

Abstract
Cancer patients often face severe organ toxicity caused by chemotherapy. Among these, chemotherapy‐induced hepatotoxicity and cardiotoxicity are the main causes of death of cancer patients. Chemotherapy‐induced cardiotoxicity even creates a new discipline termed “cardio‐oncology”. Therefore, relieving toxicities induced by chemotherapy has become a key issue for improving the survival and quality of life in cancer patients. In this work, mesenchymal stem cell exosomes with the “G‐C” abundant tetrahedral DNA nanostructure (TDN) are modified to form a decoy exosome (Exo‐TDN). Exo‐TDN reduces DOX‐induced hepatotoxicity as the “G‐C” base pairs scavenge DOX. Furthermore, Exo‐TDN with cardiomyopathic peptide (Exo‐TDN‐PCM) is engineered for specific targeting to cardiomyocytes. Injection of Exo‐TDN‐PCM significantly reduces DOX‐induced cardiotoxicity. Interestingly, Exo‐TDN‐PCM can also promote macrophage polarization into the M2 type for tissue repair. In addition, those decoy exosomes do not affect the anticancer effects of DOX. This decoy exosome strategy serves as a promising therapy to reduce chemo‐induced toxicity.
Keywords: cardio‐oncology, cardiotoxicity, chemotherapy, exosome, tetrahedral DNA nanostructure
Chemotherapy is effective but can induce severe liver damage and cardiotoxicity. A new discipline called “cardio‐oncology” is developed rapidly to address those concerns. In this work, exosomes are used as a carrier for tetrahedral DNA nanostructure (TDN) to form a decoy exosome (Exo‐TDN) that can absorb excess DOX and mitigate cardiac and liver toxicity caused by DOX therapy.

1. Introduction
Chemotherapy is still one of the most commonly used cancer therapies providing beneficial outcomes to patients. The remarkable progress of chemotherapy has enabled the prolonged survival time of cancer patients, which has led to the definition of cancer as a chronic disease.[ 1 ] However, due to the nonspecific distribution of chemotherapeutic drugs, the organ toxicity of anticancer drugs has become a primary cause of treatment‐related deaths of cancer survivors.[ 2 ] Liver plays a central role in drug metabolism and clearance. Studies have shown that >60% of chemotherapeutic drugs concentrate in the liver area, causing severe liver toxicity. Drug‐induced liver injury (DILI) is the main cause of acute liver failure, and is also a common cause of the termination of clinical trials.[ 3 ] Researchers have designed nanoparticle drug delivery systems to increase the accumulation of chemotherapeutic drugs in tumor tissues to reduce their side effects. Half of such nano‐drugs, however, are still cleared by the reticuloendothelial system through the circulation, and thus accumulate in liver and cause serious hepatotoxicity.[ 4 ] In addition, many chemotherapeutic drugs cause specific organ damage. For example, doxorubicin (DOX) is a chemotherapeutic drug that has strong cardiotoxicity.[ 5 ] Studies have shown that >400 mg m−2 dose of DOX would induce heart failure.[ 6 ] In addition to DOX, other anticancer drugs such as alkylating agents (cyclophosphamide, cisplatin, etc.) and antimetabolic drugs (fluorouracil) are also widely recognized to induce a strong cardiotoxicity. Moreover, owing to the extensive attention to chemotherapy‐induced cardiotoxicity, a new discipline called “cardio‐oncology” has emerged since the late 1990s owing to the high morbidity and mortality in the oncologic population. As such, reducing the toxicity of major organs throughout the treatment of cancer patients has become a key issue for improving the survival and quality of life of cancer patients.
Exosomes are sac‐like vesicles secreted by living cells that contain the same genetic material as parental cells, such as proteins, DNAs, and RNAs.[ 7 , 8 , 9 , 10 ] Studies have shown that exosomes derived from stem cells have the same repairing functions for damaged tissue.[ 11 , 12 , 13 ] By analyzing the composition of exosomes, studies have found that a variety of miRNA strands in exosomes, such as miR‐223, miR‐21, and miR‐210, can inhibit apoptosis.[ 14 , 15 , 16 ] Furthermore, stem cell derived exosomes have been reported to inhibit inflammation by polarizing M1‐type macrophages into the M2 type.[ 17 , 18 ] In this work, we used mesenchymal stem cell exosomes as a repairing carrier in combination with the “G‐C” abundant DNA nanostructure to synthesize a “detoxification repair agent”. GC‐rich DNA nanostructure has been widely used for DOX delivery systems owing to its high‐binding efficiency. Particularly, tetrahedral DNA with more G‐C sequences is more stable than bare double‐stranded DNA (dsDNA).[ 3 , 19 ] Therefore, our Exo‐TDN enters liver tissue through metabolic routes, thereby relieving DOX‐induced liver toxicity. In addition to protecting the liver from damage, the liver detoxicant agent would also promote the repair of a damaged liver owing to the presence of exosome carriers.[ 20 , 21 ] Importantly, the detoxification repair agent has been used to relieve cardiotoxicity by engineering myocardial targeted peptides on TDN. Exosomes from mesenchymal stem cells have also been shown to play an anti‐inflammatory role by inducing macrophage polarization, thereby promoting healing of damaged heart tissue.[ 22 , 23 ] In addition, by changing the target peptide, the system can be promoted to reduce the toxicity to multiple organs caused by DOX. The development of detoxification repair agents reduce the organ toxicity caused by chemotherapy and improve the survival time of cancer patients, which thus gives them important research significance (Scheme 1 ).
Scheme 1.

