

Abstract
Nonalcoholic liver disease is a component of metabolic syndrome associated with obesity, insulin resistance, and hyperlipidemia. Excessive alcohol consumption may accelerate the progression of steatosis, steatohepatitis, and fibrosis. While simple steatosis is considered a benign condition, nonalcoholic steatohepatitis with inflammation and fibrosis may progress to cirrhosis, liver failure, and hepatocellular cancer. Studies in rodent experimental models and primary cell cultures have demonstrated several common cellular and molecular mechanisms in the pathogenesis and regression of liver fibrosis. Chronic injury and death of hepatocytes cause the recruitment of myeloid cells, secretion of inflammatory and fibrogenic cytokines, and activation of myofibroblasts, resulting in liver fibrosis. In this review, we discuss the role of metabolically-injured hepatocytes in the pathogenesis of nonalcoholic steatohepatitis and alcohol-associated liver disease. Specifically, the role of chemokine production and de novo lipogenesis in the development of steatotic hepatocytes and the pathways of steatosis regulation will be discussed.
Keywords: Liver fibrosis, steatotic hepatocytes, de novo lipogenesis, ER stress, UPR
INTRODUCTION
Liver fibrosis results from the excessive deposition of extracellular matrix proteins that form a fibrous scar in response to chronic liver injury.1 Toxic liver fibrosis is caused by hepatitis B (HBV) or C (HCV) infection, alcohol-associated liver disease (AALD), and nonalcoholic steatohepatitis (NASH).2 Inflammation plays a key role in the pathogenesis of liver fibrosis.3 Myeloid cells are the main source of fibrogenic cytokines, including the critical activator of hepatic myofibroblasts TGFβ1, which are not present in normal liver.3 Hepatic stellate cells (HSCs) are the major source of collagen type I–producing hepatic myofibroblasts in response to toxic liver injury.4,5
Until recently, HBV and HCV were the most common causes of liver fibrosis and cirrhosis. With the development of vaccines and highly effective antiviral treatments, the incidence of HBV- and HCV-related liver diseases has declined, while NASH-associated fibrosis and HCC are increasing.6,7 AALD does not develop in thin or cachectic individuals, occurring most often in obese patients.2 Increased alcohol intake in patients with high body mass index (BMI> 27) leads to more severe liver disease. Histopathologically, both NASH and AALD can be distinguished from nonalcoholic fatty liver (NAFL) by the development in the latter of steatohepatitis, centrilobular ballooning degeneration of hepatocytes and Mallory–Denk hyaline inclusions,8,9 neutrophilic infiltration, inflammation, and activation of hepatic myofibroblasts.10 NASH is driven by ER stress and the associated activation of inflammatory responses that further exacerbate metabolic injury and activate fibroproliferative responses in the liver.11
NAFL is characterized by hepatic steatosis and is reversible;2 however, approximately 20%–24% of NAFL patients develop NASH. Whether steatosis is a benign or pre-condition that makes obese individuals more susceptible to metabolic syndrome, insulin resistance, and inflammation remains controversial.3 This review summarizes the molecular mechanisms underlying the development of hepatic steatosis and the role of de novo lipogenesis in the pathogenesis of NASH- and AALD-induced liver injury.2
1. The development of NASH and AALD liver fibrosis
1.1. NASH-induced metabolic liver injury
The pathogenesis of NASH is often explained by a “two hit” theory: obesity and insulin resistance results in metabolic injury to hepatocytes, activation of de novo lipogenesis, lipid accumulation, and lipotoxicity that further exacerbate hepatocyte damage.2 Adipose tissue contributes to insulin resistance by secreting adipokines and cytokines (e.g., leptin and adiponectin).12 Endoplasmic reticulum (ER) stress constitutes a potential “second hit” that causes the secretion of inflammatory and fibrogenic cytokines and chemokines (e.g., IL-6, TNFα, IL-1β, TGFβ1).3 ER stress is associated with changes in the gut microbiota (prevalence of Firmicutes over Bacteroidetes13), increased gut permeability, the release of bacterial products such as LPS into the circulation, activation of Toll-like-receptor (TLR)–dependent signaling pathways (specifically TLR4), and the recruitment and activation of inflammatory cells and myofibroblasts in the injured liver.14
1.2. AALD-associated liver injury
As with NAFLD, alcohol-induced steatosis can progress to alcohol-induced steatohepatitis (ASH) and AALD.3 AALD results from a chronic imbalance in hepatocyte metabolism due to direct injury by alcohol and alcohol-derived metabolites. Hepatocyte injury occurs via release of acetaldehyde, a toxic ethanol metabolite produced by hepatocytes, or upregulation of cytochrome P450 2E1, a critical enzyme involved in alcohol metabolism.15,16 Toxic alcohol metabolites, changes in the gut microbiota composition,17,18 increased intestinal permeability, and the leak of bacterial products into circulation result in inflammation and fibrogenesis.14
Despite the etiological differences between NASH and AALD-induced liver injury,3,14 the mechanisms underlying the pathogenesis of metabolic liver injury are similar, especially at the onset of metabolic injury.2 The pathogenetic mechanisms in common between these conditions are discussed.
2. Pathogenesis of liver fibrosis in NASH and AALD
2.1. Inflammation drives NASH and AALD progression
Both NAFL and alcohol-associated fatty liver are considered to be benign and reversible conditions.3 Fatty liver is characterized by the accumulation of fat droplets (mainly triglycerides and phospholipids) in hepatocytes, and this process is regulated at the level of de novo lipid synthesis, lipid secretion (VLDL), and inhibition of β-oxidation.19,20
Chronic injury to hepatocytes and hepatocyte apoptosis induce ER stress, reactive oxygen species (ROS) production, and mitochondrial dysfunction, causing the activation of inflammatory responses, including the secretion of the key cytokines/chemokines by myeloid cells.15,16 Neutrophils are first responders that enter the liver to phagocytose and clear apoptotic cells and cell debris and further facilitate recruitment and activation of other myeloid cells into the damaged liver.2 Although the specific roles of liver resident Kupffer cells versus bone-marrow–derived macrophages are uncertain, both populations are believed to contribute to liver inflammation; the secretion of IL-6, TNFα, IL-1β, and TGFβ1; and the activation of inflammatory responses that lead to liver fibrosis.2 ER stress caused by misfolded proteins induces the activation of fatty acid and cholesterol synthesis in metabolically-injured hepatocytes. IL-6 signaling induces inflammatory responses in hepatocytes, including the secretion of IL-6, CXCL1, and CCL2. IL-6, TNFα, and TGFβ1 drive HSC activation into collagen type I–producing myofibroblasts. In addition to neutrophils and macrophages, T and B lymphocytes recruited to the damaged liver mediate the adaptive immune response and contribute to metabolic liver damage, inflammation, and the formation of fibrous scar tissue by activated myofibroblasts (Figure 1).
Figure 1. Pathogenesis of toxic liver fibrosis and therapeutic implications.

Hepatocyte damage triggers the inflammatory response, leading to activation of macrophages, release of ROS and TGFβ1, and activation of quiescent HSCs into activated HSCs/myofibroblasts that produce collagen type I resulting in liver fibrosis.
2.2. Contribution of T and B cells to NASH and AALD progression
Macrophage-derived TGFβ1 and IL-6 are critical regulators of naive T-cell differentiation into T helper 17 (TH17) cells, while IL-23 regulates Th17 expansion and proliferation.21–23 Mouse Th17 cells also produce anti-inflammatory IL-22. In contrast to IL-17, IL-22 acts as a survival factor for hepatocytes,24 suggesting that the activation of specific T-cell subsets might reduce liver injury by releasing the hepatoprotective IL-22.25 In addition, IL-22 can signal through the IL-22 or IL-10 receptors on HSCs to induce their senescence.26 Overexpression of IL-22 in mice is reported to increase HSC senescence and attenuate liver fibrosis.26
Alterations in the intestinal microbiota composition strongly affect the production of IL-17,27 suggesting a correlation between dysbiosis, the immune response, and liver fibrosis.14 IL-17A facilitates the activation of myeloid cells and directly activates HSC conversion into fibrogenic myofibroblasts in experimental models of liver fibrosis.2 IL-17A increases de novo lipogenesis and TNFα -TNFRI signaling in metabolically-injured hepatocytes. Unlike IL-17A-secreting T helper 17 (Th17) CD4+ T cells, which exhibit a fibrogenic effect, CD8+ T cells mediate hepatoprotective effects. In support of this observation, ablation of CD8+ T cells in mice was found to exacerbate NASH-induced liver fibrosis, whereas genetic or pharmacological suppression of IgA+ cells attenuated NASH-induced liver fibrosis, perhaps through upregulation of IFN-producing T cells.28
2.3. Do metabolically-injured hepatocytes contribute to inflammation?
Hepatocytes constitute 60% of the total liver cells and mediate the detoxifying, metabolic, and secretory functions of the liver. Chronic liver injury causes ER stress in damaged hepatocytes, the release of ROS, and hepatocyte apoptosis.2 Apoptotic hepatocytes release damage-associated molecular patterns (DAMP), TGFβ1, and exosomes containing biologically active factors (such as chemokines/receptors, metabolites, proteases) that can rapidly deliver “stress signals” into the intracellular compartment to mediate intercellular communications.29–35 Metabolically-injured hepatocytes serve as a source of chemokines, including CXCL1, CCL2, CCL5, TGFβ1/3, IL-6, and TNFα.2 Although their contribution to inflammation is less than that of inflammatory/myeloid cells, hepatocytes can secrete chemokines and growth factors locally (into the space of Disse) that regulate crosstalk between HSCs and hepatic myeloid cells (liver resident Kupffer cells and bone-marrow–derived inflammatory cells) and endothelial cells. Furthermore, damaged hepatocytes release DAMPs and extracellular vesicles to communicate between hepatocytes and neighboring cells, thereby, promoting liver fibrosis via the activation of HSCs and Kupffer cells.36 Extracellular vesicles, including microvesicles and exosomes or exosome-like vesicles, transport large quantities of bioactive molecules that are released into the microenvironment and circulation. Hepatocyte-derived extracellular vesicle miRNA (miR-128–3P) contributes to HSC activation and liver fibrosis through downregulation of PPARγ.37–40
Damaged hepatocytes are a major source of systemic angiotensinogen, the precursor of angiotensin (Ang) II,41 which facilitates inflammatory responses in the damaged liver and potentiates TGFβ signaling and fibrosis. AngII drives the release of the cytokines TGF-β, IL-1β, and MCP1 by inflammatory cells and induces the contraction and proliferation of HSCs.42,43 The release of chemokines (such as MCP-1, MIP-1a, MIP-1b) by steatotic hepatocytes facilitates the recruitment of bone-marrow–derived inflammatory cells into the injured liver.2 Recent studies have shown cross talk between steatotic hepatocytes and the activation of fibrogenic HSCs/myofibroblasts. Two molecules that are elevated in metabolically-injured hepatocytes are cholesterol, for which the mechanistic link to NASH remains incompletely understood, and TAZ, a transcriptional regulator that promotes NASH fibrosis.44 Under physiological conditions, internalization of plasma membrane cholesterol activates soluble adenylyl cyclase (ADCY10), triggering calcium-RhoA–mediated proteasome-mediated TAZ degradation.44 In response to chronic metabolic injury, elevated hepatocyte cholesterol upregulates TAZ and promotes fibrotic NASH. Increased levels of hepatocyte-derived TAZ result in increased TAZ-TEAD–dependent hepatocyte Indian hedgehog transcription and secretion, leading to the transcription of NASH-specific genes that encode proteins responsible for HSC activation, liver fibrosis, and inflammation.44,45 TAZ silencing can suppress liver fibrosis and partially reverse NASH.46
3. The role of de novo lipogenesis in hepatocytes in the pathogenesis of NASH- and AALD-associated liver fibrosis
3.1. The mechanism underlying hepatic steatosis development in metabolically-injured liver
Many patients with obesity and insulin resistance develop hepatic steatosis. Lipid droplets of steatotic hepatocytes consist mainly of triglycerides and cholesterol.47,48 Hepatic triglycerides and cholesterol are derived from serum non-esterified fatty acids stored in adipose tissue (59%), de novo lipogenesis (26%),49 and the diet (15%).50 De novo lipid biosynthesis occurs when excessive carbohydrates are consumed or when circulating insulin levels are high51 (Figure 2). Carbohydrates undergo glycolysis to generate acetyl-CoA molecules which serve as a substrate to fuel fatty acid and cholesterol synthesis.52 Under fasting conditions, wherein insulin levels are low and glucagon levels are high, metabolic processes are shifted to fatty acid oxidation or lipolysis that allows fatty acid/cholesterol mobilization from adipose tissues into circulation, followed by uptake by the liver.47 The degree of hepatic steatosis fluctuates in both lean and healthy obese individuals depending on the circadian rhythm, diet and food composition, age, pattern of alcohol consumption (binge drinking vs social drinking), and use of specific medications.53 Until recently, chronic steatosis (NAFL) was considered to be a benign, reversible condition, and the progression of steatohepatitis to NASH was thought to be driven by inflammatory responses.54 The critical role of lipotoxicity in the pathogenesis of NASH has been recognized recently. Here we summarize evidence implicating de novo lipogenesis in the development of metabolic injury and NASH (Figure 2).
