

Abstract
Pyramidal cells and GABAergic interneurons fire together in balanced cortical networks. In contrast to this general rule, we describe a distinct neuron type in mice and rats whose spiking activity is anti-correlated with all principal cells and interneurons in all brain states but, most prevalently, during the down state of non-REM (NREM) sleep. We identify these down state-active (DSA) neurons as deep-layer neocortical neurogliaform cells that express ID2 and Nkx2.1 and are weakly immunoreactive to neuronal nitric oxide synthase. DSA neurons are weakly excited by deep-layer pyramidal cells and strongly inhibited by several other GABAergic cell types. Spiking of DSA neurons modified the sequential firing order of other neurons at down–up transitions. Optogenetic activation of ID2+Nkx2.1+ interneurons in the posterior parietal cortex during NREM sleep, but not during waking, interfered with consolidation of cue discrimination memory. Despite their sparsity, DSA neurons perform critical physiological functions.
The brain often ‘reboots’ itself during sleep from transient silence. The silent epochs, known as delta waves or down states, are interleaved among spiking activity-rich up states during NREM sleep, and the toggling between these states is known as cortical slow oscillation1–6. NREM, and particularly slow oscillations, have been implicated in the development of cortical circuits, homeostatic regulation of network dynamics and memory consolidation5–10.
Neuronal nitric oxide synthase (nNOS)-expressing neurons show elevated activity during sleep, as measured by the increased expression of the inmediate early gene cFos11–13, and might play a special role in controlling slow oscillations. Deep-layer neurons of the cortex3,4,14, a variety of somato-dendritic regenerative currents in cortical pyramidal cells and the excitatory recurrents among neurons have been postulated to be the source of stochastic metastability and persistent up state activity15. In turn, spike after-hyperpolarization and synaptic depression, combined with decreasing excitation–inhibition balance, have been suggested to be responsible for switching from up to down state3. The mechanism of the down state is less understood. In addition to the absence of spikes, synaptic activity is also reduced during the down cortical state, leaving both principal cells and inhibitory interneurons transiently in a dis-excited and dis-inhibited state. During the down–up transition, pyramidal neurons become active rapidly and sequentially10. Although some pyramidal neurons occasionally emit action potentials during the down state8, it is a generally held view that no cortical neuron fires selectively during the down state.
Against this currently held scenario, we describe a small set of deep-layer neurons in three neocortical areas of mice and rats whose spiking activity is inversely correlated with all principal cells and all other types of interneurons in both waking and sleeping states, with the highest firing rate displayed during the down state. We identify down state-active (DSA) neurons as ID2+Nkx2.1+ (ID2/Nkx2.1) neurogliaform interneurons.
Results
In the first set of experiments, we used multiple-shank silicon probes to record local field potential (LFP) and spiking activity in the posterior parietal cortex (PPC) of 11 mice (Fig. 1a–c). The depth distribution of LFPs, current sinks and sources and unit firing were used to identify cortical layers16. The most prominent landmark was the largest power in the high-frequency band (high-pass filtered >120 Hz), defining mid-layer 5. A current source in layer 5 identified the down state of slow oscillation (Fig. 1c and ref. 16). Single units were separated into putative excitatory and inhibitory classes (n = 1,457 and n = 822, respectively) using a multistep approach16 (Extended Data Fig. 1a and Methods).
Fig. 1 |. Identification and physiological properties of DSA neurons in the PPC.

a, histological verification of probe shank locations (dashed white lines) in PPC (left). Immunoreactivity for VGLUT2 delineates layers (reverse contrast) on a coronal section of the PPC (right). b, LFP depth profile (gray) recorded with one of the shanks in PPC during a 6-s slow-wave sleep epoch and a 4-s waking epoch during waking quiescence (QWake) and walking, with simultaneously recorded spikes of isolated units (each dot represents a single spike; red, pyramidal cells; blue, interneurons; black, a single DSA neuron). Light-gray LFP segments correspond to identified down states. c, Average CSD map and superimposed LFP of all the detected down states from the recording session illustrated in b. Depth distribution of high-frequency (>120-hz) spectral power identifies layer 5. Vertical line, layer 5 source peak. d, Peri-down state Z-scored firing raster plot for all putative principal cells (PYR, left), putative interneurons (INT, bottom middle) and DSA neurons (top middle), as ranked according to their magnitude of event response (right). Dashed lines delimit the ±50-ms window used to estimate the unit responses. Note that DSA neurons increase their rate selectively during the down state. e, As in d but for the average spike CCG between a reference neuron and all other simultaneously recorded units in NREM sleep (left) and quiet waking (QWake, right). Dashed lines delimit the ± 35-ms window used to quantify neuron CCG responses (right). f, Distributions of down state/up state firing rate ratio for putative pyramidal cells (PYR, red), interneurons (INT, blue) and DSA neurons (gray) (P < 10−80, KW test). g, Magnitude of down state response as a function of the CCG response. Gray dots show 47 units (out of 2,279) defined as DSA neurons. Marginal CCG (top; P < 10−34, KW test) and down state response (right; P < 10−41, KW test) distributions for pyramidal cells, interneurons and DSA neurons (top) in each group (right). h, Top: average firing auto-correlogram (mean ± 95% confidence interval (IC95)) and median burst index (medians, interquartile ranges, maxima and minima; P < 10−19, KW test) for each group. Bottom: average spike waveform (mean ± IC95) and through-to-peak spike duration (spk dur; P < 10−306, KW test) for each group (n = 1,457 pyramidal cells, 775 interneurons and 47 DSA neurons from 11 mice). i, Average high-frequency (>120-Hz) spectral power depth profile (mean ± IC95) was used to align all sessions, relative to mid-layer 5 as reference. Depth distribution for all units (right; dots are scattered in the x axis to allow better visibility). DSA neurons are located in the deep cortical layers (P < 10−15, KW test). ***P < 0.001.
Physiological characterization of DSA neurons.
Down states were identified using LFP and spiking criteria (Fig. 1b and Methods). As expected, firing rates of most putative pyramidal neurons across all layers were suppressed during down states (Fig. 1d and Extended Data Fig. 1b; P < 10−237 between up and down states; Wilcoxon paired signed-rank test). Similarly, cells classified as putative interneurons were also mostly silenced (P < 10−128). However, we also identified a small subset of neurons (n = 47 in PPC) that were specifically and dominantly active during the down state of NREM (DSA neurons; Fig. 1d; 5.60 ± 1.54 Hz during down, 2.17 ± 1.42 Hz during up; P < 10−9; Wilcoxon paired signed-rank test). Average cross-correlograms (CCG) between DSA neurons and all principal cells and all interneurons showed an inverse relationship during both NREM and waking states (Fig. 1e; for REM sleep and walk, see Extended Data Fig. 1c). The firing rate ratio between down and up states effectively segregated DSA neurons from other neurons (Fig. 1f and Extended Data Fig. 1b). Similar segregation was obtained by plotting the relationship between spikes of DSA neurons and down state epochs against cross-correlation features with other neurons (Fig. 1g and Extended Data Fig. 1d). DSA neurons had lower burst indices (defined as fraction of spikes with ≤6-ms window to all spikes), wider auto-correlograms and waveforms than other interneurons and pyramidal cells, and had longer duration waveforms than other interneurons (Fig. 1h). All DSA neurons were found in deep cortical layers (Fig. 1i).
In a second set of experiments, DSA neurons with similar features were also identified in the visual cortex (n = 6 DSA neurons of 1,326 neurons in 19 mice; Extended Data Fig. 1e and ref. 16) and prefrontal cortex in rats (n = 2 DSA neurons of 1,121 neurons; Extended Data Fig. 1e and ref. 17).
Local circuit control of DSA neurons by pyramidal cells and interneurons.
The population firing rates of pyramidal cells and interneurons varied moderately across behavioral states (PYR: 8.46% average change across all states, P < 10−41; INT: 12.08%, P < 10−75; Friedman test). In contrast, DSA neurons were most active during NREM sleep and quiet waking, coinciding with transiently reduced firing of the remaining neuronal population, and their rate decreased more than four-fold during walking and REM sleep (Fig. 2a and Extended Data Fig. 2a; 22% change across all states, P < 10−7; Friedman test). DSA neurons were negatively correlated with other neurons during their high firing rate states (NREM and quiet wakefulness), whereas population cross-correlations between pyramidal neurons and interneurons were positive and larger during NREM sleep (Fig. 2b, Extended Data Fig. 2b,c and ref. 18).
Fig. 2 |. Network properties of the DSA neurons.

a, Log distribution of average firing rates (top, P < 10−191, KW test) and median firing rates (circles; lines, interquartile ranges) of pyramidal cells, interneurons (n = 1,457, 775 and 47 cells, respectively, from 11 mice) and DSA neurons during waking quiescence (QWake), NREM sleep, rEM sleep and active behavior (Walk) (bottom). b, Average unit CCGs as a function of brain states. Top insets: mean CCG (mean ± IC95) for all groups across brain states. c, Average joint Z-score rate density among pyramidal cells, interneurons and DSA neurons. Top: firing rates of DSA neurons are more strongly anticorrelated with interneuron rates than with firing rates of pyramidal cells (mean ± IC95; n = 32 session with at least one DSA neuron, P < 10−18, F1,564 = 78.75, repeated-measures ANOVA and Spearman correlation (ρ) tested using a Student’s t-distribution). d, Average (mean ± IC95) partial correlation values of the Z-scored average rate for all groups (blue for ρDSA,INT controlling for PYR; red for ρDSA,PYr controlling for INT; magenta for ρPYr,INT controlling for DSA, n = 32 sessions, P < 10−27, F2,2787 = 61.23, repeated-measures ANOVA). e, Example cross-correlation histogram of a putative monosynaptic pyramidal cell–interneuron pair (top; red, pyramidal cells; blue, interneurons) and a pyramidal–DSA neuron pair (bottom; gray star, DSA neuron). Insets show autocorrelogram for each pair member (±20 ms). Green bins depict 1–3-ms monosynaptic window. f, Z-scored CCGs for all identified monosynaptic pairs among PYR–DSA, PYR–INT and PYR–PYR. Bottom, average spike transmission probability (trans prob, mean ± IC95) for each group. g, Group difference (medians, interquartile ranges, maxima and minima) of spike transmission probability for all detected putative monosynaptic pairs in f (n = 149, 957 and 17 for PYR–PYR, PYR–INT and PYR–DSA, respectively; P < 10−6, KW test). h, Top: down state responses (left) and average CCG (right) for all simultaneously recorded units in a representative session with two DSA neurons (arrowhead). Bottom: Z-scored within-cell type CCGs (mean ± IC95, all cell pairs from the same group were recorded simultaneously: n = 82,972 PYR–PYR; n = 17,392 INT–INT; and n = 18 DSA neuron–DSA neuron pairs, P < 10−292, KW test). *P < 0.05, **P < 0.01 and ***P < 0.001.
To gain insight on the nature of interactions of DSA neurons with the rest of the PPC population, we constructed joint co-activity histograms of pyramidal cells and putative interneurons with DSA neurons (Fig. 2c and Extended Data Fig. 2d). The firing rates of DSA neurons were more negatively correlated with the activity of interneurons (Fig. 2c, y axis and blue trace in top panel) than with those of pyramidal cells (x axis, red trace), suggesting that DSA neurons are inhibited by GABAergic cells. Moreover, partial correlation of the firing rates between DSA neurons and interneurons was more negative than the partial correlation of the firing rate between DSA neurons and pyramidal cells for all time bins tested (Fig. 2d), suggesting that DSA neurons are disinhibited during down states. Firing rate differences between pyramidal cells and interneurons could not explain these effects (Extended Data Fig. 2e). In contrast, firing rates of pyramidal neurons and interneurons were positively correlated from 10-ms to 1-s time bins (Fig. 2d). To further support these findings, we quantified putative monosynaptic connections and spike transmission probabilities between pairs of neurons16,19. As expected, we found stronger spike transmission between pyramidal cells and interneurons compared to pyramidal–pyramidal pairs (Fig. 2e-g, Extended Data Fig. 2f and ref. 16). Spike transmission between pyramidal–DSA neuron pairs was weakest (Fig. 2f,g) and less probable than between pyramidal cell–interneuron pairs (Fig. 2f and Extended Data Fig. 2f; P < 10−190, Fisher exact test), supporting the population co-activity results. Finally, DSA neurons were strongly co-active with other DSA neurons, and this synchrony was stronger than observed within interneuron and pyramidal neuron populations, respectively (Fig. 2h).
To account for the unusual firing patterns of DSA neurons, we adopted a model of slow oscillations15 to provide a proof of principle regarding how the observed spike correlations among pyramidal cells, interneurons and DSA neurons might be related to their physiological connectivity (Extended Data Fig. 3). In line with our experimental observations, the model predicted that DSA neurons are strongly inhibited by other interneurons and weakly excited by nearby deep-layer pyramidal cells.
DSA neurons comprise deep-layer ID2/Nkx2.1 interneurons.
In the search for the cellular identity of DSA neurons as well as the interneuron types that might inhibit them, we optogenetically tested four promoter-specific transgenic mouse lines expressing light-sensitive opsins (ChR or CatCh) in GABAergic subpopulations (Fig. 3a–f), including nNOS-expressing neurons11. None of the DSA neurons recorded in these mice were somatostatin (Sst)-expressing (n = 0/7 units from two Sst:ChR mice) or parvalbumin (PV)-expressing (n = 0/5 V1 units from four PV::ChR mice16) interneurons, which together account for ~90% of the infragranular layer inhibitory neurons in mice20. Two of 25 light-responding nNOS-expressing cells (from four Nos1::ChR mice) were DSA neurons (Fig. 3d) and were located below layer 5 (Extended Data Fig. 4d). Based on this clue, we searched further by screening lines that marked sparse subpopulations of nNOS-expressing interneurons preferentially in deep cortical layers. A small subpopulation of GABAergic neurons defined by Lamp5 and Lhx6 co-expression are found in both Nos1-CreERT2 and Nkx2–1-CreERT2 driver lines, with most neurons located in deep cortical layers21,22. By cross-breeding three mouse lines (ID2-CreER, Nkx2.1-Flpo and the Cre-Flpo-dependent reporter line Ai80), we created an intersectional line expressing the channelrhodopsin CatCh in neurons expressing ID2 and Nkx2.1, corresponding to Lamp5 and Lhx6, respectively23. We found that most of the recorded DSA neurons (66%, six of nine DSA neurons from five ID2/Nkx2.1 mice) were of the light-responding ID2/Nkx2.1 type (Fig. 3e,f; 2.98 ± 1.41 Hz baseline; 13.13 ± 1.73 Hz stimulation; P < 10−3 for all units, Wilcoxon paired signed-rank test). Conversely, six of eight light-responding ID2/Nkx2.1 neurons (75%) were DSA cells (Extended Data Fig. 4e).
Fig. 3 |. cell-type identity of DSA neurons and their inhibitory inputs.