Schematic showing the synthesis of tetrahedral DNA with/without myocardial targeted peptides engineered exosomes (Exo‐TDN/Exo‐TDN‐PCM) and their detoxifying ability to DOX‐induced hepatotoxicity and cardiotoxicity.
2. Results
2.1. The Construction of Exo‐TDN
The detoxification repair agent is composed of two parts: mesenchymal stem cell exosomes and TDN. We first successfully extracted and identified bone marrow mesenchymal stem cells (BMSCs) (Figure S1, Supporting Information). The exosomes were then isolated and purified from the medium of the BMSCs. Transmission electron microscopy (TEM) and nanosight analysis (NTA) results showed that the exosomes were spherical vesicles with a particle size of 100 nm (Figure 1A; Figure S2, Supporting Information). Meanwhile, western blot results confirmed the presence of specific proteins TSG101, CD81, and CD63, indicating that the extracted vesicles belonged to the exosome (Figure 1B). Next, we synthesized cholesterol‐modified TDNs and grafted them onto the exosomes. Agarose gel electrophoresis showed the successful synthesis of TDN (Figure 1C). Dynamic light scattering (DLS) and TEM results showed that the particle size of the TDN was ≈10 nm (Figures S3 and S4, Supporting Information). Atomic Force Microscopy (AFM) results showed that the height of the TDN was ≈2.5 nm (Figure 1D). As shown in Figure S5 (Supporting Information), even after adding TDN to a medium containing 10% serum, the agarose gel band was still present after 24 h, indicating the stability of TDN. The TDN was then modified on the surface of the exosome (Exo‐TDN). Exo did not generate a band in the agarose electrophoresis experiment, while TDN did. Thus, the agarose electrophoresis result could demonstrate successful loading of TDN (Figure 1E). The difference in the migration rates of TDN and Exo‐TDN was mainly due to the slow migration of exosomes with large molecular weights. Furthermore, immunogold staining followed by TEM imaging provided direct visual evidence that TDN was present on the Exo (Figure 1F). NTA and zeta potential analysis also proved the successful modification of TDN on exosomes (Figure 1G,H). When Exo and TDN reacted with a mass ratio of 1:3, the grafting efficiency of TDN reached the maximum rate of 66.2% (Figure 1I).
Figure 1.

Characterization of Exo‐TDN. A) TEM picture of exosomes. Scale bar, 100 nm. B) Western blot analysis of exosomes. CD81, CD63, and TSG101 were exosome markers. C) Agarose electrophoresis of TDN (T1: S1; T2: S1+S2; T3: T2+S3; T4: T3+S4). D) AFM image of TDN and height distribution histograms. E) Agarose electrophoresis analysis of the synthesis of Exo‐TDN. F) TEM image of Exo‐TDN with immunogold labeling of TDN. Scale bar, 100 nm. G,H) NTA (G) and Zeta potential (H) analysis of TDN, Exo, and Exo‐TDN. I) The grafting efficiency of TDN on the exosome (n = 3). J) The fluorescence sp of 1 µmol L−1 DOX solution after adding different amounts of Exo‐TDN. K) The capture efficiency of DOX by Exo‐TDN (n = 3). Data are presented as the mean ± SD. *p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001.
2.2. Exo‐TDN Captured DOX and Protected Cells
To prove the detoxification effect of Exo‐TDN, the capture efficiency of Exo‐TDN to DOX was first studied. According to previous reports, DOX is embedded in a “G‐C” base pair of double‐stranded DNA that quenches its fluorescence. As shown in Figure 1J, the introduction of Exo‐TDN significantly reduced the fluorescence intensity of DOX and the capture efficiency of Exo‐TDN reached up to 80% (Figure 1K). Next, we used the MTT method to verify the killing effect of DOX on cancer cells and their toxicity to normal cells. The results indicated that DOX showed significant proliferation inhibition effects on BRL‐3A cells (Figure 2A). Next, we verified the biocompatibility of Exo and TDN. The result showed that Exo exhibited a certain promotional effect on cell proliferation (Figure 2A). Meanwhile, various concentrations of TDN showed no significant toxic effect on cells (Figure 2A). However, before administering chemotherapy drugs, the co‐incubation of cells with TDN, Exo and Exo‐TDN effectively reduced the toxicity of DOX (Figure 2A; Figure S6, Supporting Information). The LIVE–DEAD cell‐staining results also proved that the introduction of Exo‐TDN effectively inhibited apoptosis caused by DOX (Figure 2B; Figure S7, Supporting Information). To further explore the mechanism of detoxification, we observed the distribution of Exo‐TDN and DOX within cells through a confocal laser microscope (CLSM). The CLSM and flow cytometry (FC) results showed that Exo‐TDN was taken up by BRL‐3A cells and stably present in the cytoplasm (Figure 2C; Figure S8, Supporting Information). It is known that DOX inhibits cell replication and induces apoptosis by entering the nucleus and binding to DNA. The CLSM results confirmed the enrichment of free DOX in the nucleus (Figure 2D). However, Exo‐TDN in cytoplasm efficiently bound to DOX and prevented it from entering the nucleus, thereby further protecting cells from DOX damage (Figure 2D).
Figure 2.

