

Abstract
Navtemadlin is an orally bioavailable small molecule that blocks the protein-protein interaction between MDM2 and the tumor suppressor protein p53, leading to p53-mediated cell cycle arrest and apoptosis. It is being evaluated in clinical trials for a variety of malignancies, both as a single agent and in combination regimens. A sensitive, robust LC-MS/MS method was developed to quantitate navtemadlin in plasma, and this method was also validated using brain tissue homogenate. Sample preparation involved protein precipitation of plasma or brain tissue homogenate using acetonitrile. Navtemadlin, navtemadlin glucuronide, and the internal standard, D6-navtemadlin, were separated from microsomal incubation extracts using gradient elution and a Zorbax XDB C18 column. Analytes were detected using a SCIEX 5500 triple quadrupole mass spectrometer in positive electrospray ionization mode. The assay range of 1–1,000 ng/mL was shown to be accurate (96.1–102.0% and 95.7–104%) and precise (CV ≤ 10.6% and ≤ 6.6%) in plasma and brain tissue homogenate, respectively. An 8,000 ng/mL navtemadlin sample diluted 1:10 (v/v) with plasma was also accurately quantitated. Navtemadlin has been stable in frozen plasma at −70°C for at least 20 months. This validated LC-MS/MS method was applied to determine navtemadlin concentrations in plasma and brain tissue samples from two separate patients receiving 120 mg/day navtemadlin on protocol ABTC1604.
Keywords: Assay, Tandem mass spectrometry, Navtemadlin, Validation
1. Introduction
The tumor suppressor protein p53, encoded by the TP53 gene, plays an essential role in preventing cancer development through activating the transcription of genes involved in cell cycle arrest, DNA repair, and apoptosis [1, 2]. Inactivation of p53 provides a growth advantage and is often considered a necessary step in carcinogenesis [3, 4]. Approximately 50% of all human cancers harbor TP53 mutations resulting in attenuation or loss of p53 function [5]. The remaining tumors with wild-type TP53 frequently overexpress the murine double minute 2 protein (MDM2), which negatively regulates p53 [2, 3]. MDM2 inhibits p53 activity by: 1) directly blocking transcriptional activation of p53, 2) promoting export of p53 from the nucleus to the cytoplasm, and 3) inducing degradation of p53 via ubiquitination through its E3 ligase activity [3, 5]. Disruption of binding of MDM2 to p53 blocks all three of these mechanisms and leads to p53-mediated cell cycle arrest and apoptosis. Therefore, MDM2 has become an attractive therapeutic target for the treatment of TP53 wild-type cancers [6, 7].
Navtemadlin (previously KRT232 and AMG232) is an orally bioavailable, selective small molecule inhibitor of MDM2 that blocks the protein-protein interaction between MDM2 and p53 [5, 8], with an in vitro half-maximal inhibitory concentration (IC50) of 1.0 nM in a homogeneous time resolved fluorescence based assay [8]. Navtemadlin also demonstrated robust cell-based activity, with IC50 of 9.1 nM in SJSA-1 human osteosarcoma cells, and in vivo activity, with a median effective dose (ED50) of 9.1 mg/kg in a SJSA-1 xenograft mouse model following once daily dosing for 14 days. Based on preclinical pharmacokinetic and toxicity studies, a starting dose of 15 mg daily was selected for a first-in-human study of navtemadlin monotherapy in patients with solid tumors or multiple myeloma, and the maximum tolerated dose (MTD) was determined to be 240 mg daily [9]. At the MTD, day 1 mean Cmax and mean AUC24h were 1350 ng/mL and 8480 ng*h/mL, respectively. Higher doses of navtemadlin have been used in acute myeloid leukemia (AML) studies. In a phase 1b study of navtemadlin in relapsed/refractory AML, the MTD was determined to be 360 mg daily, and the day 1 mean Cmax and mean AUC24h were 5090 ng/mL and 27900 ng*h/mL, respectively, at the MTD (NCT02016729) [10]. The PK profile of the major metabolite navtemadlin-acyl-glucuronide was ~40% of the parent compound but with a similar profile. Navtemadlin is now being evaluated in the treatment of a variety of malignancies, including leukemia, myeloproliferative neoplasms, myeloma, sarcoma, lung cancer, and brain cancer, both as a single agent and as a part of combination regimens.
