

Abstract
Background
Individuals with chronic obstructive pulmonary disease (COPD) or chronic bronchitis may experience recurrent exacerbations, which negatively impact prognosis and quality of life, and can impose a significant socioeconomic burden on the individual and wider society. Immunostimulants are a broad category of therapies that may theoretically enhance non‐specific immunity against several respiratory insults, thereby reducing exacerbation risk and severity. However, evidence to date for their use in this population is limited.
Objectives
To determine the efficacy of immunostimulants in preventing respiratory exacerbations in adults with chronic obstructive pulmonary disease, chronic bronchitis, or both.
Search methods
We used standard, extensive Cochrane search methods. The latest literature search was conducted on 25 January 2022.
Selection criteria
We included parallel randomised controlled trials (RCTs) that compared immunostimulant therapy, administered by any method and with the intention of preventing (rather than treating) exacerbations, with placebo for a minimum treatment duration of one month in adults with chronic bronchitis or COPD, or both. We excluded participants with other respiratory conditions.
Data collection and analysis
We used standard Cochrane methods. Our primary outcomes were number of participants with no exacerbations during the study period and all‐cause mortality, secondary outcomes were respiratory‐related mortality, quality of life, number of participants requiring antibiotics, exacerbation duration, respiratory‐related hospitalisation duration and adverse events/side effects. We used GRADE to assess certainty of evidence for each outcome.
Main results
This review included 36 studies involving 6192 participants. Studies were published between 1981 and 2015. Duration ranged from three to 14 months. The mean age of study participants varied between 35.2 and 82 years. Twelve studies examined participants with COPD only. Seventeen studies reported baseline lung function values; most indicated a moderate‐to‐severe degree of airflow limitation. Nineteen studies indicated inclusion of participants with a mean baseline exacerbation frequency of two or more in the preceding year. Immunostimulants investigated were OM‐85, AM3, RU41740 (Biostim), Ismigen, Diribiotine CK, thymomodulin, pidotimod, D53 (Ribomunyl), Lantigen B, Symbioflor, and hyaluronan; routes of administration were oral, sublingual, and subcutaneous. The risk of bias of the included studies was mostly low or unclear.
Participants receiving immunostimulants for a mean duration of six months were slightly more likely to be free of exacerbations during that time (odds ratio (OR) 1.48, 95% confidence interval (CI) 1.15 to 1.90; 15 RCTs, 2961 participants; moderate‐certainty evidence). The overall number needed to treat with immunostimulants for a mean of six months, to prevent one participant from experiencing an exacerbation, was 11 (95% CI 7 to 29). This outcome was associated with a moderate degree of unexplained heterogeneity (I2 = 53%). Type of immunostimulant, baseline lung function, baseline exacerbation frequency, treatment duration, and follow‐up duration did not modify the effect size, although due to heterogeneity and limited study and participant numbers within some subgroups, the validity of the subgroup treatment effect estimates were uncertain.
Immunostimulants probably result in little to no difference in all‐cause mortality (OR 0.64, 95% CI 0.37 to 1.10; 5 RCTs, 1558 participants; moderate‐certainty evidence) and respiratory‐related mortality (OR 0.40, 95% CI 0.15 to 1.07; 2 RCTs, 735 participants; low‐certainty evidence) compared to placebo; however, the effects were imprecise and data quality limited the certainty of these results.
There was a small improvement in health‐related quality of life, as measured by the St George's Respiratory Questionnaire (SGRQ), with immunostimulant compared to placebo (mean difference −4.59, 95% CI −7.59 to −1.59; 2 RCTs, 617 participants; very‐low certainty evidence). The effect estimate just met the minimum clinically important difference (MCID) score of 4 units; however, the CI width means the possibility of a non‐meaningful difference cannot be excluded.
The pooled result from five studies indicated that immunostimulants likely reduce the number of participants requiring antibiotics over a mean duration of six months (OR 0.34, 95% CI 0.18 to 0.63; 542 participants; moderate‐certainty evidence). This outcome had a low‐to‐moderate degree of heterogeneity (I2 = 38%), but the direction of effect was consistent across all studies.
There was no evidence of a difference in the odds of experiencing an adverse event with immunostimulant compared to placebo, over a mean duration of six months (OR 1.01, 95% CI 0.84 to 1.21; 20 RCTs, 3780 participants; high‐certainty evidence). The CI limits for the associated risk ratio (RR) did not cross thresholds for appreciable harm or benefit (RR 1.02, 95% CI 0.92 to 1.13). An additional seven studies reported no events rates in either study arm.
Meta‐analyses were not performed for the outcomes of exacerbation duration and respiratory‐related hospitalisation duration, due to high levels of heterogeneity across the included studies (exacerbation duration: I2 = 92%; respiratory‐related hospitalisation duration: I2 = 83%). Results from an effect direction plot and binomial probability test for exacerbation duration indicated that a significant proportion of studies (94% (95% CI 73% to 99%); P = 0.0002) favoured intervention, possibly indicating that immunostimulants are efficacious in reducing the mean exacerbation duration compared to placebo. However, the degree of uncertainty associated with this estimate remained high due to data quality and heterogeneity. Three studies reported mean duration of respiratory‐related hospitalisation, two of which demonstrated a direction of effect that favoured immunostimulant over placebo.
Authors' conclusions
In participants with chronic bronchitis or COPD, we are moderately confident that treatment with immunostimulants is associated with a small reduction in the likelihood of having an exacerbation and a moderate reduction in the requirement for antibiotics. Low numbers of events limit interpretation of the effect of immunostimulants on all‐cause and respiratory‐related mortality. We are uncertain whether immunostimulants improve quality of life, and whether they are associated with a reduction in exacerbation and respiratory‐related hospitalisation durations, although immunostimulants were generally associated with a positive effect direction in the studies that examined these outcomes. Immunostimulants appear to be safe and well‐tolerated, and are not associated with an increased risk of adverse events.
Plain language summary
Is taking an immunostimulant on top of standard medications beneficial for people with chronic obstructive pulmonary disease, chronic bronchitis, or both?
Key messages
1. In people with chronic bronchitis or COPD, immunostimulants probably reduce the likelihood of a person having an exacerbation and of requiring antibiotics for an exacerbation.
2. We are uncertain about the effect of immunostimulants on reducing the risk of death, improving quality of life, or on reducing the duration of flare‐ups or hospital stays.
3. Immunostimulants are not associated with an increased risk of side effects.
What are chronic obstructive pulmonary disease and chronic bronchitis?
Chronic obstructive pulmonary disease (COPD) and chronic bronchitis are common conditions that permanently affect the airways of the lungs. They are mainly caused by exposure to cigarette smoke or other air pollutants. People with COPD or chronic bronchitis may develop persistent symptoms of breathlessness, cough, and phlegm production, and are prone to flare‐ups (exacerbations) of these symptoms. Flare‐ups can be debilitating, worsen lung function over time, and cause further exacerbations. Several standard treatments exist to help prevent flare‐ups, which are recommended in almost all people who have a diagnosis of COPD. These mainly include quitting smoking, participating in exercise programmes, obtaining vaccinations to prevent infection, and using specific medications through an inhaler device.
What are immunostimulants?
Immunostimulants are a type of medication that are not widely used for the long‐term management of COPD or chronic bronchitis. Some scientists and doctors have suggested that immunostimulants, added to standard treatment, may help reduce the frequency and severity of flare‐ups in this patient group, by boosting the immune system response to triggers for exacerbations (such as infection with viruses or bacteria).
What did we want to find out?
We wanted to find out whether giving an immunostimulant medication, on top of standard treatment, reduced the frequency of flare‐ups in adults with COPD or chronic bronchitis. We also wanted to know whether immunostimulants reduced the risk of death, improved quality of life, and reduced the duration and severity of flare‐ups.
What did we do?
We searched for studies involving adults with COPD or chronic bronchitis or both, that took place over at least 12 weeks. Studies must have randomly divided participants into receiving either an immunostimulant or placebo (inactive replacement for a medicine), and directly compared the two groups. We included immunostimulants of any type and route of administration, although we did have prespecified criteria as to what constituted an 'immunostimulant' due to this term being relatively broad.
What did we find?
We found 36 studies involving 6192 participants. These looked at a variety of immunostimulants over durations that ranged from three to 14 months.
Results showed that participants receiving immunostimulants were slightly more likely to be free of exacerbations over an average duration of six months, compared to those who had received placebo. Treating 11 people with immunostimulants for six months would prevent one person from experiencing a flare‐up. We also found that immunostimulants likely reduced the number of participants requiring antibiotics for a flare‐up. Immunostimulants appeared to be safe, well‐tolerated, and not associated with an increased risk of side effects.
The impact of immunostimulants on death, quality of life, duration of flare‐ups, and the duration of hospital stays due to a flare‐up was unclear. Immunostimulants were generally favoured over placebo in most studies that looked at these outcomes.
What were the limitations of the evidence?
Overall, we are moderately confident in these results. Our confidence was reduced by how different some results looked between studies, the small number of studies included in some analyses, and in some instances by not having enough data or participant numbers to determine whether immunostimulants were truly better or the same as placebo.
How up‐to‐date is this evidence?
The evidence is up‐to‐date to January 2022.
Summary of findings
Summary of findings 1. Summary of findings table ‐ immunostimulant vs. placebo for adults with chronic bronchitis or chronic obstructive pulmonary disease.
| Patient or population: adults with chronic bronchitis or chronic obstructive pulmonary disease Setting: outpatients Intervention: immunostimulant Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with immunostimulant | |||||
| Number of participants with no exacerbations during the study period follow‐up: mean 6.1 months | Low | OR 1.48 (1.15 to 1.90) | 2961 (15 RCTs) | ⊕⊕⊕⊝ Moderateb,c,d | Immunostimulants likely result in a slight increase in the number of participants with no exacerbations (or inversely, result in a reduction in the number of participants with ≥ 1 exacerbations). | |
| 5 per 100a | 7 per 100 (6 to 9) | |||||
| High | ||||||
| 68 per 100a | 76 per 100 (71 to 80) | |||||
| Mortality (all‐cause) follow‐up: mean 8.4 months | Low | OR 0.64 (0.37 to 1.10) | 1558 (5 RCTs) | ⊕⊕⊕⊝ Moderatee,f | Immunostimulants probably result in little to no difference in all‐cause mortality. | |
| 21 per 1000a | 14 per 1000 (8 to 23) | |||||
| High | ||||||
| 58 per 1000a | 38 per 1000 (22 to 63) | |||||
| Mortality (respiratory‐related) follow‐up: mean 6 months | 4 per 100 | 2 per 100 (1 to 4) | OR 0.40 (0.15 to 1.07) | 735 (2 RCTs) | ⊕⊕⊝⊝ Lowf,g | Immunostimulants may result in little to no difference in respiratory‐related mortality. |
| Quality of life assessed with: St George's Respiratory Questionnaire (SGRQ) Scale from: 0 to 100 follow‐up: mean 4.5 monthsh | The mean quality of life was 37.5 points | MD 4.59 points lower (7.59 lower to 1.59 lower) | ‐ | 617 (2 RCTs) | ⊕⊝⊝⊝ Very lowi,j,k | Immunostimulants may be associated with improvement in health‐related quality‐of‐life scores, but the evidence is very uncertain. |
| Number of participants requiring antibiotics follow‐up: mean 6.6 months | Low | OR 0.34 (0.18 to 0.63) | 542 (5 RCTs) | ⊕⊕⊕⊝ Moderateb,l,m,n | Immunostimulants likely result in a reduction in the number of participants requiring antibiotics. | |
| 60 per 100a | 34 per 100 (21 to 48) | |||||
| High | ||||||
| 73 per 100a | 48 per 100 (33 to 63) | |||||
| Adverse events/side effects follow‐up: mean 6.8 months | Low | OR 1.01 (0.84 to 1.21) | 3780 (20 RCTs) | ⊕⊕⊕⊕ Higho | Immunostimulants do not increase the number of participants with an adverse event. | |
| 6 per 100a | 6 per 100 (5 to 7) | |||||
| High | ||||||
| 44 per 100a | 44 per 100 (39 to 48) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; MD: mean difference; OR: odds ratio | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
| See interactive version of this table: https://gdt.gradepro.org/presentations/#/isof/isof_question_revman_web_429628585692116898. | ||||||
a The lowest and highest risk values are the second‐lowest and second‐highest proportions of participants with no exacerbations in the control groups from the studies included in this review. b Risk of bias across most sectors for included studies was 'low' or 'unclear'. Exclusion of the studies in sensitivity analysis that were classified as 'high' risk of bias for attrition or selective reporting bias (or both) had little effect on the pooled effect estimate. No downgrade for risk of bias. c Moderate clinical and statistical heterogeneity identified (I² = 53%). Heterogeneity could not be explained within subgroups. May partly be explained by variations in treatment regimens. Downgraded once. d Funnel plot was asymmetrical, with several small studies demonstrating a positive effect (Figure 4). However, removal of the five smaller, positive studies by sensitivity analysis demonstrated no impact on the pooled estimate. Not downgraded. e Study contributing the most weight in the analysis involved an elderly population with significant comorbidity, which may limit applicability of results to a general COPD/chronic bronchitis population. However, no impact on the pooled effect estimate when excluded in sensitivity analysis. No downgrade. f Small number of events. Confidence intervals included the null effect and limited suggest that intervention may decrease or increase mortality. Downgraded once for imprecision. g Study contributing the most weight in analysis involved an elderly population with significant comorbidity, which may limit applicability of results to a general COPD/chronic bronchitis population. Given there are only two studies in this meta‐analysis, downgraded once for indirectness. h Lower score indicates better quality of life. i One of two studies presented as abstract only and judged to be 'high' risk for attrition bias. Downgraded once for risk of bias. j The minimally clinically important difference (MCID) for SGRQ is 4 points. The confidence interval did not include the null effect but the lower limit did not clear the MCID. Downgraded once for imprecision. k Two studies included in meta‐analysis, from the same author groups. One study was presented as an abstract. Uncertain that there was a large enough body of evidence to affirm this as a true result. Downgraded once for risk of publication bias. l Moderate clinical heterogeneity (I² = 38%) likely due to clinical and methodological diversity; however, uniform direction of effect estimate across individual studies. Downgraded once. m One included study involved participants with COPD and 'borderline immune deficiency'. Exclusion of this study by sensitivity analysis did not have an effect on the pooled effect estimate. Not downgraded. n Confidence interval limit for risk ratio crossed the 25% relative risk reduction threshold; however, did not include the null effect and optimal information size (OIS) criteria were met. Not downgraded. o The confidence interval included the null effect, but the limits did not cross the 25% relative risk threshold for appreciable benefit or harm and OIS criteria were met. Not downgraded.
Background
Description of the condition
Chronic obstructive pulmonary disease (COPD) is defined by the Global Initiative for Chronic Obstructive Lung Disease (GOLD) as "a common, preventable and treatable disease which is characterised by persistent respiratory symptoms and airflow limitation that is due to airway and/or alveolar abnormalities, usually caused by significant exposure to noxious particles or gases" (GOLD 2022). Globally, COPD is a major cause of morbidity and mortality. In 2015, there was an estimated prevalence of 174 million cases, with three million deaths attributable to COPD (GBD 2015). Currently, COPD is considered the third leading cause of death worldwide (WHO GHE 2016). These numbers are projected to increase further over the next 30 years due to a combination of population ageing and ongoing exposure to COPD risk factors (Lopez 2006; WHO 2018). The economic burden associated with COPD is also substantial, with direct and indirect costs placing significant financial strain on individuals, their families, wider society, and healthcare systems worldwide (ATS Foundation 2014; Jinjuvadia 2017).
The symptoms of COPD include dyspnoea (breathlessness), chronic cough, and sputum production. COPD encompasses a range of clinical phenotypes, including emphysema and chronic bronchitis, with chronic bronchitis classically being defined as chronic cough and sputum production for at least three months per year for two consecutive years (Ferris 1978). Alternative definitions of chronic bronchitis exist, including cough and phlegm almost every day or several times a week (Kim 2015). Whilst chronic bronchitis is not technically defined by airflow limitation, it may precede the development of this, and is still thought to be associated with airway disease and inflammation, an increased risk in the total number and severity of respiratory exacerbations, and functional limitations (Kim 2011; Woodruff 2016).
A COPD exacerbation is defined as an acute worsening of respiratory symptoms that results in additional treatment (Wedzicha 2007). Exacerbations are often associated with increased airway inflammation, gas trapping, and mucous production (GOLD 2022); these changes typically lead to symptoms of increased dyspnoea, alteration in sputum colour or volume, increased cough and wheeze, or a combination of these. Most COPD exacerbations are triggered by viral or bacterial respiratory infections (or both); however, environmental changes and air pollution may also play a role in either causing or worsening exacerbations (GOLD 2022; Woodhead 2011). Studies have suggested that viruses are the causative pathogen in 34% to 56% of COPD exacerbations (Mohan 2010; Papi 2006; Rohde 2003), with bacterial infections reportedly associated with up to 50% of exacerbations (Papi 2006). Additionally, viral and bacterial coinfection is common, and has been shown to correlate with an increased severity of exacerbations and longer duration of hospitalisation (Papi 2006; Singanayagam 2012).
It is widely known that respiratory exacerbations in COPD are associated with increased mortality, accelerated decline in lung function, increased hospitalisation and readmission rates, and decreased quality of life (Kanner 2001; Soler‐Cataluña 2005). In addition, a history of previous exacerbations is said to be the single biggest risk factor for future exacerbations (Hurst 2010). Some people with COPD are more prone to frequent exacerbations (defined as two or more exacerbations per year) and this group has been shown to have worse outcomes and morbidity than those who experience less‐frequent exacerbations (Seemungal 1998). Aside from impacting the health status and prognosis of individuals, exacerbations also impose a significant socioeconomic burden on society, particularly those that necessitate hospital admission.
A number of evidence‐based therapies exist to reduce symptoms and exacerbations, and improve lung function, exercise tolerance, and quality of life, in people with COPD. Key aspects of COPD management include smoking cessation, exercise, pulmonary rehabilitation, and regular vaccinations for both influenza and pneumococcal infections (GOLD 2022). Other non‐pharmacological options for some people include treatment of hypoxaemia with long‐term oxygen therapy (Cranston 2005), treatment of hypercapnia with long‐term non‐invasive ventilation (Köhnlein 2014), and surgical or bronchoscopic lung volume reduction procedures (Marruchella 2018). Pharmacologically, the mainstay of treatment in stable COPD involves inhaled bronchodilators, including beta‐agonists and anti‐muscarinic agents (GOLD 2022; Kew 2013; Tashkin 2008). If people still have a high symptom or exacerbation burden, the addition of inhaled corticosteroids to a long‐acting beta‐agonist is recommended (Nannini 2012). A number of oral anti‐inflammatory agents also reduce exacerbations in COPD, and are currently recommended for use in some people, including phosphodiesterase‐4 inhibitors, mucolytic agents, and macrolide antibiotics (Chong 2013; Ni 2015; Poole 2015).
Description of the intervention
A glossary of the main immunological terms used is provided in Appendix 1.
The 2022 GOLD guidelines have specifically mentioned use of immunostimulant or immunoregulatory agents for preventing exacerbations in people with COPD (GOLD 2022). Immunostimulants are defined as agents that create a state of non‐specific immunity and enhance the immune response towards infection or malignancy (Hadden 1993). They have existed for many years, and have long been suggested to have efficacy in preventing or reducing the severity of acute respiratory tract infections (ARTIs).
In some countries, immunostimulants are regularly used for the prevention of ARTIs in children, and for reducing the frequency and severity of exacerbations in adults with COPD or chronic bronchitis (Del‐Rio‐Navarro 2007). However, their widespread and routine use has been limited due to a shortage of high‐quality data regarding their efficacy, and a lack of understanding of their mechanisms of action and long‐term safety profiles. The 2022 GOLD guidelines acknowledge that, whilst older studies have demonstrated the efficacy of immunostimulants in reducing the severity and frequency of COPD exacerbations, further studies are needed to examine their effects in people who are receiving current 'gold‐standard' COPD maintenance therapy (Collet 1997; Li 2004).
The immunostimulant agents that have been studied and used for the purpose of preventing ARTIs fall into three main categories: bacteria‐derived agents, synthetic agents, and thymic extracts (Del‐Rio‐Navarro 2007). Most immunostimulants used in the prevention of ARTIs are orally or sublingually administered bacteria‐derived agents. These can be further categorised into those that are inactivated whole‐cell formulations, those that contain a mixture of antigenic fragments derived from several bacterial strains (bacterial lysates), and those that consist of a specific immunogenic component of a bacterium, such as ribosomal fractions or glycoproteins (Cazzola 2008; Del‐Rio‐Navarro 2007; Giovannini 2014).
Bacterial lysates are composed of constituted fragments of bacterial antigens, obtained through the chemical or mechanical lysis (breakdown) of multiple inactivated bacterial strains that are commonly associated with respiratory infections, such as Staphylococcus aureus, Streptococcus pneumoniae, Haemophilus influenzae, Klebsiella pneumoniae, Moraxella catarrhalis, Streptococcus pyogenes, and Streptococcus viridans (De Benedetto 2013).
The commercial availability of these agents varies by country, region, and clinical indication for use. As examples, a range of bacterial lysates have been approved for use for the prevention of recurrent respiratory tract infections in several European countries, including OM‐85, Ismigen, Ribomunyl, Lantigen B, and LW50020 (EMA 2019). OM‐85 is available and used across Central and South America for the treatment and prophylaxis of respiratory infections in children and adults (Pivniouk 2022). In China, several immunostimulant agents are approved for use including OM‐85 and pidotimod (a synthetic immunostimulant agent) (CMDD 2016; Pivniouk 2022). Conversely, immunostimulants are not currently approved for use in North America or Australasia.
How the intervention might work
Immunostimulants used for the purpose of preventing respiratory tract infections in people with COPD or chronic bronchitis aim to heighten the host immune response against infective insults that may subsequently trigger an exacerbation. However, despite there being much research and knowledge gained about the effects of individual immunostimulatory agents on the immune system, the exact mechanism of action of both synthetic and bacteria‐derived agents at a molecular level is not completely understood (De Benedetto 2013; Del‐Rio‐Navarro 2007).
OM‐85, a bacterial lysate derived by the chemical lysis of a number of the aforementioned bacterial strains, is thought to exert its effects through both cell‐mediated and humoral immune system pathways (De Benedetto 2013; Rozy 2008). These include augmentation of the T helper cell lymphocyte (Th1) response (Huber 2005), induction of specific immunoglobulin A antibody secretion by B lymphocyte cells (Rial 2004; Rossi 2003), direct activation of lung macrophages and monocytes (Mauel 1989; Rozy 2008), upregulation of adhesion molecules (Duchow 1992), and stimulation of phagocytic cell activity (Rozy 2008). It is thought that the immunostimulant components of OM‐85 bind to toll‐like receptors (TLRs), triggering signalling pathways that lead to activation and potentiation of the innate immune response (Alyanakian 2006; Huber 2005; Navarro 2011; Nikolova 2009).
Mechanical bacterial lysates, specifically polyvalent mechanical bacterial lysates (PMBLs), have been demonstrated to stimulate dendritic cell maturation (Morandi 2011), increase the number of circulating natural killer (NK) cells (Lanzilli 2013), increase specific immunoglobulin A antibody secretion by B cells, (Rossi 2003), and activate both memory B lymphocytes and regulatory T lymphocytes (Lanzilli 2013). Studies have shown that the degree of the immune response created by the administration of mechanical bacterial lysates to patients directly correlates with positive clinical outcomes, such as reduced exacerbation frequency (Braido 2011; Lanzilli 2006; Ricci 2014).
Other bacterial extracts, made up of bacterial proteins or ribosomal fragments, also have specific immunomodulatory effects. For example, the immunostimulant RU41740 (Biostim), made up of Klebsiella pneumoniae glycoproteins and membrane fragments, activates macrophages, stimulates the B lymphocyte cell response (Boissier 1988), and enhances antigen presentation (Pedraza‐Sánchez 2006). Like bacterial lysates, RU41740 is thought to initiate an immune response through binding of its molecular components (such as lipopolysaccharide) to TLRs (Miller 2005).
The immunostimulants containing thymic extracts also appear to interact with other TLRs and precursor T lymphocytes to increase dendritic cell, NK cell, and T cell activity, thus enhancing both innate and cell‐mediated immune responses (Del‐Rio‐Navarro 2007; Tuthill 2013). Synthetic immunostimulant compounds are reportedly better understood in terms of their molecular mechanism of action (Del‐Rio‐Navarro 2007). Examples of synthetic immunostimulants include: tucaresol, which acts by promoting the interaction between antigen‐presenting cells and T cells (Rhodes 1996); imiquimod, which acts through TLR7 and TLR8 (Spaner 2005); and pidotimod, which acts by enhancing cell‐mediated immunity (Benetti 1994).
A number of trials and systematic reviews have analysed the use of immunostimulant in preventing ARTIs in children and adults, and in preventing exacerbations in adults with chronic bronchitis or COPD. One previous Cochrane Review evaluated immunostimulants for preventing respiratory tract infections in children. The review authors included 34 placebo‐controlled trials, and reported that immunostimulants were associated with approximately 40% fewer acute respiratory infections compared with placebo. However, they also commented that "trial quality was generally poor and a high level of statistical heterogeneity was evident" (Del‐Rio‐Navarro 2012). One meta‐analysis examining the efficacy of PMBLs in preventing respiratory tract infections in children and adults looked at data across 15 randomised controlled trials. Treatment with PMBLs was associated with a significant reduction in respiratory infections compared with placebo (Cazzola 2012).
One systematic review found that the bacterial extracts OM‐85, LW‐50020, and SL‐04 were associated with improved symptoms in people with COPD, chronic bronchitis, or both, and the meta‐analysis suggested a lessened exacerbation duration. However, there was no evidence of a difference between the extracts and placebo in preventing exacerbations (Steurer‐Stey 2004). Another systematic review examined the efficacy of OM‐85 in preventing exacerbations in people with COPD or chronic bronchitis (or both). There was a non‐significant trend in favour of OM‐85; however, benefit was not clearly demonstrated across a range of important clinical outcomes (Sprenkle 2005). In 2015, one meta‐analysis and systematic review examined the effects of OM‐85 in people with COPD on exacerbation rate, in addition to several other minor clinical end points. OM‐85 was associated with a 20% reduction in exacerbation rate and 39% reduction in the incidence rate of people using antibiotics compared with placebo (Pan 2015). However, the authors concluded that there was insufficient evidence to support the routine use of OM‐85 in people with COPD, suggesting that further larger‐scale trials needed to be undertaken.
The cost‐effectiveness of the immunostimulant OM‐85 for preventing respiratory exacerbations in adults with COPD or chronic bronchitis (or both) has previously been examined. Prospective cost‐effectiveness and cost–benefit analyses were conducted alongside a randomised controlled trial comparing OM‐85 to placebo in adults with COPD (Collet 1997). Authors found that its use was associated with a reduction in the direct and indirect costs of a severe exacerbation warranting hospitalisation (Collet 2001). Other studies have also found that OM‐85 appears to be cost‐effective for the prevention of acute respiratory exacerbations in adults with chronic bronchitis (Bergemann 1994; Xuan 2014).
Why it is important to do this review
Immunostimulant agents in COPD or chronic bronchitis could theoretically enhance non‐specific immunity against a number of respiratory insults, which is an important concept given the number of pathogens, including myriad viruses, that can precipitate an exacerbation. However, the use of immunostimulants in this population thus far has been controversial. This is largely due to concerns about quantity and quality of evidence in the past, a lack of understanding of their long‐term effects, and existing uncertainty around their exact mechanisms of action. As mentioned, GOLD guidelines recognise that some older studies have reported a decrease in the severity and frequency of COPD exacerbations, but more trials are needed in people receiving currently recommended maintenance therapy (GOLD 2022). Overall, there have been mixed results from previous trials and systematic reviews regarding clinical efficacy of immunostimulants in adults with COPD or chronic bronchitis; many have reported positive impacts on exacerbation rates (or a trend towards this) and other clinically relevant outcomes, but have been unable to suggest an overall benefit in these patients due to limited availability of data, poor trial quality, or both.
This systematic review aims to critically appraise all available data regarding the efficacy and use of immunostimulants as a preventive therapy in adults with stable COPD or chronic bronchitis (or both). This may help to further understand their clinical value and safety, and may highlight areas requiring further research and development.
Objectives
To determine the efficacy of immunostimulants in preventing respiratory exacerbations in adults with chronic obstructive pulmonary disease, chronic bronchitis, or both.
Methods
Criteria for considering studies for this review
Types of studies
We included parallel randomised controlled trials (RCTs) comparing immunostimulant therapy, administered by any route, with placebo. We included studies reported in full text, those published as an abstract only, and unpublished data. We excluded cross‐over trials due to the nature of COPD as a progressive disease and due to the possibility of a carry‐over effect from the first treatment period. However, if these studies presented separate comparison data prior to any cross‐over occurring then this data set was considered for review inclusion.
Types of participants
We included studies of adults (older than 18 years of age) with a diagnosis of COPD (defined by a postbronchodilator forced expiratory volume in one second (FEV1)/forced vital capacity (FVC) ratio less than 0.7) or chronic bronchitis (defined by either the classic definition of chronic cough and sputum production for at least three months per year for two consecutive years (Ferris 1978), or alternative definitions such as cough and phlegm almost every day or several times a week (Kim 2015)), or both COPD and chronic bronchitis. An underlying principle was that study participants met well‐established criteria at the time, whether for COPD or chronic bronchitis.
We excluded studies of participants with asthma, bronchiectasis, or genetic/other lung conditions that predispose or lead to chronic airflow obstruction, such as cystic fibrosis, or people with known specific immunodeficiencies. However, if a study included these types of participants and additionally included people with COPD or chronic bronchitis (or both), we analysed data for these subsets of participants if presented separately.
Types of interventions
Participants must have received immunostimulant therapy or placebo, administered by any route (oral, sublingual, subcutaneous, or intravenous), for at least one month. Where a study involved an intermittent dosing regimen, in which the cumulative treatment days totalled less than one month, but the overall treatment duration was at least one month, we elected to include this study in the review.
We excluded studies of specific immunostimulants, such as influenza or pneumococcal vaccines, immunotherapy used to treat cancer (either directly, or the immune deficiency resulting from chemotherapy), allergic disease, and treatment to replace immunoglobulins in known specific immune deficiency disorders. We excluded trials referring to vitamins, nutritional supplements, herbal extracts, or homeopathic remedies. We excluded studies that focussed on the treatment of acute exacerbations with immunostimulants (as opposed to prevention and prophylaxis). We did not consider studies that focussed on improvement in immunological parameters as the sole outcome.
Types of outcome measures
We only included studies where the primary outcomes were measured for at least 12 weeks.
Reporting of one or more of the listed outcomes was not an inclusion criterion for the review.
Exacerbations were chosen as the primary outcome as immunostimulant therapy is most commonly administered as a preventive therapy, and thus far exacerbations have represented the main clinically relevant end point used for efficacy of immunostimulant agents and many other anti‐inflammatory drugs used in COPD or chronic bronchitis (GOLD 2022). As mentioned, exacerbations significantly affect patient morbidity and mortality and have a significant impact on overall disease burden; therefore, we considered that exacerbations and mortality were important as primary outcomes in this review.
Primary outcomes
Number of participants with no exacerbations during the study period.
Mortality (all‐cause).
There was considerable variability in the degree of reporting and description of what constituted a respiratory exacerbation between included studies; hence, we accepted the study authors' definition of an exacerbation.
As outlined in our review protocol (Fraser 2019), we had intended to look at and delineate the number of participants with exacerbations of COPD or chronic bronchitis (or both) that would be considered moderate or severe in accordance with GOLD criteria definitions (GOLD 2022), and use this information to inform our primary outcomes along with all‐cause mortality. However, following the data extraction process and prior to performing any statistical analysis, it was apparent that these outcomes could not be determined from the information and data presented in the included studies.
In order to allow us to combine and interpret our review outcomes, a deviation from our original intended primary outcomes was considered necessary. Therefore, we chose the dichotomous outcome of 'number of participants with no exacerbations during the study period.'
Secondary outcomes
Mortality (respiratory‐related).
Quality of life (participant‐reported, measured by a validated scale, such as the St George's Respiratory Questionnaire (SGRQ) (Jones 1992) or Chronic Respiratory Diseases Questionnaire (CRQ) (Guyatt 1987)).
Number of participants requiring antibiotics.
Exacerbation duration.
Hospitalisation duration (respiratory‐related).
Adverse events/side effects.
The secondary outcomes are other important measures of the efficacy and safety of immunostimulant agents.
Our protocol outlined our intentions to include the 'total number of exacerbations' as a secondary outcome; however, including this was no longer necessary given the reformulation of the primary outcomes that would capture this information. Analysing exacerbation duration was considered to provide indirect information regarding the potential socioeconomic and quality‐of‐life impacts of the intervention. Quality‐of‐life assessments highlight information regarding the impact of the intervention on the objective and subjective wellbeing of the patient, and are an important outcome in any chronic disease. Given that little is understood about the long‐term safety of these agents and concerns around this have previously limited recommendation for widespread use, adverse events were included as a secondary outcome for this review.
We added 'Number of participants requiring antibiotics' and 'hospitalisation duration (respiratory‐related)' as secondary outcome measures following data extraction and prior to data synthesis occurring. These were metrics reported in several included studies, and were considered surrogate markers of exacerbation severity in lieu of being able to obtain clear data around how many of the exacerbations could be considered 'moderate' or 'severe' (or both) by GOLD definitions (GOLD 2022). Several studies reported 'Antibiotic duration'; however, we considered that a dichotomous outcome regarding antibiotic use would be more statistically robust. Conversely, the 'number of participants requiring hospitalisation' was rarely reported, and inclusion as an outcome would have contributed little to overall comparison.
Search methods for identification of studies
Electronic searches
We identified studies from searches of the following databases and trial registries:
Cochrane Airways Trials Register (airways.cochrane.org/trials-register), via the Cochrane Register of Studies, all years to 25 January 2022;
Cochrane Central Register of Controlled Trials (CENTRAL), via the Cochrane Register of Studies, all years to 25 January 2022;
MEDLINE (OvidSP) ALL 1946 to 25 January 2022;
Embase (OvidSP) 1974 to 25 January 2022;
US National Institutes of Health Ongoing Trials Register ClinicalTrials.gov (www.clinicaltrials.gov);
World Health Organization International Clinical Trials Registry Platform (apps.who.int/trialsearch).
The database search strategies are detailed in Appendix 2. The Cochrane Airways Information Specialist in collaboration with the review authors wrote the search strategies and executed the searches.
We searched all databases and trials registries from their inception to 25 January 2022, with no restrictions on language or type of publication. We handsearched conference abstracts and grey literature identified through the Cochrane Airways Trials Register and the CENTRAL database.
Searching other resources
We checked the reference lists of all primary studies and review articles for additional references. We searched relevant manufacturers' websites for study information.
Data collection and analysis
Selection of studies
We used Cochrane's Screen4Me workflow to help assess the search results. Screen4Me comprises three components: known assessments – a service that matches records in the search results to records that have already been screened in Cochrane Crowd and been labelled as an RCT or as Not an RCT; the RCT classifier – a machine learning model that distinguishes RCTs from non‐RCTs, and if appropriate, Cochrane Crowd – Cochrane's citizen science platform where the Crowd help to identify and describe health evidence (Thomas 2020). We used the first two components.
Following this initial assessment, two review authors (AF and PP) independently screened the titles and abstracts of the search results and coded them as 'retrieve' (eligible or potentially eligible/unclear) or 'do not retrieve.' We retrieved the full‐text study reports of all potentially eligible studies and two review authors (AF and PP) independently screened them for inclusion, recording the reasons for exclusion of ineligible studies. Where we could not obtain the full‐text study reports, we marked these studies as 'awaiting classification.' We resolved any disagreements through discussion. We identified and excluded duplicates and collated multiple reports of the same study so that each study, rather than each report, was the unit of interest in the review. Seventeen translators reviewed papers published in languages other than English; if studies met initial screening criteria and were subsequently marked for inclusion, the translators completed formal data extraction sheets for individual studies. We recorded the selection process in sufficient detail to complete a PRISMA flow diagram (Moher 2009), and documented justification for each study exclusion (see Characteristics of excluded studies table and Excluded studies).
Data extraction and management
We extracted and collated data using the online software tool Covidence (Covidence), and exported online data extraction forms for individual studies to Excel worksheets. We extracted the following study characteristics from included studies using the Covidence data extraction process as a template.
Methods: study design, total duration of study, number of study centres and location, study setting, date of study.
Participants: number (n), mean age, age range, gender, severity of condition, diagnostic criteria, baseline lung function, smoking history, baseline exacerbation frequency, inclusion criteria and exclusion criteria.
Interventions: intervention, comparison, dosing regimen, type of compound, route of administration, duration of therapy.
Outcomes: primary and secondary outcomes specified and collected, and time points reported.
Notes: funding for studies and notable conflicts of interest of trial authors.
Two review authors (AF and PP) independently extracted outcome data from included studies. These data were then cross‐checked between the two review authors and a consensus decision reached by discussion for each included study. We extracted data if it was deemed to be potentially relevant to protocol‐specified outcomes; the Characteristics of included studies table and Table 2 identify the reported study outcomes for which we extracted data. One review author (AF) transferred data into the Review Manager 5 (Review Manager 2014), and double‐checked data against the data presented within the original study reports. The second review author (PP) spot‐checked study characteristics for accuracy against the study report.
1. Characteristics of included studies.
| Study ID | Total na | Study duration (weeks) | Mean age (years) | Participant type | Presence of acute exacerbation as an inclusion criteria | Intervention (total duration) | Category | Route | Relevant outcomes measured |
| Alvarez‐Mon 2005 | 344 | 26 | 67.7 | COPD | No | AM3 1 g 3 times daily (6 months) |
Candida utilis polysaccharide/protein compound | Oral | Participants with/without an exacerbation, number of exacerbations, SGRQ score, AEs |
| Alvarez‐Sala 2003 | 364 | 13 | 57.7 | COPD | No | AM3 3 g daily (3 months) |
Candida utilis polysaccharide/protein compound | Oral | SGRQ score |
| Anthoine 1985 | 110 | 26 | 62.9 | CB/COPD (data extracted limited to COPD patient subset) |
No | RU41740 (Biostim) 2 mg daily for 8 consecutive days in first month; 1 mg daily for 8 consecutive days in second/third months (3 months) |
Bacteria‐derived | Oral | Participants with/without an exacerbation, number of exacerbations, exacerbation duration, number of participants requiring antibiotics, AEs |
| Bisetti 1994 | 181 | 17 | 62.3 | CB | No | Pidotimod 800 mg daily (2 months) |
Synthetic agent | Oral | Participants with/without an exacerbation, AEs |
| Blaive 1982 | 184 | 52 | 69.2 | COPD (asthma patient subset excluded from analysis) |
No | D53 (Ribomunyl) 4 sequences of 15 days of aerosol treatment separated by 1‐week intervals. Subcutaneous injections days 7 and 14 of first sequence and day 14 of following sequences (2.7 months) |
Bacteria‐derived | Aerosol and subcutaneous | Mean number of exacerbations per participant, exacerbation duration, participants with no or a reduction in antibiotic therapy, AEs |
| Bonde 1986 | 172 | 26 | 60.5 | CB/COPD | No | RU41740 (Biostim) 2 mg daily or 8 mg daily (2 intervention groups) for 1 week, alternate weeks (3 months) |
Bacteria‐derived | Oral | Participants with/without an exacerbation, exacerbation duration, duration of antibiotic therapy, AEs, mortality (all‐cause) |
| Bongiorno 1989 | 40 | 17 | 70.0 | CB/COPD | No | AM3 500 mg 3 times daily (4 months) |
Candida utilis polysaccharide/protein compound | Oral | Participants with/without an exacerbation, mean number of exacerbations per participant, number of exacerbations, exacerbation duration, AEs |
| Braido 2015 | 288 | 52 | 69.0 | COPD | No | Ismigen 50 mg daily for 10 consecutive days/month for 3 months, then 3 months without treatment, then repeat of the initial regimen (9 months) |
Polyvalent mechanical bacterial lysate | Sublingual | Participants with/without an exacerbation, exacerbation rate, days to first exacerbation, hospitalisation days (respiratory and all‐cause), participants requiring concomitant medications, QoL scale scores, AEs, mortality (all‐cause) |
| Carlo 1990 | 40 | 13 | 65.0 | COPD | No | AM3 500 mg 3 times daily (3 months) |
Candida utilis polysaccharide/protein compound | Oral | Mean number of exacerbations per participant, AEs |
| Catena 1992 | 236 | 13 | 64.9 | COPD plus "cell‐mediated immune deficiency" | No | Thymomodulin 60 mg twice daily (3 months) |
Thymic extract | Oral | Exacerbation rate, QoL scale scores, AEs |
| Cazzola 2006 | 178 | 13 | 66.5 | COPD | No | Ismigen 50 mg daily for 10 consecutive days/month for 3 months (3 months) |
Polyvalent mechanical bacterial lysate | Sublingual | Exacerbation rate, exacerbation duration, duration of antibiotic therapy, hospitalisation rate (respiratory), hospitalisation duration (respiratory), hospitalisation days (respiratory), AEs, mortality (all‐cause) |
| Ciaccia 1994 | 494 | 22 | 65.7 | CB/COPD | No | Pidotimod 800 mg daily (2 months) |
Synthetic agent | Oral | Exacerbation rate, days to first exacerbation, exacerbation duration, duration of antibiotic therapy, AEs |
| Collet 1997 | 381 | 26 | 66.1 | COPD | No | OM‐85 7 mg daily for 1 month, then 7 mg daily for 10 consecutive days/month for 3 months (4 months) |
Bacterial lysate | Oral | Participants with/without an exacerbation, mean number of exacerbations per participant, hospitalisation rate (respiratory and all‐cause), participants requiring hospitalisation (respiratory and all‐cause), hospitalisation duration (respiratory), hospitalisation days (respiratory and all‐cause), mean number of hospital days per participant (respiratory), change in SF‐36 scale scores, AEs, mortality (respiratory and all‐cause) |
| Cvoriscec 1989 | 104 | 26 | 48.2 | CB/COPD | Yes | OM‐85 7 mg daily for 1 month, no treatment for 1 month, then 7 mg daily for 10 consecutive days/month for 3 months (5 months) |
Bacterial lysate | Oral | Exacerbation duration, total exacerbation days, participants requiring antibiotics, participants requiring bronchodilator therapy, FEV1, AEs |
| Debbas 1990 | 265 | 26 | 81.8 | CB/COPD | Yes | OM‐85 7 mg daily for 10 consecutive days/month for 3 months (3 months) |
Bacterial lysate | Oral | Participants with/without an exacerbation, number of exacerbations, participants requiring antibiotic therapy, AEs |
| De Bernardi 1992 | 60 | 17 | 62.7 | CB plus "borderline immune deficiency" | No | Lantigen B (2 intervention groups) 15 drops twice daily for 1 month, then 1 month without treatment, then a 15 days of initial regimen (2.5 months) |
Bacterial lysate | Sublingual | Mean number of exacerbations per participant, exacerbation duration, participants requiring antibiotics, AEs |
| Djuric 1989 | 59 | 26 | 45.5 | CB | Yes | OM‐85 7 mg daily for 1 month, no treatment for 1 month, then 7 mg daily for 10 consecutive days/month for 3 months (5 months) |
Bacterial lysate | Oral | Mean number of exacerbations per participant, exacerbation duration, duration of concomitant therapy, FEV1 |
| EUCTR2007‐004702‐27‐DE | 357 | 26 | Not reported | COPD | No | OM‐85 7 mg daily for 1 month, no treatment for 1 month, then 7 mg daily for 10 consecutive days/month for 3 months (5 months) |
Bacterial lysate | Oral | Participants with/without an exacerbation, number of exacerbations, exacerbation duration, days to first exacerbation, duration of concomitant therapy, hospitalisation rate and duration (all‐cause), SGRQ scores, FEV1, AEs/serious AEs, mortality (all‐cause) |
| Fietta 1988 | 29 | 39 | 57.0 | CB/COPD | No | RU41740 (Biostim) 2 mg daily for 8 consecutive days in first month; 1 mg daily for 8 consecutive days in second/third months (3 months) |
Bacteria‐derived | Oral | Participants with/without an exacerbation, mean number of exacerbations per participant, exacerbation duration, AEs |
| Foschino 1995 | 64 | 26 | 46.0 | CB | No | D53 (Ribomunyl) 1 tablet (dose not specified) daily for 4 consecutive days/week for 3 weeks, then 1 tablet daily for 4 consecutive days/month for 5 months (5.75 months) |
Bacteria‐derived | Oral | Mean number of exacerbations per participant, exacerbation duration, participants requiring antibiotics, AEs |
| Habermann 2001 | 136 | 60 | 47.3 | CB | No | Symbioflor 30 drops 3 times daily (6 months) |
Bacteria‐derived | Oral vs sublingual (liquid preparation) | Participants with/without an exacerbation, number of exacerbations, days to first exacerbation, participants requiring antibiotics, AEs |
| Hutas 1994 | 114 | 26 | 51.7 | CB/COPD | No | OM‐85 7 mg daily for 1 month, no treatment for 1 month, then 7 mg daily for 10 consecutive days/month for 3 months (5 months) |
Bacterial lysate | Oral | Exacerbation duration, duration of antibiotic therapy |
| Keller 1984 | 81 | 26 | 57.0 | CB/COPD | Yes | OM‐85 7 mg daily for 1 month, no treatment for 1 month, then 7 mg daily for 10 consecutive days/month for 3 months (5 months) |
Bacterial lysate | Oral | Number of exacerbations, duration of antibiotic therapy, duration of corticosteroid therapy, hospitalisation rate (respiratory) |
| Li 2004 | 90 | 52 | 66.0 | CB/COPD | No | OM‐85 7 mg daily for 10 consecutive days/month for 3 months (3 months) |
Bacterial lysate | Oral | Mean number of exacerbations per participant, exacerbation duration, duration of antibiotic therapy, AEs |
| Menardo 1985 | 44 | 26 | 47.7 | COPD | No | Diribiotine CK 10 mL daily for 20 consecutive days/month for 3 months (3 months) |
Bacterial lysate | Oral (liquid preparation) | Participants with/without an exacerbation, mean number of exacerbations per participant, exacerbation duration, participants requiring antibiotics, AEs |
| Messerli 1981 | 79 | 26 | 55.1 | CB | Yes | OM‐85 7 mg daily for 10 consecutive days/month for 3 months (3 months) |
Bacterial lysate | Oral | AEs |
| Olivieri 2011 | 340 | 22 | Not reported | COPD | No | OM‐85 7 mg daily for 1 month, no treatment for 1 month, then 7 mg daily for 10 consecutive days/month for 3 months (5 months) |
Bacterial lysate | Oral | Exacerbation rate, exacerbation duration, AEs |
| Orcel 1994 | 354 | 26 | 82.0 | CB/COPD | No | OM‐85 7 mg daily for 10 consecutive days/month for 3 months (3 months) |
Bacterial lysate | Oral | Participants with/without an exacerbation, number of exacerbations, mean number of antibiotic courses, participants requiring bronchodilator therapy, participants requiring corticosteroids, AEs, mortality (respiratory and all‐cause) |
| Orlandi 1983 | 19 | 17 | 52.8 | CB | No | OM‐85 7 mg daily for 1 month, then 7 mg daily for 10 consecutive days/month for 3 months (4 months) |
Bacterial lysate | Oral | Mean number of exacerbations per participant, exacerbation duration, hospitalisation duration (respiratory), mean number of hospitalisations per participant (respiratory), AEs |
| Rico 1997 | 88 | 52 | Not reported | CB/COPD | No | Thymomodulin 80 mg 3 times daily (3 months) |
Thymic extract | Oral | Mean number of exacerbations per participant, exacerbation duration, hospitalisation duration (all‐cause), mean number of concomitant medication courses, duration of concomitant therapies, AEs |
| Rochemaure 1988 | 55 | 26 | 55.7 | CB | No | Diribiotine CK 10 mL daily for 20 consecutive days/month for 3 months (3 months) |
Bacterial lysate | Oral (liquid preparation) | Mean number of exacerbations per participant, exacerbation duration, mean number of antibiotic courses, duration of antibiotic therapy, AEs |
| Soler 2007 | 273 | 26 | 57.6 | CB/COPD | Yes | OM‐85 7 mg daily for 1 month, no treatment for 1 month, then 7 mg daily for 10 consecutive days/month for 3 months (5 months) |
Bacterial lysate | Oral | Participants with/without an exacerbation, mean number of exacerbations per participant, number of exacerbations, participants requiring concomitant medication, AEs, mortality (all‐cause) |
| Tag 1993 | 50 | 26 | 35.2 | CB | No | OM‐85 7 mg daily for 1 month, no treatment for 1 month, then 7 mg daily for 10 consecutive days/month for 3 months (5 months) |
Bacterial lysate | Oral | Mean number of exacerbations per participant, exacerbation duration, duration of antibiotic therapy, duration of bronchodilator therapy, AEs |
| Tang 2015 | 428 | 22 | 63.1 | CB/COPD | Yes | OM‐85 7 mg daily for 10 consecutive days/month for 3 months (3 months) |
Bacterial lysate | Oral | Participants with/without an exacerbation, participants requiring concomitant medication, AEs |
| Venge 1996 | 29 | 35 | 61.0 | CB/COPD | No | Hyaluronan 7.5 mg subcutaneously weekly (6 months) |
Synthetic agent | Subcutaneous | Participants with/without an exacerbation, number of exacerbations, total exacerbation days, total antibiotic days, AEs |
| Xinogalos 1993 | 62 | 26 | 57.9 | COPD | No | OM‐85 7 mg daily for 1 month, no treatment for 1 month, then 7 mg daily for 10 consecutive days/month for 3 months (5 months) |
Bacterial lysate | Oral | Number of exacerbations, exacerbation duration, participants requiring antibiotics, participants requiring bronchodilator therapy |
AE: adverse events; CB: chronic bronchitis; COPD: chronic obstructive pulmonary disease; QoL: quality of life; SF‐36: 36‐item Short Form Survey; SGRQ: St George's Respiratory Questionnaire. aTotal n: number of participants for whom outcome data were available.
Assessment of risk of bias in included studies
Two review authors (AF and PP) independently assessed risk of bias for each study using the RoB 1 tool for RCTs according to criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We resolved any disagreements by discussion. We assessed risk of bias according to the following domains.
Random sequence generation.
Allocation concealment.
Blinding of participants and personnel.
Blinding of outcome assessment.
Incomplete outcome data.
Selective outcome reporting.
Other bias.
We judged each potential source of bias as high, low, or unclear, and provided a quote where relevant from the study report together with a justification for our judgement. We summarised the risk of bias judgements across different studies for each of the domains listed. When assessing attrition bias, we used an approximate cut‐off of 20% dropout for 'high' risk; however, the type of analysis performed (e.g. intention‐to‐treat (ITT)), the balance between trial arms, and reasons given for dropout were also taken into account. When considering treatment effects, we considered the risk of bias for the studies that contributed to that outcome.
Assessment of bias in conducting the systematic review
Generally, we conducted the review in accordance with the published protocol (Fraser 2019). We reported and justified deviations from the protocol in the Differences between protocol and review section of the review.
Measures of treatment effect
We analysed dichotomous data as odds ratios (ORs) with a 95% confidence interval (CI), and continuous data as mean differences (MD) with a 95% CI. For dichotomous outcomes, we intended to calculate the number needed to treat for an additional beneficial outcome (NNTB) and number needed to treat for an additional harmful outcome (NNTH).
Meta‐analyses were undertaken only where this was meaningful; that is, if the treatments, participants, and the underlying clinical question were similar enough for pooling to make sense.
We planned to describe skewed data narratively (e.g. as medians and interquartile ranges for each group).
Where multiple trial arms were reported in a single study, we included only the relevant arms. All treatment arms are specified in the Included studies section and Table 2. If two comparisons (e.g. drug A versus placebo and drug B versus placebo) were combined in the same meta‐analysis, we combined the active arms.
Several studies reported outcomes measured over discrete time intervals. Where this occurred, and the outcome was measured over a period of less than 12 weeks, we excluded these data from a meta‐analysis. If the data were analysed over at least 12 weeks, we elected to include the data that had been measured from the commencement of the study in a meta‐analysis (i.e. from baseline, as opposed to from a later time point within the study). This earlier period was chosen as it was considered to best encapsulate participants who were truly randomised, with no likelihood of a carry‐over treatment effect from earlier in the study potentially affecting outcome measures. The results from later time intervals of the relevant studies, whilst not included in meta‐analyses, are discussed within the text of the review.
We used the difference between end point scores for studies that reported quality of life using a validated scale.
We used ITT or 'full analysis set' analyses where they were reported (i.e. those where data had been imputed for participants who were randomly assigned but did not complete the study), instead of completer or per‐protocol analyses.
Unit of analysis issues
For dichotomous outcomes, we used participants, rather than events, as the unit of analysis (e.g. the number of participants with an exacerbation, rather than the number of exacerbations per participant). We avoided treating count data as dichotomous data to avoid unit‐of‐analysis error in recurring events.
Dealing with missing data
When missing data were thought to introduce serious bias, this was taken into consideration in the GRADE rating for affected outcomes.
We recorded any assumptions made around the reason or nature of missing data within the review. We performed sensitivity analyses to assess how variable the results may have been to any assumptions made about missing data.
Assessment of heterogeneity
We used the I2 statistic to measure heterogeneity among the studies in each analysis. Where there was substantial heterogeneity, we explored possible causes by performing prespecified subgroup analyses. As per Chapter 10 of the Cochrane Handbook for Systematic Reviews of Interventions (Deeks 2022), we considered the following ranges for assessing heterogeneity.
0% to 40%: might not be important.
30% to 60%: may represent moderate heterogeneity.
50% to 90%: may represent substantial heterogeneity.
75% to 100%: may show considerable heterogeneity.
Assessment of reporting biases
When we were able to pool more than 10 studies, we created and examined a funnel plot to explore possible small‐study and publication biases.
Data synthesis
We used a random‐effects model. Sensitivity analyses were performed using a fixed‐effect model.
Subgroup analysis and investigation of heterogeneity
Subgroup analyses were performed based on measures of disease severity (baseline FEV1 and exacerbation frequency).
Most studies that reported the primary outcomes included orally delivered immunostimulant agents, with only two studies analysing sublingually and subcutaneously delivered immunostimulants. Therefore, we considered it less relevant to include 'mode of delivery' as a subgroup, as originally outlined in our protocol (Fraser 2019). We considered further differentiation of the immunostimulant agents based on dose and treatment regimen; however, there was much variability between studies limiting the ability to subgroup by these categories. Therefore, we only performed subgroup analyses based on the immunostimulant type.
Because of the variation seen in treatment course lengths and follow‐up periods across all studies, we used the point of longest follow‐up for assessment of outcomes overall, and added subgroup analyses following the data extraction phase to investigate for heterogeneity that may have been explained by treatment or overall study duration (or both).
We made these above alterations by consensus discussion following data extraction, but prior to any data synthesis and analysis occurring.
We performed the following subgroup analyses.
Type of immunostimulant agent.
Severity of COPD based on lung function testing: mild or moderate (defined by FEV1 50% or greater predicted) versus severe or very severe (FEV1 less than 50% predicted) (GOLD 2022).
Mean baseline exacerbation rate two or more in the preceding year versus fewer than two in the preceding year or unspecified.
Treatment duration (three months or less versus greater than three months).
Study duration (three to less than six months versus six months to less than 12 months versus 12 months or greater).
We used the following outcomes in subgroup analyses.
Number of participants with no exacerbations during the study period.
Mortality (all‐cause).
We used the formal test for subgroup interactions in Review Manager 5 (Review Manager 2014).
Sensitivity analysis
We carried out the following sensitivity analyses, removing from the primary outcome analyses:
trials judged in the risk of bias table at high risk of bias for any of the six domains;
trials where decisions or assumptions had been made around missing data.
We compared the results from a fixed‐effect model with the random‐effects model.
Summary of findings and assessment of the certainty of the evidence
The summary of findings table contained the following outcomes.
Number of participants with no exacerbations during the study period.
Mortality (all‐cause).
Mortality (respiratory‐related).
Quality of life.
Number of participants requiring antibiotics.
Adverse events/side effects.
We used the five GRADE considerations (risk of bias, consistency of effect, imprecision, indirectness, and publication bias) to assess the certainty of the body of evidence as it related to the studies that contributed data for the prespecified outcomes. We used the methods and recommendations described in Chapter 14 of the Cochrane Handbook for Systematic Reviews of Interventions (Schünemann 2022), using GRADEpro GDT software (GRADEpro GDT). We outlined all decisions to downgrade the certainty of the evidence using footnotes and comments to aid in readers' understanding of the review.
Results
Description of studies
This review is based on a published protocol (Fraser 2019).
Results of the search
For details of the search history, see Appendix 2, and for the PRISMA study flow diagram, see Figure 1.
1.
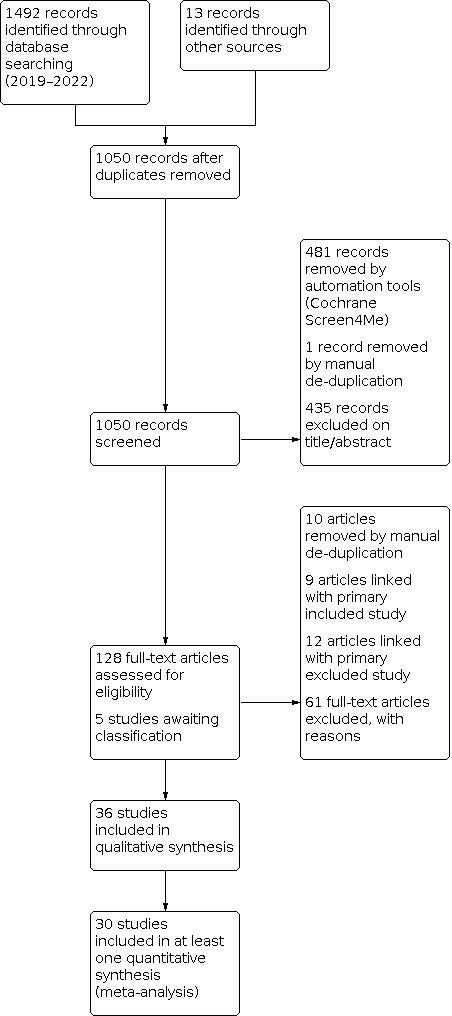
Following deduplication, the database and trial registry searches run on 24 May 2019 and 25 January 2022 retrieved 1037 references, and searches of included study and related systematic review reference lists identified a further 13 records (totalling 1050 references). The original 1037 references were additionally screened by Cochrane's Screen4Me services, which analyses and removes references from the results set before the manual screening stage using the Classifier and known assessments, and this process excluded 481 references. We then manually screened the 569 remaining records, excluding 435 on the basis of title and abstract, removing one by manual deduplication, and marking 133 for full‐text review. For five of these references, abstracts or full text (or both) were unobtainable through local library and Cochrane interlibrary loan requests, and have been marked as 'awaiting classification' (see Studies awaiting classification). Therefore, 128 studies underwent full‐text screening. Following manual deduplication and grouping of multiple study reports with the primary reference, we excluded a further 61 records, with reasons specified (see Excluded studies). The remaining 36 records were eligible for inclusion.
Included studies
The details of all included studies can be reviewed in the Characteristics of included studies table, with an overview of studies provided in Table 2.
Thirty‐six studies met the inclusion criteria; 35 were double‐blind, randomised, placebo‐controlled trials of a parallel‐group design, including adults classified as having COPD or chronic bronchitis (or both). One trial was of a randomised crossover design; however, outcomes were measured following the initial treatment and follow‐up period, prior to crossover occurring, and, therefore, we included the initial study period in this review (Venge 1996). Two studies were multi‐arm, comparing two intervention groups and placebo (Bonde 1986; De Bernardi 1992). In Bonde 1986, the two intervention groups consisted of different doses of the same immunostimulant agent; in De Bernardi 1992, the two intervention groups consisted of the same immunostimulant agent and dose, but differed by the manufacturing country. Two studies separately examined several participant subsets versus placebo following initial randomisation, respectively creating four and six total comparison groups based on the nature or severity (or both) of the underlying airways disease (Anthoine 1985; Blaive 1982). Two included studies present data in abstract form only; full reports were unable to be obtained (Alvarez‐Sala 2003; Olivieri 2011).
The 36 RCTs recruited 6192 participants. Study duration ranged from three to 14 months, with a mean duration of 6.6 months.
Twelve studies examined the use of immunostimulants in participants with COPD only (Alvarez‐Mon 2005; Alvarez‐Sala 2003; Blaive 1982; Braido 2015; Carlo 1990; Catena 1992; Cazzola 2006; Collet 1997; EUCTR2007‐004702‐27‐DE; Menardo 1985; Olivieri 2011; Xinogalos 1993). The remaining studies included participants with chronic bronchitis or COPD (or both), or chronic bronchitis alone. In studies where included participants were defined as having chronic bronchitis, it was assumed that some or all of the participants had COPD if it was documented in the inclusion criteria or text that they had a degree of airflow limitation or obstruction, or if mean baseline lung function values indicated this.
Two studies indicated that the included participants concurrently had a degree of immunodeficiency (Catena 1992; De Bernardi 1992). Catena 1992 described participants to have 'cell‐mediated immune deficiency' translated to be defined as "Multitest‐Merieux positive with no more than 2 antigens." De Bernardi 1992 indicated presence of 'borderline immunodeficiency' as an inclusion criterion, translated to be diagnosed by Merieux‐Multitest, chemotaxis, phagocytic activity of bronchoalveolar lavage macrophages, or sputum immunoglobulin A (IgA) concentration.
Where studies considered multiple participant groups based on the nature or severity (or both) of airways disease, we only extracted data for the relevant participant subsets. For example, Blaive 1982 presented data separately for participants with asthma, emphysema, and chronic bronchitis; we only extracted data for the emphysema and chronic bronchitis groups. In Anthoine 1985, where participants with chronic bronchitis were grouped into those with (Group II) or without (Group I) airflow limitation, we only extracted data for Group II participants as outcomes for this group were more completely reported by the study authors and deemed of greater relevance to this review.
Inclusion and exclusion criteria
All studies involved adults who fulfilled criteria for chronic bronchitis, COPD, or both. Inclusion and exclusion criteria for each study were variable. Eight studies did not specify any exclusion criteria (Alvarez‐Sala 2003; Blaive 1982; Debbas 1990; Keller 1984; Messerli 1981; Olivieri 2011; Orlandi 1983; Tag 1993), noting that Alvarez‐Sala 2003 and Olivieri 2011 were available in abstract form only. Several did not clarify whether participants with other concomitant respiratory illnesses were excluded. Seven studies specified the inclusion criterion of participants experiencing an acute exacerbation at the time of study enrolment; however, all examined the preventive medium‐ to long‐term (rather than acute treatment) effects of the immunostimulant agent and hence were deemed to be acceptable to include in this review (Cvoriscec 1989; Debbas 1990; Djuric 1989; Keller 1984; Messerli 1981; Soler 2007; Tang 2015).
Lung function
Seventeen studies reported baseline lung function values using FEV1 or FEV1 % predicted. Five of these indicated that the mean FEV1 % predicted of the included participants was less than 50% predicted; eight suggested that on average participants had moderate airflow limitation (mean FEV1 50% or greater and less than 80% predicted); one indicated participants on average had mild airflow limitation (mean FEV1 80% predicted or greater). Three studies reported absolute FEV1 volumes only. Of the remaining studies that did not specify baseline lung function, and excluding Olivieri 2011 (abstract only), 10 included participants with either COPD alone or COPD or chronic bronchitis (or both). The remaining studies were those where included participants had chronic bronchitis, but it was not specified by study authors whether some or all participants may have had a degree of chronic, fixed airflow limitation.
Age
The mean age of participants ranged from 35.2 years (Tag 1993) to 82 years (Orcel 1994). Three studies did not report age (EUCTR2007‐004702‐27‐DE; Olivieri 2011; Rico 1997). Across the remainder of included studies, the mean age of participants was 60.0 years. Of these, 90% of studies included participant groups with a mean age between 45 and 70 years.
Smoking status
Twenty studies reported baseline smoking status. Of the studies that analysed the number of participants who were either active or ex‐smokers, proportions ranged from 27.7% (Menardo 1985) to 95.9% (Bonde 1986). Several studies only reported the number of active smokers, and did not specifically indicate the presence or prevalence of ex‐smokers.
Baseline exacerbation frequency
Twenty‐six studies reported the baseline mean exacerbation frequency, specified a history of frequent respiratory exacerbations as an inclusion criterion, or both.
Thirteen studies reported the baseline mean exacerbation frequency of study participants. Of these, 10 studies indicated participants had a mean exacerbation frequency of two or more in the preceding year (Alvarez‐Mon 2005; Bisetti 1994; Cazzola 2006; Cvoriscec 1989; Fietta 1988; Keller 1984; Menardo 1985; Rochemaure 1988; Soler 2007; Tang 2015). Of the remaining three studies, one reported a mean exacerbation frequency over the preceding two years (Braido 2015), and two reported a mean exacerbation frequency but did not specify the timeframe over which this was measured (Carlo 1990; De Bernardi 1992). However, Carlo 1990 listed an exacerbation frequency of five or greater in the preceding year as an inclusion criterion for enrolment.
A further 13 studies specified a history of respiratory exacerbations as an inclusion criterion but did not report the baseline mean exacerbation frequency. Of these, nine studies stipulated that a history of two or more exacerbations in the preceding year was required for study inclusion (Bonde 1986; Catena 1992; Debbas 1990; EUCTR2007‐004702‐27‐DE; Foschino 1995; Olivieri 2011; Orcel 1994; Venge 1996; Xinogalos 1993). The other four studies listed an inclusion criterion of participants having a history of respiratory exacerbations, but did not specify an exacerbation rate threshold (Anthoine 1985; Ciaccia 1994; Hutas 1994; Li 2004).
Two studies reported variations on the metric of baseline mean exacerbation frequency in the 'baseline characteristics' table. Ciaccia 1994 reported the proportion of participants who had experienced three or fewer or more than three exacerbations over the year preceding the study. Bongiorno 1989 reported the number of acute exacerbation events (rather than participants with events) over the four months preceding the study.
There were no studies that indicated a significant difference in baseline exacerbation frequency between the included intervention and placebo groups.
Immunostimulants and dose
Summary tables regarding variations on the immunostimulant type, dose regimens, and total treatment durations are outlined in Table 2 and Table 3.
2. Immunostimulants included in this review.
| Trade name | Generic name | Active entity |
| Adimod | Pidotimod | Synthetic agent |
| Biostim | RU41740 | Bacteria‐derived (glycoproteins and membrane fractions of Klebsiella pneumoniae) |
| Broncho‐Vaxom, Broncho‐Munal, Ommunal, Paxoral, Vaxoral | OM‐85 | Bacterial lysate |
| Diribiotine CK | Not available | Bacterial lysate |
| Hymovis, Monovisc, Orthovisc | Hyaluronan | Synthetic agent |
| Immunoferon, Inmunol | AM3 | Glycophosphopeptical (polysaccharide and protein compounds of Candida utilis) |
| Ismigen | Not available | Polyvalent mechanical bacterial lysate |
| Lantigen B | Not available | Bacterial lysate |
| Ribomunyl, Ribovac, Immucytal | D53 | Bacteria‐derived (proteoglycans of Klebsiella pneumoniae and ribosomal fragments from a range of bacterial pathogens) |
| Thymolin, Leucotrofina | Thymomodulin | Thymic extract |
| Symbioflor | Not available | Bacteria‐derived (components of Enterococcus faecalis) |
In 16 studies, the immunostimulant used was OM‐85, an oral bacterial lysate. Other immunostimulant agents studied included: AM3, an oral polysaccharide/protein compound isolated from Candida utilis (four studies); RU41740 (Biostim), an oral bacterially derived agent consisting of glycoproteins and membrane fractions of Klebsiella pneumoniae (three studies); Ismigen, a sublingual PMBL (two studies); Diribiotine CK, an oral/liquid bacterial lysate (two studies); thymomodulin, an oral thymic extract (two studies); pidotimod, an oral synthetic agent (two studies); D53 (Ribomunyl), a bacterially derived agent consisting of proteoglycans from Klebsiella pneumoniae and various ribosomal fractions, administered in aerosol and subcutaneous form (one study) and orally (one study); Lantigen B, a sublingual bacterial lysate (one study); Symbioflor, an oral/sublingual bacterial lysate (one study); and hyaluronan, a subcutaneous synthetic agent (one study).
Twenty‐six studies included intermittent dosing regimens across the total treatment duration period, and these appeared to be specific to the type of immunostimulant agent. Immunostimulants associated with continuous regimens were AM3 (Alvarez‐Mon 2005; Alvarez‐Sala 2003; Bongiorno 1989; Carlo 1990), thymomodulin (Catena 1992; Rico 1997), pidotimod (Bisetti 1994; Ciaccia 1994), Symbioflor (Habermann 2001), and hyaluronan (Venge 1996).
Across the sixteen studies that analysed OM‐85, there were no variations on the dose of 7 mg/day; however, there were differences in dose pattern and treatment duration, with five studies using a three‐month regimen (Debbas 1990; Li 2004; Messerli 1981; Orcel 1994; Tang 2015), two studies using a four‐month regimen (Collet 1997; Orlandi 1983), and nine studies using a five‐month regimen (Cvoriscec 1989; Djuric 1989; EUCTR2007‐004702‐27‐DE; Hutas 1994; Keller 1984; Olivieri 2011; Soler 2007; Tag 1993; Xinogalos 1993).
In the four studies that included AM3, two used a total daily dose of 3 g/day (Alvarez‐Mon 2005; Alvarez‐Sala 2003), and two used a total daily dose of 1.5 g/day (Bongiorno 1989; Carlo 1990).
For the three studies that included RU41740 (Biostim), two used a dose of 2 mg/day for an initial interval followed by 1 mg/day for subsequent intervals (Anthoine 1985; Fietta 1988). One was multi‐arm and used 2 mg/day for the first intervention group and 8 mg/day for the second intervention group (Bonde 1986).
For the two studies that included thymomodulin, one used a total daily dose of 120 mg/day (Catena 1992), and the other used a total daily dose of 240 mg/day (Rico 1997).
For the two studies that included Ismigen, both used a total daily dose of 50 mg/day; however, one study used an intermittent dosing regimen over nine months' total duration (Braido 2015), and the other over three months' total duration (Cazzola 2006).
In the two studies that included Ribomunyl, the individual medication doses were not specified and there was variation between both the dose regimens and the method of administration, with Blaive 1982 using a combination of aerosol and subcutaneous routes and Foschino 1995 using an oral form.
Study size and duration
Study size ranged from 19 participants (Orlandi 1983) to 494 participants (Ciaccia 1994). Total study duration ranged from three months (Alvarez‐Sala 2003; Carlo 1990; Catena 1992) to 14 months (Habermann 2001) with a mean duration of 6.6 months. Total treatment duration, which included the time intervals between periods of treatment where intermittent dosing regimens were used, ranged from two months (Bisetti 1994; Ciaccia 1994) to nine months (Braido 2015) with a mean duration of four months.
Countries
Ten studies were conducted in Italy; five in France; three in Switzerland; two in Spain; two in countries of the former Yugoslavia; two in China; and one each in Canada, Mexico, Egypt, Greece, Hungary and Germany. Six studies were conducted across two or more European/Scandinavian countries.
Funding
Nine studies reported pharmaceutical sponsorship (Alvarez‐Mon 2005; Blaive 1982; Braido 2015; Collet 1997; Cvoriscec 1989; EUCTR2007‐004702‐27‐DE; Hutas 1994; Tang 2015; Venge 1996). Five studies did not specifically state pharmaceutical sponsorship; however, authorship details or the listed study address (or both) suggest pharmaceutical company associations (Bonde 1986; Menardo 1985; Messerli 1981; Rico 1997; Soler 2007). Fietta 1988 reported partial sponsorship from public grant funding. The remaining 21 studies did not specify any sponsorship or funding sources.
Excluded studies
We excluded 92 studies after full‐text screening. See Characteristics of excluded studies table for reasons for exclusion.
Risk of bias in included studies
Details of our risk of bias judgements are presented in the risk of bias section of the Characteristics of included studies table, with an overview in Figure 2.
2.

