

Abstract
Molecular property prediction is an essential but challenging task in drug discovery. The recurrent neural network (RNN) and Transformer are the mainstream methods for sequence modeling, and both have been successfully applied independently for molecular property prediction. As the local information and global information of molecules are very important for molecular properties, we aim to integrate the bi-directional gated recurrent unit (BiGRU) into the original Transformer encoder, together with self-attention to better capture local and global molecular information simultaneously. To this end, we propose the TranGRU approach, which encodes the local and global information of molecules by using the BiGRU and self-attention, respectively. Then, we use a gate mechanism to reasonably fuse the two molecular representations. In this way, we enhance the ability of the proposed model to encode both local and global molecular information. Compared to the baselines and state-of-the-art methods when treating each task as a single-task classification on Tox21, the proposed approach outperforms the baselines on 9 out of 12 tasks and state-of-the-art methods on 5 out of 12 tasks. TranGRU also obtains the best ROC-AUC scores on BBBP, FDA, LogP, and Tox21 (multitask classification) and has a comparable performance on ToxCast, BACE, and ecoli. On the whole, TranGRU achieves better performance for molecular property prediction. The source code is available in GitHub: https://github.com/Jiangjing0122/TranGRU.
Keywords: Local information, Global information, Fusion model, Molecular property prediction
Introduction
Molecular property prediction is one of the key tasks in the drug screening process. Deep learning-based methods have achieved great success in drug discovery, material science, and related fields [1–3], and they further optimize the drug screening process and improve the speed and efficiency of drug discovery, such as the discovery of anti-SARS-CoV-2 drugs [4]. Rational drug discovery involves a series of molecular properties, including binding affinity, toxicity, solubility, and so on. Many successful applications of machine learning and deep learning methods have been successfully adopted for molecular property prediction.
A neural network can learn molecular features from molecular representation data more directly, efficiently, and concisely [5], such as Simplified Molecular-Input Line-Entry System (SMILES) [6, 7] strings. Compared with natural language processing (NLP), modeling a suitable representation of the biological or chemical structures of molecules remains a challenging task. Molecules can be represented as SMILES strings or molecular graphs. Considering the similarity between the molecular language and natural language, some NLP-based models have been successfully applied to encode useful features of molecules from the complex structure of SMILES strings for molecular property prediction [8–12]. We focus on SMILES-based methods in this paper.
The recurrent neural network (RNN) [13] and Transformer [14] are two mainstream methods of extracting molecular representations from SMILES. RNN is usually used as an independent feature extractor for molecular property prediction [3, 15–18]; these methods only use RNN or its variants. Transformer effectively alleviates the sequence dependencies in RNN, and it has achieved great success in no matter whether 1, 2 and 3 dimensions for molecular property prediction [19–21]. However, the self-attention mechanism of Transformer makes it good at capturing global information, but weak at capturing the local information (such as structural information) from SMILES sequences [22, 23]. In summary, RNN and Transformer tend to focus on different types of features in the process of feature extraction [24], and both of them have their tendencies in molecular property prediction. However, there has been little work integrating both models for molecular property prediction.
As is well known, molecules are usually composed of a series of atomic groups, and molecular properties are often determined by several closely related atoms. Sub-sequences in SMILES strings are not just a flat sequence of atoms [25, 26]. For example, the binding between a molecule and any of its targets essentially involves the interactions between some specific atomic groups and the target protein [27]. These atomic groups are called functional groups, which are an important feature, and they usually appear in adjacent positions of the SMILES string. Usually, the local information of the molecule is expressed by functional groups, while the effective global information of the molecule needs to be extracted from the whole molecule. As mentioned above, both the local and global information of molecules is essential for molecular property prediction.
Considering the different behaviors of the bidirectional gated recurrent unit (BiGRU) [28] and Transformer in extracting molecular features, we aim to integrate the BiGRU into the encoder layer of the original Transformer, as well as self-attention to better capture local and global molecular information simultaneously. To this end, we propose a new deep neural network model called TranGRU. Specifically, TranGRU enhances the ability of the Transformer model with BiGRU to encode the local and global information of molecules, respectively. Then, a gate mechanism is used to reasonably fuse the two molecular representations. That is to say, we aim for the BiGRU to strengthen the ability to capture the local features of molecules while the original self-attention captures the global features. We adjust the features encoded by the BiGRU and Transformer through the gate mechanism to obtain a better molecular representation adaptively. The experimental results of single- and multi-task classification show strong performance on a wide range of tasks for molecular property prediction.
Overall, there are three main contributions of our paper:
We propose a deep neural network integrating the BiGRU and self-attention into the Transformer architecture, yielding a new model called TranGRU for molecular property prediction.
We explicitly model both the local and global information of the molecule and effectively fuse them via the gate mechanism to capture both local and global molecular representations simultaneously.
We carry out a series of experiments on benchmark datasets of single- and multi-task classification. When treating each property as a single-task classification on Tox21, the proposed approach outperforms the baselines on 9 out of 12 tasks and state-of-the-art methods on 5 out of 12 tasks. The approach also obtains the best performance on BBBP, and FDA, and has comparable performance on BACE, LogP, Tox21 (multi-task classification), ToxCast, and ecoli.
The rest of the paper is organized as follows: In Section 2, we give a brief literature review of the SMILES language and tokenization, then review models of RNN and Transformers used for molecular property prediction. In Section 3, we introduce TranGRU in detail. The performance of TranGRU is presented in Section 4, and we compare it with baselines and state-of-the-art methods. Finally, we summarize the achievements and highlights of our paper in Section 5.
Related work
SMILES language and tokenization
SMILES is a line notation that represents the atoms, bonds, and rings that make up molecules as a string. Each atom is represented in the alphabet of the element symbols. The bonds are represented by different symbols for the single bond (-), the double bond (=), the triple bond (#), and the quadruple bond ($); a detailed specification of SMILES can be found in OpenSMILES.1 The types of bonds that can be easily inferred through the atoms or the ring structure of the surrounding atom are generally omitted. For example, the 6-carbon ringed molecule benzene can be represented by ‘C1=CC=CC=C1’, where the ‘=’ represents a double bond and ‘1’ represents the start or the closing of a cycle or ring. SMILES strings are tokenized before encoding; for example, atom-level tokenization and SPE [25] are common methods. Figure 1 shows SMILES representations of two molecules from Tox21. SMILES is a common method for representing molecules in recent deep learning models [3, 17, 25, 26, 29, 30]. In this paper, we use canonical SMILES to represent molecules, which ensures that a molecule corresponds to a single SMILES string.
