

Abstract
Glioblastoma multiforme (GBM) is the most malignant type of brain tumor and has an extremely poor prognosis. Current treatment protocols lack favorable outcomes, and alternative treatments with superior efficacy are needed. In this study, we demonstrate that loading paclitaxel (PTX) in a polymeric, nanoparticulate delivery system is capable of improving its brain accumulation and therapeutic activity. We independently incorporated two different positively charged surface modifiers, poly(amidoamine) (PAMAM) and poly(ethylenimine) (PEI), onto poly(lactic-co-glycolic acid) (PLGA) - polyethylene glycol (PEG), PLGA-PEG, nanoparticles (NPs) using a modified nanoprecipitation technique that assures the formation of nanosized particles, while exposing the positively charged polymer on the surface. The prepared NPs underwent comprehensive analyses of their size, charge, in vitro permeability against a BBB cell line, and in vivo biodistribution. Our results demonstrated the successful fabrication of positively charged NPs using PAMAM or PEI. Importantly, significant improvement in brain accumulation (in vivo) was associated with NPs containing PAMAM compared to unmodified NPs or NPs containing PEI. Finally, the efficacy of PAMAM-modified NPs loaded with PTX was evaluated with orthotopic human GBM xenografts in a mouse model, and the data demonstrated improved survival and equivalent safety compared to soluble PTX. Our data substantiate the importance of surface chemistry on the magnitude of NP accumulation in the brain and pave the way for further in vivo evaluation of chemotherapeutic drugs against GBM that have previously been overlooked because of their limited ability to cross the BBB.
Keywords: Glioblastoma multiforme (GBM), paclitaxel (PTX), glioblastoma treatments, nanoparticle-based therapy, poly(amidoamine) (PAMAM)
Graphical Abstract
Introduction
Glioblastoma multiforme (GBM) is considered the most aggressive and is the most common primary malignant brain tumor in adults. Most GBM (~90%) originate de novo from normal glial cells with no evidence of low-grade glioma and are clinically classified as primary GBM, while secondary GBM develop from low-grade gliomas 1, 2. Both types of GBM share similar morphological features and lead to similar clinical symptoms 3. Worldwide, in 2017 GBM caused more than 100,000 deaths per year 4, with a higher incidence rate in men than women 5. Treating GBM has been and continues to be an enormous challenge. The standard treatment for GBM involves surgery followed by radiotherapy and/or chemotherapy. There are several reasons why standard GBM treatments are not particularly efficacious in the clinic including: (1) approximately 50% of patients have tumors that are non-responsive to temozolomide (TMZ), the primary gold standard, mostly due to over-expression of O6-methylguanine-DNA methyltransferase which limits the cytotoxic effects of TMZ 6; (2) TMZ only moderately penetrates the brain 7. TMZ is also rapidly hydrolyzed under physiological conditions 8; (3) invasive GBM cells can reduce the chances of complete surgical removal of GBM tissue making recurrence highly probable 9; and (4) the function of the blood brain barrier (BBB) results in most chemotherapeutic agents and targeted agents being ineffective 10, 11. For these reasons, researchers have investigated novel therapeutic agents and procedures to enhance the quality and duration of life of GBM patients 9, 12–15.
Paclitaxel (PTX), a potent antineoplastic agent, has been commonly used over the past two decades for the treatment of lung, ovarian and breast cancers 16. PTX is a microtubule-stabilizing drug that targets the well-characterized taxoid binding site of the β-tubulin subunit resulting in mitotic arrest and cell death 17. PTX, unlike TMZ, is not a standard of care agent but has been, and is still, used in clinical trials for GBM 18, 19. One drawback of PTX is that its therapeutic efficacy is limited due to a low capacity to cross the BBB 20, 21. This may be remedied to some degree by loading PTX into nanoparticles (NPs) that are capable of crossing the BBB, thereby improving the therapeutic efficacy. Thus, we sought to develop a novel drug delivery nanocarrier to deliver PTX across the BBB and effectively treat GBM.
NPs have been widely used to overcome the current barriers associated with drug delivery to the brain with great success, where controlling the size and the surface charge were found to be the two most important parameters that facilitate crossing the BBB. NPs of less than 200 nm in size and that possess a net positive charge have shown great potential in crossing the BBB 22–25. Poly(amidoamine) (PAMAM) and poly(ethylenimine) (PEI) are positively charged polymers that have been widely used in the literature to facilitate crossing the BBB and improve accumulation of therapeutics at brain tissue 23, 25–28. In addition, studies have demonstrated that even though PAMAM is toxic to red blood cells 22–25, and PEI is toxic to neurons and other cells in the nervous system 29, the cellular toxicity of PAMAM and PEI can be reduced when PAMAM or PEI are conjugated to polyethylene glycol (PEG) 25, 30. Thus, PEG molecules used in our formulations may also help to mitigate the potential toxicity of PAMAM and PEI.
More modern analyses that focus on covalent tracking of polymer constituents mostly reach a consensus that not all NPs are able to cross the BBB 31. Multiple factors need to be considered when developing a nanoparticle formulation for crossing the BBB. Factors potentially influencing transport of NPs across the BBB, such as particle size, surface charge, nanoparticle materials, and surface modifiers are being investigated so as to develop NPs with enhanced capacity to cross the BBB 15, 31, 32. However, to date the impact of the differences in NP chemistries on their abilities to cross the BBB has not been thoroughly evaluated and has been a subject of controversy.
In this study we investigated how modifying the surface chemistry, while fixing the size and charge of NPs, affects their ability to cross the BBB. To do so, we used a modified nanoprecipitation technique to prepare three different batches of NPs with equivalent hydrodynamic diameters. Our modified technique resulted in NPs with the positively charged polymer being exposed at the surface, thus evaluating its direct impact on facilitating the crossing of the BBB could be easily assessed. We used the biodegradable, biocompatible polymer poly(lactic-co-glycolic acid) (PLGA) - PEG, PLGA-PEG, to prepare the core of these NPs (PG), and then we modified the surface with either PAMAM (PGM) or PEI (PGI) to evaluate how differing the surface chemistry of these positively charged polymers affected the ability of these NPs to cross the BBB. The prepared NPs were first evaluated in vitro against a BBB cell culture model, and then their biodistribution was evaluated in a healthy mouse model to estimate the magnitude of their brain accumulation, and finally in vivo activity and safety against an orthotopic GBM tumor mouse model was investigated.
Results and discussion
NPs preparation and characterization
Our first aim was to prepare NPs of equivalent sizes and different surface chemistries and study their ability to penetrate the BBB using an in vitro cell culture model. To do so, we initially conjugated PEG to the FDA-approved biocompatible and biodegradable polymer, PLGA, and the resulting copolymer, PLGA-b-PEG, was characterized using 1H NMR (Supplementary Fig. S1) 33. The prepared PLGA-b-PEG copolymer was used downstream for NP fabrication. The PEGylation process was performed in an attempt to modify the biodistribution of drug-loaded NPs, prolong the NPs blood circulation half-life, and importantly, mitigate the toxic side effects of the positively charged modifiers, PAMAM and PEI 15, 25, 34.
NPs were prepared using a slight modification to a previously published nanoprecipitation method (Fig. 1a) 33, where the positively charged modifier, PAMAM or PEI, was co-dissolved in acetone with PLGA-b-PEG and added drop-wise to a stirred aqueous solution containing a low concentration of surfactant. Our modified method allowed for the formation of double layered NPs with the more hydrophobic polymer, PLGA-b-PEG, constituting the core of the NPs and the more hydrophilic positively charged modifiers forming the coat. This orientation is achieved because the nanoprecipitation method utilizes low energy input during fabrication of the NPs, giving the polymers the opportunity to orient themselves in a way that minimizes their free energy, thus the hydrophobic and hydrophilic polymers will orient themselves towards the core and the surface, respectively 35–37. The presence of the positively charged surface modifier on the surface of the NPs can measurably affect the ability of NPs to cross the BBB, once the size and the magnitude of the charge are kept constant.
Figure 1.
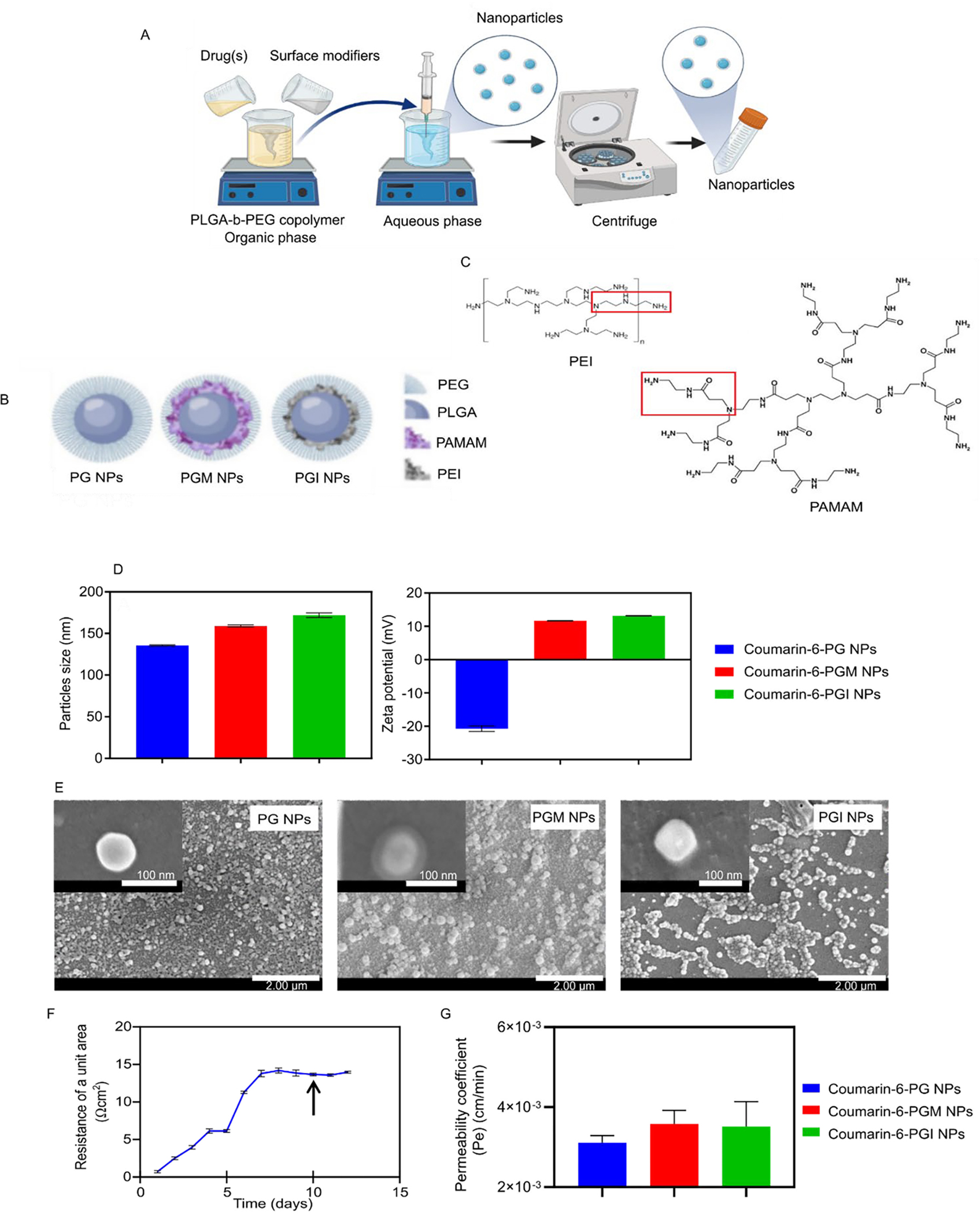
NP manufacture and properties. Schematics showing the nanoprecipitation method used for NP preparation (A), the composition of the PG, PGM and PGI NPs (B), and the structures of PAMAM and PEI (C). The average hydrodynamic diameter and ζ-potential of the coumarin-6-loaded NPs (D) and SEM images of the PG, PGM and PGI NPs (E). The TEER values of the hCMEC/D3 monocultures representing monolayer of human BBB (F) and the permeability of the coumarin-6-loaded NPs (G)
In this study three different batches of NPs were prepared: PLGA-PEG NPs (PG), PLGA-PEG + PAMAM (PGM), and PLGA-PEG + PEI (PGI) (Fig. 1b). PG NPs possessed exposed free carboxylic groups on their surface, while both PGM and PGI NPs possessed multiple exposed amine groups on their surfaces. Chemistry-wise, PGM NPs prepared with 5th generation PAMAM exhibited an extra amide group (−NCH2CH2CONHCH2CH2NH2) on their surface as compared to PGI NPs prepared with branched PEI and exhibiting a secondary amine group (−NCH2CH2CONHCH2CH2NH2) on their surface (Fig. 1c).
