

Abstract
Systemic iron homeostasis is regulated by the hepatic hormone hepcidin to balance meeting iron requirements while limiting toxicity from iron excess. Iron-mediated induction of bone morphogenetic protein (BMP) 6 is a central mechanism for regulating hepcidin production. Liver endothelial cells are the main source of endogenous BMP6, but how they sense iron to modulate BMP6 transcription and thereby hepcidin is uncertain. Here, we investigate the role of endothelial cell transferrin receptor 1 (TFR1) in iron uptake, BMP6 regulation, and systemic iron homeostasis using primary liver endothelial cell cultures and endothelial Tfrc (encoding TFR1) knockout mice. We show that intracellular iron regulates Bmp6 expression in a cell-autonomous manner, and TFR1 mediates iron uptake and Bmp6 expression by holo-transferrin in primary liver endothelial cell cultures. Additionally, endothelial Tfrc knockout mice exhibit altered iron homeostasis compared with littermate controls when fed a limited iron diet, as evidenced by increased liver iron and inappropriately low Bmp6 and hepcidin expression relative to liver iron. However, endothelial Tfrc knockout mice have a similar iron phenotype compared to littermate controls when fed an iron-rich standard diet. Finally, ferritin and non-transferrin bound iron (NTBI) are additional sources of iron that mediate Bmp6 induction in primary liver endothelial cell cultures via TFR1-independent mechanisms. Together, our data demonstrate a minor functional role for endothelial cell TFR1 in iron uptake, BMP6 regulation, and hepatocyte hepcidin regulation under iron limiting conditions, and suggest that ferritin and/or NTBI uptake by other transporters have a dominant role when iron availability is high.
Keywords: Iron sensing, hepcidin, liver, transport, hemochromatosis, anemia
Introduction
Systemic iron homeostasis is regulated by the hormone hepcidin to provide sufficient iron for erythropoiesis and other vital cellular processes, while limiting toxicity from iron excess. Produced mainly by hepatocytes, hepcidin causes degradation of its receptor, ferroportin,1 to block iron export to plasma from intestinal cells, iron-recycling macrophages, and other sources, thereby lowering plasma iron levels. Hepcidin is suppressed by iron deficiency, erythropoiesis, and pregnancy to increase iron availability,2,3 while it is induced by iron, infection, and inflammation2,4,5 to prevent iron overload and accessibility to pathogens. Many of the signals that regulate hepcidin, including iron, act by modulating the bone morphogenetic protein (BMP)-small mothers against decapentaplegic (SMAD) pathway.6–8
Mechanistically, iron loading induces the production of BMP6 and, to a lesser extent, BMP2,9–12 which bind to the BMP receptor complex on hepatocytes to activate downstream SMAD signaling and induce hepcidin transcription. The induction of BMP6 and BMP2 expression by iron overload occurs only in the liver, as a high iron diet does not induce BMP6 or BMP2 expression in other organs.9,11,13–16 Endothelial cells are the predominant source of endogenous BMP ligands for hepcidin regulation, since mice with an endothelial knockout of Bmp6 or Bmp2 exhibit hepcidin deficiency and iron overload.9,13,15 Notably, mice with a combined endothelial knockout of Bmp6 and Bmp2 exhibit an identical iron phenotype as single knockout mice,12 suggesting that BMP6 and BMP2 predominantly function together in hepcidin and iron homeostasis regulation, possibly as a heterodimeric ligand. Quantitative copy number analysis demonstrates that BMP6 is much less abundant than BMP2 in liver endothelial cells (LECs)9 suggesting that regulation of BMP6 is the rate limiting factor.
Despite its central role, how iron is sensed by LECs to regulate BMP ligand production is incompletely understood. Recent data suggest that under iron excess conditions, mitochondrial reactive oxygen species activate the antioxidant response transcription factor NRF2,17 contributing to BMP6 regulation in LECs. The mechanisms by which LECs take up iron to regulate NRF2 activity and other BMP regulatory pathways remain unknown. In physiologic conditions, plasma iron circulates bound to the iron carrier protein transferrin and is taken up by transferrin receptor 1 (TFR1)-mediated endocytosis. TFR1 is ubiquitously expressed and post-transcriptionally regulated by intracellular iron status via the iron-regulatory protein system,18 resulting in increased TFR1 under low iron conditions and decreased TFR1 under high iron conditions. Under high iron conditions, quantities of iron may exceed the iron binding capacity of transferrin, leading to the presence of non-transferrin bound iron (NTBI), which can be taken up by cells via different uptake pathways.19 The iron storage protein ferritin has also been proposed to act as a delivery system for iron under some biological conditions,20 but the uptake mechanisms are less well understood.
In this study, we used primary LECs to investigate whether iron has a cell-autonomous role to regulate Bmp6 expression, whether transferrin-bound iron is utilized by LECs to control Bmp6 expression, and the functional role of endothelial TFR1. We also generated mice that lack Tfrc in endothelial cells to study the role of endothelial TFR1 in iron sensing and hepcidin regulation in vivo. Finally, we investigated whether NTBI and ferritin also function as iron sources to regulate Bmp6 expression in LECs.
Methods
Animals
Animal experiments were approved by the Institutional Animal Care and Use Committee at Massachusetts General Hospital (MGH). To generate mice with deletion of Tfrc in endothelial cells, C57BL/6J mice harboring LoxP-flanked alleles of Tfrc (Jackson Laboratory 028363) were bred to C57BL/6N mice expressing Cre recombinase under the control of an endothelial cell-specific Stabilin-2 (Stab2) promoter21 (generously provided by Cyrill Géraud, Mannheim, Germany) to produce Tfrcfl/fl;Stab2-Cre+ and Tfrcfl/fl;Stab2-Cre−littermate controls. For Cre reporter studies, Stab2-Cre+ mice were bred to B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J (Jackson Laboratory 007909) Cre reporter mice.
Mice were housed in a temperature and humidity-controlled barrier facility on a 12-hour light-dark schedule and fed ad libitum with standard rodent chow (Prolab RMH 3000; LabDiet, 380 ppm iron). To model limited iron availability, mice were fed purified iron-limited diet (Envigo-Teklad TD190546, 20 ppm iron from ferric citrate) from 3–12 weeks of age. Iron content of the purified diets was confirmed by ICP-MS (Veterinary Diagnostic Laboratory). For ferritin injections, juvenile 5-week-old mice received a single intraperitoneal injection of 50 μg/g holo-ferritin (Sigma F4503) or solvent (PBS) for 6 hours.
Cell culture
Freshly isolated primary LECs were purchased from Cell Biologics (CD1017) and cultured on gelatin in complete growth media containing 5% fetal calf serum (Cell Biologics M1168) at 37 °C in a 5% CO2 95% air atmosphere. BMP6 induction experiments were performed up to passage 4–8 to maintain iron-mediated BMP6 responsiveness.
For iron depletion experiments, cells were treated with complete media supplemented with 100 μM deferoxamine (Sigma D9533) for 24 hours. For iron loading experiments, cells were serum starved for 8–16 hours prior to treatment with 200 μg/mL ferric ammonium citrate (Sigma F5879) for 6–24 hours, 30 μM holo-transferrin (Sigma T0665) or apo-transferrin (Sigma T1147) for 24 hours, or 100 nM holo-ferritin (Sigma F4503) or apo-ferritin (Sigma 178440) for 24 hours.
For gene knockdown experiments, cells were transfected with 40 nM non-targeting control siRNA (Invitrogen 4390846) or siRNA targeting Tfrc (Invitrogen s75457) using Lipofectamine RNAiMAX (Invitrogen) per the manufacturer’s instructions for 48 hours prior to iron treatment as described above. Gene knockdown was confirmed by qRT-PCR and immunoblotting.
Nonheme iron measurements
Serum was collected by centrifugation of clotted blood in microtainer tubes (Beckton Dickinson). Nonheme iron and unsaturated iron binding capacity were measured by colorimetric assay (Pointe Scientific 17504) to calculate transferrin saturation per the manufacturer’s instructions. Tissue nonheme iron concentrations (in μg/g wet weight) were determined as described previously.22
Complete blood counts and ELISA
Complete blood counts were measured on whole blood collected in K2-EDTA coated microtainer tubes (Beckton Dickinson) at the MGH Center for Comparative Medicine Clinical Pathology Laboratory. Serum erythropoietin (R&D Systems MEP00B) and erythroferrone (Intrinsic LifeSciences ERF-200) were measured by ELISA per the manufacturer’s instructions.
