

Abstract
Context:
Diethylene glycol (DEG) is an organic compound found in household products but also as a counterfeit solvent in medicines. DEG poisonings are characterized by acute kidney injury (AKI) and by neurological sequelae such as decreased reflexes or face and limb weakness. Previous studies in male rats have demonstrated that neurotoxic effects develop only with the establishment of AKI, but the dose sensitivity of females to DEG toxicity is unknown.
Objectives:
Assessing whether subacute administration of DEG in female rats would delineate any sex-differences in neuropathy or in kidney injury.
Methods:
Female Wistar-Han rats were orally administered doses of 4–6g/kg DEG every 12 h and monitored for 7 days. Urine was collected every 12 h and endpoint blood and cerebrospinal fluid (CSF) were collected for renal plasma parameters and total protein estimation, respectively. Motor function tests were conducted before and after treatment. Kidney and brain tissue were analyzed for metabolite content.
Results:
Of 12 animals treated with DEG, 3 developed AKI as confirmed by increased BUN and creatinine concentrations. Renal and brain DGA contents were increased in animals that developed AKI compared to animals without AKI. Total CSF protein content in animals with AKI was markedly elevated compared to control and to treated animals without AKI. Decreases in forelimb grip strength and in locomotor and rearing activity were observed in animals with AKI compared to control and to animals without AKI.
Discussion:
Repeated dosing with DEG in a female model produced nephrotoxic effects at a dose similar to that in males. The decrease in motor function and increase in CSF protein were only present in females that developed AKI. However, kidney and neurologic effects were assessed only at the end of the treatments, thus limiting determination of which effect occurs first. Limb function and coordination were measured globally and more sensitive tests such as nerve conduction studies might offer a detailed neurotoxicity assessment of the effects of DEG.
Conclusions:
These studies show that DEG toxicity does not appear to be sex-specific and that, in males and females, neurological symptoms are present only when DGA accumulation and kidney injury also occur.
Keywords: Diethylene glycol, diglycolic acid, nephrotoxicity, neurotoxicity, cerebrospinal fluid, motor function, metabolite toxicity, sex differences, female
Introduction
Diethylene glycol (DEG) has been implicated in mass poisonings throughout the world due to accidental ingestion of contaminated drugs or liquids [1–4]. DEG is also a constituent of common consumer products such as brake fluid and fog machine fluid, ingestion of which can lead to individual poisonings [5]. The typical clinical indicators of human DEG poisoning are acute kidney injury (AKI) [3] and a peripheral neuropathy characterized by decreased reflexes and coordination of movement, weakness in the arms, legs, and face. The neurological damage usually manifests about 2–7 days after ingestion. Forty out of 46 patients included in a case study of the Panama DEG epidemic reported neurotoxic symptoms [3], with over 60% experiencing limb weakness and decreased or absent reflexes. Thirteen of 14 cases that showed an increase in protein concentration in the cerebrospinal fluid (CSF) later developed neurological signs, suggesting that CSF protein elevations were an early indication of neurotoxicity. Case studies in South Africa [6] and Colorado [7] also noted elevated CSF protein concentrations.
DEG is initially metabolized by alcohol dehydrogenase to 2-hydroxyethoxyacetaldehyde [8], to 2-hydroxyethoxyacetic acid (2-HEAA), and then to diglycolic acid (DGA). HEAA is the main metabolite in the blood and urine and is responsible for the metabolic acidosis [9]. However, unlike the parent compound or other metabolites, DGA accumulates in the kidney and produces proximal tubule cell necrosis at toxic doses of DEG [10,11]. DGA accumulation has been shown to be the nephrotoxic agent in DEG poisoning [11,12].
The mechanism for the neurotoxicity of DEG is not fully understood at this time, although we have recently developed an animal model of neurotoxicity in male rats. In that study [13], neurological dysfunction from DEG ingestion only presented in animals with AKI, suggesting a link between these pathologies as has been proposed in human poisonings [14]. Furthermore, nephrotoxicity and neurotoxicity only developed in DEG-treated animals that showed DGA accumulation in kidney and brain tissue. Hence during DEG poisonings, DGA is accumulated in the target organs and appears to contribute to neurological sequelae.