a, Schematic of the opto-tagging experiments. Combined light fiber-recording probes were implanted in promoter-specific transgenic mice expressing excitatory (Chr2) opsins. b, Peristimulus histogram (PSTh) for all cells recorded in the Sst::ChR animals (n = 2 mice). Each row is a rate-normalized color-coded PSTH of a unit. Top middle: 21% of the the interneurons were classified as Sst::ChR-expressing cells. Bar graph (bottom right), probability (prob) of optogenetically di(poly) synaptically silenced neurons (mod, from modulated). c, Same as in b for PV::Chr mice (n = 4 mice in V1). d, Same as in b for Nos1::Chr mice (n = 3 mice). e, Same as in b for the ID2/Nkx2.1::CatCh mice (n = 6). f, Average optogenetic response for all recorded DSA neurons in the different promoter-specific transgenic mouse lines. Most of the DSA neurons were suppressed (−Mod) upon Sst:Chr and PV::Chr stimulation (P < 0.01 for all units, Wilcoxon paired signed-rank test). Most DSA neurons were recruited with light pulses in the ID2/Nkx2.1::CatCh line. g, Spike autocorrelogram (left) and spike waveform (right) from ID2/Nkx2.1-expressing DSA neurons are similar to those of DSA neurons observed in other mice (mean ± IC95 in black; P = 0.38 and P = 0.49, respectively, KW test).
Optogenetic stimulation of Sst- and PV-expressing neurons suppressed firing rates of most DSA neurons (Fig. 3f; Sst stimulation: 6/7 DSA units decreased; PV stimulation: 4/5 DSA units decreased significantly), in addition to inhibiting other interneurons and pyramidal cells (Fig. 3b,c; Sst: 67% of the pyramidal cells and 46% of the interneurons decreased; PV: 81% of the pyramidal cells and 48% of the interneurons decreased). Upon release from optogenetically induced suppression by PV and Sst interneurons, DSA neurons responded with excess spiking lasting 70–200 ms (Extended Data Fig. 4a,b). In Nos1::ChR mice, in addition to the two light-excited DSA neurons, optogenetic stimulation of nNOS-expressing neurons signicantly suppressed firing of ten of 25 DSA units (Fig. 3d,f). The same light intensity in ID2/Nkx2.1::Ai80 mice was less effective in suppressing other interneurons and pyramidal cells (Fig. 3e; 9.7% of the pyramidal cells and 24% of the interneurons), possibly due to sparseness of the light-stimulated ID2/Nkx2.1 neurons. The auto-correlograms and spike waveforms of opto-tagged ID2/Nkx2.1 neurons were indistinguishable from those of other DSA neurons (Fig. 3g and Extended Data Fig. 4c), indicating that all or most of DSA neurons likely belong to the ID2/Nkx2.1 type.
In vitro electrophysiological characterization of ID2/Nkx2.1 neurons (n = 22 tdTomato+ cells in seven ID2/Nkx2.1::Ai65 mice) identified them as ‘late-spiking’ neurons, with >0.6 s of delay to the first spike at near-threshold current steps, large spike after-hyperpolarization (15–20 mV) and regular, non-adapting spikes in response to supra-threshold current steps (Fig. 4a,b). Because these electrophysiological features of ID2/Nkx2.1 neurons are similar to neurogliaform cells24–28, we compared six measured parameters with those of layer 1 neurogliaform cells26. Although several parameters of the two groups differed quantitatively from each other (Fig. 4b), the delayed ‘late-spiking’ nature of both groups classifies deep-layer ID2/Nkx2.1 neurons as neurogliaform cells26,29. Only one other neuron type, the delayed fast-spiking cell, shows late spiking, but its other features differ substantially from both deep-layer ID2/Nkx2.1 and layer 1 neurogliaform cells (Extended Data Fig. 5a and ref. 30). ID2/Nkx2.1 neurons (n = 11) did not show hyperpolarization-induced rebound spikes (Extended Data Fig. 5b).
Fig. 4 |. Intrinsic electrophysiological features of L5/6 ID2/Nkx2.1 neurons.

a, Example voltage traces for a L5/6 ID2/Nkx2.1 cell (gray) and a L1 neurogliaform cell (L1 NGFC, from ref. 26, green) during supra-threshold (top), just supra-threshold, just sub-threshold (middle) and hyperpolarizing (bottom) current injections (currents shown in black). b, Six electrophysiological parameters for both L5/6 Id2;Nkx2.1 (gray) and L1 neurogliaform (green) cells. All plots show individual cells as circles (time to first spike: n = 22 and n = 27 for ID2/Nkx2.1 and L1 NGFC, respectively, P = 0.004; adaptation: n = 20 and n = 26, P = 0.001; spike width: n = 22 and n = 26, P < 10−6; sag amplitude: n = 20 and n = 22, P = 0.48; input resistance: n = 22 and n = 25, P = 0.002; resting potential: n = 18 and n = 27, P < 10−6; two-sided Wilcoxon rank-sum test). Mean values as bars, and s.e.m. values as small bars on the right. **P < 0.01 and ***P < 0.001.
Anatomical features of DSA ID2/Nkx2.1 interneurons.
Our physiological and optogenetic findings indicated that DSA neurons resided mainly below layer 5, and they were weakly excited by local pyramidal cells and strongly inhibited by Sst- and PV-expressing interneurons, compared to other neurons. Commensurate with these results, we found that the layer distribution of DSA neurons in the PPC (Fig. 1i) matched that of tdTomato-expressing ID2/Nkx2.1 interneurons (n = 54 neurons, three mice), with most neurons residing in layer 6 (~66%, Fig. 5a; P < 10−4, across-layer comparion, Kruskal–Wallis (KW) test), similar to the distribution of Lamp5–Lhx6 neurons in other areas of the mouse cortex (Extended Data Fig. 6a,b)21,22. Most (~70%) of tdTomato-expressing neurons were weakly immunoreactive for nNOS (0.33 ± 0.20 relative nNOS intensity for n = 12 tdTomato+/nNOS+ cells; 0.85 ± 0.09 for n = 9 Sst+/nNOS+ cells; P < 10−8, KW test; Fig 5b), mainly in deep layers (Fig. 5b,c; 83% in layer 6; P < 10−31, Fisher exact test; n = 3 ID2/Nkx2.1 mice), and lacked immunoreactivity for Sst and PV (n = 0/54 for both PV and Sst; Fig. 5b and Extended Data Fig. 6c), and most of them lacked neuropeptide Y (n = 11/64 immunopositive neurons; Extended Data Fig. 6e) and calretinin (n = 2/54 immunopositive neurons; Extended Data Fig. 6f). nNOS+ deep-layer tdTomato-expressing interneurons (118 cells from three ID2/Nkx2.1::Ai65 mice) had small somata, compact dendritic fields and dense local axon arborizations (inter-varicosities median distance average = 1.5 ± 0.14 μm, n = 2 neurons; Fig. 5c,d and Extended Data Fig. 6d). These features are similar to those described for neurogliaform cells in layers 2 and 3 (refs. 24,31), layer 1 (ref. 26) and layer 6 (ref. 23). We also observed PV-immunoreactive puncta in close apposition to the somata of ID2/Nkx2.1 cells, supporting the physiology-based connectivity measures (Extended Data Fig. 6c). In superficial layers, ID2/Nkx2.1 neurons immunonegative for nNOS (58% from all tdTomato+ cells in layers 1 and 2/3) were axo-axonic cells and other kinds of neurons, including PV+ cells with basket-like axon endings (Extended Data Fig. 6g,h and ref. 21), indicating that they are distinct from deep-layer ID2/Nkx2.1 cells.
Fig. 5 |. Anatomical features of ID2/Nkx2.1 interneurons in the PPc.

a, Left: sparse distribution of tdTomato+ somata in the PPC of an ID2/Nkx2.1::Ai65 mouse (70-μm-thick coronal section, low-power wide-field epifluorescence image, reverse contrast). Two of them are marked by arrowheads. Top right: depth distribution of ID2/Nkx2.1 neurons (mean ± s.e.). Gray shaded area on the top graph shows depth distribution of DSA (same as Fig. 1i, DSA). Note that most tdTomato+ neurons were found below layer 5 (overlaid as blue dots; three sections per mouse for three mice, nine samples in total) and were immunoreactive for nNOS (bottom). b, Two tdTomato+ ID2/Nkx2.1 neurons in L6, weakly immunoreactive for nNOS (white arrowheads; n = 54 tdTomato+ neurons from three mice were tested). Note also a strongly nNOS-immunopositive neuron (hollow arrowhead), which is immunoreactive for both calretinin (Cr) and Sst but does not express tdTomato (20.8-μm-thick confocal maximum-intensity Z-projection; n = 12 tdTomato+nNos+ cells). c, Axon distribution in deep cortical layers (wide-field epifluorescence, reverse contrast) from three tdTomato+ ID2/Nkx2.1 neurons. Note higher axon density in L6. right: detail of L6b. Two somata are indicated by asterisks. d, reconstruction of soma, dendrites and axonal arborization of an intracellularly filled tdTomato+ lD2/Nkx2.1 neuron (from a 183.3-μm-thick confocal stack). Inset: detail of the axonal varicosities; three additional filled tdTomato+ lD2/Nkx2.1 cells, one of them reconstructed, in Extended Data Fig. 5d. wm, white matter.
Targets of DSA ID2/Nkx2.1 interneurons.
An important question that we addressed next is the effect of DSA neurons on their targets. We identified pyramidal cells and interneurons as potential targets by their significantly decreased firing rates in response to optogenetic stimulation of ID2/Nkx2.1 interneurons (Fig. 4e; negatively modulated or −Mod; Methods). Those whose rates did not change were designated as non-modulated (Non-Mod). Both −Mod pyramidal cells and −Mod interneurons were located, on average, deeper in the cortex (below layer 5) than the Non-Mod pyramidal cells and interneurons (Fig. 6a). Both −Mod pyramidal cells and −Mod interneurons fired later during the down–up transition than Non-Mod cells (Fig. 6b and Extended Data Fig. 7a), with minor effects on the response magnitude (Fig. 6b).
Fig. 6 |. Effector role of DSA neurons.

