

Keywords: focal segmental glomerulosclerosis, glomerular filtration rate, KCa1.1 channels, NMDA receptors, store-operated calcium channels, TRPC channels
Abstract
An essential step in renal function entails the formation of an ultrafiltrate that is delivered to the renal tubules for subsequent processing. This process, known as glomerular filtration, is controlled by intrinsic regulatory systems and by paracrine, neuronal, and endocrine signals that converge onto glomerular cells. In addition, the characteristics of glomerular fluid flow, such as the glomerular filtration rate and the glomerular filtration fraction, play an important role in determining blood flow to the rest of the kidney. Consequently, disease processes that initially affect glomeruli are the most likely to lead to end-stage kidney failure. The cells that comprise the glomerular filter, especially podocytes and mesangial cells, express many different types of ion channels that regulate intrinsic aspects of cell function and cellular responses to the local environment, such as changes in glomerular capillary pressure. Dysregulation of glomerular ion channels, such as changes in TRPC6, can lead to devastating glomerular diseases, and a number of channels, including TRPC6, TRPC5, and various ionotropic receptors, are promising targets for drug development. This review discusses glomerular structure and glomerular disease processes. It also describes the types of plasma membrane ion channels that have been identified in glomerular cells, the physiological and pathophysiological contexts in which they operate, and the pathways by which they are regulated and dysregulated. The contributions of these channels to glomerular disease processes, such as focal segmental glomerulosclerosis (FSGS) and diabetic nephropathy, as well as the development of drugs that target these channels are also discussed.
CLINICAL HIGHLIGHTS
Ion channels in glomerular cells play an important role in the overall regulation of renal function. Their dysregulation has been tied to severe kidney diseases that can progress to end-stage kidney failure. For this reason, ion channels represent potential therapeutic targets for these disorders. This article reviews the literature on the characteristics, regulation, and dysregulation of plasma membrane ion channels in glomerular cells. We discuss the nature of ion channel mutations that have been implicated in glomerular disease. We also discuss other disease mechanisms that converge on ion channels, as well as how the output pathways of ion channels lead to dysfunction in glomerular cells. Potential new research directions and therapeutic strategies are also covered.
1. INTRODUCTION
The vertebrate kidneys are a pair of highly vascularized organs that play many essential roles in homeostasis. The kidneys function to maintain appropriate concentrations of electrolytes, sugars, amino acids, and enzyme cofactors, primarily within extracellular fluids (ECFs) but also over time within intracellular compartments (1–4). The kidneys play crucial roles in regulating sodium (5, 6), potassium (2, 7, 8), calcium (9), and phosphate (10, 11) excretion as well as in the regulation of acid-base balance (12). In general, the more abundant a given substance is within ECF, the more critical the kidneys are in maintaining the concentrations of that substance within allowable limits. The kidneys also allow the excretion of metabolic waste products, known as uremic toxins, that are generated by all mammalian cells as well as by microorganisms living within vertebrates (13–15). An accumulation of uremic toxins can produce adverse effects on all cells, especially within the nervous system (15, 16). The kidneys also regulate whole body water homeostasis and osmolarity and are able to control water homeostasis in large measure independently of changes in the excretion of electrolytes or uremic toxins (17). The ability of the kidneys to excrete metabolic wastes and certain ions without excessive water loss is essential for terrestrial vertebrates to avoid desiccation. The kidneys are normally able to recapture essential metabolites such as sugars and amino acids, as well as important vitamins and cofactors, and inappropriate excretion of these substances in urine is often a sign of kidney disease (18–20). These various functions of the kidney are essential for life, and some form of renal replacement therapy; hemodialysis, peritoneal dialysis, or transplantation, is required once kidney function is sufficiently compromised.