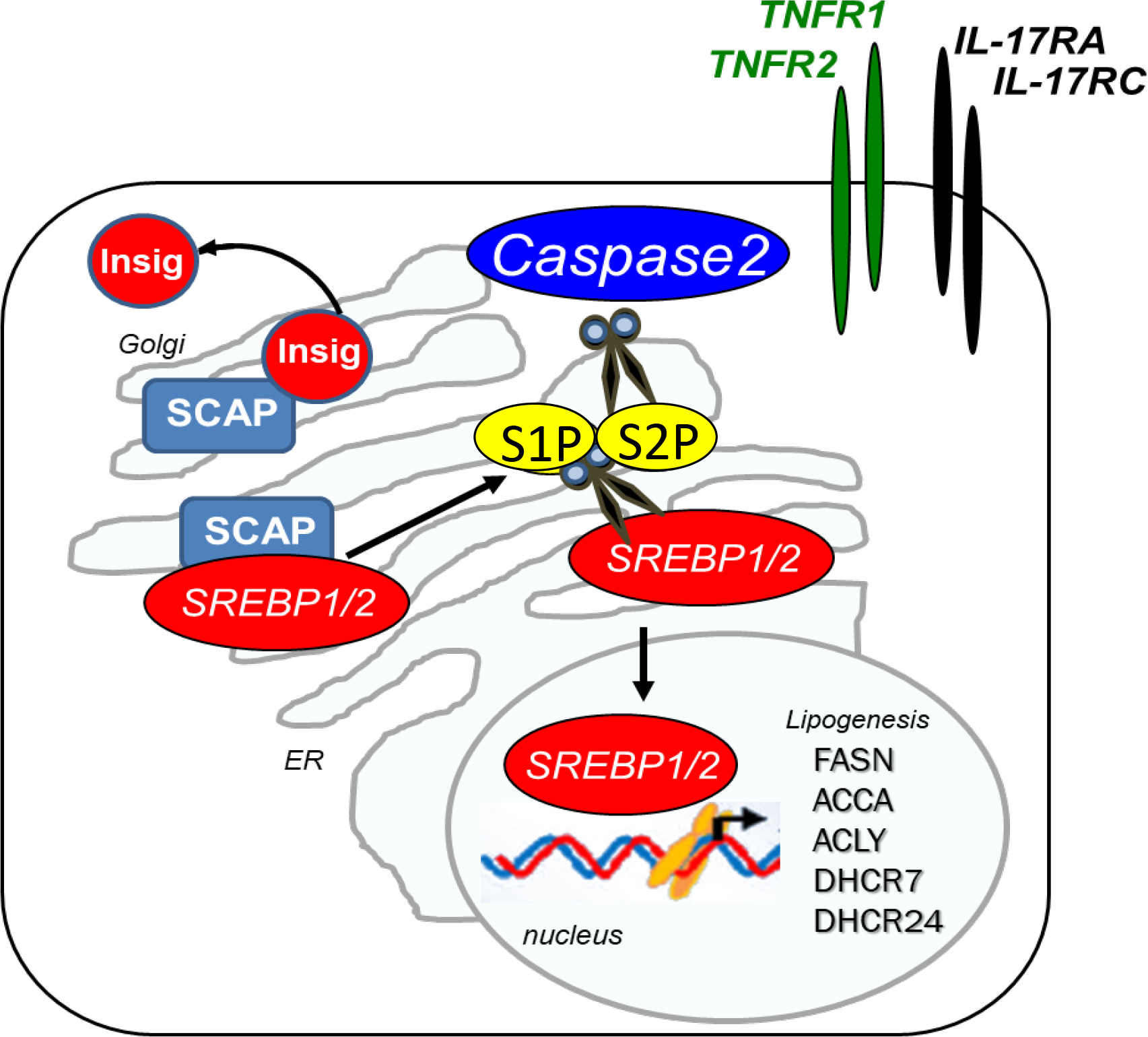
Figure 2. The caspase-2–dependent activation of de novo lipogenesis.

ER and IL-17A facilitate TNFα/TNFR-mediated lipogenesis in alcohol-damaged hepatocytes via activation of the caspase 2-SP1-SREBP1/2-DHCR7 pathway. Schematic representation of cholesterol and fatty acid synthesis pathways.
3.2. Progression of NAFL to NASH
Approximately 20% of patients with NAFL progress to NASH.55 To distinguish NAFL from NASH, a scoring system was developed and published in Hepatology, 2005.56 Now in use worldwide, this score includes the degree of steatosis, chronic steatohepatitis, inflammation, and fibrosis. The grading of biopsies ≥5 was found to correlate with a diagnosis of NASH (see Table 1). Biopsies/tissues with scores <3 are diagnosed as “not NASH.” A brief summary of the criteria used to diagnose NAFL versus NASH in the human liver is shown in Table 1.
Table 1. NASC/CRN grading criteria.
NASH diagnosis correlates with steatosis, related chronic steatohepatitis, and fibrosis. Biopsies of grade ≥5 were diagnosed as NASH; those of grade <3 were diagnosed as “not NASH.”
| Grade: | Criteria: |
|---|---|
|
| |
| Steatosis grade: | 0 <5%; |
| 1 - 5–33%; | |
| 2 - 34–66%; | |
| 3 >66% | |
|
| |
| Steatosis distribution: | centrilobular vs diffuse |
|
| |
| Lobular inflammation: | 0 - none; |
| 1 < 2 foci/20x field; | |
| 2 - 2–4 foci/20x field; | |
| 3 > 4 foci/20x field | |
|
| |
| Hepatocellular ballooning: | 0 - none; |
| 1 - mild, few; | |
| 2 - moderate-marked, many | |
|
| |
| Portal inflammation: | 0 - none; |
|
1 - mild, | |
| 2 > mild, | |
| 3 - severe | |
|
| |
| Fibrosis (Trichrome stain): | 0 - none |
| 1a - mild zone 3 perisinusoidal fibrosis, requires trichrome stain to identify | |
| 1b - moderate zone 3 perisinusoidal fibrosis, also noticeable by H&E | |
| 1c - portal fibrosis only; | |
| 2 - zone 3 perisinusoidal fibrosis and periportal fibrosis | |
| 3 - bridging fibrosis | |
| 4 - cirrhosis | |
3.3. Contribution of de novo lipogenesis to the progression of NAFL to NASH and AALD
Obesity and insulin resistance lead to the development of metabolic syndrome. Excessive hepatic fatty acid synthesis, inhibition of hepatic lipid β-oxidation, and accumulation of lipid droplets (mainly triglycerides and phospholipids) in hepatocytes results. One key metabolic process implicated in triggering NASH progression is de novo lipogenesis of cholesterol and fatty acids.51,55 The rate of de novo lipogenesis in NASH patients is elevated 3-fold over that of NAFL patients,51,57 underscoring the importance of de novo lipogenesis in the metabolically-injured liver.11,48 The rapid synthesis and excessive accumulation of fatty acids and cholesterol has a lipotoxic effect on hepatocytes.58,59 Most lipid synthesis takes place in the ER. De novo lipogenesis is critical for triggering the ER stress responses under physiological conditions and in response to chronic liver injury.55
3.4. Lipogenesis is mediated by sterol regulatory element-binding proteins.
De novo lipogenesis is regulated by sterol regulatory element-binding proteins (SREBP) 1 and 2,60 transcription factors that control production of the key enzymes that regulate fatty acid and cholesterol synthesis, respectively. Two conditions that mediate the transcription of SREBP1/2-dependent lipogenic genes have been identified: the energy-depleted state and energy-abundant state. In the energy-depleted state, the regulation of SREBP1/2 function is attributed to activation of an autonomous feedback system that senses the lack of sterol products in the microenvironment. In the “energy-abundant state,” the transcriptional activity of SREBP1/2 is driven by protein accumulation and protein misfolding in the ER, leading to the development of endoplasmic reticulum (ER) stress and subsequent induction of the adaptive unfolded protein response (UPR) system.61 The UPR is activated to restore homeostasis. If the UPR system fails to repair the underlying problem, prolonged UPR activity increases the transcriptional capability of SREBP1/2, increasing de novo lipogenesis, thereby driving steatosis, inflammation, and fibrosis (Figure 2).62
4. The pathways of SREBP1/2 activation in hepatocytes
4.1. De novo lipogenesis in metabolically-injured hepatocytes
The progression of steatosis to steatohepatitis is associated with the activation of inflammatory responses and ROS production.2 The ER is the major site of lipid synthesis in hepatocytes. Cholesterol synthesis and uptake pathways are regulated through transcriptional regulation of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, the rate-limiting enzyme for cholesterol biosynthesis, via LDLR. Promoters of these genes contain the (5′-ATCACCCCAC-3′) sterol regulatory element (SRE).63 SRE sequences are recognized by the ER membrane-localized transcription factors SRE binding protein (SREBP)1 and 2. SREBP1 plays a critical role in triglyceride synthesis via transcriptional regulation of fatty acid synthase, stearoyl-CoA desaturase, and ATP citrate lyase, while SREBP2 is mainly responsible for mediating cholesterol metabolism by regulating genes such as HMG-CoA reductase and low-density lipoprotein receptor.64 SREBP2 mediates sterol regulation in all tissues.64 Two isoforms, SREBP1a and SREBP1b, are arise from transcription of the SREBF1 gene from different promoters. SREBP1c is expressed in most tissues and regulates homeostasis of fatty acids and triglycerides in lipogenic organs such as the liver.60 Compared to SREBP1a, SREBP1c lacks 24 amino acid residues in the N-terminal CREB1-binding transactivation domain and exhibits low transcriptional activity.65 SREBP1a is highly expressed only in specific tissues and cells66 and stimulates expression of lipogenic and cholesterogenic genes needed to construct membrane lipids in growing cells (Figure 2).65,67
4.2. Regulation of SREBP1/2-dependent transcription of lipogenic genes
SREBPs are produced as inactive ER membrane-bound proteins that require post-translational modifications to function as transcription factors that translocate to the nucleus and initiate lipogenic gene transcription. Synthesized as intrinsic ER membrane proteins, SREBP1/2 are transported from the ER to the Golgi for proteolytic cleavage and processing.68 Sterol levels regulate SREBP1/2 activity by controlling SREBP1/2 transport from the ER to the Golgi, where they undergo proteolytic cleavage before translocating to the nucleus. Several independent mechanisms release SREBP1/2 from the ER. The INSIG:SCAP-S1P/S2P pathway is preferentially activated during the energy-depleted state (activated when low levels of sterols and other lipid levels are detected by specific sensor proteins in the ER), while caspase 2-S1P/S2P are primarily induced during the energy-abundant state (associated with ER stress and insulin resistance activated by TNF signaling),60 thereby regulating fatty acid and cholesterol synthesis (Figure 4).60
Figure 4. ER stress drives NAFLD progression.

