

ABSTRACT
Solid tumors consist of malignant and nonmalignant cells that together create the local tumor microenvironment (TME). Additionally, the TME is characterized by the expression of numerous soluble factors such as TGF-β. TGF-β plays an important role in the TME by suppressing T cell effector function and promoting tumor invasiveness. Up to now CAR T cells exclusively target tumor-associated antigens (TAA) located on the cell membrane. Thus, strategies to exploit soluble antigens as CAR targets within the TME are needed. This study demonstrates a novel approach using Adapter CAR (AdCAR) T cells for the detection of soluble latent TGF-β within the TME of a pancreatic tumor model. We show that AdCARs in combination with the respective adapter can be used to sense soluble tumor-derived latent TGF-β, both in vitro and in vivo. Sensing of the soluble antigen induced cellular activation and effector cytokine production in AdCAR T cells. Moreover, we evaluated AdCAR T cells for the combined targeting of soluble latent TGF-β and tumor cell killing by targeting CD66c as TAA in vivo. In sum, our study broadens the spectrum of targetable moieties for AdCAR T cells by soluble latent TGF-β.
KEYWORDS: Immunotherapy, CAR T cells, adapter CAR, TGF-β
Introduction
After the first report on CAR-expressing T cells, this emerging therapeutic concept has revolutionized cancer treatment.1 All CAR T cell therapies that are approved by the U.S. Food and Drug Administration target hematologic diseases. However, the translation for solid cancer treatment remains challenging due to the complex and immunosuppressive tumor microenvironment (TME). Physical barriers, cellular factors, and signaling molecules have been described to dampen CAR T cell efficacy.2 Thus, harnessing CAR T cell responses in the suppressive TME is highly relevant to improve the therapeutic outcome of CAR T cells in solid cancers. One strategy to enhance therapeutic efficacy is to target the tumor stroma or neovasculature. CARs have been developed against cell surface-expressed proteins, e.g. EIIIB or the fibroblast activation protein (FAP). Targeting the stroma with CAR T cells showed anti-tumor effects in several murine tumor models and was already translated into a clinical trial without severe side effects.3–7 These data stress the feasibility of this therapeutic concept. All of these approaches have in common that engineered lymphocytes are directed against cell-bound antigens, neglecting the huge repertoire of soluble targets associated with a pro-tumorigenic environment, e.g. chemokines or cytokines. Soluble molecules like chemokines were already shown to be potential candidates to enhance CAR T cell function in solid tumors. CAR T cells co-expressing the CXCR6 receptor were able to revert disease-promoting CXCL16 into an attraction signal for CAR T cells.8
The current knowledge of how to use soluble ligands as functional CAR targets is limited. So far it has been shown that soluble CAR ligands like CEA, CD30, Lewis-Y antigen, or glypican-3 either do not induce CAR signaling or in the worst case interfere with the targeting of cell-bound antigens.9–12 However, it was demonstrated in a proof of concept study that CAR T cells could be stimulated by soluble antigens directly via the CAR.13
One major immunosuppressive factor in the TME is TGF-β. This molecule is a multifunctional cytokine frequently found in the TME of various solid cancer types, e.g. gliomas or pancreatic ductal adenocarcinoma (PDAC).14,15 It is expressed in a latent form as a complex with the latency-associated peptide (LAP). First, TGF-β is synthesized as a pre-pro-peptide in the endoplasmic reticulum. The pre-pro-peptide consists of the N-terminal LAP and the C-terminal TGF-β. Next, LAP and TGF-β are cleaved by furin proteases and assembled non-covalently as dimers to build the small latent complex (SLC). LAP is essential to allow for the correct folding of active TGF-β.16–18 Furthermore, LAP surrounds active TGF-β and masks the receptor-binding sites of TGF-β keeping TGF-β in a latent form.19 In addition, the SLC can assemble with extracellular matrix (ECM) anchoring proteins in the endoplasmic reticulum.20 The most frequent binding partners of the SLC are latent TGF-β binding proteins (LTBPs). The SLC is attached to LTBPs covalently via disulfide bonds forming the large latent complex (LLC). The secreted LLC can be tethered to the ECM by e.g. fibronectin or collagen. The tethering of the LLC to the ECM is of major importance for the activation of TGF-β.
Although LTBP is the most prevalent binding partner of latent TGF-β, specialized binding proteins exist for certain cell types, e.g. for regulatory T cells (Tregs) and macrophages. The glycoprotein A repetitions predominant protein (GARP) assembles with the SLC on Tregs and is a measure for the level of immunosuppression. On macrophages or microglia, the SLC assembles with the Leucine-Rich Repeat-Containing Protein 33 (LRRC33). It is assumed that multiple ECM binding proteins allow differential spatial activation of TGF-β.21,22
Besides its membrane-bound form anchored to the ECM or immunosuppressive immune cells like Tregs, soluble latent TGF-β can be found in serum as well.23–25 In addition, the soluble version of LAP, independently of TGF- β signaling, was shown to alter migration and MMP9 secretion of oral squamous carcinoma cells, thus further highlighting the relevance of this soluble cue in the TME.26
We hypothesized that the latent TGF-β complex is an attractive clinical target moiety for sensing the TME. Therefore, we applied AdCAR T cells to sense soluble LAP and soluble latent TGF-β to broaden the spectrum of TME-related CAR targets. In contrast to conventional CARs, the specificity of an AdCAR is determined via a tagged adapter molecule, more specifically a tagged antibody, which links the AdCAR T cell to its target.27 In the study presented here, AdCAR T cells sensed soluble LAP as well as latent TGF-β in a concentration-dependent manner, and sensing of the soluble ligand was characterized both in vitro and in vivo. Moreover, the AdCAR system was tested using the same CAR as a sensor for soluble molecules and for direct tumor antigen targeting by combining different adapters in vivo. This work contributes to expand the field of CAR T cell applications to include the possibility to sense immunosuppressive signaling molecules within the TME and converting them into a pro-inflammatory response.
Material and methods
Cell lines and culture conditions
Green fluorescent protein and firefly luciferase-positive AsPC-1 cells (AsPC-1 WT) were generated in-house as described previously by Schaefer et al.28 Luciferase expressing Raji cells were generated as described previously by Seitz et al.27 AsPC-1 WT secreting latent TGF-β (AsPC-1-TGF) were generated via transduction of AsPC-1 WT with lentiviral vectors (LVV) encoding human TGF-β (UniProtKB-A0A024R0P8-1). The plasmid encoding human TGF-β was kindly provided by Stefan Edelburg (Miltenyi Biotec, Bergisch-Gladbach, Germany). Secretion of TGF-β was confirmed by ELISA (BioLegend, catalog no.: 432907). Human embryonic kidney-293 T cells (HEK293T, DSMZ no: Acc635) were obtained from DSMZ.
AsPC-1 WT and AsPC-1-TGF were cultured in RPMI 1640 (Biowest catalog no.: L0501) supplemented with 2 mM glutamine (Lonza, catalog no.: BE17-605E) and 10% fetal bovine serum (EXIMUS Maximus FBS, Catus Biotech, catalog no.: BS-2020-500). HEK293T cells were cultured in Dulbecco’s modified Eagle medium (Biowest, catalog no.: L0104) supplemented with 10% FBS. Cell lines and primary human cells were cultured at 37°C and 5% CO2.
Adapter conjugation
For the generation of anti-LAP adapter molecules (aLAP) full-length IgG1 antibodies (Miltenyi Biotec, clone: REA1214) were used. For the generation of anti-CD66c adapter molecules (aCD66c) full-length IgG1 antibodies (Miltenyi Biotec, clone: REA414) were used. For the generation of an anti-CD20 control adapter (aCD20) Rituximab/MabThera (Roche Pharma, PZN 8709896) was used. Conjugation of antibodies with EZ-Link™ NHS-LC-LC-Biotin (Thermo Fisher Scientific, catalog no.: 11841215) was done in PBS buffer at 20°C with a 3-fold molar excess of biotin for 1 h. Free LC-LC-biotin was removed via a Sephadex PD-10 G-25 column (Cytiva, catalog no.: 17085101). Conjugation was confirmed by LC-MS analysis.
Lentiviral vector construction
The lentiviral transfer plasmid encoding for the direct CD66c xs-spacer CAR (CD66c) was described previously by Schaefer et al.28 The transfer plasmid encoding the AdCAR was generated by cloning the anti-linker-label-epitope (LLE) specific scFv hBio3-3.18E7 (AdCAR1) or the anti-LLE specific scFv Bio2 4G10 (AdCAR2) into the coding sequence of a third-generation CAR backbone comprising an hIgG4 hinge, hCD8 transmembrane domain, cytoplasmatic domain of CD28, 4-1BB and CD3ζ. AdCAR1 was followed by a truncated LNGFR as a transduction marker (LNGFR+) separated via a furin P2A site. The pGFP-NFAT plasmid (System Bioscience, catalog no.: TR051-PA-1) was used for the generation of LVVs encoding the reporter cassette. The cassette contained four consecutive NFAT-AP-1 binding sites 5’ of a copGFP sequence. LVVs were manufactured as described previously by Kotter et al.29
CAR T cell manufacturing
For in vitro assays, buffy coats from healthy donors were obtained from DRK Dortmund. Density gradient centrifugation with Pancoll human (PAN-Biotech, catalog no.: P04-601000) was used for PBMC isolation. T cells were isolated from PBMCs with the Pan T Cell Isolation Kit, human (Miltenyi Biotec, catalog no.: 130-096-535). Isolated T cells were cultured in TexMACS™ Medium (Miltenyi Biotec, catalog no.: 130-097-196) supplemented with 12.5 ng/mL IL-7 (Miltenyi Biotec, catalog no.: 170-076-111) and 12.5 ng/mL IL-15 (Miltenyi Biotec, catalog no.: 170-076-114). For activation before transduction, T cells were seeded in a 24-well plate at a density of 1 × 106 cells/mL and T Cell TransAct™ (Miltenyi Biotec, catalog no.: 130-111-160) was added. After 24 h, the cells were transduced with an MOI of 10. After 3 d, the culture medium was exchanged completely to remove residual LVV particles. T cells were cultured for 10–14 d with the regular addition of fresh culture medium every 2 d. CAR T cells for in vivo studies were manufactured using the CliniMACS Prodigy™ (Miltenyi Biotec) as described previously by Lock et al.30 As starting material leukapheresis obtained from “Institut für klinische Transfusionsmedizin und Immungenetik Ulm” was used. The enriched T cells were transduced at an MOI of 10 and cultured until day 10 for harvesting.
Co-culture assays with soluble target
Soluble recombinant human LAP (hLAP) was reconstituted according to the manufacturer’s recommendations (R&D Systems, catalog no.: 246-LP-025). Latent TGF-β was produced by AsPC-1-TGF. The supernatant of these cells was harvested and centrifuged at 300 g for 10 min to deplete cellular debris. 5 × 104 AdCAR1 T cells were co-cultured with the soluble target and the adapter molecule at indicated concentrations in 96-well flat-bottom plates. After 24 h, the supernatant was removed for cytokine multiplex analysis (Miltenyi Biotec, catalog no.: 130-099-169). The expression of activation markers was determined by flow cytometric analysis using the respective antibody conjugate.