Sex-differences regarding toxicity of drugs or compounds have been well documented [15–18]. Probably the most relevant example is the chemically-related nephrotoxicant ethylene glycol, where repeated doses given in the diet or in water produce much higher mortality and morbidity (kidney damage) in male rats than in female rats [15,16]. As another example, women are more likely to develop liver disease such as cirrhosis, cardiac, and brain damage when consuming a smaller amount of ethanol compared to men [17]. Possible explanations for this discrepancy include a smaller distribution volume due to a smaller body water content, lower alcohol elimination rates due to slower metabolism, as well as estrogen exacerbating the endotoxin-lead release of inflammatory cytokines [17]. Analysis of reports on mass DEG poisonings [3,19,20] indicate that cases of DEG poisoning have been reported in both male and female humans, but the sensitivity of males compared to females to DEG poisoning was not analyzed per se in these reports. Because of the lack of direct evidence from human studies on DEG poisoning and because of the overt sex difference in dose sensitivity to the related compound ethylene glycol in rats, we wanted to evaluate this concept of sexual differences in sensitivity to toxicity with DEG, using the dosing regimen and parameters of kidney and motor function as in our male study [13]. We hypothesized that the female rat would be susceptible to DEG poisoning, although with a lower dose sensitivity than previously observed in males, based on literature showing this trend with ethylene glycol.
Materials and methods
Materials
DEG (≥99% purity by gas chromatography analysis) for gavage was obtained by Sigma-Aldrich Corporation (St. Louis, MO) and prepared in deionized water.
Animal protocol
Sixteen adult female Wistar-Han rats (Envigo, Indianapolis, IN), (200–230 g) were randomly placed into treatment groups (Table 1) and administered DEG doses by oral gavage for up to 7 days. Rats were maintained in metabolic chambers under standard conditions of humidity, temperature (25 °C±2°C) and light (12:12 h light:dark) and were allowed free access to normal rat chow (Envigo Teklad Diet) and water. The DEG doses were based on previous studies in male rats [13], which showed that oral doses of 4 g/kg – 6 g/kg administered every 12 h induced nephrotoxicity in male rats starting at about 48 h. The animal protocols were approved by the Institutional Animal Care and Use Committee (Louisiana State University Health Sciences Center, Shreveport (LSUHSC-S)) and were in accordance with the National Institutes of Health’s Guide for the Care and Use of Laboratory Animals.
Table 1.
Total number of animals and acute kidney injury (AKI) development by DEG dose schedule.
| Treatment | Number of animals | Developed AKIa | No AKI |
|---|---|---|---|
| Water control | 4 | 0 | 4 |
| DEG 4 g/kg every 12 h | 2 | 0 | 2 |
| DEG 5 g/kg every 12 h | 4 | 0 | 4 |
| DEG 6 g/kg every 12 h | 6 | 3 | 3 |
| Total | 16 | 3 | 13 |
Animals included in the group that developed acute kidney injury had elevated blood urea nitrogen (BUN) and creatinine endpoint concentrations.
Animal observations
Throughout the study, urine output was examined in all animals. Animals were removed from the dosing schedule, if there was a > 50% decrease in urine volume in a 12 h period, and were evaluated for motor function prior to euthanasia.
Urine, blood, CSF and tissue collection and analysis
As previously described in detail [13], blood, urine, CSF and tissue samples were collected for analysis.
For blood, CSF and tissue collection, animals were anesthetized at 168 h, or sooner using criteria described above, using isoflurane induction, followed by sodium pentobarbital (50 mg/kg, i.p.). Plasma from the collected blood was analyzed for concentrations of blood urea nitrogen (BUN), creatinine, glucose, sodium [Na+], potassium [K+] and chloride [Cl−] by the Ochsner LSU Health – Shreveport Clinical Laboratory. Anion gap was calculated as [(Na+ + K+) − (Cl− + HCO3−)]. Statistically increased values of plasma BUN and creatinine over values in concurrently treated control rats and over historical ranges at the endpoint were used to substantiate that the animal had AKI. Such animals were categorized as the “kidney injury” group for data analysis. Other animals that were treated with DEG for 7 days, without evidence of kidney injury, were then categorized as the “no kidney injury” group.
CSF was stored at 4 °C until analyzed for total protein content via a bicinchoninic acid assay.
One kidney and one hemisphere of the brain were collected, weighed and stored at −80 °C until further analysis.
DGA concentrations in tissue
Kidney and brain tissues were analyzed for DGA concentration using high-performance liquid chromatographic (HPLC) analysis with a two-step solid-phase extraction protocol [11,21].
Motor function tests
Motor function tests were conducted according to protocols previously described [13,22,23] before rats were placed into their metabolic chambers on day 0 and on day 7 or sooner using the early termination criteria described above. The motor function tests conducted were the accelerating rotarod (Rota-rod/RS; Letica Scientific Instruments, Barcelona, Spain), open field locomotor testing (Truscan 2.0; Coulbourn Instruments, Whitehall, PA) and forearm grip strength using a 1 kg hanging scale (American Weight Scales, Inc., Norcross, GA).
Statistics
Values in the text represent the group mean value ± SEM. Comparisons between multiple groups were performed using a one-way ANOVA with a Bonferroni post hoc analysis to determine treatment group differences. Two-way ANOVA was used for multiple comparisons, with Bonferroni’s post hoc analysis used for significance. All analyses were performed using GraphPad Prism 5 or Prism 7.05 (La Jolla, Ca) for Windows. Tests were considered significant if p<0.05.