In order to assess the ability of NPs to cross a well-established in vitro BBB cell culture model (monoculture of human brain microvascular endothelial cells, hCMEC/D3 in Transwells), we loaded our three different NP formulations with the fluorescent probe, coumarin-6, in order to facilitate tracking of the NPs. All three batches of the coumarin-6-loaded NPs were of equivalent sizes with hydrodynamic diameters that ranged from 135 to 172 nm. PG NPs exhibited a negative charge of −20.7 mV due to the carboxylic groups of PLGA, while both PGM NPs and PGI NPs both exhibited positive charges of approximately +13.2 mV, indicating that the amine-containing functional groups were exposed on the surface (Fig. 1d). Furthermore, the loading of coumarin-6 (~0.8 – 1.3 μg/mg NPs) and its encapsulation efficiency (~25 – 38%) are shown for the three NP formulations tested (supplementary Table S1). SEM images of the three NP formulations demonstrated the successful fabrication of spherical particles with smooth surface morphology. Actual NP sizes measured by SEM were found to be approximately 100 nm, which proved to be smaller than their hydrodynamic diameters; a discrepancy likely due to the presence of hydrophilic polymers on the surface of the NPs 38 such as PEG and the surface modifiers (Fig. 1e). Interestingly, PGM NPs were the only NPs that showed a clear double layered structure, even though PGI NPs exhibited equivalent positive charges. This could be related to the rigid structure of PAMAM as compared to the flexible structure of branched PEI, thus uniform arrangement was achieved on the surface when PAMAM was used, as compared to irregular distribution of PEI on the surface. Furthermore, the presence of the amide groups in PAMAM might have interacted more efficiently with PEG chains as compared to PEI amine groups, and this may have subsequently resulted in a more uniform distribution of PAMAM on the surface as compared to PEI 39.
In vitro permeability studies of coumarin-6-loaded NPs using a model BBB cell line (hCMEC/D3)
We next tested the ability of the NP formulations to cross an in vitro cell culture BBB model as demonstrated in Fig 1f. BBB hCMEC/D3 cells were seeded as a monolayer in a Transwell insert, and TEER values were measured daily in order to estimate the optimum time to start the permeability experiment. Full coverage of the membrane of the Transwell insert by the BBB cells and the formation of tight junctions are reflected by a plateau value for the measured TEER. 40–42. We started our experiment when hCMEC/D3 monolayers reached a plateau TEER value of ~ 15 Ω/cm2 (after 10 days, Fig. 1f), where different initial concentrations of coumarin-6-loaded NPs were added to the donor compartment and the flux of each concentration was measured over a 6 h period. Finally, the permeability coefficient (Pe) was calculated for each formulation by dividing the flux by the initial concentration in the donor compartment. The average Pe of coumarin-6-loaded NPs was found to be 3.1 × 10−3, 3.6 × 10−3, and 3.5 × 10−3 cm/minute for PG, PGM, and PGI NPs, respectively (Fig. 1g). Those values demonstrate a very marginal, but non-significant, increased ability of the positively charged NPs (PGM (p-value = 0.3) & PGI (p-value = 0.4)) over the negatively charged PG NPs to cross the in vitro model BBB. We expected a more profound difference because it has been previously demonstrated in the literature that the superior interaction between the positively charged NPs and the negatively charged cell membrane can improve their ability to be endocytosed and thus facilitate their permeability as compared to their negatively charged counterparts 26, 43, 44. In addition, Jallouli et al. proposed that cationic porous NPs could traverse an in vitro model BBB via transcytosis and this was shown to correlate well with in vivo findings 45. However, the degree of cellular uptake (of NPs) by both phagocytic and non-phagocytic cells has been shown to depend upon the magnitude of the positive charge 46, 47, and given that both our PGM & PGI NP formulations loaded with coumarin-6 exhibited a ζ-potential of just +13.2 mV, this could explain why the enhanced uptake of the positively charged NPs (compared to PG NPs) did not achieve statistical significance.
In vivo biodistribution studies of DIR-loaded NPs in healthy mice
W next tested our NP formulations in vivo by evaluating their ability to cross the BBB and target the brain tissues in healthy mice. This time we prepared the three NPs formulations loaded with the near-infrared fluorescent dye, DiR, to facilitate tracking their in vivo fate following IV administration. All of the prepared NPs were of equivalent hydrodynamic diameters (averaging ~161 nm; supplementary Table S1). PG NPs carried a negative charge of −39 mV and both PGM and PGI exhibited equivalent positive charges of about +37.5 mV (Fig. 2a). Furthermore, DIR loading was almost equivalent in all of the formulations with an average DIR loading of 1.57 μg/mg particles (supplementary Table S1). Interestingly, the magnitude of the charge of the DIR-loaded NPs was much higher than the coumarin-6-loaded NPs (Fig. 1d), which could be attributed to the adsorption of coumarin-6 on the surface of the NPs that subsequently masks the charge and results in a reduction in its magnitude. This occurs with coumarin-6 and not with DIR, because coumarin-6 is a smaller more hydrophilic molecule as compared to DIR, thus during NP fabrication more courmain-6 is expected to be released from the NPs and become adsorbed on the surface, as compared to DIR molecules that are inclined to remain within the more hydrophobic core 48–50. SEM images of the DIR-loaded PGM NPs confirmed the formation of double layered NPs (Fig. 2b).
Figure 2.
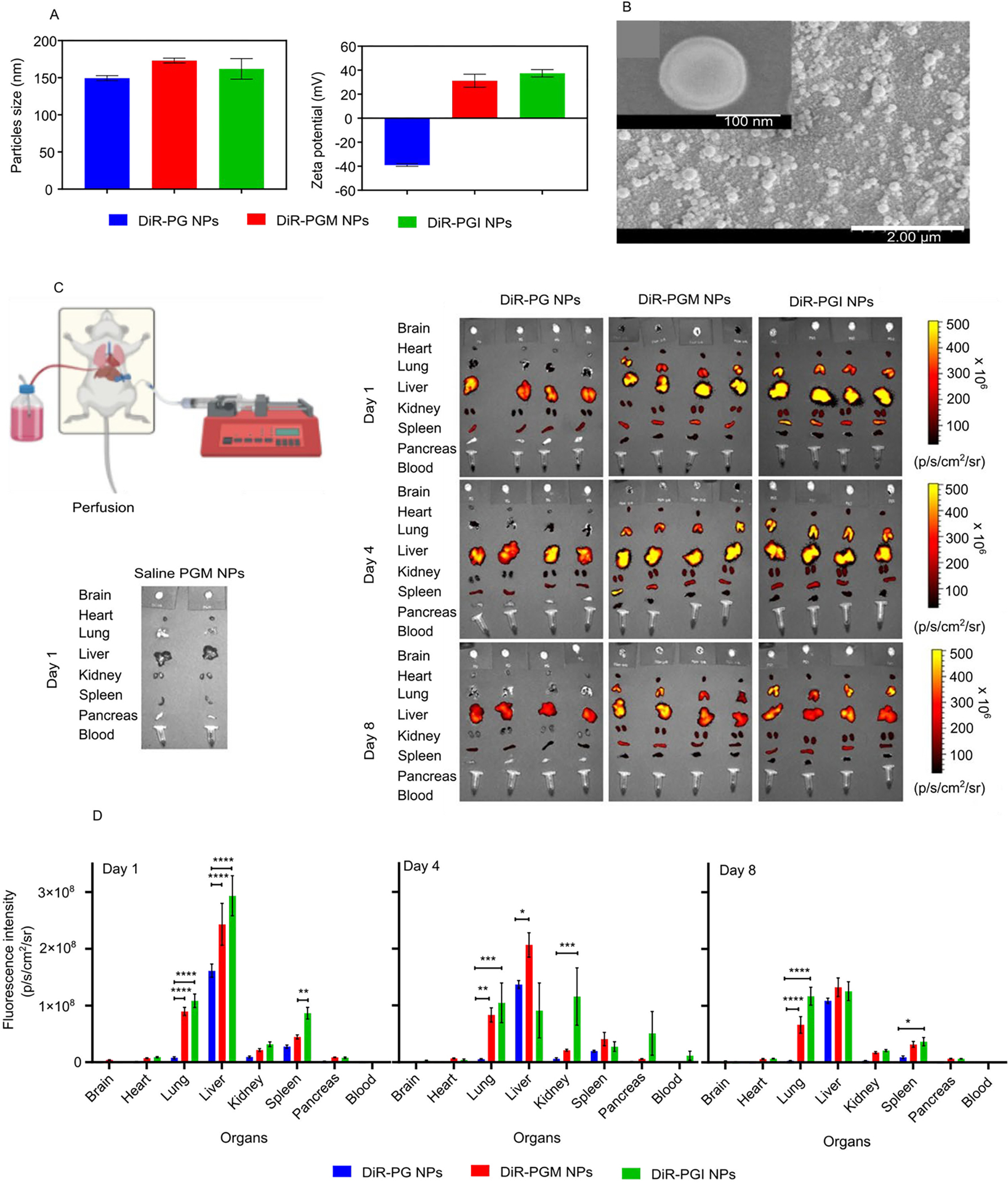
The average hydrodynamic diameter and ζ-potential of the DiR-loaded NPs (A) and SEM images of the DIR-loaded PGM NPs (B). Demonstrative illustration showing the perfusion technique to remove the blood from the organs (C, top left diagram). Fluorescence intensity of the blood and organs from mice injected with either saline, blank PGM, DiR-PG, DiR-PGM or DiR-PGI NPs 1, 4 and 8 days after injection (C, left and right panel). Biodistribution data obtained from the fluorescence intensity of DiR in mouse blood and organs 1, 4 and 8 days after IV injection of either DiR-PG (blue), DiR-PGM (red) or DiR-PGI (green) NPs. IVIS-200 was used to measure the fluorescence intensity in the indicated organs and blood samples (D), * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001. Results are reported as mean ± S.D. (n = 4).