The principal functional unit of the kidney is referred to as the nephron. Each normal human kidney contains on average around a million nephrons, although this number can vary considerably, depending on factors such as the gestational age at birth (21). A nephron is a complex structure that interacts at nearly every level with a highly organized microvasculature, and the interactions between nephrons and surrounding microvascular elements are essential for normal renal function (22–24). Nephrons can be divided into two main components. The first is the renal corpuscle, which is always located within the renal cortex. This structure comprises convoluted parallel capillary loops, known as glomerular capillaries, that are confined with an epithelial capsule known as Bowman’s capsule that comprises renal parietal cells attached to a basement membrane (25, 26). Blood is delivered to the glomerular capillaries by afferent arterioles that branch off the cortical radiate arteries. The glomerular capillaries are selectively permeable and function to produce an ultrafiltrate that is delivered to the rest of the nephron for subsequent processing. Blood leaves the glomerular capillaries through the efferent arterioles. Both the afferent and efferent arterioles contain vascular smooth muscle and are subjected to neural, endocrine, and paracrine modulation. The efferent arterioles give rise to a second set of capillary beds that surround either the proximal and distal convoluted tubules within the cortex (the peritubular capillaries) or other portions of the nephron that penetrate into the renal medulla (the vasa recta). Thus, within the kidney, there are two distinct sets of capillary beds arranged in series, which are separated by the efferent arteriole. For this reason, loss or damage to renal corpuscles results in marked disruptions of renal blood flow that can produce secondary effects throughout the kidney (27–29).
The second functional component of the nephron is a looped tubular structure comprised of specialized epithelial cells whose function and properties depend on where along the tubule they are located. The tubular epithelium of the most proximal portions of the renal tubules is continuous with the parietal cells that form the outer margins of Bowman’s capsule (30). In addition, a few glomerular cells and parietal cells are also in contact (31). The ultrafiltrate that enters renal tubules is processed as molecules are either reabsorbed, secreted into the filtrate, or both. Urine, the final product, subsequently passes into the lower urinary tract and is stored until it is eliminated from the body. Regulation of transport in the tubules normally ensures that the urine composition is appropriate for the current physiological status. For example, the urine will be more concentrated if the organism is dehydrated or will contain more Na+ after a high-Na+ meal, etc. In this review, we confine our attention to the cells within the renal corpuscle that collectively function to form the initial ultrafiltrate: mesangial cells (MCs), endothelial cells, and podocytes.
Because of the structural organization of the nephron and its associated vasculature, diseases affecting glomeruli often have devastating consequences for renal function (26, 32, 33). Clearly, a nephron with little or no ultrafiltrate delivered to the renal tubule cannot function. Similarly, if blood cannot flow through a glomerular capillary, the blood supply to the rest of that nephron and into portions of the renal medulla will also be compromised. If a sufficient number of glomeruli are functionally compromised, end-stage kidney failure (ESKF) will follow. Many of the most common causes of ESKF are associated with early pathological processes that affect glomeruli. This occurs, for example, in kidney disease secondary to diabetes or chronic hypertension and in various forms of glomerulonephritis and glomerulosclerosis (27, 34). Proteinuria, one of the cardinal symptoms of glomerular disease, remains a mainstay in the staging of chronic kidney disease (CKD) (35).
In 2005, a pair of landmark articles demonstrated profound glomerulopathies in people with mutations in TRPC6, the gene encoding transient receptor potential canonical type 6 (TRPC6) channels (36, 37). These mutations resulted in severe albuminuria and glomerulosclerosis, usually with an adult onset and in most cases progressing to ESKF. The disease associated with these mutations exhibited an autosomal dominant inheritance pattern and high penetrance. Functional analyses of these mutant TRPC6 channels showed that most of them were associated with a marked gain of function, at least when characterized in heterologous expression systems. In the intervening years, several other TRPC6 mutations were discovered and associated with renal disease, and many aspects of the regulation and dysregulation of TRPC6 in glomerular cells have been established. The TRPC6 mutations therefore represent quintessential examples of “channelopathies,” diseases driven by ion channel dysfunction. There is now evidence from humans and animal models that wild-type TRPC6 channels can be dysregulated in nongenetic forms of glomerular disease (38, 39), and therefore for purposes of this review we consider that situation to also be a channelopathy. In addition to TRPC6, other plasma membrane ion channels have been characterized in glomerular cells, and three of them, transient receptor potential canonical type 5 (TRPC5) channels, large-conductance calcium-activated potassium channels (BKs; KCa1.1), and store-operated channels (SOCs), have been suggested to play a role in glomerular disease mechanisms (40–42). Ionotropic receptors such as the N-methyl-d-aspartate (NMDA) receptor and purinergic P2X receptors function as extracellular ligand-gated ion channels and have also been implicated in the pathogenesis of glomerular disease (43, 44). There is a current unmet need for new therapies to slow or prevent the progression of glomerular diseases. Because ion channels have been effectively targeted in many disease conditions, there is now considerable interest in the ion channels of glomerular cells as potential targets for drug development.