a, Depth distribution of pyramidal cells (red, right abscissa) and interneurons (blue, left), separated into groups of negatively modulated cells (−Mod, light red and light blue) or non-modulated neurons (Non-Mod, dark colors) upon optogenetic stimulation of ID2/Nkx2.1::CatCh neurons (PYR: n = 44 and n = 410, respectively, P < 10−6; INT: n = 74 and n = 238, respectively, P < 10−15; KW test). ID2/Nkx2.1::CatCh modulated cells are located in deep layers. b, Temporal dynamics of the Z-scored down–up events responses for −Mod and Non-Mod pyramidal cells and interneurons (top left for PYR, bottom left for INT; mean ± IC95). right: timing (P < 10−18, KW test) and amplitude (P < 10−5, KW test) response group differences, respectively (medians, interquartile ranges, maxima and minima). c, Down–up events were classified as ‘low DSA rate events’ or ‘high DSA rate events’ using the average DSA neuron rate during the down epoch of the down–up event as the separatrix (top: medians, interquartile ranges, maxima and minima; bottom, mean ± IC95; 32 sessions with at least one DSA neuron from 11 mice, P < 10−7, Wilcoxon paired signed-rank test). d, Down–up temporal group response (top; medians, interquartile ranges, maxima and minima, P < 10−12, KW test) and high–low DSA neuron firing events difference distributions (bottom; P < 10−7, KW test) for −Mod (yellow, n = 51 units from nine session with at least one DSA neuron from six ID2/Nkx2.1::CatCh mice) and Non-Mod (green, n = 195 units) groups. e, Illustration of mean rank order values for ‘high’ and ‘low’ rate events, used as templates against remaining down–up events (n, n + 1,….). f, Top: Z-scored sequential activity example of a simultaneously recorded population during down–up events with low DSA rate (session mean template), vertically arranged by latency (two DSA neurons in the bottom two rows). Bottom: similar session mean template for high DSA rate events, but arranged according to low DSA rate classification. g, rank correlation values within high and low DSA events, high versus low events, randomly chosen events and slow oscillation events in the first versus the second halves of NREM sleep episodes (mean ± s.d.; n = 32 sessions with at least one DSA neuron from 11 mice; P < 10−20, Friedman test; P < 10−7 and P < 10−8 for ‘within-low’ against ‘high versus low’ and ‘within-high’ against ‘high versus low’, respectively, Tukey test). *P < 0.05, **P < 0.01 and ***P < 0.001.
The above analysis suggested that activity level of DSA neurons in the down state can delay the timing of their targets during the emerging up state and, thus, their firing order at the down–up transition32,33. To test this hypothesis, we classified each slow oscillation event (down–up pairs) based on the level of DSA neuronal activity. In each sleep session, we calculated the mean firing rate of DSA neurons and classified each down state as ‘low’ (below session mean) or ‘high’ (above mean) contribution of DSA spiking (Fig. 6c). With such separation, we re-investigated the relationship between −Mod and Non-Mod neurons (pyramidal cells and interneurons combined). We found that, when DSA neurons were active, the recruitment of the −Mod neurons was significantly delayed during down–up transitions, whereas no such difference was present for the Non–Mod neurons (Fig. 6d and Extended Data Fig. 7b), supporting the hypothesis that activity of the DSA neurons affects spike timing of their putative targets. In further support of this view, high down–up events transitioned slightly, but significantly, slower than low events (20–80% rise time (mean ± s.d.): 0.10 ± 0.08 and 0.11 ± 0.08, for low and high events, respectively; P = 0.0036, Wilcoxon paired signed-rank test; Extended Data Fig. 7c). Conversely, delta power peak frequency was significantly lower during optogenetically stimulated NREM epochs than during non-stimulated ones (1.70 ± 0.32 and 1.96 ± 0.18, respectively; P = 0.0144, Wilcoxon paired signed-rank test; Extended Data Fig. 7d).
In addition, we also computed the session mean sequence rank order of all neurons for the down–up events separately for the high and low groups. In turn, these low DSA and high DSA session sequence templates were correlated with every individual slow oscillation event (Fig. 6e; the tested event was not used for the template generation). We found that, when the high session mean DSA template was correlated with high individual DSA events, or when the low session mean DSA template was correlated with low DSA events, the correlation coefficients were significantly higher than comparisons across group events (high template with low DSA events and low template with high DSA events; Fig. 6f,g and Extended Data Fig. 7e). As further controls, within-group (high–high and low–low) rank order correlations were also higher than the correlation of rank orders between events of the first and second halves of sleep sessions (irrespective of DSA activity level) or the correlations between randomly shuffled events (average of 100 shufflings). In summary, DSA neurons can modify the firing order sequences of their target neurons in down–up transitions.
Behavioral effects of DSA ID2/Nkx2.1 interneurons.
Our second approach to examine the effect of DSA neurons on their targets was based on a behavioral readout. Recent imaging work has demonstrated the importance of the PPC in visual pattern discrimination and memory-guided behavior34, and we adopted that paradigm to test the potential role of DSA neurons in the consolidation of cue discrimination memory. Five mice, implanted bilaterally with optic fibers in deep layers of the PPC, were trained to perform a visual cue-guided choice task (Fig. 7a). The mice first learned the correspondence between the correct visual pattern and correct choice in the T maze. Two identical sessions were performed each day, session 1 in the morning and session 2 in the afternoon, so that the animals could use the information learned in the morning in their afternoon session. After the animals had learned the task, the visual discrimination patterns were varied from day to day, thus requiring new learning each day. Between sessions, animals were allowed to sleep in their home cage either unperturbed or with bilateral activation of PPC ID2/Nkx2.1 neurons during NREM sleep or waking. Non-stimulation and optogenetic stimulation days varied in a semi-random manner within and across mice. The same intensity and duration pulses (100 ms) that were found effective in driving ID2/Nkx2.1 neurons in the physiological experiments were randomly (every 500 ± 400 ms) delivered during the entire NREM sleep in the home cage after the morning session (7,496 ± 1,600 pulses).
Fig. 7 |. Perturbation of DSA neurons during NREM sleep impairs memory consolidation.