Allocation
All included studies were reported to be randomised. However, for most studies the potential for allocation bias was unclear for both random sequence generation and allocation concealment, in that the authors did not state in sufficient detail the method of randomisation, where this took place, and how it was concealed. One study was at low risk of bias for both sequence generation and allocation concealment (Braido 2015). One study was at low risk for allocation concealment, but unclear for sequence generation (Orcel 1994). One study was at high risk for both domains (Orlandi 1983).
Of the studies that were unclear regarding potential for allocation bias, 21 did not provide any information regarding the randomisation process or how concealment was achieved. Five studies used random‐draw, randomly planned sequences, or randomisation codes, but gave no further details (Anthoine 1985; Blaive 1982; Bongiorno 1989; Messerli 1981; Rochemaure 1988). Six studies used block permutation or stratified randomisation methods (or both), but it was unclear how the sequences were generated (Alvarez‐Mon 2005; Bonde 1986; Ciaccia 1994; Collet 1997; Orcel 1994; Soler 2007). One study indicated randomisation by a computer‐generated sequence without further detail (Ciaccia 1994). One study did not specify methods within the published report, but following further contact with the study author it was indicated computer‐generation may have been used (Hutas 1994).
Two studies involved unbalanced randomisation in the intervention versus placebo group (Catena 1992: 2:1; Tag 1993: 3:2).
Most studies reported the baseline characteristics of treatment groups, which appeared well‐matched. The authors of several studies only reported characteristics for the overall study population (Bonde 1986; Fietta 1988; Orlandi 1983), or sparse numerical data regarding baseline characteristics (Djuric 1989; Messerli 1981), but commented that the comparison groups were homogeneous for a range of metrics. One study provided data for its overall study population but did not specify whether the groups were well‐matched (Venge 1996). Two studies did not report on population baseline characteristics (EUCTR2007‐004702‐27‐DE; Olivieri 2011), although Olivieri 2011 was presented as an abstract only.
Blinding
All included studies were reported to be double‐blind. However, most did not provide sufficient information regarding the methods used to achieve blinding or the groups that were blinded; therefore, the potential of bias for these domains was unclear for 34 studies. One study was at low risk of bias for blinding of participants and personnel, but whether outcome assessors were blinded was not specified (Braido 2015). One study was at low risk of blinding of outcome assessors, but unclear for blinding of participants and personnel (Collet 1997).
Authors of eight studies indicated that the intervention and placebo formulations (tablets, capsules, or drops) were identical in appearance (Carlo 1990; Collet 1997; De Bernardi 1992; Djuric 1989; Keller 1984; Messerli 1981; Orcel 1994; Xinogalos 1993). Whilst noted as an important feature of ensuring adequate participant and personnel blinding, if there was no elaboration on methodology to ensure blinding or the groups that were blinded (or both) were not specified, then this information alone was deemed insufficient to create confidence that there was adequate protection against performance and detection bias.
Incomplete outcome data
Study dropout rate varied from a reported 0% (Carlo 1990) to an estimated, but not explicitly stated, 72% (Bisetti 1994). When the proportion of dropouts was more than 20% of the total study population, we considered a 'high risk' rating; however, it was taken into account whether the dropouts were balanced in number and had occurred for similar reasons between study arms. The proportion of dropouts relative to study duration, and whether an ITT versus completer analysis had been performed, were also considerations.
Five studies were at high risk of attrition bias (Alvarez‐Sala 2003; Bisetti 1994; EUCTR2007‐004702‐27‐DE; Menardo 1985; Xinogalos 1993), noting that Alvarez‐Sala 2003 was presented in abstract form only. In EUCTR2007‐004702‐27‐DE, study authors reported their concerns about significant bias in this domain due to large amounts of missing data and flaws in analysis contributed to by invalid and inaccurate assessments of outcome events. As a result, the study authors and an independent group of experts deemed the study to be flawed and efficacy conclusions unable to be made based on the available study results. It may be relevant that this was a pharmaceutical company‐sponsored trial where the results for the primary outcome were unfavourable for the intervention group compared to placebo. Further information about the reported processes that led to this conclusion can be viewed in the Characteristics of included studies table.
Seven studies were at low risk of attrition bias, as dropout rates were low or had been adequately described such that they were unlikely to have contributed to the quality and interpretation of study outcomes (Anthoine 1985; Braido 2015; Carlo 1990; Cazzola 2006; Collet 1997; Orlandi 1983; Tang 2015).
The remaining studies were at unclear risk. An 'unclear' rating was given if dropout rates, reasons, distribution across the comparison groups, or a combination of these were not well‐described or if dropouts were unbalanced between groups. If dropout rates were high and per‐protocol analysis was undertaken, even if well‐balanced between groups and reasons provided, these studies were also deemed at unclear risk.
Six studies performed an ITT analysis (Anthoine 1985; Braido 2015; Cazzola 2006; Collet 1997; Debbas 1990; Tang 2015), and one study analysed the full participant population due to a reported 100% completion rate (Carlo 1990). Several studies did not describe the presence or absence of dropouts, but the number of participants analysed at study completion appeared to match those randomised at study commencement; therefore, it was not clear whether an ITT analysis was incorporated with dropouts or exclusions having occurred, or whether all participants had completed the study (Cvoriscec 1989; De Bernardi 1992; Djuric 1989; Habermann 2001; Li 2004; Rico 1997; Venge 1996). The remaining 22 studies used a per‐protocol analysis. Of these, one study indicated the allocation of an ITT sample based on its study flow diagram; however, they used a per‐protocol analysis for the primary study outcomes (Soler 2007).
Selective reporting
One study was at high risk for bias in this domain, due to methodological and outcome assessment flaws that were identified and reported by the study authors and an independent review panel (EUCTR2007‐004702‐27‐DE; see Characteristics of included studies table). Excluding the two studies presented as abstract only (Alvarez‐Sala 2003; Olivieri 2011), nine studies were at unclear risk, because not all intended outcomes were reported, there was possible skew towards reporting of positive outcomes, or data had been presented in such a way that may have positively influenced the significance of the findings (Anthoine 1985; Bonde 1986; Ciaccia 1994; Debbas 1990; Foschino 1995; Menardo 1985; Orlandi 1983; Rochemaure 1988; Xinogalos 1993). The remaining 24 studies appear to have reported sufficiently on all intended outcomes and were, therefore, judged at low risk of bias in this domain.
Other potential sources of bias
One study indicated that baseline data for outcomes measuring the efficacy of the intervention was obtained through retrospective questionnaire, which may have introduced hindsight bias (Rico 1997). The impact of this on study outcomes was unclear, as baseline data did not appear to be relevant to primary or secondary outcome analysis.
Effects of interventions
See: Table 1
See: Table 1 for an overview of the main results together with a summary of our confidence in the evidence per outcome.
Primary outcome: number of participants with no exacerbations during the study period
Sixteen studies reported this outcome directly or indirectly (by reporting the number of participants with an exacerbation as a number of proportion of the total group population, from which the number of participants with no exacerbations could be inferred). Fifteen were included in meta‐analysis; one study was not included, as outcomes were analysed over two discrete eight‐week time intervals and this duration was shorter than had been specified in our protocol criteria (Bisetti 1994).
Immunostimulant medication increased the overall odds of not experiencing an exacerbation over the study period compared to placebo (OR 1.48, 95% CI 1.15 to 1.90; I2 = 53%; 15 studies, 2961 participants; Analysis 1.1; Figure 3; moderate‐certainty evidence). For the mean comparator risk of 0.52, this corresponded to an NNTB of 11 (95% CI 7 to 29). For the extremes of low (0.05) comparator risks for this outcome, the NNTB was 46 (95% CI 25 to 143) and for high (0.68) comparator risks was 13 (95% CI 9 to 34).
1.1. Analysis.