The in vitro coumarin-6 and DiR release profiles of PG, PGM and PGI NP formulations (supplementary Fig. S3) demonstrated ~5% of the dyes being released in the first 5 – 10 hours (burst release) followed by very limited release over the subsequent 672 hours. Dye-loaded PGM and PGI formulations did not reveal significant differences in release kinetics. These results suggest that all three NP formulations have the potential to be used for in vivo biodistribution studies. We next injected healthy mice intravenously through a retro orbital injection with either saline, 5 mg of blank PGM NPs, 5 mg of DiR-PG NPs (equivalent to ~8.3 μg DIR), 5 mg of DiR-PGM NPs (equivalent to ~8.2 μg DIR) or 6 mg of DiR-PGI NPs (equivalent to ~8.5 μg DIR). At 1-, 4-, and 8-days post injection, blood samples were collected from the mice and then, after being sacrificed, they underwent whole body perfusion (Fig. 2c, top left diagram) in order to clear all the organs of blood, making it possible to accurately evaluate the ability of NPs to cross the BBB and accumulate in the tissues of the brain as well as other organs 51. IVIS was then used to determine the magnitude of NP accumulation in different organs. Over the three time points, whole mice images showed that mice treated with positively charged NPs (PGM and PGI), retained higher signals compared mice treated with the negatively charged PG NPs (Supplementary Fig. S2). This could be explained by the higher interaction between the positive charged NPs and the negatively charged membranes of endothelial cells 23, 25, 45.
The negatively charged PG NPs were only distributed to the liver, kidneys and spleen, while the positively charged NPs (PGM and PGI) were distributed to almost all of the collected organs: liver, kidneys, spleen, heart, lungs and pancreas. Interestingly, PGM NPs were the only NPs that exhibited significant brain accumulation relative to PGI and PG, implying that the surface chemistry plays a major role in crossing the BBB (Fig. 3b; p-value < 0.05). Quantitative assessment of the IVIS data showed that over the three time points there was a superior accumulation of both PGM NPs and PGI NPs in the lungs, liver, kidneys, spleen, and pancreas, when compared to PG NPs (Fig. 2d). And in all of those cases the superiority of accumulation seems to be charge specific, with no significant impact of surface chemistry. For example, at the day 1 time point, both PGM NPs and PGI NPs exhibited ~12.5-fold increase in their lung accumulation as compared to PG NPs, with almost no significant difference in the signal between both positively charged NPs, indicating that NPs surface charge is the major driving force for lung accumulation, and that the surface chemistry does not have any apparent impact. Both PGM NPs and PGI NPs were readily entrapped in the lungs immediately after IV injection possibly due to: (1) their surface modifiers (PEGylation and positive charge), and/or (2) lung capillary filtration effects 52. Furthermore, studies have also shown that positively charged NPs are highly susceptible to being taken up by macrophages residing in the lung, liver and spleen 46. The same charge specific distribution was also observed for the pancreas, as both PGM NPs and PGI NPs exhibited an almost 4.6-fold increase in the pancreas compared to PG NPs. Given that our major focus was to estimate brain accumulation of NPs, we reimaged the brains using different IVIS settings, where we increased the laser intensity in order to amplify the signal so as to decipher the differences between the groups. The results showed that PGM NPs exhibited much greater accumulation in the brain compared to PG NPs and PGI NPs, with PGI NPs accumulating slightly more than PG NPs (Fig. 3a, upper panel). Quantitatively, at the day 1 time point, PGM NPs exhibited a 2.4- and 5.2-fold increase in brain accumulation as compared to PGI NPs and PG NPs, respectively (Fig. 3a, lower panel), suggesting the importance of surface chemistry in promoting the crossing of the BBB and achieving significant brain accumulation. Furthermore, and driven by those findings, we reimaged the brains at day 8 using a smaller field of view in order to further improve the resolution and sensitivity of the IVIS, and obtain an improved quantification of the signal, and the results further confirmed the superiority of PGM NPs for brain delivery (Fig. 3b). As described earlier, an in vitro DiR release study demonstrated that there was no significant difference in release rates of the dye from PGM NPs versus PGI NPs (supplementary Fig. S3). Thus, the finding from the in vivo biodistribution showing higher fluorescence intensity in the brain in mice administered DiR-loaded PGM NPs suggests enhanced localization of these PGM NPs rather than the enhanced fluorescence being due to increased release of DiR from the formulation post administration. In addition, our previous research, using an in vivo healthy mouse model, demonstrated that at almost all time points the drug concentration in the brain from drug-loaded PGM NPs was higher than that due to free drug solution 53.
Figure 3.
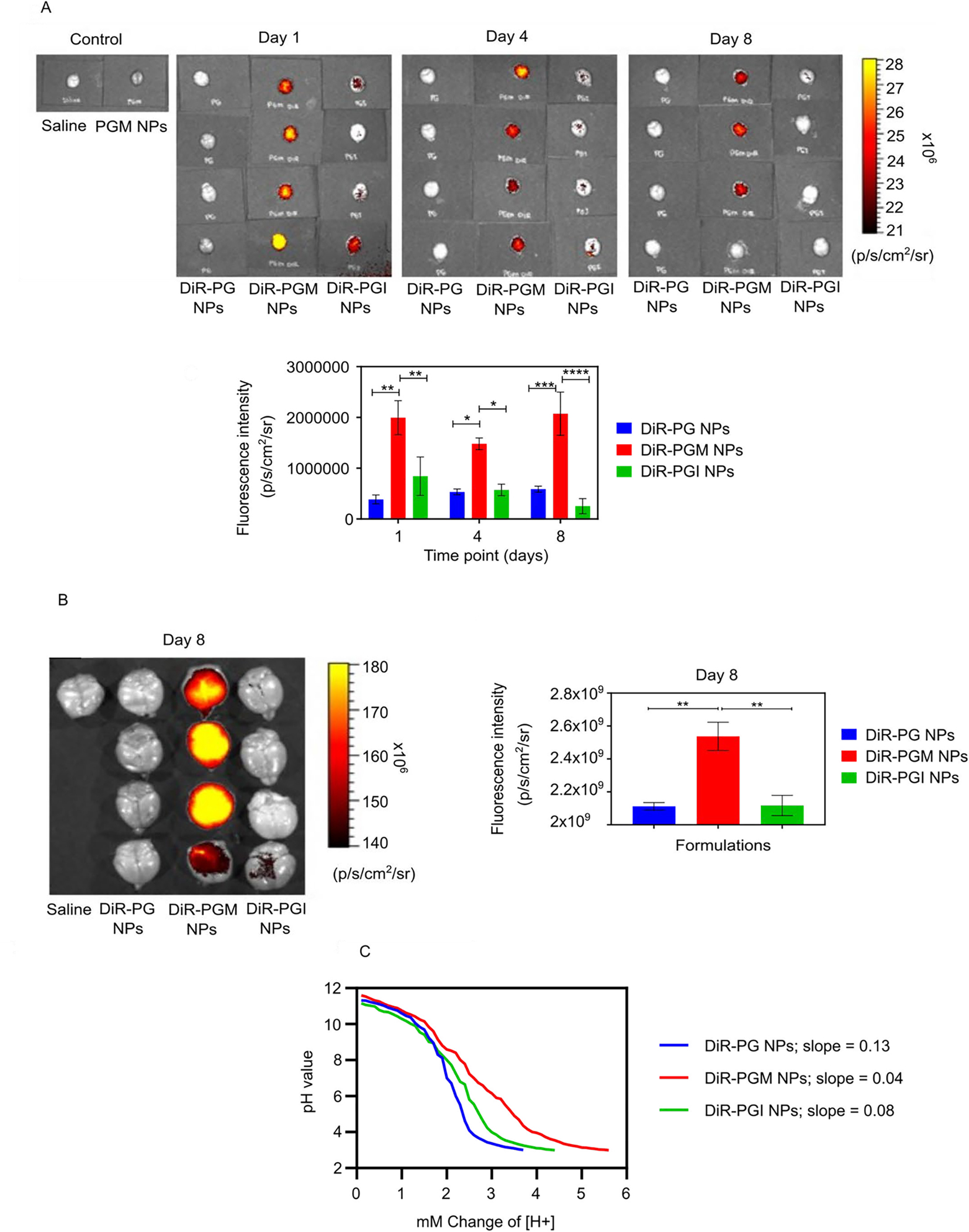
Comparing NP formulations for their ability to accumulate in the brain and for their buffering capacity. IVIS-200 visualization of the fluorescence intensity in mouse brains 1, 4, and 8 days after IV injection of either DiR-PG, DiR-PGM or DiR-PGI NPs and graphical representation of the fluorescence intensity data versus time (A). Further analysis of the day 8 post IV injection timepoint only, showing fluorescence intensity images in the brains of mice injected with the indicated DiR-loaded NPs and graphical representation (B), * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001. Results are reported as mean ± S.D. (n = 4). Acid titration experiment with 0.1 N HCl demonstrate the buffering capacity of NP formulations: PG NPs, PGM NPs and PGI NPs (C).
Both PGM NPs and PGI NPs exhibited equivalent accumulation in all organs except for the brain where PGM NPs significantly outperformed PGI NPs, despite both formulations possessing the same size and surface charge. One potential explanation for the enhanced accumulation of PGM NPs in the brain (over PGI NPs) may stem from our finding that these two formulations possessed different buffering capacities (Fig 3c). Buffering capacity affects the fate of endocytosed nanocarriers upon endocytosis 54–56, and thus differences in buffering capacity may affect the overall fate of the NPs such that PGM NPs (which displayed the higher buffering capacity) had an advantage over PGI NPs in terms of crossing the BBB versus being degraded by lysosomal components 25, 55, 57–60. Healthy BBB endothelial cells are more restricted in terms of the modes of endocytosis used compared to the endothelia of other tissues 61–63, and these restrictions may have resulted in an advantage of PGM NPs over PGI NPs specifically in brain tissue. Such a possibility nevertheless remains speculative at this stage and further investigation would be required for validation.
In addition, when NPs are dispersed into biological fluids, proteins can readily adsorb onto the NPs forming a protein corona (PC). The PC may significantly affect NP uptake efficacy, and potentially block their targeting capabilities 64. A study by Sakulkhu et al., demonstrated that positively charged NPs adsorbed 32 types of proteins, with low molecular weight (<30 kDa) proteins being the more abundant protein type to be absorbed onto NPs in vivo 65. PC formation on NPs impacts on a number of NP properties in vivo including blood circulation half-life, biodistribution, metabolism of the NPs, and toxicity 66, 67. Limiting PC formation around NPs can prolong the blood circulation time, improve the efficacy of NPs for targeted organs and optimize the rate of biodegradation of NP. PC formation on both PGM NPs and PGI NPs might impact upon the biodistribution and pharmacokinetics of NPs. Thus, in vivo experiments assessing PC formation on PG NP, PGM NP, and PGI NP formulations needs to be further investigated.
In vitro cytotoxicity against human derived glioblastoma U87MG-Red-Fluc cells
PGM NPs were selected as our lead formulation, and our next step was to load this NP formulation with PTX and evaluate its in vivo efficacy against a human derived GBM mouse xenograft model. Initially however, we aimed to confirm the superiority of PTX over the GBM standard of care drug, TMZ, in an in vitro human derived glioblastoma cell culture model (U87MG-Red-Fluc cell line), by comparing the cytotoxicity of soluble PTX to TMZ, and two other widely used anticancer drugs, 5-fluorouracil (5-FU), and doxorubicin (DOX). Drug concentrations ranging from 0.1 μM – 30 mM were incubated with the U87MG-Red-Fluc cells for 24 hours, and the results indicated that the cell viability was concentration dependent and dramatically decreased in the presence of the soluble drugs as shown in Supplementary Fig. S4. The IC50 values for all the treatments are shown in Table 1 and were lowest for PTX solution. Importantly, the IC50 value for PTX was ~12-fold lower than that of TMZ, indicating that loading PTX in a delivery system that achieves equivalent brain accumulation to that of TMZ would result in a better response against a lethal brain tumor.