a, Diode probes were implanted bilaterally in the PPC in ID2/Nkx2.1::CatCh mice. They performed in a goal-associated visual cue discrimination task (for example, blue and yellow stripe patterns) twice daily (morning and afternoon sessions). b, Performance of each mouse in the morning (session 1) and afternoon (session 2). On randomly chosen days, mice were left unperturbed in the home cage between sessions (gray symbols) or ID2/Nkx2.1 neurons were optogenetically stimulated (100-ms duration light pulses every 500 ± 400 ms) during NREM sleep (purple). As further controls, the same optogenetic stimulation during wake in the home cage (green) or during the task in the central arm continuously (yellow) did not interfere with sleep-induced memory improvement. Each symbol depicts a comparison between morning (session 1) and afternoon (session 2) sessions (3-d per condition). c, Only optogenetic perturbation of ID2/Nkx2.1 neurons during NREM sleep affected memory consolidation (mean ± s.d., P = 0.001, KW test; P < 10−4 for ‘No stim’ against ‘NREM stim’, Tukey test). d, Same stimulation protocol in a different cohort of animals that lacked Chr (‘photostimulation control’) did not affect memory consolidation (mean ± s.d.; n = 3; P = 0.8946, KW test). e, Example of rate correlation matrices for all neuron pairs from pre-NREM sleep, maze (first session) and post-NREM sleep in an example mouse (non-stimulation, control day). These matrices were used to calculate the EV and its control value, the reverse explained variance (REV), in both non-stimulated and optogenetically stimulated sessions. f, Pearson correlation coefficients for all neuron pairs for pre-sleep versus maze learning (left) and post-sleep versus maze learning (right) comparison (same example session as in e). g, Group statistics (medians, interquartile ranges, maxima and minima; n = 5 mice, at least one session per animal) of the EV and REV on non-stimulated (control) and NREM-stimulated (P = 0.002, KW test) days. EV on non-stimulated days was significantly higher than its corresponding REV (chance, P = 0.003, Tukey test) and also significantly higher than EV on NREM-stimulated days (P = 0.012). *P < 0.05 and ***P < 0.001.
As expected, on days with no perturbation in the PPC, mice showed a significant improvement of discrimination performance (probability of correct trials, computed as correct trials/all trials) in the afternoon session (Fig. 7b,c, gray color; from 0.57 ± 0.09 in the morning session to 0.68 ± 0.06 in the afternoon session, P = 6.10 × 10−5, Wilcoxon paired signed-rank test). In contrast, the memory boost from the morning session was abolished by stimulation of ID2/Nkx2.1 neurons during NREM (Fig. 7b). However, when optogenetic stimulation was applied during waking (mostly quiet wake state) in the home cage (500 ± 400-ms random pulses; 7,487 ± 1,333 pulses; P = 0.35 between NREM and awake pulses number, KW test), memory gain from sleep was not affected (Fig. 7b; P = 0.94, Tukey–Kramer range test). As a further control, when ID2/Nkx2.1 neurons were activated while the mouse ran through the center arm during the morning task, memory gain from the morning to afternoon session was not affected (Fig. 7b,c; P = 0.62, Tukey–Kramer range test).
Differences between morning session performance across days (Fig. 7b; P = 0.57, KW test) or time spent in NREM state (Extended Data Fig. 8a–e; P = 0.37, KW test) or changes in other brain states (REM and waking behavior; Extended Data Fig. 8f) could not explain these results. Light stimulation at the same intensity in a second group of mice (n = 3), which lacked ChR expression in ID2/Nkx2.1 (‘photostimulation control’), did not prevent memory improvement from morning to afternoon sessions, excluding potential non-specific effects (Fig. 7d). Our stimulation protocol affected approximately 20% and 5% of all detected up and down states, respectively (Extended Data Fig. 8g), which is largely proportional to the duration distributions of up and down states.
Finally, we explored the effect of optogenetic stimulation of ID2/Nkx2.1 cells on sleep replay of neuronal correlations using the explained variance (EV) method (Fig. 7e,f and Extended Methods)35. We found significant reactivation for non-stimulated days (Fig. 7g), an effect that was abolished by optogenetic stimulation of ID2/Nkx2.1 cells during NREM. Moreover, EV was significantly higher on non-stimulated days than on optogenetic stimulation days. In summary, artificial activation of ID2/Nkx2.1 neurons during NREM sleep eliminated the benefit of sleep-assisted memory consolidation in the PPC, likely interfering with the physiological structure of up and down states.
Discussion
We described a unique set of deep-layer neurons in PPC, V1 and frontal neocortical areas whose spiking activity is inversely correlated with all principal cells and all other types of interneurons across all brain states. This anti-correlation was most prevalent during the down state of NREM sleep where these neurons fired specifically and selectively (DSA neurons). We demonstrated that DSA neurons belong to a special subset of neurogliaform, weakly immunoreactive for nNOS with dense local axon arbors, which we identified as ID2/Nkx2.1 cells. They were inhibited by PV-, Sst- and nNOS-expressing interneurons and weakly excited by deep-layer pyramidal cells, relative to other interneurons. Spiking of DSA neurons during the down state delayed the sequential order of their target neurons at the down–up transition and prolonged the down state. Finally, we found that optogenetic activation of ID2/Nkx2.1 interneurons in the PPC during NREM sleep interfered with consolidation of cue discrimination memory and learning-induced sleep replay of neuron pairs.
Unique features of deep-layer ID2/Nkx2.1 neurogliaform interneurons.
We identified DSA neurons as deep-layer neocortical ID2/Nkx2.1 interneurons, also known as Lamp5+Lhx6+ neurons21,23. Although only a subset of the physiologically characterized DSA neurons were optogentically identified as ID2/Nkx2.1 interneurons, all DSA neurons shared similar physiological features, including spike waveform, spike auto-correlation dynamics, brain state-dependent firing rates, layer distribution and anticorrelation with spiking activity of all other neuron types. A small subset of DSA cells in Nos1::ChR mice were nNOS-expressing neurons. Anatomically, ID2/Nkx2.1 interneurons had weak nNOS expression but did not expess PV or Sst. Neurons expressing nNOS were previously classified in two major types: ‘NOS-type 1’, with strong NOS expression, large somata and Sst co-expression; and ‘NOS-type 2’, a more diverse group, with smaller-sized somata and low level of NOS expression. This latter group includes neurogliaform, vasoactive intestinal peptide (VIP), PV and Sst interneurons36,37. Thus, ID2/Nkx2.1 interneurons are within the NOS type 2 group.
In earlier experiments, nNOS-expressing neurons in multiple cortical regions of mice, rats and golden hamsters showed increased c-Fos expression after sleep deprivation, and the magnitude of nNOS expression correlated with delta power of NREM sleep11. Non-selective nNOS knockout mice show reduced NREM sleep time, shorter NREM bouts, decreased slow oscillation power and impaired NREM compensation after sleep deprivation12. In contrast, when nNOS deletion is confined to Sst-expressing neurons in the neocortex and striatum, these mice show blunted slow oscillation power in response to sleep deprivation but without altered NREM duration or sleep structure13, suggesting that, whereas power in the slow oscillation band is primarily regulated by cortical circuits2–4, control of sleep state choreography is mainly subcortical13. Mice lacking nNOS expression in Sst-positive neurons exhibit impairment in recognition memory but not in a hippocampal-dependent object recognition task13. In all these studies, the critical c-Fos-expressing neurons were of the nNOS type 1.
In our experiements, most nNOS neurons fired together with other interneurons and pyramidal cells during the up state of slow oscillation. Thus, whereas type 1 nNOS neurons, present in all cortical layers, and deep-layer ID2/Nkx2.1 interneurons are quite distinct, they appear to complement their contributions to NREM. Type 1 nNOS neurons might release nitric oxide13 and bring about deepening of NREM sleep by increasing the power of slow oscillations. ID2/Nkx2.1 interneurons, on the other hand, become selectively active during the down state, prolong the duration of the down state and bias the sequential organization of pyramidal neurons during the down–up transition and might contribute to other, yet to be discovered, functions.
ID2/Nkx2.1 interneurons showed typical features of neurogliaform cells, including small somata and dense local arborizations with small boutons24–26. The physiological features of neurogliaform neurons in the superficial cortical layers were characterized previously. Uniquely among all known GABAergic interneurons, single pre-synaptic action potentials of neurogliaform neurons can activate post-synaptic metabotropic GABAB receptors24,38 and halt activity of principal cells through dendritic calcium channels39. In addition, they affect their targets via slow inhibitory post-synaptic currents mediated by GABAA receptors localized on the dendrites of pyramidal neurons29. In response to near threshold current steps, they fire after a noticeable delay, whereas stronger depolarizing pulses induce sustained non-accommodating spike trains with large spike after-hyperpolarizations26–28. ID2/Nkx2.1 interneurons shared these biophysical features with neurogliaform neurons in superficial cortical layers, although the two groups differed in quantitative terms.
In contrast to other forebrain GABAergic interneuron types (for example, PV, Sst and VIP subtypes), neurogliaform neurons originate from several distinct embryonic brain regions. Most hippocampal neurogliaform cells arise from Nkx2.1+ lineages (Lhx6+), whereas the vast majority of neocortical neurogliaform cells have a non-Nkx2.1 caudal ganglionic eminence/preoptic area origin25,40,41, the exception being the ID2/Nkx2.1 type described here. Neurogliaform cells are present in all cortical layers but are most abundant in layer 1 and layer 2/3 (ref. 20). We observed ID2/Nkx2.1 interneurons, but not DSA neurons, scattered in all layers. Some of these superficial neurons were chandelier (axo-axonic) cells, which have been shown to derive from late-born Nkx2.1-expressing cells in a restricted region of the median ganglionic eminence42. Another unique and important feature of ID2/Nkx2.1 interneurons is that their relative prevalence, compared to other interneurons, is ten-fold higher in the primate neocortex compared to the mouse23, suggesting an expanded role for this neuron type in a more complex primate neocortical mantle.
DSA neurons were more weakly excited by neighboring pyramidal cells, compared to other interneurons, but they might receive excitatory inputs from other sources as well. At the same time, they were more strongly inhibited by PV, Sst and, to a lesser extent, NOS-expressing interneurons, relative to other interneurons. During the down state of slow oscillations, all other neurons are silent, with exceptional sporadic spiking of some pyramidal cells8. Therefore, the source or mechanisms that sustains spiking of DSA neurons remains unknown. One possibility is that, upon release from inhibition, ID2/Nkx2.1 interneurons can induce rebound spikes. Although ID2/Nkx2.1 interneurons did not display biophysical properties of rebound spiking, in our optogenetic experiments, DSA neurons emitted excess spikes for 50–200 ms upon release from PV- and Sst-induced silencing, a duration that typically corresponds to that of typical down states. A second option is that intrinsic properties of ID2/Nkx2.1 interneurons maintain sustained spiking at resting membrane potential. Our intracellular recordings in vitro did not support this scenario, but it remains to be examined whether ID2/Nkx2.1 interneurons possess membrane channels that endow them with special properties to generate action potentials at rest. Yet another explanation is that hitherto unidentified subcortical sources, specifically innervating deep-layer ID2/Nkx2.1 interneurons, are also selectively active during the down state. One such potential source could be some of the intralaminar and midline nuclei of the thalamus, several of which preferentially innervate deep cortical layers and have been related to arousal and awareness43. Another subcortical source is the cholinergic nucleus basalis, whose neurons display low-level but phasic activity during NREM sleep1. ID2/Nkx2.1 cells might respond differently to acetylcholine, which is known to suppress the activity of superficial layer neurogliaform cells44.
Effector characteristics of ID2/Nkx2.1 interneurons.
Our optogenetic activation of ID2/Nkx2.1 interneurons exerted relatively modest effects on the activity of nearby recorded neurons. This is perhaps not surpising given their ten-fold to 20-fold less density compared to PV and Sst interneurons in deep cortical layers20,22. Therefore, focal light delivery likely excited relatively few ID2/Nkx2.1 cells, which might explain why their optogenetic excitation exerted a relatively weak effect on other cells compared to activation of other interneurons.
However, a clear indication of the important contribution of DSA neurons to the down–up transition is illustrated by our observation that the spike timing of layer 6 neurons suppressed by optogenetic activation of ID2/Nkx2.1 neurons occurred later during the transition from down to up state compared to those neurons that were not affected by the optogenetic pulses. This effect might also explain why, during optogenetic activation of ID2/Nkx2.1 neurons, the delta frequency peak decreased, an indication of prolonged down states. Furthermore, within the ID2/Nkx2.1 neuron-suppressed group, neurons spiked later in the down–up transition specifically for cases when, in the preceding down state, spiking activity of DSA neuron was high.
A simple mechanism that could account for the effect of DSA neurons on the sequential organization of neurons in the wake from the down state10 is a non-uniform innervation of their targets and/or the different biophysical properties of their deep-layer pyramidal cell targets. This latter hypothesis is supported by previous observations showing that the rank order of pyramidal neuron firing during the down–up transition correlates with their baseline firing rates32. The potential targeting of thalamus-projecting layer 6 pyramidal cells by ID2/Nkx2.1 neurons might be of special significance. Previous work showed that these layer 6 neurons play a critical role in cortical gain control by affecting activity in all other layers directly and indirectly through the corticothalamic circuit45.
Additional support for the importance of DSA neurons in affecting populations of neurons comes from our optogenetic manipulation of these neurons in a memory task. Although remembering cue discrimination benefited from intervening rest/sleep between morning and afternoon sessions, this advantage was abolished by bi-hemispheric optogenetic activation of ID2/Nkx2.1 interneurons in the PPC, specifically during NREM sleep. Non-specific effects of light stimulation, such as activation of the retina or heat activation of nearby neurons, are not likely to explain these results because the same intensity of light stimulation on the ID2/Nkx2.1-GFP mice led to a similar lack of behavioral effect as the non-stimulated controls. One possible explanation for abolishing the memory consolidation benefit of sleep is that optogenetic perturbation of down states in the PCC, a region specifically involved in visual discrimination learning34, interfered with the physiological patterning of neural firing. Support for this contention was provided by the finding that replay of neuron pairs active during maze learning was significantly stronger during control days than on days when ID2/Nkx2.1 neurons were optogenetically stimulated during NREM. Although it is tempting to suggest that perturbation of the sequential order of firing of pyramidal neurons, brought about by artificial activation of ID2/Nkx2.1 interneurons, is related to the memory deficit35, further experiments are needed to firmly establish this potential link. The modest goal of our behavioral experiment was to show that artificial activation of DSA neurons can perturb memory consolidation and, thus, demonstrate the physiological effectivenes of ID2/Nkx2.1 interneurons. It is expected that the physiological consequences of ID2/Nkx2.1 neuron perturbation in different cortical regions will depend on the specific circuit function of the perturbed region.
Conclusions
We demonstrate that deep-layer neocortical ID2/Nkx2.1 interneurons comprise a distinct neuron type, characterized by an inverse activity relationship with all other cortical neurons. They share physiological features of the broader class of neurogliaform neurons, but they possess unique and distinct features. ID2/Nkx2.1 neurogliaform cells are strongly inhibited by several other interneuron types. High activity of DSA neurons delayed the onset of down–up transition and affected the sequential ordering of neurons, and their optogenetic activation in the PPC during NREM sleep interfered with consolidation of cue discrimination memory. It might be of special significance that the prevalence of ID2/Nkx2.1 interneurons increases with the evolution of neocortex. Thus, despite their small numbers, the physiological importance of ID2/Nkx2.1 interneurons is high.
Methods
Subjects and electrode implantation.
All experiments were approved by the Institutional Animal Care and Use Committee of New York University Medical Center. We used several lines of transgenic mice (n = 15 male mice) for optogenetic tagging of the recorded units: n = 2 Sst-Cre (Ssttm2.1(cre)Zjh/J, Jax stock no. 013044); n = 4 PV-Cre::Ai32 (from B6.129P2-Pvalbtm1(cre)Arbr/J, Jax stock no. 017320 and B6.Cg-Gt(ROSA)26Sortm32(CAG-COP4*H134R/EYFP)Hze/J, Jax stock no. 024109); n = 3 Nos1-Cre (B6.129-Nos1tm1(cre)Mgmj/J, Jax stock no. 017526); and n = 6 ID2-CreER/Nkx2.1-Flpo::Ai80 (from B6.129 S(Cg)-Id2tm1.1(cre/ERT2)Blh/ZhuJ, Jax stock no. 016222, Nkx2–1tm2.1(flpo)Zjh/J, Jax stock no. 028577 and B6.Cg-Gt(ROSA)26Sortm80.1(CAG-COP4*L132C/EYFP)Hze/J, Jax stock no. 025109) for optogenetic tagging of the recorded units. Tamoxifen (20 mg ml−1 in corn oil) was administered to ID2/Nkx2.1::Ai80 (or Ai65, for the histology section) animals (3 × 5 mg by oral gavage over 5 d) between P30 and P40 to activate the CreER. Sst-Cre and Nos1-Cre mice were injected with AAV-FLEX-ChR2-EYFP to express channelrhodopsin-2 in Cre-expressing neurons. Three ID2/Nkx2.1::Ai80 mice administered only with corn oil (without tamoxifen) were used as behavioral photostimulation controls.
All animals (28–35 g, 3–10 months old) were implanted with 64-site silicon probes (NeuroNexus or Cambridge NeuroTech) in the PPC (AP 2.0 mm, ML 1.75 mm, DL 0.6 mm) or V1 (AP 1.0 mm, ML 2.5 mm, DL 1 mm, with a 21° angle from the dorso-ventral axis and 10° from the mediolateral axis)46. Ground and reference wires were implanted in the skull above the cerebellum, and a grounded copper mesh hat was constructed, shielding the probes. Probes were mounted on microdrives that were advanced to layer 6 over the course of 5–8 d after surgery. For most of the experiments, a 100–200-μm fiber optic was attached to one of the shanks of the silicon probe. For V1 recordings, an optic fiber was placed right above the skull over the implantation site. The back end of the fiber was coupled to a laser diode (450 nm blue, Osram). For ID2/Nkx2.1::Ai80 recordings, a second fiber was implanted in the contralateral hemisphere at the same coordinates. The probe locations were verified histologically for all mice. Animals were allowed to recover for at least 1 week before testing. Mice were housed under standard conditions (71–73 °F and 40–50% relative humidity) in the animal facility and kept on a 12-h reverse light/dark cycle.
Recordings and behavior.
Recording sessions started 1–2 h after the onset of the dark phase of the light/dark cycle. We recorded the mice while they slept or walked freely in the home cage. Electrophysiological data were acquired using an Intan RHD2000 system (Intan Technologies) digitized with a 30-kHz rate. The wide-band signal was downsampled to 1.25 kHz and used as the LFP signal.
For optogenetic tagging of specific neuron types, blue laser light (450 nm, Osram) pulses were delivered in the PPC or above the V1. The maximum light power at the tip of the optic fiber was 1–4 mW. Then, 10–100-ms light pulses with 40%, 70% and 100% of the maximum power were delivered (n = 500–1,000 times at each intensity, at 1 Hz or at 400 ± 200-ms random intervals).
For behavior experiments, animals were handled daily and accommodated to the experimenter, recording room and cables for 1 week before the start of the experiments. Animals were also water restricted before the start of the experiments. Two (morning and afternoon) 100-trial (30–60-minute-long) behavior sessions were conducted daily, separated by 5 h, and preceded and followed by 1 h-long sleep sessions. In the goal-associated visual cue discrimination task, mice ran in a 70-cm × 40-cm (1 m above the floor) figure-eight maze to collect a water reward each time they reached the correct (left or right) corner (Fig. 7). Visual cues were arbitrary patterns delivered by two (left and right) 8 × 8 LED matrices (Adafruit) at the initial section of the central arm. For each trial, the visual cue/reward position was randomly assigned, whereas a different arbitrary pattern (distractor) was delivered in the LED matrix on the opposite side. Visual cues and distractor patterns were chosen to activate a similar number of single LEDs from the matrices and were randomly assigned to the different experimental subjects so that the visual cue for one animal was the distractor of a different animal. The same pair visual cue/distractor was maintained during training, but, once the animals learned the task, the visual patterns varied from day to day, demanding new learning each day. All subjects underwent the same number of ‘No stim’ and ‘NREM’ stimulation sessions in a randomly assigned fashion (and the same for Maze stim and Awake stim, in addition to No stim and NREM, for mice 1–3). Performance was quantified as the correct choice probability: correct choices/total number of trials. Maze doors (to prevent backward displacement of the mice), water delivery, visual patterns and optogenetic stimuli were controlled by a custom-made Arduino-based circuit (https://github.com/valegarman).
Unit clustering and neuron classification.
Spike sorting was performed semi-automatically with Kilosort47 (https://github.com/cortex-lab/KiloSort), using our own pipeline KilosortWrapper (a wrapper for KiloSort, https://github.com/brendonw1/KilosortWrapper). This was followed by manual adjustment of the waveform clusters using the software Phy (https://github.com/kwikteam/phy) and plugins for Phy designed in the laboratory (https://github.com/petersenpeter/phy-plugins). The following parameters were used for the Kilosort clustering: ops. Nfilt: 6 * numberChannels; ops.nt0: 64; ops.whitening: ‘full’; ops.nSkipCov: 1; ops.whiteningRange: 64; ops.criterionNoiseChannels: 0.00001; ops.Nrank: 3; ops. nfullpasses: 6; ops.maxFR: 20000; ops.fshigh: 300; ops.ntbuff: 64; ops.scaleproc: 200; ops.Th: [4 10 10]; ops.lam: [5 20 20]; ops.nannealpasses: 4; ops.momentum: 1./[20 800]; ops.shuffle_clusters: 1.
Unit clustering generated two separable groups (Extended Data Fig. 1a) based on their auto-correlograms, waveform characteristics16 and firing rate. Putative pyramidal cells and interneurons were tentatively separated based by these two clusters. Definitive cell identity was assigned after inspection of all features, assisted by monosynaptic excitatory and inhibitory interactions between simultaneously recorded, well-isolated units and optogenetic responses. DSA neurons were identified by the Z-scored ± 50-ms down state and ±35-ms CCG response (Fig. 1g). Another method of separation of the DSA neurons was obtained by the firing rate ratios during down and up states (Fig. 1f).
Unit activity analysis.
Units were defined as optically tagged16,46, using a P value cutoff of 10−3. Negatively modulated cells (−Mod) were identified by an average firing suppression less than 1 s.d. for the entire window of light stimulation (50–100 ms) and a P value cutoff of 10−2. Average unit CCG was computed as the mean from all Z-scored CCGs (5-ms binned, 0.6-s window) in different brain states. To calculate the depth coordinates of the units, we aligned depth profiles of electrophysiological landmarks. These included the largest amplitude peak of high-frequency LFP power (500 Hz–5 kHz) corresponding to mid-layer 5 and four prominent sinks and sources from the averaged down–up current-source density (CSD) maps for each animal (Fig. 1c).
To investigate the rate relationship among unit groups, we used joint probability density of the Z-scored rate of a given group (DSA neurons in Fig. 2c and 100 shuffling average of the 10% pyramidal cells or interneurons in Extended Data Fig. 2c) between Z-scored firing of the pyramidal cells and interneurons in 0.25-s.d. bins. We also computed the partial Spearman correlation of the Z-scored spike counts across groups for different bin sizes (0.01, 0.15, 0.20… 1.01; ref. 48).
Rank-order tests were used to identify sequences of units repeating consistently across down–up events as tested against chance order49. The timing from multiple units recorded simultaneously were transformed in a normalized sequence order from 0 to 1 within a down–up event. Only events with at least six participating units were included. Only the first spike from each unit in a cycle was considered for the rank order. Rank distribution of the correlation values was tested against shuffle correlations (500 shufflings, significance at P = 0.05) using Pearson correlations. Only events with more significant correlations than expected by chance were considered for further analysis. Finally, significant events were compared against the average rank order obtained by grouping the remaining events according to some rules (templates for DSA neuron rate higher than average and DSA neuron rate lower than average; or first half of the recording and second half of the recording session; or randomly chosen events) using, again, Pearson correlation.
EV and REV were calculated only in sessions with more than 20 simultaneously recorded neurons. Pairwise correlations for EV and REV were calculated using the Pearson correlation coefficient in 100-ms-binned spike trains. The coefficients were separately calculated for pre-sleep (only NREM), maze (only first session) and post-sleep (only NREM) epochs as correlation matrices (Fig. 7a). The correlations between these three matrices were used to assess the percentage of variance in the post-sleep period that could be explained by the patterns established during the maze experience while accounting for pre-existing correlations in the pre-sleep session (EV), according to the following equation:
where R variables are the correlation coefficients between maze (M), pre-sleep (P1) and post-sleep (P2) pairwise correlation matrices35. The control value (REV) was obtained by switching the temporal order of the pre- and post-sleep session.
Detection of monosynaptic connections.
CCG peaks can reflect monosynaptic connections19. This takes the form of short time lag (1–4 ms) positive deviation from CCG baseline, indicating putative excitatory connections. Such detection is based on testing the null hypothesis of a homogeneous baseline at short time scale50. To detect putative monosynaptic connections, 0.5-ms-binned CCGs were convolved with a 7-ms s.d. Gaussian window resulting in a predictor of the baseline rate. At each time bin, the 99.9999th percentile of the cumulative Poisson distribution (at the predicted rate) was used as the statistical threshold for significant detection of outliers from baseline. A putative connection was considered significant when at least two consecutive bins in the CCGs within +1.5 to +4 ms passed the statistical threshold19. All putative connections were visually inspected. Corrected spike transmission probability was calculated as described in English et al.19.
Brain state scoring.
Brain state scoring was performed as in Watson et al.17 with the MATLAB resource SleepScoreMaster from the repository of the laboratory (https://github.com/buzsakilab/buzcode/tree/master/detectors/detectStates/SleepScoreMaster). In short, spectrograms were constructed with a 1-s sliding 10-s window fast Fourier transform of 1,250-Hz data at log-spaced frequencies between 1 and 100 Hz. Three types of signals were used for score state: broadband LFP, narrowband theta frequency LFP and electromyogram (EMG). For broadband LFP signal, principal component analysis was applied to the Z-transformed (1–100-Hz) spectrogram. The first principal component in all cases was based on power in the low (<20-Hz) frequency range and had oppositely weighted power in the higher (>32-Hz) frequencies. Theta dominance was taken to be the ratio of the power at 5–10 Hz and 2–16 Hz from the spectrogram. EMG was extracted from the intracranially recorded signals by detecting the zero time lag correlation coefficients (r) between 300 and 600 Hz filtered signals (using a Butterworth filter at 300–600 Hz with filter shoulders spanning to 275–625 Hz) recorded at all sites. All states were reviewed by the experimenter, and corrections were made when discrepancies between automated scoring and user assessment occurred.
Detection of up and down states.
Slow waves were detected using the coincidence of a three-stage threshold crossing in three signals: a drop in high gamma power (30–600 Hz), a peak in the delta band filtered signal (0.5–8 Hz) and drop in unit-normalized firing rate. The gamma power signal was smoothed using a sliding 100-ms window and locally normalized to account for non-stationaries in the data. Time epochs in which the delta filtered signal crossed a peak threshold (0.5 s.d.) and coincided with gamma power drops (−0.5 s.d.) were considered as candidate delta peaks and binned by peak magnitude, with start and end times at the nearest crossing of the delta band threshold with a minimum duration of 40 ms. Time epochs with gamma power above 1 s.d. and lasting 0.2–1.4 s were considered putative up events. The peri-event time histogram for spikes from all cells was calculated around delta peaks in each magnitude bin and Z-score normalized. Only candidate events with a rate drop below 2 s.d. in a ±50-ms window around the delta peak were considered down states. Up states were defined as candidate epochs that immediately followed and were truncated by down states. Down state responses were aligned by the delta peak or the end of the down epoch (down–up transition), as indicated.
Spiking network simulations.
We built a network model of leaky integrate-and-fire neurons based on Jercog et al.15 and Barrio et al.51, using the Python-based simulator Brian2 (ref. 52). We include nPYR = 4,000 excitatory, nINT = 1,000 inhibitory and nDSA = 100 neurons with the following connection probability (c): cPYR-PYR = 0.1, cPYR-INT = 0.1, cINT-INT = 0.2, cINT-PYR = 0.3, cPYR-DSA = 0.05, cINT-DSA = 0.3, cDSA-PYR = 0.1 and cDSA-INT = 0.1. In our model, the membrane potential of a neuron (i) obeys:
Where τi is the membrane time constant, EL is the leak potential and Irec(t) and Iext(t) are the recurrent and external input terms, respectively. The following values were used for the τ and EL constants: τPYR = 20 ms, τINT = 10 ms, τDSA = 50 ms, ELPYR = −55.6 mV, ELINT = −55.6 mV, ELDSA = 40.0 mV. Whenever the membrane potential of the neuron i reached the threshold VTh, a spike is simulated, and the membrane potential was reset to VRes with 1 ms of refractory period. The threshold and reset values were VThPYR = −47 mV, VThINT = −37 mV, VThDSA = −40 mV and VResPYR = −51 mV, VResINT = −49.9 mV, VResDSA = −57 mV.
The recurrent input term consisted of the sum of all inhibitory and excitatory synaptic drives experienced by a given cell according to the rate of its pre-synaptic partners and governed by:
Where τX are the response time constants and gPRE,POST are the synaptic weights, with the following values for all combinations of synapses: gPYR,PYR = 0.97 mV, gPYR,INT = 3.5 mV, gPYR,DSA = 0.5 mV, gINT,PYR = −0.3, gINT,INT = −1, gINT,DSA = 0.0 to −3.0, gDSA,PYR = −0.3, gDSA,INT = −1, τE = 8.0 ms, τI = 2.0 ms and τDSA = 20.0 ms.
The external input term is composed of two terms: a Gaussian white noise (ζi) term with σ = 3 mV and a time constant τGWN equal to the membrane time constant (τi) and a source of Poisson (2-Hz) input pulses with 20-ms duration and 45-mV amplitude, called kicks (Ikicks), affecting 10% of the population (so pi = 0.1) of pyramidal cells and interneurons15:
We ran five simulations of 100 s each with a gINT,DSA of 1.5 mV to compare with data obtained from in vivo recordings. For inhibitory drive parametric experiments (gINT,DSA from 0 to 3 mV; Extended Data Fig. 3c,e), we averaged data from two simulations for each gINT,DSA value (−3.0, −2.5, −2 … 0). Code is available at https://senselab.med.yale.edu/modeldb/.
In vitro recordings.
Whole-cell patch-clamp recordings of 22 tdTomato+ cells from seven ID2/Nk2.1::Ai65 adult (post-natal days 37–198, B6.129S(Cg)-Id2tm1.1(cre/ERT2)Blh/ZhuJ, Jax stock no. 016222, Nkx2–1tm2.1(flpo)Zjh/J, Jax stock no. 028577 and B6;129S-Gt(ROSA)26Sortm65.1(CAG-tdTomato)Hze/J, Jax stock no. 021875, respectively) mice of either sex were performed as previously described26. In short, mice were killed using pentobarbital sodium and phenytoin sodium. Once unresponsive, mice were perfused with ice-cold sucrose-ACSF containing the following (in mm): 87 NaCl, 75 sucrose, 2.5 KCl, 26 NaHCO3, 1.25 NaH2PO4, 10 glucose, 1.0 CaCl2 and 2.0 MgCl2, saturated with 95% O2/5% CO2. Then, 300-μm-thick coronal brain sections were made using a Leica VT1200S vibratome and incubated at 35 °C for 30 min in ACSF containing the following (in mm): 125 NaCl, 2.5 KCl, 26 NaHCO3, 1.25 NaH2PO4, 10 glucose, 2.0 CaCl2 and 1.0 MgCl2. Slices were then kept in this solution for at least 30 min at room temperature before recording. For in vitro recordings, slices were continuously perfused with ACSF saturated with 95% O2/5% CO2 and kept at 29–32 °C. All whole-cell recordings were performed in the current-clamp configuration; the liquid junction potential was not corrected. Pipettes with resistances of 2–6 MΩ were made (Sutter Instrument) using borosilicate glass and filled with potassium gluconate intracellular solution containing the following (in mm): 130 K-gluconate, 10 KCl, 10 HEPES, 0.2 EGTA, 4 Mg-ATP and 0.3 Na-GTP, adjusted to a pH of 7.3 with KOH. Biocytin was dissolved into the intracellular solution (final concentration 0.3–0.5%) for later staining and morphological recovery (n = 11 neurons attempted). Before gaining whole-cell access, a gigaseal was obtained, and the pipette capacitance was compensated. Access resistances were monitored throughout recordings and completely compensated; cells with access resistances >35 MΩ were not analyzed. All data were collected using a Multiclamp 700B amplifier (Molecular Devices), a Digidata 1322A digitizer (Molecular Devices) and Clampex version 10.3 software (Molecular Devices); data were sampled at 20 kHz and low-pass filtered at 10 kHz.
Data were analyzed using Clampex version 10.7 software (Molecular Devices), custom-written MATLAB software (MathWorks) and Excel (Microsoft). All statistical analysis (only Mann–Whitney U tests were used) was performed using custom-written MATLAB software. Six electrophysiological properties were analyzed and compared to the same six properties of layer 1 neurogliaform cells from ref. 26. The adaption index was calculated as the ratio of the number of spikes in the last 500 ms over the number of spikes in the first 500 ms of a positive current injection that elicited ∼30-Hz firing. The time to first spike (in milliseconds) was calculated as the difference in time between the peak of the first AP and the start of the positive current injection. The AP spike width (in milliseconds) was calculated as the difference in time between the ascending and descending phases of a spike at the voltage found at the time between spike peak and spike threshold. The amplitude of voltage sags (%) were measured using hyperpolarizations to ~−100 mV from a holding potential of ~−70 mV and calculated as 100 × (Vsag minimum − Vsteady-state)/(Vsag minimum − Vholding). The input resistance (in MΩ) was calculated using Ohm’s law from averaged traces of 150-ms-long negative current injections of −20 pA. The resting membrane potential (in millivolts) was measured with 0-pA current injection soon after gaining whole-cell access. Data from recorded Id2/Nkx2.1 cells were compared to previously published neurogliaform cells in L1 (ref. 26).
Histological processing and microscopy.
ID2-CreER/Nkx2.1-Flpo::Ai65 mice (n = 5) were overdosed with pentobarbital injection (100 mg kg−1 body weight) and perfused with saline and 4% paraformaldehyde before their brains were rapidly removed. After overnight post-fixation in 4% paraformaldehyde solution, the brain was washed three times in 0.01 M phosphate-buffered saline (PBS). Coronal sections of 70–100 μm thickness were cut using a Leica Microsystems VT 1000S vibratome, washed 3–4 times in 0.01 M PBS and stored in 0.01 M PBS with 0.05% sodium azide at 4 °C. For initial visualization and documentation of tdTomato-expressing neurons, sections were mounted on glass slides in VECTASHIELD (Vector Laboratories) under a coverslip and sealed with nail polish and then observed with epifluorescence on a wide-field Leitz DMRB microscope (Leica, Openlab version 5.5.0). For the identification of tdTomato+ or GFP+ neuron molecular profiles, layer markers and putative synaptic inputs, immunohistochemistry was performed as described previously53,54. Briefly, sections were permeabilized in Tris-buffered saline (TBS) with 0.1% Triton X-100 (Tx) or subjected to 2–3 rounds of freeze–thaw (FT) over liquid nitrogen after cryoprotecting in 20% sucrose. For FT sections, a TBS base solution was used throughout. Sections were blocked in 20% normal horse serum (NHS) in TBS/TBS-Tx for at least 1 h at room temperature and then incubated in primary antibody solution (containing 1% NHS in TBS/TBS-Tx) for at least three nights at 4 °C. Control sections from C57BL/6J mice that lacked the primary antibodies were used throughout. We used the following primary antibodies (species, dilution, company, code): Sst, rat clone YC7 antibody (monoclonal), 1:500, EMD Millipore, MAB354; nNOS, rabbit, 1:1,000, EMD Millipore, AB5380; calretinin, goat, 1:1,000, Swant, CG1; RFP, rat, 1:500, ChromoTek, 5f8–100; vesicular glutamate transporter 2 (VGLUT2), guinea pig, 1:2,000, Synaptic Systems, 135 404; PV, goat, 1:1,000, Swant, PVG-214; PV, guinea pig, 1:5,000, Synaptic Systems, 195 004; GFP, rabbit, 1:500, Thermo Fisher Scientific, A-11122; and neuropeptide Y, sheep, 1:1,000, EMD Millipore, AB1583. Specificity information is provided in ref. 55. Sections were washed three times in TBS/TBS-Tx and incubated in secondary antibody solution in 1% NHS in TBS/TBS-Tx for at least 3 h at room temperature or overnight at 4 °C. Secondary antibodies raised in donkey were as previously described53,54: donkey anti-rabbit Alexa Fluor 405, 1:250, Invitrogen, A31556; donkey anti-mouse, anti-goat or anti-guinea pig DyLight405, 1:250, Jackson ImmunoResearch, 715-475-151, 705-475-147 and 706-475-148, respectively; donkey anti-rabbit or anti-goat Alexa Fluor 488, 1:1,000, Invitrogen, A21206 and A11055, respectively; donkey anti-rabbit, anti-goat, anti-sheep, anti-mouse or anti–guinea pig Cy3, 1:400, Jackson ImmunoResearch; 711-165-152, 705-165-147, 713-165-147, 715-165-151 and 706-165-148, respectively; and donkey anti-rabbit, anti-goat, anti-mouse or anti-guinea pig Cy5, 1:250, Jackson ImmunoResearch; 711-175-152, 705-175-147, 713-175-147, 715-175-151 and 706-175-148, respectively. Overviews of multi-channel immunofluorescence were acquired using wide-field epifluorescence on either an AxioImager.Z1 (Carl Zeiss, AxioVision 2009 software version Rel.4.8.1) or a Leitz DMRB microscope. Confocal microscopy (LSM 710; Carl Zeiss, ZEN 2008 version 5.0) was used to document tdTomato+ neurons and their putative inputs, as described previously54. Native tdTomato fluorescence was superior to that of the RFP antibody. For the quantification of tdTomato+ cell density, PPC layers in reference sections were defined by immunoreactivity for PV and VGLUT2 (ref. 56). Cell density was also measured in the anterior/rostral V1 (−2.6 to −2.8 mm posterior of bregma), caudal/posterior V1 (−3.4 to −3.8 mm posterior of bregma) and medial prefontal cortex (mPFC). For mPFC, counts were in range of 1.9–2.1 mm anterior of bregma, measuring in anterior cingulate, prelimbic and infralimbic and medial orbital cortex.
In vitro labeling and morphological imaging of intracellularly recorded neurons were described previously26. In short, cells (n = 11 attempted) were recorded with an intracellular K-gluconate solution containing 0.3–0.5% biocytin. After recording, slices were immediately placed into a 4% paraformaldehyde solution and stored at 4 °C overnight and then moved to 30% sucrose solution in 0.1 M PBS at 4 °C until further processing. Slices were thoroughly washed with 0.1 M PBS and then placed in CUBIC #1 solution for 2 d for tissue clearing. Then, slices were washed again thoroughly with 0.1 M PBS and treated overnight with a 1:500 dilution of streptavidin conjugated to Alexa Fluor 633 in 0.1 M PBS. After another round of washing with 0.1 M PBS, slices were treated with CUBIC #2 solution for ~30 min and then mounted in CUBIC #2 solution for confocal imaging (Zeiss) using a ×20 objective or a ×63 oil-immersion objective. The morphology of recorded cells was reconstructed using NeuTube57.
Statistical analysis.
All data presented were obtained from experimental replicates with at least three independent experimental repeats for each assay. All attempts of replication were successful. Data collection was not performed blinded to the subject conditions, except for the behavior experiments and scoring. Data analysis was performed blinded to the scorer or did not require manual scoring. Visual cues for the behavior experiments were randomly assigned to the different experimental subjects so that the visual cue for one animal was the distractor of a different animal. All subjects underwent the same number of conditions (unless stated otherwise) in a randomly assigned fashion. All statistical analyses were performed with standard MATLAB functions. No specific analysis was used to estimate minimal population sample, but the number of animals, trials and recorded cells were larger than or similar to those employed in previous studies (refs. 16,17,19,26,48,49). Unless otherwise noted, for all tests, non-parametric two-tailed Wilcoxon paired signed-rank tests, KW one-way analysis of variance (ANOVA) and Friedman tests were used. When parametric tests were used, the data satisfied the criteria for normality (Kolmogorov–Smirnov test) and equality of variance (Bartlett test for equal variance). For multiple comparisons, the Tukey honest significant differences post hoc test was employed, and the corrected *P < 0.05, **P < 0.01 and ***P < 0.001 are indicated, two-sided. NS indicates not significant. P values for Pearson correlations or Spearman correlations are computed using a Student’s t distribution for a transformation of the correlation. Results are displayed as mean ± 95% confidence interval (IC95) unless indicated otherwise. Box plots represent the median and 25th and 75th percentiles, and their whiskers represent the data range. In some of the plots, outlier values are not shown for clarity of presentation, but all data points and animals were always included in the statistical analysis. The exact number of replications for each experiment is detailed in the text and figures.
Extended Data
Extended Data Fig. 1 |. Unit classification and physiological properties of DOWN-State active (DSA) neurons.