Erba and colleagues previously reported an LC-MS/MS bioanalytical method for the pharmacokinetic analysis of navtemadlin in NCT02016729 [10]; this method has subsequently been used in other clinical studies [9, 11]. However, these prior studies lack sufficient details of the bioanalytical method, including but not limited to sample preparation, chromatography, and stability information, and they have also not validated the method using brain tissue. Herein, we describe an LC-MS/MS method for the bioanalysis of navtemadlin in plasma over the range of 1–1000 ng/mL in detail as well as a fit-for-purpose validation using monkey brain tissue which enabled the quantification of navtemadlin concentrations in human brain tissue. Our analytical method will enable the assessment of navtemadlin exposure-response relationships with various pharmacodynamic endpoints, including MIC-1 changes, toxicity, and efficacy, and will ultimately support the clinical development of navtemadlin in multiple ongoing clinical studies.
2. Materials and Methods
2.1. Chemicals and reagents
All analytes had a purity greater than 98%. Navtemadlin was purchased from MedChemExpress (Monmouth Junction, NJ), and the stable isotope D6-navtemadlin was obtained from Moravek (Brea, CA). Authentic navtemadlin glucuronide reference standard was unavailable. Navtemadlin glucuronide was therefore generated from a human liver microsome experiment using standard methods [12]. HPLC grade acetonitrile and formic acid were purchased from EMD Chemical Inc. (Billerica, MA). Molecular biological grade dimethyl sulfoxide (DMSO) was purchased from Sigma Aldrich (St. Louis, MO). Deionized water was obtained from Millipore Milli-Q-UF filtration system (Milford, MA). Pooled human liver microsomes and cofactors for the incubations were purchased from Corning (Glendale, AZ). Drug-free sodium EDTA human plasma was purchased from Biological Specialty Company (Colmar, PA, USA).
2.2. Chromatography
The LC system was a Waters Acquity UPLC system (Milford, MA). The autosampler was maintained at 10°C or below. Analytes were separated using a Zorbax XDB C18 column (50 mm × 2.1 mm, 3.5 μm, Agilent Technologies, Santa Clara, CA) at room temperature using gradient elution. Mobile phase A was water with 0.1% formic acid (v/v), and mobile phase B was acetonitrile with 0.1% formic acid (v/v). The flow rate was 0.3 mL/min. The gradient started with 50% mobile phase B and was held for 0.5 minutes, then was increased to 100% mobile phase B over 0.5 minutes, held for 1 minute, then returned back to 50% mobile phase B over 0.1 minutes and allowed to equilibrate for 0.9 minutes. Total run time was 3 minutes. The autosampler needle was washed with 0.8 mL of acetonitrile:water (50:50, v/v) and 0.7 mL of acetonitrile.
2.3. Mass spectrometry
The mass spectrometric detection was carried out using a SCIEX 5500 triple quadrupole mass spectrometer in positive electrospray ionization and multiple reaction monitoring (MRM) mode. The LC and the mass spectrometer were controlled by Analyst software (version 1.6.3 and greater).
The settings were: curtain gas 20 psi, collision gas 7 psi, ion spray voltage 5500 V, probe temperature 450°C, ion source gas one 40 psi, ion source gas two 50 psi, and exit potential 10. The collision cell exit potentials were 10 for both navtemadlin and the internal standard. The declustering potential was 150. The collision energies were 27 and 35 for navtemadlin and internal standard, respectively. MRM m/z transitions were the following: 569.2 → 194.2 for navtemadlin, 575.2 → 200.2 for the IS. Dwell time was 150 ms.
2.4. Preparation of calibration standards and quality control (QC) samples
Stock solutions for navtemadlin and the internal standard, D6-navtemadlin, were prepared at 1 mg/mL in DMSO and stored at −20°C. All working solutions, standards, and quality controls (QCs) were prepared fresh daily unless otherwise stated. Working solutions were made in acetonitrile-water (1:1, v/v) and spiked into blank human plasma to make the calibration curve and QC samples. The calibration curve consisted of eight calibrators at the following concentrations: 1, 5, 10, 25, 50, 100, 500, and 1,000 ng/mL. Blank and zero calibrators were also part of the calibration curve and were made from blank human plasma. The QC samples were made at 4 different concentrations for the validation: 1 ng/mL (lower limit of quantitation (LLOQ)), 3, 80, and 800 ng/mL. An additional above the upper limit of quantitation (AULQ) QC was prepared at 8,000 ng/mL and diluted 1:10 (v/v) in plasma for quantitation. Additional QC samples were batched and stored at −70°C for long-term and freeze-thaw stability. Homogenized monkey brain tissue was also used to prepare QCs that were run against the plasma standard curve.