Liver cell isolation and genomic analysis
LECs and Kupffer cells were sorted from 8-week-old mice by fluorescence-activated cell sorting (FACS) as described previously15 using the monoclonal antibodies listed in Supplemental Table 1. Cell sorting was performed on a FACSAria II (BD Biosciences) by the Harvard Stem Cell Institute-Center for Regenerative Medicine Flow Cytometry Core Facility at MGH.
Hepatocytes were isolated by density gradient centrifugation as described previously.15
Genomic DNA was extracted from isolated cells using a DNeasy Blood and Tissue Kit (Qiagen) and analyzed by PCR using the primers listed in Supplemental Table 2.
Magnetic-activated cell sorting (MACS)
Whole liver was incubated in digestion buffer (450 U/ml collagenase I, 125 U/ml collagenase XI, 60 U/ml Dnase I, 60 U/ml hyaluronidase in DMEM) for 30 min at 37°C with constant shaking. Single cell suspension was prepared by manual dissociation using fine scissors and filtration through 70- and 40-μm cell strainers. LECs were isolated by magnetic separation using CD31 microbeads (Miltenyi 130-097-418) following the manufacturer’s instructions.
Gene expression quantification by RT-PCR
Frozen mouse tissues or cell cultures were homogenized using Qiashredder (Qiagen) and total RNA was isolated using Pure-Link RNA Mini Kit (Invitrogen) or RNeasy Micro Kit (Qiagen) following the manufacturer’s instructions. One μg (mouse tissue) and 2 μg (cell cultures) of RNA was reverse transcribed using the High-Capacity RNA-to-cDNA Kit (Applied Biosystems). In FACS experiments, total RNA was isolated using the PicoPure RNA Isolation Kit (Thermo Fisher Scientific), and cDNA was synthesized using the SuperScript VILO cDNA Synthesis Kit (Invitrogen). Quantitative real-time PCR was performed on cDNA using the PowerUp SYBR Green Master Mix on the QuantStudio3 real-time PCR system (Applied Biosystems). Relative quantities were determined by the standard curve method, transcript levels were normalized to Rpl19, and the average of Cre− male mice (for in vivo experiments) or control groups (for in vitro experiments) were set to 1. The primer sequences are listed in Supplemental Table 2.
Western blotting
Liver pieces and cells were homogenized in RIPA lysis buffer with protease and phosphatase inhibitor cocktail (Santa Cruz 24948) and protein concentration was measured by DC-protein assay (Bio-Rad). Proteins were separated by SDS-PAGE and transferred to nitrocellulose membranes, which were blocked in 5% w/v dried nonfat milk or bovine serum albumin in TBS with 0.05% Tween-20 and incubated with primary antibodies in blocking buffer overnight at 4 °C, followed by HRP-conjugated secondary antibodies (Supplemental Table 1). Protein blots were visualized by chemiluminescence using the GeneSys G:Box Mini 6 imaging system and quantification software (Syngene).
Immunofluorescence microscopy
Livers from 8-week-old mice were fixed in periodate-lysine-paraformaldehyde overnight at 4°C, embedded in optimal cutting temperature media (Tissue Tek), and cryosectioned into seven μm frozen sections. Sections were rehydrated in PBS, blocked with 2% donkey serum with 0.5% Tween 20 and 1% BSA, and stained overnight with CD31, F4/80, or desmin antibodies, followed by FITC or AF488 secondary antibodies (Supplemental Table 1). Sections were mounted with antifade medium with DAPI (Vectashield). Images were acquired by Nikon A1R confocal laser scanning microscope at the MGH Program in Membrane Biology Microscopy Core and adjusted using Volocity software. Recombination efficiency was quantitated by counting the percent of stellate cells, Kupffer cells, and endothelial cells (determined by desmin, F4/80, or CD31 staining, respectively) that were positive for tomato fluorescence in Rosa26tdTomato;Stab2-iCre reporter mice. In total, 45 to 600 cells were counted from 3 mice per group.
Statistical analyses
Statistical analysis was performed using Prism 9 (GraphPad). Data are presented as individual values with bars representing the mean ± SEM. Shapiro-Wilk test was used to test normality. For comparisons between two groups, two-tailed student’s t-test was used for normally distributed values or Mann–Whitney U test for non-normally distributed values. For comparisons among more than two groups, statistical differences were determined by one- or two-way ANOVA followed by Holm-Sidak test. A P-value less than 0.05 was considered significant.
Results
Intracellular iron controls Bmp6 expression in primary liver endothelial cells
To investigate iron-dependent regulation of Bmp6 expression in LECs, we used primary mouse LEC cultures and manipulated their intracellular iron content. To induce iron deficiency, LECs were treated with the iron chelator deferoxamine (DFO) compared with solvent alone. To model iron loading, LECs were treated with physiologic levels of transferrin-bound iron (holo-TF) compared with iron-poor transferrin (apo-TF). Changes in cellular iron status were assessed by expression of iron storage protein ferritin (induced by iron) and Tfrc mRNA (reduced by iron). As expected, ferritin expression was lower in cells treated with DFO and higher in cells treated with holo-TF (Figure 1A), whereas Tfrc expression was increased by DFO and decreased by holo-TF (Figure 1B) compared with their respective controls. Importantly, iron depletion by DFO suppressed Bmp6 expression, and iron loading by holo-TF induced Bmp6 expression (Figure 1C). No significant changes in Bmp2 expression were detected in this model (Supplemental Figure 1). These results demonstrate that intracellular iron content plays a cell-autonomous role in regulating Bmp6 expression and that holo-TF functions as an iron source for Bmp6 regulation in primary mouse LECs.
Figure 1. Intracellular iron regulates Bmp6 transcription in primary mouse liver endothelial cells.
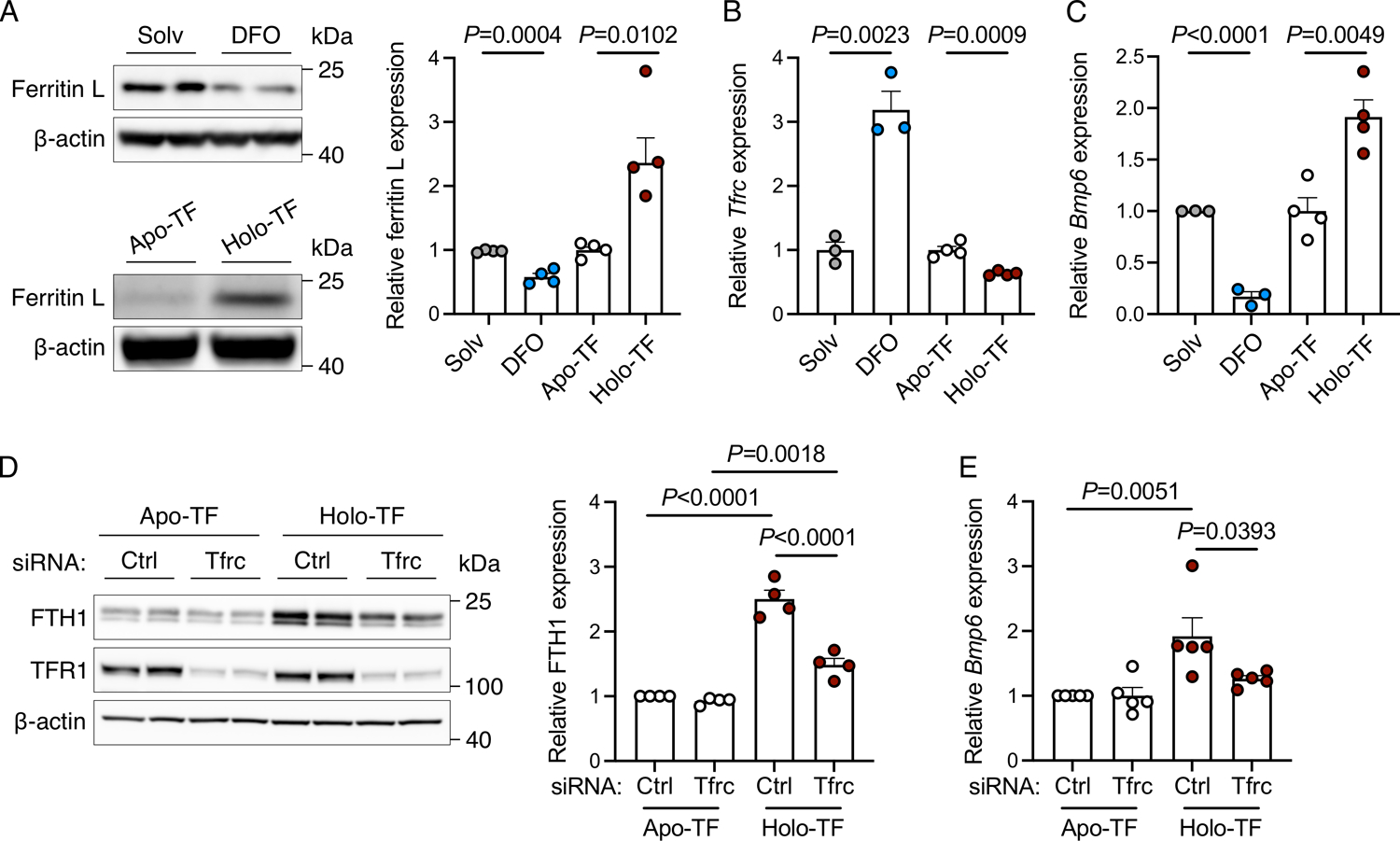
(A-C) Primary liver endothelial cells were treated with solvent (Solv) or iron-chelator deferoxamine (DFO, 100 μM) for 24 hours, or serum-starved for 8 hours before treatment with apo-or holo-transferrin (TF, 30 μM) for 24 hours and analyzed for: (A) Western blot and quantitation of iron storage protein ferritin light (L) chain relative to ß-actin as a loading control; (B-C) qRT-PCR of Tfrc and Bmp6 relative to Rpl19 expression. (D-E) Primary mouse liver endothelial cells were treated with 40 nM control (Ctrl) siRNA or siRNA targeting Tfrc for 48 hours, serum-starved for 8 hours, treated with apo- or holo-TF in serum-free media for 24 hours, and analyzed for: (D) Western blot and quantitation of ferritin heavy chain (FTH1) relative to ß-actin; (E) qRT-PCR of Bmp6 relative to Rpl19 expression. Representative blots from N=4 independent experiments shown. Bar graphs represent mean ± SEM with individual points indicating the number of replicates. Statistical differences were determined by (A-C) two-tailed Student’s t-test or (D-E) one-way ANOVA with Holm-Sidak method of multiple comparisons.