Table 1.
The IC50 values for TMZ, 5-FU, DOX or PTX when incubated with U87MG-Red-Fluc cells for 24 hours. Results are reported as mean ± S.D.
| TMZ | PTX | 5-FU | DOX | |
|---|---|---|---|---|
| IC50 values (mM) | 0.53 ± 0.16 | 0.042 ± 0.192 | 1.69 ± 0.12 | 0.050 ± 0.229 |
Physicochemical characterization and in vitro release study of PTX-loaded PGM NPs
We next loaded PTX into PGM NPs using our modified nanoprecipitation technique, and the results showed that PTX-loaded PGM NPs exhibited an average diameter of 185 nm, with a positive ζ-potential of 16.6 mV. PTX loading and encapsulation efficiency were 8.8 μg/mg NPs and 24.3%, respectively. Drug release from PLGA NPs is known to be dictated by several mechanisms including surface erosion, degradation, diffusion, and desorption 68, 69. The in vitro PTX release profile from PTX-loaded PGM NPs (Fig. 4a) was biphasic, consisting of an initial burst release phase within 100 hours followed by a gradual release phase over 480 hours. The burst release is likely to be primarily due to the rate of drug desorption of PTX from the surface of the NPs 70, 71. The slow release phase may be due to both drug diffusion and polymer degradation 71. These results suggest PGM NPs are a promising candidate for providing sustained PTX release in vivo; possessing the potential to provide sustained drug release at sufficient quantities for antitumor activity once they have reached the target tissue following IV injection.
Figure 4.
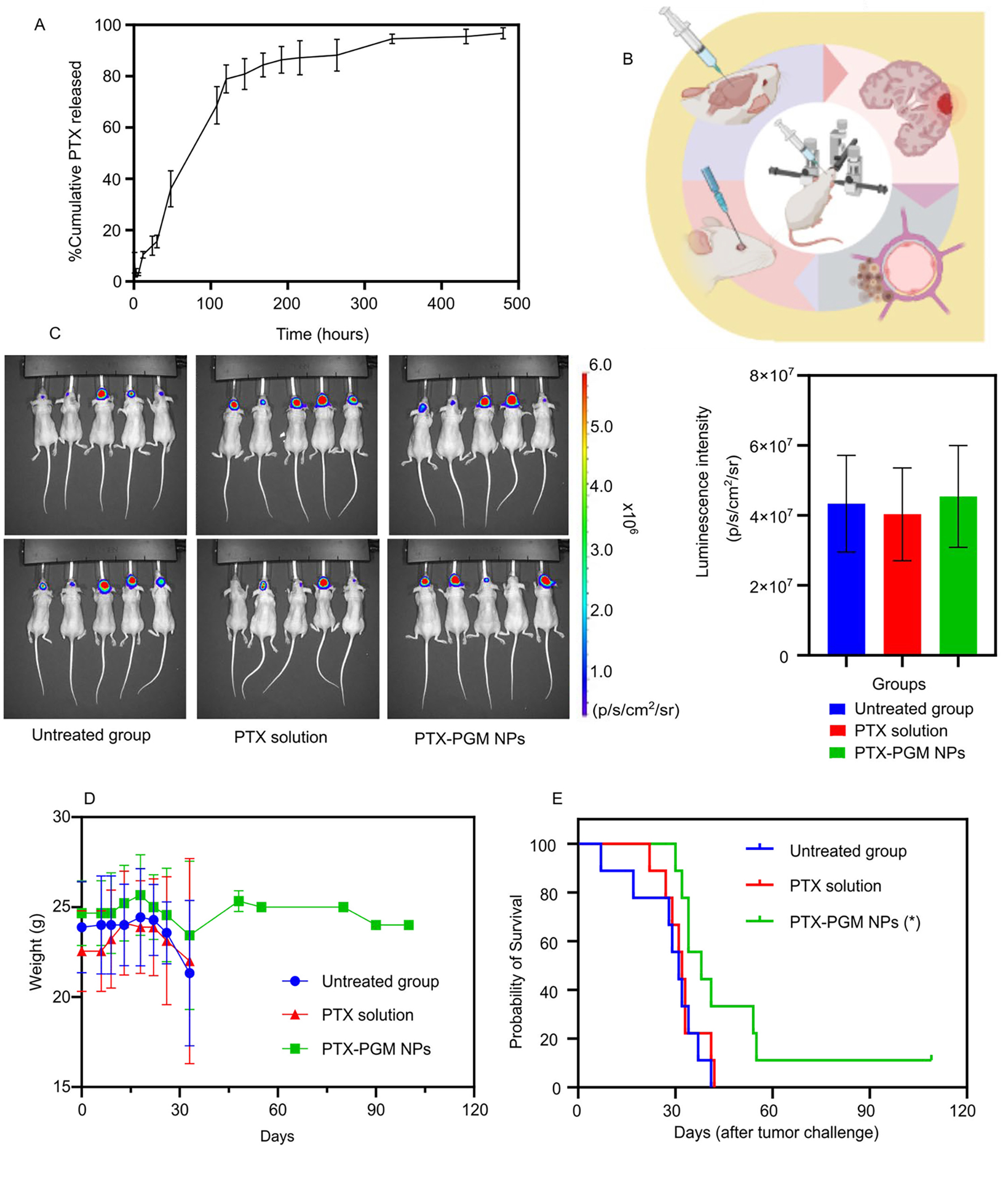
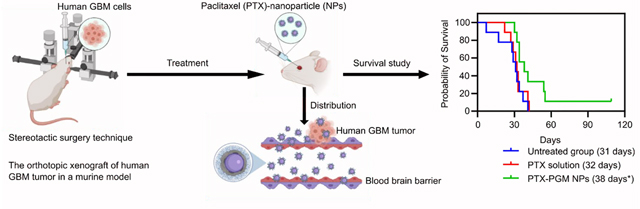
In vitro release profile of PTX from PGM NPs over 480 hours reported as mean ± S.D. (n = 3) (A). Demonstrative illustration showing the orthotopic xenograft of human GBM tumor in a murine model where the human GBM cells are intracerebrally injected into the right cerebral hemisphere to establish xenografts, using stereotactic surgery technique (B). The luminescence intensity measurements (IVIS-200) in GBM tumor challenged mice 3 days after tumor challenge (C, left panel) and graphical representation of the luminescence intensity data versus groups (C, right panel). The treatments occurred 7 days after tumor challenge, and the luminescence intensity was measured 7 days after treatments. Recorded body weights of GBM tumor challenged mice receiving the indicated treatment. Results are reported as mean ± S.D. (n = 10/group) (D). Survival curve of GBM tumor challenged mice receiving the indicated treatment (n = 10/group) (E). The PTX-PGM NP treatment was significantly different from the untreated mice and mice treated with PTX solution (* p < 0.05). Results are reported as mean ± S.D. (n = 10).
In vivo antitumor efficacy against a human derived GBM mouse xenograft tumor model
Finally, we evaluated the efficacy of PTX-loaded PGM NPs in an orthotopic human GBM tumor xenograft model in mice. Athymic NCI-nu/nu female mice were intracerebrally injected with red shifted luciferase-expressing human GBM cells, U87MG-Red-Fluc (Fig. 4b). Seven days following tumor challenge, the mice bearing GBM tumors (as determined by IVIS) were randomly divided into three treatment groups (Fig. 4c, left panel), where the mean and the standard deviation of the captured luminescence was almost equivalent in the three groups (Fig. 4c, right panel), indicating equivalent tumor sizes between the three groups. Starting on the 8th day following tumor challenge, mice were treated IV with either saline (untreated group), 75 μg of soluble PTX, or PTX loaded PGM NPs (equivalent to 75 μg of PTX), every three days, for a total of five doses. Mice weights and survival data were calculated to evaluate the safety and efficacy of our lead formulation.
None of the provided treatments exhibited toxicity as demonstrated by stable mice weights over the time course of the experiment (Fig. 4d). The Kaplan-Meier survival plot showed that the median survival of the mice (Table 2) treated with PTX-loaded PGM NPs (38 days) was significantly greater than mice receiving PTX solution (32 days, p < 0.05) or untreated mice (31 days, p < 0.05). Thus, loading PTX into our lead PGM formulation significantly improved median survival by 18.75% and 21.88% compared to soluble PTX and the untreated group, respectively (Fig. 4e). In addition, PTX solution did not significantly improve the survival of mice compared to the untreated group, supporting the reports of the limited ability of PTX to cross the BBB 20, 21. The PGM NPs played a significant role in increasing survival time of mice challenged with GBM tumors which may be explained by the NPs ability to cross the BBB and subsequently increase accumulation of PTX in the brain compared to when soluble PTX was administered.
Table 2.
Statistical analysis of antitumor efficacy in GBM tumor challenged mice after receiving the indicated treatment.
| Groups | Median survival time (days) | P-valuesΨ | P-valuesΦ |
|---|---|---|---|
| Untreated | 31 | N/A | 0.50 |
| PTX solution | 32 | 0.50 | N/A |
| PTX-PGM NPs | 38 | 0.02 | 0.04 |
N/A: not applicable
compared to untreated group and
compared to PTX solution treatment group.
Results were analyzed using RStudio software (log-rank test).
In vivo acute toxicity and safety in healthy mice
Since it is known that NP formulations can be toxic, we assayed for toxicity using serum biomarkers (Fig. 5a) including ALT, AST, ALP, and total bilirubin on day 14 post PTX administration. IV administration of PTX solution, PTX-PGM NPs and PGM NPs significantly induced a marked decrease in ALP in comparison to the untreated group, however, none of the tested biomarkers significantly increased, indicating a lack of toxicity for all treatments tested. The reason for the significantly lower levels of ALP seen in the treatment groups versus the control cannot be readily explained and would require further investigation. Additionally, weight changes (Fig 5b) were not significantly different between the untreated group and the treatment groups. These findings are further supported by there being no signs of tissue damage in any of the tested organs (brain, heart, kidney, liver, lung, spleen) as evaluated by H&E staining (Fig. 5c). Our results indicated that NP formulations were safe in vivo and could be used as a drug delivery system for GBM treatment; however, long term toxicity studies of our PGM NPs need to be carried out to ensure that they do not have toxic effects on the organs where they accumulate and potentially remain for extended periods when compared to soluble PTX.
Figure 5.