Results
DEG subacute dosing led to DGA accumulation and acute kidney injury
Renal injury
In total, 3 out of 12 animals treated with DEG developed AKI, with all being in the 6 g/kg group (Table 1). BUN and plasma creatinine values were significantly and markedly increased (6 to 10-fold higher, respectively) in animals that developed kidney injury compared to controls and to animals that were treated with DEG but did not develop kidney injury (p<0.05) (Figure 1(A,B)).
Figure 1.

Subacute DEG oral administration produced kidney injury in female rats. Kidney injury was assessed by blood urea nitrogen (BUN) (A, top) and plasma creatinine (B, bottom). DEG doses ranged from 4 g/kg to 6 g/kg, administered twice daily. Data are represented as means ± SEM (n=4 for controls, n=3 for animals that developed kidney injury, n=9 for animals that were administered DEG but did not develop kidney injury). Asterisk (*) indicates significant difference from control (one-way ANOVA followed by Bonferroni post hoc test, p<0.05). Pound sign (#) indicates significant difference between DEG-treated animals that did and did not develop kidney injury (one-way ANOVA followed by Bonferroni post hoc test, p<0.05).
DGA accumulation in tissue
Animals that developed kidney injury showed significantly increased DGA concentrations in the kidney compared to controls and to animals that did not develop kidney injury (p<0.05) (Figure 2(A)). Kidney DGA concentrations in animals with kidney injury averaged 8.9 μmol/g, while animals that did not develop kidney injury averaged 0.34 μmol/g. Additionally, animals that developed kidney injury had significantly increased DGA concentrations in the brain compared to controls and to animals that did not develop kidney injury (p<0.05) (Figure 2(B)). Brain DGA concentrations in animals with kidney injury averaged 0.65 μmol/g and animals that did not develop kidney injury averaged 0 μmol/g.
Figure 2.

Diglycolic Acid (DGA) concentrations in kidney (A) and brain (B) tissue at termination of study. Data are represented as means ± SEM or range (n=2 for controls, n=2 for animals that developed kidney injury, n=6 for animals that were administered DEG but did not develop kidney injury). Asterisk (*) indicates significant difference from control (one-way ANOVA followed by Bonferroni post hoc test, p<0.05). Pound sign (#) indicates significant difference between DEG-treated animals that did and did not develop kidney injury (one-way ANOVA followed by Bonferroni post hoc test, p<0.05).
Metabolic acidosis
DEG-treated animals that developed kidney injury displayed significant decreases in plasma bicarbonate concentrations (2 ± 0 mmol/L) compared to controls (25 ± 1.7 mmol/L) and to animals that did not develop kidney injury (27.6 ± 0.8 mmol/L) (p<0.05) (Figure 3(A)). The anion gap in animals with kidney injury was significantly increased (35.7 ± 5.7 mmol/L) compared to controls (11 ± 1.4 mmol/L) and to animals that did not develop kidney injury (11.2 ± 1 mmol/L) (p<0.05) (Figure 3(B)).
Figure 3.
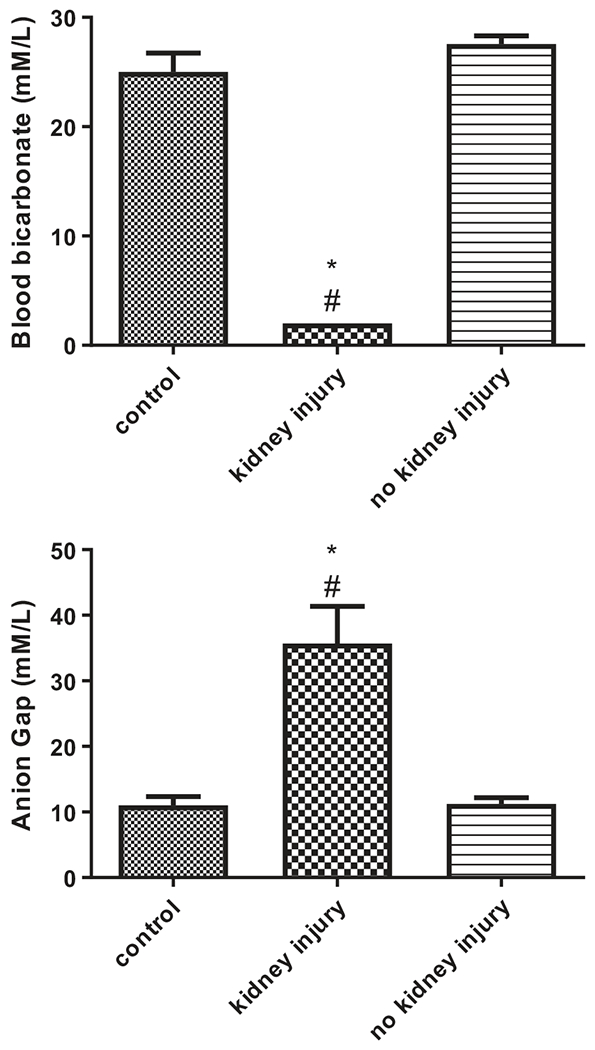
Subacute DEG oral administration produced metabolic acidosis in female rats. Metabolic acidosis was assessed using blood bicarbonate (A, top), and anion gap (B, bottom) at termination of study. Anion gap was calculated as [(Na+ + K+) – (Cl− + HCO3−)]. Data are represented as means ± SEM (n=4 for controls, n=3 for animals that developed kidney injury, n=9 for animals that were administered DEG but did not develop kidney injury). Asterisk (*) indicates significant difference from control (one-way ANOVA followed by Bonferroni post hoc test, p<0.05). Pound sign (#) indicates significant difference between DEG-treated animals that did and did not develop kidney injury (one-way ANOVA followed by Bonferroni post hoc test, p<0.05).