2.5. Sample preparation
Brain samples were homogenized in 400 μL of human EDTA plasma immediately prior to analysis. 50 μL aliquots of either plasma or brain homogenate were then added to borosilicate glass test tubes and mixed with 200 μL of acetonitrile containing the internal standard (10 ng/mL of D6-navtemadlin). For blank samples, 200 μL of acetonitrile was added in lieu of internal standard. Samples were vortex-mixed, centrifuged (1200 ×g for 5 minutes at ambient temperature), and transferred to an autosampler vial. Then 10 μL was injected onto the LC system using an autosampling device at approximately 5°C.
2.6. Method validation
The validation of this method included sensitivity, selectivity, precision and accuracy, working solution stability, sample stability, stock stability, freeze/thaw stability, long-term sample stability, carryover, and matrix equivalency. The FDA guidelines were followed for all acceptance criteria [13].
2.7. Sensitivity
Sensitivity was determined by using the signal to noise ratio of the lower limit of quantitation (LLOQ) QC from three precision and accuracy validation runs. Signal to noise values greater than 10 were deemed acceptable.
2.8. Selectivity
Selectivity was determined by assessing for the presence of endogenous or exogenous interfering peaks in six different blank EDTA plasma lots. Any interfering peak area should be less than 20% of the peak area of the analytes at the LLOQ in blank plasma. The plasma was spiked at the low QC concentration in the same six lots. Each lot was analyzed in triplicate. Acceptance criteria specify that at least 50% of the QCs in each lot must be within 85–115% of the nominal concentration.
2.9. Precision and accuracy
Precision and accuracy validation runs included a single blank and zero-level standard (blank with internal standard), a calibration curve in duplicate, and QC samples at five different concentrations (LLOQ, low, medium, high QC, and 10x dilution QC) in quintuplicate. Runs were performed on three separate occasions. Inter-day precision, intra-day precision and accuracy estimates were obtained.
2.10. Matrix Equivalency
In order to determine the differences between extracted human plasma and extracted monkey brain tissue homogenized in human plasma, precision and accuracy validation runs were performed using calibration standards in plasma and QC samples prepared using both plasma and homogenized monkey brain tissue. If the homogenized brain tissue QC samples passed using the plasma standard curve, then this served as a surrogate for matrix equivalency, and brain tissue samples would be run using plasma curve standards and QCs.
2.11. Stability
Short-term benchtop stability of the navtemadlin stock solution was evaluated at room temperature over 6 hours. Long-term stability at −20°C was determined by injecting the test stock solution and comparing to freshly prepared stock solution. Long-term stock stability was evaluated at approximately 3 and 6 months. Long-term stability in EDTA plasma at −70°C was also evaluated for up to approximately 20 months. Processed sample stability was determined by using the freshly prepared standard curve and comparing the calculated concentrations of fresh low and high QCs to the same QCs reinjected after storing at 5°C (autosampler temperature) for approximately 3 days. Freeze/thaw stability was determined by preparing four sets of QCs and subjecting each to a varying number of freeze/thaw cycles (1–3 cycles). All experiments were performed in triplicate, and mean values were compared to the initial nominal concentrations for short-term, long-term, and freeze-thaw stability.
2.12. Carryover
In order to determine if carryover was present after an injection of a sample with high navtemadlin concentration, a zero-level standard (blank with internal standard) was injected following a 1,000 ng/mL (upper limit of quantitation (ULOQ)) standard in replicates of six. Then, a lower limit of quantitation (LLOQ) standard was injected following a ULOQ standard in replicates of six. Carryover was deemed not present if the zero-level standards were below the limit of quantitation and LLOQ samples were within 20% of 1 ng/mL.
2.13. Assay application
The method was applied to two patients who received navtemadlin administered orally in an ongoing Phase I clinical trial evaluating navtemadlin in brain cancer. One patient received two doses prior to a planned brain tumor resection, and the second patient was part of dose finding studies. The protocol ABTC1604 (Clinicaltrials.gov identifier NCT03107780) was approved by the Institutional Review Board at Dana-Farber Cancer Institute, the lead institution, as well as other Adult Brain Tumor Consortium sites.
The patients provided written informed consent. Brain biopsies were obtained at the time of surgery on day 2 of treatment with 120 mg navtemadlin, as close to 3–6 hours after dosing as possible, but not more than 8 hours after dosing. Biopsy specimens (0.05–0.10 cm3 (50–100 mg)) were taken from intact sections of tumor tissue in both the non-contrast enhancing and contrast-enhacing regions, then were rinsed with ice-cold phosphate buffered saline and gently blotted on Whatman filter paper to remove surface fluid. Tissue was then placed in cryogenic tubes and then on crushed dry ice. Blood samples were collected in EDTA tubes at baseline (pre-dose) and 1, 3, 5, 8, and 24 hours post-dose after a single dose of 120 mg navtemadlin. Samples were processed immediately by refrigerated (4°C) centrifugation for 10 minutes at 1500 ×g. All specimens were stored at −70°C until analysis.