Exo‐TDN inhibited DOX‐induced apoptosis. A) The cell survival rates of BRL‐3A cell when treated with different concentrations of DOX, TDN, Exo, and Exo‐TDN (n = 3). B) LIVE–DEAD cell‐staining of BRL‐3A cell after treating with DOX, Exo+DOX, TDN+DOX, and Exo‐TDN +DOX (Blue: Live, Green: Dead). Scale bars, 100 µm. C) CLSM imaging of BRL‐3A cell uptake Exo‐TDN, and fluorescence quantification of Exo‐TDN in cytoplasm (n = 3). Scale bars, 50 µm. D) CLSM imaging of the inhibition of DOX from entering the nucleus by Exo‐TDN, as well as fluorescence quantification of DOX in the nucleus (n = 3). Scale bars, 50 µm. Data are presented as the mean ± SD. *p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001.
2.3. Anti‐Inflammatory Effects of Exo‐TDN
Since eliminating inflammation promotes healing of damaged tissue, we further studied the anti‐inflammatory effects of Exo‐TDN. CLSM and FC results showed that Exo‐TDN was taken up by macrophages (Figure 3A,B). Using CLSM and FC analysis, we evaluated the expression of two macrophage markers, CD80 (M1 marker) and CD206 (M2 marker), after incubation with exosomes. Compared to the control group or the LPS‐treated group, the Exo‐TDN group showed a lower expression of CD80 and higher expression of CD206 after 24 h treatment (Figure 3C–E). In addition, we analyzed the expression of intracellular proteins iNOS and Arg‐1 in macrophages. The western blot results showed higher expression of iNOS but lower expression of Arg‐1 compared to the control group, indicating that Exo‐TDN polarized M0 or M1 macrophages to the M2 phenotype (Figure 3F,G). These results confirmed that exosomes played an anti‐inflammatory role, which would further promote the repair of damaged tissue.
Figure 3.

Exo‐TDN polarized macrophages to the M2 phenotype. A) CLSM imaging and B) FC results of RAW 264.7 cell uptake of Exo‐TDN. Scale bars, 20 µm. C,D) FC results of the expression of CD80 (C) and CD206 (D) in RAW 264.7 cells. E) CLSM imaging and fluorescence quantification of CD80 and CD206 expression in RAW 264.7 cells (n = 3). Scale bars, 20 µm. F) Western blot results of the expression of Arg‐1 and iNOS in RAW 264.7 cells. G) Grayscale statistics for different protein bands (n = 3). Data are presented as the mean ± SD. *p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001.
2.4. The Biodistribution of Exo‐TDN or Exo‐TDN‐PCM In Vivo
By grafting the target peptides on the TDN, the multi‐organ applicability of the detoxification repair agent can be expanded. We first synthesized a maleimide‐including TDN and engineered it on the Exo surface (Exo‐TDN‐MAL). Cysteine‐modified cardiomyopathic peptide PCM (Cys‐PCM) was grafted on the surface of Exo‐TDN‐MAL through a click reaction, generating the heart detoxification repair agent of Exo‐TDN‐PCM (Figure 4A). As shown in Figure 4B, the nano‐flow cytometry result showed that Cys‐PCM‐FITC was successfully grafted on Exo‐TDN. Furthermore, modifying the PCM did not affect capture efficiency of Exo‐TDN to DOX, which prevented it from entering the nucleus (Figure S9, Supporting Information). Next, we i.v. injected DiD (1,1“‐Dioctadecyl‐3,3,3”,3'‐Tetramethylindodicarbocyanine,4‐Chlorobenzenesulfonate Salt)‐labeled Exo‐TDN or Exo‐TDN‐PCM, and then use the small animal in vivo imaging system (IVIS) to verify their targeting capabilities (Figure 4C). As shown in Figure 4D, 4 h post‐injection, Exo‐TDN was completely gathered in the liver area, while Exo‐TDN‐PCM was partially gathered in the myocardial area. We also observed the corresponding tissues using CLSM to provide additional evidence for the distribution of Exo‐TDN and Exo‐TDN‐PCM (Figure 4E; Figure S10, Supporting Information). Exo‐TDN or Exo‐TDN‐PCM was still entirely accumulated in the liver or heart area 24 h post‐injection. They mainly failed to reach the tumor area, thus indicating that they would not adsorb DOX in tumor tissue and would have no effect on the tumor‐killing effect of DOX (Figure 4F; Figure S11, Supporting Information). Furthermore, the results of Figure 4F also demonstrated that modifying PCM could significantly increase the enrichment of Exo‐TDN‐PCM to the heart.
Figure 4.
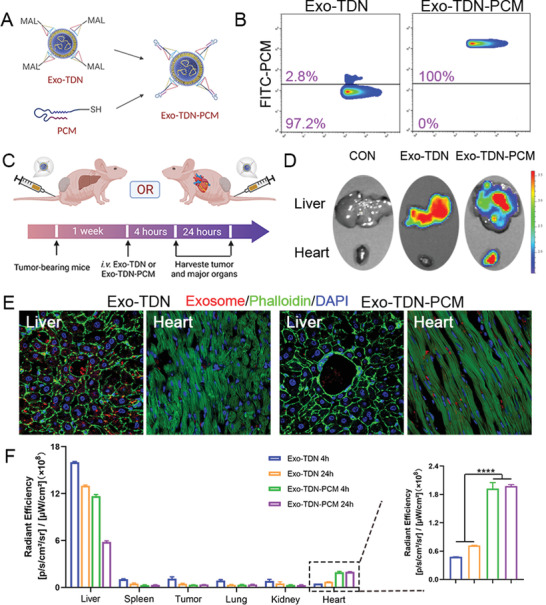
The biodistribution of Exo‐TDN or Exo‐TDN‐PCM in tumor‐bearing mice. A) Schematic of the synthesis of Exo‐TDN‐PCM. B) Nano‐flow cytometry results of Exo‐TDN and Exo‐TDN‐PCM. C) Schematic of the in vivo distribution experiment to test targeting capabilities of Exo‐TDN or Exo‐TDN‐PCM. D) Ex vivo fluorescence imaging to detect the biodistribution of DiD‐labeled Exo‐TDN and Exo‐TDN‐PCM after 4 h of in vivo injection. E) CLSM images showing the distribution of DiD‐labeled Exo‐TDN and Exo‐TDN‐PCM. F) Quantitative analysis of fluorescent intensity in the different tissues after injection of Exo‐TDN or Exo‐TDN‐PCM for 4 and 24 h (n = 3). Data are presented as the mean ± SD. *p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001.