Since TFR1 is the main transporter for holo-TF uptake, we evaluated the impact of siRNA knockdown of endogenous TFR1 compared with control siRNA on ferritin and Bmp6 induction by holo-TF versus apo-TF in primary LECs. As validation of the knockdown, treatment with Tfrc siRNA reduced both Tfrc mRNA levels (Supplemental Figure 2) and TFR1 protein expression (Figure 1D). As expected, in cells treated with control siRNA, holo-TF treatment induced ferritin protein expression (Figure 1D) and reduced Tfrc mRNA expression (Supplemental Figure 2), indicative of cellular loading. However, holo-TF treatment did not significantly reduce TFR1 protein expression at the time point studied (Figure 1D), likely due to the long half-life of TFR1.23,24 Knocking down TFR1 blunted the ferritin induction response (Figure 1D), demonstrating that TFR1 knockdown inhibited iron uptake from holo-TF. However, there was still a modest residual ferritin upregulation by holo-TF in TFR1 knockdown cells, likely attributable to incomplete (~80%) TFR1 knockdown (Figure 1D and Supplemental Figure 2). Importantly, TFR1 knockdown blunted not only ferritin induction, but also the induction of Bmp6 expression by holo-TF (Figure 1E). Taken together, these data show that TFR1 controls iron uptake from holo-TF and ultimately Bmp6 expression.
Validation of endothelial Tfrc knockout mice
In order to assess the contribution of TFR1 to endothelial iron sensing and hepcidin regulation in mouse models, we generated endothelial Tfrc knockout mice (Tfrcfl/fl;Stab2-Cre+) and littermate controls (Tfrcfl/fl;Cre-). To validate the model, we analyzed Tfrc mRNA and genomic DNA in isolated hepatocytes, LECs, and Kupffer cells from Tfrcfl/fl;Stab2-Cre+ and Tfrcfl/fl;Cre− mice. Isolation of LECs and Kupffer cells was confirmed by ~200-fold enrichment of Cd45 mRNA in the Kupffer cell fraction and ~40-fold enrichment of Cd146 mRNA in the LEC fraction (Figure 2A–B). Tfrc mRNA was reduced by >80% in LECs, but only ~25% in Kupffer cells, and was not significantly changed in isolated hepatocytes or total liver (Figure 2C). Analysis of genomic DNA showed robust Cre-mediated recombination in LECs as reflected by strong expression of the recombined Tfrc allele and loss of the floxed Tfrc allele (Figure 2D, EC). Only partial Cre-mediated recombination was seen in Kupffer cells where the recombined allele was also detected, but weaker than in LECs, and the floxed allele persisted (Figure 2D, KC). No Cre-mediated recombination was seen in hepatocytes where only the floxed allele was detected (Figure 2D, HC). Given the potential of contamination with other cell types to influence results from the FACS analysis, efficiency and specificity of Cre-mediated recombination was also assessed using Rosa26tdTomato reporter mice, which express endogenous tdTomato fluorescence in cells with Cre activity. Liver sections from Rosa26tdTomato;Stab2-Cre+ mice were stained for cell specific markers (CD31, LECs; F4/80, Kupffer cells; desmin, stellate cells) and analyzed for colocalization with tomato fluorescence signal. Hepatocytes were identified by morphology. Consistent with the results in our isolated liver cell populations, recombination occurred in >99% of LECs, with robust tomato florescence observed throughout the liver endothelium (Figure 2E). In contrast, recombination was observed in only ~25% of Kupffer cells, 0% of stellate cells, and 0% of hepatocytes (Figure 2E). Together, these results validate the efficiency and specificity of the Stab2-Cre-mediated recombination primarily to endothelial cells among liver cell types.
Figure 2. Validation of conditional endothelial-specific Tfrc knockout mice.
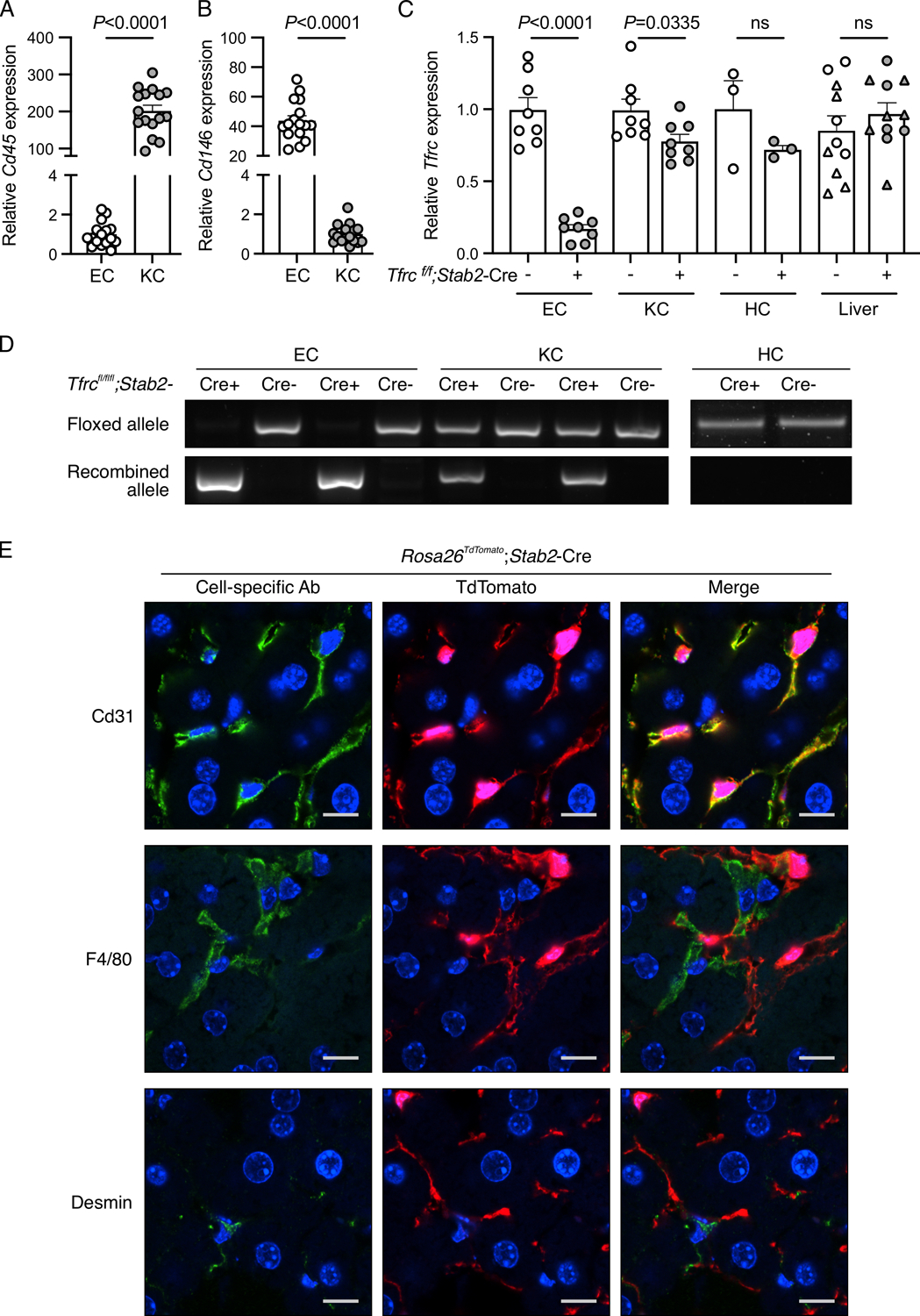
(A-D) Liver endothelial cells (EC) and Kupffer cells (KC) isolated by fluorescence-activated cell sorting, hepatocytes (HC) isolated by density-gradient centrifugation, and total livers from 8–16-week-old Tfrcfl/fl;Stab2-Cre+ and Cre− mice were analyzed for: (A-B) expression of Cd45 and Cd146 relative to Rpl19 by qRT-PCR to assess EC and KC purity; (C) Tfrc relative to Rpl19 by qRT-PCR; and (D) floxed and recombined Tfrc alleles by PCR. For panels (A-B), cells from Cre- and Cre+ mice were combined for analysis. For panel (C), whole liver analysis includes male (circles) and female (triangles) mice. For panel (D), representative gel images from N=8/group (EC, KC) and N=3/group (HC) are shown. Graphs represent mean ± SEM with individual points indicating the number of animals per group. Statistical differences were determined by two-tailed Student’s t-test. (E) Confocal immunofluorescence microscopy images depicting colocalization of cell-specific markers with tdTomato in livers from Rosa26Sortm9-tdTomato;Stab2-Cre+ reporter mice. Liver sections were stained for specific cell populations using CD31 (endothelial cells), F4/80 (Kupffer cells), or desmin (stellate cells) antibodies (green). Cells with Cre recombinase activity are endogenously red because of the removal of a stop codon upstream of the tdTomato fluorescent protein. Nuclei are labeled blue with 4′,6-diamidino-2-phenylindole. Scale bars represent 10 μm. Representative images from N=3 mice/group are shown.