Progression of steatosis (NAFL) to steatohepatitis and fibrosis (NASH) is associated with the development of ER stress, lipid accumulation, inflammation, and activation of hepatic myofibroblasts.
4.3. INSIG:SCAP-mediated regulation of SREBP1/2
Inactive ER-anchored SREBP1/2 proteins remain in the ER and are processed by Golgi enzymes by binding to the INSIG (precursor bound by insulin-induced gene 1): SCAP (SREBP cleavage-activating protein) complex.69,70 The specific mechanism that controls release of SREBP1/2 from the INSIG:SCAP is complex and is regulated by sensor proteins responding to the changing levels of insulin, oxysterols, unsaturated FA, and food intake composition.71–74 SCAP is an ER-sterol–sensing protein that binds to SREBP1 and SREBP2 via a WD40 repeat domain and chaperones both proteins from the ER to the Golgi.68 Under sterol-rich conditions, SREBPs are held in the ER through their interaction with SCAP, an anchoring molecule, and INSIG, an ER transmembrane protein. Specifically, when cholesterol in ER membranes exceeds a threshold, the sterol binds to SCAP, triggering several conformational changes75 that prevent the SCAP-SREBP complex from leaving the ER. INSIGs bind SCAP, thereby preventing SCAP-SREBP movement from the ER.76
When sterols are depleted, INSIG1 dissociates from SCAP, thereby allowing SCAP to move to the Golgi. Activation of the ER membrane-bound INSIG:SCAP chaperone drives SREBP maturation and activity. The SCAP protein forms a homotetramer with its membrane region to form a stable complex with SREBP1 or SREBP2 through its C-terminal cytoplasmic domain.77 The SREBP-SCAP complex is released from INSIG upon depletion of sterol in the environment. SCAP assists in the transport of SREBP in coat protein II (COPII ) vesicles from the ER to the Golgi.68
Translocation of the SCAP-SREBP complex from the ER to the Golgi leads to sequential proteolytic cleavage of SREBPs by active forms of site-1 membrane-bound serine proteases S1P and S2P.64 The first cleavage occurs within the 50-amino acid luminal loop, separating the SREBP into two halves. The NH2-terminal half remains attached to the membrane by its single transmembrane helix. The second cleavage occurs within this helix, releasing the bHLH-Zip domain so that it can enter the nucleus. These sequential proteolytic cleavages activate S1P and S2P.78,79 S1P cleaves the ER luminal loop of SREBPs only in cholesterol-depleted cells, and site-1 cleavage requires previous cleavage at site-2.68 The N-terminal region of SREBPs then is cleaved off by S2P. A model has been proposed in which cleavage by S1P allows the first transmembrane segment to unwind, thereby pushing the S2P cleavage site to the membrane surface, where it becomes accessible.80 Following cleavage, SREBP1/2 NH2-terminal fragments are released from the Golgi, dimerize with importin β via the SREBP helix–loop–helix leucine zipper domain,81,82 and translocate to the nucleus, where they initiate target gene transcription.83 SREBPs are responsible for the transcription of more than 30 genes needed for the uptake and synthesis of cholesterol, fatty acids, triglycerides, and phospholipids. A cell-penetrating nuclear transport modifier cSN50.1 interacts with importin β and reduces nuclear translocation of SREBP1/2 induced by lipid depletion in cells.84 The nuclear concentration of SREBP1c is regulated by circulating insulin via the PI3K–AKT–mTOR– SREBP pathway. Mammalian target of rapamycin complex 1 (mTORC1) regulates SREBP by controlling the nuclear entry of lipin 1, a phosphatidic acid phosphatase. Dephosphorylated, nuclear, catalytically active lipin 1 promotes nuclear remodeling and mediates the effects of mTORC1 on SREBP target genes, SREBP promoter activity, and nuclear SREBP protein abundance. Specifically, the lack of mTORC1-mediated lipin 1 phosphorylation promotes nuclear entry of lipin 1 and promotes the downregulation of nuclear SREBP protein. Whether lipin 1 can directly interact with SERBPs remains unknown.85
Nuclear SREBPs are rapidly degraded by the ubiquitin and proteasome pathways.86 Proteasome degradation of SCAP precedes SREBP degradation. Increased SCAP degradation is linked to downregulation of its chaperon, heat shock protein 90 (Hsp90),87 which stabilizes SCAP in the ER and Golgi. After dissociating from SCAP, INSIG1 is ubiquitinated and degraded88. SCAP is either recycled or proteolytically degraded.89 SREBP2 directly regulates transcription of the INSIG1 gene. INSIG1 protein is rapidly degraded unless needed. Feedback regulation of cholesterol synthesis requires a sufficient amount of nuclear SREBP2 for INSIG1 transcription and restoration of ER cholesterol, a regulatory mechanism known as “convergent feedback inhibition.”88,90 An additional ER-retention membrane protein, the INSIG2 isoform, was identified.91 Both isoforms can simultaneously interact with SCAP to mediate retention of the SCAP-SREBP complex in the ER membrane.92
4.4. Regulation of INSIG:SCAP-dependent SREBP1/2 activation
Chronic hyperinsulinemia produces overactive hepatic SREBP1 and lipogenesis despite insulin resistance, often referred to as “selective insulin resistance.”60,93 Proteolytic cleavage and activation of SREBP1/2 can be stimulated by insulin94 via signaling through insulin receptor substrate-1 (IRS1) and its downstream targets protein kinase B (PKB/Akt) and mTORC1. mTORC1 regulates activation of hepatic p70 S6 kinase (S6K), the major downstream effector of mTORC1, which in turn can cleave and activate SREBP1/2 and stimulate lipogenesis under conditions of insulin resistance,95,96 suggesting that selective insulin resistance depends upon mTORC1– S6K1 interaction.60 SREBP1c activity may also be induced through the nuclear hormone receptor peroxisome-proliferator-activated receptor-γ (PPARγ)97 as well as liver X receptor (LXR) activity,98 each of which plays a critical role in lipogenesis. The LXRs are members of the nuclear hormone receptor superfamily that are bound and activated by oxysterols. These receptors serve as sterol sensors to regulate the transcription of gene products that control intracellular cholesterol homeostasis through catabolism and transport. Ligand-activated nuclear PPARγ heterodimerizes with retinoid X receptors (RXRs) resulting in expression of its target genes such as CD36, a fatty acid transport protein involved in the transport and metabolism of intracellular FA.99 SREBP1c expression was shown to be upregulated in mouse tissues in an LXR-dependent manner by dietary cholesterol and synthetic agonists for both LXR and its heterodimer partner, the retinoid X receptor (RXR),98 which did not increase expression of the related gene products SREBP1a and SREBP2.98 SREBP1a and SREBP2 but not SREBP1c bind to and are stabilized by CBP and P300 as co-activators to recruit the Mediator complex (Figure 3).100,101
Figure 3. Canonical and non-canonical activation of SREBP1/2 transcription factors that drive de novo lipogenesis in steatotic hepatocytes.
SCAP-dependent lipogenesis is subject to negative feedback. Cholesterol buildup in ER membranes causes sterol binding to SCAP, which triggers a conformational change that causes SCAP to bind to insulin-induced gene, prohibiting SCAP binding to SREBPs. Conversely, when cells are sterol-deprived, SCAP escorts SREBPs from the ER to the Golgi, where S1P and S2P proteases cleave SREBPs, allowing their translocation to the nucleus to activate de novo lipogenesis and cholesterol target gene transcription. During ER stress, lipogenesis is driven in a SCAP-independent manner. The simultaneous activation of TNFα and IL-17 pathways increases caspase 2 gene expression, and its activation is dependent on IRE1α. Caspase 2 cleaves S1P into a soluble form that reaches the ER to cleave SREBPs, allowing their translocation to the Golgi, where they are further cleaved by S2P and translocate to the nucleus to activate the lipogenic gene transcription. SCAP-dependent lipogenesis is prevented by cholesterol increase, while SCAP-independent lipogenesis is a non-homeostatic mechanism subject to a positive regulatory loop. The activation of SREBP2 also increases caspase 2 levels, serving as an additional input.
4.5. Non-canonical (SCAP-independent) caspase 2-mediated activation of SREBP1/2
Despite the existence of several negative feedback loops associated with sterol/insulin-INSIG:SCAP-dependent SREBP1/2 regulation, chronic metabolic injury causes constitutive SREBP activation102,103 via (SCAP)-independent SREBP activation.11 NASH progression is associated with the lipotoxic effects of excessive accumulation of free fatty acids and free cholesterol58 on mitochondrial dysfunction,104 and the induction of TNF signaling in metabolically-injured hepatocytes, resulting in ER stress and insulin resistance. ER stress, defined as a chronic perturbation affecting ER homeostasis, is characterized by the accumulation of aberrant proteins, which disturbs the balance of the protein folding capacity of the ER to keep up with cellular demand.105 The hepatic ER plays a critical role in the maintenance of lipid membrane composition and regulation of the intrahepatic and plasma lipids (Figure 3).
Specifically, binding of macrophage-derived TNFα to the hepatic TNF receptor 1 (TNFR1) and ER stress cause persistent activation of SREBP1/2 in metabolically-injured hepatocytes via non-apoptotic caspase 2-dependent constitutive activation of S1), which initiates SREBP-activating cleavage.11 In turn, the development of ER stress inhibits INSIG expression via the PERK-mediated eIF2α signaling pathway,106,107 shifting toward TNF/TNFR1-caspase 2-S1P/S2P-driven cholesterol synthesis.108 Recent studies suggest that IL-17 signaling in fatty hepatocytes also regulates TNF-TNFRI-S1P-caspase 2-SREBP1/2 activation.48 Consistent with this finding, the inhibition of TNF- or IL-17 signaling suppresses caspase 2-dependent SREBP1/2 maturation.11,48 Although caspase activity is usually increased in apoptotic cells, caspase 2 does not exhibit apoptotic functions in metabolically damaged hepatocytes but instead acts as an enzyme-cleaving protease. Despite its ability to cleave S1P/S2P, caspase 2 cannot cleave SREBP1/2.11 These observations are consistent with the notion that progression of NAFL to NASH may depend on a second hit, such as ER stress.11 caspase 2 activates SREBP1/2 through a mechanism that, although not fully understood, is not regulated by feedback inhibition by sterols or unsaturated FA, as observed in normal SCAP-dependent SREBP activation.11,63 Although caspase 2 does not trigger hepatocyte apoptosis, caspaseexcessive caspase 2-dependent cholesterol accumulation can increase hepatocyte susceptibility to TNF-induced mitochondrial dysfunction and death.104
4.6. Alternative activation of SREBP1/2
Apoptotic responses to TNFα in hepatocytes activate pro-apoptotic caspase-3,109 which mediates the release of SREBP from the ER membrane in an S1P-independent manner, leading to nuclear translocation of SREBP1/2 and transcriptional activation of multiple lipogenic genes. In addition, non-specific SREBP1/2 cleavage by caspases-4 and -12 was observed in alcohol-exposed cells.110
5. ER stress critically regulates de novo lipogenesis in hepatocytes.
Although the pathogenesis of NASH and ALD differs, the metabolic injury of hepatocytes is quite similar. ER stress and UPR activation play a critical role in the development of hepatic steatosis, inflammation, and fibrosis. The role of ER stress in de novo lipogenesis is discussed below.
