

Abstract
Background
Among people with a diagnosis of borderline personality disorder (BPD) who are engaged in clinical care, prescription rates of psychotropic medications are high, despite the fact that medication use is off‐label as a treatment for BPD. Nevertheless, people with BPD often receive several psychotropic drugs at a time for sustained periods.
Objectives
To assess the effects of pharmacological treatment for people with BPD.
Search methods
For this update, we searched CENTRAL, MEDLINE, Embase, 14 other databases and four trials registers up to February 2022. We contacted researchers working in the field to ask for additional data from published and unpublished trials, and handsearched relevant journals. We did not restrict the search by year of publication, language or type of publication.
Selection criteria
Randomised controlled trials comparing pharmacological treatment to placebo, other pharmacologic treatments or a combination of pharmacologic treatments in people of all ages with a formal diagnosis of BPD. The primary outcomes were BPD symptom severity, self‐harm, suicide‐related outcomes, and psychosocial functioning. Secondary outcomes were individual BPD symptoms, depression, attrition and adverse events.
Data collection and analysis
At least two review authors independently selected trials, extracted data, assessed risk of bias using Cochrane's risk of bias tool and assessed the certainty of the evidence using the GRADE approach. We performed data analysis using Review Manager 5 and quantified the statistical reliability of the data using Trial Sequential Analysis.
Main results
We included 46 randomised controlled trials (2769 participants) in this review, 45 of which were eligible for quantitative analysis and comprised 2752 participants with BPD in total. This is 18 more trials than the 2010 review on this topic. Participants were predominantly female except for one trial that included men only. The mean age ranged from 16.2 to 39.7 years across the included trials. Twenty‐nine different types of medications compared to placebo or other medications were included in the analyses. Seventeen trials were funded or partially funded by the pharmaceutical industry, 10 were funded by universities or research foundations, eight received no funding, and 11 had unclear funding.
For all reported effect sizes, negative effect estimates indicate beneficial effects by active medication. Compared with placebo, no difference in effects were observed on any of the primary outcomes at the end of treatment for any medication.
Compared with placebo, medication may have little to no effect on BPD symptom severity, although the evidence is of very low certainty (antipsychotics: SMD ‐0.18, 95% confidence interval (CI) ‐0.45 to 0.08; 8 trials, 951 participants; antidepressants: SMD −0.27, 95% CI −0.65 to 1.18; 2 trials, 87 participants; mood stabilisers: SMD −0.07, 95% CI −0.43 to 0.57; 4 trials, 265 participants).
The evidence is very uncertain about the effect of medication compared with placebo on self‐harm, indicating little to no effect (antipsychotics: RR 0.66, 95% CI 0.15 to 2.84; 2 trials, 76 participants; antidepressants: MD 0.45 points on the Overt Aggression Scale‐Modified‐Self‐Injury item (0‐5 points), 95% CI −10.55 to 11.45; 1 trial, 20 participants; mood stabilisers: RR 1.08, 95% CI 0.79 to 1.48; 1 trial, 276 participants).
The evidence is also very uncertain about the effect of medication compared with placebo on suicide‐related outcomes, with little to no effect (antipsychotics: SMD 0.05, 95 % CI −0.18 to 0.29; 7 trials, 854 participants; antidepressants: SMD −0.26, 95% CI −1.62 to 1.09; 2 trials, 45 participants; mood stabilisers: SMD −0.36, 95% CI −1.96 to 1.25; 2 trials, 44 participants).
Very low‐certainty evidence shows little to no difference between medication and placebo on psychosocial functioning (antipsychotics: SMD −0.16, 95% CI −0.33 to 0.00; 7 trials, 904 participants; antidepressants: SMD −0.25, 95% CI ‐0.57 to 0.06; 4 trials, 161 participants; mood stabilisers: SMD −0.01, 95% CI ‐0.28 to 0.26; 2 trials, 214 participants).
Low‐certainty evidence suggests that antipsychotics may slightly reduce interpersonal problems (SMD −0.21, 95% CI −0.34 to ‐0.08; 8 trials, 907 participants), and that mood stabilisers may result in a reduction in this outcome (SMD −0.58, 95% CI ‐1.14 to ‐0.02; 4 trials, 300 participants). Antidepressants may have little to no effect on interpersonal problems, but the corresponding evidence is very uncertain (SMD −0.07, 95% CI ‐0.69 to 0.55; 2 trials, 119 participants).
The evidence is very uncertain about dropout rates compared with placebo by antipsychotics (RR 1.11, 95% CI 0.89 to 1.38; 13 trials, 1216 participants). Low‐certainty evidence suggests there may be no difference in dropout rates between antidepressants (RR 1.07, 95% CI 0.65 to 1.76; 6 trials, 289 participants) and mood stabilisers (RR 0.89, 95% CI 0.69 to 1.15; 9 trials, 530 participants), compared to placebo.
Reporting on adverse events was poor and mostly non‐standardised. The available evidence on non‐serious adverse events was of very low certainty for antipsychotics (RR 1.07, 95% CI 0.90 to 1.29; 5 trials, 814 participants) and mood stabilisers (RR 0.84, 95% CI 0.70 to 1.01; 1 trial, 276 participants). For antidepressants, no data on adverse events were identified.
Authors' conclusions
This review included 18 more trials than the 2010 version, so larger meta‐analyses with more statistical power were feasible. We found mostly very low‐certainty evidence that medication may result in no difference in any primary outcome. The rest of the secondary outcomes were inconclusive. Very limited data were available for serious adverse events. The review supports the continued understanding that no pharmacological therapy seems effective in specifically treating BPD pathology. More research is needed to understand the underlying pathophysiologic mechanisms of BPD better. Also, more trials including comorbidities such as trauma‐related disorders, major depression, substance use disorders, or eating disorders are needed. Additionally, more focus should be put on male and adolescent samples.
Plain language summary
What are the benefits and risks of medication for people with borderline personality disorder?
Key messages
This review is an update of a previous review on the same topic published in 2010. Although this review includes an additional 18 studies, the conclusions remain the same: there are probably no benefits and risks of medications for borderline personality disorder (BDP), but the evidence is unclear.
Better and larger studies comparing the effects of medication with placebo are needed. Such studies should focus on men, adolescents and those with additional psychiatric diagnoses.
What is BPD?
BPD affects how a person interacts with others and understands one's self. Although its exact causes are unclear, it is thought to result from a combination of genetic and environmental factors (e.g. stressful or traumatic life events when growing up). Approximately 2% of adults and 3% of adolescents are affected.
The symptoms of BPD can be grouped into four categories.
Instability in mood: People with BPD may experience intense feelings that change rapidly and are difficult to control. They may also feel empty and abandoned much of the time.
Cognitive distortions (disturbed patterns of thinking): People with BPD often have upsetting thoughts (e.g. they may think that they are a terrible person). They can have brief episodes of strange experiences (e.g. paranoid ideations or stress‐induced dissociative experiences (i.e. feeling detached from the world around them).
Impulsive behaviour: People with BPD may act impulsively and do things that could harm themselves (e.g. when sad and depressed, they may self‐harm or have suicidal feelings). They might also engage in reckless behaviour (e.g. drug misuse).
Intense but unstable relationships: People with BPD may find it difficult to keep stable relationships (e.g. they may feel very worried about being abandoned and might constantly text or call, or make threats to harm or kill themselves if the person leaves them).
A person only needs to experience five out of nine criteria across these categories to be given a diagnosis of BPD.
How is BPD treated?
No medication has been approved for the treatment of BPD. Nonetheless, a large proportion of people with BPD are given medications for sustained periods of time to alleviate their symptoms. The type of medication given is chosen based on its known effects on other disorders with similar symptoms.
Review question
We wanted to find out whether medications to treat BPD work better or worse than placebo; whether one medication works better than another; or whether one combination of medications work better than another combination of medications.
We wanted to look at how well medications worked on BPD severity, self‐harm, suicide‐related outcomes, and functioning (how well a person performs in everyday life).
We also wanted to find out if medications are associated with any unwanted side effects.
What did we do?
We searched for studies that compared the effects of different medications with placebo, another medication, or a combination of medications, in people diagnosed with BPD.
We compared and summarised the results and rated our confidence in the evidence based on factors such as sample size and methods used. Below, we present the findings from our key comparison: medication versus placebo.
What did we find?
We found 46 studies that involved 2769 people with BPD. The smallest study had 13 participants and the largest 451 participants. There were four studies with more than 100 participants. Except for one study that included men only, all studies included women. The average age of the participants ranged from 16 years to 39 years. Most studies were conducted in outpatient settings (31 studies) in Europe (20 studies) and lasted between four and 52 weeks. Pharmaceutical companies fully or partially funded 16 studies.
The studies looked at the effects of 27 different medications, mostly classified as: 1) antipsychotics (drugs to treat psychosis where a person’s thoughts and mood are so impaired that the person has lost contact with reality); 2) antidepressants (drugs to treat depression); or 3) mood stabilisers (drugs to control and even out mood swings, reducing both high moods (mania) and low moods (depression)).
Compared with placebo, medications seem to make little to no difference to BPD severity, self‐harm, suicide‐related outcomes, and psychosocial functioning. They may make little to no difference as to whether a person continues in a study or drops out. Compared to placebo, antipsychotics and mood stabilisers may make little to no difference to the occurrence of unwanted or harmful effects. No study reported on the side effects of antidepressants.
What are the limitations of the evidence?
Our confidence in the evidence varied between very low and low. The results of future research could differ from the results of this review. Four main factors reduced our confidence in the evidence. First, not all of the studies provided data about everything that we were interested in. Second, the results were very inconsistent across the different studies. Third, there were not enough studies to be certain about the results of our outcomes. Fourth, many studies did not clearly report how they were conducted.
How up‐to‐date is this evidence?
The evidence is up‐to‐date to February 2022.
Summary of findings
Summary of findings 1. Antipsychotics compared with placebo for people with borderline personality disorder.
| Antipsychotics compared with placebo for people with borderline personality disorder | ||||||
|
Patient or population: people with borderline personality disorder Settings: inpatient and outpatient Intervention: antipsychotics Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Antipsychotics | |||||
|
BPD symptom severity Timing of outcome assessment: end of treatment (5 to 12 weeks treatment duration) |
‐ | The mean score in the intervention groups was 0.18 SD lower (0.45 lower to 0.08 higher, I2 = 70%) than in the placebo group | ‐ | 951 (8 trials) | ⊕⊝⊝⊝ Very Lowa,b | An SMD of 0.18 represents a small effect. **TSA‐adjusted CI is −3.27 to 0.86 on the Zanarini BPD scale. Z value in futility area TSA DARIS = 951 TSA in futility area |
|
Self‐harm Timing of outcome assessment: end of treatment (8 to 24 weeks treatment duration) |
310 per 1000 | 208 per 1000 (211 less to 127 more self‐harm incidents than in the placebo group) | RR 0.66 (95% CI 0.15 to 2.84, I2 = 67%) | 76 (2 trials) | ⊕⊝⊝⊝ Very Lowc,d | ‐ |
|
Suicide‐related outcomes Timing of outcome assessment: end of treatment (6 to 12 weeks treatment duration) |
‐ | The mean score in the intervention groups was 0.05 SD higher (0.18 lower to 0.29 higher, I2 = 55%) than in the placebo group | ‐ | 854 (7 trials) | ⊕⊝⊝⊝ Very Lowa,e | A SMD of 0.05 represents a marginal effect. RR 0.73 (95% CI 0.31 to 1.73; 2 trials, 61 participants), low‐certainty evidence |
|
Psychosocial functioning Timing of outcome assessment: end of treatment (5 to 12 weeks treatment duration) |
‐ | The mean score in the intervention groups was 0.16 SD lower (0.33 lower to 0.00 lower, I2 = 75%) than in the placebo group | ‐ | 904 (7 trials) | ⊕⊝⊝⊝ Very Lowa,f,g | A SMD of 0.16 represents a small effect. |
|
Interpersonal problems Timing of outcome assessment: end of treatment (5 to 12 weeks treatment duration) |
‐ | The mean score in the intervention groups was 0.21 SD lower (0.34 lower to 0.08 lower, I2 = 0%) than in the placebo group | ‐ | 907 (8 trials) | ⊕⊕⊝⊝ Lowa | A SMD of 0.21 represents a small effect. TSA‐adjusted CI is −0.60 to 0.08 on Zanarini BPD Scale ‐Interpersonal Problem Index TSA DARIS = 386 |
|
Attrition Timing of outcome assessment: end of treatment (5 weeks to 6 months treatment duration) |
325 per 1000 | 361 per 1000 (36 less to 123 more than in the placebo group dropped out) | RR 1.11 (0.89 to 1.38; I2 = 35%) | 1216 (13 trials) | ⊕⊝⊝⊝ Very Lowa,h | TSA‐adjusted CI is 0.74 to 2.13 TSA DARIS = 2008 |
|
Non‐serious adverse events Timing of outcome assessment: end of treatment (8 to 12 weeks treatment duration) |
560 per 1000 |
599 per 1000 (56 less to 162 more than in the placebo group dropped out) |
RR 1.07 (0.90 to 1.29; I2 = 57%) | 814 (5 trials) | ⊕⊝⊝⊝ Very Lowa,i | TSA‐adjusted CI is 0.79 to 1.47 TSA DARIS = 1250 TSA in futility area |
| *The basis for the assumed risk (e.g. the median control group risk across trials) is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). **TSA did not include cross‐over trials. CI: Confidence interval; RR: Risk Ratio; BPD: Borderline Personality Disorder; SD: standard deviation; SMD: Standardised mean difference TSA Trial Sequential Analysis, DARIS: Diversity‐adjusted required information size | ||||||
| GRADE Working Group grades of evidence High certainty: Further research is very unlikely to change our confidence in the estimate of effect. Moderate certainty: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low certainty: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low certainty: We are very uncertain about the estimate. | ||||||
aWe downgraded the evidence two levels due to risk of bias of the included studies (indication of selective outcome reporting, indication of incomplete outcome data presented in the included studies) (rated by HEC and OJS (E))
bWe downgraded the evidence one level due to inconsistency (I2= 70%) (rated by HEC and OJS)
c We downgraded the evidence two levels due to imprecision (wide CI around the pooled effect estimate suggest both an appreciable effect and no effect; few participants included in the studies) (rated by HEC and OJS)
dWe downgraded the evidence one level due to inconsistency (I2 = 67%) (rated by HEC and OJS (D))
eWe downgraded the evidence one level due to inconsistency (I2 = 55%) (rated by HEC and OJS)
fWe downgraded the evidence one level due to inconsistency (I2 = 75%) (rated by HEC and OJS)
gWe downgraded the evidence one level due to imprecision (wide CI around the pooled effect estimate suggest both an appreciable effect and no effect) (rated by HEC and OJS)
h We downgraded the evidence one level due to imprecision (wide CI around the pooled effect estimate suggest both an appreciable effect and no effect; the TSA that did not reach the required information size (RIS)) (rated by HEC and OJS)
i We downgraded the evidence one level due to inconsistency (I2= 57%) (rated by HEC and OJS)
Summary of findings 2. Antidepressants compared with placebo for people with borderline personality disorder.
| Antidepressants compared with placebo for people with borderline personality disorder | ||||||
|
Patient or population: people with borderline personality disorder Settings: inpatient, outpatient, partial inpatient and partial outpatient Intervention: antidepressants Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Antidepressant | |||||
|
BPD symptom severity Timing of outcome assessment: end of treatment (5 to 6 weeks treatment duration) |
‐ | The mean score in the intervention group was 0.27 SD lower (0.65 lower to 1.18 higher, I2= 73) than in the placebo group | ‐ | 87 (2 trials) | ⊕⊝⊝⊝ Very lowa,b | ‐ |
|
Self‐harm Assessed with: OAS‐M (Self‐injury; scale range: 0‐40 (0 = no aggression, 40 = maximum grade of aggression) Timing of outcome assessment: end of treatment (12 weeks treatment duration) |
The mean reduction in self‐harm was 6.55 in the control group | The mean score in the intervention group was0.45 points higher (10.55 lower to 11.45 higher) than in the placebo group | ‐ | 20 (1 trial) | ⊕⊝⊝⊝ Very lowa,c | ‐ |
|
Suicide‐related outcomes Timing of outcome assessment: end of treatment (6 to 12 weeks treatment duration) |
‐ | The mean score in the intervention groups was 0.26 SD lower (1.62 lower to 1.09 higher, I2 = 80%) than in the placebo group | ‐ | 45 (2 trials) | ⊕⊝⊝⊝ Very lowa,c,d | ‐RR 1.00 (95% CI 0.71 to 1.4; 1 trial, 49 participants). Very low‐certainty evidence |
|
Psychosocial functioning Timing of outcome assessment: end of treatment (5 to 12 weeks treatment duration) |
‐ | The mean score in the intervention groups was 0.25 SD lower (0.57 lower to 0.06 higher, I2 = 0%) than in the placebo group | ‐ | 161 (4 trials) | ⊕⊝⊝⊝ Very lowa,e | ‐ |
|
Interpersonal problems Timing of outcome assessment: end of treatment (5 weeks treatment duration) |
‐ | The mean score in the intervention groups was 0.07 SD lower (0.69 lower to 0.55 higher, I2 = 66%) than in the placebo group | ‐ | 119 (2 trials) | ⊕⊝⊝⊝ Very lowa,f | ‐ |
|
Attrition Timing of outcome assessment: end of treatment (5 weeks to 6 months treatment duration) |
170 per 1000 | 182 per 1000 (59 less to 129 more than in the placebo group dropped out ) | RR 1.07 (95% CI 0.65 to 1.76; I2 = 0%) | 289 (6 trials) | ⊕⊕⊝⊝ Lowc, g | TSA‐adjusted CI is 0.07 to 14.98 TSA DARIS = 1816 |
|
Non‐serious adverse events Timing of outcome assessment: end of treatment |
no data available | no data available | no data available | no data available | no data available | no data available |
| *The basis for the assumed risk (e.g. the median control group risk across trials) is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk Ratio; BPD: Borderline Personality Disorder;OAS‐M: Modified Overt Agression Scale; SD: standard deviation; TSA Trial Sequential Analysis, DARIS: Diversity‐adjusted required information size | ||||||
| GRADE Working Group grades of evidence High certainty: Further research is very unlikely to change our confidence in the estimate of effect. Moderate certainty: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low certainty: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low certainty: We are very uncertain about the estimate. | ||||||
aWe downgraded the evidence by two levels due to imprecision (wide CI around the pooled effect estimate suggest both an appreciable effect and no effect; few patients included) (rated by HEC and OJS)
bWe downgraded the evidence one level due to inconsistency (a high I2 score of 73%) (rated by HEC and OJS)
cWe downgraded the evidence one level due to risk of bias (indication selective outcome reporting in the included study) (rated by HEC and OJS)
dWe downgraded the evidence one level due to inconsistency (a high I2 score of 80%) (rated by HEC and OJS)
eWe downgraded the evidence one level due to risk of bias (indication of incomplete outcome data) (rated by JSW and OJS) fWe downgraded the evidence one level due to inconsistency (a high I2 score of 66%) (rated by HEC and OJS) g We downgraded the evidence one level due to imprecision (wide CI around the pooled effect estimate suggest both an appreciable effect and no effect; TSA not reaching required information size (RIS)) (rated by HEC and OJS)
Summary of findings 3. Mood stabilisers compared with placebo for people with borderline personality disorder.
| Mood stabilisers compared with placebo for people with borderline personality disorder | ||||||
|
Patient or population: people with borderline personality disorder Settings: inpatient, outpatient, inpatient and outpatient Intervention: mood stabilisers Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Mood stabilisers | |||||
|
BPD symptom severity Timing of outcome assessment: end of treatment (6 to 52 weeks treatment duration) |
‐ | The mean score in the intervention groups was 0.07 SD lower than in the placebo group (0.43 lower to 0.57 higher, I2 = 55%) | ‐ | 265 (4 trials) | ⊕⊝⊝⊝ Very lowa,b,c | A SMD of 0.07 represents a marginal effect. |
|
Self‐harm Timing of outcome assessment: end of treatment (52 weeks treatment duration) |
345 per 1000 |
373 per 1000 (72 less to 166 more self‐harm incidents than in the placebo group) |
RR 1.08 (95% CI 0.79 to 1.48) | 276 (1 trial) | ⊕⊝⊝⊝ Very lowd,e | ‐ |
|
Suicide‐related outcomes Timing of outcome assessment: end of treatment (6 to 10 weeks treatment duration) |
‐ | The mean score in the intervention group was 0.36 SD lower than in the placebo group (1.96 lower to 1.25 higher, I2 = 81%) | ‐ | 44 (2 trials) | ⊕⊝⊝⊝ Very lowd,e,f | ‐ |
|
Psychosocial functioning Timing of outcome assessment: end of treatment (32 days to 6 weeks treatment duration) |
‐ | The mean score in the intervention groups was 0.01 SD lower than in the placebo group (‐0.28 lower to 0.26 higher, I2 = 0%) | ‐ | 214 (2 trials) | ⊕⊝⊝⊝ Verylowd,e,g | A SMD of 0.01 represents a marginal effect. RR 0.64 (95% CI 0.37 to 1.11; 1 trial, 16 participants).Very low‐certainty evidence |
|
Interpersonal problems Timing of outcome assessment: end of treatment (32 days to 24 weeks) |
‐ | The mean score in the intervention groups was 0.58 SD lower than in the placebo group (1.14 lower to 0.02 lower, I2 = 73%) | ‐ | 300 (4 trials) | ⊕⊕⊝⊝ Lowg,h | A SMD of 0.58 represents a moderate effect. |
|
Attrition Timing of outcome assessment: end of treatment (32 days to 24 weeks treatment duration) |
260 per 1000 | 208 per 1000 (6 less to 154 more than in the placebo group dropped out ) | RR 0.89 (95% CI 0.69 to 1.15; I2 = 0%) | 530 (9 trials) | ⊕⊕⊝⊝ Lowa,i | TSA‐adjusted CI is 0.37 to 2.23 TSA DARIS = 1300 |
|
Non‐serious adverse events Timing of outcome assessment: end of treatment (6 weeks treatment duration) |
670 per 1000 |
563 per 1000 (201 less to 7 more than in the placebo group dropped out ) |
RR 0.84 (95% CI 0.70 to 1.01; I2 = 0%) | 276 (1 trial) | ⊕⊝⊝⊝ Very lowa,e | ‐ |
| *The basis for the assumed risk (e.g. the median control group risk across trials) is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk Ratio; BPD: Borderline Personality Disorder; NA: not applicable; OAS‐M: Modified Overt Agression Scale; CGI‐I: Clinical Global Impression scale ‐ Improvement SD: standard deviation; SMD: Standardised mean difference; TSA Trial Sequential Analysis, DARIS: Diversity adjusted required information size | ||||||
| GRADE Working Group grades of evidence High certainty: Further research is very unlikely to change our confidence in the estimate of effect. Moderate certainty: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low certainty: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low certainty: We are very uncertain about the estimate. | ||||||
a We downgraded the evidence one level due to risk of bias in the included studies (indication of incomplete outcome data in the included studies) (rated by HEC and OJS) b We downgraded the evidence one level due to imprecision (wide CI around the pooled effect estimate suggest both an appreciable effect and no effect) (rated by HEC and OJS)
cWe downgraded the evidence one level due to inconsistency (a high I2 score of 55%) (rated by HEC and OJS)
dWe downgraded the evidence one level due to risk of bias (indication of selective outcome reporting in the included study) (rated by HEC and OJS) eWe downgraded the evidence two levels due to imprecision (wide CI around the pooled effect estimate suggest both an appreciable effect and no effects; results based on 1 study or few participants) (rated by HEC and OJS)
fWe downgraded the evidence one level due to inconsistency (a high I2 score of 81%) (rated by HEC and OJS)
gWe downgraded the evidence one level due to inconsistency (a high I2 score of 73%) (rated by HEC and OJS)
hWe downgraded the evidence one level due to risk of bias (indication of incomplete outcome data and vested interests)
iWe downgraded the evidence one level due to imprecision (wide CI around the pooled effect estimate suggest both an appreciable effect and no effects; TSA not reaching required information size (RIS)) (rated by HEC and OJS)
Background
Description of the condition
According to current diagnostic criteria, borderline personality disorder (BPD) is characterised by a pervasive pattern of instability in affect regulation, impulse control, interpersonal relationships, and self‐image (APA 2013; WHO 1993). Clinical hallmarks include emotional dysregulation, impulsive aggression, repeated self‐injury, and chronic suicidal tendencies (Bohus 2021; Fonagy 2009; Lieb 2004). BPD is being widely researched in order to better understand and treat the disorder‐specific symptoms. Its importance stems from the large amount of suffering of the persons concerned (Bohus 2021; Stiglmayr 2005; Zanarini 1998), debilitating functional impairments (Gunderson 2011a; Gunderson 2011b; Niesten 2016; Skodol 2002; Soetmann 2008b), and from the significant impact it has on mental health services (Cailhol 2015; Hörz 2010; Soetmann 2008a; Tyrer 2015; Zanarini 2004a; Zanarini 2012). The problem of deliberate self‐harm is also a particular issue within this group (Ayodeji 2015; Kongerslev 2015; Linehan 1997; Ose 2021; Rossouw 2012). In medical settings, people with BPD often present after self‐harming behaviour or in suicidal crisis and are treated in emergency settings, often involving repeated psychiatric hospitalisations (Bender 2006; Cailhol 2015). It is estimated that about 60% to 78% of people with BPD attempt suicide (Links 2009), though the rate of completed suicides is far less (Bohus 2021). Zanarini and colleagues found suicide rates of 4.5% during 16 years of follow‐up (Zanarini 2015), whereas Stone 1993 reported a suicide rate of 8.5% after 16.5 years. Study estimates of the lifetime risk of suicide among people with BPD range from 3% to 10% (Links 2009). Suicidal behaviour (e.g. behaviour that could cause death such as medication overdosing and purposely crashing in traffic) is reported to occur in up to 84% of people with BPD (Goodman 2012; Soloff 2002). Common risk factors associated with completed suicide are low socioeconomic status, poor psychosocial adjustment, family history of suicide, previous psychiatric hospitalisation, and absence of any outpatient treatment before the attempt (Soloff 2012).
The definition of BPD in the Diagnostic and Statistical Manual of Mental Disorders (DSM) Fifth Edition (DSM‐5; APA 2013), Fourth Edition Text Revision (DMS‐IV‐TR; APA 2000) and Fourth Edition (DSM‐IV; APA 1994) comprises nine criteria that cover the features mentioned above. At least five criteria should be met for a definite categorical BPD diagnosis to be made, and four criteria for probable diagnosis (see Appendix 1). In the alternative diagnostic classification system of the World Health Organization (WHO), the International Classification of Diseases, which is currently in its tenth edition (ICD‐10; WHO 1993), the relating condition is referred to as "Emotionally unstable personality disorder (F60.3)", of which there is an impulsive type (F60.30) and a borderline type (F60.31: see Appendix 2). The latter essentially overlaps with the DSM‐IV definition. There are 10 possible criteria in the ICD‐10 diagnosis for BPD that very closely resembles the DSM‐IV/5 criteria (Ottosson 2002), with the exception of one criterion which is not included in the DSM ("4. Difficulty in maintaining any course of action that offers no immediate reward"; WHO 1993). Out of the 10 ICD‐10 criteria, at least five must be met, one of which must be "a marked tendency to quarrelsome behaviour and to conflicts with others, especially when impulsive acts are thwarted or criticised". Currently, the successive version of the ICD (ICD‐11) is being finalised (WHO 2021). It will abolish any personality disorder categories for the sake of a general description of personality disorder in terms of severity (mild, moderate, severe) and five personality trait domains (negative affectivity, dissociality, anankastia, detachment, and disinhibition). In addition, it will include a "borderline specifier" (6D11.5), which can additionally be diagnosed and which will essentially consist of all nine BPD criteria defined in DSM‐5 (Mulder 2021; WHO 2021; see Appendix 2).
Overall, the prevalence of BPD in the general population is estimated to be about 1.5% (Torgersen 2012), with findings of single epidemiological studies ranging between 0.6% (Coid 2006) and 2.7% (Trull 2010). In clinical populations, BPD occurs frequently (Munk‐Jørgensen 2010), with studies reporting a prevalence ranging from 9.3% to 46.3% and a mean point prevalence across studies of 28.5% (Torgersen 2012). Though BPD is predominantly diagnosed in women (75%; APA 2000, APA 2013, Widiger 1993), it is estimated to be equally prevalent in men by representative community studies (Coid 2006; Grant 2008; Lenzenweger 2007; Ten Have 2016; Tomko 2014; Torgersen 2001; Torgersen 2012). Reasons for this obvious gender bias are discussed as: bias in the diagnostic criteria, bias in the application of the diagnostic criteria by clinicians, bias in diagnostic thresholds across disorders more prevalent in one gender or another, biased population sampling, bias in the assessment instruments, and bias in the diagnostic construct itself (Widiger 1998).
BPD commonly co‐occurs with mood disorders, substance misuse, eating disorders, post‐traumatic stress disorder (PTSD), attention‐deficit hyperactivity disorder (ADHD), and is also associated with other personality disorders (Coid 2006; Lenzenweger 2007; Stepp 2012; Storebø 2014; Tomko 2014).
Although the short‐ to medium‐term outcome of BPD is poor, symptomatic remission rates of about 85% to 88% have been reported within 10 years (Gunderson 2011b; Zanarini 2007). Another, smaller prospective longitudinal study reported a diagnostic remission rate of 55% after 10 years (Alvarez‐Tomas 2017). However, remission only means that diagnostic criteria are no longer fulfilled; it does not indicate the absence of any symptoms. Indeed, whereas acute symptoms — such as self‐mutilation, help‐seeking suicide threats or attempts and impulsivity — in most cases decrease with time, affective symptoms reflecting areas of chronic dysphoria, such as chronic feelings of emptiness, intense anger or a profound sense abandonment, largely remain (Zanarini 2007). Therefore, the majority of people with BPD still have significant levels of symptoms and experience severe and persistent impairment in social functioning, high unemployment rates, physical ill‐health, and a substantially reduced life expectancy (Bohus 2021; Kongerslev 2015; Ng 2016; Schneider 2019). 'Good recovery', defined as being in remission of BPD for a minimum of two years, good social functioning in terms of having at least one emotionally sustaining relationship, and good vocational functioning (working or attending school consistently on a full‐time basis) was only achieved by 50% of individuals with BPD after 10 years of prospective follow‐up, and 59% after 20 years of follow‐up (Zanarini 2012; Zanarini 2018). Younger age seems to be associated with a higher probability of diagnostic remission in the long term (Alvarez‐Tomas 2019), indicating the importance of early diagnosis and intervention (Chanen 2017).
BPD onsets happen in young people, i.e. between puberty and emerging adulthood (Chanen 2013). Today, the diagnosis is regarded reliable and valid also in adolescents, as a similarity in prevalence, phenomenology, and stability has been observed, as well as a markedly different course and outcome as compared to other disorders (Chanen 2017) or ordinary problems. For a long time, clinicians were reluctant to diagnose BPD (not only, but specifically) in adolescents due to the assumed stigma, and wanting to avoid giving young people a diagnosis believed to be an 'uncurable' disorder. However, due to emerging evidence, there is now consensus among the scientific community that the disorder should be detected and diagnosed as early as possible, given that helpful treatments do exist, and that later interventions tend to reinforce functional impairment, disability and therapeutic nihilism (Chanen 2013; Chanen 2017).
Risk factors for a poorer long‐term outcome are comorbid substance use disorders, PTSD, and anxiety cluster disorders (Zanarini 2005; Zanarini 2007), a family history of psychiatric disorder (especially mood disorders and substance use disorders), as well as individual factors such as older age, longer treatment history, pathological childhood experiences, and adult psychosocial functioning (Bohus 2021; Chanen 2012; Kongerslev 2015; Zanarini 2007).
People with BPD often have difficulties achieving and maintaining vocational and social functioning over time (Hastrup 2019b; Zanarini 2010). Furthermore, treatment‐seeking people with personality disorders, such as BPD, pose a high economic burden on society due to a frequent and often long‐term utilisation of both psychiatric and emergency services as well as loss of occupational function and income (Hastrup 2019a; Van Asselt 2007). Effective treatments could potentially decrease the high costs associated with the condition (Soetmann 2008a).
In summary, BPD is a condition that has been extensively studied and has a major impact on health services (Bode 2017; Jacobi 2021; Meuldijk 2017). The recovery from symptoms or functional impairment (or both) was previously considered likely for only a low percentage of people diagnosed with BPD. However, the long‐term course is better than what was previously assumed, due to more favourable symptomatic recoveries (Zanarini 2012). Nonetheless, people with BPD continue to have considerable interpersonal and functional problems, and sustainable recovery appears difficult to attain (Biskin 2015; Kongerslev 2015; Rossouw 2012).
Description of the intervention
To date, all major treatment guidelines consider psychotherapy as the treatment of choice for BPD and assign medications an adjunctive role (e.g. APA 2001; Bateman 2015; Cristea 2017; DGPPN 2009; Herpertz 2007; NHMRC 2013; NICE 2009; Simonsen 2019; Storebø 2020). However, the large majority of people with BPD are prescribed psychotropic medications during the course of their illness. This may be the case in times of crisis, when people with BPD present to mental health services with raised suicidality or parasuicidality, impulsivity‐associated outbreaks, psychotic‐like exacerbations, severe dissociations or aggravations of comorbid conditions (e.g. mood disorders), and so medications are used to achieve short‐term stabilisation (NHMRC 2013; NICE 2009; Riffer 2019). Such crisis interventions will not be considered in this review, but are subject to another Cochrane Review, which is currently being updated (Borschmann 2012).
In contrast to short‐term crisis medication, up to 84.1% of people with BPD who are engaged in treatment have been reported to use standing (i.e. long‐term) psychotropic medications (Bender 2001; Zanarini 2015), and as many as 92% have been reported to use any psychotropic medication for a non‐specified period of time (Paton 2015). Indeed, it is a common finding that people with BPD are more likely to use psychotropic medications than people with other psychiatric conditions such as major depressive disorder (Bender 2001; Bender 2006), mood or anxiety disorders in general (Ansell 2007), or other personality disorders (Zanarini 2004a).
Studies across different countries show that antidepressants are the class of medication most often prescribed to people with BPD (Bender 2001; Knappich 2014; Makela 2006; Paton 2015; Sansone 2003; Zanarini 2015). Zanarini and colleagues found that 79.7% of people with BPD were taking antidepressant medication, followed by anxiolytics (46.6%), neuroleptics (38.6%), and mood stabilisers (35.9%) (Zanarini 2015). They also found that about 71% of people with BPD were using medications at six‐year follow‐up (Zanarini 2004a; Zanarini 2004b), and that they were still more likely to be using antidepressants, mood stabilisers, antipsychotics or anxiolytics than Axis‐II comparison participants at 16‐year follow‐up (Zanarini 2015). Additionally, polypharmacy is common, with reports of people with BPD taking, on average, 2.02 psychotropic medications at a time (Ansell 2007), and up to 28.6% taking four or more medications (Zanarini 2004a; Zanarini 2004b).
There is no stringent or binding classification of psychotropic medications. In routine clinical care, the most commonly used classification is likely to be that which builds on the Anatomical Therapeutic Chemical (ATC) classification (WHO Collaborating Center for Drug Statistics.), where medications are grouped primarily by indication, such as antidepressants, antipsychotics, antidementive drugs, etc. This classification has been criticised for various reasons, including: not considering the fact that many psychotropic medications are not only used for one, but several indications, which may confuse consumers and lower adherence; the grouping of too heterogeneous kinds of medications into the same group; and having too close connection to marketing claims of the pharmaceutical industry (names may be mistaken to imply that the drugs definitely are effective as regards the respective outcomes; Brühl 2017). In order to target these shortcomings, the Neuroscience‐based Nomenclature has been developed (Brühl 2017; NbN3 2021), with the aim of moving from a disease‐based classification system to one that is pharmacologically driven, focusing on modes of actions rather than symptoms. This review use traditional categories, such as antipsychotics, antidepressants, mood stabilisers, antidementive medications, etc., as these might be most familiar to consumers and clinicians in the field as well as healthcare professionals (who might not have a background in psychiatry or psychology). A translation of these terms into the Neuroscience‐based Nomenclature (NBN) (NbN3 2021) can be found in Appendix 3.
In summary, prescription rates of psychotropic medications are high among people with BPD and these medications are often administered for sustained periods of time, even though medication is only advised as an adjunctive to psychotherapy (Bateman 2015; Bohus 2021; NHMRC 2013; NICE 2018). Different classes of medication are used, with antidepressants being the most frequent, but there is no standard medication treatment. Currently, any medication use in BPD is off‐label (if not used to target associated psychopathology, such as depression or anxiety, for which there is evidence for use), but up to 82% of people with BPD without comorbid conditions still receive medications to directly target BPD symptoms (Paton 2015).
How the intervention might work
The most important pharmacological domains and modes of action of each included medication are presented in Appendix 3. Essentially, the psychotropic medications included in this review are thought to work ‐ at least partially ‐ in ways described below:
The serotonergic system has been associated with mood (depressive mood, anxiety), impulsiveness and aggressiveness (Herpertz 2007). Antidepressants effectuate a higher concentration and longer availability of serotonin in the synaptic cleft by preventing its reuptake (e.g. selective serotonin reuptake inhibitors (SSRIs)), or degradation (e.g. monoamin oxidase inhibitors (MAOIs)). As an inverse relationship between serotonin levels in the brain and low mood, as well as increased impulsive behaviour, has been demonstrated, substances increasing serotonin availability might improve mood and lower impulsive behaviour.
The monoamines norepinephrine and dopamine have also been implicated in emotion and reward. Norepinephrine is related to stress as it helps the brain and body to prepare for immediate action ('fight‐flight‐response'). It increases arousal, alertness and attention, but is also related to restlessness and anxiety. Dopamine is a neurotransmitter which not only plays a major role in schizophrenia but is also related to executive functions and arousal, along with reward and motivation. Substances which increase the level of these monoamines might therefore have an impact on perception and behavioural choices.
GABA (gamma‐aminobutyric acid) is an inhibitory neurotransmitter that reduces neural excitability in the central nervous system. Heightened GABA levels have been shown to be associated with relaxing, anti‐anxiety, and anti‐convulsive effects.
Glutamate is an excitatory neurotransmitter in the central nervous system which is involved in e.g. motor‐executive functions, as well as learning and memory.
Opioid peptides bind as agonists to opioid receptors and act as endogenous analgesics. They have modulating effects on mood and have a role in the aetiology and maintenance of substance use disorders. Opioid antagonists are used in the treatment of opioid and alcohol use disorders, and opioid substances in the treatment of substance withdrawal.
Psychotropic medications may target one or several of these pharmacology domains and comprise one or more modes of action. However, it is unlikely that direct and immediate effects at the synapses can explain changes in mood, anxiety etc., since such changes only occur after weeks of treatment. Much more likely modes of actions are secondary effects on plastic brain adaptation processes, which are not well understood yet, especially in BPD.
Why it is important to do this review
BPD poses a major burden, both personal (Soetmann 2008b) and financial (Soetmann 2008a) on those directly affected, their relatives, as well as for society at large. Despite the frequent use of pharmacological interventions in clinical practice, and in research over the last three decades, any medication used in the treatment of BPD is off‐label. Given the absence of reliable evidence being supportive of medication use, prescription is often undertaken based upon known effects on symptom clusters in other disorders, out of habit, ignorance, passed‐on clinical rules of thumb or desperation (Aguglia 2019; Paris 2015; Pascual 2021; Riffer 2019; Zanarini 2004a). Clinicians and consumers, however, must be able to make informed decisions on the basis of up‐to‐date evidence (Ingenhoven 2015; Paris 2015; Silk 2015), allowing them to weigh up potential benefits and harms. This Cochrane Review aims to systematically identify, investigate, and present the current state of evidence on the topic of medications in BPD, and make suggestions about the directions for future trials.
This review supersedes a previous Cochrane Review on pharmacological interventions for BPD (Stoffers 2010). In addition to updating the former Cochrane Review, our study also seeks to address some of the methodological limitations of the preceding two reviews (Binks 2006; Stoffers 2010) by using updated methods, and including a more comprehensive search strategy. In order to do this transparently, we developed and published a new protocol prior to conducting this review (Stoffers‐Winterling 2018). The Stoffers 2010 review came to the conclusion that the evidence available at that time indicated beneficial effects for some medications (i.e. second‐generation antipsychotics, mood stabilisers and omega‐3 fatty acids), but that the overall quality of the evidence was not robust enough to draw any reliable conclusions. In the proceeding 11 years, research has continued in the field, and new findings may change conclusions of the previous review. Therefore, an update of this review seems both appropriate and timely (Garner 2016).
Objectives
To assess the effects of pharmacological treatment for people with BPD.
Methods
Criteria for considering studies for this review
Types of studies
Randomised controlled trials (RCTs).
Types of participants
Individuals of all ages, in any setting, with a formal diagnosis of BPD according to the Diagnostic and Statistical Manual of Mental Disorders (DSM) Third Edition (DSM‐III; APA 1980), Third Edition Revised (DSM‐III R; APA 1987), Fourth Edition (DSM‐IV; APA 1994), Fourth Edition Text Revision (DSM‐IV‐TR; APA 2000), and Fifth Edition (DSM‐5; APA 2013)), with or without comorbid conditions.
We required at least 70% of trial participants to have a formal diagnosis of BPD. We also included trials involving subsamples of people with BPD if data on these were available separately. We did not include trials that focused on people with mental impairment, organic brain disorder, dementia, or other severe neurologic diseases.
Types of interventions
Any medication or defined combination of medications administered at any dosage, prescribed to treat the disorder or its symptoms, compared to a placebo or active comparator medication(s) was eligible. We included trials that paired medications with an adjunctive intervention (e.g. psychological therapies), providing this was given to participants in both the intervention and control arm, and the pharmacological intervention was unique to the treatment group.
Medication should have been prescribed continuously for a minimum duration of two weeks for the trial to be eligible for our review. In addition, we judged the actual duration required for inclusion in light of the specific mode of action of the medical agent. Medication should have been used to treat the disorder or symptoms thereof.
Types of outcome measures
Outcomes could either be self‐rated or observer‐rated by clinicians. We included only adequately validated measures (plus spontaneous reporting of adverse events).
Primary outcomes
BPD severity, as assessed by, for example, the Zanarini Rating Scale for Borderline Personality Disorder (Zan‐BPD; Zanarini 2003), the Borderline Personality Disorder Severity Index (BPDSI‐IV; Arntz 2003), or the Clinical Global Impression Scale for Borderline Personality Disorder Patients (CGI‐BPD; Perez 2007).
Self‐harm, in terms of the proportion of participants with self‐harming behaviour, or as assessed by, for example, the Deliberate Self‐harm Inventory (DSHI; Gratz 2001) or the Self‐harm Behavior Questionnaire (SHBQ; Guttierez 2001).
Suicide‐related outcomes, as assessed by, for example, the Suicidal Behaviours Questionnaire (SBQ; Osman 2001) or the Beck Scale for Suicidal Ideation (BSSI; Beck 1979) or in terms of the proportion of people with BPD with suicidal acts.
Functioning, as assessed by, for example, the Global Assessment Scale (GAS; Endicott 1976), the Global Assessment of Functioning Scale (GAF; APA 1987) or the Social Functioning Questionnaire (SFQ; Tyrer 2005).
Secondary outcomes
Anger, as assessed by, for example, the 'Hostility' subscale of the Symptom Checklist‐90‐Revised (SCL‐90‐R; Derogatis 1994), or the State‐Trait Anger Expression Inventory (STAXI; Spielberger 1988).
Affective instability, as assessed by, for example, the relevant item or subscale on the Zan‐BPD (Zanarini 2003), CGI‐BPD (Perez 2007) or BPDSI‐IV (Arntz 2003).
Chronic feelings of emptiness, as assessed by, for example, the relevant item or subscale on the Zan‐BPD (Zanarini 2003), CGI‐BPD (Perez 2007) or BPDSI‐IV (Arntz 2003).
Impulsivity, as assessed by, for example, the Barrett Impulsiveness Scale (BIS; Barrett 1995), or the Anger, Irritability and Assault Questionnaire (AIAQ; Coccaro 1991).
Interpersonal problems, as assessed by, for example, the Inventory of Interpersonal Problems (IIP; Horowitz 1988), or the relevant item or subscale of the Zan‐BPD (Zanarini 2003), CGI‐BPD (Perez 2007), BPDSI‐IV (Arntz 2003), or SCL‐90‐R (Derogatis 1994).
Abandonment, as assessed by, for example, the relevant item or subscale on the Zan‐BPD (Zanarini 2003), CGI‐BPD (Perez 2007) or BPDSI‐IV (Arntz 2003).
Identity disturbance, as assessed by, for example, the relevant item or subscale on the Zan‐BPD (Zanarini 2003), CGI‐BPD (Perez 2007) or BPDSI‐IV (Arntz 2003).
Dissociation and psychotic‐like symptoms, as assessed by, for example, the Dissociative Experience Scale (DES; Bernstein 1986), or the Brief Psychiatric Rating Scale (BPRS; Overall 1962).
Depression, as assessed by, for example, the Beck Depression Inventory (BDI; Beck 1961), or the Montgomery Åsberg Depression Rating Scale (MADRS; Montgomery 1979).
Attrition, in terms of participants lost after randomisation in each group.
Adverse effects, as measured by use of standardised psychometric rating scales such as the Systematic Assessment for Treatment Emergent Events (SAFTEE; Levine 1986), laboratory values or spontaneous reporting.
Search methods for identification of studies
This current review is part of a series of reviews on interventions for BPD (Stoffers‐Winterling 2018; Storebø 2018; Storebø 2020; Stoffers 2010). Therefore, the search strategy is very comprehensive and covers all psychotherapeutic or pharmacological treatment (or both) of BPD. The search has been run four times in all databases over the years. A full search in all databases was carried out in 2017 and then additional top‐up searches were used to update search hits until the most recent search on 21 February 2022. Trial registries were handsearched several times during the work with this update, most recently on individual dates as close to the submission date of this manuscript as possible.
Electronic searches
We searched the electronic databases and trials registers listed below to identify relevant trials.
Cochrane Central Register of Controlled Trials (CENTRAL; 2022, Issue 2), in the Cochrane Library, which includes the Cochrane Developmental, Psychosocial and Learning Problems Specialised Register (searched 21 February 2022).
MEDLINE Ovid (1948 to 21 February 2022).
Embase Ovid (1974 to 21 February 2022).
CINAHL EBSCOhost (Cumulative Index to Nursing and Allied Health Literature; 1980 to 21 February 2022).
PsycINFO Ovid (1806 to 21 February 2022).
ERIC EBSCOhost (Education Resources Information Center; 1966 to 21 February 2022).
BIOSIS Previews Web of Science Clarivate Analytics (1969 to 29 June 2022).
Web of Science Clarivate Analytics (Science Citation Index Expanded and Social Sciences Citation Index 2002 to 21 February 2022).
Sociological Abstracts ProQuest (1952 to 21 February 2022).
LILACS (Latin American and Caribbean Health Science Information database (lilacs.bvsalud.org/en; searched 21 February 2022)).
Library Hub Discover (previously Copac National, Academic and Specialist Library Catalogue; copac.jisc.ac.uk, searched 21 February 2022).
ProQuest Dissertations and Theses A&I (1973 to 21 February 2022).
OpenGrey; searched 28 December 2020 (OpenGrey was shut down before top‐up searches).
DART Europe E‐Theses Portal (www.dart-europe.eu/basic-search.php, searched 27 June 2022).
Networked Digital Library of Theses and Dissertations (NDLTD; ndltd.org, searched 27 June 2022).
Australian New Zealand Clinical Trials Registry (ANZCTR; anzctr.org.au/TrialSearch.aspx, searched 25 February 2022).
ClinicalTrials.gov (clinicaltrials.gov/, searched 21 February 2022).
EU Clinical Trials Register (www.clinicaltrialsregister.eu/ctr-search/search, searched 25 February 2022).
ISRCTN Registry (www.isrctn.com/, searched 23 June 2022).
WHO International Clinical Trials Registry Platform (ICTRP; trialsearch.who.int/, searched 22 June 2022).
The search strategies for each source are available in Appendix 4. We did not limit our searches by language, year of publication, or type of publication. We sought translation of the relevant sections of non‐English language articles.
Searching other resources
On 10 and 11 March 2022, we searched for unpublished data on the websites of the United States Food and Drug Administration (FDA; www.fda.gov) and the European Medicines Agency (EMA; www.ema.europa.eu/ema) (see Appendix 4). We also handsearched relevant journals: the Journal of Personality Disorders; the American Journal of Psychiatry; JAMA Psychiatry; British Journal of Psychiatry; ACTA Psychiatrica Scandinavica; Journal of the American Academy of Child and Adolescent Psychiatry; Personality Disorders: Theory, Research and Treatment; and the Journal of Clinical Psychiatry. Additionally, we contacted researchers working in the field by email, to ask for unpublished data,and traced cross‐references from relevant literature.
Data collection and analysis
We conducted this review according to guidelines set out in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2022), and performed the analyses using the latest version of Review Manager Web (RevMan Web), Cochrane's statistical software (RevMan Web 2020).
We were not able to use all of our methods as planned (Stoffers‐Winterling 2018). We report the unused methods in Table 4.
1. Key demographic characteristics of the included studies.
Selection of studies
Thirteen reviewers (JMSW, OJS, AT, EF, BAV, MLK, MTK, CPS, JTM, JPR, HEC, SSN, JPS) worked in pairs and independently screened titles and abstracts of all records retrieved by the searches; we resolved uncertainty or disagreement by consensus. For records that could be eligible RCTs, we obtained the full‐text reports and assessed them for eligibility based on the inclusion criteria (Criteria for considering studies for this review). Review authors discussed disagreements and, if they could not reach an agreement, they consulted a third review author (ES or KL). We listed potentially relevant RCTs that did not fulfil the inclusion criteria with reasons for exclusion in the 'Characteristics of excluded studies' tables. We used Covidence software to keep track of appraised trials and decisions. To ensure transparency of study selection, we provided a flow chart in accordance with the PRISMA statement (Moher 1999), to show how many records had been excluded and for what reason (Figure 1).
1.
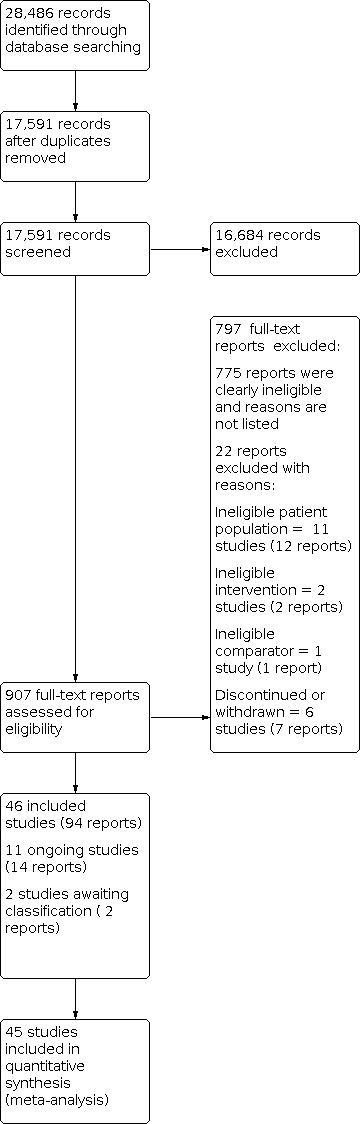
PRISMA flow diagram of study selection
Data extraction and management
We developed data extraction forms to facilitate standardisation of data extraction. All review authors extracted data. Review authors worked in pairs and completed the data collection form independently to ensure accuracy. We resolved disagreements by discussion or used an arbiter (ES) if required. JMSW, JRP, and OJS entered the data into RevMan 5 (RevMan Web 2020). In those cases where there were not enough data or data were unclear in the published trial reports, we contacted the trial authors, requesting them to supply the missing information.
Assessment of risk of bias in included studies
Using the Cochrane tool for assessing risk of bias (Higgins 2017), all review authors assessed the risk of bias in each included study across the following domains: random sequence generation, allocation concealment, performance bias, detection bias, attrition bias, reporting bias and other potential sources of bias. We included vested interest in this last domain. Andreas Lundh and colleagues have illustrated the many subtle mechanisms through which sponsorship and conflict of interest may influence intervention effects on outcomes. For more information, please see editorials by Bero 2013 and Sterne 2013, and the commentary by Gøtzsche 2015.
For each included trial, data extractors independently evaluated each risk of bias domain as being at low, unclear (uncertain) or high risk of bias, resolving disagreements by discussion. We categorised trials that had a low risk of bias in all domains as being at low risk of bias overall, and we considered trials with one or more unclear or high risk of bias domains as trials at high risk of bias overall. Given the risk of overestimation of beneficial intervention effects and underestimation of harmful intervention effects in RCTs with unclear or inadequate methodological quality (Kjaergard 2001; Lundh 2017; Moher 1998; Savović 2012a; Savović 2012b; Schulz 1995; Wood 2008), we assessed the influence of risk of bias on our results (see Sensitivity analysis).
Measures of treatment effect
Dichotomous data
We summarised dichotomous data as risk ratios (RRs) with 95% CIs. The RR is the ratio of the risk of an event in the two groups. We decided to use the RR as it may be easier to interpret than odds ratios (ORs).
Continuous data
For continuous data, we compared the mean score between the two groups to give a mean difference (MD) and presented this with 95% CIs. We used the overall MD, where possible, to compare the outcome measures from trials. We estimated the standardised MD (SMD) where different outcome measures were used to measure the same construct in the trials. We calculated SMDs using end‐scores at post‐treatment results. Where the direction of a scale was opposite to most of the other scales, we multiplied the corresponding mean values by −1 to ensure adjusted values. If the trials did not report means and standard deviations but reported other values like t‐tests and P values, we tried to transform these into standard deviations.
Our first choice was to calculate effect sizes on the basis of intention‐to‐treat (ITT) data. If means and standard deviations from an ITT analysis and missing values that were replaced were available, we used these data. In other cases, we conducted the analysis using only the available data.
We performed all calculations using the latest release of RevMan 5 software (RevMan Web 2020).
To identify the minimum relevant clinical difference (MIREDIF), we transformed the SMD to MD, using the scale with the best validity and reliability for the given outcome. For the analyses of the primary outcome, BPD symptom severity, in the comparison of psychotherapy versus treatment‐as‐usual (TAU), we transformed SMDs into MDs on the following scale, to assess whether results exceeded the MIREDIF: ZAN‐BPD Scale, We identified a MIREDIF of −3.0 points on the ZAN‐BPD, ranging from 0 to 36 points, based on a trial by Zanarini 2007. We used a MIREDIF of 0.61 in the interpersonal problems outcome, which corresponds to ½ SD based on a trial by Schulz 2007. For attrition, we used a relative risk reduction of 40%, and for non‐serious adverse events, we used a relative risk reduction of 20%.
Unit of analysis issues
Repeated observations
We calculated study estimates on the basis of post‐treatment group results. We did not use interim observations.
Cross‐over trials
We included data from four randomised cross‐over trials (Cowdry 1988; Schmahl 2012a; Schmahl 2012b; Ziegenhorn 2009). We were not able to obtain first‐period data from these cross‐over trials (by writing to the study authors) and therefore used end of period data (Curtin 2002; Elbourne 2002) because the data were unavailable. Cross‐over trials are more prone to bias from carryover effects, period effects and unit of analysis errors, but it is still possible to use the end of period data when first‐period data are not available (Curtin 2002; Elbourne 2002). This approach might however introduce a risk of a unit of analyses error, the confidence intervals will probably be too wide, and also the trial will receive too little weight. There is also a possibility of overlooking important heterogeneity. The Cochrane Handbook states, however, that this approach is conservative as the studies are under‐weighted instead of over‐weighted (Higgins 2019). Some argue that the unit of analyses errors introduced by doing this might be regarded as less serious than other types of unit of analysis error. The trial by Cowdry (Cowdry 1988) was the only one pooled with parallel‐group trials. Cowdry and colleagues reported a washout period of one week before cross‐over. When excluding the Cowdry study from the parallel‐group trials, we found no differences in the result of the analyses. The other cross‐over trials (Schmahl 2012a; Schmahl 2012b; Ziegenhorn 2009) were reported separately in the analyses.
Studies with multiple treatment groups
If a trial compared more than two intervention groups, we included all pairwise comparisons as long as they were not subject to the same meta‐analysis. If a trial included two arms at different doses of a certain medication that were tested against placebo, we combined the experimental groups into a single group, as recommended in the Cochrane Handbook for Systematic Reviews of Interventions, making a single, pairwise comparison (Higgins 2022). We hereby avoided including the same group of participants twice in the same meta‐analysis.
Adjustment for multiplicity
Multiplicity reflects the concern that performing multiple comparisons increases the risk of falsely rejecting the null hypothesis. Multiplicity, therefore, may affect the results found within a systematic review and, as a result, needs to be adjusted for. We adjusted the P values and CIs of the primary outcomes (BPD severity) and secondary outcomes (interpersonal problems, non‐serious adverse events and attrition) for multiplicity using the method described by Jakobsen 2014. We have made a conservative estimation of the anticipated intervention effect to control the risk of type 1 error (Jakobsen 2014). We have four primary outcomes, and therefore we considered a P value of 0.02 or less as the threshold for evidence of a difference for the primary outcomes. We have 11 secondary outcomes, and therefore we considered a P value of 0.008 or less as the threshold for evidence of a difference for the secondary outcomes (Jakobsen 2014).
Dealing with missing data
We tried to obtain any missing data, including incomplete outcome data, by contacting trial authors. We reported this information in the risk of bias tables.
We evaluated the methods used to handle the missing data in the publications and to what extent it was likely that the missing data influenced the results of outcomes of interest. For preference, we calculated effect sizes on the basis of ITT data. If only available data were reported, we calculated effect sizes on this basis. Where dichotomous data were not presented on the basis of ITT data, we added the number of participants lost in each group to the participants with unfavourable results, acting on the assumption that most people with BPD did not get lost at random. For continuous outcomes, we discussed each trial’s methodology for dealing with missing continuous data (e.g. last‐observation‐carried‐forward or modified ITT approach). We used per protocol analysis, as available from the trial reports (that is, results were based on the number of participants at follow‐up).
If data were not reported in an immediately usable way, we consulted a statistician.
We assessed results derived from statistically processed data in sensitivity analyses (see Sensitivity analysis).
Assessment of heterogeneity
We assessed trials for clinical homogeneity with respect to type of pharmacological interventions, setting and control groups. We took into account the number of trials and trial characteristics, such as duration, dose and participants, to judge if heterogeneity was more probable due to clinical (i.e. explainable factors) or unknown factors. In case of substantial heterogeneity, we divided analyses into subgroups according to trial characteristics, such as trial size, duration, dose or participants, and discussed the most apparent sources of heterogeneity. We evaluated methodological heterogeneity by comparing the design of trials (see Subgroup analysis and investigation of heterogeneity).
We investigated statistical heterogeneity within a certain comparison by visual inspection of the graphs and the I² statistic (Higgins 2003). We judged I² values between 0% and 40% to indicate little heterogeneity, between 30% and 60% to indicate moderate heterogeneity, between 50% and 90% to indicate substantial heterogeneity, and between 75% and 100% to indicate considerable heterogeneity (Deeks 2021). We also assessed statistical heterogeneity by the Chi² test (P < 0.10) and tau² ‒ an estimate of between‐trial variability.
Assessment of reporting biases
We produced funnel plots for comparisons with sufficient primary studies i.e. 10 or more trials and we performed Egger's statistical test for small‐trial effects (Egger 1997). We did not use a visual inspection of the funnel plot if there were fewer than 10 trials in the meta‐analysis, as recommended in the Cochrane Handbook for Systematic Reviews of Interventions (Sterne 2017).
Data synthesis
We performed statistical analyses according to recommendations in the latest version of the Cochrane Handbook for Systematic Reviews of Interventions (Deeks 2021). In carrying out the meta‐analysis, we used the inverse‐variance method for continuous data, in order to give more weight to more precise estimates from trials with less variance (mostly larger trials). For dichotomous data we used the Mantel‐Haenszel analysis method. We used the random‐effects model for meta‐analysis when there were two or more trials, since we expected some degree of clinical heterogeneity to be present in most cases, though not so substantial as to prevent pooling in principle. Where only one trial was included in an analysis, we used the fixed‐effect model, and where different models led to different results, we reported the results of both models (Sensitivity analysis). For trials with a high level of statistical heterogeneity, and where the amount of clinical heterogeneity made it inappropriate to use these trials in meta‐analyses, we provided a narrative description of the trial results. If we considered data pooling to be feasible, we pooled the primary trials effects and calculated their 95% CIs. If a trial provided more than one measure for the same outcome construct (e.g. several questionnaires for the assessment of depression), we selected the one used most often in the whole pool of included trials for effect size calculation, in order to minimise heterogeneity of outcomes in form and content. If a trial reported data of two assessment instruments that were equally frequently used, two review authors discussed the issue and chose the one which was, in its content, most appropriate for assessing people with BPD. We preferred observer‐rated measures as the primary analysis measure.
Subgroup analysis and investigation of heterogeneity
We conducted subgroup analyses to investigate the subgroups mentioned below.
Types of medication (comparing pharmacological classes as well as active medications)
Setting (outpatient compared to inpatient)
We attempted to undergo subgroup analyses for all primary outcomes; however, data were not available for all subgroups and so we investigated subgroups for the primary outcomes of BPD symptom severity, psychosocial functioning, suicide‐related outcome, and the secondary outcome anger (as this was the only outcome where it was possible to perform subgroup analyses).
We added the following subgroup analyses post hoc.
Funding (funded or partially funded by pharmaceutical industry compared to no funding received compared to funded by universities or research foundations)
Psychosocial functioning at baseline as measured by GAS, GAF or SFQ15 (comparing groups of participants with low, moderate and high impairment)
Trial size (trial size ≤ 50 compared to trial size ≤ 100 compared to trial size ≥ 100)
Type of screening (referrals compared to advertisements)
Heterogeneity‐adjusted required information size and Trial Sequential Analysis
Sequential methods like Trial Sequential Analysis (TSA) are not in general recommended to be used in Cochrane reviews. They can be used as a secondary analyses, to give an additional interpretation of the data from a specific perspective. We have used the TSA in this review as a secondary analysis testing the imprecision of some of the most important outcomes (Thomas 2021). We only performed a TSA for BPD symptom severity, interpersonal problems, non‐serious adverse events and attrition as these were the outcomes in the SoF tables where we needed to test our GRADE assessment of imprecision.
TSA is a methodology that combines a required information size (RIS) calculation for a meta‐analysis with the threshold for statistical significance (Brok 2008; Brok 2009; Thorlund 2009; Wetterslev 2008). TSA is a tool for quantifying the statistical reliability of the data in cumulative meta‐analysis, adjusting P values for sparse data and for repetitive testing on accumulating data (Brok 2008; Brok 2009; Thorlund 2009; Wetterslev 2008).
Comparable to the a priori sample size estimation in a single randomised trial, a meta‐analysis should include a RIS calculation at least as large as the sample size of an adequately powered single trial to reduce the risk of random error. TSA calculates the RIS in a meta‐analysis and provides an alpha‐spending boundary to adjust the significance level for sparse data and repetitive testing on accumulating data (CTU 2011; Wetterslev 2008), and consequently the risk of random error can be assessed. Multiple analysis of accumulating data when new trials emerge leads to repeated significant testing and hence introduces multiplicity, thus use of a conventional P value is prone to exacerbate the risk of random error (Berkey 1996; Lau 1995). Meta‐analyses not reaching the RIS are analysed with trial sequential alpha‐spending monitoring boundaries analogous to interim monitoring boundaries in a single trial (Wetterslev 2008). This approach will be crucial in future updates of the review.
If a TSA does not reveal significant findings (no crossing of the alpha‐spending boundary and no crossing of the conventional boundary of P = 0.05) before the RIS has been reached, then the conclusion should either be that more trials are needed to reject or accept an intervention effect that was used for calculation of the required sample size or — in case the cumulated Z‐curve enters the futility area — the anticipated effect can be rejected.
We used a minimally relevant clinical difference (MIREDIF) from trials defining this or, where we could not find this, we used an assumption that the minimal relevant clinical intervention effect was approximately ½ SD on the used scale, which can be used as a MIREDIF (Norman 2003).
We calculated the diversity‐adjusted required information size (DARIS; that is, the number of participants required to detect or reject a specific intervention effect in a meta‐analysis), and performed a TSA for the primary outcome, BPD symptom severity, at the end of treatment for the main comparison versus placebo, based on the following a priori assumptions:
the SD of the primary outcome;
an anticipated MIREDIF defined in a trial reporting on this or we used a ½ SD on the used scale;
a maximum type I error of 2.0% (due to four primary outcomes; Jakobsen 2014);
a maximum type II error of 10% (minimum 90% power; Castellini 2018); and
the diversity observed in the meta‐analysis.
We furthermore performed a TSA for the secondary outcome, interpersonal problems (for the main comparison versus placebo), based on the following priori assumptions:
the SD of the secondary outcome;
an anticipated MIREDIF defined in a trial reporting on this or we used a ½ SD on the used scale;
a maximum type I error of 0.8% (due to 11 secondary outcomes; Jakobsen 2014);
a maximum type II error of 10% (minimum 90% power; Castellini 2018); and
the diversity observed in the meta‐analysis.
For the secondary outcomes, 'attrition', and 'non‐serious adverse events', we calculated the a priori diversity‐adjusted required information size (DARIS; i.e. number of participants in the meta‐analysis required to detect or reject a specific intervention effect) and performed a Trial Sequential Analysis for these outcomes based on the following assumptions (Brok 2008; Brok 2009; Thorlund 2009; Wetterslev 2008; Wetterslev 2009):
Proportion of participants in the control group with adverse events;
Relative risk reduction of 40% (20% on 'non‐serious adverse events');
Type I error of 0.8% (due to 11 secondary outcomes; Jakobsen 2014);
Type II error of 10%;
Observed diversity of the meta‐analysis.
Sensitivity analysis
We assessed the impact of heterogeneity on the overall pooled effect estimate. First, we visually inspected the forest plot for 'outliers' that might contribute to heterogeneity and then removed them one‐by‐one to assess their impact on the overall outcome. Overall results were reported in the main analysis with outliers excluded, while we also conducted sensitivity analyses including these outliers.
We conducted sensitivity analyses to determine whether findings were sensitive to the following.
Imprecision, as assessed by GRADE, by conducting TSAs on the primary outcomes and for the secondary outcomes (for the main comparison versus placebo) where we were uncertain about the assessment of imprecision.
Differences when adding data from end‐of‐period data from cross‐over trials to the analyses.
-
Decisions made during the review process in relation to the:
choice of data (differences between data from trial registers and data from peer reviewed sources);
exclusion of outliers; and
type of model used for analysis (repeating the analysis using the fixed‐effect model to test the robustness of the results).
Summary of findings and assessment of the certainty of the evidence
We used Gradepro (GRADEpro 2021) to construct summary of findings tables. We reported the four primary outcomes (BPD severity, self‐harm, suicide‐related outcomes, and psychosocial functioning) and three secondary outcomes (interpersonal problems, attrition, and adverse events). We produced three summary of findings tables; one focusing on the comparison between antipsychotics and placebo at end of treatment, one on antidepressants compared with placebo at end of treatment, and one on mood stabilisers compared with placebo at end of treatment.
We used the GRADE approach to assess the quality of the body of evidence based on the extent to which one can be confident that an estimate of effect or association reflects the item being assessed. Two authors independently assessed the certainty of the evidence according to study limitations (risk of bias), indirectness of evidence, inconsistency of results, imprecision and publication bias (Atkins 2004; Andrews 2013a; Andrews 2013b; Balshem 2011; Brunetti 2013; Guyatt 2011a; Guyatt 2011b; Guyatt 2011c; Guyatt 2011d; Guyatt 2011e; Guyatt 2011f; Guyatt 2011g; Guyatt 2011h; Guyatt 2013a; Guyatt 2013b; Guyatt 2013c; Mustafa 2013). Any differences of opinion were resolved by consulting a third author. When possible, we used the MD or the RR, and we used TSA to rate the imprecision (Jakobsen 2014). Two authors (HEC and OJS) downgraded imprecision in GRADE by two levels if the accumulated number of participants was below 50% of the DARIS, and one level if between 50 and 100% of DARIS. We did not downgrade when the cumulative Z‐curve crossed the monitoring boundaries for benefit, harm, or futility, or DARIS was reached. We justified all decisions to downgrade the quality of evidence in footnotes (Korang 2020).
Results
Description of studies
For a full description of trials, see Characteristics of included studies, Characteristics of excluded studies, Characteristics of studies awaiting classification and Characteristics of ongoing studies tables.
Results of the search
Combined, all searches generated 28,486 records, of which 10,895 were duplicates, leaving 17,591 records for title and abstract screening. Of these, 16,684 records were deemed irrelevant based on title and abstract screening, leaving 907 records for full‐text inspection. Of the full‐text reports, 797 were excluded as they clearly did not match the inclusion criteria. Reasons for exclusion at this stage included not being a clinical trial or a pharmacological intervention (due to the comprehensive search) or similar. The remaining reports were examined closely to determine their eligibility. Of these, 22 reports (20 trials) were excluded for the reasons reported in Characteristics of excluded studies. We also identified 11 ongoing studies (14 reports) and two studies (2 reports) are awaiting classification (see Characteristics of studies awaiting classification and Characteristics of ongoing studies). This left 46 studies (from 94 reports ) which were eligible for inclusion in the review. Data were unavailable for effect size calculations in one of these trials, leaving 93 reports from 45 trials to be included in the quantitative synthesis. This is 18 more studies than in the 2010 review (Stoffers 2010). The 46 trials are thoroughly described in Characteristics of included studies.
Included studies
Design
We included 46 randomised controlled trials. Four of these were cross‐over trials (Cowdry 1988; Schmahl 2012a; Schmahl 2012b; Ziegenhorn 2009); six were multi‐armed (Black 2014; Cowdry 1988: Soloff 1989; Soloff 1993; Zanarini 2004; Zanarini 2007), and three had a randomised open‐label design (Bellino 2014; Bozzatello 2017; Shafti 2014).
Sample size
The total number of participants with BPD in the included trials varied considerably from 13 in the smallest trial (Schmahl 2012a) to 451 in the largest (Zanarini 2007). There were four trials with more than 100 participants (Jariani 2010; Schulz 2007; Soloff 1993; Zanarini 2001).
Setting
Thirty‐two of the included trials took place in an outpatient setting, while nine trials took place in an inpatient setting (De la Fuente 1994; Markovitz 1995a; Moen 2012; NCT00533117; Schmahl 2012a; Shafti 2010; Shafti 2014; Simpson 2004; Ziegenhorn 2009). Four trials included both inpatients and outpatients or allowed inpatients to complete as outpatients if discharged from the hospital (Crawford 2018; Schmahl 2012b; Soloff 1989; Soloff 1993), and one trial did not state whether it took place in an in‐ or outpatient setting (AstraZeneca 2007).
Country
Twenty trials were from Europe. Of these, eight were from Germany (Loew 2006; Nickel 2004; Nickel 2005; Nickel 2006; Schmahl 2012a; Schmahl 2012b; Tritt 2005; Ziegenhorn 2009), one was from Austria (Amminger 2013), one was from Belgium (De la Fuente 1994), two were from the Netherlands (AstraZeneca 2007; Rinne 2002), two were from Italy (Bellino 2014; Bozzatello 2017), two were from Spain (Pascual 2008; Soler 2005), three were from the UK (Crawford 2018; Montgomery 1982a; Montgomery 1982b), and one was from Ireland (Hallahan 2007). Three trials were from Southwest Asia (Middle East); all of them from Iran: Jariani 2010; Shafti 2010; Shafti 2014. One trial was from Australia: Kulkarni 2018. Two trials were multi‐country and were carried out in nine countries each: Schulz 2007 (Belgium, France, Germany, Norway, Portugal, Spain, Sweden, UK and US), and Zanarini 2007 (US, Italy Poland, Romania, Turkey, Chile, Peru, Argentina and Venezuela). The remaining 20 trials were from the US (Black 2014; Bogenschutz 2004; Cowdry 1988; Frankenburg 2002; Goldberg 1986; Grant 2022; Hollander 2001; Leone 1982; Linehan 2008; Markovitz 1995a; Moen 2012; NCT00533117; Reich 2009; Salzman 1995; Simpson 2004; Soloff 1989; Soloff 1993; Zanarini 2001; Zanarini 2003; Zanarini 2004).
Funding
Seventeen trials were funded or partially funded by the pharmaceutical industry (AstraZeneca 2007; Black 2014; Bogenschutz 2004; Frankenburg 2002; Grant 2022; Hollander 2001; Leone 1982; Linehan 2008; Moen 2012; Pascual 2008; Reich 2009; Schulz 2007; Simpson 2004; Soler 2005; Zanarini 2001; Zanarini 2004; Zanarini 2007), 10 where funded by universities or research foundations (Amminger 2013; Crawford 2018; Hallahan 2007; Kulkarni 2018; NCT00533117; Rinne 2002; Shafti 2014; Soloff 1989; Soloff 1993; Zanarini 2003), eight received no funding (Bellino 2014; Bozzatello 2017; Loew 2006; Nickel 2005; Nickel 2006; Shafti 2010; Tritt 2005; Ziegenhorn 2009), and 11 had unclear funding (Cowdry 1988; De la Fuente 1994; Goldberg 1986; Jariani 2010; Markovitz 1995a; Montgomery 1982a; Montgomery 1982b; Nickel 2004; Salzman 1995; Schmahl 2012a; Schmahl 2012b).
Participants
The 46 trials included a total of 2769 participants with BPD. One trial, Markovitz 1995a, was not included in the quantitative analysis due to insufficient reporting for effect size calculations, leaving a total of 45 trials (2752 participants) in the quantitative analysis. Two trials reported on completers only (Montgomery 1982b; Salzman 1995), and thus the number of participants was probably higher. The mean age ranged from 16.2 (Amminger 2013) to 39.7 years (Grant 2022). One trial did not report on demographics at baseline, leaving sex and mean age unknown (Markovitz 1995a). Fifteen trials included only females (Cowdry 1988; Frankenburg 2002; Linehan 2008; Loew 2006; Nickel 2004; Rinne 2002; Schmahl 2012a; Schmahl 2012b; Shafti 2010; Shafti 2014; Simpson 2004; Tritt 2005; Zanarini 2001; Zanarini 2003; Zanarini 2004); and one trial included only males (Nickel 2005). All remaining trials included both sexes, though predominantly females.
Diagnostic criteria
Participants were diagnosed with BPD according to criteria from DSM III, DSM III R, DSM IV, DSM IV R or ICD‐10. BPD diagnoses were confirmed by one or more standardised means of assessment. Four trials used the Diagnostic Interview for Borderline personality disorder (DIB) (Cowdry 1988; De la Fuente 1994; Soloff 1989; Soloff 1993), while four used the revised version (DIB‐R) (Reich 2009; Zanarini 2001; Zanarini 2003; Zanarini 2004). Two of the included trials used the International Personality Disorder Examination scale (IPDE) (Schmahl 2012a; Schmahl 2012b) while one used IPDE in addition to SCID‐I (Crawford 2018). One trial used the Scales of independent behaviour (SIB) (Goldberg 1986), and two trials used the Zanarini Rating Scale for Borderline Personality Disorder (ZAN‐BPD) (Grant 2022; Kulkarni 2018). One trial used the DIPD‐IV scale (Frankenburg 2002) while two used both DIPD‐IV and ZAN‐BPD (Schulz 2007; Zanarini 2007). Three trials used an unspecified clinical interview (Jariani 2010; Montgomery 1982a; Montgomery 1982b). One trial did not use a structured instrument but confirmed BPD diagnosis through consensus (Amminger 2013). Four trials stated that BPD was diagnosed according to the formal DSM criteria but did not specify if a standardised assessment instrument was used to confirm the diagnosis (Leone 1982; NCT00533117; Shafti 2010; Shafti 2014). Eleven of the trials used the Structured Clinical Interview for DSM‐IV axis II disorders (SCID II): AstraZeneca 2007; Bogenschutz 2004; Hallahan 2007; Hollander 2001; Linehan 2008; Loew 2006; Nickel 2004; Nickel 2005; Nickel 2006; Simpson 2004; Tritt 2005. In addition to SCID‐II, nine trials used one or more additional means of assessment (Bellino 2014; Bozzatello 2017; Markovitz 1995a; Moen 2012; Pascual 2008; Rinne 2002; Salzman 1995; Soler 2005; Ziegenhorn 2009). One trial used SCID but did not specify which version (Black 2014). An overview of assessment instruments used by the individual trials is found in Table 4 and Characteristics of included studies.
Participant exclusion criteria
Most trials had several exclusion criteria, with the most common one used in 38 trials, the exclusion of participants with current major depression, bipolar affective disorder or psychotic disorder. Thirty of the trials also excluded participants with alcohol or substance abuse or dependence. Other common exclusion criteria were organic illness, mental retardation, cognitive disorder or impairment, severe somatic illness, chronic medical conditions, and pregnancy or breastfeeding. For exclusion criteria in all trials, see Table 4 and Characteristics of included studies.
Interventions
Duration of interventions
Trial duration lasted from four weeks (Ziegenhorn 2009) to 52 weeks (Crawford 2018; NCT00533117). Thirty‐five of the trials had a duration of three months or less, leaving only 11 trials with a duration between three months and one year.
Format of intervention
The majority of trial interventions tested one type of medication to alleviate borderline symptoms. Two trials had more than one treatment arm investigating the effect of various doses or treatment durations of the same medication (Black 2014; Zanarini 2007). Two trials compared a medication in addition to treatment‐as‐usual with a placebo intervention in addition to treatment‐as‐usual (Crawford 2018; Kulkarni 2018), and one trial intervention specifically combined Dialectic Behaviour Therapy (DBT) with the selective serotonin reuptake inhibitor fluoxetine (NCT00533117). Ten trials were head‐to‐head trials comparing active medications to each other (Cowdry 1988; Bellino 2014; Bozzatello 2017; Jariani 2010: Leone 1982; Shafti 2010; Shafti 2014; Soloff 1989; Soloff 1993; Zanarini 2004); four of which were multiple‐arm trials, with an additional placebo comparator (Cowdry 1988; Soloff 1989; Soloff 1993; Zanarini 2004).
Type of intervention
The trials included in this review investigated the following 27 medications:
Amitriptyline
Alprazolam
Aripiprazole
Asenapine
Brexpiprazole
Carbamazepine
Clonidine
Fluoxetine
Flupenthixol
Fluvoxamine
Haloperidol
Lamotrigine
Loxapine
Memantine hydrochloride
Mianserin
Naltrexone
Olanzapine
Omega‐3 fatty acids
Phenelzine
Quetiapine
Sertraline
Thiothixene
Topiramate
Tranylcypromine
Trifluoperazine
Valporate semi sodium
Ziprasidone
Antipsychotics
First‐generation antipsychotics were used in six trials, two of which had haloperidol interventions (Soloff 1993; Shafti 2010). The other four had a thiothixene intervention (Goldberg 1986), a loxapine intervention (Leone 1982), a flupenthixol intervention (Montgomery 1982a) and a trifluoperazine hydrochloride intervention (Cowdry 1988).
Second‐generation antipsychotics were used in 16 trials, of which ten had an olanzapine intervention (Bogenschutz 2004; Bozzatello 2017; Jariani 2010; Linehan 2008; Schulz 2007; Shafti 2010; Shafti 2014; Soler 2005; Zanarini 2001; Zanarini 2007). Two trials had an aripiprazole intervention (Nickel 2006; Shafti 2014), and one trial each had a quetiapine (Black 2014), asenapine (Bozzatello 2017) ziprasidone (Pascual 2008) and brexpiprazole (Grant 2022) intervention.
Antidepressants
Seven trials had selective serotonin reuptake inhibitor (SSRI) interventions. Of these, five had a fluoxetine intervention (Markovitz 1995a; NCT00533117; Salzman 1995; Simpson 2004; Zanarini 2004). one had a fluvoxamine intervention (Rinne 2002), and one head‐to‐head trial had a sertraline intervention (Jariani 2010).
One multiple‐arm cross‐over trial had a monoamine oxidase inhibitor (MAO‐I) intervention of tranylcypromine sulfate (Cowdry 1988).
One trial had an intervention with the noradrenergic and specific serotonergic antidepressant (NaSSA) mianserin (Montgomery 1982b).
One trial had an intervention with the tricyclic antidepressant (TCA) amitriptyline (Soloff 1989).
Mood stabilisers
Eleven trials had interventions with anticonvulsants. Three trials had a lamotrigine intervention (Crawford 2018; Reich 2009; Tritt 2005) while another three used topiramate (Loew 2006; Nickel 2004; Nickel 2005). Two trials had an intervention of valproate semi sodium (Frankenburg 2002; Hollander 2001), while two others had a carbamazepine intervention and a valproate intervention (De la Fuente 1994; Moen 2012, respectively). One trial, Cowdry 1988, had a carbamazepine intervention.
Miscellaneous medications
The antidementia drug, memantine hydrochloride, was used in one trial (Kulkarni 2018). The opioid antagonist naltrexone was used in two trials (Schmahl 2012a; Schmahl 2012b). Omega‐3 fatty acids were used in four trials (Amminger 2013; Bellino 2014; Hallahan 2007; Zanarini 2003). An antihypertensive/α2‐adrenoceptor agonist was used in one trial with a clonidine intervention (Ziegenhorn 2009), and the benzodiazepine alprazolam was used in one trial (Cowdry 1988).
Concomitant medication
Eight trials did not allow concomitant medication (Bellino 2014; Bozzatello 2017; Moen 2012; Nickel 2004; Nickel 2006; Salzman 1995; Shafti 2014; Soloff 1993). Another seven trials gave specific information that psychotropics were not allowed (Frankenburg 2002; Loew 2006, Nickel 2005; Rinne 2002; Shafti 2010; Tritt 2005; Zanarini 2001). Thirteen trials did not allow for psychotropic co‐medication with the exception of benzodiazepines, SSRIs or both (Amminger 2013; AstraZeneca 2007; Black 2014; Kulkarni 2018; Leone 1982; NCT00533117; Pascual 2008; Reich 2009; Schulz 2007; Simpson 2004; Soler 2005; Soloff 1989; Ziegenhorn 2009). However, many of these had restrictions and limitations regarding dosage, stability and drug initiation of concomitant medication prior to and during the trial. One trial allowed for patients to continue with standard psychiatric care and have changes to their psychotropic medication as prescribed, which 53.1% of participants did (Hallahan 2007). In another trial, 100% of participants used methadone, but there was no further mention of co‐medication (Jariani 2010). One trial allowed antidepressants, mood stabilisers, and stimulants, but the use of any new psychotropic medication was an exclusion criterion (Grant 2022). Lastly, one trial specifically allowed medication for stable, chronic medical conditions such as hypertension but gave no further mention of other admissible concomitant medications (Bogenschutz 2004). Twelve trials did not give any information on concomitant medication (Cowdry 1988; Goldberg 1986; Hollander 2001; Linehan 2008; Markovitz 1995a; Montgomery 1982a; Montgomery 1982b; Schmahl 2012a; Schmahl 2012b; Zanarini 2003, Zanarini 2004; Zanarini 2007). Further, two trials similarly gave no details on permissible medications in the full report; however, Crawford 2018 excluded patients that were already prescribed a mood stabiliser within the past four weeks. Additionally, both arms received usual care. Another had a psychotropic medication washout for at least 10 days, a 15‐day washout for antidepressants (TCAs and MAOIs), and no patient had taken neuroleptics two months prior to the trial (De la Fuente 1994).
Concomitant treatment
Twelve trials did not allow for concomitant psychotherapeutic treatment or treatment‐as‐usual (Black 2014; Bozzatello 2017; Frankenburg 2002; Hallahan 2007; Loew 2006; Nickel 2004; Nickel 2005; Nickel 2006; Reich 2009; Rinne 2002; Shafti 2010; Tritt 2005). Five trials offered or allowed for Dialectic Behavioural therapy (DBT) (Linehan 2008; Moen 2012; NCT00533117; Simpson 2004; Soler 2005) and another eight trials allowed or provided unspecified psychotherapy, psychological and psychosocial interventions concomitant with trial intervention or supportive atheoretical psychotherapy: Amminger 2013; De la Fuente 1994; Kulkarni 2018; Montgomery 1982a; Montgomery 1982b; Pascual 2008; Schmahl 2012a; Schmahl 2012b. One trial allowed psychotherapy if initiated a minimum of three months prior to randomisation (Bogenschutz 2004). The remaining 20 trials, did not state whether any type of concomitant treatment was offered or allowed.
Comparators
The review included 22 different comparators. Nine had a placebo control intervention, and 13 were head‐to‐head trials with an active comparison intervention that was either another single medication or a combination of medications.
Control intervention
The control intervention was a placebo.
Active medication versus placebo
Antidepressants SSRIs, NaSSAs, TCAs and MAOIs versus placebo: tranylcypromine sulfate (Cowdry 1988), fluoxetine (Markovitz 1995a; NCT00533117; Salzman 1995; Simpson 2004), mianserin (Montgomery 1982b), fluvoxamine (Rinne 2002), amitriptyline (Soloff 1989), phenelzine sulfate (Soloff 1993);
First‐generation antipsychotics versus placebo: trifluoperazine hydrochloride (Cowdry 1988), thiothixene (Goldberg 1986), loxapine (Leone 1982), flupenthixole (Montgomery 1982a), haloperidol (Soloff 1989; Soloff 1993);
Second generation antipsychotics versus placebo: olanzapine (Bogenschutz 2004; Linehan 2008; Schulz 2007; Soler 2005; Zanarini 2001; Zanarini 2007), aripiprazole (Nickel 2006), ziprasidone (Pascual 2008), quetiapine (Black 2014), brexpiprazole (Grant 2022);
Anticonvulsants versus placebo: lamotrigine (Crawford 2018; Reich 2009; Tritt 2005), topiramate (Loew 2006; Nickel 2004; Nickel 2005), valproate semi sodium (Frankenburg 2002; Hollander 2001; Moen 2012), carbamazepine (Cowdry 1988; De la Fuente 1994), divalproex (Moen 2012);
Omega‐3 fatty acids versus placebo: (Amminger 2013; Hallahan 2007; Zanarini 2003);
Antidementia drug versus placebo: memantine hydrochloride (Kulkarni 2018);
Opiod antagonist versus placebo: naltrexone (Schmahl 2012a; Schmahl 2012b);
Antihypertensives/alpha‐2 adrenoceptor agonist versus placebo: clonidine (Ziegenhorn 2009);
Benzodiazepine versus placebo: alprazolam (Cowdry 1988).
Active medication versus active comparator medication
First‐generation antipsychotic versus first‐generation antipsychotic: loxapine versus chlorpromazine (Leone 1982);
Second‐generation antipsychotic versus second‐generation antipsychotic: olanzapine versus aripiprazole (Shafti 2014), and olanzapine versus asenapine (Bozzatello 2017);
Second‐generation antipsychotic versus first‐generation antipsychotic: olanzapine versus haloperidol (Shafti 2010);
First‐generation antipsychotic versus antidepressant: trifluoperazine hydrochloride versus tranylcypromine sulfate (Cowdry 1988), haloperidol versus amitriptyline (Soloff 1989), and haloperidol versus phenelzine sulfate (Soloff 1993);
Second‐generation antipsychotic versus antidepressant: olanzapine versus sertraline (Jariani 2010), and olanzapine versus fluoxetine (Zanarini 2004);
Benzodiazepine versus mood stabiliser: alprazolam versus carbamazepine (Cowdry 1988);
Benzodiazepine versus first‐generation antipsychotic: alprazolam versus trifluoperazine hydrochloride (Cowdry 1988);
Benzodiazepine versus antidepressant: alprazolam versus tranylcypromine sulfate (Cowdry 1988);
Mood stabiliser versus first‐generation antipsychotic: carbamazepine versus trifluoperazine hydrochloride (Cowdry 1988);
Mood stabiliser versus antidepressant: carbamazepine versus tranylcypromine sulfate (Cowdry 1988).
Active medication versus combination of medications
Second generation antipsychotic versus second generation antipsychotic plus antidepressant: olanzapine versus olanzapine plus fluoxetine (Zanarini 2004);
Antidepressant versus antidepressant plus second‐generation antipsychotic: fluoxetine versus fluoxetine plus olanzapine (Zanarini 2004);
Mood stabiliser versus mood stabiliser plus miscellaneous (omega‐3 fatty acid): valproic acid versus valproic acid plus eicosapentaenoic acid (EPA) plus docosahexaenoic acid (DHA) (Bellino 2014).
Outcomes
See Appendix 5 for information on outcome measurements in each trial.
Adverse effects
Adverse effects were registered in all but seven trials. Adverse effects were, in most cases, measured spontaneously, but some trials also used anthropometric and laboratory values as measurement. Five trials used the Abnormal Involuntary Movement Scale (AIMS) and the Simpson‐Angus Scale (SAS), and a few used the Side Effect Rating Scale for extrapyramidal side effects. See Appendix 5 for specifications on scales used in each trial.
Excluded studies
We excluded a total of 20 trials (from 22 reports) from this review (see Figure 1). We excluded 11 due to an ineligible population; two included people with PD but none had BPD and in nine < 70% had a BPD diagnosis. For these nine, we tried to retrieve subsample data, but the subsample data were unavailable. We also excluded two studies due to an ineligible intervention (intervention was less than two weeks and not continuous) and one included an ineligible comparator. The remaining six trials were withdrawn or discontinued. For further information, see the table of Characteristics of excluded studies.
Studies awaiting classification
We assessed two trials as awaiting classification. One trial used an omega‐3 fatty acids intervention, comparing omacor to placebo (NCT00437099), while the other investigated the antidepressant selegiline and compared it to placebo (NCT01912391). Both trials included a study population of adults with BPD and were registered, with start dates in 2009 (NCT00437099) and 2012 (NCT01912391). See Characteristics of studies awaiting classification tables for further information.
Despite rigorous searches, we were unable to find any corresponding publications or reports of trial results for either of these. Contact information was provided for the omacor trial (NCT00437099); however, we were unable to get in contact with the principal investigator, despite multiple attempts. No contact information was provided for the selegiline trial (NCT01912391), and we were unable to obtain this information. Judging from the trial registrations, both trials seem eligible for inclusion in this review. However, we are unaware of what has happened to the trials and if their results can be obtained. For this reason, we have categorised them as awaiting classification.
Ongoing studies
We included 11 ongoing trials that are assessing different pharmacological interventions for the treatment of BPD, and for which the outcome data are not yet available (see Characteristics of ongoing studies tables). All ongoing trials have a parallel design with a placebo or inert control intervention. Four of these are investigating antipsychotics, of which one has a brexpiprazole intervention (NCT04100096), one has a clozapine intervention (EudraCT 2018‐002471‐18‐GB), one has a lumateperone intervention (NCT05356013) and another is using aripiprazole (Chanen 2019). The remaining ongoing trials are investigating miscellaneous medications; one trial is investigating the female hormone, estradiol (ACTRN12617001317381), one has a vafidemstat intervention (inhibitor of Lysine Demethylase 1) (EUCTR2020‐003469‐20‐ES), one has a memantine intervention (NMDA‐receptor antagonist) (IRCT20210106049948N1), one is investigating omega‐3 fatty acids (IRCT20210531051453N1), one is a phase two trial from Boehringer Ingelheim with a medication labelled BI 1358894 (NCT04566601),one is investigating the effects of a stellate ganglion block (DRKS00015817) and one has a randomised, open‐label design and is investigating a probiotic intervention (Arteaga‐Henríquez 2020).
One trial, Chanen 2019, has a patient population of adolescents and young adults (15 to 25 years old), with auditory verbal hallucinations and BPD or ADHD. Another has a minimum age of 16 and, therefore, also include adolescents (IRCT20210106049948N1). All remaining trials are examining adults with BPD. One trial is investigating females only (ACTRN12617001317381), while the rest are including both men and women. Five trials have a duration of 12 weeks (ACTRN12617001317381; Chanen 2019; IRCT20210106049948N1; NCT04100096; NCT04566601). The duration in the remaining six trials are six months (EudraCT 2018‐002471‐18‐GB), 14 weeks (EUCTR2020‐003469‐20‐ES), 10 weeks (Arteaga‐Henríquez 2020), eight weeks (DRKS00015817; NCT05356013) and six weeks (IRCT20210531051453N1).
Risk of bias in included studies
Figure 2 and Figure 3 portray the review authors' judgements on the risk of bias across the included trials and for each trial. Extended information about the individual trials can be found in the Characteristics of included studies tables.
2.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies
3.