DEG subacute dosing produced neurotoxic effects
Cerebrospinal fluid protein content
Total protein in the CSF was significantly increased in DEG-treated animals with kidney injury (684 ± 2.3 μg/mL) compared to controls (242 ± 32.4 μg/mL) and to animals that did not develop kidney injury (234 ± 16.2 μmg/mL) (p<0.05) (Figure 4).
Figure 4.

Total protein in cerebrospinal fluid of female rats treated with DEG. Data are represented as means ± SEM or range (n=4 for controls, n=2 for animals that developed kidney injury, n=9 for animals that were administered DEG but did not develop kidney injury). Asterisk (*) indicates significant difference from control (one-way ANOVA followed by Bonferroni post hoc test, p<0.05). Pound sign (#) indicates significant difference between DEG-treated animals that did and did not develop kidney injury (one-way ANOVA followed by Bonferroni post hoc test, p<0.05).
Motor function
General motor function tests were used to evaluate changes in limb strength and coordination. The total distance traveled during open field exploration was significantly decreased in DEG-treated animals with kidney injury (2633 ± 1864 cm) compared to control (11898 ± 686 cm) and to animals without kidney injury (12113 ± 725 cm) (p<0.05) (Figure 5(A)). DEG-treated animals with kidney injury had significantly decreased rearing time (110 ± 1 s) compared to control (712 ± 90 s), and to animals without kidney injury (506 ± 56 s) (p<0.05) (Figure 5(B)). There was no difference in distance spent in the center or edges of the open field chamber along any of the groups; all groups spent approximately half of their time between both zones of the chamber (data not shown). After DEG dose administration, forelimb grip strength was decreased from baseline in animals that developed kidney injury (237 ± 4.5 g) compared to controls (374 ± 41.4 g) and to animals that did not develop kidney injury (328 ± 28.5 g) (p<0.05) (Figure 5(D)), but this difference did not reach statistical significance. Latency to fall during rotarod acceleration showed a decreasing, but not significant trend in DEG-treated animals that developed AKI (data not shown).
Figure 5.
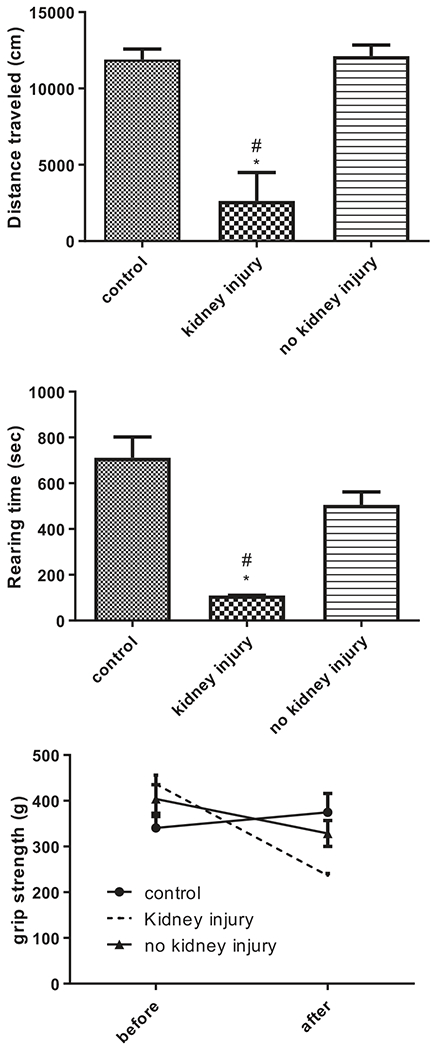
Motor function was altered in female rats treated with DEG that developed kidney injury. Motor function was evaluated by open field total distance traveled (A, top), open field rearing (B, middle), and forelimb grip strength (C, bottom). Data are represented as means ± SEM or range (n=4 for controls, n=2 for animals that developed kidney injury, n=9 for animals that were administered DEG but did not develop kidney injury). Asterisk (*) indicates significant difference from control (one-way (A and B) or two-way (C) ANOVA followed by Bonferroni post hoc test, p<0.05). Pound sign (#) indicates significant difference between DEG-treated animals that did and did not develop kidney injury (one-way (A and B) or two-way (C) ANOVA followed by Bonferroni post hoc test, p<0.05).