a, Units (n = 2,279 from 11 mice) were first classified based on trough-to-peak latency and firing rate. Each dot corresponds to one unit. Units with trough-to-peak latency < 0.55 ms were tentatively classified as narrow-waveform putative interneurons (INT; blue). Wide waveform units were grouped into putative excitatory cells (PYR cells; red) and inhibitory interneurons (INT; blue), on the basis of the bimodality of the marginal distribution of trough-to-peak latencies (top histogram), firing rate (right histogram) and burstiness (dot size) (Methods). Optogenetically identified neurons (Sst, PV, nNOS and ID2/Nkx2.1) are superimposed on the clouds. PV cells were recorded from V1. b, DOWN-state rate (P < 10−101, KW test), DOWN-state participation (P < 10−69, KW test) and UP-state rate (P < 10−136, KW test) distributions for putative pyramidal cells (PYR, red), interneurons (INT, blue) and DSA neurons (gray). c, Similar layout as in C but for rEM sleep (left) and walking behavior (right). d, Peri-DOWN-state Z scored firing raster plot for all putative principal cells (PYR, left) and putative interneurons (INT, middle) for events detected during quiet waking state, as ranked according to Fig. 1D. Dashed lines delimit the ±50 ms window used to estimate unit responses. e, DSA neurons in the rat PFC (left) and in mouse primary visual cortex, V1 (right). Left (top to bottom): spike auto-correlogram and spike-waveform, peri-DOWN-state raster plot and average spike cross-correlation (CCG) between a reference neuron and all other simultaneously recorded units in a single session (each row corresponds to spikes of a target neuron referenced to the spike of DSA neuron at time 0. Black line is average). right, autocorrelogram of 6 V1 DSA neurons. Middle panel: cross-correlograms between DOWN-state (time 0 is the trough of DOWN state) and 6 DSA neurons. Bottom: average spike cross-correlation (CCG) between a reference DSA neuron (time zero) and all other simultaneously recorded units in that session. ***P < 0.001.
Extended Data Fig. 2 |. Network property statistics of DSA interneurons.