Navtemadlin concentration was determined in plasma from one patient, and brain tissue samples from a second patient, using the validated method described above. The pharmacokinetic parameters for navtemadlin were estimated using noncompartmental analysis as implemented in Phoenix WinNonlin version 8.3 (Pharsight A Certara Company, Cary, NC).
2.14. Incurred sample reanalysis
Incurred sample reanalysis (ISR) was performed on plasma samples from ABTC1604 according to the FDA guidelines [13].
3. Results and Discussion
3.1. Chromatographic separation and detection
The LC-MS/MS method was developed and validated to quantitate navtemadlin in human EDTA plasma and brain tissue. Due to concerns of in-source fragmentation of the navtemadlin glucuronide to navtemadlin in the mass spectrometer, initial separation experiments proceeded with microsomal incubation extracts until sufficient chromatographic separation was achieved, with subsequent experiments using plasma. Stable isotope labeled navtemadlin (D6-navtemadlin) was used as an internal standard. Navtemadlin, navtemadlin glucuronide, and the internal standard were efficiently separated in microsomal incubation extracts using a Zorbax XDB C18 column with a 3 minute run time. The retention time of navtemadlin, navtemadlin glucuronide, and internal standard were 1.77, 1.41, and 1.76 minutes, respectively (Figure 1). The column eluent was diverted to wash for the first and last minute of each injection. Study samples were monitored for sufficient chromatographic separation of navtemadlin and the in-source fragmented navtemadlin glucuronide (Figure 2).
Figure 1.
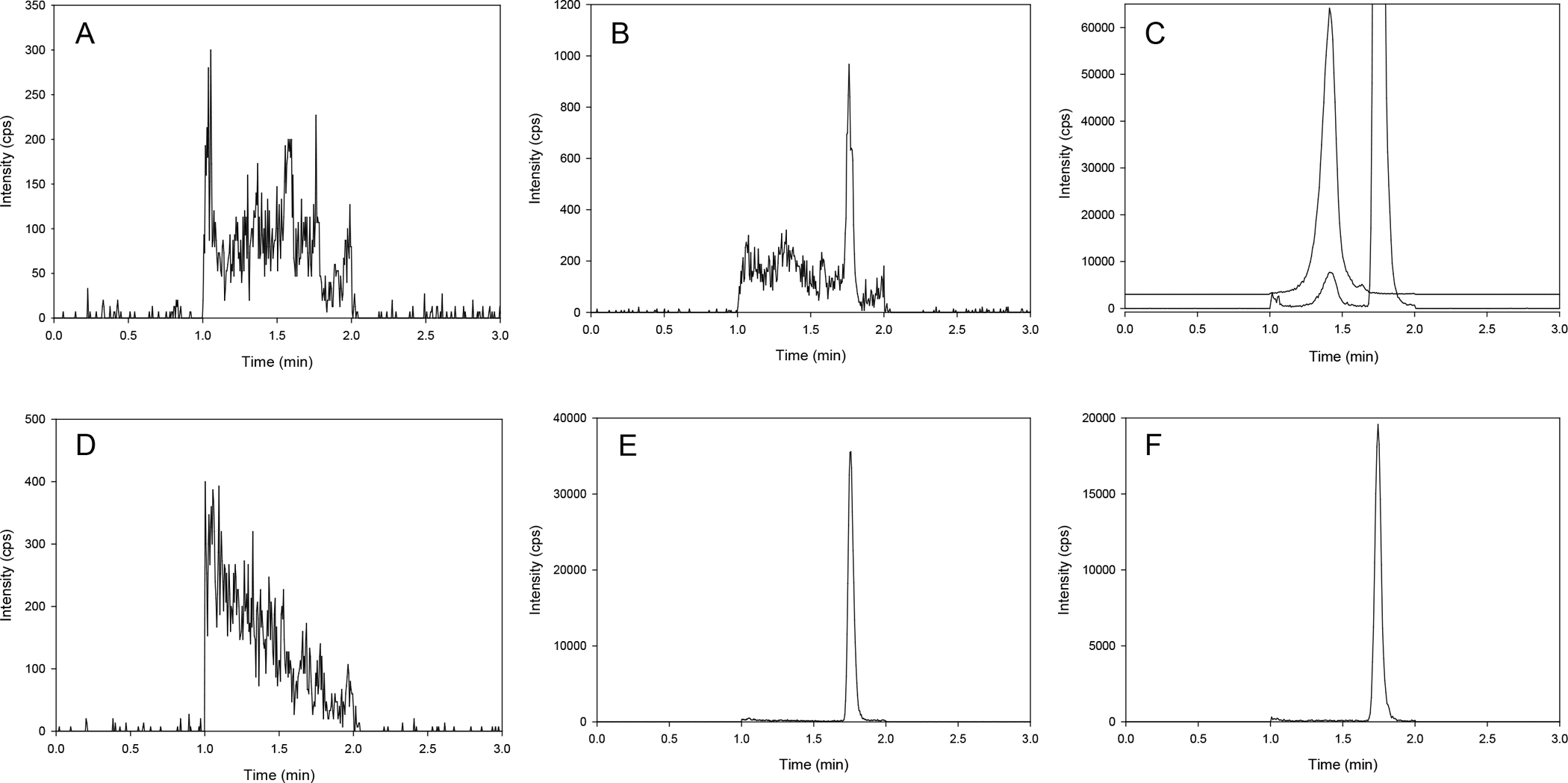
Chromatograms of navtemadlin (A, B), navtemadlin and navtemadlin glucuronide (C), internal standard (D6-navtemadlin; D, E, F). (A, D) blank human plasma; (B, E) LLOQ (1 ng/mL) in human plasma; (C, F) navtemadlin following 60 minute incubation with human liver microsomes. In order to demonstrate the separation of navtemadlin and navtemadlin glucuronide (C), the navtemadlin glucuronide trace is offset by 3000 counts (cps).
Figure 2.

Chromatograms for navtemadlin (A, B) and internal standard (D6-navtemadlin; C, D): (A, C) patient plasma sample with 404 ng/mL of navtemadlin; (B, D) patient brain tissue homogenate with 65.3 ng/g navtemadin.