Discussion
Previous studies in male rats have established that a repeated-dose DEG regimen was able to produce AKI as well as a neurotoxic syndrome that had characteristics similar to those observed in human poisoning cases [13]. The close correlation of the results in these female rats with those in the males suggests that there is likely no sex difference in DEG-related kidney and nervous system injury. No human clinical study on DEG cases has reported sex-specific toxic effects other than delineating the basic demographic characteristics of the patients such as sex. For example, 52% of the DEG patients in one Panama case study [3] and 36% in the other case control study [20] were female. However, the resulting analysis of the degree of toxicity was not differentiated by sex, so it cannot be determined if one sex was more sensitive. Similarly, 35% of patients in a pediatric case study from an outbreak in Haiti [19] and 35% in the 2008 outbreak in Nigeria [24] were female, but again no direct comparison was made. Thus, in several human outbreaks, the patient population has included a widespread distribution of males and females (>35% of the affected cases were female). These studies suggest the likelihood that DEG-toxicity in humans may not be sex-dependent, as is similar to what we observed in our animal studies.
The only reported sex difference in DEG-treated animals involved chronic studies (14 and 32-week) with low doses that are environmentally but not clinically relevant. The sex differences included urine volume, body weight, oxalate crystal formation, fertility, and tubular necrosis development, but not overall morbidity or mortality [25]. Based on the present and previous results [13], the morbidity of an overdose DEG-ingestion appears to affect male and female animals identically. This lack of sex difference is an interesting contrast to the well-known sex difference in sensitivity to ethylene glycol, where male rats are more greatly affected at lower doses than females. The sex difference in ethylene glycol toxicity is related to a difference in accumulation of oxalate in kidney tissue [15,16]. We can speculate that the lack of a sex difference with DEG as compared to that with ethylene glycol is that DEG is metabolized differently so the kidney toxicity from DEG is related to DGA accumulation, not to oxalate accumulation [9,11].
Nerve conduction studies in the Panama cases showed unexcitable motor and sensory responses, decreased motor and sensory amplitudes, and prolongation or absence of F waves, which indicated a peripheral sensorimotor neuropathy [3]. The grip strength test was used in our studies to evaluate forelimb function, while the quantification of rearing gauges the ability of the rat to lift itself up, so assesses general limb movement. The open field test assesses overall movement and the rotarod performance test measures coordination. The pattern of decreased forelimb strength, movement, and rearing in females rats that developed AKI was also observed in males and supports the hypothesis that DEG induces functional neurological impairment in this animal model [3]. The movement in the center and edge regions of the open field chamber were both reduced in females, which indicates a generalized motor impairment not a reduced motivation to move. From these studies, there is not enough evidence to propose a direct connection between AKI-development and motor dysfunction. However, only animals that developed AKI also showed neurological dysfunction. The timing for the occurrence of nephrotoxicity and neurotoxicity could not be evaluated in this study, because rats were tested for neurotoxicity only at the end of the study or after renal dysfunction was already present. To determine the chronological order of nephrotoxicity and neurotoxicity, studies using timed interval testing could answer which pathway proceeded the other. Regardless, these studies did observe that neurotoxicity was only present in female rats that also had AKI, thus suggesting a connection.
An increased CSF protein has been documented in human DEG poisonings and has been shown to occur prior to the development of overt neurological damage [3]. In DEG-treated female rats that showed neurological signs and symptoms similar to those reported in humans and male rats, an increased total CSF protein was also observed. The increase in total CSF protein (3-fold over control values) was similar to that in the male rat (5-fold over controls) [13] and to that reported in human cases in Panama [3]. DGA concentrations in the CSF were elevated in 7 out of 8 patients that developed neurotoxic effects from DEG-poisoning in the Panama outbreak [26]. DGA also accumulated in the brain of DEG-treated female rats to an average concentration of 0.65 ± 0.08 μmol/g of tissue, which was somewhat higher than that in DEG-treated males (0.23 ± 0.09 μmol/g). In comparison to the changes in males, the brain DGA concentrations were 3-fold higher in females, whereas the elevated CSF protein concentrations were 5-fold higher in males. We are not sure of the significance of these differences, in particular because the increases in CSF protein and in brain DGA concentrations in males and females both represent a huge increase over their respective control values. Overall, these changes in both female and male animals suggest that an accumulation of the postulated toxic metabolite may be a factor in the neurological changes.