Risk of bias summary: review authors' judgements about each risk of bias item for each included trial
Allocation
Sequence generation
We judged 16 trials as having a low risk of bias, where the randomisation method was described and adequate (e.g. using computer‐generated random numbers) (Amminger 2013; Bellino 2014; Bozzatello 2017; Crawford 2018; Frankenburg 2002; Grant 2022; Hallahan 2007; Kulkarni 2018; Linehan 2008; Pascual 2008; Reich 2009; Schmahl 2012a; Schmahl 2012b; Schulz 2007; Zanarini 2001; Zanarini 2007). The remaining 30 trials did not contain an exact description of how treatment allocation had been conducted, so we rated these trials as having unclear risk of bias.
Allocation concealment
We assessed eight trials as having a low risk of bias (e.g. central third party randomisation) (Amminger 2013; Crawford 2018; Frankenburg 2002; Grant 2022; Hallahan 2007; Kulkarni 2018; Schmahl 2012a; Schmahl 2012b). Three trials were rated as having a high risk of bias due to inadequate (e.g. based on day of admission or case record number) or no allocation concealment (Bellino 2014; Bozzatello 2017; Shafti 2014). The remaining 35 of the trials did not have adequate information to enable a judgement about the allocation concealment, thus they were rated as having an unclear risk of bias.
Blinding
Blinding of participants and personnel
We rated 18 trials as having a low risk of bias as they reported how the participants and personnel were kept blind to the treatment allocation (Amminger 2013; Black 2014; Bogenschutz 2004; Cowdry 1988; Crawford 2018; Frankenburg 2002; Goldberg 1986; Grant 2022; Hallahan 2007; Leone 1982; Montgomery 1982a; Nickel 2004; Nickel 2005; Soloff 1993; Tritt 2005; Zanarini 2001; Zanarini 2004). We rated three trials as having a high risk of bias due to no blinding of participants and personnel (Bellino 2014; Bozzatello 2017; Shafti 2014). The remaining 25 trials were judged as having an unclear risk of bias due to inadequate description of the blinding of participants and personnel.
Blinding of outcome assessors
We judged 15 trials as having a low risk of bias due to adequate blinding of outcome assessors (i.e. described efforts to ensure the blinding of outcome assessment and due to these efforts blinding was deemed unlikely to be broken) (Amminger 2013; Bellino 2014; Crawford 2018; De la Fuente 1994; Hallahan 2007; Hollander 2001; Linehan 2008; Moen 2012; Reich 2009; Salzman 1995; Simpson 2004; Soloff 1989; Soloff 1993; Zanarini 2004; Ziegenhorn 2009). Two trials were rated as having a high risk of bias due to no blinding of outcome assessors (Bozzatello 2017; Shafti 2014). The remaining 29 trials were rated as having an unclear risk of bias due to inadequate description (e.g. insufficient information to permit judgement of low or high risk) of the blinding of outcome assessors.
Incomplete outcome data
We judged 12 trials as having a low risk of bias due to no indication of incomplete outcome reporting (e.g. due to no missing outcome data or appropriate methods to handle missing data were described) (Amminger 2013; Goldberg 1986; Kulkarni 2018; Leone 1982; Montgomery 1982a; Montgomery 1982b; Nickel 2006; Rinne 2002; Shafti 2010; Shafti 2014; Soloff 1989; Soloff 1993). Thirteen trials were considered to be at high risk of bias due to the inadequate descriptions of possible reasons for missing data (e.g. by inappropriate application of simple imputation or the likelihood of missing data being related to the true outcome) (Bellino 2014; Black 2014; Bozzatello 2017; Cowdry 1988; Crawford 2018; Grant 2022; Hollander 2001; Markovitz 1995a; Moen 2012; Pascual 2008; Simpson 2004; Tritt 2005; Ziegenhorn 2009). The remaining 21 trials did not provide descriptions of possible reasons for missing data, so we rated these trials as having an unclear risk of bias.
Selective reporting
We considered eight trials to be at low risk of bias, as all prespecified outcomes were reported according to the published protocol or the registration of the trial, which was registered prior to conducting the trial (Bellino 2014; Bozzatello 2017; Crawford 2018; NCT00533117; Pascual 2008; Reich 2009; Soler 2005; Zanarini 2007). We rated eight trials to be at high risk of bias due to the trials not explicitly reporting data for the prespecified outcomes, even when they had initially planned to report them (Amminger 2013; AstraZeneca 2007; Black 2014; Cowdry 1988; Kulkarni 2018; Schulz 2007; Tritt 2005; Zanarini 2001). We rated the remaining 30 trials as having an unclear risk of bias due to either not having a published protocol prior to initiating the trial or not providing sufficient information in the report to assess the extent of reporting bias.
Other potential sources of bias
Vested interest
Fifteen trials were rated as having a low risk of bias in terms of funding or author affiliations (Amminger 2013; Bellino 2014; Bozzatello 2017; Crawford 2018; Hallahan 2007; Kulkarni 2018; NCT00533117; Schmahl 2012a; Schmahl 2012b; Shafti 2010; Shafti 2014; Soloff 1989; Soloff 1993; Tritt 2005; Ziegenhorn 2009). We rated 19 trials to be at high risk of bias due to author affiliations with or funding from pharmaceutical companies: AstraZeneca 2007; Black 2014; Bogenschutz 2004; Frankenburg 2002; Grant 2022; Hollander 2001; Leone 1982; Linehan 2008; Moen 2012; Pascual 2008; Reich 2009; Rinne 2002; Schulz 2007; Simpson 2004; Soler 2005; Zanarini 2001; Zanarini 2003; Zanarini 2004; Zanarini 2007. Twelve trials did not provide sufficient information about funding or affiliations to permit a judgement of low or high risk, so these trials have been rated as having an unclear risk of bias (Cowdry 1988; De la Fuente 1994; Goldberg 1986; Jariani 2010; Loew 2006; Markovitz 1995a; Montgomery 1982a; Montgomery 1982b; Nickel 2004; Nickel 2005; Nickel 2006; Salzman 1995).
Other risk of bias
We had intended to use this domain as a default category of bias that might not have been covered by the remaining categories, and could potentially be a threat to validity. Only one of the included trials, Cowdry 1988, was rated as having unclear risk of bias in this domain due to an obvious carry‐over effect between medication phases. The remaining trials had no apparent other sources of bias and were rated as low risk.
Overall risk of bias
All trials were assessed as being at high risk of bias because at least one domain was rated as being at high or unclear risk of bias.
Effects of interventions
See: Table 1; Table 2; Table 3
We present the results for each of the primary and secondary outcomes connected to the three comparisons.
Medications compared to placebo, which covers analyses of medications and a placebo comparator. All medications have been divided into the four drug classifications: antipsychotics, antidepressants, mood stabilisers, and miscellaneous medications that have been reported by name (omega‐3 fatty acids, naltrexone, clonidine, memantine hydrochloride and alprazolam).
Medication compared to another medication, which covers head‐to‐head analyses (where the comparator is an active medication instead of a placebo).
Medication compared to a combination of medications, which covers analyses of a medication compared to the combination of the same medication and another active medication.
Where a meta‐analysis involved two or more different instruments to measure the same construct, we reported effect sizes as SMD, otherwise we reported the MD. To identify the MIREDIF, we transformed the SMD to MD for the scale with best validity and reliability for that outcome. Where we could not find this, we used an assumption that the minimal relevant clinical intervention effect was approximately ½ SD on the used scale, which can be used as a MIREDIF (Norman 2004). An overview of the specific instruments used to measure each outcome by the individual trials can be found in Appendix 5.
We contacted authors of 25 trials with unclear or missing data and requested the necessary information. Authors of seven trials replied that their registered trials had never been started or had to be discontinued early due to recruiting problems, personal changes, or no funding. We retrieved additional information by email from five authors of included trials (Bellino 2014; Black 2014; Crawford 2018; Kulkarni 2018; Schmahl 2012a; Schmahl 2012b). We received no reply from authors of 17 trials (Amminger 2013; AstraZeneca 2007; Bogenschutz 2004; Coccaro 1997; Cowdry 1988; De la Fuente 1994; Jariani 2010; Koenigsberg 2003; La Malfa 2003; Markovitz 1995a; NCT00437099; NCT00533117; Shafti 2010; Shafti 2014; Serban 1984; Verkes 1998; Ziegenhorn 2009).
We performed TSA on relevant primary outcomes and the relevant secondary outcomes, interpersonal problems, attrition, and adverse events at end of treatment for the three comparisons in our summary of findings tables, adjusting for multiplicity and sparse data. We used the TSA for our rating of imprecision where we were uncertain about our ratings. We considered all trials as being at high risk of bias overall. However, we used all eligible trials in the meta‐analyses, as the Cochrane Handbook for Systematic Reviews of Interventions recommends doing when all trials are assigned the same risk of bias. We took account of our risk of bias assessment when considering the quality of the evidence using the GRADE approach, to ensure that judgements about risk of bias and other factors affecting the quality of the evidence were taken into account when interpreting the results of the review (Higgins 2022).
Negative effect estimates indicate beneficial effects by the active medication, in terms of a reduction of burden. If scales were used by the primary studies where higher scores were the preferable outcome, e.g. in terms of a better level of psychosocial functioning, scores were multiplied by (‐1) before entering them for effect size calculation, to ensure that a negative direction of effect indicated a beneficial effect throughout the whole review.
Comparison 1: Medication compared with placebo
Primary outcomes
1.1 BPD symptom severity
A total of 16 trials comparing medication with placebo reported data for BPD symptom severity.
Eight trials compared antipsychotics to placebo (Black 2014; Cowdry 1988; Goldberg 1986; Grant 2022; Pascual 2008; Schulz 2007; Soloff 1993; Zanarini 2007). The evidence indicates little to no difference but is very uncertain about the effect of antipsychotics compared with placebo on BPD symptom severity at the end of treatment (SMD ‐0.18, 95% CI ‐0.45 to 0.08; P = 0.18; I2 = 70%; 8 trials, 951 participants; very low‐certainty evidence; Analysis 1.1). The TSA showed that the z‐curve ended in the futility area, which means that the anticipated effect can be rejected. See Figure 4 in Appendix 6.
1.1. Analysis.

Comparison 1: Medications compared with placebo, Outcome 1: Primary: BPD symptom severity at end of treatment (continuous outcomes, SMDs)
4.
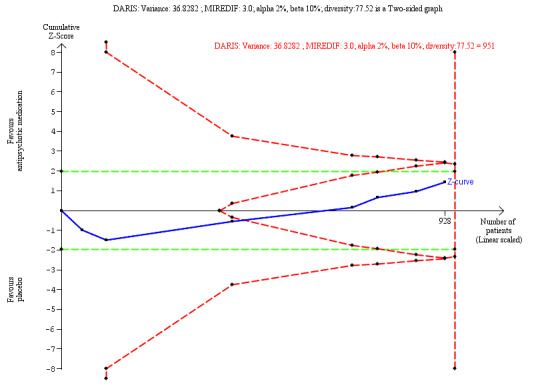
TSA borderline severity: antipsychotics
DARIS: Diversity‐adjusted required information size; MIREDIF: Minimal relevant difference; TSA: Trial Sequential Analysis
Two trials compared antidepressants to placebo post‐treatment (Cowdry 1988; Soloff 1993). The evidence indicates little to no difference but is very uncertain about the effect of antidepressants compared with placebo regarding BPD symptom severity (SMD 0.27, 95% CI −0.65 to 1.18; P = 0.57, I2 = 73%; 2 trials, 87 participants; very low‐certainty evidence; Analysis 1.1).
Four trials compared mood stabilisers to placebo (Cowdry 1988; Crawford 2018; Moen 2012; Reich 2009). The evidence indicates little to no difference but is very uncertain about the effect of mood stabilisers compared with placebo at end of treatment (SMD 0.07, 95% CI −0.43 to 0.57; P = 0.78, I2 = 55%; 4 trials, 256 participants; very low‐certainty evidence; Analysis 1.1).
Four trials compared miscellaneous pharmacological therapies to placebo: Cowdry 1988 compared alprazolam to placebo; Kulkarni 2018 compared memantin‐hydrochloride to placebo; Schmahl 2012b compared naltrexone to placebo; and Ziegenhorn 2009 compared clonidine to placebo. There was no evidence that any of these miscellaneous medications had an effect on BPD symptom severity at the end of treatment compared to placebo; Cowdry 1988: MD −0.58, 95% CI −1.63 to 0.47; P = 0.28; 1 trial, 25 participants; Analysis 1.2; Kulkarni 2018: MD 2.00, 95% CI −1.62 to 5.62; P = 0.28; 1 trial, 33 participants; Analysis 1.2; Schmahl 2012b: MD 0.10, 95% CI −0.29 to 0.49; P = 0.62; 1 trial, 32 participants; Analysis 1.2; and Ziegenhorn 2009: MD −13.11, 95% CI −65.36 to 39.14; P = 0.62; 1 trial, 34 participants; Analysis 1.2.
1.2. Analysis.

Comparison 1: Medications compared with placebo, Outcome 2: Primary: BPD symptom severity at end of treatment (continuous outcomes, MDs)
1.2 Self‐harm
Five trials comparing medications with placebo reported data for self‐harm.
Two trials compared antipsychotics to placebo (Linehan 2008; Nickel 2006). The evidence is very uncertain about the effect of antipsychotics compared with placebo at end of treatment (RR 0.66, 95% CI 0.15 to 2.84; P = 0.57, I2 = 67%; 2 trials, 76 participants; very low‐certainty evidence; Analysis 1.4).
1.4. Analysis.

Comparison 1: Medications compared with placebo, Outcome 4: Primary: Self‐harm at end of treatment (dichotomous outcomes, RRs)
One trial compared an antidepressant to placebo (Simpson 2004). It may have little to no effect but the evidence is very uncertain about the effect of the antidepressant compared with placebo at the end of treatment (MD 0.45, 95% CI −10.55 to 11.45; P = 0.94; 1 trial, 20 participants; very low‐certainty evidence; Analysis 1.3).
1.3. Analysis.

Comparison 1: Medications compared with placebo, Outcome 3: Primary: Self‐harm at end of treatment (continuous outcomes, MDs)
One trial compared a mood stabiliser to placebo (Crawford 2018). The evidence is very uncertain about the effect of mood stabiliser lamotrigine compared with placebo at the end of treatment (RR 1.08, 95% CI 0.79 to 1.48; P = 0.64; 1 trial, 276 participants; very low‐certainty evidence; Analysis 1.4).
One trial compared omega‐3 fatty acids to placebo (Hallahan 2007). There was no clear evidence of a difference between omega‐3 fatty acids and placebo regarding self‐harm at the end of treatment (RR 1.23, 95% CI 0.51 to 2.97; P = 0.65; 1 trial, 49 participants; Analysis 1.4).
1.3 Suicide‐related outcomes
Thirteen trials comparing medications with placebo reported data for suicide‐related outcomes.
Eight trials compared antipsychotics to placebo (Bogenschutz 2004; Cowdry 1988; Grant 2022; Linehan 2008; Montgomery 1982a; Pascual 2008; Soler 2005; Schulz 2007; Zanarini 2007). The evidence indicates little to no effect but is very uncertain about the effect of antipsychotics compared with placebo at the end of treatment either with continuous outcome data (SMD 0.05, 95% CI ‐0.18 to 0.29; P = 0.67; I2 = 55%; 7 trials, 854 participants; very low‐certainty evidence; Analysis 1.5) or with dichotomous outcome data (RR 0.73, 95% CI 0.31 to 1.73; P = 0.47, I2 = 62%; 2 trials, 61 participants; very low‐certainty evidence; Analysis 1.7).
1.5. Analysis.

Comparison 1: Medications compared with placebo, Outcome 5: Primary: Suicide‐related outcomes at end of treatment (continuous outcomes, SMDs)
1.7. Analysis.

Comparison 1: Medications compared with placebo, Outcome 7: Primary: Suicide‐related outcomes at end of treatment (dichotomous outcomes, RRs)
Three trials compared antidepressants to placebo (Cowdry 1988; Montgomery 1982b; Simpson 2004). The evidence indicates little to no effect but is very uncertain about the effect of antidepressants at end of treatment compared with placebo either with continuous data (SMD −0.26, 95% CI −1.62 to 1.09; P = 0.70, I2 = 80%; 2 trials, 45 participants; very low‐certainty evidence; Analysis 1.5), or with dichotomous data (RR 1.00, 95% CI 0.71 to 1.41; P = 1.00; 1 trial, 58 participants; very low‐certainty evidence; Analysis 1.7).
Two trials compared mood stabilisers to placebo (Cowdry 1988; Hollander 2001). The evidence indicates little to no effect but is very uncertain about the effect of mood stabilisers compared with placebo at the end of treatment (SMD ‐0.36, 95% CI ‐1.96 to 1.25; P = 0.66, I2 = 81%; 2 trials, 44 participants; very low‐certainty evidence; Analysis 1.5).
One trial compared omega‐3 fatty acids to placebo (Hallahan 2007). There was evidence that omega‐3 fatty acids may reduce suicide‐related outcomes more than placebo at the end of treatment (RR 0.52, 95% CI 0.28 to 0.95; P = 0.03; 1 trial, 49 participants; Analysis 1.7).
One trial also compared the benzodiazepine alprazolam to placebo (Cowdry 1988). There was no clear evidence of a difference between alprazolam and placebo on suicide‐related outcomes at end of treatment (MD 0.75, 95% CI ‐0.18 to 1.68; P = 0.11; 1 trial, 25 participants; Analysis 1.6).
1.6. Analysis.

Comparison 1: Medications compared with placebo, Outcome 6: Primary: Suicide‐related outcomes at end of treatment (continuous outcomes, MDs)
1.4 Psychosocial functioning
Seventeen trials comparing medications to placebo reported data for psychosocial functioning. For consistency, negative values indicate favourable results for the active medication as for the remaining outcomes.
Seven trials compared antipsychotics to placebo (Black 2014; Goldberg 1986; Schulz 2007; Soler 2005; Soloff 1989; Soloff 1993; Zanarini 2007). The evidence indicates little to no difference in outcome but is very uncertain about the effect of antipsychotics compared with placebo on psychosocial functioning at end of treatment (SMD −0.16, 95% CI −0.33 to 0.00; P = 0.05, I2 = 23%; 7 trials, 904 participants; very low‐certainty evidence; Analysis 1.8).
1.8. Analysis.

Comparison 1: Medications compared with placebo, Outcome 8: Primary: Psychosocial functioning at end of treatment (continuous outcomes, SMDs)
Four trials compared antidepressants to placebo (Salzman 1995; Simpson 2004; Soloff 1989; Soloff 1993). The evidence indicates little to no difference between antidepressants and placebo regarding psychosocial functioning at end of treatment, but the evidence is very uncertain (SMD ‐0.25, 95% CI −0.57 to 0.06; P = 0.11, I2 = 0%; 4 trials, 161 participants; very low‐certainty evidence; Analysis 1.8).
Two trials compared mood stabilisers to placebo (Crawford 2018; De la Fuente 1994) with little to no difference in psychosocial functioning at the end of treatment as compared to placebo (SMD −0.01, 95% CI −0.28 to 0.26; P = 0.94, I2 = 0%; 2 trials, 214 participants; very low‐certainty evidence; Analysis 1.8). Hollander 2001 obtained a similar result (RR 0.64, 95% CI 0.37 to 1.11; P = 0.11; 1 trial, 16 participants; very low‐certainty evidence; Analysis 1.10).
1.10. Analysis.

Comparison 1: Medications compared with placebo, Outcome 10: Primary: Psychosocial functioning at end of treatment (dichtotomous outcomes, RRs)
One trial compared omega‐3 fatty acids to placebo (Amminger 2013). There was evidence of a difference between mood stabilisers and placebo regarding psychosocial function at the end of treatment favouring placebo (MD 19.90, 95% CI 7.11 to 32.69; P = 0.002; 1 trial, 15 participants; Analysis 1.9).
1.9. Analysis.

Comparison 1: Medications compared with placebo, Outcome 9: Primary: Psychosocial functioning at end of treatment (continuous outcomes, MDs)
Secondary outcomes
1.5 Anger
Twenty trials comparing medications with placebo reported data for anger at the end of treatment.
Ten trials compared antipsychotics to placebo (Black 2014; Bogenschutz 2004; Cowdry 1988; Goldberg 1986; Nickel 2006; Pascual 2008; Schulz 2007; Soloff 1989; Soloff 1993; Zanarini 2007). There was evidence of a difference between treatments regarding anger at the end of treatment favouring antipsychotics (SMD −0.37, 95% CI −0.55 to −0.18; P = 0.0001, I2 = 48%; 10 trials, 1025 participants; Analysis 1.11). Inspection of the funnel plot suggested potential bias (small asymmetry; see Figure 5; Appendix 6), but we found no evidence of possible significant publication bias: Egger’s regression intercept (bias) 2.10 (two tailed, P = 0.069).
1.11. Analysis.

Comparison 1: Medications compared with placebo, Outcome 11: Secondary: Anger at end of treatment (continuous outcomes, SMDs)
5.

Funnel plot of comparison: 1 Medications compared with placebo, outcome: 1.11 Secondary: Anger at end of treatment (continuous outcomes, SMDs)
Six trials compared antidepressants to placebo (Cowdry 1988; Rinne 2002; Salzman 1995; Soloff 1989; Soloff 1993). There was evidence of a difference between treatments regarding anger at the end of treatment favouring antidepressants (SMD −0.37, 95% CI −0.64 to −0.11; P = 0.006, I2 = 0%; 6 trials, 224 participants; Analysis 1.11).
Eight trials compared mood stabilisers to placebo (Cowdry 1988; De la Fuente 1994; Frankenburg 2002; Hollander 2001; Loew 2006; Nickel 2004; Nickel 2005; Tritt 2005). Three of these trials reported extraordinarily large SMDs (Tritt 2005: SMD −1.69, 95% CI −2.62 to −0.75; Loew 2006: SMD −3.10, 95% CI −3.89 to −2.30; Nickel 2004: SMD −2.80, 95% CI −3.89 to −1.71). We decided to exclude the outliers one by one from the primary analyses, until low heterogeneity was reached (see Analysis 26.1 for sensitivity analysis). After excluding outliers, there was evidence of a difference between treatments regarding anger at the end of treatment favouring mood stabilisers (SMD −0.67, 95% CI −1.10 to −0.24; P = 0.002, I2 = 26%; 5 trials, 135 participants; Analysis 1.11).
26.1. Analysis.

Comparison 26: Sensitivity analysis: mood stabiliser trials outliers SMD > 1.5 included, Outcome 1: Secondary: Anger at end of treatment
Two trials compared omega‐3 fatty acids to placebo (Hallahan 2007; Zanarini 2003). There was a difference between treatments regarding anger at the end of treatment favouring omega‐3 fatty acids (SMD −0.48, 95% CI −0.95 to −0.01; P = 0.04, I2 = 0%; 2 trials, 76 participants; Analysis 1.11).
One cross‐over trial compared naltrexone to placebo (Schmahl 2012b). There was no evidence of a difference between treatments regarding anger at the end of treatment (MD 1.65, 95% CI −4.54 to 7.84; P = 0.60; 1 trial, 32 participants; Analysis 1.12).
1.12. Analysis.

Comparison 1: Medications compared with placebo, Outcome 12: Secondary: Anger at end of treatment (continuous outcomes, MDs)
Another cross‐over trial compared alprazolam to placebo (Cowdry 1988). There was no evidence of a difference between treatments regarding anger at the end of treatment (MD −0.57, 95% CI −1.48 to 0.34; P = 0.22; 1 trial, 25 participants; Analysis 1.12).
1.6 Affective instability
Seven trials comparing pharmacological treatments to placebo reported data for affective instability.
Four trials compared antipsychotics to placebo (Bogenschutz 2004; Pascual 2008; Schulz 2007; Zanarini 2007). There was a small difference between treatments regarding affective instability at the end of treatment favouring antipsychotics (SMD ‐0.16, 95% CI ‐0.31 to ‐0.01; P = 0.04; I2 = 0%; 4 trials, 691 participants; Analysis 1.13).
1.13. Analysis.

Comparison 1: Medications compared with placebo, Outcome 13: Secondary: Affective instability at end of treatment (continuous outcomes, SMDs)
One trial (Rinne 2002) compared an antidepressant to placebo. There was a small difference between treatments regarding affective instability at the end of treatment favouring the antidepressant (MD ‐1.66, 95% CI ‐3.26 to ‐0.06; P = 0.04; 1 trial, 38 participants; Analysis 1.14).
1.14. Analysis.

Comparison 1: Medications compared with placebo, Outcome 14: Secondary: Affective instability at end of treatment (continous outcomes, MDs)
Two trials (Reich 2009 and Crawford 2018) compared mood stabilisers to placebo. There was no difference between treatments regarding affective instability at the end of treatment (SMD −0.21, 95% CI −0.68 to 0.26; P = 0.38, I2 = 39%; 2 trials, 222 participants; Analysis 1.13).
1.7 Chronic feelings of emptiness
Four trials comparing pharmacological treatments to placebo reported data for chronic feelings of emptiness. All four trials compared antipsychotics to placebo (Bogenschutz 2004; Pascual 2008; Schulz 2007; Zanarini 2007). There was no difference between treatments regarding chronic feelings of emptiness at the end of treatment (SMD −0.00, 95% CI −0.16 to 0.15; P = 0.96, I2 = 6%; 4 trials, 691 participants; Analysis 1.15).
1.15. Analysis.

Comparison 1: Medications compared with placebo, Outcome 15: Secondary: Chronic feelings of emptiness at end of treatment (continuous outcomes, SMDs)
1.8 Impulsivity
Sixteen trials comparing pharmacological treatments to placebo reported data for impulsivity.
Ten trials compared antipsychotics to placebo (Black 2014; Bogenschutz 2004; Cowdry 1988; Grant 2022; Pascual 2008; Schulz 2007; Soler 2005; Soloff 1989; Soloff 1993; Zanarini 2007). There was no difference between treatments regarding impulsivity at end of treatment (SMD ‐0.08, 95% CI ‐0.20 to 0.04; P = 0.21; I2 = 0%: 10 trials, 1038 participants; Analysis 1.16). Inspection of the funnel plot suggested potential bias (small asymmetry; see Figure 6; Appendix 6), but we found no evidence of possible significant publication bias: Egger’s regression intercept (bias) 2.10 (two tailed, P = 0.069).
1.16. Analysis.

Comparison 1: Medications compared with placebo, Outcome 16: Secondary: Impulsivity at end of treatment (continuous outcomes, SMDs)
6.

Funnel plot of comparison: 1 Medications compared with placebo, outcome: 1.16 Secondary: Impulsivity at end of treatment (continuous outcomes, SMDs)
Four trials compared antidepressants to placebo (Cowdry 1988; Rinne 2002; Soloff 1989; Soloff 1993). There was no clear difference between treatments regarding impulsivity at end of treatment (SMD ‐0.17, 95% CI ‐0.49 to 0.15; P = 0.29; I2 = 13%; 4 trials, 182 participants; Analysis 1.16).
Five trials compared mood stabilisers to placebo (Cowdry 1988; Crawford 2018; De la Fuente 1994; Moen 2012; Reich 2009). In the analysis using dichotomous data, there was no difference between treatments regarding impulsivity at the end of treatment (RR 0.88, 95% CI 0.53 to 1.46; P = 0.61; 1 trial, 20 participants; Analysis 1.18). In the analysis using continuous data, there was little to no difference between treatments regarding impulsivity at the end of treatment favouring mood stabilisers (SMD ‐0.56, 95% CI ‐1.46 to 0.35; P = 0.23; I2 = 84%; 4 trials, 265 participants; Analysis 1.16).
1.18. Analysis.

Comparison 1: Medications compared with placebo, Outcome 18: Secondary: Impulsivity at end of treatment (dichtotomous outcomes, RRs)
One trial also compared the benzodiazepine alprazolam to placebo (Cowdry 1988). There was no difference between treatments regarding impulsivity at the end of treatment (MD 0.67, 95% CI −0.36 to 1.70; P = 0.20; 1 trial, 25 participants; Analysis 1.17).
1.17. Analysis.

Comparison 1: Medications compared with placebo, Outcome 17: Secondary: Impulsivity at end of treatment (continuous outcomes, MDs)
1.9 Interpersonal problems
Fourteen trials comparing pharmacological treatments to placebo reported data for interpersonal problems.
Eight trials compared antipsychotics to placebo (Bogenschutz 2004; Goldberg 1986; Nickel 2006; Pascual 2008; Schulz 2007; Soloff 1989; Soloff 1993; Zanarini 2007). Low‐certainty evidence suggests that antipsychotics may slightly reduce interpersonal problems (SMD −0.21, 95% CI −0.34 to −0.08; P = 0.002, I2 = 0%; 8 trials, 907 participants; low‐certainty evidence; Analysis 1.19). The TSA analysis showed that the DARIS was reached (n = 386), and that there was no risk of type 1 error (TSA adjusted CI −0.60 to 0.08). See Figure 7; Appendix 6.
1.19. Analysis.

Comparison 1: Medications compared with placebo, Outcome 19: Secondary: Interpersonal problems at end of treatment (continuous outcomes, SMDs)
7.
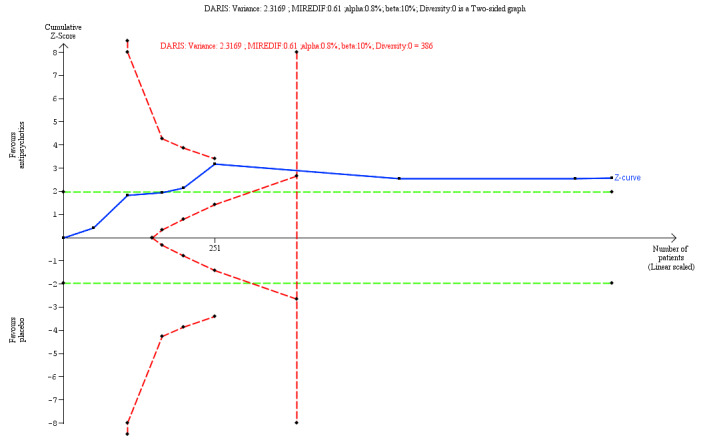
TSA interpersonal problems: antipsychotics
DARIS: Diversity‐adjusted required information size; MIREDIF: Minimal relevant difference; TSA: Trial Sequential Analysis
Two trials compared antidepressants to placebo (Soloff 1989; Soloff 1993). There was little to no difference between treatments regarding interpersonal problems at the end of treatment (SMD −0.07, 95% CI −0.69 to 0.55; P = 0.82, I2 = 66%; 2 trials, 119 participants; low‐certainty evidence; Analysis 1.19).
Four trials compared mood stabilisers with placebo (Crawford 2018; De la Fuente 1994; Frankenburg 2002; Loew 2006). This low‐certainty evidence suggested that mood stabilisers may result in a reduction of interpersonal problems, compared with placebo at end of treatment on interpersonal (SMD −0.58, 95% CI −1.14 to −0.02; P = 0.04, I2 = 73%; 4 trials, 300 participants; low‐certainty evidence; Analysis 1.19).
1.10 Abandonment
Four trials comparing pharmacological treatments to placebo reported continuous data for fear of abandonment. All four trials compared antipsychotics to placebo (Bogenschutz 2004; Pascual 2008; Schulz 2007; Zanarini 2007). There was no difference between treatments regarding fear of abandonment at the end of treatment (SMD −0.01, 95% CI −0.17 to 0.14; P = 0.88, I2 = 5%; 4 trials, 691 participants; Analysis 1.20).
1.20. Analysis.

Comparison 1: Medications compared with placebo, Outcome 20: Secondary: Abandonment at end of treatment (continuous outcomes, SMDs)
1.11 Identity disturbance
Four trials comparing pharmacological treatments to placebo reported continuous data for identity disturbance. All four trials compared antipsychotics to placebo (Bogenschutz 2004; Pascual 2008; Schulz 2007; Zanarini 2007). There was no difference between treatments regarding identity disturbance at the end of treatment (SMD −0.09, 95% CI −0.25 to 0.07; P = 0.28, I2 = 7%; 4 trials, 691 participants; Analysis 1.21).
1.21. Analysis.

Comparison 1: Medications compared with placebo, Outcome 21: Secondary: Identity disturbance at end of treatment (continuous outcomes, SMDs)
1.12 Dissociation and psychotic‐like symptoms
Thirteen trials comparing pharmacological treatments to placebo reported data for dissociation and psychotic‐like symptoms.
Eight trials compared antipsychotics to placebo (Bogenschutz 2004; Goldberg 1986; Nickel 2006; Pascual 2008; Schulz 2007; Soloff 1989; Soloff 1993; Zanarini 2007). There was a small difference between treatments regarding dissociation and psychotic‐like symptoms at the end of treatment favouring antipsychotics (SMD −0.28, 95% CI −0.50 to −0.06; P = 0.01, I2 = 55%; 8 trials, 907 participants; Analysis 1.22).
1.22. Analysis.

Comparison 1: Medications compared with placebo, Outcome 22: Secondary: Dissociation and psychotic‐like symptoms at end of treatment (continuous outcomes, SMDs)
Three trials compared antidepressants to placebo (Simpson 2004; Soloff 1989; Soloff 1993). There was no difference between treatments regarding dissociation and psychotic‐like symptoms at the end of treatment (SMD −0.22, 95% CI −0.62 to 0.18; P = 0.29, I2 = 25%; 3 trials, 139 participants; Analysis 1.22).
Three trials compared mood stabilisers to placebo (Crawford 2018; De la Fuente 1994; Loew 2006). There was no difference between treatments regarding dissociation and psychotic‐like symptoms at the end of treatment (SMD −0.23, 95% CI −0.66 to 0.20; P = 0.30; I2 = 51%; 3 trials, 270 participants; Analysis 1.22).
One trial compared omega‐3 fatty acids to placebo (Amminger 2013). There was no difference between treatments regarding dissociation and psychotic‐like symptoms at the end of treatment (MD −2.80, 95% CI −5.70 to 0.10; P = 0.09; 1 trial, 15 participants; Analysis 1.23).
1.23. Analysis.

Comparison 1: Medications compared with placebo, Outcome 23: Secondary: Dissociation and psychotic‐like symptoms at end of treatment (continuous outcomes, MDs)
1.13 Depression
Twenty‐six trials comparing pharmacological treatments to placebo reported data for depression.
Twelve trials compared antipsychotics to placebo (Black 2014; Cowdry 1988; Goldberg 1986; Grant 2022; Linehan 2008; Nickel 2006; Pascual 2008; Schulz 2007; Soler 2005; Soloff 1989; Soloff 1993; Zanarini 2007). There was a difference between treatments regarding depression at end of treatment favouring antipsychotics (SMD ‐0.22, 95% CI ‐0.42 to ‐0.01; P = 0.04; I2 = 59; 12 trials, 1138 participants; Analysis 1.24).
1.24. Analysis.

Comparison 1: Medications compared with placebo, Outcome 24: Secondary: Depression at end of treatment (continuous outcomes, SMDs)
Five trials compared antidepressants to placebo (Cowdry 1988; Salzman 1995; Simpson 2004; Soloff 1989; Soloff 1993). There was no clear difference between treatments regarding depression at end of treatment (SMD −0.37, 95% CI −0.82 to 0.08; P = 0.11, I2 = 52%; 5 trials, 187 participants; Analysis 1.24).
Six trials compared mood stabilisers to placebo (Cowdry 1988; Crawford 2018; De la Fuente 1994; Frankenburg 2002; Hollander 2001; Loew 2006). There was a difference between treatments regarding depression at the end of treatment favouring mood stabilisers (SMD −0.44, 95% CI −0.80 to −0.08; P = 0.02, I2 = 46%; 6 trials, 344 participants: Analysis 1.24).
Six trials compared miscellaneous pharmacological treatments to placebo.
Three of these trials used a parallel design and compared omega‐3 fatty acids to placebo (Amminger 2013; Hallahan 2007; Zanarini 2003). The analysis with dichotomous data showed that there was a difference between omega‐3 fatty acids and placebo regarding depression at the end of treatment favouring omega‐3 fatty acids (RR 0.48, 95% CI 0.28 to 0.81; P = 0.006; 1 trial, 49 participants; Analysis 1.26), while in the analysis with continuous data there was no difference between treatments regarding depression at end of treatment (SMD −0.54, 95% CI −1.18 to 0.11; P = 0.10, I2 = 0%; 2 trials, 42 participants; Analysis 1.24).
1.26. Analysis.

Comparison 1: Medications compared with placebo, Outcome 26: Secondary: Depression at end of treatment (dichotomous outcomes, RRs)
The other three trials used cross‐over designs. The first trial compared clonidine to placebo (Ziegenhorn 2009), and found no difference between clonidine and placebo regarding depression at the end of treatment (MD −2.54, 95% CI −10.27 to 5.19; P = 0.52; 1 trial, 34 participants; Analysis 1.25). The second trial compared naltrexone to placebo (Schmahl 2012b), and found no difference between naltrexone and placebo regarding depression at the end of treatment (MD 2.50, 95% CI −4.22 to 9.22; P = 0.47; 1 trial, 32 participants; Analysis 1.25). The third trial compared the benzodiazepine alprazolam to placebo (Cowdry 1988), and found no difference between alprazolam and placebo on depression at the end of treatment (MD 0.27, 95% CI −0.73 to 1.27; P = 0.60; 1 trial, 25 participants; Analysis 1.25).
1.25. Analysis.

Comparison 1: Medications compared with placebo, Outcome 25: Secondary: Depression at end of treatment (continuous outcomes, MDs)
1.14 Attrition
Thirty‐two trials comparing medications to placebo reported data on attrition.
Thirteen trials compared antipsychotics to placebo (Black 2014: Bogenschutz 2004; Goldberg 1986; Grant 2022; Linehan 2008; Montgomery 1982a; Pascual 2008; Schulz 2007; Soler 2005; Soloff 1989; Soloff 1993; Zanarini 2001; Zanarini 2007). There was no clear difference between treatments regarding attrition at the end of treatment (RR 1.11, 95% CI 0.89 to 1.38; P = 0.34; I2 = 35%; 13 trials, 1216 participants; low‐certainty evidence ; Analysis 1.27). However, the TSA showed that the DARIS was not reached (n = 2008), and that there was a potential risk of type 1 error (TSA‐adjusted CI 0.74 to 2.13). See Figure 8 in Appendix 6. Inspection of the funnel plot suggested potential bias (small asymmetry; see in Figure 9 in Appendix 6), but we found no evidence of possible significant publication bias: Egger’s regression intercept (bias) 1.89 (two tailed, P = 0.089).
1.27. Analysis.

Comparison 1: Medications compared with placebo, Outcome 27: Secondary: Attrition at end of treatment (dichotomous outcomes, RRs)
8.
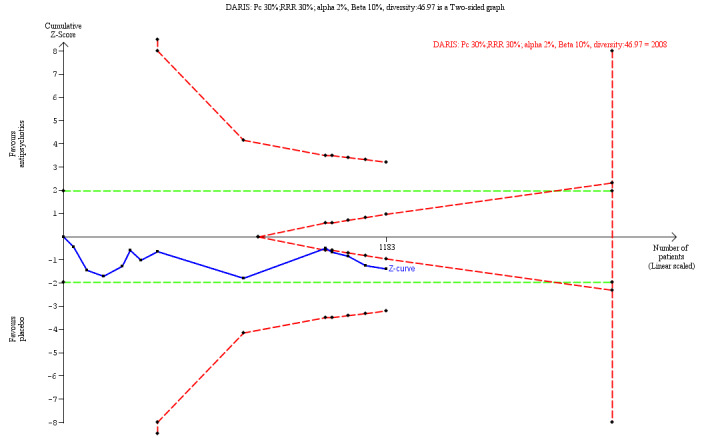
TSA attrition: antipsychotics
DARIS: Diversity‐adjusted required information size; Pc: proportion with an outcome in the control group; RRR: Relative risk ratio; TSA: Trial Sequential Analysis
9.

Funnel plot of comparison: 1 Pharmacotherapies compared with placebo, outcome: 1.19 Secondary: Attrition at end of treatment
RR: Relative Risk; SE(log[RR]): Standard Error of the logarithmic Risk Ratio
Six trials compared antidepressants to placebo (Montgomery 1982b; NCT00533117; Rinne 2002; Simpson 2004; Soloff 1989; Soloff 1993). There was no difference between treatments regarding attrition at the end of treatment (RR 1.07, 95% CI 0.65 to 1.76; P = 0.78, I2 = 0%; 6 trials, 289 participants; low‐certainty evidence; Analysis 1.27). The TSA, however, showed that the DARIS was not reached (n = 1816), and that there was a potential risk of type 1 error (TSA‐adjusted CI 0.07 to 14.98). See Figure 10 in Appendix 6.
10.
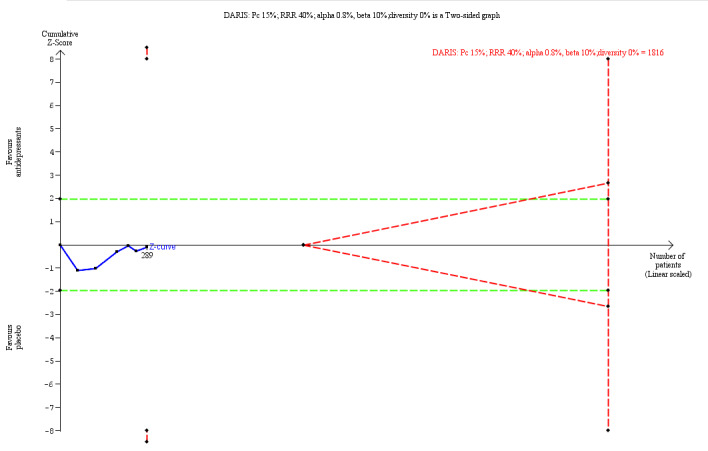
TSA attrition: antidepressants
DARIS: Diversity‐adjusted required information size;Pc: proportion with an outcome in the control group; RRR: Relative risk ratio; TSA: Trial Sequential Analysis
Nine trials compared mood stabilisers to placebo (Crawford 2018; De la Fuente 1994; Frankenburg 2002; Hollander 2001; Loew 2006; Nickel 2004; Nickel 2005; Reich 2009; Tritt 2005). The evidence is very uncertain about the effect of mood stabilisers compared with placebo on attrition at the end of treatment (RR 0.89, 95% CI 0.69 to 1.15; P = 0.37, I2 = 0%; 9 trials, 530 participants; very low‐certainty evidence; Analysis 1.27). The TSA showed that the DARIS was not reached (n = 1300), and that there was a potential risk of type 1 error (TSA‐adjusted CI 0.37 to 2.23). See Figure 11 in Appendix 6.
11.
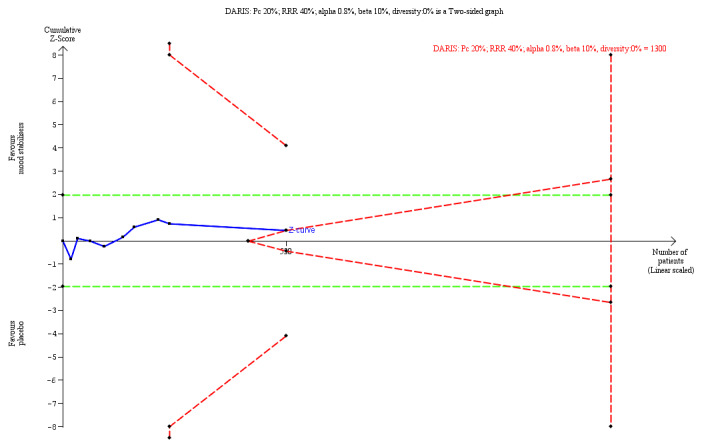
TSA attrition: mood stabilisers
DARIS: Diversity‐adjusted required information size; Pc: proportion with an outcome in the control group; RRR: Relative risk ratio; TSA: Trial Sequential Analysis
Four trials compared miscellaneous medications to placebo. Of these, two trials compared omega‐3 fatty acids to placebo (Hallahan 2007; Zanarini 2003). There was no difference between omega‐3 fatty acids and placebo regarding omega attrition at end of treatment (RR 0.61, 95% CI 0.21 to 1.79; P = 0.37, I2 = 0%; 2 trials, 79 participants; Analysis 1.27). One trial compared memantine hydrochloride to placebo (Kulkarni 2018). There was no difference between memantine hydrochloride and placebo regarding attrition at end of treatment (RR 1.57, 95% CI 0.45 to 5.52; P = 0.48; 1 trial, 33 participants; Analysis 1.27). Another trial compared clonidine to placebo (Ziegenhorn 2009). There was no difference between clonidine and placebo regarding attrition at end of treatment (RR 0.67, 95% CI 0.13 to 3.50; P = 0.63; 1 trial, 34 participants; Analysis 1.27).
1.15 Adverse events
Seven trials comparing medications to placebo reported dichotomous data on total non‐serious adverse events at end of treatment. Five trials compared antipsychotics to placebo (Black 2014; Grant 2022; Pascual 2008; Schulz 2007; Zanarini 2007). There was no clear difference in the presence of adverse events between the two groups receiving antipsychotics and placebo (RR 1.07, 95% CI 0.90 to 1.29; P = 0.43; I2 = 57%; 5 trials, 814 participants; very low‐certainty evidence; Analysis 1.28). The TSA showed that the z‐curve ended in the fidelity area. See Figure 12 in Appendix 6.
1.28. Analysis.

Comparison 1: Medications compared with placebo, Outcome 28: Secondary: Non‐serious adverse events at end of treatment (dichotomous outcomes, RRs)
12.
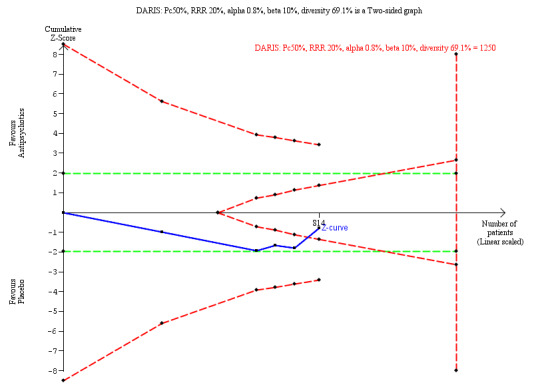
TSA non‐serious adverse events: antipsychotics
DARIS: Diversity‐adjusted required information size; Pc: proportion with an outcome in the control group; RRR: Relative risk ratio; TSA: Trial Sequential Analysis
One trial compared a mood stabiliser to placebo (Crawford 2018). There was no difference in the presence of adverse events between the two groups (RR 0.84, 95% CI 0.70 to 1.01; P = 0.07; 1 trial, 276 participants; very low‐certainty evidence; Analysis 1.28).
Another trial compared memantine hydrochloride to placebo (Kulkarni 2018). There was no difference in any adverse events between the two groups (RR 1.41, 95% CI 0.79 to 2.52; P = 0.24; 1 trial, 33 participants; Analysis 1.28).
Two trials compared pharmacological treatment to placebo and reported dichotomous data on total serious adverse events. Kulkarni 2018 compared memantine hydrochloride to placebo and reported no serious adverse events in either the experimental or the placebo group (Analysis 1.29). Grant 2022 compared brexpiprazole to placebo. One serious adverse event (mild suicidal ideation without a plan, considered serious as it necessitated hospitalisation) occurred in the placebo group. There was no statistical difference in the occurrence of serious adverse events between the two groups (RR 3.00, 95% CI 0.13 to 71.51; 1 trial, 80 participants; Analysis 1.29). No other trials specifically reported data on serious adverse events.
1.29. Analysis.

Comparison 1: Medications compared with placebo, Outcome 29: Secondary: Serious adverse events at end of treatment (dichotomous outcomes, RRs)
Please refer to Table 5 for an overview of adverse events.
2. Adverse events of pharmacotherapies versus placebo.
| Outcome or subgroup title | Pharmacotherapy | No. of studies | No. of participants | Statistical method | Effect estimate | Study ID |
| Central nervous system | ||||||
| Headache | Antipsychotics | 4 | 754 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 (0.63 to 1.62) | Black 2014; Grant 2022; Schulz 2007; Zanarini 2007 |
| Mood stabilisers | 1 | 56 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.00 (0.15 to 6.61) | Loew 2006 | |
| Memantine hydrochloride | 1 | 33 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.29 (0.71 to 2.36) | Kulkarni 2018 | |
| Dizziness | Antipsychotics | 2 | 68 | Risk Ratio (M‐H, Random, 95% CI) | 3.07 (0.40 to 23.45) | Black 2014; Pascual 2008 |
| Mood stabilisers | 1 | 56 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.50 (0.27 to 8.30) | Loew 2006 | |
| Memantine hydrochloride | 1 | 33 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.69 (0.72 to 3.98) | Kulkarni 2018 | |
| Fatigue | Antipsychotics | 3 | 692 | Risk Ratio (M‐H, Random, 95% CI) | 1.50 (0.58 to 3.89) | Grant 2022; Schulz 2007; Zanarini 2007 |
| Mood stabilisers | 1 | 56 | Risk Ratio (M‐H Fixed, 95% CI) | 2.00 (0.40 to 10.05) | Loew 2006 | |
| Memantine hydrochloride | 1 | 33 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.32 (0.52 to 3.31) | Kulkarni 2018 | |
| Somnolence | Antipsychotics | 2 | 615 | Risk Ratio (M‐H, Random, 95% CI) | 2.97 (1.75 to 5.03) | Schulz 2007; Zanarini 2007 |
| Memantine hydrochloride | 1 | 33 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.65 (0.59to 4.57) | Kulkarni 2018 | |
| Sedation | Antipsychotics | 4 | 445 | Risk Ratio (M‐H, Random, 95% CI) | 2.66 (0.99 to 7.12) | Black 2014; Pascual 2008; Schulz 2007; Zanarini 2001 |
| Anxiety | Antipsychotics | 1 | 314 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.90 (0.33 to 2.42) | Schulz 2007 |
| Insomnia | Antipsychotics | 2 | 615 | Risk Ratio (M‐H, Random, 95% CI) | 0.68 (0.33 to 1.37) | Schulz 2007; Zanarini 2007 |
| Hyperinsomnia | Antipsychotics | 1 | 62 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.34 (0.69 to 8.01) | Black 2014 |
| Increased appetite | Antipsychotics | 3 | 692 | Risk Ratio (M‐H, Random, 95% CI) | 2.68 (1.71 to 4.19) | Grant 2022; Schulz 2007; Zanarini 2007 |
| Change in appetite | Antipsychotics | 1 | 17 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.65 (0.10 to 4.06) | Black 2014 |
| Forgetfulness or confusion | Antipsychotics | 1 | 17 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.46 (0.38 to 5.60) | Black 2014 |
| Disturbances in attention | Antipsychotics | 1 | 301 | Risk Ratio (M‐H, Fixed, 95% CI) | 11.37 (0.63 to 203.81) | Zanarini 2007 |
| Restlessness | Antipsychotics | 1 | 77 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.93 (0.20 to 4.30) | Grant 2022 |
| Hallucinations | Antipsychotics | 1 | 77 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.19 (0.01 to 3.74) | Grant 2022 |
| Sleep problems | Antipsychotics | 1 | 77 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.19 (0.01 to 3.74) | Grant 2022 |
| Tremor | Antipsychotics | 1 | 77 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.31 (0.01 to 7.36) | Grant 2022 |
| Memory problems | Mood stabilisers | 1 | 56 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.00 (0.55 to 7.22) | Loew 2006 |
| Paraesthesia | Mood stabilisers | 1 | 56 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.00 (0.33 to 27.12) | Loew 2006 |
| Gait/balance disturbances | Memantine hydrochloride | 1 | 33 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.35 (0.53 to 10.45) | Kulkarni 2018 |
| Nervous system disorders | Mood stabilisers | 1 | 276 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.05 (0.68 to 1.62) | Crawford 2018 |
| Psychiatric disorders | Mood stabilisers | 1 | 276 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.94 (0.64 to 1.37) | Crawford 2018 |
| Cardiovascular and respiratory system | ||||||
| Cold/flu symptoms | Antipsychotics | 1 | 62 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.54 (0.50 to 4.73) | Black 2014 |
| Nasopharyngitis | Antipsychotics | 1 | 301 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.62 (0.23 to 1.66) | Schulz 2007 |
| Sweating | Antipsychotics | 1 | 77 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.31 (0.01 to 7.36) | Grant 2022 |
| Blood and lymphatic system disorders | Mood stabilisers | 1 | 276 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.68 (0.11 to 3.99) | Crawford 2018 |
| Cardiac disorders | Mood stabilisers | 1 | 276 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.34 (0.01 to 8.23) | Crawford 2018 |
| Endocrine disorders | Mood stabilisers | 1 | 276 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.34 (0.01 to 8.23) | Crawford 2018 |
| Respiratory, thoracic and mediastinal disorders | Mood stabilisers | 1 | 276 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.80 (0.83 to 3.94) | Crawford 2018 |
| Diastolic blood pressure in standing position (mean change from baseline to endpoint) | Antipsychotics | 1 | 290 | Mean Difference (IV, Fixed, 95% CI) | ‐0.28 (‐2.29 to 1.73) | Zanarini 2007 |
| Diastolic blood pressure in supine position (mean change from baseline to endpoint) | Antipsychotics | 1 | 290 | Mean Difference (IV, Fixed, 95% CI) | ‐0.11 (‐2.28 to 2.06) | Zanarini 2007 |
| Systolic blood pressure in standing position (mean change from baseline to endpoint) | Antipsychotics | 1 | 290 | Mean Difference (IV, Fixed, 95% CI) | 0.35 (‐2.39 to 3.09) | Zanarini 2007 |
| Systolic blood pressure in supine position (mean change from baseline to endpoint) | Antipsychotics | 1 | 290 | Mean Difference (IV, Fixed, 95% CI) | ‐1.31 (‐4.00 to 1.38) | Zanarini 2007 |
| Pulse in standing position (mean change from baseline to endpoint) | Antipsychotics | 1 | 290 | Mean Difference (IV, Fixed, 95% CI) | 0.85 (‐1.65 to 3.35) | Zanarini 2007 |
| Pulse in supine position (mean change from baseline to endpoint) | Antipsychotics | 1 | 290 | Mean Difference (IV, Fixed, 95% CI) | ‐0.11 (‐2.28 to 2.06) | Zanarini 2007 |
| Gastrointestinal system | ||||||
| Nausea | Antipsychotics | 4 | 754 | Risk Ratio (M‐H, Random, 95% CI) | 0.80 (0.49 to 1.29) | Black 2014; Grant 2022; Schulz 2007; Zanarini 2007 |
| Memantine hydrochloride | 1 | 33 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.00 (0.45 to 2.23) | Kulkarni 2018 | |
| Uneasy feeling | Antipsychotics | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 7.00 (0.38 to 129.93) | Pascual 2008 |
| Constipation | Antipsychotics | 1 | 28 | Risk Ratio (M‐H, Fixed, 95% CI) | 6.50 (0.41 to 104.20) | Zanarini 2001 |
| Memantine hydrochloride | 1 | 33 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.65 (0.59 to 4.57) | Kulkarni 2018 | |
| Dry mouth | Antipsychotics | 4 | 754 | Risk Ratio (M‐H, Random, 95% CI) | 2.60 (1.46 to 4.64) | Black 2014; Grant 2022; Schulz 2007; Zanarini 2007 |
| Gastrointestinal disorders | Mood stabilisers | 1 | 276 | Risk Ratio (M‐H, Random, 95% CI) | 0.70 (0.50 to 0.98) | Crawford 2018 |
| General disorders and administration site conditions | Mood stabilisers | 1 | 276 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 (0.50 to 2.05) | Crawford 2018 |
| Hepatobiliary disorders | Mood stabilisers | 1 | 276 | Risk Ratio (M‐H, Random, 95% CI) | 3.04 (0.13 to 74.07) | Crawford 2018 |
| Metabolism and nutrition disorders | Mood stabilisers | 1 | 276 | Risk Ratio (M‐H, Random, 95% CI) | 2.03 (0.19 to 22.12) | Crawford 2018 |
| Liver function: ALT/SGPT baseline to endpoint mean change (U/L) | Antipsychotics | 2 | 530 | Std. Mean Difference (IV, Random, 95% CI) | 0.46 (0.29 to 0.63) | Schulz 2007; Zanarini 2007 |
| Liver function: AST/SGOT baseline to endpoint mean change (U/L) | Antipsychotics | 2 | 526 | Std. Mean Difference (IV, Random, 95% CI) | 0.35 (0.18 to 0.52) | Schulz 2007; Zanarini 2007 |
| Liver function: total bilirubin baseline to endpoint mean change (μmol/L) | Antipsychotics | 1 | 264 | Mean Difference (IV, Fixed, 95% CI) | ‐0.98 (‐1.80 to ‐0.16) | Schulz 2007 |
| Liver function: direct bilirubin baseline to endpoint mean change (μmol/L) | Antipsychotics | 1 | 258 | Mean Difference (IV, Fixed, 95% CI) | ‐0.30 (‐0.51 to ‐0.09) | Schulz 2007 |
| Liver function: Gamma‐Glutamyl Transferase (GGT) baseline to endpoint mean change | Antipsychotics | 1 | 268 | Mean Difference (IV, Fixed, 95% CI) | 2.96 (0.22 to 5.70) | Zanarini 2007 |
| Lipids: total cholesterol baseline to endpoint change (mmol/L) | Antipsychotics | 2 | 327 | Std. Mean Difference (IV, Random, 95% CI) | 0.42 (0.20 to 0.64) | Schulz 2007; Soler 2005 |
| Lipids: Low‐density lipoprotein (LDL) cholesterol baseline to endpoint mean change (mmol/L) | Antipsychotics | 1 | 259 | Mean Difference (IV, Fixed, 95% CI) | 0.21 (0.06 to 0.36) | Schulz 2007 |
| Lipids: High‐density lipoprotein (HDL) cholesterol (dextran precip.) baseline to endpoint mean change (mmol/L) | Antipsychotics | 1 | 269 | Mean Difference (IV, Fixed, 95% CI) | ‐0.06 [‐0.11 to ‐0.01) | Zanarini 2007 |
| Lipids: triglycerides, fasting, baseline to endpoint mean change (mmol/L) | Antipsychotics | 1 | 203 | Mean Difference (IV, Fixed, 95% CI) | 0.27 (0.07 to 0.47) | Zanarini 2007 |
| Prolactin: baseline to endpoint mean change (μg/L) | Antipsychotics | 1 | 259 | Mean Difference (IV, Fixed, 95% CI) | 7.10 (1.64 to 12.56) | Schulz 2007 |
| Platelet count baseline to endpoint mean change (GI/L) | Antipsychotics | 2 | 517 | Std. Mean Difference (IV, Random, 95% CI) | 0.03 (‐0.53 to 0.59) | Schulz 2007; Zanarini 2007 |
| Erythrocyte count baseline to endpoint mean change (TI/L) | Antipsychotics | 1 | 262 | Mean Difference (IV, Fixed, 95% CI) | ‐0.05 (‐0.12 to 0.02) | Zanarini 2007 |
| Leukocyte count baseline to endpoint mean change (GI/L) | Antipsychotics | 1 | 262 | Mean Difference (IV, Fixed, 95% CI) | ‐0.70, (‐1.12 to ‐0.28) | Zanarini 2007 |
| Neutrophils, segmented, baseline to endpoint mean change (GI/L) | Antipsychotics | 1 | 262 | Mean Difference (IV, Fixed, 95% CI) | ‐0.60 (‐0.97 to ‐0.23) | Zanarini 2007 |
| Basophils baseline to endpoint mean change (GI/L) | Antipsychotics | 1 | 262 | Mean Difference (IV, Fixed, 95% CI) | ‐0.01 (‐0.02 to ‐0.00) | Zanarini 2007 |
| Monocytes baseline to endpoint mean change (GI/L) | Antipsychotics | 1 | 262 | Mean Difference (IV, Fixed, 95% CI) | ‐0.04, (‐0.07 to ‐0.01) | Zanarini 2007 |
| Haemoglobin baseline to endpoint mean change (mml/L‐F) | Antipsychotics | 1 | 262 | Mean Difference (IV, Fixed, 95% CI) | ‐0.11 (‐0.24 to 0.02) | Zanarini 2007 |
| Mean cell haemoglobin concentration (MCHC) baseline to endpoint mean change (mml/L‐F) | Antipsychotics | 1 | 260 | Mean Difference (IV, Fixed, 95% CI) | 0.02 (‐0.17 to 0.21) | Zanarini 2007 |
| Calcium baseline to endpoint mean change (mmol/L) | Antipsychotics | 1 | 268 | Mean Difference (IV, Fixed, 95% CI) | ‐0.03 (‐0.05 to ‐0.01) | Schulz 2007 |
| Albumin baseline to endpoint mean change (g/L) | Antipsychotics | 1 | 269 | Mean Difference (IV, Fixed, 95% CI) | ‐0.67 (‐1.42 to 0.08) | Zanarini 2007 |
| Creatine phosphokinase baseline to endpoint mean change (U/L) | Antipsychotics | 1 | 268 | Mean Difference (IV, Fixed, 95% CI) | ‐44.81 (‐95.39 to 5.77) | Zanarini 2007 |
| Urea nitrogen baseline to endpoint mean change (mmol/L) | Antipsychotics | 1 | 269 | Mean Difference (IV, Fixed, 95% CI) | ‐0.17 (‐0.46 to 0.12) | Zanarini 2007 |
| Musculoskeletal system | ||||||
| Bodily pain | Antipsychotics | 1 | 62 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.88 (0.47 to 1.64) | Black 2014 |
| Musculoskeletal and connective tissue disorders | Mood stabilisers | 1 | 276 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.16 (0.43 to 3.11) | Crawford 2018 |
| Body weight change | Antipsychotics | 7 | 810 | Std. Mean Difference (IV, Random, 95% CI) | 0.78 (0.44 to 1.12) | Bogenschutz 2004; Linehan 2008; Schulz 2007; Soler 2005; Soloff 1993; Zanarini 2001; Zanarini 2007 |
| Antidepressants | 1 | 62 | Mean Difference (IV, Fixed, 95% CI) | 0.09 (‐0.31 to 0.49) | Soloff 1993 | |
| Mood stabilisers | 5 | 184 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.26 (‐0.72 to 0.20) | Frankenburg 2002; Loew 2006; Nickel 2004; Nickel 2005; Tritt 2005 | |
| Sensory system | ||||||
| Eye disorders | Mood stabilisers | 1 | 276 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.17 (0.02 to 1.39) | Crawford 2018 |
| Reproductive system | ||||||
| Pregnancy, puerperium and perinatal conditions | Mood stabilisers | 1 | 276 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.52 (0.26 to 8.97) | Crawford 2018 |
| Reproductive system and breast disorders | Mood stabilisers | 1 | 276 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.04 (0.32 to 28.90) | Crawford 2018 |
| Menstrual pain | Mood stabilisers | 1 | 56 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.67 (0.44 to 6.31) | Loew 2006 |
| Other | ||||||
| Injury, poisoning or procedural complications | Mood stabilisers | 1 | 276 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.44 (0.26 to 0.74) | Crawford 2018 |
| Skin and subcutaneous tissue disorders | Mood stabilisers | 1 | 276 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.15 (0.75 to 1.75) | Crawford 2018 |
| Social circumstances | Mood stabilisers | 1 | 276 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.01 (0.06 to 16.06) | Crawford 2018 |
| Surgical and medical procedures | Mood stabilisers | 1 | 276 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.06 (0.46 to 35.85) | Crawford 2018 |
ALT: alanine aminotransferase. AST: aspartate transaminase. CI: confidence interval. ID: identifier. IV: inverse variance. M‐H: Mantel‐Haenszel. No.: number. SGOT: Serum glutamic‐oxaloacetic transaminase SGPT: Serum glutamic‐pyruvic transaminase
Central nervous system
Headache
Six trials comparing pharmacological treatment to placebo reported data on headache as a non‐serious adverse event.
Four trials compared antipsychotics to placebo (Black 2014; Grant 2022; Schulz 2007; Zanarini 2007). There was no difference in the presence of headache between the two groups (RR 1.01, 95% CI 0.63 to 1.62; P = 0.97; I2 = 32%; 4 trials, 754 participants; Analysis 2.1).
2.1. Analysis.

Comparison 2: Medications compared with placebo ‐ non‐serious adverse events ‐ central nervous system, Outcome 1: Antipsychotics
One trial compared a mood stabiliser to placebo (Loew 2006). There was no difference in the presence of headache between the two groups (RR 1.00, 95% CI 0.15 to 6.61; P = 1.00; 1 trial, 56 participants; Analysis 2.2).
2.2. Analysis.

Comparison 2: Medications compared with placebo ‐ non‐serious adverse events ‐ central nervous system, Outcome 2: Mood stabilisers
One trial compared memantine hydrochloride to placebo (Kulkarni 2018). There was no difference in the presence of headache between the two groups (RR 1.29, 95% CI 0.71 to 2.36; P = 0.40; 1 trial, 33 participants; Analysis 2.3)
2.3. Analysis.

Comparison 2: Medications compared with placebo ‐ non‐serious adverse events ‐ central nervous system, Outcome 3: Memantine hydrochloride
Dizziness
Four trials comparing pharmacological treatment to placebo reported data on dizziness as a non‐serious adverse event.
Two trials compared antipsychotics to placebo (Black 2014; Pascual 2008). There was no difference in the presence of dizziness between the two groups (RR 3.07, 95% CI 0.40 to 23.45; P = 0.28, I2 = 9%; 2 trials, 68 participants; Analysis 2.1).
One trial compared a mood stabiliser to placebo (Loew 2006). There was no difference in the presence of dizziness between the two groups (RR 1.50, 95% CI 0.27 to 8.30; P = 0.64; 1 trial, 56 participants; Analysis 2.2).
One trial compared memantine hydrochloride to placebo (Kulkarni 2018). There was no difference in the presence of dizziness between the two groups (RR 1.69, 95% CI 0.72 to 3.98; P = 0.23; 1 trial, 33 participants; Analysis 2.3).
Fatigue
Five trials comparing pharmacological treatment to placebo reported data on fatigue as a non‐serious adverse event.
Three trials compared antipsychotics to placebo (Grant 2022; Schulz 2007; Zanarini 2007). There was no difference in the presence of fatigue between the two groups (RR 1.50, 95% CI 0.58 to 3.89; P = 0.40; I2 = 56%; 3 trials, 692 participants; Analysis 2.1).
One trial compared a mood stabiliser to placebo (Loew 2006). There was no difference in the presence of fatigue between the two groups (RR 2.00, 95% CI 0.40 to 10.05; P = 0.40; 1 trial, 56 participants; Analysis 2.2).
One trial compared memantine hydrochloride to placebo (Kulkarni 2018). There was no difference in the presence of fatigue between the two groups (RR 1.32, 95% CI 0.52 to 3.31; P = 0.56; 1 trial, 33 participants; Analysis 2.3).
Somnolence
Three trials comparing pharmacological treatment to placebo reported data on somnolence as a non‐serious adverse event.
Two trials compared antipsychotics to placebo (Schulz 2007; Zanarini 2007). There was a difference between the groups on somnolence, with an increased risk of somnolence using antipsychotics (RR 2.97, 95% CI 1.75 to 5.03; P < 0.0001, I2 = 0%; 2 trials, 615 participants; Analysis 2.1).
One trial compared memantine hydrochloride to placebo (Kulkarni 2018). There was no difference in the presence of somnolence between the two groups (RR 1.65, 95% CI 0.59 to 4.57; P = 0.34; 1 trial, 33 participants; Analysis 2.3).
Sedation
Four trials comparing pharmacological treatment to placebo reported continuous data on sedation as a non‐serious adverse event. All four trials compared antipsychotics to placebo (Black 2014; Pascual 2008; Schulz 2007; Zanarini 2001). There was a very slight difference in sedation between the two groups in favour of placebo (RR 2.66, 95% CI 0.99 to 7.12; P = 0.05, I2 = 67%; 4 trials, 445 participants; Analysis 2.1).
Anxiety
One trial comparing pharmacological treatment to placebo reported data on anxiety as a non‐serious adverse event. Schulz 2007 compared an antipsychotic to placebo. There was no difference in anxiety between the two groups (RR 0.90, 95% CI 0.33 to 2.42; P = 0.83; 1 trial, 314 participants; Analysis 2.1).
Insomnia
Two trials comparing pharmacological treatment to placebo reported data on insomnia as a non‐serious adverse event. Both trials compared antipsychotics to placebo (Schulz 2007; Zanarini 2007). There was no difference in effects on insomnia between the two groups (RR 0.68, 95% CI 0.33 to 1.37; P = 0.28, I2 = 15%; 2 trials, 615 participants; Analysis 2.1).
Hyperinsomnia
One trial comparing pharmacological treatment to placebo reported data on hyperinsomnia as a non‐serious adverse event. Black 2014 compared an antipsychotic to placebo. There was no difference in hyperinsomnia between the two groups (RR 2.34, 95% CI 0.69 to 8.01; P = 0.17; 1 trial, 62 participants; Analysis 2.1).
Increased appetite
Three trials comparing pharmacological treatment to placebo reported data on increased appetite as a non‐serious adverse event. Grant 2022, Schulz 2007 and Zanarini 2007 compared antipsychotics to placebo. There was an effect on increased appetite in favour of placebo, (RR 2.68, 95% CI 1.71 to 4.19; P < 0.0001; I2 = 0%, 3 trials, 692 participants; Analysis 2.1).
Change in appetite
One trial comparing pharmacological treatment to placebo reported dichotomous data on changes in appetite as a non‐serious adverse event. Black 2014 compared an antipsychotic to placebo. There was no difference in changes in appetite between the two groups (RR 0.65, 95% CI 0.10 to 4.06; P = 0.64; 1 trial, 17 participants; Analysis 2.1).
Forgetfulness or confusion
One trial comparing pharmacological treatment to placebo reported data on forgetfulness/confusion as a non‐serious adverse event. Black 2014 compared an antipsychotic to placebo. There was no difference in forgetfulness/confusion between the two groups (RR 1.46, 95% CI 0.38 to 5.60; P = 0.58; 1 trial, 62 participants; Analysis 2.1).
Disturbances in attention
One trial comparing pharmacological treatment to placebo reported data on disturbances in attention as a non‐serious adverse event. Zanarini 2007 compared an antipsychotic to placebo. There was no difference in disturbances in attention between the two groups (RR 11.37, 95% CI 0.63 to 203.81; P = 0.10; 1 trial, 301 participants; Analysis 2.1).
Restlessness
One trial comparing pharmacological treatment to placebo reported data on restlessness as a non‐serious adverse event. Grant 2022 compared an antipsychotic to placebo. There was no difference in restlessness between the two groups (RR 0.93, 95% CI 0.20 to 4.30; P = 0.92; 1 trial, 77 participants; Analysis 2.1).
Hallucinations
One trial comparing pharmacological treatment to placebo reported data on hallucinations as a non‐serious adverse event. Grant 2022 compared an antipsychotic to placebo. There was no difference in hallucinations between the two groups (RR 0.19, 95% CI 0.01 to 3.74; P = 0.27; 1 trial, 77 participants; Analysis 2.1).
Sleep problems
One trial comparing pharmacological treatment to placebo reported data on sleep problems as a non‐serious adverse event. Grant 2022 compared an antipsychotic to placebo. There was no difference in sleep problems between the two groups (RR 0.19, 95% CI 0.01 to 3.74; P = 0.27; 1 trial, 77 participants; Analysis 2.1).
Tremor
One trial comparing pharmacological treatment to placebo reported data on tremor as a non‐serious adverse event. Grant 2022 compared an antipsychotic to placebo. There was no difference in tremor between the two groups (RR 0.31, 95% CI 0.01 to 7.36; P = 0.47; 1 trial, 77 participants; Analysis 2.1).
Memory problems
One trial comparing pharmacological treatment to placebo reported dichotomous data on memory problems as a non‐serious adverse event. Loew 2006 compared a mood stabiliser to placebo. There was no difference in memory problems between the two groups (RR 2.00, 95% CI 0.55 to 7.22; P = 0.29; 1 trial, 56 participants; Analysis 2.2).
Paraesthesia
One trial comparing pharmacological treatment to placebo reported dichotomous data on paraesthesia as a non‐serious adverse event. Loew 2006 compared a mood stabiliser to placebo. There was no difference in paraesthesia between the two groups (RR 3.00, 95% CI 0.33 to 27.12; P = 0.33; 1 trial, 56 participants; Analysis 2.2).
Gait/balance disturbances
One trial comparing pharmacological treatment to placebo reported dichotomous data on gait/balance disturbances. Kulkarni 2018 compared memantine hydrochloride to placebo. There was no difference in gait/balance disturbances between the two groups (RR 2.35, 95% CI 0.53 to 10.45; P = 0.26; 1 trial, 33 participants; Analysis 2.3).
Nervous system disorders
One trial comparing pharmacological treatment to placebo reported dichotomous data on nervous system disorders as a non‐serious adverse event. Crawford 2018 compared a mood stabiliser to placebo. There was no difference in the appearance of nervous system disorders between the two groups (RR 1.05, 95% CI 0.68 to 1.62; P = 0.83; 1 trial, 276 participants; Analysis 2.2).
Psychiatric disorders
One trial comparing pharmacological treatment to placebo reported data on psychiatric disorders as a non‐serious adverse event. Crawford 2018 compared a mood stabiliser to placebo. There was no difference in the appearance of psychiatric disorders between the two groups (RR 0.94, 95% CI 0.64 to 1.37; P = 0.74; 1 trial, 276 participants; Analysis 2.2).
Cardiovascular and respiratory system
Cold/flu symptoms
One trial comparing pharmacological treatment to placebo reported dichotomous data on cold/flu symptoms as a non‐serious adverse event. Black 2014 compared an antipsychotic to placebo. There was no difference in cold/flu symptoms between the two groups (RR 1.54, 95% CI 0.50 to 4.73; P = 0.45; 1 trial, 62 participants; Analysis 3.1).
3.1. Analysis.

Comparison 3: Medications compared with placebo ‐ non‐serious adverse events ‐ cardiovascular and respiratory system, Outcome 1: Antipsychotics
Nasopharyngitis
One trial comparing pharmacological treatment to placebo reported dichotomous data on nasopharyngitis as a non‐serious adverse event. Schulz 2007 compared an antipsychotic to placebo. There was no difference in nasopharyngitis between the two groups (RR 0.62, 95% CI 0.23 to 1.66; P = 0.34; 1 trial, 301 participants; Analysis 3.1).
Sweating
One trial comparing pharmacological treatment to placebo reported dichotomous data on sweating as a non‐serious adverse event. Grant 2022 compared an antipsychotic to placebo. There was no difference in sweating between the two groups (RR 0.31, 95% CI 0.01 to 7.36; P = 0.47; 1 trial, 77 participants; Analysis 3.1).
Blood and lymphatic system disorders
One trial comparing pharmacological treatment to placebo reported dichotomous data on blood and lymphatic system disorders as a non‐serious adverse event. Crawford 2018 compared a mood stabiliser to placebo. There was no difference in blood and lymphatic system disorders between the two groups (RR 0.68, 95% CI 0.11 to 3.99; P = 0.67; 1 trial, 276 participants; Analysis 3.3).
3.3. Analysis.

Comparison 3: Medications compared with placebo ‐ non‐serious adverse events ‐ cardiovascular and respiratory system, Outcome 3: Mood stabilisers
Cardiac disorders
One trial comparing pharmacological treatment to placebo reported dichotomous data on cardiac disorders as a non‐serious adverse event. Crawford 2018 compared a mood stabiliser to placebo. There was no difference in cardiac disorders between the two groups (RR 0.34, 95% CI 0.01 to 8.23; P = 0.51; 1 trial, 276 participants; Analysis 3.3).
Endocrine disorders
One trial comparing pharmacological treatment to placebo reported dichotomous data on endocrine disorders as a non‐serious adverse event. Crawford 2018 compared a mood stabiliser to placebo. There was no difference in endocrine disorders between the two groups (RR 0.34, 95% CI 0.01 to 8.23; P = 0.51; 1 trial, 276 participants; Analysis 3.3).
Respiratory, thoracic and mediastinal disorders
One trial comparing pharmacological treatment to placebo reported dichotomous data on respiratory, thoracic and mediastinal disorders as a non‐serious adverse event. Crawford 2018 compared a mood stabiliser to placebo. There was no difference in respiratory, thoracic and mediastinal disorders between the two groups (RR 1.80, 95% CI 0.83 to 3.94; P = 0.14; 1 trial, 276 participants; Analysis 3.3).
Diastolic blood pressure in standing position (mean change from baseline to endpoint)
One trial comparing pharmacological treatment to placebo reported continuous data on diastolic blood pressure in standing position (mean change from baseline to endpoint) as a non‐serious adverse event. Zanarini 2007 compared an antipsychotic to placebo. There was no difference in mean change between the two groups (MD −0.28, 95% CI −2.29 to 1.73; P = 0.78; 1 trial, 290 participants; Analysis 3.2).
3.2. Analysis.

Comparison 3: Medications compared with placebo ‐ non‐serious adverse events ‐ cardiovascular and respiratory system, Outcome 2: Antipsychotics
Diastolic blood pressure in supine position (mean change from baseline to endpoint)
One trial comparing pharmacological treatment to placebo reported continuous data on diastolic blood pressure in supine position (mean change from baseline to endpoint) as a non‐serious adverse event. Zanarini 2007 compared an antipsychotic to placebo. There was no difference in mean change between the two groups (MD −0.11, 95% CI −2.28 to 2.06; P = 0.92; 1 trial, 290 participants; Analysis 3.2).
Systolic blood pressure in standing position (mean change from baseline to endpoint)
One trial comparing pharmacological treatment to placebo reported continuous data on systolic blood pressure in standing position (mean change from baseline to endpoint) as a non‐serious adverse event. Zanarini 2007 compared an antipsychotic to placebo. There was no difference in mean change between the two groups (MD 0.35, 95% CI −2.39 to 3.09; P = 0.80; 1 trial, 290 participants; Analysis 3.2).
Systolic blood pressure in supine position (mean change from baseline to endpoint)
One trial comparing pharmacological treatment to placebo reported continuous data on systolic blood pressure in supine position (mean change from baseline to endpoint) as a non‐serious adverse event. Zanarini 2007 compared an antipsychotic to placebo. There was no difference in mean change between the two groups (MD −1.31, 95% CI −4.00 to 1.38; P = 0.34; 1 trial, 290 participants; Analysis 3.2).
Pulse in standing position (mean change from baseline to endpoint)
One trial comparing pharmacological treatment to placebo reported continuous data on pulse in standing position (mean change from baseline to endpoint) as a non‐serious adverse event. Zanarini 2007 compared an antipsychotic to placebo. There was no difference in mean change between the two groups (MD 0.85, 95% CI −1.65 to 3.35; P = 0.50; 1 trial, 290 participants; Analysis 3.2).
Pulse in supine position (mean change from baseline to endpoint)
One trial comparing pharmacological treatment to placebo reported continuous data on pulse in supine position (mean change from baseline to endpoint) as a non‐serious adverse event. Zanarini 2007 compared an antipsychotic to placebo. There was no difference in mean change between the two groups (MD −0.11, 95% CI −2.28 to 2.06; P = 0.92; 1 trial, 290 participants; Analysis 3.2).
Gastrointestinal system
Nausea
Five trials comparing pharmacological treatment to placebo reported dichotomous data on nausea and vomiting as a non‐serious adverse event.
Four trials compared antipsychotics to placebo (Black 2014; Grant 2022; Schulz 2007; Zanarini 2007). There was no difference in nausea and vomiting between the two groups ((RR 0.80, 95% CI 0.49 to 1.29; P = 0.36; I2 = 0%; 4 trials, 754 participants; Analysis 4.1).
4.1. Analysis.

Comparison 4: Medications compared with placebo – non‐serious adverse events ‐ metabolic and gastro‐intestinal system, Outcome 1: Antipsychotics
One trial compared memantine hydrochloride to placebo (Kulkarni 2018). There was no difference in nausea and vomiting between the two groups (RR 1.00, 95% CI 0.45 to 2.23; P = 1.00; 1 trial, 34 participants; Analysis 4.4).
4.4. Analysis.

Comparison 4: Medications compared with placebo – non‐serious adverse events ‐ metabolic and gastro‐intestinal system, Outcome 4: Memantine hydrochloride
Uneasy feeling
One trial comparing pharmacological treatment to placebo reported dichotomous data on feelings of uneasiness as a non‐serious adverse event. Pascual 2008 compared an antipsychotic to placebo. There was no difference in the feeling of uneasiness between the two groups (RR 7.00, 95% CI 0.38 to 129.93; P = 0.19; 1 trial, 60 participants; Analysis 4.1).
Constipation
Two trials comparing pharmacological treatment to placebo reported dichotomous data on constipation as a non‐serious adverse event. One trial compared an antipsychotic to placebo (Zanarini 2001). There was no difference in constipation between the two groups (RR 6.50, 95% CI 0.41 to 104.20; P = 0.19; 1 trial, 28 participants; Analysis 4.1). Another trial compared memantine hydrochloride to placebo (Kulkarni 2018). There was no difference in constipation between the two groups (RR 1.65, 95% CI 0.59 to 4.57; P = 0.34; 1 trial, 33 participants; Analysis 4.4).
Dry mouth
Four trials comparing pharmacological treatment to placebo reported dichotomous data on dry mouth as a non‐serious adverse event. They all compared antipsychotics to placebo (Black 2014; Grant 2022; Schulz 2007; Zanarini 2007). There was a difference between the two groups, with an increased risk of dry mouth for antipsychotics (RR 2.60, 95% CI 1.46 to 4.64; P = 0.001; I2 = 0%; 4 trials, 754 participants; Analysis 4.1).
Gastrointestinal disorders
One trial comparing pharmacological treatment to placebo reported dichotomous data on gastrointestinal disorders as a non‐serious adverse event. Crawford 2018 compared a mood stabiliser to placebo. There was a slight difference between the two groups, with an increased risk of gastrointestinal disorders for placebo (RR 0.70, 95% CI 0.50 to 0.98; P = 0.04; 1 trial, 276 participants; Analysis 4.3).
4.3. Analysis.