3.2. Sensitivity and selectivity
The LLOQ was determined to be 1 ng/mL for navtemadlin in three precision and accuracy runs (Table 1). No major interferences were noted at the retention times for navtemadlin, navtemadlin glucuronide, or the internal standard (Figure 1). The average signal to noise ratio of the LLOQ was 170.2.
Table 1.
Validation characteristics of navtemadlin
| LLOQ (1 ng/mL) | Low (3 ng/mL) | Medium (80 ng/mL) | High (800 ng/mL) | AULQ (8000 ng/mL) | |
|---|---|---|---|---|---|
|
| |||||
| Plasma * | |||||
| Average | 1.02 | 2.89 | 76.9 | 778.3 | 7490.7 |
| Standard Deviation | 0.10 | 0.30 | 3.9 | 56.4 | 561.5 |
| Accuracy (%) | 102.0 | 96.5 | 96.1 | 97.3 | 93.6 |
| Intra-run Precision (%) | 10.6 | 7.9 | 2.7 | 2.7 | 2.5 |
| Inter-run Precision (%) | 10.4 | 11.1 | 5.0 | 7.2 | 7.5 |
| Brain * | |||||
| Average | 1.00 | 2.90 | 78.4 | 832.3 | 8137 |
| Standard Deviation | 0.10 | 0.20 | 7.80 | 26.9 | 53.8 |
| Accuracy (%) | 99.1 | 95.7 | 97.9 | 104.0 | 101.7 |
| Intra-run Precision (%) | 6.6 | 4.1 | 2.3 | 3.1 | 3.3 |
| Inter-run Precision (%) | 12.8 | 6.7 | 9.9 | 3.2 | 6.6 |
|
| |||||
| Plasma Stability (−70°C) (20 months) (%) | 100.3 | 96.6 | 95.0 | ||
| Plasma Freeze/Thaw Stability (−70°C) (3 cycles) (%) | 103.7 | 102.5 | |||
| Processed Sample Stability (4°C) (76 hours) (%) | 92.7 | 93.6 | |||
n=15, quintuplicate results, each in 3 separate runs
3.3. Precision and accuracy
3.3.1. Plasma
The calibration curve for navtemadlin was constructed from the ratio of the peak area of the analyte to that of the internal standard (D6-navtemadlin) using the least-squares quadratic regression analysis with 1/x2 weight. Validation experiments yielded excellent goodness of fit, with r2 ≥ 0.99 over a calibration range of 1–1,000 ng/mL. The intra-day coefficient of variation (%CV) or precision, inter-day precision, and percent accuracy of the LLOQ in plasma were 10.6%, 10.4%, and 102.0%, respectively (Table 1). The average intra-day precision for low, medium and high QCs ranged from 2.7% to 7.9%. The inter-day precision for low, medium and high QCs ranged from 5.0% to 11.1%. The accuracy for low, medium and high QCs ranged from 96.1% to 97.3%. The intra-day precision, inter-day precision, and percent accuracy for the AULQ QCs were 2.5%, 7.5%, and 93.6%, respectively.