Transferrin receptor 2 (TFR2) is homologous to TFR1 and has also been shown to bind holo-transferrin, albeit at lower affinity.25,26 Although TFR2 is thought to have a predominantly regulatory function to control hepcidin expression and erythropoietin response to holo-TF levels,27–30 TFR2 has also been reported to mediate iron uptake in cell culture systems in the absence of TFR1.31 Thus, we determined if there was compensatory upregulation of TFR2 in the absence of TFR1. We confirmed by immunoblotting that TFR2 is not detected in magnetically sorted LECs from Tfrcfl/fl;Stab2-Cre+ or Tfrcfl/fl-Cre− mouse livers (Supplemental Figure 3).
Endothelial TFR1 does not contribute to iron sensing, BMP6 regulation, and hepcidin regulation in iron-replete conditions
To evaluate the role of TFR1 in endothelial iron sensing and hepcidin regulation in iron-replete conditions, littermate male and female Tfrcfl/fl;Stab2-Cre+ and Tfrcfl/f-Cre− mice were maintained on standard chow (380 ppm iron) and analyzed at 8 weeks. Although many endothelial Cre models also impact hematopoietic cells,15,32 Tfrcfl/fl;Stab2-Cre+ mice did not exhibit anemia or other significant changes in hematologic parameters with the exception of a mild increase in red cell distribution width (RDW) in females (Supplemental Table 3). We also did not observe any differences in serum erythropoietin or splenic expression of GypA or Tfrc between groups (Supplemental Figure 4A–C). Serum erythroferrone levels were below the level of detection. Taken together, these data suggest minimal to no impact on TFR1 activity in erythroid cells in Tfrcfl/fl;Stab2-Cre+ mice. We also did not detect any differences in serum transferrin saturation; liver iron; hepatic expression of Bmp6, Bmp2, Hamp, Id1, Smad7 transcript levels; or gene expression relative to liver iron content (Supplemental Figure 5A–G). Overall, these data show that in young adult mice, endothelial TFR1 does not play a major role in iron sensing, BMP6 regulation or hepcidin regulation in iron-replete conditions.
Functional role of endothelial TFR1 in iron sensing, BMP6 regulation, and hepcidin regulation in iron-limited conditions
Considering that Tfrc expression is suppressed by iron-loading and induced by iron-depletion (Figure 1B), we modeled conditions where iron availability is limited by feeding littermate male and female Tfrcfl/fl;Stab2-Cre+ and Tfrcfl/fl-Cre− mice a purified iron-limited diet (20 ppm iron) until 12 weeks of age. We did not observe major changes in hematologic parameters, with only mild increases in RDW in both sexes of Tfrcfl/fl;Stab2-Cre+ mice, mild reduction in mean cell volume in Tfrcfl/fl;Stab2-Cre+ males, and mild reduction in mean cell hemoglobin in Tfrcfl/fl;Stab2-Cre+ females compared with sex-matched Cre−controls (Supplemental Table 4). Importantly, we did not observe any changes in serum erythropoietin levels (Supplemental Figure 6A) or serum erythroferrone levels, which were below the limit of detection in all sexes and genotypes on the iron-limited diet. We likewise did not detect any differences in spleen or bone marrow GypA or Tfrc (Supplemental Figure 6B–E). Collectively, these data are consistent with minimal impact on erythropoiesis and no impact on known hepcidin erythroid regulators in Tfrcfl/fl;Stab2-Cre+ mice.
When maintained on an iron-limited diet, Tfrcfl/fl;Stab2-Cre+ mice did not exhibit differences in serum transferrin saturation compared with Cre− mice (Supplemental Figure 7A). However, Tfrcfl/fl;Stab2-Cre+ male mice had higher liver iron content, with a trend for higher liver iron in Tfrcfl/fl;Stab2-Cre+ females compared to their respective sex-matched controls (Figure 3A). In agreement, Tfrcfl/fl;Stab2-Cre+ male mice also had higher liver ferritin expression (Figure 3B and H). Iron concentration in male Tfrcfl/fl;Stab2-Cre+ mice was also mildly increased in other organs including the spleen and kidney, but not heart or pancreas (Supplemental Figure 7B–E). Despite increased liver iron in Tfrcfl/fl;Stab2-Cre+ male mice, we did not detect significant differences in hepatic expression of Bmp6, Bmp2, Hamp, BMP-SMAD target genes Id1 and Smad7, or phosphorylated SMAD5 (pSMAD) (Figure 3C–G, left panels), which are all typically induced by iron loading.9–15 These data suggest that liver BMP6/2 ligand expression, SMAD signaling, and hepcidin expression are all inappropriately low relative to liver iron loading in male mice lacking endothelial TFR1. Indeed, when normalized to the degree of iron loading by analyzing the ratio of gene expression relative to liver iron content as previously described,33–38 Bmp6, Bmp2, and Hamp were all lower in Tfrcfl/fl;Stab2-Cre+ male mice compared with Cre− mice (Figure 3C–E, right panels). We also observed lower expression of Id1 and Smad7 relative to liver iron content in both sexes of Tfrcfl/fl;Stab2-Cre+ mice compared to Cre− controls (Figure 3F–G). Finally, we observed less phosphorylation of SMAD5 relative to ferritin as a protein expression indicator of liver iron content in Tfrcfl/fl;Stab2-Cre+ male mice (Figure 3H). Together, these results suggest a functional role, albeit modest, of endothelial TFR1 in BMP6, hepcidin, and iron homeostasis regulation when iron availability is limited.
Figure 3. Functional role of endothelial TFR1 in iron sensing and hepcidin regulation in iron-limited conditions.