The ER in hepatocytes has a remarkable capacity to adapt to extracellular and intracellular changes, ensuring that vital hepatic metabolic functions are preserved. However, hyperlipidemia and inflammation (specifically high levels of circulating TNFα) can perturb hepatocyte ER homeostasis, contributing to the dysregulation of hepatic lipid metabolism via activation of non-canonical TNF/TNFRI-caspase 2-S1P-dependent pathway of SERBP1/2 activation. ER stress leads to constitutive activation of SREBP1/2 and increased production of toxic lipids, including cholesterol, triglycerides, and fatty acids (Figure 4).
5.1. Role of the ER in cellular homeostasis
5.1.1. ER Functions.
The ER is a cellular organelle consisting of a continuous membrane system, tubules, sheets, and a nuclear envelope with enclosed sacs. The ER mediates many essential cell functions, including protein synthesis and processing, protein transport, lipid synthesis, and calcium storage.111 The ER is enriched in hepatocytes due to their unique metabolic functions such as lipogenesis and production of secretory proteins including albumin, alpha-1 antitrypsin, and lipoproteins.112 ER stress is caused by glucose starvation, depletion of calcium in the ER lumen, inhibition of glycosylation, reduction of disulfide bonds, or excessive accumulation of unfolded and misfolded proteins.113
5.1.2. Chaperones that regulate ER folding.
Chronic metabolic injury affects proper protein folding in the ER, leading to the accumulation of protein aggregates, cellular dysfunction, and programmed cell death. Inflammatory mediators, including free radicals such as nitric oxide (NO) and ROS, TNFα and other cytokines, and metabolic dysregulation can contribute to protein misfolding. In turn, improper protein folding can cause improper degradation, mislocalization, dominant-negative mutations, and structural alterations that establish novel toxic functions, which can cause disease. The UPR is an evolutionary conserved system that coordinates cellular responses to stress or injury to limit the accumulation of misfolded proteins in the ER and prevent cell death.113 Proper protein folding in the ER114 is controlled by a high concentration of chaperones.113,115 The first chaperone uses the ability of UDPglucose/glycoprotein glucosyltransferase (UGGT) to add a single glucose to misfolded proteins, making them accessible for binding to the lectin-like chaperones CNX and CRT that repair protein folding.116 The second ER chaperone system, GRP78/BiP, binds to hydrophobic residues of unfolded proteins and mediates their retrograde translocation and proteasomal degradation.113,117–119
5.2. UPR signaling is activated to reduce ER stress
The ER engages the UPR to control hepatic protein and lipid homeostasis.55,120 Although the initial UPR activation maintains tissue homeostasis and regulates lipogenesis, chronic UPR activation leads to dysregulation of the ER regulatory system, often resulting in increased production of misfolded proteins and uncontrolled lipogenesis. Recent studies report that chronic exhaustion of the UPR plays a critical role the pathogenesis of NASH.
Activation of the UPR signaling system restores ER homeostasis via: (a) increasing ER protein folding capacity through expansion of the ER and increased expression of chaperones (such as GRP78/BiP), (b) inhibition of protein translation to limit production of misfolded proteins121, and (c) activation of autophagy and/or ER-associated protein degradation (ERAD) system that reduces ER stress122 by re-directing misfolded proteins from the ER back into the cytosol for degradation by the 26S proteasome.114,123 ER stress and UPR activation regulate cellular processes beyond ER protein folding and play crucial roles in lipid metabolism.51,106,124,125
6. The UPR signaling pathways
The UPR is adaptive response to ER stress.112,126 UPR activation comprises 3 arms, each regulated by one of three transmembrane ER-located stress sensors: a) inositol-requiring enzyme 1 alpha (IRE1α), b) double-stranded RNA-activated protein kinase-like (PKR)-like ER kinase (PERK), and c) activating transcription factor (ATF6).55 While IRE1α and ATF6 are transcription factors, PERK is a global suppresser of protein synthesis. The N-terminus of these proteins is positioned in the ER lumen and the C-terminus in the cytosol, thus connecting the two cellular compartments. Each of these proteins controls their specific downstream signaling cascades through the transcription of UPR target genes, including SREBP. Under physiological conditions, the UPR is inactive (due to inhibitory binding of GRP78/BiP to IRE1α, PERK, and ATF6) to maintain normal proteostasis in healthy hepatocytes. Upon GRP78 dissociation, all branches of the UPR are activated.127,128 Protein disulfide isomerases (PDIs) regulate UPR stress sensors. In turn, transient UPR activation prevents sudden hepatotoxic injury and promotes cell survival.129 Thus, when the threshold of misfolded protein accumulation reaches a critical point, GRP78 dissociates from the ER stress sensors, leading to activation of UPR-specific sensors. Transient activation of UPR stress sensors is also controlled by PDIs,127,128,130 suggesting that multiple factors regulate transient stress sensor activation.55In contrast, chronic UPR activation131–133 leads to protein misfolding, imbalance of calcium homeostasis, and lipid biogenesis (Figure 5),55
Figure 5. ER stress activates three UPR arms.

Progression of steatosis (NAFLD) to steatohepatitis and fibrosis (NASH) is associated with development of ER stress and activation of the unfolded protein response (UPR) to control hepatic protein and lipid homeostasis. Under physiological conditions, UPR is inactive (due to inhibitory binding of GRP78/BiP to IRE1, PERK, and ATF6) to maintain normal proteostasis in healthy hepatocytes. Upon GRP78 dissociation, all 3 arms of the UPR are activated. In response to chronic injury, all arms of the UPR contribute to NASH and, to different extents, support de novo lipogenesis.
6.1. The IRE1a-XBP1 arm
IRE1, the most conserved ER stress sensor, has two isoforms: IRE1α and IRE1b.134 IREα is the most abundant and biologically important Type I ER transmembrane protein and exhibits dual enzymatic activities: serine/threonine kinase activity and endoribonuclease (RNase) activity on its cytosolic tail.62 IRE1α activation is triggered by the binding of ER chaperone Hsp47 or by the direct binding of unfolded proteins to IRE1α,135,136 triggering IRE1α dimerization through its luminal N-terminal domain and oligomerization and trans-autophosphorylation. Subsequent conformational changes in IRE1αinitiate activation of its RNase domain.137
The endoribonuclease (RNase) activity of IRE1α degrades many ER-bound mRNAs, including mIRE1a itself via the regulated IRE1α -dependent decay (RIDD) pathway (in collaboration with RTCB RNA ligases), and acts as an RNA splicing/repair enzyme.138,139 Upon activation of the tRNA ligase RTCB pathway, IRE1α RNase mediates unconventional splicing of Xbox binding protein 1 (XBP1) messenger RNA (by removing a 26-nucleotide sequence from XBP1 unspliced (XBP1u) mRNA, causing a translational frameshift to produce transcriptionally active XBP1 spliced (XBP1s). IRE1α phosphorylates and activates the XBP1 transcription factor XBP1 via its kinase activity. XBP1 translocates to the nucleus and induces transcription of its downstream target genes, including ER chaperones and genes involved in ERAD.140
IRE1α recruits TNF receptor associated factor 2 (TRAF2) and apoptosis signal-regulating kinase 1 (ASK1) to mediate the phosphorylation of c-jun N-terminal kinase and nuclear factor kappa B (NF-kB) pathways that transcriptionally activate inflammatory and apoptotic pathways. XBP1 directly regulates transcription of specific genes responsible for the regulation of lipid metabolism (such as farnesyl diphosphate synthase, hydroxysteroid 17-beta dehydrogenase 7,141 and fibroblast growth factor 21) to protect from ER stress-induced hepatic steatosis.142 Depletion of XBP1 results in rapid feedback activation of IREα.112 The IRE1α -XBP1 arm of the UPR plays a critical role in hepatic lipid metabolism through regulation of VLDL secretion and lipogenesis (Figure 5).143–145PERK-eIF2a-ATF4 arm
PERK is a Type I ER-resident transmembrane serine/threonine protein kinase consisting of an ER luminal stress-sensing domain and a cytosolic kinase domain. Upon oligomerization, PERK phosphorylates the subunit of eukaryotic translation initiation factor 2 (eIF2a) in response to ER stress. eIF2α serves as a major substrate of PERK146 and functions to relieve the protein overload in the ER by suppressing the formation of translation initiation complexes to prevent protein translation. Phospho-eIF2α facilitates translation and expression of transcription factor ATF4, which positively regulates transcription of UPR target genes involved in protein folding and autophagy. ATF4 also transcriptionally activates CCAAT-enhancer binding protein (C/EBP) homologous protein (CHOP), which is critical for ER stress mediated apoptosis, DNA damage-inducible protein GADD34, and ATF3.147–149 The PERK-eIF2α -ATF4 arm of the UPR regulates lipogenesis and steatosis. Phosphorylation of eIF2α is regulated on several levels. In a negative feedback mechanism, ATF4 induces expression of GADD34 and constitutive repressor of eIF2α phosphorylation (CReP), which interact with protein phosphatase 1 (PP1) to promote PP1-mediated de-phosphorylation of eIF2α.150 Consequently, ATF4 translation resumes, and ATF4 transactivates UPR target genes involved in protein folding, autophagy, redox homeostasis, amino acid metabolism, and apoptosis (Figure 5).147–149
6.2. ATF6 arm
ATF6, a type II transmembrane protein, contains a cytosolic bZip domain and possesses leucine zipper transcription factor activity.112 Full and truncated forms of ATF6 have been identified (ATF6a and ATF6b, respectively).151,152 Upon ER stress-induced activation, ATF6a (p90) is released from the inhibitory BiP protein and transported from the ER to the Golgi where it is cleaved by S1P and S2P proteases. Proteolytic cleavage of the full length ATF6 results in release of the N-terminal cytosolic transcription factor ATF6b (p50), which translocates to the nucleus and initiates the transcription of genes involved in protein folding and ERAD. ATF6 was also shown to activate the transcription of XBP1, CHOP, and BiP.153 ATF6 also forms heterodimers with XBP1 to induce transcription of multiple genes involved in ERAD.154 The ATF6a arm may provide responses that prevent excessive lipogenesis (Figure 5).