3.3.2. Brain tissue homogenate
For monkey brain tissue homogenate in human plasma, the intra-day precision, inter-day precision, and percent accuracy were 6.6%, 12.8%, and 99.1%, respectively (Table 1). The intra-day precision for low, medium, and high QCs ranged from 2.3% to 4.1%. The inter-day precision for low, medium, and high QCs ranged from 3.2% to 9.9%. The accuracy for low, medium, and high QCs ranged from 95.7% to 104.0%. The intra-day precision, inter-day precision, and percent accuracy for the AULQ QCs were 3.3%, 6.6%, and 101.7%, respectively.
3.4. Matrix Equivalency
Based on the three precision and accuracy runs noted above, the monkey brain tissue homogenate was deemed equivalent to human plasma since the accuracy was within 85–115% and the precision was within 15%. Therefore, patient brain tissue samples were subsequently analyzed using plasma standard curves and QCs.
3.5. Stability
Navtemadlin stock solution in DMSO was stable at room temperature up to 1 hour but failed at 2 hours. Navtemadlin was stable in plasma at room temperature for up to 6 hours, for three freeze-thaw cycles at −70°C, and for 20 months at −70°C (Table 1). Processed samples were stable at 5°C for up to 76 hours (Table 1).
3.6. Carryover
No carryover peaks were detected in blank plasma following injection of the ULOQ standard. LLOQ standards injected after ULOQ standards also met acceptance criteria. Thus no carryover was observed for navtemadlin using the chromatographic conditions described (data not shown).
3.7. Assay application
The LC-MS/MS method was applied to plasma and brain tissue samples from two separate patients receiving 120 mg/day navtemadlin on protocol ABTC1604 (Figure 2). The first patient underwent brain tumor resection on day 2 of navtemadlin treatment, and navtemadlin concentration was 132.8 ng/g in non-contrast enhancing tumor and 65.3 ng/g in contrast enhancing tumor. Figure 3 represents the plasma concentration-time profile for navtemadlin from the second patient. In this patient, the maximum total plasma concentration (Cmax) of 404 ng/mL occurred at 2.87 h, and AUCINF was 2922 ng*h/mL, after a single dose of navtemadlin 120 mg.
Figure 3.
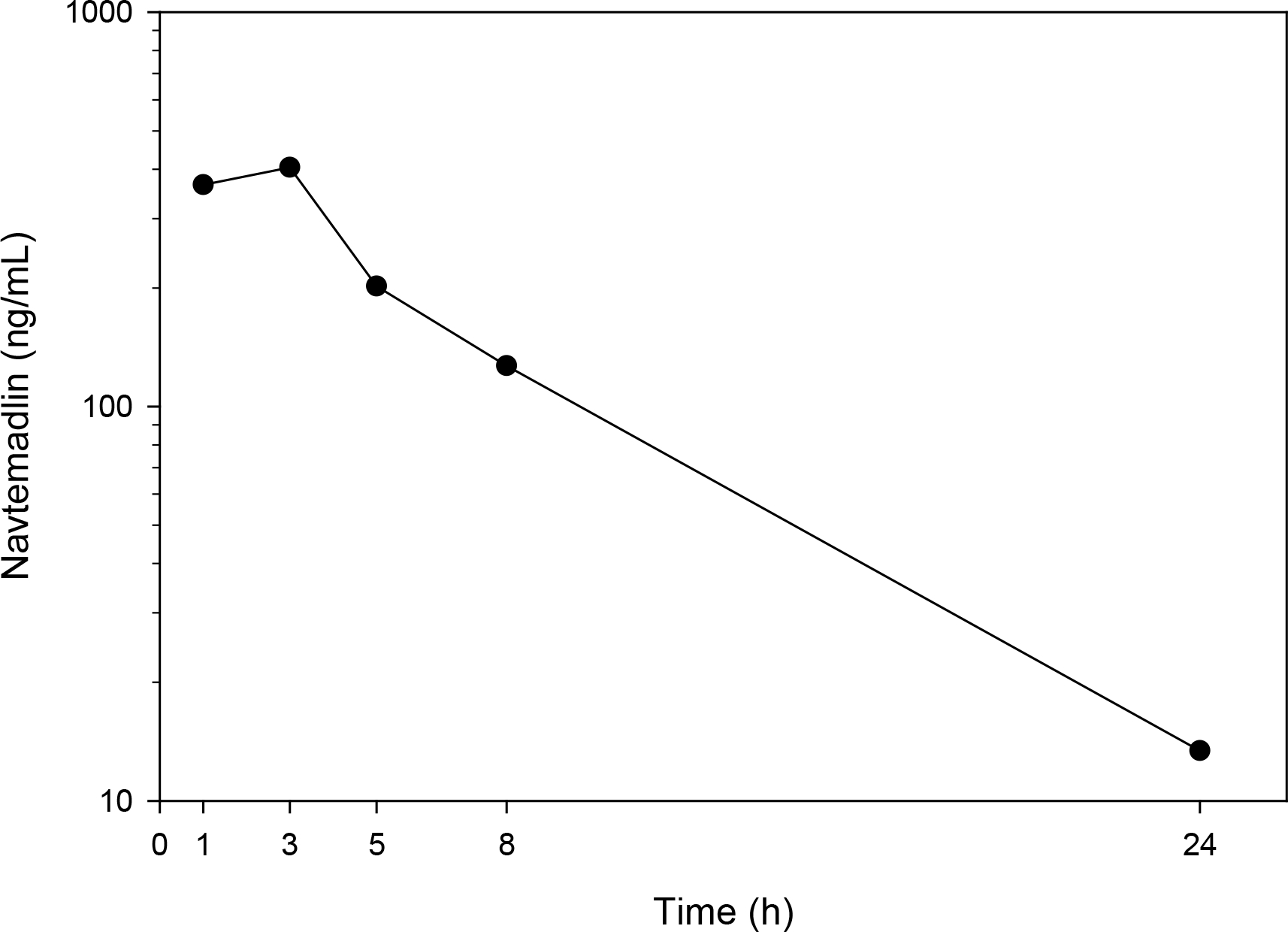
Navtemadlin concentration-time profile from a single patient following a single dose of 120 mg on a semi-logarithmic scale. The time points are the times at which plasma samples were collected relative to dose.
