

Abstract
A population pharmacokinetic (PopPK) model was previously developed for cemiplimab in patients with solid tumors, including advanced cutaneous squamous cell carcinoma (CSCC). Here, we update the existing PopPK model and characterize exposure‐response relationships using efficacy and safety data obtained in patients with recurrent or metastatic cervical cancer (R/M CC). To improve model stability and robustness of the existing PopPK model in 1062 patients, the random‐effect error model was revised, and structural covariates were removed from the base model to be tested in the covariate analysis. The updated model was used for external validation of cemiplimab pharmacokinetics (PK) in patients with R/M CC on cemiplimab monotherapy (350 mg every 3 weeks intravenously) from a phase III study (NCT03257267). Exposure‐response relationships for cemiplimab efficacy (overall survival [OS], progression‐free survival [PFS], duration of response [DOR], objective response rate [ORR]), and safety (immune‐related adverse events [irAEs]) were analyzed in 295 patients with R/M CC from the aforementioned study. The updated PopPK model showed improved stability with 94.8% successful bootstrap runs vs. 47.6% in the prior model. Cemiplimab exposure was similar across tumor types, including basal cell carcinoma, CSCC, and non‐small cell lung cancer. External validation showed the updated model adequately described cemiplimab PK in patients with R/M CC. In exposure‐response efficacy analyses, Cox proportional hazard modeling (CPHM) showed no trend between exposure and OS, Kaplan–Meier plots showed no trend between exposure and PFS or DOR, and logistic regression analyses conducted on ORR showed no exposure‐response relationship. In exposure‐response safety analyses, CPHM showed no trend between exposure and irAEs.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Population pharmacokinetic (PopPK) modeling demonstrated that a two‐compartment model with first‐order elimination, zero‐order intravenous [i.v.] infusion rate, and time‐varying change in clearance adequately described the concentration of cemiplimab in patients with advanced malignancies.
WHAT QUESTION DID THIS STUDY ADDRESS?
A more robust PopPK model of cemiplimab is developed and further validated in patients with recurrent or metastatic cervical cancer (R/M CC) on cemiplimab monotherapy (350 mg every 3 weeks [Q3W] i.v.) from EMPOWER‐Cervical 1/GOG‐3016/ENGOT‐cx9 to assess consistency in exposures across solid tumor types. Exposure‐response analyses were performed in patients with R/M CC to determine whether a relationship between cemiplimab exposure and various efficacy and safety end points exists.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Patients with R/M CC have cemiplimab exposures comparable to other tumor types, including cutaneous squamous cell carcinoma, basal cell carcinoma, and non‐small cell lung cancer. Exposure‐response analyses in patients with R/M CC demonstrated no relationship between cemiplimab exposure and evaluated efficacy or safety end points.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
The presented analyses may be supportive of the cemiplimab regimen (350 mg Q3W i.v.) used in EMPOWER‐Cervical 1/GOG‐3016/ENGOT‐cx9 for second‐line treatment of R/M CC.
INTRODUCTION
Globally, cervical cancer is the fourth most common type of cancer in terms of incidence and cancer‐related mortality in women, with an estimated 604,127 new cases and 341,831 deaths in 2020. 1 Approximately 99.7% of cervical cancer cases are attributable to persistent infection with high‐risk oncogenic types of human papillomavirus (HPV). 2 Although prophylactic vaccination against oncogenic HPV strains is a promising strategy to reduce cervical cancer incidence, a significant impact of vaccination on cervical cancer morbidity and mortality is yet to be demonstrated. 3
Patients diagnosed with early‐stage invasive disease (International Federation of Obstetrics and Gynecology [FIGO] stage IB1–IB2) can be treated effectively with radical hysterectomy plus bilateral pelvic lymphadenectomy. 4 However, most patients at diagnosis present with locally advanced disease (FIGO stage IB3–IVA), for which the preferred treatment modality is concurrent chemoradiotherapy plus intracavitary brachytherapy. 4 Patients with persistent or recurrent disease post‐radiotherapy who are not candidates for pelvic exenteration and those who present with metastatic disease (FIGO stage IVB) are treated with platinum‐containing chemotherapy, often in combination with bevacizumab, an angiogenesis inhibitor. 5 This first‐line treatment with platinum‐containing chemotherapy (±bevacizumab) in patients with recurrent or metastatic cervical cancer (R/M CC) is associated with a median overall survival (OS) of 13.3–18.3 months. 6 , 7 After progression on first‐line platinum‐containing chemotherapy for R/M CC, treatment options are limited, and prognosis is poor with a median OS of ~7–8 months in the second‐line setting. 8 , 9
Cemiplimab is a fully human, hinge‐stabilized, immunoglobulin G4 (IgG4) monoclonal antibody directed against programmed cell death‐1 (PD‐1), and derived using VelocImmune technology. 10 , 11 , 12 Cemiplimab is approved in the United States and Europe for the treatment of advanced cutaneous squamous cell carcinoma (CSCC). 13 , 14 It is also approved in the United States for the treatment of advanced basal cell carcinoma (BCC) and first‐line treatment of advanced non‐small cell lung cancer (NSCLC) with programmed cell death‐ligand 1 (PD‐L1) expression ≥50%. 14
In the phase III EMPOWER‐Cervical 1/Gynecologic Oncology Group (GOG)‐3016/European Network of Gynecological Oncological Trial groups (ENGOT)‐cx9 study (NCT03257267), cemiplimab monotherapy (350 mg every 3 weeks [Q3W] intravenously [i.v.]) was compared with the investigator's choice of single‐agent chemotherapy for treatment of patients with R/M CC who progressed after first‐line platinum‐containing chemotherapy. 15 Patients (N = 608) were enrolled regardless of PD‐L1 expression status, and included squamous cell carcinoma (n = 477) or adenocarcinoma/adenosquamous carcinoma (n = 131) histologic subtypes. 15 Overall, cemiplimab (n = 304) demonstrated a significant survival benefit with a median OS of 12.0 months vs. 8.5 months with chemotherapy (n = 304). 15
A population pharmacokinetic (PopPK) model was developed previously using pharmacokinetic (PK) observations from 548 patients pooled from a first‐in‐human study (NCT02383212) in patients with advanced solid tumors and a phase II study (NCT02760498) in patients with advanced CSCC. 16 PopPK modeling demonstrated that a two‐compartment model with first‐order elimination, zero‐order i.v. infusion rate, and time‐varying change in clearance (CL) adequately described the concentration of cemiplimab in patients. 16 Covariate analysis showed that baseline body weight, serum albumin, and IgG levels had a modest impact on cemiplimab exposure, without clinical relevance. Simulated concentration‐time profiles and corresponding exposure metrics supported approval of the cemiplimab fixed‐dose i.v. regimen of 350 mg Q3W by the US Food and Drug Administration (cemiplimab‐rwlc) and the European Commission for CSCC. 16 This initial PopPK model was subsequently updated to incorporate PK data from clinical trials of cemiplimab 350 mg Q3W in patients with advanced BCC or NSCLC. Here, the latter PopPK model, developed for cemiplimab in an overall population of 1062 patients, was updated with appropriate modifications intended to improve model stability and robustness vs. that of previous models, and an external validation of cemiplimab PK in patients with R/M CC was performed using data from the EMPOWER‐Cervical 1/GOG‐3016/ENGOT‐cx9 study. Moreover, we characterized the exposure‐response relationship of cemiplimab on efficacy and safety in patients with R/M CC from this same study.