Comparison 4: Medications compared with placebo – non‐serious adverse events ‐ metabolic and gastro‐intestinal system, Outcome 3: Mood stabilisers
General disorders and administration site conditions
One trial comparing pharmacological treatment to placebo reported dichotomous data on general disorders and administration site conditions as a non‐serious adverse event. Crawford 2018 compared a mood stabiliser to placebo. There was no difference in general disorders and administration site conditions between the two groups (RR 1.01, 95% CI 0.50 to 2.05; P = 0.97; 1 trial, 276 participants; Analysis 4.3).
Hepatobiliary disorders
One trial comparing pharmacological treatment to placebo reported dichotomous data on hepatobiliary disorders as a non‐serious adverse event. Crawford 2018 compared a mood stabiliser to placebo. There was no difference in hepatobiliary disorders between the two groups (RR 3.04, 95% CI 0.13 to 74.07; P = 0.49; 1 trial, 276 participants; Analysis 4.3).
Metabolism and nutrition disorders
One trial that compared a pharmacological treatment to placebo reported dichotomous data on metabolism and nutrition disorders as a non‐serious adverse event. Crawford 2018 compared a mood stabiliser to placebo. There was no difference in metabolism and nutrition disorders between the two groups (RR 2.03, 95% CI 0.19 to 22.12; P = 0.56; 1 trial, 276 participants; Analysis 4.3).
Liver function: alanine aminotransferase (ALT)/serum glutamic‐pyruvic transaminase (SGPT) baseline to endpoint mean change (u/L)
Two trials that compared a pharmacological treatment to placebo reported continuous data on mean change in ALT/SGPT as a non‐serious adverse event. Schulz 2007 and Zanarini 2007 compared antipsychotics to placebo. There was a difference between the two groups regarding mean change in ALT/SGPT favouring placebo (SMD 0.46, 95% CI 0.29 to 0.63; P < 0.001, I2 = 0%; 2 trials, 530 participants; Analysis 4.2).
4.2. Analysis.

Comparison 4: Medications compared with placebo – non‐serious adverse events ‐ metabolic and gastro‐intestinal system, Outcome 2: Antipsychotics
Liver function: aspartate aminotransferase (AST)/serum glutamic‐oxaloacetic transaminase (SGOT) baseline to endpoint mean change (u/L)
Two trials that compared a pharmacological treatment to placebo reported continuous data on mean change in AST/SGOT as a non‐serious adverse event. Schulz 2007 and Zanarini 2007 compared antipsychotics to placebo. There was a difference between the two groups regarding mean change in AST/SGOT favouring placebo (SMD 0.35, 95% CI 0.18 to 0.52; P < 0.001, I2 = 0%; 2 trials, 526 participants; Analysis 4.2).
Liver function: total bilirubin baseline to endpoint mean change (μmol/L)
One trial that compared a pharmacological treatment to placebo reported continuous data on mean change in total bilirubin as a non‐serious adverse event. Schulz 2007 compared an antipsychotic to placebo. There was a difference between the two groups regarding mean change in total bilirubin favouring the antipsychotic (MD −0.98, 95% CI −1.80 to −0.16; P = 0.02; 1 trial, 264 participants; Analysis 4.2).
Liver function: direct bilirubin baseline to endpoint mean change (μmol/L)
One trial that compared a pharmacological treatment to placebo reported continuous data on mean change in direct bilirubin as a non‐serious adverse event. Schulz 2007 compared an antipsychotic to placebo. There was a difference between the two groups regarding mean change in direct bilirubin favouring the antipsychotic (MD −0.30, 95% CI −0.51 to −0.09; P = 0.004; 1 trial, 258 participants; Analysis 4.2).
Liver function: gamma‐glutamyl transferase (GGT) baseline to endpoint mean change
One trial comparing a pharmacological treatment to placebo reported continuous data on mean change in GGT as a non‐serious adverse event. Zanarini 2007 compared an antipsychotic to placebo. There was a difference between the two groups regarding mean change in GGT favouring placebo (MD 2.96, 95% CI 0.22 to 5.70; P = 0.03; 1 trial, 268 participants; Analysis 4.2).
Lipids: total cholesterol baseline to endpoint change (mmol/L)
Two trials comparing pharmacological treatment to placebo reported continuous data on mean change in total cholesterol as a non‐serious adverse event. Both trials compared antipsychotics to placebo (Schulz 2007; Soler 2005). There was a difference between the two groups regarding mean change in total cholesterol favouring placebo (SMD 0.42, 95% CI 0.20 to 0.64; P = 0.0002, I2 = 0%; 2 trials, 327 participants; Analysis 4.2).
Lipids: low‐density lipoprotein (LDL) cholesterol baseline to endpoint mean change (mmol/L)
One trial comparing a pharmacological treatment to placebo reported continuous data on mean change in LDL cholesterol as a non‐serious adverse event. Schulz 2007 compared an antipsychotic to placebo. There was a difference between the two groups regarding mean change in LDL cholesterol favouring placebo (MD 0.21, 95% CI 0.06 to 0.36; P = 0.005; 1 trial, 259 participants; Analysis 4.2).
Lipids: high‐density lipoprotein (HDL) cholesterol (dextran precip.) baseline to endpoint mean change (mmol/L)
One trial comparing a pharmacological treatment to placebo reported continuous data on mean change in HDL cholesterol as a non‐serious adverse event. Zanarini 2007 compared an antipsychotic to placebo. There was a difference between the two groups regarding mean change in HDL cholesterol favouring the antipsychotic (MD −0.06, 95% CI −0.11 to −0.01; P = 0.02; 1 trial, 269 participants; Analysis 4.2).
Lipids: triglycerides, fasting, baseline to endpoint mean change (mmol/L)
One trial compared a pharmacological treatment to placebo reported continuous data on mean change in triglycerides as a non‐serious adverse event. Zanarini 2007 compared an antipsychotic to placebo. There was a difference between the two groups regarding mean change in triglycerides favouring placebo (MD 0.27, 95% CI 0.07 to 0.47; P = 0.009; 1 trial, 203 participants; Analysis 4.2).
Prolactin: baseline to endpoint mean change (μg/L)
One trial comparing a pharmacological treatment to placebo reported continuous data on mean change in prolactin as a non‐serious adverse event. Schulz 2007 compared an antipsychotic to placebo. There was a difference between the two groups regarding mean change in prolactin favouring placebo (MD 7.10, 95% CI 1.64 to 12.56; P = 0.01; 1 trial, 259 participants; Analysis 4.2).
Platelet count baseline to endpoint mean change (GI/L)
Two trials comparing a pharmacological treatment to placebo reported continuous data on mean change in platelet count as a non‐serious adverse event. Both trials compared antipsychotics to placebo (Schulz 2007; Zanarini 2007). There was no difference in platelet count between the two groups (SMD 0.03, 95% CI −0.53 to 0.59; P = 0.91, I2 = 90%; 2 trials, 517 participants; Analysis 4.2).
Erythrocyte count baseline to endpoint mean change (TI/L)
One trial comparing a pharmacological treatment to placebo reported continuous data on mean change in erythrocyte count as a non‐serious adverse event. Zanarini 2007 compared an antipsychotic to placebo. There was no difference in erythrocyte count between the two groups (MD −0.05, 95% CI −0.12 to 0.02; P = 0.14; 1 trial, 262 participants; Analysis 4.2).
Leukocyte count baseline to endpoint mean change (GI/L)
One trial comparing a pharmacological treatment to placebo reported continuous data on mean change in leukocyte count as a non‐serious adverse event. Zanarini 2007 compared an antipsychotic to placebo. There was a difference between the two groups regarding mean change in leukocyte count favouring the antipsychotic (MD −0.70, 95% CI −1.12 to −0.28; P = 0.001; 1 trial, 262 participants; Analysis 4.2).
Neutrophils, segmented, baseline to endpoint mean change (GI/L)
One trial comparing a pharmacological treatment to placebo reported continuous data on mean change in neutrophils as a non‐serious adverse event. Zanarini 2007 compared an antipsychotic to placebo. There was a difference between the two groups regarding mean change in neutrophils favouring the antipsychotic (MD −0.60, 95% CI −0.97 to −0.23; P = 0.002; 1 trial, 262 participants; Analysis 4.2).
Basophils baseline to endpoint mean change (GI/L)
One trial comparing a pharmacological treatment to placebo reported continuous data on mean change in basophils as a non‐serious adverse event. Zanarini 2007 compared an antipsychotic to placebo. There was a difference between the two groups regarding mean change in basophils favouring the antipsychotic (MD −0.01, 95% CI −0.02 to −0.00; P = 0.02; 1 trial, 262 participants; Analysis 4.2).
Monocytes baseline to endpoint mean change (GI/L)
One trial comparing a pharmacological treatment to placebo reported continuous data on mean change in monocytes as a non‐serious adverse event. Zanarini 2007 compared an antipsychotic to placebo. There was a difference between the two groups regarding mean change in monocytes favouring the antipsychotic (MD −0.04, 95% CI −0.07 to −0.01; P = 0.02; 1 trial, 262 participants; Analysis 4.2).
Haemoglobin baseline to endpoint mean change (mml/L‐F)
One trial comparing a pharmacological treatment to placebo reported continuous data on mean change in haemoglobin as a non‐serious adverse event. Zanarini 2007 compared an antipsychotic to placebo. There was no difference between the two groups regarding mean change in haemoglobin (MD −0.11, 95% CI −0.24 to 0.02; P = 0.09; 1 trial, 262 participants; Analysis 4.2).
Mean cell haemoglobin concentration (MCHC) baseline to endpoint mean change (mml/L‐F)
One trial comparing a pharmacological treatment to placebo reported continuous data on mean change in MCHC as a non‐serious adverse event. Zanarini 2007 compared an antipsychotic to placebo. There was no difference between the two groups regarding MCHC (MD 0.02, 95% CI −0.17 to 0.21; P = 0.84; 1 trial, 260 participants; Analysis 4.2).
Calcium baseline to endpoint mean change (mmol/L)
One trial comparing a pharmacological treatment to placebo reported continuous data on mean change in calcium as a non‐serious adverse event. Schulz 2007 compared an antipsychotic to placebo. There was a difference between the two groups regarding mean change in calcium favouring the antipsychotic (MD −0.03, 95% CI −0.05 to −0.01; P = 0.006; 1 trial, 268 participants; Analysis 4.2).
Albumin baseline to endpoint mean change (g/L)
One trial comparing a pharmacological treatment to placebo reported continuous data on mean change in albumin as a non‐serious adverse event. Zanarini 2007 compared an antipsychotic to placebo. There was no difference between the two groups regarding mean change in albumin (MD −0.67, 95% CI −1.42 to 0.08; P = 0.08; 1 trial, 269 participants; Analysis 4.2).
Creatine phosphokinase baseline to endpoint mean change (u/L)
One trial comparing a pharmacological treatment to placebo reported continuous data on mean change in creatine phosphokinase as a non‐serious adverse event. Zanarini 2007 compared an antipsychotic to placebo. There was no difference between the two groups regarding mean change in creatine phosphokinase (MD −44.81, 95% CI −95.39 to 5.77; P = 0.08; 1 trial, 268 participants; Analysis 4.2).
Urea nitrogen baseline to endpoint mean change (mmol/L)
One trial comparing a pharmacological treatment to placebo reported continuous data on mean change in urea nitrogen as a non‐serious adverse event. Zanarini 2007 compared an antipsychotic to placebo. There was no difference between the two groups regarding mean change in urea nitrogen (MD −0.17, 95% CI −0.46 to 0.12; P = 0.25; 1 trial, 269 participants Analysis 4.2).
Musculoskeletal system
Bodily pain
One trial comparing a pharmacological treatment to placebo reported dichotomous data on bodily pain as a non‐serious adverse event. Black 2014 compared an antipsychotic to placebo. There was no difference in bodily pain between the two groups (RR 0.88, 95% CI 0.47 to 1.64; P = 0.69; 1 trial, 62 participants; Analysis 5.1).
5.1. Analysis.

Comparison 5: Medications compared with placebo ‐ non‐serious adverse events ‐ musculoskeletal system, Outcome 1: Antipsychotics
Musculoskeletal and connective tissue disorders
One trial comparing a pharmacological treatment to placebo reported dichotomous data on musculoskeletal and connective tissue disorders as a non‐serious adverse event. Crawford 2018 compared a mood stabiliser to placebo. There was no difference in musculoskeletal and connective tissue disorders between the two groups (RR 1.16, 95% CI 0.43 to 3.11; P = 0.77; 1 trial; 276 participants; Analysis 5.4).
5.4. Analysis.

Comparison 5: Medications compared with placebo ‐ non‐serious adverse events ‐ musculoskeletal system, Outcome 4: Mood stabilisers
Body weight change
Twelve trials comparing a pharmacological treatment to placebo reported continuous data on body weight change as a non‐serious adverse event.
Seven trials compared antipsychotics to placebo (Bogenschutz 2004; Linehan 2008; Schulz 2007; Soler 2005; Soloff 1993; Zanarini 2001; Zanarini 2007). There was a difference between the two groups regarding body weight change favouring placebo (SMD 0.78, 95% CI 0.44 to 1.12; P < 0.001, I2 = 74%; 7 trials, 810 participants; Analysis 5.2).
5.2. Analysis.

Comparison 5: Medications compared with placebo ‐ non‐serious adverse events ‐ musculoskeletal system, Outcome 2: Antipsychotics
Soloff 1993 also compared an antidepressant to placebo. There was no difference between the two groups regarding body weight change (MD 0.09, 95% CI −0.31 to 0.49; P = 0.66; 1 trial, 62 participants; Analysis 5.3).
5.3. Analysis.

Comparison 5: Medications compared with placebo ‐ non‐serious adverse events ‐ musculoskeletal system, Outcome 3: Antidepressants
Five trials compared mood stabilisers to placebo (Frankenburg 2002; Loew 2006; Nickel 2004; Nickel 2005; Tritt 2005). There was no difference between the two groups regarding body weight change (SMD −0.26, 95% CI −0.72 to 0.20; P = 0.27, I2 = 55%; 5 trials, 184 participants; Analysis 5.5).
5.5. Analysis.

Comparison 5: Medications compared with placebo ‐ non‐serious adverse events ‐ musculoskeletal system, Outcome 5: Mood stabilisers
Sensory system
Eye disorders
One trial comparing a pharmacological treatment to placebo reported dichotomous data on eye disorders as a non‐serious adverse event. Crawford 2018 compared a mood stabiliser to placebo. There was no difference in eye disorders between the two groups (RR 0.17, 95% CI 0.02 to 1.39; P = 0.10; 1 trial, 276 participants; Analysis 6.1).
6.1. Analysis.

Comparison 6: Medications compared with placebo ‐ non‐serious adverse events ‐ sensory system, Outcome 1: Mood stabilisers
Reproductive system
Pregnancy, puerperium and perinatal conditions
One trial comparing a pharmacological treatment to placebo reported dichotomous data on pregnancy, puerperium and perinatal conditions as a non‐serious adverse event. Crawford 2018 compared a mood stabiliser to placebo. There was no difference in pregnancy, puerperium and perinatal conditions between the two groups (RR 1.52, 95% CI 0.26 to 8.97; P = 0.64; 1 trial, 276 participants; Analysis 7.1).
7.1. Analysis.

Comparison 7: Medications compared with placebo ‐ non‐serious adverse events ‐ reproductive system, Outcome 1: Mood stabilisers
Reproductive system and breast disorders
One trial comparing pharmacological treatment to placebo reported dichotomous data on the reproductive system and breast disorders as a non‐serious adverse event. Crawford 2018 compared a mood stabiliser to placebo. There was no difference in the reproductive system and breast disorders between the two groups (RR 3.04, 95% CI 0.32 to 28.90; P = 0.33; 1 trial, 276 participants; Analysis 7.1).
Menstrual pain
One trial comparing a pharmacological treatment to placebo reported dichotomous data on menstrual pain as a non‐serious adverse event. Loew 2006 compared a mood stabiliser to placebo. There was no difference in menstrual pain between the two groups (RR 1.67, 95% CI 0.44 to 6.31; P = 0.45; 1 trial, 56 participants; Analysis 7.1).
Other
Injury, poisoning or procedural complications
One trial comparing a pharmacological treatment to placebo reported dichotomous data on injuries, poisonings and procedural complications as a non‐serious adverse event. Crawford 2018 compared a mood stabiliser to placebo. There was a difference between the two groups regarding injuries, poisonings and procedural complications, with an increased risk of injuries, poisonings and procedural complications in the placebo group (RR 0.44, 95% CI 0.26 to 0.74; P = 0.002; 1 trial, 276 participants; Analysis 8.1).
8.1. Analysis.

Comparison 8: Medications compared with placebo ‐ non‐serious adverse events ‐ other, Outcome 1: Mood stabilisers
Skin and subcutaneous tissue disorders
One trial comparing a pharmacological treatment to placebo reported dichotomous data on skin and subcutaneous tissue disorders as a non‐serious adverse event. Crawford 2018 compared a mood stabiliser to placebo. There was no difference in skin and subcutaneous tissue disorders between the two groups (RR 1.15, 95% CI 0.75 to 1.75; P = 0.53; 1 trial, 276 participants; Analysis 8.1).
Social circumstances
One trial comparing a pharmacological treatment to placebo reported dichotomous data on social circumstances as a non‐serious adverse event. Crawford 2018 compared a mood stabiliser to placebo. There was no evidence of a difference in social circumstances between the two groups (RR 1.01, 95% CI 0.06 to 16.06; P = 0.99; 1 trial, 279 participants; Analysis 8.1).
Surgical and medical procedures
One trial comparing a pharmacological treatment to placebo reported dichotomous data on surgical and medical procedures as a non‐serious adverse event. Crawford 2018 compared a mood stabiliser to placebo. There was no difference in surgical and medical procedures between the two groups (RR 4.06, 95% CI 0.46 to 35.85; P = 0.21; 1 trial, 276 participants; Analysis 8.1).
Withdrawal due to adverse events
One trial comparing pharmacological treatment to placebo reported dichotomous data on withdrawal due to adverse events. Kulkarni 2018 compared memantine hydrochloride to placebo. There was no difference in withdrawal due to adverse events between the two groups (RR 2.82, 95% CI 0.33 to 24.43; P = 0.35; 1 trial, 33 participants; Analysis 9.1).
9.1. Analysis.

Comparison 9: Medications compared with placebo ‐ withdrew due to adverse events, Outcome 1: Memantine hydrochloride
2 Single medications compared with alternate single medications
Primary outcomes
2.1 BPD symptom severity
Three trials compared one medication with another medication and reported continuous data for BPD symptom severity.
Bozzatello 2017 compared two second‐generation antipsychotics, olanzapine and asenapine. There was no clear difference between the two treatments regarding the effects on BPD symptom severity (MD −2.23, 95% CI −8.04 to 3.58; P = 0.45; 1 trial, 51 participants; Analysis 10.1).
10.1. Analysis.

Comparison 10: Single medication compared with alternate single medication , Outcome 1: Primary: BPD symptom severity at end of treatment (continuous outcomes, MDs)
Soloff 1993 compared the antipsychotic haloperidol with the antidepressant phenelzine sulfate. There was no clear difference between the two treatments regarding BPD symptom severity (MD 5.73, 95% CI −0.33 to 11.79; P = 0.06; 1 trial, 64 participants; Analysis 10.1).
Cowdry 1988 had a cross‐over design with four active treatment arms: an arm with a benzodiazepine (alprazolam), an arm with a mood stabiliser (carbamazepine), an arm with an antipsychotic (trifluoperazine hydrochloride) and an arm with an antidepressant (tranylcypromine sulfate). From these, it was possible to make six comparisons: 1) alprazolam compared to carbamazepine; 2) alprazolam compared to trifluoperazine hydrochloride; 3) alprazolam compared to tranylcypromine sulfate; 4) carbamazepine compared to trifluoperazine hydrochloride; 5) carbamazepine compared to tranylcypromine sulfate; and 6) trifluoperazine hydrochloride compared to tranylcypromine sulfate.
There was a difference in BPD symptoms at end of treatment between alprazolam and carbamazepine, favouring alprazolam (MD −1.64, 95% CI −2.71 to −0.57; P = 0.003; 1 trial, 27 participants; Analysis 10.1), as well as between alprazolam and tranylcypromine sulfate, favouring alprazolam (MD −1.58, 95% CI −2.76 to −0.40; P = 0.009; 1 trial, 24 participants; Analysis 10.1).
There were no differences between treatments on BPD symptom severity at end of treatment for any of the other comparisons (alprazolam compared to trifluoperazine hydrochloride: MD −0.80, 95% CI −2.21 to 0.61; P = 0.27; 1 trial, 22 participants; Analysis 10.1; carbamazepine compared to trifluoperazine hydrochloride: MD 0.84, 95% CI −0.41 to 2.09; P = 0.19 ; 1 trial, 25 participants; Analysis 10.1; carbamazepine compared to tranylcypromine sulfate: MD 0.06, 95% CI −0.92 to 1.04; P = 0.90; 1 trial, 27 participants; Analysis 10.1; trifluoperazine hydrochloride compared to tranylcypromine sulfate: MD −0.78, 95% CI −2.13 to 0.57; P = 0.26; 1 trial, 22 participants; Analysis 10.1).
2.2 Self‐harm
One trial compared a medication with another medication and reported continuous data for self‐harm. Bozzatello 2017 compared two antipsychotics: olanzapine and asenapine. There was no evidence of a difference between the two antipsychotics regarding self‐harm (MD 0.21, 95% CI −0.58 to 1.00; P = 0.60; 1 trial, 51 participants; Analysis 10.2).
10.2. Analysis.

Comparison 10: Single medication compared with alternate single medication , Outcome 2: Primary: Self‐harm at end of treatment (continuous outcomes, MDs)
There were no other trials comparing medications to another medication that reported data on self‐harm.
2.3 Suicide‐related outcomes
One trial comparing medications to other medications reported continuous data on suicide‐related outcomes.
Cowdry 1988 had a cross‐over design with four active treatment arms (see above in 2.1).
There was a difference in suicide‐related outcomes at end of treatment between alprazolam and carbamazepine, favouring carbamazepine (MD 2.12, 95% CI 1.06 to 3.18; P < 0.001; 1 trial, 27 participants; Analysis 10.3); between alprazolam and trifluoperazine hydrochloride, favouring trifluoperazine hydrochloride (MD 1.73, 95% CI 0.62 to 2.84; P = 0.002; 1 trial, 22 participants; Analysis 10.3); and between alprazolam and tranylcypromine sulfate, favouring tranylcypromine (MD 2.00, 95% CI 0.89 to 3.11; P = 0.0004; 1 trial, 24 participants; Analysis 10.3).
10.3. Analysis.

Comparison 10: Single medication compared with alternate single medication , Outcome 3: Primary: Suicide‐related outcomes at end of treatment (continuous outcomes, MDs)
There was no difference between treatments on suicide‐related outcomes at end of treatment for any of the other comparisons (carbamazepine compared to trifluoperazine hydrochloride: MD −0.39, 95% CI −1.53 to 0.75; P = 0.50; 1 trial, 25 participants; Analysis 10.3; carbamazepine compared to tranylcypromine sulfate: MD −0.12, 95% CI −1.26 to 1.02; P = 0.84; 1 trial, 27 participants; Analysis 10.3; trifluoperazine hydrochloride compared to tranylcypromine sulfate: MD 0.27, 95% CI −1.00 to 1.54; P = 0.68; 1 trial, 22 participants; Analysis 10.3).
2.4 Psychosocial functioning
Five trials compared a medication with another medication and reported continuous data for psychosocial functioning.
Three trials compared one antipsychotic with another antipsychotic (Bozzatello 2017; Shafti 2010; Shafti 2014). There was no difference between the treatments for any of the comparisons regarding psychosocial functioning; for Bozzatello 2017 comparing olanzapine to asenapine (MD = 0.20, 95% CI −0.23 to 0.63, P = 0.36; 1 trial, 51 participants; Analysis 10.4); for Shafti 2010 comparing olanzapine to haloperidol (MD 0.35, 95% CI −0.45 to 1.15; P = 0.39; 1 trial, 28 participants; Analysis 10.4); or for Shafti 2014 comparing olanzapine to aripiprazole (MD 0.12, 95% CI −0.58 to 0.82; P = 0.74; 1 trial, 24 participants; Analysis 10.4).
10.4. Analysis.

Comparison 10: Single medication compared with alternate single medication , Outcome 4: Primary: Psychosocial functioning (continuous outcomes, MDs)
Two trials compared an antipsychotic to an antidepressant (Soloff 1989; Soloff 1993). Soloff 1989 compared haloperidol to amitriptyline. There was no difference between the treatments regarding psychosocial functioning (MD −3.87, 95% CI −10.67 to 2.93; P = 0.26; 1 trial, 57 participants; Analysis 10.4). One study, Soloff 1993, compared haloperidol to phenelzine sulfate and found a difference in psychosocial functioning following treatment that favoured phenelzine sulfate (MD 5.15, 95% CI 0.29 to 10.01; P = 0.04; 1 trial, 64 participants; Analysis 10.4).
Secondary outcomes
2.5 Anger
Six trials compared a medication with another medication and reported continuous data for anger.
Three trials compared an antipsychotic to another antipsychotic: Shafti 2010 compared olanzapine to haloperidol, Shafti 2014 compared olanzapine to aripiprazole, and Bozzatello 2017 compared olanzapine to asenapine. There was no difference regarding anger at the end of treatment between olanzapine and haloperidol (MD 0.21, 95% CI −8.90 to 9.32; P = 0.96; 1 trial, 28 participants; Analysis 10.5) or between olanzapine and aripiprazole (MD −0.40, 95% CI −8.05 to 7.25; P = 0.92, 1 trial, 24 participants; Analysis 10.5). However, there was a difference between asenapine and olanzapine regarding anger at end of treatment, favouring asenapine (MD 1.14, 95% CI 0.31 to 1.97; P = 0.007; 1 trial, 51 participants; Analysis 10.5).
10.5. Analysis.

Comparison 10: Single medication compared with alternate single medication , Outcome 5: Secondary: Anger at end of treatment (continuous outcomes, MDs)
Three trials compared an antipsychotic to an antidepressant. Jariani 2010 compared olanzapine to sertraline, Soloff 1989 compared haloperidol to amitriptyline, and Soloff 1993 compared haloperidol to phenelzine sulfate. There was no difference regarding anger at end of treatment between haloperidol and amitriptyline (MD −0.34, 95% CI −0.82 to 0.14; P = 0.16; 1 trial, 57 participants; Analysis 10.5), or between haloperidol and phenelzine sulfate (MD 0.06, 95% CI −0.31 to 0.43; P = 0.75; 1 trial, 64 participants; Analysis 10.5). However, there was a superior effect of olanzapine compared to sertraline regarding anger at the end of treatment (MD −0.33, 95% CI −0.48 to −0.18; P < 0.001; 1 trial, 120 participants; Analysis 10.5).
Cowdry 1988 had a cross‐over design with four active treatment arms (see above in 2.1).
There was a difference in anger at end of treatment between alprazolam and carbamazepine, favouring carbamazepine (MD 1.65, 95% CI 0.80 to 2.50; P = 0.0001; 1 trial, 27 participants; Analysis 10.5), as well as between alprazolam and tranylcypromine sulfate, favouring tranylcypromine sulfate (MD 1.41, 95% CI 0.24 to 2.58; P = 0.02; 1 trial, 24 participants; Analysis 10.5).
There was no difference between treatments in anger at the end of treatment for any of the other comparisons: alprazolam compared to trifluoperazine hydrochloride (MD 0.58, 95% CI −0.77 to 1.93; P = 0.40; 1 trial, 22 participants; Analysis 10.5); carbamazepine compared to trifluoperazine hydrochloride (MD −1.07, 95% CI −2.28 to 0.14; P = 0.08; 1 trial, 25 participants; Analysis 10.5); carbamazepine compared to tranylcypromine sulfate (MD −0.24, 95% CI −1.23 to 0.75; P = 0.64; 1 trial, 27 participants; Analysis 10.5); and trifluoperazine hydrochloride compared to tranylcypromine sulfate (MD 0.83, 95% CI −0.62 to 2.28; P = 0.26; 1 trial, 22 participants; Analysis 10.5).
2.6 Affective instability
One trial compared a medication with another medication and reported continuous data for affective instability. Bozzatello 2017 compared two antipsychotics, olanzapine and asenapine. There was a difference in effects between the two antipsychotics regarding affective instability favouring asenapine (MD 2.28, 95% CI 1.51 to 3.05; P < 0.001; 1 trial, 51 participants; Analysis 10.6).
10.6. Analysis.

Comparison 10: Single medication compared with alternate single medication , Outcome 6: Secondary: Affective instability at end of treatment (continuous outcomes, MDs)
No other trials comparing medications to alternate medications reported data on affective instability.
2.7 Chronic feelings of emptiness
One trial compared a medication with another medication and reported continuous data for chronic feelings of emptiness. Bozzatello 2017 compared the two antipsychotics, olanzapine and asenapine. There was no difference in effects between the two antipsychotics regarding chronic feelings of emptiness (MD −0.54, 95% CI −1.29 to 0.21; P = 0.16, 1 trial, 51 participants; Analysis 10.7).
10.7. Analysis.

Comparison 10: Single medication compared with alternate single medication , Outcome 7: Secondary: Chronic feelings of emptiness at end of treatment (continuous outcomes, MDs)
No other trials comparing a medication with another medication reported data on chronic feelings of emptiness.
2.8 Impulsivity
Five trials compared a medication with another medication and reported continuous data for impulsivity.
One trial compared two antipsychotics, olanzapine and asenapine (Bozzatello 2017). There was no difference between olanzapine and asenapine regarding impulsivity at the end of treatment (MD −0.78, 95% CI −1.59 to 0.03, P = 0.06; 1 trial, 51 participants; Analysis 10.8).
10.8. Analysis.

Comparison 10: Single medication compared with alternate single medication , Outcome 8: Secondary: Impulsivity at end of treatment (continuous outcomes, MDs)
Three trials compared an antipsychotic with an antidepressant. Soloff 1989 compared haloperidol to amitriptyline, Soloff 1993 compared haloperidol to phenelzine sulfate, and Zanarini 2004 compared olanzapine to fluoxetine. There was no difference between treatments regarding impulsivity at the end of treatment for any of the comparisons (haloperidol versus amitriptyline: MD 3.52, 95% CI −5.52 to 12.56; P = 0.45; 1 trial, 57 participants; Analysis 10.8; haloperidol versus phenelzine sulfate: MD 3.29, 95% CI −14.52 to 21.10; P = 0.72; 1 trial, 64 participants; Analysis 10.8; and olanzapine versus fluoxetine: MD −4.31, 95% CI −19.72 to 11.10; P = 0.58; 1 trial, 29 participants; Analysis 10.8).
Cowdry 1988 had a cross‐over design with four active treatment arms (see above in 2.1).
There was a difference in impulsivity at end of treatment in four of the comparisons, specifically between: alprazolam and carbamazepine, favouring carbamazepine (MD 2.18, 95% CI 1.20 to 3.16; P < 0.001; 1 trial, 27 participants; Analysis 10.8); alprazolam and tranylcypromine sulfate, favouring tranylcypromine sulfate (MD 1.83, 95% CI 0.66 to 3.00; P = 0.002; 1 trial, 24 participants; Analysis 10.8); carbamazepine and trifluoperazine hydrochloride, favouring carbamazepine (MD −1.73, 95% CI −2.87 to −0.59; P = 0.003; 1 trial, 25 participants; Analysis 10.8); and trifluoperazine hydrochloride and tranylcypromine sulfate, favouring tranylcypromine sulfate (MD 1.38, 95% CI 0.08 to 2.68; P = 0.04; 1 trial, 22 participants; Analysis 10.8).
There was no difference between treatments in impulsivity at end of treatment for any of the other two comparisons (alprazolam compared to trifluoperazine hydrochloride: MD 0.45, 95% CI −0.87 to 1.77; P = 0.50; 1 trial, 22 participants; Analysis 10.8; and carbamazepine compared to tranylcypromine sulfate: MD −0.35, 95% CI −1.31 to 0.61; P = 0.48; 1 trial, 27 participants; Analysis 10.8).
2.9 Interpersonal problems
Three trials compared a medication with another medication and reported continuous data for interpersonal problems.
One trial (Bozzatello 2017) compared two antipsychotics, olanzapine and asenapine. There was no difference between olanzapine and asenapine regarding interpersonal problems at the end of treatment (MD 0.40, 95% CI −0.35 to 1.15, P = 0.29; 1 trial, 51 participants; Analysis 10.9).
10.9. Analysis.

Comparison 10: Single medication compared with alternate single medication , Outcome 9: Secondary: Interpersonal problems at end of treatment (continuous outcomes, MDs)
Two trials compared an antipsychotic with antidepressants. Soloff 1989 compared haloperidol to amitriptyline and Soloff 1993 compared haloperidol to phenelzine sulfate. There was no difference between the two treatments regarding interpersonal problems at end of treatment, either when comparing haloperidol to amitriptyline (MD −0.13, 95% CI −0.62 to 0.36; P = 0.60; 1 trial, 57 participants; Analysis 10.9), or to phenelzine sulfate (MD −0.33, 95% CI −0.68 to 0.02; P = 0.06; 1 trial, 64 participants; Analysis 10.9).
2.10 Abandonment
One trial compared a medication with another medication and reported continuous data for fear of abandonment. Bozzatello 2017 compared two antipsychotics. There was no difference between the two antipsychotics (olanzapine and asenapine) regarding avoidance of abandonment (MD −0.40, 95% CI −1.07 to 0.27, P = 0.24; 1 trial, 51 participants; Analysis 10.10).
10.10. Analysis.

Comparison 10: Single medication compared with alternate single medication , Outcome 10: Secondary: Abandonment at end of treatment (continuous outcomes, MDs)
No other trials comparing a medication with another medication reported data on fear of abandonment.
2.11 Identity disturbance
One trial compared a medication with another medication and reported continuous data for identity disturbance. Bozzatello 2017 compared the two antipsychotics olanzapine and asenapine. There was no difference in effects between olanzapine and asenapine regarding identity disturbance (MD 0.68, 95% CI −0.12 to 1.48, P = 0.10; 1 trial, 51 participants; Analysis 10.11).
10.11. Analysis.

Comparison 10: Single medication compared with alternate single medication , Outcome 11: Secondary: Identity disturbance at end of treatment (continuous outcomes, MDs)
No other trials comparing a medication with another medication reported data on identity disturbance.
2.12 Dissociation and psychotic‐like symptoms
Five trials compared a medication with another medication and reported continuous data for dissociation and psychotic‐like symptoms.
Three trials compared an antipsychotic to another antipsychotic. Bozzatello 2017 compared olanzapine to asenapine, Shafti 2010 olanzapine to haloperidol, and Shafti 2014 olanzapine to aripiprazole. There was no difference between treatments regarding dissociation and psychotic‐like symptoms at the end of treatment for any of the comparisons: olanzapine and asenapine (MD −0.69, 95% CI −1.53 to 0.15; P = 0.11; 1 trial, 51 participants; Analysis 10.12), olanzapine and haloperidol (MD −2.30, 95% CI −10.15 to 5.55; P = 0.57; 1 trial, 28 participants; Analysis 10.12), and olanzapine and aripiprazole (MD −3.30, 95% CI −10.63 to 4.03; P = 0.38; 1 trial, 24 participants; Analysis 10.12).
10.12. Analysis.

Comparison 10: Single medication compared with alternate single medication , Outcome 12: Secondary: Dissociation and psychotic‐like symptoms at end of treatment (continuous outcomes, MDs)
Two trials compared an antipsychotic with antidepressants. Soloff 1989 compared haloperidol to amitriptyline and Soloff 1993 compared haloperidol to phenelzine sulfate. There was no difference between treatments for any of the comparisons regarding dissociation and psychotic‐like symptoms at the end of treatment (haloperidol versus amitriptyline: MD −0.28, 95% CI −0.69 to 0.13; P = 0.18; 1 trial, 57 participants; Analysis 10.12; and haloperidol versus phenelzine sulfate: MD 0.14, 95% CI −0.31 to 0.59; P = 0.54; 1 trial, 64 participants; Analysis 10.12).
2.13 Depression
Six trials compared a medication with another medication and reported continuous data for depression.
Bozzatello 2017 compared the two antipsychotics olanzapine and asenapine. There was a difference in effects between olanzapine and asenapine regarding depression, favouring asenapine (MD 2.90, 95% CI 0.88 to 4.92; P = 0.005; 1 trial, 51 participants; Analysis 10.13).
10.13. Analysis.

Comparison 10: Single medication compared with alternate single medication , Outcome 13: Secondary: Depression at end of treatment (continuous outcomes, MDs)
Four trials compared an antipsychotic with an antidepressant. Jariani 2010 compared olanzapine to sertraline, Soloff 1989 compared haloperidol to amitriptyline, Soloff 1993 compared haloperidol to phenelzine sulfate, and Zanarini 2004 compared olanzapine to fluoxetine. There was no difference between haloperidol and amitriptyline regarding depression at end of treatment (MD 0.88, 95% CI −5.12 to 6.88; P = 0.77; 1 trial, 57 participants; Analysis 10.13); however, a difference between treatments was observed in the comparisons between olanzapine and sertraline, favouring sertraline (MD 0.37, 95% CI 0.22 to 0.52; P < 0.001; 1 trial, 120 participants; Analysis 10.13); haloperidol and phenelzine sulfate, favouring phenelzine sulfate (MD 7.81, 95% CI 2.13 to 13.49; P = 0.007; 1 trial, 64 participants; Analysis 10.13); and olanzapine and fluoxetine, favouring olanzapine (MD −5.40, 95% CI −10.68 to −0.12; P = 0.04; 1 trial, 29 participants; Analysis 10.13).
Cowdry 1988 had a cross‐over design with four active treatment arms (see above in 2.1).
There was a difference in depression at the end of treatment between alprazolam and carbamazepine, favouring carbamazepine (MD 1.36, 95% CI 0.37 to 2.35; P = 0.007; 27 participants, 1 trial; Analysis 10.13), as well as between alprazolam and tranylcypromine sulfate, favouring tranylcypromine sulfate (MD 1.67, 95% CI 0.48 to 2.86; P = 0.006; 24 participants, 1 trial; Analysis 10.13).
There was no difference between treatments in terms of depression at end of treatment for any of the other comparisons (alprazolam compared to trifluoperazine hydrochloride: MD 0.90, 95% CI −0.49 to 2.29; P = 0.20; 1 trial, 22 participants; Analysis 10.13; carbamazepine compared to trifluoperazine hydrochloride: MD −0.46, 95% CI −1.81 to 0.89; P = 0.50; 1 trial, 25 participants; Analysis 10.13; carbamazepine compared to tranylcypromine sulfate: MD 0.31, 95% CI −0.82 to 1.44; P = 0.59; 1 trial, 27 participants; Analysis 10.13; and trifluoperazine hydrochloride compared to tranylcypromine sulfate: MD 0.77, 95% CI −0.73 to 2.27; P = 0.31; 1 trial, 22 participants; Analysis 10.13).
2.14 Attrition
Eight trials compared a medication with another medication and reported dichotomous data on attrition.
Four trials compared an antipsychotic to another antipsychotic. Bozzatello 2017 compared olanzapine to asenapine, Leone 1982 compared loxapine to chlorpromazine, Shafti 2010 compared olanzapine to haloperidol, and Shafti 2014 compared olanzapine to aripiprazole. There was no difference between treatments for any of the comparisons on attrition (olanzapine versus asenapine: RR 0.80, 95% CI 0.28 to 2.29; P = 0.68; 1 trial, 51 participants; Analysis 10.14; loxapine versus chlorpromazine: RR 1.14, 95% CI 0.46 to 2.85; P = 0.77; 1 trial, 80 participants; Analysis 10.14; olanzapine versus haloperidol: no events occurred in any of the treatment groups; Analysis 10.14; and olanzapine versus aripiprazole: no events occurred in any of the treatment groups; Analysis 10.14).
10.14. Analysis.

Comparison 10: Single medication compared with alternate single medication , Outcome 14: Secondary: Attrition at end of treatment (dichotomous outcomes, RRs)
Four trials compared an antipsychotic to an antidepressant. Jariani 2010 compared olanzapine to sertraline, Soloff 1989 compared haloperidol to amitriptyline, Soloff 1993 compared haloperidol to phenelzine sulfate, and Zanarini 2004 compared olanzapine to fluoxetine. There was no difference between treatments for any of the comparisons on attrition (olanzapine versus sertraline: no events occurred in any of the treatment groups; Analysis 10.14; haloperidol versus amitriptyline: RR 2.90, 95% CI 0.32 to 26.38; P = 0.34; 1 trial, 61 participants; Analysis 10.14; haloperidol versus phenelzine sulfate: RR 1.58, 95% CI 0.49 to 5.15; P = 0.45; 1 trial, 75 participants; Analysis 10.14; and olanzapine versus fluoxetine: RR 0.29, 95% CI 0.01 to 6.69; P = 0.44; 1 trial, 30 participants; Analysis 10.14).
2.15 Adverse events
No trials comparing a medication with another medication reported data on non‐serious adverse events of the gastrointestinal system, the sensory system or the reproductive system; or on any other adverse events than those suggested in our analysis plan.
Three trials comparing a medication with another medication reported dichotomous data on total non‐serious adverse events.
All trials compared an antipsychotic to another antipsychotic. Leone 1982 compared loxapine to chlorpromazine, Shafti 2010 compared olanzapine to haloperidol, and Shafti 2014 compared olanzapine to aripiprazole. There was no difference between treatments regarding adverse events in any of the comparisons i.e. loxapine versus chlorpromazine (RR 1.27, 95% CI 0.66 to 2.45; P = 0.47; 1 trial, 80 participants, Analysis 10.15; olanzapine versus haloperidol (RR 0.75, 95% CI 0.35 to 1.60; P = 0.46; 1 trial, 28 participants; Analysis 10.15; and olanzapine versus aripiprazole (RR 0.75, 95% CI 0.38 to 1.50; P = 0.42; 1 trial, 24 participants; Analysis 10.15). No trials comparing a medication with another medication reported data on serious adverse events.
10.15. Analysis.

Comparison 10: Single medication compared with alternate single medication , Outcome 15: Secondary: Adverse events at end of treatment (dichotomous outcomes, RRs)
Withdrawal due to adverse events
One trial comparing a medication with another medication reported dichotomous data on withdrawal due to adverse events. Bozzatello 2017 compared the antipsychotics olanzapine and asenapine. There was no difference between the treatment groups in withdrawal due to adverse events (RR 0.96, 95% CI 0.15 to 6.31; P = 0.97; 1 trial, 51 participants; Analysis 11.1).
11.1. Analysis.

Comparison 11: Single medication compared with alternate single medication ‐ withdrew due to adverse events (AE), Outcome 1: Withdrew due to AE
Central nervous system
Sedation
One trial comparing a medication with another medication reported dichotomous data on sedation as a non‐serious adverse event. Zanarini 2004 compared an antipsychotic with an antidepressant. There was an increased risk of sedation with antipsychotics compared to antidepressants (RR 3.50, 95% CI 1.23 to 9.92; P = 0.02; 1 trial, 30 participants; Analysis 12.1).
12.1. Analysis.

Comparison 12: Single medication compared with alternate single medication ‐ non‐serious adverse events ‐ central nervous system, Outcome 1: Sedation
Restlessness
Two trials comparing a medication with another medication reported dichotomous data on restlessness as a non‐serious adverse event. Leone 1982 compared the antipsychotics loxapine and chlorpromazine, while Zanarini 2004 compared the antipsychotic olanzapine to the antidepressant fluoxetine. None of the analyses showed a difference in restlessness between the groups (loxapine versus chlorpromazine: (RR 1.50, 95% CI 0.26 to 8.50; P = 0.65; 1; I2 = 0%; 1 trial, 80 participants; Analysis 12.2; and olanzapine versus fluoxetine: RR 0.70, 95% CI 0.23 to 2.11; P = 0.53; 1 trial, 30 participants; Analysis 12.2).
12.2. Analysis.

Comparison 12: Single medication compared with alternate single medication ‐ non‐serious adverse events ‐ central nervous system, Outcome 2: Restlessness
Restlessness/anxiety
One trial comparing a medication with another medication reported dichotomous data on restlessness/anxiety as a non‐serious adverse event. Bozzatello 2017 compared two antipsychotics, olanzapine and asenapine. There was no difference in restlessness/anxiety between the two groups (RR 0.19, 95% CI 0.01 to 3.82; P = 0.28; 1 trial, 51 participants; Analysis 12.3).
12.3. Analysis.

Comparison 12: Single medication compared with alternate single medication ‐ non‐serious adverse events ‐ central nervous system, Outcome 3: Restlessness/anxiety
Sleepiness/drowsiness
Two trials comparing a medication with another medication reported dichotomous data on sleepiness/drowsiness as a non‐serious adverse event. Leone 1982 compared the two antipsychotics loxapine and chlorpromazine. There was no difference in sleepiness/drowsiness between the two groups (RR 0.80, 95% CI 0.23 to 2.76; P = 0.72; 1 trial, 80 participants; Analysis 12.4). Bozzatello 2017 compared two second‐generation antipsychotics olanzapine and asenapine. There was no difference in sleepiness/drowsiness between the two groups (RR 6.74, 95% CI 0.37 to 124.21; P = 0.20; 1 trial, 51 participants; Analysis 12.4).
12.4. Analysis.

Comparison 12: Single medication compared with alternate single medication ‐ non‐serious adverse events ‐ central nervous system, Outcome 4: Sleepiness/drowsiness
Fainting spells
One trial comparing a medication with another medication reported dichotomous data on fainting spells as a non‐serious adverse event. Leone 1982 compared the two antipsychotics loxapine and chlorpromazine. There was no difference in fainting spells between the groups (RR 0.14, 95% CI 0.01 to 2.68; P = 0.19; 1 trial, 80 participants; Analysis 12.5).
12.5. Analysis.

Comparison 12: Single medication compared with alternate single medication ‐ non‐serious adverse events ‐ central nervous system, Outcome 5: Fainting spells
Akathisia
One trial comparing a medication with another medication reported dichotomous data on akathisia as a non‐serious adverse event. Bozzatello 2017 compared the antipsychotics olanzapine and asenapine. There was no difference in akathisia between the two groups (RR 0.19, 95% CI 0.01 to 3.82; P = 0.28; 1 trial, 51 participants; Analysis 12.6).
12.6. Analysis.

Comparison 12: Single medication compared with alternate single medication ‐ non‐serious adverse events ‐ central nervous system, Outcome 6: Akhatisia
Moderate anxiety
One trial comparing a medication with another medication reported dichotomous data on moderate anxiety as a non‐serious adverse event. Bozzatello 2017 compared the antipsychotics olanzapine and asenapine. There was no difference in moderate anxiety between the two groups (RR 0.32, 95% CI 0.01 to 7.53; P = 0.48; 1 trial, 51 participants; Analysis 12.7).
12.7. Analysis.

Comparison 12: Single medication compared with alternate single medication ‐ non‐serious adverse events ‐ central nervous system, Outcome 7: Moderate anxiety
Fatigue
One trial comparing a medication with another medication reported dichotomous data on fatigue as a non‐serious adverse event. Bozzatello 2017 compared the antipsychotics olanzapine and asenapine. There was no difference in fatigue between groups (RR 4.81, 95% CI 0.24 to 95.58; P = 0.30; 1 trial, 51 participants; Analysis 12.8).
12.8. Analysis.

Comparison 12: Single medication compared with alternate single medication ‐ non‐serious adverse events ‐ central nervous system, Outcome 8: Fatigue
Cardiovascular and respiratory system
Oral hypoaesthesia
One trial comparing a medication with another medication reported dichotomous data on oral hypoaesthesia as a non‐serious adverse event. Bozzatello 2017 compared the antipsychotics olanzapine and asenapine. There was no difference in oral hypoaesthesia between the two groups (RR 0.32, 95% CI 0.01 to 7.53; P = 0.48; 1 trial, 51 participants; Analysis 13.1).
13.1. Analysis.

Comparison 13: Single medication compared with alternate single medication ‐ cardiovascular and respiratory system, Outcome 1: Oral hypoaesthesia
Musculoskeletal system
Muscle spasms
One trial comparing a medication with another medication reported dichotomous data on muscle spasms as a non‐serious adverse event. Leone 1982 compared the two antipsychotics loxapine and chlorpromazine. There was no difference in muscle spasms between the groups (RR 3.00, 95% CI 0.33 to 27.63; P = 0.33; 1 trial, 80 participants; Analysis 14.1).
14.1. Analysis.

Comparison 14: Single medication compared with alternate single medication ‐ non‐serious adverse events ‐ musculoskeletal system, Outcome 1: Muscle spasm
Body weight change
Two trials comparing a medication with another medication reported continuous data on body weight change as a non‐serious adverse event. Both compared an antipsychotic to an antidepressant. Soloff 1993 compared haloperidol to phenelzine sulfate and Zanarini 2004 compared olanzapine to fluoxetine. There was no difference between haloperidol and phenelzine sulfate on body weight change (MD −0.22, 95% CI −0.59 to 0.15; P = 0.25; 1 trial, 64 participants; Analysis 14.2). However, there was a difference between olanzapine and fluoxetine on body weight change favouring fluoxetine (MD 2.50, 95% CI 0.72 to 4.28; P = 0.006; 1 trial, 29 participants; Analysis 14.2).
14.2. Analysis.

Comparison 14: Single medication compared with alternate single medication ‐ non‐serious adverse events ‐ musculoskeletal system, Outcome 2: Body weight change
Weight gain
One trial comparing a medication with another medication reported dichotomous data on weight gain as a non‐serious adverse event. Bozzatello 2017 compared the antipsychotics olanzapine and asenapine. There was no difference in weight gain between groups (RR 4.81, 95% CI 0.24 to 95.58; P = 0.30; 1 trial, 51 participants; Analysis 14.3).
14.3. Analysis.

Comparison 14: Single medication compared with alternate single medication ‐ non‐serious adverse events ‐ musculoskeletal system, Outcome 3: Weight gain (3 or more kg within 4 weeks)
3 Single medication compared with combination of medications
Primary outcomes
3.1 BPD symptom severity
One trial compared one medication with a combination of medications and reported continuous data on BPD symptom severity. Bellino 2014 compared the mood stabiliser valproic acid to valproic acid plus omega‐3 fatty acids combined. There was a difference between valproic acid alone and valproic acid plus omega‐3 fatty acids regarding BPD symptom severity at the end of treatment favouring the mood stabiliser alone (MD 8.48, 95% CI 3.39 to 13.57; P = 0.001; 1 trial, 34 participants; Analysis 15.1).
15.1. Analysis.

Comparison 15: Single medication compared with combination of medications, Outcome 1: Primary: BPD symptom severity at end of treatment (continuous outcome, MDs)
3.2 Self‐harm
One trial compared a pharmacotherapy with a combination of pharmacotherapies and reported continuous data on self‐harm. Bellino 2014 compared the mood stabiliser valproic acid to valproic acid plus omega‐3 fatty acids combined. There was a difference between valproic acid alone and valproic acid plus omega‐3 fatty acids regarding self‐harm at the end of treatment favouring the mood stabiliser alone (MD 2.55, 95% CI 0.98 to 4.12; P = 0.001; 1 trial, 34 participants; Analysis 15.2).
15.2. Analysis.

Comparison 15: Single medication compared with combination of medications, Outcome 2: Primary: Self‐harm at end of treatment (continuous outcome, MDs)
3.3 Suicide‐related outcomes
One trial compared a pharmacotherapy with a combination of pharmacotherapies and reported continuous data on suicide‐related outcomes. Bellino 2014 compared the mood stabiliser valproic acid to valproic acid plus omega‐3 fatty acids combined. There was no difference between valproic acid alone and valproic acid plus omega‐3 fatty acids regarding suicide‐related outcomes at the end of treatment (MD 0.23, 95% CI −0.74 to 1.20; P = 0.64; 1 trial, 34 participants; Analysis 15.3).
15.3. Analysis.

Comparison 15: Single medication compared with combination of medications, Outcome 3: Primary: Suicide‐related outcomes at end of treatment (continuous outcome, MDs)
3.4 Psychosocial functioning
One trial compared a pharmacotherapy with a combination of pharmacotherapies and reported continuous data on psychosocial functioning. Bellino 2014 compared the mood stabiliser valproic acid to valproic acid plus omega‐3 fatty acids combined. There was no difference between valproic acid alone and valproic acid plus omega‐3 fatty acids regarding psychosocial functioning at the end of treatment (MD 0.88, 95% CI −6.21 to 7.97; P = 0.81; 1 trial, 34 participants; Analysis 15.4).
15.4. Analysis.

Comparison 15: Single medication compared with combination of medications, Outcome 4: Primary: Psychosocial functioning at end of treatment (continuous outcome, MDs)
Secondary outcomes
3.5 Anger
Two trials compared a pharmacotherapy to a combination of pharmacotherapies and reported continuous data on anger.
Zanarini 2004 compared the antipsychotic olanzapine to olanzapine plus the antidepressant fluoxetine combined. There was no difference between the groups regarding impulsivity at the end of treatment (MD 0.46, 95% CI −12.93 to 13.85; P = 0.95; 1 trial, 29 participants; Analysis 15.5). Zanarini 2004 also compared fluoxetine to fluoxetine plus olanzapine. There was no difference between the groups at the end of treatment (MD 4.77, 95% CI −9.67 to 19.21; P = 0.52; 1 trial, 26 participants; Analysis 15.5).
15.5. Analysis.

Comparison 15: Single medication compared with combination of medications, Outcome 5: Secondary: Anger at end of treatment (continuous outcome, MDs)
Bellino 2014 compared the mood stabiliser valproic acid to valproic acid plus omega‐3 fatty acids combined. There was no difference between valproic acid alone and valproic acid plus omega‐3 fatty acids regarding anger at end of treatment (MD 0.60, 95% CI −0.82 to 2.02; P = 0.41; 1 trial, 34 participants; Analysis 15.5).
3.6 Affective instability
One trial compared a medication with a combination of medications and reported continuous data on affective instability. Bellino 2014 compared the mood stabiliser valproic acid to valproic acid plus omega‐3 fatty acids combined. There was a difference between valproic acid alone and valproic acid plus omega‐3 fatty acids regarding affective instability at the end of treatment favouring valproic acid combined with omega‐3 fatty acids (MD 1.72, 95% CI 0.68 to 2.76; P = 0.001; 1 trial, 34 participants; Analysis 15.6)
15.6. Analysis.

Comparison 15: Single medication compared with combination of medications, Outcome 6: Secondary: Affective instability at end of treatment (continuous outcome, MDs)
3.7 Chronic feelings of emptiness
One trial compared a medication with a combination of medications and reported continuous data on chronic feelings of emptiness. Bellino 2014 compared the mood stabiliser valproic acid to valproic acid plus omega‐3 fatty acids combined. There was no difference between valproic acid alone and valproic acid plus omega‐3 fatty acids regarding anger at the end of treatment (MD 0.03, 95% CI −0.97 to 1.03; P = 0.95; 1 trial, 34 participants; Analysis 15.7).
15.7. Analysis.

Comparison 15: Single medication compared with combination of medications, Outcome 7: Secondary: Chronic feelings of emptiness at end of treatment (continuous outcome, MDs)
3.8 Impulsivity
Two trials compared a medication with a combination of medications and reported continuous data on impulsivity.
One trial (Zanarini 2004) compared the antipsychotic olanzapine to olanzapine plus the antidepressant fluoxetine combined. There was no difference between treatments regarding impulsivity at the end of treatment (MD 0.46, 95% CI −12.93 to 13.85; P = 0.95; 1 trial, 29 participants; Analysis 15.8). Zanarini 2004 also compared fluoxetine to fluoxetine plus olanzapine. There was no difference between the groups regarding impulsivity at the end of treatment (MD 4.77, 95% CI −9.67 to 19.21; P = 0.52; 1 trial, 26 participants; Analysis 15.8).
15.8. Analysis.

Comparison 15: Single medication compared with combination of medications, Outcome 8: Secondary: Impulsivity at end of treatment (continuous outcome, MDs)
Another trial (Bellino 2014), compared the mood stabiliser valproic acid to valproic acid plus omega‐3 fatty acids combined. There was a difference between valproic acid alone and valproic acid plus omega‐3 fatty acids regarding impulsivity at the end of treatment, favouring valproic acid plus omega‐3 fatty acids combined (MD 12.59, 95% CI 6.11 to 19.07; P = 0.0001; 1 trial, 34 participants; Analysis 15.8).
3.9 Interpersonal problems
One trial compared a medication with a combination of medications and reported continuous data on interpersonal problems. Bellino 2014 compared the mood stabiliser valproic acid to valproic acid plus omega‐3 fatty acids combined. There was no difference between valproic acid alone and valproic acid plus omega‐3 fatty acids regarding interpersonal problems at the end of treatment (MD 0.47, 95% CI −0.41 to 1.35; P = 0.29; 1 trial, 34 participants; Analysis 15.9).
15.9. Analysis.

Comparison 15: Single medication compared with combination of medications, Outcome 9: Secondary: Interpersonal problems at end of treatment (continuous outcome, MDs)
3.10 Abandonment
One trial compared a medication with a combination of medications and reported continuous data on fear of abandonment. Bellino 2014 compared the mood stabiliser valproic acid to valproic acid plus omega‐3 fatty acids combined. There was no difference between valproic acid alone and valproic acid plus omega‐3 fatty acids regarding fear of abandonment at the end of treatment (MD 0.01, 95% CI −0.83 to 0.85; P = 0.98; 1 trial, 34 participants; Analysis 15.10).
15.10. Analysis.

Comparison 15: Single medication compared with combination of medications, Outcome 10: Secondary: Abandonment at end of treatment (continuous outcome, MDs)
3.11 Identity disturbance
One trial compared a medication with a combination of medications and reported continuous data on identity disturbance. Bellino 2014 compared the mood stabiliser valproic acid to valproic acid plus omega‐3 fatty acids combined. There was no difference between valproic acid alone and valproic acid plus omega‐3 fatty acids regarding identity disturbances at the end of treatment (MD 0.70, 95% CI −0.34 to 1.74; P = 0.19; 1 trial, 34 participants; Analysis 15.11).
15.11. Analysis.

Comparison 15: Single medication compared with combination of medications, Outcome 11: Secondary: Identity disturbance at end of treatment (continuous outcome, MDs)
3.12 Dissociation and psychotic‐like symptoms
One trial compared a medication with a combination of medications and reported continuous data on dissociation and psychotic‐like symptoms. Bellino 2014 compared the mood stabiliser valproic acid to valproic acid plus omega‐3 fatty acids combined. There was no difference between valproic acid alone and valproic acid plus omega‐3 fatty acids regarding dissociation and psychotic‐like symptoms at the end of treatment (MD 0.03, 95% CI −1.09 to 1.15; P = 0.96; 1 trial, 34 participants; Analysis 15.12).
15.12. Analysis.

Comparison 15: Single medication compared with combination of medications, Outcome 12: Secondary: Dissociation and psychotic‐like symptoms at end of treatment (continuous outcome, MDs)
3.13 Depression
Two trials compared pharmacotherapies with combinations of other pharmacotherapies and reported continuous data on depression.
Zanarini 2004 compared the antipsychotic olanzapine to olanzapine plus the antidepressant fluoxetine. There was no difference between olanzapine alone and olanzapine plus fluoxetine regarding depression at the end of treatment (MD −1.78, 95% CI −6.48 to 2.92; P = 0.46; 1 trial, 29 participants; Analysis 15.13). Zanarini 2004 also compared fluoxetine to fluoxetine combined with olanzapine. There was no difference between fluoxetine alone and fluoxetine plus olanzapine regarding depression at the end of treatment (MD 3.62, 95% CI −1.36 to 8.60; P = 0.15; 1 trial, 25 participants; Analysis 15.13).
15.13. Analysis.

Comparison 15: Single medication compared with combination of medications, Outcome 13: Secondary: Depression at end of treatment (continuous outcome, MDs)
Bellino 2014 compared the mood stabiliser valproic acid to valproic acid plus omega‐3 fatty acids combined. There was no difference between valproic acid alone and valproic acid plus omega‐3 fatty acids regarding depression at end of treatment (MD 1.30, 95% CI 0.00 to 2.60; P = 0.05; 1 trial, 34 participants; Analysis 15.13).
3.14 Attrition
Two trials compared a medication with a combination of medications and reported dichotomous data on attrition.
One trial compared the antipsychotic olanzapine to olanzapine plus the antidepressant fluoxetine (Zanarini 2004). There was no difference between olanzapine alone and olanzapine combined with fluoxetine regarding attrition (RR 0.19, 95% CI 0.01 to 3.63; P = 0.27; 1 trial, 31 participants; Analysis 15.14). Zanarini 2004 also compared fluoxetine to fluoxetine plus olanzapine. There was no difference between fluoxetine alone and fluoxetine combined with olanzapine regarding attrition (RR 0.54, 95% CI 0.05 to 5.28; P = 0.59; 1 trial, 29 participants; Analysis 15.14).
15.14. Analysis.

Comparison 15: Single medication compared with combination of medications, Outcome 14: Secondary: Attrition at end of treatment (dichotomous outcomes, RRs)
One trial compared the mood stabiliser valproic acid to valproic acid plus omega‐3 fatty acids combined (Bellino 2014). There was no difference between valproic acid alone and valproic acid combined with omega‐3 fatty acids regarding attrition (RR 0.92, 95% CI 0.29 to 2.97; P = 0.89; 1 trial, 43 participants; Analysis 15.14).
3.15 Adverse events
No trials comparing pharmacotherapies to a combination of other pharmacotherapies reported data on total serious or non‐serious adverse events; non‐serious adverse events of the cardiovascular and respiratory system, the sensory system or the reproductive system; or any other non‐serious adverse events than those suggested in our analysis plan. Nor did they report data on withdrawal due to adverse events.
Central nervous system
Sedation
One trial comparing a medication with a combination of medications reported dichotomous data on sedation as a non‐serious adverse event. Zanarini 2004 compared the antipsychotic olanzapine to olanzapine plus the antidepressant fluoxetine. There was no difference in sedation between olanzapine alone and olanzapine plus fluoxetine combined (RR 1.61, 95% CI 0.87 to 2.96; P = 0.13; 1 trial, 31 participants; Analysis 16.1). Zanarini 2004 also compared fluoxetine to fluoxetine plus olanzapine. There was no difference between fluoxetine compared to fluoxetine plus olanzapine (RR 0.46, 95% CI 0.15 to 1.44; P = 0.18; 1 trial, 29 participants; Analysis 16.1).
16.1. Analysis.

Comparison 16: Single medication compared with combination of medications ‐ non‐serious adverse events ‐ central nervous system, Outcome 1: Sedation
Gastrointestinal system
Nausea
One trial comparing a medication with a combination of medications reported dichotomous data on nausea as a non‐serious adverse event. Bellino 2014 compared the mood stabiliser valproic acid to valproic acid plus omega‐3 fatty acids combined. There was no difference between valproic acid alone and valproic acid combined with omega‐3 fatty acids regarding nausea (RR 0.22, 95% CI 0.01 to 4.34; P = 0.32; 1 trial, 34 participants; Analysis 17.1).
17.1. Analysis.

Comparison 17: Single medication compared with combination of medications ‐ non‐serious adverse events ‐ gastro‐intestinal system, Outcome 1: Nausea
Dyspepsia
One trial comparing a medication with a combination of medications reported dichotomous data on dyspepsia as a non‐serious adverse event. Bellino 2014 compared the mood stabiliser valproic acid to valproic acid plus omega‐3 fatty acids combined. There was no difference between valproic acid alone and valproic acid combined with omega‐3 fatty acids regarding dyspepsia (RR 1.13, 95% CI 0.08 to 16.55; P = 0.44; 1 trial, 34 participants; Analysis 17.2).
17.2. Analysis.

Comparison 17: Single medication compared with combination of medications ‐ non‐serious adverse events ‐ gastro‐intestinal system, Outcome 2: Dyspepsia
Muscloskeletal system
Akathisia
One trial comparing a medication with a combination of medications reported dichotomous data on akathisia as a non‐serious adverse event. Zanarini 2004 compared the antipsychotic olanzapine to olanzapine plus the antidepressant fluoxetine. There was no difference in akathisia between olanzapine alone and olanzapine plus fluoxetine combined (RR 0.75, 95% CI 0.25 to 2.28; P = 0.61; 1 trial, 31 participants; Analysis 18.1). Zanarini 2004 also compared fluoxetine to fluoxetine plus olanzapine. There was no difference in akathisia between fluoxetine alone and the combination of fluoxetine and olanzapine (RR 1.07, 95% CI 0.39 to 2.92; P = 0.89; 1 trial, 29 participants; Analysis 18.1).
18.1. Analysis.

Comparison 18: Single medication compared with combination of medications ‐ musculoskeletal system, Outcome 1: Akathisia
Body weight change
Two trials comparing a medication with a combination of medications reported continuous data on body weight change as a non‐serious adverse event.
One trial compared the antipsychotic olanzapine to olanzapine plus the antidepressant fluoxetine (Zanarini 2004). There was a difference in body weight change between olanzapine alone and olanzapine plus fluoxetine combined, which favoured the combination of olanzapine and fluoxetine (MD 1.50, 95% CI 0.09 to 2.91; P = 0.04; 1 trial, 29 participants; Analysis 18.3). Zanarini 2004 also compared fluoxetine to fluoxetine plus olanzapine. There was no difference between fluoxetine alone and the combination of fluoxetine and olanzapine in body weight change (MD −1.00, 95% CI −2.39 to 0.39; P = 0.16; 1 trial, 26 participants; Analysis 18.3).
18.3. Analysis.

Comparison 18: Single medication compared with combination of medications ‐ musculoskeletal system, Outcome 3: Body weight change
Bellino 2014 compared the mood stabiliser valproic acid to valproic acid plus omega‐3 fatty acids combined. There was no difference between valproic acid alone and valproic acid plus omega‐3 fatty acids combined for change in body weight (RR 1.13, 95% CI 0.26 to 4.80; P = 0.87; 1 trial, 34 participants; Analysis 18.2).
18.2. Analysis.

Comparison 18: Single medication compared with combination of medications ‐ musculoskeletal system, Outcome 2: Body weight change
4. Subgroup analyses
We performed subgroup analyses on substances within each medication type (and type of antipsychotics). Further, we investigated if any subgroup differences between types of medication were present before pooling them into subgroup analyses on psychosocial functioning at baseline (mild, moderate, and seriously impaired), setting (inpatient, outpatient, and mixed setting), funding (publicly funded, and industry funded), trial size (≤ 50 participants, ≤ 99 participants, and > 99 participants) and method of recruitment (advertisement, and referral). There were no differences between antipsychotics, antidepressants, and mood stabilisers in the intervention effects of outcomes for which subgroup analyses were feasible: BPD severity (test for subgroup differences: Chi² = 1.42, df = 2 (P = 0.49), I² = 0%; Analysis 1.1), suicide‐related outcomes (test for subgroup differences: Chi² = 0.44, df = 2 (P = 0.80), I² = 0%; Analysis 1.5), psychosocial functioning (test for subgroup differences: Chi² = 1.48, df = 2 (P = 0.48), I² = 0%; Analysis 1.8), and anger (test for subgroup differences: Chi² = 6.50, df = 3 (P = 0.09), I² = 53.8%; Analysis 1.11).
4.1 Types of medication
4.1.1 Antipsychotics
4.1.1.1 First‐ versus second‐generation antipsychotics
Subgroup analyses were feasible for the primary outcomes of BPD severity, suicide‐related outcomes, and psychosocial functioning. The intervention effect varied according to subgroups of antipsychotics indicating inferiority of first‐generation antipsychotics (SMD 0.29, 95% CI −0.09 to 0.67; I2 = 0%; 3 trials, 108 participants) compared to second‐generation antipsychotics (SMD ‐0.36, 95% CI ‐0.66 to ‐0.05; I2 = 74%; 5 trials, 820 participants) regarding BPD severity (test for subgroup differences: Chi² = 6.74, df = 1 (P = 0.009), I² = 85.2%, Analysis 20.1). There was no indication that intervention effects varied according to subgroups of first‐ and second‐generation antipsychotics neither regarding suicide‐related outcomes (test for subgroup differences: Chi² = 0.24, df = 1 (P = 0.63), I² = 0%; Analysis 20.2), nor psychosocial functioning (test for subgroup differences: Chi² = 0.03, df = 1 (P = 0.86), I² = 0% Analysis 20.3).
20.1. Analysis.

Comparison 20: Subgroup analysis: types of medication, Outcome 1: BPD symptom severity ‐ antipsychotics vs placebo by class (1st vs 2nd generation)
20.2. Analysis.

Comparison 20: Subgroup analysis: types of medication, Outcome 2: Suicide‐related outcomes ‐ antipsychotics vs placebo by class (1st vs 2nd generation)
20.3. Analysis.

Comparison 20: Subgroup analysis: types of medication, Outcome 3: Psychosocial functioning ‐ antipsychotics vs placebo by class (1st vs 2nd generation)
4.1.1.2 Antipsychotics by substance
Subgroup analyses were feasible for the primary outcomes of BPD severity, suicide‐related outcomes, and psychosocial functioning.
For BPD severity, subgroups included one trial each (haloperidol, thiothixene, quetiapine, ziprasidone and brexpiprazole) except for olanzapine (two trials). The subgroup analysis revealed that the intervention effect varied according to type of substance (haloperidol; MD 3.95, 95% CI ‐2.66 to 10.56; 1 trial, 58 participants; olanzapine; SMD ‐0.15, 95% CI ‐0.41 to 0.10; I2 = 60%; 2 trials, 596 participants; thiothixene; MD 1.20, 95% CI ‐1.19 to 3.60; 1 trial, 50 participants; quetiapine; MD ‐0.70, 95% CI ‐2.80 to 1.40; 1 trial, 95 participants; ziprasidone; MD ‐0.42, 95% CI ‐0.87 to 0.03; 1 trial, 60 participants; brexpiprazole; MD ‐5.30, 95% CI ‐7.56 to ‐3.04; 1 trial, 69 participants). The test for subgroup differences indicated: Chi² = 19.72, df = 5 (P = 0.001), I² = 74.6%, Analysis 20.4.
20.4. Analysis.

Comparison 20: Subgroup analysis: types of medication, Outcome 4: BPD symptom severity ‐ antipsychotics vs placebo by substance
There was no indication that intervention effects varied according to subgroups of substances, olanzapine, ziprasidone, brexpiprazole and alprazolam, either regarding suicide‐related outcomes at end of treatment (test for subgroup differences: Chi² = 4.30, df = 3 (P = 0.23), I² = 30.2%; Analysis 20.6), or between subgroups of haloperidol, olanzapine, quetiapine and thiothixene regarding the outcome of psychosocial functioning (test for subgroup differences: Chi² = 1.05, df = 3 (P = 0.79), I² = 0%; Analysis 20.7).
20.6. Analysis.

Comparison 20: Subgroup analysis: types of medication, Outcome 6: Suicide‐related outcomes ‐ antipsychotics vs placebo by substance
20.7. Analysis.

Comparison 20: Subgroup analysis: types of medication, Outcome 7: Psychosocial functioning ‐ antipsychotics vs placebo by substance
4.1.2 Antidepressants
A subgroup analysis of class and substance of antidepressants was only feasible for the primary outcome of psychosocial functioning. There was no indication that the intervention effect on psychosocial functioning varied according to antidepressant class or substance for the subgroups tricyclic antidepressant (amitriptyline) and selective serotonin reuptake inhibitor (fluoxetine). The test for subgroup differences indicated: Chi² = 0.25, df = 1 (P = 0.61), I² = 0%; Analysis 20.8.
20.8. Analysis.

Comparison 20: Subgroup analysis: types of medication, Outcome 8: Psychosocial functioning ‐ antidepressants vs placebo by class/substance
4.1.3 Mood stabilisers
Data permitted a subgroup analysis only for the primary outcome of BPD severity. There was no indication that intervention effects varied according to mood stabiliser substances divalproex and lamotrigine (test for subgroup differences: Chi² = 0.01, df = 1 (P = 0.91), I² = 0%; Analysis 20.5).
20.5. Analysis.

Comparison 20: Subgroup analysis: types of medication, Outcome 5: BPD symptom severity ‐ mood stabiliser vs placebo by substance
4.2 Psychosocial functioning at baseline
No subgroup differences were observed for any primary outcome between cohorts of mildly, moderate or seriously impaired individuals (BPD severity: test for subgroup differences: Chi² = 1.65, df = 2 (P = 0.44), I² = 0%; Analysis 21.1; suicide‐related outcomes: test for subgroup differences: Chi² = 0.05, df = 1 (P = 0.82), I² = 0%; Analysis 21.2; or psychosocial functioning: test for subgroup differences: Chi² = 1.66, df = 2 (P = 0.44), I² = 0%; Analysis 21.3).
21.1. Analysis.

Comparison 21: Subgroup analysis: psychosocial functioning at baseline, Outcome 1: Primary: BPD symptom severity by severity of impairment at baseline
21.2. Analysis.

Comparison 21: Subgroup analysis: psychosocial functioning at baseline, Outcome 2: Primary: Suicide‐related outcomes by severity of impairment at baseline
21.3. Analysis.

Comparison 21: Subgroup analysis: psychosocial functioning at baseline, Outcome 3: Primary: Psychosocial functioning by severity of impairment at baseline
4.3 Setting
The majority of trials included individuals who underwent outpatient treatment. Eight trials were conducted in an inpatient setting, and four trials in mixed in‐ and outpatient settings (see Table 4). Subgroup analyses were feasible for the primary outcomes of BPD severity, suicide‐related outcomes and psychosocial functioning. No differences were observed between setting subgroups (BPD severity: test for subgroup differences: Chi² = 1.02, df = 2 (P = 0.60), I² = 0%; Analysis 22.1; suicide‐related outcomes: test for subgroup differences:Test for subgroup differences: Chi² = 0.55, df = 1 (P = 0.46), I² = 0%; Analysis 22.2; or psychosocial functioning: test for subgroup differences: Chi² = 0.01, df = 2 (P = 1.00), I² = 0%; Analysis 22.3).
22.1. Analysis.

Comparison 22: Subgroup analysis: setting, Outcome 1: BPD symptom severity by setting
22.2. Analysis.

Comparison 22: Subgroup analysis: setting, Outcome 2: Primary: Suicide‐related outcomes at end of treatment
22.3. Analysis.

Comparison 22: Subgroup analysis: setting, Outcome 3: Primary: Psychosocial functioning at end of treatment
4.4 Funding
Most trials (16 out of 45), were funded (full or partly) by the pharmaceutical industry. Ten were publicly funded, i.e. by grants from universities, authorities or research foundations, funding for 11 trials was unclear, and eight declared that no funding was received. For BPD severity, the intervention effect varied according to subgroups of funding with a seemingly higher effect of trials partially or fully funded by the pharmaceutical industry (SMD ‐0.34, 95% CI ‐0.61 to ‐0.08; I2 = 62%, 7 trials, 862 participants) compared to publicly funded trials (SMD 0.04, 95% CI ‐0.17 to 0.25; I2 = 0%, 4 trials, 348 participants) and trials with unclear funding (SMD 0.24, 95% CI ‐0.23 to 0.70; I2 = 0%, 2 trials, 73 participants). The test for subgroup differences indicated: Chi² = 7.00, df = 2 (P = 0.03), I² = 71.4%; Analysis 23.1. Intervention effects did not vary according to subgroups of funding sources for the outcome of psychosocial functioning (test for subgroup differences: Chi² = 0.23, df = 2 (P = 0.89), I² = 0%, Analysis 23.2).
23.1. Analysis.

Comparison 23: Subgroup analysis: funding, Outcome 1: BPD symptom severity by funding
23.2. Analysis.

Comparison 23: Subgroup analysis: funding, Outcome 2: Primary: Psychosocial functioning at end of treatment
4.5 Trial size
Intervention effects varied according to trial size for the outcome of anger with an equally superior effect from trials with 50 participants or fewer (SMD ‐0.67, 95% CI ‐1.00 to ‐0.34; I2 = 55%; 13 trials, 377 participants), and with 99 participants or fewer (SMD ‐0.67, 95% CI ‐1.14 to ‐0.20; I2 = 86%; 9 trials, 546 participants), compared to larger trials with 100 participants or more (SMD ‐0.25, 95% CI ‐0.41 to ‐0.09; I2 = 0%; 2 trials, 596 participants). The test for subgroup differences indicated: Chi² = 6.78, df = 2 (P = 0.03), I² = 70.5%; Analysis 24.2 (See Figure 13). Intervention effects did not vary for these groups regarding the outcome of psychosocial functioning (test for subgroup differences: Chi² = 2.59, df = 2 (P = 0.27), I² = 22.7%, Analysis 24.1) (See Figure 14 ).
24.2. Analysis.

Comparison 24: Subgroup analysis: trial size, Outcome 2: Secondary: Anger at end of treatment
13.

Funnel plot of comparison: 26 Subgroup analyses: trial size, outcome: 26.2 Secondary: Anger at end of treatment
SE(SMD): Standard error of the standardised mean difference; SMD: Standardised mean difference
24.1. Analysis.

Comparison 24: Subgroup analysis: trial size, Outcome 1: Primary: Psychosocial functioning at end of treatment
14.

Funnel plot of comparison: 26 Subgroup analyses: trial size, outcome: 26.1 Primary: Psychosocial functioning at end of treatment
SE(SMD): Standard error of the standardised mean difference; SMD: Standardised mean difference
4.6 Recruitment
Regarding the effect of antipsychotics on anger at the end of treatment, there were no subgroup differences in intervention effects between trials including participants through referrals or advertisements. The test for subgroup differences indicated: Chi² = 0.31, df = 1 (P = 0.58), I² = 0%; Analysis 25.1).
25.1. Analysis.