3.8. Incurred sample reanalysis
Incurred sample reanalysis of 32 samples resulted in one sample (3%) having a difference larger than 20%. Therefore, the method is robust since greater than 67% of results having a difference were within 20% of the original value [13].
4. Conclusion
In summary, the herein described LC-MS/MS assay method for quantifying navtemadlin in human EDTA plasma and homogenized brain tissue has demonstrated the fundamental parameters of accuracy, precision, selectivity, sensitivity, and range of reliable response. This affirms the assay method is accurate, reproducible, reliable, and suitable for its intended use. The range of reliable response determined during the assay validation for navtemadlin was 1–1,000 ng/mL. The method has been successfully applied to determine plasma pharmacokinetics of navtemadlin after oral administration in a patient, and was modified to achieve a fit-for-purpose quantitation of navtemadlin in brain tissue from another patient. Quantitation of navtemadlin in brain tissue has not previously been reported, thus indicating a novel application of our method that will be particularly useful in clinical trials studying navtemadlin in patients with brain cancer—both primary and metastatic—moving forward. The assay is currently being used to support navtemadlin development in multiple clinical trials, including one in multiple myeloma (NCT03031730), one in soft tissue sarcoma (NCT03217266), and two in leukemia (NCT03041688 and NCT04190550).
Acknowledgments
We would like to thank Linping Xu for her quality assurance review of the data. The project described was supported by the NCI Experimental Therapeutics Clinical Trials Network (grants UM1CA186691 and U24CA247648) and the Clinical Pharmacology Training Program grant (ABK, NIH T32GM066691). The project described was also supported by the Analytical Pharmacology Core of the Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins [NIH grants P30CA006973 and UL1TR003098, and the Shared Instrument Grant S10RR026824]. The project described was also supported by grant number UL1TR003098 from the National Center for Advancing Translational Sciences (NCATS), a component of the National Institutes of Health (NIH), and the NIH Roadmap for Medical Research. Its contents are solely the responsibility of the authors and do not necessarily represent the official view of the NCATS or NIH.
