

Abstract
Technologies for cysteine disulfide detection and conjugation are pivotal to understanding protein functions and developing disulfide-derived therapeutic agents. Currently, disulfide modification requires reductive cleavage prior to functionalization, posing challenges to differentiating disulfides from free thiols. We describe herein Redox-assisted Disulfide Direct Conjugation (RDDC) as a new method to enable disulfide rebridging without cross-reacting with free thiols.
Cysteine disulfide is a post-translational modification (PTM) that plays important structural and functional roles in proteins.1, 2 Its occurrence is tightly regulated by thiol-disulfide oxidoreductase and protein disulfide isomerases to keep most cysteines in the free sulfhydryl form in the cytoplasm.3, 4 Proteins containing regulatory disulfides can alter their functions or aggregation states in response to redox changes. Chemistry that detects and selectively labels cysteine disulfides is therefore pivotal to studying protein functions and developing disulfide-derived therapeutic agents. However, all current methods require reductive cleavage of the disulfides prior to conjugation (Fig. 1A). A three-step process that involves masking the free cysteines, converting the disulfides to free thiols, and labelling the released thiols by nucleophilic chemistry5–8 or cross-coupling9 is needed to differentiate disulfides from free cysteines (Fig. 1B). In addition to introducing operational complexity, it erases connectivity information of the protein. Furthermore, basic conditions are frequently used to ensure efficient nucleophilic conjugation. Whereas cysteine is generally more nucleophilic than lysine, significant lysine cross-reaction occurs at high pH. To overcome these issues, we took advantage of the weak oxidative power of disulfides (E0 0.25 V at pH 7)10 to develop Redox-assisted Disulfide Direct Conjugation (RDDC) as a simple method to modify disulfides in aqueous buffers under aerobic conditions (Fig. 1C). By retaining disulfide connectivity, it complements the existing disulfide rebridging methods,11–19 and may find use in peptide stapling20, 21 and antibody-drug conjugate (ADC) synthesis.22–24
Figure 1. Strategies for disulfide bioconjugation and the design of RDDC.
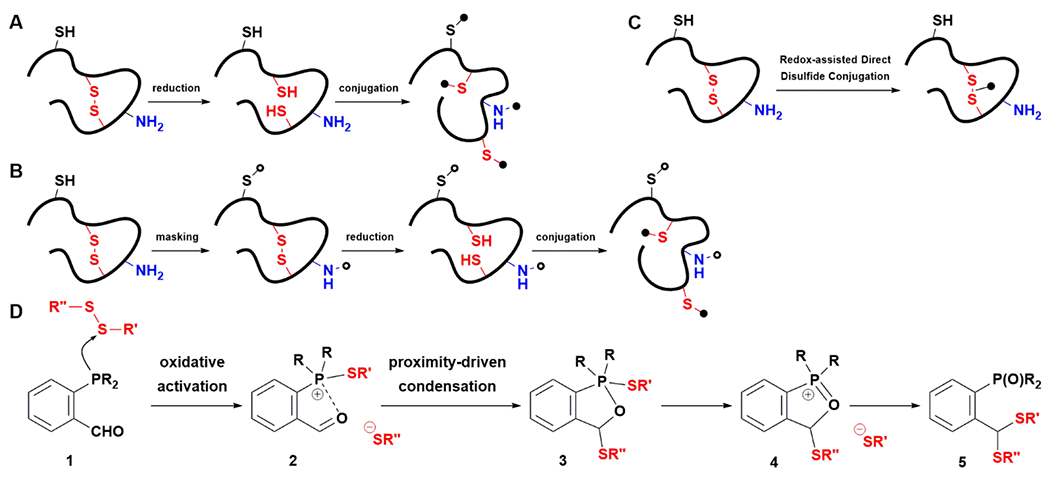
A) Traditional reduction-conjugation methods do not differentiate disulfides from free thiols. B) To achieve selective disulfide conjugation, free thiols are masked prior to disulfide reduction and conjugation. C) Redox-assisted Direct Disulfide Conjugation (RDDC) allows for selective disulfide conjugation in one-step. D) Mechanistic rationale for RDDC. Oxidation of phosphine 1 by a disulfide would activate itsaldehyde group. Trapping the released thiolate by 2 would generate 3 that rearranges through oxonium 4 to form dithioacetal 5.