Comparison 25: Subgroup analysis: recruitment, Outcome 1: Antipsychotics ‐ anger at end of treatment
5. Sensitivity analyses
5.1 Imprecision
Where we were uncertain about the assessment of imprecision, as assessed by GRADE, we tested for it, by conducting TSAs on the primary and secondary outcomes (for the main comparison versus placebo).
In order to investigate the impact of imprecision, we also drew funnel plots for the primary and secondary outcomes involving the highest number of primary trials comparing active medications to placebo, i.e. psychosocial functioning and anger. Funnel plots revealed a clear trend of more precise estimates by larger trials, and indicated that smaller trials with unfavourable outcomes might be lacking, either due to publication bias or an overestimation of effects by smaller trials (Figure 13; Figure 14).
5.2 Cross‐over trials
We found no differences when adding end‐of‐period data from one cross‐over trial to the parallel‐group analyses (Cowdry 1988).
5.3 Decisions during the review process
5.3.1 Choice of data sources for Black 2014
For one trial, results were available from a journal publication (Black 2014a) as well as a trial registry entry (NCT00880919). We decided to use the data from the peer‐reviewed journal publication, whenever possible. However, for one outcome (Zan‐BPD total), data were available from both sources. While those reported in the journal article resulted in a small effect of SMD −0.14 (95% CI −0.58 to 0.29), the data from the trial registry entry would have yielded a very large, clearly outlying large effect of SMD −1.95 (95% CI −2.47 to −1.43). This finding reinforced our decision to primarily use data from peer‐reviewed sources, whenever possible.
5.3.2 Exclusion of outliers
For the secondary outcome of anger, heterogeneity was high (87%) due to several trials reporting outstandingly high effect estimates, as the visual inspection of the forest plot indicated. We decided to exclude the outliers one‐by‐one, until heterogeneity was low. Therefore, primary analyses resulted in an effect estimate of mood stabilisers compared to placebo of SMD −0.67 (95% CI −1.10 to −0.24; I2 = 26%; 5 trials, 135 participants), once Loew 2006, Nickel 2004 and Tritt 2005 had been excluded. If all outliers were included, the effect estimate would be more than twice as high, with SMD −1.39 (95% CI −2.16 to −0.61; I2 = 84%; 8 trials, 247 participants; Analysis 26.1).
The substantial reduction of high statistical heterogeneity from 84% to 27% by excluding those trials with outstanding, exceptionally high effect estimates and the bisection of the pooled effect supports our decision not to include the outliers in the primary analyses in order to avoid an overestimation of intervention effects. All these trials originate from the same working group and had been discussed for possibly biased results previously (NICE 2009, p. 217: "The GDG [guideline development group] [...] took the decision not to consider these trials when drawing up their conclusions."
5.4 Type of model used for analyses
We repeated all analyses of primary outcomes using the fixed‐effect model to test the robustness of results. The only relevant difference was that the CI of analysis 1.6.1 (psychosocial functioning, antipsychotics versus placebo) would no longer include zero (95% CI −0.30 to −0.03 instead of 95% CI −0.33 to 0.00).
Discussion
Summary of main results
Comparisons of active medications with placebo
We included 42 trials with a parallel design and 4 trials with a cross‐over design (Cowdry 1988; Schmahl 2012a; Schmahl 2012b; Ziegenhorn 2009) in this review. Altogether, these trials randomised 2769 participants, and were reported in 98 publications. The trials compared different types of antipsychotics, antidepressants, mood stabilisers, and miscellaneous medications to placebo, another active treatment or a combination of active treatments. The duration of the trials ranged from four to 52 weeks. The majority of the trials had a duration of three months or less and most of them took place in an outpatient setting. Nineteen trials were from Europe, 17 were from the USA, three trials were from Southwest Asia (the Middle East, all of them from Iran), one trial was from Australia, and two were multi‐country trials from nine transcontinental countries.
The mean age of the participants ranged from 16.2 to 39.7 years. Fourteen trials included women only and one trial included only men. All remaining trials included both sexes, but predominantly women. All included trials were assessed to be at high risk of bias overall.
The aim of this review was to determine the effects of pharmacological treatment for individuals with borderline personality disorder (BPD) of any age. We pooled the different types of antipsychotics, antidepressants and mood stabilisers into the respective medication classes.
Compared with placebo, antipsychotics may have little to no effect on the primary outcomes of BPD symptoms severity, self‐harm, suicide‐related outcomes and psychosocial functioning, or the secondary outcomes of attrition or total non‐serious adverse events. All primary outcomes are of very low certainty (see Table 1). Notably, all but one of the RCTs comparing olanzapine to placebo and reporting on suicidality‐related outcomes observed heightened suicidality in the olanzapine‐treated groups. The evidence is very uncertain about dropout rates and adverse events as compared to placebo.
Antidepressants, when compared to placebo, may result in little to no difference in the primary outcomes of BPD symptom severity, psychosocial functioning, self‐harm and suicide‐related outcomes, but the evidence is very uncertain. Antidepressants may have no effect on the secondary outcome of interpersonal problems (very low‐certainty evidence) or the dropout rate compared with placebo (low‐certainty evidence); see Table 2. Low‐certainty evidence suggests that mood stabilisers are not associated with heightened dropout rates. No data are available on non‐serious adverse events by antidepressants.
Mood stabilisers, compared with placebo, may result in little to no difference in the primary outcomes of BPD symptom severity, self‐harm, suicide‐related outcomes, and psychosocial functioning (all very low‐certainty evidence). Mood stabilisers may reduce interpersonal problems, but the evidence is uncertain again (low‐certainty). Low‐certainty evidence suggests that mood stabilisers are not associated with heightened dropout rates. Regarding non‐serious adverse events, the evidence is very uncertain (very low‐certainty evidence, see Table 3).
We also included trials with miscellaneous medications (i.e. those not classified as antipsychotics, antidepressants or mood stabilisers). With the exception of omega‐3 fatty acids, none of these drugs (the antidementia drug memantine hydrochloride, the opioid antagonist naltrexone, the antihypertensive alpha‐2A adrenergic agonist clonidine, or the opioid antagonist naltrexone) were observed to have an effect on any of the primary or secondary outcomes compared to placebo. Omega‐3 fatty acids might have an effect on suicide‐related outcomes compared to placebo. Furthermore, in the analysis of omega‐3 fatty acids on psychosocial functioning, there might be an increase in psychosocial function at the end of treatment in favour of placebo.
We did not find any differences in the presence of total adverse events between groups for any of the medications that reported adverse events. Only one trial reported total serious adverse events. Kulkarni 2018 compared memantine hydrochloride to placebo and reported no serious adverse events in the experimental or the placebo group (Analysis 1.29). No other trial specifically reported data on serious adverse events. However, we did find evidence of a difference between groups for some individual non‐serious adverse events; for example, antipsychotics on somnolence, increased appetite, dry mouth and body weight change; and mood stabilisers on gastrointestinal disorders. We also found evidence of a difference between groups on a range of laboratory values when comparing antipsychotics to placebo; for example, mean change in AST/SGOT and ALT/SGPT as well as LDL and HDL cholesterol. Please refer to the section, Effects of interventions, for a full overview of adverse events.
Head‐to‐head comparisons of active medications
Head‐to‐head comparisons of active medications compared antipsychotics to antidepressants (Jariani 2010; Soloff 1989; Soloff 1993; Zanarini 2004) or different antipsychotics (Bozzatello 2017; Leone 1982; Shafti 2010; Shafti 2014). When comparing the two antipsychotics, olanzapine and asenapine, there was a difference in effects on the secondary outcomes of anger and depression, favouring asenapine over olanzapine. No differences in effects were observed for any other comparison of any primary or secondary outcome. Regarding adverse events, the only difference between any two treatments was observed in the comparison of an antipsychotic (olanzapine) and an antidepressant (fluoxetine), where the comparison showed a higher risk for somnolence with olanzapine than with fluoxetine.
Comparison of active medications to combinations of medications
The combination of an antidepressant (fluoxetine) and an antipsychotic (olanzapine) was compared to each of its two individual components in one trial, with the evidence indicating little to no difference between single and combined treatment for any clinical outcome (Zanarini 2004). However, higher body weight gain was observed in those who were treated with olanzapine, compared to those who received olanzapine plus fluoxetine.
Another trial compared valproic acid alone against valproic acid plus omega‐3 fatty acids (Bellino 2014). Effects were observed for the combined treatment of valproic acid plus omega‐3 fatty acids for the primary outcomes of BPD severity and self‐harm, as well as for the secondary outcomes of affective instability and impulsivity.
Subgroup analyses
We conducted subgroup analyses on types of medication, substances, psychosocial functioning at baseline, setting, funding and trial size. The evidence indicated inferiority of first‐generation antipsychotics compared to second‐generation antipsychotics regarding the outcome of BPD severity. The intervention effect on BPD severity was also influenced by the type of substance with a seemingly superior effect of brexpiprazole; however, this analysis contained sparse data in each subgroup and for this reason caution should be given to the reliability of its results.
For BPD severity, the intervention effect also varied according to subgroups of funding, with a seemingly higher effect of trials partially or fully funded by the pharmaceutical industry.
For the outcome of anger, trial size affected the intervention effect, with an unsurprising superior effect of smaller trials (those with 50 participants or less and those with 99 participants of less) compared to larger trials (those with 100 or more participants). No other subgroup analyses showed a difference in intervention effects on the investigated outcomes.
Overall completeness and applicability of evidence
Participants
A third of the included trials consisted of female participants only, while just one trial solely included males. For the trials that did include both sexes, the majority of participants were female. This situation resembles the situation in clinical settings, where higher rates of BPD are found in women than men (Ellison 2018; Newton‐Howes 2021). However, evidence from representative community trials suggest that the true prevalence is even in men and women, though findings are mixed (Eaton 2018; Ellison 2018). It has been suggested that neurobiological differences between men and women may lead to different manifestations, with men tending to show more externalised patterns (explosive, aggressive, antisocial, disruptive, impulsive behaviour, novelty seeking) and different co‐occurring disorders (men: substance use, intermittent explosive disorder, paranoid, passive‐aggressive, narcissistic, sadistic, and antisocial PDs; women: eating, mood, anxiety, somatoform and post‐traumatic stress disorders, histrionic PD) (Bohus 2021; Neacsiu 2017; Newton‐Howes 2021). Therefore, the generalisability of the findings to men with BPD may be limited. Furthermore, clinical characteristics in men may be displayed differently than in women (e.g. with antisocial features; Mancke 2015; Sher 2019), and the under‐representation of male participants in the included trials could potentially have affected the effects of medications on some outcomes. All included trials took binary approaches to the gender variable, which restrains applicability to those that are gender diverse. Additionally, it is not uncommon for people affected by BPD to experience gender dysphoria (Neacsiu 2017).
Only one trial had a mean age below 18 years (Amminger 2013: 16.2 years); the remaining trials included adult populations. Six of these included populations with a mean age below 26 years (Bellino 2014; Bozzatello 2017; Loew 2006; Nickel 2006; Soloff 1989; Zanarini 2004). Therefore, around one‐sixth (16%) of all trials reporting the mean age of their samples were conducted in young people below 26 years old, and five‐sixths in mature adult populations. Taking into account the early onset of BPD symptoms and the symptom threshold for an official BPD diagnosis, these results may not be applicable for adolescent populations.
Generally, the impairment of psychosocial functioning at baseline was moderate to mild in most trials. Notably, individuals with actively suicidal behaviour were not included in around one third of all primary trials. Therefore, the applicability of findings to people with more severe BPD, highly impaired psychosocial functioning, or suicidal ideation and/or behaviour is limited.
Comorbid disorders and substance use dependence are highly prevalent in people with BPD. However, the applicability of findings to routine settings is limited, given that the majority of trials excluded individuals with concurrent major depression, bipolar affective disorders or psychotic disorders, as well as those with alcohol or substance abuse or dependence. Similarly, in many included trials, co‐medication was not allowed. However, clinically polypharmacy is common for many people with BPD and often for substantial periods of time.
Interventions
Antidepressants and antipsychotics are the most prevalent medications in inpatients with BPD (Bridler 2015). Both medications are prescribed to around 70% of individuals each (which also illustrates the high prevalence of polypharmacy in this group). According to Bridler 2015, the most frequently prescribed antidepressants are SSRIs. However, neither the corresponding number of trials nor the pooled findings of the trials support the high use of these medications. The only possible effect of (any) antidepressants compared to placebo was found for anger (small effect size: SMD −0.33, 95% CI −0.61 to −0.05; 5 trials, 199 participants; I2 = 0%). Confining this analysis to SSRIs alone, the effect estimate would be SMD −0.38 (95% CI −0.82 to 0.07; 3 trials, 80 participants; I2 = 0%). Among antipsychotics (and psychotropic medications in general), quetiapine is the medication most often used in BPD inpatients in Europe (Bridler 2015). In contrast, only one RCT evaluating the effects of quetiapine in individuals with BPD has ever been published in full (Black 2014), while others (e.g. AstraZeneca 2007) have been completed but not published (Stoffers 2015; Stoffers‐Winterling 2020). Therefore, the findings on quetiapine use in BPD are very uncertain.
Mood stabilisers are prescribed to one‐third (33%) of European BPD inpatients (Bridler 2015). In this review, we found moderate effects on two secondary outcomes (interpersonal problems and anger), but not on any of the primary outcomes. Another 30% receive tranquillisers, 30% benzodiazepines, and still another 16% hypnotics (Bridler 2015). We have only identified one RCT testing benzodiazepine medication (Cowdry 1988). This may be due to the eligibility criteria of this review, which focuses on continuous, not crisis medication.
Outcomes
The most commonly assessed primary outcomes were BPD severity and psychosocial functioning, and the most prevalently assessed secondary outcomes were anger and depression. The use of BPD‐specific assessment scales (e.g. Zan‐BPD, BPDSI‐IV, CGI‐BPD) has increased in the last decade, allowing for differentiated assessment of BPD symptoms. Oftentimes, if BPD‐specific outcome scales have been assessed, outcome reporting is limited to general BPD severity, while more specific reporting on individual BPD symptoms is not given. BPD‐specific criteria have been found, however, to be significant predictors of the long‐term course of the disorder, and should be considered more thoroughly. Specifically, identity disturbance, chronic feelings of emptiness, or frantic efforts to avoid abandonment have emerged as significant, independent factors associated with suicide attempts (Yen 2021), as they tend to persist, even after more impulsivity‐related symptoms such as impulsivity, self‐harm, or anger have diminished, and individuals no longer meet criteria for a BPD diagnosis. However, the suffering remains (Ng 2016), and psychosocial functioning, specifically vocational functioning, remains substantially impaired. Therefore, important outcomes were not considered by the majority of trials that reported only generic, BPD‐non‐specific psychiatric scales. Regarding individual outcomes, it is apparent that there are few data on the primary outcome of self‐harm: only seven out of 45 included RCTs reported on this outcome. Individuals with active self‐harm were often ineligible for study inclusion. However, these behaviours are highly prevalent in individuals with BPD (around 80% report suicidal and self‐harming behaviour; Soloff 2012), and they constitute important treatment outcomes. In addition, some of the evidence was indirect, coming from surrogate outcomes. For example, many of the included RCTs used the Overt Aggression Scale‐Modified (OAS‐M; Coccaro 2020) to measure anger; however, this instrument actually assesses impulsive‐aggressive behaviour. The SCL‐90‐R (Derogatis 1994) subscale 'anger/hostility' covers irritability, general mental imbalance and aggressiveness. The DSM, however, specifies the corresponding diagnostic criterion as "inappropriate, intense anger OR difficulty controlling anger" (APA 2013). The 'appropriateness' of anger and facets of self‐inflicted anger, therefore, are not adequately reflected by non‐BPD‐specific anger outcome scales.
This review is based on the idea of covering all DSM‐defined BPD criteria, as they represent the 'least common denominator' among the heterogeneous population of individuals with a diagnosis of BPD. Still, each of the nine criteria include diverse facets, and so a review like this requires the use of a somewhat 'broader lens' when synthesising study outcomes; thus, the pooled findings are always composite measures of individual scales measuring somewhat heterogeneous outcomes. Pooling the data in the way that we did seemed appropriate to us as it directly reflects symptoms covered by the DSM IV‐5 criterion 9. Notably, older trials, in the absence of BPD‐specific scales like the BPDSI‐IV or the Zan‐BPD, used assessment instruments that are usually used to measure psychotic symptoms, like the BPRS (Overall 1962) or the PANSS (Kay 1987), and somewhat exceed what is covered by the DSM criterion 9. However, recent research has found that psychotic phenomena in BPD actually equal those that people with 'traditional' psychotic disorders experience, as they are stress‐reactive, but more persisting and enduring than previously thought (e.g. Cavelti 2019; Cavelti 2021; Herpertz 2007; Thompson 2019). Future versions of this review should consider analysing these outcomes separately (see Appendix 7).
Observation periods in the included trials did not fully match clinical routines, where individuals with BPD usually take (several) medications for sustained periods of time. In 34 out of 46 included RCTs (74%), treatment effects were assessed after just three months or less.
Quality of the evidence
We rated the certainty of the evidence using the GRADE approach, giving considerations to each of the following: risk of bias, inconsistency, indirectness, imprecision and publication bias. The results of this assessment are provided in Table 1, Table 2, and Table 3. Mostly, the evidence was of very low certainty, with few exemptions of low‐certainty evidence.
We assessed the risk of bias in all trials in accordance with the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2019). The majority of trials were prone to selection bias as well as attrition, reporting bias, and vested interests. High risk of bias in randomised clinical trials has been shown to overestimate benefits and underestimate harms (Kjaergard 2001; Lundh 2012; Moher 1998; Savović 2012a; Savović 2012b; Schulz 1995; Wood 2008). We considered all trials to be at high risk of bias overall. However, we used all eligible trials in the meta‐analyses, as the Cochrane Handbook for Systematic Reviews of Interventions recommends doing when all trials are assigned the same risk of bias.
The results are based on 46 trials, 45 of which were eligible for quantitative analysis, and involved 2769 participants with BPD. Most trials included small sample sizes. The total number of included participants ranged from 13 to 451 in the individual trials.
We tested imprecision, assessed as part of the GRADE approach, by conducting TSAs on the primary outcomes and for some of the secondary outcomes (interpersonal problems, attrition and non‐serious adverse events) for the main comparison versus placebo where we were uncertain about the assessment of imprecision. The result of the TSAs did not change any imprecision rated by GRADE on any of the outcomes. In order to investigate the impact of imprecision, we also drew funnel plots for the primary and secondary outcomes involving the highest number of primary trials comparing active agents to placebo (i.e. psychosocial functioning and anger). Funnel plots revealed a clear trend of more precise estimates by larger trials, and indicated that smaller trials without statistically significant effects might be lacking, either due to publication bias or an overestimation of effects by smaller trials.
Potential biases in the review process
This review was conducted in a way where we tried to minimise the risk of potential biases. We developed a protocol, Stoffers‐Winterling 2018, for this review in accordance with the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2022). We conducted extensive searches of relevant databases. There were no restrictions on publication formats, publication periods or language. In order to assure a comprehensive selection of relevant RCTs, based on the pre‐defined eligibility criteria, guarantee a fair and consistent rating of bias, and collection of any relevant study results for use in the synthesis, any steps from study selection to rating of the overall quality of the evidence were done in duplicate by two reviewers each. All analyses had been pre‐defined (Stoffers‐Winterling 2018), and any methods that could not be used are reported in Appendix 8, with reasons why. We also went to a great effort to try and obtain unpublished data pertaining to BPD subsamples where the full sample included fewer than 70% of participants with full BPD. We were able to obtain such subsample data from some trials, but in other cases, trial authors did not respond. We do not know why they did not respond, and it is possible that the subsample data we received are biased in a positive way (i.e. being associated with desired findings). We included four cross‐over trials using end‐of‐period data as we did not have data from the first period. It is possible to use end‐of‐period data from cross‐over trials and to include these data as if they were parallel‐group data (Curtin 2002), but doing so might increase the risk of carry‐over effects and a unit of analysis error. We did, however, test this in a sensitivity analysis. Some might take issue with this, but we believe this was a sensible approach. We conducted Trial Sequential Analyses to control the risk of type I errors and to estimate how far we were from obtaining the DARIS to detect or reject a certain plausible intervention effect. Lastly, as we wished to investigate differences between new subgroups post hoc, we conducted some exploratory subgroup analyses where we pooled different classes of medication because of the limited data available. As this highly inflates the risk of bias in these analyses, we advise caution when interpreting these results; they serve mainly as exploratory analyses.
Agreements and disagreements with other studies or reviews
There are three other meta‐analyses of medications for BPD available. Two of them date back 10 years now (Ingenhoven 2011; Vita 2011) and one is from 2021 (Gartlehner 2021a).
Regarding the review by Ingenhoven 2011, which focused on the effects of antipsychotics in BPD, only one more placebo‐controlled RCT has become available since (Black 2014, on quetiapine). The findings presented by Ingenhoven 2011 and the findings of this review regarding the effects on BPD symptoms are similar; however, the evidence differs with regards to global functioning. Ingenhoven 2011 classified SCL‐90 GSI scores as measures of functioning, while we did not do so in this review. From our point of view, the SCL‐90 is a measure of general psychopathology, but does not detail how severely these symptoms actually impair a person´s functioning. Therefore, we did not replicate the finding of Ingenhoven 2011 of a small effect of antipsychotics on global functioning. However, we agree that the "wide and long‐term use of antipsychotics in these patients remains controversial" (Ingenhoven 2011, p. 489).
Vita 2011 investigated the effects of antipsychotics, antidepressants and mood stabilisers by means of a meta‐analysis of randomised controlled and open‐label trials. Outcomes were pooled into three domains; affective dysregulation, impulsive‐behavioural dyscontrol, and cognitive‐perceptual symptoms. Due to the inclusion of other study designs that are more prone to risk of bias, the pooling of measures into broader outcome categories and not excluding statistical outliers with SMD > 1.5, Vita 2011 found more favourable results, with small‐ to medium‐size effects of antipsychotics on all three outcome domains, and moderate effects (antidepressants) and very large effects (mood stabilisers) on affective dysregulation.
Gartlehner 2021a investigated the effects of various pharmacological treatments for BPD on the basis of RCT evidence. Their work was conducted for APA in connection to the development of clinical practice guidelines on treatment for BPD. Gartlehner and colleagues included data on nine different medications (second‐generation antipsychotics, anticonvulsants, and second‐generation antidepressants) from 21 RCTs. They concluded that the evidence does not support the use of these medications alone to reduce the severity of BPD. This conclusion by Gartlehner and colleagues is in agreement with the findings of this review; however, there are some methodological issues that we do not agree with (Gartlehner 2021b; Pereira Ribeiro 2021). Most prominent is the exclusion of studies with a duration less than eight weeks. Such an exclusion criterion effectively omits many trials that could have provided valuable information and does not, in return, provide insight on long‐term effects, especially considering that some patients with BPD continuously receive pharmacological treatment for their symptomatology for years.
Three more recent systematic reviews have been published that outline and evaluate the published and unpublished evidence from RCTs on this topic (Hancock‐Johnson 2017; Stoffers 2015; Stoffers‐Winterling 2020). All three have pointed to the dearth of trials and concluded that the evidence did not support the use of any medication for BPD treatment. However, none of these systematic reviews included quantitative meta‐analyses of outcomes.
This review is an update and replaces the previous versions (Binks 2006; Stoffers 2010). The current review includes 18 more RCTs compared to Stoffers 2010. The new findings, however, do not lead to substantially different conclusions, and the evidence is still of low and very low quality. The authors of the 2010 review concluded that there were some beneficial effects of antipsychotics, mood stabilisers and dietary supplementation by omega‐3 fatty acids for people with BPD, and that antidepressants may be helpful for comorbid conditions. However, they also concluded that none of these medications had an effect on total BPD severity, and that there seemed to be no promising results for the BPD core symptoms: chronic feelings of emptiness, identity disturbance and fear of abandonment. The results from this current review are in accordance with conclusions from Stoffers 2010.
Authors' conclusions
Implications for practice.
Conclusions about the usefulness of medications in the continued treatment of BPD require a trade‐off between estimated benefits and harms.
We assessed the beneficial and harmful effects of medications in the continued treatment of borderline personality disorder regarding the BPD symptoms, psychosocial functioning, depression, tolerability in terms of attrition, and adverse events. The evidence was very uncertain about the beneficial effects of antipsychotics, antidepressants and mood stabilisers for all primary outcomes of this review (borderline symptoms severity, self‐harm, suicide‐related outcomes, and psychosocial functioning). As regards secondary outcomes, the evidence suggested small (antipsychotics, antidepressants) to medium‐size effects (mood stabilisers, dietary supplementation with omega‐3 fatty acids) by medication, but these effects were of very low certainty and confined to few outcomes (anger: mood stabilisers, omega‐3 fatty acids, antipsychotics, antidepressants; interpersonal problems: mood stabilisers, antipsychotics; brief psychotic‐like symptoms and dissociative phenomena: antipsychotics, omega‐3 fatty acids). On the side of potential harms, adverse effects, especially serious events, were scarcely and unsystematically reported across included trials. The only exception was olanzapine, which had (among others) been tested in two large‐scale clinical studies intended for marketing authorisation (Schulz 2007; Zanarini 2007), and for which detailed marketing authorisation reports were available. For remaining medications, clinicians and consumers will need to consider the adverse effects of medications observed in other conditions.
As the evidence is very uncertain, the findings of this review do not allow for a trade‐off of benefits and harms of medications in BPD in general. Informed decisions whether to start or continue drug treatment must (as usual in practical evidence‐based medicine) consider the individual context of a person, the somatic condition (given substantially heightened rates of physical illness in BPD; Schneider 2019), and the treatment targets, values and preferences. Still, the person who is about to undergo medical treatment must be informed about the certainty of the evidence.
Implications for research.
The current review supports the continued understanding that no pharmacological therapy seems effective in specifically treating BPD pathology. As long as there is no clear evidence of superiority of any medication over placebo, head‐to‐head comparisons of active medications or combinations of medications do not add much to the current understanding of medication effects.
Placebo‐controlled trials of medication in combination with psychotherapeutic interventions could be interesting to investigate, to assess the potential additive effect of medication to psychotherapy or psychosocial interventions, given the encouraging evidence of the beneficial effects of psychotherapy (Andrews 2013a; Cristea 2017; NHMRC 2013; Oud 2017; Storebø 2020) and guidelines recommending disorder‐specific psychotherapies as first‐line treatment for BPD (NHMRC 2013; NICE 2009; NICE 2018). Outcomes should include BPD symptoms but also psychosocial functioning, which has been found to be severely affected in the long run. There is also a need to extend observation periods in order to rule out time effects and reflect the situation of individuals with BPD adequately, as many take medications for sustained periods of time (Zanarini 2015). BPD‐specific measurement scales should be used to assess the impact of treatments on individual BPD symptoms. Individuals with lived experience should also be involved in the design of new trials in order to consider outcomes that are of importance to those who are directly affected by BPD.
When future medication trials are planned, they should also assess effects on males, adolescents, and individuals with defined comorbid mental disorders. To arrive at better drug treatments, it is necessary to understand the pathophysiology of BPD in order to develop drugs specifically acting at crucial pathophysiological pathways. Given the urge to detect and treat BPD early (Chanen 2017), more trials in young populations, including adolescents, are needed. Also, treatment targets should be considered when defining inclusion criteria for pharmacologic intervention trials, given that BPD in itself is a very heterogeneous disorder; for example, if impulsivity is the main target of a medication, it should be tested in individuals with BPD plus pronounced such symptoms. In particular, trials investigating the effects of medications in samples with, for example, comorbid depression and substance use are lacking. Since comorbidity is highly prevalent in individuals with BPD, there is a clear need to enhance the applicability of findings at this point. At least, individuals with common comorbid disorders or symptoms such as aggressive or suicidal symptoms, which are core symptoms of BPD, should not be excluded from trial participation.
We need more high‐quality trials, at low risk of bias and with sufficient sample sizes (at least 200 participants) to investigate pharmacological treatment versus placebo for people with BPD. Such trials should aim to include adolescents, since only one study in this review assessed this age group. Lastly, future trials should be based on pre‐published protocols to combat the problem of publication bias.
There might be new evidence on the way in the future giving a larger evidence base as there are some large ongoing trials in process (See Characteristics of ongoing studies).
History
Protocol first published: Issue 2, 2018
Notes
This review supersedes a previously published review on this topic: Stoffers J, Völlm BA, Rücker G, Timmer A, Huband N, Lieb K. Pharmacological interventions for borderline personality disorder. Cochrane Database of Systematic Reviews 2010, Issue 6. Art. No.: CD005653. DOI: 10.1002/14651858.CD005653.pub2. The protocol of this current review was published in 2018 (Stoffers‐Winterling 2018).
Acknowledgements
We thank Signe Sofie Nielsen and Maja Lærke Kielsholm for their contribution wíth screening titles and abstracts of records. We thank the Cochrane Developmental, Psychosocial and Learning Problems (CDPLP) Editorial Team, Joanne Duffield (Managing Editor), Sarah Davies (Deputy Managing Editor), Margaret Anderson (Information Specialist), and Geraldine Macdonald (Coordinating Editor).
The CRG Editorial Team are grateful to the following peer reviewers for their time and comments: Dr Nicholas Bendit, Staff Specialist Psychiatrist, Hunter New England Area Health, Newcastle, Australia; Andrew Chanen, Orygen, Melbourne, Australia, and The University of Melbourne, Australia; David Alejandro Pérez Ferrara, Mexico; and Katharine J Nelson MD, Associate Professor, Department of Psychiatry & Behavioral Sciences, University of Minnesota Medical School, Minneapolis, MN, USA. They also thank Anne Lethaby for copyediting the review.
We thank professor Christian Gluud, Copenhagen Trial Unit, Centre for Clinical Intervention Research, The Capital Region, Copenhagen University Hospital ─ Rigshospitalet, Copenhagen, Denmark for his kind support with the TSA analyses.
Cochrane DPLP supported the authors in the development of this intervention review.
The following people conducted the editorial process for this article:
Sign‐off Editor (final editorial decision): Professor Geraldine Macdonald, Cochrane DPLP; University of Bristol, UK; Managing Editor (selected peer reviewers, collated peer‐reviewer comments, provided editorial guidance to authors, edited the article): Dr Joanne Duffield, Cochrane DPLP; Queen's University Belfast, NI; Deputy Managing Editor (conducted editorial policy checks, provided editorial guidance to authors, edited the article): Dr Sarah Davies, Cochrane DPLP; University of Bristol, UK; Copy Editor (copy editing and production): Anne Lethaby; Peer‐reviewers (provided comments and recommended an editorial decision): Dr Nicholas Bendit, Staff Specialist Psychiatrist, Hunter New England Area Health, Newcastle, Australia (clinical/content review); Andrew Chanen, Orygen, Melbourne, Australia, and The University of Melbourne, Australia (clinical/content review); David Alejandro Pérez Ferrara, Mexico (consumer review); Katharine J Nelson MD, Associate Professor, Department of Psychiatry & Behavioral Sciences, University of Minnesota Medical School, Minneapolis, MN, USA (clinical/content review); and Margaret Anderson, Cochrane DPLP; Queen's University Belfast, NI (search review). One additional peer reviewer provided statistical peer review, but chose not to be publicly acknowledged.
Appendices
Appendix 1. DSM diagnostic criteria for BPD (301.83)
|
DSM Third Edition (DSM‐III;APA 1980) 301.83 BPD |
DSM Fourth Edition Text Revision (DSM‐IV‐TR;APA 2000) 301.83 BPD |
DSM Fifth Edition (DSM‐5;APA 2013) 301.83 BPD |
|
Diagnostic criterion A 5 of the following are required: 1. Impulsivity or unpredictability in at least 2 areas that are potentially self‐damaging (e.g. spending, sex, substance use, shoplifting, overeating, physically self‐damaging acts) 2. A pattern of unstable and intense interpersonal relationships (e.g. marked shifts of attitude, idealisation, devaluation, manipulation (consistently using others for one's own ends)) 3. Inappropriate, intense anger or lack of control of anger (e.g. frequent displays of temper, constant anger) 4. Identity disturbance manifested by uncertainty about several issues relating to identity, such as self‐image, gender identity, long‐term goals or career choice, friendship patterns, values, and loyalties (e.g. 'Who am I', 'I feel like I am my sister when I am good') 5. Affective instability: marked shifts from normal mood to depression, irritability, or anxiety, usually lasting a few hours and only rarely more than a few days, with a return to normal mood 6. Intolerance of being alone (e.g. frantic efforts to avoid being alone, depressed when alone) 7. Physically self‐damaging acts (e.g. suicidal gestures, self‐mutilation, recurrent accidents or physical fights) 8. Chronic feelings of emptiness or boredom |
Diagnostic criterion A A pervasive pattern of instability of interpersonal relationships, self‐image, and affects, and marked impulsivity beginning by early adulthood and present in a variety of contexts, as indicated by 5 (or more) of the following: 1. Frantic efforts to avoid real or imagined abandonment (note: do not include suicidal or self‐mutilating behaviour covered in criterion 5) 2. A pattern of unstable and intense interpersonal relationships characterised by alternating between extremes of idealisation and devaluation 3. Identity disturbance: markedly and persistently unstable self‐image or sense of self 4. Impulsivity in at least 2 areas that are potentially self‐damaging (e.g. spending, sex, substance abuse, reckless driving, binge eating) (note: do not include suicidal or self‐mutilating behaviour covered in criterion 5) 5. Recurrent suicidal behaviour, gestures, or threats, or self‐mutilating behaviour 6. Affective instability due to a marked reactivity of mood (e.g. intense episodic dysphoria, instability, or anxiety usually lasting a few hours and only rarely more than a few days) 7. Chronic feelings of emptiness 8. Inappropriate, intense anger or difficulty controlling anger (e.g. frequent displays of temper, constant anger, recurrent physical fights) 9. Transient, stress‐related paranoid ideation or severe dissociative symptoms |
Diagnostic criterion A A pervasive pattern of instability of interpersonal relationships, self‐image, and affects, and marked impulsivity, beginning by early adulthood and present in a variety of contexts, as indicated by 5 (or more) of the following: 1. Frantic efforts to avoid real or imagined abandonment (note: do not include suicidal or self‐mutilating behaviour covered in criterion 5) 2. A pattern of unstable and intense interpersonal relationships characterised by alternating between extremes of idealisation and devaluation 3. Identity disturbance: markedly and persistently unstable self‐image or sense of self 4. Impulsivity in at least 2 areas that are potentially self‐damaging (e.g. spending, sex, substance abuse, reckless driving, binge eating) (note: do not include suicidal or self‐mutilating behaviour covered in criterion 5) 5. Recurrent suicidal behaviour, gestures or threats, or self‐mutilating behaviour 6. Affective instability due to a marked reactivity of mood (e.g. intense episodic dysphoria, irritability, or anxiety of mood) usually lasting a few hours and only rarely more than a few days 7. Chronic feelings of emptiness 8. Inappropriate, intense anger or difficulty controlling anger (e.g. frequent displays of temper, constant anger, recurrent physical fights) 9. Transient, stress‐related paranoid ideation or severe dissociative symptoms |
|
Diagnostic criterion B If under 18, does not meet the criteria for Identity Disorder |
||
| BPD: Borderline personality disorder; DSM: Diagnostic and Statistical Manual of Mental Disorders | ||
Appendix 2. ICD‐10 research criteria for emotionally unstable personality disorder (F60.3)
| F 60.30: ICD‐10 Emotionally unstable personality disorder, impulsive type | F 60.31: ICD‐10 Emotionally unstable personality disorder, borderline type | ICD‐11: 6D11.5 Borderline pattern |
|
Diagnostic criterion A The general criteria of personality disorder (F60) must be met |
Diagnostic criterion A The general criteria of personality disorder (F60) must be met |
General criteria for personality disorders + borderline pattern: pervasive pattern of instability of: ‐ interpersonal relationships, ‐ self‐image, and ‐ affects, and ‐ marked impulsivity, as indicated by many of the following 9 criteria: |
|
Diagnostic criterion B At least 3 of the following must be present, 1 of which is 2: 1. Marked tendency to act unexpectedly and without consideration of the consequences 2. Marked tendency to quarrelsome behaviour and to conflicts with others, especially when impulsive acts are thwarted or criticised 3. Liability of outbursts of anger or violence, with inability to control the resulting behavioural explosions 4. Difficulty in maintaining any course of action that offers no immediate reward 5. Unstable and capricious mood |
Diagnostic criterion B At least 3 of the symptoms mentioned in criterion B (F60.30) must be present, and in addition at least 2 of the following: 6. Disturbances in and uncertainty about self‐image, aims and internal preferences (including sexual) 7. Liability to become involved in intense and unstable relationships, often leading to emotional crises 8. Excessive efforts to avoid abandonment 9. Recurrent threats or acts of self‐harm 10. Chronic feelings of emptiness |
Borderline pattern:
|
| ICD‐10: International Classification of Diseases ‐ Tenth Edition | ||
Appendix 3. Neuroscience‐based nomenclature
| Substance/current nomenclature | Current nomenclature | ATC classification | Neuroscience‐based nomenclature I: pharmacology domain | Neuroscience‐based nomenclature II: mode of action | Side effects1 |
| Antidepressants | |||||
| Amitriptyline | antidepressant | psychoanaleptic ‐ antidepressant ‐ monoamine reuptake inhibitor, non‐selective | serotonin, norepinephrine | reuptake inhibitor, antagonist at serotonergic receptors | e.g. dry mouth, blurry vision, urinary hesitancy, constipation, orthostatic hypotension, sedation; toxic (potentially lethal) in overdosage |
| Fluoxetine | antidepressant | psychoanaleptic ‐ antidepressant ‐ selective serotonin reuptake inhibitor | serotonin | reuptake inhibitor | e.g. GI symptoms, anxiety, changes in sleep early in treatment, sexual dysfunction. No need for down titration upon discontinuation as has very long half‐life1 CAVE: Increased risk of drug‐drug interactions by inhibition of CYP2D6 enzymes |
| Fluvoxamine | antidepressant | psychoanaleptic ‐ antidepressant ‐ selective serotonin reuptake inhibitor | serotonin | reuptake inhibitor | e.g. GI symptoms, anxiety and/or changes in sleep early in treatment, sexual dysfunction. Must be gradually decreased on discontinuation1 CAVE: Increased risk of drug‐drug interactions by inhibition of CYP2D6 enzymes. |
| Mianserin | antidepressant | psychoanaleptic ‐ antidepressant ‐ other | serotonin, norepinephrine | multimodal, alpha2‐receptor antagonist | e.g. sedation, dizziness, dry mouth, rarely granulocytopenia or agranulocytosis1 |
| Phenelzine sulfate | antidepressant | psychoanaleptic ‐ antidepressant ‐ monoamine oxidase inhibitors, non‐selective | serotonin, norepinephrine, dopamine | enzyme (MAOI) inhibitor | e.g. high probability of producing orthostatic hypotension; foods containing tyramine must be avoided; must not be used with medications inhibiting 5‐HT reuptake1 |
| Sertraline | antidepressant | psychoanaleptic ‐ antidepressant ‐ selective serotonin reuptake inhibitor, non‐selective | serotonin | reuptake inhibitor | e.g. GI symptoms, anxiety, changes in sleep early in treatment, sexual dysfunction. Must be gradually decreased on discontinuation1 |
| Tranylcypromine sulfate | antidepressant | psychoanaleptic ‐ antidepressant ‐ monoamine oxidase reuptake inhibitor, non‐selective | serotonin, norepinephrine, dopamine | multimodal | e.g. high probability of producing orthostatic hypotension; foods containing tyramine must be avoided; must not be used with medications inhibiting 5‐HT reuptake |
| Antipsychotics | |||||
| Flupenthixol | antipsychotic, first‐generation | psycholeptic ‐ antipsychotic ‐ thioxanthene derivative | dopamine, serotonin | antagonist | e.g. EPS, galactorrhoea, sedation, dizziness, weight gain. Risk of tardive dyskinesia, NMS1 |
| Haloperidol | antipsychotic, first‐generation | psycholeptic ‐ antipsychotic ‐ butyrophenone derivative | dopamine | antagonist | e.g. EPS, galactorrhoea, sedation, dizziness, weight gain. Risk of tardive dyskinesia, NMS1 |
| Loxapine | antipsychotic, first‐generation | psycholeptic ‐ antipsychotic ‐ diazepines, oxazepines, thiazepines and oxepines | dopamine, serotonin | antagonist | EPS, galactorrhoea, sedation, dizziness, weight gain. Risk of tardive dyskinesia, NMS1 |
| Thiothixene2 | antipsychotic, first‐generation | psycholeptic ‐ antipsychotic ‐ thioxanthene derivates | dopamine, serotonin | antagonist | e.g. potentially severe cardiac side effects |
| Trifluoperazine | antipsychotic, first‐generation | psycholeptic ‐ antipsychotic ‐ phenothiazines with piperazine structure | dopamine, serotonin | antagonist | e.g. EPS (low), galactorrhoea, sedation, dizziness, weight gain. Risk of tardive dyskinesia, NMS |
| Aripiprazole | antipsychotic, second‐generation | psycholeptic ‐ antipsychotic ‐ other | dopamine, serotonin | partial agonist and antagonist | e.g. agitation, anxiety, insomnia, akathisia. Weight gain and risk of diabetes low but monitoring recommended as a class warning. Class warning for increased mortality in elderly dementia patients1 |
| Asenapine | antipsychotic, second‐generation | psycholeptic ‐ antipsychotic ‐ diazepines, oxazepines, thiazepines and oxepines | dopamine, serotonin, norepinephrine | antagonist | e.g. sedation, dizziness, weight gain, EPS, galactorrhoea. Risk of tardive dyskinesia, NMS. Risk of diabetes low but monitoring recommended as a class warning. Class warning for increased mortality in elderly dementia patients1 |
| Brexpiprazole | antipsychotic, second‐generation | psycholeptic ‐ antipsychotic ‐ other | dopamine, serotonin | partial agonist and antagonist |
e.g. akathisia (less than aripiprazole) weight gain and risk of diabetes low but monitoring recommended as a class warning. Class warning for increased mortality in elderly dementia patients1 |
| Olanzapine | antipsychotic, second‐generation | psycholeptic ‐ antipsychotic ‐ diazepines, oxazepines, thiazepines and oxepines | dopamine, serotonin | antagonist | e.g. weight gain, metabolic syndrome, EPS, sedation, galactorrhoea (low), dizziness, risk of tardive dyskinesia, NMS (low). Risk of diabetes; monitoring recommended as a class warning. Class warning for increased mortality in elderly dementia patients1 |
| Quetiapine | antipsychotic, second‐generation | psycholeptic ‐ antipsychotic ‐ diazepines, oxazepines, thiazepines and oxepines | dopamine, serotonin, norepinephrine | multimodal | e.g. sedation, dizziness, weight gain; galactorrhoea (low), EPS (low); Risk of tardive dyskinesia, NMS (low). Clearance reduced in elderly; Risk of diabetes; monitoring recommended as a class warning. Class warning for increased mortality in elderly dementia patients1 |
| Ziprasidone | antipsychotic, second‐generation | psycholeptic ‐ antipsychotic ‐ indole derivative | dopamine, serotonin | antagonist | e.g. EPS, galactorrhoea, sedation, dizziness, weight gain (low), QTc issues. Risk of tardive dyskinesia, NMS. Risk of diabetes low but monitoring recommended as a class warning. Class warning for increased mortality in elderly dementia patients1 |
| Mood stabilisers | |||||
| Carbamazepine | mood stabiliser/anticonvulsant | antiepileptic ‐ carboxamide derivative | glutamate | channel blocker | e.g. dizziness, somnolence, leukopenia1 CAVE: must not be used during pregnancy |
| Valproate | mood stabiliser/anticonvulsant | antiepileptic ‐ fatty acids derivative | glutamate | unclear | e.g. weight gain, sedation, elevated liver enzymes, hair loss1 CAVE: Must not be used by women of childbearing age without effective contraception |
| Lamotrigine | mood stabiliser/anticonvulsant | antiepileptic ‐ other | glutamate | channel blocker | e.g. dizziness, rash1 |
| Topiramate | mood stabiliser/anticonvulsant | antiepileptic ‐ other | GABA, glutamate | unclear | e.g. dizziness, weight loss, paraesthesiae, somnolence, nausea, diarrhoea, fatigue, depression. Rarely acute myopia and secondary angle closure glaucoma. Pregnancy category D (positive evidence of human foetal risk)1 |
| Miscellaneous | |||||
| Clonidine | antihypertensive | antihypertensive ‐ antiadrenergic agent, analgesic ‐ antimigraine preparation | norepinephrine | agonist | e.g. hypotension, somnolence, fatigue1 |
| Memantine hydrochloride | antidementia drug | psychoanaleptics ‐ antidementia drugs ‐ other | glutamate | antagonist (NMDA) | e.g. sleepiness, dizziness and balance problems, restlessness, nausea, other GI symptoms1 |
| Naltrexone | opioid antagonist | analgesics ‐ opioids ‐ natural opium alkaloid; other nervous system drugs ‐ drugs used in addictive disorders ‐ drugs used in alcohol dependence | opioid | antagonist | e.g. nonspecific GI symptoms, can cause liver damage in high doses1 |
| Omega‐3 fatty aids | miscellaneous omega‐3 fatty acids | cardiovascular system, lipid modifying agents ‐ other | n/a | n/a | e.g. fishy taste, eructation, dyspepsia, diarrhoea, gas, nausea, and arthralgia3 |
| Alprazolam | benzodiazepine | psycholeptic ‐ anxiolytic ‐ benzodiazepine derivative | GABA | Benzodiazepine receptor agonist (non selective GABA‐A receptor positive allosteric modulator‐PAM) | e.g. sedation, somnolence, ataxia, muscle relaxation, memory deficit |
|
1reference: nbn2r.com/ (accessed 21 November 2021) 2this drug has been withdrawn from the market by its manufacturer due to severe cardiac side effects. 3reference: Novotny K, Fritz K, Parmar M. Omega‐3 Fatty Acids. [Updated 2021 Oct 1]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2021 Jan. Available from: www.ncbi.nlm.nih.gov/books/NBK564314/ Abbreviations: ATC: Anatomical Therapeutic Chemical; EPS: extrapyramidal syndrome; GABA‐A: gamma‐aminobutyric acid‐A; GI: gastrointestinal; MAOI: monoaminoxidase inhibitors; NMS: neuroleptic malignant syndrome; PAM: positive allosteric modulator; QTc: corrected QT interval | |||||
Appendix 4. Search strategies
Cochrane Central Register of Controlled Trials, in the Cochrane Library
#1 MeSH descriptor: [Borderline Personality Disorder] explode all trees #2 borderline next state* #3 borderline next personalit* #4 "axis II" or "cluster B" #5 idealization next devaluation #6 (vulnerable or hyperbolic) next temper* #7 (((unstab* or instab* or poor or disturb* or fail* or weak* or dysregulat*) next (self* or impuls* or interperson* or identit* or relation* or emotion* or affect*)) and (person* or character or PD)) #8 impulsiv* near personalit* #9 (self next (injur* or damag* or destruct* or harm* or hurt* or mutilat*)) #10 suicidal next behavio?r #11 (feel* next (empt* or bored*)) #12 (anger next control*) #13 (risk‐taking next (behavior or behaviour)) #14 #1 or #2 or #3 or #4 or #5 or #6 or #7 or #8 or #9 or #10 or #11 or #12 or #13
MEDLINE Ovid
1 Borderline Personality Disorder/ 2 ((borderline or border‐line) adj3 (state* or personalit*)).kf,tw. 3 ("Axis II" or "Cluster B" or flamboyant or "F60.3" or "F60.30" or "F60.31").kf,tw. 4 (idealization adj5 devaluation).kf,tw. 5 ((vulnerable or hyperbolic) adj3 temperament).kf,tw. 6 (((unstab* or instab* or poor or disturb* or fail* or weak or dysregulat*) adj3 (self* or impuls* or interperson* or identit* or relationship* or emotion* or affect*)) and (personality or character or PD)).kf,tw. 7 (impulsiv* adj5 (behavio?r or character or personalit*)).kf,tw. 8 (self adj3 (injur* or damag* or destruct* or harm* or hurt* or mutilat*)).kf,tw. 9 (suicidal adj3 behavio?r).kf,tw. 10 (feel* adj3 (empt* or bored*)).kf,tw. 11 (anger adj5 control*).kf,tw. 12 (risk‐taking adj3 behavio?r).kf,tw. 13 or/1‐12 14 randomised controlled trial.pt. 15 controlled clinical trial.pt. 16 randomi#ed.ab. 17 placebo.ab. 18 randomly.ab. 19 trial.ab. 20 groups.ab. 21 drug therapy.fs. 22 or/14‐21 23 exp Animals/ not Humans/ 24 22 not 23 25 13 and 24
Embase Ovid
1 borderline state/ 2 ((borderline or border‐line) adj3 (personalit* or state*)).kw,tw. 3 ("Axis II" or "Cluster B" or flamboyant or "F60.3" or "F60.30" or "F60.31").kw,tw.) 4 (idealization adj5 devaluation).kw,tw. 5 ((vulnerable or hyberbolic) adj3 temperament).kw,tw. 6 (((unstab* or instab* or poor or disturb* or fail* or weak or dysregulat*) adj3 (self* or impuls* or interperson* or identit* or relationship* or emotion* or affect*)) and (personality or character or PD)).kw,tw. 7 (impulsiv* adj5 (behavio?r or character or personalit*)).kw,tw. 8 (self adj3 (injur* or damag* or destruct* or harm* or hurt* or mutilat*)).kw,tw. 9 (suicidal adj3 (behavior or behaviour)).kw,tw. 10 (feel* adj3 (empt* or bored*)).kw,tw. 11 "anger adj5 control*".kw,tw. 12 (risk‐taking adj3 (behavior or behaviour)).kw,tw. 13 or/1‐12 14 randomised controlled trial/ 15 double blind procedure/ 16 crossover procedure/ 17 single blind procedure/ 18 (random* or factorial* or crossover* or cross‐over* or placebo* or double‐blind* or doubleblind* or single‐blind* or singleblind* or assign* or allocat* or volunteer*).ab,pt,sh,de,ti. 19 or/14‐18 20 13 and 19
CINAHL EBSCOhost (Cumulative Index to Nursing and Allied Health Literature)
S1 (MH "Borderline Personality Disorder") S2 TX borderline N3 (state* or personalit*) S3 TX "Axis II" OR "Cluster B" S4 TX idealization N3 devaluation S5 TX ((vulnerable OR hyperbolic) N3 temperament) S6 TX (((unstab* or instab* or poor or disturb* or fail* or weak or dysregulat*) N3 (self or impuls* or interperson* or identit* or relationship* or emotion* or affect*)) AND (person* or character or PD)) S7 TX (impulsiv* N3 (behavio?r OR character or personalit*)) S8 TX (feel* N3 (empt* OR bored*)) S9 S1 OR S2 OR S3 OR S4 OR S5 OR S6 OR S7 OR S8 S10 (MH "randomised Controlled Trials") OR (MH "Random Assignment") OR (MH "Random Sample+") S11 TX random* N4 (trial* OR study OR studies) S12 TX random* N4 (allocat* OR allot* OR assign* OR basis OR divid* OR order) S13 AB placebo* S14 AB trial S15 (MH "Drug Therapy+") S16 S10 OR S11 OR S12 OR S13 OR S14 OR S15 S17 S9 AND S16
PsycINFO Ovid
1 exp Borderline Personality Disorder/ 2 borderline adj3 (personalit* or state*).id,ti,ab. 3 ("Axis II" or "Cluster B").id,ti,ab. 4 (idealization adj5 devaluation).ab,id,ti. 5 ((vulnerable or hyperbolic) adj3 temperament).id,ab,ti. 6 (((unstab* or instab* or poor or disturb* or fail* or weak or dysregulat*) adj3 (self* or impuls* or interperson* or identit* or relationship* or emotion* or affect*)) and (personality or character or PD)).id,ab,ti. 7 (impulsiv* adj5 (behavio?r or character or personalit*)).id,ab,ti. 8 (self adj3 (injur* or damag* or destruct* or harm* or hurt* or mutilat*)).id,ab,ti. 9 (suicidal adj3 behavio?r).id,ab,ti. 10 (feel* adj3 (empt* or bored*)).ab,id,ti. 11 "anger adj5 control*".ab,id,ti. 12 (risk‐taking adj3 behavio?r).id,ab,ti. 13 or/1‐12 14 exp Clinical Trials/ ( 15 (random* adj allocat*).ab. 16 randomi?ed.ab. 17 placebo.ab. 18 randomly.ab. 19 trial.ab. 20 groups.ab. 21 drug therapy.sh. 22 exp Animals/ not Humans/ 23 or/14‐21 24 23 not 22 25 13 and 24
ERIC EBSCOhost (Education Resources Information Center)
S23 S11 OR S12 OR S13 OR S14 OR S15 OR S16 OR S17 OR S18 OR S19 OR S20 OR S21 OR S22 S22 TX risk‐taking N5 behaviour S21 TX anger N5 control* S20 TX feel* N3 (empt* or bored*) S19 TX suicidal N3 behavior S18 TX ( (unstab* or instab* or poor or disturb* or fail* or weak or dysregulat*) N3 TX (self or impuls* or interperson* or identit* or relationship* or emotion* or affect*)) AND TX ( personality OR character OR PD ) S17 AB (self AND (injur* or damag* or destruct* or harm or hurt* or mutilat*)) S16 AB impulsivity S15 TI impulsivity S14 TX impulsiv* N3 person* S13 TX "Axis II" OR "Cluster B" S12 TX borderline N3 state S11 TX borderline personality S10 S1 OR S2 OR S3 OR S4 OR S5 OR S6 OR S7 OR S8 OR S9 S9 AB drug S8 AB trial S7 AB randomly S6 AB placebo S5 AB randomi?ed S4 AB controlled clinical trial* S3 SU controlled clinical trial S2 TX controlled clinical trial S1 DE "randomised Controlled Trials"
BIOSIS Previews Web of Science Clarivate Analytics
#1 TOPIC: (borderline personality disorder) #2 TOPIC: ((borderline NEAR/3 (state)) #3 TOPIC: ((borderline NEAR/3 personalit*)) #4 TOPIC: (("Axis II" OR "Cluster B")) #5 TOPIC: (idealization NEAR/5 devaluation) #6 TOPIC: ((vulnerable OR hyperbolic) NEAR/3 temperament*) #7 TOPIC: (impulsiv* NEAR/5 personalit*) #8 TOPIC: ((self NEAR/3 (injur* OR damag* OR destruct* OR harm* OR hurt* OR mutilat*))) #9 TOPIC: ((((unstab* OR instab* OR poor OR disturb* OR fail* OR weak OR dysregulat*) NEAR/3 (self* OR impuls* OR interperson* OR identit* OR relationship* OR emotion* OR affect*)) AND (personality OR character OR PD))) #10 TOPIC: (suicidal NEAR/3 behavio?r) #11 TOPIC: (((feel* NEAR/3 (empt* OR bored*)))) #12 TOPIC: ((anger NEAR/5 control*)) #13 TOPIC: (risk‐taking NEAR/3 behavio?r) #14 #13 OR #12 OR #11 OR #10 OR #9 OR #8 OR #7 OR #6 OR #5 OR #4 OR #3 OR #2 OR #1 #15 TOPIC: (controlled clinical trial) #16 TOPIC: (randomised controlled trial) #17 #16 OR #15 #18 #17 AND #14
Science Citation Index and Social Science Citation Index Web of Science Clarivate Analytics
#18 #17 AND #14 #17 #16 OR #15 #16 TOPIC: (controlled clinical trial) #15 TOPIC: (randomised controlled trial) #14 #13 OR #12 OR #11 OR #10 OR #9 OR #8 OR #7 OR #6 OR #5 OR #4 OR #3 OR #2 OR #1 #13 TITLE: ((risk‐taking NEAR/3 behavio?r)) #12 TOPIC: ((risk‐taking NEAR/3 behavio?r)) #11 TITLE: ((anger NEAR/5 control*)) #10 TOPIC: ((feel* NEAR/3 (empt* OR bored*))) #9 TITLE: ((feel* NEAR/3 (empt* OR bored*))) #8 TITLE: (suicidal NEAR/3 behavio?r) #7 TITLE: (impulsivity) #6 TOPIC: ((((unstab* OR instab* OR poor OR disturb* OR fail* OR weak OR dysregulat*) NEAR/3 (self* OR impuls* OR interperson* OR identit* OR relationship* OR emotion* OR affect*)) AND (personality OR character OR PD))) #5 TOPIC: ((vulnerable or hyperbolic) NEAR/3 temperament) #4 TOPIC: ((idealization NEAR/5 devaluation)) #3 TOPIC: ("axis II" OR "Cluster B") #2 TOPIC: (borderline NEAR/3 state) #1 TOPIC: (borderline personality disorder)
Sociological Abstracts ProQuest
(((randomised controlled trial) OR (controlled clinical trial) OR SU.exact("CLINICAL TRIALS")) OR AB(randomi?ed) OR AB(randomly) OR AB(placebo) OR AB(trial)) AND ((borderline personality) OR "axis II" OR "Cluster B" OR (idealization AND devaluation) OR ((vulnerable OR hyperbolic) AND temperament) OR (((unstab* OR instab* or poor or disturb* or fail* or weak or dysregulat*) AND (self* or impuls* or interperson* or identit* or relationship* or emotion* or affect*)) AND (personality OR character OR PD)) OR (self AND (injur* OR damag* OR destruct* OR harm OR hurt* OR mutilat*)) OR "suicidal behavio?r" OR "self destructive behavio?r" OR (feel* AND (empt* OR bored*)))
LILACS (Latin American and Caribbean Health Science Information Database)
“Borderline personality disorder”, limits: Controlled clinical study
Library Hub Discover (previously COPAC)
“Borderline personality disorder”
ProQuest Dissertations A&I
(SU(borderline personality disorder) OR AB("Axis II") OR AB("Cluster B")) AND (("randomised controlled study" OR "controlled clinical study") OR AB(randomi?ed) OR AB(placebo) OR AB(randomly))
OpenGrey
“Borderline personality disorder”
DART Europe E‐theses portal
“Borderline personality disorder”
Networked Digital Library of Theses and Dissertations (NDLTD)
“Borderline personality disorder”
Australian New Zealand Clinical Trials Registry (ANZCTR)
“Borderline personality disorder”
ClinicalTrials.gov
“Borderline personality disorder”
EU Clinical Trials Register
“Borderline personality disorder”
ISRCTN Registry
“Borderline personality disorder”
UK Clinical Trials Gateway
“Borderline personality disorder”
WHO International Clinical Trials Registry Platform
“Borderline personality disorder”
US Food and Drugs Administration FDA
“Borderline personality disorder”
European Medicines Agency EMA
“Borderline personality disorder”
Appendix 5. Outcomes
Primary outcomes
1. BPD severity
| Name of scale/means of assessment | Abbreviation | Clinician‐rated (CR)/self‐rated (SR) | Study |
| Adapted Clinical Global Impression ‐ Improvement scale | ‐ | Unclear | Cowdry 1988 |
| Borderline Evaluation of Severity over Time | BEST | SR | Grant 2022; Moen 2012 |
| Borderline Personality Disorder Severity Index | BPDSI‐IV | CR | Bellino 2014; Bozzatello 2017 |
| Borderline Syndrome Index | BSI | SR | Soloff 1993 |
| Borderline Symptom List | BSL | Unclear | Ziegenhorn 2009 |
| Borderline Symptom List 95 | BSL‐95 | Unclear | Schmahl 2012b |
| Clinical Global Impression scale for Borderline Personality Disorder patients | CGI‐BPD‐Global | Unclear | Pascual 2008 |
| Schedule of Interviewing Schizotypal Personalities ‐ Borderline score | SIB‐Borderline score | CR | Goldberg 1986 |
| Zanarini Rating Scale for Borderline Personality Disorder | ZAN‐BPD | CR | Black 2014; Crawford 2018; Grant 2022; Kulkarni 2018; Reich 2009; Schulz 2007; Zanarini 2007 |
2. Self‐harm
| Name of scale/means of assessment | Abbreviation | Clinician‐rated (CR)/self‐rated (SR) | Study |
| Borderline Personality Disorder Symptom Index | BPDSI‐IV‐Parasuicidal behaviour | CR | Bozzatello 2017 |
| Deliberate Self‐harm Inventory | DSHI | CR | Crawford 2018 |
| Overt Aggression Scale Modified ‐ Auto aggression | OAS‐M‐Auto aggression | Unclear | Simpson 2004 |
| Retrospective reporting | ‐ | ‐ | Schmahl 2012b |
| Self‐Harm Inventory | SHI | SR | Bellino 2014 |
| Spontaneous reporting | ‐ | ‐ | Grant 2022: Hallahan 2007; Linehan 2008; Nickel 2006 |
3. Suicide‐related outcomes
| Name of scale/means of assessment | Abbreviation | Clinician‐rated (CR)/self‐rated (SR) | Study |
| Adapted Clinical Global Impression ‐ Improvement scale | ‐ | Unclear | Cowdry 1988 |
| Behavioural reports of numbers of episodes of self‐injuring behaviour/suicide attempts | ‐ | ‐ | Soler 2005 |
| Borderline Personality Disorder Severity Index | BPDSI‐Parasuicidal behaviours | CR | Bellino 2014 |
| Clinical Global Impression scale for Borderline Personality Disorder patients | CGI‐BPD | CR | Pascual 2008 |
| Clinical Global Impression scale for Borderline Personality Disorder patients | CGI‐BPD‐Recurrent suicidal ideation | CR | Bogenschutz 2004; Pascual 2008 |
| Columbia Suicide Severity Rating Scale | C‐SSRS | CR | Grant 2022 |
| Overt Aggression Scale Modified ‐ Suicidal ideation | OAS‐M‐Suicidal ideation | CR | Schulz 2007; Zanarini 2007 |
| Overt Aggression Scale Modified ‐ Suicidality | OAS‐M‐Suicidality | CR | Hallahan 2007; Hollander 2001; Linehan 2008; Simpson 2004 |
| Spontaneous reporting | ‐ | ‐ | Montgomery 1982a; Montgomery 1982b |
| Zanarini Rating Scale for Borderline Personality Disorder | ZAN‐BPD‐Suicidal or self‐mutilating behaviour | CR | Schulz 2007; Zanarini 2007 |
4. Psychosocial functioning
| Name of scale/means of assessment | Abbreviation | Clinician‐rated (CR)/self‐rated (SR) | Study |
| Adapted Clinical Global Impression ‐ Improvement scale | ‐ | Unclear | Cowdry 1988 |
| Clinical Global Impression Scale ‐ Severity1 | CGI‐S | CR | Bellino 2014; Bogenschutz 2004; Bozzatello 2017; Shafti 2010; Shafti 2014; Soler 2005 |
| Clinical Global Impression Scale ‐ Improvement1 | CGI‐I | CR | Hollander 2001 |
| Global Assessment of Functioning Scale2 | GAF | CR | Amminger 2013; Black 2014; Schulz 2007; Simpson 2004; Zanarini 2001; Zanarini 2007 |
| Global Assessment Scale2 | GAS | CR | De la Fuente 1994; Goldberg 1986; Markovitz 1995a; Salzman 1995; Soloff 1989; Soloff 1993 |
| Sheehan Disability Scale1 | SDS | SR | Grant 2022; Schulz 2007; Zanarini 2007 |
| Social Functioning Questionnaire1 | SFQ | SR | Crawford 2018 |
| Systematic Nurses' Observation of Psychopathology1 | SNOOP | CR | Leone 1982 |
1For the following scales, higher scores indicate a worse situation: CGI‐I, CGI‐S, SDS, SFQ, SNOOP
2 For the following scales, lower scores indicate a worse situation: GAF, GAS. To fit effect sizes of the remaining pathology outcomes, where negative effect sizes indicate an amelioration, the scores of these scales were multiplied by (‐1), meaning that negative effect estimates indicate beneficial effects.
Secondary outcomes
1. Anger
| Name of scale/means of assessment | Abbreviation | Clinician‐rated (CR)/self‐rated (SR) | Study |
| Adapted Clinical Global Impression ‐ Improvement scale | ‐ | Unclear | Cowdry 1988 |
| Anger, Irritability, and Assault Questionnaire | AIAQ | Unclear | Bogenschutz 2004 |
| Borderline Personality Disorder Severity Index ‐ Anger | BPDSI‐Anger | CR | Bellino 2014; Bozzatello 2017; Rinne 2002 |
| Buss‐Durkee Hostility Inventory | BDHI | Unclear | Pascual 2008; Soloff 1989; Soloff 1993; Shafti 2014; Shafti 2010 |
| Clinical Global Impression scale for Borderline Personality Disorder patients ‐ Inappropriate anger | CGI‐BPD‐Inappropriate anger | CR | Bogenschutz 2004; Pascual 2008 |
| Modified Overt Aggression Scale | OAS‐M | CR | Bellino 2014; Black 2014; Zanarini 2003 |
| Modified Bunney‐Hamburg rating scale | ‐ | Unclear | Cowdry 1988 |
| Overt Aggression Scale Modified ‐ Total | OAS‐M‐Total | SR | Hallahan 2007 |
| Overt Aggression Scale Modified ‐ Aggression | OAS‐M‐Aggression | SR | Hollander 2001; Simpson 2004 |
| Overt Aggression Scale Modified ‐ Anger against objects | OAS‐M‐Anger against objects | SR | Salzman 1995 |
| Overt Aggression Scale Modified ‐ Irritability | OAS‐M‐Irritability | SR | Schulz 2007; Zanarini 2007 |
| Personality Disorder Rating Scale ‐ Anger | PDRS‐Anger | CR | Salzman 1995 |
| Spielberger State‐Trait Anger Expression Inventory ‐ Anger | STAXI‐trait‐Anger | SR | Nickel 2004; Nickel 2005; Nickel 2006; Tritt 2005 |
| Symptom Check List‐90 ‐ Hostility | SCL‐90‐HOS | SR | De la Fuente 1994; Frankenburg 2002; Goldberg 1986; Jariani 2010; Loew 2006; Nickel 2006; Soloff 1993; Soloff 1989, Zanarini 2001 |
| Zanarini Rating Scale for Borderline Personality Disorder ‐ Intense anger | ZAN‐BPD‐Intense anger | CR | Schulz 2007; Zanarini 2007 |
2. Affective instability
| Name of scale/means of assessment | Abbreviation | Clinician‐rated (CR)/self‐rated (SR) | Study |
| Affective Lability Scale | ALS | SR | Reich 2009 |
| Borderline Personality Disorder Severity Index ‐ Rapid mood shifts | BPDSI‐Rapid mood shifts | CR | Rinne 2002 |
| Borderline Personality Disorder Severity Index ‐ Affective Instability | BPDSI‐Affective instability | CR | Bellino 2014; Bozzatello 2017 |
| Clinical Global Impression scale for Borderline Personality Disorder patients ‐ Affective instability | CGI‐BPD‐Affective instability | CR | Bogenschutz 2004; Pascual 2008 |
| Profile of Mood States Scale | POMS | SR | Leone 1982 |
| Zanarini Rating Scale for Borderline Personality Disorder ‐ Affective disturbance | ZAN‐BPD‐Affective disturbance | CR | Crawford 2018 |
| Zanarini Rating Scale for Borderline Personality Disorder ‐ Affective instability Score | ZAN‐BPD‐Affective instability score | CR | Reich 2009; Schulz 2007; Zanarini 2007 |
3. Chronic feeling of emptiness
| Name of scale/means of assessment | Abbreviation | Clinician‐rated (CR)/self‐rated (SR) | Study |
| Borderline Personality Disorder Severity index ‐ Affective Instability | BPDSI‐IV‐Chronic feelings of emptiness | CR | Bellino 2014; Bozzatello 2017 |
| Clinical Global Impression scale for Borderline Personality Disorder patients ‐ Chronic feelings of emptiness | CGI‐BPD‐Chronic feelings of emptiness | CR | Bogenschutz 2004; Pascual 2008 |
| Zanarini Rating Scale for Borderline Personality Disorder ‐ Chronic feeling of emptiness | ZAN‐BPD‐Chronic feelings of emptiness | CR | Schulz 2007; Zanarini 2007 |
4. Impulsivity
| Name of scale/means of assessment | Abbreviation | Clinician‐rated (CR)/self‐rated (SR) | Study |
| Adapted Clinical Global Impression ‐ Improvement scale | ‐ | Unclear | Cowdry 1988 |
| Acting‐Out Scale | AOS | CR | De la Fuente 1994 |
| Barrett Impulsiveness Scale | BIS | CR, SR | Bellino 2014; Black 2014; Grant 2022; Pascual 2008; Soloff 1989; Soloff 1993 |
| Behavioural biweekly reports of episodes of impulsivity/aggressive behaviour | ‐ | CR | Soler 2005 |
| Borderline Personality Disorder Severity Index ‐ Impulsivity | BPDSI‐Impulsivity | CR | Bellino 2014; Bozzatello 2017; Rinne 2002 |
| Clinical Global Impression scale for Borderline Personality Disorder patients ‐ Impulsivity | CGI‐BPD‐Impulsivity | CR | Bogenschutz 2004; Pascual 2008 |
| Positive and Negative Syndrome Scale ‐ Poor Impulse Control | PANSS‐Poor impulse control | CR | Amminger 2013 |
| Zanarini Rating Scale for Borderline Personality Disorder ‐ Impulsivity | ZAN‐BPD‐Impulsivity | CR | Crawford 2018; Reich 2009; Schulz 2007; Zanarini 2007 |
5. Interpersonal problems
| Name of scale/means of assessment | Abbreviation | Clinician‐rated (CR)/self‐rated (SR) | Study |
| Atypical Depression Diagnostic Scale ‐ Rejection sensitivity | ADDS‐Rejection sensitivity | SR | Soloff 1993 |
| Borderline Personality Disorder Severity Index ‐ Interpersonal problems | BPDSI‐IV‐Interpersonal problems | CR | Bellino 2014; Bozzatello 2017 |
| Clinical Global Impression scale for Borderline Personality Disorder patient ‐ Unstable interpersonal relationships | CGI‐BPD‐Unstable interpersonal relationships | CR | Bogenschutz 2004; Pascual 2008 |
| Hopkins Symptoms check List ‐ Interpersonal sensitivity | HSCL‐INT | SR | Goldberg 1986 |
| Symptom Check List‐90 ‐ Interpersonal sensitivity | SCL‐90‐INT | SR | De la Fuente 1994; Frankenburg 2002; Soloff 1989; Zanarini 2001; Zanarini 2007 |
| Symptom Check List‐90 ‐ Revised ‐ Interpersonal sensitivity | SCL‐90‐R‐INT | SR | Loew 2006; Nickel 2006 |
| Zanarini Rating Scale for Borderline Personality Disorder ‐ Unstable interpersonal relationships | ZAN‐BPD‐Unstable interpersonal relationships | CR | Schulz 2007; Zanarini 2007 |
| Zanarini Rating Scale for Borderline Personality Disorder ‐ Disturbed relationships | ZAN‐BPD‐Disturbed relationships | CR | Bellino 2014; Crawford 2018 |
6. Abandonment
| Name of scale/means of assessment | Abbreviation | Clinician‐rated (CR)/self‐rated (SR) | Study |
| Borderline Personality Disorder Severity Index ‐ Abandonment | BPDSI‐Abandonment | CR | Bellino 2014; Bozzatello 2017 |
| Clinical Global Impression scale for Borderline Personality Disorder patient ‐ Abandonment | CGI‐BPD‐Abandonment | CR | Bogenschutz 2004; Pascual 2008 |
| Zanarini Rating Scale for Borderline Personality Disorder ‐ Frantic efforts to avoid abandonment | ZAN‐BPD‐Frantic efforts to avoid abandonment | CR | Schulz 2007; Zanarini 2007 |
7. Identity disturbance
| Name of scale/means of assessment | Abbreviation | Clinician‐rated (CR)/self‐rated (SR) | Study |
| Borderline Personality Disorder Severity Index ‐ Identity | BPDSI‐Identity | CR | Bellino 2014; Bozzatello 2017 |
| Clinical Global Impression scale for Borderline Personality Disorder patient ‐ Identity disturbance | CGI‐BPD‐Identity disturbance | CR | Bogenschutz 2004; Pascual 2008 |
| Zanarini Rating Scale for Borderline Personality Disorder ‐ Identity disturbance | ZAN‐BPD‐Identity disturbance | CR | Schulz 2007; Zanarini 2007 |
8. Dissociation and psychotic‐like symptoms
| Name of scale/means of assessment | Abbreviation | Clinician‐rated (CR)/self‐rated (SR) | Study |
| Borderline Personality Disorder Severity Index ‐ Dissociation/paranoid ideation | BPDSI‐Dissociation/paranoid ideation | CR | Bellino 2014; Bozzatello 2017 |
| Brief Psychiatric Rating Scale | BPRS | Unclear | AstraZeneca 2007; Leone 1982; Pascual 2008; Shafti 2010; Shafti 2014 |
| Clinical Global Impression scale for Borderline Personality Disorder patient ‐ Paranoid ideation | CGI‐BPD‐Paranoid ideation | CR | Pascual 2008 |
| Clinical Global Impression scale for Borderline Personality Disorder patient ‐ Transient paranoia or dissociation | CGI‐BPD‐Transient paranoia or dissociation | CR | Bogenschutz 2004; Pascual 2008 |
| Dissociation State Scales | DSS | SR | Schmahl 2012a; Schmahl 2012b |
| Dissociation Questionnaire | DIS‐Q | SR | AstraZeneca 2007 |
| Dissociative Experiences Scale | DES | SR | Simpson 2004; Zanarini 2001 |
| Positive and Negative Symptoms Scale | PANNS | CR | Amminger 2013; AstraZeneca 2007; Zanarini 2001 |
| Schedule of Interviewing Schizotypal Personalities ‐ Borderline score ‐ Suspicious/paranoid | SIB‐Suspicious/paranoid | CR | Goldberg 1986 |
| Symptom Check List‐90 ‐ Revised ‐ Paranoid ideation | SCL‐90‐R‐PAR | SR | De la Fuente 1994; Loew 2006; Nickel 2006; Soloff 1989; Soloff 1993; Zanarini 2001; Zanarini 2007 |
| Symptom Check List‐90 ‐ Revised ‐ Psychoticism | SCL‐90‐R‐PSY | SR | Loew 2006; Nickel 2006; Soloff 1989; Soloff 1993; Zanarini 2001 |
| Zanarini Rating Scale for Borderline Personality Disorder ‐ Paranoid ideation or dissociation | ZAN‐BPD‐Paranoid ideation or dissociation | CR | Schulz 2007; Zanarini 2007 |
| Zanarini Rating Scale for Borderline Personality Disorder ‐ Cognitive Disturbance | ZAN‐BPD‐Cognitive disturbance | CR | Crawford 2018 |
9. Depression
| Name of scale/means of assessment | Abbreviation | Clinician‐rated (CR)/self‐rated (SR) | Study |
| Adapted Clinical Global Impression ‐ Improvement scale | ‐ | Unclear | Cowdry 1988 |
| Beck Depression Inventory | BDI | SR | Crawford 2018; Hallahan 2007; Hollander 2001; Schmahl 2012b; Simpson 2004; Soloff 1989; Soloff 1993; Ziegenhorn 2009 |
| Hamiltons 24‐item Depression Rating scale | HDRS‐24 | Unclear | De la Fuente 1994 |
| Hamilton Rating scale for Depression | Ham‐D | SR, CR | Bellino 2014; Bozzatello 2017; Grant 2022; Linehan 2008; Markovitz 1995a; Moen 2012; Nickel 2006; Salzman 1995, Schmahl 2012b, Soler 2005; Soloff 1989; Soloff 1993; Zanarini 2001 |
| Hamilton Rating scale for Depression | Ham‐D‐17 | Unclear | Pascual 2008 |
| Hopkins Symptoms check List ‐ Depression | HSCL‐DEP | SR | Goldberg 1986 |
| Modified Bunney‐Hamburg rating scale | ‐ | Unclear | Cowdry 1988 |
| Montgomery Åsberg Depression Rating Scale | MADRS | CR | Amminger 2013; Black 2014; Moen 2012; Schulz 2007; Zanarini 2003; Zanarini 2004; Zanarini 2007 |
| Personality Disorder Rating Scale ‐ Depression | PDRS‐Depression | CR | Salzman 1995 |
| Positive and Negative Syndrome Scale ‐ Depression | PANSS‐Depression | Unclear | Amminger 2013 |
| Profile of Mood States ‐ Depression | POMS‐Depression | SR | Salzman 1995 |
| Symptom Checklist‐90 ‐ Depression | SCL‐90‐DEP | SR | De la Fuente 1994; Frankenburg 2002; Soloff 1989; Soloff 1993; Zanarini 2001 |
| Symptom Checklist‐90 ‐ Revised ‐ Depression | SCL‐90‐R‐DEP | SR | Jariani 2010; Loew 2006; Nickel 2006; Zanarini 2007 |
10. Attrition
| Name of scale/means of assessment | Abbreviation | Clinician‐rated (CR)/self‐rated (SR) | Study |
| Number of patients lost after randomisation | ‐ | ‐ | Amminger 2013; AstraZeneca 2007; Bellino 2014; Black 2014; Bogenschutz 2004; Bozzatello 2017; Cowdry 1988; Crawford 2018; De la Fuente 1994; Frankenburg 2002; Goldberg 1986; Grant 2022; Hallahan 2007; Hollander 2001; Kulkarni 2018; Leone 1982; Linehan 2008; Loew 2006; Markovitz 1995a; Montgomery 1982b; NCT00533117; Nickel 2004; Nickel 2005; Pascual 2008; Reich 2009; Rinne 2002; Schmahl 2012a; Schmahl 2012b; Schulz 2007; Shafti 2014, Shafti 2010; Simpson 2004; Soler 2005; Soloff 1989; Soloff 1993; Tritt 2005; Zanarini 2001; Zanarini 2003; Zanarini 2004; Zanarini 2007; Ziegenhorn 2009 |
11. Adverse effects
Appendix 6. Trial Sequential Analysis (TSA) and funnel plot figures
Primary outcomes
BPD symptom severity
We performed a TSA on the primary outcome of borderline symptom severity at end of treatment for the antipsychotics. The analysis shows that the z‐curve ended in the futility area. See Figure 4 below.
Secondary outcomes
Anger
We drew a funnel plot for the comparison between antipsychotics and placebo for the secondary outcome of anger. The funnel plot shows a small asymmetry. See Figure 5 below.
Impulsivity
We drew a funnel plot for the comparison between antipsychotics and placebo for the secondary outcome of impulsivity. The funnel plot shows a small asymmetry. See Figure 6 below.
Interpersonal problems
We performed a TSA on the secondary outcome of interpersonal problems at end of treatment for the antipsychotics. The analysis shows that the required information size was reached. See Figure 7 below.
Attrition
We performed a TSA on the secondary outcome attrition at end of treatment for the antispychotics. The analysis shows that the z‐curve ended in the futility area. See Figure 8 below.
We drew a funnel plot for the comparison between antipsychotics and placebo for the secondary outcome of attrition. The funnel plot shows a small asymmetry. See Figure 9 below.
Non‐serious adverse events
We performed a TSA on the secondary outcome of non‐serious adverse events at end of treatment for the antipsychotics. The analysis shows that the z‐curve ended in the futility area. See Figure 10 below.
Attrition
We performed a TSA on the secondary outcome attrition at end of treatment for the antidepressants. The analysis shows that the required information size was not reached. See Figure 11 below.
We performed a TSA on the secondary outcome attrition at end of treatment for the mood stabilisers. The analysis shows that the required information size was not reached.See Figure 12 below.
Appendix 7. Methods for future versions of this review
| Issue | Methods |
| Interventions | Consider classifying medications according to neuroscience‐based nomenclature (NbN is not well‐established among potential consumers of this review as yet; see Appendix 3 for guidance how to translate conventional terms into the NbN nomenclature). |
| Outcomes | Consider analysing psychotic symptoms separately from DSM‐IV/5 critierion 9 ("transient, stress‐related paranoid ideation or severe dissociative symptoms"; analyses were conducted in accordance with the review protocol in this version; Stoffers‐Winterling 2018) |
Footnotes DSM‐IV/5: Diagnostic and Statistical Manual of Mental Disorders‐Fourth/‐Fifth Edition.
Appendix 8. Unused methods
| Section | Protocol (Stoffers‐Winterling 2018) | Review |
| Search methods for identification of studies |
Electronic searches We intended to search the UK Clinical Trials Gateway database for relevant trials. |
We did not search the UK Clinical Trials Gateway (now included in Be Part of Research) because records are fed in there from both ISRCTN and CT.gov, both of which were searched separately. |
| Unit of analysis issues |
Cluster‐randomised trials Had trials used cluster randomisation, we would have anticipated that investigators would have presented their results after appropriately controlling for clustering effects (robust standard errors or hierarchical linear models). If it had been unclear whether a cluster‐randomised trial had used appropriate controls for clustering, we would have contacted the investigators for further information. We would have requested and re‐analysed individual patient data using multilevel models that controlled for clustering, if appropriate controls had not been used. Following this, we would have analysed effect sizes and standard errors in RevMan 5 (RevMan Web 2020), using the generic inverse method (Higgins 2019). If there had been insufficient information to control for clustering, we would have entered outcome data using individuals as the units of analysis, and then conducted a sensitivity analysis to assess the potential biasing effects of inadequately controlled cluster‐randomised trials (Donner 2002). If individual participant data had not been available, we would have looked for information on intra‐class correlation coefficients to adjust for the potential clustering effects. |
We did not include any cluster‐randomised trial. |
| Dealing with missing data | Had dichotomous data not been presented on the basis of ITT data, we would have added the number of participants lost in each group to the participants with unfavourable results, acting on the assumption that most people with BPD do not get lost at random. | We were unable to perform this analysis due to insufficient information. |
| Subgroup analysis and investigation of heterogeneity | We intended to conduct subgroup analyses to make hypotheses about the subgroups mentioned below.
|
We did not conduct these preplanned analyses because of a lack of data. |
| Sensitivity analysis | We intended to assess the impact of heterogeneity on the overall pooled effect estimate by removing trials ('outliers') that contributed to heterogeneity. We intended to remove outliers one by one and assess the impact on the overall outcome.
|
We were not able to perform these analyses due to a lack of sufficient data. |
| TSA | We intended to calculate post hoc, low bias, risk diversity‐adjusted required information size TSA analyses for the primary outcomes. | We were not able to perform these analyses with low risk of bias trials as no trials were assessed as being at low risk of bias overall. |
Data and analyses
Comparison 1. Medications compared with placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 Primary: BPD symptom severity at end of treatment (continuous outcomes, SMDs) | 11 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.1.1 Antipsychotics | 8 | 951 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.18 [‐0.45, 0.08] |
| 1.1.2 Antidepressants | 2 | 87 | Std. Mean Difference (IV, Random, 95% CI) | 0.27 [‐0.65, 1.18] |
| 1.1.3 Mood stabilisers | 4 | 265 | Std. Mean Difference (IV, Random, 95% CI) | 0.07 [‐0.43, 0.57] |
| 1.2 Primary: BPD symptom severity at end of treatment (continuous outcomes, MDs) | 4 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.2.1 Clonidine | 1 | 34 | Mean Difference (IV, Fixed, 95% CI) | ‐13.11 [‐65.36, 39.14] |
| 1.2.2 Naltrexone | 1 | 32 | Mean Difference (IV, Fixed, 95% CI) | 0.10 [‐0.29, 0.49] |
| 1.2.3 Memantine hydrochloride | 1 | 33 | Mean Difference (IV, Fixed, 95% CI) | 2.00 [‐1.62, 5.62] |
| 1.2.4 Alprazolam | 1 | 25 | Mean Difference (IV, Fixed, 95% CI) | ‐0.58 [‐1.63, 0.47] |
| 1.3 Primary: Self‐harm at end of treatment (continuous outcomes, MDs) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.3.1 Antidepressants | 1 | 20 | Mean Difference (IV, Fixed, 95% CI) | 0.45 [‐10.55, 11.45] |
| 1.4 Primary: Self‐harm at end of treatment (dichotomous outcomes, RRs) | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.4.1 Antipsychotics | 2 | 76 | Risk Ratio (M‐H, Random, 95% CI) | 0.66 [0.15, 2.84] |
| 1.4.2 Mood stabilisers | 1 | 276 | Risk Ratio (M‐H, Random, 95% CI) | 1.08 [0.79, 1.48] |
| 1.4.3 Omega‐3 fatty acids | 1 | 49 | Risk Ratio (M‐H, Random, 95% CI) | 1.23 [0.51, 2.97] |
| 1.5 Primary: Suicide‐related outcomes at end of treatment (continuous outcomes, SMDs) | 9 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.5.1 Antipsychotics | 7 | 854 | Std. Mean Difference (IV, Random, 95% CI) | 0.05 [‐0.18, 0.29] |
| 1.5.2 Antidepressants | 2 | 45 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.26 [‐1.62, 1.09] |
| 1.5.3 Mood stabilisers | 2 | 44 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.36 [‐1.96, 1.25] |
| 1.6 Primary: Suicide‐related outcomes at end of treatment (continuous outcomes, MDs) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.6.1 Alprazolam | 1 | 25 | Mean Difference (IV, Fixed, 95% CI) | 0.75 [‐0.18, 1.68] |
| 1.7 Primary: Suicide‐related outcomes at end of treatment (dichotomous outcomes, RRs) | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.7.1 Antipsychotics | 2 | 61 | Risk Ratio (M‐H, Random, 95% CI) | 0.73 [0.31, 1.73] |
| 1.7.2 Antidepressants | 1 | 58 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.71, 1.41] |
| 1.7.3 Omega‐3 fatty acids | 1 | 49 | Risk Ratio (M‐H, Random, 95% CI) | 0.52 [0.28, 0.95] |
| 1.8 Primary: Psychosocial functioning at end of treatment (continuous outcomes, SMDs) | 11 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.8.1 Antipsychotics | 7 | 904 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.16 [‐0.33, ‐0.00] |
| 1.8.2 Antidepressants | 4 | 161 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.25 [‐0.57, 0.06] |
| 1.8.3 Mood stabilisers | 2 | 214 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.01 [‐0.28, 0.26] |
| 1.9 Primary: Psychosocial functioning at end of treatment (continuous outcomes, MDs) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.9.1 Omega‐3 fatty acids | 1 | 15 | Mean Difference (IV, Fixed, 95% CI) | ‐19.90 [‐32.69, ‐7.11] |
| 1.10 Primary: Psychosocial functioning at end of treatment (dichtotomous outcomes, RRs) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.10.1 Mood stabilisers | 1 | 16 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.64 [0.37, 1.11] |
| 1.11 Secondary: Anger at end of treatment (continuous outcomes, SMDs) | 19 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.11.1 Antipsychotics | 10 | 1025 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.37 [‐0.55, ‐0.18] |
| 1.11.2 Antidepressants | 6 | 224 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.37 [‐0.64, ‐0.11] |
| 1.11.3 Mood stabilisers | 5 | 135 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.67 [‐1.10, ‐0.24] |
| 1.11.4 Omega‐3 fatty acids | 2 | 76 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.48 [‐0.95, ‐0.01] |
| 1.12 Secondary: Anger at end of treatment (continuous outcomes, MDs) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.12.1 Naltrexone | 1 | 32 | Mean Difference (IV, Fixed, 95% CI) | 1.65 [‐4.54, 7.84] |
| 1.12.2 Alprazolam | 1 | 25 | Mean Difference (IV, Fixed, 95% CI) | ‐0.57 [‐1.48, 0.34] |
| 1.13 Secondary: Affective instability at end of treatment (continuous outcomes, SMDs) | 6 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.13.1 Antipsychotics | 4 | 691 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.16 [‐0.31, ‐0.01] |
| 1.13.2 Mood stabilisers | 2 | 222 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.21 [‐0.68, 0.26] |
| 1.14 Secondary: Affective instability at end of treatment (continous outcomes, MDs) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.14.1 Antidepressants | 1 | 38 | Mean Difference (IV, Fixed, 95% CI) | ‐1.66 [‐3.26, ‐0.06] |
| 1.15 Secondary: Chronic feelings of emptiness at end of treatment (continuous outcomes, SMDs) | 4 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.15.1 Antipsychotics | 4 | 691 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.00 [‐0.16, 0.15] |
| 1.16 Secondary: Impulsivity at end of treatment (continuous outcomes, SMDs) | 14 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.16.1 Antipsychotics | 10 | 1038 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.08 [‐0.20, 0.04] |
| 1.16.2 Antidepressants | 4 | 182 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.17 [‐0.49, 0.15] |
| 1.16.3 Mood stabilisers | 4 | 265 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.56 [‐1.46, 0.35] |
| 1.17 Secondary: Impulsivity at end of treatment (continuous outcomes, MDs) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.17.1 Alprazolam | 1 | 25 | Mean Difference (IV, Fixed, 95% CI) | 0.67 [‐0.36, 1.70] |
| 1.18 Secondary: Impulsivity at end of treatment (dichtotomous outcomes, RRs) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.18.1 Mood stabilisers | 1 | 20 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.88 [0.53, 1.46] |
| 1.19 Secondary: Interpersonal problems at end of treatment (continuous outcomes, SMDs) | 12 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.19.1 Antipsychotics | 8 | 907 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.21 [‐0.34, ‐0.08] |
| 1.19.2 Antidepressants | 2 | 119 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.07 [‐0.69, 0.55] |
| 1.19.3 Mood stabilisers | 4 | 300 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.58 [‐1.14, ‐0.02] |
| 1.20 Secondary: Abandonment at end of treatment (continuous outcomes, SMDs) | 4 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.20.1 Antipsychotics | 4 | 691 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.01 [‐0.17, 0.14] |
| 1.21 Secondary: Identity disturbance at end of treatment (continuous outcomes, SMDs) | 4 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.21.1 Antipsychotics | 4 | 691 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.09 [‐0.25, 0.07] |
| 1.22 Secondary: Dissociation and psychotic‐like symptoms at end of treatment (continuous outcomes, SMDs) | 12 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.22.1 Antipsychotics | 8 | 907 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.28 [‐0.50, ‐0.06] |
| 1.22.2 Antidepressants | 3 | 139 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.22 [‐0.62, 0.18] |
| 1.22.3 Mood stabilisers | 3 | 270 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.23 [‐0.66, 0.20] |
| 1.23 Secondary: Dissociation and psychotic‐like symptoms at end of treatment (continuous outcomes, MDs) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.23.1 Omega‐3 fatty acids | 1 | 15 | Mean Difference (IV, Fixed, 95% CI) | ‐2.80 [‐5.70, 0.10] |
| 1.24 Secondary: Depression at end of treatment (continuous outcomes, SMDs) | 21 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.24.1 Antipsychotics | 12 | 1138 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.22 [‐0.42, ‐0.01] |
| 1.24.2 Antidepressants | 5 | 187 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.37 [‐0.82, 0.08] |
| 1.24.3 Mood stabilisers | 6 | 344 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.44 [‐0.80, ‐0.08] |
| 1.24.4 Omega‐3 fatty acids | 2 | 42 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.54 [‐1.18, 0.11] |
| 1.25 Secondary: Depression at end of treatment (continuous outcomes, MDs) | 3 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.25.1 Clonidine | 1 | 34 | Mean Difference (IV, Fixed, 95% CI) | ‐2.54 [‐10.27, 5.19] |
| 1.25.2 Naltrexone | 1 | 32 | Mean Difference (IV, Fixed, 95% CI) | 2.50 [‐4.22, 9.22] |
| 1.25.3 Alprazolam | 1 | 25 | Mean Difference (IV, Fixed, 95% CI) | 0.27 [‐0.73, 1.27] |
| 1.26 Secondary: Depression at end of treatment (dichotomous outcomes, RRs) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.26.1 Omega‐3 fatty acids | 1 | 49 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.48 [0.28, 0.81] |
| 1.27 Secondary: Attrition at end of treatment (dichotomous outcomes, RRs) | 30 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.27.1 Antipsychotics | 13 | 1216 | Risk Ratio (M‐H, Random, 95% CI) | 1.11 [0.89, 1.38] |
| 1.27.2 Antidepressants | 6 | 289 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.65, 1.76] |
| 1.27.3 Mood stabilisers | 9 | 530 | Risk Ratio (M‐H, Random, 95% CI) | 0.89 [0.69, 1.15] |
| 1.27.4 Omega‐3 fatty acids | 2 | 79 | Risk Ratio (M‐H, Random, 95% CI) | 0.61 [0.21, 1.79] |
| 1.27.5 Memantine hydrochloride | 1 | 33 | Risk Ratio (M‐H, Random, 95% CI) | 1.57 [0.45, 5.52] |
| 1.27.6 Clonidine | 1 | 34 | Risk Ratio (M‐H, Random, 95% CI) | 0.67 [0.13, 3.50] |
| 1.28 Secondary: Non‐serious adverse events at end of treatment (dichotomous outcomes, RRs) | 7 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.28.1 Antipsychotics | 5 | 814 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.90, 1.29] |
| 1.28.2 Mood stabilisers | 1 | 276 | Risk Ratio (M‐H, Random, 95% CI) | 0.84 [0.70, 1.01] |
| 1.28.3 Memantine hydrochloride | 1 | 33 | Risk Ratio (M‐H, Random, 95% CI) | 1.41 [0.79, 2.52] |
| 1.29 Secondary: Serious adverse events at end of treatment (dichotomous outcomes, RRs) | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.29.1 Memantine hydrochloride | 1 | 33 | Risk Ratio (M‐H, Fixed, 95% CI) | Not estimable |
| 1.29.2 Brexpiprazole | 1 | 80 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.00 [0.13, 71.51] |
Comparison 2. Medications compared with placebo ‐ non‐serious adverse events ‐ central nervous system.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 2.1 Antipsychotics | 6 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1.1 Headache | 4 | 754 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.63, 1.62] |
| 2.1.2 Dizziness | 2 | 68 | Risk Ratio (M‐H, Random, 95% CI) | 3.07 [0.40, 23.45] |
| 2.1.3 Fatigue | 3 | 692 | Risk Ratio (M‐H, Random, 95% CI) | 1.50 [0.58, 3.89] |
| 2.1.4 Somnolence | 2 | 615 | Risk Ratio (M‐H, Random, 95% CI) | 2.97 [1.75, 5.03] |
| 2.1.5 Sedation | 4 | 445 | Risk Ratio (M‐H, Random, 95% CI) | 2.66 [0.99, 7.12] |
| 2.1.6 Anxiety | 1 | 314 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.33, 2.42] |
| 2.1.7 Insomnia | 2 | 615 | Risk Ratio (M‐H, Random, 95% CI) | 0.68 [0.33, 1.37] |
| 2.1.8 Hypersomnia | 1 | 62 | Risk Ratio (M‐H, Random, 95% CI) | 2.34 [0.69, 8.01] |
| 2.1.9 Forgetful or confusion | 1 | 62 | Risk Ratio (M‐H, Random, 95% CI) | 1.46 [0.38, 5.60] |
| 2.1.10 Disturbance in attention | 1 | 301 | Risk Ratio (M‐H, Random, 95% CI) | 11.37 [0.63, 203.81] |
| 2.1.11 Increased appetite | 3 | 692 | Risk Ratio (M‐H, Random, 95% CI) | 2.68 [1.71, 4.19] |
| 2.1.12 Change in appetite | 1 | 17 | Risk Ratio (M‐H, Random, 95% CI) | 0.65 [0.10, 4.06] |
| 2.1.13 Restlessness | 1 | 77 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.20, 4.30] |
| 2.1.14 Hallucinations | 1 | 77 | Risk Ratio (M‐H, Random, 95% CI) | 0.19 [0.01, 3.74] |
| 2.1.15 Sleep problems | 1 | 77 | Risk Ratio (M‐H, Random, 95% CI) | 0.19 [0.01, 3.74] |
| 2.1.16 Tremor | 1 | 77 | Risk Ratio (M‐H, Random, 95% CI) | 0.31 [0.01, 7.36] |
| 2.2 Mood stabilisers | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.2.1 Paraesthesia | 1 | 56 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.00 [0.33, 27.12] |
| 2.2.2 Headache | 1 | 56 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.00 [0.15, 6.61] |
| 2.2.3 Dizziness | 1 | 56 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.50 [0.27, 8.30] |
| 2.2.4 Fatigue | 1 | 56 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.00 [0.40, 10.05] |
| 2.2.5 Memory problems | 1 | 56 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.00 [0.55, 7.22] |
| 2.2.6 Psychiatric disorders | 1 | 276 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.64, 1.37] |
| 2.2.7 Nervous system disorders | 1 | 276 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.05 [0.68, 1.62] |
| 2.3 Memantine hydrochloride | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.3.1 Somnolence | 1 | 33 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.65 [0.59, 4.57] |
| 2.3.2 Headache | 1 | 33 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.29 [0.71, 2.36] |
| 2.3.3 Fatigue | 1 | 33 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.32 [0.52, 3.31] |
| 2.3.4 Dizziness | 1 | 33 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.69 [0.72, 3.98] |
| 2.3.5 Gait/balance disturbances | 1 | 33 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.35 [0.53, 10.45] |
Comparison 3. Medications compared with placebo ‐ non‐serious adverse events ‐ cardiovascular and respiratory system.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 3.1 Antipsychotics | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1.1 Cold/flu symptoms | 1 | 62 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.54 [0.50, 4.73] |
| 3.1.2 Nasopharyngitis | 1 | 301 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.62 [0.23, 1.66] |
| 3.1.3 Sweating | 1 | 77 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.31 [0.01, 7.36] |
| 3.2 Antipsychotics | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 3.2.1 Diastolic blood pressure, standing, baseline to endpoint mean change | 1 | 290 | Mean Difference (IV, Fixed, 95% CI) | ‐0.28 [‐2.29, 1.73] |
| 3.2.2 Diastolic blood pressure, supine, baseline to endpoint mean change | 1 | 290 | Mean Difference (IV, Fixed, 95% CI) | ‐0.11 [‐2.28, 2.06] |
| 3.2.3 Systolic blood pressure, supine, baseline to endpoint mean change | 1 | 290 | Mean Difference (IV, Fixed, 95% CI) | ‐1.31 [‐4.00, 1.38] |
| 3.2.4 Systolic blood pressure, standing, baseline to endpoint mean change | 1 | 290 | Mean Difference (IV, Fixed, 95% CI) | 0.35 [‐2.39, 3.09] |
| 3.2.5 Pulse, supine, baseline to endpoint mean change | 1 | 290 | Mean Difference (IV, Fixed, 95% CI) | ‐0.11 [‐2.28, 2.06] |
| 3.2.6 Pulse, standing, baseline to endpoint mean change | 1 | 290 | Mean Difference (IV, Fixed, 95% CI) | 0.85 [‐1.65, 3.35] |
| 3.3 Mood stabilisers | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.3.1 Blood and lymphatic system disorders | 1 | 276 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.68 [0.11, 3.99] |
| 3.3.2 Cardiac disorders | 1 | 276 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.34 [0.01, 8.23] |
| 3.3.3 Endocrine disorders | 1 | 276 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.34 [0.01, 8.23] |
| 3.3.4 Respiratory, thoracic, and mediastinal disorders | 1 | 276 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.80 [0.83, 3.94] |
Comparison 4. Medications compared with placebo – non‐serious adverse events ‐ metabolic and gastro‐intestinal system.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 4.1 Antipsychotics | 6 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1.1 Nausea | 4 | 754 | Risk Ratio (M‐H, Random, 95% CI) | 0.80 [0.49, 1.29] |
| 4.1.2 Uneasy feeling | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 7.00 [0.38, 129.93] |
| 4.1.3 Constipation | 1 | 28 | Risk Ratio (M‐H, Random, 95% CI) | 6.50 [0.41, 104.20] |
| 4.1.4 Dry mouth | 4 | 754 | Risk Ratio (M‐H, Random, 95% CI) | 2.60 [1.46, 4.64] |
| 4.2 Antipsychotics | 3 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4.2.1 liver function: ALT/SGPT baseline to endpoint mean change (U/L) | 2 | 530 | Std. Mean Difference (IV, Random, 95% CI) | 0.46 [0.29, 0.63] |
| 4.2.2 liver function: AST/SGOT baseline to endpoint mean change (U/L) | 2 | 526 | Std. Mean Difference (IV, Random, 95% CI) | 0.35 [0.18, 0.52] |
| 4.2.3 liver function: total bilirubin baseline to endpoint mean change (μmol/L) | 1 | 264 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.29 [‐0.53, ‐0.05] |
| 4.2.4 liver function: direct bilirubin baseline to endpoint mean change (μmol/L) | 1 | 258 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.35 [‐0.60, ‐0.11] |
| 4.2.5 liver function: GGT (GGPT/SGGT/YGGT) baseline to endpoint mean change | 1 | 268 | Std. Mean Difference (IV, Random, 95% CI) | 0.26 [0.02, 0.50] |
| 4.2.6 lipids: total cholesterol baseline to endpoint change (mmol/L) | 2 | 327 | Std. Mean Difference (IV, Random, 95% CI) | 0.42 [0.20, 0.64] |
| 4.2.7 lipids: LDL cholesterol baseline to endpoint mean change (mmol/L) | 1 | 259 | Std. Mean Difference (IV, Random, 95% CI) | 0.35 [0.10, 0.59] |
| 4.2.8 lipids: HDL cholesterol (dextran precip.) baseline to endpoint mean change (mmol/L) | 1 | 269 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.28 [‐0.52, ‐0.04] |
| 4.2.9 lipids: triglycerides, fasting, baseline to endpoint mean change (mmol/L) | 1 | 203 | Std. Mean Difference (IV, Random, 95% CI) | 0.37 [0.09, 0.64] |
| 4.2.10 prolactin: baseline to endpoint mean change (μg/L) | 1 | 259 | Std. Mean Difference (IV, Random, 95% CI) | 0.32 [0.07, 0.56] |
| 4.2.11 platelet count baseline to endpoint mean change (GI/L) | 2 | 517 | Std. Mean Difference (IV, Random, 95% CI) | 0.03 [‐0.53, 0.59] |
| 4.2.12 erythrocyte count baseline to endpoint mean change (TI/L) | 1 | 262 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.18 [‐0.42, 0.06] |
| 4.2.13 leukocyte count baseline to endpoint mean change (GI/L) | 1 | 262 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.40 [‐0.65, ‐0.16] |
| 4.2.14 neutrophils, segmented, baseline to endpoint mean change (GI/L) | 1 | 262 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.39 [‐0.63, ‐0.14] |
| 4.2.15 basophils baseline to endpoint mean change (GI/L) | 1 | 262 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.28 [‐0.53, ‐0.04] |
| 4.2.16 monocytes baseline to endpoint mean change (GI/L) | 1 | 262 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.28 [‐0.53, ‐0.04] |
| 4.2.17 haemoglobin baseline to endpoint mean change (mml/L‐F) | 1 | 262 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.21 [‐0.45, 0.03] |
| 4.2.18 mean cell haemoglobin concentration (MCHC) baseline to endpoint mean change (mml/L‐F) | 1 | 260 | Std. Mean Difference (IV, Random, 95% CI) | 0.03 [‐0.22, 0.27] |
| 4.2.19 calcium baseline to endpoint mean change (mmol/L) | 1 | 268 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.33 [‐0.57, ‐0.09] |
| 4.2.20 albumin baseline to endpoint mean change (g/L) | 1 | 269 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.21 [‐0.45, 0.03] |
| 4.2.21 creatine phosphokinase baseline to endpoint mean change (U/L) | 1 | 268 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.21 [‐0.45, 0.03] |
| 4.2.22 urea nitrogen baseline to endpoint mean change (mmol/L) | 1 | 269 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.14 [‐0.38, 0.10] |
| 4.3 Mood stabilisers | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4.3.1 gastrointestinal disorders | 1 | 276 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.70 [0.50, 0.98] |
| 4.3.2 general disorders and administration site conditions | 1 | 276 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.01 [0.50, 2.05] |
| 4.3.3 hepatobiliary disorders | 1 | 276 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.04 [0.13, 74.07] |
| 4.3.4 metabolism and nutrition disorders | 1 | 276 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.03 [0.19, 22.12] |
| 4.4 Memantine hydrochloride | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4.4.1 constipation | 1 | 33 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.65 [0.59, 4.57] |
| 4.4.2 nausea/vomiting | 1 | 34 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.00 [0.45, 2.23] |
Comparison 5. Medications compared with placebo ‐ non‐serious adverse events ‐ musculoskeletal system.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 5.1 Antipsychotics | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.1.1 Bodily pain | 1 | 62 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.88 [0.47, 1.64] |
| 5.2 Antipsychotics | 7 | 810 | Std. Mean Difference (IV, Random, 95% CI) | 0.78 [0.44, 1.12] |
| 5.2.1 Body weight change | 7 | 810 | Std. Mean Difference (IV, Random, 95% CI) | 0.78 [0.44, 1.12] |
| 5.3 Antidepressants | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 5.3.1 Body weight change | 1 | 62 | Mean Difference (IV, Fixed, 95% CI) | 0.09 [‐0.31, 0.49] |
| 5.4 Mood stabilisers | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.4.1 Musculoskeletal and connective tissue disorders | 1 | 276 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.16 [0.43, 3.11] |
| 5.5 Mood stabilisers | 5 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 5.5.1 Body weight change | 5 | 184 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.26 [‐0.72, 0.20] |
Comparison 6. Medications compared with placebo ‐ non‐serious adverse events ‐ sensory system.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 6.1 Mood stabilisers | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 6.1.1 Eye disorders | 1 | 276 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.17 [0.02, 1.39] |
Comparison 7. Medications compared with placebo ‐ non‐serious adverse events ‐ reproductive system.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 7.1 Mood stabilisers | 2 | 608 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.85 [0.71, 4.82] |
| 7.1.1 Pregnancy, puerperium, and perinatal conditions | 1 | 276 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.52 [0.26, 8.97] |
| 7.1.2 Reproductive system and breast disorders | 1 | 276 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.04 [0.32, 28.90] |
| 7.1.3 Menstrual pain | 1 | 56 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.67 [0.44, 6.31] |
Comparison 8. Medications compared with placebo ‐ non‐serious adverse events ‐ other.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 8.1 Mood stabilisers | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 8.1.1 Injury, poisoning, and procedural complications | 1 | 276 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.44 [0.26, 0.74] |
| 8.1.2 Skin and subcutaneous tissue disorder | 1 | 276 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.15 [0.75, 1.75] |
| 8.1.3 Social circumstances | 1 | 276 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.01 [0.06, 16.06] |
| 8.1.4 Surgical and medical procedures | 1 | 276 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.06 [0.46, 35.85] |
Comparison 9. Medications compared with placebo ‐ withdrew due to adverse events.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 9.1 Memantine hydrochloride | 1 | 33 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.82 [0.33, 24.43] |
Comparison 10. Single medication compared with alternate single medication .
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 10.1 Primary: BPD symptom severity at end of treatment (continuous outcomes, MDs) | 3 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 10.1.1 Olanzapine versus asenapine | 1 | 51 | Mean Difference (IV, Fixed, 95% CI) | ‐2.23 [‐8.04, 3.58] |
| 10.1.2 Haloperidol versus phenelzine sulfate | 1 | 64 | Mean Difference (IV, Fixed, 95% CI) | 5.73 [‐0.33, 11.79] |
| 10.1.3 Alprazolam versus carbamazepine | 1 | 27 | Mean Difference (IV, Fixed, 95% CI) | ‐1.64 [‐2.71, ‐0.57] |
| 10.1.4 Alprazolam versus trifluoperazine hydrochloride | 1 | 22 | Mean Difference (IV, Fixed, 95% CI) | ‐0.80 [‐2.21, 0.61] |
| 10.1.5 Alprazolam versus tranylcypromine sulfate | 1 | 24 | Mean Difference (IV, Fixed, 95% CI) | ‐1.58 [‐2.76, ‐0.40] |
| 10.1.6 Carbamazepine versus trifluoperazine hydrochloride | 1 | 25 | Mean Difference (IV, Fixed, 95% CI) | 0.84 [‐0.41, 2.09] |
| 10.1.7 Carbamazepine versus tranylcypromine sulfate | 1 | 27 | Mean Difference (IV, Fixed, 95% CI) | 0.06 [‐0.92, 1.04] |
| 10.1.8 Trifluoperazine hydrochloride versus tranylcypromine sulfate | 1 | 22 | Mean Difference (IV, Fixed, 95% CI) | ‐0.78 [‐2.13, 0.57] |
| 10.2 Primary: Self‐harm at end of treatment (continuous outcomes, MDs) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 10.2.1 Olanzapine versus asenapine | 1 | 51 | Mean Difference (IV, Fixed, 95% CI) | 0.21 [‐0.58, 1.00] |
| 10.3 Primary: Suicide‐related outcomes at end of treatment (continuous outcomes, MDs) | 1 | 147 | Mean Difference (IV, Fixed, 95% CI) | 1.01 [0.54, 1.47] |
| 10.3.1 Alprazolam versus carbamazepine | 1 | 27 | Mean Difference (IV, Fixed, 95% CI) | 2.12 [1.06, 3.18] |
| 10.3.2 Alprazolam versus trifluoperazine hydrochloride | 1 | 22 | Mean Difference (IV, Fixed, 95% CI) | 1.73 [0.62, 2.84] |
| 10.3.3 Alprazolam versus tranylcypromine sulfate | 1 | 24 | Mean Difference (IV, Fixed, 95% CI) | 2.00 [0.89, 3.11] |
| 10.3.4 Carbamazepine versus trifluoperazine hydrochloride | 1 | 25 | Mean Difference (IV, Fixed, 95% CI) | ‐0.39 [‐1.53, 0.75] |
| 10.3.5 Carbamazepine versus tranylcypromine sulfate | 1 | 27 | Mean Difference (IV, Fixed, 95% CI) | ‐0.12 [‐1.26, 1.02] |
| 10.3.6 Trifluoperazine hydrochloride versus tranylcypromine sulfate | 1 | 22 | Mean Difference (IV, Fixed, 95% CI) | 0.27 [‐1.00, 1.54] |
| 10.4 Primary: Psychosocial functioning (continuous outcomes, MDs) | 5 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 10.4.1 Olanzapine versus asenapine | 1 | 51 | Mean Difference (IV, Fixed, 95% CI) | 0.20 [‐0.23, 0.63] |
| 10.4.2 Olanzapine versus haloperidol | 1 | 28 | Mean Difference (IV, Fixed, 95% CI) | 0.35 [‐0.45, 1.15] |
| 10.4.3 Olanzapine versus aripiprazole | 1 | 24 | Mean Difference (IV, Fixed, 95% CI) | 0.12 [‐0.58, 0.82] |
| 10.4.4 Haloperidol versus amitriptyline | 1 | 57 | Mean Difference (IV, Fixed, 95% CI) | ‐3.87 [‐10.67, 2.93] |
| 10.4.5 Haloperidol versus phenelzine sulfate | 1 | 64 | Mean Difference (IV, Fixed, 95% CI) | 5.15 [0.29, 10.01] |
| 10.5 Secondary: Anger at end of treatment (continuous outcomes, MDs) | 7 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 10.5.1 Olanzapine versus asenapine | 1 | 51 | Mean Difference (IV, Fixed, 95% CI) | 1.14 [0.31, 1.97] |
| 10.5.2 Olanzapine versus haloperidol | 1 | 28 | Mean Difference (IV, Fixed, 95% CI) | 0.21 [‐8.90, 9.32] |
| 10.5.3 Olanzapine versus aripiprazole | 1 | 24 | Mean Difference (IV, Fixed, 95% CI) | ‐0.40 [‐8.05, 7.25] |
| 10.5.4 Olanzapine versus sertraline | 1 | 120 | Mean Difference (IV, Fixed, 95% CI) | ‐0.33 [‐0.48, ‐0.18] |
| 10.5.5 Haloperidol versus amitriptyline | 1 | 57 | Mean Difference (IV, Fixed, 95% CI) | ‐0.34 [‐0.82, 0.14] |
| 10.5.6 Haloperidol versus phenelzine sulfate | 1 | 64 | Mean Difference (IV, Fixed, 95% CI) | 0.06 [‐0.31, 0.43] |
| 10.5.7 Alprazolam versus carbamazepine | 1 | 27 | Mean Difference (IV, Fixed, 95% CI) | 1.65 [0.80, 2.50] |
| 10.5.8 Alprazolam versus trifluoperazine hydrochloride | 1 | 22 | Mean Difference (IV, Fixed, 95% CI) | 0.58 [‐0.77, 1.93] |
| 10.5.9 Alprazolam versus tranylcypromine sulfate | 1 | 24 | Mean Difference (IV, Fixed, 95% CI) | 1.41 [0.24, 2.58] |
| 10.5.10 Carbamazepine versus trifluoperazine hydrochloride | 1 | 25 | Mean Difference (IV, Fixed, 95% CI) | ‐1.07 [‐2.28, 0.14] |
| 10.5.11 Carbamazepine versus tranylcypromine sulfate | 1 | 27 | Mean Difference (IV, Fixed, 95% CI) | ‐0.24 [‐1.23, 0.75] |
| 10.5.12 Trifluoperazine hydrochloride versus tranylcypromine sulfate | 1 | 22 | Mean Difference (IV, Fixed, 95% CI) | 0.83 [‐0.62, 2.28] |
| 10.6 Secondary: Affective instability at end of treatment (continuous outcomes, MDs) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 10.6.1 Olanzapine versus asenapine | 1 | 51 | Mean Difference (IV, Fixed, 95% CI) | 2.28 [1.51, 3.05] |
| 10.7 Secondary: Chronic feelings of emptiness at end of treatment (continuous outcomes, MDs) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 10.7.1 Olanzapine versus asenapine | 1 | 51 | Mean Difference (IV, Fixed, 95% CI) | ‐0.54 [‐1.29, 0.21] |
| 10.8 Secondary: Impulsivity at end of treatment (continuous outcomes, MDs) | 5 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 10.8.1 Olanzapine versus asenapine | 1 | 51 | Mean Difference (IV, Fixed, 95% CI) | ‐0.78 [‐1.59, 0.03] |
| 10.8.2 Haloperidol versus amitriptyline | 1 | 57 | Mean Difference (IV, Fixed, 95% CI) | 3.52 [‐5.52, 12.56] |
| 10.8.3 Haloperidol versus phenelzine sulfate | 1 | 64 | Mean Difference (IV, Fixed, 95% CI) | 3.29 [‐14.52, 21.10] |
| 10.8.4 Olanzapine versus fluoxetine | 1 | 29 | Mean Difference (IV, Fixed, 95% CI) | ‐4.31 [‐19.72, 11.10] |
| 10.8.5 Alprazolam versus carbamazepine | 1 | 27 | Mean Difference (IV, Fixed, 95% CI) | 2.18 [1.20, 3.16] |
| 10.8.6 Alprazolam versus trifluoperazine hydrochloride | 1 | 22 | Mean Difference (IV, Fixed, 95% CI) | 0.45 [‐0.87, 1.77] |
| 10.8.7 Alprazolam versus tranylcypromine sulfate | 1 | 24 | Mean Difference (IV, Fixed, 95% CI) | 1.83 [0.66, 3.00] |
| 10.8.8 Carbamazepine versus trifluoperazine hydrochloride | 1 | 25 | Mean Difference (IV, Fixed, 95% CI) | ‐1.73 [‐2.87, ‐0.59] |
| 10.8.9 Carbamazepine versus tranylcypromine sulfate | 1 | 27 | Mean Difference (IV, Fixed, 95% CI) | ‐0.35 [‐1.31, 0.61] |
| 10.8.10 Trifluoperazine hydrochloride versus tranylcypromine sulfate | 1 | 22 | Mean Difference (IV, Fixed, 95% CI) | 1.38 [0.08, 2.68] |
| 10.9 Secondary: Interpersonal problems at end of treatment (continuous outcomes, MDs) | 3 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 10.9.1 Olanzapine versus asenapine | 1 | 51 | Mean Difference (IV, Fixed, 95% CI) | 0.40 [‐0.35, 1.15] |
| 10.9.2 Haloperidol versus amitriptyline | 1 | 57 | Mean Difference (IV, Fixed, 95% CI) | ‐0.13 [‐0.62, 0.36] |
| 10.9.3 Haloperidol versus phenelzine sulfate | 1 | 64 | Mean Difference (IV, Fixed, 95% CI) | ‐0.33 [‐0.68, 0.02] |
| 10.10 Secondary: Abandonment at end of treatment (continuous outcomes, MDs) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 10.10.1 Olanzapine versus asenapine | 1 | 51 | Mean Difference (IV, Fixed, 95% CI) | ‐0.40 [‐1.07, 0.27] |
| 10.11 Secondary: Identity disturbance at end of treatment (continuous outcomes, MDs) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 10.11.1 Olanzapine versus asenapine | 1 | 51 | Mean Difference (IV, Fixed, 95% CI) | 0.68 [‐0.12, 1.48] |
| 10.12 Secondary: Dissociation and psychotic‐like symptoms at end of treatment (continuous outcomes, MDs) | 5 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 10.12.1 Olanzapine versus asenapine | 1 | 51 | Mean Difference (IV, Fixed, 95% CI) | ‐0.69 [‐1.53, 0.15] |
| 10.12.2 Olanzapine versus haloperidol | 1 | 28 | Mean Difference (IV, Fixed, 95% CI) | ‐2.30 [‐10.15, 5.55] |
| 10.12.3 Olanzapine versus aripiprazole | 1 | 24 | Mean Difference (IV, Fixed, 95% CI) | ‐3.30 [‐10.63, 4.03] |
| 10.12.4 Haloperidol versus amitriptyline | 1 | 57 | Mean Difference (IV, Fixed, 95% CI) | ‐0.28 [‐0.69, 0.13] |
| 10.12.5 Haloperidol versus phenelzine sulfate | 1 | 64 | Mean Difference (IV, Fixed, 95% CI) | 0.14 [‐0.31, 0.59] |
| 10.13 Secondary: Depression at end of treatment (continuous outcomes, MDs) | 6 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 10.13.1 Olanzapine versus asenapine | 1 | 51 | Mean Difference (IV, Fixed, 95% CI) | 2.90 [0.88, 4.92] |
| 10.13.2 Olanzapine versus sertraline | 1 | 120 | Mean Difference (IV, Fixed, 95% CI) | 0.37 [0.22, 0.52] |
| 10.13.3 Haloperidol versus amitriptyline | 1 | 57 | Mean Difference (IV, Fixed, 95% CI) | 0.88 [‐5.12, 6.88] |
| 10.13.4 Haloperidol versus phenelzine sulfate | 1 | 64 | Mean Difference (IV, Fixed, 95% CI) | 7.81 [2.13, 13.49] |
| 10.13.5 Olanzapine versus fluoxetine | 1 | 29 | Mean Difference (IV, Fixed, 95% CI) | ‐5.40 [‐10.68, ‐0.12] |
| 10.13.6 Alprazolam versus carbamazepine | 1 | 27 | Mean Difference (IV, Fixed, 95% CI) | 1.36 [0.37, 2.35] |
| 10.13.7 Alprazolam versus trifluoperazine hydrochloride | 1 | 22 | Mean Difference (IV, Fixed, 95% CI) | 0.90 [‐0.49, 2.29] |
| 10.13.8 Alprazolam versus tranylcypromine sulfate | 1 | 24 | Mean Difference (IV, Fixed, 95% CI) | 1.67 [0.48, 2.86] |
| 10.13.9 Carbamazepine versus trifluoperazine hydrochloride | 1 | 25 | Mean Difference (IV, Fixed, 95% CI) | ‐0.46 [‐1.81, 0.89] |
| 10.13.10 Carbamazepine versus tranylcypromine sulfate | 1 | 27 | Mean Difference (IV, Fixed, 95% CI) | 0.31 [‐0.82, 1.44] |
| 10.13.11 Trifluoperazine hydrochloride versus tranylcypromine sulfate | 1 | 22 | Mean Difference (IV, Fixed, 95% CI) | 0.77 [‐0.73, 2.27] |
| 10.14 Secondary: Attrition at end of treatment (dichotomous outcomes, RRs) | 8 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 10.14.1 Olanzapine versus asenapine | 1 | 51 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.80 [0.28, 2.29] |
| 10.14.2 Olanzapine versus haloperidol | 1 | 28 | Risk Ratio (M‐H, Fixed, 95% CI) | Not estimable |
| 10.14.3 Olanzapine versus aripiprazole | 1 | 24 | Risk Ratio (M‐H, Fixed, 95% CI) | Not estimable |
| 10.14.4 Loxapine versus chlorpromazine | 1 | 80 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.14 [0.46, 2.85] |
| 10.14.5 Olanzapine versus sertraline | 1 | 120 | Risk Ratio (M‐H, Fixed, 95% CI) | Not estimable |
| 10.14.6 Olanzapine versus fluoxetine | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.29 [0.01, 6.69] |
| 10.14.7 Haloperidol versus phenelzine sulfate | 1 | 74 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.58 [0.49, 5.15] |
| 10.14.8 Haloperidol versus amitriptyline | 1 | 61 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.90 [0.32, 26.38] |
| 10.15 Secondary: Adverse events at end of treatment (dichotomous outcomes, RRs) | 3 | 132 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.96 [0.64, 1.45] |
| 10.15.1 Adverse events total: olanzapine versus haloperidol | 1 | 28 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.75 [0.35, 1.60] |
| 10.15.2 Adverse events total: olanzapine versus aripiprazole | 1 | 24 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.75 [0.38, 1.50] |
| 10.15.3 Adverse events total: loxapine versus chlorpromazine | 1 | 80 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.27 [0.66, 2.45] |
Comparison 11. Single medication compared with alternate single medication ‐ withdrew due to adverse events (AE).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 11.1 Withdrew due to AE | 1 | 51 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.96 [0.15, 6.31] |
| 11.1.1 Olanzapine1 versus asenapine2 | 1 | 51 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.96 [0.15, 6.31] |
Comparison 12. Single medication compared with alternate single medication ‐ non‐serious adverse events ‐ central nervous system.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 12.1 Sedation | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.50 [1.23, 9.92] |
| 12.1.1 Olanzapine versus fluoxetine | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.50 [1.23, 9.92] |
| 12.2 Restlessness | 2 | 110 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.92 [0.36, 2.32] |
| 12.2.1 Loxapine versus chlorpromazine | 1 | 80 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.50 [0.26, 8.50] |
| 12.2.2 Olanzapine versus fluoxetine | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.70 [0.23, 2.11] |
| 12.3 Restlessness/anxiety | 1 | 51 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.19 [0.01, 3.82] |
| 12.3.1 Olanzapine versus asenapine | 1 | 51 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.19 [0.01, 3.82] |
| 12.4 Sleepiness/drowsiness | 2 | 131 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.35 [0.47, 3.88] |
| 12.4.1 Olanzapine versus asenapine | 1 | 51 | Risk Ratio (M‐H, Fixed, 95% CI) | 6.74 [0.37, 124.21] |
| 12.4.2 Loxapine versus chlorpromazine | 1 | 80 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.80 [0.23, 2.76] |
| 12.5 Fainting spells | 1 | 80 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.14 [0.01, 2.68] |
| 12.5.1 Loxapine versus chlorpromazine | 1 | 80 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.14 [0.01, 2.68] |
| 12.6 Akhatisia | 1 | 51 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.19 [0.01, 3.82] |
| 12.6.1 Olanzapine versus asenapine | 1 | 51 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.19 [0.01, 3.82] |
| 12.7 Moderate anxiety | 1 | 51 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.32 [0.01, 7.53] |
| 12.7.1 Olanzapine versus asenapine | 1 | 51 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.32 [0.01, 7.53] |
| 12.8 Fatigue | 1 | 51 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.81 [0.24, 95.58] |
| 12.8.1 Olanzapine versus asenapine | 1 | 51 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.81 [0.24, 95.58] |
Comparison 13. Single medication compared with alternate single medication ‐ cardiovascular and respiratory system.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 13.1 Oral hypoaesthesia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 13.1.1 Olanzapine versus asenapine | 1 | 51 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.32 [0.01, 7.53] |
Comparison 14. Single medication compared with alternate single medication ‐ non‐serious adverse events ‐ musculoskeletal system.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 14.1 Muscle spasm | 1 | 80 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.00 [0.33, 27.63] |
| 14.1.1 Loxapine (medication 1) versus chlorpromazine (medication 2) | 1 | 80 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.00 [0.33, 27.63] |
| 14.2 Body weight change | 2 | 93 | Mean Difference (IV, Fixed, 95% CI) | ‐0.11 [‐0.47, 0.25] |
| 14.2.1 Haloperidol versus phenelzine sulfate | 1 | 64 | Mean Difference (IV, Fixed, 95% CI) | ‐0.22 [‐0.59, 0.15] |
| 14.2.2 Olanzapine versus fluoxetine | 1 | 29 | Mean Difference (IV, Fixed, 95% CI) | 2.50 [0.72, 4.28] |
| 14.3 Weight gain (3 or more kg within 4 weeks) | 1 | 51 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.81 [0.24, 95.58] |
| 14.3.1 Olanzapine versus asenapine | 1 | 51 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.81 [0.24, 95.58] |
Comparison 15. Single medication compared with combination of medications.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 15.1 Primary: BPD symptom severity at end of treatment (continuous outcome, MDs) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 15.1.1 Valproic acid versus valproic acid plus eicosapentaenoic acid and docosahexaenoic acid | 1 | 34 | Mean Difference (IV, Fixed, 95% CI) | 8.48 [3.39, 13.57] |
| 15.2 Primary: Self‐harm at end of treatment (continuous outcome, MDs) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 15.2.1 Valproic acid versus valproic acid plus eicosapentaenoic acid and docosahexaenoic acid | 1 | 34 | Mean Difference (IV, Fixed, 95% CI) | 2.55 [0.98, 4.12] |
| 15.3 Primary: Suicide‐related outcomes at end of treatment (continuous outcome, MDs) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 15.3.1 Valproic acid versus valproic acid plus eicosapentaenoic acid and docosahexaenoic acid | 1 | 34 | Mean Difference (IV, Fixed, 95% CI) | 0.23 [‐0.74, 1.20] |
| 15.4 Primary: Psychosocial functioning at end of treatment (continuous outcome, MDs) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 15.4.1 Valproic acid versus valproic acid plus eicosapentaenoic acid and docosahexaenoic acid | 1 | 34 | Mean Difference (IV, Fixed, 95% CI) | 0.88 [‐6.21, 7.97] |
| 15.5 Secondary: Anger at end of treatment (continuous outcome, MDs) | 2 | 89 | Mean Difference (IV, Fixed, 95% CI) | 0.64 [‐0.77, 2.04] |
| 15.5.1 Olanzapine versus olanzapine plus fluoxetine | 1 | 29 | Mean Difference (IV, Fixed, 95% CI) | 0.46 [‐12.93, 13.85] |
| 15.5.2 Fluoxetine versus fluoxetine plus olanzapine | 1 | 26 | Mean Difference (IV, Fixed, 95% CI) | 4.77 [‐9.67, 19.21] |
| 15.5.3 Valproic acid versus valproic acid plus eicosapentaenoic acid and docosahexaenoic acid | 1 | 34 | Mean Difference (IV, Fixed, 95% CI) | 0.60 [‐0.82, 2.02] |
| 15.6 Secondary: Affective instability at end of treatment (continuous outcome, MDs) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 15.6.1 Valproic acid versus valproic acid plus eicosapentaenoic acid and docosahexaenoic acid | 1 | 34 | Mean Difference (IV, Fixed, 95% CI) | 1.72 [0.68, 2.76] |
| 15.7 Secondary: Chronic feelings of emptiness at end of treatment (continuous outcome, MDs) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 15.7.1 Valproic acid versus valproic acid plus eicosapentaenoic acid and docosahexaenoic acid | 1 | 34 | Mean Difference (IV, Fixed, 95% CI) | 0.03 [‐0.97, 1.03] |
| 15.8 Secondary: Impulsivity at end of treatment (continuous outcome, MDs) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 15.8.1 Olanzapine versus olanzapine plus fluoxetine | 1 | 29 | Mean Difference (IV, Fixed, 95% CI) | 0.46 [‐12.93, 13.85] |
| 15.8.2 Fluoxetinge versus fluoxetine plus olanzapine | 1 | 26 | Mean Difference (IV, Fixed, 95% CI) | 4.77 [‐9.67, 19.21] |
| 15.8.3 Valproic acid versus valproic acid plus eicosapentaenoic acid and docosahexaenoic acid | 1 | 34 | Mean Difference (IV, Fixed, 95% CI) | 12.59 [6.11, 19.07] |
| 15.9 Secondary: Interpersonal problems at end of treatment (continuous outcome, MDs) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 15.9.1 Valproic acid versus valproic acid plus eicosapentaenoic acid and docosahexaenoic acid | 1 | 34 | Mean Difference (IV, Fixed, 95% CI) | 0.47 [‐0.41, 1.35] |
| 15.10 Secondary: Abandonment at end of treatment (continuous outcome, MDs) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 15.10.1 Valproic acid versus valproic acid plus eicosapentaenoic acid and docosahexaenoic acid | 1 | 34 | Mean Difference (IV, Fixed, 95% CI) | 0.01 [‐0.83, 0.85] |
| 15.11 Secondary: Identity disturbance at end of treatment (continuous outcome, MDs) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 15.11.1 Valproic acid versus valproic acid plus eicosapentaenoic acid and docosahexaenoic acid | 1 | 34 | Mean Difference (IV, Fixed, 95% CI) | 0.70 [‐0.34, 1.74] |
| 15.12 Secondary: Dissociation and psychotic‐like symptoms at end of treatment (continuous outcome, MDs) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 15.12.1 Valproic acid versus valproic acid plus eicosapentaenoic acid and docosahexaenoic acid | 1 | 34 | Mean Difference (IV, Fixed, 95% CI) | 0.03 [‐1.09, 1.15] |
| 15.13 Secondary: Depression at end of treatment (continuous outcome, MDs) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 15.13.1 Olanzapine versus olanzapine plus fluoxetine | 1 | 29 | Mean Difference (IV, Fixed, 95% CI) | ‐1.78 [‐6.48, 2.92] |
| 15.13.2 Fluoxetine versus fluoxetine plus olanzapine | 1 | 26 | Mean Difference (IV, Fixed, 95% CI) | 3.62 [‐1.36, 8.60] |
| 15.13.3 Valproic acid versus valproic acid plus eicosapentaenoic acid and docosahexaenoic acid | 1 | 34 | Mean Difference (IV, Fixed, 95% CI) | 1.30 [0.00, 2.60] |
| 15.14 Secondary: Attrition at end of treatment (dichotomous outcomes, RRs) | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 15.14.1 Olanzapine versus olanzapine plus fluoxetine | 1 | 31 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.19 [0.01, 3.63] |
| 15.14.2 Fluoxetine versus fluoxetine plus olanzapine | 1 | 29 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.54 [0.05, 5.28] |
| 15.14.3 Valproic acid versus valproic acid plus eicosapentaenoic acid and docosahexaenoic acid | 1 | 43 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.92 [0.29, 2.97] |
Comparison 16. Single medication compared with combination of medications ‐ non‐serious adverse events ‐ central nervous system.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 16.1 Sedation | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 16.1.1 Olanzapine versus olanzapine plus fluoxetine | 1 | 31 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.61 [0.87, 2.96] |
| 16.1.2 Fluoxetine versus fluoxetine plus olanzapine | 1 | 29 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.46 [0.15, 1.44] |
Comparison 17. Single medication compared with combination of medications ‐ non‐serious adverse events ‐ gastro‐intestinal system.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 17.1 Nausea | 1 | 34 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.22 [0.01, 4.34] |
| 17.1.1 Valproic acid versus valproic acid plus eicosapentaenoic acid and docosahexaenoic acid | 1 | 34 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.22 [0.01, 4.34] |
| 17.2 Dyspepsia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 17.2.1 Valproic acid versus valproic acid plus eicosapentaenoic acid and docosahexaenoic acid | 1 | 34 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.12 [0.08, 16.55] |
Comparison 18. Single medication compared with combination of medications ‐ musculoskeletal system.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 18.1 Akathisia | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.91 [0.43, 1.90] |
| 18.1.1 Olanzapine versus olanzapine plus fluoxetine | 1 | 31 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.75 [0.25, 2.28] |
| 18.1.2 Fluoxetine versus fluoxetine plus olanzapine | 1 | 29 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.07 [0.39, 2.92] |
| 18.2 Body weight change | 1 | 34 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.12 [0.26, 4.80] |
| 18.2.1 Valproic acid versus valproic acid plus eicosapentaenoic acid and docosahexaenoic acid | 1 | 34 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.12 [0.26, 4.80] |
| 18.3 Body weight change | 1 | 55 | Mean Difference (IV, Fixed, 95% CI) | 0.23 [‐0.76, 1.22] |
| 18.3.1 Olanzapine versus olanzapine plus fluoxetine | 1 | 29 | Mean Difference (IV, Fixed, 95% CI) | 1.50 [0.09, 2.91] |
| 18.3.2 Fluoxetine versus fluoxetine plus olanzapine | 1 | 26 | Mean Difference (IV, Fixed, 95% CI) | ‐1.00 [‐2.39, 0.39] |
Comparison 19. Medication compared with placebo ‐ TSA sensitivity analyses.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 19.1 BPD symptom severity at end of treatment | 6 | 859 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.10 [‐0.30, 0.10] |
| 19.1.1 Antipsychotics | 6 | 859 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.10 [‐0.30, 0.10] |
| 19.2 Secondary: interpersonal problems at end of treatment | 7 | 616 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.21 [‐0.38, ‐0.05] |
| 19.2.1 Antipsychotics | 7 | 616 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.21 [‐0.38, ‐0.05] |
| 19.3 Attrition at end of treatment | 22 | 1550 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.84, 1.17] |
| 19.3.1 Antipsychotics | 11 | 1044 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.84, 1.30] |
| 19.3.2 Antidepressants | 5 | 252 | Risk Ratio (M‐H, Random, 95% CI) | 1.14 [0.65, 1.97] |
| 19.3.3 Mood stabiliser | 8 | 254 | Risk Ratio (M‐H, Random, 95% CI) | 0.80 [0.55, 1.15] |
19.1. Analysis.

