

Keywords: cell death signaling, granuloma, host-directed therapy, immune cells, inflammation
Abstract
Mycobacterium tuberculosis (Mtb) is the pathogen that causes tuberculosis (TB), a leading infectious disease of humans worldwide. One of the main histopathological hallmarks of TB is the formation of granulomas comprised of elaborately organized aggregates of immune cells containing the pathogen. Dissemination of Mtb from infected cells in the granulomas due to host and mycobacterial factors induces multiple cell death modalities in infected cells. Based on molecular mechanism, morphological characteristics, and signal dependency, there are two main categories of cell death: programmed and nonprogrammed. Programmed cell death (PCD), such as apoptosis and autophagy, is associated with a protective response to Mtb by keeping the bacteria encased within dead macrophages that can be readily phagocytosed by arriving in uninfected or neighboring cells. In contrast, non-PCD necrotic cell death favors the pathogen, resulting in bacterial release into the extracellular environment. Multiple types of cell death in the PCD category, including pyroptosis, necroptosis, ferroptosis, ETosis, parthanatos, and PANoptosis, may be involved in Mtb infection. Since PCD pathways are essential for host immunity to Mtb, therapeutic compounds targeting cell death signaling pathways have been experimentally tested for TB treatment. This review summarizes different modalities of Mtb-mediated host cell deaths, the molecular mechanisms underpinning host cell death during Mtb infection, and its potential implications for host immunity. In addition, targeting host cell death pathways as potential therapeutic and preventive approaches against Mtb infection is also discussed.
INTRODUCTION
Mycobacterium tuberculosis (Mtb) is the etiological agent of tuberculosis (TB), which remains a leading cause of human infectious death worldwide, taking the lives of 1.5 million people in 2021 (1). One-fourth of the world’s population, or approximately two billion people, is estimated to be infected with Mtb, with more than 10 million developing active TB each year (1, 2). Mtb is a highly adapted intracellular pathogen that has evolved multiple mechanisms to manipulate host immune responses. Host cell death pathways are one of the main attributes of host-pathogen interaction in Mtb infection.
Cell death is fundamental to the interaction between Mtb and the phagocytes it infects, including macrophages (MΦ) and other types of myeloid cells, such as dendritic cells (DCs) and neutrophils. Cell death was originally divided into three major groups: type 1 (apoptosis), type II (autophagy), and type III (necrosis) (3). In recent years, several novel types of cell death have been characterized regarding their corresponding stimuli, molecular mechanisms, and morphologies. Some of these types of cell death share overlapping, although not identical, signal pathways and are not categorized into the type I–III groups. The Nomenclature Committee on Cell Death (NCCD) devised guidelines for cell death from morphological, biochemical, and functional perspectives in 2018 (4). Tang et al. (5) provide detailed historical origins and concepts used throughout cell death research development and a comprehensive summary of the molecular machinery involved in regulated cell death (RCD). Furthermore, Yan et al. (6) summarize different key features of different cell death modalities and categorize them into programmed cell death (PCD) and nonprogrammed cell death based on signal dependency.
Several studies have shown multiple and distinct types of cell deaths occurring during Mtb infection. Mtb can manipulate the kinetics and mode of cell death in infected macrophages (7, 8) to evade host immunity, shielding the bacteria from destruction by the host immune systems while allowing the bacteria to disseminate (9–11). Mtb infection of macrophages can result in multiple outcomes, including apoptosis, necrosis, or survival/maintenance of infected cells (8). The role of Mtb virulence factors in the induction of different modes of cell death remains controversial (12–14). Although macrophage apoptosis is required for antigen presentation to T cells and consequent elimination of the pathogen, necrosis can lead to a slow generation of host adaptive responses that result in Mtb dissemination and further infection of alveolar macrophages (14–16). Hence, a detailed understanding of how this successful intracellular pathogen regulates host cell death to promote its survival is crucial for developing alternative therapeutic strategies to target host proteins involved in cell death pathways (17). This article summarizes cell death modalities during in vitro and in vivo Mtb infection, characterizing the implications and consequences for host immunity, and suggests potential therapeutic opportunities and preventive approaches to combat TB.
DIFFERENT MODALITIES OF HOST CELL DEATH IN M. TUBERCULOSIS INFECTION
The interaction between Mtb and macrophages plays a crucial role in TB pathogenesis (18). Mtb grows and replicates within the infected host cells as an intracellular pathogen, mainly within the macrophages (19). Following the establishment of pulmonary infection, the transmission of the bacilli to new hosts requires a transition to the extracellular space and host cell death. Numerous studies have identified many different forms of cell death in the Mtb infection scenario, both in vitro and in vivo. A review on the topic has also listed spectrums of death modes linked to TB, including extrinsic, intrinsic, perforin/granzyme-mediated apoptosis, and several types of necrosis (20). This review discusses recent developments in multiple cell death modalities resulting directly from Mtb infection and the consequences to the host cells. It will also identify mycobacterial and host factors as potential targets for future interventions against TB.
Apoptosis
Apoptosis is an energy-dependent cascade of a highly regulated process for cellular deconstruction that encloses the cytoplasmic contents of dying cells within membrane-bound vesicles called apoptotic bodies (21). These apoptotic bodies express “eat me” signals and therefore are recognized and engulfed by the phagocytic cells via multiple specific surface receptors in a process called efferocytosis, which removes apoptotic/dying cells without causing inflammatory responses (22). Upon activation by physiological and pathological conditions, apoptosis occurs in two major pathways: the extrinsic/ligand and granzyme B-mediated pathway and the intrinsic/mitochondrial pathway (21). The critical players in these apoptotic pathways are caspases (a family of cysteine-dependent aspartate-directed proteases constitutively expressed as zymogens), adaptor proteins, tumor necrosis factor (TNF) receptor (TNF-R) family, and B cell lymphoma-2 (Bcl-2) family proteins (23). Cells that undergo apoptosis exhibit several biochemical modifications, such as protein cleavage and cross linking, DNA breakdown, and phagocytic recognition, resulting in distinctive structural pathology and morphological features (24). Another biochemical characteristic of apoptotic cells is the expression of cell surface markers leading to early phagocytic recognition by neighboring cells, allowing efficient phagocytosis of debris without compromising the surrounding tissue (21).
Host cell apoptosis during M. tuberculosis infection.
Host apoptosis signaling occurs through two major pathways, the intrinsic and the extrinsic pathways, depending on the source (intracellular vs. extracellular) of the activator, as reviewed extensively elsewhere before (22, 25–28). In this review, we discuss the factors underpinning the alterations in apoptotic cascades upon Mtb infection. These include host apoptosis-associated proteins induced following mycobacterial infection (Table 1) and mycobacterial proteins/factors that modulate host cell apoptosis pathways (Table 2).
Table 1.
Host factors modulated by Mtb in various cell death modalities
| Cell Death Pathways | Host Factor | Remarks | Cell Type | Reference |
|---|---|---|---|---|
| Apoptosis | Bcl-2 (B-cell lymphoma-2) | Integral outer mitochondrial membrane protein that blocks apoptosis; upregulated over 2.5-fold following 48 h infection of Mtb | Human alveolar MΦ, alveolar epithelial cells, murine models | (29, 30) |
| Bax (Bcl-2 associated X) | Apoptosis regulator protein; downregulated > 2-fold following Mtb infection | Human alveolar MΦ, alveolar epithelial cells, murine models | (29, 30) | |
| Bad (Bcl-2 agonist of cell death) | Promotes apoptosis by forming heterodimers with Bcl-xl and Bcl-2; downregulated > 2-fold following Mtb infection | Human alveolar MΦ and alveolar epithelial cells | (29) | |
| Bim (Bcl-2 interacting mediator) | Proapoptotic Bcl-2 family protein; upregulated after 24, 48, and 72 h after Mtb infection | Mouse MΦ J774 | (31) | |
| Bcl-w (Bcl-2l2) | Acts as anti- and proapoptotic regulator; upregulated 1.8–2-fold following Mtb infection | Human alveolar MΦ | (32) | |
| Calreticulin | Modulates Mtb survival through endoplasmic reticulum stress-mediated apoptosis; upregulated following Mtb infection | THP-1 MΦ | (33) | |
| Elongation factor 1-alpha (eEF1α) | Modulates TNF-α dependent apoptosis; down-regulated > 2-fold following pathogenic Mtb infection | Human alveolar MΦ | (32) | |
| Interferon gamma (IFN-γ) | Activates nitric oxide-induced apoptosis; upregulated following mycobacterial infection | Mouse MΦ | (34) | |
| Tumor necrosis factor-alpha (TNF-α) | Modulates apoptosis and other cell death pathways; expression is inversely proportional to mycobacterial virulence | BALB/c mouse MΦ | (35) | |
| Myeloid leukemia-1 (Mcl-1) | Provide resistance to apoptosis; upregulated as early as 4 h and peaked (∼6-fold) at 24 h after infection | THP-1 MΦ | (36) | |
| Mitochondrial mortalin | Modulates macrophage apoptosis signaling by interaction with mitochondrial mortalin; upregulated during Mtb infection. | THP-1 and RAW 264.7 MΦ | (37) | |
| Nuclear factor kappa-B (NFκB) | Regulates apoptosis following Mtb- antigen stimulation; upregulated during Mtb infection. | HeLa cells | (38) | |
| RAD23 homolog B | Modulates TNF-α dependent apoptosis; downregulated > 2-fold following pathogenic Mtb infection. | Human alveolar MΦ | (32) | |
| Retinoblastoma (Rb) | Regulates apoptosis; upregulated over 2.5-fold after 48 h of Mtb infection | Human alveolar MΦ and alveolar epithelial cells | (29) | |
| Superoxide dismutase-2 (SOD2) | Modulates TNF-α dependent apoptosis; downregulated 1.8 to 2.0-fold after 48 h post-Mtb infection | Human alveolar MΦ | (32) | |
| Toll-like receptor 2 (TLR-2) | Induces macrophage death; upregulated in Mtb infection | Mouse MΦ | (39) | |
| Lymphocyte activation gene-3 (LAG-3) | Immunomodulator that affects host cell apoptosis; induced nearly 100-fold following Mtb infection. | Macaque blood derived MΦ/T cell co-culture | (40) | |
| Kruppel-type zinc finger (ZK1) protein | Modulates TNF-α dependent apoptosis; downregulated > 2-fold following Mtb infection. | Human alveolar MΦ | (32) | |
| Gasdermin-D (GSDMD) | Mediates apoptosis but not IL-1β secretion in Mtb-infected macrophages | Human MΦ, PBMCs | (41) | |
| Pyroptosis | NOD-, LRR-. and pyrin domain-containing protein 3 (NLRP3) | Activation of the NLRP3 inflammasome to modulate inflammatory disease pathogenicity | Immune cells (T cells) | (42) |
| Caspase-1 (CASP-1) | CASP-1 activation leads to chromosomal DNA damage through an endonuclease | H9C2 cardiomyocytes | (43) | |
| Necroptosis | Receptor-interacting serine/threonine-protein kinase 3 (RIPK3) | RIPK3 complex recruits and phosphorylates MLKL to form the necrosome | Mouse embryonic fibroblasts (MEFs) | (44, 45) |
| Mixed lineage kinase domain-like (MLKL) | Initiates necroptotic cell death by allowing ion influx, cell swelling, and membrane lysis followed by the uncontrollable release of intracellular material | Mouse embryonic fibroblasts (MEFs) | (45, 46) |
Several host factors/proteins regulated following Mtb infection in different cell death modes and their fold expression are listed. MΦ, macrophages; Mtb, Mycobacterium tuberculosis; NOD, nucleotide binding oligomerization domain; PBMCs, peripheral blood mononuclear cells.
Table 2.
List of Mtb factors associated with host cell death
| Actions on host | Mtb Protein/Factor | Remarks | Cell Type | Reference |
|---|---|---|---|---|
| Apoptosis inducers | Lipomannan (LM); lipoarabinomannan (LAM); mannosylated lipoarabinomannan (manLAM) | Induces apoptosis and IL-12 secretion | Human THP-1 | (47) |
| Glycolipoprotein (p19; LpqH) | Induces apoptosis through TLR-2 and mitochondrial apoptosis-inducing factor | THP-1/CHO expressing TLR2 | (48) | |
| Mycolactone | Causes apoptosis in guinea pig ulcers and tissue culture cells | macrophage cells J774A.1/guinea pig model | (49) | |
| Periplasmic phosphate-specific transporter; 38-kDa lipoprotein (Pst-S1) | Causes TNF-α and FasL-mediated caspase-dependent apoptosis and upregulation of cell-death receptors TNFR1, TNFR2, and Fas and TLR2 | THP-1 macrophages | (50) | |
| Early Secreting Antigen Target-6kDa (ESAT-6)-family (esxA, esxT and esxL) | Induces apoptosis through activation of caspase gene expression | THP-1 macrophages | (51) | |
| PE-PGRS33 | Induces apoptosis through TLR2 pathway | RAW 264.7 and HEK293 cells | (52) | |
| Rv0183 | Induces inflammation and apoptosis in RAW macrophages | Macrophage RAW264.7 cells | (53) | |
| Rv0901 | Overexpression induces macrophage apoptosis | THP-1 macrophages | (54) | |
| Rv3499c (Mce4A) | Induces apoptosis mediated by TNF-α | THP-1 macrophages | (55) | |
| PPE37 | C-terminal domain induces apoptosis | THP-1 macrophages | (56) | |
| PE9 (Rv1088)/PE10 (Rv1089) | Induces apoptosis by TLR4 pathway | THP-1 macrophages | (57) | |
| PE_PGRS41 | Enhances intracellular survival | THP-1 macrophages | (58) | |
| PE13/Rv1195 | Increases intracellular survival by activating the p38-ERK pathway | THP-1 macrophages | (59) | |
| PE_PGRS18 | Enhances intracellular survival | THP-1 macrophages | (60) | |
| Apoptosis inhibitors | NADH-quinone oxidoreductase subunit G (NuoG) | Inhibits apoptosis of infected host cells | BALB/c or SCID/Ncr (BALB/c mice) | (61) |
| Protein Phosphatase 2 Phosphatase Activator (PtpA ) | Inhibits phagosome acidification and maturation. Interacts with tripartite motif containing 27 (TRIM27) and suppresses apoptosis |
THP-1 macrophages | (62) | |
| DUF732 domain-containing protein kinase (Rv3354) | Targets the metalloprotease resulting in suppression of apoptosis; destabilizes regulatory cullin-RING ubiquitin E3 enzymatic activity; inhibits constitutive photomorphogenesis 9 (cop9) signaling | THP-1 macrophages | (63) | |
| MPT64 (Rv1980c) | Inhibits apoptosis of macrophages through the NF-kB-miRNA21-Bcl-2 pathway | RAW264.7 macrophages | (64) | |
| Protein Kinase E (pknE) | Mutation increases the expression of genes involved in apoptosis | THP-1 macrophages | (65) | |
| Rv2456c | Inhibits apoptosis through NF-κB and extends the survival of Mtb. | Human epithelial cells (A549) & THP-1 macrophages | (66) | |
| GroEL2 (Rv0440) | Blocks macrophage apoptosis via interaction with mitochondrial mortalin | THP-1 and RAW 264.7 cell lines | (37) | |
| SecA2 (Rv1821) | Controls(?) secretion of a specific subset of proteins and blocks macrophage apoptosis | C57BL/6 mice | (67) | |
| Oligopeptide transporter subunit (OppD) | Transports Mtb and induces apoptosis | THP-1 macrophages | (68) | |
| Rv3654c/Rv3655c | Interacts with AL017 and blocks the extrinsic pathway | U937 monocyte-derived macrophage | (69) | |
| Necrosis inducers | ESAT-6 | Results in necrosis of human macrophages mediated by pyrin domain-containing protein-3 (NLRP3) | THP-1 macrophages | (70) |
| Channel protein with necrosis-inducing toxin (CpnT /Rv3903c) | Induce necrosis of human macrophages through its secreted C-terminal domain known as tuberculosis necrotizing toxin (TNT) | RAW 264.7 macrophages | (71) | |
| THP-1 macrophages | ||||
| PE25/PPE41 | Recombinant form induces necrosis | RAW 264.7 macrophages | (72) | |
| Rv2626c | High levels of necrosis were observed in THP-1 macrophages infected with MtbH37Rv over-expressing Rv2626c | THP-1 macrophages | (73) | |
| PPE68 | Inactivation decreases necrosis | THP-1 macrophages | (73) | |
| PPE31 | Shortens the life of macrophages infected by Mtb over-expressing PPE31 and promotes mycobacterial survival | RAW264.7 macrophages | (74) | |
| Pyroptosis inhibitors | Rv3364c | Binds to the cathepsin G, inhibiting its enzymatic activity and the downstream signal caspase-1. | Human monocytic cell line U937-derived macrophages | (75) |
| Inhibits host pyroptosis | ||||
| Pyroptosis inducers | Rv1579c/EST12 | Binds to RACK1 to recruit the deubiquitinase UCHL5 leading to NLRP3 inflammasome activation that promotes pyroptosis | Human monocytic THP-1 and mouse BMDMs | (76) |
| Zinc metalloprotease 1 (Zmp-1/Rv0198c) | Deletion triggers activation of caspase-1/IL-1β inflammasome leading to pyroptosis signaling and improved mycobacterial clearance | J774A and RAW 264.7 macrophages | (77) | |
| Autophagy inhibitors | Enhanced intracellular survival (Eis) | Infection with Mtb-Δeis elevates ROS generation and renders the cells highly sensitive to autophagy activation | THP-1 macrophages Mouse BMDM |
(78) |
| ESAT-6 | Transient expression of the ESAT-6/CFP-10 fusion inhibits autophagosome formation by decreasing expression levels of ATG genes | RAW264.7 macrophages | (79) | |
| PtpA | Interacts with active-phosphorylated vacuolar protein sorting 33B (VPS33B) and inhibits phagosome-lysosome fusion, impacting autophagy | THP-1 | (80) | |
| PknG | Inhibits phagosome-lysosome fusion, impacting autophagy | J774 macrophage cells | (81) | |
| ETosis | ESAT-6 | Induces a high level of intracellular Ca2+ leading to the formation of NETs characterized by extracellular DNA and myeloperoxidase | Neutrophils from healthy volunteers | (82) |
Mycobacterium tuberculosis (Mtb) modulates several types of host cell death upon infection. A list of different Mtb factors and their regulatory role in host cell death are tabulated. TLR2, Toll-like receptor 2; TNFR, tumor necrosis factor-alpha receptor.
Intrinsic apoptosis is engaged by cells that are deprived of growth factors, damaged, or infected. These diverse stimuli can tip the balance between different groups of the Bcl‐2 (B‐cell lymphoma 2) proteins leading to the activation of cell death (83). The Bcl‐2 protein family can be divided into three subfamilies: the antiapoptotic Bcl‐2 proteins, the proapoptotic BH3‐only (BH: Bcl‐2 homology) proteins, and the death effectors Bax (Bcl‐2‐associated X protein), Bak (Bcl‐2 homologous antagonist/killer), and Bok (Bcl‐2‐related ovarian killer) (84). The BH3‐only members can inhibit the anti‐apoptotic Bcl‐2 proteins, or, in some cases, directly engage Bax and Bak (e.g., Bim) (85, 86). Tipping the equilibrium in favor of proapoptotic Bcl‐2 proteins leads to the activation of Bax and Bak and results in mitochondrial outer membrane permeabilization (MOMP) (87). Some BH3‐only proteins are regulated by transcriptional regulation (the p-53 upregulated modulator of apoptosis; PUMA, is regulated by p53) (88) or by post‐translational modifications (Bim, Bid) (89). Bok, which can constitutively induce MOMP, is regulated differently by proteasomal degradation pathways (90, 91). MOMP causes the release of the key mediators of intrinsic apoptosis, cytochrome c (92), an endogenous inhibitor of apoptosis (IAP) antagonist, and SMAC/Diablo (second mitochondria-derived activator of caspases/direct IAP binding protein) (93, 94). Cytochrome c‐bound apoptotic protease activating factor 1 (Apaf1) recruits initiator caspase, caspase‐9, to form apoptosome (a multimeric scaffold protein complex) for the activation of the executioner caspases, caspase‐3 and ‐7 (95). Caspases‐3, ‐7, and ‐9 can be blocked by the major endogenous caspase inhibitor, XIAP (X chromosome‐linked IAP) (96). SMAC can antagonize XIAP and other IAPs, allowing full caspase activation and apoptosis initiation (84, 93). Caspases cleave various cellular proteins to induce characteristic changes of apoptotic death (cellular and nuclear fragmentation, DNA laddering, etc.). For example, an inhibitor of caspase‐activated DNase (ICAD) cleavage leads to the activation of caspase‐activated DNase (CAD) that induces genome fragmentation (84).
Extrinsic apoptosis is triggered by TNF family ligand‐receptor interactions, most prominently by TNF family ligands: TNF, FasL, TRAIL, and TL1A. The receptor complexes either recruit Fas‐associated protein with death domain (FADD) or TNFRSF1A‐associated via death domain (TRADD) to the oligomerized complex (84, 97). FasL‐mediated signaling will be used to describe extrinsic apoptotic signaling, and TNF signaling will be described for necroptotic signaling. FasL binds to its transmembrane receptor Fas, which recruits FADD via death domain (DD) interactions (98). The proximity of multiple caspase‐8 molecules induces the transactivation by proteolytic cleavage, resulting in the p18 and p10 fragments, which activate caspase‐3 and caspase‐7 (type I apoptosis) (84, 99). Insufficient activation of caspase‐3 leads to type II apoptosis in which caspase‐8 cleaves the BH3‐only protein Bid (BH3 interacting domain death agonist) to generate its activated form: truncated Bid (tB499) (100). tB499 stimulates the intrinsic apoptotic pathway by binding directly to Bax/Bak, inducing MOMP (type II apoptosis) (101).
Cytotoxic lymphoid cells (predominantly NK cells and cytotoxic T cells) can induce apoptosis via death receptor ligands (extrinsic apoptosis) or via the granzyme/perforin system (102, 103). After recognizing transformed or infected cells, cytotoxic cells release secretory granules containing perforin and granzyme B. These secreted factors are taken up by endocytosis and released to the cytosol by the perforin‐dependent or ‐independent pathways (104). Once released to the cytosol, granzyme B cleaves caspases and Bid (105), activating the apoptotic pathways described earlier. However, human granzyme B can also directly cleave ICAD, a known caspase‐3 target, to induce DNA fragmentation, thereby evading the need for caspases (106).
Following the encounter with Mtb, the host releases its antimicrobial defenses (Fig. 1). During this host-pathogen interaction, several host proteins/factors are induced due to either host defense mechanisms or bacterial manipulation (7). The apoptosis signaling is mainly regulated by multiple host apoptotic factors including receptor proteins [Toll-like receptor, TLRs (107) and Fas/APO-1 (apoptosis antigen 1) CD95 receptor (108, 109)], effector proteins [caspases (51), TNF-α (110), IFN-γ (111), IL-10 (112, 113), and CCL20 (chemokine C-C motif ligand 20) (114)], Bcl-2 family proteins (29, 115), as well as transcription factors (NFκB) (116).
Figure 1.

Intrinsic and extrinsic host apoptosis pathways in Mycobacterium tuberculosis infection. Apoptosis is induced by the host’s innate immune response against infection by either intrinsic or extrinsic pathways. A range of mycobacterial proteins inhibits host-induced apoptosis during successful infection. Pathogen-associated molecular patterns (PAMPs) are recognized by host signaling proteins that drive the cell toward apoptosis. Specific bacterial factors cause mitochondrial membrane depolarization and lead to the release of mitochondrial factors that trigger apoptosis by the intrinsic pathway. Various stimuli can induce intrinsic apoptosis by shifting the equilibrium of prosurvival B-cell lymphoma-2 (Bcl-2) and Bcl‐2 homology 3 (BH3) proteins. Extrinsic apoptosis can be induced by the binding of a select group of tumor necrosis factor (TNF) family ligands to their receptors leading to death-inducing signaling complex (DISC) formation by recruitment of adapter Fas‐associated protein with death domain (FADD)/TNFRSF1A‐associated via death domain (TRADD) and caspase-8. Caspase-8 auto-processes itself (cCasp8—cleaved/activated caspase-8) and can directly activate caspase-3 or cleave Bid to generate tBid and triggers intrinsic apoptosis. Apaf-1, apoptotic peptidase activating factor 1; Bax, B-cell leukemia/lymphoma-2 associated-X, Bak, B-cell leukemia/lymphoma-2 antagonist/killer; BID, BH3 interacting domain; Bok, B-cell leukemia/lymphoma-2 associated related ovarian killer; CAD, intracellular caspase-activated deoxyribonuclease; CARD, caspase recruitment domain; Casp, caspase; CytC, cytochrome C; FAD, flavin adenine dinucleotide; FASL, FAS ligand; iFAS, FS-7-associated surface antigen; LAM, lipoarabinomannan; SMAC, second mitochondria-derived activator of caspases; tBID, truncated BID; TNFα, tumor necrosis factor-alpha; TNFR, tumor necrosis factor-alpha receptor; XIAP, X-linked inhibitor of apoptosis protein. Figure created with images from smart.servier.com.
Mtb proteins are involved in both inhibition and induction of host cell apoptosis signaling, depending on the stage of infection. The following sections discuss Mtb proteins/factors involved in host apoptotic pathways. Induction of apoptosis, which occurs following mycobacterial infection, can be considered either as a host defense mechanism to eliminate the bacteria or as a pathogen-induced response to evade the host defense mechanism, which facilitates bacterial survival (117). However, the stage at which host apoptosis is induced influences the progression of the infection (7).
M. tuberculosis factors promote host apoptosis.
Several Mtb factors have been shown to induce host cell apoptosis (Table 2), including secreted proteins [19-kDa protein/LpqH (118) and ESAT-1/RD1 (56)] and surface proteins [PE_PGRS33/Rv1818c (52) and 38-kDa lipoprotein/PstS-1 (68)]. 19-kDa secreted protein is a cell wall-associated and secreted virulence factor. Recombinant p19 protein induces apoptosis in PMA (phorbal 12-myristate 13-acetate)-differentiated THP-1 macrophages in a TLR-dependent pathway (118). Recombinant p19 also appeared to induce both extrinsic and intrinsic pathways of host cell apoptosis (119). In addition, this secreted protein was reported to induce IL-1β in monocyte-derived macrophages infected with knockout (KO) Mtb strain (120).
Early secreted antigenic target of 6 kDa (ESAT-6) (Rv3875) is a well-characterized secreted virulent protein within the RD1 region of Mtb (121). Expression of this antigen is known to be induced in macrophages following Mtb infection, which inhibits T cell immune responses (51, 122). ESAT-6, as an early secretory antigenic target EsxA and phthioceroldimycocerosates (DIM), was shown to cause phagosomal rupture leading to cell death induced by virulent mycobacteria inside host phagocytes (123). ESAT-6 is known to intercept the host defenses at different stages of infection, including autophagosome formation (124), disruption of TLR signaling (125), and inhibition of IFN-γ (126). It has also been shown that ESAT-6 interferes with the NF-κB pathway and autophagy (125, 127). ESAT-6 stimulated A549 cells (human epithelial cells) are known to induce the endoplasmic reticulum (ER) stress response, causing an increase in intracellular Ca2+ concentration, which results in ROS accumulation, and therefore inducing the onset of ER stress-induced apoptosis (128). However, it is known that Mycobacterium bovis BCG (Bacillus Calmette-Guerin), with no virulent factors in its RD1 region, can induce greater host cell apoptotic signaling compared with Mtb (129); this leads to an unclear role of ESAT-6 in host cell apoptosis.
PE_PGRS33 is a surface antigen protein of pathogenic mycobacteria, including Mtb, and one of the most studied PE_PGRS proteins (130). This surface protein is known to induce apoptosis in macrophages (52, 131); it interacts with Toll-like receptor 2 (TLR2), leading to inflammatory chemokine and cytokine secretion, which is key in the immunopathogenesis of Mtb infection (130). Another Mtb surface protein involved in apoptosis is PstS-1, also known as 38-kDa lipoprotein. PstS-1 is a mannosylated glycolipoprotein of Mtb that binds to the mannose receptor in macrophages, promoting phagocytosis (132). This cell wall-associated protein acts as an immunodominant antigen that induces macrophage apoptosis in a caspase-dependent manner involving TNF-α and FasL (50). The same study also shows that PstS-1 can induce both intrinsic and extrinsic pathways in apoptotic death of human monocyte-derived macrophages (MDM) by TLR2 activation following Mtb infection (50). Since apoptosis most likely results in bacterial killing, the host cell signaling through the PstS-1 pathway might be detrimental to the intracellular survival of Mtb.