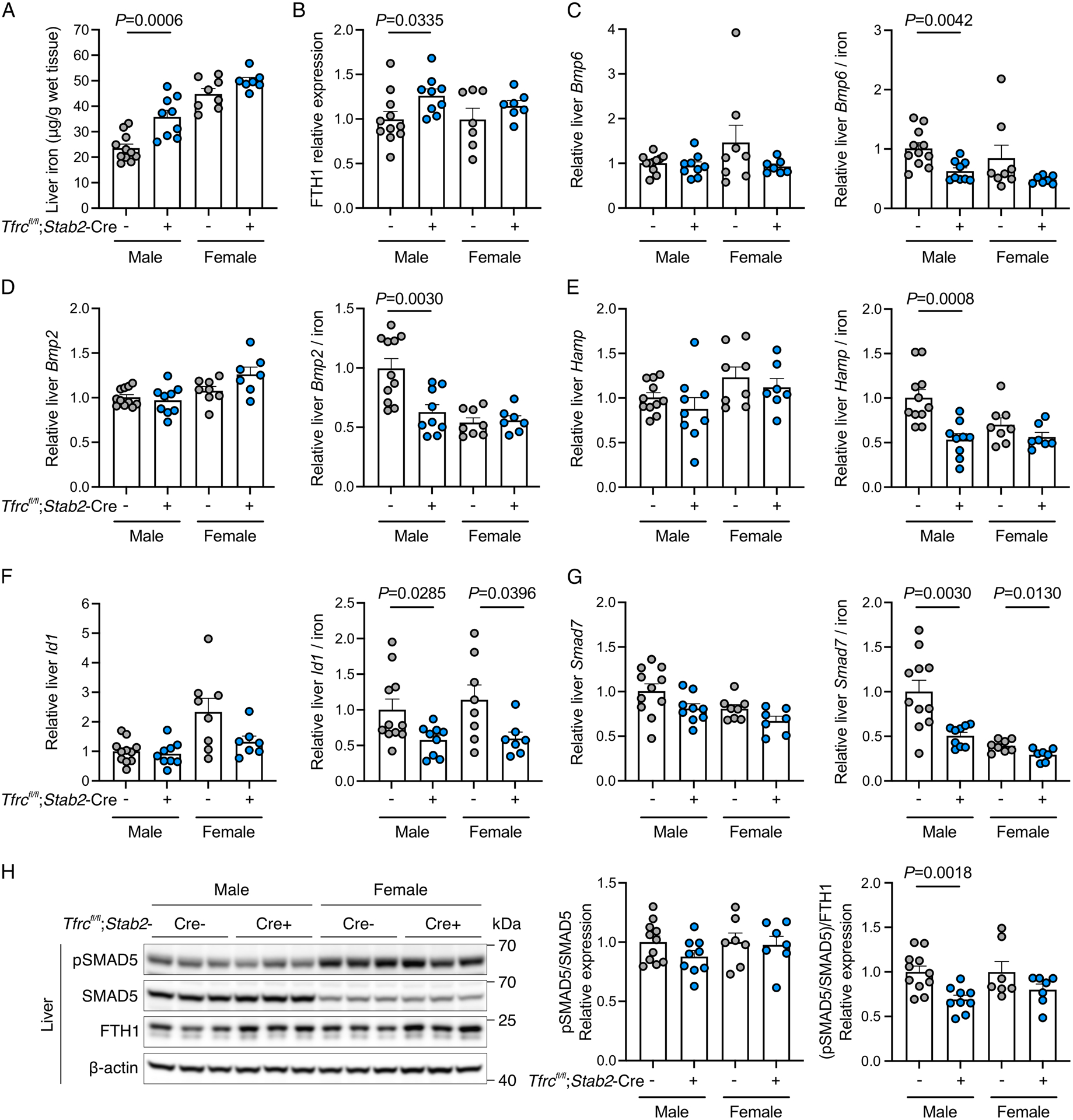
Three-week-old male (N=9–11/group) and female (N=7–8/group) Tfrcfl/fl;Stab2-Cre+ and littermate Cre− mice were fed a limited iron diet (20 ppm iron) for 9 weeks and analyzed at 12 weeks for: (A) liver iron concentration; (B) liver ferritin heavy chain (FTH1) relative to β-actin protein expression by Western blot and chemiluminescence quantitation; (C-G) hepatic Bmp6, Bmp2, Hamp, Id1, and Smad7 transcript levels relative to Rpl19 by qRT-PCR (left panels) and divided by liver iron concentration to control for iron-mediated regulation of these genes (right panels); (H) Western blot and quantitation of phosphorylated SMAD5 (pSMAD) relative to total SMAD5 (left panel) and divided by FTH1 levels to control for iron-mediated regulation (right panel). A representative blot is shown for the quantitation in panels (B) and (H). Graphs represent mean ± SEM with individual points indicating the number of animals per group. Statistical differences between sex-matched Tfrcfl/fl;Stab2-Cre+ and Cre- mice were determined by two-tailed Student’s t-test or Mann-Whitney U test for non-normally distributed values.
NTBI and holo-ferritin are sources of iron-mediated Bmp6 induction in LECs independent of TFR1
To determine if additional biological forms of iron can serve as signals for Bmp6 expression under high iron conditions, we treated primary mouse LECs with ferric ammonium citrate (FAC), a common form of NTBI in patients with iron overload,39,40 and iron-containing ferritin (holo-FTN). We confirmed that primary LECs take up these iron forms as reflected by higher expression of ferritin in cells treated with FAC and holo-FTN (Figure 4A) compared to solvent or iron-poor ferritin (apo-FTN). Similarly, we observed the expected decrease in Tfrc with FAC and holo-FTN, indicative of cellular iron loading (Figure 4B). Importantly, both forms of iron induced Bmp6 expression (Figure 4C). Taken together, these data demonstrate that both NTBI and iron-containing ferritin are additional iron signals that drive Bmp6 expression in LECs.
Figure 4. NTBI and holo-ferritin are sources of iron-mediated Bmp6 induction in primary liver endothelial cells independent of TFR1.
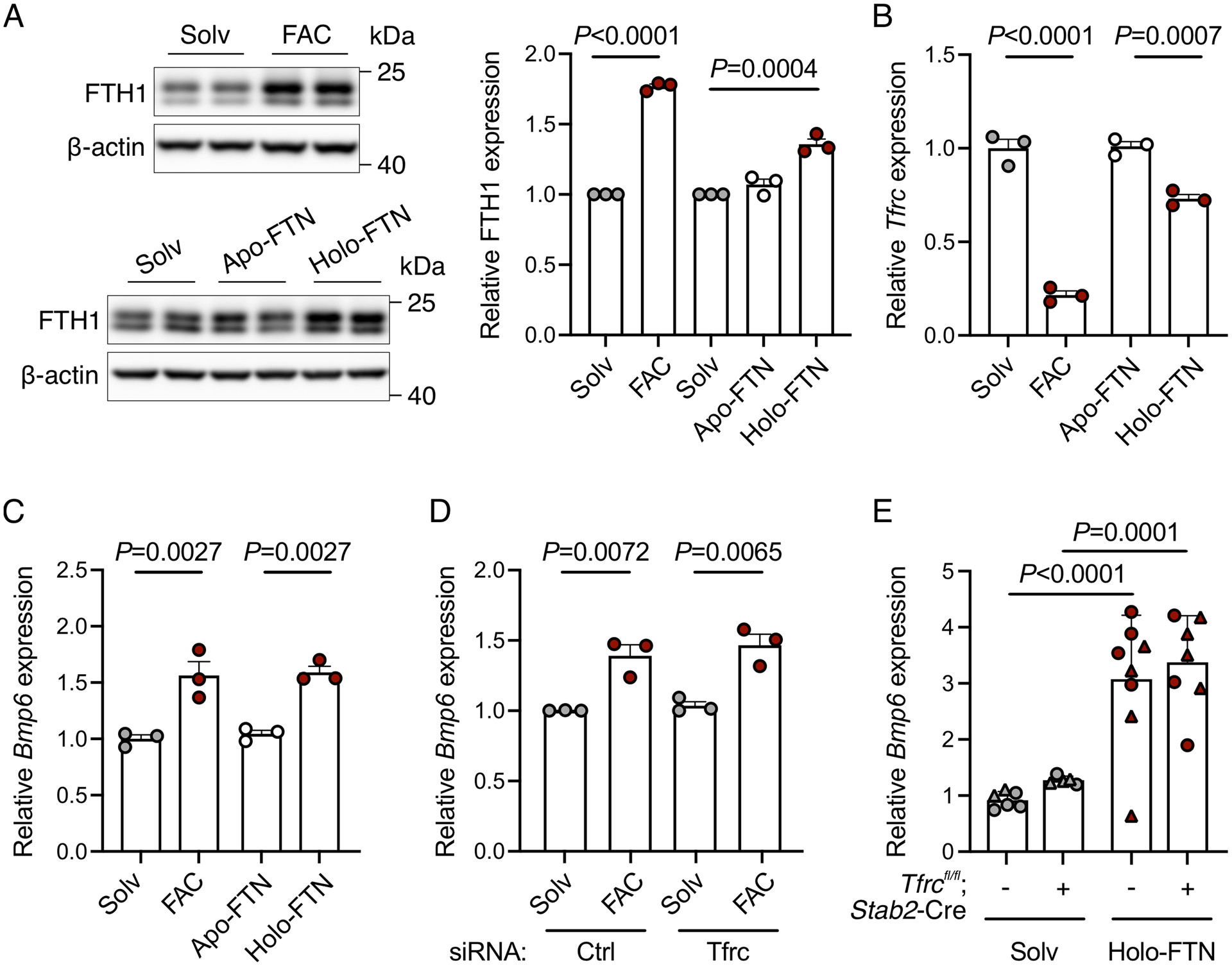
(A-C) Primary liver endothelial cells were serum starved for 12–16 hours, treated with solvent or ferric ammonium citrate (FAC, 200 μg/ml) or solvent, apo- or holo-ferritin (FTN, 100 nM) for 6–24 hours, and analyzed for: (A) Western blot and quantitation of ferritin heavy chain (FTH1) relative to ß-actin as a loading control; (B-C) qRT-PCR expression of (B) Tfrc and (C) Bmp6 relative to Rpl19. Representative blots from N=3 independent experiments are shown. (D) Primary mouse liver endothelial cells were treated with 40 nM control siRNA or siRNA targeting Tfrc for 36 hours, serum starved for 16 hours, treated with FAC for 6 hours, and analyzed by qRT-PCR for Bmp6 relative to Rpl19 expression. (E) Juvenile 5-week-old male (circles) and female (triangles) Tfrcfl/fl;Stab2-Cre+ and littermate Cre− mice received a single intraperitoneal injection of solvent or holo-ferritin (50 μg/g) for 6 hours. Livers were analyzed by qRT-PCR for Bmp6 relative to Rpl19 expression. Bar graphs represent mean ± SEM with individual points indicating the number of replicates or animals. Statistical differences were determined by (A) one-way ANOVA with Holm-Sidak method of multiple comparisons (for holo-FTN experiment), (A-D) two-tailed Student’s t-test, or (E) two-way ANOVA with Holm-Sidak method of multiple comparisons.