2.5. Study of Liver Protection by Exo‐TDN In Vivo
To further demonstrate the detoxification effect of Exo‐TDN, the MCF‐7 tumor‐bearing mice model was constructed and subsequently chemotherapy was performed (Figure 5A). After 21 days of treatment, we collected blood from different groups and conducted blood biochemical tests. The results showed that enzymes associated with liver damage, such as ALP, AST, and ALT, were significantly reduced (Figure 5B; Table S1, Supporting Information). Subsequently, we collected liver tissue from mice and performed pathological analysis. H&E and Masson staining results showed that DOX‐induced liver damage was significantly reduced in the Exo‐TDN group compared to the control group and other therapeutic groups (Figures S12 and S13, Supporting Information). Furthermore, we also performed immunohistochemical (IHC) analysis to study the liver‐protecting effect (Figure 5C). TUNEL staining results showed that Exo‐TDN significantly reduced DOX‐induced liver cell apoptosis compared to other groups (Figure 5D; Figure S14, Supporting Information). Furthermore, the liver protection mechanism of Exo‐TDN was verified by analyzing apoptosis‐related proteins Casp3 and Bcl‐2. IHC results showed that Exo‐TDN effectively attenuated the expression of apoptosis protein Casp3 and increased the expression of the anti‐apoptotic protein Bcl‐2, thus reducing DOX‐induced hepatocellular apoptosis, which was consistent with our in vitro results described above (Figure 5E,F; Figure S14, Supporting Information).
Figure 5.

Study of liver protection effect of Exo‐TDN. A) Schematic of the overall design of the animal experiments to test the liver protection of Exo‐TDN. B) Blood biochemical analysis of mice after 21 days of treatment (n = 4). C) TUNEL and apoptosis‐related proteins staining analysis of liver tissue in the treatment of different groups. Scale bars, 100 µm. D–F) Quantitative analysis of TUNEL staining images (D), Casp3 staining images (E), and Bcl‐2 staining images (F) (n = 3). G) Tumor volume changes of mice in different treatment groups (n = 8). H) Quantitative analysis of TUNEL staining images of tumor tissue in the treatment of different groups (n = 3). Data are presented as the mean ± SD. *p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001.
It was important to study if our Exo‐TDN had an effect on DOX‐induced cancer therapy. We verified the effect of Exo‐TDN on DOX‐induced tumor growth inhibition. The results of tumor volume change showed that the injection of DOX effectively inhibited tumor growth compared to the control group (Figure 5G; Figure S15, Supporting Information). After 21 days of treatment, we collected tumor tissue and performed pathological analysis. The results showed that DOX effectively promoted tumor apoptosis, indicating that the introduction of Exo, TDN, or Exo‐TDN did not affect the killing effect of DOX (Figure 5H; Figure S16, Supporting Information). In addition, we monitored and recorded the weight of the mice. Compared to the DOX, Exo+DOX, and TDN+DOX groups, the Exo‐TDN+DOX group showed a slow increase in mouse weight (Figure S17, Supporting Information). This result proved that the introduction of Exo‐TDN reduced the toxic effects of chemotherapy drugs.
2.6. Study of Heart Protection of Exo‐TDN‐PCM
After successfully demonstrating the liver‐protecting effect of Exo‐TDN and its slight effect on chemotherapy, we next broadened their application in relieving DOX‐induced cardiotoxicity by engineering Exo‐TDN with a myocardium‐targeting peptide (Figure 6A). DOX‐induced cardiotoxicity has been widely known to result in the death of cancer patients. We first detected the serum markers of myocardial injury after treatment. We collected blood from different groups and detected the content of BNP and cTnI in serum. The results showed that the content of BNP and cTnI in the Exo‐TDN‐PCM group was significantly reduced after 42 days of treatment, indicating the reduction of DOX‐induced cardiotoxicity (Figure 6B). Subsequently, we collected heart tissue and performed pathological analysis. Both Masson's trichrome staining and H&E staining results showed that the damage to heart tissue caused by DOX was significantly attenuated in the Exo‐TDN‐PCM group compared to the control group and other therapeutic groups (Figure 6C; Figures S18 and S19, Supporting Information). In addition, TUNEL staining results showed that Exo‐TDN‐PCM could effectively mitigate the DOX‐caused heart toxicity (Figure 6D; Figure S20, Supporting Information).
Figure 6.

The study of heart protection of Exo‐TDN‐PCM. A) Schematic of the overall design of the animal experiments to test the heart protection of Exo‐TDN‐PCM. B) The content of BNP and cTnI in serum after 42 days of treatment (n = 3). C) Masson staining images of heart tissue in different treatment groups. Scale bars, 100 µm. D) Quantitative analysis of TUNEL staining images (n = 3). E) Quantitative analysis of immunohistochemical staining images of heart tissue showing, Casp3, Bcl‐2 expression (n = 3). Data are presented as the mean ± SD. *p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001.