Comparison 1: Immunostimulant versus placebo, Outcome 1: Number of participants with no exacerbations during the study period
3.

The heterogeneity across the 15 studies included in this analysis was moderate to high (I2 = 53%), and was explored using preplanned subgroup analyses. Subgrouping for immunostimulant type was not associated with a consistent reduction of heterogeneity within subgroups, and the test for subgroup differences indicated that there was no evidence of a subgroup treatment effect (Chi2 = 2.82, degrees of freedom (df) = 3 (P = 0.42), I2 = 0%; Analysis 1.2). Preplanned subgroup analyses were also undertaken to assess for any variations in treatment effect related to baseline lung function and exacerbation frequency. Results of the subgroup and sensitivity analyses performed are elaborated on in 'Subgroup and sensitivity analyses' below.
1.2. Analysis.

Comparison 1: Immunostimulant versus placebo, Outcome 2: Participants with no exacerbations, by immunostimulant type
The funnel plot indicated the absence of small, neutral, or negative studies, and the likely presence of publication bias which may have skewed the pooled effect estimate towards a more positive result (Figure 4). However, exclusion of the five small, positive studies by sensitivity analysis did not significantly lessen the effect estimate.
4.

Two included studies measured this outcome in discrete time intervals (Habermann 2001; Menardo 1985). Unlike Bisetti 1994 however, the data sets from the initial time period were incorporated into meta‐analysis as they had been measured over longer intervals of three months (Menardo 1985) and six months (Habermann 2001). The later data sets were not included in meta‐analysis. The data set for the first, rather than the last, measurement period from baseline was preferentially incorporated, as it was considered that the study participants were more likely to be truly randomised at study commencement and there would be less risk of a carry‐over effect from earlier treatment exposure, which may potentially confound outcome data.
Bisetti 1994 (pidotimod) measured outcomes over four months in discrete two‐month intervals. Authors reported that fewer participants experienced exacerbations in the intervention group compared to placebo for both the zero‐ to two‐month interval (9/88 participants with intervention versus 57/75 participants with placebo) and the three‐ to four‐month interval (0/25 participants with intervention versus 13/26 participants with placebo) (reported P < 0.001 for both time intervals). However, concerns around methodology including significantly high and unexplained dropout rates led to a 'high risk' judgement for attrition bias, and the calculated ORs and CIs for the inverse of these results (i.e. the number of participants with no exacerbations) also suggested data imprecision for both the zero‐ to two‐month (OR 27.80, 95% CI 11.65 to 66.32; 163 participants) and three‐ to four‐month (OR 51.0, 95% CI 2.81 to 925.71; 51 participants) study intervals.
Menardo 1985 (Diribiotine CK), measured number of participants with no exacerbations over six months in two discrete three‐month intervals. The zero‐ to three‐month data set was included in meta‐analyses. Due to concerns regarding attrition bias, this study was at 'high risk' for this domain; however, exclusion by sensitivity analysis did not lead to augmentation of the pooled effect estimate. The four‐ to six‐month data set was not included, but for this later time period the study authors reported a difference in the number of participants without an exacerbation in the intervention group compared to placebo (5/10 participants with intervention versus 2/10 participants with placebo; OR 3.5, 95% CI 0.47 to 25.90; P = 0.22). Data imprecision lowers the certainty in this effect estimate.
Habermann 2001 (Symbioflor) measured number of participants with no exacerbations over 14 months in discrete six‐ and eight‐month intervals. The zero‐ to six‐month data set was included in meta‐analysis. Results for the seven‐ to 14‐month, post‐treatment follow‐up period indicated that the number of participants with an exacerbation during this time was less in the intervention group compared to placebo (P = 0.013, as reported by the study authors). The associated point estimate of effect suggested an increased odds of experiencing no exacerbations with intervention compared to placebo (OR 1.83, 95% CI 0.9 to 3.7; 136 participants); however, the estimate is imprecise.
Sixteen other studies that did not report the number of participants with or without an exacerbation, instead reported on alternative exacerbation metrics such as the number of exacerbation events or the mean exacerbation rate for the study period. The studies varied in the way they presented count data in terms of method of presentation, reporting of variance, and rate time frames. Therefore, this information was not extracted or pooled for the purposes of this systematic review.
Primary outcome: mortality (all‐cause)
Seven studies (2003 participants) reported mortality data (Bonde 1986; Braido 2015; Cazzola 2006; Collet 1997; EUCTR2007‐004702‐27‐DE; Orcel 1994; Soler 2007). Five were combined in meta‐analysis. We did not include Bonde 1986 as they reported a 1.2% overall mortality rate (172 participants), but it was not specified which comparison groups the deaths were associated with, and Soler 2007 reported zero‐event rates in both arms (273 participants).
Immunostimulants probably result in little to no difference in all‐cause mortality compared to placebo, measured over a mean follow‐up period of eight months (OR 0.64, 95% CI 0.37 to 1.10; I2 = 0%; 5 studies, 1558 participants; Analysis 1.8; Figure 5; moderate‐certainty evidence); however, CIs were wide and included the potential for both a clinically important difference and no difference.
1.8. Analysis.

Comparison 1: Immunostimulant versus placebo, Outcome 8: Mortality (all‐cause)
5.

The studies that were meta‐analysed only included two immunostimulant agents; OM‐85 (three studies) and Ismigen (two studies).
Orcel 1994 contributed most of the weight in the meta‐analysis for all‐cause mortality and recruited elderly participants from residential care facilities (mean age 82 years) with a diagnosis of chronic bronchitis. Reported baseline characteristics data suggested high rates of comorbidity in both groups. Therefore, the results from this study may be less applicable to the general chronic bronchitis or COPD (or both) population; however, there was little impact on the pooled effect estimate when this study was excluded in sensitivity analysis.
Secondary outcome: mortality (respiratory‐related)
Two larger studies that had reported on all‐cause mortality also reported respiratory‐related mortality (Collet 1997; Orcel 1994). Both analysed the immunostimulant OM‐85 over six months. Results from pooled analysis indicated there may be little to no difference in respiratory‐related mortality (OR 0.40, 95% CI 0.15 to 1.07; I2 = 0%; 2 studies, 735 participants; Analysis 1.14; low‐certainty evidence). However, data availability, imprecision, and indirectness lower the certainty in this effect estimate. The concerns regarding the applicability and generalisability of the data presented in Orcel 1994, with its inclusion of an elderly, comorbid study population, carried greater weight for this outcome considering this meta‐analysis included only two studies.
1.14. Analysis.

Comparison 1: Immunostimulant versus placebo, Outcome 14: Mortality (respiratory‐related)
Secondary outcome: quality of life
Although several studies reported subjective participant or physician (or both) estimations of well‐being, only six explored the impact of immunostimulants on health‐related quality of life (HRQoL) compared to placebo using validated assessment tools. The tools used included the SGRQ (Jones 1992), the 36‐item Short Form Health Survey (SF‐36), the 12‐item Short Form Health Survey (SF‐12), and the Chronic Cough Impact Questionnaire (CCIQ) (Baiardini 2005).
In three studies, investigators assessed the impact of the intervention on quality of life using the SGRQ (Alvarez‐Mon 2005; Alvarez‐Sala 2003; EUCTR2007‐004702‐27‐DE). Collet 1997 used the SF‐36. Braido 2015 used both the SF‐12 and CCIQ. Catena 1992 used an unnamed, 9‐point 'Index of improvement for quality of life' scale that referenced an earlier study in which the scale appeared to have been validated (Grossi 1989); the original publication of this article could not be obtained.
The SGRQ is made up of three subscales – symptoms, activities, and impacts – to yield a total score ranging from 0 to 100 (Jones 1992). A lower score indicates a better quality of life. Total SGRQ scores from two studies, both of which assessed the immunostimulant AM3, were combined in meta‐analysis (Alvarez‐Mon 2005; Alvarez‐Sala 2003); scores from EUCTR2007‐004702‐27‐DE could not be incorporated as there were no numerical data presented, with authors reporting that there were no differences between study arms.
There was a reduction in total SGRQ scores for participants receiving immunostimulant compared to placebo (MD −4.59, 95% CI −7.59 to −1.59; I2 = 0%; 2 studies, 617 participants; Analysis 1.15; very low‐certainty evidence). This effect met the minimum clinically important difference (MCID) of a reduction of 4 units on the SGRQ scale (Jones 2005). However, the upper limit of the CI for this effect did not clear the MCID and the possibility of a non‐meaningful difference could not be excluded. Several other factors contributed to a lowering of the certainty of this effect estimate; Alvarez‐Sala 2003 was presented as an abstract only, had a short duration of follow‐up (three months), and, due to inconsistencies in the reported number of participants for each arm, was at high risk for attrition bias. Additionally, there were no standard deviations for the total scores in each study arm reported and, for the purposes of meta‐analysis these were instead calculated from the reported P value for the MD between comparison groups.
1.15. Analysis.

Comparison 1: Immunostimulant versus placebo, Outcome 15: Quality of life (total score St George's Respiratory Questionnaire)
Alvarez‐Mon 2005 reported both mean total SGRQ scores and mean change‐from‐baseline scores over the six‐month study period for 253 participants. There was a difference in the total SGRQ scores (MD −4.6) and total 'activity' subcomponent scores (MD −7.1) described between the comparison groups at six months, favouring the intervention (P < 0.05 for both outcomes, as reported by study authors). The differences between study arms with respect to the total 'symptom' (MD −3.4) and 'impact' (MD −4.1) subcomponent scores, although favouring intervention, were reportedly non‐significant. In assessing change in mean scores from baseline, there were no differences for total SGRQ (MD −3.3; reported P = 0.076), 'activity' (MD −1.7), and 'impact' (MD −3.1) subcomponent scores, comparing intervention to placebo. However, there was a meaningful difference favouring intervention between study arms in the change of mean 'symptom' subcomponent scores from baseline (MD −5.7; reported P < 0.05).
For the three studies that analysed the effect of an immunostimulant agent on quality of life using alternative assessment tools, there were no numerical data were presented by trialists to enable qualitative or quantitative analysis in this review (Braido 2015; Catena 1992; Collet 1997). In Braido 2015 (Ismigen; 12 months' follow‐up; 288 participants) and Collet 1997 (OM‐85; six months' follow‐up; 381 participants), the study authors reported no differences in the quality‐of‐life scale scores between intervention and placebo groups, with data not shown. In Catena 1992 (thymomodulin; three months' follow‐up; 236 participants) authors provided a graphical representation of nine quality‐of‐life domains and presented data per domain for both intervention and placebo groups as a ratio of percentage of improved participants to percentage of participants who were 'worse' at the completion of the study per domain. They reported improvement in all nine domains for the intervention group and in four domains for the placebo group (reported P < 0.05). There was no comparison between the intervention and placebo groups for each domain or overall reported.
Secondary outcome: number of participants requiring antibiotics
Seven studies reported number of participants requiring antibiotics; five were combined in meta‐analysis (Anthoine 1985; Debbas 1990; De Bernardi 1992; Habermann 2001; Menardo 1985). Two studies could not be included as outcomes were analysed in discrete monthly time intervals, and this duration was shorter than our specified protocol criteria (Cvoriscec 1989; Xinogalos 1993). The studies combined in meta‐analysis included the immunostimulant agents RU41740 (Biostim) (one study), OM‐85 (one study), Lantigen B (one study), Symbioflor (one study), and Diribiotine CK (one study). The two studies not meta‐analysed both examined the effects of the immunostimulant OM‐85 BV.
Immunostimulants likely reduced the overall odds of receiving antibiotic treatment for an exacerbation over the study period compared to placebo (OR 0.34, 95% CI 0.18 to 0.63; I2 = 38%; 5 studies, 542 participants; Analysis 1.16; Figure 6; moderate‐certainty evidence). Although the upper limit for the risk ratio CI crossed the 25% relative risk threshold for appreciable benefit (RR 0.64, 95% CI 0.5 to 0.82), the optimal information size criteria was met and the CI did not include the null effect.
1.16. Analysis.

Comparison 1: Immunostimulant versus placebo, Outcome 16: Number of participants requiring antibiotics
6.

The heterogeneity of these five studies was low to moderate, and likely due to the clinical and methodological diversity between studies, noting that each included study examined a different immunostimulant agent. However, the direction of effect was consistent across studies. One studies included in the analysis was at high risk for attrition bias (Menardo 1985). Another study included participants with COPD and concurrent 'borderline immune deficiency' and there were concerns about the applicability of these data as a result (De Bernardi 1992). Another examined an elderly population (mean age 81.8 years), which may affect applicability and generalisability of these data to a general chronic bronchitis/COPD population (Debbas 1990). However, individual exclusion of these studies by sensitivity analyses had little impact on the pooled effect estimate.
One study included in the meta‐analysis measured number of participants requiring antibiotics over six months in two discrete three‐month intervals (Menardo 1985). We incorporated only the initial zero‐ to three‐month data set. The study authors also presented data on the number of participants not requiring antibiotics over the four‐ to six‐month period. The inverse of the reported results for this interval (i.e. the number of participants requiring antibiotics) corresponded to an OR of 0.64 (95% CI 0.11 to 3.92; P = 0.65; 20 participants); however, this estimate is imprecise.
Two studies analysed the effect of immunostimulant on the number of participants requiring antibiotics compared to placebo over six months in discrete monthly intervals (Cvoriscec 1989; Xinogalos 1993). Cvoriscec 1989 (104 participants) presented data in graphical form, and authors only reported on the reduction in the proportion of participants requiring antibiotics compared to study entry in the intervention group (reported P < 0.05). There were no statistical comparisons of the intervention and placebo groups presented for this outcome at any of the study time points. Xinogalos 1993 (62 participants) reported that for the first three months of the trial there were no differences in the number of participants requiring antibiotics between the study arms, with numerical data not shown. They reported data for the four‐ and six‐month time intervals, with differences noted in the proportion of participants requiring antibiotics in the immunostimulant group compared to placebo (four months: P < 0.001; six months: P < 0.002; reported by the authors). For both studies, it was difficult to draw conclusions about the impact of immunostimulants on the requirement for antibiotic use, as the short time intervals over which this outcome was measured were considered likely to overestimate treatment effect. Additionally, the high risk of attrition bias in Xinogalos 1993 impacted interpretation of data quality and certainty regarding the effect estimates.
Several studies that did not report this outcome presented data regarding antibiotic use by alternative metrics; for example, as duration of antibiotic therapy (eight studies) or the incidence of antibiotic use (three studies).
Secondary outcome: exacerbation duration
Twenty‐one studies analysed exacerbation duration. Seventeen studies were initially included in meta‐analysis; three studies reported results over discrete intervals that did not meet our protocol criteria for minimum duration of outcome measurement (Bongiorno 1989; Ciaccia 1994; Xinogalos 1993). One study reported that there were no differences in this outcome between comparison groups, with no numerical data shown (EUCTR2007‐004702‐27‐DE).
Of the studies included in the meta‐analysis, one measured outcomes over 12 months in discrete three‐month intervals (Rico 1997); we incorporated only the first zero‐ to three‐month period from baseline.
The meta‐analysis performed for these 17 studies (1693 participants) indicated a pooled effect estimate that favoured intervention, measured over a mean duration of 6.4 months; however, this result was associated with an unacceptably high level of heterogeneity (I2 = 92%; Analysis 1.17). Therefore, the mean effect was difficult to interpret and draw conclusions from, and the likelihood of data skew was also considered to make parametric analysis less robust. Consensus decision was made to disable the forest plot totals, although the graph has still been included in this review for transparency and reference purposes.
1.17. Analysis.

Comparison 1: Immunostimulant versus placebo, Outcome 17: Exacerbation duration
The degree of heterogeneity seen was interpreted to be due to methodological and clinical diversity related to study interventions, design, population (noting variability in smoking prevalence and mean participant age between included studies), potentially variable outcome definitions and measures of effect, and the use of a continuous variable. The variability in absolute values occurring between studies may indicate that results could have been presented by authors as either the mean duration of an exacerbation episode, or as the total or cumulative number of exacerbation days averaged over the number of participants for the study duration. For several studies, this differentiation was unclear. In five studies measures of variance had not been reported by the trialists; these were instead calculated and assumed from the reported MDs and P values (Cazzola 2006; Foschino 1995; Olivieri 2011; Orlandi 1983; Tag 1993). A sensitivity analysis, excluding these studies and separately those that that were at high risk of bias for any domain, did not significantly modify the pooled effect estimate.
In lieu of formally meta‐analysing this outcome, we performed a post hoc synthesis of data by vote‐counting based on direction of effect, whereby the effect estimate for each study was categorised as a binary metric (beneficial versus harmful) and a binomial probability test applied to determine the probability of observing the result if the intervention was truly ineffective (Figure 7). Of the 18 available studies, one provided no information about direction of effect and was excluded (EUCTR2007‐004702‐27‐DE). By this method, there was evidence that immunostimulant agents had an effect on reducing exacerbation duration, with 16/17 included studies favouring the intervention (94%, 95% CI 73% to 99%; P = 0.0002). Sensitivity analyses that excluded studies at high risk of bias in any domain or for which missing variance data had been imputed did not impact significantly on these results.
7.

Effect direction plot and sign test for the outcome of exacerbation duration.
Rico 1997 (thymomodulin; 88 participants) measured the impact of exacerbation duration over 12 months in discrete three‐month intervals. We included the zero‐ to three‐month data set in the above analyses, but not the later time periods. Authors reported reductions in exacerbation duration in the intervention group compared to placebo during the four‐ to six‐month (MD −2.09; reported P = 0.04), seven‐ to nine‐month (MD −1.26; reported P = 0.15) and 10‐ to 12‐month (MD −0.49; reported P = 0.44) intervals.
Bongiorno 1989 (AM3; 40 participants) analysed exacerbation duration over four months in discrete two‐month intervals. They reported a longer exacerbation duration in the intervention group versus placebo in the zero‐ to two‐month period (MD 1.65), and conversely a reduction in duration favouring immunostimulant over the three‐ to four‐month period (MD −2.45; reported P < 0.005).
Ciaccia 1994 (pidotimod) measured exacerbation duration over five months in two‐ and three‐month discrete intervals. Authors presented data for all participants (492 participants at two months; 481 at five months), and also for those with a history of three or fewer exacerbations in the preceding year (242 participants two months; 240 at five months). For all participants, there was a reduced exacerbation duration in the intervention group compared to placebo for both time periods, but authors reported this difference was only significant in the second, three‐month period (MD −2.2; reported P < 0.01). For those who were less‐frequent exacerbators, there was a difference favouring intervention across both time periods (MD −1.6 for zero to two months; and MD −2.2 for three to five months; reported P < 0.05 for both intervals). There were no data for those who were more‐frequent exacerbators (greater than three exacerbations in the preceding year).
Xinogalos 1993 (OM‐85; 62 participants) analysed the impact of immunostimulant on exacerbation duration over six months, in one‐month intervals. They reported numerical data for all time periods. In the first and third months, exacerbation duration was less in the intervention groups compared to placebo, but this difference was reported to be non‐significant. In the second month, the mean exacerbation duration was higher in the intervention group (MD +0.79). For the remainder of the study, authors reported a reduction in the mean exacerbation duration in the intervention group compared to placebo when measured over the fourth (MD −1.14; reported P < 0.001), fifth (MD −0.52; reported P < 0.05), and sixth months (MD −0.16; P < 0.002).
Secondary outcome: hospitalisation duration (respiratory‐related)
Seven studies analysed the effect of immunostimulant on hospitalisations (either all‐cause or respiratory‐related) compared to placebo (Braido 2015; Cazzola 2006; Collet 1997; EUCTR2007‐004702‐27‐DE; Keller 1984; Orlandi 1983; Rico 1997). Three of these reported mean respiratory‐related hospitalisation duration; however, data were unable to be meaningfully combined in meta‐analysis (Cazzola 2006; Collet 1997; Orlandi 1983). A forest plot was initially created combining the data on 559 participants from Cazzola 2006 and Collet 1997 (Analysis 1.18), but heterogeneity was high (I2 = 83%) and the pooled effect estimate difficult to interpret, therefore, totals were disabled. Orlandi 1983 could not be included as the trialists did not report measures of variance, and these were not estimable using alternative statistical methods from the data provided.
1.18. Analysis.

Comparison 1: Immunostimulant versus placebo, Outcome 18: Hospitalisation duration (respiratory‐related)
Collet 1997 (OM‐85; 381 participants) reported that the mean duration of respiratory‐related hospitalisation was shorter in the immunostimulant group (6.4 days) than in the placebo group (11.3 days) over six‐month follow‐up, although this difference was not significant (MD −4.9, 95% CI −7.47 to −2.33; reported P = 0.058). Cazzola 2006 (Ismigen; 178 participants) reported a reduced mean respiratory‐related hospitalisation duration in the immunostimulant group compared to placebo over 12‐month follow‐up (MD −1.6, 95% CI −2.5 to −0.7; reported P < 0.05). Orlandi 1983 (OM‐85; 19 participants) reported a mean hospitalisation duration of 0.8 days in the intervention group over four months versus 1 day in the placebo group, but did not report measures of variance or statistical significance.
Seven studies measured the impact of immunostimulant therapy on hospitalisations using a range of other metrics. These included the number of respiratory‐related hospitalisation events (three studies), number of all‐cause hospitalisation events (two studies), number of participants requiring hospitalisation for a respiratory cause (one study), number of participants requiring hospitalisation for any cause (one study), total respiratory‐related hospitalisation days (three studies), total all‐cause hospitalisation days (two studies), and mean number of hospitalisation events per participant (one study). The three studies (847 participants) that reviewed total respiratory‐related hospitalisation days were larger trials conducted over a mean follow‐up of 10 months and examined the immunostimulants OM‐85 and Ismigen. All reported decreases in total hospitalisation time in the intervention groups compared to placebo (percentage difference: Braido 2015: Ismigen, −60%; Cazzola 2006: Ismigen, −53%; Collet 1997: OM‐85, −55%).
Secondary outcome: adverse events/side effects
Twenty‐seven studies reported the proportion of participants who experienced an adverse event over the study period. Seven, involving a total of 955 participants, reported no events in either study arm (Carlo 1990; Cazzola 2006; Debbas 1990; De Bernardi 1992; Fietta 1988; Orcel 1994; Venge 1996). The remaining 20 studies were included in meta‐analysis.
There was no evidence of a difference in the odds of experiencing an adverse event when receiving immunostimulant compared to placebo (OR 1.01, 95% CI 0.84 to 1.21; I2 = 0%; 20 studies, 3780 participants; Analysis 1.19; high‐certainty evidence). The CIs included the null effect, but optimal information size criteria were met and the risk ratio upper limit did not cross the relative risk threshold for appreciable harm (RR 1.02, 95% CI 0.92 to 1.13).
1.19. Analysis.

Comparison 1: Immunostimulant versus placebo, Outcome 19: Adverse events/side effects
Of the remaining studies, Collet 1997 reported the number of adverse event occurrences between comparison groups (138 events in 191 participants with intervention versus 151 events in 190 participants with placebo). Blaive 1982 reported treatment intolerance in 2% of 184 participants overall but did not specify which groups the affected participants belonged to. Bonde 1986 reported 51 cases of 'mild' adverse reaction events in 172 participants overall and did not specify in which groups these events occurred, although authors stated that there were no significant differences between the study arms. Six studies did not report adverse events (Alvarez‐Sala 2003; Bongiorno 1989; Djuric 1989; Hutas 1994; Keller 1984; Xinogalos 1993).
The nature of reported adverse events varied between studies, but in both intervention and placebo groups more frequently included gastrointestinal upset (nausea, dyspepsia, diarrhoea, constipation, abdominal pain), skin itch or rash, upper or lower respiratory tract symptoms, and fever. Eight studies reported study withdrawals that were related to the development of adverse events/side effects; however, these were balanced across the comparison groups (Alvarez‐Mon 2005; Bisetti 1994; Braido 2015; Catena 1992; Ciaccia 1994; Messerli 1981; Rochemaure 1988; Soler 2007). In the remaining studies, there were either no dropouts that occurred due to adverse events or the trialists did not report this.
Subgroup and sensitivity analyses
Subgroup analyses
Planned subgroup analyses were undertaken for the primary outcomes only; that is, participants with no exacerbations and mortality (all‐cause), to investigate for any potential heterogeneity encountered and differences in treatment effect. Subgrouping was performed by immunostimulant type, severity of COPD based on lung function testing (FEV1 50% or greater versus less than 50%), mean baseline exacerbation frequency (two or more in the preceding year versus fewer than two in the prespecified year or unspecified), treatment duration (three months or less versus greater than three months), and study duration (three to less than six months versus six months to less than 12 months versus 12 months or greater).
For the number of participants with no exacerbations, the heterogeneity of the 15 studies included in meta‐analysis was moderate (I2 = 53%). Heterogeneity did not consistently appear to be lower within any of the subgroups than across all studies (Analysis 1.2; Analysis 1.3; Analysis 1.4; Analysis 1.5; Analysis 1.6); where subgroups had no or low levels of heterogeneity suggested by the I2 statistic, the associated P value for the Chi2 test was > 0.1.
1.3. Analysis.

Comparison 1: Immunostimulant versus placebo, Outcome 3: Participants with no exacerbations, by baseline FEV1
1.4. Analysis.

Comparison 1: Immunostimulant versus placebo, Outcome 4: Participants with no exacerbations, by baseline exacerbation frequency
1.5. Analysis.

Comparison 1: Immunostimulant versus placebo, Outcome 5: Participants with no exacerbations, by treatment duration
1.6. Analysis.

Comparison 1: Immunostimulant versus placebo, Outcome 6: Participants with no exacerbations, by total study duration
To further explore possible sources of heterogeneity associated with this outcome, we performed a post hoc subgroup analysis differentiating studies by the decade of publication (Analysis 1.7). Moderate‐to‐high levels of heterogeneity remained within the subgroups, particularly across the studies published between 1990 and 1999 (I2 = 70%) and 2000 to 2009 (I2 = 66%).
1.7. Analysis.