Redox-based bioconjugation is rare. Whereas oxidative coupling has been used to cross-link proteins and modify tyrosine,25–28 there is only a single example of oxidative protein functionalization wherein oxaziridine was used to convert methionine into sulfimide.29 We reasoned that 1 would not react with free cysteine but could be activated upon reducing a disulfide30 to induce dithioacetal formation with the newly released pair of thiols through proximity-driven condensation (Fig. 1D). Mechanistically, the reduction of disulfide by 1 would generate 2 with a cationic phosphonium center that serves as a strong Lewis acid to enhance the polarization of its neighboring carbonyl group. Subsequently, trapping of the newly released thiolate would give rise to thioacetal 3 that rearranges through 4 to give dithioacetal 5. We selected to use aldehyde as the disulfide reactive group because, except for the highly reactive formaldehyde and glutaraldehyde, it does not react with proteins in a productive manner. Imine formation with lysine is also reversible. Reductive trapping is necessary to form a stable adduct with lysine, or introducing a stabilizing group is needed to increase resident time.31 Aldehyde condensation with an N-terminal cysteine or threonine is generally slow32–34, and proteins are frequently N-acetylated or methionine-capped. However, despite a 15 kcal/mol driving force, dithioacetal has not been used in bioconjugation due to the harsh conditions required to promote its formation.
To explore the feasibility of this strategy, we first studied the ability of phosphine to promote intermolecular coupling of disulfide and aldehyde. Previously, Tazaki and Takagi reported that tributylphosphine induced the condensation of neat 6 and 7 to give 8 rapidly.35 We found that the reaction also proceeded well in toluene or diethyl ether at 0.1 M under argon. A survey of the phosphine promoters in deuterated benzene led to the conclusion that small, electron-rich phosphines are more effective in promoting dithioacetal formation (Fig. 2). The reaction was first order in phosphine and proceeded extremely fast with trimethylphosphine, reaching 80% yield in 5 min and >95% yield in 30 min when performed at 0.2 M in deuterated benzene at 23 °C (see ESI). While triphenylphosphine was essentially ineffective, the more electronically rich and less sterically hindered diethylphenylphosphine slowly promoted the formation of 8. Switching the solvent to the more polar diethyl ether improved the conversion from 3% to 12% at 1 h. We next tested the performance of this reaction in toluene across a range of substrates at 1 mmol scale using 1.5 equiv of disulfide and trimethylphosphine (Fig. 3). Aromatic aldehydes reacted with disulfides easily to form dithioacetals, with diaryl sulfides being more reactive than dialky sulfides and electron-rich aromatic aldehydes giving higher yields than electron-deficient aromatic aldehydes. Aliphatic aldehydes were less reactive than aromatic ones. For the reaction of aliphatic aldehyde with dialkyl disulfide, it was necessary to increase the amount of disulfide and phosphine to 3 equiv to obtain a useful yield of dithioacetal.
Figure 2. The efficiencies of different phosphines in promoting disulfide–aldehyde condensation in benzene.