2.7. Heart Protection Mechanism of Exo‐TDN‐PCM
Furthermore, the heart protection mechanism of Exo‐TDN‐PCM was verified. Immunostaining of apoptosis‐related proteins showed that Exo‐TDN‐PCM effectively reduced myocardial apoptosis, thereby reducing DOX‐induced myocardial damage (Figure 6E; Figure S21, Supporting Information). Subsequently, we evaluated the ability of Exo‐TDN‐PCM to promote myocardial tissue repair. After injection of Exo‐TDN‐PCM, the number of Ki67‐positive cells was significantly improved in myocardial tissue (Figure 7A,B). In addition, we analyzed the infiltration and phenotype of macrophages in myocardial tissue by immunohistochemical staining. The results showed lower expression of CD80 and higher expression of CD206 in the Exo‐TDN‐PCM group (Figure 7C; Figure S22, Supporting Information). The results indicated that Exo‐TDN‐PCM could effectively reduce inflammation of myocardial tissue by promoting the M1 to M2 transition of macrophages. The reduction of inflammation could further promote the repair of myocardial tissue. In line with the morphological benefits, injection of Exo‐TDN‐PCM augmented left ventricular ejection fraction (LVEF) and fractional shortening (FS) (Figure 7D). In short, Exo‐TDN‐PCM could effectively reduce myocardial apoptosis, as well as relieve inflammation of myocardial tissue and further promote tissue repair, thereby reducing DOX‐induced cardiomyopathy (Figure 7E). We then verified the effect of Exo‐TDN‐PCM on DOX‐induced tumor growth inhibition. The results of tumor volume change and TUNEL staining showed that the injection of DOX effectively inhibited tumor growth compared to the control group (Figure 7F–H; Figure S23, Supporting Information).
Figure 7.

The study of heart protection mechanisms of Exo‐TDN‐PCM. A) CLSM images of Ki67 staining of heart sections 42 days after injection. Scale bars, 100 µm. B) Quantitative data corresponding to Ki67 staining (n = 3). n per HPF is a unit of fluorescence brightness. n refers to the quantified fluorescence intensity. HPF refers to the high power field. C) Histochemistry score of immunohistochemical staining images of CD80 and CD206 (n = 3). D) Left ventricular ejection fraction (LVEF) and fractional shortening (FS) 42 days after treatment (n = 5). E) Schematic of the heart protection mechanism of Exo‐TDN‐PCM. F) Representative photographs of tumor after 22 days of treatment. G) Tumor volume changes of mice in different treatment groups (n = 6). H) Histochemistry score of TUNEL staining images of tumor tissue in the treatment of different groups (n = 3). Data are presented as the mean ± SD. *p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001.
3. Conclusion
In summary, we synthesized Exo‐TDN for detoxification and repair of major organs. We first extracted rat BMSCs and characterized them by FC and differentiation induction. We then extracted BMSC exosomes and functionalized them with tetrahedral DNA abundant in G‐C bases. FC and CLSM results proved that Exo‐TDN could indeed capture DOX in cells and prevented it from entering the nucleus. In this work, organ damage caused by DOX during cancer treatment was reversed by synthesis of tetrahedral DNA engineered exosomes. Tumor‐bearing mice were first protected by our Exo‐TDN before administrating DOX for chemotherapy. Our in vivo experiments showed that Exo‐TDN was effectively enriched in liver tissue and significantly reduced DOX‐induced liver damage while having only a slight effect on DOX‐induced chemotherapy. Furthermore, chemotherapy‐related cardiac dysfunction is known as one of the most notorious short‐term side effects of anticancer treatment. To relieve DOX‐induced cardiotoxicity, we further structured a detoxification repair agent to attenuate cardiotoxicity by modifying heart‐targeting peptides on the surface of Exo‐TDN. Exo‐TDN‐PCM had been shown to significantly reduce DOX‐induced cardiotoxicity through anti‐apoptosis effects. Moreover, as exosomes have been widely used for injured tissue repair, we have demonstrated that our Exo‐TDN‐PCM showed enhanced cardiomyocyte proliferation and anti‐inflammatory effects. Therefore, we believe that Exo‐TDN can be used to detoxify and repair multiple organs, and thus improve the survival time of cancer patients.
4. Experimental Section
Isolation of Bone Marrow Mesenchymal Stem Cells (BMSCs)
Three‐week‐old rats were sacrificed by dislocation; the femur and tibia were taken out using forceps and scissors, after which they were placed in PBS containing 1% penicillin and streptomycin. The epiphyses at both ends of the femur and tibia were excised in PBS to completely allow leaking from the bone cavity, after which they were placed in DMEM containing 10% FBS and 1% penicillin and streptomycin. A 1 mL syringe was used to flush out the bone marrow with culture medium. The bone marrow was then transferred to a culture flask and placed in an incubator at 37 °C containing 5% CO2. After 3 days of cultivation, the medium was changed. The cells obtained at this time were P0 generation cells.[ 24 ] Every 2 days, the medium was discarded and the cells were washed with PBS and fresh medium was added. After the cell confluence reached 80–90%, they were digested with trypsin and then sub‐cultured. P3 generation cells were selected to identify mesenchymal stem cells, and P3‐P5 generation BMSCs were selected to extract exosomes.