AdCAR T cell reporter assay
Primary human T cells were co-transduced with LVVs encoding AdCAR1 (MOI 10) and LVVs encoding GFP under NFAT-AP-1 expression control (50 µL). Cells were cultured as described previously. 5 × 104 AdCAR1 T cells were co-cultured with 125 ng/mL hLAP and 10 ng/mL aLAP for 90 h. The GFP expression was analyzed by live-cell imaging (IncuCyte® S3, Sartorius). Integrated values of GFP intensities were normalized to start values.
Transwell assay
2 × 104 AsPC-1-TGF were seeded into an HTS Transwell-96 Permeable Support with 0.4 μm pores (Merck, catalog no: CLS3381-1EA). After 24 h, 5 × 104 AdCAR1 T cells were seeded into the respective compartment of the chamber. After 24 h supernatants were removed for cytokine multiplex analysis (Miltenyi Biotec, catalog no.: 130-099-169), and the activation levels of CAR T cells were quantified by flow cytometry using the respective antibody conjugates.
In vitro cytotoxicity assay
1 × 104 AsPC-1 WT or 1 × 104 AsPC-1-TGF were seeded in 96-well flat-bottom plates. After 24 h 2.5 × 104 CAR-expressing cells, as well as aCD66c and aLAP, were added. The assay was performed in 200 µL TexMACS™ Medium (Miltenyi Biotec, catalog no.: 130-097-196) without cytokines. Tumor growth was monitored by live-cell imaging (IncuCyte® S3, Sartorius). The area per well covered by GFP+ target cells was used as a metric to measure AdCAR T cell efficacy. All values were normalized to the starting point.
Co-culture assay with tumor-derived latent TGF-β
2.5 × 104 CAR-expressing cells were seeded in 96 well flat-bottom plates. The supernatant of AsPC-1 WT and AsPC-1-TGF was harvested and processed as described previously. 50 µL tumor-derived supernatant was added together with aCD66c and aLAP. The assay was performed in 200 µL TexMACS™ Medium (Miltenyi Biotec, catalog no.: 130-097-196) without cytokines for 24 h. Cytokine concentration was measured by cytokine multiplex analysis (Miltenyi Biotec, catalog no.: 130-099-169). The expression of activation markers was determined by flow cytometric analysis.
Immunohistochemistry
Tissue specimens were embedded into OCT mounting medium (VWR, catalog no.: 00411243) and stored at −80°C. Sections of 8 μm were cut on a CM3050 S cryostat (Leica) and stored at −80°C. Before staining, tissue sections were thawed and washed with PBS, followed by fixation with 4% PFA for 10 min. After washing with PBS, sections were incubated with permeabilization solution (Miltenyi Biotec, catalog no.: 130-126-719) for 10 min. Antibodies were diluted according to the manufacturer’s protocol in antibody staining solution (Miltenyi Biotec, catalog no.: 130-126-719). Antibodies used for IHC: EPCAM-FITC (clone: REA764, Miltenyi Biotec), CD31-APC (clone: REA730, Miltenyi Biotec), LAP-PE (clone: REA1214, Miltenyi Biotec), LAP-APC (clone: REA1214, Miltenyi Biotec), fibronectin polyclonal (RRID: AB_2547054, Thermo Fisher Scientific), Rabbit IgG (H + L)-Alexa Fluor 488 (RRID: AB_2633280), REA Control Antibody (clone: REA293, Miltenyi Biotec). Staining was performed overnight at 4°C. Images were acquired with EVOS M5000 (Thermo Fisher Scientific). Images were analyzed with ImageJ 1.53e.
MACSima™ imaging cyclic staining (MICS)
Human cancer tissue specimens from ovaries were collected at the Department of Obstetrics and Gynecology at the University Hospital of Cologne. The freshly isolated tissue was embedded into OCT mounting medium (VWR, catalog no.: 00411243) and snap-frozen in liquid nitrogen. MICS was performed as described by Kinkhabwala, A., Herbel, C., Pankratz, J. et al.31
Flow cytometry
Data were acquired on a MACSQuant® Analyzer 10 or MACSQuant® X (Miltenyi Biotec) and analyzed with FlowLogic™ V.8 (Inivai Technologies) by either frequency or median fluorescence intensity (MFI). Cells were stained in CliniMACS® PBS/EDTA Buffer (Miltenyi Biotec, catalog no.: 200-070-025) with 0.5% BSA at 4°C for 10 min. Antibodies used for flow cytometry: CD137-APC (clone: REA765, Miltenyi Biotec), CD25-PE-Vio770 (clone: REA570, Miltenyi Biotec), CD69-VioBlue (clone: REA824, Miltenyi Biotec), LNGFR-PE (clone: REA844, Miltenyi Biotec), CD4-VioGreen (clone: REA623), CD8-APC-Vio770 (clone: REA734, Miltenyi Biotec), CD3-FITC (clone: REA613, Miltenyi Biotec), CD45-VioBlue (clone: REA747, Miltenyi Biotec), CD279-PE-Vio770 (clone: REA1165, Miltenyi Biotec), CD366-APC (clone: REA635, Miltenyi Biotec), CD223-FITC (clone: REA351, Miltenyi Biotec). Antibodies were used according to the manufacturer’s protocol.
In vivo mouse studies
Four- to six-week-old female NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) were purchased from Charles River Laboratories. All studies were performed according to institutional guidelines and regulations (approval numbers: 81–02.04.2018.A096, 81–02.04.2020.A255, 84–02.04.2017.A021). For the AsPC-1 WT tumor model 1 × 106 AsPC-1 WT cells expressing luciferase were injected subcutaneously (s.c.). For the AsPC-1-TGF tumor model 1 × 106 AsPC-1 TGF-β expressing luciferase were s.c. injected. For the Burkitt lymphoma model 4 × 105 Raji cells were intravenously (i.v.) injected. Before i.v. injection of T cells, mice were randomized based on tumor burden. Tumor growth was monitored twice a week via an IVIS Lumina III instrument in vivo imaging system (PerkinElmer). Images were taken 10 min after intraperitoneal (i.p.) injection of 100 µL (30 mg/mL) D-Luciferin (Gold Biotechnology, catalog no.: LUCK-1 G, PubChem Chemical ID: 44134804). Whole-body bioluminescence was measured (p/s). Adapter molecules (diluted in 100 µL PBS) and 10 mg/animal human IVIG (Takeda: Gammagard S/D 10 g, PZN: 06198486) were injected i.p. 1 d before CAR T cell injection. Adapters were injected twice a week. All CAR T cells were freshly i.v. injected. Autologous PBMCs were cryopreserved and injected directly after thawing. The health status of the animals was monitored daily. Euthanasia was applied when endpoint criteria were reached as defined by institutional regulations.
Ex vivo tissue analysis
All tissues were homogenized with the gentle MACS Octo Dissociator (Miltenyi Biotec). Spleens were dissociated in autoMACS® Running Buffer (Miltenyi Biotec, catalog no.: 130-091-221) with 0.5% BSA using the program: m_spleen_01. The lysate was filtered through Pre-Separation Filters, 30 µm (Miltenyi Biotec, catalog no.: 130-041-407). Afterwards, the cells were incubated with 1 mL red blood cell lysis buffer (Miltenyi Biotec, catalog no.: 130-094-183). Cells were washed and re-suspended in autoMACS® Running Buffer and used for downstream applications. Tumor tissue was processed with the Tumor Dissociation Kit, human (Miltenyi Biotec, catalog no.: 130-095-929) according to the manufacturer’s protocol.
Statistical analysis
Data analysis was performed with GraphPad Prism 8.1.2 software (GraphPad, USA). Tests used for statistical data analysis are indicated in corresponding figure legends. Significance was defined by the following criteria: *p < .05; ** p < .01; *** p < .001; **** p < .0001.
Results
The latency-associated peptide is related to desmoplasia in solid human cancer tissue
To evaluate the spatial distribution and co-localization of latent TGF-β with markers of the TME, ovarian cancer tissue was chosen and analyzed via MICS. The expression pattern of LAP, as a surrogate for latent TGF-β, was evaluated on ovarian tissue specimens of two cancer patients. Both patients suffered from cancer with tumor cells being characterized by EpCAM expression. By comparing tumor-rich (Figure 1) and tumor-free regions (S.1, 2 B), we were able to analyze the co-localization of LAP with the expression pattern of 94 surface markers (Table S.1). Areas invaded by tumor cells showed clear visual segregation from tumor-free tissue parts. Areas surrounding the tumor were characterized by dense CD49a expression in conjunction with CD90. LAP expression was strongly overlapping with CD90 and CD49a in desmoplastic areas surrounding EpCAM+ tumor cells. By calculating the Pearson correlation coefficient (PCC) of three regions of interest (ROIs) showing EpCAM+ tumor cells we could show that CD90 (PCC: 0.32 ± 0,08) and CD49a expression (PCC: 0.35 ± 0,04) was linearly increasing with LAP expression in these areas (Figure 1C). In contrast, in three ROIs without EpCAM expression these markers showed a markedly reduced linear correlation with LAP expression (CD49a: 0.11 ± 0,04; CD90: 0.13 ± 0,05) (S.1 C). These findings indicate that latent TGF-β is a potential antigen for CAR T cells to target desmoplastic areas of solid tumors.
Figure 1.