Comparison 19: Medication compared with placebo ‐ TSA sensitivity analyses, Outcome 1: BPD symptom severity at end of treatment
19.2. Analysis.

Comparison 19: Medication compared with placebo ‐ TSA sensitivity analyses, Outcome 2: Secondary: interpersonal problems at end of treatment
19.3. Analysis.

Comparison 19: Medication compared with placebo ‐ TSA sensitivity analyses, Outcome 3: Attrition at end of treatment
Comparison 20. Subgroup analysis: types of medication.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 20.1 BPD symptom severity ‐ antipsychotics vs placebo by class (1st vs 2nd generation) | 7 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 20.1.1 1st generation | 2 | 108 | Std. Mean Difference (IV, Random, 95% CI) | 0.29 [‐0.09, 0.67] |
| 20.1.2 2nd generation | 5 | 820 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.36 [‐0.66, ‐0.05] |
| 20.2 Suicide‐related outcomes ‐ antipsychotics vs placebo by class (1st vs 2nd generation) | 7 | 854 | Std. Mean Difference (IV, Random, 95% CI) | 0.05 [‐0.18, 0.29] |
| 20.2.1 1st generation | 2 | 103 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.20 [‐1.26, 0.86] |
| 20.2.2 2nd generation | 5 | 751 | Std. Mean Difference (IV, Random, 95% CI) | 0.07 [‐0.17, 0.31] |
| 20.3 Psychosocial functioning ‐ antipsychotics vs placebo by class (1st vs 2nd generation) | 7 | 904 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.16 [‐0.33, ‐0.00] |
| 20.3.1 1st generation | 3 | 164 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.12 [‐0.67, 0.42] |
| 20.3.2 2nd generation | 4 | 740 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.17 [‐0.32, ‐0.03] |
| 20.4 BPD symptom severity ‐ antipsychotics vs placebo by substance | 7 | 928 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.21 [‐0.49, 0.08] |
| 20.4.1 Haloperidol | 1 | 58 | Std. Mean Difference (IV, Random, 95% CI) | 0.30 [‐0.22, 0.82] |
| 20.4.2 Olanzapine | 2 | 596 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.15 [‐0.41, 0.10] |
| 20.4.3 Thiothixene | 1 | 50 | Std. Mean Difference (IV, Random, 95% CI) | 0.28 [‐0.28, 0.83] |
| 20.4.4 Quetiapine | 1 | 95 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.14 [‐0.58, 0.29] |
| 20.4.5 Ziprasidone | 1 | 60 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.47 [‐0.98, 0.05] |
| 20.4.6 Brexpiprazole | 1 | 69 | Std. Mean Difference (IV, Random, 95% CI) | ‐1.10 [‐1.61, ‐0.59] |
| 20.5 BPD symptom severity ‐ mood stabiliser vs placebo by substance | 3 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 20.5.1 Divalproex semisodium | 1 | 15 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.14 [‐1.22, 0.93] |
| 20.5.2 Lamotrigine | 2 | 222 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.08 [‐0.34, 0.19] |
| 20.6 Suicide‐related outcomes ‐ antipsychotics vs placebo by substance | 7 | 856 | Std. Mean Difference (IV, Random, 95% CI) | 0.13 [‐0.08, 0.34] |
| 20.6.1 Olanzapine | 4 | 691 | Std. Mean Difference (IV, Random, 95% CI) | 0.13 [‐0.13, 0.39] |
| 20.6.2 Ziprasidone | 1 | 60 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.27 [‐0.78, 0.23] |
| 20.6.3 Brexpiprazole | 1 | 80 | Std. Mean Difference (IV, Random, 95% CI) | 0.28 [‐0.16, 0.72] |
| 20.6.4 Alprazolam | 1 | 25 | Std. Mean Difference (IV, Random, 95% CI) | 0.62 [‐0.18, 1.43] |
| 20.7 Psychosocial functioning ‐ antipsychotics vs placebo by substance | 7 | 904 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.16 [‐0.33, ‐0.00] |
| 20.7.1 Haloperidol | 2 | 114 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.16 [‐1.08, 0.77] |
| 20.7.2 Olanzapine | 3 | 645 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.15 [‐0.30, 0.01] |
| 20.7.3 Quetiapine | 1 | 95 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.37 [‐0.81, 0.07] |
| 20.7.4 Thiothixene | 1 | 50 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.06 [‐0.61, 0.50] |
| 20.8 Psychosocial functioning ‐ antidepressants vs placebo by class/substance | 4 | 161 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.25 [‐0.57, 0.06] |
| 20.8.1 Tricyclic antidepressants (TCA)/amitriptyline | 2 | 119 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.20 [‐0.56, 0.16] |
| 20.8.2 Selective serotonin reuptake inhibitors (SSRIs)/fluoxetine | 2 | 42 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.40 [‐1.07, 0.27] |
Comparison 21. Subgroup analysis: psychosocial functioning at baseline.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 21.1 Primary: BPD symptom severity by severity of impairment at baseline | 8 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 21.1.1 Mild or slightly impaired (GAS/GAF 61 to 80) | 2 | 145 | Std. Mean Difference (IV, Random, 95% CI) | 0.03 [‐0.38, 0.43] |
| 21.1.2 Moderate severity of illness (GAS/GAF 51‐60, CGI 4) | 3 | 623 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.17 [‐0.39, 0.04] |
| 21.1.3 Marked or serious severity of illness (GAS/GAF 41‐50, CGI 5), SFQ 15 | 3 | 375 | Std. Mean Difference (IV, Random, 95% CI) | 0.00 [‐0.20, 0.21] |
| 21.2 Primary: Suicide‐related outcomes by severity of impairment at baseline | 6 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 21.2.1 Moderate severity of illness (GAS/GAF 51‐60, CGI 4) | 4 | 647 | Std. Mean Difference (IV, Random, 95% CI) | 0.16 [‐0.14, 0.46] |
| 21.2.2 Marked or serious severity of illness (GAS/GAF 41‐50, CGI 5), SFQ 15 | 2 | 80 | Std. Mean Difference (IV, Random, 95% CI) | 0.22 [‐0.22, 0.66] |
| 21.3 Primary: Psychosocial functioning by severity of impairment at baseline | 12 | 1294 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.17 [‐0.31, ‐0.04] |
| 21.3.1 Mild or slightly impaired | 3 | 167 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.32 [‐0.64, 0.00] |
| 21.3.2 Moderate severity of illness (GAS/GAF 51‐60, CGI 4) | 4 | 619 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.24 [‐0.50, 0.03] |
| 21.3.3 Marked or serious severity of illness (GAS/GAF 41‐50, CGI 5), SFQ 15 | 5 | 508 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.09 [‐0.29, 0.11] |
Comparison 22. Subgroup analysis: setting.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 22.1 BPD symptom severity by setting | 12 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 22.1.1 Inpatient | 1 | 15 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.14 [‐1.22, 0.93] |
| 22.1.2 Outpatient | 10 | 1148 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.18 [‐0.40, 0.03] |
| 22.1.3 Mixed | 1 | 120 | Std. Mean Difference (IV, Random, 95% CI) | 0.07 [‐0.37, 0.51] |
| 22.2 Primary: Suicide‐related outcomes at end of treatment | 7 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 22.2.1 Inpatient | 1 | 20 | Std. Mean Difference (IV, Random, 95% CI) | 0.44 [‐0.46, 1.33] |
| 22.2.2 Outpatient | 6 | 767 | Std. Mean Difference (IV, Random, 95% CI) | 0.09 [‐0.14, 0.32] |
| 22.3 Primary: Psychosocial functioning at end of treatment | 12 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 22.3.1 Inpatient | 2 | 39 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.19 [‐0.83, 0.44] |
| 22.3.2 Outpatient | 8 | 1022 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.17 [‐0.34, ‐0.01] |
| 22.3.3 Mixed | 2 | 233 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.18 [‐0.56, 0.20] |
Comparison 23. Subgroup analysis: funding.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 23.1 BPD symptom severity by funding | 12 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 23.1.1 Publicly (grants from universities, authorities, research foundations) | 3 | 348 | Std. Mean Difference (IV, Random, 95% CI) | 0.04 [‐0.17, 0.25] |
| 23.1.2 Funded or partially funded by pharmaceutical industry | 7 | 862 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.34 [‐0.61, ‐0.08] |
| 23.1.3 Unclear funding | 2 | 73 | Std. Mean Difference (IV, Random, 95% CI) | 0.24 [‐0.23, 0.70] |
| 23.2 Primary: Psychosocial functioning at end of treatment | 12 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 23.2.1 Publicly (grants from universities, authorities, research foundations) | 4 | 443 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.21 [‐0.55, 0.12] |
| 23.2.2 Funded or partially funded by pharmaceutical industry | 5 | 760 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.17 [‐0.31, ‐0.03] |
| 23.2.3 Unclear funding | 3 | 91 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.27 [‐0.69, 0.15] |
Comparison 24. Subgroup analysis: trial size.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 24.1 Primary: Psychosocial functioning at end of treatment | 12 | 1294 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.17 [‐0.31, ‐0.04] |
| 24.1.1 ≤ 50 | 4 | 76 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.58 [‐1.14, ‐0.02] |
| 24.1.2 ≤ 100 | 5 | 438 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.18 [‐0.39, 0.04] |
| 24.1.3 ≥ 100 | 3 | 780 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.12 [‐0.26, 0.02] |
| 24.2 Secondary: Anger at end of treatment | 22 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 24.2.1 ≤ 50 | 13 | 377 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.67 [‐1.00, ‐0.34] |
| 24.2.2 ≤ 100 | 7 | 546 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.67 [‐1.14, ‐0.20] |
| 24.2.3 ≥ 100 | 2 | 596 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.25 [‐0.41, ‐0.09] |
Comparison 25. Subgroup analysis: recruitment.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 25.1 Antipsychotics ‐ anger at end of treatment | 10 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 25.1.1 Referral | 8 | 923 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.30 [‐0.44, ‐0.16] |
| 25.1.2 Advertisement | 2 | 102 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.60 [‐1.65, 0.45] |
Comparison 26. Sensitivity analysis: mood stabiliser trials outliers SMD > 1.5 included.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 26.1 Secondary: Anger at end of treatment | 8 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 26.1.1 Mood stabilisers, outliers (SMD > 1.5) excluded | 5 | 135 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.67 [‐1.10, ‐0.24] |
| 26.1.2 Mood stabilisers, outlier (SMD >1.5) included | 8 | 247 | Std. Mean Difference (IV, Random, 95% CI) | ‐1.39 [‐2.16, ‐0.61] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Amminger 2013.
| Study characteristics | ||
| Methods | 12‐week trial with 2 arms:
Duration of trial: 12 weeks Country: Austria Setting: outpatient |
|
| Participants |
Method of recruitment of participants: "All patients were consecutive referrals to a psychosis detection unit at the Department of Child and Adolescent Psychiatry, Medical University of Vienna between May 2004 and May 2006" (Amminger 2013, p. 404) Overall sample size: 15 Diagnosis of borderline personality disorder: DSM‐IV Means of assessment: no structured interview used, consensus Mean age: 16.2 years (SD 2.1; range = 13‐25) Sex: 93.33% women Comorbidity: no information Inclusion criteria
Exclusion criteria
|
|
| Interventions |
Experimental group
Treatment name: concentrated fish oil (long‐chain omega‐3 polyunsaturated fatty acids)
Number randomised to group: 8
Duration: 12 weeks Control/comparison group Comparison name: placebo Number randomised to group: 7 Duration: 12 weeks Both groups Concomitant psychotherapy: "All patients were offered 7 sessions of needs‐based psychological and psychosocial interventions, concurrently with the research follow‐up interviews at baseline, 1, 2, 3, 4, 8, and 12 weeks" (Amminger 2013, p. 405) Concomitant pharmacotherapy: Antidepressants and benzodiazepines were allowed. Proportions of participants taking standing medication during trial observation period: antidepressants = 25% of treatment group, 14.3% of placebo group; benzodiazepines = 12.5% of treatment group, 14.3% of placebo group |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
Sample size calculation: no information Ethics approval: yes Funding source: funded by grants from universities, authorities or research foundations Conflicts of interest: No conflicts of interest reported. Comments from trial authors (limitations)
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "A computer‐generated random sequence based on a block randomized design (2 strata with block size of 4 within each stratum) was kept in a remote secure location and administered by an independent third party until all study data were collected and verified. Participants, parents, and those involved in administering interventions, assessing outcomes, data entry, and/or data analyses were blind to group assignments." (Amminger 2010, p. 148) |
| Allocation concealment (selection bias) | Low risk | Quote: “was kept in a remote secure location and administered by an independent third party until all study data were collected and verified” (Amminger 2010, p. 148) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: “Placebo capsules were carefully matched in appearance and flavour with the active treatment; they also contained the same amount of vitamin E as the n‐3 capsules, and 1% fish oil to mimic taste” (Amminger 2013, p. 405). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk |
Quote: “Participants, parents, and those involved in administering interventions, assessing outcomes, data entry and/or data analyses were blind to the group assignments” (Amminger 2010, p. 148). Comment: The intervention treatment was not associated with specific adverse effects that would allow for singling out participants with active treatment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "All analyses were performed on an intent‐to‐treat basis" (Amminger 2010, p. 149). |
| Selective reporting (reporting bias) | High risk | Comment: NCT00396643 ‐ The protocol had lipid metabolism as a secondary outcome but this was not mentioned in Amminger 2010 or Amminger 2013. No outcome on adverse effects included in the post hoc analysis |
| Vested Interest (funding and/or author affiliations) | Low risk | Comment: grant from the Stanley Medical Research Institute; grant from the National Health and Medical Research Council Australia; career development fellowship. All authors reported no biomedical financial interest or potential conflicts of interest. |
| Other bias | Low risk | Comment: no indication of other bias |
AstraZeneca 2007.
| Study characteristics | ||
| Methods | 8‐week trial with 2 arms:
Duration of trial: 8 weeks + 10 weeks follow‐up Country: The Netherlands Setting: no information |
|
| Participants |
Methods of recruitment of patients: no information Overall sample size: 24 Diagnosis of borderline personality disorder: DSM‐IV Means of assessment: SCID‐II Mean age: no information Sex: no information Comorbidity: no information Inclusion criteria
Exclusion criteria
|
|
| Interventions |
Experimental group
Treatment name: quetiapine fumarate
Number randomised to group: 13
Duration: 8 weeks Control/comparison group Comparison name: placebo Number randomised to group: 11 Duration: 8 weeks Both groups Concomitant psychotherapy: no information Concomitant pharmacotherapy: not allowed, except for SSRIs and benzodiazepines, with doses stabilised 8 weeks before start of trial period Proportions of participants taking standing medication during trial observation period: no information |
|
| Outcomes |
Primary outcomes: none Secondary outcomes
|
|
| Notes |
Sample size calculation: no information Ethic approval: no information Funding source: funded or partially funded by pharmaceutical industry Conflicts of interest: The trial director is affiliated with AstraZeneca who produces the medication tested. Comments from review authors (limitations)
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: no information |
| Allocation concealment (selection bias) | Unclear risk | Comment: no information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: Trial registration stated that the study had triple blinding (participant, care provider and investigator), however, there was no information on how the blinding was secured or if it was adequately maintained throughout the study to permit a judgement of low or high risk of bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: Trial registration stated that the study had triple blinding (participant, care provider and investigator), however, there was no information on how the blinding was secured or if it was adequately maintained throughout the study to permit a judgement of low or high risk of bias. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: dropout 5/13 in experimental intervention and 3/11 in placebo group. No information on reasons for dropout or imputation method |
| Selective reporting (reporting bias) | High risk | Comment: The protocol mentioned psychotic‐like symptoms and severity of psychiatric symptoms + mood, anger, impulsiveness, hostility and anxiety in patients with BPD. Only psychotic and dissociative symptoms were reported. |
| Vested Interest (funding and/or author affiliations) | High risk | Comment: Trial director is from AstraZeneca and the medication tested is from AstraZeneca. |
| Other bias | Low risk | Comment: no indication of other bias |
Bellino 2014.
| Study characteristics | ||
| Methods | 12‐week trial with 2 arms:
Duration of trial: 12 weeks + 24 weeks follow‐up Country: Italy Setting: outpatient |
|
| Participants |
Methods of recruitment of patients: patients with BPD attending the conducting clinic Overall sample size: 43 Diagnosis of borderline personality disorder: DSM‐IV‐TR Means of assessment: clinical expert; SCID‐I and SCID‐II Mean age: 25.2 years (SD 6.4; range = no information) Sex: 76.47% women Comorbidity: no comorbid axis I or II disorders Inclusion criteria
Exclusion criteria
|
|
| Interventions |
Experimental group
Treatment name: omega‐3 fatty acids: eicosapentaenoic acid (EPA) + docosahexaenoic acid (DHA) + valproic acid
Number randomised to group: 23
Duration: 12 weeks Control/comparison group Comparison name: valproic acid Number randomised to group: 20 Duration: 12 weeks Both groups Concomitant psychotherapy: not allowed Concomitant pharmacotherapy: not allowed Proportions of participants taking standing medication during trial observation period: none |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
Sample calculation: no information Ethics approval: yes Funding source: no funding received Conflicts of interest: No conflicts of interests reported Comments from trial authors (limitations): "The present study suffers from some limitations that should be considered: (a) the small sample size; (b) the lack of a placebo controlled group; and (c) the exclusion of patients with an Axis I co‐diagnosis" (Bellino 2014, p. 131). Comments from review authors: supplemental information received through personal email correspondence with the authors in January 2019 (Bellino 2019 [pers comm]) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: ”Research Randomizer (Urbaniak and Plous, Social Psychology Network Wesleyan University, Middletown, CT), a free web‐based service for randomization” (Bellino 2019 [pers comm]) |
| Allocation concealment (selection bias) | High risk | Quote: ”Allocation was not concealed as the study was not blind” (Bellino 2019 [pers comm]). |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: ”(...) the study was not blind” (Bellino 2019 [pers comm]). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk |
Quote: "In this study, assessment was performed by an investigator who received a training session on psychometric instruments, prior to starting the investigation" (Bellino 2014, p. 127). Quote: ”Only the investigator who performed patients' assessment was blind to the treatment they received” (Bellino 2019 [pers comm]). |
| Incomplete outcome data (attrition bias) All outcomes | High risk |
Quote: "We had a rather high rate of dropouts (almost 20%) in our study" (Bellino 2014 p. 130). Quote: "We used statistical analysis to evaluate patients completing the 12‐week trial" (Bellino 2014 p. 127). Comment: High dropout rate. Efficacy analysis on completers only |
| Selective reporting (reporting bias) | Low risk | Comment: no deviations from protocol |
| Vested Interest (funding and/or author affiliations) | Low risk | Comment: The author declared that there was no conflict of interest. |
| Other bias | Low risk | Comment: no indication of other bias |
Black 2014.
| Study characteristics | ||
| Methods | 8‐week trial with 3 arms:
Duration of trial: 8 weeks Country: USA Setting: outpatient |
|
| Participants |
Methods of recruitment of patients: referral, advertisements and word of mouth Overall sample size: 95 Diagnosis of borderline personality disorder: DSM (no information on version) Means of assessment: SCID (no information on version) Mean age: 29.5 years (SD no information; range = 18‐45) Sex: 70.53% women Comorbidity: mood disorders, anxiety disorders, substance abuse, axis I disorders Inclusion criteria
Exclusion criteria
|
|
| Interventions |
Experimental group
Treatment name: quetiapine 300 mg/day (moderate dosage)
Number randomised to group: 33
Duration: 8 weeks Comparison group Comparison name: quetiapine 150 mg/day (low dosage) Number randomised to group: 33 Duration: 8 weeks Control group Control name: placebo Number randomised to group: 29 Duration: 8 weeks All groups Concomitant psychotherapy: Participants entering the trial could not begin any type of psychotherapy during the trial. Concomitant pharmacotherapy: No other psychotropic medication than benzodiazepines and anticholinergics was permitted. Proportions of participants taking standing medication during trial observation period: Only one participant (in the placebo group) took benzodiazepines during the trial. |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
Sample calculation: yes Ethics approval: yes Funding source: funded or partially funded by pharmaceutical industry Conflicts of interest: Dr Schulz has served as a consultant to Eli Lilly, Forum, Genentech, and Teva. Comments from trial authors (limitations)
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: insufficient information on how random sequence was generated to permit a judgement of low or high risk of bias |
| Allocation concealment (selection bias) | Unclear risk | Comment: Insufficient information on concealment of random sequence generation to permit a judgement of low or high risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk |
Quote: "Participants, site personnel, and investigators were blind to treatment group assignment." Quote: "To preserve blinding, all participants received one bottle of 150 mg quetiapine (or placebo) tablets initially, and then after 4 weeks received two bottles; the second bottle contained either 150 mg quetiapine tablets (for the moderate‐dosage group) or placebo tablets (for the low‐dosage and placebo groups)." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk |
Quote: "Participants, site personnel, and investigators were blind to treatment group assignment." Comment: The article provided inadequate information on who assessed the outcomes and whether blinding was maintained to permit a judgement of low or high risk of bias. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: Dropout was 33%. ITT analysis was performed. Adverse events predicted dropout. |
| Selective reporting (reporting bias) | High risk | Comment: All outcomes from 1‐9 were defined beforehand as primary outcomes. However, in the trial, only Zan‐BPD was stated as the primary outcome. |
| Vested Interest (funding and/or author affiliations) | High risk | Quote: “Supported by a grant from AstraZeneca to Dr. Schulz, with subcontracts to Drs. Black and Zanarini” (Black 2014, p. 1181). |
| Other bias | Low risk | Comment: no indication of other bias |
Bogenschutz 2004.
| Study characteristics | ||
| Methods | 12‐week trial with 2 arms:
Duration of trial: 12 weeks (patients had to be free of mood stabilisers, antipsychotics, benzodiazepines, and antidepressants for at least 2 weeks prior to participation) Country: no information Setting: outpatient |
|
| Participants |
Methods of recruitment of patients: Patients were recruited from the community and from outpatient clinics at a university psychiatric hospital. Overall sample size: 40 Diagnosis of borderline personality disorder: DSM‐IV Means of assessment: SCID‐II Mean age: 32.6 years (SD 10.3; range = 18‐54) Sex: 62.50% women Comorbidity: "Patients met criteria for a mean of 2.9 SCID‐II personality disorders (including BPD) and a mean of 2.2 Axis I diagnoses from the MINI" (Bogenschutz 2004, p. 106). Inclusion criteria: no information Exclusion criteria
|
|
| Interventions |
Experimental group
Treatment name: olanzapine
Number randomised to group: 20
Duration: no information Control/comparison group Comparison name: placebo Number randomised to group: 20 Duration: no information Both groups Concomitant psychotherapy: "Patients were allowed to continue ongoing psychotherapy (if initiated more than 3 months prior to randomisation)" (Bogenschutz 2004, p. 105). Concomitant pharmacotherapy: medication for stable, chronic medical conditions such as hypertension Proportions of participants taking standing medication during trial observation period: no information |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
Sample calculation: no information Ethics approval: no information Funding source: funded or partially funded by pharmaceutical industry Conflicts of interest: No other conflicts of interest were reported besides funding from the pharmaceutical industry. Comments from trial authors (limitations)
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: The article referred to the trial as being randomised, however inadequate information on random sequence generation was provided to permit a judgement of low or high risk of bias. |
| Allocation concealment (selection bias) | Unclear risk | Comment: no information on allocation concealment to permit a judgement of low or high risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "double‐blind trial", "pseudo‐dose [...] for patients receiving placebo" (Bogenschutz 2004, p. 105). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: The article referred to the trial being double‐blind, however, there was insufficient information on how blinding of outcome assessors was carried out and maintained to permit a judgement of low or high risk of bias. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk |
Comment: "evaluable patients" refers to all patients remaining at least 2 weeks in the study, i.e. who attended both baseline and preliminary assessment after two weeks; reasons for early termination specified (Bogenschutz 2004, p. 106). However, 2 patients dropped out of the olanzapine group due to "violation of protocol" (Bogenschutz 2004, p. 106). Of the 40 patients enrolled, 23 completed the full 12 weeks of the trial (10 in olanzapine group, 13 in placebo group). Reasons for early termination: Lost to follow‐up: 2 in the olanzapine group, 5 in the placebo group Lack of efficacy: 2/2 Weight gain: 2/0 Sedation: 2/0 Violation of protocol: 2/0 Continuous data based on LOCF of patients remaining at least 2 weeks in the trial Dichotomous data based on ITT sample |
| Selective reporting (reporting bias) | Unclear risk | Comment: No protocol available |
| Vested Interest (funding and/or author affiliations) | High risk | Quote: "Supported by a grant from Eli Lilly and Co, Indianalpolis, Ind." (Bogenschutz 2004, p. 104) |
| Other bias | Low risk | Comment: No other apparent sources of bias were found. |
Bozzatello 2017.
| Study characteristics | ||
| Methods | 51 participants; 12‐week open‐label trial with 2 arms:
Duration of trial: 12 weeks Country: Italy Setting: outpatient |
|
| Participants |
Methods of recruitment of patients: "Patients attended the Centre for Personality Disorders of the Psychiatric Clinic, Department of Neuroscience, University of Turin, Italy." (Bozzatello 2017, p. 6) Overall sample size: 51 Diagnosis of borderline personality disorder: DSM‐V Means of assessment: SCID‐I and SCID‐II Mean age: 24.7 years (SD 5.3; range = no information) Sex: 62.5% women Comorbidity: no information Inclusion criteria
Exclusion criteria
|
|
| Interventions |
Experimental group
Treatment name: asenapine 5‐10 mg/day (dose range = 5‐10 mg/day)
Number randomised to group: 25
Duration: 12 weeks Control/comparison group Comparison name: olanzapine 5‐10 mg/day Number randomised to group: 26 Duration: 12 weeks Both groups Concomitant psychotherapy: Psychotherapy was not allowed during the trial (exclusion criterion). Concomitant pharmacotherapy: Psychothropics were not allowed during the trial (exclusion criterion). Proportions of participants taking standing medication during trial observation period: no information |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
Sample calculation: no information. Post hoc power calculations were performed. Ethics approval: yes, “Approval was obtained from the ethics committee of the University Hospital “Città della Salute e della Scienza – Ospedale dell’Ordine Mauriziano” of Turin.” (Bozzatello 2017, p. 811) Funding source: no funding received Conflicts of interest: No conflicts of interests were reported. Comments from trial authors (limitations)
Comments from review authors: dropouts due to “lack of compliance” (Bozzatello 2017, p. 809), i.e. n = 4 in asenapine group and n = 3 in olanzapine group; this introduces risk of bias. Unclear how “lack of compliance” was defined |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “Research Randomizer (Urbaniak and Plous, Social Psychology Network Wesleyan University, Middletown, CT), a free web‐based service for randomization, was used”. (Bozzatello 2017, p. 811) |
| Allocation concealment (selection bias) | High risk | Comment: no mention of allocation concealment. Unlikely that sequence generation was concealed due to open‐label design |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: open‐label trial. Participants and personnel were not blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk |
Comment: open‐label trial. Outcome assessor was not blinded. Quote: “Assessment was performed by an investigator (P.B.) who had received a training session on psychometric instruments prior to the study.” (Bozzatello 2017, p. 812) |
| Incomplete outcome data (attrition bias) All outcomes | High risk |
Quote: “Statistical analysis were performed both in the group of patients who completed the trial and in the whole group of patients who were randomized including drop‐outs. In the second group, intention to treat (ITT) analysis was performed with the last observation carried forward (LOCF).” (Bozzatello 2017, p. 812) Quote: “The ITT‐LOCF analysis was performed on the entire sample of 51 patients recruited." (Bozzatello 2017, p. 812) Quote: “We had a rather high drop‐out rate in our study (21.7%) […] intention to treat analysis with last observation carried forward was performed to analyze data in the whole group of patients who entered the trial, and the significant effects of the two drugs found with the ANOVA were the same obtained in the group of completers.” (Bozzatello 2017, p. 817) Quote: "Drop‐outs due to “lack of compliance” " (Bozzatello 2017, p. 809), i.e. n = 4 in asenapine group and n = 3 in olanzapine group; this introduces risk of bias. Unclear how “lack of compliance” was defined |
| Selective reporting (reporting bias) | Low risk | Comment: reporting according to protocol |
| Vested Interest (funding and/or author affiliations) | Low risk | Quote: “This research received no sources of funding to assist with conducting the study and preparing the manuscript”. (Bozzatello 2017, p. 817) |
| Other bias | Low risk | Comment: no apparent other biases detected |
Cowdry 1988.
| Study characteristics | ||
| Methods | A 6‐week trial with 5 arms:
Duration of trial: 6 weeks x 5 Country: USA Setting: outpatient |
|
| Participants |
Methods of recruitment of patients: referral by private psychotherapist Overall sample size: 16 Diagnosis of borderline personality disorder: DSM‐III Means of assessment: DIB Mean age: 31.6 years (range = 23‐42) Sex: 100% women Comorbidity: other DSM‐III Axis II diagnoses: dependent personality = 15 (94%); avoidant personality = 13 (81%), histrionic personality = 10 (63%), schizotypal personality = 6 (37%), narcissistic personality = 3 (19%), paranoid personality = 2 (12%); DSM‐III Axis I affective disorder diagnoses of previous episodes: atypical bipolar disorder = 1 (6%), major depressive episode with melancholia = 4 (25%), major depressive episode without melancholia = 3 (19%), dysthymic disorder only = 5 (31%) Inclusion criteria
Exclusion criteria: none mentioned |
|
| Interventions |
Experimental group
Treatment name: alprazolam
Number randomised to group: 16
Duration: 6 weeks (a two‐week dosage adjustment period, four weeks of constant dosage, a week of tapering the medication, and at least one medication‐free week before beginning the next drug trial (intervention)) Experimental group Treatment name: carbamazepine Number randomised to group: 16 Duration: 6 weeks (a two‐week dosage adjustment period, four weeks of constant dosage, a week of tapering the medication, and at least one medication‐free week before beginning the next drug trial (intervention)) Experimental group Treatment name: trifluoperazine hydrochloride Number randomised to group: 16 Duration: 6 weeks (a two‐week dosage adjustment period, four weeks of constant dosage, a week of tapering the medication, and at least one medication‐free week before beginning the next drug trial (intervention)) Experimental group Treatment name: tranylcypromine sulfate Number randomised to group: 16 Duration: 6 weeks (a two‐week dosage adjustment period, four weeks of constant dosage, a week of tapering the medication, and at least one medication‐free week before beginning the next drug trial (intervention)) Control/comparison group Comparison name: placebo Number randomised to group: 16 Duration: 6 weeks (a two‐week dosage adjustment period, four weeks of constant dosage, a week of tapering the medication, and at least one medication‐free week before beginning the next drug trial (intervention)) Both groups Concomitant psychotherapy: no information Concomitant pharmacotherapy: no information Proportions of participants taking standing medication during trial observation period: no information |
|
| Outcomes |
Primary
Secondary
|
|
| Notes |
Sample calculation: no information Ethics approval: no information Funding source: no information Conflicts of interest: no information Comments from trial authors (limitations): The number of patients was small, the medication trials were brief, and the outcome measures had to be adapted to the special needs of this population and therefore were not well validated. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: The study was referred to as randomised; however, the method used to generate the allocation sequence was not described in sufficient detail to allow an assessment of whether it produced comparable groups. |
| Allocation concealment (selection bias) | Unclear risk | Comment: Allocation concealment and the method used to conceal the allocation sequence was not described. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quotes: "The laboratory results were examined by the non‐blinded physician, and, regardless of whether the patient was actually receiving carbamazepine, a new "target dosage" was provided to the blinded physician, to be reached if side effects did not prevent dosage increases. During the final two randomized trials (trifluoperazine and tranylcypromine), the patient adhered to the MAOI diet; an initial target dosage of four capsules was established, adjusted by the blinded physician at the end of the first week (based largely on side effects), and thereafter was held constant unless major side effects necessitated a dosage decrease." (Cowdry 1988, p. 112) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: The article referred to the trial being double‐blind, however, there was insufficient information on how blinding of outcome assessors was carried out and maintained to permit a judgement of low or high risk of bias. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: large attrition rates and carry‐over effects (11 out of 16 participants dropped out). Patients who dropped out of one trial were unavailable for the following trials. |
| Selective reporting (reporting bias) | High risk |
Comments: Several outcomes were not reported e.g. adverse events, which was mentioned to have been assessed by “a 36‐item checklist during the final week of each medication trial” (Cowdry 1988, p. 188, 113), spontaneous reporting of self‐harm is not reported. Subscales of adapted CGI scale and modified Bunney‐Hamburg scales were not reported. Unclear reporting of which scale was used for each outcome and at which time point each outcome was assessed. |
| Vested Interest (funding and/or author affiliations) | Unclear risk | Comment: insufficient information on funding and conflict of interest to permit judgement of high or low risk of bias |
| Other bias | Unclear risk | Comment: There was obvious carry‐over effects despite washout periods between each medication trial. |
Crawford 2018.
| Study characteristics | ||
| Methods | 52‐weeks trial with 2 arms:
Duration of trial: 52 weeks Country: UK Setting: inpatient and outpatient |
|
| Participants |
Methods of recruitment of patients: “Potential participants were initially approached about the trial by any healthcare professional who was involved in their care, providing that the consultant psychiatrist for the team had agreed in principle to patients under their care taking part in the study. If a psychiatrist or other healthcare professional had a patient under their care who they believed met the eligibility criteria, they then introduced the patient to the trial and provided them with an information sheet. When the patient provided verbal agreement to discuss their eligibility and possible enrolment into the trial with a member of the research team, a screening number was assigned and contact details passed on to the research team to discuss consent.” (Crawford 2018, p. 6) Overall sample size: 276 Diagnosis of borderline personality disorder: DSM‐IV Means of assessment: IPDE and SCID‐I Mean age: 36.1 years (SD 11.0; range = no information) Sex: 75.36% women Comorbidity: no information; however, patients with a coexisting diagnosis of bipolar, psychotic disorders were excluded. Inclusion criteria
Exclusion criteria
|
|
| Interventions |
Experimental group
Treatment name: lamotrigine + usual care
Number randomised to group: 137
Duration: 52 weeks Control/comparison group Comparison name: inert placebo + usual care Number randomised to group: 139 Duration: 52 weeks Both groups Concomitant psychotherapy: no information Concomitant pharmacotherapy: no information. Patients were excluded if they already were prescribed a mood stabiliser within the past 4 weeks. Both groups received usual care. Proportions of participants taking standing medication during trial observation period: no information |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
Sample calculation: yes Ethics approval: "The trial was approved by the London‐Central Research Ethics Committee (reference number 2/LO/1514)". (Crawford 2018, p. 12) Funding source: funded by grants from universities, authorities or research foundations Conflicts of interest: No conflicts of interest were reported. Comments from trial authors (limitations): "Levels of adherence in this pragmatic trial were low, but greater adherence was not associated with better mental health". (Crawford 2018, p. viii) Comments from review authors: Supplemental information regarding data received through personal email correspondence with the authors in November 2020 (Crawford 2020 [pers comm]) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: ”Study participants were then randomised centrally by the Nottingham Clinical Trials Unit using a remote web‐based system. We used permuted stacked blocks stratified by study centre, severity of personality disorder and extent of hypomanic symptoms. The block size was randomly assigned between 4 and 6.” (Crawford 2018, p. xx) |
| Allocation concealment (selection bias) | Low risk | Quote: "Site pharmacies were unblinded to trial arm allocation and were provided with a list of the randomisation codes and corresponding trial arm allocation for that site. The trial medication was produced with tear‐off labels that identified it as being lamotrigine or placebo in a coded format, so that pharmacy staff could dispense the appropriate medication for a participant. Pharmacy procedures required that the tear‐off label was removed during dispensing and added to trial documents for accountability. The need to maintain the blinding of researchers and other individuals at the site was made clear to those delegated to work on the trial within the pharmacy." (Crawford 2018 p. 5‐6) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk |
Quote: "The trial medication was produced with tear‐off labels that identified it as being lamotrigine or placebo in a coded format, so that pharmacy staff could dispense the appropriate medication for a participant. Pharmacy procedures required that the tear‐off label was removed during dispensing and added to trial documents for accountability. The need to maintain the blinding of researchers and other individuals at the site was made clear to those delegated to work on the trial within the pharmacy." (Crawford 2018 p. 5‐6) Quote: “All patients, carers and referring psychiatrists were blinded to treatment assignment until the participant had left the trial or until 52 weeks post‐randomisation (whichever was the longer).” (Crawford 2018, p. 5) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: “Blinding of investigators, researchers, the trial manager and the trial statistician was maintained until all data were entered, the database was locked and initial analyses of trial data were complete. The exception to this was for participants whose referring psychiatrist was also the principal investigator, in which case the allocation for that particular participant was revealed following the final assessment.” (Crawford 2018, p. 5) |
| Incomplete outcome data (attrition bias) All outcomes | High risk |
Quote: ”The primary analysis was performed according to the intention‐to‐treat principle, without imputation of missing data.” (Crawford 2018, p. 4) "For the main analysis, complete‐case analysis was used in which participants with missing data were excluded." (Crawford 2018, HTA‐report, p. 10) |
| Selective reporting (reporting bias) | Low risk | Comment: The published protocol matched with the full report. |
| Vested Interest (funding and/or author affiliations) | Low risk | Quote: "Funding for this trial was provided by the Health Technology Assessment programme of the National Institute for Health Research (NIHR) and will be published in full in Health Technology Assessment ;Vol. 22, No. 17. See the NIHR Journals Library website for further project information. The Imperial Biomedical Research Centre Facility, which is funded by NIHR, also provided support that has contributed to the research results reported within this paper. Part of Richard Morris’ salary during the project was paid by NIHR Collaboration for Leadership in Applied Health Research and Care East Midlands". (Crawford 2018, p. viii) |
| Other bias | Low risk | Comment: no indication of other bias |
De la Fuente 1994.
| Study characteristics | ||
| Methods | 32‐day trial with 2 arms:
Duration of trial: 32 days (after at least 10 days psychotropic drug washout, 15 days for TCAs and MAOIs; no patient had taken neuroleptics in the 2‐month period before the trial) Country: Belgium Setting: inpatient |
|
| Participants |
Methods of recruitment of patients: no information Overall sample size: 20 Diagnosis of borderline personality disorder: DSM‐III‐R Means of assessment: DIB Mean age: 32.7 years (SD = no information; range = 22‐45) Sex: 70% women Comorbidity: no information Inclusion criteria
Exclusion criteria
|
|
| Interventions |
Experimental group
Treatment name: carbamazepine
Number randomised to group: 10
Duration: 32 days Control/comparison group Comparison name: placebo Number randomised to group: 10 Duration: 32 days Both groups Concomitant psychotherapy: Supportive atheoretical psychotherapy was administered to all patients. Concomitant pharmacotherapy: There was a 10‐day psychotropic drug washout prior to the trial and a 15‐day drug washout for TCAs and MAOIs. "No patient had taken neuroleptics in the 2‐month period before the study." (De la Fuente 1994, p. 480) Proportions of participants taking standing medication during trial observation period: no further details |
|
| Outcomes |
Primary outcomes: none Secondary outcomes
|
|
| Notes |
Sample calculation: no information Ethics approval: no information Funding source: unclear funding Conflicts of interest: No conflicts of interest were reported. Comments from trial authors (limitations)
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: Insufficient information on the method used for random sequence generation to permit a judgement of low or high risk of bias |
| Allocation concealment (selection bias) | Unclear risk | Comment: Insufficient information to permit judgement of high or low risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: Insufficient information on how blinding of patients was carried out and maintained to permit a judgement of low or high risk of bias |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "The study was carried out by two clinicians. One of them [...] was blind to the drug treatment and performed all the clinical and psychometric assessments. [...] We instructed the patients to not communicate side effects to the blind clinician." (De la Fuente 1994, p. 480) |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk |
Quote: "Of the 20 patients enrolled in the trial, we randomized 10 into the CBZ group and 10 received PLC. [...] Two patients receiving CBZ dropped out. [...] No patient on PLC dropped out." (De la Fuente 1994, p. 481) Comment: Reasons for early termination specified (De la Fuente 1994, p. 481): dramatic increase in frequency and intensity of aggression (against self and others): 2 in carbamazepine group, 0 in placebo group However, it remained unclear if the reported continuous outcomes were measured by LOCF analyses. We decided to use the same numbers of patients for which dichotomous outcomes were reported. For dichotomous outcomes, lacking numbers of patients were imputed as having the unfavourable results. |
| Selective reporting (reporting bias) | Unclear risk | Comment: no protocol found |
| Vested Interest (funding and/or author affiliations) | Unclear risk | Comment: authors affiliated with Department of Psychiatry, Erasme Hospital, Free University, Brussels. No details about funding or sponsoring provided |
| Other bias | Low risk | Comment: No apparent other sources of bias found |
Frankenburg 2002.
| Study characteristics | ||
| Methods | 24‐week trial with 2 arms:
Duration of trial: 24 weeks Country: USA Setting: outpatient |
|
| Participants |
Methods of recruitment of patients: advertisement in newspapers in Boston Overall sample size: 30 Diagnosis of borderline personality disorder: DSM‐IV Means of assessment: Diagnostic Interview for DSM‐IV Personality Disorders (DIPD‐IV borderline module) Mean age: 26.85 years (SD = no information; range = no information) Sex: 100% women Comorbidity: bipolar II disorder Inclusion criteria
Exclusion criteria
|
|
| Interventions |
Experimental group
Treatment name: valproate semisodium
Number randomised to group: 20
Duration: 6 months Control/comparison group Comparison name: placebo Number randomised to group: 10 Duration: 6 months Both groups Concomitant psychotherapy: no other psychotropic medication allowed Concomitant pharmacotherapy: no patient was in psychotherapy Proportions of participants taking standing medication during trial observation period: no information |
|
| Outcomes |
Primary outcomes: none Secondary outcomes
|
|
| Notes |
Sample calculation: no information Ethics approval: no information Funding source: funded or partially funded by pharmaceutical industry Conflicts of interest: No other conflicts of interest were reported besides funding from the pharmaceutical industry. Comments from trial authors (limitations)
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Prearranged random number sequence" (Frankenburg 2002, p. 443) |
| Allocation concealment (selection bias) | Low risk |
Quote: "Tablets were supplied in numbered bottles containing drug or placebo as determined by a prearranged random number sequence". (Frankenburg 2002, p. 443) Comment: Participants and investigators enrolling participants could not foresee assignment because of sequentially numbered drug containers of identical appearance. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Tablets were supplied in numbered bottles [...] Each tablet contained either 250 mg of valproate semisodium or matching inert placebo. [...] One of the investigators [...] was given either the real or a sham level (if the subject was receiving placebo). This same investigator met with the subjects for [...] medication checks and adjusted the dose according to perceived response, reported or sham level, and side effects." (Frankenburg 2002, p. 443) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: no information given on who exactly assessed outcomes, or how outcome assessor was blinded |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk |
Quote: "Endpoint values [...] are based on last observation carried forward." (Frankenburg 2002, p. 444) Comments: reasons for early termination specified (Frankenburg 2002, p. 444): Lost to follow‐up: 9 in the valproate semisodium group, 3 in the placebo group Moved out of the area: 1/0 Inability to use reliable forms of contraception: 1/0 Withdrawal of consent: 1/0 Diarrhoea and tremors: 1/0 Development of a major depressive episode: 0/2 Hair loss: 0/1 For dichotomous outcomes, lacking numbers of patients were imputed as having the unfavourable result. Of the 30 patients enrolled, 11 completed the full 24 weeks of the trial (7 in valproate semisodium group, 4 in placebo group). |
| Selective reporting (reporting bias) | Unclear risk | Comment: no protocol found |
| Vested Interest (funding and/or author affiliations) | High risk | Quote: "Supported by a grant from Abbott Laboratories, Chicago, Ill." (Frankenburg 2002, p. 442) |
| Other bias | Low risk | Comment: No apparent other sources of bias found |
Goldberg 1986.
| Study characteristics | ||
| Methods | 12‐week trial with 2 arms:
Duration of trial: 12 weeks (after 1 week placebo washout) Country: USA Setting: outpatient |
|
| Participants |
Methods of recruitment of patients: "A short version of the SIB was placed as an advertisement in the local newspaper to recruit patients". (Goldberg 1986, p. 681) Overall sample size: 40 Diagnosis of borderline personality disorder: DSM‐III Means of assessment: Schedule of Interviewing Schizotypal Personalities (SIB) Mean age: 32 years (SD = no information; range = no information) Sex: 58% women Comorbidity: schizotypal personality disorder and having at least one psychotic symptom Inclusion criteria: no information Exclusion criteria
|
|
| Interventions |
Experimental group
Treatment name: thiothixene
Number randomised to group: 24
Duration: 12 weeks Control/comparison group Comparison name: placebo Number randomised to group: 26 Duration: 12 weeks Both groups Concomitant psychotherapy: no information Concomitant pharmacotherapy: participants had to pass one week placebo washout, no further details Proportions of participants taking standing medication during trial observation period: no information |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
Sample calculation: no information Ethics approval: no information Funding source: unclear funding Conflicts of interest: No conflicts of interest were reported. Comments from trial authors (limitations)
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: Insufficient information on the method used for random sequence generation to permit a judgement of low or high risk of bias |
| Allocation concealment (selection bias) | Unclear risk | Comment: no information given on allocation concealment to permit a judgement of low or high risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Both agents were provided in identical‐appearing capsules containing 5 mg of thiothixene hydrochloride or an equivalent amount of lactose for placebo. The initial dose for all patients was one capsule [...] and on each succeeding visit the dose was increased by one capsule unless side‐effects or marked improvement intervened. A maximum dose of 40 mg, or eight capsules, was to be allowed [...]." (Goldberg 1986, p. 682) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: The article referred to the trial being double‐blind, however, there was insufficient information on how blinding of outcome assessors was carried out and maintained to permit a judgement of low or high risk of bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk |
Quote: "Patients who terminated their participation early were assessed at that point and those assessments were taken as their endpoints." (Goldberg 1986, p. 682) Comment: Of the 50 patients enrolled, 40 completed treatment (17 in thiothixene group, 23 in placebo group). Reasons for early termination: Adverse effects: 7 in thiothixene group, 0 in placebo group Lack of efficacy: 0/3 Continuous data based on LOCF |
| Selective reporting (reporting bias) | Unclear risk | Comment: no protocol found |
| Vested Interest (funding and/or author affiliations) | Unclear risk | Comment: no details on sponsoring or funding. Authors affiliated with the Department of Psychiatry, Medical College of Virginia/Virginia Commonwealth University, Richmond |
| Other bias | Low risk | Comment: No apparent other sources of bias found |
Grant 2022.
| Study characteristics | ||
| Methods | A 12‐week parallel trial with 2 arms:
Duration of trial: 12 weeks + a 13th week of tapering/safety Country: US Setting: Outpatient |
|
| Participants |
Method of recruitment of participants: Recruitment from clinics and local advertisements Overall sample size: 80 Diagnosis of borderline personality disorder: DSM‐5 Means of assessment: Zan‐BPD Mean age: mean age 39.7 ± 11.6 (range: 19‐61) Sex: 56.3% women Comorbidity: Brexpiprazole group: N = 25 (62.5%), including anxiety disorder (N = 19), mood disorder (N = 15), ADHD (N = 4), and eating disorder (N = 4). Placebo group: N = 25 (65%), including anxiety disorder (N = 19), mood disorder (N = 13), ADHD (N = 1), and eating disorder (N = 3). Inclusion criteria
Exclusion criteria
|
|
| Interventions |
Experimental group
Treatment name: Brexpiporazole 1 mg/day for 1 week, then 2 mg/day for 10 weeks, then 1 mg/day for 1 week (taper period)
Number randomised to group: 40
Duration: 12 weeks + 1 tapering week Control/comparison group Comparison name: Placebo in the same administration scheme as the experimental group Number randomised to group: 40 Duration: 12 weeks + 1 tapering week Both groups Concomitant psychotherapy: No information Concomitant pharmacotherapy: In the brexpiprazole group, eight were on antidepressants, five were on antiepileptics and three were on stimulants. In the placebo group, ten were on antidepressants, seven were on antiepileptics and three were on stimulants. Proportions of participants taking standing medication during trial observation period: 12 out of 40 (30%) in the brexipiprazole group took psychotropics and 14 out of 40 (35%) in the placebo group took psychotropics. |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
Sample size calculation: Yes. It was determined that 35 participants were needed in each treatment group to detect a difference with an overall 5% type I error risk. Ethics approval: Yes. Approval by the University of Chicago Institutional Review Board Funding source: Funded or partially funded by pharmaceutical industry Conflicts of interest: The first author has received grants from pharmaceutical industry; last author has worked as a consultant in pharmaceutical industry. Comments from trial authors (limitations)
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “The University of Chicago’s investigational pharmacy, which was independent of the research team, randomised all participants (block sizes of eight, using computer‐generated randomisation with no clinical information) to either the brexpiprazole or matching placebo in a 1:1 fashion”. (Grant 2022 p. 59) |
| Allocation concealment (selection bias) | Low risk |
Quote: “The University of Chicago’s investigational pharmacy, which was independent of the research team, randomised all participants." (Grant 2022 p. 59) Comment: trial medication was handled by an independent pharmacy. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: “The study blind was maintained by having placebo and active treatments of identical size, weight, shape and colour, as confirmed by the independent pharmacy.” (Grant 2022 p. 59) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: No information given on who performed the outcome assessment and how their blinding was maintained |
| Incomplete outcome data (attrition bias) All outcomes | High risk |
Comment: Participants were included in efficacy analyses if they completed at least one post‐randomisation visit. Imputation was not undertaken for missing data. Of the 80 patients enrolled, 55 completed the full 12 weeks of the trial (30 in the brexpiprazole group and 25 in placebo group). 69 completed at least one post‐randomisation visit (35 in the brexpiprazole group and 34 in placebo group). Reasons for early termination: Discontinued post‐randomisation but before first visit: 5 in the brexpiprazole group, 3 in the placebo group Lost to follow‐up: 5 in the brexpiprazole group, 3 in the placebo group |
| Selective reporting (reporting bias) | Unclear risk | Comment: Data have been posted in full (with the exception of two scales but a reasonable explanation for this has been given). However, this posting lacks a thorough quality control as of yet, and it is unclear why these data have not been included in the full paper. |
| Vested Interest (funding and/or author affiliations) | High risk | Quote: “This study was funded by an investigator initiated grant from Otsuka Pharmaceuticals. S.R.C.’s role in this study was funded by a Wellcome Trust Clinical Fellowship (110049/Z/15/Z and 110049/Z/15/A).” (Grant 2022 p. 63) |
| Other bias | Low risk | Comment: No other apparent sources of bias found |
Hallahan 2007.
| Study characteristics | ||
| Methods | 12‐week trial with 2 arms:
Duration of trial: 12 weeks Country: Ireland Setting: outpatient |
|
| Participants |
Methods of recruitment of patients: "All patients were recruited from the Accident and Emergency (A&E) Department of Beaumont Hospital, an academic teaching hospital in Dublin, Ireland." (Hallahan 2007, p. 118) Overall sample size: 49 Diagnosis of borderline personality disorder: DSM‐IV Means of assessment: SCID‐II Mean age: 30.6 years (SD = no information; range = no information) Sex: 65.31% women Comorbidity: recurrent self‐harm Inclusion criteria: no information Exclusion criteria
|
|
| Interventions |
Experimental group
Treatment name: omega‐3 fatty acid
Number randomised to group: 22
Duration: 12 weeks Control/comparison group Comparison name: placebo Number randomised to group: 27 Duration: 12 weeks Both groups Concomitant psychotherapy: "During the course of the study patients continued to receive standard psychiatric care and had changes to their psychotropic medication as prescribed." (Hallahan 2007, p. 118). Patients with changes to or introduction of psychotropic medication during the 6 weeks prior to screening were not eligible. Concomitant pharmacotherapy: not allowed Proportions of participants taking standing medication during trial observation period: 53.1% of participants continued to receive standard psychiatric care and had changes to their psychotropic medication as prescribed. |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
Sample calculation: yes Ethics approval: yes Funding source: funded by grants from universities, authorities or research foundations Conflicts of interest: Trial medication was provided by a pharmaceutical company. Comments from trial authors (limitations): "Although 14 patients reported episodes of self‐harm during the study, it was known a priori that the study was insufficiently powered to detect significant differences between groups". (Hallahan 2007, p. 122) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "computer‐generated list" (Hallahan 2007, p. 119) |
| Allocation concealment (selection bias) | Low risk |
Quote: "An independent colleague dispensed either active or placebo capsules according to a computer‐generated list. The code was only revealed to the researchers once data collection was complete". (Hallahan 2007, p. 119) Comment: Participants and investigators enrolling participants could not foresee assignment because of central allocation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Participants were prescribed four identical capsules of either active agent or placebo [...] Placebo ensured a degree of equality in the incidence of 'fishy breath', the most frequent side‐effect of taking active treatment." (Hallahan 2007, p. 119) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "identical capsules [...] Placebo ensured a degree of equality in the incidence of 'fishy breath' [...] An independent colleague dispensed [...] capsules according to a computer‐generated list. The code was only revealed to the researchers once data collection was complete." (Hallahan 2007, p. 119) |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk |
Comments: LOCF used, reasons for early termination specified (Hallahan 2007 p. 120):
Left district: 1 in active group, 2 in placebo group
Lost to follow‐up: 2 in active group, 2 in placebo group
Admitted to psychiatric hospital: 0 in active group, 2 in placebo group
Refused to continue treatment: 0 in active group, 1 in placebo group
Dichotomous outcomes calculated on basis of the ITT sample Of the 49 patients enrolled, 39 completed treatment (19 of the 22 allocated to active treatment, 20 of the 27 allocated to placebo). |
| Selective reporting (reporting bias) | Unclear risk | Comment: no protocol found |
| Vested Interest (funding and/or author affiliations) | Low risk |
Quotes: "Pronova (now Epax) AS, Lysaker, Norway, provided the active preparation and placebo but were not otherwise involved in the study." (Hallahan 2007, p. 118) "B.H. [i.e. first author] received salary support from the Department of Psychiatry, University of Illinois at Chicago, USA." (Hallahan 2007, p. 122) |
| Other bias | Low risk | Comment: No apparent other sources of bias found |
Hollander 2001.
| Study characteristics | ||
| Methods | 10‐week trial with 2 arms:
Duration: 10 weeks Country: USA Setting: outpatient |
|
| Participants |
Methods of recruitment of patients: "[...] referral from private psychiatrists and mental health professionals in the community, self‐help groups, outpatient clinics at Mount Sinai Medical Center and the Bronx Veterans Affairs medical Center (New York, N.Y.), advertisement, and the media." (Hollander 2001, p. 200) Overall sample size: 21 Diagnosis of borderline personality disorder: DSM‐IV Means of assessment: SCID‐II Mean age: 38.6 years (SD 10.37; range = 18‐62) Sex: 52.38% women Comorbidity: no information Inclusion criteria: no information Exclusion criteria
|
|
| Interventions |
Experimental group
Treatment name: valproate semisodium
Number randomised to group: 12*
Duration: 10 weeks Control/comparison group Comparison name: placebo Number randomised to group: 4* Duration: 10 weeks Both groups Concomitant psychotherapy: no information Concomitant pharmacotherapy: no information Proportions of participants taking standing medication during trial observation period: no information |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
Sample calculation: no information Ethics approval: no information Funding source: funded or partially funded by pharmaceutical industry Conflicts of interests: No conflicts of interest were reported besides partial funding from the pharmaceutical industry. Comments from trial authors (limitations)
Comments from review authors
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: Insufficient information on method for random sequence generation provided to permit a judgement of low or high risk of bias |
| Allocation concealment (selection bias) | Unclear risk | Comment: insufficient information to permit judgement of high or low risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk |
Quote: "The treating psychiatrist was kept blind to patient medication; blood valproate levels were read and dose adjustments to both valproate semisodium and placebo were determined by a psychiatrist not seeing patients for this study." (Hollander 2001, p. 201) Comment: no information given if opaque capsules were used, and if the placebo pseudo‐dose was also "adjusted" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "clinician‐rated outcome measures [...] based on the average of the ratings of the treating psychiatrist and independent evaluator (a psychologist blind to side effects as well as to medication group)" (Hollander 2001, p. 201) |
| Incomplete outcome data (attrition bias) All outcomes | High risk |
Quote: "Patients taking valproate semisodium had a 50% dropout rate [...] versus 100% dropout in the placebo group. [...] No patients dropped out owing to side effects; all dropped out owing to either lack of efficacy or impulsive decisions. [...]" (Hollander 2001, p. 201) Comments: LOCF used (Hollander 2001, p. 202) Initially 21 participants entered the trial; only 16 were randomised to a treatment group without giving reasons. Of the 16 patients randomised, 6 completed treatment (6 in divalproex group, 0 in placebo group). Reasons for early termination: "No patients dropped out owing to side effects; all dropped out owing to either lack of efficacy or impulsive decisions. [...]" (Hollander 2001, p. 201) |
| Selective reporting (reporting bias) | Unclear risk | Comment: no protocol found |
| Vested Interest (funding and/or author affiliations) | High risk | Quote: "Supported in part by grants from the National Institute of Mental Health (1 RO3 MH58168‐01A1), Richville, Md. (Dr. Hollander); Abbott Laboratories, Abbott Park, Ill. (Dr. Hollander); the National Center for Research Resources, National Institutes of Health (5 MO1 RR00071), Rockville, Md., for the Mount Sinai General Clinical Research Center; and the Seaver Foundation and the PBO Foundation, New York, N.Y." (Hollander 2001, p. 199) |
| Other bias | Low risk | Comment: no indication of other bias |
Jariani 2010.
| Study characteristics | ||
| Methods | 12‐week trial with 2 arms:
Duration of trial: 12 weeks Country: Iran Setting: outpatient |
|
| Participants |
Methods of recruitment of patients: no information other than patients on methadone maintenance treatment with BPD diagnosis Overall sample size: 120 Diagnosis of borderline personality disorder: DSM‐IV‐TR Means of assessment: clinical interview Mean age: 27 years (SD = no information; range = no information) Sex: no information on percentage for overall sample size Comorbidity: 100% had a substance use disorder. Inclusion criteria: no information Exclusion criteria: no information; however, stated that patients did not suffer from any axis I disorders or other somatic disorders such as hepatitis or AIDS |
|
| Interventions |
Experimental group
Treatment name: olanzapine (5‐10 mg/d, exact mean final dose unclear)
Number randomised to group: no information*
Duration: 12 weeks Control/comparison group Comparison name: sertraline (50‐100 mg/d, exact mean final dose unclear) Number randomised to group: no information* Duration: 12 weeks Both groups Concomitant psychotherapy: no information Concomitant pharmacotherapy: methadone Proportions of participants taking standing medication during trial observation period: 100% of participants were concomitantly treated with methadone. |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
Sample calculation: yes Ethics approval: yes, "This clinical trial was granted an approval from Medical Ethics Committee at Lorestan Medical University on 12/04/2007". (Jariani 2010, p. 545) Funding source: unclear funding Conflicts of interest: No conflicts of interest were reported. Comments from trial authors (limitations): none mentioned Comments from review authors: *No information provided for the number of participants in each group and not possible to contact the authors |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: According to the sample size formula, 120 males and females on MMT with a diagnosis of BPD were chosen and randomly placed in two groups in which they received either olanzapine (5‐10 mg daily) or sertraline (50‐100 mg daily) (Jariani 2010, p. 545). |
| Allocation concealment (selection bias) | Unclear risk | Comment: no information on allocation concealment reported to permit a judgement of low or high risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: no information on blinding reported to permit a judgement of low or high risk of bias |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: no information on blinding reported to permit a judgement of low or high risk of bias |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: The complete numbers of participants in each group were not provided for analysis results. No details given about patient flow after randomisation |
| Selective reporting (reporting bias) | Unclear risk | Comment: no protocol found |
| Vested Interest (funding and/or author affiliations) | Unclear risk | Comment: no information |
| Other bias | Low risk | Comment: no other sources found |
Kulkarni 2018.
| Study characteristics | ||
| Methods | 8‐week trial with 2 arms:
Duration of trial: 8 weeks + 4 weeks for washout Country: Australia Setting: outpatient |
|
| Participants |
Methods of recruitment of patients: "Participants were recruited primarily via doctor referral, and via printed and electronic advertisements on noticeboards at various sites of The Alfred Hospital (Melbourne, VIC, Australia), and were primarily from Alfred Psychiatry outpatient units and community clinics". (Kulkarni 2018, p. 182) Overall sample size: 34 Diagnosis of borderline personality disorder: DSM‐IV Means of assessment: Zan‐BPD Mean age: 34.4 years (SD = no information; range = no information) Sex: 85.29% women Comorbidity: bipolar II disorder Inclusion criteria
Exclusion criteria
|
|
| Interventions |
Experimental group
Treatment name: memantine‐hydrochloride + treatment‐as‐usual
Number randomised to group: 17
Duration: 8 weeks Control/comparison group Comparison name: placebo + treatment‐as‐usual Number randomised to group: 17 Duration: 8 weeks Both groups Concomitant psychotherapy: psychotherapy and other psychosocial interventions as usual treatment Concomitant pharmacotherapy: Treatment‐as‐usual consisted of medications of antidepressants (selective serotonin reuptake inhibitors, tricyclics, monoamine oxidase inhibitors noradrenergic and specific serotonin antagonist and serotonin noradrenaline reuptake inhibitors), mood stabilisers and antipsychotics, as well as psychotherapy and other psychosocial interventions. Proportions of participants taking standing medication during trial observation period: no information |
|
| Outcomes |
Primary outcomes:
Secondary outcomes
|
|
| Notes |
Sample calculation: yes Ethics approval: yes Funding source: funded by grants from universities, authorities or research foundations Conflicts of interest: No conflicts of interest were reported. Comments from trial authors (limitations)
Comments from review authors: supplemental information provided by trial author through email correspondence (Kulkarni 2020 [pers comm]) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “All participants were individually randomized by The Alfred Clinical Trials Pharmacy to receive either a 10 mg “run‐in dose” for 7 days followed by oral daily memantine hydrochloride 20 mg, or oral placebo according to a computer‐generated randomization list”. (Kulkarni 2018, p. 182) |
| Allocation concealment (selection bias) | Low risk | Comment: central allocation by pharmacy‐controlled randomisation |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk |
Quote: “All study personnel and participants remained blinded to treatment assignment for the duration of the study”. (Kulkarni 2018, p. 182) Comment: The article referred to the trial as being double‐blind but did not provide information on how blinding was secured. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: The article referred to the trial being double‐blind, however, there was insufficient information on how blinding of outcome assessors was carried out and maintained to permit a judgement of low or high risk of detection bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: Intent‐to‐treat imputation method mentioned in abstract. 26.5% discontinued the intervention. From the flow diagram – 8 discontinued from 34 ‐ still 33 analysed |
| Selective reporting (reporting bias) | High risk | Comment: NCT02097706 ‐ Two secondary outcomes (Cogstate (cognitive assessment) & Borderline Evaluation of Severity over Time) were mentioned in the protocol but not included in the full report. No mention of adverse effects as a secondary outcome in protocol |
| Vested Interest (funding and/or author affiliations) | Low risk | Comment: reported no conflicts of interest |
| Other bias | Low risk | Comment: no others sources found |
Leone 1982.
| Study characteristics | ||
| Methods | 6‐week trial with 2 arms:
Duration: 6 weeks Country: USA Setting: outpatient |
|
| Participants |
Methods of recruitment of patients: no information Overall sample size: 80 Diagnosis of borderline personality disorder: DSM‐III Means of assessment: no information Mean age: 30.75 years (SD = no information; range = 16‐59) Sex: 60% women Comorbidity: no information Inclusion criteria: no information Exclusion criteria
|
|
| Interventions |
Experimental group
Treatment name: loxapine
Number randomised to group: 40 (as randomised)
Duration: 6 weeks Control/comparison group Comparison name: chlorpromazine Number randomised to group: 40 (as randomised) Duration: 6 weeks Both groups Concomitant psychotherapy: no information Concomitant pharmacotherapy: "Patients did not receive any other psychotropic medication during the study. Night‐time sedatives were limited to flurazepam and chloral hydrate." (Leone 1982, p. 148) Proportions of participants taking standing medication during trial observation period: no information |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
Sample calculation: no information Ethics approval: no information Funding source: funded or partially funded by pharmaceutical industry Conflicts of interest: No conflicts of interest were reported besides from funding from the pharmaceutical industry. Comments from trial authors (limitations): "Controlled studies are needed which use competing diagnostic schemes and which systematically evaluate drug response over longer periods of time". (Leone 1982, p. 159) Comments from review authors: unable to use outcome data (except for attrition) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk |
Quote: "Matched groups [...] Subjects [...] were selected randomly to receive loxapine or chlorpromazine. [...] There were 24 women and 16 men in each treatment group." (Leone 1982, p. 148) Comment: probably matching procedure used |
| Allocation concealment (selection bias) | Unclear risk | Comment: no information given on allocation concealment to permit a judgement of low or high risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "drugs were supplied in identical opaque capsules". (Leone 1982, p. 148) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: no information given. Within this review, only the outcomes of attrition and adverse effects, that were "recorded upon appearance" (Leone 1982, p. 148), were used. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk |
Comments: continuous outcomes based on available cases:
of the 80 patients enrolled, 69 completed at least 3 weeks of treatment and were included (34 in loxapine group, 35 in placebo group) Reasons for early termination: Did not follow trial procedures: 4 in loxapine group, 4 in chlorpromazine group Had to be admitted to hospital within 3 days: 2 in loxapine group, 1 in chlorpromazine group Only dichotomous outcomes used here, for which patients who had dropped out were imputed as having the negative outcome. |
| Selective reporting (reporting bias) | Unclear risk | Comment: no protocol found |
| Vested Interest (funding and/or author affiliations) | High risk | Quote: "This study was supported by a grant from Lederle Laboratories, Pear River, New York." (Leone 1982, p. 148) |
| Other bias | Low risk | Comment: no indication of other bias |
Linehan 2008.
| Study characteristics | ||
| Methods | 24‐week trial with 2 arms:
Duration: 24 weeks Country: USA Setting: outpatient |
|
| Participants |
Methods of recruitment of patients: no information Overall sample size: 24 Diagnosis of borderline personality disorder: DSM‐IV Means of assessment: SCID‐II Mean age: 36.8 years (SD 9.0; range = no information) Sex: 100% women Comorbidity: no information Inclusion criteria
Exclusion criteria
|
|
| Interventions |
Experimental group
Treatment name: olanzapine (2.5‐15 mg/d, mean final dose 4.46, SD 1.16)
Number randomised to group: 12
Duration: 6 months Control/comparison group Comparison name: placebo Number randomised to group: 12 Duration: 6 months Both groups Concomitant psychotherapy: All participants received DBT. Concomitant pharmacotherapy: no information Proportions of participants taking standing medication during trial observation period: no information |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
Sample calculation: no information Ethics approval: no information Funding source: funded or partially funded by pharmaceutical industry Conflicts of interest: Dr Linehan is a consultant for Eli Lilly and is a member of their speakers/advisory board. Comments from trial authors (limitations): "A limitation to this study is the small sample size". (Linehan 2008, p. 1004) Comments from review authors: data refer to the intention‐to‐treat sample. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "random number sequence" (Linehan 2008, p. e2) |
| Allocation concealment (selection bias) | Unclear risk | Comment: Insufficient information on allocation concealment to permit judgement of low or high risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: Insufficient information on how blinding of participants and personnel was secured and maintained (e.g. packaging of trial medication) to permit judgement of low or high risk of bias |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Patients, psychotherapists, pharmacotherapist, and assessment interviewers were kept naive to medication assignment. At the end of the study, the pharmacotherapist and interviewers were unable to guess group assignment above chance." (Linehan 2008, p. e2) |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk |
Quote: "Outcomes were intent‐to‐treat analyses". (Linehan 2008, p. e3) Comment: Reasons for early termination specified (Linehan 2008, p. e4); patients who had dropped out were imputed as having the negative outcome. |
| Selective reporting (reporting bias) | Unclear risk | Comment: no protocol found |
| Vested Interest (funding and/or author affiliations) | High risk | Quote: "This research was supported by a grant from Eli Lilly and Co., Protocol F1D‐US‐X173, to Dr. Linehan; by Remind Rx Medication Compliance Systems; and by a contribution of electronic pill bottles from IBV Technologies, Seattle, Wash. [...]. Dr. Linehan is a consultant for, has received grant/research support and honoraria from, and is a member of the speakers/advisory board for Eli Lilly. Drs. McDavid, Brown, Sayrs, and Gallop report no additional financial or other relationships relevant to the subject of this article." (Linehan 2008, p. 999) |
| Other bias | Low risk | Comment: no indication of other bias |
Loew 2006.
| Study characteristics | ||
| Methods | 10‐week trial with 2 arms:
Duration of trial: 10 weeks Country: Germany Setting: outpatient |
|
| Participants |
Methods of recruitment of patients: Patients were primarily recruited through advertisements. Overall sample size: 56 Diagnosis of borderline personality disorder: DSM (edition not mentioned) Means of assessment: SCID‐II Mean age: 25.25 years (SD = no information; range = no information) Sex: 100% women Comorbidity: no information Inclusion criteria: no information Exclusion criteria
|
|
| Interventions |
Experimental group
Treatment name: topiramate
Number randomised to group: 28*
Duration: 10 weeks Control/comparison group Comparison name: placebo Number randomised to group: 28* Duration: 10 weeks Both groups Concomitant psychotherapy: not allowed Concomitant pharmacotherapy: any other psychotropic medication not allowed Proportions of participants taking standing medication during trial observation period: no information |
|
| Outcomes |
Primary outcomes: general psychiatric pathology, measured by SCL‐90‐R‐GSI Secondary outcomes
|
|
| Notes |
Sample calculation: yes Ethics approval: yes Funding source: no funding received Conflicts of interest: No conflicts of interest were reported Comments from trial authors (limitations): "This analysis is limited, in part, because the sample size was (despite a valid power analysis) relatively small and consisted only of moderately ill women with BPD." (Loew 2006, p. 65). "The placebo effect proved to be relatively small between the first and last evaluations. Whether these results could also be replicated to men meeting the criteria for BPD, and/or with severe cases of BPD, and/or with patients abusing substances or taking concurrent medications is unknown". (Loew 2006, p. 65). "The length of this trial was only 10 weeks, which possibly reduced the dropout rate." (Loew 2006, p. 65) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: Trial was referred to as randomised, however, there was not sufficient information on how randomisation was carried out to permit a judgement of low or high risk of bias. |
| Allocation concealment (selection bias) | Unclear risk |