Exosome Extraction
When the confluence of P3‐P5 BMSCs reached 70–80%, the medium was removed and replaced with a serum‐free medium. After culturing for 12 h, the supernatant was collected and centrifuged at 10000 rpm for 30 min. Cell debris was then removed, after which a 0.22 µm filter was used to remove large, extracellular vesicles. Then, the steps in the exosome extraction kit instructions were followed: 1/3 of the volume of the cell culture supernatant exosome extraction reagent was added, followed by 4 °C incubation overnight and then centrifuging at 10 000 × g for 30 min at 4 °C. Finally, the resulting pellet was obtained, resuspended in PBS, and stored at −80 °C.[ 25 ]
The Synthesis of DNA with Different Structures
S1‐cho‐two strands (S1‐cho and S2‐cho), three strands (S1‐cho, S2‐cho and S3‐cho), and four strands (S1‐cho, S2‐cho, S3‐cho and S4) in equimolar amounts (for a final concentration of 2 × 10−6 m) was mixed in a buffer (20 × 10−3 m Tris and 16 × 10−3 m MgCl2 at a pH of 8.0) then kept at 95 °C for 10 min, after which it was slowly cooled to 4 °C. Different structures of DNA were obtained, in which equimolar amounts of four‐stranded strands were reacted to obtain TDN.[ 26 ] The sequence of each single strand of DNA is shown in Table 1.
Table 1.
The sequence of each single strand of DNA
| ssDNA | Sequence (5“‐3”) |
|---|---|
| S1 | GAGCGTTAGCCACACACACAGTC |
| S2 | TTAGGCGAGTGTGGCAGAGGTGT |
| S3 | CGCCTAAACAAGTGGAGACTGTG |
| S4 | AACGCTCACCACTTGAACACCTC |
The Stability of Tetrahedral DNA
To verify the stability of the tetrahedral DNA, synthetic tetrahedral DNA was added to 1640 medium (10% FBS) and incubated at 37 °C multiple times. Then, 10 µL was taken, into which 2 µL of 6 × DNA loading buffer was added. The samples were mixed well and then loaded into the gel well, where they were run at 90 V for 30 min. Then, a gel imager was used to take pictures.
The Synthesis of Exo‐TDN
A total of 10 µg of exosomes was taken and dispersed into PBS, after which 30 µg of the aforementioned synthetic tetrahedral DNA was added. The samples were incubated at 37 °C for 1 h and then quickly put on ice and shaken for 1 h. Then, a 100 KDa ultrafiltration tube was used to centrifuge the samples at 4500 × g for 15 min to remove excess tetrahedral DNA, after which exosomes engineered with tetrahedral DNA were obtained.[ 27 ]
Exo‐TDN Captures DOX In Vitro
To verify the ability of Exo‐TDN to capture DOX, 1 × 10−6 m DOX was incubated with increasing concentrations of Exo‐TDN at 37 °C for 1 h, after which the fluorescence change at a wavelength of 480 nm was measured under maximum excitation. The captured DOX was then calculated from the standard DOX curve.
The Synthesis of Exo‐TDN‐PCM
First, a maleimide‐including TDN synthesized and engineered on the Exo surface (Exo‐TDN‐MAL). An appropriate amount of cysteine‐modified cardiomyopathic peptide PCM was added to the Exo‐TDN‐MAL solution. After continuous oscillation at 4 °C for 12 h, the heart detoxification repair agent EXO‐TDN‐PCM was obtained.
According to the previous method,[ 26 , 27 , 28 , 29 ] Exo‐PCM acted as a control group. An appropriate amount of DSPE‐PEG‐MAL was added to the Cys‐PCM solution. After continuous oscillation at 4 °C for 24 h, the DSPE‐modified PCM (DSPE‐PEG‐PCM) was obtained. Subsequently, an appropriate amount of DSPE‐PEG‐PCM was added to the Exo solution. After continuous oscillation at 4 °C for 4 h, the PCM‐modified Exo (Exo‐PCM) was obtained.
Cell Protection of Exo‐TDN
MTT methods were used to evaluate the protective effect of the materials on the cells. In a 96‐well plate, BRL‐3A cells were seeded at a density of 1 × 103–2 × 103 cells per well. They were then incubated overnight to make the cells adhere to the well. Different concentrations of TDN and Exo were added to the cell plate. After 4 h, DOX was added to make the final concentration 0.2–1 × 10−6 m. MTT was added after 24 h of incubation in an incubator at 37 °C and 5% CO2. Absorbance was then detected by a microplate reader, after which the survival rates of different groups of cells were calculated.
In addition, LIVE–DEAD cell staining was used to analyze cell protection of Exo‐TDN. Cells were seeded in a 48‐well plate at a density of 1 × 104 cells per well and cultured overnight. Then, 50 µL of PBS, Exo (60 µg), TDN (60 µg), or Exo‐TDN (60 µg) were added for 4 h of incubation, after which 2 × 10−6 m of DOX was added, followed by another 24 h of incubation. Then, one drop of NucBlue Live reagent and NucGreen Dead reagent was added to each well, followed by incubation for 20 min. Finally, the dye was washed away with PBS, and the samples were transferred to a fluorescence microscope for observation.
Exo‐TDN Inhibited DOX from Entering Nuclei
The BRL‐3A cells were inoculated in a 30 mm glass‐bottom culture dish at a density of 1 × 104 cells per well. After the cells adhered to the bottom, 60 µg of Exo‐TDN was added, followed by 4 h of incubation. Then, 2 × 10−6 m of DOX was added, followed by another 6, 12, or 24 h of incubation. The medium was cultured and washed three times with PBS, after which the nuclear dye Hoechst 33 342 was added, followed by incubation for 10 min. This was followed by three washings with PBS. The samples were then observed via CLSM. Three parallel wells were set up at each time point. Three fields of view were selected for shooting in each well, and then the average of red fluorescence intensity of the nuclei in the three fields of view was calculated. Finally, the average of the three wells was counted to get a histogram.