6.3. UPR proteins differentially regulate de novo lipogenesis
6.4.1. IRE1a-XBP1 pathway.
The IRE1α -XBP1 pathway directly drives hepatic steatosis, metabolic liver damage, and hypercholesterolemia (Table 2). XBP1 is a critical pro-lipogenic transcription factor155 that targets Lipin genes (LPIN1 and LPIN3), OSBP, LSS, and GPAT4. OSBP encodes a sterol–sensing protein that modulates SREBP activity in response to sterol PECR, an enzyme involved in fatty acid elongation.156,157 LSS catalyzes the formation of lanosterol from squalene, and GPAT4 adds a fatty acid to glycerol during lipogenesis.154,158,159 XBP1 ablation leads to a compensatory upregulation of its upstream enzyme IRE1α (but not PERK, ATF6, or other UPR proteins). These findings further support the proposed role of IRE1α in lipid metabolism and indicate that IRE1α activity is regulated by a feedback mechanism activated by low abundance of XBP1 (IRE1α substrate).154,160,161 Moreover, the IRE1α-regulated XBP1 and RIDD pathways have opposing effects on the expression of lipogenic genes, with the RIDD pathway promoting lipid hydrolysis and preventing lipid storage by reducing the expression of lipogenic genes.160 The silencing of lipid metabolism genes through the IRE1α -regulated mRNA decay RIDD system lowers plasma lipid concentrations.155,160 In addition, IRE1α RNase activity (but not kinase activity) increases the decay of select microRNAs (miR-17, -34a, -96, -125b) that repress translation of caspase 2 mRNA, thereby promoting caspase 2 expression. Targeting of either IRE1α or XBP1 might become a strategy for blocking de novo lipogenesis.162
Table 2: ER stress pathways are activated in response to metabolic injury induced by NASH or AALD.
The role of UPR-activated proteins in NASH and AALD is demonstrated.
| Liver Disease | UPR Component | Functions |
|---|---|---|
|
| ||
| NAFL/NASH | IRE1α | Regulates Hepatic lipogenesis through RIDD |
| Facilitates diet induced steatosis | ||
| Promotes the progression of NAFL to NASH | ||
|
|
||
| XBP1 | Promotes steatosis in metabolically injured hepatocytes | |
| Promotes diet-induced liver injury | ||
| Regulates Hepatic lipogenesis | ||
| Contributes to diet induced liver injury | ||
|
|
||
| ATF4 | Contributes to diet-induced steatosis | |
|
|
||
| GADD34 | Protects from diet-induced steatosis | |
|
|
||
| ATF6 | Protects from diet-induced steatosis | |
|
| ||
| AALD | BiP | Protects from alcohol-induced liver injury |
|
|
||
| ATF4 | Responsible for alcohol-induced steatosis | |
|
|
||
| CHOP | Promotes alcohol-induced liver injury | |
|
|
||
| ATF6 | Promotes alcohol-induced steatosis | |
Abbreviations: NAFLD, nonalcoholic fatty liver; NASH, nonalcoholic steatohepatitis; IRE1α, inositol-requiring enzyme 1α; XBP1, Xbox binding protein 1; PERK, double-stranded RNA-dependent protein kinase (PKR)-like ER kinase; ATF4, activating transcription factor 4; eIF2α, eukaryotic initiation factor 2a; GADD34, growth arrest and DNA damage-inducible protein 34; ATF6, activating transcription factor 6; RIDD, regulated IRE1a-dependent decay of mRNA; ALD, alcoholic liver disease; BiP, binding immunoglobulin protein; CHOP, CCAAT-enhancer-binding protein (C/EBP) homologous protein
6. 4.2. PERK-eIF2a-ATF4 pathway.
PERK positively regulates lipid synthesis via its downstream targets eIF2α, ATF4, and CHOP. The absence of PERK is associated with the downregulation of triglyceride and fatty acid production. In response to ER stress, PERK phosphorylates eIF2α, causing subsequent caspase 2-dependent SREBP1/2 cleavage/maturation in immune cells.163 Phosphorylated eIF2α facilitates translation of the transcription factor ATF4, which also increases de novo lipogenesis. The genes encoding the lipogenic enzymes Acac, Scd1, Fas, and Gpat are ATF4 targets.164,165 These findings indicate that the IREα -XBP1 and PERK-eIF2α -ATF4 pathways can be targeted to suppress caspase for NASH therapy.
2-SREBP1/2-dependent cholesterol and fatty acid synthesis
6.4.3. ATF6 pathway.
The role of ATF6 in de novo lipogenesis is poorly understood. The ATF6 pathway was originally implicated in the suppression of lipid metabolism because of its ability to induce ER expansion in an XBP1-independent manner.154,166,167 Further, ATF6 was reported to inhibit cholesterol synthesis via interaction with cleaved/activated SREBP2 and transcription inhibitor HDAC1, leading to the downregulation of HMGCR, HMGCS, FDFT1 (squalene synthase), and LDLR expression.168 ATF6 can also suppress hepatic triglyceride accumulation via regulation of transcriptional activity of PPARα/RXRα (retinoid X receptor alpha) heterodimers and activation of fatty acid oxidation (Cpt1, Cpt2, Acox1 and Ppara) and VLDL formation (Mttp, PDI and Apob).154,155 In another study, ATF6 activation was shown to upregulate the transcription and expression of XBP1 as well as genes involved in protein folding that support ERAD machinery, mediate ER homeostasis, and stabilize ER and Golgi biogenesis.153,169 ATF6 and XBP1s can form heterodimers that promote the expression of select genes involved in ERAD biologic functions.154,170,171
7. De novo lipogenesis in inflammatory cells and fibrogenic myofibroblasts facilitates NASH progression
7.1. Lipogenesis contributes to activation of inflammatory cells and hepatic myofibroblasts
De novo lipogenesis plays a similar role in other cells, including immune, inflammatory, and mesenchymal cells.2 Steatosis–inflammation–fibrosis mediated by lipid accumulation in different cell types and lipotoxicity is a final common pathway to the organ pathologies of immunometabolic disorders such as obesity, atherosclerosis, diabetes mellitus, NASH, chronic kidney disease, and neurological disorders. Severe cell stressors induce apoptosis through a terminal UPR. In energy-depleted states, lipids in lipid droplets are degraded via lipophagy to restore energy levels.
7.2. Lipogenesis promotes myeloid cell activation
Lipid metabolism is critical for the activation of myeloid cells, induction of inflammatory responses, the host defense mechanism, phagocytosis, and autophagy.2 Thus, macrophages internalize oxidized low-density lipoproteins and lipids from the environment, leading to the formation of foam cells with an inflammatory phenotype.172 Metabolic-sensing pathways coordinate shifts in lipid metabolism and regulate macrophage activation.173 LDLR expression is regulated by LXR, which acts as a cellular free-cholesterol–concentration sensor and mediates the expression of SREBP1c in myeloid cells174. SREBP1c was also shown to activate genes encoding inflammasome subunits in macrophages.66 SREBP2 is an important regulator of LDLR and SREBP1c expression. In addition, SREBP2 is necessary to produce an LXR ligand required for normal SREBP1c expression.175 174
7.3. Hepatic stellate cells
Lipid metabolism in myofibroblasts and HSCs is not fully understood.2 Activation of the vitamin A– retinoic acid signaling pathway, the presence of lipid droplets, and downregulation of PPARγ are involved in maintenance of the quiescent HSC phenotype.176,177. Quiescence-associated transcription factor ETS1 regulates PPARγ expression levels in qHSCs.178 In contrast, binding of methyl-CpG binding protein 2 (MeCP2) represses PPARγ transcription, leading to HSC activation into myofibroblasts.177 Emerging evidence indicates that excessive lipid accumulation facilitates fibrogenic activation of hepatic myofibroblasts. HSCs are sensitive to intracellular cholesterol levels, which causes their activation. Increased SREBP2 and microRNA-33a signaling was observed in activated HSCs and was linked to PPARγ suppression in activated HSCs. In turn, cholesterol accumulation in HSCs increases Toll-like receptor 4 protein (TLR4) levels through suppression of TLR4 endosomal-lysosomal degradation, thereby facilitating LPS and TGFβ signaling in HSCs.179 Curcumin suppresses LDLR and SREBP expression in activated HSCs by activating PPARγ, reducing cellular cholesterol, and attenuating HSC activation.180 In addition, curcumin directly regulates SREBP2 expression by suppressing specificity protein 1 (SP-1) transcription factor. The SREBP2 promoter contains an SP-1 binding GC-box, and SP-1 is implicated in elevated SREBP gene transcription.181 PPARγ and LXR play critical roles in the regulation of de novo lipogenesis and cholesterol homeostasis in HSCs. Crosstalk between PPARγ and LXR is modulated by expression of a mutant PNPLA3 allele (linked to accelerated NASH progression). HSCs carrying I148M PNPLA3 show impaired LXR signaling, leading to cholesterol accumulation and HSC activation182.
8. Concluding remarks
Lipids play a critical role in the maintenance of body homeostasis, as they serve as a source of energy and provide building blocks for cell membranes.2 The production of cholesterol and fatty acids also plays a role in the development of hepatic steatosis in response to metabolic injury. De novo lipogenesis contributes to the NASH pathogenesis and is triggered by obesity, insulin resistance, ROS production, ER stress, and TNFα -induced signaling. The synthesis of cholesterol and fatty acids is controlled on multiple levels, including the activation of non-canonical caspase 2-dependent S1P/S2P-induced processing and activation of SREBP1/2, the transcription factors that play a key role in triggering expression of the major lipogenic genes. Therefore, caspase 2 and S1P/S2P proteins are targets for the therapeutic suppression of de novo lipogenesis.2 Blocking of the upstream activators TNFα and IL-17 effectively suppresses the caspase 2-S1P/S2P-DHCR7 pathways, preventing cholesterol and fatty acid production.2 Other components of the UPR system directly or indirectly affect cholesterol synthesis. Blocking of the IRE1α -XBP1 and PERK-eIF2α -ATF4 pathways may suppress steatosis, while stimulation of ATF6 and the ERAD system can reduce steatosis by decreasing ER stress.
Grant support:
Supported by the National Institutes of Health R01DK101737, U01AA022614, R01DK099205, R01DK111866, R01AA028550, P50AA011999, U01AA018663, P30 DK120515, 5U01AA029019, R01DK091183, R01DK09920 (TK), P42ES010337 and R44DK115242 (DB). R01 AA24726, R37 AA020703, U01 AA026939, U01 AA026939–04S1, P30 DK120515 and P50 AA011999 (BS). RL receives funding support from NIEHS (5P42ES010337), NCATS (5UL1TR001442), DOD PRCRP (W81XWH-18–2-0026), NIDDK (U01DK061734, R01DK106419, R01DK121378, R01DK124318, P30DK120515), NHLBI (P01HL147835), and NIAAA (U01AA029019).
Abbreviations:
- NASH
nonalcoholic steatohepatitis
- AALD
alcohol-associated liver disease
- aHSCs
activated Hepatic Stellate Cells
- DHCR7
7-dehydrocholesterol reductase
- HMGCS1
cytoplasmic hydroxymethylglutaryl-CoA synthase
- SREBP
sterol regulatory element-binding protein
Footnotes
Conflict of interests: RL serves as a consultant for Aardvark Therapeutics, Altimmune, Anylam/Regeneron, Amgen, Arrowhead Pharmaceuticals, AstraZeneca, Bristol-Myer Squibb, CohBar, Eli Lilly, Galmed, Gilead, Glympse bio, Hightide, Inipharm, Intercept, Inventiva, Ionis, Janssen Inc., Madrigal, Metacrine, Inc., NGM Biopharmaceuticals, Novartis, Novo Nordisk, Merck, Pfizer, Sagimet, Theratechnologies, 89 bio, and Viking Therapeutics. In addition, his institution has received grant support from Allergan, Astrazeneca, Boehringer-Ingelheim, Bristol-Myers Squibb, Eli Lilly, Galectin Therapeutics, Galmed Pharmaceuticals, Genfit, Gilead, Intercept, Inventiva, Janssen, Madrigal Pharmaceuticals, Merck, NGM Biopharmaceuticals, Pfizer, and Sonic Incytes. He is also co-founder of Liponexus, Inc. BS has been consulting for Ferring Research Institute, Gelesis, HOST Therabiomics, Intercept Pharmaceuticals, Mabwell Therapeutics, Patara Pharmaceuticals and Takeda. BS is founder of Nterica Bio. UC San Diego has filed several patents with BS as inventor related to this work. BS’s institution UC San Diego has received research support from Axial Biotherapeutics, BiomX, CymaBay Therapeutics, NGM Biopharmaceuticals, Prodigy Biotech and Synlogic Operating Company.