To determine whether TFR1 has a functional role in NTBI or holo-FTN-mediated Bmp6 regulation in LECs, we tested the impact of siRNA knockdown of endogenous TFR1 compared with control siRNA in primary LEC cultures. Since holo-FTN was previously reported to induce liver Bmp6 in mice,41 and TFR1 was reported to function as a receptor for FTN uptake,42 we also tested the impact of holo-FTN treatment in Tfrcfl/fl;Stab2-Cre+ mice compared with Cre- controls. In primary LEC cultures, TFR1 knockdown did not inhibit the induction of Bmp6 expression by FAC (Figure 4D). In mice, holo-FTN similarly induced liver Bmp6 expression in Tfrcfl/fl;Stab2-Cre+ mice and Cre- controls (Figure 4E). Together, these data demonstrate that NTBI and holo-FTN regulate liver endothelial Bmp6 expression through TFR1-independent mechanisms.
Discussion
The BMP-SMAD pathway is the major signaling pathway responsible for regulating hepcidin transcription in response to body iron levels to provide adequate iron for erythropoiesis, but to limit the toxic effects of excess iron.6 Intracellular communication between endothelial cells, as the source of BMP6 and BMP2 ligands, and hepatocytes, as the main source of hepcidin, is critical for adequate sensing of iron to control hepcidin expression and systemic iron homeostasis. However, the specific contribution of iron transporters to endothelial iron uptake and BMP ligand regulation remains poorly understood.
In this study, we utilized primary mouse LECs to investigate the iron signals that control BMP ligand expression. We focused on BMP6 since BMP6 and BMP2 work together in hepcidin regulation, BMP6 is the rate limiting ligand, and BMP6 is more strongly induced by iron.9,12 We demonstrated that LECs take up iron from multiple sources, including holo-TF, NTBI, and holo-FTN, and respond by inducing Bmp6 expression in a cell-autonomous manner. Furthermore, we determined that iron loading and Bmp6 induction by holo-TF was mediated by TFR1, whereas Bmp6 induction by NTBI and holo-FTN were TFR1-independent. Finally, we demonstrated that endothelial cell TFR1 has a minor functional role in iron homeostasis regulation under iron limited conditions since Tfrcfl/fl;Stab2-Cre+ mice maintained on an iron limited diet exhibit altered systemic iron homeostasis, as reflected by higher liver iron concentration and ferritin expression.
These effects were more prominent in male mice, which could be related to the known sex-related dimorphisms in iron homeostasis between male and female mice.43,44 This is not likely related to any sex differences in Cre activity, as we detected a similar level of recombination and reduction in Tfrc expression in female and male mice.
We hypothesize that the finding of elevated liver iron levels in Tfrcfl/fl;Stab2-Cre+ mice was caused by a relative deficiency of liver BMP-SMAD signaling and hepcidin expression given the findings of a lower ratio of Bmp6, Bmp2, phosphorylated SMAD5, Id1, and Hamp to liver iron content in these mice compared to Cre− controls. In this model, endothelial cell Tfrc ablation would result in decreased iron uptake into LECs, resulting in a relative deficiency of Bmp6, similar to the findings in our LEC culture model, and potentially also Bmp2. This relative BMP ligand deficiency would result in relative hepcidin deficiency, leading to increased dietary iron absorption, which ultimately induces Bmp6, Bmp2, and hepcidin through endothelial TFR1-independent mechanisms to levels comparable to Cre- mice. However, this occurs at a higher setpoint of iron loading resulting in lower ratio of Bmp6, Bmp2, phosphorylated SMAD5, Id1, and Hamp to liver iron content. In support of this hypothesis, similar findings of altered liver iron levels with inappropriately normal expression of hepcidin and BMP-SMAD pathway components, and an altered ratio of hepcidin and BMP-SMAD pathway targets relative to liver iron or ferritin have been reported in other models with mutations in other known BMP-SMAD pathway and hepcidin regulators, including Hfe and Erfe.33–35,37,38 However, more definitive proof of this hypothesis would require more detailed characterization of the iron content and gene expression of LECs relative to other liver cell populations over time, which is precluded by technical limitations, including an inability to visualize liver iron in mice maintained on the iron-limited diet by enhanced Prussian blue staining and insufficient material and/or insufficiently pure isolated liver cell populations to quantitate iron by other methods. Another explanation for the findings of increased liver iron in endothelial Tfrc knockout mice is that less iron uptake in endothelial cells and some macrophages could lead to more locally available iron that is taken up by hepatocytes and macrophages with normal TFR1 expression. However, whereas this may change the cellular distribution of iron in the liver, this would not be expected to increase the overall amount of liver iron.
Commonly used endothelial Cre mouse models are not fully specific to endothelial cells and can also recombine in the hematopoietic compartment, including macrophages and red cell precursors.15,32 To minimize the impact of Tfrc ablation on the hematopoietic compartment, which would be expected to cause anemia,45,46 we utilized the Stab2-Cre model that is reported to have a more restricted expression pattern to sinusoidal endothelial cells, including the liver, in developing embryos and in adult mice.13,21 Although we found that the Stab2-Cre model also displayed recombination in ~25% of Kupffer cells, this model showed improved specificity compared to our prior analysis of Tek-Cre mice, which displayed 100% recombination in Kupffer cells.15 The specificity of the Stab2-Cre mouse is further demonstrated by the fact that endothelial Tfrc-deficient mice survived late into adulthood and showed normal growth characteristics. Furthermore, we did not observe any obvious major hematologic abnormalities, including anemia or increased erythropoietic drive, suggesting that cross-reactivity with the erythroid compartment is negligible. Nevertheless, it is difficult to definitively rule out a functional contribution of Tfrc deletion in a subset of Kupffer cells or other hematopoietic cell populations in the Tfrcfl/fl;Stab2-Cre+ mice. Prior analysis suggests that the liver is the main organ of endothelial-derived BMP ligands since BMP6 and BMP2 induction by iron overload does not occur in other organs including the heart or spleen.9,11,13–16 However, the Stab2-Cre model is not fully specific for liver endothelial cells and does not formally exclude the contribution of endothelial cells from other organs. Within the liver, endothelial cells lining the sinusoid are the most proximal cell type exposed to portal blood and are therefore well-positioned to sense changes in iron levels as a consequence of dietary absorption. However, the specific role for liver sinusoidal endothelial cells versus the general liver endothelial cell population in BMP6 production has not been definitively established in vivo. These questions will be resolved by the development of more specific Cre models or other techniques that specifically target liver sinusoidal endothelial cells.