Fig. 1.

Examples of the process of generating SMILES representations. Both molecules are chosen from Tox21
RNN for molecular property prediction
The bi-directional long short-term memory (BiLSTM) [31, 32] and BiGRU are two variants of RNN, which have widely used for molecular property prediction [33, 34]. Methods of RNN and its variants used for molecular property prediction can be categorized into two main groups: 1) encoding the molecular representations, and 2) extraction of key features of molecules. For the first group of methods, BiLSTM is utilized to encode a representation of each node and learn the contextual information of a molecule [15–18, 30, 35]. For example, Lv et al. [18] propose Mol2Context-vec, which adopts the deep Bi-LSTM to model the local information and semantic information of molecules, which represents the high-dimensional features of the interactions between atomic groups. Specifically, the lower-level LSTM state model the local information inside atomic groups, and the higher-level LSTM state capture the semantic information. For the second group of methods, merging the merits of LSTM [36], or combining a multi-step attention mechanism to BiLSTM [3] to facilitate the extraction of key features of neighborhoods from SMILES strings. For example, SMILES2vec [17] is a deep RNN that automatically learns features from SMILES strings to predict a broad range of chemical properties, including toxicity, activity, solubility, and solvation energy.
All the above works leveraged RNN and its variants to extract molecular information. In practice, RNN is good at capturing the local information of a sequence [23], except for the related fields of molecular property prediction, RNN is widely used in other fields [37–39]. Therefore, we continue to make full use of the tendency of the BiGRU to capture the local information of molecules for molecular property prediction. In contrast to the above work, we aim to extract molecules’ local and global information, simultaneously.
Transformer for molecular property prediction
Compared with RNN, Transformer [14] is another powerful feature extractor [24, 40], which makes use of self-attention networks [41] to extract global information. The related work of Transformer used for molecular property prediction is mainly focused on capturing the global features of molecules [23, 42] by self-attention. For example, Transformer obtains the feature representations to learn the graph-structured data [19], or are used to extract self-supervised features in MolCloZe [20]. Except capture the general features of molecules, Transformer is used to extract the structural features of molecules. For example, Transformer uses an attention mechanism to understand molecular structures beyond the limitations of the chemical language itself, which causes semantic discontinuity by paying attention to characters sparsely [43].
Considering the advantages of the existing methods, RNN tends to capture the neighborhood information of SMILES strings, while Transformer tends to extract global information. Unlike the above methods, we propose an appropriate method, which focuses on modeling both the local and global information of molecules by integrating an RNN into a Transformer encoder. TranGRU is a simple yet effective method that leans local and global molecular representations simultaneously.
TranGRU for molecular property prediction
The details of TranGRU for molecular property prediction are presented in this section. First, we give the representation of molecules used in our approach in the pre-processing step. Second, we introduce the original BiGRU [13] and Transformer [14] briefly since our model mainly relies on both of them and we also introduce the proposed TranGRU in detail. Third, we show the method of molecular property prediction used in this paper. Last, we give the model training and inference of the proposed approach. An overview of the TranGRU model is shown in Fig. 2, the tokenized molecular sequences are fed into TranGRU to encode the molecular representation, in which the outputs of both the BiGRU and self-attention are fused via a gated mechanism to adjust the weights. Finally, we obtain the predictions of the molecular property via the softmax function.
Fig. 2.
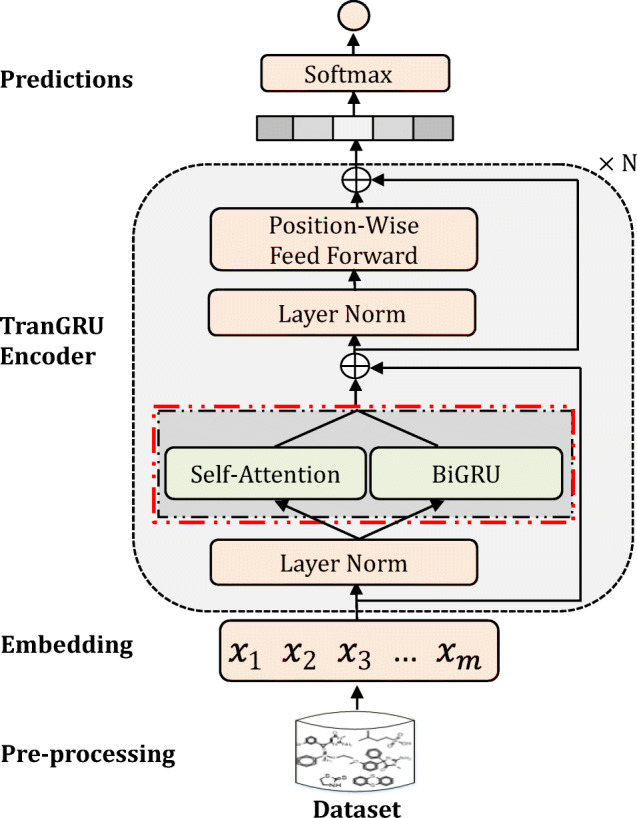
Overall framework of the proposed approach for molecular property prediction
Pre-processing
Formally, given a molecule X represented by a SMILES sequence from a dataset, X = (x1,⋯ ,xt,⋯ ,xT). xt represents a token of the SMILES sequence, and T represents the length of the sequence. Note that tokens can be atom-level or substring-level tokens. First, the SMILES strings of a molecule are pre-processed into atom-level tokens or substring-level tokens by RDKit.Chem or SPE, respectively. Then, the tokenized string is converted into a set of embeddings. Next, the embeddings are fed into the first layer of TranGRU to encode feature representation.
The TranGRU model
The TranGRU model receives the molecular embeddings from the pre-processing step. Since the TranGRU has n identical encoder layers, the outputs of the current layer will be fed into the next layer iteratively. Finally, the outputs of the top layer are considered as the molecular representation for molecular property prediction. Specifically, for a TranGRU layer that receives the inputs from the previous layer, followed by layer normalization, the outputs are fed into self-attention and BiGRU sub-layers, respectively. The two output states from the self-attention and BiGRU with the same length are fused by the gated mechanism, and followed by a layer normalization operation, then the fused states are fed into the position-wise feed-forward layer to generate the current layer outputs. We present the details of TranGRU in the following. Note that BiGRU in Section 3.2.1 and Transformer in Section 3.2.2 are the original models, which will be used in the proposed TranGRU. Section 3.2.3 presents the detail of TranGRU integrating BiGRU into Transformer encoder.