In rats, normal serum creatinine and BUN concentrations range from 0.4–0.8 mg/dL and 15–22 mg/dL respectively [27], which are similar to those measured in our female control and no AKI animals. The female AKI animals averaged 4 mg/dL for creatinine and 116 mg/dL for BUN, which vastly surpass the normal ranges and are similar to the concentrations in male AKI animals (5 mg/dL and 159 mg/dL respectively) [13]. Hence, AKI is developed in female rats treated with DEG similarly to that in male rats. The kidney effects of DEG stem from the renal accumulation of the metabolite DGA [9] as confirmed by similar kidney damage in DGA-treated animals [11]. DGA accumulation in the kidney of injured female rats, about 8.9 μmol/g, in this study was comparable to that in the male repeated-dose study [13] (10 μmol/g). The female animals received 5 to 6 doses of DEG (2.5–3 d) before developing AKI, which is similar to the time frame in male rats. Overall, the repeatedly dosed females received a total DEG dose of about 30–36 g/kg, which closely resembles the male repeat-dose data. The toxic human dose is reported to be 0.36 g/kg in the Panama study [3] and 1–1.5 g/kg in the Haiti study [19], although such estimates are generally suspect because recall of ingestion amounts is often imprecise. Hence even though higher doses of DEG are required to produce toxicity in rats than in humans, the characteristics and timing of the development of kidney toxicity and neurotoxicity are similar in rats and in humans.
In both males and females treated with DEG, only animals that had marked accumulation of DGA in the kidney and brain showed evidence of kidney injury and neurological effects. Intriguingly, the same doses that caused the majority of toxicity in males, 4–6 g/kg twice a day, elicited a comparable progression of toxicity in females dosed twice per day. Only 3 out of 12, or 25%, of the DEG-treated female animals developed AKI, while 9 out of 25 male rats (36%) dosed twice per day developed AKI [13]. Also at 6 g/kg twice per day, 50% of females had AKI, while 60% of males had AKI [13]. Thus, one might conclude that females are somewhat less sensitive at the same dose as in males, but this small difference in frequency of toxicity is not likely significant. Although similarly-dosed rats showed a variable degree of renal and neurological toxicity, dysfunction was only observed in animals where DGA accumulated in target tissues. Such variability in toxicity also occurs in humans, as has been reported in multiple epidemiological studies [19,20], where numerous people have ingested the DEG-contaminated preparation, yet only about half are reported as being poisoned. Therefore, variability in susceptibility to DEG toxicity is likely related to some factor in DGA kinetics, either in its production from DEG or its uptake and accumulation by target organ cells. Although rat strains exhibit differences in alcohol and aldehyde dehydrogenase activities [28,29], there does not appear to be much variation within the Wister-Han rat used in these studies, so differences in DEG metabolism to DGA are unlikely. DGA is a four carbon dicarboxylate acid, so it has a structure similar to that of succinate [10]. Thus, sodium dicarboxylate transporters are likely involved in DGA uptake into the proximal tubule cell. Dicarboxylic acids are also effluxed from the proximal tubule cell in exchange for organic acids via the organic acid transporters (OATs), so DGA could be effluxed by these same transporters. An increased activity in the uptake transporters or a decreased activity in the efflux transporters could produce an enhanced accumulation of DGA by the target organ cell and either variation could explain the individual susceptibility to DEG toxicity.
The metabolic acidosis in female animals that developed kidney injury was comparable to the acidosis in males. DEG-treated female animals with AKI in this study also had an average anion gap of 35.7 mmol/L compared to 36.2 mmol/L in males (~11 mmol/L in control rats). The acidosis likely results from accumulation of 2-HEAA, which is known to be elevated in the blood [9], and from increased lactate levels due to the kidney injury itself. Additionally, mitochondrial dysfunction can be caused by DGA, which could further contribute to metabolic acidosis because a decrease in ATP production would increase lactate anions in the blood [11,30,31].
