

Abstract
Adaptable and consistent neural function relies at least in part on the ongoing repair of oxidative damage that can accumulate in the brain over a lifespan. To determine whether forebrain neuron-targeted knockout of AP endonuclease 1 (APE1), a critical enzyme in the base excision DNA repair pathway, contributes to neuronal impairments, we generated APE1 conditional knockout mice under the control of the CamKIIα promotor (APE1 cKO). Spatial learning and memory were tested using the Morris water maze. Synaptic markers, including synapsin, vGLUT, GABA1, and GAD were immunostained and quantified. Dendritic morphology and number were characterized using Golgi staining. Long-term potentiation (LTP) was measured in slices from the 6-month-old brain. APE1 cKO mice did not significantly differ from WT mice in the learning phase of the Morris water maze, but performed significantly worse during the memory phase of the Morris water maze. vGLUT, GABA1, and GAD immunostaining was significantly decreased in APE1 cKO mice without concomitant changes in the number of synapsin-positive structures, suggesting that neural networks may be impaired but not at the level of total presynaptic structures. Dendrites were reduced both in number and length of spines in APE1 cKO mice. APE1 cKO brain slices exhibited decreased LTP induction compared to WT brain slices. Together, these data indicate that the conditional loss of APE1 in forebrain neurons leads to a phenotype consistent with expedited brain aging
Keywords: DNA repair, DNA damage, apurinic/apyrimidinic endonuclease-1, aging
Aging-related cognitive impairments are emerging as a global health concern with the increase in human lifespan [1, 2], emphasizing the need to understand both normal and pathological effects of age on cognitive tasks. Cognitive function declines steadily with aging, with or without amyloid deposition [3, 4], and recent evidence suggests that aspects of age-related cognitive decline may begin in healthy adults as early as the third decade of human life [5]. Impaired cognition decreases the quality of life for both the patient and their family [6, 7]. Therefore, combating age-related cognitive impairment is an important and timely research topic.
During normal aging, pathological changes appear gradually and are often associated with prolonged events such as sustained inflammation and oxidative stress [8-11]. The failure of neurons to adapt to age-related stress results in neuronal dysfunction, including decreased synaptic transmission [12, 13], dendritic spine loss [14] and suppressed neuronal network activity [15]. These and other factors are thought to underlie aging-related cognitive impairment. As an example, DNA damage is a significant consequence of oxidative stress and is closely associated with aging-related cognitive decline [10, 16]. Although endogenous mechanisms exist to repair DNA damage, these repair mechanisms become less effective with aging, leading to an accumulation of DNA lesions, loss of genome stability, and altered gene expression [17]. Such alterations in DNA structure may contribute to age-related neurodegenerative diseases [18] and cognitive dysfunction [19-21].
The enzyme apurinic/apyrimidinic endonuclease-1 (APE1) participates in both the base-excision repair (BER) pathway, one of the main DNA repair pathways responsive to oxidative DNA damage, as well as in transcription and cellular functions through its redox activity [22]. Overexpression of APE1 protein in cultured cells or animals is neuroprotective against oxidative damage and ischemic brain injuries [23-25], whereas conditional knockout of APE1 exacerbates ischemic brain injury [26]. Polymorphisms in DNA BER enzymes, including APE1, are associated with age-related cognitive impairments in healthy elderly individuals and reduced performance on mini-mental state examinations in AD patients [27, 28].
In the current study, we generated calcium/calmodulin-dependent protein kinase II alpha (CaMKIIα) promotor-driven APE1 knockout mice, in which Cre recombinase promoter activity deletes APE1 in neurons of the forebrain, including in the selectively vulnerable CA1 pyramidal cell layer of the hippocampus [29, 30]. Although many regions of the brain may be susceptible to aging-related changes in either structure or function, the somatosensory cortex and hippocampus have been well studied in terms of both cellular and associated functional changes related to aging, including loss of dendritic spines [31-33], as well as high CaMKIIα promotor activity [30]. Targeted knockout of APE1 allowed us to attribute aging-related changes in neuronal function to the absence of APE1 in CaMKIIα-expressing neurons, which are enriched in the cortex and hippocampus, and their associated behavioral and functional outcomes.
MATERIALS AND METHODS
Generation of and breeding of mutant mice
APEfl/fl mice were generated and characterized as previously described [26]. To achieve APE1 gene knockout predominantly in forebrain neurons, we crossed APEfl/fl mice with a Camk2a-cre transgenic line expressing Cre recombinase under the control of the calcium/calmodulin-dependent protein kinase II alpha (CaMKIIα) promotor (Stock No. 027310, The Jackson Laboratory). Expression controlled by this promotor is enriched in neurons in the CA1 pyramidal cell layer in the hippocampus as well as the dentate gyrus and approximately one-third of the neurons in layers II/III and VI of the neocortex, and to a lesser extent in subcortical regions [30] [34]. These mice will be referred to throughout the paper as APE cKO mice. As genotype controls, we used littermate mice lacking Cre recombinase expression (referred to as WT controls). All mice were on a C57BL/6J genetic background. Only male mice (n=50) were used for this study in order to properly power the statistical analyses, draw on the extensive literature in aging males, and, for this initial study, avoid confounds of hormone fluctuations in aging female mice.
All animal procedures were approved by the Institutional Animal Care and Use Committee at the University of Pittsburgh, and in compliance with the National Institute of Health guidelines. The animal data reporting of the current study follows the ARRIVE 2.0 guidelines [35].