M. tuberculosis interferes with host cell apoptosis signaling.
A striking contradiction exists in the literature regarding the role of apoptosis in TB disease; in some reports, virulent Mtb is described to induce apoptotic host cell death, whereas, in others, the pathogen is thought to inhibit this process (133). These differences can partly be due to the complexity of the experimental system, which relies on the interaction of viable bacteria and host cells. In the early stage of infection, apoptosis inhibition is crucial for the bacteria to survive and multiply within the host cells. Mtb has adopted several mechanisms to evade the host-protective responses (Table 1). Multiple host-induced apoptosis triggers culminate in host cell death resulting in pathogen clearance. These triggers include host-induced-reactive oxygen species (ROS), leading to oxidative stress, cytokines, damage sensors, apoptotic mediators, and cell surface receptors (7). Different strains of mycobacteria have varied abilities to modulate apoptosis resulting in different scenarios of disease outcomes. In addition, host susceptibility factors that regulate host-pathogen interactions determine the clinical outcome of the disease (134–136). Mycobacteria-induced mechanisms that inhibit apoptosis of host cells include those that interfere with pathways of ROS/RNS generation, induction of anti-apoptotic genes (137, 138), TNF-α production (139), and eicosanoid regulation (8).
Several proteins secreted by Mtb, such as superoxide dismutase, heme-dependent catalase-peroxidase KatG, serine/threonine protein kinase PknE, type I NADH dehydrogenase NuoG, Rv3654c, and Rv3655c putative proteins are known to inhibit macrophage apoptosis (69, 140). These proteins regulate the production of NO and proinflammatory cytokines to interfere with TLR mechanisms leading to the inhibition of TNF-α-induced apoptosis (69). Various secreted virulence factors of Mtb interfere with the caspase (86), JAK2/STAT1 (141), TNF-α (142), and Bcl-2 pathways to suppress macrophage apoptosis and increase the survival rate of pathogens (143, 144).
Mtb infection can also inhibit host apoptosis through the blockage of eicosanoids (8). Eicosanoids are locally acting bioactive signaling lipids derived from arachidonic acid and related polyunsaturated fatty acids categorized as prostaglandins (PGE) that regulate a diverse set of homeostatic and inflammatory processes (145). Mtb infection leads to the formation of foamy macrophages with accumulated lipid droplets (146). The lipid droplets support the formation of eicosanoids and contribute toward host defense against Mtb. Thus, Mtb-mediated modulation of eicosanoid production determines the infected macrophage cell death signaling, resulting in a substantial impact on the outcome of infection and skewing the host cell death mode from apoptosis to necrosis (8).
In summary, both intrinsic and extrinsic pathways of host cell apoptosis can be triggered and/or suppressed at various stages of Mtb infection. When apoptosis occurs as an early response to the infection, it contributes primarily to the pro-host response that eliminates the bacterial pathogen. However, apoptosis that occurs at a later stage of infection gravitates toward pro-pathogenic mode, facilitating bacterial survival and dissemination, and averting proinflammatory host immune responses. Apoptosis in Mtb infection has been extensively studied; for more comprehensive reviews on apoptosis signaling, readers may refer to these articles by Briken and Miller (133), Behar et al. (147), Xu et al. (148), Lam et al. (149), Galluzzi et al. (4), Mohareer et al. (7), and Zhai et al. (143).
Necrosis
The definition and concept of necrosis, which was previously viewed as a nonprogrammed and proinflammatory mode of cell death, has expanded over time (150). Traditional and classical necrosis was classified as accidental cell death (ACD) according to NCCD-2018, which defined ACD as a “virtually instantaneous and uncontrollable form of cell death corresponding to the physical disassembly of the plasma membrane caused by extreme physical, chemical, or mechanical cues” (4). Necrosis has also been later categorized into “mitochondrial permeability transition-driven necrosis” (MPT-necrosis) and programmed necrosis, also known as necroptosis (4, 151). Programmed necrosis is tightly interconnected with innate inflammatory responses and is active during infections and tissue injury and repair (152).
Recently, there is a paradigm shift in our understanding of cellular necrosis. Contrary to the earlier conception that necrosis is accidental and unregulated, recent evidence suggests that necrosis may be regulated (4, 6, 153). The ability of specific receptors, such as tumor necrosis factor-α (TNF-α), to trigger necrosis suggests that this is a regulated event with a structured signaling pathway (4, 154). Other signals, including physical and chemical stressors such as ischemia-reperfusion (IR) injury, oxidative stress, calcium (Ca2+) overload, extensive DNA damage, and irradiation, can induce necrotic cell death (155). ROS generation, as a consequence of mitochondrial membrane potential loss, plays a central role in the progression of necrosis to osmotic swelling and, ultimately, cell lysis (156). Various host factors, including MPT (MPT-driven necrosis), receptor-interacting protein (RIP) kinases 1 and 3 (RIPK1/RIPK3, mostly involved in necrosis, pyroptosis, necroptosis), mixed lineage kinase domain-like (MLKL, mainly involved in necroptosis), caspase-1 and caspase 11 (primarily involved in pyroptosis), and cathepsin B (mostly involved in pyronecrosis) have been shown to regulate necrosis (151, 157, 158). Furthermore, chemical mediators have been shown to inhibit different necrotic signals, suggesting that necrosis can be regulated by extrinsic factors, such as therapeutic drugs (151). The other forms of regulated necrosis, as per NCCD-2018, include parthanatos, oxytosis (cell death by the accumulation of ROS), ferroptosis, and ETosis, all of which lead to inflammation (4). All these necrotic cell death modes have specific characteristics and overlapping pathways, which are highly relevant to the pathogenesis and treatment of TB.
M. tuberculosis infection promotes host cell necrosis.
Necrosis induction, associated with the granulomatous response during Mtb infection, is central to bacterial survival and dissemination and contributes to the severity and morbidity of the disease (7). Recent evidence shows that necrotic macrophages provide a niche for Mtb growth and survival before entering the extracellular milieu (159). In addition, neutrophils also undergo necroptosis following Mtb infection (160).
The cells that are frequently observed to participate in inflammatory pathology in actively necrotic granulomas are thought to be neutrophils (82). Intra-alveolar neutrophil infiltration and an excessive inflammatory response play critical roles in the progression to active TB, which is characterized by initial caseous and later liquefactive necrosis in the lung (161, 162). Neutrophil necrosis induced by Mtb contributes to bacterial growth in host cells, thus sustaining the infection (160). Mtb infection renders human macrophages necrotic, favoring bacterial replication (159). Mtb has also been shown to induce neutrophil extracellular traps (NETs) that promote the recruitment and activation of effector cells (163).
Several Mtb proteins activate the necrosis of host macrophages in vivo (164). ESAT-6 protein induces intracellular Ca2+ influx, triggering neutrophil necrosis and production of NETs, ultimately contributing to necrotic pathology and TB transmission (82). This resulted in ESAT-6 becoming an important therapeutic target (165). In addition, the Mtb PPE11 (Rv0453) protein, found in infected guinea pig lungs, promoted mycobacterial survival under stressful conditions by enhancing inflammation, organ pathology, and host-cell death (166). Recombinant PE17 (Rv1646) inhibits the production of proinflammatory cytokines (IL-6, IL-12, and TNF-α) and enhances macrophage necrosis (167). The degree of tissue necrosis and lung inflammation may be Mtb strain-specific; TLR2-deficient mice infected with Mtb W-Beijing exhibited increased neutrophil infiltration (168). The molecular mechanisms by which Mtb and its effectors aggravate host cell necrosis during TB remain unclear. Further characterization of Mtb protein and lipid functions in inducing host cell necrosis would certainly provide insight into its virulence mechanisms. Understanding the mechanisms of induction of host cell necrosis following Mtb infection would facilitate the development of novel therapeutic approaches.
Necroptosis as a prototype of regulated necrosis with proinflammatory functions.
Necroptosis, considered a link between host cell death and inflammation, is initiated by perturbations of the intracellular or extracellular microenvironment detected by specific death receptors such as TNF receptor, Fas receptor, pattern recognition receptors (PRRs) (TLR3, TLR4, Z-DNA binding domain protein-1), adaptor proteins, including MLKL, and RIPK1/RIPK3 (169). Necroptosis is activated downstream of death domain receptors, such as TNFR and Fas (170). Activation of these receptors recruits adapter proteins, including FADD, TRADD, and Toll/interleukin-1 receptor (TIR)-domain-containing adapter inducing interferon-β (TRIF), which interact with RIPK1 and caspase-8 or -10 (171). Necroptosis starts with the ubiquitylation of RIPK1 by IAPs, keeping it nonfunctional and enabling proinflammatory downstream activity via NFκB (170). Following the detection of cell death signaling, RIPK1 is deubiquitylated by CYLD and can thus recruit and phosphorylate RIPK3, forming a complex called ripoptosome (172, 173). The RIPK1/RIPK3 complex further recruits and phosphorylates MLKL. The oligomerization of phosphorylated MLKL occurs in the presence of highly phosphorylated inositol phosphate (IP6), which leads to the formation of the necrosome (44, 45). The MLKL oligomers translocate to regions in the plasma membrane enriched with phosphatidylinositol phosphate (PIP), forming pores. These processes ultimately lead to necroptotic cell death due to ion influx, cell swelling, and membrane lysis, followed by the dysregulated release of intracellular material (170, 174). The cytosolic nucleic acid sensors, such as RIG-I and cyclic GMP-AMP synthase (cGAS)/stimulator of interferon genes (STING), also contribute to necroptotic cell death, as they induce the production of type 1 interferons (IFN-I) and TNF-α and thus promote necroptosis via an autocrine feedback loop (170, 175, 176). In macrophages, downstream of TNFR or TLR engagement, active caspase-8 cleaves the cytokine blocker N4BP1, thus promoting an increase in cytokine release (177). Following activation, RIPK3 phosphorylates the pyruvate dehydrogenase complex (PDC) in mitochondria and promotes aerobic respiration and mitochondrial ROS production (170). The DNA-dependent activator of IFN regulatory factor (DAI) recruits RIPK3 in the presence of cytosolic microbial DNA released during infection, which bypasses RIPK1-dependent activation of MLKL and leads to the formation of necrosome complex (45, 170). Necroptosis can also be triggered by the activation of pattern recognition receptors (PRRs), such as TLR3 and TLR4, intracellular sensing proteins including DAI, RIG-I, and MDA-5, and interferon signaling (178).
Several types of mycobacterial proteins have been reported to induce necroptosis in human cells, including THP-1 cells and peripheral blood mononuclear cells (PBMCs) based on propidium iodide (PI) staining and lactate dehydrogenase (LDH) assays (72–74). Mtb infection promotes necroptosis as it is a favorable outcome as necrosis for mycobacterial survival (7). Necroptosis is linked to apoptosis with caspase-8 activity, which functions as a switch between apoptosis and necroptosis. Active caspase-8 cleaves RIP1K/RIP3K, inhibiting necroptosis, whereas inactive caspase-8 promotes necroptosis (179). Mtb induces the secretion of TNF-α and at high bacterial load can signal toward necroptosis of infected host cells. Other Mtb factors that have been reported to induce necroptosis include early secretion system-1 (ESX-1) and CpnT (180). The activation of RIP1K/RIP3K/MLKL has been established in Mtb infection. Following Mtb infection, the depletion of bacterial nicotinamide adenine dinucleotide (NAD+) activates RIPK3 and MLKL, the key mediators of necroptosis, leading to the death of host macrophages in a RIPK3- and MLKL-dependent manner (180).
Pyroptosis as microbicidal and caspase-dependent necrosis and regulator of inflammation.
Pyroptosis is a distinct type of cell death culminating in the loss of plasma membrane integrity and induced by activation of inflammasome sensors that includes the nucleotide-binding and oligomerization domain (NOD)-like receptor (NLR) family, the DNA receptor absent in melanoma 2 (AIM2), and the pyrin receptor (170). This cell death mechanism depends on caspase-1 and can cause plasma membrane rupture (181, 182). The formation of a supramolecular assembly pyroptosome, also known as an inflammasome, is the hallmark of pyroptosis (183). The inflammatory caspase-1 acts on the substrate gasdermin and releases a 31 kDa N-terminal domain of the protein that has a pore-forming activity, which is the final and direct executor of pyroptotic cell death (43). The plasma membrane rupture causes cellular swelling and osmotic lysis releasing cytoplasmic contents resulting in inflammation. Caspase-1 activation leads to chromosomal DNA damage through an endonuclease but does not cause the characteristic DNA fragmentation. Apart from caspase-1, other caspases involved in pyroptosis include caspase-4, -5, and -11, whose mechanism of action is poorly characterized (43).
Upon internalization of Mtb in the phagosome, phagosomal pores are formed through which bacterial factors, including mycobacterial extracellular DNA, trigger the cGAS/STING pathway leading to IFN-γ response (184) inducing inflammasome formation. Extensive studies have shown the importance of the inflammasome in TB pathogenesis using pyrin domain-containing protein-3 (NLRP3) deficient mice, which effectively controls the disease progress (26, 185). The role of inflammasomes as antipathogen or antihost responses depends on the delicate balance of its activity between pyroptosis induction and the production of proinflammatory cytokines, such as IL-1β and IL-18, which contribute to inflammation and cell death (181, 186, 187). The induction of host inflammasomes depends on the nature of infecting Mtb strain and the activation status of the macrophages (188). In general, macrophages activated with IFN-γ induce greater inflammasomes, and IL-1β, IL-18, and IL-1R deficient mice with defective inflammasome activation develop more significant pathological symptoms upon Mtb infection (185–187). Mtb mutants lacking the early secreted antigenic target of 6 kDa (ESAT-6) and early secretion system-1 (ESX-1) fail to induce inflammasomes suggesting the association of mycobacterial factors such as ESAT-6 and extracellular DNA with inflammasome formation and activity (189). In addition, recent studies have found that the molecular mechanisms of NLRP3 inflammasome sensing of Mtb involve type VII secretion system ESX-1, cell surface lipids (trehalose-6,6-dimycolate/trehalose-6,6-dibehenate;TDM/TDB), secreted effector proteins (LpqH, PPE13, EST12, EsxA), and double-stranded RNA acting on the priming and/or activation steps of inflammasome activation (188, 190). Thus, it appears that ESAT-6 and ESX-1 systems-mediated activation of inflammasome causes more disease that favors Mtb survival and transmission since ESAT-6 mutants are attenuated for intracellular survival. In contrast, Mtb also mediates inhibition of the NLRP3 inflammasome by limiting exposure of cell surface ligands via Hip1 hydrolase by inhibiting the host cell cathepsin G protease through the secreted Mtb effector Rv3364c and by limiting intracellular triggers (K+ and Cl− efflux and cytosolic reactive oxygen species production) using serine/threonine kinase PknF (188).
Pyroptosis serves as an efficient antimicrobial defense pathway. As in other microbial infections, mycobacteria induce inflammasome by recognizing pathogen-associated molecular patterns (PAMPs) (191). Pyroptosis plays a crucial role in reducing the bacterial burden. The role of inflammasome formation and IL-β processing in bacterial clearance and its inhibition by Mtb was demonstrated in a murine infection model (77, 192). Secretion of IL-1β and IL-18 by macrophages infected with Mtb was reported to be dependent on NLRP3 and ASC but not NLRC4 (193). IL-1r1−/− and IL-18−/− mice were very susceptible to Mtb infection (194–196). Casp1−/− and Asc−/− mice were also more susceptible than WT mice due to defective granuloma formation (197). However, the resistance of Nlrp3−/− mice to Mtb infection was not significantly different from that of WT mice (197). This observation suggests the involvement of other inflammasome activation pathways in TB pathogenesis. Surprisingly, the production of IL-1β during Mtb infection was reported to occur also in a caspase-1-independent manner (198). Pathogenic Mtb counteracts macrophage pyroptosis through its secreted protein Rv3364c, inhibiting the activity of cathepsin G, a serine protease downstream of caspase-1 (158). Another secreted factor of Mtb, zinc metalloprotease-1 (Zmp-1), inhibits inflammasome and IL-β activation, thus contributing to bacterial defense against host antimicrobial responses during infection (8, 77).
Pyronecrosis is another form of pyroptosis involving inflammasome activity, which is caspase-1 independent but cathepsin B-dependent cell death pathway (199, 200). The pyronecrosis signaling pathway overlaps with pyroptosis up to the stage of inflammasome formation utilizing the NOD, LRR (leucine-rich repeat protein), NLRP3, and apoptosis-associated speck-like protein containing a C-terminal caspase recruitment domain-D(ASC) proteins (201). The caspase independence of pyronecrosis was demonstrated in a caspase-1-deficient mouse model (200, 202, 203). However, the role of this host cell death pathway in the host-pathogen interactions during TB is poorly characterized.
Other types of regulated necrosis involved in M. tuberculosis infection.
Ferroptosis.
Ferroptosis is another example of RCD pathways, driven by perturbed intracellular iron homeostasis resulting in excessive accumulation of iron-dependent ROS and lipid peroxides, surpassing cellular antioxidation capacity and damaging the membrane structure (204–206). The imbalance of oxidation stress and antioxidant status leads to lipid peroxidation causing lethal damage to nucleic acids, proteins, and lipids, which leads to ferroptosis with shrunken cell volume, condensed mitochondrial membrane densities, and reduction or absence of mitochondria crista that differ from apoptosis and necrosis morphologically (204, 207, 208).
Recent reports provide the first evidence that ferroptosis, a newly described form of regulated cell death, is detrimental for the host during Mtb infection, that the finding has important implications for the development of host-directed therapies for TB (209). Upon macrophage infection by Mtb, the increased labile iron levels promote ROS-dependent lipid peroxidation. Accumulation of lipid peroxides induces plasma membrane destabilization, leading to ferroptosis-mediated cell death. Ferroptosis drives tissue necrosis and allows Mtb to thrive and spread, probably due to iron availability and the lack of an efficient macrophage antimicrobial immune response (209, 210).
ETosis.
ETosis refers to a cell death mode involving the formation of extracellular traps (ETs) in response to bacterial and viral infections (211). Mtb induces ETs through ETosis in different host cells, including neutrophils (NETosis), macrophages (METosis), and eosinophils (EETosis) (212–214).
ETosis was first described in neutrophils, which differs from apoptosis or necrosis in releasing decondensed chromatin filled with antimicrobial peptides, microbicidal enzymes, and proteinases (215). These neutrophil extracellular traps (NETs) contain eliminated microorganisms, from viruses and bacteria to yeasts and parasites, during infection of neutrophils by respective microbe (212). Upon infection, Mtb can induce NETs in vitro, as demonstrated in isolated human neutrophils and in vivo in guinea pig lungs (163, 216). Induction of NETs by Mtb is ROS- and phagocytosis-dependent (217). However, NETs were able to trap Mtb, though it is unclear if these NETs can effectively clear the bacteria (163). The NETs had Hsp72 molecule interspersed, which were suggested to transfer danger signals from neutrophils to macrophages, thus stimulating these cells for a more aggressive innate response (217). In addition to neutrophils, macrophages generate extracellular traps called METs, which constitute a phagocytosis-independent mode of antimicrobial mechanism (213). There is limited information on mycobacterium-induced METosis. However, recent studies show that only the cord-forming phenotype of Mtb induces METosis; notably, METosis is dependent on bacterial ESAT-6 expression (218).
The eosinophil-mediated extracellular traps (EETs) are structurally different from neutrophil-induced traps and can also induce cell death in a process referred to as EETosis (214). EETosis forms thicker fibers with globular structures containing intact eosinophil granules and presents lesser susceptibility to proteolytic degradation (214). EETosis presents a more complex pathophysiology than NETosis in patients with chronic obstructive pulmonary disease, as the debris is less likely to be degraded by proteolytic activity (219). Notably, the role of EETosis in the pathogenesis of mycobacterial infection remains unknown.
Another mode of antimicrobial defense mediated by inflammatory cell death includes the generation of pore-induced intracellular traps (PITs) analogous to extracellular traps. PITs trap viable intracellular bacteria within macrophages that are killed by efferocytosis and subsequently phagocytosed by neutrophils (220).
Parthanatos.
Parthanatos is a modality of RCD initiated by hyperactivation of a specific component of the DNA damage response (DDR) machinery called poly(ADP-ribose) polymerase 1 (PARP1) and precipitated by the consequent bioenergetic catastrophe coupled to apoptosis-inducing factor mitochondria associated 1 (AIFM1, also known as AIF)-dependent and macrophage migration inhibitory factor (MIF)-dependent DNA degradation (4). Notably, parthanatos appears to occur not only as a consequence of severe/prolonged alkylating DNA damage but also in response to oxidative stress, hypoxia, hypoglycemia, and inflammatory cues (221–223).
One of the key processes of parthanatos is the binding of PARP1 to AIF, which promotes the release of AIF into the cytosol and its translocation into the nucleus, where it mediates large-scale DNA fragmentation and chromatin condensation (221, 224–226). Recent studies show that MIF functions as the main nuclease precipitating parthanatos in a recent screening for AIF-binding proteins (227). Thus, cytosolic AIF reportedly promotes the translocation of MIF into the nucleus, where MIF precipitates parthanatos by catalyzing DNA cleavage. In addition, MIF depletion or specific mutations in its nuclease domain confer protection against parthanatos in vitro and in vivo (227).
Inhibitors of PARP1, such as BMN 673 (talazoparib), ABT-888 (veliparib), and AG-014699 (rucaparib), are often used to inhibit parthanatos (228). Following Mtb infection, treatment with these three compounds failed to inhibit Mtb-induced macrophage cell death, suggesting that parthanatos is unlikely to account for the death of the Mtb-infected macrophages and is not the dominant cell death mode or that the Mtb-activated parthanatos pathway is not amenable for inhibition by these drugs (229). In Mtb infection, intracellular stress can activate PARP1, which signals mitochondrial dysfunction potentially involving receptor-interacting protein kinases 1 (RIPK1), highlighting a connection between death receptors and TLR signaling of host cells (11, 155, 230).
Autophagy
Autophagy is a fundamental, innate cellular homeostasis process that recycles defective or malfunctioning cellular organelles and proteins into an energy conservation mechanism (231). Currently, 32 different autophagy-related genes (Atg) have been identified by genetic screening, with many of these genes conserved in multiple organisms, emphasizing the importance of the autophagic process in responses to starvation across phylogeny (232). The three primary types of autophagy occurring in mammalian cells are microautophagy, macroautophagy, and chaperone-mediated autophagy (CMA) (233). Among these three modes, macroautophagy is the most studied and synonymous with autophagy. Macroautophagy involves the formation of a cargo lined by phagophore, a double-layered membrane derived from the endoplasmic reticulum (ER) that isolates the autophagic contents from the cytoplasm, which then fuses with lysosome (233, 234).
Macroautophagy occurs constitutively at a low level but can further be induced under stress conditions, such as nutrient or energy starvation, to degrade cytoplasmic materials into metabolites that can be used in biosynthetic processes or energy production, allowing for cell survival (235). Under normal conditions, macroautophagy assists in cellular maintenance by specifically degrading damaged or superfluous organelles (236). Macroautophagy eliminates intracellular pathogens as part of host defense and is called “xenophagy,” whereby bacteria are engulfed by autophagosomes and degraded after fusion with lysosomes to form autolysosomes (237–239). Recent studies have demonstrated that xenophagy is essential in multiple diseases such as cancer, diabetes, cardiomyopathy, neurodegeneration, liver disease, autoimmune diseases, and pathogen infections, including Mtb (240). In this review, we focus on xenophagy, which will hereafter be referred to as “autophagy,” and its roles in host defense against Mtb.
Autophagy as an innate immune defense mechanism against M. tuberculosis infection.
As an immune mechanism, autophagy controls inflammation and acts as a cell-autonomous defense against intracellular microbes, including Mtb. Another significant role of autophagy is its anti-inflammatory and tissue-sparing function. This combination of antimicrobial and anti-inflammatory actions prevents active disease in animal models (241). In humans, genetic links between autophagy and susceptibility to TB provide further support for these combined roles of autophagy (241, 242). The formation of a double membrane phagosome, which fuses with a lysosome, is a hallmark of autophagy (233). It is a constitutive cellular process induced under stress conditions such as nutrient starvation, oxidative stress, pathogen infection, and hypoxia, which degrades cytoplasmic material into metabolites and degrades cytoplasmic foreign bodies (243, 244).
Autophagy is activated by microbial pattern receptors, including the Toll-like receptors (TLRs), a retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs), and nucleotide-binding and oligomerization domain (NOD)-like receptors (NLRs), which activate the downstream signaling pathways like p38 mitogen-activated protein kinase (p38MAPK), RIP1/extracellular signal-regulated kinase (ERK), and subsequent activation of the vacuolar protein sorting 34 (VPS-34) complex (245, 246), as illustrated in Fig. 2. TLR4 was the first PRR linked to autophagy through Toll/interleukin-1 receptor (TIR)-domain-containing adapter inducing interferon-β (TRIF)-p38 rather than myeloid differentiation primary response 88 (MyD88) (247–249). The innate and adaptive immune responses converge at autophagy to successfully eliminate pathogens. Cytokines such as IL-1β and IFN-γ induce autophagy and are host-protective (250, 251). Autophagy is also induced by immune cell-cell contact between specific T-cells and Mtb-infected macrophages (252). The autophagy signaling ultimately kills mycobacteria through lytic and antimicrobial properties, which are more robust than antimicrobial compartments of the conventional phagosomes (253). A fraction of bacteria escape the phagosome into the cytosol through permeabilization of the phagosomal membrane and are targeted for selective autophagic process, wherein the bacteria are ubiquitinated (248).
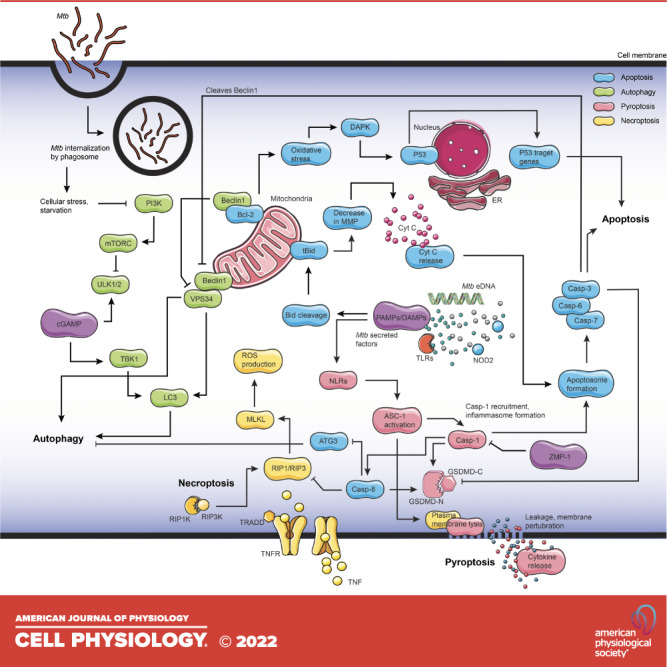
Figure. 2.

Mycobacterium tuberculosis (Mtb) modulation of host autophagy signaling. Multiple surface receptors and intracellular stress signals, including Mtb factors, modulate autophagy signaling. Microbial surface pattern receptors such as Toll-like receptors (TLRs), RIG-I like receptors (RLRs), and NOD-like receptors (NLRs) recognize mycobacterial secreted factors, which further activate p38 mitogen-activated protein kinases (MAPK) and receptor-interacting serine/threonine-protein kinase 1 (RIP1)/extracellular signal-regulated kinases (ERK) that phosphorylate phosphoinositide 3-kinases (PI3K) and signal Beclin-1 phosphorylation to trigger autophagy. Autophagy culminates in the fusion of lysosomes with autophagosomes. Early secreted antigenic target of 6 kDa (ESAT-6), lipoarabinomannan (LAM), enhanced intracellular survival protein 2 (Eis), serine/threonine-protein phosphatase 2 A activator (PtpA), protein kinase G (PknG), early secretion system-1 (Esx-1), LAM/mannose-capped lipoarabinomannan (ManLAM) inhibit autophagy. P in the blue circle represents forward signaling by phosphorylation, while P in the red circle represents inhibition of phosphorylation and, thereby, inhibition of downstream signaling. Following phagocytosis, Mtb resides in the phagosome and blocks phagosome maturation. Mtb secretes Esx-1, promoting phagosome damages that trigger ubiquitination, recruitment of autophagic adaptors and mycobacterial capture via stimulator of interferon genes (STING). NOD, nucleotide binding and oligomerization domain; RIG-I, retinoic acid-inducible gene-I. Figure created with images from smart.servier.com.