BiGRU encoder
We adopt BiGRU to capture the local information of molecules. For simplicity, we use the function BiGRU(⋅) to represent the transformation from xt to ht via a BiGRU encoder:
| 1 |
After encoding, we get a sequence of hidden states, H = (h1,⋯ ,hT). H denotes the representation of a molecule.
The BiGRU gradually encodes the whole molecular representation in a sequential way, which usually captures more local molecular features. BiGRU tends to capture local information, paying more attention to encoding information close to the current token and less attention to encoding information far from the current token. Consequently, we integrate the BiGRU into Transformer to enhance the ability to extract local molecular information.
Transformer encoder
Transformer [14] encoder consists of a stack of identical layers, whereas a Transformer encoder layer consists of a multi-head self-attention sub-layer followed by a position-wise feed-forward sub-layer. For a molecular representation X, which will be fed into the encoder to extract features for molecular property prediction. The i-th encoder layer receives a sequence state Xi as input and computes a new sequence state Hi of the same length, note that when i = 1, Xi = X.
Self-attention encodes the inputs, then gets the intermediate state Zi. Zi is computed as follows:
| 2 |
where , T is the sequence length of a molecule and D is the hidden state size. LN(⋅) denotes layer normalization [44]. Note that, we apply pre-normalization to each sub-layer to alleviate gradient problems. SA(⋅) represents the multi-head self-attention network. The matrices WQ,WK,andWV ∈ RD×D linearly map Xi into query, and key-value sets via linear transformation.
Zi is the intermediate state of the outputs of the encoder with residual connections. Then, Zi is fed into the feed-forward sub-layer. Hi is the output of the feed-forward sub-layer, which can be represented by:
| 3 |
where , and FFN(⋅) represents the feed-forward neural network. Residual connection [45] and layer normalization are applied in both sub-layers to alleviate gradient problems [46].
For simplicity, we use the function Trans(⋅) to represent the transformation from Xi to Hi via a Transformer encoder layer. The computation is as follows:
| 4 |
Because there are multiple identical layers in Transformer, Xi = Hi − 1, where 1 ≤ i ≤ I. The output of the current layer is considered as the input of the next layer.
Although Transformer has achieved satisfactory performance for molecular property prediction [21], we argue that the self-attention mechanism may not be sufficient to encode both local- and global-level molecular features. To improve the ability to capture molecular information, we propose explicitly modeling the local and global information for molecular property prediction.
The TranGRU encoder
To improve the ability of the model to extract both local and global features at the same time, we adopt both BiGRU and self-attention as the main components of the encoder to encode both local and global information for molecular property prediction.
A TranGRU encoder layer receives the inputs from the previous layer, and then followed by a layer-norm operation. Next, the outputs of the layer-norm will be fed into BiGRU and self-attention respectively to encode feature representations. Specifically, we integrate BiGRU into the Transformer layer to enhance the ability to aggregate the local information of molecules. For the i-th layer, we use and as the first hidden state outputs of self-attention and BiGRU, respectively. Therefore, (2) is updated as follows:
| 5 |
| 6 |
Next, we obtain the new intermediate state Zi via the gated mechanism to fuse the outputs of the BiGRU and self-attention . The computation is shown as (7).
| 7 |
where ∘ is element-wise multiplication.
Then, (8) is the detailed computation of gate gi.
| 8 |
where σ(⋅) is a logistic sigmoid function, and are learned model parameters.
Zi is the hidden state that fuses the outputs of BiGRU and self-attention. Zi is fed into the feed-forward sub-layer which outputs Hi by (9).
| 9 |
Since a TranGRU encoder consists of I stacked identical layers, the output of i-th layer will be used as the input of the (i + 1)-th layer. The top layer output HI is considered as the representation of the input molecule, and it is fed to the molecular property prediction module.
Molecular property prediction
After encoding of TranGRU encoder, we obtain the final encoder output states HI . The final molecular representation H will be obtained by the mean-pooling operation over Hi. Finally, we obtain the molecular property prediction via the softmax function.
| 10 |
where , U is a model parameter, L is the property number, and D is the hidden state size. P(H) is the prediction of a property. It should be noted that the proposed TranGRU model is a general molecular features encoder, and the different molecular property prediction tasks can be adapted by the softmax function.
TranGRU is a general method for extracting molecular representations on molecular property prediction. Specifically, TranGRU relies on BiGRU and self-attention to encode the inputs, as BiGRU tends to capture the local information of molecules, and self-attention tends to extract the global information of the molecule. Both the local and global information of a molecule is very important for the molecular properties. Therefore, we explicitly model the local and global information simultaneously to better predict the molecular properties.
Training and inference
We formulate the optimization function (i.e.,training loss) as label loss. Label loss is defined as negative likelihood of predicting correct labels of multiple downstream tasks l ∈ L:
| 11 |
where M is the number of molecules, and L is the number of property. Note that the processing in the inference is the same with the trainning stage.
Experiments and discussions
Datasets
The datasets in the paper are chosen from the widely used benchmarks MoleculeNets [1] and ZINC [47]. BACE, BBBP, HIV, Tox21, and ToxCast are from MoleculeNet. LogP and FDA are from ZINC. Table 1 presents a brief introduction to the datasets used in the experiments. Table 10 presents more details of the datasets used in the experiments.
Table 1.
Details of the datasets used in the experiments
| Dataset | Tasks | Type | Size | Metric | Actives |
|---|---|---|---|---|---|
| BACE [48] | 1 | Classification | 1513 | ROC-AUC | 691 |
| BBBP [49] | 1 | Classification | 2037 | ROC-AUC | 1567 |
| HIV [1] | 1 | Classification | 41127 | ROC-AUC | 1443 |
| Tox21 [50] | 12 | Classification | 7831 | ROC-AUC | ∥ |
| ToxCast [51] | 617 | Classification | 7818 | ROC-AUC | ∥ |
| LogP [47] | 1 | Classification | 9000 | ROC-AUC | 4502 |
| FDA [47] | 1 | Classification | 2907 | ROC-AUC | 1467 |
BACE, BBBP, HIV, LogP, and FDA are the single binary classification tasks, and the number of each task is 1. Tox21 contains 12 tasks, ToxCast contains 617 tasks, and ClinTox contains 2 tasks. Each task is treated as a binary classification task. Note that some problems in stereochemistry are not taken into account
Table 10.