METHODS
Patients
The PopPK dataset used to update the existing PopPK model was pooled from phase I (NCT02383212), phase II (NCT02760498, NCT03132636), and phase III (NCT03088540) trials of cemiplimab across solid tumor types. The updated PopPK model was used to perform an external validation of cemiplimab PK in patients with R/M CC from the phase III EMPOWER‐Cervical 1/GOG‐3016/ENGOT‐cx9 study (NCT03257267). Exposure‐response relationships for efficacy and safety of cemiplimab were also characterized in patients with R/M CC from EMPOWER‐Cervical 1/GOG‐3016/ENGOT‐cx9. A summary of cemiplimab doses, dosing regimens, and PK sampling schemes of the studies included in this analysis of PK and exposure‐response data are provided in Table S1. All studies were conducted in accordance with ethical principles of the Declaration of Helsinki, and were consistent with International Conference on Harmonisation, Good Clinical Practices, and applicable regulatory requirements.
Modeling software
The PopPK of cemiplimab was characterized by nonlinear mixed‐effects modeling using NONMEM version 7.4 (ICON Development Solutions). A pooled NONMEM‐ready dataset was constructed using SAS version 9.4 (SAS Institute). For the PopPK analysis, R software version 4.0.2 (R Development Core Team; http://www.r‐project.org/) was used for pre‐ and post‐processing of NONMEM output and preparation of figures. The NONMEM model code for the PopPK analyses is provided in the Appendix S1. Exposure‐response analyses were conducted with R software version 3.6.2 using the “survival,” “survminer,” and “tidyverse” packages.
Bioanalytical assays
Serum samples for quantification of functional cemiplimab were analyzed using a validated enzyme‐linked immunosorbent assay. The assay utilized the recombinant human PD‐1 extracellular domain as the capture reagent. Captured cemiplimab was detected using a biotinylated anti‐human IgG4 monoclonal antibody. The lower limit of quantification was 0.078 mg/L of cemiplimab in human serum.
Data exclusion and below‐limit‐of‐quantification records handling
Data excluded from the PopPK analysis included predose samples collected prior to the first cemiplimab dose, inversion samples defined as predose concentrations higher than the corresponding end‐of‐infusion concentrations, and postdose sample concentrations below the limit of quantification (BLOQ). If the proportion of postbaseline BLOQ observations accounted for <10% of the sampling observations, and visual assessment of data distribution showed no systematic trends, Beal's M1 approach was implemented, and BLOQ observations were omitted from the PopPK analysis. 17 Outliers were identified using the modeling outputted absolute conditional weighted residuals (CWRES) or absolute individual weighted residuals (IWRES). Observations with |CWRES| > 5 or |IWRES| > 5 were classified as potential outliers. To evaluate the influence of outliers, a sensitivity analysis was conducted in which the updated PopPK model was run with and without outliers. If a change of >20% was observed in ≥1 of the key fixed‐effects parameters, such as CL and central volume of distribution (V1), outliers were considered influential in the model.
PopPK model building
Previously, a PopPK model was developed for cemiplimab in an overall population of 1062 patients. An update of the existing PopPK model was performed using PK data from 1063 patients with solid tumors, including CSCC, BCC, NSCLC, and R/M CC (Table S2). In contrast to the analysis dataset of the existing PopPK model, PK samples previously identified as concentration outliers were included in the dataset for the model update. The existing PopPK model was updated using the approach shown in Figure S1. First, the random‐effects structure in the existing base model (excluding structural covariates) was re‐evaluated. Following incorporation of a revised random‐effect error model and selection of an appropriate base model, potential intrinsic and extrinsic covariates predictive of cemiplimab PK variability were evaluated for significance. Covariates evaluated as baseline parameters included age, sex, race, ethnicity, body weight, albumin, alanine aminotransferase (ALT), aspartate aminotransferase, total bilirubin, predicted creatinine CL, serum creatinine, Eastern Cooperative Oncology Group (ECOG) performance status, and tumor types. Albumin was also assessed as a time‐dependent covariate. As IgG and baseline PD‐L1 expression level were only available for a limited number of patients and studies, these covariates were evaluated in post hoc analyses. A full covariate model was constructed using a forward selection procedure, with covariates that contributed to a change in objective function value (OFV) of >6.63 units considered statistically significant (p < 0.01). This was followed by a backward elimination process, in which each covariate was removed from the full covariate model separately. A change in OFV of >10.83 units upon removal of a covariate was considered statistically significant (p < 0.001).
PopPK model evaluation
Model evaluation for the updated final base and covariate models was via graphical examination of standard diagnostic and population analysis goodness‐of‐fit plots. Model stability was assessed using the condition number of the correlation matrix of the parameter estimates, calculated as the ratio of the largest to smallest eigenvalue. A condition number >1000 is indicative of a severely ill‐conditioned model. The robustness of the updated model and precision in parameter estimates was assessed using a bootstrap approach. To verify predictive adequacy of the updated final model, prediction‐corrected visual predictive checks (pcVPCs) were performed. For the pcVPCs, 500 datasets were generated using the final PopPK model, which replicated the design, dose regimens, sample sizes, and covariate distributions from the observed dataset. Predicted cemiplimab concentrations at each nominal PK timepoint were normalized by the typical predicted concentration. Prediction‐corrected observed cemiplimab concentrations were binned by time and overlaid with the 5th and 95th percentiles (90% confidence interval [CI]) of the simulated summary measures at the corresponding percentiles (5th, 50th, and 95th percentiles).
Predictive performance of the PK model for patients with R/M CC
In EMPOWER‐Cervical 1/GOG‐3016/ENGOT‐cx9, 304 patients with R/M CC were treated with cemiplimab monotherapy (350 mg Q3W i.v.). At the time of this analysis, data from 295 of 304 cemiplimab‐treated patients were available. However, three patients had no quantifiable cemiplimab concentrations after the first dose. Thus, the predictive performance of the updated final PopPK model was assessed for 292 patients with R/M CC from EMPOWER‐Cervical 1/GOG‐3016/ENGOT‐cx9 using an external posterior predictive check (PPC). External validation was possible, as PK data from EMPOWER‐Cervical 1/GOG‐3016/ENGOT‐cx9 were not included in the dataset for the PopPK model. Parameter estimates from the PopPK model were assumed to have a multivariate normal distribution. The multivariate normal distribution was used as an approximate posterior distribution to generate 500 sets of population parameter values. Each set of these population parameter values was used to simulate a dataset conditioned upon the observed study design. The observed cemiplimab concentrations were overlaid with the 5th and 95th percentiles (90% CI) of the simulated summary measures at the corresponding percentiles (5th, 50th, and 95th). Post hoc PK parameters corresponding to the period after the first cemiplimab dose and at steady‐state were generated from the typical PK profiles for each patient.