Mtb replicates in macrophages (MΦ) in part by inhibiting phagosome-lysosome fusion until IFN-γ activates the MΦ to traffic Mtb to the lysosome (254, 255). The role of autophagy in host cell death is mainly inferred from studies performed in cultured MΦ or dendritic cells (DC) where Mtb colocalizes with autophagy-related (ATG) proteins, such as ATG5, ATG12, ATG16L1, p62 (SQSTM1), NDP52, calcium binding and coiled-coil domain 2 (CALCOCO2/NDP52), Beclin-1, and microtubule-associated protein 1A/1B-light chain 3 (LC3) (248, 256–259), stimulation of autophagy increases bacterial killing (259–261), and inhibition of autophagy allows for increased bacterial survival (241, 248, 256, 259, 260). To understand the role of autophagy in Mtb elimination in vivo, individual Atg genes were genetically deleted in mice using the Cre-loxP system, which knocked out the concerned gene in a specific cell type and was used for infection studies (262). Following the Mtb challenge, of all the atg gene knockouts, only atg5 significantly altered mycobacterial persistence, suggesting the dispensable nature of ATG-associated proteins for the progression of TB (262). Furthermore, ATG5 has a crucial role in autophagosome formation, and atg5 knockout (KO) mice (atg5fl/fl-LysM-Cre) showed increased Mtb burden and succumbed to infection at 30 days postinfection (248, 262, 263). Thus, autophagy confers host cell protection by activating several antimycobacterial responses in the infected cells. In addition, atg5fl/fl-LysM-Cre mice lacking ATG5 in monocyte-derived cells and neutrophils (polymorphic mononuclear cells, PMN) have an extremely severe phenotype similar to mice lacking IFN-γ signaling (264).
Among the various ATG factors involved in autophagy, ATG5 is the most well studied during Mtb infection; further, autophagy-independent functions of ATG5 have also been reported (265–270). A genetic approach was utilized to determine the role of Atg and the requirement of autophagy in the host resistance to Mtb infection in vivo. Recent studies have discovered that, contrary to expectation, autophagic capacity does not correlate with the outcome of Mtb infection (262). Instead, ATG5 is involved in preventing PMN-mediated immunopathology and protects against Mtb infection (262). By analyzing different Cre-mediated deletion strains, it is revealed that loss of Atg5 in PMN, but not alveolar MΦ or DC, can result in loss of control of Mtb infection, but the severe susceptibility of the Atg5fl/fl-LysM-Cre mice relies on the deletion of atg5 in multiple LysM+ cell types. These findings also show a PMN-intrinsic role for ATG5 during Mtb infection. Notably, the reversal of all phenotypes in the Atg5fl/fl-LysM-Cre mice upon PMN depletion positions PMN as a significant driver in the dysfunctional response in these mice. These results point to a model where infection with Mtb induces a proinflammatory response that leads to the recruitment of PMN to the lung. The absence of Atg5 expression within the responding myeloid cells leads to uncontrolled accumulation of PMN in the lung, which causes increased pathology and likely provides an expanded niche for bacterial infection. The animal then succumbs to infection before the adaptive immune response can control the inflammation and bacterial replication. Together, the in vivo genetic analyses presented here shift the understanding of the role of ATG5 during Mtb infection, revealing a new outcome of ATG5 activity in early events of the innate immunity required to regulate TB disease pathology and Mtb replication (262).
M. tuberculosis inhibits autophagy to promote survival in host cells.
The formation of the autophagosome following Mtb infection and the fusion to a lysosome is broken down into five main steps: 1) initiation, 2) elongation, 3) maturation, 4) fusion, and 5) degradation (271). Autophagy initiation is regulated by the master regulator, the mammalian/mechanistic target of Rapamycin (mTOR). It is a negative regulator of autophagy, meaning its dephosphorylation is responsible for autophagy induction. Dephosphorylation of mTOR results in the translocation of the Unc-51-like autophagy activating kinase (Ulk1/2)-ATG13-FAK family-interacting protein (FIP200)-ATG101 complex to the endoplasmic reticulum (272, 273). Class III phosphatidylinositol-3-kinase (PI3K) activates the VPS34-Beclin1-VPS15-ATG14 complex. The PI3K complex induces phosphatidylinositol-3-phosphate, which then recruits double Fab1, YOTB/ZK632.12, Vac1, and EEA1 (FYVE)-containing protein 1 (DFCP1) and WD-repeat domain phosphoinositide-interacting (WIPI) family proteins to initiate the omegasome/phagophore formation (272, 274–276). Elongation of the phagophore into the autophagosome is conducted by the ATG7–ATG10 complex and then conjugated to ATG12-ATG5-ATG16L1 on the phagophore membrane (277). ATG4 cleaves LC3 into LC3-I, while the ATG7–ATG3 complex lipidates LC3-I into LC3-II by conjugating phosphatidyl-ethanolamine (PE). The completed autophagosome fuses with the lysosome to degrade the autophagosome cargo for subsequent metabolite recycling or antigen presentation (Fig. 2) (278–282).
Various bacterial effector proteins are known to modulate autophagy. Many of these effectors are secreted through the type I to type VII and type IX secretion systems (283). Mycobacteria have numerous Type VII secretion systems (Esx1–Esx5). Mtb Esx1 is responsible for the puncture of the phagosome, allowing for mycobacterial escape (284). Mtb cytosolic DNA is recognized by the cytosolic DNA sensor, cyclic GMP-AMP synthase (cGAS), resulting in the release of cyclic guanosine monophosphate (cGAMP). cGAMP is recognized by the stimulator of interferon genes (STING), leading to type I IFN release and the recruitment of autophagy receptors p62, NDP52, and optineurin (248, 285). These receptors are recruited to the ubiquitinated pathogen, thereby allowing for specific targeting by the autophagosome (Fig. 2). The receptors contain an LC3 interaction region (LIR) to bind the LC3 autophagy protein (286–288).
EspB is a part of the Esx1 secretory apparatus responsible for the secretion of early secretory antigenic target-6 (ESAT-6). Treatment of macrophages with EspB protein demonstrates downregulation of the IFN-γ receptor IFN-γR1, resulting in the inhibition of STAT-1 activation even in the presence of IFN-γ (289). EspB and ESAT-6 are not the only Mtb proteins linked to the inhibition of autophagy. The “enhanced intracellular survival” (eis) gene of Mtb confers enhanced survival of nonpathogenic Mycobacterium smegmatis in macrophages, although it is not required for the persistence of pathogenic Mtb in these cells (78, 290). During Mtb infection, eis significantly inhibits the activation of c-jun N-terminal kinase (JNK), which prevents the induction of non-canonical autophagy through ATG7. JNK activation also induced reactive oxygen species (ROS) generation and significantly increased type 2 macrophage cell death by Mtb eis deletion mutant (78). Studies have revealed that Mtb eis is an efficient N(ε)-acetyltransferase that acetylates Lys55 of dual-specificity protein phosphatase 16 (DUSP16)/MAPK phosphatase-7 (MKP-7), a JNK-specific phosphatase, thus inhibiting JNK phosphorylation and inflammatory cytokine generation (291). Mtb eis was also found to substantially inhibit the production of TNF-α, IL-4, and IL-6, while simultaneously stimulating IFN-γ and IL-10 secretion (78, 292, 293).
Mtb inhibits autophagy to protect against bacterial clearance and host cell death, which impedes antigen presentation (271). The Mtb PE_PGRS47 protein inhibits autophagy and limits MHC (major histocompatibility complex) class II antigen presentation (281). Other Mtb PE/PPE proteins are also known to inhibit autophagy, including PE_PGRS41 (58) and PE_PGRS29 (294), by interacting with autophagy machinery. Mtb also secretes a probable ligase (CpsA) to inhibit the noncanonical autophagy pathway designated as LC3-associated phagocytosis (LAP) and NADPH oxidase (295). In contrast to canonical autophagosomes, LAP does not result in double-membrane structures and instead promotes rapid phagosome maturation (296). This cellular process limits the phagocytosed ability of the pathogen to replicate by accelerating phagosome maturation while regulating the IFN pathway and antigen presentation (271). In addition, the maturation of Mtb-containing autophagosomes into autolysosomes was inhibited by blocking the recruitment of the late endosome marker Rab7 (124). Inhibition of Rab5 conversion to Rab7 in endosomes is a well-established method in which mycobacteria inhibit lysosomal fusion (297).
An interplay between autophagy and innate immunity following M. tuberculosis infection.
TLR signaling and autophagy are linked; while TLR signaling recognizes pathogen-associated molecular patterns through pattern-recognition receptors (PRRs), the autophagic pathways eliminate harmful pathogens (298). Previous studies show that many Mtb molecules act as ligands for TLRs and non-TLRs, and the recognition of mycobacterial molecules may initiate or regulate autophagy (299–301). Multiple TLR stimuli, in addition to NOD-like receptor-induced signals or CD40, can activate autophagic responses to induce host innate immunity and overcome a mycobacterial phagosome arrest (247, 302–305). It is of interest that, regardless of the TLR ligand stimulus, host autophagy activation can lead to the clearance of mycobacteria-containing phagosomes through the recruitment of autophagosomes and induction of phagosomal maturation (247, 304). Furthermore, several crucial components of TLR signaling, such as MyD88, TIR-domain-containing adapter-inducing interferon-β (TRIF), and mitogen-activated protein kinase (MAPK) are involved in the regulation of autophagy activation (247, 304, 306).
Several mycobacterial factors can activate or modulate host autophagy. The well-known mycobacterial TLR2 ligand, 19-kDa lipoprotein, activates autophagic responses in monocytes/macrophages. This only occurs with enough active vitamin D metabolites or with Cyp27b1 gene activation, followed by functional vitamin D signaling (307). In non-TLR-mediated autophagy, muramyl dipeptide (MDP) leads to the formation of autophagosomes through receptor-interacting serine-threonine kinase-2 (RIPK-2), ATG5, ATG7, and ATG16L1 (308, 309). Both NOD1 and NOD2 are involved in autophagy activation and bactericidal responses by recruitment of ATG16L1 to the plasma membrane and bacterial wrapping by autophagosomes (309). Data show that NOD2 activation leads to the expression of autophagy proteins such as LRGM, LC3, and ATG16L1 in human alveolar macrophages, which contribute to improved intracellular control of virulent Mtb (257). In addition, mutant mice harboring variants of NOD2 (3020insC) showed elevated NF-κB activation in response to MDP and upregulated IL-1β secretion (310). Mutant NOD2 (Crohn’s disease-associated NOD2 frameshift mutation) mice also failed to recruit ATG16L1 to the plasma membrane and induce autophagy, suggesting that NOD2 and autophagic pathways are functionally linked to increased bacterial killing and maintaining host immune homeostasis (298).
The mycobacterial NOD2 ligand MDP, which is N-glycosylated by N-acetyl muramic acid hydroxylase, shows more potent NOD2-stimulating immune activation in mouse models (311). Thus, mycobacterial N-glycosylated MDP may induce autophagy to a greater extent than N-acetylated MDP, the most common form in bacteria. It remains unclear whether mycobacterial signaling induced by the C-type lectin receptor Dectin-1 or Mincle and by scavenger receptors is involved in activating or regulating the autophagic pathway during infection (298). Understanding the receptor-specific regulation and induction of autophagy by individual mycobacterial ligands or antigens may facilitate the development of effective therapeutic and vaccine strategies against human TB.
Autophagy modulates the M. tuberculosis-induced cytokine response.
Multiple studies have provided insight into the roles of various protective cytokines in activating autophagy, thus promoting antimycobacterial immune defenses. IFN-γ enhances the autophagic control of Mtb, whereas the Th2 cytokines IL-4 and IL-13 inhibit those effects (312). IFN-γ and its effector mechanisms are mediated through multiple pathways: the induction of antimicrobial effectors, including inducible nitric oxide synthase 2, which may produce nitric oxide; the IFN-inducible GTPase family LRG47 (Irgm) in mice (313); and upregulated expression of MHC class II molecules, which play a central role in antigen processing and presentation (314). Autophagy activation promotes antigen processing and presentation by MHC class II molecules and results in the bridging of innate and adaptive immune responses. In this aspect, IFN-γ may facilitate host protective immune responses through the connection of autophagy activation and innate immune activation for controlling Mtb infection (298).
Proinflammatory TNF-α plays an important role in the activation of phagosome maturation because TNF-α neutralization suppresses IFN-γ-induced phagosome maturation in primary human peripheral blood monocyte-derived macrophages (BMDMs) (315, 316). These results indicate that TNF-α may synergize IFN-γ-induced antimicrobial and autophagic responses against intracellular mycobacteria (317). Other cytokines, including IL-2, CCL2, IL-6, and TGF-β, play key roles in positively regulating autophagy in various cell types, such as IL-2 in CD4+ T cells, CCL2 and IL-6 in CD11b+ peripheral blood mononuclear cells (PBMCs), and TGF-β in hepatocarcinoma cell lines (317). Various innate cytokines, including CCL2, IL-6, and TNF-α, are increased during the initiation of mycobacterial infection. These cytokines play a role in granuloma formation, pathophysiology, and local immunity in the lungs during Mtb infection (318). Thus, it is crucial to determine the roles of each and/or combined cytokines in controlling the progression of Mtb infection.
Mtb-induced IL-12p40 and IL-23p19 expression are negatively regulated in human macrophages by the mammalian targets of rapamycin (mTOR)/70-kDa ribosomal S6 kinase 1 (S6K1) pathway (319). These findings suggest that IL-12/IL-23 is likely induced during autophagic pathway activation in mycobacterial infection. In addition, IL-23 production by Mtb in dendritic cells is mediated by the combined sensing of NOD2 and TLR2, whereas IL-12p70 production requires IFN-γ or the TLR7/8 ligand R848 (320), suggesting a mechanistic link between protective cytokines and the induction of autophagy.
Autophagy negatively regulates the activation of transcription, processing, and secretion of several proinflammatory cytokines, including IL-1α, IL-1β, and IL-18 (321–325). Negative regulation of the autophagy pathway in controlling IL-1β production involves two mechanisms: targeting pro-IL-1β for degradation in lysosomal compartments and inhibiting NLRP3 inflammasome activation (323, 326). In contrast, it was revealed that autophagy played an important role in the induction of TNF-α transcription and secretion (322). Another finding showed an under-appreciated contribution of autophagy to the synthesis and secretion of the proinflammatory cytokine IL-1β (327). The cross talk between autophagic pathways and various activating/inhibiting cytokine stimuli may be influenced by the microenvironment, which affects the behavior of immune cells, or by functional properties of the innate receptors or signaling molecules that contribute to cytokine generation and the autophagic process (298).
PANoptosis
PANoptosis is an inflammatory programmed cell death (PCD) pathway activated by specific triggers and regulated by the PANoptosome complex that has key features of pyroptosis, apoptosis, and/or necroptosis; hence the term PANoptosis. However, it cannot be classified in any of the PCD pathways mentioned earlier (328). The PANoptosome provides a molecular scaffold for the contemporaneous engagement of key molecules from pyroptosis, apoptosis, and/or necroptosis (329–331). PANoptosis has been observed in various viral, bacterial, and fungal infections, autoinflammatory diseases, and cancer. This mode of PCD has very significant pathophysiological relevance to TB since this pathway can module the host cell response and plays a crucial role in determining the outcome of host-pathogen interactions (332). The characterization of PANoptosis was also central to the molecular understanding of cytokine storm, a life-threatening condition caused by excessive cytokine production due to inflammatory cell death (333). In addition to pathogenic and inflammatory disease triggers, cytokines can induce PANoptosis (334).
Mtb infection results in Esx-1-mediated plasma membrane damage responses that cause potassium ion (K+) efflux, leading to the activation of caspase-1/NLRP3/Gasdermin-D (GSDMD)-mediated pyroptosis in human monocytes and macrophages (335). Phospholipids, platelet-activating factor (PAF), and PAF-like lipids can activate the canonical NLRP3 inflammasome through mechanisms involving K+ efflux and Ca2+ influx independent of the PAF receptor (336). Although K+ efflux is required for the NEK7-NLRP3 association, the mechanisms underlying K+ efflux-mediated NLRP3 inflammasome activation are not fully understood (337). Thus, more studies are needed to elucidate how K+ efflux triggered by multiple signals activates the assembly of the NLRP3 inflammasome complex.
CROSS TALK BETWEEN DIFFERENT TYPES OF HOST CELL DEATH FOLLOWING M. TUBERCULOSIS INFECTION
Various reports have demonstrated that virulent Mtb can evade the immune response by modulating cell death mechanisms. Caspase-8 was initially identified as a component of extrinsic apoptotic signaling platform death-inducing signaling complex (DISC) (338, 339) and later discovered as part of the cytosolic TNF‐induced complex II (340). Soon, it became apparent that caspase‐8 has a more complex role in regulating multiple cell death pathways (84). The enzymatic activity of caspase‐8 determines if cells survive or die via apoptosis or necrosis. Pharmacological inhibition of caspase‐8 (e.g., by zVAD‐FMK or Emricasan) or inhibition by FLIP(S/R) leads to necroptosis and RIP1 activation (341). Interestingly, RIP1 also restricts caspase‐8‐mediated cell death as the loss of RIP1 is sensitized to TNF‐induced apoptosis (46, 342). The interplay of apoptosis and necrosis is apparent in many inflammatory in vivo models where they are frequently simultaneously activated (343), given that the majority of signaling proteins are common to both pathways, and the balance of expression or activation of critical factors can tip the balance in favor of apoptosis or necrosis (84).
The overlapping mechanisms of cell death between apoptosis and necrosis are evidence for the cross talk between the two different pathways of immune responses. Other types of cell death, such as pyroptosis and necroptosis, have overlapping characteristics of apoptosis and necrosis and are the pinnacle of cross talk between these pathways (4, 7, 344). Several host factors interconnect the cell death pathways by interacting with molecules and regulators involved in all these different pathways (345). TNF-α is known for its pleiotropic effects, leading to various host cell death modalities, including apoptosis, necrosis, and necroptosis (7, 346). cFLIP, an inhibitor of caspase-8 (hence inhibitor of extrinsic apoptosis), can interact with ATG3 and inhibit the promotion of autophagy. Interaction of cFLIPL (cellular Fas-associated death domain-like interleukin-1-beta-converting enzyme), one of the isoforms of cFLIP, with caspase-8 leads to caspase-8 inactivation, but its ability to cleave RIP1 is retained and can therefore inhibit necroptosis (347). In addition, Bcl-2 interacts with Beclin-1, a crucial player in the autophagy pathway. However, Bcl-2 is also known to be upregulated in mycobacterial infection, which not only inhibits apoptosis but also inhibits autophagy. Other factors such as caspases-3, -7, and -8 cleave Beclin-1 and class III phosphoinositol-3-kinase (PI3K) no longer signal the autophagy pathway. The cleaved Beclin-1 localizes to mitochondria and reinforces apoptosis, and inhibits autophagy. The autophagic protein ATG5, a calpain substrate, functions similarly by localizing to mitochondria, associates with Bcl-xL, and induces apoptosis (269). Furthermore, Mtb secretes factors such as ESAT-6, which either causes apoptosis (at lower bacterial load) or necrosis (at higher bacterial load). ESAT-6 can also induce NETosis that can be attributed to the pore-forming nature of ESAT-6 along with PDIM (Phthiocerol dimycocerosates), which causes the destabilization of membranes. At lower bacterial load, ESAT-6 induces mild inflammation, which is sufficient to induce apoptosis, whereas, at higher bacterial load, it causes loss of membrane integrity, triggering necrosis or its forms, including necroptosis and pyroptosis (7, 147, 149). Esx-1 and LAM induce most of the cell death modalities, including apoptosis, necrosis, and autophagy (19, 20). The schematic representation of the cross talk involving multiple cell death pathways in Mtb infection is presented in Fig. 3.
Figure 3.

Cross talk of mycobacterial factors during different forms of host cell death. Various cell death modalities may result from either mycobacteria-induced events or host-induced innate defense mechanisms. The dynamics of these modalities determine the outcome of Mycobacterium tuberculosis (Mtb) infection. Several host factors are involved in multiple cell death pathways, and Mtb factors can inhibit host defense mechanisms at various levels. Arrows indicate activation, deactivation, and forward signaling, the color of the signaling components defines the particular pathway as indicated. The pathways are simplified to accommodate the cross connections showing the mycobacterial factors that increase the complexity of multiple cell death networks in eliminating mycobacteria. ASC-1, alanine/serine/cysteine transporter-1; ATG3, autophagy related 3; Bcl-2, B-cell leukemia/lymphoma 2; Bid, BH3 interacting domain; Casp, caspase; cGAMP, cyclic guanosine monophosphate/adenosine monophosphate; CytC, cytochrome C; DAMPs, death associated molecular patterns; DAPK, death associated protein kinase; eDNA, extracellular DNA; ER, endoplasmic reticulum; GSDMD, Gasdermin D; LC3, light chain 3; MLKL, mixed lineage kinase domain-like pseudokinase; MMP, matrix metalloproteinase; mTORC, mammalian target of rapamycin-C; NLR, NOD-like receptor; NOD, nucleotide binding oligomerization domain; p53, protein 53; PAMPs, pathogen associated molecular patterns; PI3K, phosphatidylinositol 3-kinase; RIPK, receptor interacting serine/threonine kinase; ROS, reactive oxygen species; tBid, truncated BH3 interacting domain; TBK, Tank-binding kinase; TLR, Toll-like receptor; TNF, tumor necrosis factor-alpha; TNFR, tumor necrosis factor-alpha receptor; TRADD, TNF receptor superfamily member 1 A associated via death domain; ULK, Unc-51 like autophagy activating kinase; VPS34, vacuolar protein sorting 34; ZMP-1, zinc metalloproteinase-1. Figure created with images from smart.servier.com.
A recent study presents evidence supporting the simultaneous activation of several cell death pathways by virulent Mtb strains in infected macrophages (348). Mtb H37Ra (avirulent) and H37Rv (virulent) strains were used to infect human monocyte-derived macrophages (MDM) in vitro, and the molecules involved in apoptosis, necroptosis, and pyroptosis were measured. Results revealed that Mtb H37Rv infection increased the Bcl-2 transcript and protein, decreased the BAX transcript, and increased phosphorylated Bcl-2 at the protein level. H37Rv infection was also shown to increase the expression of necroptotic pathway molecules, including ASK1, p-38, RIPK1, RIPK3, and caspase-8. In contrast, avirulent Mtb H37Ra infection increased the expression of caspase-8 and decreased RIPK3 expression. In that study, while IL-1β expression was independent of Mtb virulence, NLRP3 and CASP1 expression was increased at a low multiplicity of infection (MOI) in both strains. These observations suggest a role for bacterial MOI in activating host cell pyroptosis. In addition, virulent Mtb inhibits apoptosis mediated by Bcl-2 family molecules; but, at the same time, increases the expression of molecules involved in apoptosis, necroptosis, and pyroptosis at the transcriptional and protein levels, which might function as a mechanism to avoid the immune response and promote bacterial survival inside the host cells (348).
It is important to note that other pathways get activated or modulated when Mtb subverts one cell death pathway. This clearly defines the host signaling toward a particular outcome. Apoptosis appears as the first mode of the host cell’s innate defense mechanism that induces cell death, whereas the autophagy-dependent cell death pathway is the final option when other death modalities are subverted. Mtb blocks several host cell death mechanisms that lead to bacterial clearance (apoptosis, autophagy, and pyroptosis) while promoting those that support mycobacterial persistence (necrosis, necroptosis, apoptosis in contextual mode), leading to successful infection. Thus, the cross talk between the various cell death modalities reflects the complex dynamics during host-pathogen interaction in TB.
THE ROLE OF MICRORNAS AND LONG NONCODING RNAs IN HOST CELL DEATH PATHWAYS FOLLOWING M. TUBERCULOSIS INFECTION
MicroRNAs (miRNAs) are small noncoding RNAs that regulate host defense mechanisms against viruses, bacteria, and fungi. It is known that bacteria modulate host miRNA expression and, thereby, the host response to innate and adaptive immunity (349). miRNAs are involved in the delicate interplay between Mtb and its host, which contributes to dynamic changes in gene expression patterns and influences host-pathogen interactions (Table 3). Differential expression of miRNAs upon infection with Mtb regulates host signaling pathways linked to inflammation, autophagy, apoptosis, and polarization of macrophages (389).
Table 3.
miRNAs influencing multiple host cell death pathways in M. tuberculosis infection
| miRNA | Remarks | Cell Death Pathway | Reference |
|---|---|---|---|
| miR-155 | Highly overexpressed in Mtb infected mice 30-day postinfection | Apoptosis | (350) |
| ESAT-6 promotes apoptosis of macrophages through miR-155-SOCS1 interaction | (351) | ||
| Inhibits apoptosis of infected cells by targeting FOXO3 | (352) | ||
| Induces apoptosis following BCG infection | (353) | ||
| miR-155-5p | Promotes the intracellular Mtb replication during early innate immune responses and helps the survival of infected macrophages | Apoptosis | (354, 355) |
| miR-196b | Increased expression in patients with active TB and functions as a marker for active TB | Apoptosis | (356) |
| Induces apoptosis by targeting c-Myc and Bcl2 | (357) | ||
| miR-325-3p | Promotes Mtb persistence and latency in the host cell | Apoptosis | (358) |
| Inhibits apoptosis through activation of STAT3 signaling | (359) | ||
| miR-20a-5p | Downregulated by Mtb infection, triggers cell apoptosis to facilitate mycobacterial clearance through targeting JNK2 in human macrophages | Apoptosis | (360) |
| miR-708 | Upregulated in sera of patients with TB | Apoptosis | (361) |
| Induces apoptosis by targeting E-cadherin regulators | (362) | ||
| miR-let-7e | Inhibits apoptosis by targeting the executioner of programmed cell death Caspase-3 | Apoptosis | (363) |
| miR-29a | Inhibits apoptosis upon mycobacterial infection via Caspase-3 and -7 | Apoptosis | (363) |
| miR-582-5p | Upregulated in patients with active TB, inhibits apoptosis of monocytes by targeting FOXO1 | Apoptosis | (364) |
| miR-21 | Inhibits apoptosis, targets several genes of both intrinsic and extrinsic apoptosis pathways | Apoptosis | (365) |
| Decreased expression in CD4 T cells and peripheral blood of patients with TB | (366) | ||
| Functions as a key switch in the inflammatory response | (367) | ||
| Inhibits apoptosome formation by targeting Bcl-2 pathway | (64) | ||
| miR-145 | Downregulation of miR-145 leads to overexpression of its targets and inhibition of apoptosis | Apoptosis | (368) |
| miR-125b | Mtb lipomannan destabilizes TNF biosynthesis by miR-125b regulation | Apoptosis | (369) |
| miR-223 | Upregulated in patients with active TB and inhibits apoptosis of macrophages by targeting FOXO3 | Apoptosis | (370) |
| miR-27b | Modulates inflammatory response and apoptosis upon Mtb infection | Apoptosis | (371) |
| miR-155 | Promotes autophagy to eliminate intracellular Mtb by targeting Rheb | Autophagy | (261) |
| miR-99b | Downregulates the production of TNF-α and TNF-α receptor | Autophagy | (372) |
| miR-26a | Impairs Mtb survival in macrophages by targeting the transcription factor Klotho-like factor-4 (KLF4) | Autophagy | (373) |
| miR-23a-5p | Overexpression of miR-23a-5p prevented Mtb-induced autophagy in macrophages by modulating TLR2/MyD88/NF-κB signaling | Autophagy | (374) |
| miR-33 | Enables Mtb intracellular survival and persistence inside the host | Autophagy | (375) |
| Targets autophagy effectors (ATG5, ATG12, LC3B and LAMP1), inhibits autophagy, and reprogram host lipid metabolism | (375) | ||
| miR-27a-5p | Downregulates Ca2+ signaling leading to inhibition of autophagosome formation and xenophagy; promotes intracellular survival of Mtb | Autophagy | (376) |
| miR-144-3p | Inhibits autophagy activation by targeting ATG4 in RAW264.7 macrophages | Autophagy | (377) |
| miR-144-5p | Inhibits phagosome maturation and T cell function | Autophagy | (378) |
| Upregulated in human MDMs upon Mtb infection | (379) | ||
| Targets the 3′-UTR of DNA damage-regulated autophagy modulator 2 (DRAM2) in PBMC-derived macrophages and THP-1 cells | (379) | ||
| miR-889-5p | Overexpression inhibits autophagy and maintains bacterial survival in granulomas | Autophagy | (380) |
| miR-125a | Increases robustly in TB and inhibits autophagy by targeting UVRAG and anti-microbial responses | Autophagy | (381) |
| miR-30a | Inhibits autophagy and promotes Mtb survival mechanism in human macrophages by targeting Beclin-1 | Autophagy | (382) |
| miR-3619-5p | Targets cathepsin S and affects autophagy in THP-1 macrophages | Autophagy | (383) |
| miR-17-5p | Involved in autophagy by targeting ULK-1, controls the survival of mycobacteria in macrophages | Autophagy | (384) |
| Regulates autophagy in Mtb-infected macrophages by targeting Mcl-1 and STAT3 | (385) | ||
| miR-20a | Inhibits autophagy by targeting ATG7 and ATG16L1 and promotes mycobacterial survival in macrophages | Autophagy | (386) |
| miR-142-3p | Inhibits autophagy upon Mtb infection, targets N-Wasp leading to reduced phagocytosis | Autophagy | (387) |
| miR-20b | Inhibits Mtb-induced inflammation in mouse lung by targeting NLRP3 | Pyroptosis | (388) |
Several miRNAs are regulated following mycobacterial infection from several in vitro and in vivo studies. ATG, autophagy related; Bcl2, B-cell leukemia/lymphoma-2; ESAT-6, early secreted antigenic target of 6 kDa; LC3, light chain 3; MDM, monocyte-derived macrophages; Mtb, Mycobacterium tuberculosis; NLRP3, NOD-, LRR- and pyrin domain-containing protein 3; PBMC, peripheral blood mononuclear cell; TB, tuberculosis.