Details of the datasets used in the experiments
| Dataset | Tasks | Actives | Description |
|---|---|---|---|
| BACE | 1 | 691 | Binding results for inhibitors of human BACE-1. |
| BBBP | 1 | 1567 | Blood-brain barrier penetration. |
| HIV | 1 | 1443 | Ability to inhibit HIV replication. |
| Tox21 | 12 | ∖ | Toxicity measurements. |
| ToxCast | 617 | ∖ | Another dataset provides toxicology data. |
| LogP | 1 | 4502 | Solubility of molecules. |
| FDA | 1 | 1467 | Approved drug compounds by FDA. |
BACE, BBBP, HIV, LogP, and FDA are binary classification tasks, and the number of the prediction task is 1. Tox21 contains 12 tasks, and ClinTox contains 2 tasks. Each task is treated as a binary classification task. Note that some problems in stereochemistry are not taken into account
Experimental settings
We employ two types of tokenizers to tokenize SMILES sequences. We use the open-source package RDKit.2 to preprocess SMILES strings from various datasets. The embedding size is 64. Early stopping is used to stop training, and the maximum training epoch number is set to 100; the optimization of the models is performed by the Adam optimizer. We split the dataset by scaffold-split or random-split following MoleculeNet [1]. For LogP and FDA, we use random-split. Table 11 gives the details of the splitting methods of the datasets used in the experiments. The detail of the parameters is shown in Table 2.
Table 11.
Details of the datasets used in the experiments
| Dataset | BACE | BBBP | Tox21 | ToxCast | HIV | LogP | FDA |
|---|---|---|---|---|---|---|---|
| Ins. | 1513 | 2037 | 7831 | 7718 | 41127 | 10000 | 2907 |
| Train | 1210 | 1626 | 6264 | 6174 | 36604 | 8000 | 2326 |
| Dev. | 151 | 205 | 783 | 772 | 2261 | 1000 | 290 |
| Test | 152 | 193 | 784 | 772 | 2262 | 1000 | 291 |
Table 2.
Some hyper-parameters used in TranGRU
| Name of hyper-parameter | Value |
|---|---|
| input dimension | 64 |
| batch size | 2 |
| epoch | 100 |
| latent size | 64 |
| learning rate | 0.001 |
| warmup | 0.15 |
| aggregation type | ‘MEAN’ |
| Model N | 6 |
| dropout | 0.1 |
Baselines and related works
We comprehensively evaluate the performance of our model against nine state-of-the-art methods including RNN and Transformer variants. The details are presented as follows:
Seq2seq [15]: Seq2seq is a typical NLP model that uses unsupervised methods to learn molecular representations.
Seq3seq [9]: It is based on the seq2seq model defining a loss function that contains both self-recovery loss and inference task loss.
SMILES-BERT [29]: It is a semi-supervised model consisting of an attention mechanism-based Transformer Layer. The pre-trained model uses large-scale unlabeled data to pre-train through a Masked SMILES Recovery task that can easily be generalized into different molecular property prediction tasks via fine-tuning.
Smi2Vec*-LSTM [36]: It is a system that merges the merits of various techniques, such as the long short-term memory (LSTM) RNN, and is designed for learning atoms and solving the classic problems in the field of drug discovery.
Smi2Vec-BiGRU [16]: It is designed for learning atoms and solving single- and multitask binary classification problems in the field of drug discovery. This approach transforms the molecule data in the SMILES format into a set of sample vectors and then feeds them into the bidirectional gated recurrent unit neural networks for training, which yields low-dimensional vector representations for the molecular drug.
GraphSAGE [52]: Through the method of sampling and aggregating the neighbor embeddings of nodes, GraphSAGE can capture graph structure information effectively.
GraSeq [30]: GraSeq joints graph and sequence for molecular property prediction. Specifically, it makes a complementary combination of GNNs and RNNs to model two types of molecular inputs, respectively.
Mol2vec [35]: Similar molecular structures have similar vector representations. Mol2vec learns vector representations of molecular structures by Word2Vec [53]. The vector representation of a compound can be obtained by combining the vectors of its molecular substructures.
Experimental results
We analyze the performance of the model by comparing the performance of baselines and state-of-the-art methods on the benchmark datasets. ROC-AUC and accuracy are adopted as the evaluation metrics in this paper.
Comparison with baselines
To verify the performance of the proposed model, we follow the work of [16, 36] in treating each property on the representative Tox21 as a single task. The Transformer represents the standard Transformer model. We adopt both BiGRU and Transformer as the baselines; the layer depth of the Transformer is set to 6, which is identical to the layer depth of the proposed TranGRU. We adopt the atom-level tokenizer from RDKit. The comparison results are shown in Table 3; our model achieves the best performance except on the tasks of SR-ARE, SR-HSE, and SR-MMP.
Table 3.
Comparison ROC-AUC between TranGRU and baseline models on Tox21. The best performance is denoted by bold
| Tasks | BiGRU | Transformer | TranGRU |
|---|---|---|---|
| NR-AR | 0.7945 | 0.8207 | 0.8241 |
| NR-AR-LBD | 0.8035 | 0.8251 | 0.8470 |
| NR-AhR | 0.8028 | 0.8184 | 0.8327 |
| NR-Aromatase | 0.7526 | 0.7758 | 0.7838 |
| NR-ER | 0.6640 | 0.6792 | 0.6907 |
| NR-ER-LBD | 0.8315 | 0.8028 | 0.8433 |
| NR-PPAR-gamma | 0.7966 | 0.8254 | 0.8375 |
| SR-ARE | 0.7252 | 0.6575 | 0.7014 |
| SR-ATAD5 | 0.7097 | 0.6970 | 0.7267 |
| SR-HSE | 0.7163 | 0.7380 | 0.7362 |
| SR-MMP | 0.8296 | 0.7697 | 0.8156 |
| SR-p53 | 0.8025 | 0.7543 | 0.8100 |
Table 4 shows the comparison results of different tokenizers on HIV. “-S” represents using RDKit⋅Chem as the tokenizer, which we also denote as atom-level tokenization. “-B” represents that the tokenizer is SPE, which we denote as substring-level. The performance gap is not significant between the results obtained by atom-level and substring-level tokenizers. TranGRU achieves the best performance in terms of ROC-AUC on HIV compared with the baselines. Although there is little difference in performance between the two tokenizers, TranGRU has the best performance.
Table 4.