Exposure‐response analysis
Exposure‐response relationships for cemiplimab efficacy and safety were analyzed in 295 of 304 patients with R/M CC on cemiplimab monotherapy (350 mg Q3W i.v.) from EMPOWER‐Cervical 1/GOG‐3016/ENGOT‐cx9. Patients with squamous and nonsquamous histology were included in the exposure‐response analysis dataset if they received at least one dose of cemiplimab and had at least one nonmissing postdose response. Individually predicted post hoc PK parameters after the first dose were derived from the final PopPK model.
Exposure‐response relationships were characterized for several efficacy and safety end points. The primary efficacy end point was OS. Secondary end points were progression‐free survival (PFS), duration of response (DOR), and objective response rate (ORR). Occurrence of immune‐related adverse events (irAEs) grade ≥3 was the primary safety end point, and irAEs of any grade was an additional safety end point. Exposure metrics of interest for the exposure‐efficacy and exposure‐safety analysis comprised the following individually predicted post hoc PK parameters: trough concentration after the first dose (Ctrough,1) and maximum concentration after the first dose (Cmax,1), respectively, and average concentration after the first dose (Cav,1). Correlations between cemiplimab exposure and OS, PFS, and DOR were explored by plotting nonparametric Kaplan–Meier curves with each exposure metric stratified by quartile. Box‐and‐whisker plots were used to compare distribution of exposure metrics between patients who achieved the best overall response (BOR) and the remaining patient population. Logistic regression analyses were used to determine whether exposure metrics were possible predictors of achieving BOR. For these analyses, ORR was based on BOR, and taken to be the proportion of individuals achieving BOR. Correlations between cemiplimab exposure and irAEs were explored by plotting nonparametric Kaplan–Meier curves with each cemiplimab exposure metric stratified by quartiles of exposure after the first dose.
If graphical analysis suggested a potential exposure‐response relationship, additional modeling approaches were used to provide further validation. This included use of Cox proportional hazards modeling (CPHM) to adjust for baseline values of albumin, PD‐L1 expression, body weight, and ECOG performance status.
RESULTS
PopPK analysis
Analysis set
The updated final PopPK model included data from 1063 patients with solid tumors, including CSCC, BCC, NSCLC, and R/M CC, contributing to 17,312 cemiplimab concentration observations. A total of 1390 PK samples were excluded from the PopPK analysis, as summarized in Table S3. Demographic characteristics and baseline values for relevant covariates in the PopPK model are summarized in Table S4.
Base model
The base model structure was a two‐compartment model, with zero‐order i.v. infusion, first‐order elimination, and time‐varying CL described by a sigmoid maximum effect (Emax) function. The previous PopPK model included interindividual variability (IIV) terms on CL and intercompartmental CL (Q), V1 and V2, Emax, and time to reach 50% of the maximum change in CL (T50). To address model instability due to overparameterization, the major model refinement vs. the previous model was removing the IIV estimates on Emax and T50. The off‐diagonal covariance between IIV random effects on CL/Q and V1/V2 were also removed. The residual error model was simplified by removing proportional error components and estimating the log‐additive error only.
Final PopPK model
Covariate analysis resulted in retention of the following relationships in the final model: baseline body weight, sex, time‐varying albumin, baseline ALT, CSCC tumor type, and BCC tumor type on CL; baseline body weight, sex, and baseline albumin on V1; BCC tumor type on Emax; and other tumor type on T50. Structural PK parameter estimates for the final PopPK model were well estimated with percent relative standard error values <8% (Table 1). The HILL parameter in the sigmoid Emax function for time‐varying CL was fixed at 2.50, consistent with the previous PopPK model. The IIV of CL and Q, and V1 and V2, and the residual error were reduced vs. the base model. Post hoc analysis of PK parameters in the updated final model are shown in Table 2. In the overall population, mean (percent coefficient of variation [CV%]) baseline CL decreased from 0.246 L/day (40.9%) at first dose to 0.218 L/day (43.7%) at steady‐state (mean decrease of 10.8%). The change in CL between first dose and steady‐state was generally similar across tumor types, except in patients with BCC, for whom minimal change in CL was demonstrated. Mean (CV%) volume of distribution (Vss) of cemiplimab was estimated at 5.89 L (28.8%), and elimination half‐life was 19.7 days at first dose and 22.3 days at steady‐state. Following multiple dosing of cemiplimab 350 mg Q3W, the accumulation ratio was approximately two‐fold for all tumor types.
TABLE 1.
Cemiplimab PK parameters after modeling with the analysis dataset or bootstrap datasets for the final PopPK model
| Parameter | Final PopPK model | Bootstrap datasets (N = 474) a | |||||
|---|---|---|---|---|---|---|---|
| Estimate | ASE | %RSE | 95% CI b | Mean | Median | 95% CI b | |
| Fixed effects | |||||||
| TVCL0, L/day | 0.254 | 0.00395 | 1.56 | 0.246, 0.262 | 0.254 | 0.254 | 0.241, 0.269 |
| TVQ, L/day | 0.652 | 0.0238 | 3.65 | 0.606, 0.699 | 0.650 | 0.651 | 0.581, 0.728 |
| TVV1, L | 3.35 | 0.0422 | 1.26 | 3.27, 3.44 | 3.35 | 3.35 | 3.27, 3.44 |
| TVV2, L | 2.52 | 0.0419 | 1.66 | 2.44, 2.60 | 2.52 | 2.51 | 2.29, 2.81 |
| TVEmax | −0.174 | 0.00838 | 4.82 | −0.190, −0.157 | −0.181 | −0.180 | −0.232, −0.136 |
| TVT50, days | 73.7 | 5.57 | 7.56 | 62.8, 84.7 | 90.8 | 74.4 | 46.2, 245 |
| HILL | 2.50 c | — | — | — | |||
| CLWGTBL | 0.539 | 0.0496 | 9.19 | 0.442, 0.637 | 0.539 | 0.539 | 0.443, 0.639 |
| VssWGTBL | 0.499 | 0.0501 | 10.0 | 0.401, 0.597 | 0.499 | 0.501 | 0.412, 0.569 |
| CLSEX | −0.137 | 0.0180 | 13.2 | −0.172, −0.101 | −0.133 | −0.134 | −0.167, −0.0925 |
| V1SEX | −0.0801 | 0.0186 | 23.3 | −0.117, −0.0435 | −0.0798 | −0.0794 | −0.115, −0.0471 |
| V1ALBBL | −0.217 | 0.0617 | 28.4 | −0.338, −0.0961 | −0.221 | −0.225 | −0.361, −0.0870 |
| CLALB | −1.11 | 0.0269 | 2.42 | −1.16, −1.06 | −1.12 | −1.11 | −1.24, −0.984 |
| CLALTBL | −0.0729 | 0.0162 | 22.2 | −0.105, −0.0411 | −0.0724 | −0.0720 | −0.108, −0.0370 |
| CLCSCC | −0.216 | 0.0206 | 9.55 | −0.256, −0.176 | −0.219 | −0.219 | −0.257, −0.181 |
| CLBCC | −0.211 | 0.0258 | 12.2 | −0.261, −0.160 | −0.209 | −0.209 | −0.261, −0.158 |
| EmaxBCC | −0.872 | 0.110 | 12.6 | −1.09, −0.657 | −0.841 | −0.870 | −1.00, −0.517 |
| T50OTHER | −0.491 | 0.0595 | 12.1 | −0.608, −0.374 | −0.537 | −0.518 | −0.987, −0.169 |
| Residual variability | |||||||
| RE | 0.241 d | 0.000366 | 0.15 | 0.240, 0.242 d | 0.241 d | 0.240 d | 0.227, 0.258 d |
| IIV | |||||||
| ETA1 – CL_Q | 0.0892 | 0.00362 | 4.05 | 0.0822, 0.0963 | 0.0886 | 0.0884 | 0.0759, 0.101 |
| ETA2 – V1_V2 | 0.0709 | 0.00144 | 2.03 | 0.0681, 0.0737 | 0.0692 | 0.0674 | 0.0459, 0.105 |
| OFV | −27,190.388 | ||||||
Note: The η‐shrinkage values were 7.8% for CL_Q and 8.5% for V1_V2.