The roles of various miRNAs have been demonstrated in mycobacterial infection (390, 391). In vitro studies to identify differentially expressed miRNAs during Mtb infection using H37Rv and H37Ra revealed nine genes that were differentially expressed (392). One of these genes includes miR-155, which is consistently induced across studies and is known to be upregulated by mycobacterial components lipomannan (LM) and ESAT-6, which are very well associated with apoptosis (351, 353). miR-155 is known to target PKA signaling, activation of caspase-3, Bak-1, and translocation of cytochrome C through MAPK signaling. Loss of miR-155 through KO in BCG caused compromised apoptosis of host cells (353). miR-155 is also known to enhance autophagy of mycobacteria-infected macrophages by targeting Ras homolog enriched in brain (Rheb) (261). A study involving differential expression of miRNAs using PBMCs from patients with TB compared with healthy individuals revealed the role of miR-582, which caused inhibition of apoptosis by downregulating FoxO1 that regulates host response to Mtb infection (364).
miR-29 targets Bcl-2, MCL-1, p85a complex (kinase), CDC42 and thereby regulates apoptosis pathway in immune cells; inhibits IFN-γ pathway and increases apoptosis of cells involved in anti-tubercular responses. miR-29 contributes to the inhibition of autophagy by downregulating the secretion of IFN-γ (393). Another miRNA molecule, miR-21, is activated upon Mtb infection mediated by ESAT-6 and inhibits the secretion of proinflammatory cytokines like IL-12, IL-6, and TNF-α. It also upregulates the production of anti-inflammatory cytokine IL-10 and promotes dendritic cell apoptosis by targeting Bcl-2 (366). Several miRNAs induced during Mtb infection and the host cell modalities affected are listed in Table 3. Recent studies suggest that miRNAs regulate innate immune response against Mtb infection. Moreover, differential miRNA expression in Mtb infection can reflect the disease progression and help distinguish between active and latent Mtb infection (LTBI) (391). These findings helped in the identification and application of miRNAs as potential biomarkers of TB (391). In addition, the active participation of miRNAs in modulating autophagy and apoptosis responses against Mtb provides a better understanding of miRNAs as host-directed therapy (HDT) against TB.
POTENTIAL THERAPEUTICS AND TRANSLATIONAL OPPORTUNITIES TARGETING HOST CELL DEATH IN M. TUBERCULOSIS INFECTION
Since cell death pathways significantly impact the outcome of host-pathogen interactions during Mtb infection, several of the molecules involved in these pathways have been tested as targets for potential intervention for TB. IFN-alfacon-1 (Infergen/IFG) is a synthetic form of IFN-α2b that has immunomodulatory activity and has been shown to enhance the maturation and activation of macrophages (394). IFG exerts bactericidal activity through autophagy and, thus, attenuates Mtb survival in human macrophages (395). Therefore, targeting autophagy through adjunctive immunotherapy with IFG may provide a novel treatment strategy to control Mtb infection. Similarly, peroxisome proliferator-activated receptor alpha (PPARα) agonists (e.g., fibrates), which regulate energy homeostasis and inflammation, promote autophagy, lysosomal biogenesis, phagosome maturation, and antimicrobial defense against Mtb and, therefore, can be used in TB treatment (396). Loperamide, an antidiarrheal drug that increases the degradation of long-lived cellular proteins, induces autophagy in macrophages, and reduces Mtb growth and burden (397, 398). Furthermore, it was shown that vitamin D supplementation in patients with pulmonary TB without cavitation could enhance autophagy in macrophages and improve innate immune function and, therefore, may help to control the intracellular growth of mycobacteria (399). Interestingly, adjunct therapy using CD40 and TLR4 agonists (C40.T4) also significantly enhanced the bactericidal potency of anti-TB drugs by increasing autophagy, thus providing a novel therapeutic avenue to target drug resistance in anti-TB therapy (394). Various TB treatments targeting host cell death mechanisms are listed in Table 4.
Table 4.
List of potential therapeutic compounds that target host cell death pathways in M. tuberculosis infection
| Drug Name | Drug Class | Mechanism of Action | References |
|---|---|---|---|
| 2-Phospho-l-ascorbic acid | Vitamin | Inhibitor of SapM via metal oxidation | (400) |
| Aspirin | Nonsteroidal anti-inflammatory drug | Adjunctive host-directed therapy for pulmonary TB | (401, 402) |
| AX20017 | Research compound | PknG inhibitor | (81) |
| AZD4462 | Research compound | PknG inhibitor | (403, 404) |
| AZD7762 | Research compound | PknG inhibitor | (405) |
| Carbamazepine | Anti-convulsant | Autophagy inducer | (406) |
| Concanavalin A | Research compound | Necrosis activator | (407) |
| Cyclosporin A | Immunosuppressant | Inhibitor of calcineurin | (408) |
| FK506 | Immunosuppressive agent | Inhibitor of calcineurin | (408) |
| Imatinib | Cancer growth blocker | Inhibitor of Abelson tyrosine kinase | (400) |
| Isoniazid | Antitubercular antibiotic | Prodrug, cell-wall growth inhibitor, an autophagy inducer | (409) |
| l-ascorbic acid | Vitamin | Competitive inhibitor of SapM, redox destabilizer | (400) |
| L335-M34 | Research compound | PtpA inhibitor | (410) |
| Levofloxacin | Fluoroquinolone antibiotic | Apoptosis inducer | (411) |
| Loperamide | Anti-motility (gut)/anti-diarrheal | HDT, autophagy inducer | (398) |
| Metformin | Type 2-diabetic | HDT, oxidative stress inducer | (412) |
| Nitazoxanide/tizodanide | Antiparasitic | Autophagy modulator | (413, 414) |
| NU-6027 | Research compound | PknG inhibitor | (415) |
| OA-NO2 | Research compound | PknG inhibitor | (416) |
| Pretomanid/PA824 | Nitroimidazole | Nitric oxide stress inducer | (417, 418) |
| Pyrazinamide | Antitubercular antibiotic | Autophagy inducer | (419) |
| R406 | Research compound | PknG inhibitor | (403) |
| Rapamycin | Macrolide antibiotic | Apoptosis inducer | (420) |
| Rifampicin | Antitubercular Agent | DNA replication inhibitor | (421) |
| Saxifragifolin D | Research compound | Accumulator of PI3K | (422) |
| Sclerotiorin | Natural product | Aldose reductase, lipoxygenase and PknG inhibitor | (423) |
| Streptomycin | Aminoglycoside antibiotic | Necrosis inhibitor | (424) |
| Vitamin D3 | Vitamin | Adjunctive host-directed therapy for pulmonary TB | (401) |
HDT, host-directed therapy; PI3K, phosphatidylinositol-3-kinase; PknG, protein kinase G.
Apoptosis as a Direct Target of Adjunctive TB Therapy
Manipulation of apoptosis in the context of vaccination holds a promising future of therapeutic goals for the early translation of knowledge about cell death in Mtb infection. The proapoptotic recombinant strain BCG ΔureC::hly+ (rBCG;BCG ΔureC::hly+) expresses the pore-forming listeriolysin of Listeria monocytogenes and lacks urease C to ensure an optimal intraphagosomal pH for listeriolysin activity (19). This strain has higher efficacy and is more immunogenic than the parental BCG in inducing Th1 and Th17 responses in preclinical models, with more profound CD4 and CD8 T cell stimulation (425, 426). Although the responsible immune mechanisms are yet to be characterized, cross priming of CD8 T cells has been suggested as a critical mechanism underpinning the protection conferred by rBCGBCG ΔureC::hly+ over the parental BCG strain (425).
Opportunities for adjunctive TB therapies that directly enhance apoptosis are less noticeable and could risk damaging necessary cells (e.g., T cells) and/or generating an excess of apoptotic bodies at risk for secondary necrosis. Nonetheless, a more comprehensive understanding of how different fates for mononuclear phagocytes (MPs) and neutrophils influence the effectiveness of host defense and the quantity and quality of immune pathology in TB is vital to understanding TB pathogenesis and new targets for treatment and diagnosis.
Targeting Autophagy for Host-Directed Therapy
Successful infection of Mtb occurs when it subverts the host-induced death programs. The mammalian target of rapamycin (mTOR), a negative regulator of autophagy, can be activated by virulent mycobacteria (427). Macrophage reprogramming causes multiple lipid biogenesis and interruption of the autophagy pathway mediated by Esx-1 (127, 260, 428, 429). IFN-γ can induce autophagy, whereas Th2 cytokines can dominantly inhibit autophagy 162, which in turn activates AMP-activated protein kinase (AMPK) (251, 419, 430). Anti-TB drugs, such as isoniazid and pyrazinamide, also induce autophagy by exposing mycobacterial products within the cell, supporting the potential of the autophagy pathway for host-directed therapy (431, 432). In addition, mTOR inhibitors, such as rapamycin and everolimus that induce autophagy, also possess antimicrobial effects in in vitro Mtb infection models. Notably, these two drugs showed promising results as an adjunctive to standard antibiotics therapy in a clinical trial of human pulmonary TB (433–436).
SUMMARY AND CONCLUSIVE REMARKS
Despite advances made in our understanding of disease pathogenesis, TB remains a global health challenge and a significant cause of morbidity and mortality. Increasing rates of multi- and extreme-drug resistance Mtb strains, coupled with extended multidrug treatment regimens and co-occurrence of other diseases such as diabetes and AIDS, have impeded efforts to control TB worldwide effectively. In recent years, extensive research in host cell death mechanisms have contributed important information on various cell death pathways during Mtb infection and their impact on TB treatment (Fig. 3 and Table 4).
Further understanding of Mtb pathology in the context of host cell death pathways and developing novel therapies are urgently needed. In this regard, targeting molecular pathways that regulate various host cell death modalities would benefit the treatment of infectious diseases such as TB. Recently, universal guidelines for using and interpreting assays to monitor host cell death by autophagy have been released (437). This would facilitate integrating and synchronizing results generated by laboratories worldwide using various autophagy-related assay platforms. Such measures may accelerate translational research from bench to bedside and contribute significantly to developing novel treatment measures to control TB.
GRANTS
This study was supported by funding from the Bill and Melinda Gates Foundation under Grant OPP1157210 (to S.S.) and the National Institute for Allergy and Infectious Diseases (NIAID) of the US National Institute of Health (NIH) under Grant R01AI161822 (to S.S. and A.K.). S.T. acknowledges support from NIGMS (SC1GM140968) and the Border Biomedical Research Center (BBRC).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.N. and S.S. conceived and designed research; A.N. prepared figures; A.N. drafted manuscript; A.N., F.C.K., D.P., S.T., A.K., and S.S. edited and revised manuscript; A.N., F.C.K., D.P., S.T., A.K., and S.S. approved final version of manuscript.
REFERENCES
- 1.World Health Organization. Global Tuberculosis Report 2021. Geneva: WHO, 2021. [Google Scholar]
- 2. Fukunaga R, Glaziou P, Harris JB, Date A, Floyd K, Kasaeva T. Epidemiology of tuberculosis and progress toward meeting global targets—worldwide, 2019. MMWR Morb Mortal Wkly Rep 70: 427–430, 2021. doi: 10.15585/mmwr.mm7012a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Green DR, Llambi F. Cell death signaling. Cold Spring Harb Perspect Biol 7: a006080, 2015. doi: 10.1101/cshperspect.a006080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 25: 486–541, 2018. doi: 10.1038/s41418-017-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G. The molecular machinery of regulated cell death. Cell Res 29: 347–364, 2019. doi: 10.1038/s41422-019-0164-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yan G, Elbadawi M, Efferth T. Multiple cell death modalities and their key features. World Acad Sci J 2: 39–48, 2020. doi: 10.3892/wasj.2020.40. [DOI] [Google Scholar]
- 7. Mohareer K, Asalla S, Banerjee S. Cell death at the cross roads of host-pathogen interaction in Mycobacterium tuberculosis infection. Tuberculosis (Edinb) 113: 99–121, 2018. doi: 10.1016/j.tube.2018.09.007. [DOI] [PubMed] [Google Scholar]
- 8. Behar SM, Divangahi M, Remold HG. Evasion of innate immunity by Mycobacterium tuberculosis: is death an exit strategy? Nat Rev Microbiol 8: 668–674, 2010. doi: 10.1038/nrmicro2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Upadhyay S, Mittal E, Philips J. Tuberculosis and the art of macrophage manipulation. Pathog Dis 76: fty037, 2018. doi: 10.1093/femspd/fty037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stutz MD, Clark MP, Doerflinger M, Pellegrini M. Mycobacterium tuberculosis: rewiring host cell signaling to promote infection. J Leukoc Biol 103: 259–268, 2018. doi: 10.1002/JLB.4MR0717-277R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Srinivasan L, Ahlbrand S, Briken V. Interaction of Mycobacterium tuberculosis with host cell death pathways. Cold Spring Harb Perspect Med 4: a022459–a022459, 2014. doi: 10.1101/cshperspect.a022459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen M, Gan H, Remold HG. A mechanism of virulence: virulent Mycobacterium tuberculosis strain H37Rv, but not attenuated H37Ra, causes significant mitochondrial inner membrane disruption in macrophages leading to necrosis. J Immunol 176: 3707–3716, 2006. doi: 10.4049/jimmunol.176.6.3707. [DOI] [PubMed] [Google Scholar]
- 13. Butler RE, Brodin P, Jang J, Jang M-S, Robertson BD, Gicquel B, Stewart GR. The balance of apoptotic and necrotic cell death in Mycobacterium tuberculosis infected macrophages is not dependent on bacterial virulence. PLoS One 7: e47573, 2012. doi: 10.1371/journal.pone.0047573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schaible UE, Winau F, Sieling PA, Fischer K, Collins HL, Hagens K, Modlin RL, Brinkmann V, Kaufmann SHE. Apoptosis facilitates antigen presentation to T lymphocytes through MHC-I and CD1 in tuberculosis. Nat Med 9: 1039–1046, 2003. doi: 10.1038/nm906. [DOI] [PubMed] [Google Scholar]
- 15. Winau F, Weber S, Sad S, de Diego J, Hoops SL, Breiden B, Sandhoff K, Brinkmann V, Kaufmann SHE, Schaible UE. Apoptotic vesicles crossprime CD8 T cells and protect against tuberculosis. Immunity 24: 105–117, 2006. doi: 10.1016/j.immuni.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 16. Flynn JL, Chan J. Immunology of tuberculosis. Annu Rev Immunol 19: 93–129, 2001. doi: 10.1146/annurev.immunol.19.1.93. [DOI] [PubMed] [Google Scholar]
- 17. Mahon RN, Hafner R. Immune cell regulatory pathways unexplored as host-directed therapeutic targets for Mycobacterium tuberculosis: an opportunity to apply precision medicine innovations to infectious diseases. Clin Infect Dis 61: S200–S216, 2015. doi: 10.1093/cid/civ621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Guirado E, Schlesinger LS, Kaplan G. Macrophages in tuberculosis: friend or foe. Semin Immunopathol 35: 563–583, 2013. doi: 10.1007/s00281-013-0388-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Moraco AH, Kornfeld H. Cell death and autophagy in tuberculosis. Semin Immunol 26: 497–511, 2014. doi: 10.1016/j.smim.2014.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Parandhaman DK, Narayanan S. Cell death paradigms in the pathogenesis of Mycobacterium tuberculosis infection. Front Cell Infect Microbiol 4: 31, 2014. doi: 10.3389/fcimb.2014.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol 35: 495–516, 2007. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee J, Hartman M, Kornfeld H. Macrophage apoptosis in tuberculosis. Yonsei Med J 50: 1–11, 2009. doi: 10.3349/ymj.2009.50.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Strasser A, O'Connor L, Dixit VM. Apoptosis signaling. Annu Rev Biochem 69: 217–245, 2000. doi: 10.1146/annurev.biochem.69.1.217. [DOI] [PubMed] [Google Scholar]
- 24. Hengartner MO. The biochemistry of apoptosis. Nature 407: 770–776, 2000. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 25. Hail N Jr., Carter BZ, Konopleva M, Andreeff M. Apoptosis effector mechanisms: a requiem performed in different keys. Apoptosis 11: 889–904, 2006. doi: 10.1007/s10495-006-6712-8. [DOI] [PubMed] [Google Scholar]
- 26. Briken V. Mycobacterium tuberculosis genes involved in regulation of host cell death. Adv Exp Med Biol 783: 93–102, 2013. doi: 10.1007/978-1-4614-6111-1_5. [DOI] [PubMed] [Google Scholar]
- 27. Galluzzi L, Maiuri MC, Vitale I, Zischka H, Castedo M, Zitvogel L, Kroemer G. Cell death modalities: classification and pathophysiological implications. Cell death Differ 14: 1237–1243, 2007. doi: 10.1038/sj.cdd.4402148. [DOI] [PubMed] [Google Scholar]
- 28. Hongmei Z. Extrinsic and intrinsic apoptosis signal pathway review. In: Apoptosis and Medicine, edited by Ntuli T. London, UK: IntechOpen, 2012, p. 3–22. [Google Scholar]
- 29. Danelishvili L, McGarvey J, Li Y-J, Bermudez LE. Mycobacterium tuberculosis infection causes different levels of apoptosis and necrosis in human macrophages and alveolar epithelial cells. Cell Microbiol 5: 649–660, 2003. doi: 10.1046/j.1462-5822.2003.00312.x. [DOI] [PubMed] [Google Scholar]
- 30. Mogga SJ, Mustafa T, Sviland L, Nilsen R. Increased Bcl‐2 and reduced Bax expression in infected macrophages in slowly progressive primary murine Mycobacterium tuberculosis infection. Scand J Immunol 56: 383–391, 2002. doi: 10.1046/j.1365-3083.2002.01140.x. [DOI] [PubMed] [Google Scholar]
- 31. Aguiló N, Uranga S, Marinova D, Martín C, Pardo J. Bim is a crucial regulator of apoptosis induced by Mycobacterium tuberculosis. Cell death Dis 5: e1343, 2014. doi: 10.1038/cddis.2014.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Spira A, Carroll JD, Liu G, Aziz Z, Shah V, Kornfeld H, Keane J. Apoptosis genes in human alveolar macrophages infected with virulent or attenuated Mycobacterium tuberculosis: a pivotal role for tumor necrosis factor. Am J Respir Cell Mol Biol 29: 545–551, 2003. doi: 10.1165/rcmb.2002-0310OC. [DOI] [PubMed] [Google Scholar]
- 33. Jo SH, Choi J-A, Lim Y-J, Lee J, Cho S-N, Oh S-M, Go D, Kim S-H, Song C-H. Calreticulin modulates the intracellular survival of mycobacteria by regulating ER-stress-mediated apoptosis. Oncotarget 8: 58686–58698, 2017. doi: 10.18632/oncotarget.17419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Herbst S, Schaible UE, Schneider BE. Interferon gamma activated macrophages kill mycobacteria by nitric oxide induced apoptosis. PLoS One 6: e19105, 2011. doi: 10.1371/journal.pone.0019105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Keane J, Shurtleff B, Kornfeld H. TNF-dependent BALB/c murine macrophage apoptosis following Mycobacterium tuberculosis infection inhibits bacillary growth in an IFN-γ independent manner. Tuberculosis (Edinb) 82: 55–61, 2002. doi: 10.1054/tube.2002.0322. [DOI] [PubMed] [Google Scholar]
- 36. Sly LM, Hingley-Wilson SM, Reiner NE, McMaster WR. Survival of Mycobacterium tuberculosis in host macrophages involves resistance to apoptosis dependent upon induction of antiapoptotic Bcl-2 family member Mcl-1. J Immunol 170: 430–437, 2003. doi: 10.4049/jimmunol.170.1.430. [DOI] [PubMed] [Google Scholar]
- 37. Joseph S, Yuen A, Singh V, Hmama Z. Mycobacterium tuberculosis Cpn60. 2 (GroEL2) blocks macrophage apoptosis via interaction with mitochondrial mortalin. Biol Open 6: 481–488, 2017. doi: 10.1242/bio.023119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shi J, Zhang H, Fang L, Xi Y, Zhou Y, Luo R, Wang D, Xiao S, Chen H. A novel firefly luciferase biosensor enhances the detection of apoptosis induced by ESAT-6 family proteins of Mycobacterium tuberculosis. Biochem Biophys Res Commun 452: 1046–1053, 2014. [Erratum in Biochem Biophys Res Commun 522: 1069–1070, 2020]. doi: 10.1016/j.bbrc.2014.09.047. [DOI] [PubMed] [Google Scholar]
- 39. Sánchez D, Rojas M, Hernández I, Radzioch D, García LF, Barrera LF. Role of TLR2-and TLR4-mediated signaling in Mycobacterium tuberculosis-induced macrophage death. Cell Immunol 260: 128–136, 2010. doi: 10.1016/j.cellimm.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 40. Phillips BL, Mehra S, Ahsan MH, Selman M, Khader SA, Kaushal D. LAG3 expression in active Mycobacterium tuberculosis infections. Am J Pathol 185: 820–833, 2015. doi: 10.1016/j.ajpath.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Theobald SJ, Gräb J, Fritsch M, Suárez I, Eisfeld HS, Winter S, Koch M, Hölscher C, Pasparakis M, Kashkar H, Rybniker J. Gasdermin D mediates host cell death but not interleukin-1β secretion in Mycobacterium tuberculosis-infected macrophages. Cell Death Discov 7: 327, 2021. doi: 10.1038/s41420-021-00716-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci 20: 3328, 2019. doi: 10.3390/ijms20133328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Qiu Z, Lei S, Zhao B, Wu Y, Su W, Liu M, Meng Q, Zhou B, Leng Y, Xia Z-y. NLRP3 inflammasome activation-mediated pyroptosis aggravates myocardial ischemia/reperfusion injury in diabetic rats. Oxid Med Cell Longev 2017: 1–17, 2017. doi: 10.1155/2017/9743280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhao J, Jitkaew S, Cai Z, Choksi S, Li Q, Luo J, Liu Z-G. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci USA 109: 5322–5327, 2012. doi: 10.1073/pnas.1200012109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang J-G, Alvarez-Diaz S, Lewis R, Lalaoui N, Metcalf D, Webb AI, Young SN, Varghese LN, Tannahill GM, Hatchell EC, Majewski IJ, Okamoto T, Dobson RCJ, Hilton DJ, Babon JJ, Nicola NA, Strasser A, Silke J, Alexander WS. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 39: 443–453, 2013. doi: 10.1016/j.immuni.2013.06.018. [DOI] [PubMed] [Google Scholar]
- 46. Rickard JA, O'Donnell JA, Evans JM, Lalaoui N, Poh AR, Rogers T, Vince JE, Lawlor KE, Ninnis RL, Anderton H, Hall C, Spall SK, Phesse TJ, Abud HE, Cengia LH, Corbin J, Mifsud S, Di Rago L, Metcalf D, Ernst M, Dewson G, Roberts AW, Alexander WS, Murphy JM, Ekert PG, Masters SL, Vaux DL, Croker BA, Gerlic M, Silke J. RIPK1 regulates RIPK3-MLKL-driven systemic inflammation and emergency hematopoiesis. Cell 157: 1175–1188, 2014. doi: 10.1016/j.cell.2014.04.019. [DOI] [PubMed] [Google Scholar]
- 47. Dao DN, Kremer L, Guérardel Y, Molano A, Jacobs WR, Porcelli SA, Briken V. Mycobacterium tuberculosis lipomannan induces apoptosis and interleukin-12 production in macrophages. Infect Immun 72: 2067–2074, 2004. doi: 10.1128/IAI.72.4.2067-2074.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol 5: 987–995, 2004. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 49. George KM, Chatterjee D, Gunawardana G, Welty D, Hayman J, Lee R, Small PL. Mycolactone: a polyketide toxin from Mycobacterium ulcerans required for virulence. Science 283: 854–857, 1999. doi: 10.1126/science.283.5403.854. [DOI] [PubMed] [Google Scholar]
- 50. Sanchez A, Espinosa P, Esparza MA, Colon M, Bernal G, Mancilla R. Mycobacterium tuberculosis 38‐kDa lipoprotein is apoptogenic for human monocyte‐derived macrophages. Scand J Immunol 69: 20–28, 2009. doi: 10.1111/j.1365-3083.2008.02193.x. [DOI] [PubMed] [Google Scholar]
- 51. Derrick SC, Morris SL. The ESAT6 protein of Mycobacterium tuberculosis induces apoptosis of macrophages by activating caspase expression. Cell Microbiol 9: 1547–1555, 2007. doi: 10.1111/j.1462-5822.2007.00892.x. [DOI] [PubMed] [Google Scholar]
- 52. Basu S, Pathak SK, Banerjee A, Pathak S, Bhattacharyya A, Yang Z, Talarico S, Kundu M, Basu J. Execution of macrophage apoptosis by PE_PGRS33 of Mycobacterium tuberculosis is mediated by Toll-like receptor 2-dependent release of tumor necrosis factor-α. J Biol Chem 282: 1039–1050, 2007. doi: 10.1074/jbc.M604379200. [DOI] [PubMed] [Google Scholar]
- 53. Xu G, Jia H, Li Y, Liu X, Li M, Wang Y. Hemolytic phospholipase Rv0183 of Mycobacterium tuberculosis induces inflammatory response and apoptosis in alveolar macrophage RAW264.7 cells. Can J Microbiol 56: 916–924, 2010. doi: 10.1139/w10-079. [DOI] [PubMed] [Google Scholar]
- 54. Zhang L, Zhong Q, Bao L, Zhang Y, Gao L, Huang B, Zhang H-D. Rv0901 from Mycobacterium tuberculosis, a possible novel virulent gene proved through the recombinant Mycobacterium smegmatis. Jpn J Infect Dis 62: 26–31, 2009. [PubMed] [Google Scholar]
- 55. Chandolia A, Rathor N, Sharma M, Saini NK, Sinha R, Malhotra P, Brahmachari V, Bose M. Functional analysis of mce4A gene of Mycobacterium tuberculosis H37Rv using antisense approach. Microbiol Res 169: 780–787, 2014. doi: 10.1016/j.micres.2013.12.008. [DOI] [PubMed] [Google Scholar]
- 56. Ahmad J, Farhana A, Pancsa R, Arora SK, Srinivasan A, Tyagi AK, Babu MM, Ehtesham NZ, Hasnain SE. Contrasting function of structured N-terminal and unstructured C-terminal segments of Mycobacterium tuberculosis PPE37 protein. MBio 9, e01712–, 2018. doi: 10.1128/mBio.01712-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tiwari B, Ramakrishnan UM, Raghunand TR. The Mycobacterium tuberculosis protein pair PE 9 (R v1088)–PE 10 (Rv 1089) forms heterodimers and induces macrophage apoptosis through Toll‐like receptor 4. Cell Microbiol 17: 1653–1669, 2015. doi: 10.1111/cmi.12462. [DOI] [PubMed] [Google Scholar]