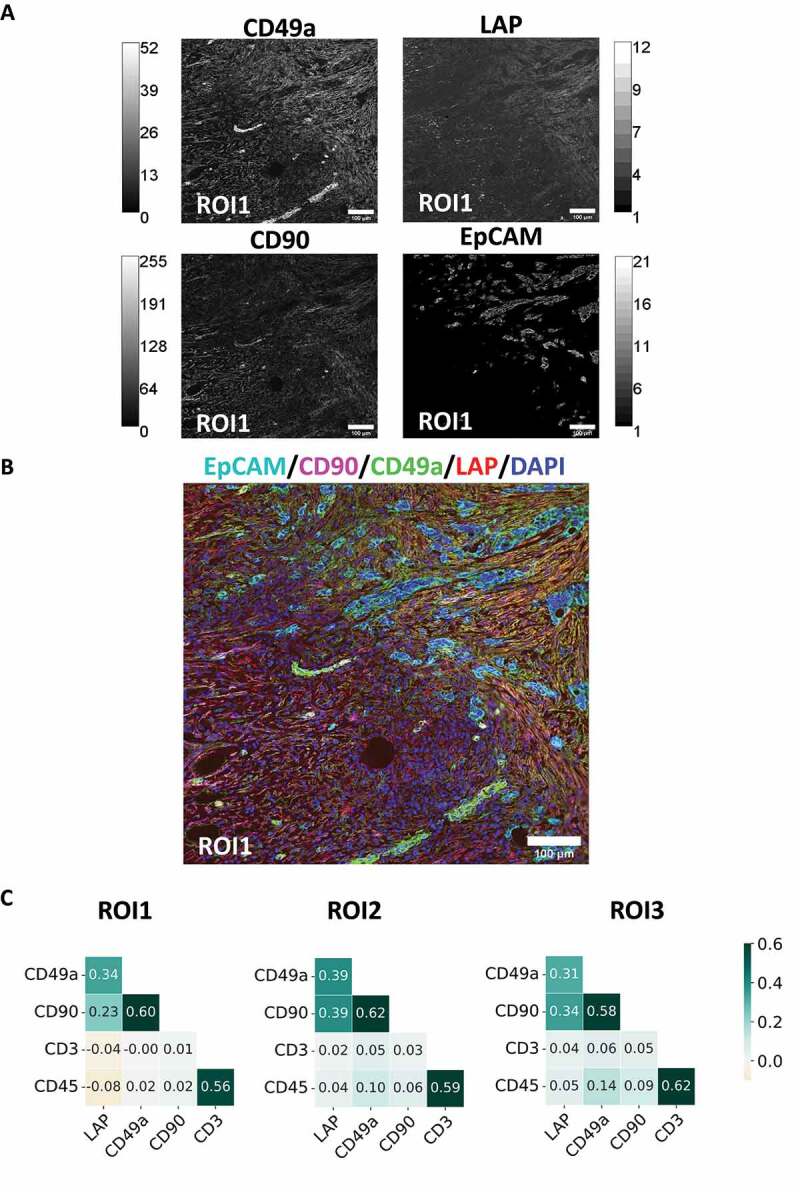
Expression pattern of LAP in presence of EpCAM+ tumor cells on a human cancer section from ovaries. A section of acetone fixed human ovarian cancer tissue was stained with 95 fluorescence-labelled antibodies by MICS. (A) Single antibody stainings of CD49a, LAP, CD90 and EpCAM with grey scale and (B) an overlay thereof are depicted for ROI1 (cyan: EpCAM, red: LAP, blue: DAPI, green: CD90). Scale bar indicates 100 µm. (C) Heatmap of PCC values of three tumor-rich tissue ROIs.
AdCAR T cells are activated by soluble LAP
Human latent TGF-β exists as a membrane-bound complex in human tumor tissue (Figure 1) but also in soluble formats. We postulated that AdCAR T cells can be used not only to target antigens anchored to the tumor membrane but also to detect soluble antigens such as soluble latent TGF-β by tagged antibodies. Theoretically, the antigen-bound adapter complex induces multimerization of AdCAR molecules specific for the adapter tag. The formation of tertiary complexes consisting of antigen, adapter, and AdCAR then triggers T cell-intrinsic signaling cascades and induces effector mechanisms, e.g. cellular activation and cytokine secretion (Figure 2A).
Figure 2.
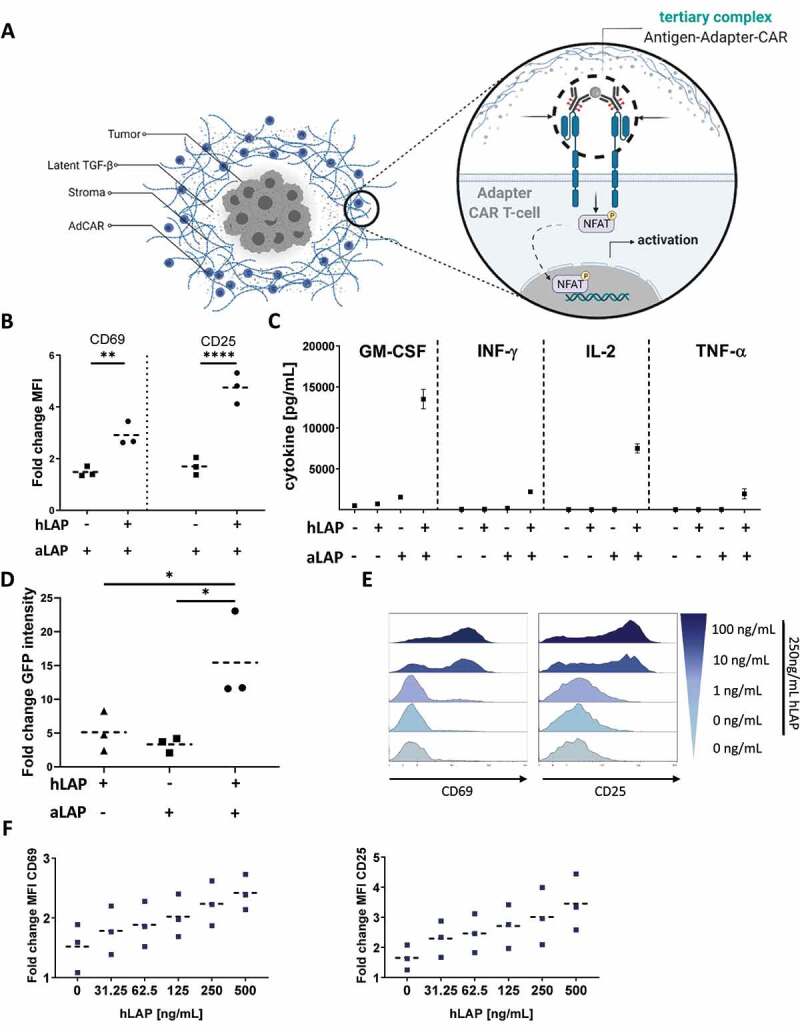
A soluble antigen is sufficient to activate AdCAR T cells by forming a tertiary complex with antigen and adapter. (A) Concept of triggering AdCAR T cells with soluble antigens within the TME of solid tumors. (B) AdCAR1 T cells co-expressing the transduction marker LNGFR (LNGFR+) were incubated for 24 h with 10 ng/mL aLAP with or without 125 ng/mL hLAP. CD69 and CD25 expression was analyzed by flow cytometry. (C) Analysis of cytokine secretion by cytokine multiplex assay. Data plotted with mean ± 1 standard deviation (SD). (D) LNGFR+ AdCAR1 T cells were incubated for 90 h with 10 ng/mL aLAP with or without 125 ng/mL hLAP. CAR signaling was analyzed by GFP expression. Integrated GFP intensity was normalized to start values. (E) LNGFR+ AdCAR1 T cells were incubated for 24 h with 250 ng/mL of soluble hLAP and indicated concentrations of aLAP. Histograms of CD69 and CD25 expression shown for one exemplary donor. (F) LNGFR+ AdCAR1 T cells were co-incubated with indicated amounts of hLAP and 100 ng/mL aLAP for 24 h. MFI values were normalized to T cell activation in absence of soluble hLAP and aLAP. Means are plotted as dashed lines. Data shown were obtained from (n=3) independent donors measured in duplicates if not indicated differently. Statistical analysis was performed with Two-way ANOVA (B) or One-way ANOVA (D). Correction for multiple comparisons was done with Holm-Sidak (D). Illustrations created with BioRender.com.
To evaluate our hypothesis, primary human T cells were engineered to express an AdCAR (AdCAR1) specific for the LLE epitope of LC-LC-biotin. AdCAR1 T cells were incubated with an LLE-tagged antibody specific for LAP (aLAP) in the presence or absence of recombinant soluble human LAP (hLAP). AdCAR1 T cells showed significantly higher expression levels of the activation markers CD69 and CD25 in presence of hLAP and aLAP (Figure 2B, S.3). AdCAR1 T cells also secreted higher levels of pro-inflammatory cytokines GM-CSF, IFN-γ, IL-2, and TNF-α (Figure 2C) when both hLAP and aLAP were available. To demonstrate soluble antigen-induced activation of the NFAT signaling pathway, AdCAR1 T cells were co-transduced with a reporter construct. The reporter contained GFP under the transcriptional control of a four time consecutive repeat of the NFAT-AP-1 multimer. GFP expression was effectively induced in presence of both, the soluble target and the adapter molecule, while only background levels were detectable in control conditions (Figure 2D). In addition, we could show that the activation level of AdCAR1 T cells was tunable by either the adapter dose (Figure 2E) or the level of hLAP (Figure 2F).
AdCAR T cells sense soluble latent TGF-β in presence of tumor cells
Next, we examined if AdCAR1 T cells not only respond to soluble hLAP but also to latent TGF-β secreted by tumor cells. This is of particular interest because tumor cells as well as the secreted immunosuppressive TGF-β have been described to dampen the activation level of T cells. To test our hypothesis, we have chosen the AsPC-1 pancreatic tumor model which is well established in our group.28 AsPC-1 were transduced with LVVs encoding latent TGF-β (AsPC-1-TGF) to establish constitutive secretion of latent TGF-β (S.4). To evaluate whether AdCAR1 T cells detect tumor-derived soluble antigens, supernatants of AsPC-1-TGF with known concentrations of latent TGF-β plus aLAP were added to AdCAR1 T cells. The expression levels of activation markers and secreted cytokines of AdCAR1 T cells were analyzed after 24 h. AdCAR1 T cells were activated by the addition of soluble latent TGF-β isolated from AsPC-1-TGF in a dose-dependent manner at concentrations of soluble latent TGF-β exceeding 2 µg/mL. The activation was characterized by the upregulation of CD69 (Figure 3A) as well as CD137 (Figure 3B) and the secretion of pro-inflammatory cytokines such as IFN-γ, GM-CSF, IL-2, and TNF-α (Figure 3C). Conditioned medium of AsPC-1-TGF containing at least 10 µg/mL of latent TGF-β was needed to trigger GM-CSF or IFN-γ release. However, the activation of AdCAR1 T cells by AsPC-1-TGF conditioned supernatant was lower compared to hLAP since 125 ng/mL hLAP was already sufficient to induce cytokine secretion (Figure 2C). Therefore, we analyzed the concentration of active TGF-β in the AsPC-1-TGF conditioned medium (S.6). The supernatant of AsPC-1-TGF contained approx. 300 pg/mL active TGF-β which might explain the decreased sensitivity of AdCAR1 T cells toward latent TGF-ß as a soluble ligand in this setting. Importantly, AdCAR1 T cells converted TGF-β into a pro-inflammatory stimulus, although the active TGF-β in the AsPC-1-TGF conditioned supernatant was a negative regulator of the cellular activation.
Figure 3.