Time‐dependent CL was modeled using a sigmoid Emax relationship: .
Abbreviations: ASE, asymptotic standard error; BCC, basal cell carcinoma; CI, confidence interval; CL, clearance; CLALB, covariate impact of time‐varying albumin on CL; CLALTBL, covariate impact of baseline alanine aminotransferase on CL; CLBCC, covariate impact of BCC tumor type on CL; CLCSCC, covariate impact of CSCC tumor type on CL; CLSEX, covariate impact of female sex on CL; CLWGTBL, covariate impact of baseline body weight on CL_Q; CSCC, cutaneous squamous cell carcinoma; Emax, maximum effect in sigmoid model; EmaxBCC, covariate impact of BCC tumor type on Emax; η, interindividual random effects; HILL, hill exponent (γ) in the sigmoid Emax function describing the change in CL with time; IIV, interindividual variability; OFV, objective function value; PK, pharmacokinetics; PopPK, population PK; Q, intercompartmental clearance between the central and peripheral compartments; RE, residual error; RSE, relative standard error; SCC, squamous cell carcinoma; T50, time to reach 50% of the maximum change in CL; T50OTHER, covariate impact of other tumor type on T50; TVCL0, typical value of clearance at baseline; TVEmax, typical value of maximum change in CL with time; TVQ, typical value of inter‐compartmental clearance; TVT50, typical value of time to reach 50% of the maximum change in CL; TVV1, typical value of central volume of distribution; TVV2, typical value of peripheral volume of distribution; V1, distribution volume of central compartment; V2, distribution volume of peripheral compartment; V1ALBBL, covariate impact of baseline albumin on V1; V1SEX, covariate impact of female sex on V1; VssWGTBL, covariate impact of baseline body weight on V1_V2.
Results are summarized for 474 of 500 bootstrap runs that converged successfully and had condition number <1000.
The 2.5th and 97.5th percentiles of parameter distributions are reported as the 95% CI.
The HILL parameter in the sigmoid Emax function for time‐varying CL was fixed at 2.50.
Residual error is represented as a positive value by calculating the square root of (estimate)2.
TABLE 2.
Post hoc cemiplimab PK parameters using the final PopPK model
| Parameter | Tumor type (study) a | Patients, N | Mean (CV%) | SD |
|---|---|---|---|---|
| Clearance at the first dose, L/day | All patients | 1062 | 0.246 (40.9) | 0.100 |
| CSCC (study 1540) | 188 | 0.214 (33.4) | 0.0715 | |
| BCC (study 1620) | 132 | 0.192 (35.0) | 0.0671 | |
| NSCLC (study 1624) | 345 | 0.265 (37.9) | 0.100 | |
| All solid tumors (study 1423) | 397 | 0.262 (42.5) | 0.111 | |
| Clearance at steady‐state, L/day | All patients | 1062 | 0.218 (43.7) | 0.0951 |
| CSCC (study 1540) | 188 | 0.187 (38.6) | 0.0721 | |
| BCC (study 1620) | 132 | 0.194 (36.0) | 0.0700 | |
| NSCLC (study 1624) | 345 | 0.227 (42.9) | 0.0972 | |
| All solid tumors (study 1423) | 397 | 0.232 (45.4) | 0.105 | |
| Change in clearance, % b | All patients | 1062 | −10.8 | 15.4 |
| CSCC (study 1540) | 188 | −12.6 | 11.9 | |
| BCC (study 1620) | 132 | 2.7 | 20.4 | |
| NSCLC (study 1624) | 345 | −14.3 | 15.4 | |
| All solid tumors (study 1423) | 397 | −11.3 | 12.3 | |
| Half‐life at the first dose, days | All patients | 1062 | 19.7 (41.4) | 8.18 |
| CSCC (study 1540) | 188 | 21.4 (29.0) | 6.22 | |
| BCC (study 1620) | 132 | 24.9 (31.6) | 7.85 | |
| NSCLC (study 1624) | 345 | 17.5 (56.5) | 9.88 | |
| All solid tumors (study 1423) | 397 | 19.2 (33.3) | 6.40 | |
| Half‐life at steady‐state, days | All patients | 1062 | 22.3 (41.9) | 9.32 |
| CSCC (study 1540) | 188 | 24.8 (31.2) | 7.74 | |
| BCC (study 1620) | 132 | 24.5 (26.9) | 6.58 | |
| NSCLC (study 1624) | 345 | 20.7 (58.5) | 12.1 | |
| All solid tumors (study 1423) | 397 | 21.8 (34.5) | 7.50 | |
| Accumulation index | All patients | 1062 | 2.05 (23.5) | 0.482 |
| CSCC (study 1540) | 188 | 2.22 (21.6) | 0.481 | |
| BCC (study 1620) | 132 | 2.07 (18.9) | 0.391 | |
| NSCLC (study 1624) | 345 | 1.98 (27.0) | 0.535 | |
| All solid tumors (study 1423) | 397 | 2.02 (21.9) | 0.441 | |
| Vss, L c | All patients | 1062 | 5.89 (28.8) | 1.70 |
| CSCC (study 1540) | 188 | 5.86 (26.8) | 1.57 | |
| BCC (study 1620) | 132 | 6.07 (27.8) | 1.68 | |
| NSCLC (study 1624) | 345 | 5.61 (33.9) | 1.90 | |
| All solid tumors (study 1423) | 397 | 6.10 (25.2) | 1.53 | |
| AUC3wks after the fifth dose, % | All patients | 1062 | 92.1 (11.6) | 10.7 |
| CSCC (study 1540) | 188 | 89.1 (8.76) | 7.80 | |
| BCC (study 1620) | 132 | 94.6 (11.5) | 10.9 | |
| NSCLC (study 1624) | 345 | 91.3 (15.2) | 13.9 | |
| All solid tumors (study 1423) | 397 | 93.3 (8.29) | 7.73 |
Note: One patient from study 1423 was excluded from the summary of post hoc parameter estimates due to extremely high predicted Vss of 183.6 L that was inconsistent with the typical distribution volume for monoclonal antibodies. Therefore, post hoc parameters are reported for 1062 patients.