- 58. Deng W, Long Q, Zeng J, Li P, Yang W, Chen X, Xie J. Mycobacterium tuberculosis PE_PGRS41 enhances the intracellular survival of M. smegmatis within macrophages via blocking innate immunity and inhibition of host defense. Sci Rep 7: 1–13, 2017. doi: 10.1038/srep46716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Li H, Li Q, Yu Z, Zhou M, Xie J. Mycobacterium tuberculosis PE13 (Rv1195) manipulates the host cell fate via p38-ERK-NF-κB axis and apoptosis. Apoptosis 21: 795–808, 2016. doi: 10.1007/s10495-016-1249-y. [DOI] [PubMed] [Google Scholar]
- 60. Yang W, Deng W, Zeng J, Ren S, Ali MK, Gu Y, Li Y, Xie J. Mycobacterium tuberculosis PE_PGRS18 enhances the intracellular survival of M. smegmatis via altering host macrophage cytokine profiling and attenuating the cell apoptosis. Apoptosis 22: 502–509, 2017. doi: 10.1007/s10495-016-1336-0. [DOI] [PubMed] [Google Scholar]
- 61. Velmurugan K, Chen B, Miller JL, Azogue S, Gurses S, Hsu T, Glickman M, Jacobs WR, Porcelli SA, Briken V. Mycobacterium tuberculosis nuoG is a virulence gene that inhibits apoptosis of infected host cells. PLoS Pathog 3: e110, 2007. doi: 10.1371/journal.ppat.0030110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wang J, Teng JLL, Zhao D, Ge P, Li B, Woo PCY, Liu CH. The ubiquitin ligase TRIM27 functions as a host restriction factor antagonized by Mycobacterium tuberculosis PtpA during mycobacterial infection. Sci Rep 6: 34827, 2016. doi: 10.1038/srep34827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Danelishvili L, Babrak L, Rose SJ, Everman J, Bermudez LE. Mycobacterium tuberculosis alters the metalloprotease activity of the COP9 signalosome. MBio 5: e01278, 2014. doi: 10.1128/mBio.01278-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wang Q, Liu S, Tang Y, Liu Q, Yao Y. MPT64 protein from Mycobacterium tuberculosis inhibits apoptosis of macrophages through NF-kB-miRNA21-Bcl-2 pathway. PLoS One 9: e100949, 2014. doi: 10.1371/journal.pone.0100949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kumar D, Narayanan S. pknE, a serine/threonine kinase of Mycobacterium tuberculosis modulates multiple apoptotic paradigms. Infect Genet Evol 12: 737–747, 2012. doi: 10.1016/j.meegid.2011.09.008. [DOI] [PubMed] [Google Scholar]
- 66. Smith KLJ, Lee S. Inhibition of apoptosis by Rv2456c through nuclear factor-κB extends the survival of Mycobacterium tuberculosis. Int J Mycobacteriol 5: 426–436, 2016. doi: 10.1016/j.ijmyco.2016.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Braunstein M, Espinosa BJ, Chan J, Belisle JT, Jacobs WR. SecA2 functions in the secretion of superoxide dismutase A and in the virulence of Mycobacterium tuberculosis. Mol Microbiol 48: 453–464, 2003. doi: 10.1046/j.1365-2958.2003.03438.x. [DOI] [PubMed] [Google Scholar]
- 68. Dasgupta A, Sureka K, Mitra D, Saha B, Sanyal S, Das AK, Chakrabarti P, Jackson M, Gicquel B, Kundu M, Basu J. An oligopeptide transporter of Mycobacterium tuberculosis regulates cytokine release and apoptosis of infected macrophages. PLoS One 5: e12225, 2010. doi: 10.1371/journal.pone.0012225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Danelishvili L, Yamazaki Y, Selker J, Bermudez LE. Secreted Mycobacterium tuberculosis Rv3654c and Rv3655c proteins participate in the suppression of macrophage apoptosis. PLoS One 5: e10474, 2010. doi: 10.1371/journal.pone.0010474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wong KW, Jacobs WR Jr.. Critical role for NLRP3 in necrotic death triggered by Mycobacterium tuberculosis. Cell Microbiol 13: 1371–1384, 2011. doi: 10.1111/j.1462-5822.2011.01625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sun J, Siroy A, Lokareddy RK, Speer A, Doornbos KS, Cingolani G, Niederweis M. The tuberculosis necrotizing toxin kills macrophages by hydrolyzing NAD. Nat Struct Mol Biol 22: 672–678, 2015. doi: 10.1038/nsmb.3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Tundup S, Mohareer K, Hasnain SE. Mycobacterium tuberculosis PE25/PPE41 protein complex induces necrosis in macrophages: role in virulence and disease reactivation? FEBS Open Bio 4: 822–828, 2014. doi: 10.1016/j.fob.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Danelishvili L, Everman J, Bermudez LE. Mycobacterium tuberculosis PPE68 and Rv2626c genes contribute to the host cell necrosis and bacterial escape from macrophages. Virulence 7: 23–32, 2016. doi: 10.1080/21505594.2015.1102832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Feng S, Hong Z, Zhang G, Li J, Tian G-B, Zhou H, Huang X. Mycobacterium PPE31 contributes to host cell death. Front Cell Infect Microbiol 11: 629836, 2021. doi: 10.3389/fcimb.2021.629836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Danelishvili L, Everman JL, McNamara MJ, Bermudez LE. Inhibition of the plasma-membrane-associated serine protease cathepsin G by Mycobacterium tuberculosis Rv3364c suppresses caspase-1 and pyroptosis in macrophages. Front Microbiol 2: 281, 2011. doi: 10.3389/fmicb.2011.00281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Qu Z, Zhou J, Zhou Y, Xie Y, Jiang Y, Wu J, Luo Z, Liu G, Yin L, Zhang X-L. Mycobacterial EST12 activates a RACK1–NLRP3–gasdermin D pyroptosis–IL-1β immune pathway. Sci Adv 6: eaba4733, 2020. doi: 10.1126/sciadv.aba4733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Master SS, Rampini SK, Davis AS, Keller C, Ehlers S, Springer B, Timmins GS, Sander P, Deretic V. Mycobacterium tuberculosis prevents inflammasome activation. Cell host Microbe 3: 224–232, 2008. doi: 10.1016/j.chom.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Shin D-M, Jeon B-Y, Lee H-M, Jin HS, Yuk J-M, Song C-H, Lee S-H, Lee Z-W, Cho S-N, Kim J-M, Friedman RL, Jo E-K. Mycobacterium tuberculosis eis regulates autophagy, inflammation, and cell death through redox-dependent signaling. PLoS Pathog 6: e1001230, 2010. doi: 10.1371/journal.ppat.1001230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Zhang L, Zhang H, Zhao Y, Mao F, Wu J, Bai B, Xu Z, Jiang Y, Shi C. Effects of Mycobacterium tuberculosis ESAT-6/CFP-10 fusion protein on the autophagy function of mouse macrophages. DNA Cell Biol 31: 171–179, 2012. doi: 10.1089/dna.2011.1290. [DOI] [PubMed] [Google Scholar]
- 80. Bach H, Papavinasasundaram KG, Wong D, Hmama Z, Av-Gay Y. Mycobacterium tuberculosis virulence is mediated by PtpA dephosphorylation of human vacuolar protein sorting 33B. Cell host Microbe 3: 316–322, 2008. doi: 10.1016/j.chom.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 81. Walburger A, Koul A, Ferrari G, Nguyen L, Prescianotto-Baschong C, Huygen K, Klebl B, Thompson C, Bacher G, Pieters J. Protein kinase G from pathogenic mycobacteria promotes survival within macrophages. Science 304: 1800–1804, 2004. doi: 10.1126/science.1099384. [DOI] [PubMed] [Google Scholar]
- 82. Francis R, Butler R, Stewart G. Mycobacterium tuberculosis ESAT-6 is a leukocidin causing Ca2+ influx, necrosis and neutrophil extracellular trap formation. Cell Death Dis 5: e1474, 2014. doi: 10.1038/cddis.2014.394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kale J, Osterlund EJ, Andrews DW. BCL-2 family proteins: changing partners in the dance towards death. Cell Death Differ 25: 65–80, 2018. doi: 10.1038/cdd.2017.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kist M, Vucic D. Cell death pathways: intricate connections and disease implications. EMBO J 40: e106700, 2021. doi: 10.15252/embj.2020106700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kim SE, Zhang L, Ma K, Riegman M, Chen F, Ingold I, Conrad M, Turker MZ, Gao M, Jiang X, Monette S, Pauliah M, Gonen M, Zanzonico P, Quinn T, Wiesner U, Bradbury MS, Overholtzer M. Ultrasmall nanoparticles induce ferroptosis in nutrient-deprived cancer cells and suppress tumour growth. Nat Nanotechnol 11: 977–985, 2016. doi: 10.1038/nnano.2016.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol 15: 49–63, 2014. doi: 10.1038/nrm3722. [DOI] [PubMed] [Google Scholar]
- 87. Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292: 727–730, 2001. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell 7: 683–694, 2001. doi: 10.1016/S1097-2765(01)00214-3. [DOI] [PubMed] [Google Scholar]
- 89. Lei K, Davis RJ. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc Natl Acad Sci USA 100: 2432–2437, 2003. doi: 10.1073/pnas.0438011100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Llambi F, Wang Y-M, Victor B, Yang M, Schneider DM, Gingras S, Parsons MJ, Zheng JH, Brown SA, Pelletier S, Moldoveanu T, Chen T, Green DR. BOK is a non-canonical BCL-2 family effector of apoptosis regulated by ER-associated degradation. Cell 165: 421–433, 2016. doi: 10.1016/j.cell.2016.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Ke FFS, Vanyai HK, Cowan AD, Delbridge ARD, Whitehead L, Grabow S, Czabotar PE, Voss AK, Strasser A. Embryogenesis and adult life in the absence of intrinsic apoptosis effectors BAX, BAK, and BOK. Cell 173: 1217–1230.e17, 2018. doi: 10.1016/j.cell.2018.04.036. [DOI] [PubMed] [Google Scholar]
- 92. Stein JC, Hansen G. Mannose induces an endonuclease responsible for DNA laddering in plant cells. Plant Physiol 121: 71–80, 1999. doi: 10.1104/pp.121.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 102: 33–42, 2000. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 94. Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, Moritz RL, Simpson RJ, Vaux DL. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 102: 43–53, 2000. doi: 10.1016/S0092-8674(00)00009-X. [DOI] [PubMed] [Google Scholar]
- 95. Chai J, Shi Y. Apoptosome and inflammasome: conserved machineries for caspase activation. Natl Sci Rev 1: 101–118, 2014. doi: 10.1093/nsr/nwt025. [DOI] [Google Scholar]
- 96. Chaudhary AK, Yadav N, Bhat TA, O'Malley J, Kumar S, Chandra D. A potential role of X-linked inhibitor of apoptosis protein in mitochondrial membrane permeabilization and its implication in cancer therapy. Drug Discov Today 21: 38–47, 2016. doi: 10.1016/j.drudis.2015.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Wilson NS, Dixit V, Ashkenazi A. Death receptor signal transducers: nodes of coordination in immune signaling networks. Nat Immunol 10: 348–355, 2009. doi: 10.1038/ni.1714. [DOI] [PubMed] [Google Scholar]
- 98. Volpe E, Sambucci M, Battistini L, Borsellino G. Fas–Fas ligand: checkpoint of T cell functions in multiple sclerosis. Front Immunol 7: 382, 2016. doi: 10.3389/fimmu.2016.00382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Zhong B, Liu M, Bai C, Ruan Y, Wang Y, Qiu L, Hong Y, Wang X, Li L, Li B. Caspase-8 induces lysosome-associated cell death in cancer cells. Mol Ther 28: 1078–1091, 2020. doi: 10.1016/j.ymthe.2020.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 94: 491–501, 1998. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- 101. Dewson G, Kluck RM. Mechanisms by which Bak and Bax permeabilise mitochondria during apoptosis. J Cell Sci 122: 2801–2808, 2009. doi: 10.1242/jcs.038166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Pardo J, Wallich R, Martin P, Urban C, Rongvaux A, Flavell RA, Müllbacher A, Borner C, Simon MM. Granzyme B-induced cell death exerted by ex vivo CTL: discriminating requirements for cell death and some of its signs. Cell Death Differ 15: 567–579, 2008. doi: 10.1038/sj.cdd.4402289. [DOI] [PubMed] [Google Scholar]
- 103. Martínez-Lostao L, Anel A, Pardo J. How do cytotoxic lymphocytes kill cancer cells? Clin Cancer Res 21: 5047–5056, 2015. doi: 10.1158/1078-0432.CCR-15-0685. [DOI] [PubMed] [Google Scholar]
- 104. Voskoboinik I, Whisstock JC, Trapani JA. Perforin and granzymes: function, dysfunction and human pathology. Nat Rev Immunol 15: 388–400, 2015. doi: 10.1038/nri3839. [DOI] [PubMed] [Google Scholar]
- 105. Boivin WA, Cooper DM, Hiebert PR, Granville DJ. Lab Invest 89: 1195–1220, 2009. doi: 10.1038/labinvest.2009.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Thomas DA, Du C, Xu M, Wang X, Ley TJ. DFF45/ICAD can be directly processed by granzyme B during the induction of apoptosis. Immunity 12: 621–632, 2000. doi: 10.1016/S1074-7613(00)80213-7. [DOI] [PubMed] [Google Scholar]
- 107. Faridgohar M, Nikoueinejad H. New findings of Toll-like receptors involved in Mycobacterium tuberculosis infection. Pathog Glob Health 111: 256–264, 2017. doi: 10.1080/20477724.2017.1351080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Oddo M, Renno T, Attinger A, Bakker T, MacDonald HR, Meylan PR. Fas ligand-induced apoptosis of infected human macrophages reduces the viability of intracellular Mycobacterium tuberculosis. J Immunol 160: 5448–5454, 1998. [PubMed] [Google Scholar]
- 109. Manfredi AA, Heltai S, Rovere P, Sciorati C, Paolucci C, Galati G, Rugarli C, Vaiani R, Clementi E, Ferrarini M. Mycobacterium tuberculosis exploits the CD95/CD95 ligand system of γ δ T cells to cause apoptosis. Eur J Immunol 28: 1798–1806, 1998. doi:. [DOI] [PubMed] [Google Scholar]
- 110. Allie N, Grivennikov SI, Keeton R, Hsu N-J, Bourigault M-L, Court N, Fremond C, Yeremeev V, Shebzukhov Y, Ryffel B, Nedospasov SA, Quesniaux VFJ, Jacobs M. Prominent role for T cell-derived tumour necrosis factor for sustained control of Mycobacterium tuberculosis infection. Sci Rep 3: 1809, 2013. doi: 10.1038/srep01809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Zhang J, Sun B, Huang Y, Kouadir M, Zhou X, Wang Y, Zhao D. IFN-γpromotes THP-1 cell apoptosis during early infection with Mycobacterium bovis by activating different apoptotic signaling. FEMS Immunol Med Microbiol 60: 191–198, 2010. doi: 10.1111/j.1574-695X.2010.00732.x. [DOI] [PubMed] [Google Scholar]
- 112. Redford PS, Murray PJ, O'Garra A. The role of IL-10 in immune regulation during M. tuberculosis infection. Mucosal Immunol 4: 261–270, 2011. doi: 10.1038/mi.2011.7. [DOI] [PubMed] [Google Scholar]
- 113. O'Leary SN, O'Sullivan MP, Keane J. IL-10 blocks phagosome maturation in Mycobacterium tuberculosis-infected human macrophages. Am J Respir Cell Mol Biol 45: 172–180, 2011. doi: 10.1165/rcmb.2010-0319OC. [DOI] [PubMed] [Google Scholar]
- 114. Rivero-Lezcano OM, González-Cortés C, Reyes-Ruvalcaba D, Diez-Tascón C. CCL20 is overexpressed in Mycobacterium tuberculosis-infected monocytes and inhibits the production of reactive oxygen species (ROS). Clin Exp Immunol 162: 289–297, 2010. doi: 10.1111/j.1365-2249.2010.04168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Tait SWG, Parsons MJ, Llambi F, Bouchier-Hayes L, Connell S, Muñoz-Pinedo C, Green DR. Resistance to caspase-independent cell death requires persistence of intact mitochondria. Dev Cell 18: 802–813, 2010. doi: 10.1016/j.devcel.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Bai X, Feldman NE, Chmura K, Ovrutsky AR, Su W-L, Griffin L, Pyeon D, McGibney MT, Strand MJ, Numata M, Murakami S, Gaido L, Honda JR, Kinney WH, Oberley-Deegan RE, Voelker DR, Ordway DJ, Chan ED. Inhibition of nuclear factor-kappa B activation decreases survival of Mycobacterium tuberculosis in human macrophages. PLoS One 8: e61925, 2013. doi: 10.1371/journal.pone.0061925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Aguilo J, Marinova D, Martín C, Pardo J. ESX-1-induced apoptosis during mycobacterial infection: to be or not to be, that is the question. Front Cell Infect Microbiol 3: 88, 2013. doi: 10.3389/fcimb.2013.00088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. López M, Sly LM, Luu Y, Young D, Cooper H, Reiner NE. The 19-kDa Mycobacterium tuberculosis protein induces macrophage apoptosis through Toll-like receptor-2. J Immunol 170: 2409–2416, 2003. doi: 10.4049/jimmunol.170.5.2409. [DOI] [PubMed] [Google Scholar]
- 119. Sánchez A, Espinosa P, García T, Mancilla R. The 19 kDa Mycobacterium tuberculosis lipoprotein (LpqH) induces macrophage apoptosis through extrinsic and intrinsic pathways: a role for the mitochondrial apoptosis-inducing factor. Clin Dev Immunol 2012: 1–11, 2012. doi: 10.1155/2012/950503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Ciaramella A, Cavone A, Santucci MB, Garg SK, Sanarico N, Bocchino M, Galati D, Martino A, Auricchio G, D'Orazio M, Stewart GR, Neyrolles O, Young DB, Colizzi V, Fraziano M. Induction of apoptosis and release of interleukin-1β by cell wall-associated 19-kDa lipoprotein during the course of mycobacterial infection. J Infect Dis 190: 1167–1176, 2004. doi: 10.1086/423850. [DOI] [PubMed] [Google Scholar]
- 121. Sreejit G, Ahmed A, Parveen N, Jha V, Valluri VL, Ghosh S, Mukhopadhyay S. The ESAT-6 protein of Mycobacterium tuberculosis interacts with beta-2-microglobulin (β2M) affecting antigen presentation function of macrophage. PLoS Pathog 10: e1004446, 2014. doi: 10.1371/journal.ppat.1004446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Samten B, Wang X, Barnes PF. Mycobacterium tuberculosis ESX-1 system-secreted protein ESAT-6 but not CFP10 inhibits human T-cell immune responses. Tuberculosis 89: S74–S76, 2009. doi: 10.1016/S1472-9792(09)70017-4. [DOI] [PubMed] [Google Scholar]
- 123. Augenstreich J, Arbues A, Simeone R, Haanappel E, Wegener A, Sayes F, Le Chevalier F, Chalut C, Malaga W, Guilhot C, Brosch R, Astarie-Dequeker C. ESX-1 and phthiocerol dimycocerosates of Mycobacterium tuberculosis act in concert to cause phagosomal rupture and host cell apoptosis. Cell Microbiol 19: e12726, 2017. doi: 10.1111/cmi.12726. [DOI] [PubMed] [Google Scholar]
- 124. Chandra P, Ghanwat S, Matta SK, Yadav SS, Mehta M, Siddiqui Z, Singh A, Kumar D. Mycobacterium tuberculosis inhibits RAB7 recruitment to selectively modulate autophagy flux in macrophages. Sci Rep 5: 1–10, 2015. doi: 10.1038/srep16320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Pathak SK, Basu S, Basu KK, Banerjee A, Pathak S, Bhattacharyya A, Kaisho T, Kundu M, Basu J. Direct extracellular interaction between the early secreted antigen ESAT-6 of Mycobacterium tuberculosis and TLR2 inhibits TLR signaling in macrophages. Nat Immunol 8: 610–618, 2007. [Erratum in Nat Immunol 16: 326, 2015]. doi: 10.1038/ni1468. [DOI] [PubMed] [Google Scholar]
- 126. Wang X, Barnes PF, Dobos-Elder KM, Townsend JC, Chung Y-T, Shams H, Weis SE, Samten B. ESAT-6 inhibits production of IFN-γ by Mycobacterium tuberculosis-responsive human T cells. J Immunol 182: 3668–3677, 2009. doi: 10.4049/jimmunol.0803579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Romagnoli A, Etna MP, Giacomini E, Pardini M, Remoli ME, Corazzari M, Falasca L, Goletti D, Gafa V, Simeone R, Delogu G, Piacentini M, Brosch R, Fimia GM, Coccia EM. ESX-1 dependent impairment of autophagic flux by Mycobacterium tuberculosis in human dendritic cells. Autophagy 8: 1357–1370, 2012. doi: 10.4161/auto.20881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Choi H-H, Shin D-M, Kang G, Kim K-H, Park JB, Hur GM, Lee H-M, Lim Y-J, Park J-K, Jo E-K, Song C-H. Endoplasmic reticulum stress response is involved in Mycobacterium tuberculosis protein ESAT-6-mediated apoptosis. FEBS Lett 584: 2445–2454, 2010. doi: 10.1016/j.febslet.2010.04.050. [DOI] [PubMed] [Google Scholar]
- 129. Guo S, Xue R, Li Y, Wang SM, Ren L, Xu JJ. The CFP10/ESAT6 complex of Mycobacterium tuberculosis may function as a regulator of macrophage cell death at different stages of tuberculosis infection. Med Hypotheses 78: 389–392, 2012. doi: 10.1016/j.mehy.2011.11.022. [DOI] [PubMed] [Google Scholar]
- 130. Kramarska E, Squeglia F, De Maio F, Delogu G, Berisio R. PE_PGRS33, an important virulence factor of Mycobacterium tuberculosis and potential target of host humoral immune response. Cells 10: 161, 2021. doi: 10.3390/cells10010161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Delogu G, Pusceddu C, Bua A, Fadda G, Brennan MJ, Zanetti S. Rv1818c‐encoded PE_PGRS protein of Mycobacterium tuberculosis is surface exposed and influences bacterial cell structure. Mol Microbiol 52: 725–733, 2004. doi: 10.1111/j.1365-2958.2004.04007.x. [DOI] [PubMed] [Google Scholar]
- 132. Esparza M, Palomares B, García T, Espinosa P, Zenteno E, Mancilla R. PstS-1, the 38-kDa Mycobacterium tuberculosis glycoprotein, is an adhesin, which binds the macrophage mannose receptor and promotes phagocytosis. Scand J Immunol 81: 46–55, 2015. doi: 10.1111/sji.12249. [DOI] [PubMed] [Google Scholar]
- 133. Briken V, Miller JL. Living on the edge: inhibition of host cell apoptosis by Mycobacterium tuberculosis. Future Microbiol 3: 415–422, 2008. doi: 10.2217/17460913.3.4.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Maliarik MJ, Iannuzzi MC. Host genetic factors in resistance and susceptibility to tuberculosis infection and disease. Semin Respir Crit Care Med 24: 223–228, 2003. doi: 10.1055/s-2003-39021. [DOI] [PubMed] [Google Scholar]
- 135. Azad AK, Sadee W, Schlesinger LS. Innate immune gene polymorphisms in tuberculosis. Infect Immun 80: 3343–3359, 2012. doi: 10.1128/IAI.00443-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. van Tong H, Velavan TP, Thye T, Meyer CG. Human genetic factors in tuberculosis: an update. Trop Med Int Health 22: 1063–1071, 2017. doi: 10.1111/tmi.12923. [DOI] [PubMed] [Google Scholar]
- 137. Kim SY, Jeong J-M, Kim SJ, Seo W, Kim M-H, Choi W-M, Yoo W, Lee J-H, Shim Y-R, Yi H-S, Lee Y-S, Eun HS, Lee BS, Chun K, Kang S-J, Kim SC, Gao B, Kunos G, Kim HM, Jeong W-I. Pro-inflammatory hepatic macrophages generate ROS through NADPH oxidase 2 via endocytosis of monomeric TLR4–MD2 complex. Nat Commun 8: 2247, 2017. doi: 10.1038/s41467-017-02325-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Divangahi M, Desjardins D, Nunes-Alves C, Remold HG, Behar SM. Eicosanoid pathways regulate adaptive immunity to Mycobacterium tuberculosis. Nat Immunol 11: 751–758, 2010. doi: 10.1038/ni.1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Balcewicz-Sablinska MK, Keane J, Kornfeld H, Remold HG. Pathogenic Mycobacterium tuberculosis evades apoptosis of host macrophages by release of TNF-R2, resulting in inactivation of TNF-α. J Immunol 161: 2636–2641, 1998. [PubMed] [Google Scholar]
- 140. Miller JL, Velmurugan K, Cowan MJ, Briken V. The type I NADH dehydrogenase of Mycobacterium tuberculosis counters phagosomal NOX2 activity to inhibit TNF-α-mediated host cell apoptosis. PLoS Pathog 6: e1000864, 2010. doi: 10.1371/journal.ppat.1000864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Rojas M, Olivier M, García LF. Activation of JAK2/STAT1-α-dependent signaling events during Mycobacterium tuberculosis-induced macrophage apoptosis. Cell Immunol 217: 58–66, 2002. doi: 10.1016/S0008-8749(02)00515-4. [DOI] [PubMed] [Google Scholar]
- 142. Clay H, Volkman HE, Ramakrishnan L., Tumor necrosis factor signaling mediates resistance to mycobacteria by inhibiting bacterial growth and macrophage death. Immunity 29: 283–294, 2008. doi: 10.1016/j.immuni.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Zhai W, Wu F, Zhang Y, Fu Y, Liu Z. The immune escape mechanisms of Mycobacterium tuberculosis. Int J Mol Sci 20: 340, 2019. doi: 10.3390/ijms20020340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Abdalla AE, Ejaz H, Mahjoob MO, Alameen AAM, Abosalif KOA, Elamir MYM, Mousa MA. Intelligent mechanisms of macrophage apoptosis subversion by Mycobacterium. Pathogens 9: 218, 2020. doi: 10.3390/pathogens9030218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Dennis EA, Norris PC., Eicosanoid storm in infection and inflammation. Nat Rev Immunol 15: 511–523, 2015. [Erratum in Nat Rev Immunol 15: 724, 2015]. doi: 10.1038/nri3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Agarwal P, Gordon S, Martinez FO. Foam cell macrophages in tuberculosis. Front Immunol 12: 775326, 2021. doi: 10.3389/fimmu.2021.775326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Behar SM, Martin CJ, Booty MG, Nishimura T, Zhao X, Gan H-X, Divangahi M, Remold HG. Apoptosis is an innate defense function of macrophages against Mycobacterium tuberculosis. Mucosal Immunol 4: 279–287, 2011. doi: 10.1038/mi.2011.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Xu G, Wang J, Gao GF, Liu CH. Insights into battles between Mycobacterium tuberculosis and macrophages. Protein Cell 5: 728–736, 2014. doi: 10.1007/s13238-014-0077-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Lam A, Prabhu R, Gross CM, Riesenberg LA, Singh V, Aggarwal S. Role of apoptosis and autophagy in tuberculosis. Am J Physiol Lung Cell Mol Physiol 313: L218–L229, 2017. doi: 10.1152/ajplung.00162.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Yang Y, Jiang G, Zhang P, Fan J. Programmed cell death and its role in inflammation. Mil Med Res 2: 12, 2015. doi: 10.1186/s40779-015-0039-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Berghe TV, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol 15: 135–147, 2014. doi: 10.1038/nrm3737. [DOI] [PubMed] [Google Scholar]
- 152. Chan FK-M, Luz NF, Moriwaki K. Programmed necrosis in the cross talk of cell death and inflammation. Annu Rev Immunol 33: 79–106, 2015. doi: 10.1146/annurev-immunol-032414-112248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Land WG. The role of damage-associated molecular patterns in human diseases: Part-1. Promoting inflammation and immunity. Sultan Qaboos Univ Med J 15: e9–21, 2015. [PMC free article] [PubMed] [Google Scholar]
- 154. Wang X, Lin Y. Tumor necrosis factor and cancer, buddies or foes? Acta Pharmacol Sin 29: 1275–1288, 2008. doi: 10.1111/j.1745-7254.2008.00889.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Vanlangenakker N, Vanden Berghe T, Vandenabeele P. Many stimuli pull the necrotic trigger, an overview. Cell Death Differ 19: 75–86, 2012. doi: 10.1038/cdd.2011.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev 94: 909–950, 2014. doi: 10.1152/physrev.00026.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Cai Z, Liu Z-G. Execution of RIPK3-regulated necrosis. Mol Cell Oncol 1: e960759, 2014. doi: 10.4161/23723548.2014.960759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Alu A, Han X, Ma X, Wu M, Wei Y, Wei X. The role of lysosome in regulated necrosis. Acta Pharm Sin B 10: 1880–1903, 2020. doi: 10.1016/j.apsb.2020.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Lerner TR, Borel S, Greenwood DJ, Repnik U, Russell MRG, Herbst S, Jones ML, Collinson LM, Griffiths G, Gutierrez MG. Mycobacterium tuberculosis replicates within necrotic human macrophages. J Cell Biol 216: 583–594, 2017. doi: 10.1083/jcb.201603040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Dallenga T, Repnik U, Corleis B, Eich J, Reimer R, Griffiths GW, Schaible UE. M. tuberculosis-induced necrosis of infected neutrophils promotes bacterial growth following phagocytosis by macrophages. Cell host Microbe 22: 519–530.e3, 2017. doi: 10.1016/j.chom.2017.09.003. [DOI] [PubMed] [Google Scholar]