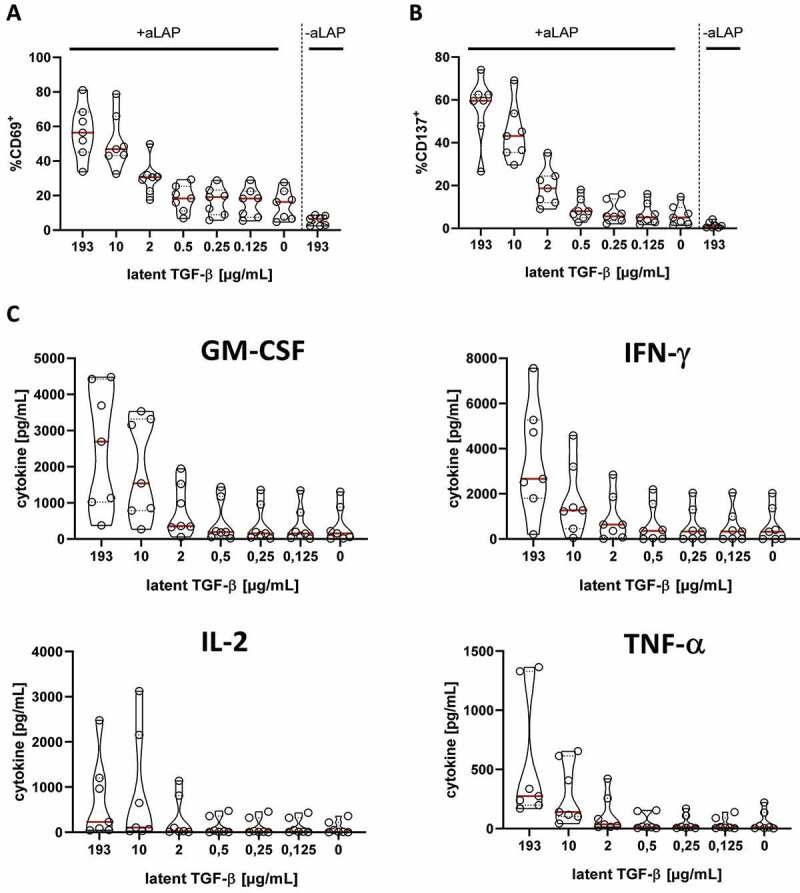
Latent TGF-β secreted by AsPC-1 cells stimulates AdCAR T cells in presence of aLAP adapter. AdCAR1 T cells were co-cultured with serial dilutions of AsPC-1-TGF secreted soluble latent TGF-β with or without 100 ng/mL aLAP for 24 h. Activation of AdCAR T cells was quantified by (A) CD69, (B) CD137 expression of LNGFR+ cells and (C) cytokine secretion. Data shown were obtained from (n=7) independent donors measured in duplicates if not indicated differently. Data plotted with medians (red lines) and quartiles (dashed lines).
To unravel the activation of AdCAR1 T cells mediated by tumor cells and/or soluble antigens, both cell types were co-cultured in transwells separated by a membrane. The membrane was permeable for soluble latent TGF-β secreted by the tumor cells but direct contact between tumor and T cells was inhibited (Figure 4A). In line with previous results, the frequency and MFI levels of CD137 were significantly increased for CD8+ and CD4+ T cells only in presence of both aLAP and tumor cells secreting soluble latent TGF-β, which confirmed the activation of AdCAR1 T cells by a soluble ligand (Figure 4B-C). In addition, AsPC-1-TGF and AdCAR1 T cells were seeded in the same compartment. In this condition, frequency and MFI levels of CD137 were significantly increased for CD8+ and CD4+ in presence of aLAP as well. The direct cell-to-cell contact improved the activation of CD4+ T cells in particular. This was depicted by the increased frequency and MFI of CD137 in presence of aLAP and tumor cells secreting the soluble target. Thus, soluble antigen sensing was not suppressed when AdCAR1 T cells got into proximity with tumor cells.
Figure 4.
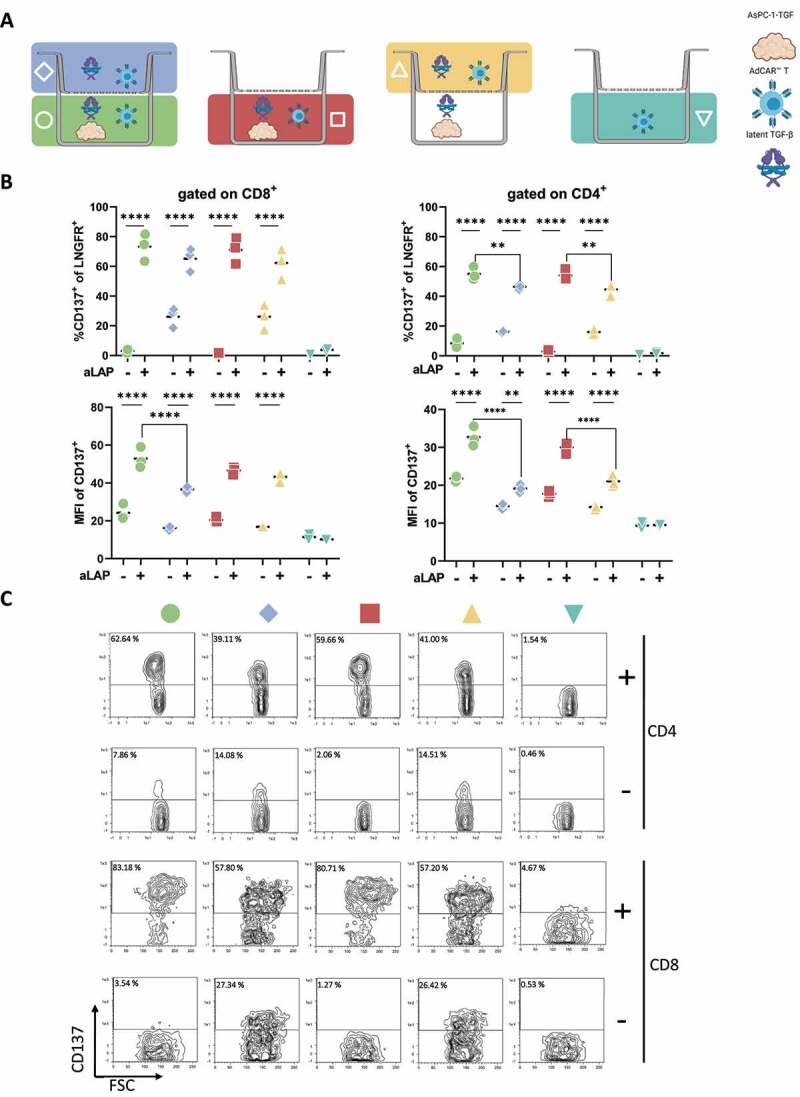
AdCAR T cells sense tumor-derived soluble latent TGF-β in presence of tumor cells. (A) Schematic representation of transwell assay conditions. (B) LNGFR+ AdCAR1 T cells were cultured in a transwell plate (pore size 0.4 µm) for 24 h. CD137 expression and MFI were analyzed for CD4+ and CD8+ cells in presence of aLAP (+) or absence (-). (C) CD137 expression of CD4+ or CD8+ AdCAR T cells after co-culture of AdCAR1 T cells and AsPC-1-TGF in the transwell assay is depicted for one exemplary donor. Data shown were obtained from (n=3) independent donors measured in duplicates plotted with mean (dashed line) if not indicated differently. Statistical analysis was performed with Two-way ANOVA. Correction for multiple comparisons was done with Holm-Sidak. Illustrations created with BioRender.com.
These results indicate that the activation of AdCAR1 T cells was dependent on adapter and target availability. Both with and without direct cell–cell contact, AdCAR1 T cells converted the presence of latent TGF-β into an activating stimulus. This indicates that sensing soluble latent TGF-β can trigger T cell activation already before the first tumor cell contact, as long as the soluble target concentration reaches the activation threshold.
AdCAR T cells can target a TAA and sense a soluble ligand simultaneously
The feasibility of combined targeting of multiple, membrane-bound TAA antigens with the same AdCAR by simultaneously applying adapters of different specificities was recently shown by Seitz et al.27 We sought to expand this concept and assess the impact of an adapter molecule specific for a TAA (CD66c) on the sensing of a soluble antigen (latent TGF-β) by AdCAR T cells.
Since the affinity of the AdCAR toward the LLE adapter may affect the combined targeting of two different antigens, we compared two AdCAR constructs which differed in their LLE-specific scFvs (Figure 5A). The scFv clone hBio3-3.18E7 of AdCAR1 showed a lower affinity in flow cytometry towards the LLE-tag compared to the Bio2 4G10 scFv which was part of the AdCAR2 construct (data not published).
Figure 5.
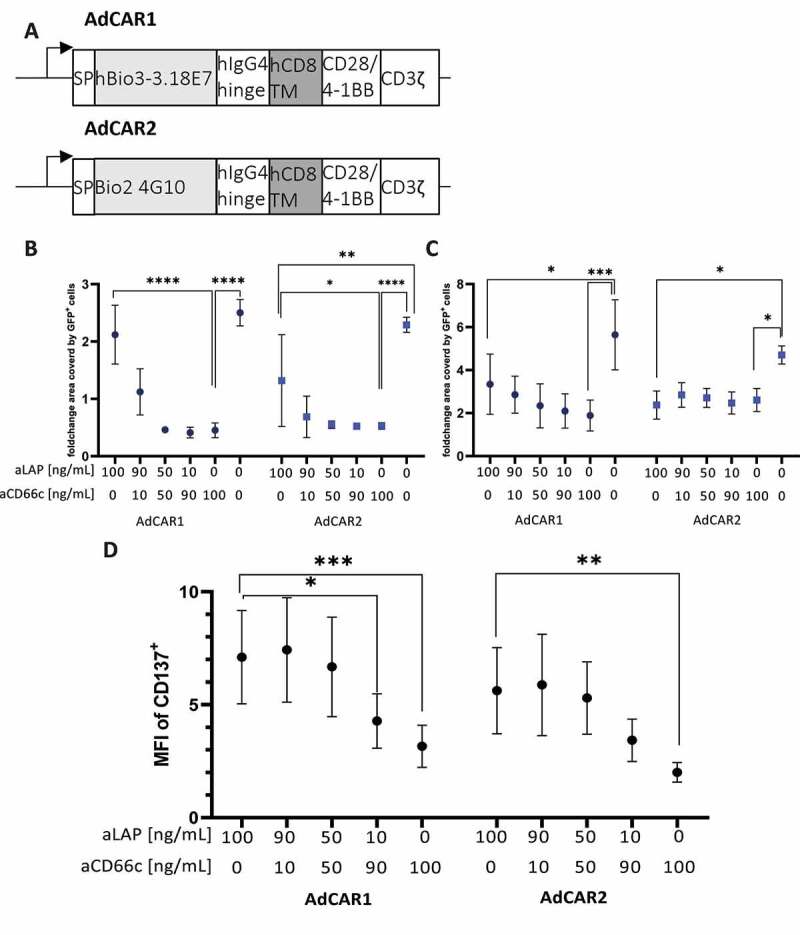
Targeting latent TGF-β of AsPC-1-TGF tumor cells induces AdCAR T cell activation and does not interfere with targeting a TAA. (A) Architecture of AdCAR1 and AdCAR2. Primary human T cells expressing AdCAR1 or AdCAR2 were either co-cultured with (B) AsPC-1 WT or (C) AsPC-1-TGF for 141 h in presence of indicated mixtures of aLAP and aCD66c. The surface area in wells covered by target cells normalized to the start value was quantified with the IncuCyte® S3 system. (D) T cells were co-cultured with supernatant of AsPC-1-TGF for 24 h in presence of indicated mixtures of aLAP and aCD66c. MFI of CD137+ cells was analyzed. Data shown were obtained from (n=3) independent donors measured in duplicates if not indicated differently and plotted with mean and ± 1 (SD). Statistical analysis was performed with Two-way ANOVA. Correction for multiple comparisons was done with Holm-Sidak correction.