Abbreviations: AUC3wks, area under the concentration time curve for a 3‐week dosing interval; BCC, basal cell carcinoma; CSCC, cutaneous squamous cell carcinoma; CV%, percentage coefficient of variation; NSCLC, non‐small cell lung cancer; PK, pharmacokinetic; PopPK, population PK; Vss, volume of distribution; V1, volume of the central compartment; V2, volume of the peripheral compartment.
ClinicalTrials.gov numbers are NCT02760498, NCT03132636, NCT03088540, and NCT02383212 for studies 1540, 1620, 1624, and 1423, respectively.
Change between initial clearance of cemiplimab at first dose and clearance of cemiplimab at steady‐state.
Vss = V1 + V2.
CL and Q, as well as V1 and V2, were found to be dependent on body weight. In the updated model, the estimated exponent for the relationship of CL and Q with body weight was 0.539 (CLWGTBL [covariate impact of baseline body weight on CL and Q]); the estimated exponent for the relationship of V1 and V2 with body weight was 0.499 (VssWGTBL [covariate impact of baseline body weight on V1 and V2]), respectively. The previous model incorporated body weight using a fixed allometric value of 0.75 for CL and Q, and a fixed allometric value of 1 for V1 and V2, respectively. Cemiplimab exposure with a fixed‐dose regimen of 350 mg Q3W decreased with increasing body weight. Based on the area under the concentration‐time curve for a 3‐week dosing interval (AUC3wks), patients with low baseline body weight (<5th percentile; mean [CV%]: 45.8 [9.1] kg) were predicted to have a 36% higher exposure compared with patients with baseline body weights between the 5th and 95th percentile (mean [CV%]: 74.9 [18.0] kg). Conversely, patients with high baseline body weight (>95th percentile; mean [CV%]: 123 [11.5] kg) had a 26% lower exposure compared with patients with baseline body weights between the 5th and 95th percentiles.
Cemiplimab CL had an inverse linear relationship with time‐varying albumin that indicated lower CL as albumin levels increased. Patients with low baseline albumin levels (<30 g/L) were predicted to have ~29% lower mean cemiplimab AUC3wks at steady‐state vs. patients with normal baseline albumin levels (>35 g/L). The predicted steady‐state mean exposure metrics (Cmax, Ctrough, and AUC3wks) in each albumin category were consistent with those from the prior model (within <18%). Baseline CL was ~20% higher in patients with NSCLC leading to lower cemiplimab exposure vs. patients with CSCC or BCC. However, exposure differences across tumor types were <20%, based on AUC3wks. Moreover, predicted mean values for each exposure metric were consistent with those from the previous model (within ≤15%). The other covariates identified as statistically significant in the updated final model were not considered to impact cemiplimab exposure, including slightly lower CL (−13.7%) and V1 (−8.0%) in female vs. male patients, and a decrease in CL with increasing ALT, with minimal change (<8%) in cemiplimab CL at the 5th and 95th percentiles of baseline ALT (8 and 56.9 IU/L, respectively), compared with the median (19 IU/L). The tornado plots shown in Figure S3 illustrate that the impact of statistically significant covariates on cemiplimab exposure is within the level of between‐subject variability reported from the PopPK model.
Post hoc analysis showed that baseline PD‐L1 expression was not a significant covariate on cemiplimab baseline CL or time‐dependent CL parameters. Although higher baseline IgG levels were associated with greater cemiplimab CL, at the 5th and 95th percentiles of baseline IgG (5.31 and 17.08 g/L, respectively), cemiplimab CL was predicted to differ by ≤15% compared with a median IgG level of 9.63 g/L.
Evaluation of the final PopPK model using a bootstrap approach showed that of 500 bootstrap runs, 474 (94.8%) converged successfully. Inspection of goodness‐of‐fit plots demonstrated alignment between observed, individual model‐predicted, and typical individual‐predicted cemiplimab concentration data over time (Figure 1). The pcVPCs show that the observed data are generally contained within the simulated 90% CIs (Figure 2). The final PopPK model is shown in Figure S4.
FIGURE 1.
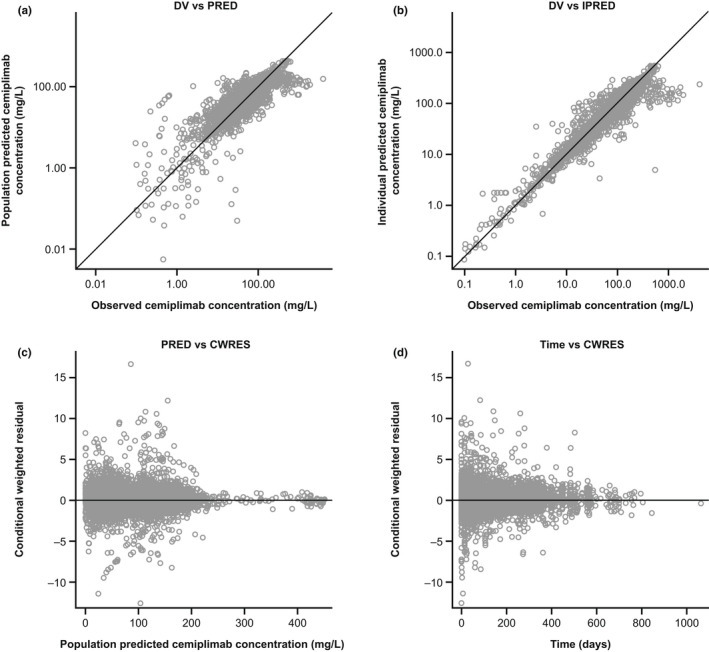
Diagnostic plots of the final covariate model. CWRES, conditional weighted residuals; DV, observed data; IPRED, individual predicted data; PK, pharmacokinetics; PRED, population predicted data.
FIGURE 2.
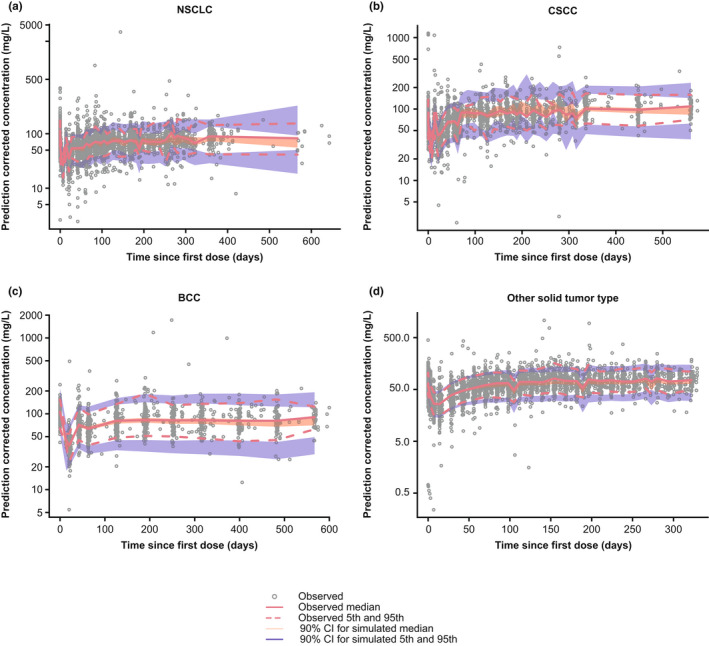
Prediction‐corrected visual predictive check plots of the final PopPK model by tumor type. Gray solid circles represent individually observed concentrations. The red solid and dashed lines represent the observed median and the 5th and 95th percentiles, respectively. The shaded areas represent the 90% CIs for the median, 5th, and 95th percentiles of the simulated data. BCC, basal cell carcinoma; CI, confidence interval; CSCC, cutaneous squamous cell carcinoma; NSCLC, non‐small cell lung cancer.