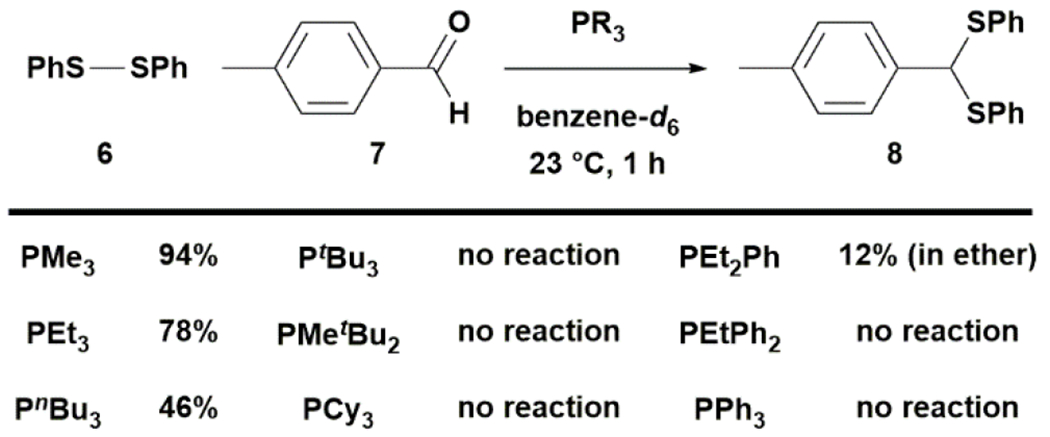
Small, electron-rich phosphines are generally more reactive. Reaction conditions: 6:7:PR3=1:1:1, substrate concentration: 0.1 M.
Figure 3. Scope of the trimethylphosphine-promoted bisthioacetal formation.
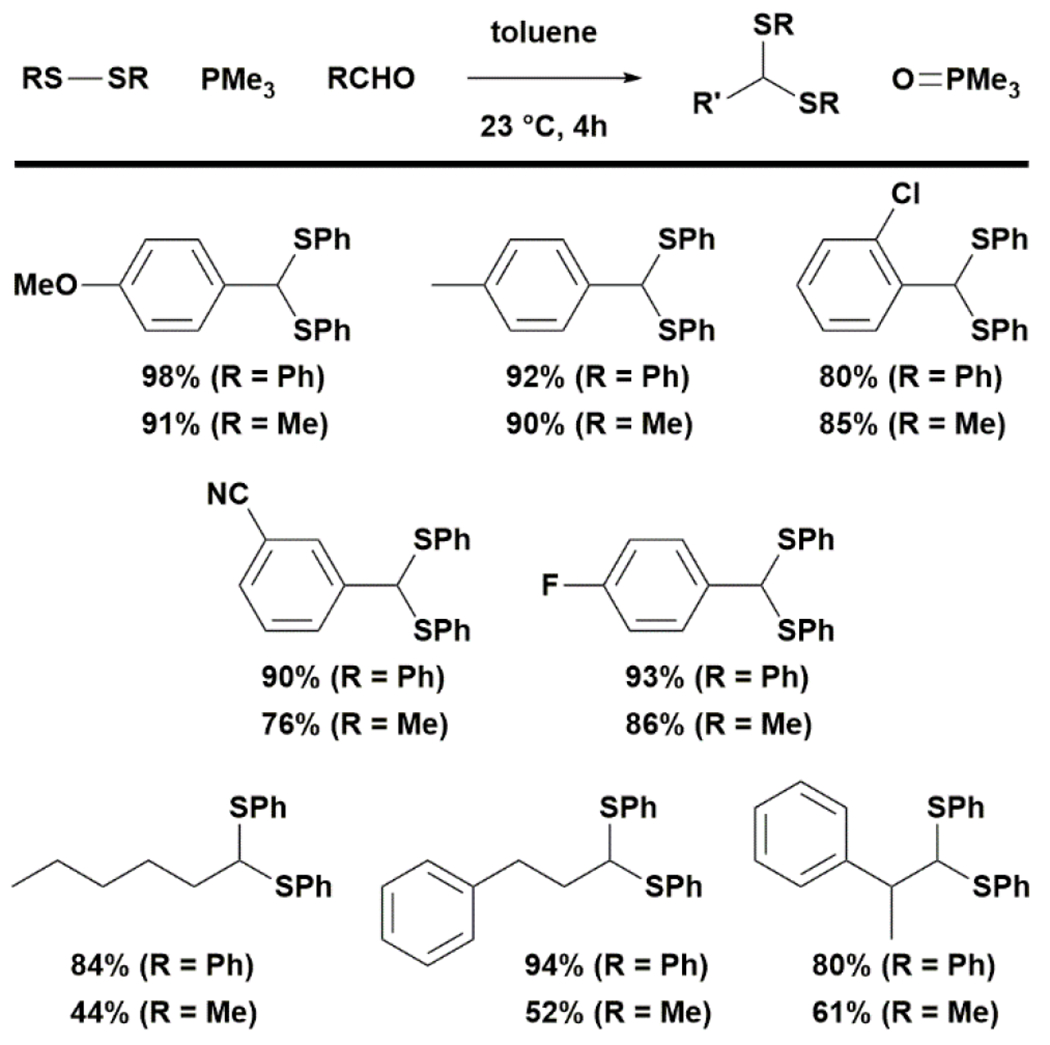
RCHO 1.0 mmol, 1.0 equiv, RSSR: 1.5 equiv, PMe3: 1.5 equiv. For aliphatic aldehyde/dialkyl disulfide: RSSR: 3.0 equiv, PMe3: 3.0 equiv. The reported yields are after isolation.