To investigate functional differences between the two AdCAR constructs AsPC-1 WT and AsPC-1-TGF were co-cultured with AdCAR T cells for 141 h at varying ratios of aCD66c and aLAP. AdCAR1 and AdCAR2 showed significant killing of AsPC-1 WT in presence of 100 ng/mL aCD66c (Figure 5B). For AdCAR1, the killing efficacy was decreased at a ratio of 90 ng/mL aLAP plus 10 ng/mL aCD66c compared to 100 ng/mL aCD66c. In contrast, AdCAR2 showed constant tumor cell killing, irrespective of the amount of added aLAP, from 10 ng/mL to 100 ng/mL aCD66c (Figure 5B). Due to the higher affinity of AdCAR2 for the tagged adapter, AdCAR2 T cells may induce target cell killing also at lower concentrations of aCD66c. In addition, AdCAR2 also significantly reduced tumor growth of AsPC-1 WT in presence of 100 ng/mL aLAP compared to the control without an adapter. This effect might have been caused by LAP expression on the surface of AsPC-1 WT.
AsPC-1-TGF proliferation was controlled in an adapter-dependent manner as well. However, both AdCARs were less effective compared to the killing of AsPC-1 WT which can be explained by the presence of active TGF-β in AsPC-1-TGF conditioned supernatant (S.6). Interestingly, the tumor cell growth inhibition was not significantly different in presence of 100 ng/ml aCD66c compared to any condition combining aCD66c with aLAP. We found that 100 ng/mL aLAP led to a similar anti-tumor effect as 100 ng/mL aCD66c. This indicates that both AdCARs can target surface-bound LAP on AsPC-1-TGF and elicit effector functions (Figure 5C).
The impact of aCD66c presence on the sensing of a soluble antigen was addressed by co-culturing AdCAR T cells with AsPC-1-TGF conditioned supernatant as well as aCD66c and aLAP at varying ratios. Both AdCAR constructs showed an aLAP concentration-dependent activation when co-cultured with supernatant derived from AsPC-1-TGF (Figure 5D).
Thus, we could show in vitro that both killing of tumor cells and the simultaneous sensing of soluble ligands are feasible.
AdCAR T cells sense soluble latent TGF-β LAP in vivo in a reconstituted mouse model
As we have demonstrated the combined targeting of soluble latent TGF-β and CD66c as TAA with one AdCAR in vitro (Figure 5B-D), we asked whether this mechanistic concept is applicable in vivo as well. In our previous in vitro study, two CARs with different LLE-specific scFvs were compared to identify the most suitable construct for the combined targeting of soluble latent TGF-β and CD66c (Figure 5). AdCAR2 outperformed the previously used AdCAR1 with regard to the killing of AsPC-1 WT tumor cells at lower adapter doses of the LLE-tagged antibody specific for CD66c at simultaneously higher concentrations of aLAP.
Recently, Seitz et al. demonstrated the functionality of AdCAR T cells in vivo in a lymphoma Raji model in combination with the FDA-approved CD20-specific monoclonal antibody rituximab.27 To verify the finding that AdCAR2 is more potent compared to AdCAR1 in vivo as well, we decided to compare both AdCAR architectures (AdCAR1 and AdCAR2) head-to-head in vivo in a similar model. To serve as an adapter for the AdCAR T cells, rituximab was conjugated with the LLE tag. NSG mice were i.v. inoculated with 4 × 105 luciferase expressing Raji cells on day −5. On day 0 we i.v. injected 6 × 106 AdCAR T cells. The rituximab adapter was injected 1 d before the AdCAR T cells, and subsequently 50 µg adapter were applied i.p. twice a week until the end of the study (S.7A). Both AdCAR constructs initially controlled the tumor compared to control cohorts missing AdCAR T cells. However, AdCAR1 failed to control the tumor until the end of the study, whereas AdCAR2 eradicated the Raji tumor in 5 out of 5 mice, leading to a significant (p = .001) improvement in tumor control compared to AdCAR1 (S.7B-C). Therefore, AdCAR2 was used for simultaneous targeting of multiple target antigens in subsequent in vivo studies.
The efficacy of AdCAR2 T cells for combined targeting of CD66c and soluble latent TGF-β was evaluated in the AsPC-1-TGF tumor model. This tumor model recapitulates facets of human solid tumors, e.g. tumor-secreted soluble and ECM-linked latent TGF-β (S.5). To strengthen the clinical relevance of our model, AdCAR2 T cells were co-injected with or without autologous PBMCs. Enhanced anti-tumor control and cytokine crosstalk between endogenous immune cells and direct CAR T cells targeting the TME has previously been shown.3,4 In this way, we additionally evaluated whether the combined targeting of soluble antigens and TAAs by AdCAR2 T cells benefits from the presence of endogenous immune cells (Figure 6A).
Figure 6.
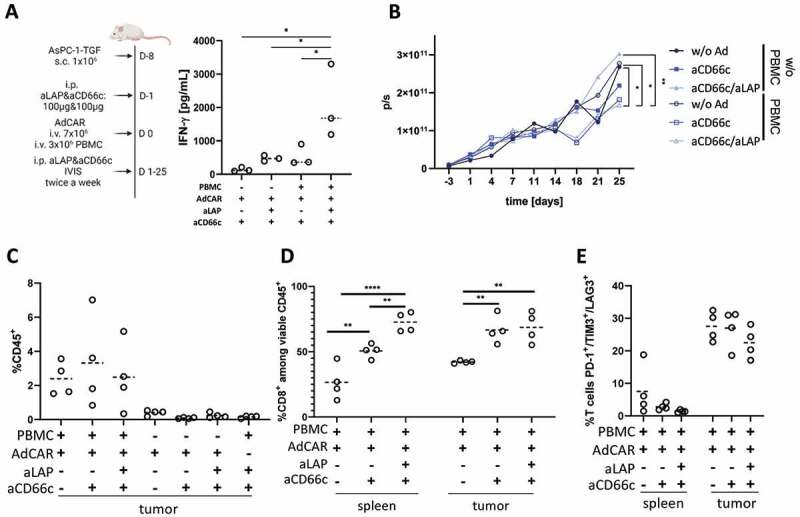
Autologous PBMCs synergized with AdCAR T cells and improved functionality in the AsPC-1-TGF tumor model. (A) Schematic representation of the study design and measurement of serum IFN-γ levels on day 4 to assess soluble antigen sensing in mice reconstituted with autologous PBMCs and inoculated with AsPC-1-TGF. (B) Quantification of AdCAR2-mediated cytotoxicity by total BLI. Median of each cohort (n≥5) is shown. (C) Ex vivo analysis of viable CD45+ cells in tumor tissue and of (D) viable CD8+ T cells as well as (E) exhaustion marker expression of T cells in spleens and tumor tissues (n=4). Ex vivo analysis was performed on day 25 for all cohorts. Statistical analysis was performed with ordinary one-way ANOVA (A) or Two-way ANOVA (B,D). Correction for multiple comparisons was done with Holm-Sidak correction. Median is plotted as dashed line. Each dot represents one animal. Experiments were performed with cells of one donor.
Since cytokine secretion has been identified as a functional outcome of sensing soluble antigens by AdCAR T cells in vitro (Figure 3), the INF-γ levels in peripheral blood were measured on day 4. As expected, IFN-γ levels were enhanced in cohorts that received the combination of aCD66c and aLAP, verifying the previous in vitro results. In presence of aCD66c alone the cytokine secretion was lower compared to cohorts treated with the combination of aCD66c and aLAP (Figure 6A). This finding indicates that the sensing of latent TGF-β via aLAP triggered the release of cytokines independent of the TAA-specific CD66c adapter. Surprisingly, compared to all other cohorts, significantly higher levels of IFN-γ were observed when CD66c and latent TGF-β were targeted by AdCAR2 T cells in NSG mice reconstituted with autologous PBMCs. For instance, aCD66c and aLAP administration resulted in IFN-γ levels that were 4.5-fold higher in mice inoculated with PBMCs compared to mice that did not receive PBMCs (Figure 6A). In summary, and independent of PBMC injection, our data indicate that AdCAR2 T cells are stimulated by soluble latent TGF-β in vivo. In contrast to targeting a TAA, targeting of a soluble antigen induced an early pro-inflammatory cytokine release. Thus, targeting of latent TGF-β, which is found in an immunosuppressive TME, resulted in an earlier onset of AdCAR T cell activation compared to targeting a TAA, in absence of soluble antigen targeting.
In addition, AdCAR2 T cells co-administered with autologous PBMCs controlled the tumor on day 18 in presence of either aCD66c or aCD66c plus aLAP, whereas non-reconstituted NSG mice with or without adapter administration showed tumor growth progression (Figure 6B). Tumor burden was significantly lower in cohorts treated with PBMC plus AdCAR2 T cells and either aCD66c (p = .0221) or CD66c and aLAP (p = .0421), compared to cohorts treated with AdCAR2 T in absence of PBMC and adapter. On day 25, a significantly lower tumor burden was observed by BLI measurement when AdCAR2 T cells were applied with autologous PBMC and in combination with aCD66c and aLAP compared to cohorts treated with PBMC but no adapter molecules (Figure 6B). In contrast, no anti-tumor effects were detectable in absence of PBMCs emphasizing the importance of endogenous immune cells for profound anti-tumor effects in this solid tumor model. Importantly, also in this in vivo setting no inhibitory effect of aLAP adapter on CAR T cell cytotoxicity was observed.
The end-point analysis of tumor composition revealed higher frequencies of CD45+ cells infiltrating the tumor in PBMC-treated cohorts, thus also pointing toward potential synergistic effects of endogenous immune cells with AdCAR2 T cells (Figure 6C). In PBMC-treated mice, both in spleens and tumor tissues an adapter-dependent stimulation of CD8+ cells was observed as significantly lower frequencies of CD8+ were found in absence of adapter. Interestingly, combined aLAP and aCD66c altered the CD4:CD8 ratio depending on the respective localization of the AdCAR2 T cells. In spleens, the frequency of CD8+ T cells was significantly increased by 1.4-fold in cohorts treated with aCD66 plus aLAP compared to cohorts treated with aCD66c alone, pointing toward an aLAP-specific activation of AdCAR2 T cells (Figure 6D). The systemic presence of soluble latent TGF-β might explain this change in CD4:CD8 ratios observed in spleens, i.e. distant from the tumors. Frequencies of CD8+ T cells in tumor tissues were comparable in mice treated with aCD66c or aCD66c plus aLAP indicating TAA-dependent activation of cytotoxic T cells. Importantly, no reduction of cytotoxic AdCAR2 T cells was observed when soluble antigen sensing and tumor targeting were combined.