Comparison 1: Immunostimulant versus placebo, Outcome 7: Participants with no exacerbations, by decade of publication
In terms of the potential impact of subgrouping on estimates of treatment effect, there were no significant subgroup effects found and the test for subgroup differences across all subgroup analyses was negative, for both primary outcomes. However, for the outcome of participants with no exacerbations, as there was moderate to substantial unexplained heterogeneity demonstrated between trials within most subgroups, the validity of the treatment effect estimate for each subgroup and the true presence or absence of subgroup differences was uncertain. Additionally, in the subgroup analyses of immunostimulant type (Analysis 1.2), lung function (Analysis 1.3), study duration (Analysis 1.6), and decade of publication (Analysis 1.7), several subgroups contained a limited number of studies; therefore, uneven covariate distribution also limits certainty of the impact of subgrouping effect. For all‐cause mortality, this is also a concern, with low minimum (two) and maximum (three) subgroup study numbers.
When the number of participants with no exacerbations was subgrouped by decade of publication, visualisation of the forest plot implied a trend towards the production of more conservative results in studies that were more recently published. Studies published between 2010 and 2019 (OR 1.08, 95% CI 0.79 to 1.48; I2 = 0%; 2 studies, 672 participants) or between 2000 and 2009 (OR 1.27, 95% CI 0.98 to 1.66; I2 = 66%; 4 studies, 1002 participants) appeared to have a lesser effect size than those published between 1990 and 1999 (OR 1.45, 95% CI 1.12 to 1.88; I2 = 70%; 4 studies, 965 participants) and before 1990 (OR 2.15, 95% CI 1.26 to 3.66; participants 322; studies 5; I2 = 25%; 5 studies, 322 participants). However, inferences about effect sizes were limited by the degree of heterogeneity within the subgroups and uneven covariate distribution.
Sensitivity analyses
In accordance with our protocol, sensitivity analyses were performed excluding studies at high risk of bias in any of the six domains, for any outcome. Where sensitivity analyses were undertaken, this is specified above under the relevant outcome headings. This type of sensitivity analysis did not lead to appreciable modification of the pooled effect estimate for any of the review outcomes.
Post hoc sensitivity analyses were also performed excluding trials that were more likely to lessen the overall data quality, to determine the impact of their absence on the effect estimate. These analyses are also specified above under the relevant outcome headings. Where these were conducted, this was little to no impact on the effect estimates observed.
Last, planned sensitivity analyses were undertaken for outcomes where there had been assumptions concerning missing data. This was largely relevant for the HRQoL and exacerbation duration outcomes, where missing variance measures for individual studies had been calculated from reported MDs and P values. As the meta‐analysis for HRQoL only contained two studies, conclusions were unable to be drawn from a sensitivity analysis that excluded the study where data assumptions had been made (Alvarez‐Sala 2003). The meta‐analysis for exacerbation duration was not undertaken for aforementioned reasons; instead, this outcome was analysed dichotomously using a direction‐of‐effect table (Figure 7). Sensitivity analyses were performed excluding the studies at high risk of bias in any domain or those where data had been assumed, both of which had little impact on the overall proportion of 'positive' studies or associated binomial probability.
Discussion
Summary of main results
The review included 36 studies involving 6192 participants. Studies were published between 1981 and 2015. Study duration ranged from three to 14 months. Immunostimulants investigated were OM‐85, AM3, RU41740 (Biostim), Ismigen, Diribiotine CK, thymomodulin, pidotimod, D53 (Ribomunyl), Lantigen B, Symbioflor, and hyaluronan, with administration including oral, sublingual, and subcutaneous routes. Twenty‐six studies included intermittent, rather than continuous, dosing regimens. The risks of bias of the included studies were mostly low or unclear.
The mean age of study participants ranged between 35.2 and 82 years. Twelve studies examined participants with COPD only. Seventeen studies reported baseline lung function values; most indicated a moderate‐to‐severe degree of airflow limitation. Nineteen studies indicated inclusion of participants with a mean baseline exacerbation frequency of two or more in the preceding year.
Primary outcomes
Participants administered an immunostimulant agent for a mean duration of six months were slightly more likely to be free of exacerbations during that time (OR 1.48, 95% CI 1.15 to 1.90; 15 studies, 2961 participants; moderate‐certainty evidence), although this result was moderately heterogeneous (I2 = 53%). Based on a mean estimate of baseline risk of 52%, 11 (95% CI 7 to 29) participants required treatment for this duration for one to be exacerbation‐free.
Five studies were included in meta‐analysis for all‐cause mortality, with the pooled result suggesting that immunostimulants probably result in little to no difference in all‐cause mortality measured over a mean duration of eight months, although the CIs were not sufficiently narrow to exclude a clinically important difference (OR 0.64, 95% CI 0.37 to 1.10; 5 studies, 1558 participants; moderate‐certainty evidence).
Immunostimulants increased the odds of participants being exacerbation‐free in all but three studies (Braido 2015; Collet 1997; EUCTR2007‐004702‐27‐DE). Braido 2015 examined the effect of the Ismigen compared to placebo over 12 months in 288 participants. The study was well‐described and at low risk of bias for half of the domains, with no high‐risk judgement in any domain. Study authors found no difference between the proportion of participants with no exacerbations in the intervention (70.55%) and placebo (71.13%) groups, postulating that the reasons for this may have been due to the low general exacerbation rate seen among study participants (greater than 70% participants did not experience an exacerbation over the study duration), and 27% dropout rate across the study (well‐described, ITT analysis used). Similarly Collet 1997, which examined the effect of OM‐85 over six months in 381 participants, was at low risk of bias for half of the domains, with no high‐risk judgement in any domain. Authors reported no difference between intervention and placebo with respect to the number of participants experiencing one or more exacerbations during the study period (P = 0.872, as reported in the text of the paper). Reported proportions equated to 55.5% of participants in the immunostimulant group and 56.3% of participants in the placebo group with no exacerbations during the study period. It was suggested by the authors that this observed result may have been related to more frequent recording of respiratory events that did not meet the criteria definition for an exacerbation in the placebo compared to the immunostimulant group. In EUCTR2007‐004702‐27‐DE, where trialists analysed the effect of OM‐85 versus placebo in 357 participants over six months, the negative direction of the point estimate of effect for the immunostimulant group was more noticeable (OR 0.81, 95% CI 0.52 to 1.24), although CIs were wide and included the possibility of no difference or a positive effect. Of note, this study was at high risk for attrition bias and selective outcome reporting; the study authors, as well as an independent group of experts, considered the study to be flawed due to the degree of ineligible and missing data, and considered that conclusions could not be drawn from this result. All three studies were associated with pharmaceutical company sponsorship.
Secondary outcomes
Immunostimulants may result in little to no difference in respiratory‐related mortality when comparing the odds in intervention and placebo groups over a mean duration of six months (OR 0.40, 95% CI 0.15 to 1.07; 2 studies, 735 participants; low‐certainty evidence); however, the upper limit of the CI for the estimate also included the possibility of no difference between groups. Furthermore, certainty of this estimate was lessened by the inclusion of only two studies in meta‐analysis, one of which included a population of participants that may not be generalisable to a broader population of participants with COPD or chronic bronchitis (or both).
Immunostimulants may be associated with an improvement in HRQoL scores, measured by the SGRQ, but the evidence was very uncertain. The pooled effect estimate from two studies indicated a small increase in the odds of improvement in scores with immunostimulant compared to placebo (MD −4.59, 95% CI −7.59 to −1.59), and met the MCID score of 4 units. However, the CI upper limit did not clear this threshold, and the possibility of no difference between groups could not be excluded.
The pooled result from five studies indicated that immunostimulants likely reduced the number of participants requiring antibiotics over a mean duration of six months (OR 0.34, 95% CI 0.18 to 0.63; 5 studies, 542 participants; moderate‐certainty evidence), with a moderate positive effect seen. This result had a low‐to‐moderate degree of heterogeneity (I2 = 38%), but the direction of effect was consistent across all studies.
The combined data from 20 studies that reported adverse events indicated no difference in the odds of experiencing an adverse event with immunostimulant compared to placebo, over a mean duration of six months (OR 1.01, 95% CI 0.84 to 1.21; 3780 participants; high‐certainty evidence). The CI limits for the associated risk ratio did not cross the 25% risk reduction threshold for appreciable harm or benefit (RR 1.02, 95% CI 0.92 to 1.13). An additional seven studies reported no events in both study arms; hence, were not incorporated into meta‐analysis.
We attempted to meta‐analyse data regarding the effect of immunostimulants from 17 studies that had reported on exacerbation duration; however, conclusions were unable to be drawn due to the high level of heterogeneity (I2 = 92%) and probable data skew. Results from an effect direction plot and binomial probability test indicated that a significant proportion of studies reported a direction of effect that favoured intervention, possibly indicating that immunostimulants are efficacious in reducing exacerbation duration compared to placebo (94%, 95% CI 73% to 99%; P = 0.0002). However, the degree of uncertainty associated with this estimate remained high.
Three studies reported mean respiratory‐related hospitalisation duration, two of which demonstrated a direction of effect that favoured immunostimulant compared to placebo; effect size and direction for the other was unclear, as measures of variance were not reported and unable to be estimated. Data were not interpretable from a meta‐analysis due to high levels of heterogeneity (I2 = 83%). The impact of immunostimulant on hospitalisations in general was reported using a wide variety of metrics across several studies. Three of the larger, more‐recent trials all reported reductions in total respiratory‐related hospital days over a mean duration of 10 months with immunostimulant compared to placebo.
For several outcomes, there was moderate‐to‐significant heterogeneity across the included studies. Therefore, results should be interpreted with caution. Outcomes where there was no heterogeneity were mortality (all‐cause), mortality (respiratory‐related), quality of life, and adverse events/side effects. Subgroup analyses were performed to investigate causes of heterogeneity for the primary outcome of number of participants with no exacerbations; however, heterogeneity remained high within subgroups. Subgroups that had low or no heterogeneity detected mostly contained small numbers of studies and participants, with non‐significant associated P values for the Chi2 test for heterogeneity.
Subgrouping did not modify the treatment effect estimate for either of the two primary outcomes; however, the validity of the estimates were limited by the degree of heterogeneity and uneven covariate distribution within some subgroups. Therefore, it was difficult to draw conclusions from these subgroup analyses.
Overall completeness and applicability of evidence
Of particular relevance to earlier trials, the definitions of chronic bronchitis or COPD (or both) in several included studies were not always well‐defined and, especially for trials involving participants with chronic bronchitis alone, were based on clinical criteria only. Only 17 studies reported lung function; of these, 13 included participants with moderate‐to‐severe airflow limitation (GOLD 2022). Although efforts were made to exclude those studies that may have included participants with other respiratory illnesses, such as asthma or bronchiectasis, it was not always clear what methods trialists had used to differentiate these conditions among the enrolled participants. Smoking prevalence appeared to vary widely across studies; although, many did not distinguish between the proportions of never‐smokers and ex‐smokers. Studies that were outliers with respect to mean participant age may also indicate a significant degree of disease heterogeneity across the included study participants, although the study with the lowest mean participant age was an outlier, only assessed one of the review outcomes, and was not included in meta‐analysis. All these factors may have impacted on the combinability of data within the review, contributed to the heterogeneity across several outcomes, and likely affected the generalisability of the evidence to a wider population of participants with COPD, as defined by current criteria (GOLD 2022). However, creating more stringent population criteria for study inclusion at the outset of this review may have excluded a significant proportion of studies, particularly older studies, which included participants based on disease definitions at the time of enrolment, as opposed to on the basis of more‐recently established and current gold‐standard diagnostic criteria.
Most studies included participants with a history of respiratory exacerbations; however, many involved short‐to‐medium follow‐up periods (mean study duration 6.6 months). Especially in studies where exacerbation outcomes were measured over periods of less than six months, any positive treatment effect in the intervention group may have been potentially exaggerated by short time frames of outcome measurement; conversely, having shorter follow‐up periods may not have allowed exacerbations to manifest in either group, contributing to negative or non‐significant results. It is similarly difficult to extrapolate evidence around mortality to a wider population when this outcome was measured over a relatively short mean duration of 8.4 months.
The applicability of the results of the earlier studies is likely to be affected by a degree of publication bias; it is probable that small, negative, or neutral immunostimulant trials were unpublished based on the results of a funnel plot performed for the primary outcome (Figure 4). The included smaller, positive trials were associated with much broader CIs and were given less weight in meta‐analysis; their exclusion did not appear to have a significant impact on the pooled effect estimate for the primary outcome. The effect of advances in COPD management over the years also influences interpretation of the results of the earlier, positive studies. It is unlikely that participants in these studies were exposed to what would currently be considered 'standard' non‐pharmacological and pharmacological management for COPD or chronic bronchitis (or both) (GOLD 2022).
The 36 included studies reported a wide range of outcomes that were direct or surrogate markers for exacerbation frequency or severity (or both). The outcome measures chosen for this review were restricted to those that were considered to enhance combinability of the data, and we preferentially used dichotomous measures for parametric analysis as they were deemed to be more statistically robust. Some other frequently reported outcomes in the included studies included count data presented as a continuous outcome, such as 'mean number of exacerbations'; these were not included in this review due to the likelihood of skew associated with this metric. Therefore, there be a range of similar or related outcome measures reported in the included studies that were not incorporated in the review analyses but which may be of clinical relevance.
Quality of the evidence
Overall, we graded the certainty of the evidence to be moderate, meaning that the true effect is probably close to the estimate of the effect, but that further research is likely to have an important impact on our confidence in the estimate and may change the estimate (see Table 1). Several considerations led to downgrades in the level of certainty in the effect estimate for most main outcomes. For individual outcomes, the level of certainty varied from very low (quality of life) to high (adverse events/side effects).
One outcome was downgraded for risk of bias (quality of life); the meta‐analysis only included two studies, one of which was at high risk for attrition bias and for which standard deviation data had also been imputed. Most included studies for other outcomes were at low or unclear risk of bias for each domain, and exclusion of the studies at high risk of bias by sensitivity analysis did not impact on the treatment effect estimate. Two outcomes were downgraded for inconsistency (participants with no exacerbations and participants requiring antibiotics), due to moderate levels of heterogeneity. Three outcomes were downgraded for imprecision (all‐cause mortality, respiratory‐related mortality, and quality of life), as the CIs were not sufficiently narrow to exclude the possibility of no or little effect. One outcome was downgraded for indirectness (respiratory‐related mortality); only two studies were included in this analysis, one of which examined a group of participants that were not considered to be representative of a broader population with COPD or chronic bronchitis (or both), with this characteristic being relevant to the measured outcome. One outcome was downgraded for publication bias (quality of life), as only two studies were included in the analysis produced by the same author groups; one of which was an abstract. It was considered that there was not a sufficiently large body of evidence included in the analysis to affirm the effect estimate as a true result. Although the funnel plot indicated a likely degree of likely publication bias for the outcome of participants with no exacerbations (Figure 4), exclusion of the smaller, positive studies in a sensitivity analysis did not significantly alter the effect estimate, therefore this outcome was not downgraded.
The quality of the data for the outcome of exacerbation duration was very low (not meta‐analysed or included in the summary of findings table). Results from pooled analysis were associated with a high level of heterogeneity, which was interpreted to be due to methodological and clinical diversity related to study interventions, participant characteristics and study design, potentially variable outcome definitions and measures of effect, and the use of a continuous variable. It was not always clear whether trialists reported the mean duration of individual exacerbation episodes, or whether total cumulative exacerbation days during the study period had been averaged over the number of participants. Additionally, imputation of data was required for several included studies due to measures of variance not being reported. Interpretation of data related to mean duration of respiratory‐related hospitalisations was also affected by heterogeneity between included studies in a pooled analysis and a limited number of studies reporting this outcome.
Potential biases in the review process
We attempted to minimise bias during the review process by completing a comprehensive electronic search of all published and unpublished data. When screening titles and abstracts, we used a conservative approach for inclusion, especially considering the number of studies published in non‐English languages that could not be adequately screened at the first stage without the assistance of a translator. Two review authors independently reviewed full‐text articles and extracted data using Covidence software (Covidence) to ensure standardised data extraction methods. Where studies required translation, and translators were able to confirm that the studies met initial screening criteria, we sent standardised data extraction documents to translators to complete. Where possible, we used two independent translators per study to minimise the risk of bias.
Not all included studies were clear regarding definitions of chronic bronchitis, COPD, or what constituted a respiratory exacerbation. We accepted the trialists' definitions of these when marking studies for inclusion, given the variability in reporting across studies. Therefore, there is the possibility that some included studies contained participants or had exacerbation criteria that were not generalisable or consistent compared to other included trials, which may have impacted on the validity and applicability of the review results.
Several study analyses imputed standard deviations. Where this occurred, we used conservative estimates of P values and performed calculations in accordance with accepted practices; however, assumptions may have impacted on CI overlap and potentially increased heterogeneity. In one study, it was unclear whether reported measures of variance were standard deviations or standard errors (Hutas 1994). We contacted the study author for clarification, but this was unable to be differentiated. Based on use of standard deviation for continuous variables elsewhere in the study, we recorded values as such. Several studies did not clearly specify the number of participants analysed at the point of relevant outcome measurements (i.e. whether the number was less than at baseline due to dropouts that had not been reported); in these cases, we used an ITT approach for assuming the number of participants at any time point of the study.
Several studies reported data at discrete time intervals. Where these time intervals met our criteria of outcomes being measured over at least 12 weeks, we included the interval from baseline (as opposed to later intervals) in meta‐analyses; the rationale was that we considered participants analysed during the time interval of longest follow‐up would no longer be truly 'randomised' and representative of a baseline sample of patients, and treatment effects might have been confounded by prior exposure to the medication and study dropouts during the earlier parts of the study. Treatment of the data in this way may have influenced the combinability of these studies with others that had instead analysed end‐of‐study outcomes over longer durations, which may have contributed to heterogeneity when meta‐analysing data.
Our primary and secondary outcomes were changed following the data extraction process. This deviation from protocol was carefully considered and occurred following consensus discussion, prior to any data synthesis or analysis occurring. While the eventual outcomes were broadly congruent with our original outcomes, the ways in which these were measured were altered somewhat to suit the availability and patterns of data seen across studies; this may have introduced bias, especially if there was a degree of selective reporting of positive outcomes across studies.
Additionally, subgroup analyses required modification following data extraction to investigate for anticipated heterogeneity related to differences in treatment and study duration, which varied broadly across included studies. Again, these changes occurred prior to data synthesis or analysis occurring. We performed a post hoc subgroup analysis involving decade of study publication following data synthesis to further investigate heterogeneity that remained unexplained by the preplanned subgroupings. Therefore, the results of this analysis should be interpreted with caution.
Agreements and disagreements with other studies or reviews
The 2022 GOLD guidelines have specifically mentioned use of immunostimulant or immunoregulatory agents for preventing exacerbations in adults with COPD (GOLD 2022). However, their widespread and routine use has been limited due to a shortage of high‐quality data regarding their efficacy, and a lack of understanding of their mechanisms of action and long‐term safety profiles. The 2022 GOLD guidelines suggest that further studies are needed to examine the effects of these agents in people who are receiving current gold‐standard COPD maintenance therapy.
One previous systematic review found that the bacterial extracts O‐85, LW‐50020, and SL‐04 were associated with improved symptoms in people with COPD, chronic bronchitis, or both, and the meta‐analysis suggested a lessened exacerbation duration (MD −2.7, 95% CI −3.5 to −1.8). However, there was no difference between the extracts and placebo in preventing exacerbations (Steurer‐Stey 2004). Another systematic review looked at the efficacy of OM‐85 in preventing exacerbations in people with COPD or chronic bronchitis (or both). For the end point of one or more exacerbations, there was a non‐significant trend in favour of OM‐85 (RR 0.83, 95% CI 0.65 to 1.05); however, there were varied results across a range of other clinically important outcomes including hospitalisations and antibiotic use (Sprenkle 2005). In 2015, one meta‐analysis and systematic review examined the effects of OM‐85 in people with COPD on exacerbation rate, in addition to several other secondary clinical end points including hospitalisation duration and antibiotic use. OM‐85 was associated with a 20% reduction in exacerbation rate (RR 0.80, 95% CI 0.65 to 0.97; P = 0.03, as reported in the text of the paper) and a 39% reduction in the incidence rate of participants using antibiotics compared with placebo (RR 0.61, 95% CI 0.48 to 0.77; reported P < 0.0001) (Pan 2015). However, the authors concluded that there was not enough evidence to support the routine use of OM‐85 in people with COPD, suggesting that further larger‐scale trials needed to be undertaken.
The findings of this review suggest that immunostimulants may be associated with a small increase in the odds of remaining exacerbation‐free, which is consistent with the direction of effect for exacerbation outcomes seen in Sprenkle 2005 and Pan 2015. The finding that immunostimulant was associated with a reduced proportion of participants requiring antibiotics compared to placebo is also congruent with the findings in Pan 2015. Although our review found a positive effect direction with respect the outcomes of exacerbation duration and respiratory‐related hospitalisation duration, data were unable to be meaningfully combined in meta‐analysis and concerns around data quality and heterogeneity impacted on our certainty of these results.
Authors' conclusions
Implications for practice.
We graded the certainty of the evidence in this review to be moderate, meaning that the true effect is probably close to the estimate of the effect, but that further research is likely to have an important impact on our confidence in the estimate and may change the estimate. Our certainty in the pooled estimates was affected by several considerations, which led to downgrades in the associated level of certainty for most main outcomes.
Immunostimulants may slightly reduce the odds of an exacerbation in adults with chronic obstructive pulmonary disease (COPD) or chronic bronchitis (or both). Approximately 1 in 11 people may avoid experiencing an exacerbation, if all were to take treatment for a mean of six months. It is likely that immunostimulants are associated with a reduction in the requirement for antibiotics, which may suggest a reduction in the severity of exacerbations, although this relationship is only assumed. Immunostimulants probably result in little to no reduction in all‐cause and respiratory‐related mortality, but estimates are imprecise and data quality is limited. It is uncertain whether immunostimulants improve quality of life, and whether they reduce exacerbation duration or respiratory‐related hospitalisation duration (or both), although immunostimulants were generally associated with a positive effect direction in the studies that examined these outcomes. Immunostimulants appear to be safe and well‐tolerated and are not associated with an increased risk of adverse events.
Immunostimulants may be able to be considered as add‐on therapy to other gold‐standard, guideline‐based COPD therapies in those who continue to experience frequent exacerbations or who are requiring frequent courses of antibiotics for exacerbations, or both. They could be considered as an alternative option to any standard, usual‐care COPD therapy if this is not tolerated or cannot be used for any reason.
Implications for research.
Future studies may consider addressing the value of immunostimulants in populations with COPD or chronic bronchitis (or both) in study populations that are more well‐characterised, so that the subsets of patients who might benefit the most (or the least) from these therapies can be better identified. There should be a focus on people who have frequent or severe respiratory exacerbations and who are currently receiving maximal guideline‐based therapy. Populations should be stratified by the severity of their COPD (GOLD 2022) – incorporating lung function and symptoms and exacerbation history – and use of concomitant medications. Study outcomes should include exacerbations, hospitalisations (COPD and all‐cause), mortality (COPD and all‐cause), health‐related quality of life scores measured using a validated tool, and adverse events. It may be useful to differentiate the baseline and post‐treatment severity of the exacerbations experienced into mild, moderate, or severe (GOLD 2022). Follow‐up duration should be of sufficient length to allow for better representation of true exacerbation and mortality incidence, and the timing of the study should be considered (i.e. whether the study spans periods of the year when exacerbations are more likely).
Based on the challenges encountered in meta‐analysing and qualitatively interpreting the continuous outcome variables presented across the studies included in this review, we would advocate for the selection of robust, dichotomous treatment effect measures where possible; for example, by measuring the number of participants with/without an exacerbation, requiring antibiotics, or requiring hospitalisation, as opposed to event rate or mean number of events per participant. If event duration is to be measured, it should be specified whether the reported unit of time is per event or added cumulatively over the total study period. Time‐to‐first exacerbation may be a useful outcome to consider in future trials or review updates (or both), with the intervention effect expressed as a hazard ratio. As treatment regimens included in this review varied considerably, including for the same immunostimulant agent, it may be worthwhile conducting further trials comparing the effects of differing regimens for individual immunostimulant agents head‐to‐head in this patient subset. Further studies incorporating a cost–benefit analysis of immunostimulant therapies are also likely to be beneficial.
History
Protocol first published: Issue 5, 2019
Acknowledgements
We acknowledge the translators who assisted with translations of non‐English language studies, including Ms Epaminondas La Bella, Ms Ursula Feuz, Mr Beat Brungger, Ms Cristiana Riboni, Dr Simona Nistor‐Grahl, Mr Nikolay Kuzmin, Ms Katharina Greulich, Ms Cholpon Tashtanbekova, Ms Ana Beatriz Pizzaro, Ms Charlotte Flahou, Ms Klara Szabo, Ms Edyta Ryczek, Ms Carolyn Hughes, Mr Ben Shaw, Prof Dr Daniel Bereczki, Ms Amelia Cava, and Ms Ilaria Terrinoni.
We acknowledge Prof György Böszörményi‐Nagy, one of the senior authors in Hutas 1994, who assisted with study translation and provided further information regarding study details.
The authors and Cochrane Airways are grateful to Blanca Estela Del‐Rio‐Navarro, Department of Allergy and Immunology, Hospital Infantil de México "Federico Gómez," Mexico City, Mexico for her peer review of the review manuscript.
The Background and Methods sections of this review are based on a standard template used by Cochrane Airways.
This project was supported by the National Institute for Health and Care Research (NIHR), via Cochrane Infrastructure funding to the Cochrane Airways Group. The views and opinions expressed herein are those of the review authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, National Health Service, or the Department of Health and Social Care.
Appendices
Appendix 1. Glossary of terms
| Term | Definition | Term | Definition |
| Adaptive immunity | The components of the immune system involved in the specific and long‐term response to a particular antigen. Includes B and T lymphocyte activity. | Immunogenic | Describes a substance able to provoke an immune response. |
| Adhesion molecule | Cell‐surface proteins that interact with T lymphocytes and enable their migration through the endothelium at sites of inflammation. | Inactivated whole‐cell formulation | A substance that contains whole micro‐organisms (e.g. bacteria, viruses) that have been killed through physical or chemical processes. |
| Antibody | A specialised protein produced by B lymphocytes in response to an antigen, in order to help eliminate it. Also known as an immunoglobulin. | Innate immunity | The rapid and non‐specific components of the immune system that are involved in the first response to an antigen. |
| Antigen | A substance that is recognised by the immune system as being foreign, and that triggers an immune response e.g. bacteria, viruses. | Macrophage | A type of white blood cell involved in the innate immune response. |
| Antigen Presentation | The process by which antigen is 'presented' to lymphocyte cells in order to activate the adaptive (i.e. specific) immune response against that antigen. | Monocyte | A type of white blood cell involved in the innate immune response. |
| B lymphocyte | A type of white blood cell involved in the adaptive immune response. B cells are responsible for antibody production and secretion. | Natural killer cell | A type of white blood cell involved in the innate immune response. |
| Cell‐mediated immunity | The component of the immune response that involves the direct action of immune cells (such as T lymphocytes) against a pathogen. | Phagocytic | Describes immune system cells that are capable of ingesting foreign particles, bacteria, or debris created by the immune response, e.g. macrophages, monocytes, dendritic cells. |
| Dendritic cell | An antigen‐presenting cell that is involved in the innate immune response. | Ribosome | A complex molecule found in the cytoplasm of all living cells, which is responsible for the production of biologic proteins. |
| Glycoprotein | A protein that has a carbohydrate group attached. Often located at the cell surface, and play an important role in the mechanisms of infection by bacteria and viruses. | T lymphocyte | A type of white blood cell that plays an important role in the cell‐mediated, adaptive immune response. |
| Humoral immunity | The component of the immune response that involves action against the antigen by the molecules found in extracellular fluids (e.g. secreted antibodies). Also called antibody‐mediated immunity. | Toll‐like receptors | A class of cell surface‐membrane proteins that recognise molecules shared by a variety of pathogens. Binding of these pathogens leads to signalling pathways that activate the innate, and subsequently the adaptive, immune responses. |
Appendix 2. Database search strategies
| Database/search platform/date of last search | Search strategy | Results |
|
Airways Register (via Cochrane Register of Studies)
Date of most recent search: 25 January 2022 |
#1 MeSH DESCRIPTOR Pulmonary Disease, Chronic Obstructive Explode All #2 MeSH DESCRIPTOR Bronchitis, Chronic #3 (obstruct*) near3 (pulmonary or lung* or airway* or airflow* or bronch* or respirat*) #4 COPD:MISC1 #5 (COPD OR COAD OR COBD OR AECOPD):TI,AB,KW #6 #1 OR #2 OR #3 OR #4 OR #5 #7 MESH DESCRIPTOR Adjuvants, Immunologic EXPLODE ALL #8 immunostimulant* #9 immunomodulat* #10 immunoadjuvant* #11 immunologic adjuvant* #12 immunobalt or lw50020 or luivac or paspat or munostin #13 OM‐85 or OM85 or "OM 85" #14 bronchovaxom or broncho‐vaxom or "broncho vaxom" #15 pulmonar‐om or "pulmonar om" or pulmonarom #16 D53 #17 ribomunyl or ribovac or immucytal #18 MESH DESCRIPTOR Lipopolysaccharides EXPLODE ALL #19 lipopolysaccharide* #20 ru41740 or ru‐41740 or "ru 41740" or biostim #21 MESH DESCRIPTOR Cell Extracts EXPLODE ALL #22 MESH DESCRIPTOR Thymus Extracts EXPLODE ALL #23 thymus extract* #24 thymic extract* or thymomodulin* #25 thymosin* #26 MESH DESCRIPTOR Pelargonium #27 pelargonium* or umckaloabo #28 am3 or imunoferon or immunoferon or inmunoferon #29 glycophosphopep* #30 pidotimod or adimod #31 MESH DESCRIPTOR Levamisole #32 levamisole #33 tucaresol #34 imiquimod #35 bacterial NEXT (lysate* OR extract* OR antigen*) #36 (PMBL or BL):ti,ab #37 Ismigen #38 Lantigen B #39 IRS19 OR "IRS 19" #40 #7 OR #8 OR #9 OR #10 OR #11 OR #12 OR #13 OR #14 OR #15 OR #16 OR #17 OR #18 OR #19 OR #20 OR #21 OR #22 OR #23 OR #24 OR #25 OR #26 OR #27 OR #28 OR #29 OR #30 OR #31 OR #32 OR #33 OR #34 OR #35 OR #36 OR #37 OR #39 #41 #40 AND #6 | May 2019 = 185 January 2022 = 18 |
|
CENTRAL (via Cochrane Register of Studies)
Date of most recent search: 25 January 2022 |
#1 MeSH DESCRIPTOR Pulmonary Disease, Chronic Obstructive Explode All #2 MeSH DESCRIPTOR Bronchitis, Chronic #3 (obstruct*) near3 (pulmonary or lung* or airway* or airflow* or bronch* or respirat*) #4 COPD:MISC1 #5 (COPD OR COAD OR COBD OR AECOPD):TI,AB,KW #6 #1 OR #2 OR #3 OR #4 OR #5 #7 MESH DESCRIPTOR Adjuvants, Immunologic EXPLODE ALL #8 immunostimulant* #9 immunomodulat* #10 immunoadjuvant* #11 immunologic adjuvant* #12 immunobalt or lw50020 or luivac or paspat or munostin #13 OM‐85 or OM85 or "OM 85" #14 bronchovaxom or broncho‐vaxom or "broncho vaxom" #15 pulmonar‐om or "pulmonar om" or pulmonarom #16 D53 #17 ribomunyl or ribovac or immucytal #18 MESH DESCRIPTOR Lipopolysaccharides EXPLODE ALL #19 lipopolysaccharide* #20 ru41740 or ru‐41740 or "ru 41740" or biostim #21 MESH DESCRIPTOR Cell Extracts EXPLODE ALL #22 MESH DESCRIPTOR Thymus Extracts EXPLODE ALL #23 thymus extract* #24 thymic extract* or thymomodulin* #25 thymosin* #26 MESH DESCRIPTOR Pelargonium #27 pelargonium* or umckaloabo #28 am3 or imunoferon or immunoferon or inmunoferon #29 glycophosphopep* #30 pidotimod or adimod #31 MESH DESCRIPTOR Levamisole #32 levamisole #33 tucaresol #34 imiquimod #35 bacterial NEXT (lysate* OR extract* OR antigen*) #36 (PMBL or BL):ti,ab #37 Ismigen #38 Lantigen B #39 IRS19 OR "IRS 19" #40 #7 OR #8 OR #9 OR #10 OR #11 OR #12 OR #13 OR #14 OR #15 OR #16 OR #17 OR #18 OR #19 OR #20 OR #21 OR #22 OR #23 OR #24 OR #25 OR #26 OR #27 OR #28 OR #29 OR #30 OR #31 OR #32 OR #33 OR #34 OR #35 OR #36 OR #37 OR #39 #41 #40 AND #6 | May 2019 = 229 January 2022 = 48 |
|
MEDLINE (Ovid) ALL
Date of most recent search: 25 January 2022 |
1. exp Pulmonary Disease, Chronic Obstructive/ 2. Bronchitis, Chronic/ 3. (obstruct* adj3 (pulmonary or lung* or airway* or airflow* or bronch* or respirat*)).ti,ab. 4. (COPD or AECOPD).ti,ab. 5. or/1‐4 6. exp Adjuvants, Immunologic/ 7. immunostimulant*.ti,ab. 8. immunomodulat*.ti,ab. 9. immunoadjuvant*.ti,ab. 10. immunologic adjuvant*.ti,ab. 11. (immunobalt or lw50020 or luivac or paspat or munostin).ti,ab. 12. (OM85 or OM 85).ti,ab. 13. OM‐85.ti,ab. 14. (bronchovaxom or "broncho vaxom").ti,ab. 15. ("pulmonar om" or pulmonarom).ti,ab. 16. D53.ti,ab. 17. (ribomunyl or ribovac or immucytal).ti,ab. 18. exp Lipopolysaccharides/ 19. lipopolysaccharide*.ti,ab. 20. (ru41740 or "ru 41740" or biostim).ti,ab. 21. exp Cell Extracts/ 22. exp Thymus Extracts/ 23. thymus extract*.ti,ab. 24. (thymic extract* or thymomodulin*).ti,ab. 25. thymosin*.ti,ab. 26. Pelargonium/ 27. (pelargonium* or umckaloabo).ti,ab. 28. (am3 or imunoferon or immunoferon or inmunoferon).ti,ab. 29. glycophosphopep*.ti,ab. 30. (pidotimod or adimod).ti,ab. 31. Levamisole/ 32. levamisole.ti,ab. 33. tucaresol.ti,ab. 34. imiquimod.ti,ab. 35. (bacterial adj2 (lysate* or extract* or antigen*)).ti,ab. 36. (PMBL or BL).ti,ab. 37. Ismigen.ti,ab. 38. Lantigen B.ti,ab. 39. (IRS19 or "IRS 19").ti,ab. 40. or/6‐39 41. 5 and 40 42. (controlled clinical trial or randomized controlled trial).pt. 43. (randomized or randomised).ab,ti. 44. placebo.ab,ti. 45. dt.fs. 46. randomly.ab,ti. 47. trial.ab,ti. 48. groups.ab,ti. 49. or/42‐48 50. Animals/ 51. Humans/ 52. 50 not (50 and 51) 53. 49 not 52 54. 41 and 53 | May 2019 = 409 January 2022 = 105 |
|
Embase (Ovid)
Date of most recent search: 25 January 2022 |
1. exp Pulmonary Disease, Chronic Obstructive/ 2. Bronchitis, Chronic/ 3. (obstruct* adj3 (pulmonary or lung* or airway* or airflow* or bronch* or respirat*)).ti,ab. 4. (COPD or AECOPD).ti,ab. 5. or/1‐4 6. exp Adjuvants, Immunologic/ 7. immunostimulant*.ti,ab. 8. immunomodulat*.ti,ab. 9. immunoadjuvant*.ti,ab. 10. immunologic adjuvant*.ti,ab. 11. (immunobalt or lw50020 or luivac or paspat or munostin).ti,ab. 12. (OM85 or OM 85).ti,ab. 13. OM‐85.ti,ab. 14. (bronchovaxom or "broncho vaxom").ti,ab. 15. ("pulmonar om" or pulmonarom).ti,ab. 16. D53.ti,ab. 17. (ribomunyl or ribovac or immucytal).ti,ab. 18. exp Lipopolysaccharides/ 19. lipopolysaccharide*.ti,ab. 20. (ru41740 or "ru 41740" or biostim).ti,ab. 21. exp Cell Extracts/ 22. exp Thymus Extracts/ 23. thymus extract*.ti,ab. 24. (thymic extract* or thymomodulin*).ti,ab. 25. thymosin*.ti,ab. 26. Pelargonium/ 27. (pelargonium* or umckaloabo).ti,ab. 28. (am3 or imunoferon or immunoferon or inmunoferon).ti,ab. 29. glycophosphopep*.ti,ab. 30. (pidotimod or adimod).ti,ab. 31. Levamisole/ 32. levamisole.ti,ab. 33. tucaresol.ti,ab. 34. imiquimod.ti,ab. 35. (bacterial adj2 (lysate* or extract* or antigen*)).ti,ab. 36. (PMBL or BL).ti,ab. 37. Ismigen.ti,ab. 38. Lantigen B.ti,ab. 39. (IRS19 or "IRS 19").ti,ab. 40. or/6‐39 41. 5 and 40 42. Randomized Controlled Trial/ 43. randomization/ 44. controlled clinical trial/ 45. Double Blind Procedure/ 46. Single Blind Procedure/ 47. Crossover Procedure/ 48. (clinica$ adj3 trial$).tw. 49. ((singl$ or doubl$ or trebl$ or tripl$) adj3 (mask$ or blind$ or method$)).tw. 50. exp Placebo/ 51. placebo$.ti,ab. 52. random$.ti,ab. 53. ((control$ or prospectiv$) adj3 (trial$ or method$ or stud$)).tw. 54. (crossover$ or cross‐over$).ti,ab. 55. or/42‐54 56. exp animals/ or exp invertebrate/ or animal experiment/ or animal model/ or animal tissue/ or animal cell/ or nonhuman/ 57. human/ or normal human/ or human cell/ 58. 56 and 57 59. 56 not 58 60. 55 not 59 61. 41 and 60 | May 2019 = 408 January 2022 = 85 |
|
ClinicalTrials.gov
Date of most recent search: 25 January 2022 |
Study type: Interventional
Condition: COPD Intervention: Immunostimulant OR immunomodulatory OR immunoadjuvant OR immunologic adjuvant OR bacterial lysate OR thymus extract |
May 2019 = 4 January 2022 = 1 |
|
WHO trials portal
Date of most recent search: 25 January 2022 |
Condition: COPD Intervention: Immunostimulant OR immunomodulatory OR immunoadjuvant OR immunologic adjuvant OR bacterial lysate OR thymus extract |
May 2019 = 0 January 2022 = 0 |
Data and analyses
Comparison 1. Immunostimulant versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 Number of participants with no exacerbations during the study period | 15 | 2961 | Odds Ratio (IV, Random, 95% CI) | 1.48 [1.15, 1.90] |
| 1.2 Participants with no exacerbations, by immunostimulant type | 15 | 2961 | Odds Ratio (IV, Random, 95% CI) | 1.48 [1.15, 1.90] |
| 1.2.1 OM‐85 | 6 | 1933 | Odds Ratio (IV, Random, 95% CI) | 1.32 [0.97, 1.79] |
| 1.2.2 AM3 | 2 | 293 | Odds Ratio (IV, Random, 95% CI) | 1.35 [0.83, 2.20] |
| 1.2.3 Biostim | 3 | 253 | Odds Ratio (IV, Random, 95% CI) | 2.39 [1.00, 5.70] |
| 1.2.4 Other | 4 | 482 | Odds Ratio (IV, Random, 95% CI) | 2.59 [0.88, 7.59] |
| 1.3 Participants with no exacerbations, by baseline FEV1 | 8 | 2070 | Odds Ratio (IV, Random, 95% CI) | 1.34 [1.06, 1.71] |
| 1.3.1 FEV1 ≥ 50% predicted | 5 | 1264 | Odds Ratio (IV, Random, 95% CI) | 1.50 [1.03, 2.17] |
| 1.3.2 FEV1 < 50% predicted | 3 | 806 | Odds Ratio (IV, Random, 95% CI) | 1.15 [0.87, 1.52] |
| 1.4 Participants with no exacerbations, by baseline exacerbation frequency | 15 | 2961 | Odds Ratio (IV, Random, 95% CI) | 1.48 [1.15, 1.90] |
| 1.4.1 Mean exacerbation rate ≥ 2 in preceding year | 10 | 2064 | Odds Ratio (IV, Random, 95% CI) | 1.53 [1.13, 2.07] |
| 1.4.2 Mean exacerbation rate < 2 in preceding year or not specified | 5 | 897 | Odds Ratio (IV, Random, 95% CI) | 1.43 [0.87, 2.35] |
| 1.5 Participants with no exacerbations, by treatment duration | 15 | 2961 | Odds Ratio (IV, Random, 95% CI) | 1.48 [1.15, 1.90] |
| 1.5.1 Duration ≤ 3 months | 7 | 1221 | Odds Ratio (IV, Random, 95% CI) | 1.73 [1.23, 2.43] |
| 1.5.2 Duration > 3 months | 8 | 1740 | Odds Ratio (IV, Random, 95% CI) | 1.30 [0.93, 1.82] |
| 1.6 Participants with no exacerbations, by total study duration | 15 | 2961 | Odds Ratio (IV, Random, 95% CI) | 1.48 [1.15, 1.90] |
| 1.6.1 Duration 3 to < 6 months | 3 | 241 | Odds Ratio (IV, Random, 95% CI) | 2.39 [0.72, 7.89] |
| 1.6.2 Duration 6 to < 12 months | 11 | 2432 | Odds Ratio (IV, Random, 95% CI) | 1.52 [1.14, 2.03] |
| 1.6.3 Duration ≥ 12 months | 1 | 288 | Odds Ratio (IV, Random, 95% CI) | 0.97 [0.58, 1.62] |
| 1.7 Participants with no exacerbations, by decade of publication | 15 | 2961 | Odds Ratio (IV, Random, 95% CI) | 1.48 [1.15, 1.90] |
| 1.7.1 Years 2010–2019 | 2 | 672 | Odds Ratio (IV, Random, 95% CI) | 1.08 [0.79, 1.48] |
| 1.7.2 Years 2000–2009 | 4 | 1002 | Odds Ratio (IV, Random, 95% CI) | 1.39 [0.87, 2.24] |
| 1.7.3 Years 1990–1999 | 4 | 965 | Odds Ratio (IV, Random, 95% CI) | 1.64 [0.96, 2.79] |
| 1.7.4 Years 1980–1989 | 5 | 322 | Odds Ratio (IV, Random, 95% CI) | 2.74 [1.27, 5.92] |
| 1.8 Mortality (all‐cause) | 5 | 1558 | Odds Ratio (IV, Random, 95% CI) | 0.64 [0.37, 1.10] |
| 1.9 Mortality (all‐cause), by immunostimulant type | 5 | 1558 | Odds Ratio (IV, Random, 95% CI) | 0.64 [0.37, 1.10] |
| 1.9.1 OM‐85 | 3 | 1092 | Odds Ratio (IV, Random, 95% CI) | 0.69 [0.38, 1.26] |
| 1.9.2 Ismigen | 2 | 466 | Odds Ratio (IV, Random, 95% CI) | 0.47 [0.14, 1.60] |
| 1.10 Mortality (all‐cause), by baseline FEV1 | 4 | 1201 | Odds Ratio (IV, Random, 95% CI) | 0.65 [0.38, 1.13] |
| 1.10.1 FEV1 ≥ 50% predicted | 2 | 642 | Odds Ratio (IV, Random, 95% CI) | 0.79 [0.42, 1.47] |
| 1.10.2 FEV1 < 50% predicted | 2 | 559 | Odds Ratio (IV, Random, 95% CI) | 0.34 [0.11, 1.09] |
| 1.11 Mortality (all‐cause), by baseline exacerbation frequency | 5 | 1558 | Odds Ratio (IV, Random, 95% CI) | 0.64 [0.37, 1.10] |
| 1.11.1 Mean exacerbation rate ≥ 2 in preceding year | 3 | 889 | Odds Ratio (IV, Random, 95% CI) | 0.70 [0.38, 1.29] |
| 1.11.2 Mean exacerbation rate < 2 in preceding year or not specified | 2 | 669 | Odds Ratio (IV, Random, 95% CI) | 0.44 [0.13, 1.46] |
| 1.12 Mortality (all‐cause), by treatment duration | 5 | 1558 | Odds Ratio (IV, Random, 95% CI) | 0.64 [0.37, 1.10] |
| 1.12.1 Duration ≤ 3 months | 2 | 532 | Odds Ratio (IV, Random, 95% CI) | 0.72 [0.39, 1.34] |
| 1.12.2 Duration > 3 months | 3 | 1026 | Odds Ratio (IV, Random, 95% CI) | 0.42 [0.14, 1.31] |
| 1.13 Mortality (all‐cause), by total study duration | 5 | 1558 | Odds Ratio (IV, Random, 95% CI) | 0.64 [0.37, 1.10] |
| 1.13.1 Duration 6 to < 12 months | 3 | 1092 | Odds Ratio (IV, Random, 95% CI) | 0.69 [0.38, 1.26] |
| 1.13.2 Duration ≥ 12 months | 2 | 466 | Odds Ratio (IV, Random, 95% CI) | 0.47 [0.14, 1.60] |
| 1.14 Mortality (respiratory‐related) | 2 | 735 | Odds Ratio (IV, Random, 95% CI) | 0.40 [0.15, 1.07] |
| 1.15 Quality of life (total score St George's Respiratory Questionnaire) | 2 | 617 | Mean Difference (IV, Random, 95% CI) | ‐4.59 [‐7.59, ‐1.59] |
| 1.16 Number of participants requiring antibiotics | 5 | 542 | Odds Ratio (IV, Random, 95% CI) | 0.34 [0.18, 0.63] |
| 1.17 Exacerbation duration | 17 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 1.18 Hospitalisation duration (respiratory‐related) | 2 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 1.19 Adverse events/side effects | 20 | 3780 | Odds Ratio (IV, Random, 95% CI) | 1.01 [0.84, 1.21] |
1.9. Analysis.

Comparison 1: Immunostimulant versus placebo, Outcome 9: Mortality (all‐cause), by immunostimulant type
1.10. Analysis.

Comparison 1: Immunostimulant versus placebo, Outcome 10: Mortality (all‐cause), by baseline FEV1
1.11. Analysis.

Comparison 1: Immunostimulant versus placebo, Outcome 11: Mortality (all‐cause), by baseline exacerbation frequency
1.12. Analysis.

Comparison 1: Immunostimulant versus placebo, Outcome 12: Mortality (all‐cause), by treatment duration
1.13. Analysis.