Based on these results, we considered dialkylarylphosphine to have a balanced activity toward disulfide cleavage and dithioacetal trapping for RDDC. It would also be less air-sensitive than trialkylphosphine. We thus prepared phosphines 11a–c from 9 by reacting the lithiated arene with chlorodimethylphosphine, chlorodiethylphosphine or chlorodiisopropylphosphine to give phosphines 10a–c. Subsequent deprotection of the aldehyde by treating with tetrafluoroboric acid yielded bench-stable phosphonium salts 11a–c in 95% purity (Fig. 4). 11a was obtained as a free-flowing solid and exhibited high water solubility. Despite an active RDDC reagent, it is less stable in basic buffer under aerobic conditions commonly used for biological studies. We thus shifted our focus to 11b (R=Et) that existed as an oil.
Figure 4. Synthesis of the RDDC reagent.

11 are air-stable tetrafluoroborate salts.
Using lipoic acid as a model substrate for RDDC, we found that 11b effectively modified its disulfide group in basic aqueous buffer to provide dithioacetal 12 smoothly (Fig. 5). The reaction proceeded in good yields even at sub-millimolar concentrations under aerobic conditions. All common biological buffers (borate, Tris, HEPES, and carbonate) with pH ranging from 8 to 10 could be used as the solvent. For example, when screened at nanomolar scale, 12 was obtained in 73% yield in pH 8.2 HEPES buffer, 77% in pH 9 borate buffer, and 82% in pH 9 Tris buffer after 1 d of reaction based on LC-MS analysis of the crude reaction mixture (see ESI). However, little product was formed in pH 7.4 PBS buffer and pH 5 acetate buffer. The lack of reactivity at lower pH was presumably due to the unfavorable equilibrium between the inactive phosphonium and the active phosphine forms. Performing RDDC of lipoic acid with 11b at 0.1 mmol scale in pH 8.2 HEPES buffer gave 12 in 80% isolated yield. The sterically hindered 11c (R=iPr) gave no appreciable amount of the desired product under the same conditions. Interestingly, 2-(diphenylphosphino)benzaldehyde had low aqueous solubility but could still react with lipoic acid very slowly in tetrahydrofuran-water (4:1) at room temperature to give a small amount of the desired products after 2 w of reaction.
Figure 5. RDDC proceeds smoothly in water.