Golgi staining
Golgi staining was performed using the FD Rapid GolgiStain™ kit (FD NeuroTechnologies, Columbia, MD), according to the manufacturer’s instructions. Briefly, fresh brain samples were immediately prepared in the impregnation solution for 2 weeks followed by immersion in solution C for 1 week at room temperature. Brain tissues were sliced into 100 µm-thick coronal sections and mounted on gelatin-coated slides. The approximate stereotactic coordinates were as follows: layers II/III of the somatosensory cortex (ML 2.20, AP -1.5, DV 0.6), hippocampal CA1 (ML 1.7, AP -2.0, DV 1.4), and hippocampal CA3 (ML 2.0, AP -2.0, DV 2.2). After rinsing in distilled water, slides were immersed in freshly prepared staining solution for 10 minutes followed by dehydration in ethanol and clearing in xylene. Dendritic spines were quantified as spine length and number of spines per 10 µm length of spine. From each area, 4-5 neurons were counted per animal, and 4-5 animals were used per genotype. Images were captured using an Olympus BX51 microscope at 4X, 10X, and 60X magnification. Average spine densities were defined as the number of protrusions divided by the length of dendritic shaft and were measured using the ImageJ software.
Immunofluorescence staining
Immunofluorescence staining was performed on 25-μm free-floating coronal brain sections as previously described [26, 36]. For reference, approximate stereotactic coordinates were as follows: layers II/III of the somatosensory cortex (ML 2.2, AP -1.5, DV 0.6), hippocampal CA1 (ML 1.7, AP -2.0, DV 1.4), and hippocampal CA3 (ML 2.3, AP -2.0, DV 2.3). Primary antibodies included the following: anti-APE1 (ab268072, abcam), anti-vGlut-1 (75-066, NeuroMab), anti-GABA-A-R-Alpha1 (GABRA1) (75-136, NeuroMab), anti-synapsin (ab1543p, Millipore), anti-GAD (ab1511, Millipore), anti-NeuN (ABN78, Sigma Aldrich).
Morris water maze
The Morris water maze test was performed at 4, 5, and 6 months of age, as previously described [23, 37]. Briefly, a circular platform (11 cm in diameter) was submerged in one quadrant of a circular pool (109 cm in diameter) of opaque water. Each mouse was put in the water in one of four quadrants and was allowed up to 60 seconds to find the platform. When each trial ended, the mouse was placed on the platform for another 30 seconds to help it remember the external spatial cues displayed around the room. During the learning phase, the time needed to find the submerged platform (latency) was recorded for every trial. Four trials per day were performed for four consecutive days. A single 60 second probe test was performed on the final day of testing, wherein the platform was removed (memory) and the time spent in the goal quadrant was recorded. Data were recorded and analyzed using AnyMaze Software (Stoelting Co., Wood Dale, IL). All behavioral tests were carried out by researchers who were blinded to group allocation.
Two-object novel object recognition
The novel object recognition (NOR) task was performed to evaluate recognition memory [38, 39]. On days that the NOR and MWM tasks overlapped, the NOR was executed first. Briefly, the task was conducted in an open field arena with two objects placed in the center area of the arena. The objects were of different shapes and appearance, but similar height and volume. On the day before testing, mice were habituated once in the empty open field by being initially placed facing the wall nearest to the experimenter and allowed to explore the open field for 5 minutes. On the testing day, two identical objects were placed in the open field, 5 cm away from the wall, and the mouse was allowed to move freely for familiarization with 20 seconds of exploration of both objects, then recorded for a 10-minute observation period. Object exploration was scored using videography when the mouse contacted or sniffed the object or approached the object (i.e., when the distance between the snout and the object was less than 2 cm). The objects and the open field were cleaned with 70% (vol/vol) ethanol to minimize olfactory cues. During the novel cue testing period (occurring 1 hour following the observation period), one object was replaced with a novel object (differing in shape and contrast color but placed in the same location) and the mouse was recorded for a 10-minute testing period. The discrimination index was defined as the exploration time of the novel object (as assessed by the AnyMaze automated video-tracking system) divided by the total exploration time.
Electrophysiology (Long term potentiation)
Mice were sacrificed with carbon dioxide inhalation and decapitated. Brains were quickly removed and transferred to oxygenated, ice-cold artificial cerebrospinal fluid (aCSF) with high magnesium (124 mM NaCl, 26 mM NaHCO3, 10 mM glucose, 3 mM KCl, 1.25 mM KH2PO4, 5 mM MgSO4, and 3.4 mM CaCl2). The brains were cut into 400 μm coronal hippocampal slices using a tissue chopper (Thermo Scientific Microm HM450, Waltham, Massachusetts, U.S) and maintained for 30 min in a recovery chamber with oxygenated aCSF at RT. The hippocampal slices were then transferred to an interface recording chamber and exposed to a warm, humidified atmosphere of 95% O2/5% CO2 with preheated (33 ± 0.5 °C) oxygenated aCSF perfused at 1 mL/min. After incubation in the recording chamber for 1 hour, a single glass pipette filled with normal aCSF was used to record field excitatory postsynaptic potentials (fEPSPs). Before each experiment, the stimulation intensity was adjusted to 40-50% of the maximum of fEPSP without spike component, which was considered the test pulse. Next, the test pulse was delivered at a 0.05Hz and the responses were recorded. After 10 mins of baseline stimulating, LTP was induced and the fEPSP was analyzed for its initial falling slope [40].
Image Processing
Images were acquired using confocal microscopy (60x magnification with z-stacks), acquiring one image in each matched anatomical region (CA1, CA3, DG, and CTX) per animal, and processed with Image J or IMARIS for unbiased counting of automatically recognized cells or synaptic spots by a blinded observer. Excitatory and inhibitory synapses were imaged and processed with Image J and three-dimensional reconstruction using IMARIS software (Bitplane). Using IMARIS tools such as surface rendering, fluorescence thresholding, and masking of unwanted immuno-labeling, we were able to obtain a representation 3D structure of an individual neuron including the number and location of its glutamatergic and GABAergic synaptic inputs [41]. Finally, the spots were quantified by imaging software as the number of positive spots per mm2.