Quote: "Randomization was carried out confidentially by the clinic administration". (Loew 2006, p. 63) Comment: unclear which measures were taken to assure confidentiality throughout the study |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "Subjects received blinded medication daily, which constituted either topiramate or a matching placebo.[…]Tablets were supplied in numbered boxes. Both subjects and clinicians were blinded regarding topiramate/placebo assignment." (Loew 2006, p. 63) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk |
Quote: "Subjects received blinded medication daily, which constituted either topiramate or a matching placebo.[…]Tablets were supplied in numbered boxes. Both subjects and clinicians were blinded regarding topiramate/placebo assignment." (Loew 2006, p. 63) Comment: Unclear if 'clinicians' also applied to outcome assessors, therefore, insufficient information to permit a judgement of low or high risk of bias |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk |
Quote: "Fifty‐nine subjects were eligible to take part in the study [...] 56 patients were required [...] randomization was carried out [...] with a 1:1 assignment to the active drug (N = 28) and placebo (N = 28)". (Loew 2006, p. 63) Comments: Of the 56 patients enrolled, 52 completed treatment (27 in topiramate group, 25 in placebo group). Reasons for early termination: Absent more than twice for weekly evaluation: 1 in the topiramate group, 3 in the placebo group LOCF used; reasons for early termination specified (Loew 2006, p. 63) Not clear why or how the 56 participants were finally chosen out of the 59 potential participants |
| Selective reporting (reporting bias) | Unclear risk | Comment: no protocol found |
| Vested Interest (funding and/or author affiliations) | Unclear risk | Quote: "The study was planned and conducted independent[ly] of any institutional influence and approved by the clinic's ethics committee in accordance with the Declaration of Helsinki and ethical laws pertaining to the medical professions." (Loew 2006, p. 63) |
| Other bias | Low risk | Comment: No apparent other sources of bias found |
Markovitz 1995a.
| Study characteristics | ||
| Methods | A 14‐week trial with 2 arms:
Duration of trial: 14 weeks Country: USA Setting: inpatient |
|
| Participants |
Methods of recruitment of patients: no information Overall sample size: 17 Diagnosis of borderline personality disorder: DSM‐III‐R Means of assessment: SCID‐II and Gunderson's DIB20 Mean age: no information Sex: no information Comorbidity: Axis I: Each patient had on average 3.0 current Axis I diagnoses and 4.7 lifetime diagnoses at the time of the study. Axis II: 100% borderline, 82% self‐defeating, 82% paranoid, 71% compulsive, 65% avoidant, 65% dependent, 59% histrionic, 59% passive‐aggressive, 53% schizotypal, 35% narcissistic, 35% antisocial. Axis III: 92% premenstrual syndrome, 47% headaches/migraines, 41% IBS, 35% fibrocytis, 29% neurodermatitis, 29% sleep apnoea Inclusion criteria: no information Exclusion criteria: no information |
|
| Interventions |
Experimental group
Treatment name: fluoxetine
Number randomised to group: 9
Duration: 14 weeks Control/comparison group Comparison name: placebo Number randomised to group: 8 Duration: 14 weeks Both groups Concomitant psychotherapy: no information Concomitant pharmacotherapy: no information Proportions of participants taking standing medication during trial observation period: no information |
|
| Outcomes |
Primary
Secondary
|
|
| Notes |
Sample calculation: no information Ethics approval: no information Funding source: no information Conflicts of interest: no information Comments from trial authors (limitations): "All of the rating instruments were continuing to show increasing improvement at 14 weeks in patients on fluoxetine, suggesting the trial may not have been long enough". (Markovitz 1995a, p. 271) Comment from review authors: This trial was not included in quantitative analyses due to data unavailability for effect size calculations. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: The study was referred to as randomised; however, the method used to generate the allocation sequence was not described in sufficient detail to allow an assessment of whether it produced comparable groups. |
| Allocation concealment (selection bias) | Unclear risk | Comment: Allocation concealment and the method used to conceal the allocation sequence was not described. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: The study was referred to as being double‐blind but there was no information on method or if the blinding was successful. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: The study was referred to as being double‐blind but there was no information on method or if the blinding was successful. |
| Incomplete outcome data (attrition bias) All outcomes | High risk |
Quote: “Dropout after 3 weeks but prior to 14 weeks and their final rating scores carried through for each subsequent time point. Seven of nine patients on fluoxetine completed the entire study, and seven of eight patients on placebo completed all 14 weeks of the trial”. (Markovitz 1995a, p. 271) Comment: last‐observation‐carried‐forward |
| Selective reporting (reporting bias) | Unclear risk | Comment: no protocol available. Insufficient information to permit judgement of high or low risk of bias |
| Vested Interest (funding and/or author affiliations) | Unclear risk | Comment: insufficient information on funding and conflict of interest to permit judgement of high or low risk of bias |
| Other bias | Low risk | Comment: The study appeared to be free of other sources of bias. |
Moen 2012.
| Study characteristics | ||
| Methods | 16‐week trial with 2 arms:
Duration of trial: 16 weeks (a 4‐week selection phase and a 12‐week experimental phase) Country: USA Setting: inpatient |
|
| Participants |
Methods of recruitment of patients: "[...] newspaper and radio advertisements in the Minneapolis area. Local psychiatric clinics and mental health centers also were notified of the study, although no clinical referrals were made". (Moen 2012, p. 256‐7) Overall sample size: 15 Diagnosis of borderline personality disorder: DSM‐IV Means of assessment: SCID‐I, SCID‐II and past clinical records to screen for other mental illnesses, and SCL‐90 Mean age: 35.5 years (SD = no information; range = 22‐51) Sex: 80% women Comorbidity: "Although current diagnosis of major depression was an exclusion criterion, a history of major depression was allowed provided it had been 12 weeks since the last major depressive episode." (Moen 2012, p. 257). No further information Inclusion criteria: BPD diagnosis Exclusion criteria
|
|
| Interventions |
Experimental group
Treatment name: divalproex
Number randomised to group: 10
Duration: 12 weeks Control/comparison group Comparison name: placebo Number randomised to group: 5 Duration: 12 weeks Both groups Concomitant psychotherapy: condensed 4 weeks of Dialectical Behaviour Therapy (DBT) prior to medication Concomitant pharmacotherapy: no information; however, there was a washout before trial initiation and each potential research patient had to be medication‐free for 2 to 4 weeks. Proportions of participants taking standing medication during trial observation period: no information |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
Sample calculation: no information Ethics approval: approved by institutional review board for Texas University Health Science Centre – Houston, USA Funding source: funded or partially funded by pharmaceutical industry Conflicts of interest: Dr Schulz is a consultant to Eli Lilly and company and Genetech. Comments from trial authors (limitations): "[...] the small sample size limited our statistical power to investigate significant treatment group differences as well as our ability to generalize from our sample". (Moen 2012, p. 259) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: insufficient information on methods used to generate random sequence to permit a judgement of low or high risk of bias |
| Allocation concealment (selection bias) | Unclear risk | Comment: no information on allocation concealment to permit judgement of low or high risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk |
Quote: "Blood samples were obtained to measure blood concentration of divalproex ER at weeks 4, 8, and 16. A study physician then adjusted the dose to maintain the medication at the therapeutic range. The study physician changed the dosing of the study medicine and placebo at pre‐determined dose‐escalation steps so as not to reveal medication assignment". (Moen 2012, p. 258) Comment: Personnel appeared to have been blinded, but there was no information regarding how participants were blinded (e.g. on concealment of medication type) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "All raters were kept blind of the serum divalproex level results. Only one study physician (A.A.), who did not participate in the ratings, was privy to the results of serum divalproex levels." (Moen 2012, p. 257) |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: last‐observation‐carried‐forward (LOCF) used. Low numbers overall |
| Selective reporting (reporting bias) | Unclear risk | Comment: no protocol found |
| Vested Interest (funding and/or author affiliations) | High risk | Quote: "This study was sponsored by a research grant from Abbott Pharmaceuticals to the principal investigator, S. Charles Schulz, MD. Dr. Schulz receives grant or research support from AstraZeneca, Myriad RBM, and Otsuka and is a consultant to Eli Lilly and Company and Genetech. Drs. Moen, Freitag, Miller, Lee, and Adityanjee and Ms. Romine and Ms. Song report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products". (Moen 2012, p. 260) |
| Other bias | Low risk | Comment: no other sources found |
Montgomery 1982a.
| Study characteristics | ||
| Methods | 6‐month trial with 2 arms:
Duration: 24 weeks Country: UK Setting: outpatient |
|
| Participants |
Methods of recruitment of patients: "[...] endogenously depressed patients who were taking part in a comparative antidepressant efficacy study of zimelidine and maprotiline were recruited". (Montgomery 1982a, p. 292) Overall sample size: 30 Diagnosis of borderline personality disorder: DSM‐III Means of assessment: clinical interview Mean age: 35.05 years (SD = no information; range = no information) Sex: 70% women Comorbidity: no information Inclusion criteria
Exclusion criteria
|
|
| Interventions |
Experimental group
Treatment name: flupenthixol
Number randomised to group: 14
Duration: 6 months Control/comparison group Comparison name: placebo Number randomised to group: 16 Duration: 6 months Both groups Concomitant psychotherapy: All patients attended the special crisis intervention clinic within two weeks of the index suicidal act. Concomitant pharmacotherapy: no information Proportions of participants taking standing medication during trial observation period: no information |
|
| Outcomes |
Primary outcomes:
Secondary outcomes: adverse effects, measured by standard reporting form |
|
| Notes |
Sample calculation: no information Ethics approval: no information Funding source: unclear funding Conflicts of interest: No conflicts of interest were reported. Comments from trial authors (limitations): none mentioned |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: insufficient information on methods used to generate random sequence to permit a judgement of low or high risk of bias |
| Allocation concealment (selection bias) | Unclear risk | Comment: no information provided on allocation concealment to permit a judgement of low or high risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "intramuscular flupenthixol decanoate or placebo drawn from identical matching ampoules" (Montgomery 1979, p. 227) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "intramuscular flupenthixol decanoate or placebo drawn from identical matching ampoules" (Montgomery 1979, p. 227) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk |
Quote: "To preserve blindness patients with significant Parkinsonian side effects were removed from the trial and counted as dropouts." (Montgomery 1979, p. 227) Comments: reported dichotomous outcomes based on the completer sample (no further details on dropout patients concerning diagnosis, sex, and age) Of the 37 patients enrolled, 30 completed treatment (4 dropouts in the active group leaving 14 completers, 3 dropouts in the placebo group leaving 16 completers) Reasons for early termination: Parkinsonian side effects: 2 in flupenthixol group/0 in placebo group No reason given: 2/3 Only dichotomous data used in this review; dropouts were imputed as having the negative outcome. |
| Selective reporting (reporting bias) | Unclear risk | Comment: no protocol found |
| Vested Interest (funding and/or author affiliations) | Unclear risk | Comment: no details about funding/sponsoring provided |
| Other bias | Low risk | Comment: no indication of other bias |
Montgomery 1982b.
| Study characteristics | ||
| Methods | 38 participants; 6‐month trial with 2 arms:
Duration of trial: 24 weeks Country: UK Setting: outpatient |
|
| Participants |
Methods of recruitment of patients: "[...] endogenously depressed patients who were taking part in a comparative antidepressant efficacy study of zimelidine and maprotiline were recruited". (Montgomery 1982a, p. 292) Overall sample size: 38 Diagnosis of borderline personality disorder: DSM‐III Means of assessment: clinical interview Mean age: 35.65 years (SD = no information; range = no information) Sex: 68.42% women Comorbidity: no information Inclusion criteria
Exclusion criteria
|
|
| Interventions |
Experimental group
Treatment name: mianserin
Number randomised to group: 29
Duration: 6 months Control/comparison group Comparison name: placebo Number randomised to group: 29 Duration: 6 months Both groups Concomitant psychotherapy: Patients were followed up in a clinic, with back‐up from social workers, community nurses and a crisis intervention team. Concomitant pharmacotherapy: no information Proportions of participants taking standing medication during trial observation period: no information |
|
| Outcomes |
Primary outcomes: suicidal behaviour, measured by the number of participants in each group with/without act of self‐harm within the 6 months of treatment. Assessed at week 4, 8, 12, 16, 20 and 24 (EOT) Secondary outcomes: attrition in terms of participants lost after randomisation |
|
| Notes |
Sample calculation: no information Ethics approval: no information Funding source: unclear funding Conflicts of interest: No conflicts of interest were reported. Comments from trial authors (limitations): none mentioned |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: insufficient information on methods used to generate random sequence to permit a judgement of low or high risk of bias |
| Allocation concealment (selection bias) | Unclear risk | Comment: no information provided on allocation concealment to permit a judgement of low or high risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: The trial was referred to as being double‐blind, however, insufficient information was given on how blinding of participants and personnel was carried out (packaging of trial medication etc.) and maintained, to permit a judgement of low or high risk of bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: The trial was referred to as being double‐blind, however, insufficient information was given on how blinding of outcome assessors was carried out and maintained, to permit a judgement of low or high risk of bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk |
Comment: dichotomous outcomes used here were based on the ITT sample; patients who had dropped out were imputed as having the negative outcome. High dropout rate (20 out of 58; Montgomery 1983, p. 787), but reasons not specified, nor to which treatment group the lost patients belonged. Therefore, dropouts could not be imputed in categorical outcomes as having the negative outcome. |
| Selective reporting (reporting bias) | Unclear risk | Comment: no protocol found |
| Vested Interest (funding and/or author affiliations) | Unclear risk | Comment: no details about funding/sponsoring provided |
| Other bias | Low risk | Comment: no indication of other bias |
NCT00533117.
| Study characteristics | ||
| Methods | 12‐month trial with 4 arms:
Duration of trial: 12 months Country: USA Setting: inpatient |
|
| Participants |
Method of recruitment of participants: "Participants were recruited from the emergency department, clinician referrals and advertisements. The recruitment period ended 6 months prior to the study end date." (NCT00533117) Overall sample size: 75 (86 participants were randomised but 11 dropped out prior to the start of treatment) Diagnosis of borderline personality disorder: DSM‐IV Means of assessment: no information Mean age: 30.2 years (SD 8.7; range = no information) Sex: 77.3% women Comorbidity: no information Inclusion criteria
Exclusion criteria
|
|
| Interventions |
Experimental group 1
Treatment name: dialectic behaviour therapy (DBT) + fluoxetine
Number randomised to group: 18
Duration: 12 months Control/comparison group 1 Comparison name: DBT + placebo Number randomised to group: 19 Duration: 12 months Experimental group 2 Treatment name: supportive therapy + fluoxetine Number randomised to group: 20 Duration: 12 months Control/comparison group 2 Comparison name: supportive therapy + placebo Number randomised to group: 18 Duration: 12 months All groups Concomitant psychotherapy: DBT or supportive therapy Concomitant pharmacotherapy: Benzodiazepines were permitted for sleep. Proportions of participants taking standing medication during trial observation period: no information |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
Sample calculation: no information Ethics approval: no information Funding source: funded by grants from universities, authorities or research foundations Conflicts of interest: No conflicts of interest were reported. Comments from trial authors (limitations): none mentioned |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: insufficient information on methods used to generate random sequence to permit a judgement of low or high risk of bias. |
| Allocation concealment (selection bias) | Unclear risk | Comment: no information provided on allocation concealment to permit a judgement of low or high risk of bias. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: Trial registration stated that the study had triple‐blinding (participant, investigator and outcome assessor), however, there was no information on how the blinding was secured or if it was adequately maintained throughout the study to permit a judgement of low or high risk of bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: Trial registration stated that the study had triple‐blinding (participant, investigator and outcome assessor), however, there was no information on how the blinding was secured or if it was adequately maintained throughout the study to permit a judgement of low or high risk of bias. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: even dropout rates, total rates around ¼ of participants, no information on ITT, but all randomised participants were analysed. |
| Selective reporting (reporting bias) | Low risk | Comment: only a trial registry. All data reported |
| Vested Interest (funding and/or author affiliations) | Low risk | Comment: Sponsored by New York State Psychiatric Institute; Collaboration with National Institute of Mental Health |
| Other bias | Low risk | Comment: no indication of other bias |
Nickel 2004.
| Study characteristics | ||
| Methods | 8‐week trial with 2 arms:
Duration of trial: 8 weeks Country: Germany Setting: outpatient |
|
| Participants |
Method of recruitment of participants: no information Overall sample size: 31 Diagnosis of borderline personality disorder: DSM‐IV Means of assessment: SCID‐II Mean age: 26.05 years (SD = no information; range = no information) Sex: 100% women Comorbidity: no information Inclusion criteria
Exclusion criteria
|
|
| Interventions |
Experimental group
Treatment name: topiramate
Number randomised to group: 21
Duration: 8 weeks Control/ comparison group Comparison name: placebo Number randomised to group: 10 Duration: 8 weeks Both groups Concomitant psychotherapy: not allowed Concomitant pharmacotherapy: not allowed Proportions of participants taking standing medication during trial observation period: no information; however, concomitant medication was not allowed. |
|
| Outcomes |
Primary outcomes: none Secondary outcomes
|
|
| Notes |
Sample calculation: yes, "According to a power analysis, 21 patients were required for a topiramate trial." (Nickel 2004, p. 1516) Ethics approval: yes, "[The trial design] was approved by the clinic's "Ethikkomision" (the German equivalent of the Committee on Human Subjects". (Nickel 2004, p. 1516) Funding source: unclear funding Conflicts of interest: No conflicts of interest were reported. Comments from trial authors (limitations)
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: The article mentioned that the trial was randomised, however, there was insufficient information on how the randomisation procedure was carried out to permit a judgement of low or high risk of bias. |
| Allocation concealment (selection bias) | Unclear risk |
Quote: "Randomization was carried out confidentially by the clinic administration." (Nickel 2004, p. 1516) Comment: Allocation sequence appeared to have been confidential, however, there was insufficient information on how this confidentiality was maintained to permit a judgement of low or high risk of bias. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Tablets were supplied in numbered boxes. Both subjects and clinicians were blinded regarding topiramate/placebo assignment." (Nickel 2004, p. 1516). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk |
Quote: "Tablets were supplied in numbered boxes. Both subjects and clinicians were blinded regarding topiramate/placebo assignment." (Nickel 2004, p. 1516). Comment: Unclear if clinicians were also outcome assessors, therefore insufficient information to permit judgement of low or high risk of bias. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk |
Quote: "Two subjects, who failed to appear 2 to 3 times for the weekly evaluations, dropped out of the study, and their data were not further analysed. Finally, data from 29 women [...] were evaluated." (Nickel 2004, p. 1516) Comment: continuous outcomes based on available case analysis. Of the 31 patients enrolled, 29 completed treatment. Reasons for early termination: Failed to appear at least 2 times for weekly evaluation, no further details: 2 in topiramate group/0 in placebo group |
| Selective reporting (reporting bias) | Unclear risk | Comment: no protocol found |
| Vested Interest (funding and/or author affiliations) | Unclear risk | Quote: "The authors report no financial affiliation or other relationship relevant to the subject matter of this article." (Nickel 2004, p. 1515) |
| Other bias | Low risk | Comment: No apparent other sources of bias found |
Nickel 2005.
| Study characteristics | ||
| Methods | 8‐week trial with 2 arms:
Duration of trial: 8 weeks Country: Germany Setting: outpatient |
|
| Participants |
Method of recruitment of participants: Patients were recruited with the assistance of colleagues in their practices and the clinic outpatient department, as well as through advertisements in the local and regional press. Overall sample size: 44 Diagnosis of borderline personality disorder: DSM‐IV Means of assessment: SCID‐II Mean age: 29.1 years (SD = no information; range = no information) Sex: 100% men Comorbidity: Comorbid mood disorders, somatoform disorders, anxiety disorders and eating disorders were ascertained in the trial sample. Inclusion criteria
Exclusion criteria
|
|
| Interventions |
Experimental group
Treatment name: topiramate
Number randomised to group: 22
Duration: 8 weeks Control/comparison group Medication name: placebo Number randomised to group: 22 Duration: 8 weeks Both groups Concomitant psychotherapy: not allowed Concomitant pharmacotherapy: psychotropic medication not allowed Proportions of participants taking standing medication during trial observation period: no information; however, concomitant psychotropic medication was not allowed. |
|
| Outcomes |
Primary outcomes: none Secondary outcomes
|
|
| Notes |
Sample calculation: no information Ethics approval: design approved by Ethikkommission der ROMED Kliniken KG Funding source: no funding received Conflicts of interest: No conflicts of interest were reported. Comments from trial authors (limitations)
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: The article mentioned that the trial was randomised, however, there was insufficient information on how the randomisation procedure was carried out to permit a judgement of low or high risk of bias. |
| Allocation concealment (selection bias) | Unclear risk |
Quote: "Randomization was carried out confidentially by the clinic administration." (Nickel 2005, p. 496) Comment: Allocation sequence appeared to have been confidential, however, there was insufficient information on how this confidentiality was maintained to permit a judgement of low or high risk of bias. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Subjects received blinded medication daily, which at the beginning amounted to either 50 mg of topiramate or of a matching placebo. [...]Tablets were supplied in numbered boxes. Both subjects and clinicians were blinded regarding topiramate or placebo assignment." (Nickel 2005, p. 496) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk |
Quote: "Subjects received blinded medication daily, which at the beginning amounted to either 50 mg of topiramate or of a matching placebo. [...]Tablets were supplied in numbered boxes. Both subjects and clinicians were blinded regarding topiramate or placebo assignment." (Nickel 2005, p. 496) Comment: Unclear if clinicians were also outcome assessors, therefore insufficient information to permit judgement of low or high risk of bias |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk |
Quotes: "Forty‐eight subjects were eligible to take part in the study [...] 44 patients were required [...] randomization was carried out confidentially by the clinical administration [...] 1:1 randomisation ratio for topiramate (TG, N = 22) versus placebo treatment (N = 22)". (Nickel 2005, p. 496) "Two subjects from the placebo group failed to appear more than twice for the weekly evaluations and dropped out of the study; their data were not further analysed. Thus, data from 42 men (42 out of 44) were evaluated." (Nickel 2004, p. 1516). Comments: unclear, why or how the 44 participants were finally chosen out of the 47 potential participants Of the 44 patients enrolled, 42 completed treatment (22 in the active group, 20 in the placebo group) Reasons for early termination: Failed to appear more than twice for weekly evaluation, no further reasons given: 0 in the active group, 2 in the placebo group Reasons for early termination not further specified. Continuous outcomes based on available case analysis. For dichotomous data, dropouts were imputed as having the negative outcome. |
| Selective reporting (reporting bias) | Unclear risk | Comment: no protocol found |
| Vested Interest (funding and/or author affiliations) | Unclear risk | Quote: "The study was planned and conducted in accordance with the Declaration of Helsinki and ethical laws pertaining to the medical professions and its design approved by Ethikkommission der ROMED Kliniken KG. All subjects gave written informed consent. The study was conducted independent of any institutional influence and was not funded, and there were no conflicts of interest." (Nickel 2004, p. 496) |
| Other bias | Low risk | Comment: No apparent other sources of bias found |
Nickel 2006.
| Study characteristics | ||
| Methods | 8‐week trial with 2 arms:
Duration of trial: 8 weeks Country: Germany Setting: outpatient |
|
| Participants |
Method of recruitment of participants: Participants were recruited through advertisements. Overall sample size: 52 Diagnosis of borderline personality disorder: DSM‐IV Means of assessment: SCID‐II Mean age: 21.65 years (SD 3.4; range = no information) Sex: 82.69% women, 17.3% men Comorbidity: Comorbidity included depressive disorders, anxiety disorders, obsessive‐compulsive disorders and somatoform disorders. Inclusion criteria: meeting criteria for borderline personality disorder Exclusion criteria
|
|
| Interventions |
Experimental group
Treatment name: aripiprazole
Number randomised to group: 26
Duration: 8 weeks Control/comparison group Comparison name: placebo Number randomised to group: 26 Duration: 8 weeks Both groups Concomitant psychotherapy: not allowed Concomitant pharmacotherapy: not allowed Proportions of participants taking standing medication during trial observation period: no information; however, concomitant medication was not allowed. |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
Sample calculation: no information Ethics approval: Yes, "The study was planned and conducted in accordance with the Declaration of Helsinki and ethical laws pertaining to the medical profession, and its design was approved by the clinic’s ethics committee". (Nickel 2006, p. 835) Funding source: no funding received Conflicts of interest: No conflicts of interest were reported Comments from trial authors (limitations)
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: The article mentioned that the trial was randomised, however, there was insufficient information on how the randomisation procedure was carried out to permit a judgement of low or high risk of bias. |
| Allocation concealment (selection bias) | Unclear risk |
Quote: "The random assignment was carried out confidentially by the clinic administration and arranged so that the same number of patients would be treated with the active drug (N = 26, 21 women and 5 men) as with a placebo (N = 26, 22 women and 4 men)". (Nickel 2006, p. 835) Comment: Allocation sequence appeared to have been confidential, however, there was insufficient information on how this confidentiality was maintained to permit a judgement of low or high risk of bias. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "the subjects received medication in a blinded manner, […] The dosage remained constant. Tablets were supplied in numbered boxes. Both the subjects and the clinicians were blinded regarding the assignment of aripiprazole or placebo." (Nickel 2006, p. 835) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk |
Quote: "the subjects received medication in a blinded manner, […] The dosage remained constant. Tablets were supplied in numbered boxes. Both the subjects and the clinicians were blinded regarding the assignment of aripiprazole or placebo." (Nickel 2006, p. 835) Comment: Unclear if clinicians were also outcome assessors, therefore insufficient information to permit judgement of low or high risk of bias |
| Incomplete outcome data (attrition bias) All outcomes | Low risk |
Quotes: "Five subjects who missed more than two weekly evaluations dropped out." (Nickel 2006, p. 835) "according to the intent‐to‐treat principle performed with the last‐observation‐carried‐forward" (Nickel 2007, p. 1025) Comments: Of the 52 patients enrolled, 47 completed treatment. Reasons for dropout not further specified. Reasons for early termination: Failed to appear more than twice for weekly evaluation, no further reasons given: 5 participants, no further details Continuous outcomes based on ITT sample (LOCF); dichotomous outcomes based on ITT sample |
| Selective reporting (reporting bias) | Unclear risk | Comment: no protocol found |
| Vested Interest (funding and/or author affiliations) | Unclear risk | Quote: "The study was planned and conducted in accordance with the Declaration of Helsinki and ethical laws pertaining to the medical profession, and its design was approved by the clinic's ethics committee. The study was conducted independently of any institutional influence and was not funded." (Nickel 2006, p. 835) |
| Other bias | Low risk | Comment: No apparent other sources of bias found |
Pascual 2008.
| Study characteristics | ||
| Methods | 12‐week trial with 2 arms:
Duration of trial: 12 weeks, following a 2‐week baseline period Country: Spain Setting: outpatient |
|
| Participants |
Method of recruitment of participants: "[...] referred from clinical service (outpatient and psychiatrics emergency services)" (Pascual 2008, p. e2) Overall sample size: 60 Diagnosis of borderline personality disorder: DSM‐IV Means of assessment: SCID‐II and DIB‐R Mean age: 29.2 years (SD = no information; range = no information) Sex: 81.67% women Comorbidity: no information Inclusion criteria
Exclusion criteria
|
|
| Interventions |
Experimental group
Treatment name: ziprasidone
Number randomised to group: 30
Duration: 12 weeks Control/comparison group Comparison name: placebo Number randomised to group: 30 Duration: 12 weeks Both groups Concomitant psychotherapy: Patients participated in weekly, 2‐hour, non‐specific group psychotherapy sessions. Concomitant pharmacotherapy: allowed to continue with benzodiazepine (maximum of 40 mg/day), antidepressants, and mood stabilisers if initiated prior to inclusion; doses could not be modified. Proportions of participants taking standing medication during trial observation period: In the ziprasidone condition, 76.7% of patients were taking benzodiazepines, 70% were taking antidepressants and 40% were taking mood stabilisers. In the placebo condition, the proportions were 83.3% benzodiazepines, 73.3% antidepressants and 40% mood stabilisers. |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
Sample calculation: no information Ethics approval: yes Funding source: funded or partially funded by pharmaceutical industry Conflicts of interest: No conflicts of interest were reported besides partial funding from the pharmaceutical industry. Comments from trial authors (limitations)
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomization was performed by blocks of 4 generated using the SPSS software package". (Pascual 2008, p. 604) |
| Allocation concealment (selection bias) | Unclear risk | Comment: no information provided on allocation concealment to permit a judgement of low or high risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: The trial was referred to as being double‐blind, however, no information was provided on how blinding was carried out or maintained. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: The trial was referred to as being double‐blind, however, no information was provided on how blinding was carried out or maintained. |
| Incomplete outcome data (attrition bias) All outcomes | High risk |
Quote: "All analyses were conducted on an intent‐to‐treat basis. [...] Patients were included in the analyses only if they had a baseline measure and at least 1 post‐baseline measure. [...] The end point was based on a last‐observation‐carried‐forward (LOCF) strategy." (Pascual 2008, p. 604 et seq.) Comment: intent‐to‐treat data referred to all participants that were randomly assigned and who initiated the experimental phase (Pascual 2008, p. 605). However, it remained unclear why 5 out of the 65 eligible participants "dropped out during the selection phase". (Pascual 2008, p. 605) Reasons for dropout specified and balanced across the two groups, including withdrawal due to "clinician decision/insufficient treatment effect" (Pascual 2008, p. 605 et seq.) Of the 60 patients enrolled, 29 completed the full 12 weeks of the trial (13 in ziprasidone group, 16 in placebo group). Reasons for early termination: Need of psychiatric hospitalisation: 4 in ziprasidone group/3 in placebo group Adverse events/patient decision: 9/4 Clinician decision/insufficient treatment effect: 3/7 Other reasons: 1/0 Continuous data based on LOCF data of the ITT sample; dichotomous data based on ITT sample |
| Selective reporting (reporting bias) | Low risk | Comment: The trial protocol was available and all of the prespecified primary and secondary outcomes that were of interest in the review were reported in the prespecified way. |
| Vested Interest (funding and/or author affiliations) | High risk | Quote: "This study was supported by grants from the Fondo de Investigación Sanitaria (Ministry of Health, Spain), the REM‐TAP Network, and Pfizer, Madrid, Spain. The authors report no additional financial or other relationships relevant to the subject of this article." (Pascual 2008, p. 603) |
| Other bias | Low risk | Comment: no indication of other bias |
Reich 2009.
| Study characteristics | ||
| Methods | 12‐week trial with 2 arms:
Duration: 12 weeks Country: USA Setting: outpatient |
|
| Participants |
Method of recruitment of participants: Patients were recruited through websites and advertising on local radio and television stations. Overall sample size: 27 Diagnosis of borderline personality disorder: DSM‐IV Means of assessment: DIB‐R score > 8 Mean age: 31.2 years (SD = no information; range = no information) Sex: 88.89% women, 11.11% men Comorbidity: Cormobid diagnoses included in the trial were major depression, PTSD, OCD, GAD, panic disorder, social phobia and specific phobia. Major depression was the most common comorbid diagnosis in both groups. Comorbid anxiety disorders were also common in both groups. For the lamotrigine group, panic disorder was the second most common comorbid diagnosis, followed by post‐traumatic stress disorder; for the placebo group, panic disorder was the second most common comorbid diagnosis, followed by social phobia. There were no significant differences in comorbidity between the two groups. Inclusion criteria
Exclusion criteria
|
|
| Interventions |
Experimental group
Treatment name: lamotrigine
Number randomised to group: 15
Duration: 12 weeks Control/comparison group Medication name: placebo Number randomised to group: 12 (one of the 13 patients assigned to placebo was disqualified because of failure to adhere to the trial protocol and was not included in analyses) Duration: 12 weeks Both groups Concomitant psychotherapy: Patients enrolled in psychotherapy in the last 30 days were not eligible. Concomitant pharmacotherapy: Patients could be taking one antidepressant, but had to have been on a stable dose of that medication for 1 month. Proportions of participants taking standing medication during trial observation period: "Significantly more patients in the placebo group than in the lamotrigine took antidepressants during the study (week 2: 4.73, df = 1, P = 0.03). In the placebo group, one patient was taking doxepin, one patient was taking bupropion, one patient was taking citalopram, and two patients were taking escitalopram. In the lamotrigine group, one patient was taking paroxetine." (Reich 2009, p. 272) |
|
| Outcomes |
Primary outcomes: BPD severity, measured by ZAN‐BPD total score. Assessed at baseline and every week for 12 weeks (EOT) Secondary outcomes
|
|
| Notes |
Sample calculation: no information Ethics approval: no information Funding source: funded or partially funded by pharmaceutical industry Conflicts of interest: No conflicts of interest were reported besides funding from the pharmaceutical industry. Comments from trial authors (limitations)
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients were randomized [...] in a 1:1 manner. This was determined by a prearranged random number sequence." (Reich 2009, p. e‐3) |
| Allocation concealment (selection bias) | Unclear risk | Comment: insufficient information to permit judgement (unclear, if the number sequence was kept confidentially or if enrolling investigators could possibly foresee assignment) |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk |
Quote: "The prescribing psychiatrist (D.B.R.) prescribed one tablet of study medication, 25 mg of lamotrigine, or matching inert placebo". (Reich 2009, p. 2) Comment: The study was described as being double‐blind, however, there was insufficient information on how the blinding was secured or if it was adequately maintained throughout the study to permit a judgement of low or high risk of bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "double‐distinction between "prescribing psychiatrist (D.B.R.)" who fixed the dose and "study staff" who made assessments" (Reich 2009, p. 3) |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk |
Quote: "One patient in the placebo group was disqualified because of failure to adhere to the study protocol." (Reich 2009, p. e‐3) Comment: not clear if the reported mean changes were based on the ITT sample or completers only |
| Selective reporting (reporting bias) | Low risk | Comment: The trial protocol was available and all of the prespecified primary and secondary outcomes that were of interest in the review that were prespecified in the protocol were reported in the text. |
| Vested Interest (funding and/or author affiliations) | High risk | Quote: "The study was supported by a grant from GlaxoSmithKline." (Reich 2009, p. e‐5) |
| Other bias | Low risk | Comment: no indication of other bias |
Rinne 2002.
| Study characteristics | ||
| Methods | 6‐week trial with 2 arms:
Duration: 6 weeks, patients had to be medication‐free for at least 2 weeks before entering the trial Country: Holland Setting: outpatients |
|
| Participants |
Method of recruitment of participants: "[Patients were] recruited from psychiatric outpatient clinics, from community mental health centres, and through advertisement in newspapers and on the Internet." (Rinne 2002, p. 2049) Overall sample size: 38 Diagnosis of borderline personality disorder: DSM‐IV Means of assessment: SCID‐II + a score of 110 or more on the Borderline Trait and Distress scale of a self‐report screener for personality disorders (ADP‐IV) + score of 20 or more on BPDSI Mean age: 29.2 years (SD 7.6 years; range = no information) Sex: 100% women Comorbidity: depression (28.95%), dysthymia (21.05%), generalised anxiety disorder (7.89%) and PTSD (31.58%) Inclusion criteria
Exclusion criteria: no information |
|
| Interventions |
Experimental group
Treatment name: fluvoxamine
Number randomised to group: 20
Duration: 6 weeks; patients had to be medication‐free for at least 2 weeks prior to entering trial Control/comparison group Comparison name: placebo Number randomised to group: 18 Duration: 6 weeks; patients had to be medication‐free for at least 2 weeks prior to entering trial Both groups Concomitant psychotherapy: Two patients who began psychotherapy dropped out of the trial; thus, psychotherapeutic treatment was likely not to have been allowed. Concomitant pharmacotherapy: Patients had to stop taking all psychoactive drugs and be medication‐free for at least 2 weeks before entering the trial (6 weeks for fluoxetine). Proportions of participants taking standing medication during trial observation period: Patients had to be free of medication 2‐6 weeks prior to entering trial; however, no further information was stated. |
|
| Outcomes |
Primary outcomes: none Secondary outcomes
|
|
| Notes |
Sample calculation: no information Ethics approval: no information Funding source: funded by grants from universities, authorities or research foundations Conflicts of interest: No conflicts of interest were reported. Comments from trial authors (limitations)
Comments from review authors: This is an RCT followed by a single‐blind half cross‐over and an open treatment phase; only the first RCT phase was regarded in this review. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: Insufficient information on methods used to generate random sequence to permit a judgement of low or high risk of bias |
| Allocation concealment (selection bias) | Unclear risk | Comment: Insufficient information provided on allocation concealment to permit a judgement of low or high risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: The trial was referred to as being double‐blind, however, no information was provided on how blinding was carried out or maintained. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: The trial was referred to as being double‐blind, however, no information was provided on how blinding was carried out or maintained. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk |
Quotes: "The final study group comprised the 38 subjects eligible for participation". (Rinne 2002, p. 2049), "an intent‐to‐treat analysis was performed" (Rinne 2002, p. 2050) Comments: Of the 38 patients enrolled, 35 completed the RCT phase (19 in active drug group, 16 in placebo group). Reasons for early termination: Serious aggravation of self‐damaging behaviours: 0 in the fluvoxamine group, 2 in the placebo group Severe side effects: 1/0 Continuous outcomes based on ITT; BMDP imputation technique used for dropouts |
| Selective reporting (reporting bias) | Unclear risk | Comment: no protocol found |
| Vested Interest (funding and/or author affiliations) | High risk | Quote: "Supported by the De Geestgronden Institute of Mental Health Care, by Stichting tot Steun of Vereiniging Bennekom, by national Fund for Mental Health grant 4820, and by Solvay Pharma." (Rinne 2002, p. 2053) |
| Other bias | Low risk | Comment: No apparent other sources of bias found |
Salzman 1995.
| Study characteristics | ||
| Methods | 12‐week trial with 2 arms:
Duration of trial: 12 weeks Country: USA Setting: outpatient |
|
| Participants |
Method of recruitment of participants: Patients were recruited through newspaper advertisement. Overall sample size: 22 Diagnosis of borderline personality disorder: DSM‐III‐R Means of assessment: DIB‐R, SCID‐II and clinical interview Mean age: 36.3 years (SD = no information; range = no information) Sex: 63.64% women* Comorbidity: Patients were excluded if they had current axis I disorders, as determined by clinical interview, or concurrent secondary axis II disorder. No further information Inclusioncriteria: no information Exclusion criteria
|
|
| Interventions |
Experimental group
Treatment name: fluoxetine
Number randomised to group: 13
Duration: 12 weeks with 1 week of placebo run‐in Control/comparison group Comparison name: placebo Number randomised to group: 9 Duration: 13 weeks, including 1 week of placebo run‐in Both groups Concomitant psychotherapy: Two patients were receiving psychotherapy; however, they did not differ in demographic variables, in entry criteria, or in response to treatment from the remaining participants. Concomitant pharmacotherapy: Other psychotropic medication was an exclusion criterion. Proportions of participants taking standing medication during trial observation period: Other psychotropic medication was an exclusion criterion. No further information provided |
|
| Outcomes |
Primary outcomes: mental health status (functioning), measured by GAS. Assessed at baseline and every week after for 13 weeks (EOT). Analyses for pre and post‐treatments only Secondary outcomes
|
|
| Notes |
Sample calculation: no information Ethics approval: no information Funding source: unclear funding Conflicts of interest: No conflicts of interest were reported. Comments from trial authors (limitations)
Comments from review authors: *reported on completers only |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk |
Quote: "random‐assignment comparison" (Salzman 1995, p. 24) Comment: Trial was referred to as randomised, however, there was not sufficient information on how randomisation was carried out to permit a judgement of low or high risk of bias. |
| Allocation concealment (selection bias) | Unclear risk | Comment: Insufficient information provided on allocation concealment to permit a judgement of low or high risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk |
Quote: "All subjects began with a single, 20 mg capsule or identical placebo, and doses were titrated up to a maximum of 60 mg/day according to the needs of the patient and in accordance with package insert guidelines." (Salzman 1995, p. 24) Comment: Participants appeared to have been blinded as medication and placebo were identical, however, there was no information about whether personnel were blind to allocation sequence. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Subjects were evaluated by independent observers". (Salzman 1995, p. 24) |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk |
Quote: "Thirty‐one subjects met criteria for this study; four decided not to enroll and were lost to follow‐up. Of 27 subjects who enrolled in the study, 22 completed the trial. One subject dropped out because she wanted assurance that she would be in the medication group; four others dropped out without explanation and were lost to follow‐up." (Salzman 1995, p. 24) Comment: Of the 27 patients enrolled, 22 completed treatment. Reasons for early termination: Wanted assurance to be in the active drug group: 1 (not specified, which group) Dropped out without explanation: 4 (not specified, which group) Continuous outcomes based on completer analysis |
| Selective reporting (reporting bias) | Unclear risk | Comment: no protocol found |
| Vested Interest (funding and/or author affiliations) | Unclear risk | Comment: no details provided |
| Other bias | Low risk | Comment: No apparent other sources of bias found |
Schmahl 2012a.
| Study characteristics | ||
| Methods | 6‐week trial with 2 arms:
Duration of trial: 8 weeks. In both Schmahl 2012a and Schmahl 2012b, the first week (‘week 0’) was without pharmaceutical intervention and served to assess a baseline level. Therapeutic interventions were carried out during the following 6 weeks (‘weeks 1–6’), split into two phases: (a) 3 weeks of treatment with naltrexone and (b) 3 weeks of treatment with placebo (cross‐over design). The sequence of the two treatment phases was randomised and concealed from both the patients and the trial personnel. In both trials, the last week (‘week 7’) was without pharmacological intervention. Country: Germany Setting: inpatient |
|
| Participants |
Methods of recruitment of patients: "Patients were recruited and treated from August 1998 to June 2001 at the Department of Psychiatry and Psychotherapy, University of Freiburg." (Schmahl 2012a, p. 63) Overall sample size: 13 Diagnosis of borderline personality disorder: DSM‐IV Means of assessment: IPDE Mean age: 28.3 years (SD 8.0; range = 18–44) Sex: 100% women Comorbidity: no information Inclusion criteria
Exclusion criteria
|
|
| Interventions |
Experimental group
Treatment name: naltrexone
Number randomised to group: 13
Duration: 3 weeks Control/comparison group Comparison name: placebo Number randomised to group: 13 (cross‐over) Duration: 3 weeks Both groups Concomitant psychotherapy: non‐specific therapeutic intervention Concomitant pharmacotherapy: Some pharmacotherapies were not allowed: no psychotropic medication permitted two weeks before and during the trial (fluoxetine = 4 weeks; lithium = 8 weeks). Concomitant treatment with opioid analgesics was an exclusion criterion. Proportions of participants taking standing medication during trial observation period: no information |
|
| Outcomes |
Primary outcomes: none Secondary outcomes
|
|
| Notes |
Sample calculation: no information Ethics approval: yes Funding source: unclear funding Conflicts of interest: No conflicts of interest were reported. Comments from trial authors (limitations): "the relatively small sample size is a major limitation, especially as the non‐significant results preclude any firm conclusions and as the possibilities of subgroup and predictor analyses are quite limited in small samples". (Schmahl 2012a, p. 67) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Comment: A computer‐generated randomisation list was used to carry out block randomisation (Schmahl 2012a, p. 62). |
| Allocation concealment (selection bias) | Low risk | Quote: "The sequence of the two treatment phases was randomized and concealed from both the patients and the study personnel". (Schmahl 2012a, p. 62). |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: The study was described as being double‐blind, however, there was no information on how the blinding was secured or if it was adequately maintained throughout the study to permit a judgement of low or high risk of bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: The study was described as being double‐blind, however, there was no information on how the blinding was secured or if it was adequately maintained throughout the study to permit a judgement of low or high risk of bias. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "We carried out both intent‐to‐treat (ITT) analyses (using the LOCF method) and analyses according to protocol (ATP). The results were very similar when using either strategy, and we decided to report the results from ATP analyses only if the results from ITT analyses were markedly different. As we found little support for assuming that the dropouts might be related to treatment, effect size estimates from ATP analyses might be less biased than ITT effect‐size estimates and were used accordingly. The pattern of dropouts was balanced across treatment condition; in both studies one patient dropped out in both treatment conditions". (Schmahl 2012a, p. 63) |
| Selective reporting (reporting bias) | Unclear risk | Comment: secondary outcome in protocol did not state mild‐moderate‐intense adverse events as did the full report. Lack of definition of serious adverse events and non‐serious adverse events |
| Vested Interest (funding and/or author affiliations) | Low risk | Quote: "There were no conflicts of interest" (Schmahl 2012a, p. 67). |
| Other bias | Low risk | Comment: no other apparent source of bias |
Schmahl 2012b.
| Study characteristics | ||
| Methods | 6‐week trial with 4 arms:
Duration of trial: 8 weeks. In both Schmahl 2012a and Schmahl 2012b, the first week (‘week 0’) was without pharmaceutical intervention and served to assess a baseline level. Therapeutic interventions were carried out during the following 6 weeks (‘weeks 1–6’), split into two phases: (a) 3 weeks of treatment with naltrexone and (b) 3 weeks of treatment with placebo (cross‐over design). The sequence of the two treatment phases was randomised and concealed from both the patients and the trial personnel. In both trials, the last week (‘week 7’) was without pharmacological intervention. Country: Germany Setting: inpatient and outpatient |
|
| Participants |
Methods of recruitment of patients: Patients were recruited from January 2006 to September 2007 at the Department of Psychosomatic Medicine and Psychotherapy, Central Institute of Mental Health, Mannheim; the Department of Psychiatry and Psychotherapy, University of Rostock; and the Center for Psychosomatic Medicine, Bad Wiessee. Overall sample size: 16 Diagnosis of borderline personality disorder: DSM‐IV Means of assessment: IPDE Mean age: 29.2 years (SD 8.9; range = 18–42) Sex: 100% women Comorbidity: pre‐existing substance misuse Inclusion criteria
Exclusion criteria
|
|
| Interventions |
Experimental groups
Treatment name: naltrexone (50 + 200 mg naltrexone and blood plasma levels)
Number randomised to group: 16 (cross‐over)
Duration: 3 weeks Control/comparison groups Comparison name: placebo Number randomised to group: 16 (cross‐over) Duration: 3 weeks Both groups Concomitant psychotherapy: non‐specific therapeutic intervention Concomitant pharmacotherapy: Some pharmacotherapies were not allowed: no psychotropic medication permitted two weeks before and during the trial (fluoxetine = 4 weeks; lithium = 8 weeks). Concomitant treatment with opioid analgesics was an exclusion criterion. Proportions of participants taking standing medication during trial observation period: no information |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes | See Schmahl 2012a | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Comment: A computer‐generated randomisation list was used to carry out block randomisation (Schmahl 2012b, p. 62). |
| Allocation concealment (selection bias) | Low risk | Quote: "The sequence of the two treatment phases was randomized and concealed from both the patients and the study personnel". (Schmahl 2012b, p. 62). |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: The study was described as being double‐blind, however, there was no information on how the blinding was secured or if it was adequately maintained throughout the study to permit a judgement of low or high risk of bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: The study was described as being double‐blind, however, there was no information on how the blinding was secured or if it was adequately maintained throughout the study to permit a judgement of low or high risk of bias. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "We carried out both intent‐to‐treat (ITT) analyses (using the LOCF method) and analyses according to protocol (ATP). The results were very similar when using either strategy, and we decided to report the results from ATP analyses only if the results from ITT analyses were markedly different. As we found little support for assuming that the dropouts might be related to treatment, effect size estimates from ATP analyses might be less biased than ITT effect size estimates and were used accordingly. The pattern of dropouts was balanced across treatment condition; in both studies one patient dropped out in both treatment conditions". (Schmahl 2012b, p. 63) |
| Selective reporting (reporting bias) | Unclear risk | Comment: secondary outcome in protocol did not state mild‐moderate‐intense adverse events as did the full report. Lack of definition of serious adverse events and non‐serious adverse events |
| Vested Interest (funding and/or author affiliations) | Low risk | Quote: "There were no conflicts of interest" (Schmahl 2012b, p. 67). |
| Other bias | Low risk | Comment: no apparent other source of bias |
Schulz 2007.
| Study characteristics | ||
| Methods | 12‐week trial with 2 arms:
Duration: 12 weeks (after screening period of 2‐14 days) Country: Belgium, France, Germany, Norway, Portugal, Spain, Sweden, UK and USA Setting: outpatient |
|
| Participants |
Method of recruitment of participants: no information Overall sample size: 314 Diagnosis of borderline personality disorder: DSM‐IV Means of assessment: Diagnostic Interview for DSM‐IV Personality Disorders (DIPD‐IV) and Zan‐BPD total score of 9 or higher Mean age: 31.81 years (SD = no information; range = no information) Sex: 71.02% women Comorbidity: no information; however, many psychiatric disorders were excluded. Inclusion criteria
Exclusion criteria
|
|
| Interventions |
Experimental group
Treatment name: olanzapine (2.5‐20 mg/d, mean final dose 7.09 mg/d, SD 5.11)
Number randomised to group: 150
Duration: 12 weeks Control/comparison group Comparison name: placebo Number randomised to group: 155 Duration: 12 weeks Both groups Concomitant psychotherapy: no information Concomitant pharmacotherapy: no medications with primarily CNS activity (except for protocol‐specified benzodiazepines and hypnotics) Proportions of participants taking standing medication during trial observation period: no information |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
Sample calculation: no information Ethics approval: yes Funding source: funded or partially funded by pharmaceutical industry Conflicts of interest: Dr Schulz has consulted for Eli Lilly, AstraZeneca and Vanda. Authors HCD, QT, YT, DL and SC are employed by Lilly Research Laboratories. Comments from trial authors (limitations): none mentioned |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk |
Quote: "patients [...] were randomly assigned to treatment". (Eli Lilly 2008, p. 15), Comment: Randomisation conducted centrally |
| Allocation concealment (selection bias) | Unclear risk |
Quote: "All participants, study site personnel and investigators were masked to randomisation codes." (Schulz 2008, p. e1) Comment: Insufficient information on how allocation sequence was concealed and maintained until randomised trial phase started to permit a judgement of low or high risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: Participants and personnel were described as being masked to randomisation codes, however, there was insufficient information about how blinding in the randomised phase was carried out and maintained (e.g. identical capsules of trial medication). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: Participants and personnel were described as being masked to randomisation codes, however, there was Insufficient information about how blinding in the randomised phase was carried out and maintained (e.g. blind to adverse events etc.). |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk |
Quote: "Analyses were done on an intent‐to‐treat basis [...] In general, LOCF mean change analyses". (Eli Lilly 2008, p. 5) Quote: "Of the 314 randomized patients, 305 had both a baseline and a non‐missing post‐baseline observation and were thus qualified for the primary efficacy analysis." (Eli Lilly 2008, p. 16) Comment: Unclear, what "non‐missing post‐baseline observation" exactly means. However, discontinuing participants were included in the 305 participants whose results were analysed using LOCF. Continuous outcomes based on LOCF/ITT 314 patients were enrolled and randomly allocated. Outcomes referred partly to all of them, partly to 310 or 305 patients. No further details given |
| Selective reporting (reporting bias) | High risk | Comment: Several outcome measures (secondary and adverse events) were reported that were not prespecified according to the trial protocol. |
| Vested Interest (funding and/or author affiliations) | High risk | Quote: "This study was sponsored by Eli Lilly. S.C.S. has received honorarium from Eli Lilly, AstraZeneca and Bristol‐Meyers Squibb; grant fees from Eli Lilly, AstraZeneca, Abbott, MIND Institute and the NIMH; and consultation fees from Eli Lilly, AstraZeneca and Vanda. H.C.D., Q.T., Y.T., D.L. and S.C. are employed by Lilly Research Laboratories." (Schulz 2008 p. e1) |
| Other bias | Low risk | Comment: No indication of other bias |
Shafti 2010.
| Study characteristics | ||
| Methods | 8‐week trial with 2 arms:
Duration of trial: 8 weeks Country: Iran Setting: inpatient |
|
| Participants |
Method of recruitment of participants: The participants were inpatients. Not otherwise specified Overall sample size: 28 Diagnosis of borderline personality disorder: DSM‐IV Means of assessment: no information Mean age: 29.49 years (SD = no information; range = no information) Sex: 100% women Comorbidity: none Inclusion criteria: female gender Exclusion criteria: any prominent comorbid mental disorder |
|
| Interventions |
Experimental group
Treatment name: olanzapine
Number randomised to group: 14
Duration: 8 weeks Control/comparison group Comparison name: haloperidol Number randomised to group: 14 Duration: 8 weeks Both groups Concomitant psychotherapy: not allowed Concomitant pharmacotherapy: not allowed Proportions of participants taking standing medication during trial observation period: no other concurrent psychotropic medication during testing |
|
| Outcomes |
Primary outcomes: mental health status (functioning), measured by CGI‐S and BPRS. Assessed at baseline and at week 8 (EOT) Secondary outcomes
|
|
| Notes |
Sample calculation: yes Ethics approval: yes Funding source: no funding received Conflicts of interest: No conflicts of interest were reported. Comments from trial authors (limitations): "Small size of the samples, short duration of the trial, and sex‐based sampling were among the weaknesses of this trial". (Shafti 2010, p. 46) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: No information on method for random sequence generation to permit a judgement of low or high risk of bias |
| Allocation concealment (selection bias) | Unclear risk | Comment: Insufficient information on concealment of random sequence allocation to permit a judgement of low or high risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: The article referred to the trial being double‐blind, however, there was insufficient information on how blinding of participants and personnel was carried out and maintained to permit a judgement of low or high risk of bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: The article referred to the trial being double‐blind, however, there was insufficient information on how blinding of outcome assessors was carried out and maintained to permit a judgement of low or high risk of bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: No attrition |
| Selective reporting (reporting bias) | Unclear risk | Comment: No protocol found |
| Vested Interest (funding and/or author affiliations) | Low risk | Comment: No conflicts of interest reported |
| Other bias | Low risk | Comment: No other sources found |
Shafti 2014.
| Study characteristics | ||
| Methods | 8‐week trial with 2 arms:
Duration of trial: 8 weeks Country: Iran Setting: inpatient |
|
| Participants |
Method of recruitment of participants: “selected from outpatients among clientele of two psychiatric clinics and also inpatients from the female wards of Razi Psychiatric Hospital.” (Shafti 2014, p. 39) Overall sample size: 24 Diagnosis of borderline personality disorder: DSM‐IV‐TR Means of assessment: no information Age: 27.4 years (SD = no information; range = no information) Sex: 100% women Comorbidity: "Patients were excluded from the trial if any prominent co‐morbid mental disorder was present.” (Shafti 2014, p. 39) Inclusion criteria
Exclusion criteria
|
|
| Interventions |
Experimental group
Treatment name: olanzapine
Number randomised to group: 12
Duration: 8 weeks Control/comparison group Comparison name: aripiprazole Number randomised to group: 12 Duration: 8 weeks Both groups Concomitant psychotherapy: not allowed Concomitant pharmacotherapy: No other concurrent psychotropic medication permitted during testing. No further information Proportions of participants taking standing medication during trial observation period: no information |
|
| Outcomes |
Primary outcomes:
Secondary outcomes
|
|
| Notes |
Sample calculation: ”Post hoc analysis showed an intermediary power = 0.46 for of this trial, which became power = 0.76 in compromise power analysis.” (Shafti 2014, p. 41) Ethics approval: ethical committee of Tehran University Funding source: funded by grants from universities, authorities or research foundations Conflicts of interest: No conflicts of interest were reported. Comments from trial authors (limitations): The open‐label procedure, small sample size, short duration of assessment, inexact comparable doses, and gender‐based sampling were among the weak points of this trial". (Shafti 2014, p. 42) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: inadequate information on method used to generate random sequence to permit judgement of low or high risk of bias |
| Allocation concealment (selection bias) | High risk | Comment: An open‐label trial. No allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: No blinding |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Comment: No blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: “analysis for efficacy was based on data from the same number of patients in both groups (n = 12 in each), because all of the patients remained in and completed the entire 8 weeks of the study.” (Shafti 2014 p. 40) |
| Selective reporting (reporting bias) | Unclear risk | Comment: No protocol found |
| Vested Interest (funding and/or author affiliations) | Low risk | Quote: "S.S.S., H.K.: The authors reported no conflict of interest related to this article. Funded by Department of Research" (Shafti 2014 p. 43) |
| Other bias | Low risk | Comment: No other sources found |
Simpson 2004.
| Study characteristics | ||
| Methods | 12‐week trial with 2 arms:
Duration: 12 weeks (after a 1‐week placebo run‐in) Country: USA Setting: partial hospitalisation |
|
| Participants |
Method of recruitment of participants: "Participants were recruited from all admissions to the Women’s Partial Program, a 5‐day DBT‐based, partial hospital program, using a brief self‐report questionnaire." (Simpson 2004, p. 380) Overall sample size: 25 Diagnosis of borderline personality disorder: DSM‐IV Means of assessment: SCID‐II Mean age: 36.26 years (SD = no information; range = no information Sex: 100% women Comorbidity: recruited patients were already hospitalised with comorbid Axis I pathology. No further information Inclusion criteria: no information Exclusion criteria
|
|
| Interventions |
Experimental group
Treatment name: fluoxetine
Number randomised to group: 12
Duration: 12 weeks Control/comparison group Medication name: placebo Number randomised to group: 13 Duration: 12 weeks Both groups Concomitant psychotherapy: Patients were recruited from a partial hospital programme and all received DBT (weekly, 1‐hour sessions of individual DBT; weekly, 2‐hour skills group; round‐the‐clock emergency consultation availability). Concomitant pharmacotherapy: The only other psychotropic allowed was 50‐100 mg/day trazodone for insomnia. Proportions of participants taking standing medication during trial observation period: no information |
|
| Outcomes |
Primary outcomes
Secondary outcomes
* "The post‐treatment assessment was conducted during week 10 to minimise the influence of "termination issues" expected to affect this population". (Simpson 2004, p. 381) |
|
| Notes |
Sample calculation: no information Ethics approval: no information Funding source: funded or partially funded by pharmaceutical industry Conflicts of interest: No conflicts of interest were reported besides funding from the pharmaceutical industry. Comments from trial authors (limitations)
Comments from review authors:
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: The article referred to the trial as being randomised, however, there was no information on how the randomisation procedure was carried out to permit a judgement of low or high risk of bias. |
| Allocation concealment (selection bias) | Unclear risk | Comment: No information given on allocation concealment to permit a judgement of low or high risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: No information given how blinding of participants was attempted, especially in light of the day clinic setting with possibly shared group therapy |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk |
Quote: "A non‐treating study psychiatrist was available to break the blind in event of a clinical emergency." (Simpson 2004 p. 381) Comment: In contrast, the treating clinician was probably blind. |
| Incomplete outcome data (attrition bias) All outcomes | High risk |
Comment: Of the 25 patients enrolled, 12 were randomised to fluoxetine and 13 to placebo. 20 completed treatment (9 in fluoxetine group, 11 in placebo group).
Reasons for early termination:
Negative experience of the placebo washout period, which led to a reversal of their willingness to tolerate a potential assignment to the placebo condition: 3 in fluoxetine group, 0 in placebo group
Sought hospitalisation at another facility: 0/1
Intolerable lack of improvement: 0/1 Comment: Reasons for early termination specified (Simpson 2004 p. 381) Continuous outcomes were only reported for trial completers, while dropouts could be imputed as having the negative outcome for dichotomous data. |
| Selective reporting (reporting bias) | Unclear risk | Comment: Insufficient information to permit judgement of 'Yes' or 'No' |
| Vested Interest (funding and/or author affiliations) | High risk | Quote: "Support for this study was provided by the Department of Psychiatry and Human Behaviour at Brown Medical School and Eli Lilly." (Simpson 2004, p. 379) |
| Other bias | Low risk |
Quote: "Diary card records of pill ingestion were reviewed, and pill counts were made as a compliance measure." (Simpson 2004 p. 381) Quote: "1‐week placebo run‐in" (Simpson 2004 p. 380), "the only other medication allowed was 50 to 100 mg/day of trazodone for insomnia." (Simpson 2004 p. 381) |
Soler 2005.
| Study characteristics | ||
| Methods | 12‐week trial with 2 arms:
Duration: 12 weeks (after a 4‐week selection phase during which the pre‐intervention baseline was established but no therapeutic intervention was given) Country: Spain Setting: outpatient |
|
| Participants |
Method of recruitment of participants: Patients were referred from clinical services. Overall sample size: 60 Diagnosis of borderline personality disorder: DSM‐IV Means of assessment: SCID‐II and DIB‐R Mean age: 40.74 years (SD = no information; range = no information) Sex: 86.57% women Comorbidity: no information; however, patients with unstable comorbid axis I disorders were excluded. Inclusion criteria
Exclusion criteria
|
|
| Interventions |
Experimental group
Treatment name: olanzapine
Number randomised to group: 30
Duration: 12 weeks Control/comparison group Comparison name: placebo (no further details) Number randomised to group: 30 Duration: 12 weeks Both groups Concomitant psychotherapy: All patients received DBT (weekly, 150‐minute skills training group sessions; phone calls). Concomitant pharmacotherapy: Participants could continue treatment with benzodiazepines, antidepressants and mood stabilisers, but doses could not be modified. Proportions of participants taking standing medication during trial observation period: Participants were taking other medications before or during the treatment in both groups. In the olanzapine group, 73.3% of participants were taking benzodiazepine, 80% were taking antidepressants and 33.3% were taking mood stabilisers. In the placebo group, 60% of participants were taking benzodiazepine, 70% were taking antidepressants and 16.7% were taking mood stabilisers. |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
Sample calculation: no information Ethics approval: no information Funding source: funded or partially funded by pharmaceutical industry Conflicts of interest: No conflicts of interest were reported besides partial funding from the pharmaceutical industry. Comments from trial authors (limitations)
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: insufficient information on methods used to generate random sequence to permit a judgement of low or high risk of bias |
| Allocation concealment (selection bias) | Unclear risk | Comment: No information given on allocation concealment to permit a judgement of low or high risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: The trial was referred to as being double‐blind, however, insufficient information was given on how blinding of participants and personnel was carried out (packaging of trial medication etc.) and maintained, to permit a judgement of low or high risk of bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: The trial was referred to as being double‐blind, however, insufficient information was given on how blinding of outcome assessors was carried out and maintained, to permit a judgement of low or high risk of bias. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk |
Quote: "All analyses were conducted on an intent‐to‐treat basis. The endpoint was based on a last‐observation‐carried‐forward strategy. Patients were included in the analyses only if they had a baseline measure and at least one post‐baseline measure." (Soler 2005 p. 1222) Quote: "Sixty subjects were randomly assigned to dialectical behaviour therapy plus olanzapine or placebo and started the experimental phase; 42 subjects (70%) completed the study.There were no between‐group differences regarding demographic variables or concomitant treatments at baseline. Neither dialectical behaviour therapy intervention time nor dropout rates differed significantly between the two groups (eight of the 30 patients who received olanzapine versus 10 of the 30 who received placebo dropped out before the end of the study." (Soler 2005 p. 1222 et seq.) Comment: reasons for dropouts given; numbers balanced across groups Continuous outcomes based on ITT (LOCF) Of the 60 patients enrolled, 42 completed treatment (22 in active drug group, 20 in placebo group) Reasons for early termination: No reasons given |
| Selective reporting (reporting bias) | Low risk | Comment: The trial protocol was available and all of the prespecified primary and secondary outcomes that were of interest in the review were reported in the way they were prespecified. |
| Vested Interest (funding and/or author affiliations) | High risk | Quote: "Supported by grants from the Fondo de Investigación Sanitaria (Ministry of Health, Spain) and from Eli Lilly and Co. Madrid." (Soler 2005 p. 1223) |
| Other bias | Low risk | Comment: No indication of other bias |
Soloff 1989.
| Study characteristics | ||
| Methods | 5‐week trial with 3 arms:
Duration: 5 weeks (after 1‐week washout) Country: USA Setting: inpatient (after 3 weeks, some allowed to complete as outpatients) |
|
| Participants |
Method of recruitment of participants: Patients were referred for trial evaluation if their primary clinician made a presumptive clinical diagnosis of borderline personality disorder, schizotypal personality disorder, or both. Overall sample size: 90 Diagnosis of borderline personality disorder: DSM‐III Means of assessment: DIB; GAS score of 50 or less; and either a score of 17 or higher on Ham‐D or 66 or greater on the Inpatient Multidimension Rating Scale (IMPS) Mean age: 25.1 years (SD = no information; range = no information) Sex: 75.56% women, 24.44% men Comorbidity: Patients meeting research diagnostic criteria by clinical history or interview on the Schedule for Affective Disorders and Schizophrenia for a current diagnosis of major depression were included but coded for separate statistical analysis. Method of recruitment suggested that patients with comorbid schizotypal personality disorder were included. No further information Inclusion criteria
Exclusion criteria
|
|
| Interventions |
Experimental group
Treatment name: amitriptyline
Number randomised to group: 29
Duration: 5 weeks Comparison group Comparison name: haloperidol Number randomised to group: 28 Duration: 5 weeks Control group Medication name: placebo (no further details) Number randomised to group: 28 Duration: 5 weeks All groups Concomitant psychotherapy: Patients were treated as psychiatric inpatients for at least 3 weeks. No further details Concomitant pharmacotherapy: Biperiden hydrochloride (2 mg) was allowed, as needed, for extrapyramidal reactions. Proportions of participants taking standing medication during trial observation period: Patients were observed free of medication for at least one week prior to beginning intervention. No further information provided |
|
| Outcomes |
Primary outcomes: mental health status (functioning), measured by GAS. Assessed at baseline and once weekly for 5 weeks (EOT) Secondary outcomes
|
|
| Notes |
Sample calculation: no information Ethics approval: no information Funding source: funded by grants from universities, authorities or research foundations Conflicts of interest: No conflicts of interest were reported. Comments from trial authors (limitations): "Since our study was restricted to acutely decompensated patients, generalisability to less‐disturbed populations remains to be demonstrated". (Soloff 1986, p. 696, in Soloff 1989) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: Article referred to the trial as being randomised, however there was no information about the randomisation procedure to permit a judgement of low or high risk of bias. |
| Allocation concealment (selection bias) | Unclear risk | Comment: Insufficient information about allocation concealment to permit a judgement of low or high risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk |
Quote: "Numbered tablets [...] were given". (Soloff 1986, p. 692) Comment: Trial was referred to as being double‐blind. Trial medication appeared to have been concealed by packaging, however, there was no information about whether blinding was maintained throughout the trial and whether personnel were blind. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Weekly ratings by two 'blind investigators', an onward psychiatrist serving as the non‐blind psychiatrist (for safety)" (Soloff 1989, p. 693) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk |
Quote: "Five patients failed to complete the minimum two weeks on medication needed for inclusion in outcome analysis, one taking amitriptyline, three taking haloperidol, and one taking placebo." (Soloff 1989, p. 242) Continuous outcomes based on LOCF/ITT A minimum of 2 weeks receiving medication was required to include data for endpoint analysis Of the 90 patients enrolled, 85 completed treatment (29 in amitriptyline group, 28 in haloperidol group, 28 in placebo group) Reasons for early termination: Failed to complete the minimum 2 weeks on medication needed for inclusion in outcome analysis (1 in amitriptyline group, 3 in haloperidol group, 1 in placebo group) Comment: Reasons for dropouts not further specified. Total number of dropouts small, though, and balanced across groups |
| Selective reporting (reporting bias) | Unclear risk | Comment: no protocol found |
| Vested Interest (funding and/or author affiliations) | Low risk | Quote: "This work was supported by NIMH grants 35392, MHCRC 30915, and MH00658." (Soloff 1989 p. 245) |
| Other bias | Low risk | Comment: no apparent other sources of bias found |
Soloff 1993.
| Study characteristics | ||
| Methods | 5‐week trial with 3 arms:
Duration: 5 weeks (after 1‐week washout) Country: USA Setting: patients in the hospital for a minimum of 2 weeks and after discharge were seen weekly as outpatients |
|
| Participants |
Method of recruitment of participants: Patients were recruited from the inpatient services of the Western Psychiatric Institute and Clinic of the University of Pittsburgh (PA). Overall sample size: 108 Diagnosis of borderline personality disorder: DSM‐III‐R Means of assessment: DIB Mean age: 26.7 years (SD 7.2; range = no information) Sex: 75.93% women, 24.07% men Comorbidity: Patients with a current diagnosis of major depressive disorder without psychosis were included and coded for separate statistical analysis. Inclusion criteria: no information Exclusion criteria
|
|
| Interventions |
Experimental group 1
Treatment name: haloperidol
Number randomised to group: 30
Duration: 5‐week acute treatment trial followed by 16 weeks continuation treatment for medication responders Experimental group 2 Treatment name: phenelzine sulphate Number randomised to group: 34 Duration: 5‐week acute treatment trial followed by 16 weeks continuation treatment for medication responders Control/comparison group Comparison name: placebo Number randomised to group: 28 Duration: 5‐week acute treatment trial followed by 16 weeks continuation treatment for medication responders All groups Concomitant psychotherapy: not specified. Patients were inpatients; some were allowed to complete as outpatients after 2 weeks. Concomitant pharmacotherapy: At the start, patients were kept free of medication for at least 7 days, in order to washout street drugs or prescribed medications. Proportions of participants taking standing medication during trial observation period: no information |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