Comparison 1: Immunostimulant versus placebo, Outcome 13: Mortality (all‐cause), by total study duration
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Alvarez‐Mon 2005.
| Study characteristics | ||
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics AM3
Placebo
Inclusion criteria: COPD, defined spirometrically as FEV1/FVC ratio < 70% and FEV1 35–70% of the predicted value; ≥ 2 episodes of acute respiratory exacerbation during previous year; clinically stable, with no history of infection or exacerbation of their illness within 4 weeks prior to trial. Exclusion criteria: significant spirometric postbronchodilator (salbutamol 200 μg) response in FEV1 (> 15% and > 200 mL); bronchiectasis, active pulmonary tuberculosis, lung cancer, cancer of any other organ, cystic fibrosis, restrictive lung disease, heart failure (NYHA functional class ≥ III), advanced kidney failure (serum creatinine > 4 mg/dL), or uncompensated liver disease (Child‐Pugh stage B or C); receiving treatment with immunosuppressant medications, immunomodulators, cimetidine, or any other drug that might modify the immune response; treated with systemic corticosteroids in 2 weeks prior to the study. Pretreatment: no significant differences between groups. |
|
| Interventions |
Intervention characteristics AM3
Placebo
|
|
| Outcomes |
Health‐related quality of life (total SGRQ score)
Adverse events
Number of participants with no exacerbations
|
|
| Identification |
Sponsorship source: I.F. Cantabria Country: Spain Setting: outpatient Author's name: Melchor Alvarez‐Mon, MD, PhD Institution: Departamento de Medicina, Universidad de Alcala, Carretera Madrid‐Barcelona Email: mams@tsai.es Address: Km 33,600, E‐28871 Alcala ´de Henares, Madrid, Spain Year: 2000 Participant type: COPD |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "A randomized, double‐blind, placebo‐controlled trial." Quote: "The study had a random four‐block design." Blocked/restricted randomisation stated; however, approach was incompletely defined and process of selecting the blocks unclear. |
| Allocation concealment (selection bias) | Unclear risk | Not specified. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double‐blind, controlled study." Reported that double‐blinding occurred, but who was blinded not explicitly stated, and blinding methods not specified. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not specified who measured the outcomes or if they were blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Per‐protocol analysis. High but comparable dropout rates between arms: 24% in treatment arm (42/176) and 26% in placebo arm (43/168). Reasons not specified for participants who "voluntarily" abandoned study. |
| Selective reporting (reporting bias) | Low risk | All main outcomes appeared to have been reported on. |
Alvarez‐Sala 2003.
| Study characteristics | ||
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics AM3
Placebo
Inclusion criteria: COPD, not otherwise specified. Exclusion criteria: not specified. Pretreatment: no significant differences. |
|
| Interventions |
Intervention characteristics AM3
Placebo
|
|
| Outcomes |
Health‐related quality of life (total score SGRQ)
|
|
| Identification |
Sponsorship source: not specified Country: not specifically stated. Authorship details identifies the research group was based in Spain. Setting: not specified, presumed to be outpatient. Author's name: JL Alvarez‐Sala, M Alvarez‐Mon Institution: Departamento de Medicina, Universidad de Alcala Address: Madrid, Spain Participant type: COPD Year: not specified. Abstract published 2003. |
|
| Notes | Abstract only. Outcome SDs calculated from reported P value for the MD in numerical scores between comparison groups. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation occurred, but methods not specified (abstract only). |
| Allocation concealment (selection bias) | Unclear risk | Not specified (abstract only). |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind study, but not specified which groups were blinded and blinding methods (abstract only). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not specified (abstract only). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Although this was only an abstract and dropout rates/exclusions/attrition were not specified, the number of participants randomised was less than the total number of participants based on the reported n for each group (authors reported 349 randomised participants total, and follow on to report 186 in the intervention group and 178 in the placebo group, i.e. 364). Number of completers not reported for any outcome. |
| Selective reporting (reporting bias) | Unclear risk | Given this is an abstract only it is difficult to make an accurate assessment. The abstract did imply that quality of life was the primary outcome, and this was reported on. However, it was referenced that other clinical, biochemical, and haematological parameters were examined during the study and these are only partially reported with no associated raw or graphical data. |
Anthoine 1985.
| Study characteristics | ||
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics RU41740 (Biostim)
Placebo
Inclusion criteria: adults with CB, who had episodes of 'super‐infection' in the year preceding the study. Exclusion criteria: if participants stopped smoking during the study; if participants were taking immune‐modifying treatment (corticosteroids, immunosuppressants, respiratory vaccines). Pretreatment: no significant differences in baseline characteristics. |
|
| Interventions |
Intervention characteristics RU41740 (Biostim)
Placebo
|
|
| Outcomes |
Number of participants with no exacerbations
Exacerbation duration
Number of participants requiring antibiotics
Adverse events
|
|
| Identification |
Sponsorship source: not specified Country: France Setting: multicentre, outpatient Author's name: D Dusser (differed from primary author) Institution: Hopital Laennec Address: 75007, Paris Participant type: all had CB, divided into 2 groups after randomisation: simple CB (group I) and chronic obstructive bronchitis, with or without respiratory insufficiency (group II). Each was compared with a separate placebo population. Most reported outcomes were for group II only, with the exception of adverse events. Year: 1982–1983 |
|
| Notes | Study published in French. Participants were initially randomised to 2 groups (intervention vs placebo), but were further divided into group I (intervention vs placebo) and group II (intervention vs placebo) based on the absence of presence of airflow limitation, resulting in 4 comparison groups. Data extracted for group II alone, as this was considered the most relevant to the review and was also associated with more reported and relevant outcomes. Adverse outcomes reported for the original intervention vs placebo comparison groups, and did not differentiate between group I and II. Therefore, data on adverse events was extracted for all 110 participants. Treatment days did not total 30 days (3 × 8‐day courses); however, the treatment spanned across 3 months and a continuous effect was assumed, therefore, the study was marked for inclusion. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation reported to be by random draw but no further details specified. |
| Allocation concealment (selection bias) | Unclear risk | Not specified. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind study, but blinded groups and methodology not specified. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not specified. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Analysis appeared to be ITT. 6 participants were excluded at the beginning of the study for not meeting study criteria. Following this, no further dropouts/exclusions/attrition were reported. |
| Selective reporting (reporting bias) | Unclear risk | There was only partial reporting of outcomes for group I, and the secondary outcomes were only reported for group II. In general there was alignment between study aims and outcomes reported, but this could have been clearer. |
Bisetti 1994.
| Study characteristics | ||
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics Pidotimod
Placebo
Inclusion criteria: CB Exclusion criteria: use of other immunostimulating medications, antibiotic prophylaxis, or receiving vaccinations during the study treatment or follow‐up period. Pretreatment: groups described as homogeneous, but no statistical analysis undertaken to determine presence of any significant differences in baseline characteristics. |
|
| Interventions |
Intervention characteristics Pidotimod
Placebo
|
|
| Outcomes |
Adverse events
Number of participants with no exacerbations
|
|
| Identification |
Sponsorship source: not specified Country: Italy Setting: multicentre, outpatient Author's name: Prof Dr A Bisetti Institution: La Clinica Tisiopneumologica Universitaria, Ospedale Forlanini Address: Piazza Forlanini, I‐00151 Rome (Italy) Year: 1994 Participant type: CB |
|
| Notes | Outcomes measured over 4 months in discrete 2‐month intervals. Data for each 2‐month interval was unable to be combined and used in meta‐analysis as this would be inconsistent with protocol criteria (specifying outcomes need to be measured over ≥ 12 weeks). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation occurred, but methods were not specified. |
| Allocation concealment (selection bias) | Unclear risk | Authors reported use of an "experimental key," which may suggest allocation concealment, but methods around ensuring allocation concealment were generally not clear. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind study, but groups and methodology not specified. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Study involved measuring clinicians' and participants' judgement regarding efficacy of either intervention or placebo. This may have occurred with both groups being blinded, but it was unclear. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Per‐protocol analysis. There was an initial low but unbalanced dropout rate between groups at 2 months; 5% (5/93) in intervention and 15% (13/88) in placebo. At 4 months, i.e. completion of the study, outcomes were only analysed for a small proportion of the original participants, and a dropout/exclusion/attrition rate of 73% (68/93) for the intervention group and 70% (62/88) for placebo group was implied. No clarification given as to the discrepancy between n at 2 months and n at 4 months. |
| Selective reporting (reporting bias) | Low risk | All intended outcomes appeared to have been reported on. |
Blaive 1982.
| Study characteristics | ||
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics D53 (Ribomunyl)
Placebo
Overall (CB and emphysema only)
Inclusion criteria: adults with chronic bronchopathies (accepted definition). Exclusion criteria: none specified. Pretreatment: no differences between groups specified. |
|
| Interventions |
Intervention characteristics D53 (Ribomunyl)
Placebo
|
|
| Outcomes |
Exacerbation duration
Adverse events
|
|
| Identification |
Sponsorship source: trial requested by Inava Laboratories Country: France Setting: not specifically stated, presumed to be outpatient Author's name: B Blaive Institution: Service de Pneumologie de l'Hopital Pasteur Address: 06031 Nice Participant type: participants with 'chronic bronchopathies' including CB, emphysema and asthma. Year: 1979–1981 |
|
| Notes | Study published in French. Asthma group excluded from analysis. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation performed by random draw, possibly with blocked randomisation as authors referred to participants being balanced for every 10 cases. However, methods not clearly specified. |
| Allocation concealment (selection bias) | Unclear risk | Products were provided by Inava laboratories anonymously, which may point to allocation concealment. However, methods not further specified. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind study, but groups and methods not specified. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not specified. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Per‐protocol analysis. 300 participants entered study; 273 completed (9% dropout rate). Although this was low ii was unclear whether dropouts were balanced across groups and not specified why these participants were excluded/lost to follow‐up. |
| Selective reporting (reporting bias) | Low risk | All outcomes appeared to have been reported on. |
Bonde 1986.
| Study characteristics | ||
| Methods |
Study design: randomised controlled trial Study grouping: parallel group. Multi‐arm trial with 3 comparison groups; 2 alternate intervention doses and placebo. |
|
| Participants |
Baseline characteristics No characteristics provided for individual comparison groups. RU41740 (Biostim) 2 mg
RU41740 (Biostim) 8 mg
Placebo
Overall
Inclusion criteria: aged > 18 years with stage 2 or 3 CB as defined by MRC criteria and who had experienced ≥ 3 exacerbations during the year prior to study. Exclusion criteria: acute infectious episode in the month prior to study; systemic medication with steroids in excess of prednisolone 10 mg/day or other immune‐suppressing or enhancing drugs; receiving antibiotic prophylaxis; vaccination against influenza or bacteria within 6 months preceding trial enrolment. Pretreatment: no significant differences between groups specified. |
|
| Interventions |
Intervention characteristics RU41740 (Biostim) 2 mg
RU41740 (Biostim) 8 mg
Placebo
|
|
| Outcomes |
Number of participants with no exacerbations
Exacerbation duration
Adverse events
Mortality (all‐cause)
|
|
| Identification |
Sponsorship source: not specified. Intervention drug was developed by Roussel‐Uclaf, and the medical department of Roussel‐Uclaf was mentioned in association with the authors' names. Country: not specifically stated. Authorship details identified hospitals in Denmark, Sweden, Finland, and France Setting: not specifically stated, presumed to be outpatient Author's name: Jan Bonde, MD Institution: Department of Anaesthesiology, KAS Herlev Address: Herlev Ringvej, DK‐2730 Herlev, Denmark Participant type: CB and COPD (latter defined as 'stage 3 CB' i.e. chronic obstructive bronchitis) Year: 1983 |
|
| Notes | Combined 2 mg and 8 mg intervention groups for the purpose of meta‐analysis. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised by balanced block randomisation (blocks of 12) for each centre; however, the process of selecting the blocks and further details of the method were not specified. |
| Allocation concealment (selection bias) | Unclear risk | Not specified. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind study, but groups and full methodology not specified. Authors mentioned the blister‐packing of tablets to ensure participant blinding, but it was not mentioned if they were identical. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not specified. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Per‐protocol analysis. There was a 13% (26/198) dropout rate from baseline at 3 months and 20% (39/198) dropout rate from baseline at 6 months. Reasons for dropout/exclusion/attrition were given; however, authors did not clearly specify dropout events per group. The 13 participants who left the study between 3 and 6 months were broadly categorised by their comparison groups, and indicated a higher dropout rate in the 8 mg intervention group (14%, 8/56) vs the 2 mg (5%, 3/55) and placebo (5%, 3/61) groups over this time interval. |
| Selective reporting (reporting bias) | Unclear risk | All main outcomes appeared to have been reported on, but little numerical or graphical data provided for the 3‐ to 6‐month follow‐up period and on some of the secondary outcomes (e.g. antibiotic consumption and duration, adverse effects, and mortality – reported overall but not by group). |
Bongiorno 1989.
| Study characteristics | ||
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics AM3
Placebo
Inclusion criteria: adults with CB and history of recurrent respiratory infections, who had been treated by the authors at least once in the preceding 3 years. Exclusion criteria: treatment with corticosteroids, immunosuppressants, immunomodulators. Pretreatment: no significant differences between groups. |
|
| Interventions |
Intervention characteristics AM3
Placebo
|
|
| Outcomes |
Number of participants with no exacerbations
Exacerbation duration
Adverse events
|
|
| Identification |
Sponsorship source: not specified Country: not specified. 2 authors are associated with an Italian institution Setting: not specifically stated, presumed to be outpatient Author's name: A Bongiorno Institution: University of Palermo Address: Italy Participant type: CB, COPD, or both Year: not specified. Study published 1989 |
|
| Notes | Study published in Spanish. The outcome of exacerbation duration was measured over 4 months in discrete 2‐month intervals. Data for each 2‐month interval were unable to be combined and used in meta‐analysis as this would be inconsistent with protocol criteria (specifying outcomes need to be measured over ≥ 12 weeks). |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation occurred according to a 'randomly planned sequence,' but further details and methodology not specified. |
| Allocation concealment (selection bias) | Unclear risk | Not specified. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blinding was not specifically mentioned; however, the study was placebo‐controlled and the assessment of efficacy at least appeared to be blinded according to authors (see below). No information specified regarding the personnel who administered the treatment during the trial and whether they were blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Translated quote: "At the end of the trial, a blinded evaluation of the efficacy of the treatment was conducted." The methods of blinding were not further specified. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Authors reported attrition of 4/20 (20%) participants in the intervention group and 5/20 (25%) participants in the placebo group. They were replaced by 9 new study participants. It was not clear at what phase of the study the attrition occurred, at what phase study replacements commenced treatment, and whether the replacements were allocated to the same intended groups as the dropouts. Given numbers were balanced between comparison groups it is unlikely this will significantly contribute towards bias, but insufficient information provided and the potential magnitude of the effect unclear. |
| Selective reporting (reporting bias) | Low risk | All intended outcomes appeared to have been reported on. |
Braido 2015.
| Study characteristics | ||
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics Ismigen
Placebo
Inclusion criteria: adults aged > 40 years with documented diagnosis of moderate, severe, or very severe COPD, according to the GOLD 2006 guidelines and based on the WHO performance grading of 0, 1, or 2; adequate haematological, renal, and liver function 14 days prior to study randomisation; women were non‐lactating and of non‐child‐bearing potential. Exclusion criteria: having received any prior antineoplastic drug therapy or immunosuppressive drugs; under continuous treatment with systemic steroids; co‐existing severe cardiac disease, including uncontrolled angina pectoris or myocardial infarction within 6 months of enrolment; had uncontrolled hypertension or any other uncontrolled severe medical condition, including active gastroduodenal ulcer, alcohol disorders (hepatitis, Korsakoff's syndrome), and diabetes; had uncontrolled infection or evolutive intracranial hypertension; were pregnant or breastfeeding at study commencement; had any psychological, familial, sociological, or geographical condition potentially hampering compliance with the study protocol and follow‐up schedule. Pretreatment: no significant differences between groups. |
|
| Interventions |
Intervention characteristics Ismigen
Placebo
|
|
| Outcomes |
Number of participants with no exacerbations
Health‐related quality of life (total score)
Adverse events
Mortality (all‐cause)
|
|
| Identification |
Sponsorship source: Lallemand Pharma SA, Lugano, Switzerland Country: Italy Setting: outpatient Author's name: G Melioli Institution: Clinica di Malattie Allergologiche e Respiratory, Padiglione Maragliano‐A.O.U. San Martino, A.O.U. San Martino Email: giovannimelioli@gmail.com Address: Largo R Benzi, 10, 16132 Genova, Italy Participant type: COPD Year: 2009–2012 |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomisation was performed by the CRO (SPRIM Italy/ALS GCP), using a validated system that automated the random assignment of treatment groups by randomised numbers in a 1:1 ratio." Randomisation performed externally by an automatic sequence generator. Well‐described. |
| Allocation concealment (selection bias) | Low risk | Quote: "randomisation was performed by the CRO (SPRIM Italy/ALS GCP), using a validated system that automated the random assignment of treatment groups by randomised numbers in a 1:1 ratio. Both the drug and the placebo were produced by Bruschettini Srl, Genova, Italy and were then sent to Doppel Farmaceutici which labelled the boxes and blisters with sequential numbers, according to the randomisation list." Allocation appeared to be computer‐generated and by a separate third party. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As above. Double‐blind study. Authors described process of prepacked medications and labels applied externally, with treatment and placebo appearing identical. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not specified. Authors did not explain how the key was broken or when, or whether the analysts were blinded when assessing outcomes for each group. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "All of the randomised patients were included in the statistical analyses, based on their assignments to the treatment or the placebo group and independent of the patient's eligibility and the treatment actually received (the intention‐to‐treat principle)." ITT analysis used. Clear study flow diagram provided. At end of first treatment period there were 25/142 (17.6%) withdrawals in placebo and 20/146 (13.7%) withdrawals in treatment group. 99/142 (69.7%) of placebo participants and 110/146 (75.3%) of treatment participants completed study. Therefore, overall dropout rate was high at 27.4%; however, this may have been due to study duration being 12 months. |
| Selective reporting (reporting bias) | Low risk | The intended primary outcome of "reduction of exacerbations by 25%" was not reached, but the authors elaborated extensively on the possible reasons for this and reported on a number of other exacerbation metrics. Other data for intended secondary outcomes (such as number of participants requiring concomitant medications, quality‐of‐life scale scores) was only partially reported as non‐significant between groups. |
Carlo 1990.
| Study characteristics | ||
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics AM3
Placebo
Inclusion criteria: adults with stable COPD, who had ≥ 5 acute respiratory infections and worsening of pulmonary symptoms (cough, dyspnoea) over the preceding year. Exclusion criteria: severe chronic heart, liver, or kidney disease; malignancies; treatment with corticosteroids or other medications with immunosuppressive properties; treatment with immunostimulants. Pretreatment: no significant differences between groups. |
|
| Interventions |
Intervention characteristics AM3
Placebo
|
|
| Outcomes |
Adverse events
|
|
| Identification |
Sponsorship source: not specified Country: not specifically stated. Authors were associated with a hospital in Switzerland Setting: not specifically stated, presumed to be outpatient Author's name: Carlo, MSr Institution: San Sisto Hospital Address: Poschiavo, Switzerland Participant type: COPD Year: not specified. Study published in 1990 |
|
| Notes | Study published in Spanish. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation occurred by personal computer generation, but otherwise details were not specified. |
| Allocation concealment (selection bias) | Unclear risk | Not specified. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Study was double‐blind, but groups and methodology not specified. Authors did state that treatment and placebo capsules appeared identical. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not specified. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants completed the study. |
| Selective reporting (reporting bias) | Low risk | All intended outcomes appeared to be reported on. |
Catena 1992.
| Study characteristics | ||
| Methods |
Study design: randomised controlled trial Study grouping: parallel group Other: unbalanced 2:1 (intervention:placebo). |
|
| Participants |
Baseline characteristics Thymomodulin
Placebo
Inclusion criteria: adults aged ≥ 40 years with COPD, defined by cough and sputum production for ≥ 3 months per year for 2 consecutive years and an FEV1 20% lower than the reference value (measured in stable conditions during the autumn of 1991). Following these initial eligibility criteria, participants were only included if they had an exacerbation history of ≥ 2 exacerbations in each of the preceding 2 years, had a score of < 10 on a semi‐quantitative scale score including 6 symptoms of cough, fever, sputum volume, sputum characteristics, dyspnoea, pulmonary findings (with each symptom rated from 0 to 3), and had a positive response at 48 hours to ≤ 2 antigens on Multitest‐Merieux testing. Exclusion criteria: recent exacerbations; new chest x‐ray changes; previous pulmonary resection; infectious and chronic inflammatory disease; neoplasm; heart, kidney, liver failure; malabsorption; pregnancy or breastfeeding; repeated antibiotic therapies. Pretreatment: no significant baseline differences between comparison groups. Groups were unbalanced (2:1 thymomodulin:placebo) |
|
| Interventions |
Intervention characteristics Thymomodulin
Placebo
|
|
| Outcomes |
Health‐related quality of life (total score)
Adverse events
|
|
| Identification |
Sponsorship source: not specified Country: Italy Setting: 30 centres, outpatient Author's name: E Catena Institution: not specified Address: not specified Participant type: COPD with concurrent cell‐mediated immune deficiency (as measured by a positive response at 48 hours to ≤ 2 antigens on Multitest‐Merieux testing) Year: December 1991 to April 1992 |
|
| Notes | Study published in Italian. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation occurred, but methods not specified. |
| Allocation concealment (selection bias) | Unclear risk | Not specified. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind study but groups and methodology not specified. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not specified. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Per‐protocol analysis. There appeared to be incongruence in the number of people excluded for protocol violation (prior to randomisation) comparing Figure 1 and Table 1 of the publication. Following study commencement, dropout rates were 8/159 (5%) for the intervention group and 8/77 (10%) for the placebo group, but no information regarding the nature/reasons for these dropouts. |
| Selective reporting (reporting bias) | Low risk | All intended outcomes appeared to have been reported on. |
Cazzola 2006.
| Study characteristics | ||
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics Ismigen
Placebo
Inclusion criteria: moderate‐to‐very severe COPD according to the GOLD severity classification (II–IV), able to understand instructions given by medical staff, able to be co‐operative and in contact with the study centre regularly. Exclusion criteria: suspected lung cancer; continuous domiciliary oxygen therapy; BMI < 19.5 and > 30.0; taking corticosteroids; azathioprine and other immunosuppressive drugs during the previous 6 months; had an episode of acute exacerbation treated with antibiotics within previous month. Pretreatment: no significant differences between groups. |
|
| Interventions |
Intervention characteristics Ismigen
Placebo
|
|
| Outcomes |
Exacerbation duration
Hospitalisation duration (respiratory)
Adverse events
Mortality (all‐cause)
|
|
| Identification |
Sponsorship source: not specified Country: Italy Setting: outpatient Author's name: Mario Cazzola Institution: Pneumology and Allergology Unit and Respiratory Clinical Pharmacology Section, Department of Respiratory Medicine, A. Cardarelli Hospital Email: mcazzola@qubisoft.it Address: Via del Parco Margherita, 2480121 Naples, Italy Participant type: COPD Year: 2003 |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "The eligible patients were randomised into two fairly uniform groups from the point of view of their demographic and baseline data." Randomisation occurred, but methods not specified. |
| Allocation concealment (selection bias) | Unclear risk | Not specified. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind study but groups and methodology not specified. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not specified. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "Twenty‐five patients dropped out of the trial by failing to return for study examinations or due to noncompliance with the trial medication." Appeared to be an ITT analysis although not explicitly stated. Similar rates of dropouts between groups. 82% completed, follow‐up process well‐documented. |
| Selective reporting (reporting bias) | Low risk | Quote: "The two primary study outcomes were the number of AECOPD [acute exacerbations of COPD] events which occurred during the trial period and the follow‐up, and the duration and severity of exacerbation episodes. Secondary outcomes included the rate and length of hospitalization due to AECOPD, and the use of antibiotics and other respiratory drugs." All outcomes appeared to have been reported at 12 months, although there were no data reported at 3 and 6 months. No adverse events reported across the total trial duration despite the length of study. |
Ciaccia 1994.
| Study characteristics | ||
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics Pidotimod
Placebo
Inclusion criteria: hospitalised or outpatient participants age > 45 years, with CB in a stable phase of ≥ 5 years' duration and history of exacerbations during the winter period prior to study. Participants must have had a reduction in FEV1 by < 40% relative to theoretical. Exclusion criteria: CB in an infectious exacerbation phase; recent exacerbation or episode of airway infection in previous 2 weeks; neoplastic disease; asthma or emphysema; severe heart failure, kidney failure, or liver failure; autoimmune disease; fall in bodyweight by ≥ 20% in last 3 months; gastrointestinal disease liable to reduce absorption of study drug; any disease with a poor short‐term prognosis; confirmed or presumptive pregnancy; intermittent or chronic treatment with immunostimulant drugs; cortisone doses > 15 mg/day during days prior to starting trial, or therapy with other immunosuppressant drugs; receiving prophylactic antibiotic therapy. Pretreatment: no significant differences between groups with reported baseline characteristics. Factors such as smoking habits, past/current medicine history, clinical status, asthenia and anorexia, and respiratory function parameters were reported by authors to have been recorded by a doctor at baseline, but were not included in the baseline characteristics table. |
|
| Interventions |
Intervention characteristics Pidotimod
Placebo
|
|
| Outcomes |
Exacerbation duration
Adverse events
|
|
| Identification |
Sponsorship source: not specified Country: Italy Setting: 49 centres, outpatients Author's name: Prof A Ciaccia Institution: Clinica de Malattie dell'Apparato Respiratorio e Tisiologia, Universita degli Studi di Ferrara (Department of Respiratory Tract Diseases and Physiology, University of Ferrara) Email: not reported Address: Via Fossato di Mortara, 23, I‐44100 Ferrara, Italy Participant type: CB or COPD, or both Year: not specified. Study published 1994 |
|
| Notes | Exacerbation duration measured over 5 months in 2 discrete intervals (0–2 months and 3–5 months). Data for the 2‐month interval from baseline was unable to be used in meta‐analysis as this would be inconsistent with protocol criteria (specifying outcomes need to be measured over ≥ 12 weeks). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Authors reported a stratified, blocked permutation randomisation scheme; however, no further information provided to ensure that this was a truly randomised process. |
| Allocation concealment (selection bias) | Unclear risk | Not specified. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind; however, groups and methodology not specified. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not specified. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Per‐protocol analysis. Reasons for dropouts were well‐documented and well‐matched between groups, with an overall dropout rate of 11.4%. |
| Selective reporting (reporting bias) | Unclear risk | Appeared to report on main outcomes but reporting appeared skewed towards data that were statistically significant. Outcomes for the group of participants with a lesser baseline exacerbation frequency (≤ 3 episodes) was more heavily reported than the outcomes for the participants with a higher baseline exacerbation frequency. |
Collet 1997.
| Study characteristics | ||
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics OM‐85
Placebo
Inclusion criteria: ≥ 20 pack‐year smoking history, FEV1 20–70% of predicted value with < 15% improvement after inhaled salbutamol 200 μg. Exclusion criteria: severe concomitant disease making follow‐up difficult or unlikely; been prescribed medications affecting the immune system (i.e. immunosuppressants or systemic corticosteroids for > 2 weeks in the last month); had an episode of acute exacerbation treated with antibiotics within previous month. Pretreatment: more participants in placebo group than in treatment group had been hospitalised before the trial (68.4% vs 62.8%); however, authors reported that following examination of the hospitalisation causes the 2 groups (quote) "did not differ in any substantive or systematic manner." |
|
| Interventions |
Intervention characteristics OM‐85
Placebo
|
|
| Outcomes |
Hospitalisation duration (respiratory)
Health‐related quality of life (total score SF‐36 survey)
Adverse events
Mortality (all‐cause)
Mortality (respiratory)
Number of participants with no exacerbations
|
|
| Identification |
Sponsorship source: funded by Jouveinal Inc; however, authors stated that the project was developed and carried out in complete independence from the sponsor. Country: Canada Setting: outpatient Author's name: Dr Jean‐Paul Collet, MD Institution: Randomized Clinical Trial Unit, SMBD Jewish General Hospital Address: 3755 Côte St Catherine, Suite A‐132, Montréal, PQ, H3T 1E2 Canada Participant type: COPD Year: 1994 |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Randomization was centralized at the SMBD Jewish General Hospital, stratified by institution and degree of ventilatory impairment (i.e. subjects were divided into those whose FEV1 was 20 to 40% of predicted and those whose FEV1 was greater than 40%). Block randomization in groups of four patients was carried out to insure a balanced treatment allocation within strata." Unclear how randomisation was generated but otherwise well‐described. |
| Allocation concealment (selection bias) | Unclear risk | Not specified. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Authors referred to placebo being identical in appearance to intervention tablet but methods of concealment not otherwise specified. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "All this work was blind to the nature of the treatment administered." Detailed information provided regarding data collection and outcome assessments. Interpretation of interim analysis results and decisions were taken by an independent committee. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ITT analysis. Very few participants lost to follow‐up and dropouts/attrition/exclusions well‐described. |
| Selective reporting (reporting bias) | Low risk | All intended outcomes appeared to have been reported on. |
Cvoriscec 1989.
| Study characteristics | ||
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics OM‐85
Placebo
Inclusion criteria: adults aged 20–69 years with diagnosis of CB, who had an acute bronchitis episode at time of enrolment. Exclusion criteria: diseases other than mild chronic obstructive bronchitis; treated concomitantly with corticosteroids or other immunotherapeutics. Pretreatment: no significant differences between groups. |
|
| Interventions |
Intervention characteristics OM‐85
Placebo
|
|
| Outcomes |
Exacerbation duration
Number of participants requiring antibiotics
Adverse events
|
|
| Identification |
Sponsorship source: OM Laboratories, Geneva (Switzerland) and Lek, Ljubljana (Yugoslavia) supplied the intervention drug and placebo. 1 author appeared to be associated with the Research and Development Department at Lek. Country: Yugoslavia (modern day Croatia and Slovenia) Setting: participants were recruited during an acute exacerbation, but assumed that remainder of study was conducted in an outpatient setting. Author's name: B Cvoriscec Institution: Dr Josip Kajfes' General Hospital, Zagreb Participant type: CB main criteria; included some participants with COPD Year: not specified. Published 1989 |
|
| Notes | The outcome of number of participants requiring antibiotics was measured over 6 months in discrete 1‐monthly intervals. Data for each interval were unable to be combined and used in meta‐analysis as this would be inconsistent with protocol criteria (specifying outcomes need to be measured over ≥ 12 weeks). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation occurred, but methods were not specified. |
| Allocation concealment (selection bias) | Unclear risk | Not specified. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind study but groups and methodology not specified. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not specified. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Reasons for exclusion or attrition, or whether this occurred at all, were not specified. The n for each outcome analysis equalled the number originally randomised but unclear if this was because the study performed an ITT analysis or whether there were no dropouts. |
| Selective reporting (reporting bias) | Low risk | All main outcomes appeared to have been reported on. Reporting of adverse events was partial and only referred to 1 participant in the intervention group experiencing what authors deemed to be an adverse reaction; however, there was no objective reporting of other events (which may or may not have been related) in either the treatment or placebo groups over the 6‐month study period. |
Debbas 1990.
| Study characteristics | ||
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics OM‐85
Placebo
Inclusion criteria: hospitalised elderly people with a clinical definition of CB (WHO definition), and ≥ 4 acute exacerbations during the previous autumn to winter period. Exclusion criteria: none specified. Pretreatment: only basic baseline characteristics shown, with no apparent significant differences. Authors reported that groups differed with respect to "the secondary diagnosis, with a significantly higher rate of cardiovascular disease in the OM‐85 BV group." Baseline characteristics were reported for the original enrolled 396 participants, rather than the 265 participants who had a confirmed diagnoses of CB and participated in the study. |
|
| Interventions |
Intervention characteristics OM‐85
Placebo
|
|
| Outcomes |
Number of participants with no exacerbations
Number of participants requiring antibiotics
Adverse events
|
|
| Identification |
Sponsorship source: not specified Country: authors are associated with centres in France Setting: participants recruited during a hospital admission, but for the remainder of the study assumed outpatient setting. Author's name: Dr N Debbas Institution: Centre Recherche des Laboratoires Fournier Address: 9 Rue Petitot, Dijon 21100, France Participant type: CB (although mention is made of stage III and IV bronchitis which may imply obstructive airways disease, although data not analysed separately for these participants) Year: not specified. Study published in 1990. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomly assigned to treatment." Little mention was made of this study being a randomised trial, but within the text authors did report that participants were randomly assigned to treatment. Methods of randomisation not specified. |
| Allocation concealment (selection bias) | Unclear risk | Not specified. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind study, but groups and methodology not specified. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not specified. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | ITT analysis stated; however, whether there were any dropouts during the study and final numbers were not reported. It seemed unusual that characteristics were analysed for the originally enrolled 396 participants (and that each of these were evenly randomised to the 2 intervention groups) before then excluding 131 participants due to an absence of the inclusion criteria diagnosis. |
| Selective reporting (reporting bias) | Unclear risk | Main outcomes appeared to have been reported on, but some outcomes were only partially reported and there were possibly discrepancies within the reporting. For example, authors stated that, "observed effect was more pronounced in III and IV stage patients," but provided no numerical data to clarify this statement. Number of participants without an exacerbation during study period was 48% in intervention group and 39% in placebo group; however, from the raw data provided in Table 2 the proportions appeared to be 50% in intervention group and 37% in placebo group. Data around adverse events and "intercurrent" events were also partially reported. |
De Bernardi 1992.
| Study characteristics | ||
| Methods |