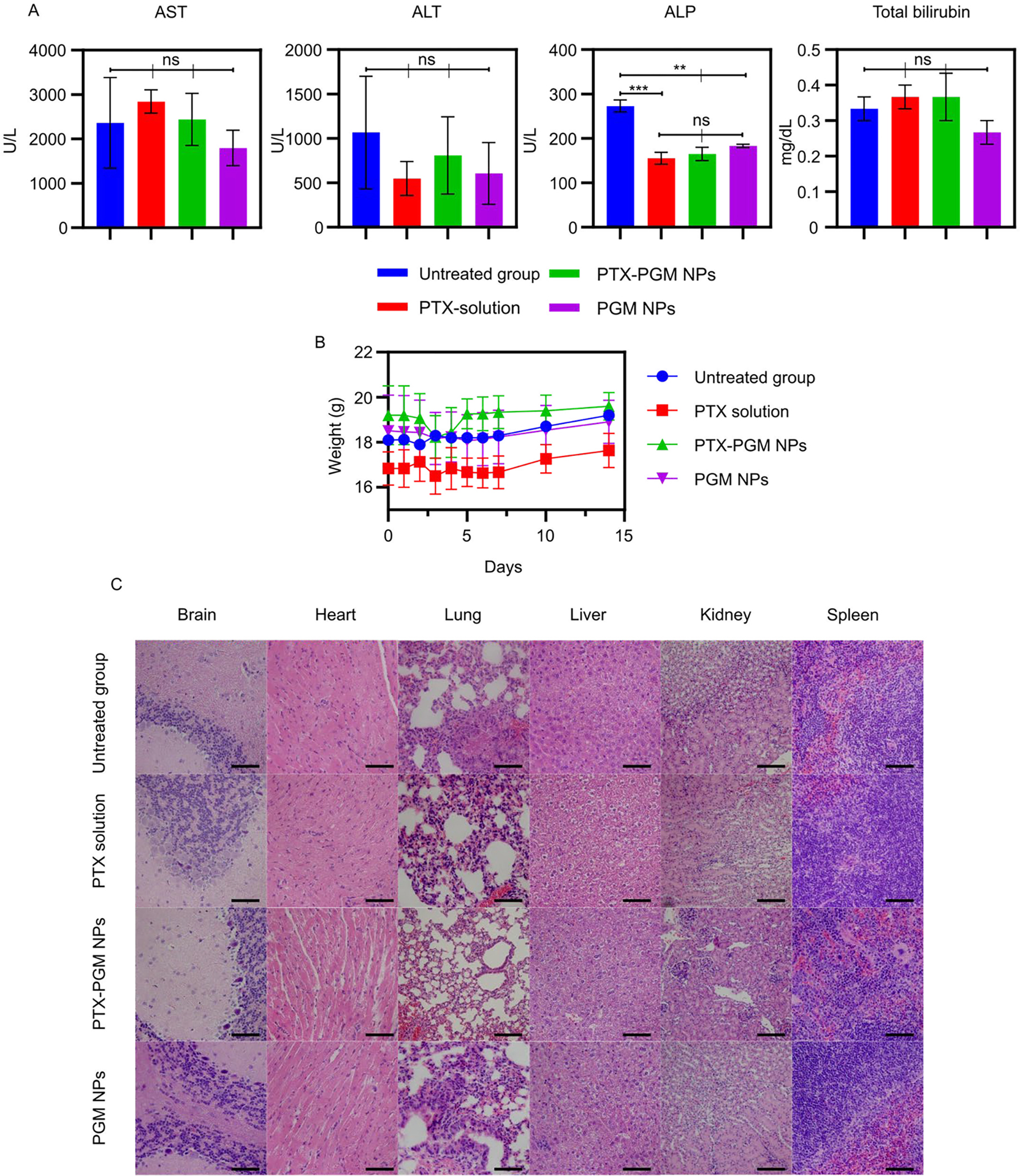
Serum biomarkers including aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase (ALP), and total bilirubin in mice receiving indicated treatments. Blood samples were taken before the mice were euthanized at day 14 subsequent to PTX administration (A). Weights of mice treated with indicated treatments (B). H&E staining of organs (brain, heart, lung, liver, kidney, and spleen) collected from mice given indicated treatments; scale bar = 200 μm (C). Statistical analysis was performed using one-way analysis of variance test. Data are expressed as mean ± SE (n = 3); ** p < 0.01, *** p < 0.001 and ns = not significant.
Conclusion
In this study we found that modifying the surface of NPs with PAMAM significantly improved their capacity to accumulate in the brain as compared to using PEI, in healthy mice. Furthermore, treating mice with PTX-loaded PGM NPs significantly improved the survival of mice bearing GBM orthotopic xenografts, compared to soluble PTX or the untreated control. Our data substantiate the ability of PGM NPs to cross the BBB and improve the efficacy of its loaded cargo, placing PGM NPs as a promising delivery system to treat brain tumors. Thus, PGM NPs may act as a delivery platform to test a myriad of anticancer therapies against brain tumors that have previously failed because of their limited ability to cross the BBB. Utilizing our novel delivery system might successfully introduce new agents to treat GBM, setting the stage for further improvement in the outcomes of treating this lethal disease.
Materials and methods
Synthesis of PLGA-block (b)-PEG-COOH copolymer
PLGA-b-PEG-COOH copolymer was synthesized using the EDC/NHS chemical reaction following our previously published protocol 33. Briefly, 250 mg PLGA-COOH (Resomer® RG 502H, Evonik, Mapleton, IL) was dissolved in 2 mL dichloromethane (DCM, Sigma Aldrich, St. Louis, MO). Three mg N-hydroxysuccinimide (NHS, Sigma Aldrich) and 4.8 mg 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC, Sigma Aldrich) were dissolved in 2.5 mL DCM, and then added to the PLGA-COOH solution. This mixture was stirred at room temperature, during which time the PLGA-COOH reacted through EDC/NHS chemistry to form PLGA-NHS (an amine-reactive sulfo-NHS ester). A washing solution composed of a 20 mL mixture of ethyl ether and methanol (50:50) was added to the previously prepared solution and centrifuged at 5000 xg for 10 minutes (Eppendorf 5810-R, Eppendorf®, Westbury, NY) to remove residual EDC/NHS. This washing step was carried out twice and then PLGA-NHS was dried under rotary evaporator vacuum (Buchi, Flawil, Switzerland) for 30 minutes. The PLGA-NHS was dissolved in DCM (4 mL) to which 11 μL N, N-diisopropylethylamine (DIPEA, Sigma Aldrich) was added. To this, 30 mg NH2-PEG-COOH (3.4 kDa, Creative PEGWorks, Durham, NC) was added and left stirring at room temperature for 24 hours to form PLGA-b-PEG-COOH. After which time, the washing solution was added to the PLGA-b-PEG-COOH solution which was then centrifuged to remove any unreacted NH2-PEG-COOH. The PLGA-b-PEG-COOH copolymer was then dried using the rotary evaporator.
Preparation of DiR-loaded NP formulations
PLGA-b-PEG-COOH NPs loaded with DiR (1,1’-dioctadecyltetramethyl indotricarbocyanine Iodide, PerkinElmer®, St. Hopkinton, MA) were modified and prepared by using a previously published nanoprecipitation method 33, 72. PLGA-b-PEG-COOH copolymer (50 mg) was dissolved in 5 mL acetone, then 100 μL of a 1 mg/mL solution of DiR in absolute ethanol was added to the copolymer solution. In contrast, when incorporating PAMAM or PEI onto the NPs, 125 μL of generation 5 (G5) PAMAM (Sigma Aldrich) solution (5 % wt/v in methanol) or 3 mg PEI (high molecular weight, water-free, Sigma Aldrich) dissolved in 2.5 mL acetone was added to the copolymer solution. The final mixture was vortexed, transferred to a 5 mL syringe fitted with a G26 needle, and added dropwise into 15 mL of an aqueous solution of 0.1% poly-(vinyl alcohol) (PVA, hydrolyzed 80% (Mw 9,000 – 10,000), Sigma Aldrich) with continuous stirring for 30 minutes. The NP suspension was collected after the organic solvents were removed by rotary evaporation over 4 hours. The suspension was then transferred to Amicon® Ultra-15 centrifugal filter units (Mw cutoff = 100 kDa) (Millipore Sigma, St. Louis, MO) and washed with Nanopure water by centrifuging at 4000 xg for 20 minutes (Eppendorf 5810-R, Eppendorf®, Westbury, NY). These DiR-loaded PG NPs were subsequently washed three more times. NPs were freshly prepared before each experiment.
Determination of DiR loading and encapsulation efficiency
The DiR loading and encapsulation efficiency were determined by fluorescence spectrophotometry (Spectra M5 Microplate Spectrophotometer (Molecular Devices, Sunnyvale, CA)). The NPs were dissolved in 1 mL dimethyl sulfoxide (DMSO, Sigma Aldrich) and vortexed until the solution became clear. This mixture was diluted with an equal volume of DMSO, and the fluorescence was measured. The DiR concentration in the samples was calculated by comparing the fluorescence values to a calibration curve made from known concentrations of DiR. Excitation and emission wavelengths of DiR were measured at 750 nm and 780 nm, respectively. DiR loading in the NPs and encapsulation efficiency were calculated using Equations 1 and 2, respectively.
| Equation 1 |
| Equation 2 |
Preparation of coumarin-6-loaded NP formulations
Coumarin-6-loaded NPs were prepared by using a nanoprecipitation method as described previously. Briefly, 50 mg of PLGA-b-PEG-COOH copolymer was dissolved in 5 mL acetone, then 500 μL of a 0.25 mg/mL solution of coumarin-6 dissolved in DMSO was added to the copolymer solution. When incorporating PAMAM or PEI on the NPs, 125 μL of generation 5 (G5) PAMAM solution (5 % wt/v in methanol) or 3 mg PEI dissolved in 2.5 mL acetone was added in the copolymer solution. This final mixture was vortexed and transferred to a 5 mL syringe fitted with a G26 needle. The same modified nanoprecipitation method including the solvent evaporation and washing steps were performed as described for the preparation of DiR-loaded NP formulations. NPs were freshly prepared before each experiment.
Determination of coumarin-6 loading and encapsulation efficiency
The coumarin-6 loading and encapsulation efficiency were determined by fluorescence spectrophotometry. The NPs were dissolved in 1 mL DMSO and vortexed until the solution became clear. This mixture was diluted with an equal volume of DMSO and the fluorescence was measured. The coumarin-6 concentration in the samples was calculated by comparing the fluorescence values to a calibration curve made from known concentrations of coumarin-6. Excitation and emission wavelengths of coumarin-6 were measured at 470 nm and 510 nm, respectively. Coumarin-6 loading in the NPs and encapsulation efficiency were calculated using Equations 3 and 4, respectively.
| Equation 3 |
| Equation 4 |
Preparation of PTX-loaded PGM NPs
PTX-loaded PGM NPs were prepared using a nanoprecipitation method as described previously 33, 72. Briefly, 1 mg PTX (LC Laboratories®, Woburn, MA) was dissolved in 1 mL acetone and vortexed. PLGA-b-PEG-COOH copolymer (50 mg) was dissolved in 4 mL acetone and combined with the PTX solution and vortexed, then 125 μL G5 PAMAM solution was added to this mixture. After vortexing, the solution was transferred to a 5 mL syringe fitted with a G26 needle. The same modified nanoprecipitation method including the solvent evaporation and washing steps were performed as described for the preparation of DiR-loaded NP formulations. NPs were freshly prepared before being used in each experiment.
Determination of PTX loading and encapsulation efficiency
The PTX loading and encapsulation efficiency were determined by high-performance liquid chromatography (HPLC) using an Agilent 1100 Series HPLC station coupled with a UV-Vis diode array detector (Agilent Technologies, Santa Clara, CA). A reverse phase C-18 column (Waters, Symmetry® 5μm, 4.6 × 150 mm) was used in the assay with a mobile phase consisting of acetonitrile (ACN): Nanopure water (60:40) using a flow rate of 0.8 mL/min. The detection wavelength was 227 nm, and the injection volume was 50 μL. Chromatographic separation of PTX was performed at room temperature. HPLC sample preparation was performed by taking approximately 1 mg PTX-loaded PGM NPs and dissolving them in 500 μL ACN, vortexing until the solution became clear, and then adding an equivalent volume of Nanopure water. This mixture was then further diluted using an equal volume of ACN: Nanopure water (50:50), sonicated until the solution became clear and centrifuged at 14000 xg for 2 minutes to separate residual NPs. The supernatant was directly injected into the HPLC and the PTX concentration was determined by comparing the area under the curve of the sample to a calibration curve made with known concentrations of PTX. The drug loading and encapsulation efficiency were calculated using Equations 5 and 6, respectively.
| Equation 5 |
| Equation 6 |
NP physicochemical characterization
The physicochemical properties of the NPs, namely hydrodynamic size and zeta (ζ) potential were measured using Dynamic Light Scattering (DLS) and Laser Doppler Velocimetry (LDV), respectively. NPs were resuspended in Nanopure water and measurements were performed using a Zetasizer Nano ZS particle analyzer (Malvern Instrument Ltd., Westborough, MA).