ROC-AUC scores on HIV. The best performance is denoted by bold
| Tokens | BiGRU | Transformer | TranGRU |
|---|---|---|---|
| -S | 0.7927 | 0.7484 | 0.8105 |
| -B | 0.7942 | 0.8079 | 0.8115 |
“-S” represents using an atom-level tokenizer to tokenize the SMILES strings, and “-B” represents using SPE as the tokenizer
Figure 3 presents comparisons of different baselines on Tox21 and ToxCast. Both are multi-task classification datasets. TranGRU outperforms the others on both datasets. Therefore, TranGRU has better performance on multi-task classification datasets.
Fig. 3.

Performance comparison with different baselines on multi-classification tasks
As a comparison with the baselines, TranGRU achieves the best performance on HIV and 9 out of 12 tasks on Tox21 for single-task classification. TranGRU outperforms the baselines on Tox21 and ToxCast, both of which are classification problems of multi-task.
Comparison with state-of-the-art methods
To Compare the performance of ROC-AUC with the most advanced methods (i.e., Smi2Vec*-LSTM [36] and Smi2Vec-BiGRU [16]) on Tox21, Table 5 shows the details. TranGRU obtains the best performance on 5 out of 12 tasks on Tox21, namely NR-AR, NR-AR-LBD, NR-Aromatase, NR-PPAR-gamma, and SR-p53.
Table 5.
Comparison with state-of-the-art methods in terms of the ROC-AUC scores of each task on Tox21. The best performance is denoted by bold
| Tasks | Smi2Vec*-LSTM | Smi2Vec-BiGRU | TranGRU |
|---|---|---|---|
| NR-AR | 0.6914 | 0.7114 | 0.8241 |
| NR-AR-LBD | 0.7477 | 0.8243 | 0.847 |
| NR-AhR | 0.678 | 0.8793 | 0.8327 |
| NR-Aromatase | 0.4964 | 0.6985 | 0.7838 |
| NR-ER | 0.6231 | 0.736 | 0.6907 |
| NR-ER-LBD | 0.5308 | 0.8675 | 0.8433 |
| NR-PPAR-gamma | 0.5659 | 0.7494 | 0.8375 |
| SR-ARE | 0.6414 | 0.761 | 0.7014 |
| SR-ATAD5 | 0.5 | 0.7632 | 0.7267 |
| SR-HSE | 0.612 | 0.7845 | 0.7362 |
| SR-MMP | 0.7425 | 0.8599 | 0.8156 |
| SR-p53 | 0.518 | 0.7321 | 0.81 |
The performance of Smi2Vec*-LSTM and Smi2Vec-BiGRU below are from the original papers
Evaluation on single- and multi-task classification
Both FDA and Logp are single-task classification problems. Figure 4 shows the comparison of different models on FDA and LogP; our model outperforms the other models on FDA and is second best on LogP. Table 6 presents the performance on different single- and multi-classification tasks. Our model has better performance on BBBP and gets the second-best performance on Tox21 and ToxCast. Pre-trained models achieve relatively better performance on BACE, so Mol2Vec has better performance. Since GraSeq is a fusion learning method of graph and sequence, which is more complicated than our model, performance on BACE, Tox21, and ToxCast of GraSeq is slightly better than that of our model.
Fig. 4.

Comparison of ROC-AUC on FDA and LogP
Table 6.
The performance comparison (ROC-AUC) with state-of-the-art methods on datasets from MoleculeNet. The best performance is denoted by bold
| Dataset | BACE | BBBP | Tox21 | ToxCast |
|---|---|---|---|---|
| Mol2Vec | 0.8137 | 0.8505 | 0.7497 | 0.6678 |
| Seq2seq | 0.7725 | 0.9073 | 0.7976 | 0.7107 |
| Seq3seqFP | 0.7725 | 0.9073 | 0.7107 | − |
| GraSeq | 0.8382 | 0.9426 | 0.8195 | 0.733 |
| TranGRU | 0.7896 | 0.9772 | 0.8126 | 0.7126 |
To evaluate the performance of TranGRU comprehensively, we also present a comparison of the metric of accuracy on LogP. We report the accuracy given in their paper in Table 7. TranGRU has the best performance among the methods.
Table 7.
Comparison of the accuracy between TranGRU and the state-of-the-art sequence models on LogP. The best performance is denoted by bold
Analysis and discussion
Analysis of the layer depth
To compare the performance of different encoder depths, we set the number of layers of TranGRU from 1 to 12. Figure 5 presents the variation of accuracy and ROC-AUC on BBBP and LogP. The change in ROC-AUC is slight on BBBP, while the variation in accuracy is relatively larger. The performance tendency on LogP is the same as that on BBBP. Figure 5 also shows that when the layer number is set to 6, TranGRU has the best performance on both BBBP and LogP. When the sizes of the datasets are relatively small, such as BBBP, the influence of the layer depth is not significant.
Fig. 5.

Variation of ROC-AUC on BBBP and LogP with deepening layers
Analysis of the parameters and performance
Table 8 shows the performance results of accuracy and ROC-AUC on BBBP and LogP. According to the trade-off between the performance and the size of the datasets, the default layer number of TranGRU is set to 6.
Table 8.
Comparison of the parameters and performance of TranGRU on BBBP and LogP
| Model | Layers | att-heads | Accuracy | ROC-AUC | Accuracy | ROC-AUC |
|---|---|---|---|---|---|---|
| BBBP | BBBP | LogP | LogP | |||
| TranGRU | 6 | 4 | 0.9534 | 0.9799 | 0.941 | 0.984 |
| TranGRU(big) | 12 | 4 | 0.9134 | 0.9746 | 0.901 | 0.974 |
Evaluation on an AI cures open challenge for drug discovery related to COVID-19
We also use ecoli, a dataset from AI Cures, which is an open challenge on drug discovery aiming at discovering new antibiotics to cure secondary lung infections caused by COVID-19. The entire dataset consists of 2,335 chemical molecules. The prediction target is to determine whether a molecule has antibacterial activity, or can inhibit Pseudomonas aeruginosa, which is a bacterium leading to secondary lung infection of COVID-19 patients [54].
Table 9 is a comparison of the test ROC-AUC values on ecoli, where the results of AdvProp and Chemprop ++ are cited from [54]. Among 27 teams participating in the open challenge, AdvProp obtains the best performance, and Chemprop ++ is the second best. Compared with the performances of the participating teams, the test ROC-AUC of our approach is better than that of Chemprop ++ but does not surpass that of AdvProp.