Study design: randomised controlled trial Study grouping: parallel group. Multi‐arm, with 3 comparison groups (2 intervention and 1 placebo). |
|
| Participants |
Baseline characteristics Lantigen B (GB)
Lantigen B (Italy)
Placebo
Inclusion criteria: CB, diagnosed using MRC criteria, with borderline immunosuppression verified using immunological tests (Multitest‐Merieux, chemotaxis, phagocytic activity of bronchoalveolar lavage macrophages, concentration of sputum IgA) Exclusion criteria: use of corticosteroid or other immunomodulatory drugs in previous 3 months, contraindications to undergoing bronchoalveolar lavage Pretreatment: no significant differences between groups. |
|
| Interventions |
Intervention characteristics Lantigen B (UK)
Lantigen B (Italy)
Placebo
|
|
| Outcomes |
Exacerbation duration
Number of participants requiring antibiotics
Adverse events
|
|
| Identification |
Sponsorship source: not specified Country: not specified. Authors are associated with institutions in Italy Setting: not specified, presumed outpatient. Author's name: M de Bernardi Institution: Istituto Farmacologia II, Universita di Pavia Address: Pavia, Italy Participant type: CB (note all participants also had a 'borderline' immunodeficiency defined by immunological testing) Year: not specified. Published 1992 |
|
| Notes | Study published in Italian. For meta‐analysis, the 2 intervention groups were combined. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation occurred, but methods not specified. |
| Allocation concealment (selection bias) | Unclear risk | Not specified. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind study and authors reported use of identical packaging for intervention groups and placebo; however, groups and methodology not specified. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not specified. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not specified whether analysis was ITT or per‐protocol, and no there was no reporting of dropouts/exclusions/attrition during the study period. As the number of participants analysed at study completion was the same as was randomised, it could either mean that there were no dropouts or that ITT analysis occurred. |
| Selective reporting (reporting bias) | Low risk | All intended outcomes appeared to have been reported on. |
Djuric 1989.
| Study characteristics | ||
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics OM‐85
Placebo
Inclusion criteria: adults with CB and mild (reversible) airways obstruction Exclusion criteria: other pulmonary diseases; concomitant treatment with corticosteroids or other immunotherapeutics Pretreatment: no significant differences between groups |
|
| Interventions |
Intervention characteristics OM‐85
Placebo
|
|
| Outcomes |
Exacerbation duration
|
|
| Identification |
Sponsorship source: not specified Country: authors are associated with institutions in Belgrade. Setting: participants were recruited during an inpatient stay; however, assumed outpatient setting for the remainder of the study. Author's name: O Djuric Institution: Institute of Pulmonary Diseases and Tuberculosis Address: Belgrade, Yugoslavia Participant type: CB with mild (reversible) airflow obstruction Year: not specified. Published 1989. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation occurred, but methods not specified. |
| Allocation concealment (selection bias) | Unclear risk | Not specified. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind study. Authors reported that placebo capsule looked identical, but otherwise the groups and methodology not specified. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not specified. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not clear whether ITT or per‐protocol analysis and no information reported on dropouts/exclusions/attrition. Number of participants analysed in outcome graphs appeared equal to number randomised at beginning of study, so it was possible that data presented were ITT or there were no dropouts. |
| Selective reporting (reporting bias) | Low risk | All main outcomes appeared to have been reported on although for some outcomes there was no presentation of raw data. No reporting on adverse outcomes. |
EUCTR2007‐004702‐27‐DE.
| Study characteristics | ||
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics None provided. Inclusion criteria: adults aged ≥ 40 years with history of documented acute exacerbations of CB ≥ 2 in previous year and COPD stage II to III, a postbronchodilator FEV1 ≥ 30% and < 80% predicted and documented within 6 months prior to enrolment in study, an active or past smoking history of ≥ 20 pack‐years, able to provide written informed consent. Exclusion criteria: participants with asthma, mucoviscidosis, bronchiectasis, known disseminated malignancy, known chronic systemic infections or inflammatory conditions (e.g. rheumatoid arthritis, systemic lupus erythematosus, or active sarcoidosis), previous solid organ transplantation, myocardial infarction or cerebrovascular accident within 6 months prior to study enrolment; treated with the following medications: corticosteroids the day of visit 1, oral vaccination with live vaccine within 4 weeks before study commencement, previous or concomitant (or both) immunosuppressive or immunostimulating therapy within 3 months before study commencement, regular oral corticosteroids of prednisolone ≥ 10 mg for > 2 weeks; known allergy or previous intolerance to study medication; pregnant, lactating, or of child‐bearing potential and no reliable contraception (oral contraceptive, intrauterine device, or a Pearl Index < 1); unable to follow instructions or unreliable (including those who were non‐compliant, had known alcoholism or drug abuse, or history of a serious psychiatric disorder) as well as those unwilling to give informed consent or to abide by requirements of protocol; any other clinical condition which, in the opinion of the investigator, would not allow safe completion of protocol and safe administration of study medication; undergone a major surgical procedure within 3 months of enrolment in study or who had participated in a drug study within 4 weeks prior to study. Pretreatment: no data regarding baseline characteristics reported. |
|
| Interventions |
Intervention characteristics OM‐85
Placebo
|
|
| Outcomes |
Adverse events
Serious adverse events
Number of participants with no exacerbations
Mortality (all‐cause)
Exacerbation duration
Health‐related quality of life (total score SGRQ)
|
|
| Identification |
Sponsorship source: OM Pharma Country: several countries in Europe (Austria, Belgium, Germany, and Italy) Setting: multicentre. Presumed to be outpatient Author's name: Prof Dario Olivieri, MD Institution: Rasori Hospital, University of Parma Address: Parma, Italy Participant type: COPD Year: not specified. Published 2007. |
|
| Notes | OM/Vifor Pharma and an independent group of experts, considered the study to be flawed. Vifor Pharma was of the opinion that efficacy conclusions on OM‐85 BV could not be made due to ineligible and missing data. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Eligible patients were randomised to either Broncho‐Vaxom ® or placebo." Randomisation occurred but methods not specified. |
| Allocation concealment (selection bias) | Unclear risk | Not specified. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind study but groups and methodology are not specified. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not specified. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: "It is important to note that study management and quality control at OM/Vifor Pharma identified several data quality issues during the analysis phase of the study. Numerous inconsistencies were found between case report forms and datasets. In addition, a large amount of missing data was identified. These quality issues were assessed to be mainly due to insufficient monitoring of the trial and poor data management by the Contract Research Organisation (CRO) tasked with study conduct and data management. A blinded re‐adjudication of all exacerbations observed during the trial was conducted by an independent expert group. Numerous cases of exacerbation were not considered to be valid events by the experts. Also, a post‐hoc analysis of the reported pre‐study acute exacerbations, an important study inclusion criterion, was conducted. Based on the protocol definition of an acute exacerbation, it appears that many patients probably did not have ≥ 2 documented exacerbations in the year before the study began. This reduced the number of valid patients in the study and thus the statistical power of the study to be able to detect a difference in the treatment arms. Based on all these above‐mentioned factors, OM/Vifor Pharma, as well as an independent group of experts, considers the study to be flawed. Vifor Pharma is of the opinion that efficacy conclusions on OM‐85 BV cannot be made based on this study." Deemed to be high risk of bias for this domain based on the above. |
| Selective reporting (reporting bias) | High risk | Data reported for number of participants with acute exacerbations, adverse events, and mortality; for all other outcomes data reported as "not significant." However, the flaws with data collection and study design were likely to have a flow‐on effect in terms of reporting bias (although not intentional, as it appeared all main outcomes were looked at). Vifor Pharma was of the opinion that efficacy conclusions on OM‐85 BV could not be made from this study. |
Fietta 1988.
| Study characteristics | ||
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics RU41740 (Biostim)
Placebo
Overall
Inclusion criteria: adults aged > 18 years with CB (Stage I–III defined by MRC criteria) with ≥ 2 exacerbations in the 12 months preceding study commencement. Exclusion criteria: acute exacerbation in month prior to study; systemic medication with more than the equivalent dose of prednisone 10 mg/day, or with other drugs that affect the immune system and vaccination against influenza virus or against bacteria within 6 months before entering trial; receiving prophylactic antibiotic regimen. Pretreatment: authors reported no significant differences between groups, although baseline characteristics were only reported for the 29 participants as a whole. |
|
| Interventions |
Intervention characteristics RU41740 (Biostim)
Placebo
|
|
| Outcomes |
Exacerbation duration
Number of participants with no exacerbations
Adverse events
|
|
| Identification |
Sponsorship source: partially supported by grants from the Consiglio Nazionale delle Ricerche and the Minestero della Publica Istruzione (Rome, Italy) Country: authors associated with institutions in Italy Setting: not specifically stated, presumed to be outpatient. Author's name: A Fietta Institution: Department of Chemotherapy, University of Pavia Address: Via Taramelli 5I‐27 100 Pavia, Italy Participant type: CB (although included stage III CB, which by current definitions would be people with COPD/airflow limitation) Year: not specified. Published 1988 |
|
| Notes | Treatment days did not total 30 days (3 × 8‐day courses); however, treatment spanned across 3 months and a continuous effect was assumed; therefore, study included. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation occurred but methods not specified. |
| Allocation concealment (selection bias) | Unclear risk | Not specified. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind, but groups and methodology not specified. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not specified. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Authors reported that 36 participants entered study and 29 were randomised to 2 groups after exclusions for not fulfilling protocol requirements. They stated that 6 participants were excluded because of missing data and 1 died during the trial; it is unclear if this a clarification of the initial 7 exclusions described (which would seem incongruent given data were collected for these participants) or whether an additional 6 dropouts/exclusions occurred. In which case these additional dropouts were only partially described. It was unclear to which comparison groups the dropouts belonged. For the above reasons, it is unclear whether analysis was ITT or per‐protocol. Number of participants analysed at trial completion equalled the number randomised; but if data were collected for the 7 described exclusions then analysis may be per‐protocol. If ITT analysis was used and the 7 exclusions occurred prior to study commencement then any further/absence of dropouts following randomisation have not been reported on. |
| Selective reporting (reporting bias) | Low risk | All main outcomes appeared to have been reported on. |
Foschino 1995.
| Study characteristics | ||
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics D53 (Ribomunyl)
Placebo
Inclusion criteria: adults with CB (ATS criteria), with ≥ 3 respiratory infective episodes during the previous winter (defined by symptoms of cough, sputum, and fever). Exclusion criteria: tubercular pathology, bronchiectasis, pulmonitis, and bronchopneumonia, or bronchial pathology due to gastric reflux; treatment with immunostimulant or immunosuppressive therapies in previous 6 months; women who were, or suspected to be, pregnant. Pretreatment: not specified |
|
| Interventions |
Intervention characteristics D53 (Ribomunyl)
Placebo
|
|
| Outcomes |
Exacerbation duration
Number of participants requiring antibiotics
Adverse events
|
|
| Identification |
Sponsorship source: not specified Country: authors were associated with institutions in Italy Setting: not specifically stated, presumed to be outpatient Author's name: Prof E Gramiccioni Institution: Cattedra Malattie Apparato Respiratorio, Policlinico di Bari Address: Piazza G. Cesare, 1 ‐ 70124 Bari Participant type: CB Year: not specified; study submitted in 1994. |
|
| Notes | Study published in Italian. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation occurred but methods not specified. |
| Allocation concealment (selection bias) | Unclear risk | Not specified. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Study was double‐blind, but groups and methodology not specified. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not specified. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 61/64 participants completed study. 1 withdrew voluntarily and 2 were excluded because they contracted bronchopulmonary pathology and abdominal herpes zoster. Authors did not specify how many participants were analysed at completion of study for efficacy (i.e. whether a per‐protocol analysis or an ITT analysis was used); for clinical outcomes n was unclear. Biomarker outcomes of study suggest that n for both intervention and placebo groups were significantly lower (17 and 18 respectively for immunoglobulin analysis; 8 and 8 for T cell analysis) |
| Selective reporting (reporting bias) | Unclear risk | Unclear from paper, abstract, or translator whether all outcomes were reported on. |
Habermann 2001.
| Study characteristics | ||
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics Symbioflor
Placebo
Inclusion criteria: confirmed medical history of chronic obstructive bronchitis (defined by productive cough for the preceding 2 years on most days for ≥ 3 months before admission to study), FEV1 > 1 L Exclusion criteria: pregnant or breastfeeding; severe allergic diathesis; coexisting severe systemic diseases (not specified); chronic emphysema; type A bronchitis; Kartagener's syndrome; cystic fibrosis Pretreatment: no differences in age, height, or weight. A greater proportion of participants in placebo group had required antibiotic treatment for a preceding exacerbation (82% in placebo group compared to 74% in intervention group). |
|
| Interventions |
Intervention characteristics Symbioflor
Placebo
|
|
| Outcomes |
Number of participants with no exacerbations
Number of participants requiring antibiotics
Adverse events
|
|
| Identification |
Sponsorship source: not specified Country: authors associated with institutions in Germany Setting: multicentre. Not specifically stated but presumed to be outpatient. Author's name: Dr Kurt Zimmerman Institution: Institut fur Mikrookologie Email: kurt.zimmerman@mikrooek.de Address: Auf den Luppen 8, Herborn‐Horbach, Germany Participant type: chronic obstructive bronchitis Year: not specified. Published 2001. |
|
| Notes | Outcome of number of participants with no exacerbations was analysed over 14 months in 2 discrete intervals (0–6 months and 7–14 months). For meta‐analysis, the first interval from baseline was used. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation occurred but methods not specified. |
| Allocation concealment (selection bias) | Unclear risk | Not specified. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind study but groups and methodology not specified. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not specified. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Analysis appeared to be ITT, as authors stated that all participants who were enrolled were finally accepted for evaluation of efficacy/tolerability; however, this is not explicitly stated/made clear. Data were not available for 2 participants but not specified to which comparison group they belonged. There was no reporting on occurrence/absence of dropouts during the 14‐month study period. |
| Selective reporting (reporting bias) | Low risk | All main outcomes appeared to have been reported on. Of note, some figures with presentation of graphical representations of the data did not match the outcomes ascribed to them (although numerical data presented within the text of study). |
Hutas 1994.
| Study characteristics | ||
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics OM‐85
Placebo
Inclusion criteria: adults aged 20–70 years with a current WHO definition of CB, who had several exacerbations in the 6‐month autumn/winter period of previous year, and had FEV1 > 50% predicted. Exclusion criteria: active pulmonary disease; long‐term steroid or immunosuppressant therapy; previous treatment with OM‐85 BV or another immunostimulant Pretreatment: higher proportion of active smokers in placebo group. |
|
| Interventions |
Intervention characteristics OM‐85
Placebo
|
|
| Outcomes |
Exacerbation duration
|
|
| Identification |
Sponsorship source: OM Laboratories, Geneva, Switzerland. Study was monitored by OM Laboratories. Case report forms were sent to OM Laboratories. Data were analysed by an independent informatic company (Biometrix). Country: Hungary Setting: 3 centres, outpatient Author's name: Dr Imre Hutas Institution: Semmelweis University Address: Budapest, Hungary Participant type: CB or COPD or both (participants defined as having mild obstruction) Year: 1992–1993 |
|
| Notes | Study published in Hungarian. Unclear whether SE or SD used. Because authors have reported "SD" for some outcomes, taken to be SD. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation occurred but methods not specified. Further information obtained from original author suggested computer generation used. |
| Allocation concealment (selection bias) | Unclear risk | Translated quote: "during the study none of the envelopes containing information on patient allocation had to be opened." This possibly indicated a low risk of selection bias, but there was insufficient information provided to make an accurate judgement. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind study but groups and methodology not specified. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not specified. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There were 6 dropouts (5%), which the study mentions, but did not state why the dropouts occurred, which comparison groups were affected or at what point in the study the dropouts occurred. Participants' baseline characteristics and all outcomes were analysed per protocol. |
| Selective reporting (reporting bias) | Low risk | All main outcomes appeared to have been reported on. |
Keller 1984.
| Study characteristics | ||
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics OM‐85
Placebo
Inclusion criteria: adults with an acute exacerbation of CB at time of recruitment. Exclusion criteria: none specified. Pretreatment: no significant differences between groups. |
|
| Interventions |
Intervention characteristics OM‐85
Placebo
|
|
| Outcomes | Study reported on outcomes related to number of acute exacerbation events, number of hospitalisations, and duration of antibiotic therapy and other concomitant medications. However, did not report outcomes that were aligned with the intended review outcomes. | |
| Identification |
Sponsorship source: not specified Country: authors associated with institutions in Switzerland Setting: participants were recruited during an acute exacerbation; however, assumed outpatient setting for remainder of study. Author's name: Dr R Keller Institution: Kantonsspital, Aarau Address: CH‐5001 Aarau, Switzerland Participant type: CB Year: 1981–1982 |
|
| Notes | Study published in German. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation occurred but method not specified. |
| Allocation concealment (selection bias) | Unclear risk | Not specified. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind trial and authors reported that study medications (quote) "could not be differentiated." However, groups and methodology not otherwise specified. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not specified. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Analysis appeared to be per‐protocol. Authors reported that following treatment commencement there were 3 participants in placebo group who ceased treatment due to perceived inefficacy, with no such type of loss in intervention group. No further comments about dropouts/exclusions during study; however tabulated results suggested a range of n for each comparison group depending on outcome analysed. Based on these ranges, it appeared there may have been 4 dropouts in intervention group and 6 in placebo group (12% overall); however, it is unclear. |
| Selective reporting (reporting bias) | Low risk | All main outcomes were reported on, but some outcomes were only partially reported as there were (quote) "no significant differences" with no associated numerical data (e.g. number of exacerbation events and hospitalisations). |
Li 2004.
| Study characteristics | ||
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics OM‐85
Placebo
Inclusion criteria: CB and COPD as defined by the Fifth National Symposium of the Chinese Respirology Society, history of cigarette smoking, had acute exacerbations of COPD requiring hospitalisation and repeated antibiotic therapy during the year prior to trial, free of acute episodes or lower respiratory tract infections in the 4 weeks prior to study commencement. Exclusion criteria: immunostimulant use in preceding 3 months; corticosteroid use in preceding 4 weeks; concurrent serious cardiopulmonary, hepatic, or renal diseases; presence of respiratory failure. Pretreatment: no significant differences between groups. |
|
| Interventions |
Intervention characteristics OM‐85
Placebo
|
|
| Outcomes |
Exacerbation duration
Adverse events
|
|
| Identification |
Sponsorship source: not specified Country: China Setting: outpatient Author's name: Dr Li Jing Institution: Guangzhou Institute of Respiratory Disease Email: jingli016@21cn.com Address: Guangzhou, 510120, China Participant type: COPD/CB Year: not specified. Published 2004 |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation occurred but methods not specified. |
| Allocation concealment (selection bias) | Unclear risk | Not specified. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind study, but groups and methodology not specified. Active and placebo capsules were both made by the pharmaceutical company, so it could be assumed that treatments looked identical, but no detail provided regarding this, the code, or distribution. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not specified. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No information provided regarding presence/absence of dropouts or whether analysis was ITT. For sputum culture analysis at 3 months, results implied that n for each comparison group equalled the number who were initially randomised. Elsewhere in the report n was not given for other outcomes/time points. |
| Selective reporting (reporting bias) | Low risk | All intended outcomes appeared to have been reported on. |
Menardo 1985.
| Study characteristics | ||
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics Diribiotine CK
Placebo
Inclusion criteria: COPD with ≥ 3 infective exacerbations in preceding year Exclusion criteria: recent other immunomodulation treatment or respiratory vaccination; prolonged treatment with steroids or antibiotics; presence of neoplasia, severe immunodeficiency; severe cardiac, renal, hepatic, haematological disease or evolving tuberculosis. Pretreatment: higher proportion of men, active/ex‐smokers in placebo group. Marginally higher baseline exacerbation rate in placebo group. |
|
| Interventions |
Intervention characteristics Diribiotine CK
Placebo
|
|
| Outcomes |
Number of participants with no exacerbations
Exacerbation duration
Number of participants requiring antibiotics
Adverse events
|
|
| Identification |
Sponsorship source: not specified. 1 authors was associated with Vevey (makers of Diribiotine) Country: authors were associated with institutions in France and Switzerland Setting: not specifically stated, presumed to be outpatient Author's name: Prof FB Michel Institution: Hopital de l'Aiguelongue Address: Av du Major‐Flandre 34059 Montpellier, France Participant type: COPD Year: December 1981 to November 1983 |
|
| Notes | Study published in French. The outcomes of number of participants with no exacerbations and number of participants requiring antibiotics were measured over 6 months in 2 discrete 3‐month intervals. The first interval from baseline was used in meta‐analysis. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation occurred, but methods not specified. |
| Allocation concealment (selection bias) | Unclear risk | Not specified. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind study, but groups and methodology not specified. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not specified. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Per‐protocol analysis. Number of participants for various study time points and outcomes varied considerably. No information provided regarding dropouts/exclusions/attrition that occurred during study. Within the text, authors reported that 56 participants were initially enrolled, 44 analysed at day 90 and 39 analysed at day 180. However, the outcome table for participants without exacerbations and number of participants receiving antibiotic therapy suggest an n of 29 at 90 days and 20 at 180 days. This may suggest a dropout rate of up to 64%. |
| Selective reporting (reporting bias) | Unclear risk | Main outcomes appeared to be reported on, although not all were provided as numerical data. Given the incongruities regarding the number of participants at various time points for the study and which seem to vary between outcomes reported, it was unclear whether a degree of selective reporting also occurred. |
Messerli 1981.
| Study characteristics | ||
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics OM‐85
Placebo
Inclusion criteria: CB, with an acute exacerbation at the time of enrolment. Exclusion criteria: not specifically stated from the outset; however, in the results section, participants were excluded from both treatment and placebo groups for using antibiotics. Pretreatment: no significant differences between groups regarding sex, proportion of smokers, and illness history (such as number and severity of episodes before treatment). However, there was a difference in the number of days absent from work before treatment, with more days off in the treatment group (P < 0.005). Aside from age and baseline symptoms, raw data were not presented for these metrics. |
|
| Interventions |
Intervention characteristics OM‐85
Placebo
|
|
| Outcomes |
Adverse events
|
|
| Identification |
Sponsorship source: not specified; however, the study address and 2 author associations were listed as the OM‐85 BV research lab. Country: authors associated with institutions in Switzerland Setting: participants appeared to have been recruited at the time of an acute exacerbation; however, assumed outpatient setting for the remainder of study. Author's name: C Messerli Institution: not stated Address: 22 rue du Bois‐du‐Lan, 1217 Meyrin 2, Geneva Participant type: CB Year: 1978–1979 |
|
| Notes | Study published in French. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation occurred using a "randomised list." Methods otherwise not specified. |
| Allocation concealment (selection bias) | Unclear risk | Not specified. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind study, and authors stated that tablets and packaging were identical for each participant. However, groups and other methodology not specified. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not specified. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Per‐protocol analysis. Reasons for dropouts/exclusions were described, and overall 71/81 (87.6%) participants completed study. Higher number of dropouts/exclusions in treatment group (13.3% in intervention vs 6% in placebo). |
| Selective reporting (reporting bias) | Low risk | All intended outcomes appeared to have been reported on. |
Olivieri 2011.
| Study characteristics | ||
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics Not specified. Assumed 2 equal groups of 170 (340 participants). Inclusion criteria: adults aged > 40 years who had a diagnosis of stage II or III COPD (FEV1 30–80% predicted), were past or active smokers, history of ≥ 2 documented exacerbations of COPD in preceding year. Exclusion criteria: not specified. Pretreatment: not specified. |
|
| Interventions |
Intervention characteristics OM‐85
Placebo
|
|
| Outcomes |
Exacerbation duration
Adverse events
|
|
| Identification |
Sponsorship source: not specified Country: 5 unspecified countries Setting: multicentre, outpatient Comments: abstract only Author's name: Dario Olivieri Institution: Department of Clinical and Experimental Medicine, Universita di Parma Address: Parma, Italy Participant type: COPD Year: not specified. Published 2011. |
|
| Notes | Abstract only. Assumed 2 equal comparison groups of 170 participants. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation occurred but methods not specified (abstract only). |
| Allocation concealment (selection bias) | Unclear risk | Not specified (abstract only). |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind study, but groups and methodology not specified (abstract only). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not specified (abstract only). |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not enough information provided to determine dropout rates or whether analysis was ITT (or both) (abstract only). |
| Selective reporting (reporting bias) | Unclear risk | Not enough information able to be obtained (abstract only). |
Orcel 1994.
| Study characteristics | ||
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics OM‐85
Placebo
Inclusion criteria: CB (history of excessive sputum expectoration occurring on most days during ≥ 3 consecutive months for > 2 consecutive years), aged ≥ 65 years and history of 4 documented acute infections of the lower respiratory tract treated with antibiotics, in reference period during the previous year (6 months between 1 October 1986 and 30 April 1987). Exclusion criteria: asthma or bullous emphysema; history of malignancy; progressive infectious or immune diseases; left heart failure; renal or hepatic insufficiency; received immunosuppressive treatment in previous 3 months, or another immunomodulating agent or antipneumococcal vaccine in previous 6 months. Pretreatment: higher total number of comorbidities in the OM‐85 BV treatment group (403 in intervention vs 331 in placebo; not statistically significant). Larger number of cardiovascular disorders (201 in intervention vs 137 in placebo) in those receiving the intervention (P < 0.01). |
|
| Interventions |
Intervention characteristics OM‐85
Placebo
|
|
| Outcomes |
Number of participants with no exacerbations
Adverse events
Mortality (all‐cause)
Mortality (respiratory)
|
|
| Identification |
Sponsorship source: not specified Country: France Setting: 25 centres, outpatient institutions for older people Author's name: JPh Derenne Institution: Service de Pneumologie et Réanimation Groupe Hospitalier Pitié‐Salpêtrière Address: 47 boulevard de l'Hôpital, 75013 Paris, France Participant type: CB Year: 1987–1988 |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "allocated to either OM‐85 BV or placebo treatment by a centralized randomization procedure, stratified for each centre." Randomisation occurred and there are some details provided regarding this process, but methods not clearly specified. |
| Allocation concealment (selection bias) | Low risk | Judged to be low due to the centralisation of the randomisation procedure (i.e. independent of the centres conducting the studies). There were also independent assessors analysing eligibility for study. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "The second group took similar capsules containing only the excipients." Double‐blind study, but groups and methodology not specified. Treatments were similar in appearance. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not specified. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "Thirty nine patients died…Twenty five patients did not complete the study for other reasons, including transfer to another hospital, failure to follow the protocol, or return to their family home (10 placebo, 15 OM‐85 BV)." There was an 18% dropout rate over 6 months. This was largely contributed to by number participants who died during trial period – likely related to the study involving a very elderly population with significant comorbidity. All participants were accounted for and dropouts between groups appeared balanced. Analysis per protocol, rather than ITT. |
| Selective reporting (reporting bias) | Low risk | All main outcomes appeared to have been reported on, although there was partial reporting/no numerical data presented for some outcomes. |
Orlandi 1983.
| Study characteristics | ||
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics OM‐85
Placebo
Overall
Inclusion criteria: adults aged > 18 years with diagnosis of CB (no definition specified). Exclusion criteria: none specified. Pretreatment: groups described as homogeneous for age, gender, and duration of disease. |
|
| Interventions |
Intervention characteristics OM‐85
Placebo
|
|
| Outcomes |
Exacerbation duration
Hospitalisation duration (respiratory)
Adverse events
|
|
| Identification |
Sponsorship source: not specified. Country: authors were associated with institutions in Italy Setting: outpatient Author's name: Prof O Orlandi Institution: San Luigi Gonzaga University Hospital Address: Sanatorio San Luigi, Italy Participant type: CB Year: 1982–1983 |
|
| Notes | Study published in Italian. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Authors reported that participants were divided into 2 groups using a "randomization code;" however, the code type was not specified in the article. Given that the total number of participants was very small (20 patients) and authors also stated that they were divided into 2 homogeneous groups (for age, sex, diagnosis, and duration of disease), it is unlikely that there was a 'true' randomisation. |
| Allocation concealment (selection bias) | High risk | As above, due to the small number of participants and participants were intentionally divided into 2 homogeneous groups (for age, sex, diagnosis, and duration of disease), there is likely to have been a degree of selection bias. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind study, but groups and methodology not specified. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not specified. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 20 participants were randomised and 19 completed the trial. 1 participant in the placebo group appeared to have left the study but no reason given as to why and no timeframe specified; otherwise all other participants were included in analysis. |
| Selective reporting (reporting bias) | Unclear risk | All main outcomes appeared to have been reported on; however, no numerical data provided. Data were only presented in graphical form where the authors ascribed a P < 0.05 for statistically significant results, although it was not clear from the text which outcomes, time frames, and comparison groups (e.g. change from baseline, or treatment vs placebo) the P values correspond to. The patient and doctor assessment of efficacy was not well‐described and it was unclear how efficacy was assessed. |
Rico 1997.
| Study characteristics | ||
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics Thymomodulin
Placebo
Inclusion criteria: CB or emphysema (ATS definition). Exclusion criteria: previous immunosuppressive or immunomodulatory treatment; viral hepatitis; cancer; acute respiratory or other infection; severe hepato‐renal or cardiovascular disease; terminal illness; malnutrition. Pretreatment: no significant differences between groups. |
|
| Interventions |
Intervention characteristics Thymomodulin
Placebo
|
|
| Outcomes |
Exacerbation duration
Adverse events
|
|
| Identification |
Sponsorship source: not specified. 3 authors are associated with a company named 'Pharmacia & Upjohn' Country: all authors are associated with institutions in Mexico Setting: outpatient Author's name: Dr Favio Gerardo Rico Mendez Institution: Departamento de Neumologia, Centro Medico Nacional La Raza, Instituto Mexicano del Seguro Social Address: Jacarandas y Vallejo. Mexico 02990, DF Participant type: COPD or chronic bronchitis, or both Year: 1994–1995 |
|
| Notes | Study published in Spanish. Outcomes measured over 12 months in discrete 3‐month intervals. The first interval from baseline was used in meta‐analysis. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation reported (not evident from the English abstract, but the authors mentioned this in study text), but methods not specified. |
| Allocation concealment (selection bias) | Unclear risk | Not specified. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind study, but groups and methods not specified. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not specified. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Dropouts/attrition/exclusions from analysis not specified and authors did not report whether ITT analysis was used. Most measured outcomes were continuous and n was not specified in outcome tables. |
| Selective reporting (reporting bias) | Low risk | All intended outcomes appeared to have been reported on. |
| Other sources of bias | Unclear risk | Baseline data obtained by retrospective collation, possibly introducing an element of hindsight bias. |
Rochemaure 1988.
| Study characteristics | ||
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics Diribiotine CK
Placebo
Inclusion criteria: CB (WHO definition), PaO2 > 50 mmHg, ≥ 2 infectious episodes in 12 months preceding trial. Exclusion criteria: active respiratory infection at time of study enrolment; presence of cancer or severe immune disease; evolutive tuberculosis or severe cardiac, kidney, or haematological disease; substance abuse or mental health concerns; had received vaccines in 5 months preceding trial (excluding influenza vaccine); receiving immunomodulatory therapy, antibiotics, or corticosteroids. Pretreatment: not specified |
|
| Interventions |
Intervention characteristics Diribiotine CK
Placebo
|
|
| Outcomes |
Exacerbation duration
Adverse events
|
|
| Identification |
Sponsorship source: not specified Country: France Setting: 3 hospitals. Initially recruited from an inpatient setting; however, assumed outpatient setting for the remainder of study. Author's name: J Rochemaure Institution: Hotel‐Dieu de Paris Address: 1 place du Paris Notre‐Dame, F‐75181 Paris Cedex 04 Participant type: CB as per the WHO definition (International Disease Classification, 491.2) Year: not specified. Abstract published 1987; study published 1988. |
|
| Notes | Study published in French. Associated English abstract linked to this study. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation occurred but methods not specified. |
| Allocation concealment (selection bias) | Unclear risk | The drug manufacturer and 1 of the authors (JR) kept the codes in sealed envelopes, but other details not specified. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind study, but groups and methodology not specified. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not specified. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Per‐protocol analysis. 9 (16%) dropouts overall; 6 (21%) in placebo group and 3 (12%) in treatment group. Reasons for dropouts well‐described and the 2 groups appeared balanced. |
| Selective reporting (reporting bias) | Unclear risk | All main outcomes appeared to have been reported on but there was a focus on those that were significant. |
Soler 2007.
| Study characteristics | ||
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics OM‐85
Placebo
Inclusion criteria: outpatients aged 40–75 years, fulfilling diagnostic criteria of CB or mild COPD (GOLD stages I or II) and with spirometry in last 12 months demonstrating an FEV1 ≥ 50% predicted, who were experiencing an acute bronchitic exacerbation with 3 or 4 of: increased cough, increased sputum, coloured sputum, or increased dyspnoea. Exclusion criteria: allergic asthma, mucoviscidosis, bronchiectasis, alpha‐1‐antitrypsin deficiency; severe cardiovascular disease, significant hepatic or renal insufficiency; cancer; autoimmune disease and other systemic diseases related to immune system disorders; allergy or previous intolerance to study medication; pregnant, breastfeeding or of child‐bearing potential without reliable contraceptive methods; unreliable patients (non‐compliance, alcoholism); major surgical procedure within 3 months of study start; participation in another clinical trial within 3 months of study start; treatment with antibiotics within 1 week before trial start; previous concomitant immunosuppressive or immunostimulant therapy during last 3 months before study entry; concomitant treatment with systemic corticosteroids (exceeding 2 weeks) or with an unregistered drug during whole trial Pretreatment: higher percentage of women in placebo group, which study authors considered alone would be unlikely to affect the outcome but it was reported that there was a larger proportion of non‐smokers among the female population and they had a better‐preserved lung function (data not shown). Otherwise, no apparent significant differences between groups. |