Statistical Analysis
Data were expressed as mean±SEM and analyzed for normality using the Shapiro-Wilk test. Students’ t-test was used to measure between-group differences and non-parametric tests were used for non-normally distributed data. Behavioral data were analyzed using two-way ANOVA for multiple comparisons followed by the Bonferroni post hoc. LTP timecourse was analyzed using two-way ANOVA followed by the Tukey post hoc. Statistical analysis and image plotting was conducted by using GraphPad Prism 8.0. A p value less than 0.05 was defined as statistically significant.
RESULTS
CaMKIIα-APE1 cKO leads to reduced expression of APE1 in forebrain neurons
The CamKIIα promotor can drive GFP expression in about 70% of granule and pyramidal neurons in the hippocampus and about 30% of neurons in the neocortex [29]. We first examined whether conditional knockout of APE1 by CaMKIIα-Cre reduced the expression of the DNA repair enzyme APE1 in the hippocampus and cortex by co-immunostaining APE1 and the mature neuron marker, NeuN. As NeuN is not exclusively localized to CaMKIIα-expressing cells and a heterogenous population of mature neurons exist in different brain structures, one would not expect complete knockout in all NeuN+ cells. Accordingly, we found that the number of APE1+NeuN+ cells per mm2 in the hippocampus and cortex was significantly reduced in CA1, CA3 and DG subregions of the hippocampus (Fig. 1A-C, E-G). The most dramatic reduction of APE1+/NeuN+ cells was observed in the DG, consistent with a wide range of prior studies that indicate the DG has a high number of CaMKIIα-expressing neurons [30]. Similarly, APE1 expression in NeuN+ cells was also reduced in layers II/III of the somatosensory cortex (Fig. 1D, H) of APE cKO mice compared to WT mice.
Figure 1.
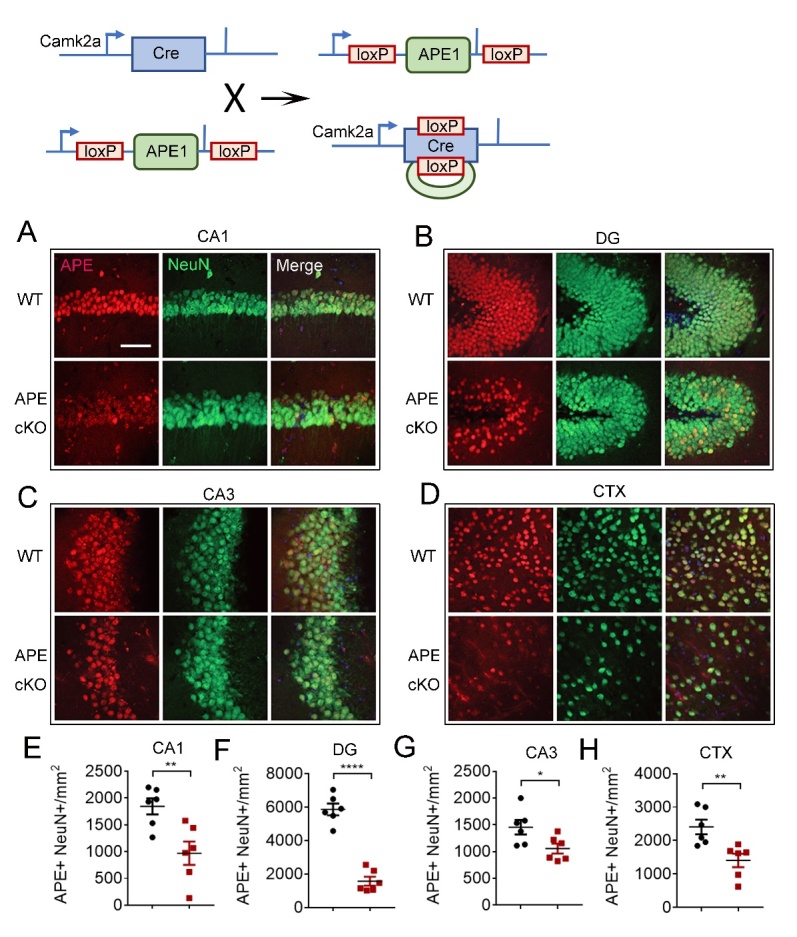
CaMKIIα-driven conditional knockout of APE1 (APE1 cKO) leads to significantly reduced APE expression in mature neurons of cortex and hippocampus. Mice with CaMKIIα promoter-driven conditional knockout of APE1 were generated by breeding APE1flox/flox mice with hemizygous CaMKIIα-cre mice, yielding CaMKIIα-Cre:APE1flox/flox mice. Immunofluorescence staining showed lack of localization of APE expression with NeuN+ cells (mature neurons) in cKO brain. Representative images of APE (red) and NeuN (green) in CA1 (A), DG (B), CA3 (C) and CTX (D). Quantification of double staining of APE1 and NeuN in CA1 (E), DG (F), CA3 (G) and CTX (H). Bar=50 µm. All data are presented as scatterplots with mean±SEM, n=6 animals/group. *P<0.05, **P<0.01 analyzed by Student’s t-tests.