Table 9.
Comparison performance on ecoli by the metric of test ROC-AUC
Conclusions
We propose TranGRU to enhance the ability to capture both the local and global information of molecules. It is a simple and effective approach for molecular property prediction that integrates BiGRU into the Transformer encoder. We adaptively fuse both features via a gate mechanism and then feed them into the next encoder layer. Experiments on different classification tasks show that TranGRU significantly outperforms state-of-the-art methods on BBBP, FDA, LogP, and Tox21 and achieves comparable performance on BACE and ToxCast. We also make a comparison on ecoli, and the experimental results show that TranGRU surpasses the second best method from the AI Cures open challenge evaluated according to the test ROC-AUC. Furthermore, the performance of TranGRU may be further improved to some extent by deepening the layers with a suitable mechanism.
TranGRU is a sequence encoder to extract molecular representations, which not only can be used for classification tasks for molecular property prediction but also can be used for regression tasks. In addition, TranGRU can be used as the feature extractor of molecules in the downstream tasks. In future work, we will apply TranGRU to these scenarios, as well as explore improving the performance of modeling three-dimensional molecular data for molecular property prediction.
Biographies
Jing Jiang
received her B.S. degree from School of Information Engineering at North China University of Water Resources and Electric Power in 2011, M.S. degree of Computer Science and Technology at Northwest Minzu University in 2014. She is currently a PhD candidate of School of Information Science & Engineering at Lanzhou University. Her research interests are concentrated in cheminformatics, complex network, and deep learning.

Ruisheng Zhang
received his B.S. degree from the Department of Mathematics at Lanzhou University in 1983, M.S. degree from the Department of Chemistry at Lanzhou University in 1990, and Ph.D degree from the department of Chemistry at Lanzhou University in 1996. He is currently a Professor of School of Information Science & Engineering at Lanzhou University. His current research interests include cheminformatics, machine learning, service computing and information security.

Jun Ma
received her B.S. degree from the College of Mathematics and Communication at Northwest Normal University in 2004. M.S. degree from School of Information Science and Engineering at Lanzhou University in 2008., and Ph.D. degree from School of Information Science & Engineering at Lanzhou University in 2019. She is currently a. engineer of School of Information Science & Engineering at Lanzhou University. Her current research interests include pattern recognition, and pharmaceutical informatics.

Yunwu Liu
received the B.S. degree from the Department School of Physics at Lanzhou University in 2005, and the M.S. degree from the Department of Electronic and Information Engineering, Lanzhou Jiaotong University in 2011, respectively. He is currently a PhD candidate of School of Information Science & Engineering at Lanzhou University. The main research fields are cheminformatics and complex networks.

Enjie Yang
received the B.S. degree from the College of Electronic and Information Engineering (CEIE) of Tongji University, Shanghai, China, in 2017. He is currently pursuing the M.S. degree in School of Information Science & Engineering, Lanzhou University, Gansu, China. The main research fields are machine learning, computing chemistry and computing biology.

Shikang Du
received the B.S. degree from the College of Computer Science and Engineering, Northwest Normal University, in 2019 and the M.S. degree from School of Information Science & Engineering, Lanzhou University, in 2022. He is currently pursuing the Ph.D. degree with College of Earth and Environmental Sciences at Lanzhou University. His current research interests include machine learning, time series forecasting, and meteorological big data.

Zhili Zhao
received his M.S. degree from School of Information Science & Engineering at Lanzhou University in 2009, and Ph.D. degree from Free University of Berlin, Germany in 2014. He is currently an Associate Professor of School of Information Science & Engineering at Lanzhou University. His current research interests include complex network analysis and machine learning.

Yongna Yuan
received her B.S. degree from the Department of Chemistry at Hebei Normal University in 2005, Ph.D degree from the Department of Chemistry at both Lanzhou University in 2010 and Mancheter University in 2012. She is currently a Associate Professor of School of Information Science & Engineering at Lanzhou University. Her current research interests include RNA force field development, machine learning.

Appendix A: Supporting information available
A.1 Details of the datasets
The datasets used in the experiments are chosen from MoleculeNet [1] and ZINC [47]. Table 10 gives the details.
Fig. 6.

The graphic abstract of TranGRU. The red dotted box indicates the main component of TranGRU. We integrate BiGRU into the Transformer encoder, as well as self-attention to focus on the local and global information of molecules
A.2 Mehtods of splitting the datasets
The details of the dataset splitting methods are given in Table 11. BACE, BBBP, Tox21, ToxCast, and HIV are obtained from MoleculeNet [1], Lop and FDA are obtained from ZINC [47]. We split the datasets into training, development, and test subsets in a ratio of 8/1/1 by random or scaffold splitting methods.
A.3 The graphic abstract of the paper
Author Contributions
Jing Jiang: Writing, Data analysis, and Research design; Ruisheng Zhang: Supervision. All authors read the final manuscript and gave some suggestions for revision.
Funding
This work has been supported by the research project funded by the Natural Science Foundation of Gansu Province, China (Grant No. 20JR10RA613, No. 21JR7RA460).
Declarations
Conflict of Interests
The authors declare that they have no conflicts of interest.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Jing Jiang, Email: jiangj2019@lzu.edu.cn.
Ruisheng Zhang, Email: zhangrs@lzu.edu.cn.
Jun Ma, Email: junma@lzu.edu.cn.
Yunwu Liu, Email: liuyw19@lzu.edu.cn.
Enjie Yang, Email: yangej20@lzu.edu.cn.
Shikang Du, Email: dushk19@lzu.edu.cn.
Zhili Zhao, Email: zhaozhl@lzu.edu.cn.
Yongna Yuan, Email: yuanyn@lzu.edu.cn.