a, Statistical contrast matrices (two-sided Tukey’s test) for firing rates of pyramidal cells (PYR), interneurons (INT) and DSA interneurons during waking quiescence (QWake), NREM sleep, REM sleep and active behavior (Walk). b, Same layout and statistical comparisons as in A but for the average unit CCGs as a function of brain state. c, Pearson correlations (tested using a Student’s t distribution) between average CCG responses of NREM sleep, QWake, rEM sleep and walking behavior for all groups. Note that spike vs population relationships are preserved across brain states. d, Top: Average joint Z-score rate density between 10% of the interneurons (100 shufflings), 90% remaining interneurons and pyramidal cells neurons (left) and between 10% of the pyramidal cells (100 shufflings), interneurons and remaining pyramidal neurons. Bottom: Spearman correlation and statistical contrast matrices (two-sided Tukey’s test) for interneuron and pyramidal neurons rate and Z-scored population for all shown joint histograms. e, Average (mean ± IC95) partial correlation values of the Z-scored rate for all groups (blue for ρDSA,INT controlling for PYR; red for ρDSA,PYR controlling for INT; magenta for ρPYR,INT controlling for DSA), after truncating high firing rate units to match median the spikes number of pyramidal cells (average from n = 32 sessions, P < 10−25, F(2, 2787) = 57.42, repeated measures ANOVA). f, Left: distribution of excitatory divergence in all groups. Only putative pyramidal units excited their postsynaptic target cells (incidence probability for all groups in top-inset; P < 10−110, χ2(2) = 504.24, χ2 test). Right: distribution of excitatory convergence for all cell groups (P < 10−67, χ2(2) = 306.15, χ2 test). DSA neurons have fewer excitatory connections than the interneurons group (P < 10−4, χ2(1) = 11.36, χ2 test). ***P < 0.001.
Extended Data Fig. 3 |. Mechanisms of DSA neuron firing during DOWN states – model results.