The morphology of the NPs was determined using Scanning Electron Microscopy (SEM) (Hitachi S- 4800 SEM, Hitachi High-Tech America, Inc., Schaumburg, IL). A suspension of NPs (0.05 mg/mL in Nanopure water) was added onto a silicon wafer mounted onto a SEM stub and incubated at room temperature for 24 hours to allow the water to evaporate. Next, the sample was sputter coated with gold-palladium using an argon beam K550 sputter coater (Emitech Ltd., Kent, England) before imaging.
In vitro release study of PTX-loaded PGM NPs
The cumulative release of PTX from PGM NPs was studied using Float-A-lyzer® dialysis (50 KD MWCO, 1-mL capacity, regenerated cellulose membrane, Spectrum Laboratories, Inc., Piscataway, NJ). Briefly, NPs were suspended in 1 mL of release medium (PBS pH 7.4 + 0.4% Tween 80), transferred into a Float-A-lyzer® device and placed in a 50 mL Falcon tube containing 12 mL of release medium. The tubes were left shaking at 300 rpm and 37°C (Eppendorf, Hauppauge, NY) for 20 days. Samples were taken at predetermined time points (0, 1, 3, 6, 12, 24, 48, and every 24 hours for up to 480 hours) and the release medium was replaced with fresh media after each sample was taken to maintain sink conditions. All release samples were carried out in triplicate. The PTX concentration was measured using the aforementioned HPLC-UV method and this data was used to construct a release profile curve by plotting the percent cumulative PTX released versus time.
Transendothelial electrical resistance (TEER)
Transendothelial electrical resistance (TEER) was measured to evaluate model BBB integrity. The cell-culture inserts were allowed to equilibrate to room temperature and TEER was measured by using an EVOM2 meter and an EndOhm-6 chamber (World Precision Instruments, Sarasota, FL). TEER values were calculated by Equations 7 and 8.
| Equations 7 |
| Equations 8 |
Where R total represents resistance of the cell cultured Transwell® insert and R blank represents resistance of the empty Transwell® insert.
Permeability
The human brain microvascular endothelial cell line (hCMEC/D3; EMD Millipore, Temecula, CA) was used in this work. The cell line was cultured in EndoGRO™-MV complete media kit (EMD Millipore, Temecula, CA) supplemented with 1 ng/mL fibroblast growth factor basic (FGF-2), in cell culture flasks that were coated with 1:20 diluted collagen type I rat tail (3.8 mg/mL stock; EMD Millipore, Temecula, CA). Cells were maintained at 37°C in a humidified 5% CO2 incubator (Sanyo Electric Biomedical Co Ltd., Japan). At 80% confluence, the cells were trypsinized and sub-cultured.
The permeability of the established in vitro BBB model was evaluated. When the TEER values were found to remain constant, in vitro experiments were started. The Transwell® inserts and basolateral section/lower chamber were washed with 1 × PBS and 300 μL and 500 μL of EndoGRO™-MV complete media was added to the apical side and basolateral side, respectively. Coumarin-6 NPs (containing 0.08 – 0.2 μg of coumarin-6; equivalent to 0.1 – 0.15 mg of NPs) were added to the apical side. At 1, 3 and 6 hours of culture, 500 μL of medium was collected from the basolateral side and replenished with 500 μL of new medium each time. At 6 hours, the 300 μL of medium from the apical side was collected to determine amount of coumarin-6. The concentration of coumarin-6 was determined by fluorescence spectrophotometry. The permeability coefficient (Pe) was obtained by using Equation 9.
| Equation 9 |
Where dQ/dt represents linear apparent transcytosis rate from apical to basolateral chambers; C0 represents solute concentration in the donor chamber (μg/mL); and A represents the surface area of the tissue available for diffusion (0.33 cm2).
Biodistribution studies
Biodistribution of the DiR-PG, DiR-PGM and DiR-PGI NPs compared to saline and blank PGM NPs was performed using 8 week-old female BALB/CJ mice with a weight range between 20 – 23 g. Animals were allowed unrestricted access to food and water. All animal procedures were conducted in accordance with the institutional animal care and use committee (IACUC). After general anesthesia with isoflurane, the 48 mice were randomly divided into three treatment groups and intravenously (IV) injected with saline, 5 mg of blank NPs, 5mg of DiR-PG NPs (equivalent to ~8.3 μg DiR), 5 mg of DiR-PGM NPs (equivalent to ~8.2 μg DiR) or 6 mg of DiR-PGI NPs (equivalent to ~8.5 μg DiR) using retro-orbital injection. Biodistribution was visually observed using an IVIS-200 instrument (Xenogen, PerkinElmer, Waltham, MA) to track the accumulation of DiR in the brain. The relative amount of DiR was calculated and graphically displayed as fluorescence intensity. The whole body of the anesthetized mice was imaged 1, 4 and 8 days after IV injection of the NPs. At the end of each time point, four mice from each treatment group were perfused by using a modified perfusion technique 51, 73, 74 while they were alive to reduce interference from NPs in the circulation and then euthanized. NP biodistribution was determined for the major organs (brain, heart, lung, liver, kidney, spleen, and pancreas) that were removed from the animals and for blood samples collected prior to perfusion and then visualizing the DiR fluorescence intensity using the IVIS-200.
Buffering capacity of NP formulations
The ability of NPs to resist acidification was evaluated using the acid titration assay as previously described by Zhang et al. 75. Briefly, 10 mg/mL of NP formulations were suspended in 150 mM NaCl. The pH was first adjusted to ~9.0 and then titrated with 10 μL of 0.1 N HCl until a pH of 3.0 was reached. The slope of the line from the graph of pH versus [HCl] provided an indication of the intrinsic buffering capacity of the delivery NPs.
In vitro cytotoxicity against U87MG-Red-Fluc cells
Human derived glioblastoma U87MG-Red-Fluc cells (Bioware® Brite cell line) were purchased from PerkinElmer, Inc. (Waltham, MA). Cells were cultured in Eagle’s Minimal Essential Medium (EMEM Cat. No. 30–2003 (ATCC, Manassas, VA)) supplemented with 10% fetal bovine serum (FBS) (Atlanta Biologicals, Lawrenceville, GA), and 0.2% (w/v) puromycin dihydrochloride (Gibco, Invitrogen, Waltham, MA). They were maintained in a humidified 5% CO2 incubator (Sanyo Scientific Autoflow, IR direct heat CO2 incubator, Wood dale, IL) at 37°C.
U87MG-Red-Fluc cells were dispensed into the wells of a 96-well flat-bottom microtiter plate at a density of 104 cells/well and allowed to incubate overnight in EMEM (+ supplements: i.e. complete medium). The U87MG-Red-Fluc cells were treated with one of four anticancer agents: TMZ solution (Sigma Aldrich), 5-fluorouracil (5-FU) solution (Sigma Aldrich), doxorubicin (DOX) solution (MCE®MedChemExpress, Monmouth, NJ), and PTX solution (LC Laboratories, Woburn, MA) at varying concentrations (30 mM – 0.1 μM) for 24 hours. All tests were performed in triplicate. After 24 hours, the media was removed and 150 μL of the MTS reagent (CellTiter 96R Aqueous One Solution Reagent (Promega Corporation, Madison, WI)) was added into each well. Media alone and untreated-U87MG-Red-Fluc cells were used as negative control groups. The absorbance at 490 nm was measured after 4 hours of incubation with the MTS reagent in a humidified 5% CO2 at 37°C. The cell viability was expressed as a percentage of the ratio of the absorbance of the test sample to the absorbance of the control. The potency of the anticancer agents was determined by comparing the concentrations needed for 50% inhibition/reduction of growth/viability (IC50).
In vivo efficacy of PTX-loaded PGM NPs
The antitumor efficacy of the PTX-PGM NPs was tested in 8 week-old female athymic NCI-nu/nu nude mice with a weight range between 20 – 23 g. All animal procedures were conducted in accordance with the IACUC. Animals were allowed unrestricted access to food and water. Intracranial tumor formation was performed following a modification to a previously reported protocol 76. The nude mice were anesthetized by IP injection of a ketamine/xylazine mixture (87.5 mg/kg ketamine/12.5 mg/kg xylazine) (Phoenix Pharmaceuticals, Burlingame, CA). For the tumor challenge, nude mice were intracerebrally injected with 3 × 105 U87MG-Red-Fluc cells into the right cerebral hemisphere to establish xenografts using a stereotaxic surgery technique 77. Briefly, a 1–1.5 cm midline sagittal incision along the superior aspect of the cranium from anterior to posterior was made using a sterile disposable scalpel. Then a small burr hole was made with a sterile microdrill bit at predetermined coordinates (2 mm posterior to the bregma, 1 mm laterally, and 2 mm deep to the dura). Seven days later, mice bearing GBM tumors were then randomly placed into one of three treatment groups (n = 10/group): untreated, PTX free drug solution, or PTX-PGM NPs. The treatments were administered IV to deliver 75 μg of PTX by retro-orbital injection once every three days (five doses total).
Animal weights were monitored to ensure the health of the mice and the safety of the formulation. Tumor growth in mice was determined by measuring luciferase expression using the IVIS-200 instrument post-IP injection of D-luciferin potassium solution (150 mg/kg) (PerkinElmer, Inc., Waltham, MA)) at 3, 7, and 27 days. Luminescence intensity was measured to represent tumor growth. Additionally, median survival time was evaluated and compared to the control group.
Euthanasia criteria were as follows: mice were monitored every day for signs of distress such as piloerection, decreased activity and motility, decreased food intake (malnutrition), and dehydration. Veterinarians were consulted as to treatment options or whether or not to euthanize and were performed as appropriate. In addition, body weights were recorded three times per week for each mouse. Mice that lost more than 20% of their body weights (from initial weight: Day 0) were euthanized. All animal procedures were conducted in accordance with the institutional animal care and use committee (IACUC).
Acute toxicity
For these studies, 8 week-old female BALB/CJ mice with a weight range 16 – 19 g were used and allowed free access to food and water. Animals were housed at the Medical Laboratories at the University of Iowa and kept on a daily 12 hr light/12 hr dark cycle. All animal experiments were performed in accordance with the University of Iowa guidelines for the care and use of laboratory animals. Mice were randomly divided into four treatment groups: untreated, PTX solution, PTX-PGM NPs, and PGM NPs. The mice were injected with saline (untreated), 75 μg of PTX solution, 6 mg of PTX-PGM NPs (equal to 75 μg PTX) or 6 mg of PGM NPs by retro-orbital injection to observe in vivo acute toxicity. Mice were anesthetized and euthanized at day 14 subsequent to PTX administration. At the end of the study, organs and blood samples were harvested to evaluate histopathological and biochemical traits. Changes in weight were also monitored.
Biochemical examination
Blood samples for serum biochemical examination were collected after anesthetization. The blood samples were centrifuged at 14,000 xg for 10 minutes within 1 hour of having been collected, and the supernatant was harvested. All the biochemical parameters were determined on a clinical automatic chemistry analyzer (IDEXX Catalyst One Chemistry Analyzer, IDEXX BioAnalytics, Columbia, MO), including aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase (ALP), and total bilirubin.