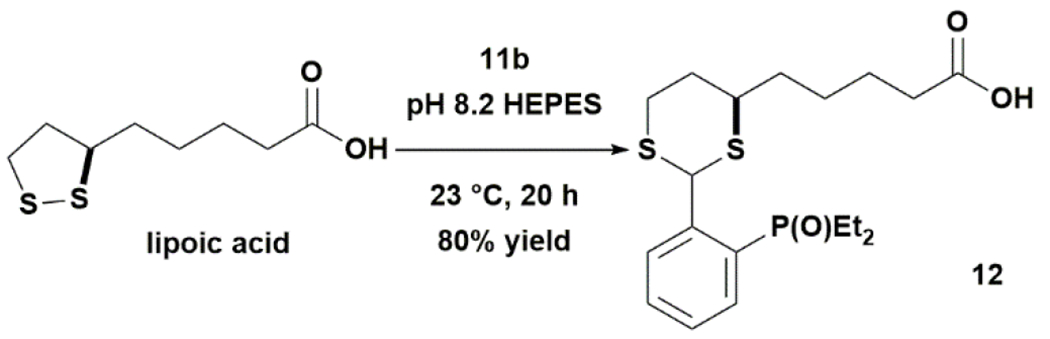
11b inserted into the disulfide of lipoic acid effectively in pH 8.2 HEPES buffer under aerobic conditions. Reaction conditions: 0.1 mmol lipoic acid, 0.02M, 10 equiv 11b.
To further study the utility of 11b in RDDC, we tested its ability to modify salmon calcitonin 13 (Fig. 6), a disulfide-containing peptide hormone that activates calcitonin receptor (CTR) and amylin receptors (AMYRs).36 The reaction proceeded with 58% conversion when treating a solution of 2 mM of 13 in pH 8.2 HEPES buffer or pure water with 3 equiv of 11b at room temperature for 18 h (see ESI). Dithioacetal 14 was formed cleanly as a mixture of two diastereomers. 14’ with a mass of two additional 11b units on 14 was observed as the only by-product (<5%). Treating 13 with a large excess of 11b increased the amount of 14’. Incubating the crude reaction mixture with (aminomethyl)polystyrene converted 14’ to 14, suggesting that 14’ was formed reversibly from imine condensation of 11b with the lysine residues of 14. Performing the reaction under argon improved the conversion to ~80%, but also increased the ratio of 14:14’ to 1:1. Adding another 3 equiv of 11b led to a complete conversion to 14’. Interestingly, we did not detect 14’ when performing this reaction in DMSO. However, 14’ rapidly formed upon diluting the reaction mixture with water. Finally, adding 2.0 equiv of cysteine did not affect the reaction. Nor did we observe any product derived from 11b and cysteine.
Figure 6. RDDC in peptide stapling.
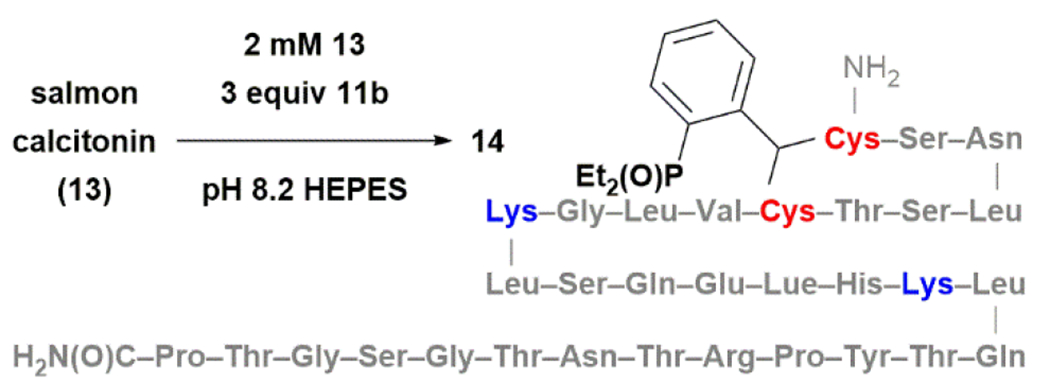
11b inserted into the disulfide of salmon calcitonin (13) under physiologically relevant conditions.