CaMKIIα-driven conditional APE1 knockout promotes neurological dysfunction
In order to examine whether a deficiency in APE1 impairs cognitive function at an earlier stage of life compared to WT littermates, we subjected WT and APE1 cKO mice to the Morris water maze, a spatial learning and memory task, at 4-6 months of age [37]. We found no difference between WT and APE cKO mice in the latency to locate the submerged platform at 4, 5 or 6 months of age (Fig. 2A-D). However, the ability to remember the location once the platform was removed (memory phase) was significantly impaired in APE1 cKO mice compared to WT mice (Fig. 2A, E). To ensure the results were not confounded by impairments in swimming, we examined the swim speed of both groups and found no differences between genotypes in any of the four quadrants of the pool, during either the learning or memory phase of the test (Fig. 2F). The novel object recognition test also helps to measure recognition memory in rodents. We found that the discrimination index (i.e, the time spent exploring the novel object divided by the total time exploring both familiar and novel objects) displayed a statistical trend (p=0.35, Mann-Whitney U test, n=7/group) toward a decrease in the APE1 cKO mice compared to WT mice (data not shown). As these two memory tasks are distinct, the novel object recognition task may require longer aging periods to reveal statistically significant differences between groups or may have been underpowered.
Figure 2.

Deficiency of APE1 results in impaired spatial memory function at 4-6 months of age. (A-F) The Morris water maze was conducted at 4, 5 and 6 months of age in WT or APE cKO mice. (A) Representative swim path traces of mice attempting to find an underwater platform (learning) or searching for the platform after its removal (memory). Latency (time needed to find the platform) was recorded from day 1 to day 4. Time in target quadrant reflects memory and was recorded on day 5. APE cKO mice did not differ in latency to find the platform at 4 months (n=13 WT, 5 APE cKO) (B), 5 months (n=14 WT, 12 APE cKO) (C), and 6 months (n=10 WT, 9 APE cKO) (D) of age, but spent less time in the target quadrant when the platform was removed (n=5 WT, 6 APE cKO) (E). (F) Swim speeds were measured on day 5 to ensure swimming abilities were comparable between the two groups. *P<0.05; **P<0.01 vs matched wild type mice, analyzed by two-way ANOVA followed by Bonferroni post-hoc. All data shown as scatterplots with the mean±SEM.
CaMKIIα-driven conditional APE1 knockout potentiates synaptic network loss in brain
To explore cellular changes associated with the early onset of cognitive decline in APE1 cKO mice compared to WT mice, we examined synapses of neuronal networks in the hippocampus and layers II/III of the somatosensory cortex at 6 months of age. We first examined the expression of synapsin, a panneuronal marker of synapses, to determine if APE1 cKO led to gross changes in the number of synapses. We found that the number of synapsin immunostained spots did not change significantly between the APE1 cKO and WT mice in any of the observed brain regions (Fig. 3C, D), indicating that the density of synpasin-positive synapses does not appear to be altered at this early timepoint. To examine an excitatory synaptic marker relevant to learning and memory, we found that the expression of vGLUT1, an excitatory glutamatergic pre-synaptic protein necessary for the packaging of glutamate into presynaptic vesicles, was significantly reduced in CA1 and CA3 of the hippocampus and the cortex of the APE1cKO mice compared to WT mice, based on IMARIS quantification of the positive spots of vGLUT1 (Fig. 3A, B). However, no significant changes between genotypes were observed in the quantification of vGLUT1 positive spots in the DG of the hippocampus (Fig. 3A, B), possibly reflective of the glutamatergic inputs from the entorhinal cortex that may not express CamKII, and thus still maintain endogenous APE1 expression (as searched on the MGI recombinase activity database).
Figure 3.

Double immunofluorescence for vGlut1-NeuN and vGlut1-Synapsin in the hippocampus and cortex. (A) Representative images demonstrating vGlut1 (red) and NeuN (green) immunofluorescence staining in CA1 and cortex (CTX). Scale bar: 50 µm. (B) Quantification of vGlut1-positive spots in CA1, dentate gyrus (DG), CA3, and cortex (CTX). (C) Representative images demonstrating vGlut1 (red) and Synapsin (green) immunofluorescence staining in CA1, DG, CA3, and CTX of the mice brain. (D) Quantification of synapsin-positive spots in CA1, DG, CA3. and CTX. All data shown as scatterplots with the mean±SEM, n=6 animals/group. *P<0.05, **P<0.01, ***P<0.001 analyzed by Student’s t-tests.
As GABAergic synaptic transmission is an important inhibitory neuronal activity, we also examined the impact of APE1 cKO on the expression of GABAergic synaptic markers, GABRA1, a postsynaptic protein, and glutamic acid decarboxylase (GAD), a pre-synaptic protein [42]. We found that the number of GABRA1 and GAD positive spots in the brain of APE1 cKO mice was significantly reduced in all the brain regions examined at the age of 4-6 months (Fig. 4A-I). Together, these data indicate that APE1 cKO driven by the CamKIIα promoter may disrupt excitatory and inhibitory circuitry, but not the overall density of synaptic structures, at 6 months of age.
Figure 4.