Histopathological examination
Brains, hearts, lungs, livers, spleens, and kidneys were harvested from mice after being euthanized. Each organ was fixed in 10% Neutral buffered formalin (RPI, Mount Prospect, IL), and then embedded in paraffin (EM-400, Surgipath, Leica Biosystems, Inc., Buffalo Groove, IL). The organs were sliced into 5 μm sections using a microtome (HM 355S Automatic Microtome, Thermo Scientific™, Kalamazoo, MI) and then stained with hematoxylin and eosin (H&E, Leica) and imaged using an Olympus BX61 microscope (Olympus, Center Valley, PA). The images were processed using cellSens software (Olympus).
Statistical analysis
To compare data between two groups, the data was analyzed using the student’s unpaired two-tailed t-test (GraphPad Prism version 7.00 for Windows, GraphPad Software, La Jolla, CA). To compare data between more than two groups, one-way analysis of variance (ANOVA) test was used followed by Tukey’s post-hoc test. Analysis of survival curves was performed using the Kaplan-Meier estimator test and the log-rank test (RStudio software, RStudio, Inc., Boston, MA). All plots were created using GraphPad Prism. A statistically significant difference was considered for p-values <0.05. Each point represents the mean ± standard deviation (S.D.) of the replicates from one representative experiment.
Supplementary Material
Acknowledgements
K.W. acknowledges support from the Government Pharmaceutical Organization (GPO) scholarship. J.C.Q. acknowledges support from the Alfred P. Sloan Foundation, the University of Iowa Graduate College, and the American Association for University Women. A.K.S acknowledges support from the Cancer Center support grant (P30 CA086862) and the Lyle and Sharon Bighley Chair of Pharmaceutical Sciences. The authors would like to acknowledge the use of the University of Iowa Central Microscopy Research Facility, a core resource supported by the University of Iowa Vice President for Research, and the Carver College of Medicine (S10 RR029274-01).
Footnotes
Supporting Information Available
References
- 1.Tso C-L; Freije WA; Day A; Chen Z; Merriman B; Perlina A; Lee Y; Dia EQ; Yoshimoto K; Mischel PS; Liau LM; Cloughesy TF; Nelson SF, Distinct Transcription Profiles of Primary and Secondary Glioblastoma Subgroups. Cancer research 2006, 66 (1), 159. [DOI] [PubMed] [Google Scholar]
- 2.Kabat GC; Etgen AM; Rohan TE, Do steroid hormones play a role in the etiology of glioma? Cancer Epidemiol Biomarkers Prev 2010, 19 (10), 2421–7. [DOI] [PubMed] [Google Scholar]
- 3.Kleihues P; Ohgaki H, Primary and secondary glioblastomas: from concept to clinical diagnosis. Neuro-oncology 1999, 1 (1), 44–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bush NAO; Chang SM; Berger MS, Current and future strategies for treatment of glioma. Neurosurgical Review 2017, 40 (1), 1–14. [DOI] [PubMed] [Google Scholar]
- 5.Mahvash M; Hugo H-H; Maslehaty H; Mehdorn HM; Stark AM, Glioblastoma Multiforme in Children: Report of 13 Cases and Review of the Literature. Pediatric Neurology 2011, 45 (3), 178–180. [DOI] [PubMed] [Google Scholar]
- 6.Hegi ME; Diserens AC; Gorlia T; Hamou MF; de Tribolet N; Weller M; Kros JM; Hainfellner JA; Mason W; Mariani L; Bromberg JE; Hau P; Mirimanoff RO; Cairncross JG; Janzer RC; Stupp R, MGMT gene silencing and benefit from temozolomide in glioblastoma. The New England journal of medicine 2005, 352 (10), 997–1003. [DOI] [PubMed] [Google Scholar]
- 7.Lee SY, Temozolomide resistance in glioblastoma multiforme. Genes Dis 2016, 3 (3), 198–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gürten B; Yenigül E; Sezer A; Malta S, Complexation and enhancement of temozolomide solubility with cyclodextrins. Brazilian Journal of Pharmaceutical Sciences 2018, 54. [Google Scholar]
- 9.Serventi J; Behr J, Surgery and Evidence-based Treatments in Patients with Newly Diagnosed High-grade Glioma. Seminars in Oncology Nursing 2018, 34 (5), 443–453. [DOI] [PubMed] [Google Scholar]
- 10.Cardoso FL; Brites D; Brito MA, Looking at the blood–brain barrier: Molecular anatomy and possible investigation approaches. Brain Research Reviews 2010, 64 (2), 328–363. [DOI] [PubMed] [Google Scholar]
- 11.Chen Y; Liu L, Modern methods for delivery of drugs across the blood–brain barrier. Advanced drug delivery reviews 2012, 64 (7), 640–665. [DOI] [PubMed] [Google Scholar]
- 12.Kessler AF; Frömbling GE; Gross F; Hahn M; Dzokou W; Ernestus R-I; Löhr M; Hagemann C, Effects of tumor treating fields (TTFields) on glioblastoma cells are augmented by mitotic checkpoint inhibition. Cell Death Discov 2018, 4, 12–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kinzel A; Ambrogi M; Varshaver M; Kirson ED, Tumor Treating Fields for Glioblastoma Treatment: Patient Satisfaction and Compliance With the Second-Generation Optune(®) System. Clin Med Insights Oncol 2019, 13, 1179554918825449–1179554918825449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Friedman HS; Prados MD; Wen PY; Mikkelsen T; Schiff D; Abrey LE; Yung WK; Paleologos N; Nicholas MK; Jensen R; Vredenburgh J; Huang J; Zheng M; Cloughesy T, Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol 2009, 27 (28), 4733–40. [DOI] [PubMed] [Google Scholar]
- 15.Wiwatchaitawee K; Quarterman JC; Geary SM; Salem AK, Enhancement of Therapies for Glioblastoma (GBM) Using Nanoparticle-based Delivery Systems. AAPS PharmSciTech 2021, 22 (2), 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vella-zarb L; Baisch U; Dinnebier RE, Small Molecule, Big Difference: The Role of Water in the Crystallization of Paclitaxel. Journal of Pharmaceutical Sciences 2013, 102 (2), 674–683. [DOI] [PubMed] [Google Scholar]
- 17.Field JJ; Kanakkanthara A; Miller JH, Microtubule-targeting agents are clinically successful due to both mitotic and interphase impairment of microtubule function. Bioorganic & Medicinal Chemistry 2014, 22 (18), 5050–5059. [DOI] [PubMed] [Google Scholar]
- 18.Zhang DY; Dmello C; Chen L; Arrieta VA; Gonzalez-Buendia E; Kane JR; Magnusson LP; Baran A; James CD; Horbinski C; Carpentier A; Desseaux C; Canney M; Muzzio M; Stupp R; Sonabend AM, Ultrasound-mediated Delivery of Paclitaxel for Glioma: A Comparative Study of Distribution, Toxicity, and Efficacy of Albumin-bound Versus Cremophor Formulations. Clin Cancer Res 2020, 26 (2), 477–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang X; Zhang Q; Lv L; Fu J; Jiang Y; Xin H; Yao Q , Glioma and microenvironment dual targeted nanocarrier for improved antiglioblastoma efficacy. Drug Delivery 2017, 24 (1), 1401–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fellner S; Bauer B; Miller DS; Schaffrik M; Fankhänel M; Spruss T; Bernhardt G; Graeff C; Färber L; Gschaidmeier H; Buschauer A; Fricker G, Transport of paclitaxel (Taxol) across the blood-brain barrier in vitro and in vivo. J Clin Invest 2002, 110 (9), 1309–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nance E; Zhang C; Shih TY; Xu Q; Schuster BS; Hanes J, Brain-penetrating nanoparticles improve paclitaxel efficacy in malignant glioma following local administration. ACS Nano 2014, 8 (10), 10655–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fang J; Nakamura H; Maeda H, The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Advanced drug delivery reviews 2011, 63 (3), 136–151. [DOI] [PubMed] [Google Scholar]
- 23.Saraiva C; Praça C; Ferreira R; Santos T; Ferreira L; Bernardino L, Nanoparticle-mediated brain drug delivery: Overcoming blood–brain barrier to treat neurodegenerative diseases. Journal of Controlled Release 2016, 235, 34–47. [DOI] [PubMed] [Google Scholar]
- 24.He C; Hu Y; Yin L; Tang C; Yin C, Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials 2010, 31 (13), 3657–3666. [DOI] [PubMed] [Google Scholar]
- 25.Santos SD; Xavier M; Leite DM; Moreira DA; Custodio B; Torrado M; Castro R; Leiro V; Rodrigues J; Tomas H; Pego AP, PAMAM dendrimers: blood-brain barrier transport and neuronal uptake after focal brain ischemia. Journal of controlled release : official journal of the Controlled Release Society 2018, 291, 65–79. [DOI] [PubMed] [Google Scholar]
- 26.Jingyan L; Cristina S, PLA/PLGA nanoparticles for delivery of drugs across the blood-brain barrier. Nanotechnology Reviews 2013, 2 (3), 241–257. [Google Scholar]
- 27.Park T-E; Singh B; Li H; Lee J-Y; Kang S-K; Choi Y-J; Cho C-S, Enhanced BBB permeability of osmotically active poly(mannitol-co-PEI) modified with rabies virus glycoprotein via selective stimulation of caveolar endocytosis for RNAi therapeutics in Alzheimer’s disease. Biomaterials 2015, 38, 61–71. [DOI] [PubMed] [Google Scholar]
- 28.Pardridge WM, Blood-Brain Barrier and Delivery of Protein and Gene Therapeutics to Brain. Front Aging Neurosci 2019, 11, 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shi L; Tang GP; Gao SJ; Ma YX; Liu BH; Li Y; Zeng JM; Ng YK; Leong KW; Wang S, Repeated intrathecal administration of plasmid DNA complexed with polyethylene glycol-grafted polyethylenimine led to prolonged transgene expression in the spinal cord. Gene Therapy 2003, 10 (14), 1179–1188. [DOI] [PubMed] [Google Scholar]
- 30.Saqafi B; Rahbarizadeh F, Effect of PEI surface modification with PEG on cytotoxicity and transfection efficiency. Micro & Nano Letters 2018, 13 (8), 1090–1095. [Google Scholar]
- 31.Lombardo SM; Schneider M; Türeli AE; Günday Türeli N, Key for crossing the BBB with nanoparticles: the rational design. Beilstein J Nanotechnol 2020, 11, 866–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ceña V; Játiva P, Nanoparticle crossing of blood–brain barrier: a road to new therapeutic approaches to central nervous system diseases. Nanomedicine 2018, 13 (13), 1513–1516. [DOI] [PubMed] [Google Scholar]
- 33.Chitphet K; Geary SM; Chan CHF; Simons AL; Weiner GJ; Salem AK, Combining Doxorubicin-Loaded PEGylated Poly(Lactide-co-glycolide) Nanoparticles with Checkpoint Inhibition Safely Enhances Therapeutic Efficacy in a Melanoma Model. ACS Biomaterials Science & Engineering 2020, 6 (5), 2659–2667. [DOI] [PubMed] [Google Scholar]