In conclusion, RDDC is a new bioconjugation method that allows for direct disulfide modification. Compared to previous disulfide rebridging methods, it does not require reductive cleavage of the disulfide prior to conjugation. RDDC selectively inserts a one-carbon unit into the disulfide linkage through redox-assisted dithioacetal formation. It complements the current peptide disulfide stapling methods that utilize alkylation37–46 or conjugate addition13, 19, 47–53 to establish a stable bridge with a two or three-carbon insertion. RDDC does not cross-react with cysteine and only reacts with lysine minimally and reversibly. It can also be used in common aqueous buffers for biological studies without an organic cosolvent. These features may render RDDC a useful method for studying disulfide PTM. However, the conversion and selectivity of peptide stapling by 11b under dilute aqueous conditions is still unsatisfactory. As the separation of 14 from unreacted 13 is challenging, further improvement is required to overcome this limitation and make this method practically useful for peptide stapling and ADC synthesis. Efforts to apply RDDC to activity-based protein profiling (ABPP)54 of disulfides in biological systems55, 56 is underway.
Supplementary Material
Acknowledgments
This work is supported by NIH (R21 GM137179 and R01 CA226419), CPRIT (RP180725). CC is a Southwestern Medical Foundation Scholar in Biomedical Research.
Footnotes
Conflicts of interest
The authors declare no conflicts of interest.
Electronic Supplementary Information (ESI) available: See DOI: 10.1039/x0xx00000x
Notes and references
- 1.Cremers CM and Jakob U, J. Biol. Chem, 2013, 288, 26489–26496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chiu J and Hogg PJ, J. Biol. Chem, 2019, 294, 2949–2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hatahet F and Ruddock LW, Antioxid. Redox Signal, 2009, 11, 2807–2850. [DOI] [PubMed] [Google Scholar]
- 4.Cook KM and Hogg PJ, Antioxid. Redox Signal, 2013, 18, 1987–2015. [DOI] [PubMed] [Google Scholar]
- 5.Gunnoo SB and Madder A, ChemBioChem, 2016, 17, 529–553. [DOI] [PubMed] [Google Scholar]
- 6.Kuan SL, Wang T and Weil T, Chem. Eur. J, 2016, 22, 17112–17129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boutureira O and Bernardes GJL, Chem. Rev, 2015, 115, 2174–2195. [DOI] [PubMed] [Google Scholar]
- 8.Koniev O and Wagner A, Chem. Soc. Rev, 2015, 44, 5495–5551. [DOI] [PubMed] [Google Scholar]
- 9.Vinogradova EV, Zhang C, Spokoyny AM, Pentelute BL and Buchwald SL, Nature, 2015, 526, 687–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scott EM, Duncan IW and Ekstrand V, J. Biol. Chem, 1963, 238, 3928–3933. [PubMed] [Google Scholar]
- 11.Liberatore FA, Comeau RD, McKearin JM, Pearson DA, Belonga BQ III, Brocchini SJ, Kath J, Phillips T, Oswell K and Lawton RG, Bioconjugate Chem, 1990, 1, 36–50. [DOI] [PubMed] [Google Scholar]
- 12.Badescu G, Bryant P, Bird M, Henseleit K, Swierkosz J, Parekh V, Tommasi R, Pawlisz E, Jurlewicz K, Farys M, Camper N, Sheng X, Fisher M, Grygorash R, Kyle A, Abhilash A, Frigerio M, Edwards J and Godwin A, Bioconjugate Chem, 2014, 25, 1124–1136. [DOI] [PubMed] [Google Scholar]