A surprising outcome was the tumor regrowth after day 18, which was further investigated. To this end, exhaustion levels of tumor resident T cells were evaluated and compared to the phenotypic representation of T cells found in spleens on day 25. The frequency of PD-1, TIM3, and LAG3 triple-positive cells was 5.5-fold higher for tumor-infiltrating T cells when compared to T cells derived from spleens, indicating high levels of exhaustion (Figure 6E). Remarkably, this was also detectable in tumor-resident T cells in the absence of an adapter pointing towards TME-mediated T cell exhaustion independent of T cell activation.
In summary, our data underline the relevance of a reconstituted mouse model to better study the complex interactions of CAR T cells and endogenous immune cells within the TME. Most importantly, AdCAR T cells can also sense soluble ligands by adapter molecules, both in vitro and in vivo, and elicit anti-tumoral activity without interference offering novel strategies for multiplexed targeting of solid tumors.
Discussion
Currently, six CAR T cell therapies are approved by the U.S. Food and Drug Administration and available to cancer patients, all of them targeting hematological malignancies. However, the majority of cancer patients suffer from solid tumors.32 One major challenge when treating patients with solid tumors is the immunosuppressive TME,33 characterized by the presence of several immunomodulatory molecules like MMPs, nutrient-depleting enzymes, or chemokines.34
To our knowledge, we provide here the first example of AdCAR T cells targeting a soluble immunomodulatory molecule in vitro and in vivo. In the approach described here, AdCAR T cells convert immunosuppressive latent TGF-β into a pro-inflammatory, controllable mechanistic outcome. Only in presence of a soluble target and adapter, cellular activation markers are expressed by AdCAR T cells, and pro-inflammatory cytokines are secreted.
One cytokine that exhibits immunosuppression in the TME is TGF-β. TGF-β is a pleiotropic cytokine and is associated with immune evasion by inhibiting cytotoxic T cell responses, interfering with antigen presentation, or by inducing a Treg phenotype.35–37 Different approaches to target TGF-β are under investigation, e.g. neutralizing antibodies, ligand traps, or small molecules.38 In addition, targeting LAP by monoclonal antibodies elicited an anti-tumor response in various tumor models by reducing, e.g. TGF-β bioavailability and immunosuppressive T cells.39 Thus, latent TGF-β could also be an attractive target for CAR T cell therapy. The clinical relevance of latent TGF-β is further supported by a study of Hawinkels et al. investigating latent TGF-β levels locally in tumor tissue. Significantly higher levels of latent TGF-β were found in homogenates of gastric cancer patients as compared to adjacent control tissue.40 In our study, LAP, as a part of latent TGF-β, was found on cancer tissue sections from ovaries. LAP was associated with desmoplastic areas invaded by EpCAM+ tumor cells. In addition, we found that LAP expression is co-localized with surface markers of fibroblasts (CD90) and integrins (CD49a).
LAP expression correlated the strongest with CD49a expression in the analyzed ovarian cancer tissue. A central function of CD49a is the binding of collagen in the ECM of malignant tissue. The expression of CD49a was reported for ovarian cancer, and the expression level is linked to drug resistance.41 In addition to ovarian cancer, CD49a expression is reported in pancreatic ductal adenocarcinoma (PDAC) as well and is linked to chemotherapy resistance of PDAC cells, cell attachment, and spreading.42 CD49 is expressed on tissue-resident memory T cells as well as on TILs. Interestingly, CD49a enhances the motility of the TILs leading to insufficient tumor recognition.43,44 Furthermore, CD49a can be found on an NK cell subset with reduced cytotoxic potential which was described as being proangiogenic in the TME of hepatocellular carcinoma patients.45 Overall, CD49a is involved in numerous mechanisms leading to immune evasion of solid tumors, i.e. ovarian and pancreatic cancer.
The second highest correlation was found for LAP and CD90. CD90 is a marker of fibroblasts and is described to be expressed on CAFs.46 CD90 expression was found to be significantly increased in PDAC patients. In addition to fibroblasts, CD90 was detected on vascular endothelial cells and activated pancreatic stellate cells of PDAC patients, which further highlights the central role of CD90 expressing cells within the TME of PDAC.47 In summary, the correlation of LAP with both CD49a and CD90 appears to be of relevance, since co-expression can be linked to the biological function of TGF-β in the TME.
CAFs are known to secrete TGF-β which leads to a remodeling of the ECM in ovarian cancer by inducing MMP secretion.48 With regard to PDAC, an interplay of CD49a and TGF-β was described as well. CD49a and TGF-β response genes, e.g. COL4A1 and ZEB1, were found to be co-expressed in samples of pancreatic cancer patients pointing toward functional relations.42
Thus, latent TGF-β is associated with central immunosuppressive processes in the TME of multiple solid tumors. Therefore, it is a clinically relevant target to demonstrate the functionality of soluble antigen sensing in conjunction with the AdCAR system.
Besides being anchored in the TME or its cell surface expression latent TGF-β can also be found as a soluble complex in the human serum.25,49 Sensing of soluble ligands with CARs was first demonstrated in the context of CAR T cells directly targeting the antigen of choice.13 Dimerization and mechanical coupling as requirements for sensing soluble ligands with CARs have been described. Although the soluble antigen is targeted indirectly by adapters in the study presented here, the AdCAR system fulfills these requirements. The receptor dimerization is mediated via the soluble target-adapter complex and leads to sufficient coupling of the CAR’s signaling domains. Our work demonstrates how the concept of rewiring CARs to soluble ligands can be converted into therapeutic use. In contrast to the currently known direct CAR approach, the AdCAR system offers several advantages: (1) AdCAR T cells are able to elicit tumor cell killing combined with sensing of soluble antigens and (2) increase the controllability of targeting soluble ligands. In this study, the AdCAR system showed a concentration-dependent response to both the soluble ligand and the adapter. This has two advantages in terms of clinical application. First, gradients of latent TGF-β may enable discrimination of healthy- and tumor-tissue. Second, in contrast to the current direct CAR approach, the AdCAR system may allow tight pharmacological control, e.g. by adapter withdrawal or discontinuation of the infusion in case of severe side effects or after tumor clearance when TGF-β levels return to normal.
In our mouse model, AdCAR2 T cells targeting soluble latent TGF-β induced a faster pro-inflammatory cytokine secretion as compared to a membrane-bound TAA targeted by AdCAR2 T cells. Moreover, altered CD4:CD8 ratios toward CD8+ T cells in the spleens were observed. Overall, this indicates soluble antigen-dependent stimulation and cytotoxic T cell engagement. For solid tumors, these mechanistic findings may overcome limited peripheral survival of infused CAR T cells until the tumor site is reached, e.g. due to their initial residence in lung and liver tissue.50 However, a more detailed analysis is needed to evaluate the consequences of repeated antigen stimulation on cellular fitness by targeting soluble ligands and/or TAAs.51
The combined targeting of soluble latent TGF-β and a TAA by AdCAR2 T cells induced only a transient control of the tumor in the AsPC1-TGF-β model overexpressing TGF-β. High levels of active TGF-β secreted by AsPC1-TGF tumors and its immunosuppressive function may explain the reduction of cytolytic activity of AdCAR2 T cells. Interestingly, the co-administration of autologous PBMC enhanced the anti-tumor effect of AdCAR2 T cells, leading to transient tumor control in our immunosuppressive AsPC1-TGF model. Here, elevated levels of systemic IFN-γ and CD45+ T cells in the tumor point toward synergistic effects of CAR T cells and endogenous immune cells. We observed better persistence and proliferation of adoptively transferred cells in the periphery when AdCAR2 T cells were combined with endogenous immune cells. It has been recently described that IFN-γ secreted by CAR T cells improves endogenous anti-tumor response within the TME. Enhanced IL-12 secretion and MHC-II upregulation were described to empower cytolytic T cell function.52 In line with our observation, the benefit of engaging innate immune cells when targeting the TME was shown for anti-EIIIB fibronectin-targeted CAR T cells and anti-FAP CAR T cells.3,4 Nevertheless, AdCAR2 T cells did not completely eradicate the tumor in both models tested. Of note, AdCAR T cells targeting latent TGF-β may elicit the killing of CAFs, Treg cells, and immunosuppressive dendritic cells expressing LAP on their surface.39,53 Therefore, an orthotopic mouse model or a fully immunocompetent mouse model could shed more light on immune cell-mediated effects elicited by anti-latent TGF-β targeting.
Insufficient recruitment of cytotoxic cells from the periphery may have contributed to the incomplete anti-tumor response. Similar effects were observed in an autochthonous prostate cancer model with OTI transgenic TCR T cells.54 In addition, Lesch et al. reported recently that expression of C-X-C chemokine receptor type 6 improved the recruitment of T cells to the tumor and anti-tumor response.8 As the penetration of adapter molecules into the TME is fundamental for the recruitment and activation of AdCARs, the distribution of the adapter needs to be analyzed in future studies. Diffusion barriers due to high antigen density leading to non-homogenous adapter distribution within the solid tumor mass can interfere with AdCAR T cell functionality.55,56 In particular, the penetration of the aLAP adapter is prone to this effect since the target moiety is present systemically and locally in the TME.
In this study, GFP was expressed as a marker gene in an activation-dependent manner upon sensing the soluble antigen. We envision that the activation-dependent expression can be also applied to therapeutically relevant transgenes. Dominant-negative TGF-β receptors or cytokines to diminish the immunosuppressive effect of TGF-β could be considered as possible candidates.57–59
Our work demonstrates that AdCAR T cells respond to immunosuppressive tumor-derived soluble antigens with a pro-inflammatory response in vivo. The versatility of the AdCAR system to target multiple antigens simultaneously through different adapter specificities also simplifies the process of CAR manufacturing. Alternatively, direct CAR T cells can be used to target multiple antigens as well.60,61 For instance, T cells could be modified with two different CAR constructs to either sense a soluble ligand or induce killing of tumor cells. In the case of conventional direct CAR T cells, either double transduction or the use of a bicistronic vector would be needed. The major limitations of these approaches, compared to the AdCAR approach, would be the undefined CAR T cell product and lower lentiviral titers due to the limited viral packing size.62
One alternative to the AdCAR system could be the use of a tandem CAR to sense the soluble ligand and recognize the TAA simultaneously. One scFv could be specific for CD66c and the second one for latent TGF-β. However, when targeting a soluble factor and TAA we consider the safety profile of the AdCAR to be superior compared to a tandem CAR approach. In particular, when targeting latent TGF-β, which could – apart from the TME – also be abundant in non-tumor regions and systemically, a safety mechanism might be required.