Study limitations
This study has attempted to compare the response of female rats to DEG to previous results in male rats similarly exposed; however, this study was not conducted at the same time such that the conditions by which the rats were housed were likely slightly different. Additionally, female rats were not dosed identically as the male rats (only being dosed with DEG twice per day). The reasons for this include that the male study indicated few responses in animals dosed once per day and so we reduced the number of animals used for ethical reasons. The overall number of subjects in this study was small, so the study may be under powered to detect a true difference in response of males and females. A close examination of the data shows a trend towards female rats being less sensitive to the effects of DEG then male rats (50% affected at the high dose compared to 60% in males; 25% affected overall versus 36% in males). While we established that AKI must develop in animals in order to detect neurological changes, motor and renal function tests at intermediate points throughout the study would have provided a chronology of the timing of renal damage relative to the neurological effects. In the Panama outbreak, CSF protein was elevated before the onset of overt neurological signs, so intermediate CSF collection would have allowed us to study CSF protein as a possible early indicator of more severe neurotoxicity [3]. Additionally, the motor function tests conducted were a global measurement of limb movement and coordination and more sensitive tests such as nerve conduction studies might offer more detailed neurotoxicity assessment following DEG-administration.
Conclusion
This study of DEG toxicity in a female animal model observed nephrotoxicity and established neurotoxic effects that paralleled those seen in males. Kidney injury biomarkers were markedly increased in DEG-treated animals, confirming that these females had developed AKI similar to that reported in males. DGA accumulated in the kidney tissue of females at concentrations comparable to previous studies, but only in the animals that had kidney injury. DGA also accumulated in the brain only in females that developed AKI and neurological injury. Motor function parameters decreased and total CSF protein increased in animals that developed AKI. Importantly, this study showed that there does not seem to be a sexual difference in toxicity profile in that males and females appeared to develop AKI and neurotoxicity at a similar frequency and severity during DEG-administration.
Acknowledgements
The authors thank Mychal Grames for his assistance with animal training. Thanks to Julie Tobin and Noel Jacquet for their technical assistance during urine and tissue collection.
Funding
This research was supported by the NIH, grant R15 ES029704.
Footnotes
Disclosure Statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- [1].Schep LJ, Slaughter RJ, Temple WA, et al. Diethylene glycol poisoning. Clin Toxicol. 2009;47(6):525–535. [DOI] [PubMed] [Google Scholar]
- [2].Schier JG, Rubin CS, Miller D, et al. Medication-associated diethylene glycol mass poisoning: a review and discussion on the origin of contamination. J Public Health Policy. 2009;30(2):127–143. [DOI] [PubMed] [Google Scholar]
- [3].Sosa NR, Rodriguez GM, Schier JG, et al. Clinical, laboratory, diagnostic, and histopathologic features of diethylene glycol poisoning-Panama, 2006. Ann Emerg Med. 2014;64(1):38–47. [DOI] [PubMed] [Google Scholar]
- [4].de Oliveira Dias M. Fatality, malpractice, or sabotage? case on craft beer poisoning in Minas Gerais Brazil. East African Scholars Multidisc Bull. 2020;3:26–31. [Google Scholar]
- [5].Gummin DD, Mowry JB, Beuhler MC, et al. 2019 Annual Report of The American Association of Poison Control Centers’ National Poison Data System (NPDS): 37th annual report. Clin Toxicol. 2020;58(12):1360–1541. [DOI] [PubMed] [Google Scholar]
- [6].Bowie MD, McKenzie D. Diethylene glycol poisoning in children. S Afr Med J. 1972;46(27):931–934. [PubMed] [Google Scholar]
- [7].Rollins YD, Filley CM, McNutt JT, et al. Fulminant ascending paralysis as a delayed sequela of diethylene glycol (Sterno) ingestion. Neurology. 2002;59(9):1460–1463. [DOI] [PubMed] [Google Scholar]
- [8].Wiener HL, Richardson KE. Metabolism of diethylene glycol in male rats. Biochem Pharmacol. 1989;38(3):539–541. [DOI] [PubMed] [Google Scholar]
- [9].Besenhofer LM, McLaren MC, Latimer B, et al. Role of tissue metabolite accumulation in the renal toxicity of diethylene glycol. Toxicol Sci. 2011;123(2):374–383. [DOI] [PubMed] [Google Scholar]
- [10].Landry GM, Martin S, McMartin KE. Diglycolic acid is the nephrotoxic metabolite in diethylene glycol poisoning inducing necrosis in human proximal tubule cells in vitro. Toxicol Sci. 2011;124(1):35–44. [DOI] [PubMed] [Google Scholar]