|
| Interventions |
Intervention characteristics OM‐85
Placebo
|
|
| Outcomes |
Number of participants with no exacerbations
Adverse events
Mortality (all‐cause)
|
|
| Identification |
Sponsorship source: not specified. 1 author was associated with OM‐Pharma and all 3 authors appeared to be associated with the Swiss‐German OM‐85 Study Group. Country: Switzerland and Germany Setting: > 44 centres. Participants were recruited at the time of an exacerbation; however, assumed outpatient setting for remainder of study. Author's name: Prof Dr med M Solèr Institution: Pneumologie St Claraspital Email: markus.soler@claraspital.ch Address: CH‐4016 Basel (Switzerland) Participant type: mild COPD or CB Year: not specified. Published 2007 |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomly assigned." Quote: "The randomization occurred in blocks of 4 per centre." Blocked randomisation used, but methods of sequence generation not specified. |
| Allocation concealment (selection bias) | Unclear risk | Not specified. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "The test medications had identical appearances." Randomisation occurred in blocks of 4 per centre, which may have made it easier to ascertain what participants may have received; however, medications reported to be identical, which may have lessened this risk. Groups that were blinded and any further methodology to ensure blinding not specified. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not specified. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | ITT sample, but per‐protocol analysis used for mean exacerbation rate/discussion of primary outcome. Dropout rate 14.6% between ITT and per‐protocol sample, but well‐described and groups well‐matched. |
| Selective reporting (reporting bias) | Low risk | All intended outcomes appeared to have been reported on. |
Tag 1993.
| Study characteristics | ||
| Methods |
Study design: randomised controlled trial Study grouping: parallel group. Unbalanced 3:2 intervention:placebo |
|
| Participants |
Baseline characteristics OM‐85
Placebo
Inclusion criteria: adults with CB. Exclusion criteria: not specified. Pretreatment: no reported significant differences between comparison groups. |
|
| Interventions |
Intervention characteristics OM‐85
Placebo
|
|
| Outcomes |
Exacerbation duration
Adverse events
|
|
| Identification |
Sponsorship source: not specified Country: authors were associated with an institution in Egypt Setting: outpatient, single centre Author's name: Prof Dr Mohamed A Tag El Din Institution: Department of Chest Diseases, Faculty of Medicine, Ain Shams University Address: 26 Aly Shalaby Street, Saint Fatima, Heliopolis, Cairo, Egypt Participant type: CB Year: not specified. Published 1993 |
|
| Notes | Unbalanced 3:2 intervention:placebo. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation 3:2 intervention:placebo. Methods not specified. |
| Allocation concealment (selection bias) | Unclear risk | Not specified. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind study, but groups and methods not specified. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not specified. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not reported whether there were any dropouts/exclusions, or whether analysis was ITT. Most measured outcomes were continuous and n was unclear. |
| Selective reporting (reporting bias) | Low risk | All intended outcomes appeared to have been reported on. |
Tang 2015.
| Study characteristics | ||
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics OM‐85
Placebo
Inclusion criteria: adults aged 40–75 years, with physician‐diagnosed CB or COPD (for duration > 6 months), who had an acute exacerbation according to GOLD definition at enrolment; had FEV1 40–70% predicted value within 6 months prior to study enrolment. Exclusion criteria: allergic asthma, mucoviscidosis, bronchiectases, alpha‐1‐antitrypsin deficiency; malignant diseases; severe cardiovascular disease, significant hepatic or renal insufficiency, any chronic disease that might impair the patient's ability to participate in study; previously (within 6 months before study entry) or concomitantly taking immunostimulating or immunosuppressive agents; undergoing treatment with high doses of corticosteroids (prednisone > 10 mg/day). Pretreatment: no significant differences between groups. |
|
| Interventions |
Intervention characteristics OM‐85
Placebo
|
|
| Outcomes |
Number of participants with no exacerbations
Adverse events
|
|
| Identification |
Sponsorship source: Sponsored by OM Pharma Country: China Setting: 13 clinical centres, outpatient Author's name: Qingyu Xiu Institution: Department of Respiratory Medicine, Changzheng Hospital, Second Military Medical University Email: xiuqing_yu@126.com Address: 415 Fengyang Rd, Shanghai 200003, China Participant type: CB or COPD, or both Year: September 2005 to February 2008 |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomly assigned, in a 1:1 ratio." Randomisation occurred but methods not further specified. |
| Allocation concealment (selection bias) | Unclear risk | Not specified. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind study, but groups and methods not specified. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not specified. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 5% of participants excluded following initial enrolment for 'missing data,' but balanced between groups. Study used ITT, per‐protocol, and safety set analyses. Between ITT and per‐protocol set, there was loss of 30 participants (11% from intervention group and 5% from placebo group, which would tend to underestimate any treatment effect). |
| Selective reporting (reporting bias) | Low risk | All intended outcomes were reported on and well‐defined. |
Venge 1996.
| Study characteristics | ||
| Methods |
Study design: randomised controlled trial Study grouping: parallel crossover study; however, only data from the first interval of trial (before crossover occurred) were used. |
|
| Participants |
Baseline characteristics Hyaluronan
Placebo
Overall
Inclusion criteria: CB (defined by 1965 British MRC criteria) with > 3 infectious exacerbations of CB during the preceding 2 winter seasons Exclusion criteria: receiving oral corticosteroids (> 10 mg/day), immunosuppressive treatment, or continuous prophylactic antibiotic treatment Pretreatment: no baseline characteristics presented for each comparison group and not specified whether there were any significant differences. |
|
| Interventions |
Intervention characteristics Hyaluronan
Placebo
|
|
| Outcomes |
Number of participants with no exacerbations
Adverse events
Number of participants requiring antibiotics
|
|
| Identification |
Sponsorship source: supported by grants from the Swedish Medical Research Council and Pharmacia AB Country: authors were associated with institutions in Sweden and Denmark Setting: not specified, presumed outpatient Author's name: Prof Per Venge, MD Institution: Department of Clinical Chemistry, University Hospital Address: S‐751 85, Uppsala, Sweden Participant type: CB or COPD, or both Year: not specified. Study received by journal in 1995 |
|
| Notes | Parallel crossover study; only extracted data from the first trial interval from baseline (before crossover occurred). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation occurred but methods not specified. |
| Allocation concealment (selection bias) | Unclear risk | Not specified. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind study. Participants likely were blinded (details reported regarding volume of liquid injected and using same apparatus to inject), but other methods to ensure blinding and which groups were blinded were s not specified. A nurse administered the injections, who was separate to the data collectors, but it was not specified whether the person administering the intervention was blinded to the contents. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not specified. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number of dropouts/exclusions/protocol violations was not reported on. It is not stated whether analysis was ITT or per‐protocol. Number of participants analysed in the first and second study periods appeared the same, which raises the question of how feasible this might have been to have no dropouts over a 2‐year period. |
| Selective reporting (reporting bias) | Low risk | For the first study period all intended outcomes appeared to have been reported on. |
Xinogalos 1993.
| Study characteristics | ||
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
|
| Participants |
Baseline characteristics OM‐85
Placebo
Inclusion criteria: diagnosis of COPD with ≥ 3 exacerbations during corresponding period of previous year. Exclusion criteria: treatment with corticosteroids, immunosuppressants, or immunostimulating agents. Pretreatment: no significant differences between groups. |
|
| Interventions |
Intervention characteristics OM‐85
Placebo
|
|
| Outcomes |
Exacerbation duration
Number of participants requiring antibiotics
|
|
| Identification |
Sponsorship source: not specified Country: authors are associated with institutions in Greece Setting: not specified, presumed outpatient Author's name: S Xinogalos Institution: Laboratory of Experimental Pharmacology, Medical School, Athens University Address: Athens, Greece Participant type: COPD Year: 1990–1991 |
|
| Notes | Outcomes measured over 6 months in discrete 1‐monthly intervals. Data for each interval were unable to be combined and used in meta‐analysis as this would be inconsistent with protocol criteria (specifying outcomes need to be measured over ≥ 12 weeks). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "patients were randomly allocated." Randomisation occurred but methods not specified. |
| Allocation concealment (selection bias) | Unclear risk | Not specified. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "The taste, size and colour of the BV capsules and placebo were identical." Double‐blind study, but groups and methodology not further specified beyond the appearance of the medications were identical. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not specified. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: "of 104 patients who were initially chosen for the trial, only 62 completed the 6‐month period of follow‐up. The rest interrupted the observations for reasons unrelated to treatment (20 in the BV and 22 in the placebo group)." Per‐protocol analysis. High dropout rates (40% overall), although well‐matched between groups. Reasons for dropouts/exclusions not specified. Much of the "significant" data were after the 4‐month period, at which point it was unclear how many remained in each group. |
| Selective reporting (reporting bias) | Unclear risk | Detailed reporting but the dropouts affected quality of the data. Also reported by month rather than over total duration of study period, which may have a favourable effect on the data. Adverse events not described. |
ATS: American Thoracic Society; BMI: body mass index; CB: chronic bronchitis; CCIQ: Chronic Cough Impact Questionnaire; CI: confidence interval; COPD: chronic obstructive pulmonary disease; FEV1: forced expiratory volume in one second; FVC: forced vital capacity; GOLD: Global Initiative for Obstructive Lung Disease; IgA: immunoglobulin A; ITT: intention to treat; MD: mean difference; MRC: Medical Research Council; n: number of participants; NYHA: New York Heart Association; PaO2: partial pressure of oxygen; PMBL: polyvalent mechanical bacterial lysate; SD: standard deviation; SE: standard error; SF‐12: 12‐Item Short Form Survey; SF‐36: 36‐Item Short Form Survey; SGRQ: St George's Respiratory Questionnaire; WHO: World Health Organization.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Ahrens 1983 | Wrong participant population |
| Anonymous 1988 | Wrong treatment intent (acute treatment study) |
| Anonymous 1993 | Review article only |
| Anonymous 2008 | Wrong intervention |
| Anthonisen 1997 | Review article only |
| Antonova 2004 | Wrong intervention |
| Antonova 2008 | Wrong participant population |
| Aparis 1987 | Wrong treatment intent (acute treatment study) |
| Banos 1997 | Wrong study design |
| Basacopol 1990 | Wrong study design |
| Braido 2011 | Wrong outcomes |
| Carre 1993 | Review article only |
| Carta 1994 | Wrong participant population |
| Catena 1991 | Wrong study design |
| Cazzola 2009 | Duplicate. Wrong study design |
| Centanni 1997 | Wrong study design |
| Cogo 2003 | Wrong study design |
| Cogo 2014 | Wrong study design |
| De Fenoyl 1986 | Review article only |
| Emanuele 1989 | Wrong study design |
| EUCTR2012‐003253‐28‐ES | Study protocol only |
| EUCTR2013‐001940‐71‐GB | Wrong study design |
| Fietta 1989 | Wrong study design |
| Fietta 1992 | Wrong study design |
| Fischer 1992 | Wrong participant population |
| Gao 2014 | Wrong participant population |
| Germouty 1986 | Wrong participant population |
| Grassi 1988 | Wrong study design |
| Jia 2015 | Wrong treatment intent (acute treatment study) |
| Kalinina 2003 | Wrong outcomes |
| Khedr 1993 | Wrong participant population |
| Koatz 2016 | Wrong participant population |
| Lacaille 1988 | Wrong participant population |
| Mahashur 2002 | Wrong intervention |
| Malolepszy 1991 | Wrong participant population |
| Marcatili 1989 | Wrong treatment intent (acute treatment study) |
| Matthys 2013 | Wrong intervention |
| Menon 2004 | Abstract only (no extractable data) |
| Moniuszko 1991 | Wrong study design |
| NCT01842360 | Study protocol only |
| NCT02417649 | Study protocol only |
| Nishantha 2014 | Wrong study design |
| Nouvet 1989 | Wrong study design |
| Obrecht 1975 | Wrong study design |
| Panchyshyna 1997 | Wrong study design |
| Pozzi 1994 | Wrong study design |
| Rekalova 1992 | Wrong treatment intent (acute treatment study) |
| Ricci 2014 | Wrong outcomes |
| Rico Mendez 1997 | Wrong study design |
| Rimoldi 1990 | Wrong study design |
| Romanski 1997 | Wrong participant population |
| Rutishauser 1998 | Wrong participant population |
| Shameen 2005 | Wrong study design |
| Shmelev 1995 | Wrong study design |
| Smirnova 2006 | Wrong study design |
| Sofia 1988 | Wrong study design |
| Spiropoulos 1995 | Wrong study design |
| Targowski 2005 | Wrong study design |
| Viallat 1983 | Wrong study design |
| Zanussi 1990 | Wrong study design |
| Zheng 2008 | Wrong study design |
Characteristics of studies awaiting classification [ordered by study ID]
Alvarez‐Sala 2004.
| Methods | Unknown |
| Participants | Participant type: COPD |
| Interventions | AM3 vs placebo |
| Outcomes | Unknown |
| Notes | No abstract or full‐text able to be located for screening. |
Berra 1988.
| Methods | Unknown |
| Participants | Participant type: unknown |
| Interventions | Biostim vs placebo |
| Outcomes | Unknown |
| Notes | No abstract or full‐text able to be located for screening. Possibly not relevant to review as title implies acute treatment (rather than preventive) intent of intervention and may include those with chronic respiratory illnesses aside from COPD/CB. |
Magyar 1985.
| Methods | Unknown |
| Participants | Participant type: CB Baseline characteristics OM‐85 BV
Placebo
Overall
Inclusion criteria: CB, otherwise unknown. Exclusion criteria: aged > 65 years; with 'severe' airflow limitation |
| Interventions | OM‐85 BV vs placebo |
| Outcomes |
Hospitalisation duration
Unclear whether there were any further outcomes relevant to this review – other outcome measures as suggested in Sprenkle 2005 included mean number of exacerbations, mean antibiotic duration, and mean steroid duration over a 6‐month period. |
| Notes | No abstract or full‐text able to be located for screening. Details obtained from previously published systematic review (Sprenkle 2005). |
Shrewsbury 2014.
| Methods |
Study design: randomised controlled trial Study grouping: parallel group |
| Participants | Participant type: COPD Baseline characteristics n = approximately 400 participants Inclusion criteria: COPD, with a recent exacerbation in 2 subsets: 1. ≥ 100 presenting for outpatient treatment and 2. ≥ 100 hospitalised for ≤ 7 days and discharged within the last 3 days. Men or women aged > 40 years; COPD for ≥ 18 months; chronic productive cough for ≥ 3 months in each of 2 years prior to screening; ≥ 2 documented exacerbations in previous 18 months; postbronchodilator FEV1/FVC ratio < 0.70 and predicted FEV1 30–80% of normal; former smoker or current smoker (≥ 10 pack‐years); contraception if sexually active. Exclusion criteria: other lung disease; treatment with roflumilast or theophylline within 1 month; lobar pneumonia within last 3 months; hospitalisation > 7 days for the current acute exacerbation, or intubation; previous exacerbations requiring > 3 weeks to stabilise. |
| Interventions |
Intervention characteristics AQX‐1125
|
| Outcomes |
Exacerbations
Health‐related quality of life
Adverse events
|
| Notes |
Sponsorship source: not specified, although author details suggest an association with Aquinox Pharmaceuticals, Inc. Country: not specified, author was associated with an institution in Canada Author's name: SB Shrewsbury Institution: Aquinox Pharmaceuticals Inc, Richmond, BC, Canada Email: sshrewsbury@aqxpharma.com Year: unknown Notes: no abstract or full‐text able to be located for screening. This may be the abstract for a protocol rather than a published review as there were no results reported. |
Soler 2001.
| Methods |
Study design: randomised controlled trial Study grouping: unknown |
| Participants | Participant type: CB or 'mild' COPD, or both Baseline characteristics OM‐85 BV
Placebo
Overall:
Inclusion criteria: history of stage I COPD or CB; current, acute exacerbation of bronchitis Exclusion criteria: FEV1 ≤ 50% |
| Interventions | OM‐85 BV vs placebo |
| Outcomes | Unclear whether there were any outcomes measured relevant to this review – outcome measures as suggested in Sprenkle 2005 included mean number of exacerbations per participant, number of participants hospitalised (respiratory and all‐cause), mean number of days of antibiotic use, number of participants receiving ≥ 1 prescription of corticosteroids, symptom scores over a 6‐month period. |
| Notes | No abstract or full‐text able to be located for screening. Details obtained from previously published systematic review (Sprenkle 2005). n and data for the primary outcome of mean exacerbation rate are congruent with that of Soler 2007; however, the outcomes measured appeared to differ between publications, therefore, it could not be confirmed whether this is an alternate publication of the same study. |
CAT: COPD Assessment Tool; CB: chronic bronchitis; COPD: chronic obstructive pulmonary disease; EXACT: Exacerbation of Chronic Pulmonary Disease Tool; FEV1: forced expiratory volume in one second; FVC: forced vital capacity; n: number of participants.
Differences between protocol and review
Several of the primary and secondary outcomes, or the way in which these were measured, differed between protocol (Fraser 2019) and review. We adapted them following data extraction, prior to any data synthesis or analysis occurring, and by consensus discussion.
During the data extraction process it was apparent that some of our original outcomes would be unable to be determined or combined, based on the way trialists had reported outcome results. For example, although we considered that differentiating between moderate and severe exacerbations was of significant clinical relevance at the commencement of this review, this information was unable to be extracted from the included studies. Because the primary and secondary outcomes were altered, this affected the content of the summary of findings table, which continues to reflect the main relevant outcomes of this review but also differed from what was outlined in our protocol. For example, 'number of participants with no exacerbations during the study period,' 'mortality (respiratory‐related),' and 'number of participants requiring antibiotics' were outcomes included in our final summary of findings table, that had not been specified in our review protocol.
The dichotomous outcome of 'number of participants with no exacerbations during the study period' was instead chosen as a primary outcome and considered more statistically robust than the often‐reported outcome of 'mean exacerbation rate,' due to the likelihood of skew associated with this metric. We added 'Number of participants requiring antibiotics' and 'hospitalisation duration (respiratory)' as secondary outcome measures. Several included studies reported these and were considered to be surrogate markers of exacerbation severity, in lieu of being able to obtain clear data around how many of the exacerbations were deemed "moderate" or "severe" (or both) by GOLD definitions (GOLD 2022). Several studies also reported 'Antibiotic duration'; however, it was again considered that a dichotomous outcome regarding antibiotic use would be more statistically robust. Conversely, 'number of participants requiring hospitalisation' was rarely reported, and inclusion as an outcome would have, therefore, contributed little to overall comparison.
For the outcome of quality of life, we meta‐analysed the difference between end point scores on a validated scale. Our protocol had outlined preferentially using change‐from‐baseline scores; however, the reporting of this metric in the included studies was limited. Where this was reported, this was qualitatively analysed in the text of this review.
The subgroup analyses performed in this review also differ from the protocol; modifications occurred following data extraction and prior to any data synthesis, and were done to investigate for anticipated heterogeneity related to the varying treatment and study durations seen across the included studies. Most studies that reported on our primary outcome measures included oral immunostimulant agents, with only two studies analysing sublingually and subcutaneously delivered immunostimulants. Therefore, we considered it less relevant to include 'mode of delivery' as a subgroup, as originally outlined in our protocol. We considered further differentiation of the immunostimulant agents based on dose and treatment regimen; however, there was much variability between studies, limiting meaningful subgroup analysis. Therefore, we performed subgroup analysis only performed for immunostimulant type.
Following data synthesis, we performed a post hoc subgroup analysis differentiating the decade of study publication for the primary outcome of 'number of participants with no exacerbations,' to further investigate the moderate degree of heterogeneity that remained despite the preplanned subgroup analyses.
We had not outlined limits on data heterogeneity in our protocol. However, to aid in understanding of the heterogeneity seen following data extraction and to optimise data synthesis and presentation, interpretations on the level of heterogeneity were made in accordance with Chapter 10 of the Cochrane Handbook for Systematic Reviews of Interventions (Deeks 2022). Following consultation with a statistician, it was determined that several of the outcomes had an associated level of heterogeneity that would make the data difficult to interpret and less robust if analysed parametrically. This was particularly seen with the outcome of 'exacerbation duration.' Instead of meta‐analysis, and in accordance with statistician suggestion, for this outcome we performed a post hoc synthesis of data by vote‐counting based on effect direction, whereby the effect estimate for each study that reported this outcome was categorised as a binary metric (beneficial versus harmful) and a binomial probability test applied to determine the probability of observing the result if the intervention was truly ineffective.
During the screening process, we contacted the authors of two studies for further information, without response (Shrewsbury 2014; Nishantha 2014). Following data extraction, we contacted one of the authors of Hutas 1994 for clarification of whether they used standard deviations or standard errors for reported continuous outcomes. Otherwise, there was no need to contact study authors for additional data. There were several reasons for this; including the age of the studies, that they were not reported in English, or author contact details were not readily available to facilitate further correspondence.
Contributions of authors
AF: drafted the protocol (lead), developed and ran the search strategy (along with Liz Stovold, Information Specialist), obtained copies of studies, selected which studies to include, extracted data from studies, entered data into Review Manager Web, carried out the analysis, interpreted the analysis, drafted the final review (lead), and plans to update the review.
PP: drafted the protocol, developed the search strategy, selected which studies to include, extracted data from studies, carried out the analysis, interpreted the analysis, drafted the final review, and plans to update the review.
Contributions of editorial team
Sally Spencer (Co‐ordinating Editor): edited the review; advised on methodology, interpretation, and content; approved the review prior to publication.
Rebecca Fortescue (Co‐ordinating Editor): checked the data in the review.
Emma Dennett (Deputy Co‐ordinating Editor): advised on methodology, interpretation, and content; edited the review.
Iain Crossingham (Contact Editor): edited the review; advised on content.
Emma Jackson (Managing Editor): conducted peer review; edited the references and other sections of the review.
Elizabeth Stovold (Information Specialist): designed the search strategy, edited the search methods and appendices.
Sources of support
Internal sources
-
Auckland, Waitemata and Counties Manukau District Health Boards, New Zealand
Ashley Fraser was employed by Auckland, Waitemata and Counties Manukau District Health Boards for the purpose of clinical work as a medical registrar.
-
Bay of Plenty District Health Board, New Zealand
Ashley Fraser was employed initially for the purpose of clinical work as a medical registrar.
-
University of Auckland, New Zealand
Phillippa Poole was employed by University of Auckland.
External sources
-
National Institute for Health and Care Research (NIHR), UK
Cochrane Airways received Cochrane infrastructure funding from NIHR
Declarations of interest
AF: none.
PP: none.
New
References
References to studies included in this review
Alvarez‐Mon 2005 {published data only}
- Alvarez-Mon M, Miravitlles M, Morera J, Callol L, Alvarez-Sala JL. Treatment with the immunomodulator AM3 improves the health-related quality of life of patients with COPD. Chest 2005;127(4):1212-8. [DOI: 10.1378/chest.127.4.1212] [DOI] [PubMed] [Google Scholar]
- Miravitlles M, Murio C, Morera J, Callol L, Alvarez-Sala JL, Alvarez-Mon M. Effect of AM3 on health-related quality of life in patients with chronic obstructive pulmonary disease belonging to risk groups. Medicina Clinica 2008;130(18):688-92. [DOI] [PubMed] [Google Scholar]
Alvarez‐Sala 2003 {published data only}
- Alvarez-Sala JL, Alvarez-Mon M. Treatment with Cellmune(TM) (AM3), an oral immunomodulator, increases the quality of life of chronic obstructive pulmonary disease (COPD) patients. Respiratory Care 2003;48(11):1071. [Google Scholar]
Anthoine 1985 {published data only}
- Anthoine D, Blaive B, Cabanieu G, Chretien J, Danrigal A, Ducreuzet C, et al. Double-blind study of Biostim in the prevention of superinfection in patients with chronic bronchopathy. Revue de Pneumologie Clinique 1985;41(3):213-7. [PubMed] [Google Scholar]
Bisetti 1994 {published data only}
- Bisetti A, Ciappi G, Bariffi F, Catena E, Rocco V, Vaccaro L, et al. Evaluation of the efficacy of pidotimod in the exacerbations in patients affected with chronic bronchitis. Arzneimittel-Forschung 1994;44(12A):1499-502. [PubMed] [Google Scholar]
Blaive 1982 {published data only}
- Blaive B, Demichelis B, Lemoigne F, Clary C, Macone F. Effectiveness of vaccination by ribosomal extracts in the preventive treatment of infections episodes in chronic bronchopathies. Medecine et Maladies Infectieuses 1982;12(8):475-9. [Google Scholar]
Bonde 1986 {published data only}
- Bonde J, Dahl R, Edelstein R, Kok-Jensen A, Lazer L, Punakivi L, et al. The effect of RU 41.740, an immune modulating compound, in the prevention of acute exacerbations in patients with chronic bronchitis. European Journal of Respiratory Diseases 1986;69(4):235-41. [PubMed] [Google Scholar]
Bongiorno 1989 {published data only}
- Bongiorno A, Cataldo MG, Mazzola G, Indovina I. Controlled versus placebo study of the efficacy and tolerance of a new immunomodulator AM3 (immunoferon) in chronic bronchitis. Ciencia Medica 1989;6(9):357-62. [Google Scholar]
Braido 2015 {published data only}
- Braido F, Melioli G, Cazzola M, Fabbri L, Blasi F, Moretta L, et al. Sub-lingual administration of a polyvalent mechanical bacterial lysate (PMBL) in patients with moderate, severe, or very severe chronic obstructive pulmonary disease (COPD) according to the GOLD spirometric classification: a multicentre, double-blind, randomised, controlled, phase IV study (AIACE study: advanced Immunological Approach in COPD Exacerbation). Pulmonary Pharmacology & Therapeutics 2015;33:75-80. [DOI: 10.1016/j.pupt.2015.03.006] [DOI] [PubMed] [Google Scholar]
- EUCTR2007-000006-67-IT. Sub-lingual administration of a polyvalent mechanical bacterial lysate (PMBL) in patients with moderate or severe and very severe chronic obstructive pulmonary disease (COPD) according to GOLD classification: a multicenter, international, double blind, randomized, controlled, phase IV study. hwww.who.int/trialsearch/Trial2.aspx?TrialID=EUCTR2007-000006-67-IT (first received 8 June 2009). [DOI] [PubMed]
Carlo 1990 {published data only}
- Carlo M Sr, Carlo M Jr. Double blind study controlled with placebo of a new immunomodulator substance. Geriatrika 1990;6(6):65-9. [Google Scholar]
Catena 1992 {published data only}
- Catena E, Grassi C, Grossi E. Efficacy of treatment with thymomodulin in the profilaxis of exacerbations in COPD patients with cell-mediated deficit. Giornale Italiano della Malattie del Torace 1992;46(3):193-202. [Google Scholar]
Cazzola 2006 {published data only}
- Cazzola M. A new bacterial lysate protects by reducing infectious exacerbations in moderate to very severe COPD: a double-blind, randomized, placebo-controlled trial. Luglio 2006;6(3):191-9. [Google Scholar]
Ciaccia 1994 {published data only}
- Ciaccia A, Zavattini G, Ferri P. Pidotimod in the treatment of chronic bronchitis exacerbations. Giornale Italiano delle Malattie del Torace 1993;47(1-2):27-33. [Google Scholar]
- Ciaccia A. Pidotimod activity against chronic bronchitis exacerbations. Arzneimittel-Forschung 1994;44(12A):1516-20. [PubMed] [Google Scholar]
Collet 1997 {published data only}
- Collet JP, Shapiro P, Ernst P, Renzi T, Ducruet T, Robinson A. Effects of an immunostimulating agent on acute exacerbations and hospitalizations in patients with chronic obstructive pulmonary disease. The PARI-IS Study Steering Committee and Research Group. Prevention of acute respiratory infection by an immunostimulant. American Journal of Respiratory and Critical Care Medicine 1997;156(6):1719-24. [DOI: 10.1164/ajrccm.156.6.9612096] [DOI] [PubMed] [Google Scholar]
Cvoriscec 1989 {published data only}
- Cvoriscec B, Ustar M, Pardon R, Palecek I, Stipic-Markovic A, Zimic B. Oral immunotherapy of chronic bronchitis: a double-blind placebo-controlled multicentre study. Respiration; International Review of Thoracic Diseases 1989;55(3):129-35. [DOI: 10.1159/000195723] [DOI] [PubMed] [Google Scholar]
Debbas 1990 {published data only}
- Debbas N, Derenne JP. Preventive effects of immunostimulating product on recurrent infections of chronic bronchitis in the elderly. Lung 1990;168:737-40. [DOI] [PubMed] [Google Scholar]
De Bernardi 1992 {published data only}
- De Bernardi M, Pedrinazzi PM, Re A, Colombo P, Fabiani A, Zanasi A. Immunostimulation induced by bacterial lysates in chronic bronchitis. A double blind placebo controlled clinical study. Acta Toxicologica et Therapeutica 1992;13(2):73-92. [Google Scholar]
Djuric 1989 {published data only}
- Djuric O, Mihailovic-Vucinic V, Stojcic V. Effect of Broncho-Vaxom on clinical and immunological parameters in patients with chronic obstructive bronchitis. A double-blind, placebo-controlled study. International Journal of Immunotherapy 1989;5(3):139-43. [Google Scholar]
EUCTR2007‐004702‐27‐DE {published data only}
- EUCTR2007-004702-27-DE. Multicentre, double-blind, placebo-controlled, randomised clinical study of Broncho-Vaxom® (Broncho-Munal®) for the protection from acute exacerbations of COPD. www.who.int/trialsearch/Trial2.aspx?TrialID=EUCTR2007-004702-27-DE (first received 7 December 2007).
Fietta 1988 {published data only}
- Fietta A, Bersani C, De Rose V, Mangiarotti P, Merlini C, Uccelli M, et al. Double-blind trial RU 41740 vs. placebo: immunological and clinical effects in a group of patients with chronic bronchitis. Respiration; International Review of Thoracic Diseases 1988;54(3):145-52. [DOI: 10.1159/000195515] [DOI] [PubMed] [Google Scholar]
Foschino 1995 {published data only}
- Foschino BM, Resta O, Cassano A, Altieri A, Guido P, Piti A, et al. Infectious exacerbations of chronic pulmonary diseases. Therapeutic effectiveness of immunomodulants. Minerva Pneumologica 1995;34(2):39-44. [Google Scholar]
Habermann 2001 {published data only}
- Habermann W, Zimmermann K, Skarabis H, Kunze R, Rusch V. The effect of a bacterial immunostimulant (human Enterococcus faecalis bacteria) on the occurrence of relapse in patients with chronic recurrent bronchitis. Arzneimittel-Forschung 2001;51(11):931-7. [DOI] [PubMed] [Google Scholar]
Hutas 1994 {published data only (unpublished sought but not used)}
- Hutas I, Kraszko P, Boszormenyi Nagy G. Immunomodulation therapy in chronic bronchitis (multicenter study). Orvosi Hetilap 1994;135(23):1251-4. [PubMed] [Google Scholar]
Keller 1984 {published data only}
- Keller R, Hinz G. Effect of an oral polyvalent bacterial lysate (Broncho-Vaxom) in chronic bronchitis. Praxis und Klinik der Pneumologie EMT 1984;38(6):225-8. [PubMed] [Google Scholar]
- Keller R. Multicenter double-blind study of the action of Broncho-Vaxom in chronic bronchitis. Schweizerische Medizinische Wochenschrift 1984;114(25):934-7. [PubMed] [Google Scholar]
Li 2004 {published data only}
- Li J, Zheng JP, Yuan JP, Yang L, Luo DF, An JY, et al. Protection of oral immunostimulant bronchovaxom against with chronic obstructive pulmonary disease. Respirology 2005;10(3):A137. [Google Scholar]
- Li J, Zheng JP, Yuan JP, Zeng GQ, Zhong NS, Lin CY. Protective effect of a bacterial extract against acute exacerbation in patients with chronic bronchitis accompanied by chronic obstructive pulmonary disease. Chinese Medical Journal 2004;117(6):828-34. [PubMed] [Google Scholar]
Menardo 1985 {published data only}
- Menardo JL, Dussol JP, Betbeder A. Immunotherapy of respiratory infection by oral immunomodulation in chronic obstructive bronchopneumopathy. Medecine et Hygiene 1985;43(1623):2722-6. [Google Scholar]
Messerli 1981 {published data only}
- Messerli C, Michetti F, Sauser-Hall P, Stäubli C, Taddei M, Weiss S, et al. Effect of a bacterial lysate (Broncho-Vaxom) in the therapy of chronic bronchitis: multi-center double-blind clinical trial. Revue Médicale de la Suisse Romande 1981;101(2):143-6. [PubMed] [Google Scholar]
Olivieri 2011 {published data only}
- Olivieri D. Reducing exacerbations in COPD with OM-85: a multicentre, double-blind, placebo-controlled trial. European Respiratory Journal 2011;38(55):9s. [Google Scholar]
Orcel 1994 {published data only}
- Orcel B, Delclaux B, Baud M, Derenne JP. Oral immunization with bacterial extracts for protection against acute bronchitis in elderly institutionalized patients with chronic bronchitis. European Respiratory Journal 1994;7(3):446-52. [DOI] [PubMed] [Google Scholar]
- Orcel B, Delclaux B, Baud M, Derenne JP. Preventive effect of an immunomodulator, OM-85 BV, on acute exacerbations of chronic bronchitis in elderly patients. Preliminary results at six months in 291 patients. Revue des Maladies Respiratoires 1993;10(1):23-8. [PubMed] [Google Scholar]
Orlandi 1983 {published data only}
- Orlandi O, Bruna S, Bagnasco G. Immunostimulation treatment with a bacteriolysate in recurring bacterial infections in chronic bronchitics. Minerva Pneumologica 1983;22:461-4. [Google Scholar]
Rico 1997 {published data only}
- Rico Mendez FG, Tellez Diaz E, Garcia Sancho C, Sanchez Diaz R, Yoma Medina J. Efficacy and prevention with thymomodulin on the numbers and duration of respiratory infectious episodes in adults with chronic obstructive lung disease. Revista Alergia Mexico 1997;44(4):93-101. [Google Scholar]
Rochemaure 1988 {published data only}
- Rochemaure J, Lehert P, Sauvaget J, Robillard M, Betbeder-Matibet A. Reduction using an immunomodulator of the level of respiratory infections in chronic bronchitis. Revue de Pneumologie Clinique 1988;44(1):43-7. [PubMed] [Google Scholar]
- Rochemaure J, Lehert P, Sauvaget J, Robillard M, Murray N, Castanier JC. Usefulness of immunomodulating agents. Lancet 1987;2(8559):101. [DOI] [PubMed] [Google Scholar]
Soler 2007 {published data only}
- Soler M, Mutterlein R, Cozma G, on behalf of the Swiss-German OM Study Group. Double-blind study of OM-85 in patients with chronic bronchitis or mild chronic obstructive pulmonary disease. Respiration 2007;74(1):26-32. [DOI: 10.1159/000093933] [DOI] [PubMed] [Google Scholar]
- Soler M, Mutterlein R, Cozma G. Prevention of exacerbations in chronic bronchitis and COPD with OM-85, a double-blind, placebo-controlled trial. American Thoracic Society 100th International Conference; 2004 May 21-26; Orlando (FL). D22[Poster 506].
Tag 1993 {published data only}
- Tag E, Ashmawi S, Emam WE. Effect of a polyvalent bacterial extract, Broncho-Vaxom, in the prophylaxis of acute exacerbations of chronic bronchitis. European Journal of Clinical Research 1993;4:99-105. [Google Scholar]
Tang 2015 {published data only}
- Tang H, Fang Z, Saborio GP, Xiu Q. Efficacy and safety of OM-85 in patients with chronic bronchitis and/or chronic obstructive pulmonary disease. Lung 2015;193(4):513-9. [DOI: 10.1007/s00408-015-9737-3] [DOI] [PubMed] [Google Scholar]
- Tang H, Fang Z, Xiu Q. Efficacy and safety of bacterial lysates in patients with chronic obstructive pulmonary disease and exacerbation. European Respiratory Journal 2011;38(55):599s. [Google Scholar]
Venge 1996 {published data only}
- Venge P, Pedersen B, Hakansson L, Hallgren R, Lindblad G, Dahl R. Subcutaneous administration of hyaluronan reduces the number of infectious exacerbations in patients with chronic bronchitis. American Journal of Respiratory and Critical Care Medicine 1996;153(1):312-6. [DOI: 10.1164/ajrccm.153.1.8542136] [DOI] [PubMed] [Google Scholar]
Xinogalos 1993 {published data only}
- Xinogalos S, Duratsos D, Varonos D. Clinical effectiveness of Broncho-Vaxom (BV) in patients with chronic obstructive pulmonary disease. International Journal of Immunotherapy 1993;9(2):135-42. [Google Scholar]
References to studies excluded from this review
Ahrens 1983 {published data only}
- Ahrens J, Wiedenbach M. Efficacy of the immunostimulant Broncho-Vaxom. Schweizerische Medizinische Wochenschrift 1984;114(25):932-4. [PubMed] [Google Scholar]
- Ahrens J. Klinische wirksamkeit eines oralen immuntherapeutikums. Atemwegs-Lungenkr 1983;9:127-33. [Google Scholar]
Anonymous 1988 {published data only}
- Italian Group for the Study of Biological Immunomodulators. Effect of a purified glycoprotein substance extracted from Klebsiella pneumoniae in the clinical course of acute infections of the airways in patients with chronic bronchitis. Recenti Progressi in Medicina 1988;79(2):73-8. [PubMed] [Google Scholar]
Anonymous 1993 {published data only}
- Anonymous. Pidotimod. Drugs of the future 1993;18(12):1163-4. [Google Scholar]
Anonymous 2008 {published data only}
- Anonymous. Proof of effectiveness of Pelargonium extract (Umckaloabo®) for the treatment of chronic bronchitis in a double-blind study. Schweizerische Zeitschrift fur Ganzheitsmedizin 2008;20(1):10-2. [Google Scholar]
Anthonisen 1997 {published data only}
- Anthonisen NR. OM-85 BV for COPD. American Journal of Respiratory and Critical Care Medicine 1997;156(6):1713-4. [DOI] [PubMed] [Google Scholar]
Antonova 2004 {published data only}
- Antonova LP, Markova TP, Kurbatova EA. Naso-subcutaneous application of the polycomponent vaccine VP-4 for the treatment of patients with bronchial asthma and chronic obstructive bronchitis. Zhurnal Mikrobiologii, Epidemiologii, i Immunobiologii 2004;6:36-40. [PubMed] [Google Scholar]
Antonova 2008 {published data only}
- Antonova LP, Romanov VV, Averbakh MM. Experience with bronchomunal used in the combined treatment of patients with bronchial asthma and chronic obstructive pulmonary disease. Problemy Tuberkuleza I Boleznej Legkih 2008;4:8-11. [PubMed] [Google Scholar]
Aparis 1987 {published data only}
- Aparis O, Berger M, Boisnic F, Cazard J, Delvaux H, Dewailly P, et al. Advantage of RU41740 associated with antibiotherapy in acute bronchial infections. A double blind versus placebo trial. Revue des Maladies Respiratoires 1987;4(6):66. [Google Scholar]
Banos 1997 {published data only}
- Banos V, Gomez J, Garcia A, Ruiz J, Alvarez R, Lorenzo M, et al. Effectiveness of immunomodulating treatment (thymostimulin) in chronic obstructive pulmonary disease. Respiration; International Review of Thoracic Diseases 1997;64(3):220-3. [DOI: 10.1159/000196674] [DOI] [PubMed] [Google Scholar]