In sum, an adapter-dependent CAR T cell activation can provide tight pharmacological control providing a safety strategy to tune the CAR activity in a clinical setting. As a consequence, exploiting soluble TME markers for CAR T cell therapy may become very attractive for clinical translation. Nevertheless, further knowledge of how targeting soluble factors with CARs can assist the anti-tumor response of host immune cells is needed. The presented approach for soluble antigen sensing may broaden the spectrum of effective CAR T cell therapies which are available to patients suffering from solid tumors.
Supplementary Material
Acknowledgments
The authors thank Stefan Edelburg for providing the plasmid encoding latent TGF-β and Sarah Schmitz for supporting the adapter conjugation. Furthermore, the authors thank Marek Wieczorek for his support in creating the illustrations.
Disclosure statement
Niels Werchau, Bettina Kotter, Elvira Criado-Moronati, Andre Gosselink, Nicole Cordes, Dominik Lock, Simon Lennartz, Carolin Kolbe, Nora Winter, Karin Teppert, Fabian Engert, Brian Webster, Joerg Mittelstaet, Daniel Schaefer, Mario Assenmacher, Thomas Schaser, Pierre Abramowski, Andrew D. Kaiser were employees of Miltenyi Biotec B.V. & Co. KG at the time the study was conducted.
Niels Werchau, Bettina Kotter, Joerg Mittelstaet, and Andrew D. Kaiser are coinventors of a patent application focusing on sensing of soluble antigens with adapter CAR technology. Joerg Mittelstaet and Andrew D. Kaiser are coinventors of a patent application focusing on adapter CAR technology.
Michael von Bergwelt-Baildon: Astellas Pharma, Bristol-Myers Squibb, Kite Gilead, Miltenyi Biotec, MOLOGEN, MSD, Novartis, Roche (Honoraria, Speakers’ Bureau, Research Funding, Travel, Accomodations, Expenses).
Authorship contributions
NW and BK developed the idea of sensing soluble antigens with AdCARs. NW, BK, ECM, NoW, BW, DS, planned and/or conducted AdCAR T- or CAR T-cell in vitro experiments. NW and AG planned and conducted in silico data analysis. PM, MM, and DR conducted human tumor isolation. NW, DL, NC, SL, CK, KT, FE planned and/or performed in vivo experiments. NW, BK, JM, PA, TS, ADK, MA, and MvB interpreted the data. NW, PA, and TS wrote the final manuscript. ADK and MvB initiated and supervised the study. All authors approved the final version of this manuscript before submission.
Data and Materials Availability
All data associated with this study are present in the paper or the supplementary materials.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/2162402X.2022.2140534
References
- 1.Gross G, Waks T, Eshhar Z.. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci. 1989;86(24):10024–16. doi: 10.1073/pnas.86.24.10024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Motz GT, Coukos G. Deciphering and reversing tumor immune suppression. Immunity. 2013;39(1):61–73. doi: 10.1016/j.immuni.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xie YJ, Dougan M, Jailkhani N, Ingram J, Fang T, Kummer L, Momin N, Pishesha N, Rickelt S, Hynes RO, et al. Nanobody-based CAR T cells that target the tumor microenvironment inhibit the growth of solid tumors in immunocompetent mice. Proc Natl Acad Sci U S A. 2019;116(33):16656. doi: 10.1073/pnas.1912487116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang L-CS, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, Antzis M, Cotner CE, Johnson LA, Durham AC, et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor t cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res. 2014;2(2):154–166. doi: 10.1158/2326-6066.CIR-13-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kakarla S, Chow KKH, Mata M, Shaffer DR, Song XT, Wu MF, Liu H, Wang LL, Rowley DR, Pfizenmaier K, et al. Antitumor effects of chimeric receptor engineered human T cells directed to tumor stroma. Mol Ther. 2013;21(8):1611–1620. doi: 10.1038/mt.2013.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pircher M, Schuberth P, Gulati P, Sulser S, Weder W, Curioni A, Renner C, Petrausch U. FAP-specific re-directed T cells first in-man study in malignant pleural mesothelioma: experience of the first patient treated. Journal for ImmunoTherapy of Cancer. 2015;3(S2):120. doi: 10.1186/2051-1426-3-S2-P120. [DOI] [Google Scholar]
- 7.Curioni A, Britschgi C, Hiltbrunner S, Bankel L, Gulati P, Weder W, Opitz I, Lauk O, Caviezel C, Knuth A, et al. A phase I clinical trial of malignant pleural mesothelioma treated with locally delivered autologous anti-FAP-targeted CAR T-cells. Ann Oncol. 2019;30. doi: 10.1093/annonc/mdz253.052. [DOI] [Google Scholar]
- 8.Lesch S, Blumenberg V, Stoiber S, Gottschlich A, Ogonek J, Cadilha BL, Dantes Z, Rataj F, Dorman K, Lutz J, et al. T cells armed with C-X-C chemokine receptor type 6 enhance adoptive cell therapy for pancreatic tumours. Nature Biomedical Engineering. 2021;5(11):1246–1260. doi: 10.1038/s41551-021-00737-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hombach A, Koch D, Sircar R, Heuser C, Diehl V, Kruis W, Pohl C, Abken H. A chimeric receptor that selectively targets membrane-bound carcinoembryonic antigen (mCEA) in the presence of soluble CEA. Gene Therapy. 1999;6(2):300–304. doi: 10.1038/sj.gt.3300813. [DOI] [PubMed] [Google Scholar]
- 10.Hombach A, Heuser C, Sircar R, Tillmann T, Diehl V, Pohl C, Abken H. An anti-CD30 chimeric receptor that mediates CD3-zeta-independent T-cell activation against Hodgkin’s lymphoma cells in the presence of soluble CD30. Cancer Res. 1998;58:1116–1119. [PubMed] [Google Scholar]
- 11.Westwood JA, Murray WK, Trivett M, Haynes NM, Solomon B, Mileshkin L, Ball D, Michael M, Burman A, Mayura-Guru P, et al. The Lewis-Y carbohydrate antigen is expressed by many human tumors and can serve as a target for genetically redirected T cells despite the presence of soluble antigen in serum. J Immunother. 2009;32(3):292–301. doi: 10.1097/CJI.0b013e31819b7c8e. [DOI] [PubMed] [Google Scholar]
- 12.Sun L, Gao F, Gao Z, Ao L, Li N, Ma S, Jia M, Li N, Lu P, Sun B, et al. Shed antigen-induced blocking effect on CAR-T cells targeting Glypican-3 in Hepatocellular Carcinoma. J Immunother Cancer. 2021;9(4):1–14. doi: 10.1136/jitc-2020-001875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chang ZL, Lorenzini MH, Chen X, Tran U, Nathanael J, Chen YY. Rewiring T-cell responses to soluble factors with chimeric antigen receptors. Nat Chem Biol. 2018;14(3):317–324. doi: 10.1038/nchembio.2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Samuels V, Barrett JM, Bockman S, Pantazis CG, Allen MB. Immunocytochemical study of transforming growth factor expression in benign and malignant gliomas. Am J Pathol. 1989;134:895–902. [PMC free article] [PubMed] [Google Scholar]
- 15.Cave DD, Di Guida M, Costa V, Sevillano M, Ferrante L, Heeschen C, Corona M, Cucciardi A, Lonardo E. TGF-β1 secreted by pancreatic stellate cells promotes stemness and tumourigenicity in pancreatic cancer cells through L1CAM downregulation. Oncogene. 2020;39(21):4271–4285. doi: 10.1038/s41388-020-1289-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miyazono K, Hellman U, Wernstedt C, Heldin CH. Latent high molecular weight complex of transforming growth factor β1. Purification from human platelets and structural characterization. J Biol Chem. 1988;263(13):6407–6415. doi: 10.1016/s0021-9258(18)68800-3. [DOI] [PubMed] [Google Scholar]
- 17.Gray AM, Mason AJ. Requirement for activin A and transforming growth factor–beta 1 pro-regions in homodimer assembly. Science. 1990;247(4948):1328–1330. doi: 10.1126/science.2315700. [DOI] [PubMed] [Google Scholar]
- 18.Dubois CM, Laprise M-H, Blanchette F, Gentry LE, Leduc R. Processing of Transforming Growth Factor β1 Precursor by Human Furin Convertase. J Biol Chem. 1995;270(18):10618–10624. doi: 10.1074/jbc.270.18.10618. [DOI] [PubMed] [Google Scholar]
- 19.Shi M, Zhu J, Wang R, Chen X, Mi L, Walz T, Springer TA. Latent TGF-β structure and activation. Nature. 2011;474(7351):343–349. doi: 10.1038/nature10152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Todorovic V, Rifkin DB. LTBPs, more than just an escort service. J Cell Biochem. 2012;113(2):410–418. doi: 10.1002/jcb.23385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qin Y, Garrison BS, Ma W, Wang R, Jiang A, Li J, Mistry M, Bronson RT, Santoro D, Franco C, et al. A milieu molecule for tgf-β required for microglia function in the nervous system. Cell. 2018;174(1):156–171. doi: 10.1016/j.cell.2018.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Batlle E, Massagué J. Transforming growth factor-β signaling in immunity and cancer. Immunity. 2019;50(4):924–940. doi: 10.1016/j.immuni.2019.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gleizes P, Munger JS, Nunes I, Harpel JG, Mazzieri R, Noguera I, Rifkin DB. TGF‐β Latency: biological Significance and Mechanisms of Activation. Stem Cells. 1997;15(3):190–197. doi: 10.1002/stem.150190. [DOI] [PubMed] [Google Scholar]
- 24.Stockis J, Colau D, Coulie PG, Lucas S. Membrane protein GARP is a receptor for latent TGF-β on the surface of activated human Treg. Eur J Immunol. 2009;39(12):3315–3322. doi: 10.1002/eji.200939684. [DOI] [PubMed] [Google Scholar]
- 25.Areström I, Zuber B, Bengtsson T, Ahlborg N. Measurement of human latent transforming growth factor-β1 using a latency associated protein-reactive ELISA. J Immunol Methods. 2012;379(1–2):23–29. doi: 10.1016/j.jim.2012.02.016. [DOI] [PubMed] [Google Scholar]