- 161. Niazi MKK, Dhulekar N, Schmidt D, Major S, Cooper R, Abeijon C, Gatti DM, Kramnik I, Yener B, Gurcan M, Beamer G. Lung necrosis and neutrophils reflect common pathways of susceptibility to Mycobacterium tuberculosis in genetically diverse, immune-competent mice. Dis Model Mech 8: 1141–1153, 2015. doi: 10.1242/dmm.020867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Marzo E, Vilaplana C, Tapia G, Diaz J, Garcia V, Cardona P-J. Damaging role of neutrophilic infiltration in a mouse model of progressive tuberculosis. Tuberculosis (Edinb) 94: 55–64, 2014. doi: 10.1016/j.tube.2013.09.004. [DOI] [PubMed] [Google Scholar]
- 163. Filio-Rodríguez G, Estrada-García I, Arce-Paredes P, Moreno-Altamirano MM, Islas-Trujillo S, Ponce-Regalado MD, Rojas-Espinosa O. In vivo induction of neutrophil extracellular traps by Mycobacterium tuberculosis in a guinea pig model. Innate Immun 23: 625–637, 2017. doi: 10.1177/1753425917732406. [DOI] [PubMed] [Google Scholar]
- 164. Kim JK, Silwal P, Jo EK. Host-pathogen dialogues in autophagy, apoptosis, and necrosis during mycobacterial infection. Immune Netw 20: e37, 2020. doi: 10.4110/in.2020.20.e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Rybniker J, Chen JM, Sala C, Hartkoorn RC, Vocat A, Benjak A, Boy-Röttger S, Zhang M, Székely R, Greff Z, Orfi L, Szabadkai I, Pató J, Kéri G, Cole ST. Anticytolytic screen identifies inhibitors of mycobacterial virulence protein secretion. Cell Host Microbe 16: 538–548, 2014. doi: 10.1016/j.chom.2014.09.008. [DOI] [PubMed] [Google Scholar]
- 166. Peng X, Luo T, Zhai X, Zhang C, Suo J, Ma P, Wang C, Bao L. PPE11 of Mycobacterium tuberculosis can alter host inflammatory response and trigger cell death. Microb Pathog 126: 45–55, 2019. doi: 10.1016/j.micpath.2018.10.031. [DOI] [PubMed] [Google Scholar]
- 167. Li Z, Liu H, Li H, Dang G, Cui Z, Song N, Wang Q, Liu S, Chen L. PE17 protein from Mycobacterium tuberculosis enhances Mycobacterium smegmatis survival in macrophages and pathogenicity in mice. Microb Pathog 126: 63–73, 2019. doi: 10.1016/j.micpath.2018.10.030. [DOI] [PubMed] [Google Scholar]
- 168. Gopalakrishnan A, Dietzold J, Verma S, Bhagavathula M, Salgame P. Toll-like receptor 2 prevents neutrophil-driven immunopathology during infection with Mycobacterium tuberculosis by curtailing CXCL5 production. Infect Immun 87: e00760-18, 2019. doi: 10.1128/IAI.00760-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature 517: 311–320, 2015. doi: 10.1038/nature14191. [DOI] [PubMed] [Google Scholar]
- 170. Bertheloot D, Latz E, Franklin BS. Necroptosis, pyroptosis and apoptosis: an intricate game of cell death. Cell Mol Immunol 18: 1106–1121, 2021. doi: 10.1038/s41423-020-00630-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, Hupe M, Cain K, MacFarlane M, Häcker G, Leverkus M. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol Cell 43: 449–463, 2011. doi: 10.1016/j.molcel.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK-M. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137: 1112–1123, 2009. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao Y-S, Damko E, Moquin D, Walz T, McDermott A, Chan FK-M, Wu H. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 150: 339–350, 2012. doi: 10.1016/j.cell.2012.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Samson AL, Zhang Y, Geoghegan ND, Gavin XJ, Davies KA, Mlodzianoski MJ, Whitehead LW, Frank D, Garnish SE, Fitzgibbon C, Hempel A, Young SN, Jacobsen AV, Cawthorne W, Petrie EJ, Faux MC, Shield-Artin K, Lalaoui N, Hildebrand JM, Silke J, Rogers KL, Lessene G, Hawkins ED, Murphy JM. MLKL trafficking and accumulation at the plasma membrane control the kinetics and threshold for necroptosis. Nat Commun 11: 3151, 2020. doi: 10.1038/s41467-020-16887-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Schock SN, Chandra NV, Sun Y, Irie T, Kitagawa Y, Gotoh B, Coscoy L, Winoto A. Induction of necroptotic cell death by viral activation of the RIG-I or STING pathway. Cell Death Differ 24: 615–625, 2017. doi: 10.1038/cdd.2016.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Brault M, Olsen TM, Martinez J, Stetson DB, Oberst A. Intracellular nucleic acid sensing triggers necroptosis through synergistic type I IFN and TNF signaling. J Immunol 200: 2748–2756, 2018. doi: 10.4049/jimmunol.1701492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Gitlin AD, Heger K, Schubert AF, Reja R, Yan D, Pham VC, Suto E, Zhang J, Kwon YC, Freund EC, Kang J, Pham A, Caothien R, Bacarro N, Hinkle T, Xu M, McKenzie BS, Haley B, Lee WP, Lill JR, Roose-Girma M, Dohse M, Webster JD, Newton K, Dixit VM. Integration of innate immune signalling by caspase-8 cleavage of N4BP1. Nature 587: 275–280, 2020. doi: 10.1038/s41586-020-2796-5. [DOI] [PubMed] [Google Scholar]
- 178. Amarante-Mendes GP, Adjemian S, Branco LM, Zanetti LC, Weinlich R, Bortoluci KR. Pattern recognition receptors and the host cell death molecular machinery. Front Immunol 9: 2379, 2018. doi: 10.3389/fimmu.2018.02379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Feltham R, Vince JE, Lawlor KE. Caspase‐8: not so silently deadly. Clin Transl Immunology 6: e124, 2017. doi: 10.1038/cti.2016.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Pajuelo D, Gonzalez-Juarbe N, Tak U, Sun J, Orihuela CJ, Niederweis M. NAD+ depletion triggers macrophage necroptosis, a cell death pathway exploited by Mycobacterium tuberculosis. Cell Rep 24: 429–440, 2018. doi: 10.1016/j.celrep.2018.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol 7: 99–109, 2009. doi: 10.1038/nrmicro2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Maltez VI, Miao EA. Reassessing the evolutionary importance of inflammasomes. J Immunol 196: 956–962, 2016. doi: 10.4049/jimmunol.1502060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Yu P, Zhang X, Liu N, Tang L, Peng C, Chen X. Pyroptosis: mechanisms and diseases. Signal Transduct Target Ther 6: 128, 2021. doi: 10.1038/s41392-021-00507-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Wassermann R, Gulen MF, Sala C, Perin SG, Lou Y, Rybniker J, Schmid-Burgk JL, Schmidt T, Hornung V, Cole ST, Ablasser A. Mycobacterium tuberculosis differentially activates cGAS-and inflammasome-dependent intracellular immune responses through ESX-1. Cell host Microbe 17: 799–810, 2015. doi: 10.1016/j.chom.2015.05.003. [DOI] [PubMed] [Google Scholar]
- 185. Dorhoi A, Nouailles G, Jörg S, Hagens K, Heinemann E, Pradl L, Oberbeck-Müller D, Duque-Correa MA, Reece ST, Ruland J, Brosch R, Tschopp J, Gross O, Kaufmann SHE. Activation of the NLRP3 inflammasome by Mycobacterium tuberculosis is uncoupled from susceptibility to active tuberculosis. Eur J Immunol 42: 374–384, 2012. doi: 10.1002/eji.201141548. [DOI] [PubMed] [Google Scholar]
- 186. Lupfer CR, Kanneganti TD. The role of inflammasome modulation in virulence. Virulence 3: 262–270, 2012. doi: 10.4161/viru.20266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Labbé K, Saleh M. Cell death in the host response to infection. Cell Death Differ 15: 1339–1349, 2008. doi: 10.1038/cdd.2008.91. [DOI] [PubMed] [Google Scholar]
- 188. Rastogi S, Briken V. Interaction of mycobacteria with host cell inflammasomes. Front Immunol 13: 791136, 2022. doi: 10.3389/fimmu.2022.791136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Briken V, Ahlbrand S, Shah S. Mycobacterium tuberculosis and the host cell inflammasome: a complex relationship. Front Cell Infect Microbiol 3: 62, 2013. doi: 10.3389/fcimb.2013.00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Mishra BB, Moura-Alves P, Sonawane A, Hacohen N, Griffiths G, Moita LF, Anes E. Mycobacterium tuberculosis protein ESAT‐6 is a potent activator of the NLRP3/ASC inflammasome. Cell Microbiol 12: 1046–1063, 2010. doi: 10.1111/j.1462-5822.2010.01450.x. [DOI] [PubMed] [Google Scholar]
- 191. Briard B, Malireddi RKS, Kanneganti TD. Role of inflammasomes/pyroptosis and PANoptosis during fungal infection. PLoS Pathog 17: e1009358, 2021. doi: 10.1371/journal.ppat.1009358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Sahoo M, Ceballos-Olvera I, del Barrio L, Re F. Role of the inflammasome, IL-1β, and IL-18 in bacterial infections. ScientificWorldJournal 11: 2037–2050, 2011. doi: 10.1100/2011/212680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Koo IC, Wang C, Raghavan S, Morisaki JH, Cox JS, Brown EJ. ESX‐1‐dependent cytolysis in lysosome secretion and inflammasome activation during mycobacterial infection. Cell Microbiol 10: 1866–1878, 2008. doi: 10.1111/j.1462-5822.2008.01177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Sugawara I, Yamada H, Kaneko H, Mizuno S, Takeda K, Akira S. Role of interleukin-18 (IL-18) in mycobacterial infection in IL-18-gene-disrupted mice. Infect Immun 67: 2585–2589, 1999. doi: 10.1128/IAI.67.5.2585-2589.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Sugawara I, Yamada H, Hua S, Mizuno S. Role of interleukin (IL)‐1 type 1 receptor in mycobacterial infection. Microbiol Immunol 45: 743–750, 2001. doi: 10.1111/j.1348-0421.2001.tb01310.x. [DOI] [PubMed] [Google Scholar]
- 196. Juffermans NP, Florquin S, Camoglio L, Verbon A, Kolk AH, Speelman P, van Deventer SJ, van Der Poll T. Interleukin-1 signaling is essential for host defense during murine pulmonary tuberculosis. J Infect Dis 182: 902–908, 2000. doi: 10.1086/315771. [DOI] [PubMed] [Google Scholar]
- 197. McElvania Tekippe E, Allen IC, Hulseberg PD, Sullivan JT, McCann JR, Sandor M, Braunstein M, Ting JP-Y. Granuloma formation and host defense in chronic Mycobacterium tuberculosis infection requires PYCARD/ASC but not NLRP3 or caspase-1. PLoS One 5: e12320, 2010. doi: 10.1371/journal.pone.0012320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Mayer-Barber KD, Barber DL, Shenderov K, White SD, Wilson MS, Cheever A, Kugler D, Hieny S, Caspar P, Núñez G, Schlueter D, Flavell RA, Sutterwala FS, Sher A. Cutting edge: caspase-1 independent IL-1β production is critical for host resistance to Mycobacterium tuberculosis and does not require TLR signaling in vivo. J Immunol 184: 3326–3330, 2010. doi: 10.4049/jimmunol.0904189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Welin A, Eklund D, Stendahl O, Lerm M. Human macrophages infected with a high burden of ESAT-6-expressing M. tuberculosis undergo caspase-1-and cathepsin B-independent necrosis. PLoS One 6: e20302, 2011. doi: 10.1371/journal.pone.0020302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Willingham SB, Allen IC, Bergstralh DT, Brickey WJ, Huang MT-H, Taxman DJ, Duncan JA, Ting JP-Y. NLRP3 (NALP3, Cryopyrin) facilitates in vivo caspase-1 activation, necrosis, and HMGB1 release via inflammasome-dependent and -independent pathways. J Immunol 183: 2008–2015, 2009. doi: 10.4049/jimmunol.0900138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Satoh T, Kambe N, Matsue H. NLRP3 activation induces ASC-dependent programmed necrotic cell death, which leads to neutrophilic inflammation. Cell Death Dis 4: e644, 2013. doi: 10.1038/cddis.2013.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Duncan JA, Gao X, Huang MT-H, O'Connor BP, Thomas CE, Willingham SB, Bergstralh DT, Jarvis GA, Sparling PF, Ting JP-Y. Neisseria gonorrhoeae activates the proteinase cathepsin B to mediate the signaling activities of the NLRP3 and ASC-containing inflammasome. J Immunol 182: 6460–6469, 2009. doi: 10.4049/jimmunol.0802696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Willingham SB, Bergstralh DT, O'Connor W, Morrison AC, Taxman DJ, Duncan JA, Barnoy S, Venkatesan MM, Flavell RA, Deshmukh M, Hoffman HM, Ting JP-Y. Microbial pathogen-induced necrotic cell death mediated by the inflammasome components CIAS1/cryopyrin/NLRP3 and ASC. Cell Host Microbe 2: 147–159, 2007. doi: 10.1016/j.chom.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B, Stockwell BR. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149: 1060–1072, 2012. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Cao JY, Dixon SJ. Mechanisms of ferroptosis. Cell Mol Life Sci 73: 2195–2209, 2016. doi: 10.1007/s00018-016-2194-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascón S, Hatzios SK, Kagan VE, Noel K, Jiang X, Linkermann A, Murphy ME, Overholtzer M, Oyagi A, Pagnussat GC, Park J, Ran Q, Rosenfeld CS, Salnikow K, Tang D, Torti FM, Torti SV, Toyokuni S, Woerpel KA, Zhang DD. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171: 273–285, 2017. doi: 10.1016/j.cell.2017.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Yu H, Guo P, Xie X, Wang Y, Chen G. Ferroptosis, a new form of cell death, and its relationships with tumourous diseases. J Cell Mol Med 21: 648–657, 2017. doi: 10.1111/jcmm.13008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, Kang R, Tang D. Ferroptosis: process and function. Cell Death Differ 23: 369–379, 2016. doi: 10.1038/cdd.2015.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Meunier E, Neyrolles O. Die another way: ferroptosis drives tuberculosis pathology. J Exp Med 216: 471–473, 2019. doi: 10.1084/jem.20190038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Amaral EP, Costa DL, Namasivayam S, Riteau N, Kamenyeva O, Mittereder L, Mayer-Barber KD, Andrade BB, Sher A. A major role for ferroptosis in Mycobacterium tuberculosis–induced cell death and tissue necrosis. J Exp Med 216: 556–570, 2019. doi: 10.1084/jem.20181776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Guimaraes-Costa AB, Nascimento MT, Wardini AB, Pinto-da-Silva LH, Saraiva EM. ETosis: a microbicidal mechanism beyond cell death. J Parasitol Res 2012: 929743, 2012. doi: 10.1155/2012/929743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Delgado-Rizo V, Martínez-Guzmán MA, Iñiguez-Gutierrez L, García-Orozco A, Alvarado-Navarro A, Fafutis-Morris M. Neutrophil extracellular traps and its implications in inflammation: an overview. Front Immunol 8: 81, 2017. doi: 10.3389/fimmu.2017.00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Wong K-W, Jacobs WR Jr.. Mycobacterium tuberculosis exploits human interferon γ to stimulate macrophage extracellular trap formation and necrosis. J Infect Dis 208: 109–119, 2013. doi: 10.1093/infdis/jit097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Ueki S, Tokunaga T, Fujieda S, Honda K, Hirokawa M, Spencer LA, Weller PF. Eosinophil ETosis and DNA traps: a new look at eosinophilic inflammation. Curr Allergy Asthma Rep 16: 54, 2016. doi: 10.1007/s11882-016-0634-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science 303: 1532–1535, 2004. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 216. Ramos-Kichik V, Mondragón-Flores R, Mondragón-Castelán M, Gonzalez-Pozos S, Muñiz-Hernandez S, Rojas-Espinosa O, Chacón-Salinas R, Estrada-Parra S, Estrada-García I. Neutrophil extracellular traps are induced by Mycobacterium tuberculosis. Tuberculosis (Edinb) 89: 29–37, 2009. doi: 10.1016/j.tube.2008.09.009. [DOI] [PubMed] [Google Scholar]
- 217. Braian C, Hogea V, Stendahl O. Mycobacterium tuberculosis-induced neutrophil extracellular traps activate human macrophages. J Innate Immun 5: 591–602, 2013. doi: 10.1159/000348676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Kalsum S, Braian C, Koeken VACM, Raffetseder J, Lindroth M, van Crevel R, Lerm M. The cording phenotype of Mycobacterium tuberculosis induces the formation of extracellular traps in human macrophages. Front Cell Infect Microbiol 7: 278, 2017. doi: 10.3389/fcimb.2017.00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Echevarría LU, Leimgruber C, García González J, Nevado A, Álvarez R, García LN, Quintar AA, Maldonado CA. Evidence of eosinophil extracellular trap cell death in COPD: does it represent the trigger that switches on the disease? Int J Chron Obstruct Pulmon Dis 12: 885, 2017. doi: 10.2147/COPD.S115969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Jorgensen I, Zhang Y, Krantz BA, Miao EA. Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. J Exp Med 213: 2113–2128, 2016. doi: 10.1084/jem.20151613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Fatokun AA, Dawson VL, Dawson TM. Parthanatos: mitochondrial‐linked mechanisms and therapeutic opportunities. Br J Pharmacol 171: 2000–2016, 2014. doi: 10.1111/bph.12416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Virág L, Robaszkiewicz A, Rodriguez-Vargas JM, Oliver FJ. Poly (ADP-ribose) signaling in cell death. Mol Aspects Med 34: 1153–1167, 2013. doi: 10.1016/j.mam.2013.01.007. [DOI] [PubMed] [Google Scholar]
- 223. David KK, Andrabi SA, Dawson TM, Dawson VL. Parthanatos, a messenger of death. Front Biosci (Landmark Ed) 14: 1116–1128, 2009. doi: 10.2741/3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Yu S-W, Andrabi SA, Wang H, Kim NS, Poirier GG, Dawson TM, Dawson VL. Apoptosis-inducing factor mediates poly (ADP-ribose)(PAR) polymer-induced cell death. Proc Natl Acad Sci USA 103: 18314–18319, 2006. doi: 10.1073/pnas.0606528103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Wang Y, Kim NS, Haince J-F, Kang HC, David KK, Andrabi SA, Poirier GG, Dawson VL, Dawson TM. Poly (ADP-ribose)(PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1–dependent cell death (parthanatos). Sci Signal 4: ra20, 2011. doi: 10.1126/scisignal.2000902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Wang H, Yu S-W, Koh DW, Lew J, Coombs C, Bowers W, Federoff HJ, Poirier GG, Dawson TM, Dawson VL. Apoptosis-inducing factor substitutes for caspase executioners in NMDA-triggered excitotoxic neuronal death. J Neurosci 24: 10963–10973, 2004. doi: 10.1523/JNEUROSCI.3461-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Wang Y, An R, Umanah GK, Park H, Nambiar K, Eacker SM, Kim BWoo, Bao L, Harraz MM, Chang C, Chen R, Wang JE, Kam T-I, Jeong JS, Xie Z, Neifert S, Qian J, Andrabi SA, Blackshaw S, Zhu H, Song H, Ming G-L, Dawson VL, Dawson TM. A nuclease that mediates cell death induced by DNA damage and poly (ADP-ribose) polymerase-1. Science 354: aad6872, 2016. doi: 10.1126/science.aad6872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Kam T-I, Mao X, Park H, Chou S-C, Karuppagounder SS, Umanah GE, Yun SP, Brahmachari S, Panicker N, Chen R, Andrabi SA, Qi C, Poirier GG, Pletnikova O, Troncoso JC, Bekris LM, Leverenz JB, Pantelyat A, Ko HS, Rosenthal LS, Dawson TM, Dawson VL. Poly(ADP-ribose) drives pathologic α-synuclein neurodegeneration in Parkinson's disease. Science 362: eaat8407, 2018. doi: 10.1126/science.aat8407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Zhang L, Jiang X, Pfau D, Ling Y, Nathan CF. Type I interferon signaling mediates Mycobacterium tuberculosis–induced macrophage death. J Exp Med 218: e20200887, 2021. doi: 10.1084/jem.20200887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Sridharan H, Upton JW. Programmed necrosis in microbial pathogenesis. Trends Microbiol 22: 199–207, 2014. doi: 10.1016/j.tim.2014.01.005. [DOI] [PubMed] [Google Scholar]
- 231. Ryter SW, Cloonan SM, Choi AM. Autophagy: a critical regulator of cellular metabolism and homeostasis. Mol Cells 36: 7–16, 2013. doi: 10.1007/s10059-013-0140-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol 10: 458–467, 2009. doi: 10.1038/nrm2708. [DOI] [PubMed] [Google Scholar]
- 233. Parzych KR, Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal 20: 460–473, 2014. doi: 10.1089/ars.2013.5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Mizushima N. Autophagy: process and function. Genes Dev 21: 2861–2873, 2007. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 235. Yorimitsu T, Klionsky DJ. Autophagy: molecular machinery for self-eating. Cell Death Differ 12: 1542–1552, 2005. doi: 10.1038/sj.cdd.4401765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol 22: 124–131, 2010. doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Paulus GL, Xavier RJ. Autophagy and checkpoints for intracellular pathogen defense. Curr Opin Gastroenterol 31: 14–23, 2015. doi: 10.1097/MOG.0000000000000134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Castrejón-Jiménez NS, Leyva-Paredes K, Hernández-González JC, Luna-Herrera J, García-Pérez BE. The role of autophagy in bacterial infections. Biosci Trends 9: 149–159, 2015. doi: 10.5582/bst.2015.01035. [DOI] [PubMed] [Google Scholar]
- 239. Tattoli I, Sorbara MT, Vuckovic D, Ling A, Soares F, Carneiro LAM, Yang C, Emili A, Philpott DJ, Girardin SE. Amino acid starvation induced by invasive bacterial pathogens triggers an innate host defense program. Cell host Microbe 11: 563–575, 2012. doi: 10.1016/j.chom.2012.04.012. [DOI] [PubMed] [Google Scholar]
- 240. Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell 176: 11–42, 2019. doi: 10.1016/j.cell.2018.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Deretic V. Autophagy in tuberculosis. Cold Spring Harb Perspect Med 4: a018481, 2014. doi: 10.1101/cshperspect.a018481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Chauhan S, Mandell MA, Deretic V. Mechanism of action of the tuberculosis and Crohn disease risk factor IRGM in autophagy. Autophagy 12: 429–431, 2016. doi: 10.1080/15548627.2015.1084457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Svenning S, Johansen T. Selective autophagy. Essays Biochem 55: 79–92, 2013. doi: 10.1042/bse0550079. [DOI] [PubMed] [Google Scholar]
- 244. He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 43: 67–93, 2009. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Kim YK, Shin JS, Nahm MH. NOD-like receptors in infection, immunity, and diseases. Yonsei Med J 57: 5–14, 2016. doi: 10.3349/ymj.2016.57.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Wicherska-Pawłowska K, Wróbel T, Rybka J. Toll-like receptors (TLRs), NOD-like receptors (NLRs), and RIG-I-like receptors (RLRs) in innate immunity. TLRs, NLRs, and RLRs ligands as immunotherapeutic agents for hematopoietic diseases. Int J Mol Sci 22: 13397, 2021. doi: 10.3390/ijms222413397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Xu Y, Jagannath C, Liu X-D, Sharafkhaneh A, Kolodziejska KE, Eissa NT. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 27: 135–144, 2007. doi: 10.1016/j.immuni.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Watson RO, Manzanillo PS, Cox JS. Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell 150: 803–815, 2012. doi: 10.1016/j.cell.2012.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Konno H, Konno K, Barber GN. Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell 155: 688–698, 2013. doi: 10.1016/j.cell.2013.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Pilli M, Arko-Mensah J, Ponpuak M, Roberts E, Master S, Mandell MA, Dupont N, Ornatowski W, Jiang S, Bradfute SB, Bruun J-A, Hansen TE, Johansen T, Deretic V. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity 37: 223–234, 2012. doi: 10.1016/j.immuni.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Klug-Micu GM, Stenger S, Sommer A, Liu PT, Krutzik SR, Modlin RL, Fabri M. CD40 ligand and interferon-γ induce an antimicrobial response against M ycobacterium tuberculosis in human monocytes. Immunology 139: 121–128, 2013. doi: 10.1111/imm.12062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Petruccioli E, Romagnoli A, Corazzari M, Coccia EM, Butera O, Delogu G, Piacentini M, Girardi E, Fimia GM, Goletti D. Specific T cells restore the autophagic flux inhibited by Mycobacterium tuberculosis in human primary macrophages. J Infect Dis 205: 1425–1435, 2012. doi: 10.1093/infdis/jis226. [DOI] [PubMed] [Google Scholar]
- 253. Singh SB, Ornatowski W, Vergne I, Naylor J, Delgado M, Roberts E, Ponpuak M, Master S, Pilli M, White E, Komatsu M, Deretic V. Human IRGM regulates autophagy and cell-autonomous immunity functions through mitochondria. Nat Cell Biol 12: 1154–1165, 2010. doi: 10.1038/ncb2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. de Martino M, Lodi L, Galli L, Chiappini E. Immune response to Mycobacterium tuberculosis: a narrative review. Front Pediatr 7: 350, 2019. doi: 10.3389/fped.2019.00350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Bussi C, Gutierrez MG. Mycobacterium tuberculosis infection of host cells in space and time. FEMS Microbiol Rev 43: 341–361, 2019. doi: 10.1093/femsre/fuz006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Dutta RK, Kathania M, Raje M, Majumdar S. IL-6 inhibits IFN-γ induced autophagy in Mycobacterium tuberculosis H37Rv infected macrophages. Int J Biochem Cell Biol 44: 942–954, 2012. doi: 10.1016/j.biocel.2012.02.021. [DOI] [PubMed] [Google Scholar]
- 257. Juárez E, Carranza C, Hernández-Sánchez F, León-Contreras JC, Hernández-Pando R, Escobedo D, Torres M, Sada E. NOD 2 enhances the innate response of alveolar macrophages to M ycobacterium tuberculosis in humans. Eur J Immunol 42: 880–889, 2012. doi: 10.1002/eji.201142105. [DOI] [PubMed] [Google Scholar]