- 34.Mitchell MJ; Billingsley MM; Haley RM; Wechsler ME; Peppas NA; Langer R, Engineering precision nanoparticles for drug delivery. Nat Rev Drug Discov 2021, 20 (2), 101–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhuang J; Fang RH; Zhang L, Preparation of particulate polymeric therapeutics for medical applications. Small Methods 2017, 1 (9), 1700147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Salatin S; Barar J; Barzegar-Jalali M; Adibkia K; Kiafar F; Jelvehgari M, Development of a nanoprecipitation method for the entrapment of a very water soluble drug into Eudragit RL nanoparticles. Res Pharm Sci 2017, 12 (1), 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kamaly N; Yameen B; Wu J; Farokhzad OC, Degradable Controlled-Release Polymers and Polymeric Nanoparticles: Mechanisms of Controlling Drug Release. Chem Rev 2016, 116 (4), 2602–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zielińska A; Carreiró F; Oliveira AM; Neves A; Pires B; Venkatesh DN; Durazzo A; Lucarini M; Eder P; Silva AM; Santini A; Souto EB, Polymeric Nanoparticles: Production, Characterization, Toxicology and Ecotoxicology. Molecules 2020, 25 (16), 3731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Seib FP; Jones AT; Duncan R, Comparison of the endocytic properties of linear and branched PEIs, and cationic PAMAM dendrimers in B16f10 melanoma cells. Journal of controlled release : official journal of the Controlled Release Society 2007, 117 (3), 291–300. [DOI] [PubMed] [Google Scholar]
- 40.Bunchongprasert K; Shao J, Impact of Media in Transport Study on Cell Monolayer Integrity and Permeability. Journal of Pharmaceutical Sciences 2020, 109 (2), 1145–1152. [DOI] [PubMed] [Google Scholar]
- 41.Hinkel S; Mattern K; Dietzel A; Reichl S; Müller-Goymann CC, Parametric investigation of static and dynamic cell culture conditions and their impact on hCMEC/D3 barrier properties. International Journal of Pharmaceutics 2019, 566, 434–444. [DOI] [PubMed] [Google Scholar]
- 42.Watson PMD; Paterson JC; Thom G; Ginman U; Lundquist S; Webster CI, Modelling the endothelial blood-CNS barriers: a method for the production of robust in vitro models of the rat blood-brain barrier and blood-spinal cord barrier. BMC Neurosci 2013, 14, 59–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ahmed KK; Geary SM; Salem AK, Surface engineering tumor cells with adjuvant-loaded particles for use as cancer vaccines. Journal of Controlled Release 2017, 248 (Supplement C), 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yildirimer L; Thanh NTK; Loizidou M; Seifalian AM, Toxicology and clinical potential of nanoparticles. Nano Today 2011, 6 (6), 585–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jallouli Y; Paillard A; Chang J; Sevin E; Betbeder D, Influence of surface charge and inner composition of porous nanoparticles to cross blood-brain barrier in vitro. Int J Pharm 2007, 344 (1–2), 103–9. [DOI] [PubMed] [Google Scholar]
- 46.Blanco E; Shen H; Ferrari M, Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol 2015, 33 (9), 941–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hauck TS; Ghazani AA; Chan WCW, Assessing the Effect of Surface Chemistry on Gold Nanorod Uptake, Toxicity, and Gene Expression in Mammalian Cells. 2008, 4 (1), 153–159. [DOI] [PubMed] [Google Scholar]
- 48.Komesli Y; Yildirim Y; Karasulu E, Visualisation of real-time oral biodistribution of fluorescent labeled self-microemulsifying drug delivery system of olmesartan medoxomil using optical imaging method. Drug Metabolism and Pharmacokinetics 2021, 36, 100365. [DOI] [PubMed] [Google Scholar]
- 49.Steele TW; Huang CL; Kumar S; Widjaja E; Chiang Boey FY; Loo JS; Venkatraman SS, High-throughput screening of PLGA thin films utilizing hydrophobic fluorescent dyes for hydrophobic drug compounds. J Pharm Sci 2011, 100 (10), 4317–29. [DOI] [PubMed] [Google Scholar]
- 50.Finke JH; Richter C; Gothsch T; Kwade A; Büttgenbach S; Müller-Goymann CC, Coumarin 6 as a fluorescent model drug: How to identify properties of lipid colloidal drug delivery systems via fluorescence spectroscopy? European Journal of Lipid Science and Technology 2014, 116 (9), 1234–1246. [Google Scholar]
- 51.Au - Gage GJ; Au - Kipke DR; Au - Shain W, Whole Animal Perfusion Fixation for Rodents. JoVE 2012, (65), e3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.E Hoyt R; V Hawkins J; B. St Clair M; J. Kennett M, Chapter 2 - Mouse Physiology. In The Mouse in Biomedical Research (Second Edition), Fox JG; Davisson MT; Quimby FW; Barthold SW; Newcomer CE; Smith AL, Eds. Academic Press: Burlington, 2007; pp 23–XVI. [Google Scholar]
- 53.Wiwatchaitawee K; Mekkawy AIAA; Quarterman JC; Naguib YW; Ebeid K; Geary SM; Salem AK, The MEK 1/2 inhibitor PD98059 exhibits synergistic anti-endometrial cancer activity with paclitaxel in vitro and enhanced tissue distribution in vivo when formulated into PAMAM-coated PLGA-PEG nanoparticles. Drug delivery and translational research 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang X-Q; Wang X-L; Huang S-W; Zhuo R-X; Liu Z-L; Mao H-Q; Leong KW, In Vitro Gene Delivery Using Polyamidoamine Dendrimers with a Trimesyl Core. Biomacromolecules 2005, 6 (1), 341–350. [DOI] [PubMed] [Google Scholar]
- 55.Freeman EC; Weiland LM; Meng WS, Modeling the proton sponge hypothesis: examining proton sponge effectiveness for enhancing intracellular gene delivery through multiscale modeling. J Biomater Sci Polym Ed 2013, 24 (4), 398–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Benjaminsen RV; Mattebjerg MA; Henriksen JR; Moghimi SM; Andresen TL, The possible “proton sponge “ effect of polyethylenimine (PEI) does not include change in lysosomal pH. Mol Ther 2013, 21 (1), 149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Donahue ND; Acar H; Wilhelm S, Concepts of nanoparticle cellular uptake, intracellular trafficking, and kinetics in nanomedicine. Advanced Drug Delivery Reviews 2019, 143, 68–96. [DOI] [PubMed] [Google Scholar]
- 58.Patel MM; Patel BM, Crossing the Blood–Brain Barrier: Recent Advances in Drug Delivery to the Brain. CNS Drugs 2017, 31 (2), 109–133. [DOI] [PubMed] [Google Scholar]
- 59.Srinageshwar B; Dils A; Sturgis J; Wedster A; Kathirvelu B; Baiyasi S; Swanson D; Sharma A; Dunbar GL; Rossignol J, Surface-Modified G4 PAMAM Dendrimers Cross the Blood–Brain Barrier Following Multiple Tail-Vein Injections in C57BL/6J Mice. ACS Chemical Neuroscience 2019, 10 (9), 4145–4150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Luzio JP; Pryor PR; Bright NA, Lysosomes: fusion and function. Nature Reviews Molecular Cell Biology 2007, 8 (8), 622–632. [DOI] [PubMed] [Google Scholar]
- 61.Hervé F; Ghinea N; Scherrmann J-M, CNS delivery via adsorptive transcytosis. AAPS J 2008, 10 (3), 455–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Preston JE; Joan Abbott N; Begley DJ, Chapter Five - Transcytosis of Macromolecules at the Blood–Brain Barrier. In Advances in Pharmacology, Davis TP ., Ed. Academic Press: 2014; Vol. 71, pp 147–163. [DOI] [PubMed] [Google Scholar]
- 63.Alberts B; Wilson JH; Hunt T, Molecular biology of the cell. 5th ed.; Garland Science: New York, 2008; p xxxiii, 1601, 90 p. [Google Scholar]
- 64.Mosquera J; García I; Liz-Marzán LM, Cellular Uptake of Nanoparticles versus Small Molecules: A Matter of Size. Accounts of Chemical Research 2018, 51 (9), 2305–2313. [DOI] [PubMed] [Google Scholar]
- 65.Sakulkhu U; Maurizi L; Mahmoudi M; Motazacker M; Vries M; Gramoun A; Ollivier Beuzelin MG; Vallée JP; Rezaee F; Hofmann H, Ex situ evaluation of the composition of protein corona of intravenously injected superparamagnetic nanoparticles in rats. Nanoscale 2014, 6 (19), 11439–50. [DOI] [PubMed] [Google Scholar]
- 66.Bai X; Wang J; Mu Q; Su G, In vivo Protein Corona Formation: Characterizations, Effects on Engineered Nanoparticles’ Biobehaviors, and Applications. Frontiers in Bioengineering and Biotechnology 2021, 9 (263). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Raoufi M; Hajipour MJ; Kamali Shahri SM; Schoen I; Linn U; Mahmoudi M, Probing fibronectin conformation on a protein corona layer around nanoparticles. Nanoscale 2018, 10 (3), 1228–1233. [DOI] [PubMed] [Google Scholar]
- 68.Jain GK; Pathan SA; Akhter S; Ahmad N; Jain N; Talegaonkar S; Khar RK; Ahmad FJ, Mechanistic study of hydrolytic erosion and drug release behaviour of PLGA nanoparticles: Influence of chitosan. Polymer Degradation and Stability 2010, 95 (12), 2360–2366. [Google Scholar]
- 69.Fredenberg S; Wahlgren M; Reslow M; Axelsson A, The mechanisms of drug release in poly(lactic-co-glycolic acid)-based drug delivery systems—A review. International Journal of Pharmaceutics 2011, 415 (1), 34–52. [DOI] [PubMed] [Google Scholar]
- 70.Yoo J; Won Y-Y, Phenomenology of the Initial Burst Release of Drugs from PLGA Microparticles. ACS Biomaterials Science & Engineering 2020, 6 (11), 6053–6062. [DOI] [PubMed] [Google Scholar]
- 71.Hines DJ; Kaplan DL , Poly(lactic-co-glycolic) acid-controlled-release systems: experimental and modeling insights. Crit Rev Ther Drug Carrier Syst 2013, 30 (3), 257–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ebeid K; Meng X; Thiel KW; Do AV; Geary SM; Morris AS; Pham EL; Wongrakpanich A; Chhonker YS; Murry DJ; Leslie KK; Salem AK, Synthetically lethal nanoparticles for treatment of endometrial cancer. Nat Nanotechnol 2018, 13 (1), 72–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Srinivasan RC; Kannisto K; Strom SC; Gramignoli R, Evaluation of different routes of administration and biodistribution of human amnion epithelial cells in mice. Cytotherapy 2019, 21 (1), 113–124. [DOI] [PubMed] [Google Scholar]
- 74.Hong W; Hu R; Huang X; Lu X; Czech T; Tang J, In vivo pharmacokinetics and biodistribution of novel all-trans retinoic acid derivative-loaded, folate-modified poly (l-amino acid) micelles. Journal of Drug Delivery Science and Technology 2017, 41, 436–443. [Google Scholar]
- 75.Zhang XQ; Intra J; Salem AK, Conjugation of polyamidoamine dendrimers on biodegradable microparticles for nonviral gene delivery. Bioconjug Chem 2007, 18 (6), 2068–76. [DOI] [PubMed] [Google Scholar]
- 76.Boyd NH; Walker K; Fried J; Hackney JR; McDonald PC; Benavides GA; Spina R; Audia A; Scott SE; Libby CJ; Tran AN; Bevensee MO; Griguer C; Nozell S; Gillespie GY; Nabors B; Bhat KP; Bar EE; Darley-Usmar V; Xu B; Gordon E; Cooper SJ; Dedhar S; Hjelmeland AB, Addition of carbonic anhydrase 9 inhibitor SLC-0111 to temozolomide treatment delays glioblastoma growth in vivo. JCI Insight 2017, 2 (24). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Poole EI; McGavin JJ; Cochkanoff NL; Crosby KM, Stereotaxic surgery for implantation of guide cannulas for microinjection into the dorsomedial hypothalamus in young rats. MethodsX 2019, 6, 1652–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.