References
- [1].Levine AJ, Oren M, The first 30 years of p53: growing ever more complex, Nat Rev Cancer 9(10) (2009) 749–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Vazquez A, Bond EE, Levine AJ, Bond GL, The genetics of the p53 pathway, apoptosis and cancer therapy, Nat Rev Drug Discov 7(12) (2008) 979–87. [DOI] [PubMed] [Google Scholar]
- [3].Canon J, Osgood T, Olson SH, Saiki AY, Robertson R, Yu D, Eksterowicz J, Ye Q, Jin L, Chen A, Zhou J, Cordover D, Kaufman S, Kendall R, Oliner JD, Coxon A, Radinsky R, The MDM2 Inhibitor AMG 232 Demonstrates Robust Antitumor Efficacy and Potentiates the Activity of p53-Inducing Cytotoxic Agents, Mol Cancer Ther 14(3) (2015) 649–58. [DOI] [PubMed] [Google Scholar]
- [4].Vogelstein B, Lane D, Levine AJ, Surfing the p53 network, Nature 408(6810) (2000) 307–10. [DOI] [PubMed] [Google Scholar]
- [5].Rew Y, Sun D, Discovery of a small molecule MDM2 inhibitor (AMG 232) for treating cancer, J Med Chem 57(15) (2014) 6332–41. [DOI] [PubMed] [Google Scholar]
- [6].Ye Q, Jiang M, Huang WT, Ling Y, Olson SH, Sun D, Xu G, Yan X, Wong BK, Jin L, Pharmacokinetics and metabolism of AMG 232, a novel orally bioavailable inhibitor of the MDM2-p53 interaction, in rats, dogs and monkeys: in vitro-in vivo correlation, Xenobiotica 45(8) (2015) 681–92. [DOI] [PubMed] [Google Scholar]
- [7].Vassilev LT, MDM2 inhibitors for cancer therapy, Trends Mol Med 13(1) (2007) 23–31. [DOI] [PubMed] [Google Scholar]
- [8].Sun D, Li Z, Rew Y, Gribble M, Bartberger MD, Beck HP, Canon J, Chen A, Chen X, Chow D, Deignan J, Duquette J, Eksterowicz J, Fisher B, Fox BM, Fu J, Gonzalez AZ, Gonzalez-Lopez De Turiso F, Houze JB, Huang X, Jiang M, Jin L, Kayser F, Liu JJ, Lo MC, Long AM, Lucas B, McGee LR, McIntosh J, Mihalic J, Oliner JD, Osgood T, Peterson ML, Roveto P, Saiki AY, Shaffer P, Toteva M, Wang Y, Wang YC, Wortman S, Yakowec P, Yan X, Ye Q, Yu D, Yu M, Zhao X, Zhou J, Zhu J, Olson SH, Medina JC, Discovery of AMG 232, a potent, selective, and orally bioavailable MDM2-p53 inhibitor in clinical development, J Med Chem 57(4) (2014) 1454–72. [DOI] [PubMed] [Google Scholar]
- [9].Gluck WL, Gounder MM, Frank R, Eskens F, Blay JY, Cassier PA, Soria JC, Chawla S, de Weger V, Wagner AJ, Siegel D, De Vos F, Rasmussen E, Henary HA, Phase 1 study of the MDM2 inhibitor AMG 232 in patients with advanced P53 wild-type solid tumors or multiple myeloma, Invest New Drugs 38(3) (2020) 831–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Erba HP, Becker PS, Shami PJ, Grunwald MR, Flesher DL, Zhu M, Rasmussen E, Henary HA, Anderson AA, Wang ES, Phase 1b study of the MDM2 inhibitor AMG 232 with or without trametinib in relapsed/refractory acute myeloid leukemia, Blood Adv 3(13) (2019) 1939–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Taylor A, Lee D, Allard M, Poland B, Greg Slatter J, Phase 1 Concentration-QTc and Cardiac Safety Analysis of the MDM2 Antagonist KRT-232 in Patients With Advanced Solid Tumors, Multiple Myeloma, or Acute Myeloid Leukemia, Clin Pharmacol Drug Dev 10(8) (2021) 918–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Rudek MA, Venitz J, Ando Y, Reed E, Pluda JM, Figg WD, Factors involved in the pharmacokinetics of COL-3, a matrix metalloproteinase inhibitor, in patients with refractory metastatic cancer: clinical and experimental studies, J Clin Pharmacol 43(10) (2003) 1124–35. [DOI] [PubMed] [Google Scholar]
- [13].Food and Drug Administration Center for Drug Evaluation and Research, U.S. Department of Health and Human Services., Guidance for industry bioanalytical method validation, May, 2018. [Google Scholar]