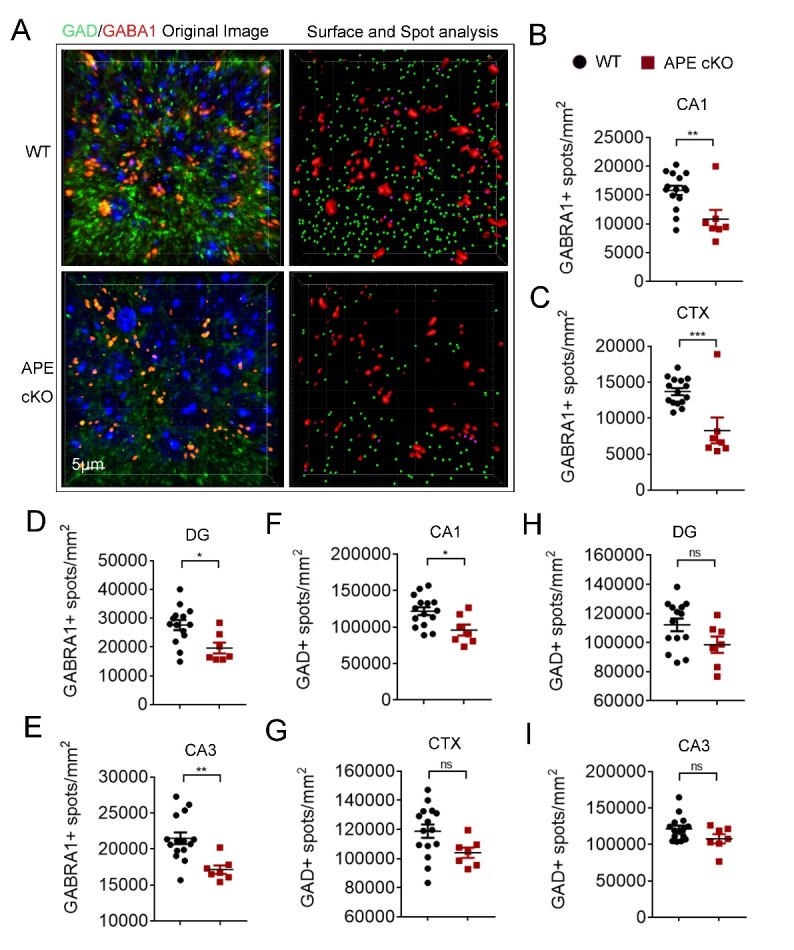
Reduced expression of GABRA1 and GAD in APE1 cKO brain. (A) Representative images demonstrating GABRA1 (red) and GAD (green) immunofluorescence staining in CA1, dentate gyrus (DG), CA3, and cortex (CTX). Scale bar: 50 µm. (B-E) Quantification of GABRA1-positive spots in CA1 (B), CTX (C), DG (D) and CA3 (E). Quantification of GAD-positive spots in CA1 (F), CTX (G), DG (H), and CA3 (I). All data shown as as scatterplots with the mean±SEM, n=6 animals/group. *P<0.05, **P<0.01,***P<0.001 analyzed by Student’s t-tests.
APE1 cKO potentiates dendritic spine shrinkage and impairs electrophysiological function of the mice brain
Dendritic spines are small protrusions from neuronal dendrites that are considered major sites of information processing and storage [43, 44]. To examine whether the APE1 cKO affects dendritic spines, we analyzed Golgi-impregnated neuronal structures from the mice at 4-6 months of age [43, 44]. APE1 cKO significantly reduced the number of apical dendritic spines as well as the length of these spines within both the CA1 and layers II/III of somatosensory cortical regions (Fig. 5A-E). As shown in the magnified images of the dendrites, APE1 cKO leads to significant shrinkage of dendritic spines compared to WT mice (Fig. 5F). In order to extend the above morphological findings into a functional level, we examined long-term potentiation (LTP) in the hippocampus. APE1 cKO decreased the percentage and the slope of the field excitatory postsynaptic potentials (fEPSPs) in CA1 compared to WT (Fig. 6A, B), indicating a loss of synaptic function in the APE1 hippocampus.
Figure 5.

APE cKO reduces dendritic spines in hippocampal CA1 and the cortex. (A) Representative images of Golgi-stained sections of the dorsal hippocampus and cortex of WT and APE cKO mice at 6 months of age. (B-E) Quantification of number of spines per unit length and spine length in the indicated brain regions of the same animals. All data shown as as scatterplots with the mean±SEM, n=5 WT, 4 APE cKO animals/group. ***P<0.001, ****P<0.0001 analyzed by Student’s t-tests.
Figure 6.

APE cKO mice showed reduced field excitatory postsynaptic potential in the hippocampal brain slice. Electrophysiological analyses of long-term potentiation (LTP) from the hippocampal region. (A) Representative traces of fEPSPs. (B) APE cKO mice showed significantly decreased level of fEPSPs after LTP induction. (C) The retention of LTP was assessed over the 50-60 min timeframe after LTP induction. n=5 WT, 7 APE cKO animals/group. Data in (B) shown as mean±SEM; data in (C) shown as mean±SD, ***P<0.001 analyzed by two-way ANOVA followed by Tukey post-hoc.
DISCUSSION
Aging is characterized by an accumulation of oxidative stress [45], which can culminate in oxidative DNA damage and a resulting decline in cognitive function [46]. The enzyme APE1 has a variety of cellular functions, including oxidative DNA repair as well as transcriptional modification and cellular activity, and has been shown to facilitate post-stroke neurological recovery [26] and prevent brain tumor formation by preserving the integrity of the neural genome [47]. However, it remains unknown whether a deficiency in APE1 in neuronal cells will accelerate aging-related cognitive decline. Therefore, we generated the APE1 conditional knockout mice under the control of CaMKIIα, a promoter driving Cre recombinase expression in the forebrain, including the CA1 pyramidal cell layer of the hippocampus [29]. The present study reveals that conditional knockout of APE1 under the control of CaMKIIα leads to loss of both excitatory and inhibitory synaptic components, dendritic spine shrinkage, impaired LTP, and memory disruption between the ages of 4-6 months, well before any of these features are observed in WT brain. Thus, this conditional knockout mouse may serve as an animal model of premature neurological decline.