Basacopol 1990 {published data only}
- Basacopol A, Carantino A, Calciu M. Broncho-Vaxom in the treatment and prevention of chronic pulmonary diseases. Clinical Trials Journal 1990;27(5):336-45. [Google Scholar]
- Basacopol A. Broncho-Vaxom in the treatment of chronic pulmonary lesions. Pneumoftiziologia 1992;41(1):25-30. [PubMed] [Google Scholar]
Braido 2011 {published data only}
- Braido F, Traggiai E, Lanzilli G, Bazorro G, Folli C, Garelli V, et al. Modification of cell mediated immune-response in patients treated with a polyvalent mechanical bacterial lysate. European Respiratory Journal 2011;38:550. [Google Scholar]
Carre 1993 {published data only}
- Carre PC, Leophonte P. Role of anti-infective treatments in the management of chronic obstructive bronchopneumopathies. Revue des Maladies Respiratoires 1993;10(2):105-13. [PubMed] [Google Scholar]
Carta 1994 {published data only}
- Carta P, Zedda S, Locci F, Lisci ML, Bande M, Del Giacco GS. Double-blind, randomized clinical trial of a bacterial immunomodulator in chronic obstructive bronchopneumonic infections. International Journal of Immunotherapy 1994;10(1):25-33. [Google Scholar]
Catena 1991 {published data only}
- Catena E. Thymopentin in the prevention of infective exacerbation in chronic bronchitis. Archivio Monaldi per le Malattie del Torace 1991;46(1-6):93-100. [PubMed] [Google Scholar]
Cazzola 2009 {published data only}
- Cazzola M, Noschese P, Di Perna F. Value of adding a polyvalent bacterial lysate to therapy of COPD patients under regular treatment with salmeterol/fluticasone (SFC) [Abstract]. European Respiratory Society Annual Congress; 2009 Sep 12-16; Vienna, Austria. [DOI] [PubMed]
- Cazzola M, Noschese P, Di Perna F. Value of adding a polyvalent mechanical bacterial lysate to therapy of COPD patients under regular treatment with salmeterol/fluticasone. Therapeutic Advances in Respiratory Disease 2009;3(2):59-63. [DOI: 10.1177/1753465809104677] [DOI] [PubMed] [Google Scholar]
Centanni 1997 {published data only}
- Centanni S, Pregliasco F, Bonfatti C, Mensi C, Tarsia P, Guarnieri R, et al. Clinical efficacy of a vaccine-immunostimulant combination in the prevention of influenza in patients with chronic obstructive pulmonary disease and chronic asthma. Journal of Chemotherapy 1997;9(4):273-8. [DOI: 10.1179/joc.1997.9.4.273] [DOI] [PubMed] [Google Scholar]
Cogo 2003 {published data only}
- Cogo R, Ramponi A, Scivoletto G, Rippoli R. Prophylaxis for acute exacerbations of chronic bronchitis using an antibacterial sublingual vaccine obtained through mechanical lysis: a clinical and pharmacoeconomic study. Acta Bio Medica 2003;74(2):81-7. [PubMed] [Google Scholar]
Cogo 2014 {published data only}
- Cogo R. Pidotimod activity in patients affected by COPD. Minerva Pneumologica 2014;53(1):21-6. [Google Scholar]
De Fenoyl 1986 {published data only}
- De Fenoyl O. An immunomodulating substance ('Biostim') obtained from Klebsiella pneumoniae. Comptes Rendus de Therapeutique et de Pharmacologie Clinique 1986;4(37):3-15. [Google Scholar]
Emanuele 1989 {published data only}
- Emanuele E, Corvaja E, Cumina T, Coppolino E. The immunomodulating effect of AM3 in the treatment of chronic bronchopathy. Rassegna di Medicina Interna 1989;10(1-2):25-31. [Google Scholar]
EUCTR2012‐003253‐28‐ES {published data only}
- EUCTR2012-003253-28-ES. Evaluation of the efficacy and safety of bactek in COPD. www.who.int/trialsearch/Trial2.aspx?TrialID=EUCTR2012-003253-28-ES (first received 29 October 2012).
EUCTR2013‐001940‐71‐GB {published data only}
- EUCTR2013-001940-71-GB. A study to investigate the effects of Broncho-Vaxom (OM-85 BV) on the immune system in patients with chronic obstructive pulmonary disease. www.who.int/trialsearch/Trial2.aspx?TrialID=EUCTR2013-001940-71-GB (first received 22 July 2013).
Fietta 1989 {published data only}
- Fietta A, Bersani C, Mangiarotti P, Merlini C, Uccelli M. Phagocytes and their pharmacological modulation in chronic bronchitis. International Journal of Immunopathology and Pharmacology 1989;2(2):115-22. [Google Scholar]
Fietta 1992 {published data only}
- Fietta AM, Merlini C, Uccelli M, Gialdroni Grassi G, Grassi C. Immunological and clinical effect of long-term oral treatment with RU 41740 in patients with chronic bronchitis: double-blind trial long-term versus standard dose regimen. Respiration; International Review of Thoracic Diseases 1992;59(5):253-8. [DOI: 10.1159/000196069] [DOI] [PubMed] [Google Scholar]
Fischer 1992 {published data only}
- Fischer H, Eckenberger HP, Aubel A, Kammereit A, Elsasser U, et al. Pravention von Infektrezidiven der oberen und unteren Luftwege. Atemw Lungenkrkh 1992;4:146-55. [DOI: ] [Google Scholar]
- Fischer H, Eckenberger HP, Aubel A, MacDonald TT, Challacombe SJ, Bland PW, et al. An oral bacterial lysate for the prevention of recurrent infections of the respiratory tract. Results of a double-blind placebo controlled 6 months study in 342 patients. In: MacDonald TT, Challacombe SJ, Bland PW, Stokes CR, Heatley RV, Mowat AM, editors(s). Advances in Mucosal Immunology. Norwell (MA): Kluwer Academic Publishers, 1990:351-2. [Google Scholar]
Gao 2014 {published data only}
- Gao J, Gao X, Kong L. To investigate the prevention of OM-85 on bronchiectasis exacerbations (iPROBE) in Chinese patients: study protocol for a randomized controlled trial. Trials 2014;15(150):1-6. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Germouty 1986 {published data only}
- Germouty J. Immunotherapy of recurrent respiratory infections. Double-blind study of a new immunomodulator in 60 patients. Revue de Pneumologie Clinique 1986;42(4):207-13. [PubMed] [Google Scholar]
Grassi 1988 {published data only}
- Grassi C, Bellero V, Donner CF, Fumagalli G, Orlandi O, Rimoldi A. Double-blind multicentre clinical study with Broncho-Vaxom in chronic bronchitis. European Respiratory Journal 1988;1(Suppl 2):252s. [Google Scholar]
Jia 2015 {published data only}
- Jia Z, Feng Z, Tian R, Wang Q, Wang L. Thymosin alpha1 plus routine treatment inhibit inflammatory reaction and improve the quality of life in AECOPD patients. Immunopharmacology and Immunotoxicology 2015;37(4):388-92. [DOI] [PubMed] [Google Scholar]
Kalinina 2003 {published data only}
- Kalinina EP, Zhuravskaia NS, Tsyvkina GI, Koziavina NV. Correction of immune disorders with neoselenium in patients with chronic bronchitis. Klinicheskaia Meditsina 2003;81(3):43-6. [PubMed] [Google Scholar]
Khedr 1993 {published data only}
- Khedr MS. Clinical and immunological efficacy of Broncho-Vaxom(TM) in chronic bronchitis. A double-blind study. Acta Therapeutica 1993;19(1):49-60. [Google Scholar]
Koatz 2016 {published data only}
- Koatz AM, Coe NA, Ciceran A, Alter AJ. Clinical and immunological benefits of OM-85 bacterial lysate in patients with allergic rhinitis, asthma, and COPD and recurrent respiratory infections. Lung 2016;194(4):687-97. [DOI: 10.1007/s00408-016-9880-5] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koatz AM, Zakin L, Ciceran A. Cost consequence of preventive treatment with OM 85 bacterial lysate compared to the same patients without OM 85 the previous year in allergic rhinitis, asthma and COPD in Argentina. Value in Health 2015;18(7):A498. [Google Scholar]
- Koatz AM. Evaluation of recurrent respiratory infections in patients with allergic rhinitis, asthma, and/or COPD pre-and post-treatment with bacterial lysate vaccines. Annals of Allergy, Asthma and Immunology 2014;113(5):A41. [Google Scholar]
Lacaille 1988 {published data only}
- Lacaille F. Administration of RU 41740, a prophylactic immunomodulator protecting against infection, during an episode of acute respiratory infection. Summarized results of 3 therapeutic trials. Presse Medicale 1988;17(28):1453-7. [PubMed] [Google Scholar]
Mahashur 2002 {published data only}
- Mahashur AA, Pal M, Parikh HS, Karkera SG, Kukreja A. Study of the efficacy of immunomodulator imunocin, as an add-on therapy in the management of patients with chronic obstructive pulmonary disorder and bronchiectasis. Indian Practitioner 2002;55(6):393-9. [Google Scholar]
Malolepszy 1991 {published data only}
- Malolepszy J, Chyrek-Borowska S, Siwinska-Golebiowska HK, Rogala E, Roszkowski W. Multicentre clinical trial of Broncho-Vaxom in chronic bronchitis and asthma. Acta Therapeutica 1991;17(3):273-82. [Google Scholar]
Marcatili 1989 {published data only}
- Marcatili S, Catena E. Treatment of infective recurrences of chronic bronchopneumopathies with Klebsiella pneumoniae extracts. Arch Monaldi Mal Torace 1989;44(4-6):765-6. [PubMed] [Google Scholar]
Matthys 2013 {published data only}
- Matthys H, Funk P. Pelargonium sidoides preparation EPS 7630 in COPD: health-related quality-of-life and other patient-reported outcomes in adults receiving add-on therapy. Current Medical Research and Opinion 2018;34(7):1245-51. [DOI: 10.1080/03007995.2017.1416344] [DOI] [PubMed] [Google Scholar]
- Matthys H, Malek FA. Antibiotic use in patients with COPD receiving EPS 7630 as an add-on treatment. Atemwegs und Lungenkrankheiten 2015;41(1):27-34. [DOI: ] [Google Scholar]
- Matthys H, Pliskevich D, Reineke T, Malek FA. Health-related quality of life (HRQL) and patient-reported outcomes (PRO) in COPD patients receiving add-on-therapy with EPS 7630. European Respiratory Journal 2011;38(55):18s. [Google Scholar]
- Matthys H, Pliskevich DA, Bondarchuk OM, Malek FA, Tribanek M, Kieser M. Randomised, double-blind, placebo-controlled trial of EPS 7630 in adults with COPD. Respiratory Medicine 2013;107(5):691-701. [DOI: 10.1016/j.rmed.2013.02.011] [DOI] [PubMed] [Google Scholar]
- Matthys H, Pliskevich DA, Malek FA, Tribanek M, Kieser M. Add-on-therapy in COPD with an herbal drug preparation. American Journal of Respiratory and Critical Care Medicine 2010;181:A4421. [Google Scholar]
- Matthys H, Pliskevich DA, Reineke T, Malek FA. Add-on therapy with EPS 7630 prolongs time to first exacerbation and improves health-related quality of life in COPD. Respiration 2013;85(6):605-6. [DOI: ] [Google Scholar]
Menon 2004 {published data only}
- Menon B. Effect of polyvalent bacterial extract (Broncho Vaxom) in the prophylaxis of acute exacerbation of COPD. Indian Journal of Allergy, Asthma and Immunology 2004;18(2):103. [Google Scholar]
Moniuszko 1991 {published data only}
- Moniuszko T, Rogalewska A, Chyrek-Borowska S. Clinical evaluation of Broncho-Vaxom efficiency. Polski Tygodnik Lekarski 1991;46(22-23):422-3. [PubMed] [Google Scholar]
NCT01842360 {published data only}
- NCT01842360. Evaluation of the efficacy and safety of MV130 in chronic obstructive pulmonary disease (COPD). clinicaltrials.gov/show/NCT01842360 (first received 29 April 2013).
NCT02417649 {published data only}
- NCT02417649. Advanced immunological approach in COPD exacerbation. clinicaltrials.gov/show/NCT02417649 (first received 15 April 2015).
Nishantha 2014 {published data only (unpublished sought but not used)}
- Nishantha KM, Gunaratne HC, Yasaratne D, Medegedara RM. Polyvalent mechanical bacterial lysate in advanced COPD; a randomized controlled trial. Respirology 2014;19(Suppl 3):121. [DOI: 10.1111/resp.12417] [DOI] [Google Scholar]
Nouvet 1989 {published data only}
- Nouvet G, Guerin JC, Leophonte P, Muir JF, Taytard A. Prevention of the recurrence of bronchial infection by ribomunyl tablet. Revue des Maladies Respiratoires 1989;6(Suppl 2):R105. [Google Scholar]
Obrecht 1975 {published data only}
- Obrecht U, Scherrer M. Diribiotine aerosols administered to patients with chronic bronchial obstruction. Double-blind study. Revue Medicale de la Suisse Romande 1975;95(5):345-59. [PubMed] [Google Scholar]
Panchyshyna 1997 {published data only}
- Panchyshyna MV, Al-Qdemat YA, Panchyshyn JM, Petruh LI, Kazmirchuk TG. Effect of flurenizide on adaptive reactions in patients with chronic obstructive pulmonary disease. International Journal of Clinical Pharmacology Research 1997;17(1):47-52. [PubMed] [Google Scholar]
Pozzi 1994 {published data only}
- Pozzi E, Dolcetti A, Orlandi O, Cirianni C, Moreo G, Piacenza G, et al. Pidotimod in the treatment of patients affected by bacterial exacerbations of chronic bronchitis. Arzneimittel-Forschung 1994;44(12A):1495-8. [PubMed] [Google Scholar]
Rekalova 1992 {published data only}
- Rekalova EM. The immunomodulator kemantan in the treatment of patients with exacerbated chronic obstructive bronchitis. Likarska Sprava 1992;4:73-6. [PubMed] [Google Scholar]
Ricci 2014 {published data only}
- Ricci R, Palmero C, Bazurro G, Riccio AM, Garelli V, Di Marco E, et al. The administration of a polyvalent mechanical bacterial lysate in elderly patients with COPD results in serological signs of an efficient immune response associated with a reduced number of acute episodes. Pulmonary Pharmacology and Therapeutics 2014;27(1):109-13. [DOI: 10.1016/j.pupt.2013.05.006] [DOI] [PubMed] [Google Scholar]
Rico Mendez 1997 {published data only}
- Rico Mendez G, Massey L, Martinez Baez J, Mugica J, Martinez L. Prevention of respiratory tract infections (RTI) in patients with chronic obstructive pulmonary disease (COPD) with the use of oral bacterial lysates (OBL). European Respiratory Journal 1997;10(Suppl 25):202S. [Google Scholar]
Rimoldi 1990 {published data only}
- Rimoldi R, Clini V, Corsico R, Corda M, Bosisio E, Giura R, et al. Prophylaxis of acute infectious episodes in patients with chronic bronchitis with a newer immunomodulating agent: a prospective, controlled, randomized clinical trial. European Respiratory Journal 1990;3(Suppl 10):86S. [Google Scholar]
Romanski 1997 {published data only}
- Romanski B, Tomeczko J, Wilewska-Kubo T, Wierciski A, Tomaszewska B, Glejzer-Kichler O, et al. Therapeutic and immunomodulating properties of Peat Preparation To-Apa (PPT) administered to patients with recurrent respiratory tract infection in the course of nonatopic bronchial asthma and chronic obstructive bronchitis. International Review of Allergology and Clinical Immunology 1997;3(2):111-23. [Google Scholar]
Rutishauser 1998 {published data only}
- Rutishauser M, Pitzke P, Grevers G, Aubel A, Elsasser U, Kämmereit A. Use of a polyvalent bacterial lysate in patients with recurrent respiratory tract infections: results of a prospective, placebo-controlled, randomized, double-blind study. Advances in Therapy 1998;15:330-41. [PubMed] [Google Scholar]
Shameen 2005 {published data only}
- Shameen M, Bhargav R, Ahmad Z, Sharma D, Shah NN. Effect of bronchovaxom on incidence and severity of upper respiratory tract infections in COPD patients. Indian Journal of Allergy Asthma and Immunology 2005;19(1):37-42. [Google Scholar]
Shmelev 1995 {published data only}
- Shmelev E, Khomenko A, Sokolova L, Philppov V, Kosmaidi G, Evguschenko G. Broncho-Vaxom in the management of chronic obstructive pulmonary diseases. European Respiratory Journal 1995;8(Suppl 19):236S. [Google Scholar]
Smirnova 2006 {published data only}
- Smirnova IaA, Trofimov VI, Shaporova NL. Efficacy of vaccine therapy with ribomunil in patients with chronic obstructive pulmonary disease. Terapevticheskii Arkhiv 2006;78(8):70-3. [PubMed] [Google Scholar]
Sofia 1988 {published data only}
- Sofia M, Molino A, Mormile M, Di Benedetto G, De Simone F, Carratu L. Chronic bronchitis: a double-blind clinical trial for the evaluation of the therapeutical effectiveness of orally administered thymomodulin in the prevention of acute attacks. Giornale Italiano delle Malattie del Torace 1988;41(6):339-42. [Google Scholar]
- Sofia M. Chronic bronchitis: a double blind clinical trial for the evaluation of the therapeutical effectiveness of orally administered thymomodulin in the prevention of acute attacks. Indian Journal of Tuberculosis 1989;36(4):255. [Google Scholar]
Spiropoulos 1995 {published data only}
- Spiropoulos K, Lymberopoulos D, Ginopoulos P, Lavranos A. Effect of long-term therapy with oral Broncho-Vaxom in spirometry of chronic bronchitis patients. Lotta Contro la Tubercolosi e le Malattie Polmonari Sociali 1995;65(1-2):167-71. [Google Scholar]
Targowski 2005 {published data only}
- Targowski T, Jahnz-Rozyk K, Plusa T, Niedzialkowski P, Rozynska R. Influence of Broncho-Vaxom treatment on serum concentration of metalloproteinase-9 in patients with chronic obstructive pulmonary disease. Polski Merkuriusz Lekarski 2005;19(113):630-3. [PubMed] [Google Scholar]
Viallat 1983 {published data only}
- Viallat JR, Costantini D, Boutin C, Farisse P. Double-blind trial of an immunomodulator of bacterial origin (Biostim) in the prevention of infectious episodes in chronic bronchitis. Poumon et le Coeur 1983;39(1):53-7. [PubMed] [Google Scholar]
Zanussi 1990 {published data only}
- Zanussi C, Balsano F, Fagiolo U. Thymopentin in the prevention of recurrent respiratory infections in aged subjects with chronic bronchitis: a multicentre controlled randomized clinical trial. International Journal of Immunotherapy 1990;6(1):53-60. [Google Scholar]
Zheng 2008 {published data only}
- Zheng BX, Cheng DY, Xu G, Fan LL, Yang Y, Yang W. The prophylactic effect of thymosin alpha 1 on the acute exacerbation of chronic obstructive pulmonary disease. Sichuan da Xue Xue Bao. Yi Xue Ban 2008;39(4):588-90. [PubMed] [Google Scholar]
References to studies awaiting assessment
Alvarez‐Sala 2004 {published data only}
- Alvarez-Sala JL, Alvarez-Mon M. Treatment with AM3 (Immunoferon), an oral immunomodulator, enhances the quality of life of patients with chronic obstructive pulmonary disease. Archivos de Bronconeumologia 2004;40(Suppl 2):37. [Google Scholar]
Berra 1988 {published data only}
- Berra A, Giella D, Galasso R, Matonti V, Lauriello G. Comparison of Klebsiella pneumoniae extract (Ru-41740) with placebo in acute infections in patients with chronic respiratory disease. Lotta Contro la Tuberculosi e le Malattie Polmonari Sociali 1988;58(3-4):727-30. [Google Scholar]
Magyar 1985 {published data only}
- Magyar P, Miskovits G, Nagy IA, Tarjan E, Zsiray M, Lantos A, et al. The therapeutic and preventive effects of BronchoVaxom, a lyophilized bacterial lysate, in chronic bronchitis: a double-blind placebo-controlled study. Pneumologia Hungarica 1985;38:293-303. [Google Scholar]
Shrewsbury 2014 {published data only (unpublished sought but not used)}
- Shrewsbury SB, Bjermer L, Vestbo J, Kuna P, Saarelainen P, Balint B, et al. The Flagship study: a 12-week phase II study to evaluate the efficacy and safety of AQX-1125 following exacerbations in patients with chronic obstructive pulmonary disease (COPD) by targeting the SHIP1 pathway [Abstract]. American Journal of Respiratory and Critical Care Medicine 2014;189:A2889. [Google Scholar]
Soler 2001 {published data only}
- Soler M, Mutterlein R. Multicentre, double-blind, placebo-controlled, randomised, clinical study of Broncho-Vaxom (OM-85) in patients suffering from acute exacerbations of chronic bronchitis or mild obstructive pulmonary disease. Clinical Study Reports 2001.
Additional references
Alyanakian 2006
- Alyanakian MA, Grela F, Aumeunier A, Chiavaroli C, Gouarin C, Bardel E, et al. Transforming growth factor-beta and natural killer T-cells are involved in the protective effect of a bacterial extract on type 1 diabetes. Diabetes 2006;55(1):179-85. [DOI: 10.2337/diabetes.55.01.06.db05-0189] [DOI] [PubMed] [Google Scholar]
ATS Foundation 2014
- American Thoracic Society Foundation. The global burden of lung disease. foundation.thoracic.org/news/global-burden.php (accessed 26 December 2018).
Baiardini 2005
- Baiardini I, Braido F, Fassio O, Tarantini F, Pasquali M, Tarchino F, et al. A new tool to assess and monitor the burden of chronic cough on quality of life: Chronic Cough Impact Questionnaire. Allergy 2005;60:482-8. [DOI] [PubMed] [Google Scholar]
Benetti 1994
- Benetti GP, Fugazza L, Stramba BM, Montalto F, Bombelli G, La Vecchia G, et al. Ex vivo evaluation of pidotimod activity on cell-mediated immunity. Drug Research 1994;44(12A):1476-9. [PMID: ] [PubMed] [Google Scholar]
Bergemann 1994
- Bergemann R, Brandt A, Zoellner U, Donner CF. Preventive treatment of chronic bronchitis: a meta-analysis of clinical trials with a bacterial extract (OM-85 BV) and a cost-effectiveness analysis. Archivio Monaldi per le Malattie del Torace 1994;49(4):302-7. [PMID: ] [PubMed] [Google Scholar]
Boissier 1988
- Boissier MC. Experimental immunopharmacology of RU 41740. La Presse Medicale 1988;17(28):1426-9. [PMID: ] [PubMed] [Google Scholar]
Braido 2011
- Braido F, Schenone G, Pallestrini E, Reggiardo G, Cangemi G, Canonica GW, et al. The relationship between mucosal immunoresponse and clinical outcome in patients with recurrent upper respiratory tract infections treated with a mechanical bacterial lysate. Journal of Biological Regulators and Homeostatic Agents 2011;25(3):477-85. [PMID: ] [PubMed] [Google Scholar]
Cazzola 2008
- Cazzola M, Rogliani P, Curradi G. Bacterial extracts for the prevention of acute exacerbations in chronic obstructive pulmonary disease: a point of view. Respiratory Medicine 2008;102(3):321-7. [DOI: 10.1016/j.rmed.2007.11.002] [DOI] [PubMed] [Google Scholar]
Cazzola 2012
- Cazzola M, Anapurapu S, Page CP. Polyvalent mechanical bacterial lysate for the prevention of recurrent respiratory infections: a meta-analysis. Pulmonary Pharmacology and Therapeutics 2012;25(1):62-8. [DOI: 10.1016/j.pupt.2011.11.002] [DOI] [PubMed] [Google Scholar]
Chong 2013
- Chong J, Leung B, Poole P. Phosphodiesterase 4 inhibitors for chronic obstructive pulmonary disease. Cochrane Database of Systematic Reviews 2013, Issue 11. Art. No: CD002309. [DOI: 10.1002/14651858.CD002309.pub4] [DOI] [PubMed] [Google Scholar]
CMDD 2016
- Chinese Marketed Drug Database. Updated 28 March 2016. www.drugfuture.com/cndrug/ (accessed prior to 12 June 2022).
Collet 1997
- Collet JP, Shapiro P, Ernst P, Renzi T, Ducruet T, Robinson A. Effects of an immunostimulating agent on acute exacerbations and hospitalizations in patients with chronic obstructive pulmonary disease. The PARI-IS Study Steering Committee and Research Group. Prevention of Acute Respiratory Infection by an Immunostimulant. American Journal of Respiratory and Critical Care Medicine 1997;156(6):1719-24. [DOI: 10.1164/ajrccm.156.6.9612096] [DOI] [PubMed] [Google Scholar]
Collet 2001
- Collet JP, Ducruet T, Haider S, Shapiro S, Robinson A, Renzi PM, et al. Economic impact of using an immunostimulating agent to prevent severe acute exacerbations in patients with chronic obstructive pulmonary disease. Canadian Respiratory Journal 2001;8(1):27-33. [DOI: 10.1155/2001/508015] [DOI] [PubMed] [Google Scholar]
Covidence [Computer program]
- Covidence. Melbourne, Australia: Veritas Health Innovation, (accessed prior to 1 September 2022). Available at covidence.org.
Cranston 2005
- Cranston JM, Crockett AJ, Moss JR, Alpers JH. Domiciliary oxygen for chronic obstructive pulmonary disease. Cochrane Database of Systematic Reviews 2005, Issue 4. Art. No: CD001744. [DOI: 10.1002/14651858.CD001744.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
De Benedetto 2013
- De Benedetto F, Sevieri G. Prevention of respiratory tract infections with bacterial lysate OM-85 bronchomunal in children and adults: a state of the art. Multidisciplinary Respiratory Medicine 2013;8(1):33. [DOI: 10.1186/2049-6958-8-33] [DOI] [PMC free article] [PubMed] [Google Scholar]
Deeks 2022
- Deeks JJ, Higgins JP, Altman DG. Chapter 10: Analysing data and undertaking meta-analyses. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.3 (updated February 2022). Cochrane, 2022. In: Available from training.cochrane.org/handbook. [Google Scholar]
Del‐Rio‐Navarro 2007
- Del-Rio-Navarro BE, González-Díaz S, Escalante-Domínguez AJ, Blandón-Vijil V. Immunostimulants in the prevention of respiratory infections. International Journal of Biotechnology 2007;9(3/4):246-60. [DOI: 10.1504/IJBT.2007.014245] [DOI] [Google Scholar]
Del‐Rio‐Navarro 2012
- Del-Rio-Navarro BE, Espinosa Rosales F, Flenady V, Sienra-Monge JJ. Immunostimulants for preventing respiratory tract infection in children. Evidence Based Child Health 2012;7(2):629-717. [DOI: 10.1002/ebch.1833] [DOI] [PubMed] [Google Scholar]
Duchow 1992
- Duchow J, Marchant A, Delville JP, Schandene L, Goldman M. Upregulation of adhesion molecules induced by Broncho-Vaxom® on phagocytic cells. International Journal of Immunopharmacology 1992;14(5):761-6. [DOI: 10.1016/0192-0561(92)90073-T] [DOI] [PubMed] [Google Scholar]
EMA 2019
- European Medicines Agency. Assessment report: bacterial lysates-containing medicinal products indicated for respiratory conditions. Published 28 June 2019. www.ema.europa.eu/en/medicines/human/referrals/bacterial-lysates-containing-medicinal-products-indicated-respiratory-conditions (accessed prior to 12 June 2022).
Ferris 1978
- Ferris BG. Epidemiology standardization project (American Thoracic Society). American Review of Respiratory Disease 1978;118:1-120. [PMID: ] [PubMed] [Google Scholar]
GBD 2015
- GBD 2015 Chronic Respiratory Disease Collaborators. Global, regional, and national deaths, prevalence, disability-adjusted life years, and years lived with disability for chronic obstructive pulmonary disease and asthma, 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Respiratory Medicine 2017;5(9):691-706. [DOI: 10.1016/S2213-2600(17)30293-X] [DOI] [PMC free article] [PubMed] [Google Scholar]
Giovannini 2014
- Giovannini M, Salvini F, Riva E. Bacterial extracts as immunomodulators for the prevention of recurrent respiratory infections in children. Journal of Medical Microbiology and Diagnosis 2014;3:136. [DOI: 10.4172/2161-0703.1000136] [DOI] [Google Scholar]
GOLD 2006
- Rabe KF, Hurd S, Anzueto A, Barnes PJ, Buist SA, Calverley P, et al. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease: GOLD Executive Summary. American Journal of Respiratory and Critical Care Medicine 2007;176:532-55. [DOI] [PubMed] [Google Scholar]
GOLD 2022
- Global Initiative for Chronic Obstructive Lung Disease (GOLD). Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: 2022 report. goldcopd.org/2022-gold-reports-2/ (accessed 6 March 2022).
GRADEpro GDT [Computer program]
- GRADEpro GDT. Version accessed prior to 17 January 2019. Hamilton (ON): McMaster University (developed by Evidence Prime), 2015. Available at gradepro.org.
Grossi 1989
- Grossi E, Blasi F, Nastri A, Montanari C. Predittivita di una scala di valutazione sintomatologica nella diagnosi delle nacutizzazioni in corso di BPCO: validazione mediante modello di analisi discriminante multivariata. Rassegna di Patologia Dell'Apparato Respiratorio 1989;IV(3):1-103. [Google Scholar]
Guyatt 1987
- Guyatt GH, Berman LB, Townsend M, Pugsley SO, Chambers LW. A measure of quality of life for clinical trials in chronic lung disease. Thorax 1987;42(10):773-8. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hadden 1993
- Hadden JW. Immunostimulants. Trends in Pharmacological Sciences 1993;14(5):169-74. [PMID: ] [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from training.cochrane.org/handbook/archive/v5.1/.
Huber 2005
- Huber M, Mossmann M, Bessler WG. Th1-orientated immunological properties of the bacterial extract OM-85 BV. European Journal of Medical Research 2005;10(5):209-17. [PMID: ] [PubMed] [Google Scholar]
Hurst 2010
- Hurst JR, Vestbo J, Anzueto A, Locantore N, Müllerova H, Tal-Singer R, et al. Susceptibility to exacerbation in chronic obstructive pulmonary disease. New England Journal of Medicine 2010;363(12):1128-38. [DOI: 10.1056/NEJMoa0909883] [DOI] [PubMed] [Google Scholar]
Jinjuvadia 2017
- Jinjuvadia C, Jinjuvadia R, Mandapakala C, Durairajan N, Liangpunsakul S, Soubani AO. Trends in outcomes, financial burden, and mortality for acute exacerbation of chronic obstructive pulmonary disease (COPD) in the United States from 2002–2010. COPD: Journal of Chronic Obstructive Pulmonary Disease 2017;14(1):72-9. [DOI: 10.1080/15412555.2016.1199669] [DOI] [PMC free article] [PubMed] [Google Scholar]
Jones 1992
- Jones PW, Quirk FH, Baveystock CM, Littlejohns P. A self-complete measure of health status for chronic airflow limitation: the St. George's Respiratory Questionnaire. American Review of Respiratory Disease 1992;145(6):1321-7. [DOI] [PubMed] [Google Scholar]
Jones 2005
- Jones PW. St. George's Respiratory Questionnaire: MCID. COPD 2005;2(1):75-9. [DOI] [PubMed] [Google Scholar]
Kanner 2001
- Kanner RE, Anthonisen NR, Connett JE, Lung Health Study Research Group. Lower respiratory illnesses promote FEV(1) decline in current smokers but not ex-smokers with mild chronic obstructive pulmonary disease: results from the lung health study. American Journal of Respiratory and Critical Care Medicine 2001;164(3):358-64. [DOI: 10.1164/ajrccm.164.3.2010017] [DOI] [PubMed] [Google Scholar]
Kew 2013
- Kew KM, Mavergames C, Walters JA. Long-acting beta2-agonists for chronic obstructive pulmonary disease. Cochrane Database of Systematic Reviews 2013, Issue 10. Art. No: CD010177. [DOI: 10.1002/14651858.CD010177.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kim 2011
- Kim V, Han MK, Vance GB, Make BJ, Newell JD, Hokanson JE. The chronic bronchitic phenotype of COPD: an analysis of the COPD Gene Study. Chest 2011;140(3):626-33. [DOI: 10.1378/chest.10-2948] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kim 2015
- Kim V, Crapo J, Zhao H, Jones PW, Silverman EK, Comellas A, et al. Comparison between an alternative and the classic definition of chronic bronchitis in COPDGene. Annals of the American Thoracic Society 2015;12(3):332-9. [DOI: 10.1513/AnnalsATS.201411-518OC] [DOI] [PMC free article] [PubMed] [Google Scholar]
Köhnlein 2014
- Köhnlein T, Windisch W, Köhler D, Drabik A, Geiseler J, Hartl S, et al. Non-invasive positive pressure ventilation for the treatment of severe stable chronic obstructive pulmonary disease: a prospective, multicentre, randomised, controlled clinical trial. Lancet Respiratory Medicine 2014;2(9):698-705. [DOI: 10.1016/S2213-2600(14)70153-5] [DOI] [PubMed] [Google Scholar]
Lanzilli 2006
- Lanzilli G, Falchetti R, Cottarelli A, Macchi A, Ungheri D, Fuggetta MP. In vivo effect of an immunostimulating bacterial lysate on human B lymphocytes. International Journal of Immunopathology and Pharmacology 2006;19(3):551-9. [DOI: 10.1177/039463200601900311] [DOI] [PubMed] [Google Scholar]
Lanzilli 2013
- Lanzilli G, Traggiai E, Braido F, Garelli V, Folli C, Chiappori A, et al. Administration of a polyvalent mechanical bacterial lysate to elderly patients with COPD: effects on circulating T, B and NK cells. Immunology Letters 2013;149(1-2):62-7. [DOI: 10.1016/j.imlet.2012.11.009] [DOI] [PubMed] [Google Scholar]
Li 2004
- Li J, Zheng JP, Yuan JP, Zeng GQ, Zhong NS, Lin CY. Protective effect of a bacterial extract against acute exacerbation in patients with chronic bronchitis accompanied by chronic obstructive pulmonary disease. Chinese Medical Journal 2004;117(6):828-34. [PMID: ] [PubMed] [Google Scholar]
Lopez 2006
- Lopez AD, Shibuya K, Rao C, Mathers CD, Hansell AL, Held LS, et al. Chronic obstructive pulmonary disease: current burden and future projections. European Respiratory Journal 2006;27(2):397-412. [DOI: 10.1183/09031936.06.00025805] [DOI] [PubMed] [Google Scholar]
Marruchella 2018
- Marruchella A, Faverio P, Bonaiti G, Pesci A. History of lung volume reduction procedures. Journal of Thoracic Disease 2018;10:S3326-34. [DOI: 10.21037/jtd.2018.04.165] [DOI] [PMC free article] [PubMed] [Google Scholar]
Mauel 1989
- Mauel J, Pham T, Kreis B, Bauer J. Stimulation by a bacterial extract (Broncho-Vaxom) of the metabolic and functional activities of murine macrophages. International Journal of Immunopharmacology 1989;11(6):637-45. [DOI: 10.1016/0192-0561(89)90149-5] [DOI] [PubMed] [Google Scholar]
Miller 2005
- Miller SI, Ernst RK, Bader MW. LPS, TLR4 and infectious disease diversity. Nature Reviews Microbiology 2005;3(1):36-46. [DOI: 10.1038/nrmicro1068] [DOI] [PubMed] [Google Scholar]
Mohan 2010
- Mohan A, Chandra S, Agarwal D, Guleria R, Broor S, Gaur B, et al. Prevalence of viral infection detected by PCR and RT-PCR in patients with acute exacerbation of COPD: a systematic review. Respirology 2010;15(3):536-42. [DOI: 10.1111/j.1440-1843.2010.01722.x] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moher 2009
- Moher D, Liberati A, Tetzlaff J, Altman D. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLOS Medicine 2009;6(7):e1000097. [DOI: 10.1371/journal.pmed.1000097] [DOI] [PMC free article] [PubMed] [Google Scholar]
Morandi 2011
- Morandi B, Agazzi A, D'Agostino A, Antonini F, Costa G, Sabatini F, et al. A mixture of bacterial mechanical lysates is more efficient than single strain lysate and of bacterial-derived soluble products for the induction of an activating phenotype in human dendritic cells. Immunology Letters 2011;138(1):86-91. [DOI: 10.1016/j.imlet.2011.03.006] [DOI] [PubMed] [Google Scholar]
Nannini 2012
- Nannini LJ, Lasserson TJ, Poole P. Combined corticosteroid and long-acting beta2-agonist in one inhaler versus long-acting beta2-agonists for chronic obstructive pulmonary disease. Cochrane Database of Systematic Reviews 2012, Issue 9. Art. No: CD006829. [DOI: 10.1002/14651858.CD006829.pub2] [DOI] [PubMed] [Google Scholar]
Navarro 2011
- Navarro S, Cossalter G, Chiavaroli C, Kanda A, Fleury S, Lazzari A, et al. The oral administration of bacterial extracts prevents asthma via the recruitment of regulatory T cells to the airways. Mucosal Immunology 2011;4:53-65. [DOI: 10.1038/mi.2010.51] [DOI] [PubMed] [Google Scholar]
Ni 2015
- Ni W, Shao X, Cai X, Wei C, Cui J, Wang R, et al. Prophylactic use of macrolide antibiotics for the prevention of chronic obstructive pulmonary disease exacerbation: a meta-analysis. PLOS One 2015;10(3):e0121257. [DOI: 10.1371/journal.pone.0121257. eCollection 2015] [DOI] [PMC free article] [PubMed] [Google Scholar]
Nikolova 2009
- Nikolova M, Stankulova D, Taskov H, Nenkov P, Maximov V, Petrunov B. Polybacterial immunomodulator respivax restores the inductive function of innate immunity in patients with recurrent respiratory infections. International Immunopharmacology 2009;9:425-32. [DOI: 10.1016/j.intimp.2009.01.004] [DOI] [PubMed] [Google Scholar]
Pan 2015
- Pan L, Jiang XG, Guo J, Tian Y, Liu CT. Effects of OM-85 BV in patients with chronic obstructive pulmonary disease: a systematic review and meta-analysis. Journal of Clinical Pharmacology 2015;55(10):1086-92. [DOI: 10.1002/jcph.518] [DOI] [PubMed] [Google Scholar]
Papi 2006
- Papi A, Bellettato CM, Braccioni F, Romagnoli M, Casolari P, Caramori G, et al. Infections and airway inflammation in chronic obstructive pulmonary disease severe exacerbations. American Journal of Respiratory and Critical Care Medicine 2006;173(10):1114-21. [DOI: 10.1164/rccm.200506-859OC] [DOI] [PubMed] [Google Scholar]
Pedraza‐Sánchez 2006
- Pedraza-Sánchez S, González-Hernández Y, Escobar-Gutiérrez A, Ramachandra L. The immunostimulant RU41740 from Klebsiella pneumoniae activates human cells in whole blood to potentially stimulate innate and adaptive immune responses. International Immunopharmacology 2006;6(4):635-46. [DOI: 10.1016/j.intimp.2005.10.006] [DOI] [PubMed] [Google Scholar]
Pivniouk 2022
- Pivniouk V, Pivniouk O, DeVries A, Uhrlaub JL, Michael A, Pivniouk D, et al. The OM-85 bacterial lysate inhibits SARS-CoV-2 infection of epithelial cells by downregulating SARS-CoV-2 receptor expression. Journal of Allergy and Clinical Immunology 2022;149(3):923-33.e6. [DOI: 10.1016/j.jaci.2021.11.019] [DOI] [PMC free article] [PubMed] [Google Scholar]
Poole 2015
- Poole P, Chong J, Cates CJ. Mucolytic agents versus placebo for chronic bronchitis or chronic obstructive pulmonary disease. Cochrane Database of Systematic Reviews 2015, Issue 7. Art. No: CD001287. [DOI: 10.1002/14651858.CD001287.pub5] [DOI] [PubMed] [Google Scholar]
Review Manager 2014 [Computer program]
- Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Rhodes 1996
- Rhodes J. Covalent chemical events in immune induction: fundamental and therapeutic aspects. Immunology Today 1996;17(9):436-41. [DOI: 10.1016/0167-5699(96)10050-5Get] [DOI] [PubMed] [Google Scholar]
Rial 2004
- Rial A, Lens D, Betancor L, Benkiel H, Silva JS, Chabalgoity JA. Intranasal immunization with a colloid-formulated bacterial extract induces an acute inflammatory response in the lungs and elicits specific immune responses. Infection and Immunity 2004;72(5):2679-88. [DOI: 10.1128/IAI.72.5.2679-2688.2004] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ricci 2014
- Ricci R, Palmero C, Bazurro G, Riccio AM, Garelli V, Di Marco E, et al. The administration of a polyvalent mechanical bacterial lysate in elderly patients with COPD results in serological signs of an efficient immune response associated with a reduced number of acute episodes. Pulmonary Pharmacology and Therapeutics 2014;27(1):109-13. [DOI: 10.1016/j.pupt.2013.05.006] [DOI] [PubMed] [Google Scholar]
Rohde 2003
- Rohde G, Wiethege A, Borg I, Kauth M, Bauer TT, Gillissen A. Respiratory viruses in exacerbations of chronic obstructive pulmonary disease requiring hospitalisation: a case-control study. Thorax 2003;58(1):37-42. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Rossi 2003
- Rossi GA, Peri C, Raynal ME, Defilippi AC, Risso FM, Schenone G, et al. Naturally occurring immune response against bacteria commonly involved in upper respiratory tract infections: analysis of the antigen-specific salivary IgA levels. Immunology Letters 2003;86(1):85-91. [PMID: ] [DOI] [PubMed] [Google Scholar]
Rozy 2008
- Rozy A, Chorostowska-Wynimko J. Bacterial immunostimulants – mechanism of action and clinical application in respiratory diseases. Advances in Respiratory Medicine 2008;76(5):353-9. [PMID: ] [PubMed] [Google Scholar]
Schünemann 2022
- Schünemann HJ, Higgins JP, Vist GE, Glasziou P, Akl EA, Skoetz N, et al. Chapter 14: Completing 'Summary of findings' tables and grading the certainty of the evidence. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.3 (updated February 2022). Cochrane, 2022. Available from www.training.cochrane.org/handbook.
Seemungal 1998
- Seemungal TA, Donaldson GC, Paul EA, Bestall JC, Jeffries DJ, Wedzicha JA. Effect of exacerbation on quality of life in patients with chronic obstructive pulmonary disease. American Journal of Respiratory and Critical Care Medicine 1998;157(5):1418-22. [DOI: 10.1164/ajrccm.157.5.9709032] [DOI] [PubMed] [Google Scholar]
Singanayagam 2012
- Singanayagam A, Joshi PV, Mallia P, Johnston SL. Viruses exacerbating chronic pulmonary disease: the role of immune modulation. BMC Medicine 2012;10:27. [DOI: 10.1186/1741-7015-10-27] [DOI] [PMC free article] [PubMed] [Google Scholar]
Soler‐Cataluña 2005
- Soler-Cataluña JJ, Martínez-García MA, Román Sánchez P, Salcedo E, Navarro M, Ochando R. Severe acute exacerbations and mortality in patients with chronic obstructive pulmonary disease. Thorax 2005;60(11):925-31. [DOI: 10.1136/thx.2005.040527] [DOI] [PMC free article] [PubMed] [Google Scholar]
Spaner 2005
- Spaner DE, Miller RL, Mena J, Grossman L, Sorrenti V, Shi Y. Regression of lymphomatous skin deposits in a chronic lymphocytic leukemia patient treated with the Toll-like receptor-7/8 agonist, imiquimod. Leukemia and Lymphoma 2005;46(6):935-9. [DOI: 10.1080/10428190500054426] [DOI] [PubMed] [Google Scholar]
Sprenkle 2005
- Sprenkle MD, Niewoehner DE, MacDonald R, Rutks I, Wilt TJ. Clinical efficacy of OM-85 BV in COPD and chronic bronchitis: a systematic review. COPD: Journal of Chronic Obstructive Pulmonary Disease 2005;2(1):167-75. [PMID: ] [DOI] [PubMed] [Google Scholar]
Steurer‐Stey 2004
- Steurer-Stey C, Bachmann LM, Steurer J, Tramèr MR. Oral purified bacterial extracts in chronic bronchitis and COPD: systematic review. Chest 2004;126(5):1645-55. [10.1378/chest.126.5.1645] [DOI] [PubMed] [Google Scholar]
Tashkin 2008
- Tashkin DP, Celli B, Senn S, Burkhart D, Kesten S, Menjoge S, et al. A 4-year trial of tiotropium in chronic obstructive pulmonary disease. New England Journal of Medicine 2008;359(15):1543-54. [DOI: 10.1056/NEJMoa0805800] [DOI] [PubMed] [Google Scholar]
Thomas 2020
- Thomas J, McDonald S, Noel-Storr A, Shemilt I, Elliott J, Mavergames C, et al. Machine learning reduced workload with minimal risk of missing studies: development and evaluation of a randomized controlled trial classifier for Cochrane Reviews. Journal of Clinical Epidemiology 2020;133:140-51. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tuthill 2013
- Tuthill CW, King RS. Thymosin alpha 1-A peptide immune modulator with a broad range of clinical applications. Journal of Clinical and Experimental Pharmacology 2013;3:133. [DOI: 10.4172/2161-1459.1000133] [DOI] [Google Scholar]
Wedzicha 2007
- Wedzicha JA, Seemungal TA. COPD exacerbations: defining their cause and prevention. Lancet 2007;370(9589):786-96. [DOI: 10.1016/S0140-6736(07)61382-8] [DOI] [PMC free article] [PubMed] [Google Scholar]
WHO 2018
- World Health Organization. Projections of mortality and causes of death, 2016–2060. www.who.int/healthinfo/global_burden_disease/projections/en/ (accessed prior to 17 January 2019).
WHO GHE 2016
- Global Health Estimates 2016: deaths by cause, age and sex, by country and by region, 2000–2016. www.who.int/healthinfo/global_burden_disease/estimates/en/ (accessed prior to 17 January 2019).
Woodhead 2011
- Woodhead M, Blasi F, Ewig S, Garau J, Huchon G, Ieven M, et al. Guidelines for the management of adult lower respiratory tract infections – full version. Clinical Microbiology and Infection 2011;17(6):E1-59. [DOI: 10.1111/j.1469-0691.2011.03672.x] [DOI] [PMC free article] [PubMed] [Google Scholar]
Woodruff 2016
- Woodruff PG, Barr RG, Bleecker E, Christenson SA, Couper D, Curtis JL, et al. Clinical significance of symptoms in smokers with preserved pulmonary function. New England Journal of Medicine 2016;374(19):1811-21. [DOI: 10.1056/NEJMoa1505971] [DOI] [PMC free article] [PubMed] [Google Scholar]
Xuan 2014
- Xuan J, Wang L, Yin H, Xuan D, Zhou Y, Hu S. The cost-effectiveness of OM-85 in managing respiratory tract infections in China. Journal of Medical Economics 2015;18(3):167-72. [DOI: 10.3111/13696998.2014.971159] [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Fraser 2019
- Fraser A, Poole P. Immunostimulants versus placebo for preventing exacerbations in adults with chronic bronchitis or chronic obstructive pulmonary disease. Cochrane Database of Systematic Reviews 2019, Issue 5. Art. No: CD013343. [DOI: 10.1002/14651858.CD013343] [DOI] [PMC free article] [PubMed] [Google Scholar]