References
- 1.Wu Z, Ramsundar B, Feinberg EN, Gomes J, Geniesse C, Pappu AS, Leswing K, Pande V. Moleculenet: a benchmark for molecular machine learning. Chem Sci. 2018;9(2):513–530. doi: 10.1039/C7SC02664A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hu R, Chen J, Zhou L (2022) A transformer-based deep neural network for arrhythmia detection using continuous ecg signals. Computers in Biology and Medicine :105325 [DOI] [PubMed]
- 3.Wu C-K, Zhang X-C, Yang Z-J, Lu A-P, Hou T-J, Cao D-S. Learning to smiles: ban-based strategies to improve latent representation learning from molecules. Brief Bioinform. 2021;22(6):327. doi: 10.1093/bib/bbab327. [DOI] [PubMed] [Google Scholar]
- 4.Xu T, Xu M, Zhu W, Chen CZ, Zhang Q, Zheng W, Huang R. Efficient identification of anti-sars-cov-2 compounds using chemical structure-and biological activity-based modeling. J Med Chem. 2022;65:4590–4599. doi: 10.1021/acs.jmedchem.1c01372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chuang KV, Gunsalus LM, Keiser MJ. Learning molecular representations for medicinal chemistry: miniperspective. J Med Chem. 2020;63(16):8705–8722. doi: 10.1021/acs.jmedchem.0c00385. [DOI] [PubMed] [Google Scholar]
- 6.Weininger D. Smiles, a chemical language and information system. 1. introduction to methodology and encoding rules. J Chem Inf Comput Sci. 1988;28(1):31–36. doi: 10.1021/ci00057a005. [DOI] [Google Scholar]
- 7.Weininger D, Weininger A, Weininger JL. Smiles. 2. algorithm for generation of unique smiles notation. J Chem Inf Comput Sci. 1989;29:97–101. doi: 10.1021/ci00062a008. [DOI] [Google Scholar]
- 8.Gilmer J, Schoenholz SS, Riley PF, Vinyals O, Dahl GE (2017) Neural message passing for quantum chemistry. In: International conference on machine learning, PMLR, pp 1263–1272
- 9.Zhang X, Wang S, Zhu F, Xu Z, Wang Y, Huang J (2018) Seq3seq fingerprint: towards end-to-end semi-supervised deep drug discovery. In: Proceedings of the 2018 ACM international conference on bioinformatics, computational biology, and health informatics, pp 404–413
- 10.Winter R, Montanari F, Noé F, Clevert D-A. Learning continuous and data-driven molecular descriptors by translating equivalent chemical representations. Chem Sci. 2019;10(6):1692–1701. doi: 10.1039/C8SC04175J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li P, Wang J, Qiao Y, Chen H, Yu Y, Yao X, Gao P, Xie G, Song S. An effective self-supervised framework for learning expressive molecular global representations to drug discovery. Brief Bioinform. 2021;22(6):109. doi: 10.1093/bib/bbab109. [DOI] [PubMed] [Google Scholar]
- 12.Li P, Li Y, Hsieh C-Y, Zhang S, Liu X, Liu H, Song S, Yao X. Trimnet: learning molecular representation from triplet messages for biomedicine. Brief Bioinform. 2021;22(4):266. doi: 10.1093/bib/bbaa266. [DOI] [PubMed] [Google Scholar]
- 13.Cho K, van Merrienboer B, Bahdanau D, Bengio Y (2014) On the properties of neural machinetranslation: encoder-decoder approaches. In: Proceedings of SSST 2014, pp 103–111
- 14.Vaswani A, Shazeer N, Parmar N, Uszkoreit J, Jones L, Gomez AN, Kaiser ł, Polosukhin I (2017) Attention is all you need. Advances in neural information processing systems 30:
- 15.Xu Z, Wang S, Zhu F, Huang J (2017) Seq2seq fingerprint: an unsupervised deep molecular embedding for drug discovery. In: Proceedings of the 8th ACM international conference on bioinformatics, computational biology, and health informatics, pp 285–294
- 16.Lin X, Quan Z, Wang Z-J, Huang H, Zeng X. A novel molecular representation with bigru neural networks for learning atom. Briefings in bioinformatics. 2020;21(6):2099–2111. doi: 10.1093/bib/bbz125. [DOI] [PubMed] [Google Scholar]
- 17.Goh GB, Hodas NO, Siegel C, Vishnu A (2018) Smiles2vec: an interpretable general-purpose deep neural network for predicting chemical properties. ICLR
- 18.Lv Q, Chen G, Zhao L, Zhong W, Yu-Chian Chen C. Mol2context-vec: learning molecular representation from context awareness for drug discovery. Brief Bioinform. 2021;22(6):317. doi: 10.1093/bib/bbab317. [DOI] [PubMed] [Google Scholar]
- 19.Ying C, Cai T, Luo S, Zheng S, Ke G, He D, Shen Y, Liu T-Y (2021) Do transformers really perform badly for graph representation? Adv Neural Inf Process Syst 34:
- 20.Wang Y, Chen X, Min Y, Wu J (2021) Molcloze: a unified cloze-style self-supervised molecular structure learning model for chemical property prediction. In: 2021 IEEE international conference on bioinformatics and biomedicine (BIBM), IEEE, pp 2896–2903
- 21.Chen D, Gao K, Nguyen DD, Chen X, Jiang Y, Wei G-W, Pan F. Algebraic graph-assisted bidirectional transformers for molecular property prediction. Nat Commun. 2021;12(1):1–9. doi: 10.1038/s41467-021-23720-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tran KM, Bisazza A, Monz C (2016) Recurrent memory networks for language modeling. In: Proceedings of the 2016 conference of the north american chapter of the association for computational linguistics: human language technologies, pp 321–331
- 23.Hao J, Wang X, Yang B, Wang L, Zhang J, Tu Z (2019) Modeling recurrence for transformer. In: Proceedings of the 2019 conference of the North American chapter of the association for computational linguistics: human language technologies, volume 1 (Long and Short Papers), pp 1198–1207
- 24.Chen MX, Firat O, Bapna A, Johnson M, Macherey W, Foster G, Jones L, Schuster M, Shazeer N, Parmar N et al (2018) The best of both worlds: combining recent advances in neural machine translation. In: Proceedings of the 56th annual meeting of the association for computational linguistics (Volume 1: Long Papers), pp 76–86