Sample calculation: no information Ethics approval: no information Funding source: funded by grants from universities, authorities or research foundations Conflicts of interest: No conflicts of interest were reported. Comments from trial authors (limitations)
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: insufficient information on methods used to generate random sequence to permit a judgement of low or high risk of bias |
| Allocation concealment (selection bias) | Unclear risk | Comment: Insufficient information to permit judgement of low or high risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote; "Average daily doses of medication, including placebo pseudo‐dose, are given". (Soloff 1993 p. 380) Comment: The measures undertaken to ensure blinding seem elaborate and were described in detail, so the blinding of participants seems to have been thoroughly ensured. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Medication could be increased up to six tablets (haloperidol, 6 mg; phenelzine sulfate, 90 mg; placebo, six tablets)" (Soloff 1993 p. 378). Average daily doses of medication, including placebo pseudo‐dose, were given (Soloff 1993 p. 380). Comment: The measures undertaken to ensure blinding seem elaborate and were described in detail, so the blinding of the rating trial personnel seems to have been thoroughly ensured. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk |
Quote: "Sixteen patients failed to complete the minimum 3 weeks of medication required for end‐point analysis". (Soloff 1993 p. 380) Comment: Reasons for these dropouts not further specified. Total number of dropouts small, though, and balanced across groups. Continuous outcomes based on all cases with a minimum of 3 weeks of medication exposure. Of the 108 patients enrolled, 92 completed treatment (30 in haloperidol group, 34 in phenelzine group, 28 in placebo group). Reasons for early termination: Relating to medication assignment (e.g. side effects), clinical worsening, factors unrelated to the protocol; not specified by group Patients failing to complete the minimum 3 weeks of medication required for endpoint analysis: 6 in the haloperidol group, 4 in the phenelzine group, 6 in the placebo group |
| Selective reporting (reporting bias) | Unclear risk | Comment: no protocol found |
| Vested Interest (funding and/or author affiliations) | Low risk |
Quote: "This study was supported by National Institute of Mental Health grant MH35392 and by Clinical Research Center grant MH30915." (p. 697) Quote: ”This work was supported by grants MH35392, MH00658, and CRC30915 from the National Institute of Mental Health,Bethesda, Md.” (Soloff 1993, p.385) |
| Other bias | Low risk | Comment: no apparent other sources of bias found |
Tritt 2005.
| Study characteristics | ||
| Methods | 8‐week trial with 2 arms:
Duration: 8 weeks Country: Germany Setting: outpatient |
|
| Participants |
Method of recruitment of participants: Recruitment of patients was accomplished primarily through family doctors’ advertisements. Overall sample size: 27 Diagnosis of borderline personality disorder: DSM‐IV Means of assessment: SCID‐II Mean age: 29.15 years (SD = no information; range = no information) Sex: 100% women Comorbidity: no information Inclusion criteria
Exclusion criteria
|
|
| Interventions |
Experimental group
Treatment name: lamotrigine
Number randomised to group: 18
Duration: 8 weeks Control/comparison group Medication name: placebo Number randomised to group: 9 Duration: 8 weeks Both groups Concomitant psychotherapy: Other psychotropic medication was not allowed. Concomitant pharmacotherapy: Other psychotropic medication was an exclusion criterion. No further information provided Proportions of participants taking standing medication during trial observation period: no information |
|
| Outcomes |
Primary outcomes: none Secondary outcomes
|
|
| Notes |
Sample calculation: no information Ethics approval: yes Funding source: no funding received Conflicts of interest: No conflicts of interest were reported. Comments from trial authors (limitations)
Comments from review authors: continuous outcomes based on ITT data (LOCF) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: insufficient information on methods used to generate random sequence to permit a judgement of low or high risk of bias |
| Allocation concealment (selection bias) | Unclear risk | Comment: There was no information on how sequence allocation was concealed to permit a judgement of low or high risk of bias. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk |
Quote: "Tablets were supplied in numbered boxes." (Tritt 2005, p. 288) Quote: "Each individual received one blinded capsule medication daily [...] Both subjects and clinicians were blinded regarding assignment." (Tritt 2005, p. 288) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: The trial was referred to as being double‐blind, however, insufficient information was given on how blinding of outcome assessors was carried out and maintained, to permit a judgement of low or high risk of bias. |
| Incomplete outcome data (attrition bias) All outcomes | High risk |
Quote: "Thirty‐eight subjects were eligible to take part in the study [...] The necessary sample size was calculated [...] This resulted in a group size of n = 27 patients [...] active drug (n = 18) compared to placebo (n = 9)". (Tritt 2005, p. 288) Comment: not clear why or how the 27 participants were finally chosen out of the 38 potential participants |
| Selective reporting (reporting bias) | High risk | Comment: All outcomes (i.e. one assessment instrument) reported as planned to be assessed were also reported. However, it seemed implausible to use only one assessment instrument in such a complex trial. There was no protocol available to check the predefined outcome measure(s). |
| Vested Interest (funding and/or author affiliations) | Low risk | Quote: "The study was conducted independently of any institutional influence and was not funded." (Tritt 2005, p. 288) |
| Other bias | Low risk | Comment: no apparent other sources of bias found |
Zanarini 2001.
| Study characteristics | ||
| Methods | 6‐month trial with 2 arms:
Duration: 6 months Country: USA Setting: outpatient |
|
| Participants |
Method of recruitment of participants: through advertisement in Boston‐area newspapers seeking women aged between 18 and 40 years, who were disturbed by moodiness, distrustfulness, impulsivity, and painful and difficult relationships Overall sample size: 28 Diagnosis of borderline personality disorder: DSM‐IV Means of assessment: DIB‐R Mean age: 26.7 years (SD = no information; range = no information) Sex: 100% women Comorbidity: no information Inclusion criteria: meeting both DIB‐R and DSM‐IV criteria for BPD and not meeting current criteria for major depression Exclusion criteria
|
|
| Interventions |
Experimental group
Treatment name: olanzapine
Number randomised to group: 19
Duration: 6 months Control/comparison group Comparison name: placebo Number randomised to group: 9 Duration: 6 months Both groups Concomitant psychotherapy: not specified Concomitant pharmacotherapy: No other psychotropic medication was allowed. Proportions of participants taking standing medication during trial observation period: Patients currently prescribed any psychotropic medication that they thought was helping to alleviate troublesome symptoms were excluded. No further information provided |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
Sample calculation: no information Ethics approval: no information Funding source: funded or partially funded by pharmaceutical industry Conflicts of interest: No conflicts of interest were reported besides partial funding from the pharmaceutical industry. Comments from trial authors (limitations)
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "random number sequence" (Zanarini 2001, p. 850) |
| Allocation concealment (selection bias) | Unclear risk | Comment: There was no information on how sequence allocation was concealed to permit a judgement of low or high risk of bias. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk |
Quote: "Tablets were supplied in numbered bottles containing drug or placebo as determined by a random number sequence." (Zanarini 2001, p. 850) Quote: "Each tablet contained either 2.5 mg of olanzapine or matching inert placebo. [...] Both subjects and clinicians were blinded to olanzapine/placebo assignment. The blind was broken after the acquisition of all endpoint data for all subjects." (Zanarini 2001, p. 850) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: The trial was referred to as being double‐blind, however, insufficient information was given on how blinding of outcome assessors was carried out and maintained, to permit a judgement of low or high risk of bias. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk |
Quote: "Thirty subjects completed all aspects of pre‐randomization assessment. However, 2 of these subjects were excluded [...] because it was determined that they were responding well to a selective serotonin reuptake inhibitor. Twenty‐eight subjects entered the trial and were randomly assigned [...] All [...] completed at least 2 post‐baseline visits and were included in all subsequent analyses." (Zanarini 2001, p. 851) Comment: Of the 28 patients enrolled, 9 completed treatment (8 in olanzapine group, 1 in placebo group) Reasons for early termination: Sedation: 1 in olanzapine group, 0 in placebo group Increased anxiety or depression: 3/2 Perceived weight gain: 2/0 Lost to follow‐up: 5/6 Continuous outcomes based on ITT sample (LOCF) Comment: high dropout rate overall, but adequately addressed |
| Selective reporting (reporting bias) | High risk | Quote: "Due to the small number of subjects, results pertaining to secondary outcome measures will not be reported." (Zanarini 2001, p. 851) |
| Vested Interest (funding and/or author affiliations) | High risk | Quote: "Supported, in part, by a grant from Eli Lilly" (Zanarini 2001, p. 849) |
| Other bias | Low risk | Comment: no apparent other sources of bias found |
Zanarini 2003.
| Study characteristics | ||
| Methods | 8‐week trial with 2 arms:
Duration: 8 weeks Country: USA Setting: outpatient |
|
| Participants |
Method of recruitment of participants: "Patients recruited through advertisements in Boston newspapers with the ads asking, “Are you extremely moody? Do you often feel out of control? Are your relationships painful and difficult?" (Zanarini 2003, p. 167) Overall sample size: 30 Diagnosis of borderline personality disorder: DSM‐IV Means of assessment: DIB‐R Mean age: 26.3 years (SD 6.2; range = no information) Sex: 100% women Comorbidity: no information; however, exclusion criteria suggest comorbid mental disorders were not allowed Inclusion criteria
Exclusion criteria
|
|
| Interventions |
Experimental group
Treatment name: ethyl‐eicosapentaenoic acid (E‐EPA)
Number randomised to group: 20
Duration: 8 weeks Control/comparison group Comparison name: placebo: mineral oil Number randomised to group: 10 Duration: 8 weeks Both groups Concomitant psychotherapy: no information Concomitant pharmacotherapy: Patients were excluded if they were currently being prescribed any psychotropic medication or taking E‐EPA supplements; however, no further information was provided. Proportions of participants taking standing medication during trial observation period: no information |
|
| Outcomes |
Primary outcomes : none Secondary outcomes
|
|
| Notes |
Sample calculation: no information Ethics approval: no information Funding source: funded by grants from universities, authorities or research foundations Conflicts of interest: Trial medication was provided by a pharmaceutical company. Comments from trial authors (limitations): "The main limitations of this study are that only women were studied and all participants were moderately ill. Whether similar results would be found for male participants or participants with a more severe symptom picture is unknown." Zanarini 2003, p. 168) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: insufficient information on methods used to generate random sequence to permit a judgement of low or high risk of bias |
| Allocation concealment (selection bias) | Unclear risk | Comment: no information given on allocation concealment to permit a judgement of low or high risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: The trial was referred to as being double‐blind, however, insufficient information was given on how blinding of participants and personnel was carried out (packaging of trial medication etc.) and was maintained, to permit a judgement of low or high risk of bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: The trial was referred to as being double‐blind, however, insufficient information was given on how blinding of outcome assessors was carried out and maintained, to permit a judgement of low or high risk of bias. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk |
Quote: "The three subjects who discontinued their participation (two taking E‐EPA and one taking placebo) did so because of life events unrelated to the study." (Zanarini 2003, p. 168) Comments: Of the 30 patients enrolled, 27 completed treatment (18 in E‐EPA group, 9 in placebo group) Reasons for early termination: Life events unrelated to the trial: 2 in E‐EPA group, 1 in placebo group Continuous outcomes were based on completers only. |
| Selective reporting (reporting bias) | Unclear risk | Comment: no protocol found |
| Vested Interest (funding and/or author affiliations) | High risk | Quotes: "Capsules were supplied by Laxdale Pharmaceuticals (Stirling, U.K.)." (Zanarini 2003, p. 167), "Supported by an Independent Investigator Award from the National Alliance for Research on Schizophrenia and Depression to Dr. Zanarini." (Zanarini 2003, p. 169) |
| Other bias | Low risk | Comment: no apparent other sources of bias found |
Zanarini 2004.
| Study characteristics | ||
| Methods | 8‐week trial with 3 arms:
Duration: 8 weeks Country: USA Setting: outpatient |
|
| Participants |
Method of recruitment of participants: "Recruitment [...] was accomplished primarily through advertisement in Boston, Mass. area newspapers." (Zanarini 2004, p. 904) Overall sample size: 45 Diagnosis of borderline personality disorder: DSM‐IV Means of assessment: DIB‐R Mean age: 23 years (SD 5.7; range = no information) Sex: 100% women Comorbidity: Current major depression, current or lifetime schizophrenia, schizoaffective disorder and bipolar disorder were exclusion criteria. In terms of Axis I disorders, 93.3% had a history of a mood disorder, 51.1% had a history of a substance use disorder, 48.9% had a history of an anxiety disorder and 44.4% had a history of an eating disorder. Inclusion criteria
Exclusion criteria
|
|
| Interventions |
Experimental group
Treatment name: fluoxetine
Number randomised to group: 14
Duration: 8 weeks Comparison group 1 Treatment name: olanzapine Number randomised to group: 16 Duration: 8 weeks Comparison group 2 Treatment name: fluoxetine + olanzapine Number randomised to group: 15 Duration: 8 weeks All groups Concomitant psychotherapy: not specified Concomitant pharmacotherapy: Current prescription to any psychotropic medication was an exclusion criterion. No other information provided Proportions of participants taking standing medication during trial observation period: no information |
|
| Outcomes |
Primary outcomes: none Secondary outcomes
|
|
| Notes |
Sample calculation: no information Ethics approval: yes Funding source: funded or partially funded by pharmaceutical industry Comments from trial authors (limitations)
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: insufficient information on methods used to generate random sequence to permit a judgement of low or high risk of bias |
| Allocation concealment (selection bias) | Unclear risk | Comment: insufficient information to permit judgement of high or low risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Dose was adjusted by an unblinded psychiatrist according to perceived response and side effects. Both subjects and raters were blinded to study assignment. The blind was broken after acquisition of all endpoint data for all subjects." (Zanarini 2004, p. 904) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Dose was adjusted by an unblinded psychiatrist according to perceived response and side effects. Both subjects and raters were blinded to study assignment. The blind was broken after acquisition of all endpoint data for all subjects." (Zanarini 2004, p. 904) |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk |
Comments: Reasons for dropout specified (Zanarini 2004 p. 905), but outcome data were only reported for completers. Of the 45 patients enrolled, 42 completed treatment (13 in fluoxetine group, 16 in olanzapine group, 13 in fluoxetine + olanzapine group) Reasons for early termination: Onset of a number of psychosocial stressors culminating in a suicide gesture: 1/0/0 Dizziness and headaches: 0/0/1 Lost to follow‐up: 0/0/1 |
| Selective reporting (reporting bias) | Unclear risk | Comment: no protocol found |
| Vested Interest (funding and/or author affiliations) | High risk | Quote: "Supported by a grant from Eli Lilly, Indianapolis, Ind." (Zanarini 2004, p. 903) |
| Other bias | Low risk | Comment: no apparent other sources of bias found |
Zanarini 2007.
| Study characteristics | ||
| Methods | 12‐week with 3 arms:
Duration: 12 weeks (after a 2‐week screening period) Country: Argentina, Chile, Italy, Peru, Poland, Romania, Turkey, USA, and Venezuela Setting: outpatient |
|
| Participants |
Method of recruitment of participants: no information Overall sample size: 451 Diagnosis of borderline personality disorder: DSM‐IV‐TR Means of assessment: Diagnostic Interview for Borderline Personality Disorders, 4th Version (DIBP‐IV) and ZAN‐BPD Age: 32.98 years (SD 10.83; range = no information) Sex: 73.61% women Comorbidity
Inclusion criteria
Exclusion criteria
|
|
| Interventions |
Experimental group
Treatment name: olanzapine (2.5 mg/day)
Number randomised to group: 150
Duration: 12 weeks Comparison group Treatment name: olanzapine (5‐10 mg/day, mean dose 6.66 mg/day) Number randomised to group: 148 (106 women, 42 men) Duration: 12 weeks Control group Comparison name: placebo Number randomised to group: 153 (117 women, 36 men) Duration: 12 weeks All groups Concomitant psychotherapy: Beginning any type of psychotherapy within the 3 months prior to visit 1 or during the acute phase of the trial was not allowed. Ongoing psychotherapy > 3 months at the time of visit 1 was allowed; however, if there was an increase in psychotherapy frequency or change in type of psychotherapy during trial periods 1 or 2, the patients were discontinued. Concomitant pharmacotherapy: The use of benzodiazepine or hypnotics or lorazepam and episodic use of anticholinergics or benztropine mesylate or biperiden or trihexyphenidyl was allowed during the trial at a specific dose; however, the use of anticholinergics as prophylaxis for extrapyramidal symptoms was not allowed. Proportions of participants taking standing medication during trial observation period: no information |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
Sample calculation: no information Ethics approval: no information Funding source: funded or partially funded by pharmaceutical industry Conflicts of interest: "Dr. Zanarini has received grant/research support from Eli Lilly. Dr. Schulz has been a consultant for Eli Lilly and has received research grants from Eli Lilly and AstraZeneca. Drs. Detke, Tanaka, Deberdt and Kryzhanovskaya are employees and stock shareholders of Eli Lily. Drs. Zhao, Lin and Corya are employees of Eli Lilly". (Zanarini 2007, p. 1361) Comments from trial authors (limitations)
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk |
Quote: "patients were randomised to 1 of 3 treatment groups." (Eli Lilly 2008, p. 3) Comment: randomisation conducted centrally |
| Allocation concealment (selection bias) | Unclear risk | Comment: Insufficient information on how allocation sequence was concealed and maintained until randomised trial phase started to permit a judgement of low or high risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: Article stated that the trial was blinded, however, there was Insufficient information about how blinding in the randomised phase was carried out and maintained (e.g. identical capsules of trial medication). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: Insufficient information about outcome assessors and how they were blinded to permit a judgement of low or high risk of bias |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk |
Quote: "451 were randomly assigned (148 [...] to olanzapine 5‐to‐10 mg/day treatment group, 150 [...] to olanzapine 2.5‐mg/day treatment group, and 153 to placebo)." (Eli Lilly 2008, p. 14), "last observation carried forward" (Eli Lilly 2008, p. 3) Comments: continuous data based on LOCF data; dichotomous data based on ITT sample Of the 451 patients enrolled, 294 completed the full 12 weeks of the double‐blind treatment phase (97/103/94). In this review, only the groups receiving olanzapine 5 to 10 mg/day or placebo group were included. |
| Selective reporting (reporting bias) | Low risk | Comment: The trial protocol was available and all of the study's prespecified (primary and secondary) outcomes that were of interest in the review were reported in the way they were prespecified. |
| Vested Interest (funding and/or author affiliations) | High risk | Comment: Eli Lilly was the trial sponsor. Most trial results used here were from the company's trial report (the remaining references were either clinical trial register entries or congress abstracts and did not provide detailed data). |
| Other bias | Low risk | Comment: no indication of other bias |
Ziegenhorn 2009.
| Study characteristics | ||
| Methods | 18‐week trial with 2 arms:
Duration of trial: 8 weeks (4 weeks + 4‐week cross‐over) Country: Germany Setting: inpatient |
|
| Participants |
Methods of recruitment of patients: patients admitted to an intensive inpatient treatment programme Overall sample size: 18 (17 were included in the analysis due to 1 nonstarter) Diagnosis of Borderline personality disorder: DSM‐IV Means of assessment: Mini International Neuropsychiatric Interview for DSM‐IV; SCID‐II Mean age: 32 years (SD 8; range = 19‐44) Sex: 94.44% women Comorbidity: 88% had comorbid disorders: PTSD, eating disorders and/or substance abuse Inclusion criteria
Exclusion criteria
|
|
| Interventions |
Experimental group
Treatment name: clonidine
Number randomised to group: 18
Duration: 4 weeks Control/comparison group Comparison name: placebo Number randomised to group: 18 Duration: 4 weeks Both groups Concomitant psychotherapy: no information Concomitant pharmacotherapy: allowed. Patients were included if they were on a stable psychotropic medication regimen of up to 3 psychotropic drugs. Proportions of participants taking standing medication during trial observation period: 10 participants took antidepressants, three took antipsychotics and one took valproate. |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
Sample calculation: no information Ethics approval: yes Funding source: no funding received Conflicts of interest: No conflicts of interest were reported. Comments from trial authors (limitations)
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk |
Quote: “After a baseline evaluation, patients were assigned to receive either clonidine or placebo capsules first using a block randomisation procedure.” (Ziegenhorn 2009, p. 170) Comment: Insufficient information about the block randomisation procedure to permit a judgement of low or high risk of bias |
| Allocation concealment (selection bias) | Unclear risk | Comment: No information provided on concealment of random sequence allocation to permit a judgement of low or high risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk |
Quote: "Unblinding of patients to the treatment condition by adverse effects might have biased results towards improvements. Nonetheless, doses were escalated slowly, and adverse effects were infrequent". (Ziegenhorn 2009, p. 173) Comment: The trial was referred to as being double‐blind, however, there was insufficient information on how the blinding was carried out and maintained to permit a judgement of low or high risk of bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Each patient was evaluated by 2 blinded researchers independently.” (Ziegenhorn 2009, p. 170) |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: 7 dropouts. 1 patient excluded before randomisation and initiation of treatment. Potential risk of bias. Last observation was carried forward and was used as an imputation method |
| Selective reporting (reporting bias) | Unclear risk | Comment: no protocol found, but the trial reported a protocol being approved by the ethics board |
| Vested Interest (funding and/or author affiliations) | Low risk | Comment: There were no sources of support, and none of the authors had any conflicts of interest to declare. |
| Other bias | Low risk | Comment: no apparent other source of bias |
ADDS: Atypical Depression Diagnostic Scale. ADHD: attention deficit hyperactivity disorder. ADP: Assessment of DSM‐IV Personality Disorders Questionnaire. A&E: accident and emergency. AIAQ: Anger, Irritability and Assault Questionnaire. AIDS: acquired immune deficiency syndrome. AIMS: Abnormal Involuntary Movements Scale. ALS: Affective Lability Scale. ANOVA: analysis of variance. ASI: Addiction Severity Index ATP: according to protocol. BARS: Barnes Akathisia Rating Scale. BDHI: Buss‐Durkee Hostility Inventory. BDI: Beck Depression Inventory. BEST: Borderline Evaluation of Severity over Time. BIS: Barrett Impulsiveness Scale. BMDP: The Biomedical Data, Program. BPD(SI): Borderline Personality Disorder (Severity Index). BPRS: Brief Psychiatric Rating Scale. BSL: Borderline SymptomList. CAPS‐D: Clinician Administered PTSD Scale. CBZ: carbamazepine. CGI‐BPD: Clinical Global Impression scale for Borderline Personality Disorder. CGI‐I: Clinical Global Impression scale ‐ Improvement. CGI‐S: Clinical Global Impression scale ‐ Severity. CI: confidence interval. CNS: central nervous system. CSSRS: Columbia Suicidal Severity Rating Scale. DBT: Dialectal Behavioural Therapy. DES: Dissociative Experiences Scale. DHA: docosahexaenoic. DIB/DIB‐R: Gunderson's Diagnostic Interview for Borderline Patients (R: Revised version). DIPD‐IV: Diagnostic Interview for DSM‐IV Personality Disorders. DIS‐Q: Dissociation Questionnaire. DOTES: Dosage Record and Emergent Treatment Symptom Scale. DSHI: Deliberate Self‐Harm Inventory. DSM‐III: Diagnostic and Statistical Manual of Mental Disorders, 3rd Edition. DSM‐III‐R: Diagnostic and Statistical Manual of Mental Disorders, 3rd Edition, Revised. DSM‐IV: Diagnostic and Statistical Manual of Mental Disorders, 4th Edition. DSM‐IV‐TR: Diagnostic and Statistical Manual of Mental Disorders, 4th Edition, Text Revision. DSM‐V: Diagnostic and Statistical Manual of Mental Disorders, 5th Edition. DSS: Dissociative States Scale. EEG: electroencephalogram. E‐EPA: ethyl eicosapentaenoic acid. EKG: electrocardiogram. EOT: end of treatment. EPA: eicosapentaenoic acid. ER: emergency room et seq: and what follows. GAD: generalised anxiety disorder. GAF: Global Assessment of Functioning Scale. GAS: Global Assessment scale. HAM‐D/HDRS‐24: Hamilton Depression Rating Scale (HDRS‐24: 24‐item version). HSCL: Hopkins Symptoms Check List. IBS: irritable bowel syndrome. IMPS: Inpatient Multidimension Rating Scale. IMS: Involuntary Movement Scale. IPDE: International Personality Disorder Examination. IQ: intelligence quotient. ITT: intention‐to‐treat. lb: pounds. LOCF: last‐observation‐carried‐forward. MADRS: Montgomery Åsberg Depression Rating Scale. MAOIs: monoamine oxidase inhibitors. MMT: methadone maintenance treatment MOAS/OAS‐M: Modified Overt Aggression Scale. NMDAR: N‐methyl‐D‐aspartate. NSSI: non‐suicidal self‐injury. OCD: obsessive compulsive disorder. OFC: olanzapine‐fluoxetine combination PANSS: Positive and Negative Syndrome Scale. PDRS: Personality Disorder Rating Scale. PLC: placebo. POMS: Profile of Mood States scale. PTSD: post‐traumatic stress disorder. SCID‐I: Structured Clinical Interview for DSM, Version 1. SCID‐II: Structured Clinical Interview for DSM, Version 2. SCL‐90/HSCL: (Hopkins) Symptom Check list‐90 (Symptom scales: DEP: Depression, GSI: Global Severity Index; HOS: hostility INT; Interpersonal sensitivity, PAR; Paranoid ideation, PSY; Psychotisism) R: Revised version. SD: standard deviation. SFQ: Social Functioning Questionnaire. SHI: Self‐Harm Inventory. SIB: Schedule of Interviewing Schizotypal Personalities ‐ Borderline Score. SNOOP: Systematic Nurses' Observation of Psychopathology. SOFAS: Social Occupational Functioning Assessment Scale. SPD: sensory processing disorder. SSI: Scale for Suicidal Ideation SSRI: selective serotonin reuptake inhibitors. STAXI: State‐Trait Anger Expression Inventory. STIC: Self Report test of Impulsive Control TCAs: tricyclic antidepressants. TG: topiramate group UKU: udvalg for kliniske undersogelser. Zan‐BPD: Zanarini Rating Scale for Borderline Personality Disorder.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| ACTRN12615000705583 | Unavailable trial: trial withdrawn due to recruitment difficulties |
| Bellino 2006b | Ineligible comparator: fluoxetine vs fluoxetine + interpersonal therapy; testing the effects of additional psychotherapy compared to pharmacotherapy alone |
| Coccaro 1997 | Subsample data unavailable (sought subsample data for BPD subpopulation (33% of overall sample; Stoffers‐Winterling J 2018 [pers comm]) but did not retrieve requested information) |
| Hollander 2005 | Subsample data unavailable (less than 70% had a full diagnosis of BPD; authors of the 2010 review approached study authors for subsample data but did not retrieve them) |
| ISRCTN11135486 | Unavailable trial: trial never started because funding provider withdrew (Malevani email reply on 17 June 2020) |
| Koenigsberg 2003 | Subsample data unavailable (less than 70% of participants had BPD; no subsample data retrieved by authors of the preceding version of this review despite contacting the study authors (Stoffers 2010)) |
| La Malfa 2003 | Subsample data unavailable (less than 70% of participants had BPD; separate data on BPD patients available but number of BPD patients allocated to each group unclear; the authors of the previous version of this review were unable to retrieve subsample data (Stoffers 2010)) |
| Links 1990 | Ineligible patient population: participants with BPD features (mean DIB score 9.47, SD = 0.75); exact number of individuals with full BPD unclear |
| Marchesi 2006 | Ineligible patient population: participants had no official BPD diagnosis |
| NCT00255554 | Unavailable trial: trial not funded and never started |
| NCT00463775 | Unavailable trial: trial withdrawn as recruitment not progressing as planned |
| NCT00633802 | Unavailable trial: trial discontinued due to personnel changes |
| NCT01103180 | Unavailable trial: trial terminated due to problems with recruitment |
| NCT03395314 | Ineligible intervention: intervention duration shorter than 2 weeks |
| Parsons 1989 | Subsample data unavailable (less than 70% participants with BPD; no subsample available) |
| Rombold 2014 | Subsample data unavailable (less than 70% participants with BPD; retrieved subsample data for PD but fewer than 5 participants in total had BPD) |
| Russell 2003 | Ineligible patient population: participants had PD but not BPD |
| Serban 1984 | Subsample data unavailable (less than 70% participants had BPD; unable to retrieve subsample data) |
| Verkes 1998 | Subsample data unavailable (participants were suicide‐attempt repeaters, not clear how many patients actually had BPD; the authors of the previous version of this review were unable to retrieve subsample data (Stoffers 2010)) |
| Wollmer 2022 | Ineligible intervention: intervention duration shorter than 2 weeks |
BPD: borderline personality disorder DIB: Diagnostic Interview for Borderline Patients PD: personality disorder SD: standard deviation vs: versus
Characteristics of studies awaiting classification [ordered by study ID]
NCT00437099.
| Methods |
Allocation: randomised Intervention model: parallel assignment (3 arms) Blinding: triple (participant, investigator and outcomes assessor) Duration of trial: 12 weeks Timing of assessment: baseline, week 2, 4, 6, 8, 10, and 12 |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions |
Experimental 1
Drug name: omega‐3‐acid ethyl esters: eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA)
Brand name: Omacor®
Drug dose: 1.680 mg/day
Route of administration: oral
Administration: capsule
Additional intervention: cognitive behaviour therapy (CBT) Experimental 2 Drug name: omega‐3‐acid ethyl esters: EPA and DHA Brand name: Omacor® Drug dose: 3.360 mg/day Route of administration: oral Administration: capsule Additional intervention: CBT Comparator Drug name: placebo Brand name: N/A Drug dose: N/A Route of administration: oral Administration: no information Additional intervention: CBT |
| Outcomes |
Primary outcomes
Secondary outcomes
|
| Notes |
Study start date: February 2009 Source of funding: Hospital Universitari Vall d'Hebron Research Institute Conflicts of interest: none reported Recruitment status: unknown, as reported on trial register; did not receive reply from PI on request and unable to identify publication |
NCT01912391.
| Methods |
Allocation: randomised Intervention model: parallel assignment (2 arms) Blinding: double (participant and investigator) Duration: 12 weeks Timing of assessment: baseline and every week for 12 weeks |
| Participants |
Inclusion criteria
Exclusion Criteria
|
| Interventions |
Experimental
Drug name: selegiline
Brand name: Emsam
Drug dose: 12 mg/day
Route of administration: transdermal patch
Administration: once daily Comparator Drug name: placebo Brand name: N/A Drug dose: N/A Route of administration: transdermal patch Administration: once daily |
| Outcomes |
Primary outcome: Hopkins Symptom Checklist 90‐Revised (SCL 90‐R) scale assessed at weeks 1‐12 Secondary outcomes
|
| Notes |
Trial start date: October 2012 Source of funding: Mood and Anxiety Research, Incorporated Conflicts of interest: The Study Director, Paul Markovitz, is the Director of Mood and Anxiety Research. Recruitment status: completed |
BDNF: brain‐derived neurotrophic factor. BIS‐11: Barratt Impulsivity Scale‐11. BPD: borderline personality disorder. BPRS: Brief Psychiatric Rating Scale. CBT: cognitive behavioural therapy. CGI: Clinical Global Impression scale. DBT: Dialectal Behavioural Therapy. DHA: docosahexaenoic acid. DIB‐R: Diagnostic Interview for Borderline Patients‐Revised. DSM‐IV: Diagnostic Manual of Mental Disorders, 4th edition EEAG: Global Activity Scale. EPA: eicosapentaenoic acid. HAM‐D: Hamilton Depression Inventory. MAOI: monoamine oxidase inhibitor. N/A: not applicable. PI: principal investigator. SASS: Social Adaptation Self‐evaluation Scale, SCL‐90‐R: Symptom Checklist 90‐Revised. SDS: Sheehan Disability Scale. STAI: State‐Trait Anxiety Inventory. STAXI: State‐Trait Anger Expression Inventory. YMRS: Young Mania Rating Scale.
Characteristics of ongoing studies [ordered by study ID]
ACTRN12617001317381.
| Study name | Public title: Estrogen for the treatment of borderline personality disorder Scientific title: A randomised placebo controlled trial of estradiol for the treatment of women with borderline personality disorder |
| Methods | Allocation: randomised Intervention model: parallel assignment Blinding: double (participant and treatment administration) Duration: 12 weeks Timing of assessment: baseline, week 2, week 4, week 6, week 8, week 10, week 12 |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions |
Experimental
Drug name: estradiol
Brand name: no information
Drug dose: 100 mcg twice weekly
Route of administration: transdermal
Administration: patch Comparator Drug name: placebo Brand name: N/A Drug dose: N/A Route of administration: transdermal Administration: patch |
| Outcomes |
Primary outcomes
Secondary outcomes
|
| Starting date | 13 January 2020 |
| Contact information | Name: Jayashri Kulkarni Address: Monash Alfred Psychiatry Research Centre, Level 4, 607 St Kilda Rd Melbourne VIC 3004, Australia Phone: +61 3 9076 6564 Email: jayashri.kulkarni@monash.edu |
| Notes | Source of funding: Monash Alfred Psychiatry Research Centre |
Arteaga‐Henríquez 2020.
| Study name | Public title: Treating impulsivity in adults with probiotics (PROBIA) Scientific title: Randomized placebo‐controlled treatment of impulsivity in adults with probiotics |
| Methods | Allocation: randomised Intervention model: parallel assignment (2 arms) Blinding: none, open‐label Duration: 10 weeks Timing of assessment: baseline, week 5, week 10 and week 11 |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions |
Experimental
Drug name: 3 LAB species known to have anti‐inflammatory effects and restoring the intestinal barrier, and 4 fermentable fibres: pediococcus pentosaceus 5‐33:3; lactobacillus paracasei subsp. paracasei 19; and lactobacillus plantarum 2362 in combination with the following four fermentable fibres: betaglucan, inulin, pectin and resistant starch
Brand name: probiotic Synbiotic 2000 Forte (SF)
Drug dose: one dose daily
Route of administration: oral
Administration: powder (to be spread on top of cold foods such as muesli, salad or yogurt) Comparator Drug name: placebo (non‐digestible carbohydrate with similar texture and flavour to SF) Brand name: N/A Drug dose: one dose daily Route of administration: oral Administration: powder (to be spread on top of cold foods such as muesli, salad or yogurt) |
| Outcomes |
Primary outcomes
Secondary outcomes
|
| Starting date | 22 February 2019 |
| Contact information | Name: Josep Antoni Ramos‐Quiroga Email:jaramos@vhir.org |
| Notes | Source of funding: no information |
Chanen 2019.
| Study name | Public title: VERBATIM: a randomised controlled trial of aripiprazole for the treatment of auditory verbal hallucinations in borderline personality disorder Scientific title: A randomised controlled trial of aripiprazole for the treatment of auditory verbal hallucinations in borderline personality disorder |
| Methods | Allocation: randomised Intervention model: parallel assignment (2 arms) Blinding: quadruple (participant, treatment administration, outcome assessors and data analysts) Duration: 12 weeks (and 2‐week follow‐up) Timing of assessment: baseline and every week for 12 weeks |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions |
Experimental
Drug name: aripiprazole
Brand name: no information
Drug dose: individual titration. Week 1 = 2 mg/day; week 2 = 5 mg/day; week 3 = 10 mg/day; week 4 to week 12 = increments to 15 mg once daily, 20 mg once daily then 30 mg (maximum allowed doses) once daily
Route of administration: oral
Administration: capsule
Additional intervention: CAT Comparator Drug name: placebo Brand name: N/A Drug dose: N/A Route of administration: oral Administration: capsule Additional intervention: CAT |
| Outcomes |
Primary outcome:
Secondary outcomes
|
| Starting date | 1 September 2016 |
| Contact information | Name: Andrew Chanen Affiliation: Orygen, The National Centre of Excellence in Youth Mental Health Address: 35 Poplar Rd, (Locked Bag 10), Parkville, Victoria, 3052 Phone: +61393422800 |
| Notes | Source of funding: National Health and Medical Research Council |
DRKS00015817.
| Study name | Public title: Stellate ganglion block in patients with borderline personality disorder and post‐traumatic stress disorder Scientific title: as above |
| Methods | Allocation: randomised Intervention model: parallel assignment Blinding: no information Duration: 8 weeks Timing of assessment: Baseline, twice a week (the Dissociations Tension Scale‐4 (DSS‐4) and Clinical Global Impression Skala (CGI)) and once a week (Beck Depression Inventory (BDI), Borderline Symptom List‐23 (BSL‐23), State Trait Anxiety Inventory‐S (STAI‐S), PTSD Checklist for DSM‐5 (PCL‐5) and the Patient and Observer Scare Assessment Scale (POSAS)) for 8 weeks, and baseline and week 8 (Questionnaire of Dissociative Symptoms (FDS) and the Symptom Checklist‐90‐R (SCL‐90‐R)). |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions |
Experimental (arm 1)
Stationary patients with post‐traumatic stress disorder and borderline personality disorder receive in addition to a standard therapy (dialectical behavorial therapy, after week 4) a series of 8 stellate ganglion blocks (2 per week) on both sides, with each 3 mL Ropivacain 1% Comparator (arm 2) Stationary patients with post‐traumatic stress disorder and borderline personality disorder receive the same procedure as in arm 1 without the stellate ganglion block |
| Outcomes |
Primary outcomes
Secondary outcome
|
| Starting date | 02 January 2019 |
| Contact information | Name: Mr. Prof. Dr. med. Christian Schmahl Affiliation: Klinik für Psychosomatik und Psychotherapeutische Medizin, Zentralinstitut für Seelische Gesundheit Address: Theodor‐Kutzer‐Ufer 1‐3, 68167 Mannheim, Germany Phone: 0621 1703‐4021 Email: christian.schmahl@zi‐mannheim.de |
| Notes | Source of funding: Klinik für Anästhesiologie und Operative Intensivmedizin, Schmerzzentrum,Universitätsmedizin Mannheim |
EUCTR2020‐003469‐20‐ES.
| Study name | Public title: A phase IIb study to evaluate the safety and efficacy of vafidemstat in an adult borderline personality disorder population Scientific title: A double‐blind, randomized, placebo‐controlled, adaptive 14‐week phase IIb trial to evaluate the efficacy and safety of vafidemstat in an adult borderline personality disorder (BPD) population (PORTICO) |
| Methods | Allocation: randomised Intervention model: parallel assignment Blinding: double‐blind Duration: 14 weeks Timing of assessment: Baseline, specific weeks (no further information), and week 14 |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions |
Experimental
Drug name: Vafidemstat
Brand name: N/A
Drug dose: 1.2 mg
Route of administration: oral
Administration: no information Comparator: placebo |
| Outcomes |
Primary outcomes
Secondary outcomes
|
| Starting date | 19 November 2020 |
| Contact information | Name: Clinical Operations Affiliation: Oryzon Genomics S.A. Address: Sant Ferran 74 08940 Cornellà de Llobregat, Barcelona Spain Phone: 34647796923 Email: sgutierrez@oryzon.com |
| Notes | Source of funding: Oryzon Genomics S.A |
EudraCT 2018‐002471‐18‐GB.
| Study name | Public title: Clozapine in the treatment of borderline personality disorder Scientific title: The clinical effectiveness and cost effectiveness of clozapine for inpatients with borderline personality disorder: randomised controlled trial ‐ CALMED |
| Methods | Allocation: randomised Intervention model: parallel assignment Blinding: double (participant and treatment administration) Duration: 6 months Timing of assessment: baseline, 3 months, 6 months, 12 months, 18 months |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions |
Experimental
Drug name: clozapine
Brand name: Clozaril
Drug dose: 12.5 mg/day to 400 mg/day
Route of administration: oral
Administration: capsule Comparator Drug name: placebo Brand name: N/A Drug dose: N/A Route of administration: oral Administration: capsule |
| Outcomes |
Primary outcome
Secondary outcomes
|
| Starting date | 18 January 2019 |
| Contact information | Name: Mike Crawford Affiliation: Imperial College London Adress: Du Cane Road W12 0NN London United Kingdom Phone: 02083834161 Email: m.crawford@imperial.ac.uk |
| Notes | Source of funding: no information |
IRCT20210106049948N1.
| Study name | Public title: Memantine and borderline personality disorder Scientific title: The effect of memantine on the symptoms of borderline personality disorder in the Iranian population |
| Methods | Allocation: randomised Intervention model: parallel assignment Blinding: double‐blinding. “The researcher does not know whether the patient being evaluated belongs to the placebo group or the memantine group. The patients also do not know if they have used placebo or memantine. Drugs and placebos are similar in appearance, such as color, shape, and so on. The patient (placebo or memantine groups) receive the drug in encoded packets. The coding is done by the psychiatrist and the evaluator and the patient is blind”. Duration: 12 weeks Timing of assessment: baseline and at the end of weeks 2, 4, 6, 8, and 12 |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions |
Experimental
Drug name: memantine
Brand name: not stated
Drug dose: 10 mg daily in week 9, 20 mg daily week 10‐12
Route of administration: oral
Administration: The treatment group will receive a placebo daily for the first 8 weeks, then memantine daily the last 4 weeks. Comparator Drug name: placebo Brand name: not stated Drug dose: not stated Route of administration: oral Administration: The placebo group take a placebo daily for 12 weeks. |
| Outcomes |
Primary outcomes
|
| Starting date | 21 April 2021 |
| Contact information | Name: Fariba Karimzadeh Address: Cellular and Molecular Research Center, Iran University of Medical Sciences, Hemmat Highway 1449614535 Tehran Iran Phone: +98 21 8670 4725 Email: fariba_karimzade@yahoo.com |
| Notes | Source of funding: Iran University of Medical Sciences |
IRCT20210531051453N1.
| Study name | Public title: Effect of omega‐3 fatty acid in borderline personality disorder Scientific title: Study the effectiveness of omega‐3 fatty acids as adjuvant treatment on depression, aggression and poor impulse control in hospitalized patients of borderline personality disorder |
| Methods | Allocation: randomised Intervention model: parallel assignment Blinding: double‐blinded Duration: 6 weeks Timing of assessment: at baseline and 12 weeks after the trial |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions |
Experimental
Drug name: omega‐3
Brand name: Actover Pharmaceutical Company
Drug dose: 2 grams per day
Route of administration: oral
Administration: daily for 6 weeks Comparator Drug name: olanzapine Brand name: Abidi Pharmaceutical Company Drug dose: 5‐15 mg per day, according to patent’s response Route of administration: oral Administration: daily for 6 weeks |
| Outcomes |
Primary outcome
Secondary outcomes
|
| Starting date | 8 January 2020 |
| Contact information | Name: Ensieh Sadri Address: No 17, South Ebrahimi Ave, East Ferdos Blvd, Tehran 1481958465 Tehran Iran (Islamic Republic of) Phone: +98 21 4405 7355 Email: sadri.ensieh@gmail.com |
| Notes | Source of funding: University of Social Welfare and Rehabilitation Sciences |
NCT04100096.
| Study name | Public title: A trial of brexpiprazole in the treatment of borderline personality disorder Scientific title: A multicenter, randomized, flexible‐dose, double‐blind trial of brexpiprazole versus placebo for the treatment of adults with borderline personality disorder |
| Methods | Allocation: randomised Intervention model: parallel assignment (2 arms) Blinding: quadruple (participant, care provider, investigator and outcomes assessor) Duration: 12 weeks Timing of assessment: baseline and up to 12 weeks |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions |
Experimental
Drug name: brexpiprazole
Brand name: Rexulti®
Drug dose: 2‐3 mg/day (flexible dose)
Route of administration: oral
Administration: tablet Comparator Drug name: placebo Brand name: N/A Drug dose: N/A Route of administration: oral Administration: tablet |
| Outcomes |
Primary outcomes
Secondary outcomes
|
| Starting date | 17 October 2019 |
| Contact information | Name: Otsuka Call Center Phone: 844‐687‐8522 Email: OtsukaRMReconciliation@rmpdc.org |
| Notes | Source of funding: Otsuka Pharmaceutical Development & Commercialization, Inc |
NCT04566601.
| Study name | Public title: A study to test different doses of BI 1358894 and find out whether they reduce symptoms in people with borderline personality disorder Scientific title: A phase II randomized, double‐blinded, placebo‐controlled parallel group trial to examine the efficacy and safety of 4 oral doses of BI 1358894 once daily over 12 week treatment period in patients with borderline personality disorder |
| Methods | Allocation: randomised Intervention model: parallel assignment (4 arms) Blinding: quadruple (participant, care provider, investigator, outcomes assessor) Duration: 12 weeks Timing of assessment: baseline and week 10 |
| Participants |
Inclusion criteria
Further inclusion criteria also apply. Exclusion criteria
Further exclusion criteria also apply. |
| Interventions |
Experimental
Drug name: BI 1358894
Brand name: N/A
Drug dose: 4 drug doses (unspecified in protocol)
Route of administration: oral
Administration: once daily Comparator Drug name: placebo Brand name: N/A Drug dose: N/A Route of administration: oral Administration: once daily |
| Outcomes |
Primary outcomes
Secondary outcomes
|
| Starting date | 13 November 2020 |
| Contact information | Name: Boehringer Ingelheim Email:clintriage.rdg@boehringer-ingelheim.com |
| Notes | Source of funding: Boehringer Ingelheim |
NCT05356013.
| Study name | Public title: Calypta in borderline personality disorder Scientific title: A Double‐Blind, Placebo‐Controlled Study of Caplyta in the Treatment of Borderline Personality Disorder |
| Methods | Allocation: randomised Intervention model: parallel assignment Blinding: quadruple (participant, care provider, investigator, outcome assessors) Duration: 8 weeks Timing of assessment: baseline and weekly for 8 weeks |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions |
Experimental: caplyta All participants who are randomised to Caplyta will receive 42 mg/day starting the first week of the study. Participants will be seen every two weeks for 8 weeks. Dosage changes and reductions will not be permitted. After study conclusion (week 8), the dose will be discontinued. Comparator: placebo All participants who are randomised to placebo will receive an identical placebo pill to the experimental drug starting the first week of the study. Participants will be seen every two weeks for 8 weeks. After study conclusion (week 8), the dose will be discontinued. |
| Outcomes |
Primary outcomes
Secondary outcomes
|
| Starting date | Not specified (first posted 2 May 2022) |
| Contact information |
Name: Jon E Grant, MD, JD, MPH, University of Chicago (Primary Investigator) Other contact information: Name: Eve K Chesivoir Phone: 7737029066 E‐mail: chesivoir@yoda.bsd.uchicago.edu Name: Stephanie Valle Phone: 7738343778 E‐mail: svalle@yoda.bsd.uchicago.edu |
| Notes | Source of funding: University of Chicago and Intra‐Cellular Therapies, Inc. |
A/A: Agitation/Aggression subscale. AAPI: Agitation‐Aggression Psychiatric Inventory. ADHD: attention deficit hyperactivity disorder. AVH: auditory verbal hallucinations. BDI: Beck Depression Inventory. BEST: Barrett Evaluation of Severity over TIme. BIS: Barrett Impulsiveness Scale. BMI: body mass index. BoPD: borderline personality disorder. BPD: borderline personality disorder. BPDSI‐IV: Borderline Personality Disorder Severity Index, 4th edition. BSL‐23: Borderline Symptom List‐23. CAT: Cognitive Analytical Therapy. CGI‐I: Clinical Global Impression‐Improvement. CGI‐S: Clinical Global Impression–Severity. CNS: Central nervous system. C‐SSRS: Columbia Suicide Severity Rating Scale. DERS‐16: Difficulties in Emotion Regulation Scale. DSM‐IV: Diagnostic Manual of Mental Disorders, 4th edition. DSM‐5: Diagnostic Manual of Mental Disorders, 5th edition. DSS‐4: Dissociation Tension Scale‐4. ECG: Electrocardiogram. ECT: Electroconvulsive therapy. EuroQoL: European Quality of Life. FDS: Fragebogen zu dissoziativen Symptomen. GCP: Good clinical practice. HIV: Human immunodeficiency virus. ICH: International Council on Harmonization. IMP: Investigational medicinal product. IQ: Intelligence quotient. LAB: Laboratory. MAO: Monoamine oxidase inhibitors. MDD: Major depressive disorder. MIDI: Minnesota Impulsive Disorders Interview. MINI: The Mini‐International Neuropsychiatric Interview. MS: Multiple sclerosis. N/A: Not applicable. PCL‐5: PTSD Checklist for DSM‐5. PGI‐S: Patient Global Impression Severity Scale. PHQ‐9: Patient Health Questionnaire‐9. PI: Principal investigator. POSAS: Patient and Observer Scare Assessment Scale. PTSD: Post‐traumatic stress disorder. SCID‐II: Structured clinical interview for DSM‐IV. SCID‐5‐PD: Structured Clinical Interview for DSM‐5 Personality Disorders. SCL‐90‐R: Symptom Check List‐90‐Revised. SDS: Sheehan Disability Scale. SF: Social functioning. SmPC: Summary of Product Characteristics. STAI‐S: State‐Trait Anxiety Inventory. T4: Thyroxine. TMS: Transcranial magnetic stimulation. WOCBP: Women of childbearing potential. Zan‐BPD: Zanarini Rating Scale for Borderline Personality Disorder.
Differences between protocol and review
Methods
Data collection and analysis
Selection of studies
The protocol specified that six review authors (JMSW, OJS, BAV, MLK, JTM, SSN) would work in pairs and independently screen titles and abstracts of all records retrieved by the searches. However, the following additional review authors also selected trials: AT, EF, MSJ, JPR, JPS, and HEC.
The following authors, who worked on the protocol for the review, selected some trials but left the author group early in the development of the review as they no longer wished to be authors: SSN and MTK.
The protocol, moreover, specified that KL and ES would act as arbiters. However, in the review, OJS and JMSW also functioned as arbiters.
Unit of analysis issues
Repeated observations
We planned to conduct separate analyses for different follow‐up time points, but we did not have data for this, so we only used end‐of‐treatment data.
Cross‐over trials
We included data from four randomised cross‐over trials (Cowdry 1988; Schmahl 2012a; Schmahl 2012b; Ziegenhorn 2009). We were not able to obtain first‐period data from these cross‐over trials and therefore used end‐of‐period data (Curtin 2002; Elbourne 2002) (we wrote to the study authors and asked for the first‐period data, but they did not have these data). This approach might introduce the risk of a unit‐of‐analysis error. In addition, the confidence intervals are likely to be too wide and the trial will receive too little weight in the analysis. There is also the possibility that we could overlook important heterogeneity too. The Cochrane Handbook, however, states that this approach is conservative, as the studies are under‐weighted instead of over‐weighted (Higgins 2022), and some might argue that the unit‐of‐analysis error introduced by doing this is less serious than some other types of unit‐of‐analysis errors. The cross‐over trial by Cowdry is the only one pooled with parallel‐group trials (Cowdry 1988). Cowdry and colleagues reported a washout period of one week before cross‐over (Cowdry 1988). When excluding the Cowdry study from the analysis with the parallel‐group trials, we found no differences in the results. The other cross‐over trials were reported separately in the analyses (Schmahl 2012a; Schmahl 2012b; Ziegenhorn 2009).
Subgroup analysis
We added a post hoc subgroup analysis on funding, as we believe it is important to conduct an empirical test of the influence of funding on the effect estimates. Andreas Lundh and colleagues have shown that there are many subtle mechanisms through which sponsorship and conflict of interest may influence intervention effects on outcomes (Lundh 2018). The AMSTAR (A MeaSurement Tool to Assess systematic Reviews) tool for the assessment of the methodological quality of systematic reviews also includes funding and conflicts of interest as a domain (Shea 2017). In addition, we added subgroup analyses on differences in psychosocial functioning at baseline, trial size and type of recruitment, as we believe it is important to conduct an empirical test of the influence of these factors on the effect estimates. Trial sizes varied considerably from 13 in the smallest trial (Schmahl 2012a) to 451 in the largest (Zanarini 2007). Since trial size is connected to the precision of effect estimates, we believe the impact of trial sizes on the findings of this review should be investigated.
Heterogeneity‐adjusted required information size and Trial Sequential Analysis
When performing the TSAs, we did not use a standard deviation (SD) for the primary outcome of 1.0, and we did not always use an anticipated intervention effect of Hedge's g of 0.5 ( ½ SD), as described in the protocol (Stoffers‐Winterling 2018). Instead, we used the SD from the trial as a basis for the transformation of SMD values to MD values. Also, we preferred to use the MIREDIF reported in articles; only if we could not find it did we use the ½ SD on the specific scale, as this can be used as a MIREDIF (Norman 2004). The MIREDIF reported in articles is the best, as these often have data from epidemiological studies, but the MIREDIFs on different rating scales are seldom reported.
Sensitivity analysis
We added one analysis post hoc: imprecision, assessed by GRADE, by conducting TSAs on the primary outcomes and the three secondary outcomes not closely connected to the BPD core symptoms (interpersonal functioning, attrition and adverse effects) for the main comparison. We conducted these sensitivity analyses only on outcomes where we were uncertain about the GRADE assessment of imprecision. These analyses tested whether the effect estimates were imprecise due to too low number of participants in the meta‐analyses and therefore not enough information size to detect or reject the intervention effect, or whether the required information size was large enough to detect or reject the intervention effect. We tested our GRADE assessment of imprecision by using TSA on one primary outcome and three secondary outcomes.
We also conducted a sensitivity analysis for the secondary outcome of anger, for the comparison of mood stabliisers versus placebo, as a high amount of heterogeneity was found (I2 = 84%), and the range of effect estimates included extraordinarily high scores (SMD ‐3.10 to ‐0.15). Therefore, we removed outliers exceeding effect estimates of SMD 1.5.
We added one sensitivity analysis testing whether including end‐of‐period data in cross‐over trials in some of the analyses changed the statistical significance. We did as we were not able to obtain first‐period data from these cross‐over trials.
In addition, we conducted a sensitivity analysis testing the impact of our choice of data sources. For one trial, Black 2014, data were available for the outcome of BPD severity from both the trial registry entry (NCT00880919) as well as from the full journal publication. We conducted a sensitivity analysis to test the impact of choosing the peer‐reviewed journal publication as our primary data source.
Contributions of authors
Jutta M Stoffers‐Winterling: protocol development; study selection; conception and design of the review; data extraction; data entry; interpretation of data; assessment of risk of bias and GRADE; data analysis; and writing of final report
Ole Jakob Storebø: protocol development; study selection; conception, design and coordination of the review; data extraction; data entry; interpretation of data; assessment of risk of bias and GRADE; data analysis; and writing of final report
Birgit A Völlm: protocol development; study selection; data extraction; and writing of final report
Mickey T Kongerslev: protocol development; study selection; data extraction; assessment of risk of bias; and writing of final report
Jessica T Mattivi: protocol development; study selection; data extraction; and writing of final report
Mie Sedoc Jørgensen: study selection; data extraction; assessment of risk of bias; and writing of final report
Johanne Marie Pereira Ribero: study selection; data extraction; data entry; interpretation of data; assessment of risk of bias; data analysis and writing of final report
Erlend Faltinsen: study selection; data extraction; data entry; assessment of risk of bias; data analysis; and writing of final report
Adnan Todorovac: study selection; data extraction; data entry; assessment of risk of bias; data analysis; and writing of final report
Christian Paul Sales: study selection; data extraction; and writing of final report
Julie Perrine Schaug: study selection; data extraction; assessment of risk of bias; and writing of final report
Henriette E Callesen: study selection, data extraction; assessment of risk of bias and GRADE; data analysis; and writing of final report
Klaus Lieb: protocol development; and writing of final report
Erik Simonsen: protocol development; and writing of final report
All authors contributed to writing the review.
Jutta Stoffers‐Winterling is the guarantor for the review.
Sources of support
Internal sources
-
Psychiatric Research Unit, Region Zealand Psychiatry, Roskilde, Denmark
Ole Jakob Storebø, Johanne Pereira Ribeiro and Erik Simonsen worked on this review during office hours.
-
University Medical Center Mainz, Germany, Denmark
Jutta Stoffers‐Winterling, Klaus Lieb and Jessica Mattivi worked on this review during office hours.
External sources
-
The Psychiatric Management Research Fund, Psychiatry Region Zealand, Denmark
This is a small fund where the management of psychiatry in Region Zealand in Denmark distributes small amounts to research projects within psychiatry.
Declarations of interest
Jutta M Stoffers‐Winterling (JSW) is a board‐certified clinical psychologist ('Psychologische Psychotherapeutin', cognitive behaviour therapy). She does not prescribe or administer medication in a clinical context.
Ole Jakob Storebø (OJS) is a clinical psychologist with the Psychiatric Research Unit, Region Zealand Psychiatry, Denmark, and does not prescribe or administer medication in a clinical context. He also reports being the trial coordinator for an ongoing study at Region Zealand investigating a new drug treatment for people with borderline personality disorder (NCT04566601), which is included in this review; the study is funded and designed by Boehringer Ingelheim. OJS did not assess the eligibility of this trial which was assessed by two independent reviewers (JPR and JSW). In addition, OJS is an editor for Cochrane Developmental, Psychosocial and Learning Problems (DPLP); however, he was not involved in the editorial process for this review. OJS is also Editor‐in‐Chief for the Scandinavian Journal of Child and Adolescent Psychiatry and Psychology.
Jessica T Mattivi (JTM) is currently working with Psychiatric Services in Meran, Italy. JTM has declared a grant from the German Federal Ministry of Education and Research, paid to University Medical Center Mainz, Germany. The grant was for a systematic review on psychosocial interventions for self‐harm in adolescents; the funder did not have any role in design, methods, data analysis and reporting of the study.
Mickey T Kongerlev is a clinical psychologist and does not prescribe or administer medication in a clinical context. He is employed as manager of a District Psychiatric Service in Region Zealand Mental Health Services, Roskilde, Denmark, and is President of the Institute of Personality Theory and Psychopathology.
Birgit A Völlm is a medical practitioner with the University of Rostock, with the authority to prescribe medication in a clinical context. She is affiliated to the Royal College of Psychiatrists and the DGPPN (Deutsche Gesellschaft für Psychiatrie, Psychotherapie, Psychosomatik und Nervenheilkunde; the German Association for Psychiatry, Psychotherapy and Psychosomatics, which is the largest scientific medical association focusing on mental health in Germany), who have declared an opinion or position on the topic.
Henriette E Callesen is a neuroscientist and does not prescribe or administer medication in a clinical context. She has declared that she has no conflicts of interest.
Adnan Todorovac is a clinical psychologist and does not prescribe or administer medication in a clinical context. He has declared that he has no conflicts of interest.
Christian P Sales is a clinical psychologist with Nottinghamshire Healthcare NHS Foundation Trust, UK, and does not prescribe or administer medication in a clinical context. He has declared that he has no conflicts of interest.
Erlend Faltinsen is a clinical psychologist and does not prescribe or administer medication in a clinical context.
Mie S Jørgensen is a clinical psychologist with Region Zealand Mental Health Services, Denmark, and does not prescribe or administer medication in a clinical context. She has declared that she has no conflicts of interest.
Johanne P Ribeiro is a registered nurse with the authority to administer prescribed medications; however, she is employed as a research assistant at Region Zealand and therefore does not administer medication in a clinical context. She has declared that she has no conflicts of interest.
Julie P Schaug is a psychologist and does not prescribe or administer medication in a clinical context. She has declared that she has no conflicts of interest.
Erik Simonsen (ES) is a psychiatrist with the authority to prescribe medication in a clinical context but does not exercise this authority as he is currently employed as a research director. ES reports that he is the Princpal Investigator of one site (Slagelse, Denmark) of an ongoing, international, multi‐site study investigating a new drug treatment for people with borderline personality disorder (NCT04566601), which is included in this review; the study is funded and designed by Boehringer Ingelheim. ES did not assess the eligibility of this trial, which was assessed by two independent reviewers (JPR and JSW). ES also reports travel and associated expenses (congress fees and accommodation) from Boehringer Ingelheim.
Klaus Lieb (KL) is a board‐certified cognitive behaviour therapist with a special interest in schema therapy. He is also a psychiatrist with the authority to prescribe medication (i.e. recommend treatments for personality disorders); however, he is employed as Director of the Department of Psychiatry and Psychotherapy, University Medical Centre Mainz, Germany and therefore does not prescribe or administer medication in a clinical context. KL is also an editor for DPLP but was not involved in the editorial process for this review.
Shares first authorship.
Shares last authorship.
New
References
References to studies included in this review
Amminger 2013 {published data only}
- Amminger GP, Chanen AM, Ohmann S, Klier CM, Mossaheb N, Bechdolf A, et al. Omega-3 fatty acid supplementation in adolescents with borderline personality disorder and ultra-high risk criteria for psychosis: a post hoc subgroup analysis of a double-blind, randomized controlled trial. Canadian Journal of Psychiatry [Revue Canadienne de Psychiatrie] 2013;58(7):402-8. [DOI: 10.1177/070674371305800705] [PMID: ] [DOI] [PubMed] [Google Scholar]
- Amminger GP, Schäfer MR, Papageorgiou K, Klier CM, Cotton SM, Harrigan SM, et al. Long-chain omega-3 fatty acids for indicated prevention of psychotic disorders: a randomized, placebo-controlled trial. Archives of General Psychiatry 2010;67(2):146-54. [DOI: 10.1001/archgenpsychiatry.2009.192] [PMID: ] [DOI] [PubMed] [Google Scholar]
AstraZeneca 2007 {published data only}
- NCT00254748. Verkes Borderline Study: the effect of quetiapine on borderline personality disordered patients [The effect of quetiapine on psychotic-like symptoms in borderline personality disordered patients: a randomised placebo-controlled trial]. clinicaltrials.gov/ct2/show/NCT00254748 (first received 15 November 2005).
- Van den Broek PJ, Penterman EJ, Hummelen JW, Verkes RJ. P.3.c.038. The effect of quetiapine on psychotic-like symptoms in borderline personality disorder. A placebo-controlled trial. European Neuropsychopharmacology 2008;18(Suppl 4):S425-6. [DOI: 10.1016/S0924-977X(08)70622-9] [DOI] [Google Scholar]
Bellino 2014 {published data only}
- Bellino S, Bozzatello P, Brunetti C, De Grandi E, Bogetto F. Omega-3 fatty acids associated with valproate in the treatment of borderline personality disorder: a controlled study [Acidi grassi omega-3 associati alvalproato nel trattamento del disturbo borderlinedi personalità: uno studio controllato]. Journal of Psychopathology 2013;19(4):296-303. [WEB PAGE: Available at www.jpsychopathol.it/wp-content/uploads/2015/07/02-Bellino-acidi1.pdf] [Google Scholar]
- Bellino S, Bozzatello P, Rocca G, Bogetto F. Efficacy of omega-3 fatty acids in the treatment of borderline personality disorder: a study of the association with valproic acid. Journal of Psychopharmacology 2014;28(2):125-32. [DOI: 10.1177/0269881113510072] [PMID: ] [DOI] [PubMed] [Google Scholar]
- Bozzatello P, Rocca P, Bellino S. Combination of omega-3 fatty acids and valproic acid in treatment of borderline personality disorder: a follow-up study. Clinical Drug Investigation 2018;38(4):367-72. [DOI: 10.1007/s40261-017-0617-x] [PMID: ] [DOI] [PubMed] [Google Scholar]
Black 2014 {published data only}
- Bellino S. Inclusion of study in Cochrane review. Email to: A Tadorovac 31 January 2019.
- Black DW, Zanarini MC, Romine A, Shaw M, Allen J, Schulz SC. Comparison of low and moderate dosages of extended-release quetiapine in borderline personality disorder: a randomized, double-blind, placebo-controlled trial. American Journal of Psychiatry 2014;171(11):1174-82. [DOI: 10.1176/appi.ajp.2014.13101348] [PMID: ] [DOI] [PubMed] [Google Scholar]
- Lee SS, Allen J, Black DW, Zanarini MC, Schulz SC. Quetiapine's effect on the SCL-90-R domains in patients with borderline personality disorder. Annals of Clinical Psychiatry 2016;28(1):4-10. [PMID: ] [PubMed] [Google Scholar]
- NCT00880919. Seroquel extended release (XR) for the management of borderline personality disorder (BPD) [Seroquel XR for the management of borderline personality disorder (BPD)]. clinicaltrials.gov/ct2/show/NCT00880919 (first received 10 April 2009).
Bogenschutz 2004 {published data only}
- Bogenschutz MP, Nurnberg HG. Olanzapine versus placebo in the treatment of borderline personality disorder. Journal of Clinical Psychiatry 2004;65(1):104-9. [DOI: 10.4088/jcp.v65n0118] [PMID: ] [DOI] [PubMed] [Google Scholar]
- Nurnberg HG, Bogenschutz MP. Atypical antipsychotic treatment of BPD: a 12-week, double-blind, placebo-control trial with olanzapine. In: 156th Annual Meeting of the American Psychiatric Association; 2003 May 17-22; San Fransisco (CA). 2003.
Bozzatello 2017 {published data only}ACTRN12614000551695
- ACTRN12614000551695. Efficacy and tolerability of asenapine compared with olanzapine in borderline personality disorder: a randomized controlled trial [Asenapine versus olanzapine in the treatment of impulsivity, aggression, and self injuries in patients with borderline personality disorder]. www.anzctr.org.au/Trial/Registration/TrialReview.aspx?id=365182 (first received 12 May 2014).
- Bozzatello B, Rocca P, Uscinska M, Bellino S. Efficacy and tolerability of asenapine compared with olanzapine in borderline personality disorder: an open-label randomized controlled trial. Central Nervous System Drugs 2017;31(9):801-19. [DOI: 10.1007/s40263-017-0458-4] [PMID: ] [DOI] [PubMed] [Google Scholar]
Cowdry 1988 {published data only}
- Cowdry RW, Gardner DL. Pharmacotherapy of borderline personality disorder. Alprazolam, carbamazepine, trifluoperazine, and tranylcypromine. Archives of General Psychiatry 1988;45(2):111-9. [DOI: 10.1001/archpsyc.1988.01800260015002] [PMID: ] [DOI] [PubMed] [Google Scholar]
- Gardner DL, Cowdry RW. Alprazolam-induced dyscontrol in borderline personality disorder. American Journal of Psychiatry 1985;142(1):98-100. [DOI: 10.1176/ajp.142.1.98] [PMID: ] [DOI] [PubMed] [Google Scholar]
- Gardner DL, Cowdry RW. Development of melancholia during carbamazepine treatment in borderline personality disorder. Journal of Clinical Psychopharmacology 1986;6(4):236-9. [PMID: ] [PubMed] [Google Scholar]
- Gardner DL, Cowdry RW. Positive effects of carbamazepine on behavioral dyscontrol in borderline personality disorder. American Journal of Psychiatry 1986;143(4):519-22. [DOI: 10.1176/ajp.143.4.519] [PMID: ] [DOI] [PubMed] [Google Scholar]
Crawford 2018 {published data only}
- Crawford MJ, Sanatinia R, Barrett B, Byford S, Cunningham G, Gakhal K, et al. Lamotrigine versus inert placebo in the treatment of borderline personality disorder: study protocol for a randomized controlled trial and economic evaluation. Trials 2015;16:308. [DOI: 10.1186/s13063-015-0823-x] [PMCID: PMC4506596] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford MJ, Sanatinia R, Barrett B, Cunningham G, Dale O, Ganguli P, et al. Lamotrigine for people with borderline personality disorder: a RCT. Health Technology Assessment 2018;22(17):1-68. [DOI: 10.3310/hta22170] [PMCID: PMC5925438] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford MJ. Inclusion of your study in Cochrane review. Email to: A Pathmanathan Ratnam 09 November 2020.
De la Fuente 1994 {published data only}
- De la Fuente JM, Lotstra F. A trial of carbamazepine in borderline personality disorder. European Neuropsychopharmacology 1994;4(4):479-86. [DOI: 10.1016/0924-977x(94)90296-8] [PMID: ] [DOI] [PubMed] [Google Scholar]
- De la Fuente JM, Lotstra F. Carbamazepine in borderline personality disorder. European Neuropsychopharmacology 1993;3(3):350. [DOI: 10.1016/0924-977X(93)90132-6] [DOI] [PubMed] [Google Scholar]
Frankenburg 2002 {published data only}
- Frankenburg FR, Zanarini MC. Divalproex sodium treatment of women with borderline personality disorder and bipolar II disorder: a double-blind placebo-controlled pilot study. Journal of Clinical Psychiatry 2002;63(5):442-6. [DOI: 10.4088/jcp.v63n0511] [PMID: ] [DOI] [PubMed] [Google Scholar]
Goldberg 1986 {published data only}
- Goldberg SC, Schulz SC, Schulz PM, Resnick RJ, Hamer RM, Friedel RO. Borderline and schizotypal personality disorders treated with low-dose thiothixene vs placebo. Archives of General Psychiatry 1986;43(7):680-6. [DOI: 10.1001/archpsyc.1986.01800070070009] [PMID: ] [DOI] [PubMed] [Google Scholar]
Grant 2022 {published data only}
- Grant J, Valle S, Chesivoir E, Ehsan D, Chamberlain S. A double-blind placebo-controlled study of brexpiprazole for the treatment of borderline personality disorder. British Journal of Psychiatry 2022;220(2):58-63. [CENTRAL: PMC7612273] [DOI: 10.1192/bjp.2021.159] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hallahan 2007 {published data only}
- Hallahan B, Hibbeln JR, Davis JM, Garland MR. Omega-3 fatty acid supplementation in patients with recurrent self-harm. Single-centre double-blind randomised controlled trial. British Journal of Psychiatry 2007;190:118-22. [DOI: 10.1192/bjp.bp.106.022707] [PMID: ] [DOI] [PubMed] [Google Scholar]
Hollander 2001 {published data only}
- Hollander E, Allen A, Lopez RP, Bienstock CA, Grossman R, Siever LJ, et al. A preliminary double-blind, placebo-controlled trial of divalproex sodium in borderline personality disorder. Journal of Clinical Psychiatry 2001;62(3):199-203. [DOI: 10.4088/jcp.v62n0311] [PMID: ] [DOI] [PubMed] [Google Scholar]
Jariani 2010 {published data only}
- Jariani M, Saaki M, Nazari H, Birjandi M. The effect of olanzapine and sertraline on personality disorder in patients with methadone maintenance therapy. Psychiatria Danubina 2010;22(4):544-7. [PMID: ] [PubMed] [Google Scholar]
Kulkarni 2018 {published data only}
- Kulkarni J, Thomas N, Hudaib AR, Gavrilidis E, Grigg J, Tan R, et al. Effect of the glutamate NMDA receptor antagonist memantine as adjunctive treatment in borderline personality disorder: an exploratory, randomised, double-blind, placebo-controlled trial. Central Nervous System Drugs 2018;32(2):179-87. [DOI: 10.1007/s40263-018-0506-8] [PMID: ] [DOI] [PubMed] [Google Scholar]
- Kulkarni J [pers comm]. Re: Your Request. Email to: A Pathmanatan Ratnam 14 December 2020.
- NCT02097706. A novel drug for borderline personality disorder [A randomised double-blind placebo controlled investigation of the efficacy of a novel drug as an adjunct in patients with borderline personality disorder]. clinicaltrials.gov/ct2/show/study/NCT02097706 (first received 25 March 2014).
Leone 1982 {published data only}
- Leone NF. Response of borderline patients to loxapine and chlorpromazine. Journal of Clinical Psychiatry 1982;43(4):148-50. [PMID: ] [PubMed] [Google Scholar]
Linehan 2008 {published data only}
- Linehan M. Update on dialectical behavioral therapy for BPD. In: 158th Annual Meeting of the American Psychiatric Association; 2005 May 21-26; Atlanta (GA). 2005.
- Linehan MM, McDavid JD, Brown MZ, Sayrs JH, Gallop RJ. Olanzapine plus dialectical behavior therapy for women with high irritability who meet criteria for borderline personality disorder: a double-blind, placebo-controlled pilot study. Journal of Clinical Psychiatry 2008;69(9):999-1005. [DOI: 10.4088/jcp.v69n0617] [PMID: ] [DOI] [PubMed] [Google Scholar]
Loew 2006 {published data only}
- Loew TH, Nickel MK, Muehlbacher M, Kaplan P, Nickel C, Kettler C, et al. Topiramate treatment for women with borderline personality disorder: a double-blind, placebo-controlled study. Journal of Clinical Psychopharmacology 2006;26(1):61-6. [DOI: 10.1097/01.jcp.0000195113.61291.48] [PMID: ] [DOI] [PubMed] [Google Scholar]
- Loew TH, Nickel MK. Topiramate treatment of women with borderline personality disorder, part II: an open 18-month follow-up. Journal of Clinical Psychopharmacology 2008;28(3):355-7. [DOI: 10.1097/JCP.0b013e318173a8fb] [PMID: ] [DOI] [PubMed] [Google Scholar]
Markovitz 1995a {published data only}
- Markovitz PJ. Pharmacotherapy of impulsivity, aggression, and related disorders. In: Hollander E, Stein DJ, editors(s). Impulsivity and Aggression. New York (NY): Wiley, 1995:263-87. [Google Scholar]
Moen 2012 {published data only}
- Moen R, Freitag M, Miller M, Lee S, Romine A, Song S, et al. Efficacy of extended-release divalproex combined with "condensed" dialectical behavior therapy for individuals with borderline personality disorder. Annals of Clinical Psychiatry 2012;24(4):255-60. [PMID: ] [PubMed] [Google Scholar]
- NCT00222482. Depakote ER in borderline personality disorder [A double-blind and placebo controlled assessment of depakote ER in borderline personality disorder]. clinicaltrials.gov/ct2/show/NCT00222482 (first received 14 September 2005).
Montgomery 1982a {published data only}
- Montgomery SA, Montgomery D. Pharmacological prevention of suicidal behaviour. Journal of Affective Disorders 1982;4(4):291-8. [DOI: 10.1016/0165-0327(82)90026-x] [PMID: ] [DOI] [PubMed] [Google Scholar]
- Montgomery SA, Montgomery DB, Janyanthi Rani S, Roy DH, Shaw PJ, McAuley R. Maintenance therapy in repeat suicidal behaviour: a placebo controlled trial. In: 10th International Congress for Suicide Prevention and Crisis Intervention; 1979 Jun 17-20; Ottawa (ON). 1979.
Montgomery 1982b {published data only}
- Montgomery D, Montgomery S, Roy D. Mianserin in the prophylaxis of suicidal behaviour: a double-blind placebo controlled trial. In: Soubrier JP, Vedrinne J, editors(s). Depression and Suicide: Aspects Medicaux, Psychologiques et Socio-Culturels. Paris (FR): Pergamon, 1983:786-90. [DOI: 10.1016/B978-0-08-027080-7.50130-0] [DOI] [Google Scholar]
- Montgomery SA, Montgomery D. Pharmacological prevention of suicidal behaviour. Journal of Affective Disorders 1982;4(4):291-8. [DOI: 10.1016/0165-0327(82)90026-x] [PMID: ] [DOI] [PubMed] [Google Scholar]
- Montgomery SA, Roy D, Montgomery DB. The prevention of recurrent suicidal acts. British Journal of Clinical Pharmacology 1983;15(Suppl 2):183-8S. [DOI: 10.1111/j.1365-2125.1983.tb05864.x] [PMCID: PMC1427879] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
NCT00533117 {published data only}
- Fertuck EA, Keilp J, Song I, Morris MC, Wilson ST, Brodsky BS, et al. Higher executive control and visual memory performance predict treatment completion in borderline personality disorder. Psychotherapy and Psychosomatics 2012;81(1):38-43. [DOI: 10.1159/000329700] [PMCID: PMC3242704] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT00533117. Treating suicidal behavior and self-mutilation in people with borderline personality disorder [Treating suicidal behavior and self-mutilation in borderline personality disorder]. clinicaltrials.gov/ct2/show/results/NCT00533117?sect=X043716&view=results#part (first received 19 September 2007).
Nickel 2004 {published data only}
- Nickel M. Topiramate reduced aggression in female patients with borderline personality disorder. European Archives of Psychiatry and Clinical Neuroscience 2007;257(7):432-3. [DOI: 10.1007/s00406-007-0735-1] [PMID: ] [DOI] [PubMed] [Google Scholar]
- Nickel MK, Nickel C, Mitterlehner FO, Tritt K, Lahmann C, Leiberich PK, et al. Topiramate treatment of aggression in female borderline personality disorder patients: a double-blind, placebo-controlled study. Journal of Clinical Psychiatry 2004;65(11):1515-9. [DOI: 10.4088/jcp.v65n1112] [PMID: ] [DOI] [PubMed] [Google Scholar]
Nickel 2005 {published data only}
- Nickel MK, Loew TH. Treatment of aggression with topiramate in male borderline patients, part II: 18-month follow-up. European Psychiatry 2008;23(2):115-7. [DOI: 10.1016/j.eurpsy.2007.09.004] [PMID: ] [DOI] [PubMed] [Google Scholar]
- Nickel MK, Nickel C, Kaplan P, Lahmann C, Mühlbacher M, Tritt K, et al. Treatment of aggression with topiramate in male borderline patients: a double-blind, placebo-controlled study. Biological Psychiatry 2005;57(5):495-9. [DOI: 10.1016/j.biopsych.2004.11.044] [PMID: ] [DOI] [PubMed] [Google Scholar]
- Nickel MK. Topiramate treatment of aggression in male borderline patients. Australian and New Zealand Journal of Psychiatry 2007;41(5):461-2. [DOI: 10.1080/00048670701261251] [PMID: ] [DOI] [PubMed] [Google Scholar]
Nickel 2006 {published data only}
- Nickel MK, Loew TH, Pedrosa Gil F. Aripiprazole in treatment of borderline patients, part II: an 18-month follow-up. Psychopharmacology 2007;191(4):1023-6. [DOI: 10.1007/s00213-007-0740-0] [PMID: ] [DOI] [PubMed] [Google Scholar]
- Nickel MK, Mühlbacher M, Nickel C, Kettler C, Pedrosa Gil F, Bachler E, et al. Aripiprazole in the treatment of patients with borderline personality disorder: a double-blind, placebo-controlled study. American Journal of Psychiatry 2006;163(5):833-8. [DOI: 10.1176/ajp.2006.163.5.833] [PMID: ] [DOI] [PubMed] [Google Scholar]
- Nickel MK. Aripiprazole treatment of patients with borderline personality disorder. Journal of Clinical Psychiatry 2007;68(11):1815-6. [DOI: 10.4088/jcp.v68n1124f] [PMID: ] [DOI] [PubMed] [Google Scholar]