Synaptic connectivity between neurons is an essential component of normal cognitive function [48]. Glutamatergic synapses are particularly important for the emergence of LTP and the formation of memories. Consistent with our observation that APE cKO reduced the expresson of the vesicular glutamate transporter vGLUT1 in the termination zone of Schaeffer collaterals (CA3 and CA1), the cKO mice also exhibited impairments in LTP and memory, but not learning, aspects of the Morris water maze. This phenotype echos that of vGLUT1 knockout mice, wherein LTP is impaired despite a lack of behavior changes in the learning arm of the Morris water maze test [49]. Our observation that the novel object discrimination index was not different between mouse genotypes requires further exploration. Novel object testing can be highly sensitive to age as well as testing paradigms [50]. Our testing protocol may have included too much time after the introduction of the novel object, and thus the exploration may have been “diluted” as the mouse became quickly familiarized. We plan to shorten the length of time analyzed for the discrimination index, as well as extend the age beyond the current period of 6 months. In addition, cognitive behaviors rely on an complex network of activity, involving both compensatory systems as well as modulation by other cell types, such as myelinating oligodendrocytes. For this reason, we avoid conclusions that relate a specific brain region to a specific behavioral outcome.
Aging-induced impairments in the presynaptic and postsynaptic components of the GABAergic system also may result in cognitive dysfunction by disturbing the balance between inhibitory and excitatory neuronal activities [51]. CaMKIIα specific depletion of APE1 resulted in loss of both pre- and post-synaptic GABAergic markers (GAD and GABRA1, respectively) in all areas examined in the hippocampus and to an extent in sensorimotor cortical layers II/III. We do not expect that the loss of GABAergic markers is directly due to the loss of APE1 within inhibitory neurons, as CaMKIIα promotor activity (and thus APE1 knockout) is not associated with GAD-positive neurons [52]. Thus, further investigation to determine how potential loss of APE1 in excitatory neurons can lead to the loss of GABAergic markers is merited. The loss of GABAergic immunoreactivity has been identified as a feature of the aging brain and is associated with cognitive impairment [51, 53]. Further studies on synaptic disinhibition or inhibitory signaling in the aging brain of APE1 cKO mice would build well upon the current study.
In addition to alterations in neurotransmitter markers, aging has been closely associated with morphological alterations of neuronal synapses and dendritic spines [54]. Although we did not observe differences in the total number of synapses in our APE1 cKO brains, we observed morphological alterations in dendritic spines that are consistent with an aging phenotype and were absent in age-matched controls. The reduction in dendritic spines of the APE1 cKO mice - both in number and in length - is likely to contribute to the observed impairments in electrophysiological function as well as worsened memory function.
Aging-related cognitive decline is emerging as a critical health concern [1, 2] and poses an enormous socioeconomic burden. Animal studies of this phenomenon are challenging, as it usually takes ~12-18 months for aging-related deficits to emerge [55]. Developing a practical animal model for aging-related studies is of great importance; in this context, it is important to note that subtle morphological or electrophysiological changes may occur even before six months of age in the brains of APE1 cKO mice. However, the current study details mild neural changes consistent with the observed cognitive impairment in APE1 cKO mice that was undetected in wildtype animals. These findings increase the potential of the APE1 cKO mice as a model for premature development of an aging phenotype, thereby avoiding survivor bias in aging colonies and mimicking the endogenous processes that contribute to aged brain phenotype.
The present report shows that CaMKIIα-driven conditional knockout of APE1 in forebrain neurons forces premature phenotypic changes associated with aged adults, which may serve as a promising model for research aging-related deficits with an accelerated timeframe of 4-6 months.
Acknowledgements
We thank Patricia Strickler for administrative support and Xuguang Gao for help with establishing the electrophysiology set up. This project was supported by the University of Pittsburgh School of Medicine. The APEfl/fl mice were generated by R.A.S under the support of NIH NS036736 (to J.C.). J.C. is the Richard King Mellon Professor of Neurology and a recipient of the VA Senior Research Career Scientist Award. J.C., S.H.H., and R.A.S. are also supported by VA Merit Review grants.
Footnotes
Conflicts of interest
The authors have declared that no conflict of interest exists.
References
- [1].Hebert LE, Scherr PA, Bienias JL, Bennett DA, Evans DA (2003). Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch Neurol, 60:1119-1122. [DOI] [PubMed] [Google Scholar]
- [2].Bishop NA, Lu T, Yankner BA (2010). Neural mechanisms of ageing and cognitive decline. Nature, 464:529-535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Satoh A, Imai SI, Guarente L (2017). The brain, sirtuins, and ageing. Nat Rev Neurosci, 18:362-374. [DOI] [PubMed] [Google Scholar]