We did not observe any differences in iron phenotype in Tfrcfl/fl;Stab2-Cre+ mice compared to Cre negative controls when fed standard chow. This is not entirely surprising considering the relatively high iron content of the diet (380 ppm). In this condition, high iron levels would suppress TFR1 expression, obscuring any differences between mice with and without endothelial TFR1. Additionally, other forms of iron may be present, including NTBI, which would be expected to be rapidly cleared from the portal blood by the liver. NTBI is known to be taken up at a quicker rate and by different transporters than transferrin-bound iron, including divalent metal-ion transporter 1 (DMT1), ZRT/IRT-like protein (ZIP) 8, and ZIP14.19,47–50 Our in vitro model suggests that NTBI is also taken up by LECs to induce Bmp6 expression in a TFR1-independent fashion. We hypothesize that NTBI uptake by different transporters contributes to appropriate BMP6 and hepcidin regulation under high iron conditions in the Tfrcfl/fl;Stab2-Cre+ mice. Future studies will be needed to determine the identity of the transporters responsible for NTBI uptake in LECs.
Interestingly, we found that FAC loaded LECs with iron faster than holo-TF, inducing ferritin protein expression and Bmp6 expression within 6 hours (Supplemental Figure 8A–B), whereas holo-TF required longer incubation of 24 hours to achieve significant increases in ferritin and Bmp6 (Supplemental Figure 8C–D). Given the shorter time needed for iron loading and Bmp6 induction by FAC compared to holo-TF, we hypothesize that NTBI-mediated regulation of Bmp6 expression is activated quickly by high iron levels that overwhelm the transferrin-binding capacity, whereas holo-TF mediated regulation of Bmp6 is activated more slowly under more iron limited conditions. This may explain why oral iron gavage, which induces a submaximal increase in transferrin saturation without increasing liver iron, was not reported to induce liver Bmp6 expression after 4–8 hours.11,16 However, a large enough dose of iron that does increase NTBI would be anticipated to induce liver Bmp6 in that time frame.
Although iron regulates BMP6 in a cell autonomous fashion in our in vitro system, our data does not exclude a contribution of paracrine signals from other cell types. Hepatocytes or macrophages, the main iron storage sites,51 could communicate their iron status to LECs by secreting iron-containing ferritin or other signals to modulate LEC iron content and BMP6 regulation. Indeed, we found that cultured LECs and mice responded to holo-FTN by inducing Bmp6 expression in a TFR1-independent fashion. This is in agreement with a prior study reporting Bmp6 induction by holo-FTN in juvenile mice.41 Although human TFR1 was previously proposed to be a receptor for ferritin uptake,42 this was only demonstrated in human cell culture systems, and remains to be validated in vivo. Interestingly, the same study reported that ferritin failed to bind mouse TFR1,42 suggesting that even if an iron uptake function for human TFR1 is confirmed, this may be a divergent function between humans and mice. Notably, global ZIP14 (Slc39a14)-deficient mice, where iron accumulates in the liver non-parenchymal cells including macrophages and endothelial cells, rather than hepatocytes, exhibited a stronger induction of Bmp6 expression with iron loading.48 This finding demonstrates that while hepatocyte iron loading may contribute to BMP6 regulation by endothelial cells, it is not required. It remains to be determined under which conditions secretion and signaling of iron-containing ferritin by hepatocytes or macrophages may act as signals for BMP6 regulation, and which transporters are responsible. Future studies will also be needed to more completely understand the intracellular mechanisms by which different forms of iron regulate BMP6 in LECs, including the contribution of the NRF2 pathway that has been demonstrated to contribute to BMP6 regulation under high iron conditions.17
Overall, our data show that multiple forms of iron and iron uptake pathways serve as signals for BMP6 regulation in liver endothelial cells, and that iron-mediated BMP6 regulation can occur in a cell-autonomous manner. Additionally, we demonstrate that endothelial TFR1 has only a minor functional role in iron sensing, BMP6 regulation, and hepcidin regulation when iron availability is limited, and is redundant under iron replete conditions. These findings are central for understanding the key question of how iron levels are sensed by endothelial cells to regulate BMP6 expression and understanding how this process is modulated in iron disorders.
Supplementary Material
Acknowledgements
The authors thank the Microscopy Core of the Program in Membrane Biology for training and use of confocal microscopes, the Harvard Stem Cell Institute-Center for Regenerative Medicine Flow Cytometry Core Facility for performing cell sorting, and the Center for Comparative Medicine Clinical Pathology Laboratory for blood analyses. This study was supported by the National Institutes of Health grants T32-DK007540 (to ALF), R01-DK087727 and R01-DK128068 (to JLB), the Patricia and Scott Eston Massachusetts General Hospital Research Scholar Award (to JLB), and Cooley’s Anemia Foundation Research Fellowship (to XX). The Nikon A1R confocal was purchased using NIH Shared Instrumentation Grant S10 RR03156. Additional support for the Program in Membrane Biology Microscopy Core came from the Boston Area Diabetes and Endocrinology Research Center (DK57521) and the MGH Center for the Study of Inflammatory Bowel Disease (DK43351).
Footnotes
Conflict of interest statement
JLB has been a consultant for Incyte Corporation and Alnylam Pharmaceuticals, and owns equity in Ferrumax Pharmaceuticals, a company focused on targeting RGM proteins (including hemojuvelin) and bone morphogenetic protein (BMP/TGF-beta) superfamily signaling as hepcidin modulating agents for the treatment of anemia and other iron disorders. Dr. Babitt’s interests were reviewed and are managed by Massachusetts General Hospital and Mass General Brigham in accordance with their conflict-of-interest policies. The other authors declare no conflict of interest.
References
- 1.Nemeth E, Tuttle MS, Powelson J, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306(5704):2090–2093. [DOI] [PubMed] [Google Scholar]
- 2.Ganz T, Olbina G, Girelli D, Nemeth E, Westerman M. Immunoassay for human serum hepcidin. Blood. 2008;112(10):4292–4297. [DOI] [PubMed] [Google Scholar]
- 3.Sangkhae V, Fisher AL, Chua KJ, Ruchala P, Ganz T, Nemeth E. Maternal hepcidin determines embryo iron homeostasis in mice. Blood. 2020;136(19):2206–2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nemeth E, Rivera S, Gabayan V, et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest. 2004;113(9):1271–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nemeth E, Valore EV, Territo M, Schiller G, Lichtenstein A, Ganz T. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood. 2003;101(7):2461–2463. [DOI] [PubMed] [Google Scholar]
- 6.Fisher AL, Babitt JL. Coordination of iron homeostasis by bone morphogenetic proteins: Current understanding and unanswered questions. Dev Dyn. 2022;251(1):26–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Camaschella C, Nai A, Silvestri L. Iron metabolism and iron disorders revisited in the hepcidin era. Haematologica. 2020;105(2):260–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sangkhae V, Nemeth E. Regulation of the Iron Homeostatic Hormone Hepcidin. Adv Nutr. 2017;8(1):126–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Canali S, Wang CY, Zumbrennen-Bullough KB, Bayer A, Babitt JL. Bone morphogenetic protein 2 controls iron homeostasis in mice independent of Bmp6. Am J Hematol. 2017;92(11):1204–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kautz L, Meynard D, Monnier A, et al. Iron regulates phosphorylation of Smad1/5/8 and gene expression of Bmp6, Smad7, Id1, and Atoh8 in the mouse liver. Blood. 2008;112(4):1503–1509. [DOI] [PubMed] [Google Scholar]