Predictive performance of the PK model for patients with R/M CC
External validation of the final PopPK model was achieved based on 292 patients with R/M CC from EMPOWER‐Cervical 1/GOG‐3016/ENGOT‐cx9, contributing 2030 PK samples. Demographic characteristics and baseline values for relevant covariates among patients in EMPOWER‐Cervical 1/GOG‐3016/ENGOT‐cx9 are summarized in Table S5. An external PPC demonstrated that a large majority of the observed cemiplimab concentrations are generally contained within the simulated 90% CIs, indicating the final PopPK model adequately described cemiplimab PK in patients with R/M CC (Figure 3). Post hoc cemiplimab PK parameters for patients with R/M CC from EMPOWER‐Cervical 1/GOG‐3016/ENGOT‐cx9 were estimated using the final PopPK model and found to be comparable to those in the overall population (Table S6). Model‐predicted typical cemiplimab exposures (Cmax, Ctrough, and AUC3wks) after first dose and at steady‐state for patients from EMPOWER‐Cervical 1/GOG‐3016/ENGOT‐cx9 were comparable to exposures in patients with other tumor types (Table S7).
FIGURE 3.
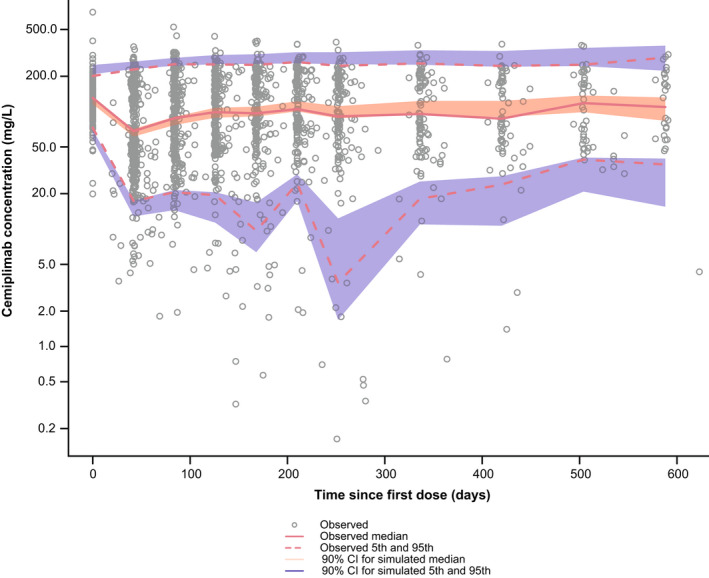
External posterior predictive check of cemiplimab exposure for patients with R/M CC from EMPOWER‐Cervical 1/GOG‐3016/ENGOT‐cx9 using the final PopPK model. Gray open circles represent individually observed concentrations. The red solid and dashed lines represent the observed median and the 5th and 95th percentiles, respectively. The shaded areas represent the 90% CIs for the median, 5th, and 95th percentiles of the simulated data. CI, confidence interval; ENGOT, European Network of Gynecological Oncological Trial; GOG, Gynecologic Oncology Group; PopPK, population pharmacokinetic; R/M CC, recurrent or metastatic cervical cancer.
Exposure‐response analysis in patients with R/M CC
Predicted exposure metrics for cemiplimab 350 mg based on simulations using individual‐predicted post hoc PK parameters are provided in Table S8, with a breakdown by exposure quartiles detailed in Table S9.
Nonparametric Kaplan–Meier curves for OS after the first cemiplimab dose suggested a relationship between predicted exposures (Ctrough,1 and Cav,1), with increased exposures associated with higher probabilities of OS (Figure 4). CPHM showed that Ctrough,1 and Cav,1 were significant as sole predictors of OS hazard in the model, but after adjusting for baseline covariates of albumin, body weight, and ECOG performance status, both exposure metrics were no longer significant because the 95% CI for the hazard ratio crossed 1 (Figure 5). Nonparametric Kaplan–Meier curves for PFS after the first cemiplimab dose did not reveal a relationship between predicted PFS and Ctrough,1 or Cav,1 (Figure S5). Moreover, the rank order of the exposure quartiles was not preserved, thus further analyses for PFS with CPHM were not undertaken. Similarly, Kaplan–Meier curves for DOR did not reveal increasing or decreasing trends in Ctrough,1 or Cav,1 after the first cemiplimab dose (Figure S6). Therefore, it is unlikely that an exposure‐response relationship for DOR exists in patients with R/M CC over the exposure range studied. Box plots comparing Ctrough,1 and Cav,1 based on BOR are shown in Figure S7. Exposure metrics between responders (i.e., patients with complete or partial response) and all other patients were unlikely to differ due to the overlapping distribution of values and comparable mean and median values. Logistic regression models were constructed to examine whether a trend existed between exposure metrics (Ctrough,1 or Cav,1) and ORR (based on BOR). These regression models with Ctrough,1 and Cav,1 fitted as continuous exposure metrics demonstrate a relatively flat fitted regression line with no increasing or decreasing trend in the cemiplimab exposure quartiles (Figure S8).
FIGURE 4.
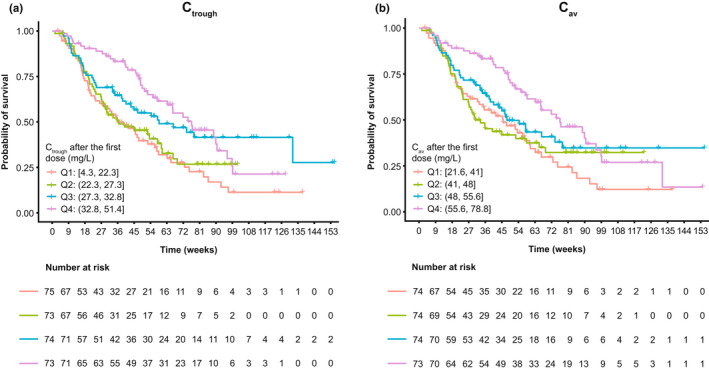
Kaplan–Meier curves of OS stratified by quartiles of individual predicted cemiplimab (a) Ctrough and (b) Cav after the first dose in patients with R/M CC from EMPOWER‐Cervical 1/GOG‐3016/ENGOT‐cx9. The vertical hatches through the Kaplan–Meier curves represent the last documented times that patients were observed to be alive. These censored patients may have dropped out of the study, or, because not all patients were enrolled in the study at the same time, they may have been censored at the time that the data cutoff occurred for this analysis. A drop in the Kaplan–Meier curve indicates the time of death due to any cause. The table provided underneath the Kaplan–Meier plot describes the number of patients alive in each quartile at each timepoint, which, for those patients, occurred before the data cutoff date. Ctrough and Cav exposure quartiles are detailed in Table S9. Cav, average concentration; Ctrough, trough serum concentration; ENGOT, European Network of Gynecological Oncological Trial; GOG, Gynecologic Oncology Group; OS, overall survival; Q, quartile; R/M CC, recurrent or metastatic cervical cancer.