- [11].Robinson CN, Latimer B, Abreo F, et al. In-vivo evidence of nephrotoxicity and altered hepatic function in rats following administration of diglycolic acid, a metabolite of diethylene glycol. Clin Toxicol. 2017;55(3):196–205. [DOI] [PubMed] [Google Scholar]
- [12].Sprando RL, Mossoba ME, Black T, et al. 28-day repeated dose response study of diglycolic acid: Renal and hepatic effects. Food Chem Toxicol. 2017;106(Pt A):558–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Jamison CN, Dayton RD, Latimer B, et al. Neurotoxic effects of nephrotoxic compound diethylene glycol. Clinical Toxicology. 2021;0(0):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Alfred S, Coleman P, Harris D, et al. Delayed neurologic sequelae resulting from epidemic diethylene glycol poisoning. Clin Toxicol (Phila)). 2005;43(3):155–159. [PubMed] [Google Scholar]
- [15].Melnick RL. Toxicities of ethylene glycol and ethylene glycol monoethyl ether in Fischer 344/N rats and B6C3F1 mice. Environ Health Perspect. 1984;57:147–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Robinson M, Pond CL, Laurie RD, et al. Subacute and subchronic toxicity of ethylene glycol administered in drinking water to Sprague-Dawley rats. Drug Chem Toxicol. 1990;13(1):43–70. [DOI] [PubMed] [Google Scholar]
- [17].Sato N, Lindros KO, Baraona E, et al. Sex difference in alcohol-related organ injury. Alcoholism Clin Exp Res. 2001;25(s1):40S–445. [DOI] [PubMed] [Google Scholar]
- [18].Liu L, Jiang Z, Liu J, et al. Sex differences in subacute toxicity and hepatic microsomal metabolism of triptolide in rats. Toxicology. 2010;271(1-2):57–63. [DOI] [PubMed] [Google Scholar]
- [19].O’Brien KL, Selanikio JD, Hecdivert C, et al. Epidemic of pediatric deaths from acute renal failure caused by diethylene glycol poisoning. Acute Renal Failure Investigation Team. Jama. 1998;279(15):1175–1180. [DOI] [PubMed] [Google Scholar]
- [20].Rentz ED, Lewis L, Mujica OJ, et al. Outbreak of acute renal failure in Panama in 2006: a case-control study. Bull World Health Organ. 2008;86(10):749–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Landry GM, Dunning CL, Abreo F, et al. Diethylene glycol-induced toxicities show marked threshold dose response in rats. Toxicol Appl Pharmacol. 2015;282(3):244–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Jackson KL, Dayton RD, Klein RL. AAV9 supports wide-scale transduction of the CNS and TDP-43 disease modeling in adult rats. Mol Ther Methods Clin Dev. 2015;2(August):15036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Jackson KL, Dayton RD, Orchard EA, et al. Preservation of forelimb function by UPF1 gene therapy in a rat model of TDP-43-induced motor paralysis. Gene Ther. 2015;22(1):20–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Abubukar A, Awosanya E, Badaru O, et al. Fatal poisoning among young children from diethylene glycol-contaminated acetaminophen– Nigeria, 2008–2009. Morb Mortal Wkly Rep. 2009;58:1345–1347. [PubMed] [Google Scholar]
- [25].Snellings WM, McMartin KE, Banton MI, et al. Human health assessment for long-term oral ingestion of diethylene glycol. Regul Toxicol Pharm. 2017;87:S1–S20. [DOI] [PubMed] [Google Scholar]
- [26].Schier JG, Hunt DR, Perala A, et al. Characterizing concentrations of diethylene glycol and suspected metabolites in human serum, urine, and cerebrospinal fluid samples from the Panama DEG mass poisoning. Clinical Toxicology. 2013;51(10):923–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Thammitiyagodage MG, Silva NR, De Rathnayake C, et al. Biochemical and histopathological changes in Wistar rats after consumption of boiled and un-boiled water from high and low disease prevalent areas for chronic kidney disease of unknown etiology (CKDu) in north Central Province (NCP) and its comparison with low disease prevalent Colombo, Sri Lanka. BMC Nephrol. 2020;21(1):38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].McCormick DL. 2017. Preclinical evaluation of carcinogenicity using standard-bred and genetically engineered rodent models. In: Faqi AS, editor. A comprehensive guide to toxicology in non-clinical drug development. 2nd edn. Amsterdam: Elsevier Inc. p. 273–292. [Google Scholar]
- [29].Lohmiller JJ, Swing SP, Hanson MM. 2019. Reproduction and breeding. In: Suckow M, Hankenson FC, Wilson R, Folery P, editors. The laboratory rat. 3rd ed. Amsterdam: Elsevier Inc. p. 157–164. [Google Scholar]
- [30].Landry GM, Dunning CL, Conrad T, et al. Diglycolic acid inhibits succinate dehydrogenase activity in human proximal tubule cells leading to mitochondrial dysfunction and cell death. Toxicol Lett. 2013;221(3):176–184. [DOI] [PubMed] [Google Scholar]
- [31].Conrad T, Landry GM, Aw TY, et al. Diglycolic acid, the toxic metabolite of diethylene glycol, chelates calcium and produces renal mitochondrial dysfunction in vitro. Clin Toxicol. 2016;54(6):501–511. [DOI] [PubMed] [Google Scholar]