- [4].Knezevic D, Verhoeff NPL, Hafizi S, Strafella AP, Graff-Guerrero A, Rajji T, et al. (2018). Imaging microglial activation and amyloid burden in amnestic mild cognitive impairment. J Cereb Blood Flow Metab, 38:1885-1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Salthouse TA (2009). When does age-related cognitive decline begin? Neurobiol Aging, 30:507-514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Santisteban MM, Iadecola C (2018). Hypertension, dietary salt and cognitive impairment. J Cereb Blood Flow Metab, 38:2112-2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wortley D, An JY, Heshmati A (2017). Tackling the Challenge of the Aging Society: Detecting and Preventing Cognitive and Physical Decline through Games and Consumer Technologies. Healthc Inform Res, 23:87-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Huang TT, Leu D, Zou Y (2015). Oxidative stress and redox regulation on hippocampal-dependent cognitive functions. Arch Biochem Biophys, 576:2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Salminen LE, Paul RH (2014). Oxidative stress and genetic markers of suboptimal antioxidant defense in the aging brain: a theoretical review. Rev Neurosci, 25:805-819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Li Y, Xie L, Huang T, Zhang Y, Zhou J, Qi B, et al. (2019). Aging Neurovascular Unit and Potential Role of DNA Damage and Repair in Combating Vascular and Neurodegenerative Disorders. Front Neurosci, 13:778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhang ZJ, Zheng XX, Zhang XY, Zhang Y, Huang BY, Luo T (2019). Aging alters Hv1-mediated microglial polarization and enhances neuroinflammation after peripheral surgery. CNS Neurosci Ther. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Barnes CA (1994). Normal aging: regionally specific changes in hippocampal synaptic transmission. Trends Neurosci, 17:13-18. [DOI] [PubMed] [Google Scholar]
- [13].Griffith WH, Dubois DW, Fincher A, Peebles KA, Bizon JL, Murchison D (2014). Characterization of age-related changes in synaptic transmission onto F344 rat basal forebrain cholinergic neurons using a reduced synaptic preparation. J Neurophysiol, 111:273-286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Pereira AC, Lambert HK, Grossman YS, Dumitriu D, Waldman R, Jannetty SK, et al. (2014). Glutamatergic regulation prevents hippocampal-dependent age-related cognitive decline through dendritic spine clustering. Proc Natl Acad Sci U S A, 111:18733-18738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Gleichmann M, Mattson MP (2010). Alzheimer's disease and neuronal network activity. Neuromolecular Med, 12:44-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Gredilla R, Bohr VA, Stevnsner T (2010). Mitochondrial DNA repair and association with aging--an update. Exp Gerontol, 45:478-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gorbunova V, Seluanov A, Mao Z, Hine C (2007). Changes in DNA repair during aging. Nucleic Acids Res, 35:7466-7474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Langie SA, Cameron KM, Ficz G, Oxley D, Tomaszewski B, Gorniak JP, et al. (2017). The Ageing Brain: Effects on DNA Repair and DNA Methylation in Mice. Genes(Basel), 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sykora P, Misiak M, Wang Y, Ghosh S, Leandro GS, Liu D, et al. (2015). DNA polymerase beta deficiency leads to neurodegeneration and exacerbates Alzheimer disease phenotypes. Nucleic Acids Res, 43:943-959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Borgesius NZ, de Waard MC, van der Pluijm I, Omrani A, Zondag GC, van der Horst GT, et al. (2011). Accelerated age-related cognitive decline and neurodegeneration, caused by deficient DNA repair. J Neurosci, 31:12543-12553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Xiao JY, Xiong BR, Zhang W, Zhou WC, Yang H, Gao F, et al. (2018). PGE2-EP3 signaling exacerbates hippocampus-dependent cognitive impairment after laparotomy by reducing expression levels of hippocampal synaptic plasticity-related proteins in aged mice. CNS Neurosci Ther, 24:917-929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Oliveira TT, Fontes-Dantas FL, de Medeiros Oliveira RK, Pinheiro DML, Coutinho LG, da Silva VL, et al. (2021). Chemical Inhibition of Apurinic-Apyrimidinic Endonuclease 1 Redox and DNA Repair Functions Affects the Inflammatory Response via Different but Overlapping Mechanisms. Front Cell Dev Biol, 9:731588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Leak RK, Li P, Zhang F, Sulaiman HH, Weng Z, Wang G, et al. (2015). Apurinic/apyrimidinic endonuclease 1 upregulation reduces oxidative DNA damage and protects hippocampal neurons from ischemic injury. Antioxid Redox Signal, 22:135-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Mantha AK, Dhiman M, Taglialatela G, Perez-Polo RJ, Mitra S (2012). Proteomic study of amyloid beta (25-35) peptide exposure to neuronal cells: Impact on APE1/Ref-1's protein-protein interaction. J Neurosci Res, 90:1230-1239. [DOI] [PubMed] [Google Scholar]
- [25].Stetler RA, Gao Y, Zukin RS, Vosler PS, Zhang L, Zhang F, et al. (2010). Apurinic/apyrimidinic endonuclease APE1 is required for PACAP-induced neuroprotection against global cerebral ischemia. Proc Natl Acad Sci U S A, 107:3204-3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Stetler RA, Gao Y, Leak RK, Weng Z, Shi Y, Zhang L, et al. (2016). APE1/Ref-1 facilitates recovery of gray and white matter and neurological function after mild stroke injury. Proc Natl Acad Sci U S A, 113:E3558-3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lillenes MS, Stoen M, Gunther CC, Selnes P, Stenset VT, Espeseth T, et al. (2017). Mitochondrial transcription factor A (TFAM) rs1937 and AP endonuclease 1 (APE1) rs1130409 alleles are associated with reduced cognitive performance. Neurosci Lett, 645:46-52. [DOI] [PubMed] [Google Scholar]
- [28].Lillenes MS, Espeseth T, Stoen M, Lundervold AJ, Frye SA, Rootwelt H, et al. (2011). DNA base excision repair gene polymorphisms modulate human cognitive performance and decline during normal life span. Mech Ageing Dev, 132:449-458. [DOI] [PubMed] [Google Scholar]
- [29].Kahl A, Anderson CJ, Qian L, Voss H, Manfredi G, Iadecola C, et al. (2018). Neuronal expression of the mitochondrial protein prohibitin confers profound neuroprotection in a mouse model of focal cerebral ischemia. J Cereb Blood Flow Metab, 38:1010-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wang X, Zhang C, Szabo G, Sun QQ (2013). Distribution of CaMKIIalpha expression in the brain in vivo, studied by CaMKIIalpha-GFP mice. Brain Res, 1518:9-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].von Bohlen und Halbach O, Zacher C, Gass P, Unsicker K (2006). Age-related alterations in hippocampal spines and deficiencies in spatial memory in mice. J Neurosci Res, 83:525-531. [DOI] [PubMed] [Google Scholar]