a, Spiking neural model containing 100 leaky DSA neurons receiving an asymmetric inhibitory/excitatory drive (top-left scheme). Bottom, UP/DOWN transitions with DOWN-selective firing DSA neurons (gray dots in the rastergram at the top). Top right, log firing rate distributions in the model corresponded to the those of the recorded neurons. b, Peri-DOWN-state Z scored firing raster plot for all simulated principal cells (PYR, left) and interneurons (INT, right), including (incl) 10% of DSAn, ranked according to their event response. Bottom, Z-scored average CCG for each model neuron group (compare with Fig. 1e). The peri-DOWN state raster plots and unit cross-correlograms (Extended Data Fig. 3b) were similar to experimental results (compare with Fig. 1d, e) for both pyramidal and interneuron populations (Supp. Fig. 3C-D; PYR rate mean ± SD: 0.2 ± 0.09 Hz during DOWN, 1.31 ± 0.41 hz during UP; P ~ 0; INT: 0 ± 0.1 Hz during DOWN, 8.38 ± 2.52 Hz during UP, P ~ 0) as well as for the DSA group (7.43 ± 0.90 Hz during DOWN, 1.29 ± 0.34 Hz during UP, P < 10−83; Kruskal-Wallis test). c, Magnitude of DOWN-state response as a function of the CCG response. Marginal CCG (top, n = 20.000, 5000 and 500 PYR, INT and DSAn; P ~ 0, KW test) and DOWN-state response (right, P ~ 0, KW test) distributions for simulated pyramidal cells, interneurons and DSA neurons in each group. d, DOWN-state/UP-state firing rate ratio (P ~ 0, KW test), DOWN-state rate (P ~ 0, KW test), DOWN-state participation (P ~ 0, KW test) and UP-state rate (P ~ 0, KW test) distributions for all simulated pyramidal cells (PYR, red), interneurons (INT, blue) and DSA neurons (gray). e, Average (mean ± IC95) partial correlation values of the Z-scored rate for DSA and INT (blue), DSA and PYR (red), and PYR and INT (magenta) for different values of inhibitory weight simulated in the INT-DSA connection (0 to −3 mV, n = 2 simulations of 5100 cells per condition; P ~ 0, F(2, 44.094) = 278.36, repeated measures ANOVA). Model neurons showed that partial correlation between pyramidal cells and interneurons was positive at all time bins but negative for DSA neurons versus pyramidal cells and interneurons (compare with Fig. 2i), and it was strongly dependent on the inhibitory drive imposed on the DSA neurons by other inhibitory neurons. f, DSA neurons Z-scored DOWN-state responses (mean ± IC95) for different values of inhibition simulated on the DSA neurons (n = 200 DSAn per condition). DSA neurons rate during UP-states, but not during DOWN-states, depends on the inhibitory strength between INT and DSA neuron pairs. g, The peak delay of DSA neurons during DOWN-states correlates with the inhibitory strength of the INT-DSA connection (mean ± IC95, n = 2 simulations of 5100 cells per conditions; Pearson Correlations correlations tested using a Student’s t distribution). h, Partial correlation spectra (mean ± IC95) of the Z-scored simulated rate for each group as in Fig. 2d (n = 5 simulations of 5100 cells using a DSA Inh Drive = 1.5 mV; P ~ 0, F(2, 31.495) = 921.33, repeated measures ANOVA). i, Temporal dynamics of the Z-scored DOWN-state responses across all model neurons from H (top-left, mean ± IC95; P < 10−104, KW test). Simulated DSA neurons reach peak firing rate ~ 40 ms after maximum rate decrease of pyramidal cells and interneurons during DOWN-states (top-right; medians, interquartile ranges and error bars). Bottom, the average CCG delay between simulated DSA neuron spikes and spikes of other cells (bottom, dashed gray line; P < 10−315, KW test) matches DOWNs states responses (~40 ms, left avCCG average curves, right, medians, interquartile ranges, maxima and minima). j, Similar displays as in I but generated from recorded neurons (mean ± IC95; n = 1457, 775 and 47 PYr, INT and DSAn, respectively, from 11 mice; P < 10−22 for the temporal dynamic of the Z-scored DOWN-state responses and P < 10−7 for the average CCG delay, KW test). The trough of the cross-correlogram of DSA neurons versus other neurons occurred earlier than the peak of the cross-correlograms of other neurons as in the model. *P < 0.05; **P < 0.01; ***P < 0.001. For model details, see Methods.
Extended Data Fig. 4 |. Mechanisms of DSA neurons firing during the DOWN-states.

a, Top: Z-scored peristimulus histogram (PSTh) for cells recorded in the Sst::Chr animals (n = 2 mice), grouped as pyramidal cells (PYR, red), interneurons (INT, blue) and DSA neurons (gray). Horizontal bar: duration of optogenetic stimulation. Middle: firing rate trajectory for the DSA – INT response (P<10−29 between baseline and rebound epochs, KW test). Bottom: Firing trajectory for the DSA – PYR response (P < 10−11, KW test). b, Same than A but for PV::ChR animals (n = 4 mice; P=0.009 and P = 0.39 for ‘INT resp vs DSA resp’ and ‘PYR resp vs DSA resp’, respectively, KW test). Non-significant, n.s. c, Spike auto-correlogram (top, mean ± IC95) and spike waveform (bottom, mean ± IC95) of ID2/Nkx2.1-expressing DSA neurons (gray, n = 6) in optogenetic experiments are similar to non-responding DSA neurons (dark yellow, n = 3, ID2-/Nkx2.1- or non-recombined neurons) as well as to DSA neurons observed in other mice (black, n = 41; KW test). d, Depth distribution of all recorded units (as in Fig. 1i), highlighting the position of the nNOS+ neurons (yellow diamonds; n = 25 cells optogenetically responding cells from 4 Nos1::Chr mice). Note the deep position of the two DSA/nNOS+ neurons (encircled). e, Same layout as in D but highlighting the position of the ID2 + /Nkx2.1+ cells in black circles (n = 8 from 6 ID2/Nkx2.1::Ai80 mice). Note that the two ID2 + /Nkx2.1+ cells, residing above layer 5 (encircled), were not DSA neurons. **P < 0.01; ***P < 0.001, after Tukey’s honesty post hoc multiple comparisons.
Extended Data Fig. 5 |. Intrinsic electrophysiological features of L5/6 ID2/Nkx2.1 neurons.

a, Group differences (mean ± SD) for three electrophysiological parameters between L1 neurogliaform cells (green, n = 25 L1 NGFC), L5/6 ID2/Nkx2.1 (gray, n = 18 cells) and delayed fast spiking cells (in blue, from ref. 30; n = 23 dFS). Note that Goldberg et al used 600 ms second square pulses on the delayed respond tests. b, Example of rebound spiking test protocol. Average traces from a L5/6 ID2/Nkx2.1 cell held at −70 mV, then hyperpolarized to −90 mV for 3 s, then depolarized to either −70 mV (black) or −50 mV (red). If present, rebound spikes should be apparent immediately following the depolarizing step (n = 11 L5/6 ID2/Nkx2.1 cells).
Extended Data Fig. 6 |. Anatomical features of deep and superficial cortical ID2/Nkx2.1 interneurons.
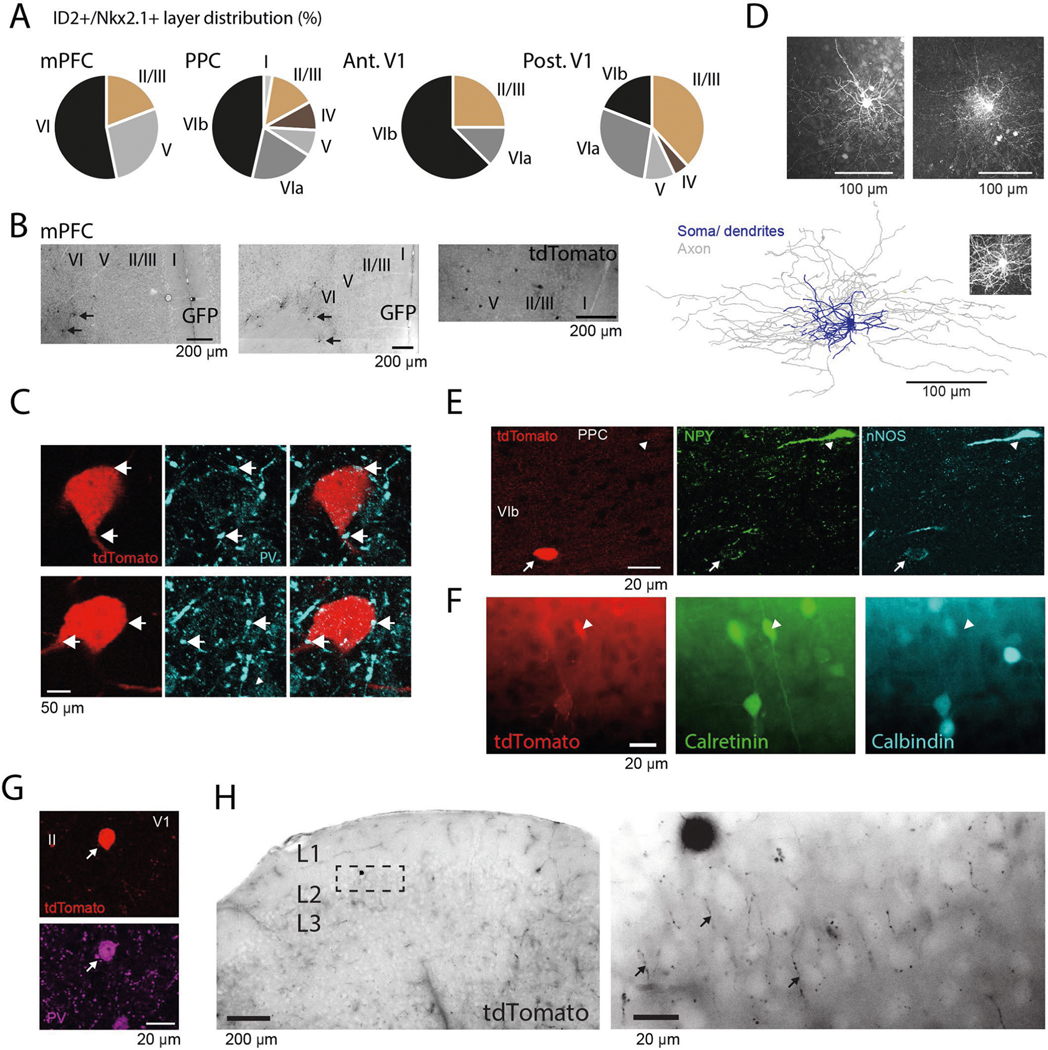
a, Depth distribution of ID2 + /Nkx2.1+ neurons (tdTomato+ or GFP+) in the medial prefrontal cortex (mPFC), PPC and anterior and posterior regions of the primary visual cortex (V1). b, Photographs showing distribution of GFP+ (or tdTomato+) neurons in the mPFC of an ID2/Nkx2.1::Ai80 mouse (Ai65) (top and middle images, reverse contrast epifluorescence; n = 47 neurons in 9 samples from 3 mice). Most neurons were found in layer 6. c, A layer 6 tdTomato+ ID2/Nkx2.1 neuron (red) at two different depth levels. Note the putative PV+ (cyan) puncta in close apposition to the tdTomato+ cell (arrows). Top, 0.38 μm-thick single optical section. Bottom, 0.76 μm-thick maximum intensity z-projection (representative micrograph, tested in 3 mice). d, Top: Z projections (confocal maximum-intensity projection) of two intracellularly-filled tdTomato+ lD2/Nkx2.1 neuron from two ID2/NKx2.1::Ai65 mouse. Bottom: reconstruction of an intracellularly-filled tdTomato+ lD2/Nkx2.1 neuron (from a 223.2 μm thick confocal stack, maximum-intensity projection as inset) of an ID2/NKx2.1::Ai65 mouse. The two reconstructions of the ID2/Nkx2.1 neurons showed similarities to neurogliaform cells in L2/324,29 and L126. Total dendritic length: 7968.5 μm and 11641 μm for the cell in Fig. 5d and the cell in Extended Data Fig. 6, respectively; total axonal length: 77873 μm and 53669 μm; membrane surface in dendrites and soma: 1.02 × 105 μm2 and 2.81 × 105 μm2; membrane surface in axons: 9.21 × 105 μm2 and 1.14 × 106 μm2; number of dendritic branches: 117 and 105; axonal branches: 747 and 314. e, ID2+/Nkx2.1+ neuron (red) in L6b weakly immunoreactive for both NPY and nNos (arrow; n = 64 tdTomato+ ID2/Nkx2.1 neuron from 3 mice were tested against NPY). Note also strongly immunoreactive NPY+ nNOS+ neuron lacking tdTomato expression (arrowhead on top). f, A tdTomato+ ID2/Nkx2.1 neuron in L5 of PPC immunoreactive for calretinin (white arrowheads) and lacking expression of Calbindin (widefield epifluorescence; n = 54 tdTomato+ ID2/Nkx2.1 neuron from 3 mice were tested, 2 of which were positive for calretinin). g, PV+ tdTomato+ neuron in L2 of V1 (representative micrograph, tested in 3 mice). h, Left: a layer 2 tdTomato+ ID2/Nkx2.1 neuron in layer 2 (widefield epifluorescence, reverse contrast). right, high magnification of the marked rectangle area shows the tdTomato+ ID2/Nkx2.1 neuron axon distribution (‘cartridges’, some of them marked by arrows) typical of axo-axonic neurons (widefield epifluorescence, reverse contrast; representative micrograph, tested in 3 mice). The ID2/Nkx2.1 cells labeled here are identical to the Lamp5/Lhx6 type described in ref. 21,22. ID2 and Lamp5 are both broadly expressed in putative neurogliaform interneurons21, and Lhx6 is expressed downstream of Nkx2.1 in cortical interneurons. The proportion of the Lamp5/Lhx6 subtype has been reported to be 1.4 ± 0.2% of total interneurons22, with the majority located in L5/6.
Extended Data Fig. 7 |. Effector role of the DSA neurons.

a, Cross-correlation (CCG) between Non-Mod (reference, n = 648 units) and –Mod (yellow, n = 118 units) compared to the CCG within only Non-Mod cells (green) (top and bottom-left; mean ± IC95; P = 0.004, KW test), and CCG asymmetry [before-after]/[before+after] (bottom-right; medians, interquartile ranges, maxima and minima; P < 10−5, KW test). b, Amplitude response differences (medians, interquartile ranges, maxima and minima) for –Mod (n = 51 units from 9 session with at least 1 DSAn from 6 ID2/Nkx2.1::CatCh mice) and Non-Mod cells (n = 195 units) in LOW and HIGH DSA neuron rate DOWN-UP events. c, Temporal dynamics of the Z-scored DOWN-UP events responses for LOW and hIGh events (n = 2.954 and 5.317 events, respectively; medians, interquartile ranges, maxima and minima). d, Averages (mean ± IC95) of the Fourier-transform spectra for non-stimulated (control, black) and stimulated (green) epochs (top) during NREM (n = 12 sessions from 6 mice). Peak frequency, peak amplitude and delta power (0.5–4 Hz) comparisons for non-stimulated and stimulated epochs (bottom). Note decreased peak frequency of in the delta band during stimulated epochs, likely reflecting the prolongation of DOWN state upon activation of ID2/Nkx2.1 neurons (see Fig. 6). e, Bottom, Z-scored sequential activity of a simultaneously recorded population during DOWN-UP events sorted for ‘High DSA rate’ events (session mean template), vertically arranged by latency. Top, session mean template for ‘Low DSA rate’ events but arranged according to ‘high DSA rate’ classification (see Fig. 6f for ‘Low DSA rate’). Note that the two types of event sequences are different when DSA spiking activity in the slow oscillation event is high or low. *p < 0.05; **p < 0.01; ***p < 0.001.
Extended Data Fig. 8 |. Stimulation of DSA neurons during NREM sleep.