REFERENCES
- 1.Friedman SL Liver fibrosis -- from bench to bedside. J Hepatol 38 Suppl 1, S38–53 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Kisseleva T & Brenner D Molecular and cellular mechanisms of liver fibrosis and its regression. Nat Rev Gastroenterol Hepatol 18, 151–166 (2021). [DOI] [PubMed] [Google Scholar]
- 3.Gao B, Ahmad MF, Nagy LE & Tsukamoto H Inflammatory pathways in alcoholic steatohepatitis. J Hepatol 70, 249–259 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iwaisako K, et al. Origin of myofibroblasts in the fibrotic liver in mice. Proc Natl Acad Sci U S A 111, E3297–3305 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Desmouliere A Hepatic stellate cells: the only cells involved in liver fibrogenesis? A dogma challenged. Gastroenterology 132, 2059–2062 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Huang DQ, El-Serag HB & Loomba R Global epidemiology of NAFLD-related HCC: trends, predictions, risk factors and prevention. Nature Reviews Gastroenterology & Hepatology 18, 223–238 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Younossi ZM, et al. Association of nonalcoholic fatty liver disease (NAFLD) with hepatocellular carcinoma (HCC) in the United States from 2004 to 2009. Hepatology 62, 1723–1730 (2015). [DOI] [PubMed] [Google Scholar]
- 8.O’Shea RS, Dasarathy S, McCullough AJ, Practice Guideline Committee of the American Association for the Study of Liver, D. & Practice Parameters Committee of the American College of, G. Alcoholic liver disease. Hepatology 51, 307–328 (2010). [DOI] [PubMed] [Google Scholar]
- 9.Lucey MR, Mathurin P & Morgan TR Alcoholic hepatitis. N Engl J Med 360, 2758–2769 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Takahashi Y & Fukusato T Histopathology of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J Gastroenterol 20, 15539–15548 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim JY, et al. ER Stress Drives Lipogenesis and Steatohepatitis via Caspase-2 Activation of S1P. Cell 175, 133–145 e115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsochatzis EA, Papatheodoridis GV & Archimandritis AJ Adipokines in nonalcoholic steatohepatitis: from pathogenesis to implications in diagnosis and therapy. Mediators Inflamm 2009, 831670 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moschen AR, Kaser S & Tilg H Non-alcoholic steatohepatitis: a microbiota-driven disease. Trends Endocrinol Metab 24, 537–545 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Avila MA, et al. Recent advances in alcohol-related liver disease (ALD): summary of a Gut round table meeting. Gut 69, 764–780 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cederbaum AI Alcohol metabolism. Clin Liver Dis 16, 667–685 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lieber CS, Rubin E & DeCarli LM Hepatic microsomal ethanol oxidizing system (MEOS): differentiation from alcohol dehydrogenase and NADPH oxidase. Biochem Biophys Res Commun 40, 858–865 (1970). [DOI] [PubMed] [Google Scholar]
- 17.Teschke R Alcoholic Liver Disease: Alcohol Metabolism, Cascade of Molecular Mechanisms, Cellular Targets, and Clinical Aspects. Biomedicines 6(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yan AW, et al. Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology 53, 96–105 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao B & Bataller R Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology 141, 1572–1585 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miller AM, Horiguchi N, Jeong WI, Radaeva S & Gao B Molecular mechanisms of alcoholic liver disease: innate immunity and cytokines. Alcohol Clin Exp Res 35, 787–793 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Giles DA, Moreno-Fernandez ME & Divanovic S IL-17 Axis Driven Inflammation in Non-Alcoholic Fatty Liver Disease Progression. Curr Drug Targets 16, 1315–1323 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McGeachy MJ, Cua DJ & Gaffen SL The IL-17 Family of Cytokines in Health and Disease. Immunity 50, 892–906 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oppmann B, et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity 13, 715–725 (2000). [DOI] [PubMed] [Google Scholar]
- 24.Zenewicz LA, et al. Interleukin-22 but not interleukin-17 provides protection to hepatocytes during acute liver inflammation. Immunity 27, 647–659 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Radaeva S, Sun R, Pan HN, Hong F & Gao B Interleukin 22 (IL-22) plays a protective role in T cell-mediated murine hepatitis: IL-22 is a survival factor for hepatocytes via STAT3 activation. Hepatology 39, 1332–1342 (2004). [DOI] [PubMed] [Google Scholar]
- 26.Kong X, Feng D, Mathews S & Gao B Hepatoprotective and anti-fibrotic functions of interleukin-22: therapeutic potential for the treatment of alcoholic liver disease. J Gastroenterol Hepatol 28 Suppl 1, 56–60 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou D, et al. Total fecal microbiota transplantation alleviates high-fat diet-induced steatohepatitis in mice via beneficial regulation of gut microbiota. Sci Rep 7, 1529 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shalapour S, et al. Inflammation-induced IgA+ cells dismantle anti-liver cancer immunity. Nature 551, 340–345 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Llacuna L, et al. Reactive oxygen species mediate liver injury through parenchymal nuclear factor-kappaB inactivation in prolonged ischemia/reperfusion. Am J Pathol 174, 1776–1785 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu RM & Desai LP Reciprocal regulation of TGF-β and reactive oxygen species: A perverse cycle for fibrosis. Redox Biol 6, 565–577 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hattori S, et al. FR-167653, a selective p38 MAPK inhibitor, exerts salutary effect on liver cirrhosis through downregulation of Runx2. Lab Invest 87, 591–601 (2007). [DOI] [PubMed] [Google Scholar]
- 32.Foo NP, Lin SH, Lee YH, Wu MJ & Wang YJ α-Lipoic acid inhibits liver fibrosis through the attenuation of ROS-triggered signaling in hepatic stellate cells activated by PDGF and TGF-β. Toxicology 282, 39–46 (2011). [DOI] [PubMed] [Google Scholar]
- 33.Canbay A, et al. Apoptotic body engulfment by a human stellate cell line is profibrogenic. Lab Invest 83, 655–663 (2003). [DOI] [PubMed] [Google Scholar]
- 34.Canbay A, et al. Kupffer cell engulfment of apoptotic bodies stimulates death ligand and cytokine expression. Hepatology 38, 1188–1198 (2003). [DOI] [PubMed] [Google Scholar]
- 35.Garcíade León Mdel C, et al. Hepatocyte production of modulators of extracellular liver matrix in normal and cirrhotic rat liver. Exp Mol Pathol 80, 97–108 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Ibrahim SH, et al. Mixed lineage kinase 3 mediates release of C-X-C motif ligand 10-bearing chemotactic extracellular vesicles from lipotoxic hepatocytes. Hepatology 63, 731–744 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Povero D, et al. Lipid-induced hepatocyte-derived extracellular vesicles regulate hepatic stellate cell via microRNAs targeting PPAR-γ. Cell Mol Gastroenterol Hepatol 1, 646–663.e644 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eguchi A, et al. Comprehensive characterization of hepatocyte-derived extracellular vesicles identifies direct miRNA-based regulation of hepatic stellate cells and DAMP-based hepatic macrophage IL-1β and IL-17 upregulation in alcoholic hepatitis mice. J Mol Med (Berl) 98, 1021–1034 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee YS, et al. Exosomes derived from palmitic acid-treated hepatocytes induce fibrotic activation of hepatic stellate cells. Sci Rep 7, 3710 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hernández A, et al. Extracellular vesicles derived from fat-laden hepatocytes undergoing chemical hypoxia promote a pro-fibrotic phenotype in hepatic stellate cells. Biochim Biophys Acta Mol Basis Dis 1866, 165857 (2020). [DOI] [PubMed] [Google Scholar]
- 41.Niimura F, Okubo S, Fogo A & Ichikawa I Temporal and spatial expression pattern of the angiotensinogen gene in mice and rats. Am J Physiol 272, R142–147 (1997). [DOI] [PubMed] [Google Scholar]
- 42.Bataller R, et al. Angiotensin II induces contraction and proliferation of human hepatic stellate cells. Gastroenterology 118, 1149–1156 (2000). [DOI] [PubMed] [Google Scholar]
- 43.Granzow M, et al. Angiotensin-II type 1 receptor-mediated Janus kinase 2 activation induces liver fibrosis. Hepatology 60, 334–348 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang X, et al. Cholesterol Stabilizes TAZ in Hepatocytes to Promote Experimental Non-alcoholic Steatohepatitis. Cell Metab 31, 969–986 e967 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang X, et al. Hepatocyte TAZ/WWTR1 Promotes Inflammation and Fibrosis in Nonalcoholic Steatohepatitis. Cell Metab 24, 848–862 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang X, et al. A Therapeutic Silencing RNA Targeting Hepatocyte TAZ Prevents and Reverses Fibrosis in Nonalcoholic Steatohepatitis in Mice. Hepatol Commun 3, 1221–1234 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Canbay A, Bechmann L & Gerken G Lipid metabolism in the liver. Z Gastroenterol 45, 35–41 (2007). [DOI] [PubMed] [Google Scholar]
- 48.Ma HY, et al. IL-17 signaling in steatotic hepatocytes and macrophages promotes hepatocellular carcinoma in alcohol-related liver disease. J Hepatol (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Donnelly KL, et al. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest 115, 1343–1351 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hager L, et al. Lecithin:cholesterol acyltransferase deficiency protects against cholesterol-induced hepatic endoplasmic reticulum stress in mice. J Biol Chem 287, 20755–20768 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Basseri S & Austin RC Endoplasmic reticulum stress and lipid metabolism: mechanisms and therapeutic potential. Biochem Res Int 2012, 841362 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Davidson NO & Shelness GS APOLIPOPROTEIN B: mRNA editing, lipoprotein assembly, and presecretory degradation. Annu Rev Nutr 20, 169–193 (2000). [DOI] [PubMed] [Google Scholar]
- 53.Saran AR, Dave S & Zarrinpar A Circadian Rhythms in the Pathogenesis and Treatment of Fatty Liver Disease. Gastroenterology 158, 1948–1966 e1941 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pouwels S, et al. Non-alcoholic fatty liver disease (NAFLD): a review of pathophysiology, clinical management and effects of weight loss. BMC Endocr Disord 22, 63 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lebeaupin C, et al. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J Hepatol 69, 927–947 (2018). [DOI] [PubMed] [Google Scholar]
- 56.Kleiner DE, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 41, 1313–1321 (2005). [DOI] [PubMed] [Google Scholar]
- 57.Loomba R & Sanyal AJ The global NAFLD epidemic. Nat Rev Gastroenterol Hepatol 10, 686–690 (2013). [DOI] [PubMed] [Google Scholar]
- 58.Caballero F, et al. Enhanced free cholesterol, SREBP-2 and StAR expression in human NASH. J Hepatol 50, 789–796 (2009). [DOI] [PubMed] [Google Scholar]
- 59.Farrell GC & van Rooyen D Liver cholesterol: is it playing possum in NASH? Am J Physiol Gastrointest Liver Physiol 303, G9–11 (2012). [DOI] [PubMed] [Google Scholar]
- 60.Shimano H & Sato R SREBP-regulated lipid metabolism: convergent physiology - divergent pathophysiology. Nat Rev Endocrinol 13, 710–730 (2017). [DOI] [PubMed] [Google Scholar]