- 13.Smith MEB, Schumacher FF, Ryan CP, Tedaldi LM, Papaioannou D, Waksman G, Caddick S and Baker JR, J. Am. Chem. Soc, 2010, 132, 1960–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maruani A, Alom S, Canavelli P, Lee MTW, Morgan RE, Chudasama V and Caddick S, Chem. Commun, 2015, 51, 5279–5282. [DOI] [PubMed] [Google Scholar]
- 15.Morais M, Nunes JPM, Karu K, Forte N, Benni I, Smith MEB, Caddick S, Chudasama V and Baker JR, Org. Biomol. Chem, 2017, 15, 2947–2952. [DOI] [PubMed] [Google Scholar]
- 16.Martin JS, MacKenzie CJ, Fletcher D and Gilbert IH, Bioorg. Med. Chem, 2019, 27, 2066–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maruani A, Smith MEB, Miranda E, Chester KA, Chudasama V and Caddick S, Nat. Commun, 2015, 6, 6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walsh SJ, Omarjee S, Galloway WRJD, Kwan TT-L, Sore HF, Parker JS, Hyvönen M, Carroll JS and Spring DR, Chem. Sci, 2019, 10, 694–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walsh SJ, Iegre J, Seki H, Bargh JD, Sore HF, Parker JS, Carroll JS and Spring DR, Org. Biomol. Chem, 2020, 18, 4224–4230. [DOI] [PubMed] [Google Scholar]
- 20.Lau YH, Andrade P. d., Wu Y and Spring DR, Chem. Soc. Rev, 2015, 44, 91–102. [DOI] [PubMed] [Google Scholar]
- 21.Li X, Chen S, Zhang W-D and Hu H-G, Chem. Rev, 2020, 120, 10079–10144. [DOI] [PubMed] [Google Scholar]
- 22.Agarwal P and Bertozzi CR, Bioconjugate Chem, 2015, 26, 176–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chudasama V, Maruani A and Caddick S, Nat. Chem, 2016, 8, 114–119. [DOI] [PubMed] [Google Scholar]
- 24.Bargh JD, Isidro-Llobet A, Parker JS and Spring DR, Chem. Soc. Rev, 2019, 48, 4361–4374. [DOI] [PubMed] [Google Scholar]
- 25.Kodadek T, Duroux-Richard I and Bonnafous J-C, Trends Pharmacol. Sci, 2005, 26, 210–217. [DOI] [PubMed] [Google Scholar]
- 26.ElSohly AM and Francis MB, Acc. Chem. Res, 2015, 48, 1971–1978. [DOI] [PubMed] [Google Scholar]
- 27.Dorta DA, Deniaud D, Mével M and Gouin SG, Chem. Eur. J, 2020, 26, 14257–14269. [DOI] [PubMed] [Google Scholar]
- 28.Ban H, Gavrilyuk J and Barbas CF III, J. Am. Chem. Soc, 2010, 132, 1523–1525. [DOI] [PubMed] [Google Scholar]
- 29.Lin S, Yang X, Jia S, Weeks AM, Hornsby M, Lee PS, Nichiporuk RV, Iavarone AT, Wells JA, Toste FD and Chang CJ, Science, 2017, 355, 597–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Overman LE and O’Connor EM, J. Am. Chem. Soc, 1976, 98, 771–775. [Google Scholar]
- 31.Yang T, Cuesta A, Wan X, Craven GB, Hirakawa B, Khamphavong P, May JR, Kath JC, Lapek JD Jr., Niessen S, Burlingame AL, Carelli JD and Taunton J, Nat. Chem. Biol, 2022, 18, 934–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bandyopadhyay A, Cambray S and Gao J, Chem. Sci, 2016, 7, 4589–4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bermejo-Velasco D, Nawale GN, Oommen OP, Hilborn J and Varghese OP, Chem. Commun, 2018, 54, 12507–12510. [DOI] [PubMed] [Google Scholar]
- 34.Rosen CB and Francis MB, Nat. Chem. Biol, 2017, 13, 697–705. [DOI] [PubMed] [Google Scholar]