Heart Protection Model
MCF‐7 cells were digested with trypsin, washed twice with PBS and resuspended with Matrigel in equal volume. The sample density was adjusted to 1 × 107 cell per mL, and 0.1 mL was inoculated under the skin of the right back of the nude mice. When the tumors reached ≈70–80 mm3, the mice were divided into six groups with six tumor‐bearing mice in each group. The experimental groups were the control group, the DOX group, the TDN+DOX group, the Exo‐PCM+DOX group, the Exo‐TDN+DOX group, and the Exo‐TDN‐PCM+DOX group. The control group was intravenously injected with 500 µL of PBS, and received a second injection of 100 µL of PBS 4 h later. The mice in the DOX group were treated with 500 µL of PBS, and 4 h later they were given 100 µL of DOX (5 mg kg−1) injected into the tail vein. In the TDN+DOX group, the mice were intravenously injected with 500 µL of PBS containing 20 µg of TDN, and then they received a second injection of 100 µL of DOX (5 mg kg−1) 4 h later. In the Exo‐PCM + DOX, Exo‐TDN + DOX, and Exo‐TDN‐PCM + DOX group, mice were injected intravenously with 500 µL of PBS containing 20 µg of Exo‐PCM, Exo‐TDN, or Exo‐TDN‐PCM, respectively, after which they received a second tail vein injection of 100 µL of DOX (5 mg kg−1) 4 h later.[ 30 ] This treatment was repeated every 3 days, and tumor sizes were measured with a caliper and mice were weighed. Cardiac function was measured by using the Vevo 2100 Imaging System. Both ejection fraction (EF) and fractional shortening (FS) were determined by measurements from the left ventricular internal diameter at end‐diastole (LVIDd) and systole (LVIDs). After 42 days, mouse myocardial tissue was collected and subjected to pathological and immunomic analysis. Subsequently, immunohistochemical images were quantified through Aipathwell. Histochemistry score (H‐Score) was used to represent semi‐quantitative results of tissue staining for each slice. H‐Score = ∑(pi × i) = (percentage of weak intensity × 1) + (percentage of moderate intensity × 2) + (percentage of strong intensity × 3).[ 31 , 32 ]
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Acknowledgements
M.F., H.L., and D.S. contributed equally to this work. This work was supported by the National Natural Science Foundation of China (21977024, 21601046, and 31971304), Science Fund for Creative Research Groups of Nature Science Foundation of Hebei Province (B2021201038), Guangdong Basic and Applied Basic Research Foundation (2021B1515120065), the Beijing‐Tianjin‐Hebei Basic Research Cooperation Project (19JCZDJC64100), Cross‐Disciplinary Project of Hebei University (DXK201916), One Hundred Talent Project of Hebei Province (E2018100002), the National Science Foundation of China (81873493 and 82170271), and Henan Talents Program (204200510017). The authors are grateful to the Medical Comprehensive Experimental Center of Hebei University for the animal experiments. All animal experiments were conducted according to the requirements of the Experimental Animal Ethics Committee of Hebei University (Approval No. 2018004).
Fan M., Li H., Shen D., Wang Z., Liu H., Zhu D., Wang Z., Li L., Popowski K. D., Ou C., Zhang K., Zhang J., Cheng K., Li Z., Decoy Exosomes Offer Protection Against Chemotherapy‐Induced Toxicity. Adv. Sci. 2022, 9, 2203505. 10.1002/advs.202203505
Contributor Information
Jinchao Zhang, Email: jczhang6970@163.com.
Ke Cheng, Email: ke_cheng@unc.edu.
Zhenhua Li, Email: zhenhuali@hbu.edu.cn.
Data Availability Statement
Research data are not shared.
References
- 1. Ward B. W., Schiller J. S., Goodman R. A., Prev. Chronic Dis. 2014, 11, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sung H., Ferlay J., Siegel R. L., Laversanne M., Soerjomataram I., Jemal A., Bray F., CA Cancer J. Clin. 2021, 71, 209. [DOI] [PubMed] [Google Scholar]
- 3. Zhao R., Liu X., Yang X., Jin B., Shao C., Kang W., Tang R., Adv. Mater. 2018, 30, 1801304. [DOI] [PubMed] [Google Scholar]
- 4. Kleiner D. E., Chalasani N. P., Lee W. M., Fontana R. J., Bonkovsky H. L., Watkins P. B., Hayashi P. H., Davern T. J., Navarro V., Reddy R., Talwalkar J. A., Stolz A., Gu J., Barnhart H., Hoofnagle J. H., Hepatology 2014, 59, 661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li L., Takemura G., Li Y., Miyata S., Esaki M., Okada H., Kanamori H., Khai N. C., Maruyama R., Ogino A., Minatoguchi S., Fujiwara T., Fujiwara H., Circulation 2006, 113, 535. [DOI] [PubMed] [Google Scholar]
- 6. Waterhouse D. N., Tardi P. G., Mayer L. D., Bally M. B., Drug Saf. 2001, 24, 903. [DOI] [PubMed] [Google Scholar]