- 258. Seto S, Tsujimura K, Horii T, Koide Y. Autophagy adaptor protein p62/SQSTM1 and autophagy-related gene Atg5 mediate autophagosome formation in response to Mycobacterium tuberculosis infection in dendritic cells. PLoS One 8: e86017, 2013. doi: 10.1371/journal.pone.0086017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Sakowski ET, Koster S, Portal Celhay C, Park HS, Shrestha E, Hetzenecker SE, Maurer K, Cadwell K, Philips JA. Ubiquilin 1 promotes IFN-γ-induced xenophagy of Mycobacterium tuberculosis. PLoS Pathog 11: e1005076, 2015. doi: 10.1371/journal.ppat.1005076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 119: 753–766, 2004. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 261. Wang J, Yang K, Zhou L, Wu Y, Zhu M, Lai X, Chen T, Feng L, Li M, Huang C, Zhong Q, Huang X. MicroRNA-155 promotes autophagy to eliminate intracellular mycobacteria by targeting Rheb. PLoS Pathog 9: e1003697, 2013. doi: 10.1371/journal.ppat.1003697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Kimmey JM, Huynh JP, Weiss LA, Park S, Kambal A, Debnath J, Virgin HW, Stallings CL. Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature 528: 565–569, 2015. doi: 10.1038/nature16451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Castillo EF, Dekonenko A, Arko-Mensah J, Mandell MA, Dupont N, Jiang S, Delgado-Vargas M, Timmins GS, Bhattacharya D, Yang H, Hutt J, Lyons CR, Dobos KM, Deretic V. Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc Natl Acad Sci USA 109: E3168–E3176, 2012. doi: 10.1073/pnas.1210500109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Nandi B, Behar SM. Regulation of neutrophils by interferon-γ limits lung inflammation during tuberculosis infection. J Exp Med 208: 2251–2262, 2011. doi: 10.1084/jem.20110919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Hwang S, Maloney NS, Bruinsma MW, Goel G, Duan E, Zhang L, Shrestha B, Diamond MS, Dani A, Sosnovtsev SV, Green KY, Lopez-Otin C, Xavier RJ, Thackray LB, Virgin HW. Nondegradative role of Atg5-Atg12/Atg16L1 autophagy protein complex in antiviral activity of interferon γ. Cell host Microbe 11: 397–409, 2012. doi: 10.1016/j.chom.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Zhao Z, Fux B, Goodwin M, Dunay IR, Strong D, Miller BC, Cadwell K, Delgado MA, Ponpuak M, Green KG, Schmidt RE, Mizushima N, Deretic V, Sibley LD, Virgin HW. Autophagosome-independent essential function for the autophagy protein Atg5 in cellular immunity to intracellular pathogens. Cell host Microbe 4: 458–469, 2008. doi: 10.1016/j.chom.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Martinez J, Malireddi RKS, Lu Q, Cunha LD, Pelletier S, Gingras S, Orchard R, Guan J-L, Tan H, Peng J, Kanneganti T-D, Virgin HW, Green DR. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat Cell Biol 17: 893–906, 2015. doi: 10.1038/ncb3192. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 268. Jounai N, Takeshita F, Kobiyama K, Sawano A, Miyawaki A, Xin K-Q, Ishii KJ, Kawai T, Akira S, Suzuki K, Okuda K. The Atg5–Atg12 conjugate associates with innate antiviral immune responses. Proc Natl Acad Sci USA 104: 14050–14055, 2007. doi: 10.1073/pnas.0704014104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, Brunner T, Simon H-U. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol 8: 1124–1132, 2006. doi: 10.1038/ncb1482. [DOI] [PubMed] [Google Scholar]
- 270. Maskey D, Yousefi S, Schmid I, Zlobec I, Perren A, Friis R, Simon H-U. ATG5 is induced by DNA-damaging agents and promotes mitotic catastrophe independent of autophagy. Nat Commun 4: 2130, 2013. doi: 10.1038/ncomms3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Strong EJ, Lee S. Targeting autophagy as a strategy for developing new vaccines and host-directed therapeutics against mycobacteria. Front Microbiol 11: 614313, 2021. doi: 10.3389/fmicb.2020.614313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 6: 764–776, 2010. doi: 10.4161/auto.6.6.12709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol 22: 132–139, 2010. doi: 10.1016/j.ceb.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 274. Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G, Ktistakis NT. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol 182: 685–701, 2008. doi: 10.1083/jcb.200803137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, Maejima I, Shirahama-Noda K, Ichimura T, Isobe T, Akira S, Noda T, Yoshimori T. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol 11: 385–396, 2009. doi: 10.1038/ncb1846. [DOI] [PubMed] [Google Scholar]
- 276. Polson HEJ, de Lartigue J, Rigden DJ, Reedijk M, Urbé S, Clague MJ, Tooze SA. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy 6: 506–522, 2010. doi: 10.4161/auto.6.4.11863. [DOI] [PubMed] [Google Scholar]
- 277. Fujita N, Itoh T, Omori H, Fukuda M, Noda T, Yoshimori T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell 19: 2092–2100, 2008. doi: 10.1091/mbc.e07-12-1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278. Knodler LA, Celli J. Eating the strangers within: host control of intracellular bacteria via xenophagy. Cell Microbiol 13: 1319–1327, 2011. doi: 10.1111/j.1462-5822.2011.01632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279. Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature 469: 323–335, 2011. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Münz C. Autophagy beyond intracellular MHC class II antigen presentation. Trends Immunol 37: 755–763, 2016. doi: 10.1016/j.it.2016.08.017. [DOI] [PubMed] [Google Scholar]
- 281. Saini NK, Baena A, Ng TW, Venkataswamy MM, Kennedy SC, Kunnath-Velayudhan S, Carreño LJ, Xu J, Chan J, Larsen MH, Jacobs WR, Porcelli SA. Suppression of autophagy and antigen presentation by Mycobacterium tuberculosis PE_PGRS47. Nat Microbiol 1: 16133, 2016. doi: 10.1038/nmicrobiol.2016.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Yu L, Chen Y, Tooze SA. Autophagy pathway: cellular and molecular mechanisms. Autophagy 14: 207–215, 2018. doi: 10.1080/15548627.2017.1378838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283. Jiao Y, Sun J. Bacterial manipulation of autophagic responses in infection and inflammation. Front Immunol 10: 2821, 2019. doi: 10.3389/fimmu.2019.02821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284. Conrad WH, Osman MM, Shanahan JK, Chu F, Takaki KK, Cameron J, Hopkinson-Woolley D, Brosch R, Ramakrishnan L. Mycobacterial ESX-1 secretion system mediates host cell lysis through bacterium contact-dependent gross membrane disruptions. Proc Natl Acad Sci USA 114: 1371–1376, 2017. doi: 10.1073/pnas.1620133114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285. Watson RO, Bell SL, MacDuff DA, Kimmey JM, Diner EJ, Olivas J, Vance RE, Stallings CL, Virgin HW, Cox JS. The cytosolic sensor cGAS detects Mycobacterium tuberculosis DNA to induce type i interferons and activate autophagy. Cell Host Microbe 17: 811–819, 2015. doi: 10.1016/j.chom.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286. Thurston TLM, Ryzhakov G, Bloor S, von Muhlinen N, Randow F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol 10: 1215–1221, 2009. doi: 10.1038/ni.1800. [DOI] [PubMed] [Google Scholar]
- 287. Zheng YT, Shahnazari S, Brech A, Lamark T, Johansen T, Brumell JH. The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J Immunol 183: 5909–5916, 2009. doi: 10.4049/jimmunol.0900441. [DOI] [PubMed] [Google Scholar]
- 288. Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, Richter B, Korac J, Waidmann O, Choudhary C, Dötsch V, Bumann D, Dikic I. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 333: 228–233, 2011. doi: 10.1126/science.1205405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289. Huang D, Bao L. Mycobacterium tuberculosis EspB protein suppresses interferon-γ-induced autophagy in murine macrophages. J Microbiol Immunol Infect 49: 859–865, 2016. doi: 10.1016/j.jmii.2014.11.008. [DOI] [PubMed] [Google Scholar]
- 290. Wei J, Dahl JL, Moulder JW, Roberts EA, O'Gaora P, Young DB, Friedman RL. Identification of a Mycobacterium tuberculosis gene that enhances mycobacterial survival in macrophages. J Bacteriol 182: 377–384, 2000. doi: 10.1128/JB.182.2.377-384.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Kim KH, An DR, Song J, Yoon JY, Kim HS, Yoon HJ, Im HN, Kim J, Kim DJ, Lee SJ, Kim K-H, Lee H-M, Kim H-J, Jo E-K, Lee JY, Suh SW. Mycobacterium tuberculosis Eis protein initiates suppression of host immune responses by acetylation of DUSP16/MKP-7. Proc Natl Acad Sci USA 109: 7729–7734, 2012. doi: 10.1073/pnas.1120251109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292. Lella RK, Sharma C. Eis (enhanced intracellular survival) protein of Mycobacterium tuberculosis disturbs the cross regulation of T-cells. J Biol Chem 282: 18671–18675, 2007. doi: 10.1074/jbc.C600280200. [DOI] [PubMed] [Google Scholar]
- 293. Samuel LP, Song C-H, Wei J, Roberts EA, Dahl JL, Barry CE, Jo E-K, Friedman RL. Expression, production and release of the Eis protein by Mycobacterium tuberculosis during infection of macrophages and its effect on cytokine secretion. Microbiology (Reading) 153: 529–540, 2007. doi: 10.1099/mic.0.2006/002642-0. [DOI] [PubMed] [Google Scholar]
- 294. Chai Q, Wang X, Qiang L, Zhang Y, Ge P, Lu Z, Zhong Y, Li B, Wang J, Zhang L, Zhou D, Li W, Dong W, Pang Y, Gao GF, Liu CH. A Mycobacterium tuberculosis surface protein recruits ubiquitin to trigger host xenophagy. Nat Commun 10: 1973, 2019. doi: 10.1038/s41467-019-09955-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295. Köster S, Upadhyay S, Chandra P, Papavinasasundaram K, Yang G, Hassan A, Grigsby SJ, Mittal E, Park HS, Jones V, Hsu F-F, Jackson M, Sassetti CM, Philips JA. Mycobacterium tuberculosis is protected from NADPH oxidase and LC3-associated phagocytosis by the LCP protein CpsA. Proc Natl Acad Sci USA 114: E8711–E8720, 2017. [Erratum in Proc Natl Acad Sci USA 114: E9752, 2017]. doi: 10.1073/pnas.1707792114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296. Fazeli G, Wehman AM. Rab GTPases mature the LC3-associated midbody phagosome. Commun Integr Biol 10: e1297349, 2017. doi: 10.1080/19420889.2017.1297349. [DOI] [Google Scholar]
- 297. Rink J, Ghigo E, Kalaidzidis Y, Zerial M. Rab conversion as a mechanism of progression from early to late endosomes. Cell 122: 735–749, 2005. doi: 10.1016/j.cell.2005.06.043. [DOI] [PubMed] [Google Scholar]
- 298. Jo E-K. Autophagy as an innate defense against mycobacteria. Pathog Dis 67: 108–118, 2013. doi: 10.1111/2049-632X.12023. [DOI] [PubMed] [Google Scholar]
- 299. Jo E-K, Yang C-S, Choi CH, Harding CV. Intracellular signalling cascades regulating innate immune responses to Mycobacteria: branching out from Toll‐like receptors. Cell Microbiol 9: 1087–1098, 2007. doi: 10.1111/j.1462-5822.2007.00914.x. [DOI] [PubMed] [Google Scholar]
- 300. Kleinnijenhuis J, Oosting M, Joosten LAB, Netea MG, Van Crevel R. Innate immune recognition of Mycobacterium tuberculosis. Clin Dev Immunol 2011: 405310, 2011. doi: 10.1155/2011/405310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301. Saiga H, Shimada Y, Takeda K. Innate immune effectors in mycobacterial infection. Clin Dev Immunol 2011: 347594, 2011. doi: 10.1155/2011/347594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302. Sanjuan MA, Dillon CP, Tait SWG, Moshiach S, Dorsey F, Connell S, Komatsu M, Tanaka K, Cleveland JL, Withoff S, Green DR. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 450: 1253–1257, 2007. doi: 10.1038/nature06421. [DOI] [PubMed] [Google Scholar]
- 303. Sanjuan MA, Milasta S, Green DR. Toll‐like receptor signaling in the lysosomal pathways. Immunol Rev 227: 203–220, 2009. doi: 10.1111/j.1600-065X.2008.00732.x. [DOI] [PubMed] [Google Scholar]
- 304. Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V. Toll‐like receptors control autophagy. EMBO J 27: 1110–1121, 2008. doi: 10.1038/emboj.2008.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305. Orvedahl A, Levine B. Eating the enemy within: autophagy in infectious diseases. Cell Death Differ 16: 57–69, 2009. doi: 10.1038/cdd.2008.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306. Shi C-S, Kehrl JH. MyD88 and Trif target Beclin 1 to trigger autophagy in macrophages. J Biol Chem 283: 33175–33182, 2008. doi: 10.1074/jbc.M804478200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307. Shin D-M, Yuk J-M, Lee H-M, Lee S-H, Son JW, Harding CV, Kim J-M, Modlin RL, Jo E-K. Mycobacterial lipoprotein activates autophagy via TLR2/1/CD14 and a functional vitamin D receptor signalling. Cell Microbiol 12: 1648–1665, 2010. doi: 10.1111/j.1462-5822.2010.01497.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308. Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P, Ferguson DJP, Campbell BJ, Jewell D, Simmons A. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med 16: 90–97, 2010. doi: 10.1038/nm.2069. [DOI] [PubMed] [Google Scholar]
- 309. Travassos LH, Carneiro LAM, Ramjeet M, Hussey S, Kim Y-G, Magalhães JG, Yuan L, Soares F, Chea E, Le Bourhis L, Boneca IG, Allaoui A, Jones NL, Nuñez G, Girardin SE, Philpott DJ. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol 11: 55–62, 2010. doi: 10.1038/ni.1823. [DOI] [PubMed] [Google Scholar]
- 310. Maeda S, Hsu L-C, Liu H, Bankston LA, Iimura M, Kagnoff MF, Eckmann L, Karin M. Nod2 mutation in Crohn's disease potentiates NF-κB activity and IL-1β processing. Science 307: 734–738, 2005. [Erratum in Science 308: 633, 2005]. doi: 10.1126/science.1103685. [DOI] [PubMed] [Google Scholar]
- 311. Coulombe F, Divangahi M, Veyrier F, de Léséleuc L, Gleason JL, Yang Y, Kelliher MA, Pandey AK, Sassetti CM, Reed MB, Behr MA. Increased NOD2-mediated recognition of N-glycolyl muramyl dipeptide. J Exp Med 206: 1709–1716, 2009. doi: 10.1084/jem.20081779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312. Harris J, De Haro SA, Master SS, Keane J, Roberts EA, Delgado M, Deretic V. T helper 2 cytokines inhibit autophagic control of intracellular Mycobacterium tuberculosis. Immunity 27: 505–517, 2007. [Erratum in Immunity 27: 685, 2007]. doi: 10.1016/j.immuni.2007.07.022. [DOI] [PubMed] [Google Scholar]
- 313. MacMicking JD, Taylor GA, McKinney JD. Immune control of tuberculosis by IFN-gamma-inducible LRG-47. Science 302: 654–659, 2003. doi: 10.1126/science.1088063. [DOI] [PubMed] [Google Scholar]
- 314. Harding CV, Boom WH. Regulation of antigen presentation by Mycobacterium tuberculosis: a role for Toll-like receptors. Nat Rev Microbiol 8: 296–307, 2010. doi: 10.1038/nrmicro2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315. Harris J, Hope JC, Keane J. Tumor necrosis factor blockers influence macrophage responses to Mycobacterium tuberculosis. J Infect Dis 198: 1842–1850, 2008. doi: 10.1086/593174. [DOI] [PubMed] [Google Scholar]
- 316. Harris J, Keane J. How tumour necrosis factor blockers interfere with tuberculosis immunity. Clin Exp Immunol 161: 1–9, 2010. doi: 10.1111/j.1365-2249.2010.04146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317. Harris J. Autophagy and cytokines. Cytokine 56: 140–144, 2011. doi: 10.1016/j.cyto.2011.08.022. [DOI] [PubMed] [Google Scholar]
- 318. Cooper AM, Mayer-Barber KD, Sher A. Role of innate cytokines in mycobacterial infection. Mucosal Immunol 4: 252–260, 2011. doi: 10.1038/mi.2011.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319. Yang C-S, Song C-H, Lee J-S, Jung S-B, Oh J-H, Park J, Kim H-J, Park J-K, Paik T-H, Jo E-K. Intracellular network of phosphatidylinositol 3-kinase, mammalian target of the rapamycin/70 kDa ribosomal S6 kinase 1, and mitogen-activated protein kinases pathways for regulating mycobacteria-induced IL-23 expression in human macrophages. Cell Microbiol 8: 1158–1171, 2006. doi: 10.1111/j.1462-5822.2006.00699.x. [DOI] [PubMed] [Google Scholar]
- 320. Gerosa F, Baldani-Guerra B, Lyakh LA, Batoni G, Esin S, Winkler-Pickett RT, Consolaro MR, De Marchi M, Giachino D, Robbiano A, Astegiano M, Sambataro A, Kastelein RA, Carra G, Trinchieri G. Differential regulation of interleukin 12 and interleukin 23 production in human dendritic cells. J Exp Med 205: 1447–1461, 2008. doi: 10.1084/jem.20071450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321. Saitoh T, Fujita N, Jang MH, Uematsu S, Yang B-G, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, Tanaka K, Kawai T, Tsujimura T, Takeuchi O, Yoshimori T, Akira S. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature 456: 264–268, 2008. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 322. Crişan TO, Plantinga TS, van de Veerdonk FL, Farcaş MF, Stoffels M, Kullberg B-J, van der Meer JWM, Joosten LAB, Netea MG. Inflammasome-independent modulation of cytokine response by autophagy in human cells. PLoS One 6: e18666, 2011. doi: 10.1371/journal.pone.0018666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323. Harris J, Hartman M, Roche C, Zeng SG, O'Shea A, Sharp FA, Lambe EM, Creagh EM, Golenbock DT, Tschopp J, Kornfeld H, Fitzgerald KA, Lavelle EC. Autophagy controls IL-1β secretion by targeting pro-IL-1β for degradation. J Biol Chem 286: 9587–9597, 2011. doi: 10.1074/jbc.M110.202911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324. Nakahira K, Haspel JA, Rathinam VAK, Lee S-J, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, Fitzgerald KA, Ryter SW, Choi AMK. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12: 222–230, 2011. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature 469: 221–225, 2011. [Erratum in Nature 475: 122, 2011]. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 326. Jo EK, Shin DM, Choi AM. Autophagy: cellular defense to excessive inflammation. Microbes Infect 14: 119–125, 2012. doi: 10.1016/j.micinf.2011.08.014. [DOI] [PubMed] [Google Scholar]
- 327. Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, Deretic V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J 30: 4701–4711, 2011. doi: 10.1038/emboj.2011.398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328. Wang Y, Kanneganti T-D. From pyroptosis, apoptosis and necroptosis to PANoptosis: A mechanistic compendium of programmed cell death pathways. Comput Struct Biotechnol J 19: 4641–4657, 2021. doi: 10.1016/j.csbj.2021.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329. Christgen S, Zheng M, Kesavardhana S, Karki R, Malireddi RKS, Banoth B, Place DE, Briard B, Sharma BR, Tuladhar S, Samir P, Burton A, Kanneganti T-D. Identification of the PANoptosome: a molecular platform triggering pyroptosis, apoptosis, and necroptosis (PANoptosis). Front Cell Infect Microbiol 10: 237, 2020. doi: 10.3389/fcimb.2020.00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330. Banoth B, Tuladhar S, Karki R, Sharma BR, Briard B, Kesavardhana S, Burton A, Kanneganti T-D. ZBP1 promotes fungi-induced inflammasome activation and pyroptosis, apoptosis, and necroptosis (PANoptosis). J Biol Chem 295: 18276–18283, 2020. doi: 10.1074/jbc.RA120.015924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331. Malireddi RKS, Kesavardhana S, Karki R, Kancharana B, Burton AR, Kanneganti T-D. RIPK1 distinctly regulates Yersinia-induced inflammatory cell death, PANoptosis. Immunohorizons 4: 789–796, 2020. doi: 10.4049/immunohorizons.2000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332. Samir P, Malireddi R, Kanneganti T-D. The PANoptosome: a deadly protein complex driving pyroptosis, apoptosis, and necroptosis (PANoptosis). Front Cell Infect Microbiol 10: 238, 2020. doi: 10.3389/fcimb.2020.00238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333. Karki R, Kanneganti T-D. The ‘cytokine storm’: molecular mechanisms and therapeutic prospects. Trends Immunol 42: 681–705, 2021. doi: 10.1016/j.it.2021.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334. Karki R, Sharma BR, Tuladhar S, Williams EP, Zalduondo L, Samir P, Zheng M, Sundaram B, Banoth B, Malireddi RKS, Schreiner P, Neale G, Vogel P, Webby R, Jonsson CB, Kanneganti T-D. Synergism of TNF-α and IFN-γ triggers inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 infection and cytokine shock syndromes. Cell 184: 149–168.e17, 2021. doi: 10.1016/j.cell.2020.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335. Beckwith KS, Beckwith MS, Ullmann S, Sætra RS, Kim H, Marstad A, Åsberg SE, Strand TA, Haug M, Niederweis M, Stenmark HA, Flo TH. Plasma membrane damage causes NLRP3 activation and pyroptosis during Mycobacterium tuberculosis infection. Nat Commun 11: 1–18, 2020. doi: 10.1038/s41467-020-16143-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336. Deng M, Guo H, Tam JW, Johnson BM, Brickey WJ, New JS, Lenox A, Shi H, Golenbock DT, Koller BH, McKinnon KP, Beutler B, Ting JP-Y. Platelet-activating factor (PAF) mediates NLRP3-NEK7 inflammasome induction independently of PAFR. J Exp Med 216: 2838–2853, 2019. doi: 10.1084/jem.20190111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337. He Y, Zeng MY, Yang D, Motro B, Núñez G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 530: 354–357, 2016. doi: 10.1038/nature16959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338. Boldin MP, Goncharov TM, Goltsev YV, Wallach D. Involvement of MACH, a novel MORT1/FADD-interacting protease, in Fas/APO-1- and TNF receptor-induced cell death. Cell 85: 803–815, 1996. doi: 10.1016/S0092-8674(00)81265-9. [DOI] [PubMed] [Google Scholar]
- 339. Muzio M, Chinnaiyan AM, Kischkel FC, O'Rourke K, Shevchenko A, Ni J, Scaffidi C, Bretz JD, Zhang M, Gentz R, Mann M, Krammer PH, Peter ME, Dixit VM. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death–inducing signaling complex. Cell 85: 817–827, 1996. doi: 10.1016/S0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]
- 340. Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114: 181–190, 2003. doi: 10.1016/S0092-8674(03)00521-X. [DOI] [PubMed] [Google Scholar]
- 341. Fricker N, Beaudouin J, Richter P, Eils R, Krammer PH, Lavrik IN. Model-based dissection of CD95 signaling dynamics reveals both a pro- and antiapoptotic role of c-FLIPL. J Cell Biol 190: 377–389, 2010. doi: 10.1083/jcb.201002060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342. Dillon CP, Weinlich R, Rodriguez DA, Cripps JG, Quarato G, Gurung P, Verbist KC, Brewer TL, Llambi F, Gong Y-N, Janke LJ, Kelliher MA, Kanneganti T-D, Green DR. RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell 157: 1189–1202, 2014. doi: 10.1016/j.cell.2014.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343. Webster JD, Vucic D. The balance of TNF mediated pathways regulates inflammatory cell death signaling in healthy and diseased tissues. Front Cell Dev Biol 8: 365, 2020. doi: 10.3389/fcell.2020.00365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344. Nikoletopoulou V, Markaki M, Palikaras K, Tavernarakis N. Crosstalk between apoptosis, necrosis and autophagy. Biochim Biophys Acta 1833: 3448–3459, 2013. doi: 10.1016/j.bbamcr.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 345. Su Z, Yang Z, Xu Y, Chen Y, Yu Q. Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol Cancer 14: 48, 2015. doi: 10.1186/s12943-015-0321-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346. Tracey KJ, Cerami A. Tumor necrosis factor: a pleiotropic cytokine and therapeutic target. Annu Rev Med 45: 491–503, 1994. doi: 10.1146/annurev.med.45.1.491. [DOI] [PubMed] [Google Scholar]
- 347. Tsuchiya Y, Nakabayashi O, Nakano H. FLIP the switch: regulation of apoptosis and necroptosis by cFLIP. Int J Mol Sci 16: 30321–30341, 2015. doi: 10.3390/ijms161226232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348. Ramon-Luing LA, Olvera Y, Flores-Gonzalez J, Palacios Y, Carranza C, Aguilar-Duran Y, Vargas MA, Gutierrez N, Medina-Quero K, Chavez-Galan L. Diverse cell death mechanisms are simultaneously activated in macrophages infected by virulent Mycobacterium tuberculosis. Pathogens 11: 492, 2022. doi: 10.3390/pathogens11050492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349. Das K, Garnica O, Dhandayuthapani S. Modulation of host miRNAs by intracellular bacterial pathogens. Front Cell Infect Microbiol 6: 79, 2016. doi: 10.3389/fcimb.2016.00079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350. Malardo T, Gardinassi LG, Moreira BP, Padilha É, Lorenzi JCC, Soares LS, Gembre AF, Fontoura IC, de Almeida LP, de Miranda Santos IKF, Silva CL, Coelho-Castelo AAM. MicroRNA expression signatures in lungs of mice infected with Mycobacterium tuberculosis. Tuberculosis (Edinb) 101: 151–159, 2016. doi: 10.1016/j.tube.2016.09.003. [DOI] [PubMed] [Google Scholar]
- 351. Yang S, Li F, Jia S, Zhang K, Jiang W, Shang Y, Chang K, Deng S, Chen M. Early secreted antigen ESAT-6 of Mycobacterium tuberculosis promotes apoptosis of macrophages via targeting the microRNA155-SOCS1 interaction. Cell Physiol Biochem 35: 1276–1288, 2015. doi: 10.1159/000373950. [DOI] [PubMed] [Google Scholar]
- 352. Huang J, Jiao J, Xu W, Zhao H, Zhang C, Shi Y, Xiao Z. MiR-155 is upregulated in patients with active tuberculosis and inhibits apoptosis of monocytes by targeting FOXO3. Mol Med Rep 12: 7102–7108, 2015. doi: 10.3892/mmr.2015.4250. [DOI] [PubMed] [Google Scholar]
- 353. Ghorpade DS, Leyland R, Kurowska-Stolarska M, Patil SA, Balaji KN. MicroRNA-155 is required for Mycobacterium bovis BCG-mediated apoptosis of macrophages. Mol Cell Biol 32: 2239–2253, 2012. doi: 10.1128/MCB.06597-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354. Rothchild AC, Sissons JR, Shafiani S, Plaisier C, Min D, Mai D, Gilchrist M, Peschon J, Larson RP, Bergthaler A, Baliga NS, Urdahl KB, Aderem A. MiR-155-regulated molecular network orchestrates cell fate in the innate and adaptive immune response to Mycobacterium tuberculosis. Proc Natl Acad Sci USA 113: E6172–E6181, 2016. doi: 10.1073/pnas.1608255113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355. Kumar R, Halder P, Sahu SK, Kumar M, Kumari M, Jana K, Ghosh Z, Sharma P, Kundu M, Basu J. Identification of a novel role of ESAT-6-dependent miR-155 induction during infection of macrophages with Mycobacterium tuberculosis. Cell Microbiol 14: 1620–1631, 2012. doi: 10.1111/j.1462-5822.2012.01827.x. [DOI] [PubMed] [Google Scholar]