FIGURE 5.
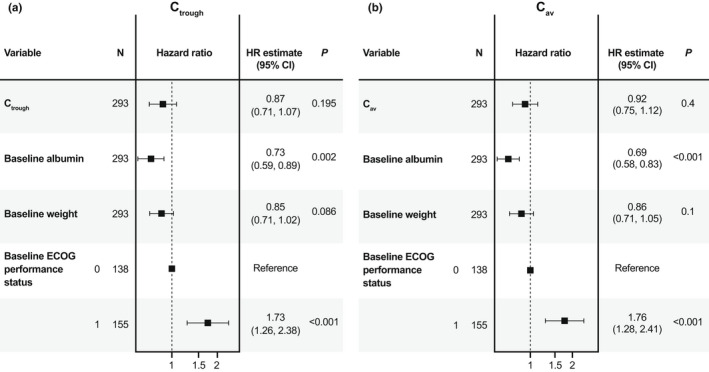
Forest plot of Cox proportional hazards model of OS with individual predicted cemiplimab (a) Ctrough and (b) Cav after the first dose and baseline covariates as model predictors in patients with R/M CC from EMPOWER‐Cervical 1/GOG‐3016/ENGOT‐cx9 (Cox proportional hazard modeling data shown for 293 out of 295 patients as baseline albumin values were not available for two patients). Ctrough and Cav were fit based on after first dose values in this model. Continuous covariates were standardized to have a mean of 0 and SD of 1 by centering values from the mean value and scaling by the SD of the covariate. Interpretation of the estimated HR is based on a change in one unit of SD from the mean value rather than a change in one unit of the covariate from the baseline value of 0. The 95% CI represents the 95% CI of the HR estimate, where a value of 1 contained within the interval indicates that the covariate is not significant. Mean values are detailed in Table S8. Cav, average concentration; CI, confidence interval; Ctrough, trough serum concentration; ECOG, Eastern Cooperative Oncology Group; ENGOT, European Network of Gynecological Oncological Trial; GOG, Gynecologic Oncology Group; HR, hazard ratio; OS, overall survival; R/M CC, recurrent or metastatic cervical cancer.
Nonparametric Kaplan–Meier curves of grade ≥3 irAEs (Figure S9) and all irAEs (Figure S10) suggested higher cemiplimab exposure quartiles correlated with lower probabilities of patients with R/M CC experiencing irAEs. Univariate CPHM for grade ≥3 irAEs (Table S10) and for all irAEs (Table S11), however, demonstrated that exposure metrics were not significant predictors of irAE hazard.
DISCUSSION
The updated PopPK model for cemiplimab showed improved model stability and robustness, as represented by 94.8% successful bootstrap runs vs. 47.6% of runs that converged successfully in the previous model using essentially the same PK dataset. Moreover, the updated model demonstrated precision of the structural model and variance parameters allowing for reliable predictions of individual PK exposures. The PopPK characteristics of cemiplimab between the two models are similar based on parameter estimates for the updated model vs. those from the previous model. Covariate analysis resulted in identification of the same general sources of PK variability as in the previous model, including body weight, albumin, baseline IgG, and tumor type. Cemiplimab exposure metrics, including Cmax, Ctrough, and AUC3wks, after the first dose and at steady‐state were similar across tumor types. Furthermore, predicted mean values for each parameter using the updated model were within ≤15% of the values predicted using the previous model.
External validation demonstrated that the updated model was suitable to predict cemiplimab concentrations in patients with R/M CC from EMPOWER‐Cervical 1/GOG‐3016/ENGOT‐cx9 receiving cemiplimab monotherapy (350 mg Q3W i.v.). The consistency of PK profiles in patients with R/M CC from EMPOWER‐Cervical 1/GOG‐3016/ENGOT‐cx9 compared to patients with CSCC, BCC, NSCLC, and all other solid tumor types, confirmed there were no meaningful differences in cemiplimab exposure across the conditions and parameters studied.
In EMPOWER‐Cervical 1/GOG‐3016/ENGOT‐cx9, cemiplimab demonstrated significant survival benefit with a median OS of 12.0 months vs. 8.5 months with chemotherapy (hazard ratio for death, 0.69; 95% CI, 0.56–0.84; 2‐sided p < 0.001) for second‐line treatment of R/M CC. 15 Moreover, ORR was higher for cemiplimab at 16.4% vs. 6.3% for chemotherapy, and the Kaplan–Meier estimated median DOR was 16.4 months for cemiplimab vs. 6.9 months for chemotherapy. Grade ≥3 treatment‐emergent adverse events, regardless of attribution, occurred in 45.0% of cemiplimab‐treated patients vs. 53.4% of chemotherapy‐treated patients.
In the present study, the relationship between cemiplimab exposures and efficacy and safety end points was analyzed in patients with R/M CC from EMPOWER‐Cervical 1/GOG‐3016/ENGOT‐cx9. Given that steady‐state metrics are often affected by treatment response, with higher exposures observed in responders than in the rest of the patient population, only exposure metrics after the first dose were used in the exposure‐response analysis in order to minimize the confounding effect of treatment response. 18 , 19 In the exposure‐efficacy analysis, Kaplan–Meier plots stratified by exposure quartiles revealed a relationship between cemiplimab exposure and OS. Subsequent CPHM, however, demonstrated that exposure metrics were not statistically significant predictors of OS hazard after adjusting for baseline albumin, body weight, and ECOG performance status. Furthermore, exposure‐response analyses for PFS and DOR showed no significant relationship with cemiplimab exposure metrics (Ctrough and Cav) after the first dose, as determined through graphical analyses, including Kaplan–Meier plots stratified by quartiles of exposure and logistic regression. Consequently, these analyses indicate that the clinical efficacy of cemiplimab in patients with R/M CC would not be enhanced with exposures (Ctrough and Cav) exceeding the range provided by a cemiplimab regimen of 350 mg Q3W. In the exposure‐response analyses for safety, CPHM demonstrated that cemiplimab exposure metrics (Cmax) were not predictive of irAE hazard after adjusting for baseline covariates. This suggests that the occurrence of irAEs is likely to be related to intrinsic immune characteristics rather than cemiplimab concentrations in serum (exposure).