- 61.Bertolio R, et al. Sterol regulatory element binding protein 1 couples mechanical cues and lipid metabolism. Nat Commun 10, 1326 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Adams CJ, Kopp MC, Larburu N, Nowak PR & Ali MMU. Structure and Molecular Mechanism of ER Stress Signaling by the Unfolded Protein Response Signal Activator IRE1. Front Mol Biosci 6, 11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brown MS & Goldstein JL The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 89, 331–340 (1997). [DOI] [PubMed] [Google Scholar]
- 64.Horton JD, Goldstein JL & Brown MS SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest 109, 1125–1131 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Toth JI, Datta S, Athanikar JN, Freedman LP & Osborne TF Selective coactivator interactions in gene activation by SREBP-1a and -1c. Mol Cell Biol 24, 8288–8300 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Im SS, et al. Linking lipid metabolism to the innate immune response in macrophages through sterol regulatory element binding protein-1a. Cell Metab 13, 540–549 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shimano H, et al. Overproduction of cholesterol and fatty acids causes massive liver enlargement in transgenic mice expressing truncated SREBP-1a. J Clin Invest 98, 1575–1584 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brown MS, Radhakrishnan A & Goldstein JL Retrospective on Cholesterol Homeostasis: The Central Role of Scap. Annu Rev Biochem 87, 783–807 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Azzu V, et al. Suppression of insulin-induced gene 1 (INSIG1) function promotes hepatic lipid remodelling and restrains NASH progression. Mol Metab 48, 101210 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee SH, Lee JH & Im SS The cellular function of SCAP in metabolic signaling. Exp Mol Med 52, 724–729 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yabe D, Komuro R, Liang G, Goldstein JL & Brown MS Liver-specific mRNA for Insig-2 down-regulated by insulin: implications for fatty acid synthesis. Proc Natl Acad Sci U S A 100, 3155–3160 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yellaturu CR, et al. Insulin enhances post-translational processing of nascent SREBP-1c by promoting its phosphorylation and association with COPII vesicles. J Biol Chem 284, 7518–7532 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hannah VC, Ou J, Luong A, Goldstein JL & Brown MS Unsaturated fatty acids down-regulate srebp isoforms 1a and 1c by two mechanisms in HEK-293 cells. J Biol Chem 276, 4365–4372 (2001). [DOI] [PubMed] [Google Scholar]
- 74.Owen JL, et al. Insulin stimulation of SREBP-1c processing in transgenic rat hepatocytes requires p70 S6-kinase. Proc Natl Acad Sci U S A 109, 16184–16189 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brown AJ, Sun L, Feramisco JD, Brown MS & Goldstein JL Cholesterol addition to ER membranes alters conformation of SCAP, the SREBP escort protein that regulates cholesterol metabolism. Mol Cell 10, 237–245 (2002). [DOI] [PubMed] [Google Scholar]
- 76.Goldstein JL, Rawson RB & Brown MS Mutant mammalian cells as tools to delineate the sterol regulatory element-binding protein pathway for feedback regulation of lipid synthesis. Arch Biochem Biophys 397, 139–148 (2002). [DOI] [PubMed] [Google Scholar]
- 77.Espenshade PJ & Hughes AL Regulation of sterol synthesis in eukaryotes. Annu Rev Genet 41, 401–427 (2007). [DOI] [PubMed] [Google Scholar]
- 78.Sakai J, et al. Sterol-regulated release of SREBP-2 from cell membranes requires two sequential cleavages, one within a transmembrane segment. Cell 85, 1037–1046 (1996). [DOI] [PubMed] [Google Scholar]
- 79.Sakai J, et al. Molecular identification of the sterol-regulated luminal protease that cleaves SREBPs and controls lipid composition of animal cells. Mol Cell 2, 505–514 (1998). [DOI] [PubMed] [Google Scholar]
- 80.Ye J, Dave UP, Grishin NV, Goldstein JL & Brown MS Asparagine-proline sequence within membrane-spanning segment of SREBP triggers intramembrane cleavage by site-2 protease. Proc Natl Acad Sci U S A 97, 5123–5128 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nagoshi E & Yoneda Y Dimerization of sterol regulatory element-binding protein 2 via the helix-loop-helix-leucine zipper domain is a prerequisite for its nuclear localization mediated by importin beta. Mol Cell Biol 21, 2779–2789 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lee SJ, et al. The structure of importin-beta bound to SREBP-2: nuclear import of a transcription factor. Science 302, 1571–1575 (2003). [DOI] [PubMed] [Google Scholar]
- 83.Shimomura I, et al. Insulin selectively increases SREBP-1c mRNA in the livers of rats with streptozotocin-induced diabetes. Proc Natl Acad Sci U S A 96, 13656–13661 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liu Y, et al. Nuclear transport modulation reduces hypercholesterolemia, atherosclerosis, and fatty liver. J Am Heart Assoc 2, e000093 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Peterson TR, et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 146, 408–420 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hirano Y, Yoshida M, Shimizu M & Sato R Direct demonstration of rapid degradation of nuclear sterol regulatory element-binding proteins by the ubiquitin-proteasome pathway. J Biol Chem 276, 36431–36437 (2001). [DOI] [PubMed] [Google Scholar]
- 87.Kuan YC, et al. Heat Shock Protein 90 Modulates Lipid Homeostasis by Regulating the Stability and Function of Sterol Regulatory Element-binding Protein (SREBP) and SREBP Cleavage-activating Protein. J Biol Chem 292, 3016–3028 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gong Y, et al. Sterol-regulated ubiquitination and degradation of Insig-1 creates a convergent mechanism for feedback control of cholesterol synthesis and uptake. Cell Metab 3, 15–24 (2006). [DOI] [PubMed] [Google Scholar]
- 89.Asano L, et al. Vitamin D Metabolite, 25-Hydroxyvitamin D, Regulates Lipid Metabolism by Inducing Degradation of SREBP/SCAP. Cell Chem Biol 24, 207–217 (2017). [DOI] [PubMed] [Google Scholar]
- 90.Goldstein JL, DeBose-Boyd RA & Brown MS Protein sensors for membrane sterols. Cell 124, 35–46 (2006). [DOI] [PubMed] [Google Scholar]
- 91.Yabe D, Brown MS & Goldstein JL Insig-2, a second endoplasmic reticulum protein that binds SCAP and blocks export of sterol regulatory element-binding proteins. Proc Natl Acad Sci U S A 99, 12753–12758 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yang T, et al. Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell 110, 489–500 (2002). [DOI] [PubMed] [Google Scholar]
- 93.Brown MS & Goldstein JL Selective versus total insulin resistance: a pathogenic paradox. Cell Metab 7, 95–96 (2008). [DOI] [PubMed] [Google Scholar]
- 94.Kohjima M, et al. SREBP-1c, regulated by the insulin and AMPK signaling pathways, plays a role in nonalcoholic fatty liver disease. Int J Mol Med 21, 507–511 (2008). [PubMed] [Google Scholar]
- 95.Li S, Brown MS & Goldstein JL Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc Natl Acad Sci U S A 107, 3441–3446 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ai D, et al. Activation of ER stress and mTORC1 suppresses hepatic sortilin-1 levels in obese mice. J Clin Invest 122, 1677–1687 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schadinger SE, Bucher NL, Schreiber BM & Farmer SR PPARgamma2 regulates lipogenesis and lipid accumulation in steatotic hepatocytes. Am J Physiol Endocrinol Metab 288, E1195–1205 (2005). [DOI] [PubMed] [Google Scholar]
- 98.Repa JJ, et al. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev 14, 2819–2830 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nguyen P, et al. Liver lipid metabolism. J Anim Physiol Anim Nutr (Berl) 92, 272–283 (2008). [DOI] [PubMed] [Google Scholar]
- 100.Oliner JD, Andresen JM, Hansen SK, Zhou S & Tjian R SREBP transcriptional activity is mediated through an interaction with the CREB-binding protein. Genes Dev 10, 2903–2911 (1996). [DOI] [PubMed] [Google Scholar]
- 101.Yang F, et al. An ARC/Mediator subunit required for SREBP control of cholesterol and lipid homeostasis. Nature 442, 700–704 (2006). [DOI] [PubMed] [Google Scholar]
- 102.Nakagawa H, et al. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 26, 331–343 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Farrell GC, van Rooyen D, Gan L & Chitturi S NASH is an Inflammatory Disorder: Pathogenic, Prognostic and Therapeutic Implications. Gut Liver 6, 149–171 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mari M, et al. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab 4, 185–198 (2006). [DOI] [PubMed] [Google Scholar]
- 105.Banhegyi G, et al. Endoplasmic reticulum stress. Ann N Y Acad Sci 1113, 58–71 (2007). [DOI] [PubMed] [Google Scholar]
- 106.Bobrovnikova-Marjon E, et al. PERK-dependent regulation of lipogenesis during mouse mammary gland development and adipocyte differentiation. Proc Natl Acad Sci U S A 105, 16314–16319 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lee JN & Ye J Proteolytic activation of sterol regulatory element-binding protein induced by cellular stress through depletion of Insig-1. J Biol Chem 279, 45257–45265 (2004). [DOI] [PubMed] [Google Scholar]
- 108.Colgan SM, Hashimi AA & Austin RC Endoplasmic reticulum stress and lipid dysregulation. Expert Rev Mol Med 13, e4 (2011). [DOI] [PubMed] [Google Scholar]
- 109.Wang X, et al. Cleavage of sterol regulatory element binding proteins (SREBPs) by CPP32 during apoptosis. EMBO J 15, 1012–1020 (1996). [PMC free article] [PubMed] [Google Scholar]
- 110.Pastorino JG & Shulga N Tumor necrosis factor-alpha can provoke cleavage and activation of sterol regulatory element-binding protein in ethanol-exposed cells via a caspase-dependent pathway that is cholesterol insensitive. J Biol Chem 283, 25638–25649 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Schwarz DS & Blower MD The endoplasmic reticulum: structure, function and response to cellular signaling. Cell Mol Life Sci 73, 79–94 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Liu X & Green RM Endoplasmic reticulum stress and liver diseases. Liver Res 3, 55–64 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Boelens J, Lust S, Offner F, Bracke ME & Vanhoecke BW Review. The endoplasmic reticulum: a target for new anticancer drugs. In Vivo 21, 215–226 (2007). [PubMed] [Google Scholar]
- 114.Gething MJ & Sambrook J Protein folding in the cell. Nature 355, 33–45 (1992). [DOI] [PubMed] [Google Scholar]
- 115.Ruddon RW & Bedows E Assisted protein folding. J Biol Chem 272, 3125–3128 (1997). [DOI] [PubMed] [Google Scholar]
- 116.Hammond C, Braakman I & Helenius A Role of N-linked oligosaccharide recognition, glucose trimming, and calnexin in glycoprotein folding and quality control. Proc Natl Acad Sci U S A 91, 913–917 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Knittler MR, Dirks S & Haas IG Molecular chaperones involved in protein degradation in the endoplasmic reticulum: quantitative interaction of the heat shock cognate protein BiP with partially folded immunoglobulin light chains that are degraded in the endoplasmic reticulum. Proc Natl Acad Sci U S A 92, 1764–1768 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Blond-Elguindi S, et al. Affinity panning of a library of peptides displayed on bacteriophages reveals the binding specificity of BiP. Cell 75, 717–728 (1993). [DOI] [PubMed] [Google Scholar]
- 119.Brodsky JL, et al. The requirement for molecular chaperones during endoplasmic reticulum-associated protein degradation demonstrates that protein export and import are mechanistically distinct. J Biol Chem 274, 3453–3460 (1999). [DOI] [PubMed] [Google Scholar]
- 120.Walter P & Ron D The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086 (2011). [DOI] [PubMed] [Google Scholar]
- 121.Harding HP, Zhang Y & Ron D Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397, 271–274 (1999). [DOI] [PubMed] [Google Scholar]