- 356. Zhang H, Sun Z, Wei W, Liu Z, Fleming J, Zhang S, Lin N, Wang M, Chen M, Xu Y, Zhou J, Li C, Bi L, Zhou G. Identification of serum microRNA biomarkers for tuberculosis using RNA-seq. PLoS One 9: e88909, 2014. doi: 10.1371/journal.pone.0088909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357. Abe W, Nasu K, Nakada C, Kawano Y, Moriyama M, Narahara H. miR-196b targets c-myc and Bcl-2 expression, inhibits proliferation and induces apoptosis in endometriotic stromal cells. Hum Reprod 28: 750–761, 2013. doi: 10.1093/humrep/des446. [DOI] [PubMed] [Google Scholar]
- 358. Sinigaglia A, Peta E, Riccetti S, Venkateswaran S, Manganelli R, Barzon L. Tuberculosis-associated microRNAs: from pathogenesis to disease biomarkers. Cells 9: 2160, 2020. doi: 10.3390/cells9102160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359. Fu B, Xue W, Zhang H, Zhang R, Feldman K, Zhao Q, Zhang S, Shi L, Pavani KC, Nian W, Lin X, Wu H. MicroRNA-325-3p facilitates immune escape of Mycobacterium tuberculosis through targeting LNX1 via NEK6 accumulation to promote anti-apoptotic STAT3 signaling. mBio 11: e00557-20, 2020. doi: 10.1128/mBio.00557-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360. Zhang G, Liu X, Wang W, Cai Y, Li S, Chen Q, Liao M, Zhang M, Zeng G, Zhou B, Feng CG, Chen X. Down-regulation of miR-20a-5p triggers cell apoptosis to facilitate mycobacterial clearance through targeting JNK2 in human macrophages. Cell Cycle 15: 2527–2538, 2016. doi: 10.1080/15384101.2016.1215386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361. Fu Y, Yi Z, Wu X, Li J, Xu F. Circulating microRNAs in patients with active pulmonary tuberculosis. J Clin Microbiol 49: 4246–4251, 2011. doi: 10.1128/JCM.05459-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362. Saini S, Yamamura S, Majid S, Shahryari V, Hirata H, Tanaka Y, Dahiya R. MicroRNA-708 induces apoptosis and suppresses tumorigenicity in renal cancer cells. Cancer Res 71: 6208–6219, 2011. doi: 10.1158/0008-5472.CAN-11-0073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363. Sharbati J, Lewin A, Kutz-Lohroff B, Kamal E, Einspanier R, Sharbati S. Integrated microRNA-mRNA-analysis of human monocyte derived macrophages upon Mycobacterium avium subsp. hominissuis infection. PLoS One 6: e20258, 2011. doi: 10.1371/journal.pone.0020258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364. Liu Y, Jiang J, Wang X, Zhai F, Cheng X. miR-582-5p is upregulated in patients with active tuberculosis and inhibits apoptosis of monocytes by targeting FOXO1. PLoS One 8: e78381, 2013. doi: 10.1371/journal.pone.0078381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365. Buscaglia LE, Li Y. Apoptosis and the target genes of microRNA-21. Chin J Cancer 30: 371–380, 2011. doi: 10.5732/cjc.30.0371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366. Kleinsteuber K, Heesch K, Schattling S, Kohns M, Sander-Jülch C, Walzl G, Hesseling A, Mayatepek E, Fleischer B, Marx FM, Jacobsen M. Decreased expression of miR-21, miR-26a, miR-29a, and miR-142-3p in CD4+ T cells and peripheral blood from tuberculosis patients. PLoS One 8: e61609, 2013. doi: 10.1371/journal.pone.0061609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367. Sheedy FJ. Turning 21: induction of miR-21 as a key switch in the inflammatory response. Front Immunol 6: 19, 2015. doi: 10.3389/fimmu.2015.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368. Starczynowski DT, Kuchenbauer F, Argiropoulos B, Sung S, Morin R, Muranyi A, Hirst M, Hogge D, Marra M, Wells RA, Buckstein R, Lam W, Humphries RK, Karsan A. Identification of miR-145 and miR-146a as mediators of the 5q– syndrome phenotype. Nat Med 16: 49–58, 2010. doi: 10.1038/nm.2054. [DOI] [PubMed] [Google Scholar]
- 369. Rajaram MVS, Ni B, Morris JD, Brooks MN, Carlson TK, Bakthavachalu B, Schoenberg DR, Torrelles JB, Schlesinger LS. Mycobacterium tuberculosis lipomannan blocks TNF biosynthesis by regulating macrophage MAPK-activated protein kinase 2 (MK2) and microRNA miR-125b. Proc Natl Acad Sci USA 108: 17408–17413, 2011. doi: 10.1073/pnas.1112660108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370. Xi X, Zhang C, Han W, Zhao H, Zhang H, Jiao J. MicroRNA-223 is upregulated in active tuberculosis patients and inhibits apoptosis of macrophages by targeting FOXO3. Genet Test Mol Biomarkers 19: 650–656, 2015. doi: 10.1089/gtmb.2015.0090. [DOI] [PubMed] [Google Scholar]
- 371. Liang S, Song Z, Wu Y, Gao Y, Gao M, Liu F, Wang F, Zhang Y. MicroRNA-27b modulates inflammatory response and apoptosis during Mycobacterium tuberculosis infection. J Immunol 200: 3506–3518, 2018. doi: 10.4049/jimmunol.1701448. [DOI] [PubMed] [Google Scholar]
- 372. Singh Y, Kaul V, Mehra A, Chatterjee S, Tousif S, Dwivedi VP, Suar M, Van Kaer L, Bishai WR, Das G. Mycobacterium tuberculosis controls microRNA-99b (miR-99b) expression in infected murine dendritic cells to modulate host immunity. J Biol Chem 288: 5056–5061, 2013. doi: 10.1074/jbc.C112.439778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373. Sahu SK, Kumar M, Chakraborty S, Banerjee SK, Kumar R, Gupta P, Jana K, Gupta UD, Ghosh Z, Kundu M, Basu J. MicroRNA 26a (miR-26a)/KLF4 and CREB-C/EBPβ regulate innate immune signaling, the polarization of macrophages and the trafficking of Mycobacterium tuberculosis to lysosomes during infection. PLoS Pathog 13: e1006410, 2017. doi: 10.1371/journal.ppat.1006410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374. Gu X, Gao Y, Mu D-G, Fu E-Q. MiR-23a-5p modulates mycobacterial survival and autophagy during mycobacterium tuberculosis infection through TLR2/MyD88/NF-κB pathway by targeting TLR2. Exp Cell Res 354: 71–77, 2017. doi: 10.1016/j.yexcr.2017.03.039. [DOI] [PubMed] [Google Scholar]
- 375. Ouimet M, Koster S, Sakowski E, Ramkhelawon B, van Solingen C, Oldebeken S, Karunakaran D, Portal-Celhay C, Sheedy FJ, Ray TD, Cecchini K, Zamore PD, Rayner KJ, Marcel YL, Philips JA, Moore KJ. Mycobacterium tuberculosis induces the miR-33 locus to reprogram autophagy and host lipid metabolism. Nat Immunol 17: 677–686, 2016. doi: 10.1038/ni.3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376. Liu F, Chen J, Wang P, Li H, Zhou Y, Liu H, Liu Z, Zheng R, Wang L, Yang H, Cui Z, Wang F, Huang X, Wang J, Sha W, Xiao H, Ge B. MicroRNA-27a controls the intracellular survival of Mycobacterium tuberculosis by regulating calcium-associated autophagy. Nat Commun 9: 4295, 2018. doi: 10.1038/s41467-018-06836-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377. Guo L, Zhou L, Gao Q, Zhang A, Wei J, Hong D, Chu Y, Duan X, Zhang Y, Xu G. MicroRNA-144-3p inhibits autophagy activation and enhances Bacillus Calmette-Guérin infection by targeting ATG4a in RAW264.7 macrophage cells. PLoS One 12: e0179772, 2017. doi: 10.1371/journal.pone.0179772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378. Liu Y, Wang X, Jiang J, Cao Z, Yang B, Cheng X. Modulation of T cell cytokine production by miR-144* with elevated expression in patients with pulmonary tuberculosis. Mol Immunol 48: 1084–1090, 2011. doi: 10.1016/j.molimm.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 379. Kim JK, Lee H-M, Park K-S, Shin D-M, Kim TS, Kim YS, Suh H-W, Kim SY, Kim IS, Kim J-M, Son J-W, Sohn KM, Jung SS, Chung C, Han S-B, Yang C-S, Jo E-K. MIR144* inhibits antimicrobial responses against Mycobacterium tuberculosis in human monocytes and macrophages by targeting the autophagy protein DRAM2. Autophagy 13: 423–441, 2017. doi: 10.1080/15548627.2016.1241922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380. Chen D-Y, Chen Y-M, Lin C-F, Lo C-M, Liu H-J, Liao T-L. MicroRNA-889 inhibits autophagy to maintain mycobacterial survival in patients with latent tuberculosis infection by targeting TWEAK. mBio 11: e03045-19, 2020. doi: 10.1128/mBio.03045-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381. Kim JK, Yuk J-M, Kim SY, Kim TS, Jin HS, Yang C-S, Jo E-K. MicroRNA-125a inhibits autophagy activation and antimicrobial responses during mycobacterial infection. J Immunol 194: 5355–5365, 2015. doi: 10.4049/jimmunol.1402557. [DOI] [PubMed] [Google Scholar]
- 382. Chen Z, Wang T, Liu Z, Zhang G, Wang J, Feng S, Liang J. Inhibition of autophagy by MiR-30A induced by Mycobacteria tuberculosis as a possible mechanism of immune escape in human macrophages. Jpn J Infect Dis 68: 420–424, 2015. doi: 10.7883/yoken.JJID.2014.466. [DOI] [PubMed] [Google Scholar]
- 383. Pawar K, Sharbati J, Einspanier R, Sharbati S. Mycobacterium bovis BCG interferes with miR-3619-5p control of Cathepsin S in the process of autophagy. Front Cell Infect Microbiol 6: 27, 2016. doi: 10.3389/fcimb.2016.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384. Duan X, Zhang T, Ding S, Wei J, Su C, Liu H, Xu G. microRNA-17-5p modulates bacille calmette-guerin growth in RAW264.7 cells by targeting ULK1. PLoS One 10: e0138011, 2015. doi: 10.1371/journal.pone.0138011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385. Kumar R, Sahu SK, Kumar M, Jana K, Gupta P, Gupta UD, Kundu M, Basu J. MicroRNA 17-5p regulates autophagy in Mycobacterium tuberculosis-infected macrophages by targeting Mcl-1 and STAT3. Cell Microbiol 18: 679–691, 2016. doi: 10.1111/cmi.12540. [DOI] [PubMed] [Google Scholar]
- 386. Guo L, Zhao J, Qu Y, Yin R, Gao Q, Ding S, Zhang Y, Wei J, Xu G. microRNA-20a inhibits autophagic process by targeting ATG7 and ATG16L1 and favors mycobacterial survival in macrophage cells. Front Cell Infect Microbiol 6: 134, 2016. doi: 10.3389/fcimb.2016.00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387. Bettencourt P, Marion S, Pires D, Santos LF, Lastrucci C, Carmo N, Blake J, Benes V, Griffiths G, Neyrolles O, Lugo-Villarino G, Anes E. Actin-binding protein regulation by microRNAs as a novel microbial strategy to modulate phagocytosis by host cells: the case of N-Wasp and miR-142-3p. Front Cell Infect Microbiol 3: 19, 2013. doi: 10.3389/fcimb.2013.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388. Lou J, Wang Y, Zhang Z, Qiu W. MiR-20b inhibits mycobacterium tuberculosis induced inflammation in the lung of mice through targeting NLRP3. Exp Cell Res 358: 120–128, 2017. doi: 10.1016/j.yexcr.2017.06.007. [DOI] [PubMed] [Google Scholar]
- 389. Kundu M, Basu J. The role of microRNAs and long non-coding RNAs in the regulation of the immune response to mycobacterium tuberculosis infection. Front Immunol 12: 687962, 2021. doi: 10.3389/fimmu.2021.687962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390. Harapan H, Fitra F, Ichsan I, Mulyadi M, Miotto P, Hasan NA, Calado M, Cirillo DM. The roles of microRNAs on tuberculosis infection: meaning or myth? Tuberculosis (Edinb) 93: 596–605, 2013. doi: 10.1016/j.tube.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391. Sabir N, Hussain T, Shah SZA, Peramo A, Zhao D, Zhou X. miRNAs in tuberculosis: new avenues for diagnosis and host-directed therapy. Front Microbiol 9: 602, 2018. doi: 10.3389/fmicb.2018.00602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392. Das K, Saikolappan S, Dhandayuthapani S. Differential expression of miRNAs by macrophages infected with virulent and avirulent Mycobacterium tuberculosis. Tuberculosis 93: S47–S50, 2013. doi: 10.1016/S1472-9792(13)70010-6. [DOI] [PubMed] [Google Scholar]
- 393. Xiong Y, Fang J-H, Yun J-P, Yang J, Zhang Y, Jia W-H, Zhuang S-M. Effects of MicroRNA-29 on apoptosis, tumorigenicity, and prognosis of hepatocellular carcinoma. Hepatology 51: 836–845, 2010. doi: 10.1002/hep.23380. [DOI] [PubMed] [Google Scholar]
- 394. Khan N, Pahari S, Vidyarthi A, Aqdas M, Agrewala JN. Stimulation through CD40 and TLR-4 is an effective host directed therapy against Mycobacterium tuberculosis. Front Immunol 7: 386, 2016. doi: 10.3389/fimmu.2016.00386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395. Pahari S, Khan N, Aqdas M, Negi S, Kaur J, Agrewala JN. Infergen stimulated macrophages restrict Mycobacterium tuberculosis growth by autophagy and release of nitric oxide. Sci Rep 6: 39492, 2016. doi: 10.1038/srep39492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396. Kim YS, Lee H-M, Kim JK, Yang C-S, Kim TS, Jung M, Jin HS, Kim S, Jang J, Oh GT, Kim J-M, Jo E-K. PPAR-α activation mediates innate host defense through induction of TFEB and lipid catabolism. J Immunol 198: 3283–3295, 2017. doi: 10.4049/jimmunol.1601920. [DOI] [PubMed] [Google Scholar]
- 397. Loetchutinat C, Saengkhae C, Marbeuf-Gueye C, Garnier-Suillerot A. New insights into the P‐glycoprotein‐mediated effluxes of rhodamines. Eur J Biochem 270: 476–485, 2003. doi: 10.1046/j.1432-1033.2003.03403.x. [DOI] [PubMed] [Google Scholar]
- 398. Juárez E, Carranza C, Sánchez G, González M, Chávez J, Sarabia C, Torres M, Sada E. Loperamide restricts intracellular growth of Mycobacterium tuberculosis in lung macrophages. Am J Respir Cell Mol Biol 55: 837–847, 2016. doi: 10.1165/rcmb.2015-0383OC. [DOI] [PubMed] [Google Scholar]
- 399. Afsal K, Selvaraj P. Effect of 1, 25-dihydroxyvitamin D3 on the expression of mannose receptor, DC-SIGN and autophagy genes in pulmonary tuberculosis. Tuberculosis 99: 1–10, 2016. doi: 10.1016/j.tube.2016.03.010. [DOI] [PubMed] [Google Scholar]
- 400. Fernandez-Soto P, Bruce AJE, Fielding AJ, Cavet JS, Tabernero L. Mechanism of catalysis and inhibition of Mycobacterium tuberculosis SapM, implications for the development of novel antivirulence drugs. Sci Rep 9: 10315, 2019. doi: 10.1038/s41598-019-46731-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401. Kolloli A, Subbian S. Host-directed therapeutic strategies for tuberculosis. Front Med (Lausanne) 4: 171, 2017. doi: 10.3389/fmed.2017.00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402. Kroesen VM, Rodríguez-Martínez P, García E, Rosales Y, Díaz J, Martín-Céspedes M, Tapia G, Sarrias MR, Cardona P-J, Vilaplana C. A beneficial effect of low-dose aspirin in a murine model of active tuberculosis. Front Immunol 9: 798, 2018. doi: 10.3389/fimmu.2018.00798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403. Kanehiro Y, Tomioka H, Pieters J, Tatano Y, Kim H, Iizasa H, Yoshiyama H. Identification of novel mycobacterial inhibitors against mycobacterial protein kinase G. Front Microbiol 9: 1517, 2018. doi: 10.3389/fmicb.2018.01517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404. Nguyen L, Walburger A, Houben E, Koul A, Muller S, Morbitzer M, Klebl B, Ferrari G, Pieters J. Role of protein kinase G in growth and glutamine metabolism of Mycobacterium bovis BCG. J Bacteriol 187: 5852–5856, 2005. [Erratum in J Bacteriol 187: 7165, 2005]. doi: 10.1128/JB.187.16.5852-5856.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405. Zabludoff SD, Deng C, Grondine MR, Sheehy AM, Ashwell S, Caleb BL, Green S, Haye HR, Horn CL, Janetka JW, Liu D, Mouchet E, Ready S, Rosenthal JL, Queva C, Schwartz GK, Taylor KJ, Tse AN, Walker GE, White AM. AZD7762, a novel checkpoint kinase inhibitor, drives checkpoint abrogation and potentiates DNA-targeted therapies. Mol Cancer Ther 7: 2955–2966, 2008. doi: 10.1158/1535-7163.MCT-08-0492. [DOI] [PubMed] [Google Scholar]
- 406. Schiebler M, Brown K, Hegyi K, Newton SM, Renna M, Hepburn L, Klapholz C, Coulter S, Obregón-Henao A, Henao Tamayo M, Basaraba R, Kampmann B, Henry KM, Burgon J, Renshaw SA, Fleming A, Kay RR, Anderson KE, Hawkins PT, Ordway DJ, Rubinsztein DC, Floto RA. Functional drug screening reveals anticonvulsants as enhancers of mTOR‐independent autophagic killing of Mycobacterium tuberculosis through inositol depletion. EMBO Mol Med 7: 127–139, 2015. doi: 10.15252/emmm.201404137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407. Bezabeh T, Mowat MRA, Jarolim L, Greenberg AH, Smith ICP. Detection of drug-induced apoptosis and necrosis in human cervical carcinoma cells using 1H NMR spectroscopy. Cell Death Differ 8: 219–224, 2001. doi: 10.1038/sj.cdd.4400802. [DOI] [PubMed] [Google Scholar]
- 408. Jayachandran R, Sundaramurthy V, Combaluzier B, Mueller P, Korf H, Huygen K, Miyazaki T, Albrecht I, Massner J, Pieters J. Survival of mycobacteria in macrophages is mediated by coronin 1-dependent activation of calcineurin. Cell 130: 37–50, 2007. doi: 10.1016/j.cell.2007.04.043. [DOI] [PubMed] [Google Scholar]
- 409. Kim S-Y, Park Y-J, Kim W-I, Lee S-H, Ludgerus Chang C, Kang S-J, Kang C-S. Molecular analysis of isoniazid resistance in Mycobacterium tuberculosis isolates recovered from South Korea. Diagn Microbiol Infect Dis 47: 497–502, 2003. doi: 10.1016/S0732-8893(03)00132-9. [DOI] [PubMed] [Google Scholar]
- 410. Dutta NK, He R, Pinn ML, He Y, Burrows F, Zhang Z-Y, Karakousis PC. Mycobacterial protein tyrosine phosphatases A and B inhibitors augment the bactericidal activity of the standard anti-tuberculosis regimen. ACS Infect Dis 2: 231–239, 2016. doi: 10.1021/acsinfecdis.5b00133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411. Serebryakova VA, Urazova OI, Novitsky VV, Vengerovskii AI, Kononova TE, Vasil'eva OA, Beresneva AE. Effects of levofloxacin on blood lymphocyte apoptosis in patients with pulmonary tuberculosis: an in vitro study. Bull Exp Biol Med 168: 109–112, 2019. doi: 10.1007/s10517-019-04659-x. [DOI] [PubMed] [Google Scholar]
- 412. Padmapriyadarsini C, Bhavani PK, Natrajan M, Ponnuraja C, Kumar H, Gomathy SN, Guleria R, Jawahar SM, Singh M, Balganesh T, Swaminathan S. Evaluation of metformin in combination with rifampicin containing antituberculosis therapy in patients with new, smear-positive pulmonary tuberculosis (METRIF): study protocol for a randomised clinical trial. BMJ Open 9: e024363, 2019. doi: 10.1136/bmjopen-2018-024363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413. Lam KKY, Zheng X, Forestieri R, Balgi AD, Nodwell M, Vollett S, Anderson HJ, Andersen RJ, Av-Gay Y, Roberge M. Nitazoxanide stimulates autophagy and inhibits mTORC1 signaling and intracellular proliferation of Mycobacterium tuberculosis. PLoS Pathog 8: e1002691, 2012. doi: 10.1371/journal.ppat.1002691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414. Williams DE, Dalisay DS, Chen J, Polishchuck EA, Patrick BO, Narula G, Ko M, Av-Gay Y, Li H, Magarvey N, Andersen RJ. Aminorifamycins and sporalactams produced in culture by a Micromonospora sp. isolated from a Northeastern-Pacific marine sediment are potent antibiotics. Org Lett 19: 766–769, 2017. doi: 10.1021/acs.orglett.6b03619. [DOI] [PubMed] [Google Scholar]
- 415. Mesguiche V, Parsons RJ, Arris CE, Bentley J, Boyle FT, Curtin NJ, Davies TG, Endicott JA, Gibson AE, Golding BT, Griffin RJ, Jewsbury P, Johnson LN, Newell DR, Noble MEM, Wang LZ, Hardcastle IR. 4-Alkoxy-2, 6-diaminopyrimidine derivatives: inhibitors of cyclin dependent kinases 1 and 2. Bioorg Med Chem Lett 13: 217–222, 2003. doi: 10.1016/S0960-894X(02)00884-3. [DOI] [PubMed] [Google Scholar]
- 416. Gil M, Graña M, Schopfer FJ, Wagner T, Denicola A, Freeman BA, Alzari PM, Batthyány C, Durán R. Inhibition of Mycobacterium tuberculosis PknG by non-catalytic rubredoxin domain specific modification: reaction of an electrophilic nitro-fatty acid with the Fe–S center. Free Radic Biol Med 65: 150–161, 2013. doi: 10.1016/j.freeradbiomed.2013.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417. Stover CK, Warrener P, VanDevanter DR, Sherman DR, Arain TM, Langhorne MH, Anderson SW, Towell JA, Yuan Y, McMurray DN, Kreiswirth BN, Barry CE, Baker WR. A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature 405: 962–966, 2000. doi: 10.1038/35016103. [DOI] [PubMed] [Google Scholar]
- 418. Barry CE, Boshoff HI, Dowd CS. Prospects for clinical introduction of nitroimidazole antibiotics for the treatment of tuberculosis. Curr Pharm Des 10: 3239–3262, 2004. doi: 10.2174/1381612043383214. [DOI] [PubMed] [Google Scholar]
- 419. Kim J-J, Lee H-M, Shin D-M, Kim W, Yuk J-M, Jin HS, Lee S-H, Cha G-H, Kim J-M, Lee Z-W, Shin SJ, Yoo H, Park YK, Park JB, Chung J, Yoshimori T, Jo E-K. Host cell autophagy activated by antibiotics is required for their effective antimycobacterial drug action. Cell Host Microbe 11: 457–468, 2012. doi: 10.1016/j.chom.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 420. Floto RA, Sarkar S, Perlstein EO, Kampmann B, Schreiber SL, Rubinsztein DC. Small molecule enhancers of rapamycin-induced TOR inhibition promote autophagy, reduce toxicity in Huntington’s disease models and enhance killing of mycobacteria by macrophages. Autophagy 3: 620–622, 2007. doi: 10.4161/auto.4898. [DOI] [PubMed] [Google Scholar]
- 421. Piccaro G, Pietraforte D, Giannoni F, Mustazzolu A, Fattorini L. Rifampin induces hydroxyl radical formation in Mycobacterium tuberculosis. Antimicrob Agents Chemother 58: 7527–7533, 2014. doi: 10.1128/AAC.03169-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422. Zhou J, Xu R, Du X-Z, Zhou X-D, Li Q. Saxifragifolin D attenuates phagosome maturation arrest in Mycobacterium tuberculosis-infected macrophages via an AMPK and VPS34-dependent pathway. AMB Express 7: 11, 2017. doi: 10.1186/s13568-016-0317-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423. Chen D, Ma S, He L, Yuan P, She Z, Lu Y. Sclerotiorin inhibits protein kinase G from Mycobacterium tuberculosis and impairs mycobacterial growth in macrophages. Tuberculosis (Edinb) 103: 37–43, 2017. doi: 10.1016/j.tube.2017.01.001. [DOI] [PubMed] [Google Scholar]
- 424. Dobos KM, Spotts EA, Quinn FD, King CH. Necrosis of lung epithelial cells during infection with Mycobacterium tuberculosis is preceded by cell permeation. Infect Immun 68: 6300–6310, 2000. doi: 10.1128/IAI.68.11.6300-6310.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425. Farinacci M, Weber S, Kaufmann SH. The recombinant tuberculosis vaccine rBCG ΔureC::hly(+) induces apoptotic vesicles for improved priming of CD4(+) and CD8(+) T cells. Vaccine 30: 7608–7614, 2012. doi: 10.1016/j.vaccine.2012.10.031. [DOI] [PubMed] [Google Scholar]
- 426. Desel C, Dorhoi A, Bandermann S, Grode L, Eisele B, Kaufmann SHE. Recombinant BCG ΔureC hly+ induces superior protection over parental BCG by stimulating a balanced combination of type 1 and type 17 cytokine responses. J Infect Dis 204: 1573–1584, 2011. doi: 10.1093/infdis/jir592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427. Zullo AJ, Lee S. Mycobacterial induction of autophagy varies by species and occurs independently of mammalian target of rapamycin inhibition. J Biol Chem 287: 12668–12678, 2012. doi: 10.1074/jbc.M111.320135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428. Singh V, Jamwal S, Jain R, Verma P, Gokhale R, Rao KVS. Mycobacterium tuberculosis-driven targeted recalibration of macrophage lipid homeostasis promotes the foamy phenotype. Cell Host Microbe 12: 669–681, 2012. doi: 10.1016/j.chom.2012.09.012. [DOI] [PubMed] [Google Scholar]
- 429. Shui W, Petzold CJ, Redding A, Liu J, Pitcher A, Sheu L, Hsieh T-Y, Keasling JD, Bertozzi CR. Organelle membrane proteomics reveals differential influence of mycobacterial lipoglycans on macrophage phagosome maturation and autophagosome accumulation. J Proteome Res 10: 339–348, 2011. doi: 10.1021/pr100688h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430. Høyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, Bianchi K, Fehrenbacher N, Elling F, Rizzuto R, Mathiasen IS, Jäättelä M. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-β, and Bcl-2. Mol Cell 25: 193–205, 2007. doi: 10.1016/j.molcel.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 431. Chandel NS. Mitochondria as signaling organelles. BMC Biol 12: 34, 2014. doi: 10.1186/1741-7007-12-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432. Singh P, Subbian S. Harnessing the mTOR pathway for tuberculosis treatment. Front Microbiol 9: 70, 2018. doi: 10.3389/fmicb.2018.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433. Ashley D, Hernandez J, Cao R, To K, Yegiazaryan A, Abrahem R, Nguyen T, Owens J, Lambros M, Subbian S, Venketaraman V. Antimycobacterial effects of everolimus in a human granuloma model. J Clin Med 9: 2043, 2020. doi: 10.3390/jcm9072043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434. Cao R, To K, Kachour N, Beever A, Owens J, Sathananthan A, Singh P, Kolloli A, Subbian S, Venketaraman V. Everolimus-induced effector mechanism in macrophages and survivability of Erdman, CDC1551 and HN878 strains of Mycobacterium tuberculosis infection. Biomol Concepts 12: 46–54, 2021. doi: 10.1515/bmc-2021-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435. Mukundan S, Bhatt R, Lucas J, Tereyek M, Chang TL, Subbian S, Parekkadan B. 3D host cell and pathogen-based bioassay development for testing anti-tuberculosis (TB) drug response and modeling immunodeficiency. Biomol Concepts 12: 117–128, 2021. doi: 10.1515/bmc-2021-0013. [DOI] [PubMed] [Google Scholar]
- 436. Wallis RS, Ginindza S, Beattie T, Arjun N, Likoti M, Edward VA, Rassool M, Ahmed K, Fielding K, Ahidjo BA, Vangu MDT, Churchyard G. Adjunctive host-directed therapies for pulmonary tuberculosis: a prospective, open-label, phase 2, randomised controlled trial. Lancet Respir Med 9: 897–908, 2021. doi: 10.1016/S2213-2600(20)30448-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437. Klionsky DJ, Abdel-Aziz AK, Abdelfatah S, Abdellatif M, Abdoli A, Abel S, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 17: 1–382, 2021. doi: 10.1080/15548627.2020.1797280. [DOI] [PMC free article] [PubMed] [Google Scholar]