Based on efficacy and safety data from clinical trials and supported by PopPK modeling and simulations, a cemiplimab fixed‐dose i.v. regimen of 350 mg Q3W is approved in the United States and Europe for the treatment of advanced CSCC. The same cemiplimab dosage regimen is also approved in the United States for the treatment of advanced BCC, and first‐line treatment of advanced NSCLC with PD‐L1 expression ≥50%. Here, we show that the updated PopPK model adequately described cemiplimab PK in patients with R/M CC on cemiplimab monotherapy (350 mg Q3W i.v.), and confirmed that cemiplimab PK are similar across tumor types. Moreover, an absence of exposure–response relationships for efficacy and safety end points was demonstrated for patients with R/M CC on cemiplimab monotherapy (350 mg Q3W i.v.). Thus, the updated PopPK model and exposure‐response analyses, together with the favorable clinical efficacy outcomes and safety findings observed in EMPOWER‐Cervical 1/GOG‐3016/ENGOT‐cx9, may be supportive of the cemiplimab regimen of 350 mg Q3W i.v. for second‐line treatment of R/M CC.
AUTHOR CONTRIBUTIONS
J.H.N., D.E., N.D., A.P., D.C., J.D.D., and N.A.‐H. wrote the manuscript. J.H.N., A.P., D.C., J.D.D., and N.A.‐H. designed the research. J.H.N., D.E., N.D., A.P., J.D.D., D.C., and N.A.‐H. performed the research. J.H.N., D.E., N.D., A.P., J.D.D., D.C., and N.A.‐H. analyzed the data.
FUNDING INFORMATION
This analysis was funded by Regeneron Pharmaceuticals, Inc., and Sanofi.
CONFLICT OF INTEREST
J.H.N., A.P., D.C., and J.D.D. are employees of and stockholders in Regeneron Pharmaceuticals, Inc. D.E., N.D., and N.A.‐H. are full‐time employees of the Ann Arbor Pharmacometrics Group (A2PG) and consultants for Regeneron Pharmaceuticals, Inc.
Supporting information
Appendix S1
ACKNOWLEDGMENTS
The authors thank the patients, their families, and all investigators involved in the studies used in this analysis. Medical writing support was provided by Atif Riaz, PhD, of Prime, Knutsford, UK, supported by Regeneron Pharmaceuticals, Inc., according to Good Publication Practice guidelines (http://annals.org/aim/article/2424869/good‐publication‐practice‐communicating‐company‐sponsored‐medical‐research‐gpp3). The sponsor was involved in the study design and collection, analysis, and interpretation of data, as well as data checking of information provided in the manuscript. The authors were responsible for all content and editorial decisions, and received no honoraria related to the development of this publication.
Nguyen J‐H, Epling D, Dolphin N, et al. Population pharmacokinetics modeling and exposure‐response analyses of cemiplimab in patients with recurrent or metastatic cervical cancer. CPT Pharmacometrics Syst Pharmacol. 2022;11:1458‐1471. doi: 10.1002/psp4.12855
REFERENCES
- 1. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209‐249. [DOI] [PubMed] [Google Scholar]
- 2. Okunade KS. Human papillomavirus and cervical cancer. J Obstet Gynaecol. 2020;40:602‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Canfell K, Kim JJ, Brisson M, et al. Mortality impact of achieving WHO cervical cancer elimination targets: a comparative modelling analysis in 78 low‐income and lower‐middle‐income countries. Lancet. 2020;395:591‐603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. National Comprehensive Cancer Network . NCCN clinical practice guidelines in oncology: cervical cancer (version 1.2022). 2021. https://www.nccn.org/professionals/physician_gls/pdf/cervical.pdf. Accessed November 20, 2021.
- 5. Cohen AC, Roane BM, Leath CA 3rd. Novel therapeutics for recurrent cervical cancer: moving towards personalized therapy. Drugs. 2020;80:217‐227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tewari KS, Sill MW, Long HJ 3rd, et al. Improved survival with bevacizumab in advanced cervical cancer. N Engl J Med. 2014;370:734‐743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kitagawa R, Katsumata N, Shibata T, et al. Paclitaxel plus carboplatin versus paclitaxel plus cisplatin in metastatic or recurrent cervical cancer: the open‐label randomized phase III trial JCOG0505. J Clin Oncol. 2015;33:2129‐2135. [DOI] [PubMed] [Google Scholar]
- 8. Lorusso D, Ferrandina G, Pignata S, et al. Evaluation of pemetrexed (Alimta, LY231514) as second‐line chemotherapy in persistent or recurrent carcinoma of the cervix: the CERVIX 1 study of the MITO (Multicentre Italian Trials in Ovarian Cancer and Gynecologic Malignancies) group. Ann Oncol. 2010;21:61‐66. [DOI] [PubMed] [Google Scholar]
- 9. Miller DS, Blessing JA, Bodurka DC, Bonebrake AJ, Schorge JO. Evaluation of pemetrexed (Alimta, LY231514) as second line chemotherapy in persistent or recurrent carcinoma of the cervix: a phase II study of the Gynecologic Oncology Group. Gynecol Oncol. 2008;110:65‐70. [DOI] [PubMed] [Google Scholar]
- 10. Burova E, Hermann A, Waite J, et al. Characterization of the anti‐PD‐1 antibody REGN2810 and its antitumor activity in human PD‐1 knock‐in mice. Mol Cancer Ther. 2017;16:861‐870. [DOI] [PubMed] [Google Scholar]
- 11. Macdonald LE, Karow M, Stevens S, et al. Precise and in situ genetic humanization of 6 Mb of mouse immunoglobulin genes. Proc Natl Acad Sci U S A. 2014;111:5147‐5152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Murphy AJ, Macdonald LE, Stevens S, et al. Mice with megabase humanization of their immunoglobulin genes generate antibodies as efficiently as normal mice. Proc Natl Acad Sci U S A. 2014;111:5153‐5158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. European Medicines Agency . LIBTAYO® EPAR. 2019. https://www.ema.europa.eu/en/medicines/human/EPAR/libtayo. Accessed December 3, 2021.
- 14. Regeneron Pharmaceuticals, Inc., Sanofi‐Aventis US LLC . LIBTAYO® (cemiplimab‐rwlc) injection, for intravenous use [prescribing information]. 2021. https://www.regeneron.com/downloads/libtayo_fpi.pdf. Accessed December 3, 2021.
- 15. Tewari KS, Monk BJ, Vergote I, et al. Survival with cemiplimab in recurrent cervical cancer. N Engl J Med. 2022;386:544‐555. [DOI] [PubMed] [Google Scholar]
- 16. Yang F, Paccaly AJ, Rippley RK, Davis JD, DiCioccio AT. Population pharmacokinetic characteristics of cemiplimab in patients with advanced malignancies. J Pharmacokinet Pharmacodyn. 2021;48:479‐494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Beal SL. Ways to fit a PK model with some data below the quantification limit. J Pharmacokinet Pharmacodyn. 2001;28:481‐504. [DOI] [PubMed] [Google Scholar]
- 18. Dai HI, Vugmeyster Y, Mangal N. Characterizing exposure‐response relationship for therapeutic monoclonal antibodies in immuno‐oncology and beyond: challenges, perspectives, and prospects. Clin Pharmacol Ther. 2020;108:1156‐1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu C, Yu J, Li H, et al. Association of time‐varying clearance of nivolumab with disease dynamics and its implications on exposure response analysis. Clin Pharmacol Ther. 2017;101:657‐666. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1