- [32].Hickmott P, Dinse H (2013). Effects of aging on properties of the local circuit in rat primary somatosensory cortex (S1) in vitro. Cereb Cortex, 23:2500-2513. [DOI] [PubMed] [Google Scholar]
- [33].Cali C, Wawrzyniak M, Becker C, Maco B, Cantoni M, Jorstad A, et al. (2018). The effects of aging on neuropil structure in mouse somatosensory cortex-A 3D electron microscopy analysis of layer 1. PLoS One, 13:e0198131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Dragatsis I, Zeitlin S (2000). CaMKIIalpha-Cre transgene expression and recombination patterns in the mouse brain. Genesis, 26:133-135. [DOI] [PubMed] [Google Scholar]
- [35].Percie du Sert N, Hurst V, Ahluwalia A, Alam S, Avey MT, Baker M, et al. (2020). The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. J Cereb Blood Flow Metab, 40:1769-1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lobo A, Launer LJ, Fratiglioni L, Andersen K, Di Carlo A, Breteler MM, et al. (2000). Prevalence of dementia and major subtypes in Europe: A collaborative study of population-based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology, 54:S4-9. [PubMed] [Google Scholar]
- [37].Zhang J, Zhang W, Gao X, Zhao Y, Chen D, Xu N, et al. (2019). Preconditioning with partial caloric restriction confers long-term protection against grey and white matter injury after transient focal ischemia. J Cereb Blood Flow Metab, 39:1394-1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Leger M, Quiedeville A, Bouet V, Haelewyn B, Boulouard M, Schumann-Bard P, et al. (2013). Object recognition test in mice. Nat Protoc, 8:2531-2537. [DOI] [PubMed] [Google Scholar]
- [39].Balbi M, Vanni MP, Vega MJ, Silasi G, Sekino Y, Boyd JD, et al. (2019). Longitudinal monitoring of mesoscopic cortical activity in a mouse model of microinfarcts reveals dissociations with behavioral and motor function. J Cereb Blood Flow Metab, 39:1486-1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Orfila JE, Grewal H, Dietz RM, Strnad F, Shimizu T, Moreno M, et al. (2019). Delayed inhibition of tonic inhibition enhances functional recovery following experimental ischemic stroke. J Cereb Blood Flow Metab, 39:1005-1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Fogarty MJ, Hammond LA, Kanjhan R, Bellingham MC, Noakes PG (2013). A method for the three-dimensional reconstruction of Neurobiotin-filled neurons and the location of their synaptic inputs. Front Neural Circuits, 7:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Micheva KD, Busse B, Weiler NC, O'Rourke N, Smith SJ (2010). Single-synapse analysis of a diverse synapse population: proteomic imaging methods and markers. Neuron, 68:639-653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Jeanneret V, Ospina JP, Diaz A, Manrique LG, Merino P, Gutierrez L, et al. (2018). Tissue-type plasminogen activator protects the postsynaptic density in the ischemic brain. J Cereb Blood Flow Metab, 38:1896-1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Yu P, Venkat P, Chopp M, Zacharek A, Shen Y, Ning R, et al. (2019). Role of microRNA-126 in vascular cognitive impairment in mice. J Cereb Blood Flow Metab, 39:2497-2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Cai W, Zhang K, Li P, Zhu L, Xu J, Yang B, et al. (2017). Dysfunction of the neurovascular unit in ischemic stroke and neurodegenerative diseases: An aging effect. Ageing Res Rev, 34:77-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Li P, Stetler RA, Leak RK, Shi Y, Li Y, Yu W, et al. (2018). Oxidative stress and DNA damage after cerebral ischemia: Potential therapeutic targets to repair the genome and improve stroke recovery. Neuropharmacology, 134:208-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Dumitrache LC, Shimada M, Downing SM, Kwak YD, Li Y, Illuzzi JL, et al. (2018). Apurinic endonuclease-1 preserves neural genome integrity to maintain homeostasis and thermoregulation and prevent brain tumors. Proc Natl Acad Sci U S A, 115:E12285-E12294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Hiratani N, Fukai T (2018). Redundancy in synaptic connections enables neurons to learn optimally. Proc Natl Acad Sci U S A, 115:E6871-E6879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Balschun D, Moechars D, Callaerts-Vegh Z, Vermaercke B, Van Acker N, Andries L, et al. (2010). Vesicular glutamate transporter VGLUT1 has a role in hippocampal long-term potentiation and spatial reversal learning. Cereb Cortex, 20:684-693. [DOI] [PubMed] [Google Scholar]
- [50].Traschutz A, Kummer MP, Schwartz S, Heneka MT (2018). Variability and temporal dynamics of novel object recognition in aging male C57BL/6 mice. Behav Processes, 157:711-716. [DOI] [PubMed] [Google Scholar]
- [51].Rozycka A, Liguz-Lecznar M (2017). The space where aging acts: focus on the GABAergic synapse. Aging Cell, 16:634-643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Liu XB, Murray KD (2012). Neuronal excitability and calcium/calmodulin-dependent protein kinase type II: location, location, location. Epilepsia, 53 Suppl 1:45-52. [DOI] [PubMed] [Google Scholar]
- [53].Stanley EM, Fadel JR, Mott DD (2012). Interneuron loss reduces dendritic inhibition and GABA release in hippocampus of aged rats. Neurobiol Aging, 33:431 e431-413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Eavri R, Shepherd J, Welsh CA, Flanders GH, Bear MF, Nedivi E (2018). Interneuron Simplification and Loss of Structural Plasticity As Markers of Aging-Related Functional Decline. J Neurosci, 38:8421-8432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Suissa L, Flachon V, Guigonis JM, Olivieri CV, Burel-Vandenbos F, Guglielmi J, et al. (2020). Urinary ketone body loss leads to degeneration of brain white matter in elderly SLC5A8-deficient mice. J Cereb Blood Flow Metab, 40(8):1709-1723. [DOI] [PMC free article] [PubMed] [Google Scholar]