- 35.Masato T and Makoto T, Chem. Lett, 1979, 8, 767–770. [Google Scholar]
- 36.Cao J, Belousoff MJ, Liang Y-L, Johnson RM, Josephs TM, Fletcher MM, Christopoulos A, Hay DL, Danev R, Wootten D and Sexton PM, Science, 2022, 375, eabm9609. [DOI] [PubMed] [Google Scholar]
- 37.Kemp DS and McNamara PE, J. Org. Chem, 1985, 50, 5834–5838. [Google Scholar]
- 38.Timmerman P, Beld J, Puijk WC and Meloen RH, ChemBioChem, 2005, 6, 821–824. [DOI] [PubMed] [Google Scholar]
- 39.Smeenk LEJ, Dailly N, Hiemstra H, Maarseveen J. H. v. and Timmerman P, Org. Lett, 2012, 14, 1194–1197. [DOI] [PubMed] [Google Scholar]
- 40.Heinis C, Rutherford T, Freund S and Winter G, Nat. Chem. Biol, 2009, 5, 502–507. [DOI] [PubMed] [Google Scholar]
- 41.Dewkar GK, Carneiro PB and Hartman MCT, Org. Lett, 2009, 11, 4708–4711. [DOI] [PubMed] [Google Scholar]
- 42.Ramos-Tomillero I, Perez-Chacon G, Somovilla-Crespo B, Sanchez-Madrid F, Domínguez JM, Cuevas C, Zapata JM, Rodríguez H and Albericio F, Bioconjugate Chem, 2018, 29, 1199–1208. [DOI] [PubMed] [Google Scholar]
- 43.Martínez-Sáez N, Sun S, Oldrini D, Sormanni P, Boutureira O, Carboni F, Compañón I, Deery MJ, Vendruscolo M, Corzana F, Adamo R and Bernardes GJL, Angew. Chem. Int. Ed, 2017, 56, 14963–14967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yin Y, Fei Q, Liu W, Li Z, Suga H and Wu C, Angew. Chem. Int. Ed, 2019, 58, 4880–4885. [DOI] [PubMed] [Google Scholar]
- 45.Murar CE, Ninomiya M, Shimura S, Karakus U, Boyman O and Bode JW, Angew. Chem. Int. Ed, 2020, 59, 8425–8429. [DOI] [PubMed] [Google Scholar]
- 46.Wu L-H, Zhou S, Luo Q-F, Tian J-S and Loh T-P, Org. Lett, 2020, 22, 8193–8197. [DOI] [PubMed] [Google Scholar]
- 47.Brocchini S, Balan S, Godwin A, Choi J-W, Zloh M and Shaunak S, Nat. Protoc, 2006, 1, 2241–2252. [DOI] [PubMed] [Google Scholar]
- 48.Wang T, Riegger A, Lamla M, Wiese S, Oeckl P, Otto M, Wu Y, Fischer S, Barth H, Kuana SL and Weil T, Chem. Sci, 2016, 7, 3234–3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li Z, Huang R, Xu H, Chen J, Zhan Y, Zhou X, Chen H and Jiang B, Org. Lett, 2017, 19, 4972–4975. [DOI] [PubMed] [Google Scholar]
- 50.Chudasama V, Smith MEB, Schumacher FF, Papaioannou D, Waksman G, Baker JR and Caddick S, Chem. Commun, 2011, 47, 8781–8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schumacher FF, Nobles M, Ryan CP, Smith MEB, Tinker A, Caddick S and Baker JR, Bioconjugate Chem, 2011, 22, 132–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jones MW, Strickland RA, Schumacher FF, Caddick S, Baker JR, Gibson MI and Haddleton DM, J. Am. Chem. Soc, 2012, 134, 1847–1852. [DOI] [PubMed] [Google Scholar]
- 53.Robertson NS, Walsh SJ, Fowler E, Yoshida M, Rowe SM, Wu Y, Sore HF, Parker JS and Spring DR, Chem. Commun, 2019, 55, 9499–9502. [DOI] [PubMed] [Google Scholar]
- 54.Cravatt BF, Wright AT and Kozarich JW, Annu. Rev. Biochem, 2008, 77, 383–414. [DOI] [PubMed] [Google Scholar]
- 55.Paulsen CE and Carroll KS, Chem. Rev, 2013, 113, 4633–4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alcock LJ, Perkins MV and Chalker JM, Chem. Soc. Rev, 2018, 47, 231–268. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.