- 122.Hosoi T & Ozawa K Endoplasmic reticulum stress in disease: mechanisms and therapeutic opportunities. Clin Sci (Lond) 118, 19–29 (2009). [DOI] [PubMed] [Google Scholar]
- 123.Meusser B, Hirsch C, Jarosch E & Sommer T ERAD: the long road to destruction. Nat Cell Biol 7, 766–772 (2005). [DOI] [PubMed] [Google Scholar]
- 124.Kammoun HL, et al. GRP78 expression inhibits insulin and ER stress-induced SREBP-1c activation and reduces hepatic steatosis in mice. J Clin Invest 119, 1201–1215 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhang K, et al. The unfolded protein response transducer IRE1alpha prevents ER stress-induced hepatic steatosis. EMBO J 30, 1357–1375 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhang K & Kaufman RJ Signaling the unfolded protein response from the endoplasmic reticulum. J Biol Chem 279, 25935–25938 (2004). [DOI] [PubMed] [Google Scholar]
- 127.Groenendyk J, et al. Interplay between the oxidoreductase PDIA6 and microRNA-322 controls the response to disrupted endoplasmic reticulum calcium homeostasis. Sci Signal 7, ra54 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Eletto D, Eletto D, Dersh D, Gidalevitz T & Argon Y Protein disulfide isomerase A6 controls the decay of IRE1alpha signaling via disulfide-dependent association. Mol Cell 53, 562–576 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hur KY, et al. IRE1alpha activation protects mice against acetaminophen-induced hepatotoxicity. J Exp Med 209, 307–318 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Higa A, et al. Endoplasmic reticulum stress-activated transcription factor ATF6alpha requires the disulfide isomerase PDIA5 to modulate chemoresistance. Mol Cell Biol 34, 1839–1849 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lake AD, et al. The adaptive endoplasmic reticulum stress response to lipotoxicity in progressive human nonalcoholic fatty liver disease. Toxicol Sci 137, 26–35 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lee S, Kim S, Hwang S, Cherrington NJ & Ryu DY Dysregulated expression of proteins associated with ER stress, autophagy and apoptosis in tissues from nonalcoholic fatty liver disease. Oncotarget 8, 63370–63381 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Puri P, et al. Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology 134, 568–576 (2008). [DOI] [PubMed] [Google Scholar]
- 134.Tirasophon W, Welihinda AA & Kaufman RJ A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev 12, 1812–1824 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sepulveda D, et al. Interactome Screening Identifies the ER Luminal Chaperone Hsp47 as a Regulator of the Unfolded Protein Response Transducer IRE1alpha. Mol Cell 69, 238–252 e237 (2018). [DOI] [PubMed] [Google Scholar]
- 136.Karagoz GE, et al. An unfolded protein-induced conformational switch activates mammalian IRE1. Elife 6(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lee KP, et al. Structure of the dual enzyme Ire1 reveals the basis for catalysis and regulation in nonconventional RNA splicing. Cell 132, 89–100 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Hollien J, et al. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J Cell Biol 186, 323–331 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hollien J & Weissman JS Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 313, 104–107 (2006). [DOI] [PubMed] [Google Scholar]
- 140.Siwecka N, et al. The Structure, Activation and Signaling of IRE1 and Its Role in Determining Cell Fate. Biomedicines 9(2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lee AH, Iwakoshi NN & Glimcher LH XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol 23, 7448–7459 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Jiang S, et al. Fibroblast growth factor 21 is regulated by the IRE1alpha-XBP1 branch of the unfolded protein response and counteracts endoplasmic reticulum stress-induced hepatic steatosis. J Biol Chem 289, 29751–29765 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Reimold AM, et al. An essential role in liver development for transcription factor XBP-1. Genes Dev 14, 152–157 (2000). [PMC free article] [PubMed] [Google Scholar]
- 144.Zhang K, et al. The unfolded protein response sensor IRE1alpha is required at 2 distinct steps in B cell lymphopoiesis. J Clin Invest 115, 268–281 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wang S, et al. IRE1alpha-XBP1s induces PDI expression to increase MTP activity for hepatic VLDL assembly and lipid homeostasis. Cell Metab 16, 473–486 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Shi Y, et al. Identification and characterization of pancreatic eukaryotic initiation factor 2 alpha-subunit kinase, PEK, involved in translational control. Mol Cell Biol 18, 7499–7509 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Harding HP, et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell 6, 1099–1108 (2000). [DOI] [PubMed] [Google Scholar]
- 148.Jiang HY, et al. Activating transcription factor 3 is integral to the eukaryotic initiation factor 2 kinase stress response. Mol Cell Biol 24, 1365–1377 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Novoa I, Zeng H, Harding HP & Ron D Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J Cell Biol 153, 1011–1022 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Harding HP, et al. Ppp1r15 gene knockout reveals an essential role for translation initiation factor 2 alpha (eIF2alpha) dephosphorylation in mammalian development. Proc Natl Acad Sci U S A 106, 1832–1837 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Adachi Y, et al. ATF6 is a transcription factor specializing in the regulation of quality control proteins in the endoplasmic reticulum. Cell Struct Funct 33, 75–89 (2008). [DOI] [PubMed] [Google Scholar]
- 152.Wu J, et al. ATF6alpha optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev Cell 13, 351–364 (2007). [DOI] [PubMed] [Google Scholar]
- 153.Yoshida H, Matsui T, Yamamoto A, Okada T & Mori K XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891 (2001). [DOI] [PubMed] [Google Scholar]
- 154.Yamamoto K, et al. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev Cell 13, 365–376 (2007). [DOI] [PubMed] [Google Scholar]
- 155.Moncan M, et al. Regulation of lipid metabolism by the unfolded protein response. J Cell Mol Med 25, 1359–1370 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Raychaudhuri S & Prinz WA The diverse functions of oxysterol-binding proteins. Annu Rev Cell Dev Biol 26, 157–177 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Das AK, Uhler MD & Hajra AK Molecular cloning and expression of mammalian peroxisomal trans-2-enoyl-coenzyme A reductase cDNAs. J Biol Chem 275, 24333–24340 (2000). [DOI] [PubMed] [Google Scholar]
- 158.Baker CH, Matsuda SP, Liu DR & Corey EJ Molecular cloning of the human gene encoding lanosterol synthase from a liver cDNA library. Biochem Biophys Res Commun 213, 154–160 (1995). [DOI] [PubMed] [Google Scholar]
- 159.Chen YQ, et al. AGPAT6 is a novel microsomal glycerol-3-phosphate acyltransferase. J Biol Chem 283, 10048–10057 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.So JS, et al. Silencing of lipid metabolism genes through IRE1alpha-mediated mRNA decay lowers plasma lipids in mice. Cell Metab 16, 487–499 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Lee AH & Glimcher LH Intersection of the unfolded protein response and hepatic lipid metabolism. Cell Mol Life Sci 66, 2835–2850 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Upton JP, et al. IRE1alpha cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase-2. Science 338, 818–822 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Moserova I, et al. Caspase-2 and oxidative stress underlie the immunogenic potential of high hydrostatic pressure-induced cancer cell death. Oncoimmunology 6, e1258505 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Li H, et al. ATF4 deficiency protects mice from high-carbohydrate-diet-induced liver steatosis. Biochem J 438, 283–289 (2011). [DOI] [PubMed] [Google Scholar]
- 165.Xiao G, et al. ATF4 protein deficiency protects against high fructose-induced hypertriglyceridemia in mice. J Biol Chem 288, 25350–25361 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Bommiasamy H, et al. ATF6alpha induces XBP1-independent expansion of the endoplasmic reticulum. J Cell Sci 122, 1626–1636 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Chen X, et al. Hepatic ATF6 Increases Fatty Acid Oxidation to Attenuate Hepatic Steatosis in Mice Through Peroxisome Proliferator-Activated Receptor alpha. Diabetes 65, 1904–1915 (2016). [DOI] [PubMed] [Google Scholar]
- 168.Zeng L, et al. ATF6 modulates SREBP2-mediated lipogenesis. EMBO J 23, 950–958 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Okada T, Yoshida H, Akazawa R, Negishi M & Mori K Distinct roles of activating transcription factor 6 (ATF6) and double-stranded RNA-activated protein kinase-like endoplasmic reticulum kinase (PERK) in transcription during the mammalian unfolded protein response. Biochem J 366, 585–594 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Shoulders MD, et al. Stress-independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell Rep 3, 1279–1292 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Glembotski CC, Rosarda JD & Wiseman RL Proteostasis and Beyond: ATF6 in Ischemic Disease. Trends Mol Med 25, 538–550 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Batista-Gonzalez A, Vidal R, Criollo A & Carreno LJ New Insights on the Role of Lipid Metabolism in the Metabolic Reprogramming of Macrophages. Front Immunol 10, 2993 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Artyomov MN, Sergushichev A & Schilling JD Integrating immunometabolism and macrophage diversity. Semin Immunol 28, 417–424 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Sukhorukov VN, et al. Lipid Metabolism in Macrophages: Focus on Atherosclerosis. Biomedicines 8(2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Rong S, et al. Expression of SREBP-1c Requires SREBP-2-mediated Generation of a Sterol Ligand for LXR in Livers of Mice. Elife 6(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Hazra S, et al. Peroxisome proliferator-activated receptor gamma induces a phenotypic switch from activated to quiescent hepatic stellate cells. J Biol Chem 279, 11392–11401 (2004). [DOI] [PubMed] [Google Scholar]
- 177.Mann J, et al. MeCP2 controls an epigenetic pathway that promotes myofibroblast transdifferentiation and fibrosis. Gastroenterology 138, 705–714, 714 e701–704 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Liu X, et al. Identification of Lineage-specific Transcription Factors That Prevent Activation of Hepatic Stellate Cells and Promote Fibrosis Resolution. Gastroenterology (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Tomita K, et al. Free cholesterol accumulation in hepatic stellate cells: mechanism of liver fibrosis aggravation in nonalcoholic steatohepatitis in mice. Hepatology 59, 154–169 (2014). [DOI] [PubMed] [Google Scholar]
- 180.Kang Q & Chen A Curcumin suppresses expression of low-density lipoprotein (LDL) receptor, leading to the inhibition of LDL-induced activation of hepatic stellate cells. Br J Pharmacol 157, 1354–1367 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Kang Q & Chen A Curcumin inhibits srebp-2 expression in activated hepatic stellate cells in vitro by reducing the activity of specificity protein-1. Endocrinology 150, 5384–5394 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Bruschi FV, et al. PNPLA3 I148M Variant Impairs Liver X Receptor Signaling and Cholesterol Homeostasis in Human Hepatic Stellate Cells. Hepatol Commun 3, 1191–1204 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]