model of myosin‐induced experimental myositis. J Neuroimmunol 2006;176:9–15. [DOI] [PubMed] [Google Scholar]
- 7. Sugihara T, Sekine C, Nakae T, Kohyama K, Harigai M, Iwakura Y, et al. A new murine model to define the critical pathologic and therapeutic mediators of polymyositis. Arthritis Rheum 2007;56:1304–14. [DOI] [PubMed] [Google Scholar]
- 8. Okiyama N, Sugihara T, Iwakura Y, Yokozeki H, Miyasaka N, Kohsaka H. Therapeutic effects of interleukin‐6 blockade in a murine model of polymyositis that does not require interleukin‐17A. Arthritis Rheum 2009;60:2505–12. [DOI] [PubMed] [Google Scholar]
- 9. Narazaki M, Hagihara K, Shima Y, Ogata A, Kishimoto T, Tanaka T. Therapeutic effect of tocilizumab on two patients with polymyositis. Rheumatology (Oxford) 2011;50:1344–6. [DOI] [PubMed] [Google Scholar]
- 10. Bilgic H, Ytterberg SR, Amin S, McNallan KT, Wilson JC, Koeuth T, et al. Interleukin‐6 and type I interferon‐regulated genes and chemokines mark disease activity in dermatomyositis. Arthritis Rheum 2009;60:3436–46. [DOI] [PubMed] [Google Scholar]
- 11. Bohan A, Peter JB, Bowman RL, Pearson CM. Computer‐assisted analysis of 153 patients with polymyositis and dermatomyositis. Medicine (Baltimore) 1977;56:255–86. [DOI] [PubMed] [Google Scholar]
- 12. Miller FW, Rider LG, Chung YL, Cooper R, Danko K, Farewell V, et al. Proposed preliminary core set measures for disease outcome assessment in adult and juvenile idiopathic inflammatory myopathies. Rheumatology (Oxford) 2001;40:1262–73. [DOI] [PubMed] [Google Scholar]
- 13. Sultan SM, Allen E, Oddis CV, Kiely P, Cooper RG, Lundberg IE, et al. Reliability and validity of the myositis disease activity assessment tool. Arthritis Rheum 2008;58:3593–9. [DOI] [PubMed] [Google Scholar]
- 14. Rider LG, Lachenbruch PA, Monroe JB, Ravelli A, Cabalar I, Feldman BM, et al. Damage extent and predictors in adult and juvenile dermatomyositis and polymyositis as determined with the myositis damage index. Arthritis Rheum 2009;60:3425–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rider LG, Giannini EH, Brunner HI, Ruperto N, James‐Newton L, Reed AM, et al. International consensus on preliminary definitions of improvement in adult and juvenile myositis. Arthritis Rheum 2004;50:2281–90. [DOI] [PubMed] [Google Scholar]
- 16. Oddis CV, Rider LG, Reed AM, Ruperto N, Brunner HI, Koneru B, et al. International consensus guidelines for trials of therapies in the idiopathic inflammatory myopathies. Arthritis Rheum 2005;52:2607–15. [DOI] [PubMed] [Google Scholar]
- 17. Rider LG, Koziol D, Giannini EH, Jain MS, Smith MR, Whitney‐Mahoney K, et al. Validation of manual muscle testing and a subset of eight muscles for adult and juvenile idiopathic inflammatory myopathies. Arthritis Care Res (Hoboken) 2010;62:465–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Concha JS, Pena S, Gaffney RG, Patel B, Tarazi M, Kushner CJ, et al. Developing classification criteria for skin‐predominant dermatomyositis: the Delphi process. Br J Dermatol 2020;182:410–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disclosureform
APPENDIX A: MMT‐8
APPENDIX B: Final myositis response criteria for minimal, moderate, and major improvement in adult dermatomyositis/polymyositis (DM/PM) and combined adult DM/PM and juvenile DM clinical trials and studies