- 25.Li X, Fourches D. Smiles pair encoding: a data-driven substructure tokenization algorithm for deep learning. J Chem Inf Model. 2021;61(4):1560–1569. doi: 10.1021/acs.jcim.0c01127. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Z, Guan J, Zhou S. Fragat: a fragment-oriented multi-scale graph attention model for molecular property prediction. Bioinformatics. 2021;37(18):2981–2987. doi: 10.1093/bioinformatics/btab195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guvench O. Computational functional group mapping for drug discovery. Drug Disc Today. 2016;21(12):1928–1931. doi: 10.1016/j.drudis.2016.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chakrabarty A, Pandit OA, Garain U (2017) Context sensitive lemmatization using two successive bidirectional gated recurrent networks. In: Proceedings of the 55th annual meeting of the association for computational linguistics (volume 1: Long Papers), pp 1481–1491
- 29.Wang S, Guo Y, Wang Y, Sun H, Huang J (2019) Smiles-bert: large scale unsupervised pre-training for molecular property prediction. In: Proceedings of the 10th ACM international conference on bioinformatics, computational biology and health informatics, pp 429–436
- 30.Guo Z, Yu W, Zhang C, Jiang M, Chawla NV (2020) Graseq: graph and sequence fusion learning for molecular property prediction. In: Proceedings of the 29th ACM international conference on information & knowledge management, pp 435–443
- 31.Hochreiter S, Schmidhuber J. Long short-term memory. Neural Comput. 1997;9(8):1735–1780. doi: 10.1162/neco.1997.9.8.1735. [DOI] [PubMed] [Google Scholar]
- 32.Zhang F, Hu C, Yin Q, Li W, Li H-C, Hong W. Multi-aspect-aware bidirectional lstm networks for synthetic aperture radar target recognition. IEEE Access. 2017;5:26880–26891. doi: 10.1109/ACCESS.2017.2773363. [DOI] [Google Scholar]
- 33.Min S, Lee B, Yoon S. Deep learning in bioinformatics. Brief Bioinforma. 2017;18(5):851–869. doi: 10.1093/bib/bbw068. [DOI] [PubMed] [Google Scholar]
- 34.Berrar D, Dubitzky W. Deep learning in bioinformatics and biomedicine. Brief Bioinforma. 2021;22(2):1513–1514. doi: 10.1093/bib/bbab087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jaeger S, Fulle S, Turk S. Mol2vec: unsupervised machine learning approach with chemical intuition. J Chem Inf Model. 2018;58(1):27–35. doi: 10.1021/acs.jcim.7b00616. [DOI] [PubMed] [Google Scholar]
- 36.Quan Z, Lin X, Wang Z-J, Liu Y, Wang F, Li K (2018) A system for learning atoms based on long short-term memory recurrent neural networks. In: 2018 IEEE international conference on bioinformatics and biomedicine (BIBM), IEEE, pp 728–733
- 37.Woźniak M, Siłka J, Wieczorek M, Alrashoud M. Recurrent neural network model for iot and networking malware threat detection. IEEE Trans Ind Inform. 2020;17(8):5583–5594. doi: 10.1109/TII.2020.3021689. [DOI] [Google Scholar]
- 38.Woźniak M, Wieczorek M, Siłka J, Połap D. Body pose prediction based on motion sensor data and recurrent neural network. IEEE Trans Ind Inform. 2020;17(3):2101–2111. doi: 10.1109/TII.2020.3015934. [DOI] [Google Scholar]
- 39.Siłka J, Wieczorek M, Woźniak M. Recurrent neural network model for high-speed train vibration prediction from time series. Neural Comput Applic. 2022;34:13305–13318. doi: 10.1007/s00521-022-06949-4. [DOI] [Google Scholar]
- 40.Tay Y, Dehghani M, Bahri D, Metzler D (2020) Efficient transformers: a survey. ACM Computing Surveys (CSUR)
- 41.Parikh AP, Täckström O, Das D, Uszkoreit J (2016) A decomposable attention model for natural language inference. In: EMNLP
- 42.Gaiński P, Maziarka ł, Danel T, Jastrzebski S (2022) Huggingmolecules: an open-source library for transformer-based molecular property prediction (student abstract). In: Proceedings of the AAAI conference on artificial intelligence, vol 36. pp 12949–12950
- 43.Kim H, Na J, Lee WB. Generative chemical transformer: neural machine learning of molecular geometric structures from chemical language via attention. J Chem Inf Model. 2021;61(12):5804–5814. doi: 10.1021/acs.jcim.1c01289. [DOI] [PubMed] [Google Scholar]
- 44.Xu J, Sun X, Zhang Z, Zhao G, Lin J (2019) Understanding and improving layer normalization. In: Proceedings of the 33rd international conference on neural information processing systems, pp 4381–4391
- 45.He K, Zhang X, Ren S, Sun J (2016) Deep residual learning for image recognition. In: Proceedings of CVPR, pp 770–778
- 46.Bengio Y, Simard P, Frasconi P. Learning long-term dependencies with gradient descent is difficult. IEEE Trans Neural Netw. 1994;5(2):157–166. doi: 10.1109/72.279181. [DOI] [PubMed] [Google Scholar]
- 47.Sterling T, Irwin JJ. Zinc 15–ligand discovery for everyone. J Chemical information and modeling. 2015;55(11):2324–2337. doi: 10.1021/acs.jcim.5b00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Subramanian G, Ramsundar B, Pande V, Denny RA. Computational modeling of β-secretase 1 (bace-1) inhibitors using ligand based approaches. J Chem Inf Model. 2016;56(10):1936–1949. doi: 10.1021/acs.jcim.6b00290. [DOI] [PubMed] [Google Scholar]
- 49.Martins IF, Teixeira AL, Pinheiro L, Falcao AO. A bayesian approach to in silico blood-brain barrier penetration modeling. J Chem Inf Model. 2012;52(6):1686–1697. doi: 10.1021/ci300124c. [DOI] [PubMed] [Google Scholar]
- 50.Tox21 (2014) Data Challenge. https://tripod.nih.gov/tox21/challenge/(Accessed:2022-07-28)
- 51.Richard AM, Judson RS, Houck KA, Grulke CM, Volarath P, Thillainadarajah I, Yang C, Rathman J, Martin MT, Wambaugh JF, et al. Toxcast chemical landscape: paving the road to 21st century toxicology. Chem Res Toxicol. 2016;29(8):1225–1251. doi: 10.1021/acs.chemrestox.6b00135. [DOI] [PubMed] [Google Scholar]
- 52.Hamilton W, Ying Z, Leskovec J (2017) Inductive representation learning on large graphs. Adv Neural Inf Process Syst 30:
- 53.Mikolov T, Sutskever I, Chen K, Corrado GS, Dean J. Distributed representations of words and phrases and their compositionality. Adv Neural Inf Process Syst. 2013;2:3111–3119. [Google Scholar]
- 54.Wang Z, Liu M, Luo Y, Xu Z, Xie Y, Wang L, Cai L, Qi Q, Yuan Z, Yang T, et al. Advanced graph and sequence neural networks for molecular property prediction and drug discovery. Bioinformatics. 2022;38(9):2579–2586. doi: 10.1093/bioinformatics/btac112. [DOI] [PubMed] [Google Scholar]