- 7. Wang Z. Z., Popowski K. D., Zhu D. S., de Juan Abad B. L., Wang X. Y., Liu M. R., Lutz H., De Naeyer N., DeMarco C. T., Denny T. N., Dinh P. C., Li Z. H., Cheng K., Nat. Biomed. Eng. 2022, 6, 791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Popowski K. D., de Juan Abad B. L., George A., Silkstone D., Belcher E., Chung J., Ghodsi A., Lutz H., Davenport J., Flanagan M., Piedrahita J., Dinh P. C., Cheng K., Extracellular Vesicle 2022, 1, 100002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li Z. H., Wang Z. Z., Dinh P. C., Zhu D. S., Popowski K. D., Lutz H., Hu S. Q., Lewis M. G., Cook A., Andersen H., Greenhouse J., Pessaint L., Lobo L. J., Cheng K., Nat. Nanotechnol. 2021, 16, 942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wen Y., Fu Q., Soliwoda A., Zhang S., Zheng M. F., Mao W. J., Wan Y., Extracellular Vesicle 2022, 1, 100004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Molinaro R., Corbo C., Martinez J. O., Taraballi F., Evangelopoulos M., Minardi S., Yazdi I. K., Zhao P., De Rosa E., Sherman M. B., De Vita A., Toledano Furman N. E., Wang X., Parodi A., Tasciotti E., Nat. Mater. 2016, 15, 1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hu S., Li Z., Shen D., Zhu D., Huang K., Su T., Dinh P.‐U., Cores J., Cheng K., Nat. Biomed. Eng. 2021, 5, 1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu M., Lutz H., Zhu D., Huang K., Li Z., Dinh P. C., Gao J., Zhang Y., Cheng K., Adv. Sci. 2020, 8, 2002127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang Y., Li L., Yu J., Zhu D., Zhang Y., Li X., Gu H., Zhang C. Y., Zen K., Biomaterials 2014, 35, 4390. [DOI] [PubMed] [Google Scholar]
- 15. Alvarez‐Erviti L., Seow Y., Yin H., Betts C., Lakhal S., Wood M. J., Nat. Biotechnol. 2011, 29, 341. [DOI] [PubMed] [Google Scholar]
- 16. van den Boorn J. G., Dassler J., Coch C., Schlee M., Hartmann G., Adv. Drug Delivery Rev. 2013, 65, 331. [DOI] [PubMed] [Google Scholar]
- 17. Wynn T. A., Chawla A., Pollard J. W., Nature 2013, 496, 445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Davies L. C., Jenkins S. J., Allen J. E., Taylor P. R., Nat. Immunol. 2013, 14, 986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhao C., Song H., Scott P., Zhao A., Tateishi‐Karimata H., Sugimoto N., Ren J., Qu X., Angew. Chem. Int. Ed. Engl. 2018, 57, 15723. [DOI] [PubMed] [Google Scholar]
- 20. Hu S. Q., Li Z. H., Shen D. L., Zhu D. S., Huang K., Su T., Dinh P.‐U., Cores J., Cheng K., Nat. Biomed. Eng. 2021, 5, 1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yao J. L., Huang K., Zhu D. S., Chen T., Jiang Y. F., Zhang J. Y., Mi L. J., Xuan H., Hu S. Q., Li J. L., Zhou Y. F., Cheng K., ACS Nano 2021, 15, 11099. [DOI] [PubMed] [Google Scholar]
- 22. Su N., Hao Y. Y., Wang F., Hou W. D., Chen H. F., Luo Y., Sci. Adv. 2021, 7, 7207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhao J. X., Li X. L., Hu J. X., Chen F., Qiao S. H., Sun X., Gao L., Xie J., Xu B., Cardiovasc. Res. 2019, 115, 1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Soleimani M., Nadri S., Nat. Protoc. 2009, 4, 102. [DOI] [PubMed] [Google Scholar]
- 25. Cheng Q. Q., Shi X. J., Han M. L., Smbatyan G., Lenz H., Zhang Y., J. Am. Chem. Soc. 2018, 140, 16413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li J., Xun K. y., Pei K., Liu X. J., Peng X. Y., Du Y. L., Qiu L. P., Tan W. H., J. Am. Chem. Soc. 2019, 141, 18013. [DOI] [PubMed] [Google Scholar]
- 27. Zhuang J. L., Tan J. Z., Wu C. L., Zhang J., Liu T., Fan C. H., Li J. P., Zhang Y. Q., Nucleic Acids Res. 2020, 48, 8870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li Z. H., Hu S. Q., Huang K., Su T., Cores J., Cheng K., Sci. Adv. 2020, 6, 589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fan M., Liu H. F., Yan H. Y., Che R. J., Jin Y., Yang X. J., Zhou X. H., Yang H., Ge K., Liang X.‐J., Zhang J. C., Li Z. H., Biomaterials 2022, 282, 121424. [DOI] [PubMed] [Google Scholar]
- 30. Zhao R. B., Liu X. Y., Yang X. Y., Jin B., Shao C. Y., Kang W. J., Tang R. K., Adv. Mater. 2018, 30, 1801304. [DOI] [PubMed] [Google Scholar]
- 31. Guo R., Berry L. D., Aisner D. L., Sheren J., Boyle T., Bunn P. A. Jr., Johnson B. E., Kwiatkowski D. J., Drilon A., Sholl L. M., Kris M. G., J. Thorac. Oncol. 2019, 14, 1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Paschalis A., Sheehan B., Riisnaes R., Rodrigues D. N., B G., Bertan C., Ferreira A., Lambros M. B. K., Seed G., Yuan W., Dolling D., Welti J. C., Neeb A., Sumanasuriya S., Rescigno P., Bianchini D., Tunariu N., Carreira S., Sharp A., Oyen W., J. S de Bono. Eur. Urol. 2019, 76, 469. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
Research data are not shared.