- 26.Thomas GJ, Hart IR, Speight PM, Marshall JF. Binding of TGF-β1 latency-associated peptide (LAP) to αvβ6 integrin modulates behaviour of squamous carcinoma cells. Br J Cancer. 2002;87(8):859–867. doi: 10.1038/sj.bjc.6600545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seitz CM, Mittelstaet J, Atar D, Hau J, Reiter S, Illi C, Kieble V, Engert F, Drees B, Bender G, et al. Novel adapter CAR-T cell technology for precisely controllable multiplex cancer targeting. Oncoimmunology. 2021;10(1). doi: 10.1080/2162402X.2021.2003532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schäfer D, Tomiuk S, Küster LN, Al RW, Henze J, Tischler-Höhle G, Agorku DJ, Brauner J, Linnartz C, Lock D, et al. Identification of CD318, TSPAN8 and CD66c as target candidates for CAR T cell based immunotherapy of pancreatic adenocarcinoma. Nat Commun. 2021;12(1):1453. doi: 10.1038/s41467-021-21774-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kotter B, Engert F, Krueger W, Roy A, Al RW, Cordes N, Drees B, Webster B, Werchau N, Lock D, et al. Titratable pharmacological regulation of CAR T cells using zinc finger-based transcription Factors. Cancers (Basel). 2021;13(19):4741. doi: 10.3390/cancers13194741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lock D, Mockel-Tenbrinck N, Drechsel K, Barth C, Mauer D, Schaser T, Kolbe C, Al Rawashdeh W, Brauner J, Hardt O, et al. Automated manufacturing of potent CD20-directed chimeric antigen receptor T cells for clinical use. Hum Gene Ther. 2017;28(10):914–925. doi: 10.1089/hum.2017.111. [DOI] [PubMed] [Google Scholar]
- 31.Kinkhabwala A, Herbel C, Pankratz J, Yushchenko DA, Rüberg S, Praveen P, Reiß S, Rodriguez FC, Schäfer D, Kollet J, et al. MACSima imaging cyclic staining (MICS) technology reveals combinatorial target pairs for CAR T cell treatment of solid tumors. Sci Rep. 2022;12(1):1911. doi: 10.1038/s41598-022-05841-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 Countries. CA Cancer J Clin. 2021;71(3):209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 33.Marofi F, Motavalli R, Safonov VA, Thangavelu L, Yumashev AV, Alexander M, Shomali N, Chartrand MS, Pathak Y, Jarahian M, et al. CAR T cells in solid tumors: challenges and opportunities. Stem Cell Res Ther. 2021;12(1):1–16. doi: 10.1186/S13287-020-02128-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Labani-Motlagh A, Ashja-Mahdavi M, Loskog A. The tumor microenvironment: a milieu hindering and obstructing antitumor immune responses. Front Immunol. 2020;11(940). doi: 10.3389/fimmu.2020.00940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen C-H, Seguin-Devaux C, Burke NA, Oriss TB, Watkins SC, Clipstone N, Ray A. transforming growth factor β blocks tec kinase phosphorylation, ca2+ influx, and nfatc translocation causing inhibition of t cell differentiation. J Exp Med. 2003;197(12):1689–1699. doi: 10.1084/jem.20021170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen W, Jin W, Hardegen N, Lei K, Li L, Marinos N, McGrady G, Wahl SM. Conversion of peripheral CD4+cd25− naive t cells to cd4+cd25+ regulatory t cells by tgf-β induction of transcription factor foxp3. J Exp Med. 2003;198(12):1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kobie JJ, Wu RS, Kurt RA, Lou S, Adelman MK, Whitesell LJ, Ramanathapuram LV, Arteaga CL, Akporiaye ET. Transforming growth factor beta inhibits the antigen-presenting functions and antitumor activity of dendritic cell vaccines. Cancer Res. 2003;63:1860–1864. [PubMed] [Google Scholar]
- 38.Kim B-G, Malek E, Choi SH, Ignatz-Hoover JJ, Driscoll JJ. Novel therapies emerging in oncology to target the TGF-β pathway. J Hematol Oncol. 2021;14(1):55–75. doi: 10.1186/s13045-021-01053-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gabriely G, da Cunha AP, Rezende RM, Kenyon B, Madi A, Vandeventer T, Skillin N, Rubino S, Garo L, Mazzola MA, et al. Targeting latency-associated peptide promotes antitumor immunity. Sci Immunol. 2017;2(11):1738. doi: 10.1126/sciimmunol.aaj1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hawinkels LJAC, Verspaget HW, van Duijn W, van der Zon JM, Zuidwijk K, Kubben FJGM, Verheijen JH, Hommes DW, Lamers CBHW, Sier CFM. Tissue level, activation and cellular localisation of TGF-β1 and association with survival in gastric cancer patients. Br J Cancer. 2007;97(3):398–404. doi: 10.1038/sj.bjc.6603877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wei L, Yin F, Zhang W, Li L. ITGA1 and cell adhesion-mediated drug resistance in ovarian cancer. Int J Clin Exp Pathol. 2017;10:5522–5529. [Google Scholar]
- 42.Gharibi A, La Kim S, Molnar J, Brambilla D, Adamian Y, Hoover M, Hong J, Lin J, Wolfenden L, Kelber JA. ITGA1 is a pre-malignant biomarker that promotes therapy resistance and metastatic potential in pancreatic cancer. Sci Rep. 2017;7(1):1–14. doi: 10.1038/s41598-017-09946-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Melssen MM, Rodriguez AB, Slingluff CL, Engelhard VH. CD49a and CD103 expression on tumor infiltrating lymphocytes in B16-OVA tumors depends on location and is correlated with a naive phenotype. J Immunol. 2016;196(1 Supplement):212.21. [Google Scholar]
- 44.Melssen MM, Lindsay RS, Stasiak K, Rodriguez AB, Briegel AM, Cyranowski S, Rutkowski MR, Conaway MR, Melief CJM, van der Burg SH, et al. Differential expression of CD49a and CD49b determines localization and function of tumor-infiltrating CD8+ T cells. Cancer Immunol Res. 2021;9(5):583. doi: 10.1158/2326-6066.CIR-20-0427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zecca A, Barili V, Rizzo D, Olivani A, Biasini E, Laccabue D, Valle RD, Ferrari C, Cariani E, Missale G. Intratumor regulatory noncytotoxic NK cells in patients with hepatocellular carcinoma. Cells. 2021;10(3):1–19. doi: 10.3390/CELLS10030614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sukowati CHC, Anfuso B, Crocé LS, Tiribelli C. The role of multipotent cancer associated fibroblasts in hepatocarcinogenesis. BMC Cancer. 2015;15(1):188. doi: 10.1186/s12885-015-1196-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhu J, Thakolwiboon S, Liu X, Zhang M, Lubman DM. Overexpression of CD90 (Thy-1) in Pancreatic Adenocarcinoma Present in the Tumor Microenvironment. PLoS One. 2014;9(12):e115507. doi: 10.1371/JOURNAL.PONE.0115507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yeung T-L, Leung CS, Wong -K-K, Samimi G, Thompson MS, Liu J, Zaid TM, Ghosh S, Birrer MJ, Mok SC. TGF-β modulates ovarian cancer invasion by upregulating caf-derived versican in the tumor microenvironment. Cancer Res. 2013;73(16):5016–5028. doi: 10.1158/0008-5472.CAN-13-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Khan SA, Joyce J, Tsuda T. Quantification of active and total transforming growth factor-β levels in serum and solid organ tissues by bioassay. BMC Res Notes. 2012;5:636. doi: 10.1186/1756-0500-5-636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Visioni A, Kim M, Wilfong C, Blum A, Powers C, Fisher D, Gabriel E, Skitzki J. Intra-arterial versus intravenous adoptive cell therapy in a mouse tumor model. J Immunother. 2018;41(7):313–318. doi: 10.1097/CJI.0000000000000235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Good CR, Aznar MA, Kuramitsu S, Samareh P, Agarwal S, Donahue G, Ishiyama K, Wellhausen N, Rennels AK, Ma Y, et al. An NK-like CAR T cell transition in CAR T cell dysfunction. Cell. 2021;184(25):6081–6100.e26. doi: 10.1016/j.cell.2021.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Boulch M, Cazaux M, Loe-Mie Y, Thibaut R, Corre B, Lemaître F, Grandjean CL, Garcia Z, Bousso P. A cross-talk between CAR T cell subsets and the tumor microenvironment is essential for sustained cytotoxic activity. Sci Immunol. 2021;6(57):1–17. doi: 10.1126/sciimmunol.abd4344. [DOI] [PubMed] [Google Scholar]
- 53.Zhuang J, Lu Q, Shen B, Huang X, Shen L, Zheng X, Huang R, Yan J, Guo H. TGFβ1 secreted by cancer-associated fibroblasts induces epithelial-mesenchymal transition of bladder cancer cells through lncRNA-ZEB2NAT. Sci Rep. 2015;5(1):11924. doi: 10.1038/srep11924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chou CK, Schietinger A, Liggitt HD, Tan X, Funk S, Freeman GJ, Ratliff TL, Greenberg NM, Greenberg PD. Cell-intrinsic abrogation of tgf-β signaling delays but does not prevent dysfunction of self/tumor-specific cd8 t cells in a murine model of autochthonous prostate cancer. J Immunol. 2012;189(8):3936–3946. doi: 10.4049/jimmunol.1201415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lu G, Nishio N, van den Berg Ns, Martin BA, Fakurnejad S, van Keulen S, Colevas AD, Thurber GM, Rosenthal EL. Co-administered antibody improves penetration of antibody–dye conjugate into human cancers with implications for antibody–drug conjugates. Nat Commun. 2020;11(1):5667. doi: 10.1038/s41467-020-19498-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lu G, Fakurnejad S, Martin BA, van den Berg NS, van Keulen S, Nishio N, Zhu AJ, Chirita SU, Zhou Q, Gao RW, et al. Predicting Therapeutic Antibody Delivery into Human Head and Neck Cancers. Clin Cancer Res. 2020;26(11):2582–2594. doi: 10.1158/1078-0432.CCR-19-3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Webster B, Xiong Y, Hu P, Wu D, Alabanza L, Orentas RJ, Dropulic B, Schneider D. Self-driving armored CAR-T cells overcome a suppressive milieu and eradicate CD19+ Raji lymphoma in preclinical models. Mol Ther. 2021;29(9):2691–2706. doi: 10.1016/j.ymthe.2021.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Alabanza LM, Xiong Y, Vu B, Webster B, Wu D, Hu P, Zhu Z, Dropulic B, Dash P, Schneider D. Armored BCMA CAR T Cells Eliminate Multiple Myeloma and Are Resistant to the Suppressive Effects of TGF-β. Front Immunol. 2022;13(832645). doi: 10.3389/fimmu.2022.832645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Markley JC, Sadelain M. IL-7 and IL-21 are superior to IL-2 and IL-15 in promoting human T cell–mediated rejection of systemic lymphoma in immunodeficient mice. Blood. 2010;115(17):3508–3519. doi: 10.1182/blood-2009-09-241398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hegde M, Corder A, Chow KK, Mukherjee M, Ashoori A, Kew Y, Zhang YJ, Baskin DS, Merchant FA, Brawley VS, et al. Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred t cells in glioblastoma. Molecular Therapy. 2013;21(11):2087–2101. doi: 10.1038/mt.2013.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schneider D, Xiong Y, Wu D, Nӧlle V, Schmitz S, Haso W, Kaiser A, Dropulic B, et al. A tandem CD19/CD20 CAR lentiviral vector drives on-target and off-target antigen modulation in leukemia cell lines. Journal for ImmunoTherapy of Cancer. 2017;5(1):42. doi: 10.1186/s40425-017-0246-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kumar M, Keller B, Makalou N, Sutton RE. Systematic Determination of the Packaging Limit of Lentiviral Vectors. Human Gene Therapy. 2001;12(15):1893–1905. doi: 10.1089/104303401753153947. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.