a, Multitaper spectrogram from 0–40 Hz of one entire recording day with NREM optogenetic stimulation (including baseline, behavioral session 1, rest period and behavioral session 2) utilized for state scoring. Middle, electromyogram (EMG) over the same session (au, arbitrary units). Bottom, state scoring of the same session (as in ref. 17). States are coded as label to the left. b, Three-dimensional plot showing state segregations. Each point corresponds to 1 s of recording time, with colors indicating the identified state during that second as labeled. c, d, Same than a-B but for a no-simulation (control) day. e, NREM sleep durations (mean ± SD) between test sessions 1 and 2 are similar in the four different conditions. f, Normalized fractions of time spent in Wake, NREM and rEM in the home cage between behavioral sessions 1 and 2. g, CCG trace (mean ± IC95) between light pulses and UP and DOWN states (left) for all NREM stimulated sessions (n = 15 sessions). Right, fraction of stimulated events over all detected events (medians, interquartile ranges, maxima and minima; P < 10−5, KW test). ***p < 0.001.
Acknowledgements
We would like to thank L. Menéndez de la Prida, K. McClain, D. Levenstein and A. Fernández-Ruiz and the rest of the members of the Buzsáki Laboratory for helpful comments on the manuscript. This work was supported by the European Molecular Biology Organization (EMBO) postdoctoral fellowship (EMBO ALTF 1161–2017), a Human Frontiers Science Program postdoctoral fellowship (LT0000717/2018) to M.V., the UK Medical Research Council (MR/R011567/1) to T.J.V., a Leon Levy Neuroscience Fellowship to I.Z., P01NS074972, R01NS110079, R01NS107257 and F31NS106793 to B.R. and NIH MH54671, NIH MH107396, NS 090583, NSF PIRE (grant no. 1545858), U19 NS107616 and U19 NS104590.
Footnotes
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41593-021-00797-6.
Competing interests
The authors declare no competing interests.
Additional information
Extended data is available for this paper at https://doi.org/10.1038/s41593-021-00797-6.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41593-021-00797-6.
Peer review information Nature Neuroscience thanks Giulio Tononi and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Code availability
All custom code is freely available on the Buzsáki Laboratory repository: https://github.com/buzsakilab/buzcode.
Data availability
The data that support the main findings of this study are publicly available in the Buzsaki Lab Databank: https://buzsakilab.com/wp/public-data/.
References
- 1.Buzsaki G. et al. Nucleus basalis and thalamic control of neocortical activity in the freely moving rat. J. Neurosci 8, 4007–4026 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Steriade M, Nunez A. & Amzica F. A novel slow (< 1 Hz) oscillation of neocortical neurons in vivo: depolarizing and hyperpolarizing components. J. Neurosci 13, 3252–3265 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sanchez-Vives MV & McCormick DA Cellular and network mechanisms of rhytmic recurrent activity in neocortex. Nat. Neurosci 3, 1027–1034 (2000). [DOI] [PubMed] [Google Scholar]
- 4.Hasenstaub A. et al. Inhibitory postsynaptic potentials carry synchronized frequency information in active cortical networks. Neuron 47, 423–435 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Steriade M. & Timofeev I. Neuronal plasticity in thalamocortical networks during sleep and waking oscillations. Neuron 37, 563–576 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Tononi G. & Cirelli C. Sleep and the price of plasticity: from synaptic and cellular homeostasis to memory consolidation and integration. Neuron 81, 12–34 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takehara-Nishiuchi K. & McNaughton BL Spontaneous changes of neocortical code for associative memory during consolidation. Science 322, 960–963 (2008). [DOI] [PubMed] [Google Scholar]
- 8.Todorova R. & Zugaro M. Isolated cortical computations during delta waves support memory consolidation. Science 366, 377–381 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Massimini M, Huber R, Ferrarelli F, Hill S. & Tononi G. The sleep slow oscillation as a traveling wave. J. Neurosci 24, 6862–6870 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luczak A, Barthó P, Marguet SL, Buzsáki G. & Harris KD Sequential structure of neocortical spontaneous activity in vivo. Proc. Natl Acad. Sci. USA 104, 347–352 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gerashchenko D. et al. Identification of a population of sleep-active cerebral cortex neurons. Proc. Natl Acad. Sci. USA 105, 10227–10232 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morairty SR et al. A role for cortical nNOS/NK1 neurons in coupling homeostatic sleep drive to EEG slow wave activity. Proc. Natl Acad. Sci. USA 110, 20272–20277 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zielinski MR et al. Somatostatin+/nNOS+ neurons are involved in delta electroencephalogram activity and cortical-dependent recognition memory. Sleep 42, 1–16 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Compte A, Sanchez-Vives MV, McCormick DA & Wang XJ Cellular and network mechanisms of slow oscillatory activity (<1 Hz) and wave propagations in a cortical network model. J. Neurophysiol 89, 2707–2725 (2003). [DOI] [PubMed] [Google Scholar]
- 15.Jercog D. et al. UP-DOWN cortical dynamics reflect state transitions in a bistable network. eLife 6, e22425 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Senzai Y, Fernandez-Ruiz A. & Buzsáki G. Layer-specific physiological features and interlaminar interactions in the primary visual cortex of the mouse. Neuron 101, 500–513(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Watson BO, Levenstein D, Greene JP, Gelinas JN & Buzsáki G. Network homeostasis and state dynamics of neocortical sleep. Neuron 90, 839–852 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Okun M. et al. Diverse coupling of neurons to populations in sensory cortex. Nature 521, 511–515 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.English DF et al. Pyramidal cell–interneuron circuit architecture and dynamics in hippocampal networks. Neuron 96, 505–520 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tremblay R, Lee S. & Rudy B. GABAergic interneurons in the neocortex: from cellular properties to circuits. Neuron 91, 260–292 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tasic B. et al. Shared and distinct transcriptomic cell types across neocortical areas. Nature 563, 72–78 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hodge RD et al. Conserved cell types with divergent features in human versus mouse cortex. Nature 573, 61–68 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krienen FM et al. Innovations present in the primate interneuron repertoire. Nature 586, 262–269 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oláh S. et al. Regulation of cortical microcircuits by unitary GABA-mediated volume transmission. Nature 461, 1278–1281 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Overstreet-Wadiche L. & McBain CJ Neurogliaform cells in cortical circuits. Nat. Rev. Neurosci 16, 458–468 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schuman B. et al. Four unique interneuron populations reside in neocortical layer 1. J. Neurosci 39, 125–139 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang X. et al. Principles of connectivity among morphologically defined cell types in adult neocortex. Science 350, aac9462 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cadwell CR et al. Electrophysiological, transcriptomic and morphologic profiling of single neurons using Patch-seq. Nat. Biotechnol 34, 199–203 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Szabadics J, Tamás G. & Soltesz I. Different transmitter transients underlie presynaptic cell type specificity of GABAA,slow and GABAA,fast. Proc. Natl Acad. Sci. USA 104, 14831–14836 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goldberg EM et al. K+ channels at the axon initial segment dampen near-threshold excitability of neocortical fast-spiking GABAergic interneurons. Neuron 58, 387–400 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karube F, Kubota Y. & Kawaguchi Y. Axon branching and synaptic bouton phenotypes in GABAergic nonpyramidal cell subtypes. J. Neurosci 24, 2853–2865 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peyrache A, Battaglia FP & Destexhe A. Inhibition recruitment in prefrontal cortex during sleep spindles and gating of hippocampal inputs. Proc. Natl Acad. Sci. USA 108, 17207–17212 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Levenstein D, Buzsáki G. & Rinzel J. NREM sleep in the rodent neocortex and hippocampus reflects excitable dynamics. Nat. Commun 10, 1–12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harvey CD, Coen P. & Tank DW Choice-specific sequences in parietal cortex during a virtual-navigation decision task. Nature 484, 62–68 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoffman KL & McNaughton BL Coordinated reactivation of distributed memory traces in primate neocortex. Science 297, 2070–2073 (2002). [DOI] [PubMed] [Google Scholar]
- 36.Kubota Y, Hattori R. & Yui Y. Three distinct subpopulations of GABAergic neurons in rat frontal agranular cortex. Brain Res. 649, 159–173 (1994). [DOI] [PubMed] [Google Scholar]
- 37.Perrenoud Q, Rossier J, Geoffroy H, Vitalis T. & Gallopin T. Diversity of GABAergic interneurons in layer VIa and VIb of mouse barrel cortex. Cereb. Cortex 23, 423–441 (2013). [DOI] [PubMed] [Google Scholar]
- 38.Tamás G, Lörincz A, Simon A. & Szabadics J. Identified sources and targets of slow inhibition in the neocortex. Science 299, 1902–1905 (2003). [DOI] [PubMed] [Google Scholar]
- 39.Craig MT & McBain CJ The emerging role of GABAB receptors as regulators of network dynamics: fast actions from a ‘slow’ receptor? Curr. Opin. Neurobiol 26, 15–21 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tricoire L. et al. Common origins of hippocampal ivy and nitric oxide synthase expressing neurogliaform cells. J. Neurosci 30, 2165–2176 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Niquille M. et al. Neurogliaform cortical interneurons derive from cells in the preoptic area. eLife 7, e32017 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Taniguchi H, Lu J. & Huang ZJ The spatial and temporal origin of chandelier cells in mouse neocortex. Science 339, 70–74 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Van Der Werf YD, Witter MP & Groenewegen HJ The intralaminar and midline nuclei of the thalamus. Anatomical and functional evidence for participation in processes of arousal and awareness. Brain Res. Rev 39, 107–140 (2002). [DOI] [PubMed] [Google Scholar]
- 44.Brombas A, Fletcher LN & Williams SR Activity-dependent modulation of layer 1 inhibitory neocortical circuits by acetylcholine. J. Neurosci 34, 1932–1941 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Olsen SR, Bortone DS, Adesnik H. & Scanziani M. Gain control by layer six in cortical circuits of vision. Nature 483, 47–54 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
References
- 46.Stark E, Koos T. & Buzsáki G. Diode probes for spatiotemporal optical control of multiple neurons in freely moving animals. J. Neurophysiol 108, 349–363 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pachitariu M, Steinmetz NA, Kadir SN, Carandini M. & Harris KD Fast and accurate spike sorting of high-channel count probes with KiloSort. Adv. Neural Inform. Process. Sys 29, 4448–4456 (2016). [Google Scholar]
- 48.Valero M. et al. Mechanisms for selective single-cell reactivation during offline sharp-wave ripples and their distortion by fast ripples. Neuron 94, 1234–1247.e7 (2017). [DOI] [PubMed] [Google Scholar]
- 49.Navas-Olive A. et al. Multimodal determinants of phase-locked dynamics across deep-superficial hippocampal sublayers during theta oscillations. Nat. ommun 11, 2217 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stark E. & Abeles M. Unbiased estimation of precise temporal correlations between spike trains. J. Neurosci. Methods 179, 90–100 (2009). [DOI] [PubMed] [Google Scholar]
- 51.Barrio-Alonso E, Fontana B, Valero M. & Frade JM Pathological aspects of neuronal hyperploidization in Alzheimer’s disease evidenced by computer simulation. Front. Genet 11, 287 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stimberg M, Brette R. & Goodman DFM Brian 2, an intuitive and efficient neural simulator. eLife 8, e47314 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Valero M. et al. Determinants of different deep and superficial CA1 pyramidal cell dynamics during sharp-wave ripples. Nat. Neurosci 18, 1281–1290 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Viney TJ et al. Network state-dependent inhibition of identified hippocampal CA3 axo-axonic cells in vivo. Nat. Neurosci 16, 1802–1811 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Salib M. et al. GABAergic medial septal neurons with low-rhythmic firing innervating the dentate gyrus and hippocampal area CA3. J. Neurosci 39, 4527–4549 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hovde K, Gianatti M, Witter MP & Whitlock JR Architecture and organization of mouse posterior parietal cortex relative to extrastriate areas. Eur. J. Neurosci 49, 1313–1329 (2019). [DOI] [PubMed] [Google Scholar]
- 57.Feng L, Zhao T. & Kim J. Neutube 1.0: a new design for efficient neuron reconstruction software based on the SWC format. eNeuro 2, ENEURO.0049–14.2014 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the main findings of this study are publicly available in the Buzsaki Lab Databank: https://buzsakilab.com/wp/public-data/.