- 11.Corradini E, Meynard D, Wu Q, et al. Serum and liver iron differently regulate the bone morphogenetic protein 6 (BMP6)-SMAD signaling pathway in mice. Hepatology. 2011;54(1):273–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xiao X, Dev S, Canali S, et al. Endothelial Bone Morphogenetic Protein 2 (Bmp2) Knockout Exacerbates Hemochromatosis in Homeostatic Iron Regulator (Hfe) Knockout Mice but not Bmp6 Knockout Mice. Hepatology. 2020;72(2):642–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koch PS, Olsavszky V, Ulbrich F, et al. Angiocrine Bmp2 signaling in murine liver controls normal iron homeostasis. Blood. 2017;129(4):415–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kautz L, Besson-Fournier C, Meynard D, Latour C, Roth MP, Coppin H. Iron overload induces BMP6 expression in the liver but not in the duodenum. Haematologica. 2011;96(2):199–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Canali S, Zumbrennen-Bullough KB, Core AB, et al. Endothelial cells produce bone morphogenetic protein 6 required for iron homeostasis in mice. Blood. 2017;129(4):405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang C-Y, Canali S, Bayer A, Dev S, Agarwal A, Babitt JL. Iron, erythropoietin, and inflammation regulate hepcidin in Bmp2-deficient mice, but serum iron fails to induce hepcidin in Bmp6-deficient mice. American Journal of Hematology. 2019;94(2):240–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lim PJ, Duarte TL, Arezes J, et al. Nrf2 controls iron homeostasis in haemochromatosis and thalassaemia via Bmp6 and hepcidin. Nat Metab. 2019;1(5):519–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilkinson N, Pantopoulos K. The IRP/IRE system in vivo: insights from mouse models. Front Pharmacol. 2014;5:176–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Knutson MD. Non-transferrin-bound iron transporters. Free Radic Biol Med. 2019;133:101–111. [DOI] [PubMed] [Google Scholar]
- 20.Truman-Rosentsvit M, Berenbaum D, Spektor L, et al. Ferritin is secreted via 2 distinct nonclassical vesicular pathways. Blood. 2018;131(3):342–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Géraud C, Koch P-S, Zierow J, et al. GATA4-dependent organ-specific endothelial differentiation controls liver development and embryonic hematopoiesis. J Clin Invest. 2017;127(3):1099–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Torrance JD, Bothwell TH. A simple technique for measuring storage iron concentrations in formalinised liver samples. S Afr J Med Sci. 1968;33(1):9–11. [PubMed] [Google Scholar]
- 23.Johnson MB, Enns CA. Diferric transferrin regulates transferrin receptor 2 protein stability. Blood. 2004;104(13):4287–4293. [DOI] [PubMed] [Google Scholar]
- 24.Chloupková M, Zhang AS, Enns CA. Stoichiometries of transferrin receptors 1 and 2 in human liver. Blood Cells Mol Dis. 2010;44(1):28–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.West AP Jr., Bennett MJ, Sellers VM, Andrews NC, Enns CA, Bjorkman PJ. Comparison of the interactions of transferrin receptor and transferrin receptor 2 with transferrin and the hereditary hemochromatosis protein HFE. J Biol Chem. 2000;275(49):38135–38138. [DOI] [PubMed] [Google Scholar]
- 26.Kleven MD, Jue S, Enns CA. Transferrin Receptors TfR1 and TfR2 Bind Transferrin through Differing Mechanisms. Biochemistry. 2018;57(9):1552–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Camaschella C, Roetto A, Calì A, et al. The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22. Nature Genetics. 2000;25(1):14–15. [DOI] [PubMed] [Google Scholar]
- 28.Nemeth E, Roetto A, Garozzo G, Ganz T, Camaschella C. Hepcidin is decreased in TFR2 hemochromatosis. Blood. 2005;105(4):1803–1806. [DOI] [PubMed] [Google Scholar]
- 29.Fleming Robert E, Ahmann John R, Migas Mary C, et al. Targeted mutagenesis of the murine transferrin receptor-2 gene produces hemochromatosis. Proceedings of the National Academy of Sciences. 2002;99(16):10653–10658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Silvestri L, Nai A, Pagani A, Camaschella C. The extrahepatic role of TFR2 in iron homeostasis. Front Pharmacol. 2014;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kawabata H, Yang R, Hirama T, et al. Molecular cloning of transferrin receptor 2. A new member of the transferrin receptor-like family. J Biol Chem. 1999;274(30):20826–20832. [DOI] [PubMed] [Google Scholar]
- 32.Payne S, De Val S, Neal A. Endothelial-Specific Cre Mouse Models. Arterioscler Thromb Vasc Biol. 2018;38(11):2550–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Coffey R, Jung G, Olivera JD, et al. Erythroid overproduction of erythroferrone causes iron overload and developmental abnormalities in mice. Blood. 2022;139(3):439–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Corradini E, Garuti C, Montosi G, et al. Bone morphogenetic protein signaling is impaired in an HFE knockout mouse model of hemochromatosis. Gastroenterology. 2009;137(4):1489–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Piperno A, Girelli D, Nemeth E, et al. Blunted hepcidin response to oral iron challenge in HFE-related hemochromatosis. Blood. 2007;110(12):4096–4100. [DOI] [PubMed] [Google Scholar]
- 36.Fillebeen C, Charlebois E, Wagner J, et al. Transferrin receptor 1 controls systemic iron homeostasis by fine-tuning hepcidin expression to hepatocellular iron load. Blood. 2019;133(4):344–355. [DOI] [PubMed] [Google Scholar]
- 37.Ryan JD, Ryan E, Fabre A, Lawless MW, Crowe J. Defective bone morphogenic protein signaling underlies hepcidin deficiency in HFE hereditary hemochromatosis. Hepatology. 2010;52(4):1266–1273. [DOI] [PubMed] [Google Scholar]
- 38.Wallace DF, Summerville L, Crampton EM, Frazer DM, Anderson GJ, Subramaniam VN. Combined deletion of Hfe and transferrin receptor 2 in mice leads to marked dysregulation of hepcidin and iron overload. Hepatology. 2009;50(6):1992–2000. [DOI] [PubMed] [Google Scholar]
- 39.Evans RW, Rafique R, Zarea A, et al. Nature of non-transferrin-bound iron: studies on iron citrate complexes and thalassemic sera. J Biol Inorg Chem. 2008;13(1):57–74. [DOI] [PubMed] [Google Scholar]
- 40.Grootveld M, Bell JD, Halliwell B, Aruoma OI, Bomford A, Sadler PJ. Non-transferrin-bound iron in plasma or serum from patients with idiopathic hemochromatosis. Characterization by high performance liquid chromatography and nuclear magnetic resonance spectroscopy. J Biol Chem. 1989;264(8):4417–4422. [PubMed] [Google Scholar]
- 41.Feng Q, Migas MC, Waheed A, Britton RS, Fleming RE. Ferritin upregulates hepatic expression of bone morphogenetic protein 6 and hepcidin in mice. American Journal of Physiology-Gastrointestinal and Liver Physiology. 2012;302(12):G1397–G1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li L, Fang CJ, Ryan JC, et al. Binding and uptake of H-ferritin are mediated by human transferrin receptor-1. Proc Natl Acad Sci U S A. 2010;107(8):3505–3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krijt J, Čmejla R, Sýkora V, Vokurka M, Vyoral D, Nečas E. Different expression pattern of hepcidin genes in the liver and pancreas of C57BL/6N and DBA/2N mice. Journal of Hepatology. 2004;40(6):891–896. [DOI] [PubMed] [Google Scholar]
- 44.Latour C, Kautz L, Besson-Fournier C, et al. Testosterone perturbs systemic iron balance through activation of epidermal growth factor receptor signaling in the liver and repression of hepcidin. Hepatology (Baltimore, Md). 2014;59(2):683–694. [DOI] [PubMed] [Google Scholar]
- 45.Levy JE, Jin O, Fujiwara Y, Kuo F, Andrews NC. Transferrin receptor is necessary for development of erythrocytes and the nervous system. Nat Genet. 1999;21(4):396–399. [DOI] [PubMed] [Google Scholar]
- 46.Shufen W, Xuyan H, Qian W, et al. Transferrin receptor 1-mediated iron uptake plays an essential role in hematopoiesis. Haematologica. 2020;105(8):2071–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang C-Y, Jenkitkasemwong S, Duarte S, et al. ZIP8 is an iron and zinc transporter whose cell-surface expression is up-regulated by cellular iron loading. The Journal of biological chemistry. 2012;287(41):34032–34043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jenkitkasemwong S, Wang CY, Coffey R, et al. SLC39A14 Is Required for the Development of Hepatocellular Iron Overload in Murine Models of Hereditary Hemochromatosis. Cell Metab. 2015;22(1):138–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gunshin H, Mackenzie B, Berger UV, et al. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature. 1997;388(6641):482–488. [DOI] [PubMed] [Google Scholar]
- 50.Liuzzi JP, Aydemir F, Nam H, Knutson MD, Cousins RJ. Zip14 (Slc39a14) mediates non-transferrin-bound iron uptake into cells. Proc Natl Acad Sci U S A. 2006;103(37):13612–13617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Muckenthaler MU, Rivella S, Hentze MW, Galy B. A Red Carpet for Iron Metabolism. Cell. 2017;168(3):344–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.