Pascual 2008 {published data only}
- NCT00635921. Ziprasidone in the treatment of borderline personality disorder [Ziprasidone in the treatment of borderline personality disorder: a double-blind, placebo-controlled, randomized study]. clinicaltrials.gov/ct2/show/NCT00635921 (accessed 9 September 2009).
- Pascual JC, Soler J, Puigdemont D, Pérez-Egea R, Tiana T, Alvarez E, et al. Ziprasidone in the treatment of borderline personality disorder: a double-blind, placebo-controlled, randomized study. Journal of Clinical Psychiatry 2008;69(4):603-8. [PMID: 10.4088/jcp.v69n0412] [DOI] [PubMed] [Google Scholar]
Reich 2009 {published data only}
- NCT00634062. Study of lamotrigine treatment of affective instability in borderline personality disorder. clinicaltrials.gov/ct2/show/NCT00634062 (first received 4 March 2008). [NCT00634062]
- Reich DB, Zanarini MC, Bieri KA. A preliminary study of lamotrigine in the treatment of affective instability in borderline personality disorder. International Clinical Psychopharmacology 2009;24(5):270-5. [DOI: 10.1097/YIC.0b013e32832d6c2f] [PMID: ] [DOI] [PubMed] [Google Scholar]
Rinne 2002 {published data only}
- Rinne T, Van den Brink W, Wouters L, Van Dyck R. SSRI treatment of borderline personality disorder: a randomized, placebo-controlled clinical trial for female patients with borderline personality disorder. American Journal of Psychiatry 2002;159(12):2048-54. [DOI: 10.1176/appi.ajp.159.12.2048] [PMID: ] [DOI] [PubMed] [Google Scholar]
Salzman 1995 {published data only}
- Salzmann C, Wolfson AN, Schatzberg A, Looper J, Henke R, Albanese M, et al. Effect of fluoxetine on anger in symptomatic volunteers with borderline personality disorder. Journal of Clinical Psychopharmacology 1995;15(1):23-9. [DOI: 10.1097/00004714-199502000-00005] [PMID: ] [DOI] [PubMed] [Google Scholar]
Schmahl 2012a {published data only}
- NCT01133301. Naltrexone in the treatment of dissociative symptoms in patients with borderline personality disorder. clinicaltrials.gov/ct2/show/NCT01133301 (first received 17 May 2010).
- Schmahl C, Kleindienst N, Limbergera M, Ludäscher P, Mauchnika J, Deiblera P, et al. Evaluation of naltrexone for dissociative symptoms in borderline personality disorder. International Clinical Psychopharmacology 2012;27(1):61-8. [DOI: 10.1097/YIC.0b013e32834d0e50] [PMID: ] [DOI] [PubMed] [Google Scholar]
Schmahl 2012b {published data only}
- NCT00124839. Naltrexone in borderline personality disorder [Evaluation of the efficacy of the opioid antagonist naltrexone on the incidence and intensity of flashbacks and dissociative states in patients with borderline personality disorder]. clinicaltrials.gov/ct2/show/NCT00124839 (first received 27 July 2005).
- NCT01133301. Naltrexone in the treatment of dissociative symptoms in patients with borderline personality disorder. clinicaltrials.gov/ct2/show/record/NCT01133301 (first received 17 May 2010).
- Schmahl C, Kleindienst N, Limbergera M, Ludäscher P, Mauchnika J, Deiblera P, et al. Evaluation of naltrexone for dissociative symptoms in borderline personality disorder. International Clinical Psychopharmacology 2012;27(1):61-8. [DOI: 10.1097/YIC.0b013e32834d0e50] [PMID: ] [DOI] [PubMed] [Google Scholar]
Schulz 2007 {published data only}
- Eli Lilly and Company. Efficacy and safety of olanzapine in patients with borderline personality disorder: a randomized, flexible-dose, double-blind comparison with placebo [Summary ID #6257. Clinical Study Summary: F1D-MC-HGKL]. www.lillytrials.com/results_files/zyprexa/zyprexa_summary_6257 (accessed 14 February 2008).
- NCT00091650. Olanzapine in patients with borderline personality disorder [Efficacy and safety of olanzapine in patients with borderline personality disorder: a randomized flexible dose double-blind comparison with placebo]. clinicaltrials.gov/ct2/show/NCT00091650 (first accessed 14 September 2004).
- Schulz SC, Zanarini M, Detke H, Trzaskoma Q, Lin D, DeBerdt W, et al. P02.101 Olanzapine for the treatment of borderline personality disorder: a flexible-dose 12-week randomized double-blind placebo-controlled study. International Journal of Neuropsychopharmacology 2006;9(Suppl 1):S191. [DOI: 10.1017/S1461145706007292] [DOI] [Google Scholar]
- Schulz SC, Zanarini MC, Bateman A, Bohus M, Detke HC, Trzaskoma Q, et al. Olanzapine for the treatment of borderline personality disorder: variable dose, 12-week randomised double-blind placebo-controlled study. British Journal of Psychiatry 2008;193(6):485-92. [DOI: 10.1192/bjp.bp.107.037903] [PMID: ] [DOI] [PubMed] [Google Scholar]
- Schulz SC, Zanarini MC, Detke HC, Trzaskoma Q, Lin D, DeBerdt W, et al. Olanzapine for the treatment of borderline personality disorder: a flexible‐dose, 12‐week, randomized, double‐blind, placebo‐controlled study. In: Association of European Psychiatrsts. 15th European Congress of Psychiatry; 2007 Mar 17‐21; Madrid, Spain. 2007.
- Schulz SC, Zanarini MC, Detke HC, Trzaskoma Q, Lin D, DeBerdt W, et al. P248 Olanzapine for the treatment of borderline personality disorder: a flexible-dose 12-week randomized double-blind placebo-controlled study. European Psychiatry 2007;22(Suppl 1):S172. [DOI: 10.1016/j.eurpsy.2007.01.564] [DOI] [PubMed] [Google Scholar]
- Zanarini MC, Schulz SC, Detke HC, Tanaka Y, Zhao F, Trzaskoma Q, et al. 80. Olanzapine for the treatment of borderline personality disorder: two 12-week randomized double-blind placebo-controlled trials. Neuropsychopharmacology 2006;31(Suppl 1):S229-30. [DOI: 10.1017/S1461145706007292] [DOI] [Google Scholar]
Shafti 2010 {published data only}
- Shafti SS, Shaveisi B. Olanzapine versus haloperidol in the management of borderline personality disorder. Journal of Clinical Psychopharmacology 2010;30(1):44-7. [DOI: 10.1097/JCP.0b013e3181c826ff] [PMID: ] [DOI] [PubMed] [Google Scholar]
Shafti 2014 {published data only}
- Shafti SS, Kaviani H. A comparative study on olanzapine and aripiprazole for symptom management in female patients with borderline personality disorder. Klinik Psikofarmakoloji Bülteni [Bulletin of Clinical Psychopharmacology] 2015;25(1):38-43. [DOI: 10.5455/bcp.20140923100030] [DOI] [Google Scholar]
- Shafti SS, Kaviani H. Olanzapine vs aripiprazole in the management of borderline personality disorder. Current Psychopharmacology 2014;3(2):132-6. [DOI: 10.2174/2211556003666140912223626] [DOI] [Google Scholar]
Simpson 2004 {published data only}
- Simpson EB, Yen S, Costello E, Rosen K, Begin A, Pistorello J, et al. Combined dialectical behavior therapy and fluoxetine in the treatment of borderline personality disorder. Journal of Clinical Psychiatry 2004;65(3):379-85. [DOI: 10.4088/jcp.v65n0314] [PMID: ] [DOI] [PubMed] [Google Scholar]
Soler 2005 {published data only}
- Pascual JC, Soler J, Barrachina J, Campins J, Puigdemont D, Alvarez E, et al. Olanzapine plus dialectical behavior therapy of patients with BPD. In: Psychotherapy and Psychopharmacology: Dissolving the Mind-Brain Barrier. 157th Annual Meeting of the American Psychiatric Association; 2004 May 1-6; New York (NY). 2004.
- Soler J, Pascual JC, Campins J, Barrachina J, Puigdemont D, Alvarez E, et al. Double-blind, placebo-controlled study of dialectical behavior therapy plus olanzapine for borderline personality disorder. American Journal of Psychiatry 2005;162(6):1221-4. [DOI: 10.1176/appi.ajp.162.6.1221] [PMID: ] [DOI] [PubMed] [Google Scholar]
Soloff 1989 {published data only}
- Soloff PH, George A, Nathan RS, Schulz PM, Perel JM. Behavioral dyscontrol in borderline patients treated with amitriptyline. Psychopharmacology Bulletin 1987;23(1):177-81. [PMID: ] [PubMed] [Google Scholar]
- Soloff PH, George A, Nathan RS, Schulz PM, Perel JM. Paradoxical effects of amitriptyline on borderline patients. American Journal of Psychiatry 1986;143(12):1603-5. [DOI: 10.1176/ajp.143.12.1603] [PMID: ] [DOI] [PubMed] [Google Scholar]
- Soloff PH, George A, Nathan RS, Schulz PM, Ulrich RF, Perel JM. Progress in pharmacotherapy of borderline disorders. A double-blind study of amitriptyline, haloperidol, and placebo. Archives of General Psychiatry 1986;43(7):691-7. [DOI: 10.1001/archpsyc.1986.01800070081010] [PMID: ] [DOI] [PubMed] [Google Scholar]
- Soloff PH, George A, Nathan S, Schulz PM, Cornelius JR, Herring J, et al. Amitriptyline versus haloperidol in borderlines: final outcomes and predictors of response. Journal of Clinical Psychopharmacology 1989;9(4):238-46. [DOI: 10.1097/00004714-198908000-00002] [PMID: ] [DOI] [PubMed] [Google Scholar]
- Soloff PH, George A, Nathan S, Schulz PM, Ulrich RF, Perel JM. Amitriptyline and haloperidol in unstable and schizotypal borderline disorders. Psychopharmacology Bulletin 1986;22(1):177-82. [PMID: ] [PubMed] [Google Scholar]
Soloff 1993 {published data only}
- Cornelius JR, Soloff PH, George A, Ulrich RF, Perel JM. Haloperidol vs phenelzine in continuation therapy of borderline disorder. Psychopharmacology Bulletin 1993;29(2):333-7. [PMID: ] [PubMed] [Google Scholar]
- Cornelius JR, Soloff PH, Perel JM, Ulrich RF. Continuation pharmacotherapy of borderline personality disorder with haloperidol and phenelzine. American Journal of Psychiatry 1993;150(12):1843-8. [DOI: 10.1176/ajp.150.12.1843] [PMID: ] [DOI] [PubMed] [Google Scholar]
- Soloff PH, Cornelius J, George A, Nathan S, Perel JM, Ulrich RF. Efficacy of phenelzine and haloperidol in borderline personality disorder. Archives of General Psychiatry 1993;50(5):377-85. [DOI: 10.1001/archpsyc.1993.01820170055007] [PMID: ] [DOI] [PubMed] [Google Scholar]
Tritt 2005 {published data only}
- Leiberich P, Nickel MK, Tritt K, Pedrosa Gil F. Lamotrigine treatment of aggression in female borderline patients, part II: an 18-month follow-up. Journal of Psychopharmacology 2008;22(7):805-8. [DOI: 10.1177/0269881107084004] [PMID: ] [DOI] [PubMed] [Google Scholar]
- Tritt K, Nickel C, Lahmann C, Leiberich PK, Rother WK, Loew TH, et al. Lamotrigine treatment of aggression in female borderline-patients: a randomized, double-blind, placebo-controlled study. Journal of Psychopharmacology 2005;19(3):287-91. [DOI: 10.1177/0269881105051540] [PMID: ] [DOI] [PubMed] [Google Scholar]
Zanarini 2001 {published data only}
- Zanarini MC, Frankenburg FR. Olanzapine treatment of female borderline personality disorder patients: a double-blind, placebo-controlled pilot study. Journal of Clinical Psychiatry 2001;62(11):849-54. [DOI: 10.4088/jcp.v62n1103] [PMID: ] [DOI] [PubMed] [Google Scholar]
Zanarini 2003 {published data only}
- Zanarini MC, Frankenburg FR. Omega-3 fatty acid treatment of women with borderline personality disorder: a double-blind, placebo-controlled pilot study. American Journal of Psychiatry 2003;160(1):167-9. [DOI: 10.1176/appi.ajp.160.1.167] [PMID: ] [DOI] [PubMed] [Google Scholar]
Zanarini 2004 {published data only}
- Zanarini MC, Frankenburg FR, Parachini EA. A preliminary, randomized trial of fluoxetine, olanzapine, and the olanzapine-fluoxetine combination in women with borderline personality disorder. Journal of Clinical Psychiatry 2004;65(7):903-7. [DOI: 10.4088/jcp.v65n0704] [PMID: ] [DOI] [PubMed] [Google Scholar]
Zanarini 2007 {published data only}
- Eli Lilly and Company. Efficacy and safety of olanzapine in patients with borderline personality disorder: a randomized double-blind comparison with placebo [Summary ID # 6253. Clinical Study Summary: Study F1D-MC-HGKK]. www.lillytrials.com/results_files/zyprexa/zyprexa_summary_6523/ (accessed 14 February 2008).
- NCT00088036. Efficacy and safety of olanzapine in patients with borderline personality disorder [Efficacy and safety of olanzapine in patients with borderline personality disorder: a randomized double-blind comparison with placebo]. clinicaltrials.gov/ct2/show/NCT00088036 (first received 19 July 2004).
- Zanarini MC, Schulz SC, Detke HC, Tanaka Y, Zhao F, Lin D, et al. A dose comparison of olanzapine for the treatment of borderline personality disorder: a 12-week randomized, double-blind, placebo controlled study. European Psychiatry 2007;22(Suppl 1):S172-3. [DOI: 10.1016/j.eurpsy.2007.01.565] [DOI] [PubMed] [Google Scholar]
- Zanarini MC, Schulz SC, Detke HC, Tanaka Y, Zhao F, Lin D, et al. A dose comparison of olanzapine for the treatment of borderline personality disorder: a 12-week randomized, double-blind, placebo-controlled study. In: Association of European Psychiatrists. 15th European Congress of Psychiatry; 2007 Mar 17-21; Madrid, Spain. 2007.
- Zanarini MC, Schulz SC, Detke HC, Tanaka Y, Zhao F, Lin D, et al. P02.102 A dose comparison of olanzapine for the treatment of borderline personality disorder: a 12-week randomized double-blind placebo-controlled study. International Journal of Neuropsychopharmacology 2006;9(Suppl 1):S191. [DOI: 10.1017/S1461145706007450] [DOI] [PubMed] [Google Scholar]
- Zanarini MC, Schulz SC, Detke HC, Tanaka Y, Zhao F, Trzaskoma Q, et al. 80. Olanzapine for the treatment of borderline personality disorder: two 12-week randomized double-blind placebo-controlled trials. Neuropsychopharmacology 2006;31(Suppl 1):S229-30. [DOI: 10.1017/S1461145706007292] [DOI] [Google Scholar]
Ziegenhorn 2009 {published data only}
- Ziegenhorn AA, Roepke S, Schommer NC, Merkl A, Danker-Hopfe H, Perschel FH, et al. Clonidine improves hyperarousal in borderline personality disorder with or without comorbid posttraumatic stress disorder: a randomized, double-blind, placebo-controlled trial. Journal of Clinical Psychopharmacology 2009;29(2):170-3. [DOI: 10.1097/JCP.0b013e31819a4bae] [PMID: ] [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
ACTRN12615000705583 {published data only}
- ACTRN12615000705583. Investigation of the benefits of using quetiapine as an adjunct treatment in addition to psychotherapy in patients diagnosed with severe Borderline Personality Disorder. www.anzctr.org.au/Trial/Registration/TrialReview.aspx?id=368589 (first received 8 July 2015).
Bellino 2006b {published data only}
- Bellino S, Zizza M, Rinaldi C, Bogetto F. Combined treatment of major depression in patients with borderline personality disorder: a comparison with pharmacotherapy. Canadian Journal of Psychiatry 2006;51(7):453-60. [DOI: 10.1177/070674370605100707] [PMID: ] [DOI] [PubMed] [Google Scholar]
Coccaro 1997 {published data only (unpublished sought but not used)}
- Coccaro EF, Kavoussi RJ. Fluoxetine and impulsive aggressive behavior in personality-disordered subjects. Archives of General Psychiatry 1997;54(12):1081-8. [DOI: 10.1001/archpsyc.1997.01830240035005] [PMID: ] [DOI] [PubMed] [Google Scholar]
Hollander 2005 {published data only}
- Hollander E, Swann AC, Coccaro EF, Jiang P, Smith TB. Impact of trait impulsivity and state aggression on divalproex versus placebo response in borderline personality disorder. American Journal of Psychiatry 2005;162(3):621-4. [DOI: 10.1176/appi.ajp.162.3.621] [PMID: ] [DOI] [PubMed] [Google Scholar]
- Hollander E, Tracy KA, Swann AC, Coccaro EF, McElroy SL, Wozniak P, et al. Divalproex in the treatment of impulsive aggression: efficacy in cluster B personality disorders. Neuropsychopharmacology 2003;28(6):1186-97. [DOI: 10.1038/sj.npp.1300153] [PMID: ] [DOI] [PubMed] [Google Scholar]
ISRCTN11135486 {published data only}11135486
- ISRCTN11135486. Quetiapine versus sertraline as the pharmacological component in a standardised psychopharmacological and psychotherapeutic treatment of borderline personality disorder: a randomised, rater-blinded study. www.isrctn.com/ISRCTN11135486 (first received 1 August 2006). [ISRCTN11135486]
- Malevani J. AW: Cochrane review: RCT zu Quetapin und Sertraline bei BPS [personal communication]. Email to: J Stoffers-Winterling 17 June 2020.
Koenigsberg 2003 {published data only}
- Koenigsberg HW, Reynolds D, Goodman M, New AS, Mitropoulou V, Trestman RL, et al. Risperidone in the treatment of schizotypal personality disorder. Journal of Clinical Psychiatry 2003;64(6):628-34. [DOI: 10.4088/jcp.v64n0602] [PMID: ] [DOI] [PubMed] [Google Scholar]
La Malfa 2003 {published data only}
- La Malfa G, Lampronti L, Bertelli M, Conte M, Cabras P. Aggressiveness and personality disorders: evaluation of efficacy and existence of predictors in the treatment with fluvoxamine and lithium [Aggressività e disturbi della personalità]. Minerva Psychiatrica 2003;44(2):75-82. [WEB PAGE: Available at www.minervamedica.it/en/journals/minerva-psychiatry/article.php?cod=R17Y2003N02A0075] [Google Scholar]
Links 1990 {published data only}
- Links PS, Steiner M, Boiago I, Irwin D. Lithium therapy for borderline patients: preliminary findings. Journal of Personality Disorders 1990;4(2):173-81. [DOI: 10.1521/pedi.1990.4.2.173] [DOI] [Google Scholar]
Marchesi 2006 {published data only}
- Marchesi C, De Panfilis C, Cantoni A, Fontò S, Giannelli MR, Maggini C. Personality disorders and response to medication treatment in panic disorder: a 1-year naturalistic study. Progress in Neuro-Psychopharmacology & Biological Psychiatry 2006;30(7):1240-5. [DOI: 10.1016/j.pnpbp.2006.03.010] [PMID: ] [DOI] [PubMed] [Google Scholar]
NCT00255554 {published data only}
- Goodman M, New AS, Koenigsberg HW, Hazlett E, Flory J, Siever L. Psychotherapy and combined treatment in BPD: possible effects. In: Psychosomatic Medicine. Integrating Psychiatry & Medicine. 158th Annual Meeting of the American Psychiatric Association; 2005 May 21-26; Atlanta (GA). 2005:126-7. [ABSTRACT #: No. 19F] [PDF: www.psychiatry.org/File%20Library/Psychiatrists/Directories/Library-and-Archive/syllabus/am_syllabus_2005.pdf]
- Goodman M. RE: [EXTERNAL] Cochrane Colllaboration review [personal communication]. Email to: J Stoffers-Winterling 16 June 2018.
- NCT00255554. DBT and escitalopram in borderline personality disorder [Effects of dialectical behavioral therapy and escitalopram on impulsive aggression, affective instability and cognitive processing in borderline personality disorder]. clinicaltrials.gov/ct2/show/NCT00255554 (first received 17 November 2005). [NCT00255554]
NCT00463775 {published data only}
- NCT00463775. Topiramate for treatment of patients with borderline personality disorder and alcohol dependence. clinicaltrials.gov/ct2/show/NCT00463775 (first received 18 April 2007).
NCT00633802 {published data only}
- Bloch M. Re: RCT of Risperidone in borderline PD [pers commun]. Email to: J. Stoffers-Winterling 15 August 2018.
- NCT00633802. Low-dose risperidone treatment for subjects suffering from borderline personality disorder. clinicaltrials.gov/ct2/show/NCT00633802 (first received 4 March 2008).
NCT01103180 {published data only}
- McCloskey M. Fwd: Cochrane Collaboration review - RCT of escitalopram in BPD. Email to: J Stoffers-Winterling 15 August 2018.
- NCT01103180. Selective serotonin reuptake inhibitors (SSRIs) in borderline personality disorder [SSRIs and self-harm in borderline personality disorder]. clinicaltrials.gov/ct2/show/NCT01103180 (first received 12 April 2010).
NCT03395314 {published data only}
- NCT03395314. Ketamine in borderline personality disorder [A randomized active placebo controlled trial of ketamine in borderline personality disorderKetamine in Borderline Personality Disorder]. clinicaltrials.gov/ct2/show/NCT03395314 (first received 10 January 2018). [NCT03395314]
Parsons 1989 {published data only}
- Parsons B, Quitkin FM, McGrath PJ, Stewart JW, Tricamo E, Ocepek-Welikson K, et al. Phenelzine, imipramine, and placebo in borderline patients meeting criteria for atypical depression. Psychopharmacology Bulletin 1989;25(4):524-34. [PMID: ] [PubMed] [Google Scholar]
Rombold 2014 {published data only}
- Rombold F, Lauterbach E, Felber W, Mueller-Oerlinghausen B, Ahrens B, Bronisch T, et al. Adjunctive lithium treatment in the prevention of suicidal behavior in patients with depression and comorbid personality disorders. International Journal of Psychiatry in Clinical Practice 2014;18(4):300-3. [DOI: 10.3109/13651501.2014.940052] [PMID: ] [DOI] [PubMed] [Google Scholar]
Russell 2003 {published data only}
- Russell JM, Kornstein SG, Shea MT, McCullough JP, Harrison WM, Hirschfeld RM, et al. Chronic depression and comorbid personality disorders: response to sertraline versus imipramine. Journal of Clinical Psychiatry 2003;64(5):554-61. [DOI: 10.4088/jcp.v64n0510] [PMID: ] [DOI] [PubMed] [Google Scholar]
Serban 1984 {published data only}
- Serban G, Siegel S. Response of borderline and schizotypal patients to small doses of thiothixene and haloperidol. American Journal of Psychiatry 1984;141(11):1455-8. [DOI: 10.1176/ajp.141.11.1455] [PMID: ] [DOI] [PubMed] [Google Scholar]
Verkes 1998 {published data only}
- Verkes RJ, Van der Mast RC, Hengeveld MW, Tuyl JP, Zwinderman AH, Van Kempen GM. Reduction by paroxetine of suicidal behavior in patients with repeated suicide attempts but not major depression. American Journal of Psychiatry 1998;155(4):543-7. [DOI: 10.1176/ajp.155.4.543] [PMID: ] [DOI] [PubMed] [Google Scholar]
Wollmer 2022 {published data only}
- Wollmer MA, Neumann I, Jung S, Bechinie A, Herrmann J, Müller A, et al. Clinical effects of glabellar botulinum toxin injections on borderline personality disorder: a randomized controlled trial. Journal of Psychopharmacology 2022;36(2):159-69. [DOI: 10.1177/02698811211069108] [PMID: ] [DOI] [PubMed] [Google Scholar]
References to studies awaiting assessment
NCT00437099 {published data only}
- NCT00437009. Efficacy of omega-3 fatty acids on borderline personality disorder: a randomised, double blind clinical trial. clinicaltrials.gov/ct2/show/NCT00437099 (first received 16 February 2007).
NCT01912391 {published data only}
- NCT01912391. Clinical research study to evaluate selegiline in the treatment of borderline personality disorder [A phase III randomized double-blind, 12 week, placebo controlled trial of transdermal selegiline in borderline personality disorder (BPD) to evaluate efficacy and safety]. clinicaltrials.gov/ct2/show/NCT01912391 (first received 16 July 2013).
References to ongoing studies
ACTRN12617001317381 {published data only}ACTRN12617001317381
- ACTRN12617001317381. Estrogen for the treatment of borderline personality disorder [A randomised placebo controlled trial of estradiol for the treatment of women with borderline personality disorder]. anzctr.org.au/Trial/Registration/TrialReview.aspx?id=373455 (first received 10 August 2017).
Arteaga‐Henríquez 2020 {published data only}
- Arteaga-Henríquez G, Rosales-Ortiz SK, Arias-Vásquez A, Bitter I, Ginsberg Y, Ibañez-Jimenez P, et al. Treating impulsivity with probiotics in adults (PROBIA): study protocol of a multicenter, double-blind, randomized, placebo-controlled trial. Trials 2020;21(1):161. [DOI: 10.1186/s13063-019-4040-x] [PMCID: PMC7014653] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT03495375. Treating impulsivity in adults with probiotics (PROBIA) [Randomized placebo-controlled treatment of impulsivity in adults with probiotics]. clinicaltrials.gov/ct2/show/NCT03495375 (first received 26 February 2018).
Chanen 2019 {published data only}ACTRN12616001192471
- ACTRN12616001192471. VERBATIM: a randomised controlled trial of aripiprazole for the treatment of auditory verbal hallucinations in borderline personality disorder [A randomised controlled trial of aripiprazole for the treatment of auditory verbal hallucinations in borderline personality disorder]. anzctr.org.au/Trial/Registration/TrialReview.aspx?id=371038 (first received 26 August 2016).
- Chanen AM, Betts J, Jackson H, McGorry P, Nelson B, Cotton SM, et al. Ripiprazole compared with placebo for auditory verbal hallucinations in youth with borderline personality disorder: protocol for the VERBATIM randomized controlled trial. Early Intervention in Psychiatry 2019;13(6):1373-81. [DOI: 10.1111/eip.12774] [PMID: ] [DOI] [PubMed] [Google Scholar]
DRKS00015817 {published data only}
- DRKS00015817. Stellate ganglion block in patients with borderline personality disorder and posttraumatic stress disorder. www.drks.de/DRKS00015817 (first received 31 October 2018).
EUCTR2020‐003469‐20‐ES {published data only}
- EUCTR2020-003469-20-ES. A phase IIb study to evaluate the safety and efficacy of vafidemstat in an adult borderline personality disorder population [A double blind, randomized, placebo-controlled, adaptive 14-week phase IIb trial to evaluate the efficacy and safety of vafidemstat in an adult borderline personality disorder (BPD) population (PORTICO)]. trialsearch.who.int/?TrialID=EUCTR2020-003469-20-ES (first received 19 November 2020). [EUCTR2020-003469-20-ES]
EudraCT 2018‐002471‐18‐GB {published data only}ISRCTN18352058
- EudraCT 2018-002471-18. Clozapine in the treatment of borderline personality disorder [The clinical effectiveness and cost effectiveness of clozapine for inpatients with borderline personality disorder: randomised controlled trial]. clinicaltrialsregister.eu/ctr-search/search?query=2018-002471-18/GB (first received 19 June 2019).
- ISRCTN18352058. Clozapine in the treatment of borderline personality disorder. The CALMED study [The clinical effectiveness and cost effectiveness of clozapine for inpatients with borderline personality disorder: randomised controlled trial]. www.isrctn.com/ISRCTN18352058 (first received 18 March 2019).
IRCT20210106049948N1 {published data only}IRCT20210106049948N1
- IRCT20210106049948N1. Memantine and borderline personality disorder [The effect of memantine on the symptoms of Borderline personality disorder in the Iranian population]. trialsearch.who.int/?TrialID=IRCT20210106049948N1 (first received 21 February 2021). [IRCT20210106049948N1]
IRCT20210531051453N1 {published data only}IRCT20210531051453N1
- IRCT20210531051453N1. Effect of omega 3 fatty acid in borderline personality disorder [Study the effectiveness of omega 3 fatty acid as adjuvant treatment on depression, aggression and poor impuls control in hospitalized patients of borderline personality disorder]. trialsearch.who.int/Trial2.aspx?TrialID=IRCT20210531051453N1 (first received 30 July 2021). [IRCT20210531051453N1]
NCT04100096 {published data only}
- NCT04100096. A trial of brexpiprazole in the treatment of borderline personality disorder [A multicenter, randomized, flexible-dose, double-blind trial of brexpiprazole versus placebo for the treatment of adults with borderline personality disorder]. clinicaltrials.gov/ct2/show/NCT04100096 (first received 20 September 2019).
NCT04566601 {published data only}
- NCT04566601. A study to test different doses of BI 1358894 and find out whether they reduce symptoms in people with borderline personality disorder [A phase II randomized, double-blinded, placebo-controlled parallel group trial to examine the efficacy and safety of 4 oral doses of BI 1358894 once daily over 12 week treatment period in patients with borderline personality disorder]. clinicaltrials.gov/ct2/show/NCT04566601 (first received 28 September 2020).
NCT05356013 {published data only}
- NCT05356013. Caplyta in borderline personality disorder. clinicaltrials.gov/ct2/show/NCT05356013 (first received 02 May 2022).
Additional references
Aguglia 2019
- Aguglia A, Serafini G, Nebbia J, Salvi V, Martinotti G, Corbo M, et al. Off-label use of second-generation antipsychotics in borderline personality disorder: a survey of Italian psychiatrists. Journal of Personality Disorders 2021;35(3):321-5. [DOI] [PubMed] [Google Scholar]
Alvarez‐Tomas 2017
- Alvarez-Tomás I, Soler J, Bados A, Martín-Blanco A, Elices M, Carmona C, et al. Long-term course of borderline personality disorder: a prospective 10-year follow-up study. Journal of Personality Disorders 2017;31(5):590-605. [DOI] [PubMed] [Google Scholar]
Alvarez‐Tomas 2019
- Álvarez-Tomás I, Ruiz J, Guilera G, Bados A. Long-term clinical and functional course of borderline personality disorder: a meta-analysis of prospective studies. European Psychiatry 2019;56:75-83. [DOI: 10.1016/j.eurpsy.2018.10.010] [PMID: ] [DOI] [PubMed] [Google Scholar]
Amminger 2010
- Amminger GP, Schäfer MR, Papageorgiou K, Klier CM, Cotton SM, Harrigan SM, et al. Long-chain omega-3 fatty acids for indicated prevention of psychotic disorders: a randomized, placebo-controlled trial. Archives of General Psychiatry 2010;67(2):146-54. [DOI: 10.1001/archgenpsychiatry.2009.192] [PMID: ] [DOI] [PubMed] [Google Scholar]
Andrews 2013a
- Andrews J, Guyatt G, Oxman AD, Alderson P, Dahm P, Falck-Ytter Y, et al. GRADE guidelines: 14. Going from evidence to recommendations: the significance and presentation of recommendations. Journal of Clinical Epidemiology 2013;66(7):719–25. [DOI: 10.1016/j.jclinepi.2012.03.013] [PMID: ] [DOI] [PubMed] [Google Scholar]
Andrews 2013b
- Andrews JC, Schünemann HJ, Oxman AD, Pottie K, Meerpohl JJ, Coello PA, et al. GRADE guidelines: 15. Going from evidence to recommendation - determinants of a recommendation’s direction and strength. Journal of Clinical Epidemiology 2013;66(7):726–35. [DOI: 10.1016/j.jclinepi.2013.02.003] [PMID: ] [DOI] [PubMed] [Google Scholar]
Ansell 2007
- Ansell EB, Sanislow CA, McGlashan TH, Grilo CM. Psychosocial impairment and treatment utilization by patients with borderline personality disorder, other personality disorders, mood and anxiety disorders, and a healthy comparison group. Comprehensive Psychiatry 2007;48(4):329–36. [DOI: 10.1016/j.comppsych.2007.02.001] [PMID: ] [DOI] [PubMed] [Google Scholar]
APA 1980
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM-III). 3rd edition. Washington (DC): American Psychiatric Association, 1980. [ISBN: 978-0-89042-0416] [Google Scholar]
APA 1987
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM-III-R). 3rd edition. Washington (DC): American Psychiatric Association, 1987. [ISBN: 978-0-89042-0195] [Google Scholar]
APA 1994
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM-IV). 4th edition. Washington (DC): American Psychiatric Association, 1994. [ISBN: 978-0-89042-0610] [Google Scholar]
APA 2000
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM-IV-TR). 4th edition. Washington (DC): American Psychiatric Association, 2000. [ISBN: 978-0-89042-0256] [Google Scholar]
APA 2001
- American Psychiatric Association. Practice guideline for the treatment of patients with borderline personality disorder. American Journal of Psychiatry 2001;158(10 Suppl):1-52. [PMID: ] [PubMed] [Google Scholar]
APA 2013
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM-5). 5 edition. Washington (DC): American Psychiatric Association, 2013. [ISBN: 978-0-89042-554-1] [Google Scholar]
Arntz 2003
- Arntz A, Van den Hoorn M, Cornelis J, Verheul R, Van den Bosch W, De Bie AJHT. Reliability and validity of the borderline personality disorder severity index. Journal of Personality Disorders 2003;17(1):45-59. [PMID: ] [DOI] [PubMed] [Google Scholar]
Atkins 2004
- Atkins D, Best D, Briss PA, Eccles M, Falck-Ytter Y, Flottorp S, et al. Grading quality of evidence and strength of recommendations. BMJ 2004;328(7454):1490-9. [DOI: 10.1136/bmj.328.7454.1490] [PMCID: PMC428525] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ayodeji 2015
- Ayodeji E, Green J, Roberts C, Trainor G, Rothwell J, Woodham A, et al. The influence of personality disorder on outcome in adolescent self-harm. British Journal of Psychiatry 2015;207(4):313-9. [DOI: 10.1192/bjp.bp.113.138941] [PMID: ] [DOI] [PubMed] [Google Scholar]
Balshem 2011
- Balshem H, Helfand M, Schünemann HJ, Oxman AD, Kunz R, Brozek J, et al. GRADE guidelines: 3. Rating the quality of evidence. Journal of Clinical Epidemiology 2011;64(4):401–6. [DOI: 10.1016/j.jclinepi.2010.07.015] [PMID: ] [DOI] [PubMed] [Google Scholar]
Barrett 1995
- Barrett ES, Stanford MS. Impulsiveness. In: Costello CG, editors(s). Personality Characteristics of the Personality Disordered. New York (NY): Wiley, 1995:91-118. [ISBN: 978-0-471-01529-1] [Google Scholar]
Bateman 2015
- Bateman AW, Gunderson J, Mulder R. Treatment of personality disorder. Lancet 2015;385(9969):735-43. [PMID: 10.1016/S0140-6736(14)61394-5] [PMID: ] [DOI] [PubMed] [Google Scholar]
Beck 1961
- Beck AT, Ward CH, Mendelson M, Mock J, Erbaugh J. An inventory for measuring depression. Archives of General Psychiatry 1961;4:561-71. [PMID: ] [DOI] [PubMed] [Google Scholar]
Beck 1979
- Beck AT, Kovacs M, Weissman A. The assessment of suicidal intention: the Scale for Suicide Ideation. Journal of Consulting and Clinical Psychology 1979;47(2):343-52. [PMID: ] [DOI] [PubMed] [Google Scholar]
Bellino 2019 [pers comm]
- Bellino S. Inclusion of study in Cochrane review. Email to: A Tadorovac 31 January 2019.
Bender 2001
- Bender DS, Dolan RT, Skodol AE, Sanislow CA, Dyck IR, McGlashan TH, et al. Treatment utilization by patients with personality disorders. American Journal of Psychiatry 2001;158(2):295–302. [DOI: 10.1176/appi.ajp.158.2.295] [PMID: ] [DOI] [PubMed] [Google Scholar]
Bender 2006
- Bender DS, Skodol AE, Pagano ME, Dyck IE, Grilo CM, Shea MT, et al. Prospective assessment of treatment use by patients with personality disorders. Psychiatric Services 2006;57(2):254–7. [DOI: 10.1176/appi.ps.57.2.254] [PMCID: PMC2705621] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Berkey 1996
- Berkey CS, Mosteller F, Lau J, Antman E. Uncertainty of the time of first significance in random effects cumulative meta-analysis. Controlled Clinical Trials 1996;17(5):357-71. [PMID: ] [DOI] [PubMed] [Google Scholar]
Bernstein 1986
- Bernstein EM, Putnam FW. Development, reliability, and validity of a dissociation scale. Journal of Nervous and Mental Disease 1986;174(12):727-35. [PMID: ] [DOI] [PubMed] [Google Scholar]
Bero 2013
- Bero L. Why the Cochrane risk of bias tool should include funding source as a standard item. Cochrane Database of Systematic Reviews 2013;(12):ED000075. [DOI: 10.1002/14651858.ED000075] [PMID: ] [DOI] [PMC free article] [PubMed]
Biskin 2015
- Biskin RS. The lifetime course of borderline personality disorder. Canadian Journal of Psychiatry 2015;60(7):303-8. [DOI: 10.1177/070674371506000702] [PMCID: PMC4500179] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Black 2014a
- Black DW, Zanarini MC, Romine A, Shaw M, Allen J, Schulz SC. Comparison of low and moderate dosages of extended-release quetiapine in borderline personality disorder: a randomized, double-blind, placebo-controlled trial. American Journal of Psychiatry 2014;171(11):1174-82. [DOI: 10.1176/appi.ajp.2014.13101348] [PMID: ] [DOI] [PubMed] [Google Scholar]
Bode 2017
- Bode K, Vogel R, Walker J, Kröger C. Health care costs of borderline personality disorder and matched controls with major depressive disorder: a comparative study based on anonymized claims data. European Journal of Health Economics 2017;18(9):1125-35. [DOI] [PubMed] [Google Scholar]
Bohus 2021
- Bohus M, Stoffers-Winterling JM, Sharp C, Krause-Utz A, Schmahl C, Lieb K. Borderline personality disorder. Lancet 2021;398:1528-40. [DOI] [PubMed] [Google Scholar]
Borschmann 2012
- Borschmann R, Henderson C, Hogg J, Phillips R, Moran P. Crisis interventions for people with borderline personality disorder. Cochrane Database of Systematic Reviews 2012, Issue 6. Art. No: CD009353. [DOI: 10.1002/14651858.CD009353.pub2] [PMID: ] [DOI] [PubMed] [Google Scholar]
Bridler 2015
- Bridler R, Häberle A, Müller ST, Cattapan K, Grohmann R, Toto S, et al. Psychopharmacological treatment of 2195 in-patients with borderline personality disorder: a comparison with other psychiatric disorders. European Neuropsychopharmacology 2015;25(6):763-72. [PMID: 10.1016/j.euroneuro.2015.03.017] [PMID: ] [DOI] [PubMed] [Google Scholar]
Brok 2008
- Brok J, Thorlund K, Gluud C, Wetterslev J. Trial sequential analysis reveals insufficient information size and potentially false positive results in many meta-analyses. Journal of Clinical Epidemiology 2008;61(8):763-9. [DOI: 10.1016/j.jclinepi.2007.10.007] [PMID: ] [DOI] [PubMed] [Google Scholar]
Brok 2009
- Brok J, Thorlund K, Wetterslev J, Gluud C. Apparently conclusive meta-analysis may be inconclusive -- trial sequential analysis adjustment of random error risk due to repetitive testing of accumulating data in apparently conclusive neonatal meta-analyses. International Journal of Epidemiology 2009;38(1):287-98. [DOI: 10.1093/ije/dyn188] [PMID: ] [DOI] [PubMed] [Google Scholar]
Brühl 2017
- Brühl AB, Sahakian BJ. Neuroscience-based nomenclature: improving clinical and scientific terminology in research and clinical psychopharmacology. Psychological Medicine 2017;47(8):1339-41. [DOI] [PubMed] [Google Scholar]
Brunetti 2013
- Brunetti M, Shemilt I, Pregno S, Vale L, Oxman AD, Lord J, et al. GRADE guidelines: 10. Considering resource use and rating the quality of economic evidence. Journal of Clinical Epidemiology 2013;66(2):140–50. [DOI: 10.1016/j.jclinepi.2012.04.012] [PMID: ] [DOI] [PubMed] [Google Scholar]
Cailhol 2015
- Cailhol L, Thalamas C, Garrido C, Birmes P, Lapeyre-Mestre M. Mental health service utilization among borderline personality disorder inpatients [Utilisation des services de soin par les patients hospitalisés, présentant un trouble de personnalité borderline en Midi-Pyrénées]. L'Encéphale 2015;41(2):115-22. [DOI: 10.1016/j.encep.2014.10.008] [PMID: ] [DOI] [PubMed] [Google Scholar]
Cavelti 2019
- Cavelti M, Thompson K, Hulbert C, Betts J, Jackson H, Francey S, et al. Preliminary evidence for the cognitive model of auditory verbal hallucinations in youth with borderline personality disorder. Front Psychiatry 2019;10:292. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Cavelti 2021
- Cavelti M, Thompson K, Chanen AM, Kaess M. Psychotic symptoms in borderline personality disorder: developmental aspects. Current Opinion in Psychology 2021;37:26-31. [PMID: ] [DOI] [PubMed] [Google Scholar]
Chanen 2012
- Chanen AM, Kaess M. Developmental pathways to borderline personality disorder. Current Psychiatry Reports 2012;14(1):45-53. [DOI: 10.1007/s11920-011-0242-y] [PMID: ] [DOI] [PubMed] [Google Scholar]
Chanen 2013
- Chanen AM, McCutcheon L. Prevention and early intervention for borderline personality disorder: current status and recent evidence. British Journal of Psychiatry 2013;Supplement(54):s24-9. [DOI] [PubMed] [Google Scholar]
Chanen 2017
- Chanen A, Sharp C, Hoffman P, Global Alliance for Prevention and Early Intervention for Borderline Personality Disorder. Prevention and early intervention for borderline personality disorder: a novel public health priority. World Psychiatry 2017;16(2):215-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Coccaro 1991
- Coccaro EF, Harvey PD, Kupsaw-Lawrence E, Herbert JL, Bernstein DP. Development of neuropharmacologically-based behavioral assessments of impulsive aggressive behavior. Journal of Neuropsychiatry and Clinical Neurosciences 1991;3(2):S44-51. [PMID: ] [PubMed] [Google Scholar]
Coccaro 2020
- Coccaro EF. The Overt Aggression Scale Modified (OAS-M) for clinical trials targeting impulsive aggression and intermittent explosive disorder: validity, reliability, and correlates. Journal of Psychiatric Research 2020;124:50-7. [DOI: 10.1016/j.jpsychires.2020.01.007] [PMID: ] [DOI] [PubMed] [Google Scholar]
Coid 2006
- Coid J, Yang M, Tyrer P, Roberts A, Ullrich S. Prevalence and correlates of personality disorder in Great Britain. British Journal of Psychiatry 2006;188:423-31. [DOI: 10.1192/bjp.188.5.423] [PMID: ] [DOI] [PubMed] [Google Scholar]
Crawford 2020 [pers comm]
- Crawford M. Inclusion of your study in Cochrane review. Email to: A Pathmanathan Ratnam 09 November 2020.
Cristea 2017
- Cristea IA, Gentili C, Cotet CD, Palomba D, Barbui C, Cuijpers P. Efficacy of psychotherapies for borderline personality disorder: a systematic review and meta-analysis. JAMA Psychiatry 2017;74(4):319-28. [DOI: 10.1001/jamapsychiatry.2016.4287] [PMID: ] [DOI] [PubMed] [Google Scholar]
CTU 2011
- Copenhagen Trial Unit. TSA - Trial Sequential Analysis. www.ctu.dk/tsa (accessed 12 December 2016).
Curtin 2002
- Curtin F, Elbourne D, Altman DG. Meta-analysis combining parallel and cross-over clinical trials. III: the issue of carry-over. Statistics in Medicine 2002;21(15):2161-73. [DOI: 10.1002/sim.1207] [PMID: ] [DOI] [PubMed] [Google Scholar]
Deeks 2021
- Deeks JJ, Higgins JP, Altman DG, editor(s). Chapter 10: Analysing data and undertaking meta-analyses. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.2 (updated February 2021), Cochrane 2021. Available from www.training.cochrane.org/handbook.
Derogatis 1994
- Derogatis LR, Lazarus L. SCL-90-R, Brief Symptom Inventory, and matching clinical rating scales. In: Maruish ME, editors(s). The Use of Psychological Testing for Treatment Planning and Outcome Assessment. 1st edition. Hillsdale (NJ): Lawrence Erlbaum Associates, 1994. [ISBN: 978-0-80581-1629] [Google Scholar]
DGPPN 2009
- Deutsche Gesellschaft für Psychiatrie und Psychotherapie Psychosomatik und Nervenheilkunde (DGPPN) [German Association for Psychiatry, Psychotherapy and Psychosomatics]. S2-Praxisleitlinien in Psychiatrie und Psychotherapie. Band 1: Behandlungsleitlinie Persönlichkeitsstörungen [S2 Practical Guidelines in Psychiatry and Psychotherapy. Treatment Guideline Personality Disorders]. Vol. 1. Heidelberg (DE): Steinkopff Verlag, 2009. [ISBN: 978-3-7985-1853-7] [Google Scholar]
Donner 2002
- Donner A, Klar N. Issues in the meta-analysis of cluster randomized trials. Statistics in Medicine 2002;21(19):2971-80. [DOI: 10.1002/sim.1301] [PMID: ] [DOI] [PubMed] [Google Scholar]
Eaton 2018
- Eaton NR, Greene AL. Personality disorders: community prevalence and socio-demographic correlates. Current Opinion in Psychology 2018;21:28-32. [DOI: 10.1016/j.copsyc.2017.09.001] [PMID: ] [DOI] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne DR, Altman DG, Higgins JPT, Curtin F, Worthington HV, Vail A. Meta-analyses involving cross-over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140-9. [DOI] [PubMed] [Google Scholar]
Eli Lilly 2008
- Eli Lilly and Company. Efficacy and safety of olanzapine in patients with borderline personality disorder: a randomized, flexible-dose, double-blind comparison with placebo [Summary ID #6257. Clinical Study Summary: F1D-MC-HGKL]. www.lillytrials.com/results_files/zyprexa/zyprexa_summary_6257 (accessed 14 February 2008).
Ellison 2018
- Ellison WD, Rosenstein LK, Morgan TA, Zimmerman M. Community and clinical epidemiology of borderline personality disorder. Psychiatric Clinics of North America 2018;41(4):561-73. [DOI: 10.1016/j.psc.2018.07.008] [PMID: ] [DOI] [PubMed] [Google Scholar]
Endicott 1976
- Endicott J, Spitzer RL, Fleiss JL, Cohen J. The Global Assessment scale: a procedure for measuring overall severity of psychiatric disturbance. Archives of General Psychiatry 1976;33(6):766-71. [PMID: ] [DOI] [PubMed] [Google Scholar]
Fonagy 2009
- Fonagy P, Luyten P. A developmental, mentalization-based approach to the understanding and treatment of borderline personality disorder. Development and Psychopathology 2009;21(4):1355-81. [DOI: 10.1017/S0954579409990198] [PMID: ] [DOI] [PubMed] [Google Scholar]
Garner 2016
- Garner P, Hopewell S, Chandler J, MacLehose H, Schünemann HJ, Akl EA, et al. When and how to update systematic reviews: consensus and checklist. BMJ 2016;354:i3507. [PMCID: PMC4955793] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Gartlehner 2021a
- Gartlehner G, Crotty K, Kennedy S, Edlund MJ, Ali R, Siddiqui M, et al. Pharmacological treatments for borderline personality disorder: a systematic review and meta-analysis. Central Nervous System Drugs 2021;35(10):1053-67. [CENTRAL: PMC8478737] [DOI: 10.1007/s40263-021-00855-4] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Gartlehner 2021b
- Gartlehner G, Crotty K, Edlund MJ, Viswanathan M. Authors’ reply to Pereira Ribeiro et al: comment on “Pharmacological Treatments for Borderline Personality Disorder: A Systematic Review and Meta‑Analysis”. Central Nervous System Drugs 2021;35:1335-6. [DOI: ] [DOI] [PubMed] [Google Scholar]
Goodman 2012
- Goodman M, Roiff T, Oakes AH, Paris J. Suicidal risk and management in borderline personality disorder. Current Psychiatry Reports 2012;14(1):79-85. [DOI: 10.1007/s11920-011-0249-4] [PMID: ] [DOI] [PubMed] [Google Scholar]
GRADEpro 2021 [Computer program]
- GRADEpro GDT (guideline development tool). Version accessed prior to 18 October 2022. Hamilton (Ontario): Grade Working Group, McMaster University and Evidence Prime, 2021.
Grant 2008
- Grant BF, Chou SP, Goldstein RB, Huang B, Stinson FS, Saha TD, et al. Prevalence, correlates, disability, and comorbidity of DSM-IV borderline personality disorder: results from the Wave 2 National Epidemiologic Survey on Alcohol and Related Conditions. Journal of Clinical Psychiatry 2008;69:533-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
Gratz 2001
- Gratz KL. Meausrement of deliberate self-harm: preliminary data on the deliberate self-harm inventory. Journal of Psychopathology and Behavioral Assessment 2001;23(4):253-63. [DOI: 10.1023/A:1012779403943] [DOI] [Google Scholar]
Gunderson 2011a
- Gunderson JG. Borderline personality disorder. New England Journal of Medicine 2011;364(21):2037-42. [DOI: 10.1056/NEJMcp1007358] [DOI] [PubMed] [Google Scholar]
Gunderson 2011b
- Gunderson JG, Stout RL, McGlashan TH, Shea MT, Morey LC, Grilo CM, et al. Ten-year course of borderline personality disorder: psychopathology and function from the Collaborative Longitudinal Personality Disorders study. Archives of General Psychiatry 2011;68(8):827-37. [DOI: 10.1001/archgenpsychiatry.2011.37] [PMCID: PMC3158489] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Guttierez 2001
- Guttierez PM, Osman A, Barrios FX, Kopper BA. Development and initial validation of the Self-Harm Behavior Questionnaire. Journal of Personality Assessment 2001;77(3):475-90. [DOI: 10.1207/S15327752JPA7703_08] [PMID: ] [DOI] [PubMed] [Google Scholar]
Guyatt 2011a
- Guyatt G, Oxman AD, Akl EA, Kunz R, Vist G, Brozek J, et al. GRADE guidelines: 1. Introduction - GRADE evidence profiles and summary of findings tables. Journal of Clinical Epidemiology 2011;64(4):383–94. [DOI: 10.1016/j.jclinepi.2010.04.026] [PMID: ] [DOI] [PubMed] [Google Scholar]
Guyatt 2011b
- Guyatt GH, Oxman AD, Kunz R, Atkins D, Brozek J, Vist G, et al. GRADE guidelines: 2. Framing the question and deciding on important outcomes. Journal of Clinical Epidemiology 2011;64(4):395–400. [DOI: 10.1016/j.jclinepi.2010.09.012] [PMID: ] [DOI] [PubMed] [Google Scholar]
Guyatt 2011c
- Guyatt GH, Oxman AD, Vist G, Kunz R, Brozek J, Alonso-Coello P, et al. GRADE guidelines: 4. Rating the quality of evidence - study limitations (risk of bias). Journal of Clinical Epidemiology 2011;64(4):407–15. [DOI: 10.1016/j.jclinepi.2010.07.017] [PMID: ] [DOI] [PubMed] [Google Scholar]
Guyatt 2011d
- Guyatt GH, Oxman AD, Montori V, Vist G, Kunz R, Brozek J, et al. GRADE guidelines: 5. Rating the quality of evidence - publication bias. Journal of Clinical Epidemiology 2011;64(12):1277–82. [DOI: 10.1016/j.jclinepi.2011.01.011] [PMID: ] [DOI] [PubMed] [Google Scholar]
Guyatt 2011e
- Guyatt GH, Oxman AD, Kunz R, Brozek J, Alonso-Coello P, Rind D, et al. GRADE guidelines 6. Rating the quality of evidence - imprecision. Journal of Clinical Epidemiology 2011;64(12):1283–93. [DOI: 10.1016/j.jclinepi.2011.01.012] [PMID: ] [DOI] [PubMed] [Google Scholar]
Guyatt 2011f
- Guyatt GH, Oxman AD, Kunz R, Woodcock J, Brozek J, Helfand M, et al. GRADE guidelines: 7. Rating the quality of evidence - inconsistency. Journal of Clinical Epidemiology 2011;64(12):1294–302. [DOI: 10.1016/j.jclinepi.2011.03.017] [PMID: ] [DOI] [PubMed] [Google Scholar]
Guyatt 2011g
- Guyatt GH, Oxman AD, Kunz R, Woodcock J, Brozek J, Helfand M, et al. GRADE guidelines: 8. Rating the quality of evidence - indirectness. Journal of Clinical Epidemiology 2011;64(12):1303–10. [DOI: 10.1016/j.jclinepi.2011.04.014] [PMID: ] [DOI] [PubMed] [Google Scholar]
Guyatt 2011h
- Guyatt GH, Oxman AD, Sultan S, Glasziou P, Akl EA, Alonso-Coello P, et al. GRADE guidelines: 9. Rating up the quality of evidence. Journal of Clinical Epidemiology 2011;64(12):1311–6. [DOI: 10.1016/j.jclinepi.2011.06.004] [PMID: ] [DOI] [PubMed] [Google Scholar]
Guyatt 2013a
- Guyatt G, Oxman AD, Sultan S, Brozek J, Glasziou P, Alonso-Coello P, et al. GRADE guidelines: 11. Making an overall rating of confidence in effect estimates for a single outcome and for all outcomes. Journal of Clinical Epidemiology 2013;66(2):151–7. [DOI: 10.1016/j.jclinepi.2012.01.006] [PMID: ] [DOI] [PubMed] [Google Scholar]
Guyatt 2013b
- Guyatt GH, Oxman AD, Santesso N, Helfand M, Vist G, Kunz R, et al. GRADE guidelines: 12. Preparing summary of findings tables - binary outcomes. Journal of Clinical Epidemiology 2013;66(2):158–72. [DOI: 10.1016/j.jclinepi.2012.01.012] [PMID: ] [DOI] [PubMed] [Google Scholar]
Guyatt 2013c
- Guyatt GH, Thorlund K, Oxman AD, Walter SD, Patrick D, Furukawa TA, et al. GRADE guidelines: 13. Preparing summary of findings tables and evidence profiles - continuous outcomes. Journal of Clinical Epidemiology 2013;66(2):173–83. [DOI: 10.1016/j.jclinepi.2012.08.001] [PMID: ] [DOI] [PubMed] [Google Scholar]
Gøtzsche 2015
- Gøtzsche P. Re: Why the Cochrane risk of bias tool should include funding source as a standard item. Response to JAC Sterne. cochranelibrary.com/editorial/10.1002/14651858.ED000076 (accessed 19 January 2018).
Hancock‐Johnson 2017
- Hancock-Johnson E, Griffiths C, Picchioni M. A focused systematic review of pharmacological treatment for borderline personality disorder. Central Nervous System Drugs 2017;31(5):345-56. [DOI: 10.1007/s40263-017-0425-0] [PMID: ] [DOI] [PubMed] [Google Scholar]
Hastrup 2019a
- Hastrup LH, Kongerslev MT, Simonsen E. Low vocational outcome among people diagnosed with borderline personality disorder during first admission to mental health services in Denmark: a nationwide 9-year register-based study. Journal of Personality Disorders 2019;33(3):326-40. [DOI: 10.1521/pedi_2018_32_344] [DOI] [PubMed] [Google Scholar]
Hastrup 2019b
- Hastrup LH, Jennum P, Ibsen R, Kjellberg J, Simonsen E. Societal costs of borderline personality disorders: a matched-controlled nationwide study of patients and spouses. Acta Psychiatrica Scandinavica 2019;140(5):458-67. [DOI: 10.1111/acps.13094] [PMID: ] [DOI] [PubMed] [Google Scholar]
Herpertz 2007
- Herpertz SC, Zanarini M, Schulz CS, Siever L, Lieb K, Möller H-J, et al. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of personality disorders. World Journal of Biological Psychiatry 2007;8(4):212-44. [DOI: 10.1080/15622970701685224] [PMID: ] [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. BMJ 2003;327(7414):557-60. [DOI: 10.1136/bmj.327.7414.557] [PMCID: PMC192859] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2017
- Higgins JPT, Altmand DG, Sterne JAC, editor(s). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Churchill R, Chandler J, Cumpston M, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.2.0 (updated June 2017). Cochrane, 2017. Available from www.training.cochrane.org/handbook/archive/v5.2.
Higgins 2019
- Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al, editor(s). Cochrane Handbook for Systematic Reviews of Interventions. 2nd edition. Chichester (UK): John Wiley & Sons, 2019. [WEB PAGE: Available from www.training.cochrane.org/handbook] [Google Scholar]
Higgins 2022
- Higgins JPT, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 6.3 (updated February 2022). Cochrane 2022. Available from www.training.cochrane.org/handbook.
Horowitz 1988
- Horowitz LM, Rosenberg SE, Baer BA, Ureño G, Villaseñor VS. Inventory of interpersonal problems: psychometric properties and clinical applications. Journal of Consulting and Clinical Psychology 1988;56(6):885-92. [PMID: ] [DOI] [PubMed] [Google Scholar]
Hörz 2010
- Hörz S, Zanarini MC, Frankenburg FR, Reich DB, Fitzmaurice G. Ten-year use of mental health services by patients with borderline personality disorder and with other axis II disorders. Psychiatric Services 2010;61(6):612-6. [DOI: 10.1176/ps.2010.61.6.612] [PMCID: PMC3889171] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ingenhoven 2011
- Ingenhoven TJ, Duivendvoorden HJ. Differential effectiveness of antipsychotics in borderline personality disorder: Meta-analyses of placebo-controlled, randomized clinical trials on symptomatic outcome domains. Journal of Clinical Psychopharmacology 2011;31(4):489-96. [DOI: 10.1097/JCP.0b013e3182217a69] [PMID: ] [DOI] [PubMed] [Google Scholar]
Ingenhoven 2015
- Ingenhoven T. Pharmacotherapy for borderline patients: business as usual or by default? Journal of Clinical Psychiatry 2015;76(4):e522-3. [DOI: 10.4088/JCP.14com09522] [PMID: ] [DOI] [PubMed] [Google Scholar]
Jacobi 2021
- Jacobi F, Grafiadeli R, Volkmann H, Schneider I. Disease burden of borderline personality disorder: cost of illness, somatic comorbidity and mortality [[Krankheitslast der Borderline-Persönlichkeitsstörung: Krankheitskosten, somatische Komorbidität und Mortalität]]. Nervenarzt 2021;92(7):660-9. [DOI] [PubMed] [Google Scholar]
Jakobsen 2014
- Jakobsen JC, Wetterslev J, Winkel P, Lange T, Gluud C. Thresholds for statistical and clinical significance in systematic reviews with meta-analytic methods. BMC Medical Research Methodology 2014;14:120. [DOI: 10.1186/1471-2288-14-120] [PMCID: PMC4251848] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kay 1987
- Kay SR, Fiszbein A, Opler LA. The positive and negative syndrome scale (PANSS) for schizophrenia. Schizophr Bull 1987;13(2):261-76. [PMID: ] [DOI] [PubMed] [Google Scholar]
Kjaergard 2001
- Kjaergard LL, Villumsen J, Gluud C. Reported methodologic quality and discrepancies between large and small randomized trials in meta-analyses. Annals of Internal Medicine 2001;135(11):982-9. [PMID: ] [DOI] [PubMed] [Google Scholar]
Knappich 2014
- Knappich M, Hörz-Sagstetter S, Schwerthöffer D, Leucht S, Rentrop M. Pharmacotherapy in the treatment of patients with borderline personality disorder: results of a survey among psychiatrists in private practices. International Clinical Psychopharmacology 2014;29(4):224-8. [DOI: 10.1097/YIC.0000000000000021] [PMCID: PMC4047315] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kongerslev 2015
- Kongerslev MT, Chanen AM, Simonsen E. Personality disorder in childhood and adolescence comes of age: a review of the current evidence and prospects for future research. Scandinavian Journal of Child and Adolescent Psychiatry and Psychology 2015;3(1):31-48. [DOI: 10.21307/sjcapp-2015-004] [DOI] [Google Scholar]
Korang 2020
- Korang Steven Kwasi, Juul Sophie, Nielsen Emil Eik, Feinberg Joshua, Siddiqui Faiza, Ong Giok, et al. Vaccines to prevent COVID-19: a protocol for a living systematic review with network meta-analysis including individual patient data (The LIVING VACCINE Project). Systematic Reviews 2020;9(1):1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kulkarni 2020 [pers comm]
- Kulkarni J. Re: Your Request. Email to: A Pathmanathan Ratnam 14 December 2020.
Lau 1995
- Lau J, Schmid CH, Chalmers TC. Cumulative meta-analysis of clinical trials builds evidence for exemplary medical care. Journal of Clinical Epidemiology 1995;48(1):45-57. [PMID: ] [DOI] [PubMed] [Google Scholar]
Lenzenweger 2007
- Lenzenweger MF, Lane MC, Loranger AW, Kessler RC. DSM-IV personality disorders in the National Comorbidity Survey Replication. Biological Psychiatry 2007;62(6):553-64. [DOI: 10.1016/j.biopsych.2006.09.019] [PMCID: PMC2044500] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Levine 1986
- Levine J, Schooler NR. SAFTEE: a technique for the systematic assessment of side effects in clinical trials. Psychopharmacology Bulletin 1986;22(2):343-81. [PMID: ] [PubMed] [Google Scholar]
Lieb 2004
- Lieb K, Zanarini MC, Schmahl C, Linehan MM, Bohus M. Borderline personality disorder. Lancet 2004;364(9432):453-61. [DOI: 10.1016/S0140-6736(04)16770-6] [PMID: ] [DOI] [PubMed] [Google Scholar]
Linehan 1997
- Linehan MM. Behavioral treatments of suicidal behaviors: definitional obfuscation and treatment outcomes. Annals of the New York Academy of Sciences 1997;836:302-28. [PMID: ] [DOI] [PubMed] [Google Scholar]
Links 2009
- Links PS, Kolla N. Assessing and managing suicide risk. In: Oldham JM, Skodol AE, Bender DS, editors(s). Essentials of Personality Disorders. 1st edition. Arlington (VA): American Psychiatric Publishing, Inc, 2009:343-60. [ISBN: 978-1-58562-358-7] [Google Scholar]
Lundh 2017
- Lundh A, Lexchin J, Mintzes B, Schroll JB, Bero L. Industry sponsorship and research outcome. Cochrane Database of Systematic Reviews 2017, Issue 2. Art. No: MR000033. [DOI: 10.1002/14651858.MR000033.pub3] [PMCID: PMC8132492] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lundh 2018
- Lundh Andreas, Lexchin Joel, Mintzes Barbara, Schroll Jeppe B, Bero Lisa. Industry sponsorship and research outcome: systematic review with meta-analysis. Intensive care medicine 2018;44(10):1603-1612. [DOI] [PubMed] [Google Scholar]
Makela 2006
- Makela EH, Moeller KE, Fullen JE, Gunel E. Medication utilization patterns and methods of suicidality in borderline personality disorder. Annals of Pharmacotherapy 2006;40(1):49-52. [DOI: 10.1345/aph.1E479] [PMID: ] [DOI] [PubMed] [Google Scholar]
Mancke 2015
- Mancke F, Bertsch K, Herpertz SC. Gender differences in aggression of borderline personality disorder. Borderline Personal Disord Emot Dysregul 2015;9(2):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Meuldijk 2017
- Meuldijk D, McCarthy A, Bourke ME, Grenyer BF. The value of psychological treatment for borderline personality disorder: Systematic review and cost offset analysis of economic evaluations. PLoS One 2017;1(12(3)):e0171592. [DOI] [PMC free article] [PubMed] [Google Scholar]
Moher 1998
- Moher D, Pham B, Jones A, Cook DJ, Jadad A, Moher M, et al. Does quality of reports of randomised trials affect estimates of intervention efficacy reported in meta-analyses? Lancet 1998;352(9128):609-13. [DOI: 10.1016/S0140-6736(98)01085-X] [PMID: ] [DOI] [PubMed] [Google Scholar]
Moher 1999
- Moher D, Cook DJ, Eastwood S, Olkin I, Rennie D, Stroup DF. Improving the quality of reports of meta-analyses of randomised controlled trials: the QUOROM statement. Quality of Reporting of Meta-analyses. Lancet 1999;354(9193):1896-900. [PMID: ] [DOI] [PubMed] [Google Scholar]
Montgomery 1979
- Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. British Journal of Psychiatry 1979;134:382-9. [DOI: 10.1192/bjp.134.4.382] [PMID: ] [DOI] [PubMed] [Google Scholar]
Montgomery 1983
- Montgomery D, Montgomery S, Roy D. Mianserin in the prophylaxis of suicidal behaviour: a double‐blind placebo controlled trial. In: Soubrier JP, Vedrinne J, editors(s). Dépression et Suicide: Aspects Medicaux, Psychologiques et Socio-Culturels. Paris (FR): Pergamon Press, 1983:786‐90. [DOI: 10.1016/B978-0-08-027080-7.50130-0] [DOI] [Google Scholar]
Mulder 2021
- Mulder RT. CD-11 personality disorders: utility and implications of the new model. Front Psychiatry 2021;12:655548. [DOI: ] [PMID: ] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Munk‐Jørgensen 2010
- Munk-Jørgensen P, Najarraq Lund M, Bertelsen A. Use of ICD-10 diagnoses in Danish psychiatric hospital-based services in 2001-2007. World Psychiatry 2010;9(3):183-4. [PMCID: PMC2948730] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Mustafa 2013
- Mustafa RA, Santesso N, Brozek J, Akl EA, Walter SD, Norman G, et al. The GRADE approach is reproducible in assessing the quality of evidence of quantitative evidence syntheses. Journal of Clinical Epidemiology 2013;66(7):736-42. [DOI: 10.1016/j.jclinepi.2013.02.004] [PMID: ] [DOI] [PubMed] [Google Scholar]
NbN3 2021
- Neuroscience-Based Nomenclature. Neuroscience-Based Nomenclature, Second Edition-reivsed. https://nbn2r.com/search (accessed 02 December 2021).
NCT00880919
- NCT00880919. Seroquel extended release (XR) for the management of borderline personality disorder (BPD) [Seroquel XR for the management of borderline personality disorder (BPD)]. clinicaltrials.gov/ct2/show/NCT00880919 (first received 10 April 2009).
Neacsiu 2017
- Neacsiu DA, Eberle WJ, Keng SL, Fang MC, Rosenthal ZM. Understanding borderline personality disorder across sociocultural groups: findings, issues, and future directions. Current Psychiatry Reviews 2017;13(3):188-223. [PMID: 10.2174/1573400513666170612122034] [DOI] [Google Scholar]
Newton‐Howes 2021
- Newton-Howes G, Cunningham R, Atkinson J. Personality disorder prevalence and correlates in a whole of nation dataset. Soc Psychiatry Psychiatr Epidemiol 2021;56(4):679-85. [DOI] [PubMed] [Google Scholar]
Ng 2016
- Ng FY, Bourke ME, Grenyer BF. Recovery from borderline personality disorder: a systematic review of the perspectives of consumers, clinicians, family and carers. PLoS One 2016;11(8):e0160515. [DOI: 10.1371/journal.pone.0160515] [PMCID: PMC4978398] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
NHMRC 2013
- National Health and Medical Research Council. Clinical Practice Guideline for the Management of Borderline Personality Disorder. Melbourne: National Health and Medical Research Council, 2013. [WEB PAGE: Available from www.nhmrc.gov.au/guidelines/publications/mh25] [Google Scholar]
NICE 2009
- National Institute for Health and Clinical Excellence. Borderline personality disorder: recognition and management. NICE clinical guideline 78; 28 January 2009. Available at www.nice.org.uk/guidance/cg78 (accessed 29 June 2021).
NICE 2018
- National Instiute for Health and Care Excellence (NICE). 2018 surveillance of personality disorders (NICE guidelines CG77 and CG78). Surveillance report. Published 19 July 2018. Available from www.nice.org.uk/guidance/cg78/resources/2018-surveillance-of-personality-disorders-nice-guidelines-cg77-and-cg78-4906490080/chapter/Surveillance-decision?tab=evidence. (accessed 05 December 2021). [PubMed]
Niesten 2016
- Niesten IJ, Karan E, Frankenburg FR, Fitzmaurice GM, Zanarini MC. Description and prediction of the income status of borderline patients over 10 years of prospective follow-up. Personality and Mental Health 2016;10(4):285-92. [DOI: 10.1002/pmh.1331] [PMID: ] [DOI] [PubMed] [Google Scholar]
Norman 2004
- Norman GR, Sloan JA, Wyrwich KW. The truly remarkable universality of half a standard deviation: confirmation through another look. Expert Review of Pharmacoeconomics & Outcomes Research 2004;4(5):581-5. [DOI: 10.1586/14737167.4.5.581] [PMID: ] [DOI] [PubMed] [Google Scholar]
Ose 2021
- Ose SO, Tveit T, Mehlum L. Non-suicidal self-injury (NSSI) in adult psychiatric outpatients - a nationwide study. Journal of Psychiatric Research 2021;133:1-9. [DOI: 10.1016/j.jpsychires.2020.11.031] [PMID: ] [DOI] [PubMed] [Google Scholar]
Osman 2001
- Osman A, Bagge CL, Gutierrez PM, Konick LC, Kopper BA, Barrios FX. The Suicidal Behaviors Questionnaire-Revised (SBQ-R): validation with clinical and nonclinical samples. Assessment 2001;8(4):443-54. [DOI: 10.1177/107319110100800409] [PMID: ] [DOI] [PubMed] [Google Scholar]
Ottosson 2002
- Ottosson HA, Ekselius LA, Grann MA, Kullgren G. Cross-system concordance of personality disorder diagnoses of DSM-IV and diagnostic criteria for research of ICD-10. Journal of Personality Disorders 2002;16(3):283-92. [DOI: 10.1521/pedi.16.3.283.22537] [PMID: ] [DOI] [PubMed] [Google Scholar]
Oud 2017
- Oud M, Arntz A, Hermens ML, Verhoef R, Kendall T. Specialized psychotherapies for adults with borderline personality disorder: A systematic review and meta-analysis. Aust N Z J Psychiatry 2018;52(10):949-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Overall 1962
- Overall JE, Gorham DR. The Brief Psychiatric Rating Scale. Psychological Reports 1962;10(3):799-812. [DOI: 10.2466/pr0.1962.10.3.799] [DOI] [Google Scholar]
Paris 2015
- Paris J. Why patients with severe personality disorders are overmedicated. Journal of Clinical Psychiatry 2015;76(4):e521. [DOI: 10.4088/JCP.14com09441] [PMID: ] [DOI] [PubMed] [Google Scholar]
Pascual 2021
- Pascual JC, Martín-Blanco A, Soler J. Twenty-Year Trends in the Psychopharmacological Treatment of Outpatients with Borderline Personality Disorder: A Cross-Sectional Naturalistic Study in Spain.. CNS Drugs 2021;35(9):1023-32. [DOI] [PubMed] [Google Scholar]
Paton 2015
- Paton C, Crawford MJ, Bhatti SF, Patel MX, Barnes TE. The use of psychotropic medication in patients with emotionally unstable personality disorder under the care of UK Mental Health Services. Journal of Clinical Psychiatry 2015;76(4):e512–8. [DOI: 10.4088/JCP.14m09228] [PMID: ] [DOI] [PubMed] [Google Scholar]
Pereira Ribeiro 2021
- Pereira Ribeiro, J, Sedoc Jørgensen, M, Storebø, OJ. Comment on: “Pharmacological Treatments for Borderline Personality Disorder: A Systematic Review and Meta‑Analysis”. CNS Drugs 2021 Nov;35:1333-1334. [https://doi.org/10.1007/s40263-021-00872-3] [DOI] [PubMed] [Google Scholar]
Perez 2007
- Perez V, Barrachina J, Soler J, Pascual JC, Campins MJ, Puigdemont D, et al. The clinical global impression scale for borderline personality disorder patients (CGI-BPD): a scale sensible to detect changes [Impresión clínica global para pacientes con trastorno límite de la personalidad (ICG-TLP): una escala sensible al cambio]. Actas Españolas de Psiquiatría 2007;35(4):229-35. [PMID: ] [PubMed] [Google Scholar]
RevMan Web 2020 [Computer program]
- Review Manager Web (RevMan Web). Version 3.2.1. The Cochrane Collaboration, 2020. Available at revman.cochrane.org.
Riffer 2019
- Riffer F, Farkas M, Streibl L, Kaiser E, Sprung M. Psychopharmacological treatment of patients with borderline personality disorder: comparing data from routine clinical care with recommended guidelines. International Journal of Psychiatry in Clinical Practice 2019;23(3):178-88. [DOI: 10.1080/13651501.2019.1576904] [PMID: ] [DOI] [PubMed] [Google Scholar]
Rossouw 2012
- Rossouw TI, Fonagy P. Mentalization-based treatment for self-harm in adolescents: a randomized controlled trial. Journal of the American Academy of Child and Adolescent Psychiatry 2012;51(12):1304-13. [DOI: 10.1016/j.jaac.2012.09.018] [PMID: ] [DOI] [PubMed] [Google Scholar]
Sansone 2003
- Sansone RA, Rytwinski D, Gaither GA. Borderline personality and psychotropic medication prescription in an outpatient psychiatry clinic. Comprehensive Psychiatry 2003;44(6):454-8. [DOI: 10.1016/S0010-440X(03)00147-0] [PMID: ] [DOI] [PubMed] [Google Scholar]
Savović 2012a
- Savović J, Jones H, Altman D, Harris R, Jüni P, Pildal J, et al. Influence of reported study design characteristics on intervention effect estimates from randomised controlled trials: combined analysis of meta-epidemiological studies. Health Technology Assessment 2012;16(35):1-82. [DOI: 10.3310/hta16350] [PMID: ] [DOI] [PubMed] [Google Scholar]
Savović 2012b
- Savović J, Jones HE, Altman DG, Harris RJ, Jüni P, Pildal J, et al. Influence of reported study design characteristics on intervention effect estimates from randomized, controlled trials. Annals of Internal Medicine 2012;157(6):429-38. [DOI: 10.7326/0003-4819-157-6-201209180-00537] [PMID: ] [DOI] [PubMed] [Google Scholar]
Schneider 2019
- Schneider F, Erhart M, Hewer W, Loeffler LA, Jacobi F. Mortality and medical comorbidity in the severely mentally ill. Deutsches Arzteblatt International 2019;116(23-24):405-11. [DOI: 10.3238/arztebl.2019.0405] [PMCID: PMC6683445] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Schulz 1995
- Schulz KF, Chalmers I, Hayes RJ, Altman DG. Empirical evidence of bias: dimensions of methodological quality associated with estimates of treatment effects in controlled trials. JAMA 1995;273(5):408-12. [DOI: 10.1001/jama.273.5.408] [PMID: ] [DOI] [PubMed] [Google Scholar]
Schulz 2008
- Schulz SC, Zanarini MC, Bateman A, Bohus M, Detke HC, Trzaskoma Q, et al. Olanzapine for the treatment of borderline personality disorder: variable dose, 12‐week, randomised double‐blind placebo‐controlled study. British Journal of Psychiatry 2008;193(6):485‐92. [DOI: 10.1192/bjp.bp.107.037903] [PMID: ] [DOI] [PubMed] [Google Scholar]
Shea 2017
- Shea Beverley J, Reeves Barnaby C, Wells George, Thuku Micere, Hamel Candyce, Moran Julian, Moher David, Tugwell Peter, Welch Vivian, Kristjansson Elizabeth. AMSTAR 2: a critical appraisal tool for systematic reviews that include randomised or non-randomised studies of healthcare interventions, or both. bmj 2017;358:j4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sher 2019
- Sher L, Rutter SB, New AS, Siever LJ, Hazlett EA. Gender differences and similarities in aggression, suicidal behaviour, and psychiatric comorbidity in borderline personality disorder. Acta Psychiatr Scand 2019;139(2):145-53. [DOI] [PubMed] [Google Scholar]
Silk 2015
- Silk KR. Management and effectiveness of psychopharmacology in emotionally unstable and borderline personality disorder. Journal of Clinical Psychiatry 2015;76(4):e524–5. [DOI: 10.4088/JCP.14com09534] [PMID: ] [DOI] [PubMed] [Google Scholar]
Simonsen 2019
- Simonsen S, Bateman A, Bohus M, Dalewijk HJ, Doering S, Kaera A, et al. European guidelines for personality disorders: past, present and future. Borderline Personality Disorder and Emotion Dysregulation 2019;6:9. [DOI: 10.1186/s40479-019-0106-3] [PMCID: PMC6530178] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Skodol 2002
- Skodol AE, Gunderson JG, McGlashan TH, Dyck IR, Stout RL, Bender DS, et al. Functional impairment in patients with schizotypal, borderline, avoidant, or obsessive-compulsive personality disorder. American Journal of Psychiatry 2002;159(2):276-83. [DOI: 10.1176/appi.ajp.159.2.276] [PMID: ] [DOI] [PubMed] [Google Scholar]
Soetmann 2008a
- Soeteman DI, Hakkaart-van Roijen L, Verheul R, Busschbach JJ. The economic burden of personality disorders in mental health care. Journal of Clinical Psychiatry 2008;69(2):259–65. [PMID: ] [DOI] [PubMed] [Google Scholar]
Soetmann 2008b
- Soeteman DI, Verheul R, Busschbach JJ. The burden of disease in personality disorders: diagnosis-specific quality of life. Journal of Personality Disorders 2008;22(3):259-68. [DOI: 10.1521/pedi.2008.22.3.259] [PMID: ] [DOI] [PubMed] [Google Scholar]
Soloff 2002
- Soloff PH, Lynch KG, Kelly TM. Childhood abuse as a risk factor for suicidal behavior in borderline personality disorder. Journal of Personality Disorders 2002;16(3):201-14. [DOI: 10.1521/pedi.16.3.201.22542] [PMID: ] [DOI] [PubMed] [Google Scholar]
Soloff 2012
- Soloff PH, Chiappetta L. Prospective predictors of suicidal behavior in borderline personality disorder at 6-year follow-up. Am J Psychiatry 2012;169(5):484-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
Spielberger 1988
- Spielberger CD. Manual for the State-Trait Anger Expression Inventory. Odessa (Fl): Psychological Assessment Resources, 1988. [Google Scholar]
Stepp 2012
- Stepp SD. Development of borderline personality disorder in adolescence and young adulthood: introduction to the special section. Journal of Abnormal Child Psychology 2012;40(1):1-5. [DOI: 10.1007/s10802-011-9594-3] [PMCID: PMC3865353] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sterne 2013
- Sterne JA. Why the Cochrane risk of bias tool should not include funding source as a standard item. Cochrane Database of Systematic Reviews 2013;12:ED000076. [DOI: 10.1002/14651858.ED000076] [PMID: ] [DOI] [PMC free article] [PubMed]
Sterne 2017
- Sterne JAC, Egger M, Moger D, Boutron I, editor(s). Chapter 10: Addressing reporting biases. In: Higgins JPT, Churchill R, Chandler J, Cumpston MS, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.2.0 (updated June 2017). Cochrane, 2017. Available from www.training.cochrane.org/handbook.
Stiglmayr 2005
- Stiglmayr CE, Grathwol T, Linehan MM, Ihorst G, Fahrenberg J, Bohus M. Aversive tension in patients with borderline personality disorder: a computer-based controlled field study. Acta Psychiatrica Scandinavica 2005;111(5):372-9. [DOI: 10.1111/j.1600-0447.2004.00466.x] [PMID: ] [DOI] [PubMed] [Google Scholar]
Stoffers 2015
- Stoffers JM, Lieb K. Pharmacotherapy for borderline personality disorder--current evidence and recent trends. Current Psychiatry Reports 2015;17(1):534. [DOI: 10.1007/s11920-014-0534-0] [PMID: ] [DOI] [PubMed] [Google Scholar]
Stoffers‐Winterling 2018
- Stoffers-Winterling JM, Storebø OJ, Völm BA, Mattivi JT, Nielsen SS, Kielsholm ML, et al. Pharmacological interventions for people with borderline personality disorder. Cochrane Database of Systematic Reviews 2018, Issue 2. Art. No: CD012956. [DOI: 10.1002/14651858.CD012956] [DOI] [PMC free article] [PubMed] [Google Scholar]
Stoffers‐Winterling 2020
- Stoffers-Winterling J, Storebø OJ, Lieb K. Pharmacotherapy for borderline personality disorder: an update of published, unpublished and ongoing studies. Current Psychiatry Reports 2020;22(8):37. [DOI: 10.1007/s11920-020-01164-1] [PMCID: PMC7275094] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Stoffers‐Winterling J 2018 [pers comm]
- Stoffers-Winterling J. Cochrane Collaboration review - RCT of escitalopram in BPD. Email to: E Coccaro 13 August 2018.
Stone 1993
- Stone MH. Long-term outcome in personality disorders. British Journal of Psychiatry 1993;162:299-313. [DOI: 10.1192/bjp.162.3.299] [PMID: ] [DOI] [PubMed] [Google Scholar]
Storebø 2014
- Storebø OJ, Simonsen E. Is ADHD an early stage in the development of borderline personality disorder? Nordic Journal of Psychiatry 2014;68(5):289-95. [DOI: 10.3109/08039488.2013.841992] [PMID: ] [DOI] [PubMed] [Google Scholar]
Storebø 2018
- Storebø¸ OJ, Stoffers-Winterling JM, Völlm BA, Kongerslev MT, Mattivi JT, Kielsholm ML, et al. Psychological therapies for people with borderline personality disorder. Cochrane Database of Systematic Reviews 2018, Issue 2. Art. No: CD012955. [DOI: 10.1002/14651858.CD012955] [DOI] [PMC free article] [PubMed] [Google Scholar]
Storebø 2020
- Storebø OJ, Stoffers-Winterling JM, Völm BA, Kongerslev MT, Mattivi JT, Jørgensen MS, et al. Psychological therapies for people with borderline personality disorder. Cochrane Database of Systematic Reviews 2020, Issue 5. Art. No: CD012955. [DOI: 10.1002/14651858.CD012955.pub2] [PMCID: PMC7199382] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ten Have 2016
- Ten Have M, Verheul R, Kaasenbrood A, Van Dorsselaer S, Tuithof M, Kleinjan M, et al. Prevalence rates of borderline personality disorder symptoms: a study based on the Netherlands Mental Health Survey and Incidence Study-2. BMC Psychiatry 2016;16:249. [DOI: 10.1186/s12888-016-0939-x] [PMCID: PMC4949762] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Thomas 2021
- Thomas J, Askie LM, Berlin JA, Elliott JH, Ghersi D, Simmonds M, Takwoingi Y, Tierney JF, Higgins HPT. Prospective approaches to accumulating evidence.. In: Higgins JPT, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA, editors(s). Cochrane 2021. Cochrane Handbook for Systematic Reviews of Interventions version 6.2. Vol. Chapter 22. Wiley. Awailable from: www.training.cochrane.org/handbook, Updated February 2021. [Google Scholar]
Thompson 2019
- Thompson KN, Cavelti M, Chanen AM. Psychotic symptoms in adolescents with borderline personality disorder features. Eur Child Adolesc Psychiatry 2019;28(7):985-92. [PMID: ] [DOI] [PubMed] [Google Scholar]
Thorlund 2009
- Thorlund K, Devereaux PJ, Wetterslev J, Guyatt G, Ioannidis JP, Thabane L, et al. Can trial sequential monitoring boundaries reduce spurious inferences from meta-analysis? International Journal of Epidemiology 2009;38(1):276-86. [DOI: 10.1093/ije/dyn179] [PMID: ] [DOI] [PubMed] [Google Scholar]
Tomko 2014
- Tomko RL, Trull TJ, Wood PK, Sher KJ. Characteristics of borderline personality disorder in a community sample: comorbidity, treatment utilization, and general functioning. Journal of Personality Disorders 2014;28(5):734-50. [DOI: 10.1521/pedi_2012_26_093] [PMCID: PMC3864176] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Torgersen 2001
- Torgersen S, Kringlen E, Cramer V. The prevalence of personality disorders in a community sample. Archives of General Psychiatry 2001;58(6):590–6. [DOI: 10.1001/archpsyc.58.6.590] [PMID: ] [DOI] [PubMed] [Google Scholar]
Torgersen 2012
- Torgersen S. Epidemiology. In: Widiger TA, editors(s). The Oxford Handbook of Personality Disorders. New York (NY): Oxford University Press, 2012:186-205. [DOI: 10.1093/oxfordhb/9780199735013.013.0009] [ISBN: 978-0-19-973501-3] [DOI] [Google Scholar]
Trull 2010
- Trull TJ, Jahng S, Tomko RL, Wood PK, Sher KJ. Revised NESARC personality disorder diagnoses: gender, prevalence, and comorbidity with substance dependence disorders. Journal of Personality Disorders 2010;24(4):412-26. [DOI: 10.1521/pedi.2010.24.4.412] [PMCID: PMC3771514] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tyrer 2005
- Tyrer P, Nur U, Crawford M, Karlsen S, McLean C, Rao B, et al. The Social Functioning Questionnaire: a rapid and robust measure of perceived functioning. International Journal of Social Psychiatry 2005;51(3):265-75. [PMID: ] [PubMed] [Google Scholar]
Tyrer 2015
- Tyrer P, Reed GM, Crawford MJ. Classification, assessment, prevalence, and effect of personality disorders. Lancet 2015;385(9969):717-26. [DOI: 10.1016/S0140-6736(14)61995-4] [DOI] [PubMed] [Google Scholar]
Van Asselt 2007
- Van Asselt AD, Dirksen CD, Arntz A, Severens JL. The cost of borderline personality disorder: societal cost of illness in BPD-patients. European Psychiatry 2007;22(6):354-61. [DOI: 10.1016/j.eurpsy.2007.04.001] [PMID: ] [DOI] [PubMed] [Google Scholar]
Vita 2011
- Vita A, De Peri L, Sacchetti E. Antipsychotics, antidepressants, anticonvulsants, and placebo on the symptom dimensions of borderline personality disorder: a meta-analysis of randomized controlled and open-label trials. Journal of Clinical Psychopharmacology 2011;31(5):613-24. [DOI: 10.1097/JCP.0b013e31822c1636] [PMID: ] [DOI] [PubMed] [Google Scholar]
Wetterslev 2008
- Wetterslev J, Thorlund K, Brok J, Gluud C. Trial sequential analysis may establish when firm evidence is reached in cumulative meta-analysis. Journal of Clinical Epidemiology 2008;61(1):64-75. [DOI: 10.1016/j.jclinepi.2007.03.013] [PMID: ] [DOI] [PubMed] [Google Scholar]
Wetterslev 2009
- Wetterslev J, Thorlund K, Brok J, Gluud C. Estimating required information size by quantifying diversity in random-effects model meta-analyses. BMC Medical Research Methodology 2009;9:86. [DOI: 10.1186/1471-2288-9-86.] [PMCID: PMC2809074] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
WHO 1993
- World Health Organisation. The ICD-10 Classification of Mental and Behavioral Disorders: Diagnostic Criteria for Research. Geneva (CH): World Health Organization, 1993. [Google Scholar]
WHO 2021
- World Health Organisation. ICD-11 for Mortality and Morbidity Statistics (Version: 05/2021). icd.who.int/browse11/l-m/en (accessed 6 June 2021).
WHO Collaborating Center for Drug Statistics.
- WHO Collaborating Center for Drug Statistics. ATC/DDD Index 2021. https://www.whocc.no/atc_ddd_index/ (accessed 03 December 2021).
Widiger 1993
- Widiger TA, Trull TJ. Borderline and Narcissistic Personality Disorders. In: PB Sutker, HE Adams, editors(s). Comprehensive Handbook of Psychopathology. New York, NY: Plenum, 1993:371-94. [Google Scholar]
Widiger 1998
- Widiger TA. Invited essay: Sex biasesin the diagnosis of personality disorders. Journal of Personality Disorders 1998;12(2):95-118. [DOI] [PubMed] [Google Scholar]
Wood 2008
- Wood L, Egger M, Gluud LL, Schulz KF, Jüni P, Altman DG, et al. Empirical evidence of bias in treatment effect estimates in controlled trials with different interventions and outcomes: meta-epidemiological study. BMJ 2008;336(7644):601-5. [DOI: 10.1136/bmj.39465.451748.AD] [PMCID: PMC2267990] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Yen 2021
- Yen S, Peters JR, Nishar S, Grilo CM, Sanislow CA, Shea MT, et al. Association of borderline personality disorder criteria with suicide attempts: findings from the Collaborative Longitudinal Study of Personality Disorders over 10 years of follow-up. JAMA Psychiatry 2021;78(2):187-94. [DOI: 10.1001/jamapsychiatry.2020.3598] [PMCID: PMC7675214] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Zanarini 1998
- Zanarini MC, Frankenburg FR, DeLuca CJ, Hennen J, Khera GS, Gunderson JG. The pain of being borderline: dysphoric states specific to borderline personality disorder. Harvard Review of Psychiatry 1998;6(4):201-7. [DOI: 10.3109/10673229809000330] [PMID: ] [DOI] [PubMed] [Google Scholar]
Zanarini 2004a
- Zanarini MC, Frankenburg FR, Hennen J, Silk KR. Mental health service utilization by borderline personality disorder patients and Axis II comparison subjects followed prospectively for 6 years. Journal of Clinical Psychiatry 2004;65(1):28-36. [DOI: 10.4088/jcp.v65n0105] [PMID: ] [DOI] [PubMed] [Google Scholar]
Zanarini 2004b
- Zanarini MC, Frankenburg FR, Hennen J, Reich DB, Silk KR. Axis I comorbidity in patients with borderline personality disorder: 6-year follow-up and prediction of time to remission. American Journal of Psychiatry 2004;161(11):2108-14. [DOI: 10.1176/appi.ajp.161.11.2108] [PMID: ] [DOI] [PubMed] [Google Scholar]
Zanarini 2005
- Zanarini MC, Frankenburg FR, Hennen J, Reich DB, Silk KR. The McLean Study of Adult Development (MSAD): overview and implications of the first six years of prospective follow-up. Journal of Personality Disorders 2005;19(5):505-23. [DOI: 10.1521/pedi.2005.19.5.505] [PMID: ] [DOI] [PubMed] [Google Scholar]
Zanarini 2010
- Zanarini MC, Frankenburg FR, Reich DB, Fitzmaurice G. The 10-year course of psychosocial functioning among patients with borderline personality disorder and axis II comparison subjects. Acta Psychiatrica Scandinavica 2010;122(2):103-9. [DOI: 10.1111/j.1600-0447.2010.01543.x] [PMCID: PMC3876887] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Zanarini 2012
- Zanarini MC, Frankenburg FR, Reich B, Fitzmaurice G. Attainment and stability of sustained symptomatic remission and recovery among patients with borderline personality disorder and axis II comparison subjects: a 16-year prospective follow-up study. American Journal of Psychiatry 2012;169(5):476-83. [DOI: 10.1176/appi.ajp.2011.11101550] [PMCID: PMC3509999] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Zanarini 2015
- Zanarini MC, Frankenburg FR, Reich DB, Harned AL, Fitzmaurice GM. Rates of psychotropic medication use reported by borderline patients and axis II comparison subjects over 16 years of prospective follow-up. Journal of Clinical Psychopharmacology 2015;35(1):63-7. [DOI: 10.1097/JCP.0000000000000232] [PMCID: PMC4276426] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Zanarini 2018
- Zanarini MC, Temes CM, Frankenburg FR, REich DB, Fitzmaurice GM. Description and prediction of time-to-attainment of excellent recovery for borderline patients followed prospectively for 20 years. Psychiatry Research 2018;262:40-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to other published versions of this review
Binks 2006
- Binks C, Fenton M, McCarthy L, Lee T, Adams CE, Duggan C. Pharmacological interventions for people with borderline personality disorder. Cochrane Database of Systematic Reviews 2006, Issue 1. Art. No: CD005653. [DOI: 10.1002/14651858.CD005653] [PMID: ] [DOI] [PubMed] [Google Scholar]
Stoffers 2010
- Stoffers J, Völlm BA, Rücker G, Timmer A, Huband N, Lieb K. Pharmacological interventions for borderline personality disorder. Cochrane Database of Systematic Reviews 2010, Issue 6. Art. No: CD005653. [DOI: 10.1002/14651858.CD005653.pub2] [PMCID: PMC4169794] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]