In the following sections, we review the current status of ion channels in glomerular cells (TABLE 1), including studies of their expression, trafficking, gating and permeation, and modulation. In addition, we discuss their dysregulation in glomerular disease models, the downstream consequences of their activation, and their potential as therapeutic targets for glomerular diseases. Because nearly all of this literature pertains to MCs and podocytes, those cells are the primary focus of this review. Before we discuss ion channel physiology in detail, we also provide a brief overview of the process of glomerular filtration and the nature of the cell types that form this structure.
Table 1.
Ion channels expressed in glomerular cells
| Ion Channels | Mesangial Cells | Podocytes |
|---|---|---|
| BK (KCa1.1) | Yes | Yes |
| Cav | Yes | |
| I Cl.Ca | Yes | |
| I Cl.vol | Yes | |
| KATP | Yes | |
| NMDA receptor | Yes | Yes |
| P2X receptor | Yes | Yes |
| Piezo2 | Yes | |
| SOC | Yes | Yes |
| TRPA1 | Yes | |
| TRPC1 | Yes | |
| TRPC3 | Yes | Yes |
| TRPC4 | Yes | |
| TRPC5 | Yes | |
| TRPC6 | Yes | Yes |
| TRPP2 | Yes |
See glossary for abbreviations.
2. OVERVIEW OF GLOMERULAR FILTRATION AND DISEASES
2.1. Cell Types within Glomeruli
The glomerular filtration barrier is a complex microvascular structure that is contained within Bowman’s capsule (25, 30). Water and certain solutes move through the filtration apparatus by a convective flow process driven by Starling’s forces. The cells that comprise the glomerular filter are organized in part by their relationship to the glomerular basement membrane (GBM) (45). The GBM is a gel-like structure consisting of type IV collagen, laminin, fibronectin, entactin, and heparan sulfate proteoglycan that is thought to have a substantial permeability to water, small solutes, and proteins (46). As with all capillaries, the interior face of the GBM is lined by endothelial cells. Glomerular endothelial cells have an unusually high density of fenestrae, and they are covered with a negatively charged glycocalyx comprised of sulfated proteoglycans and glycoproteins (46, 47). The fenestrations cover between 20% and 50% of the endothelial surface, thereby creating a substantial permeation pathway for water and small solutes (25). Fluids, cells, and solutes that fail to traverse the wall of the glomerular capillary move into the efferent arteriole and from there into the peritubular microcirculation or the vasa recta. The diameter of the endothelial fenestrations makes them permeable to albumin and many other proteins, but the endothelial glycocalyx and fenestrations nevertheless contribute substantially to the overall permselectivity of the glomerular filtration barrier, especially on the basis of the net charge of proteins (25, 47). FIGURE 1A shows a schematic of the glomerulus and the cell types within glomeruli. FIGURE 1B shows an electron micrograph of a mouse glomerulus. Several capillary loops can be seen, along with portions of the capillary lumen, and podocytes with their foot processes attached to the outer surface of the capillary. Intravital imaging of a superficial glomerulus in the rat kidney (FIGURE 1C) reveals the organization of larger vessels and smaller capillary loops.
FIGURE 1.

Organization of glomeruli. A: schematic representation of the glomerulus. The glomerulus is located in the renal cortex and consists of a network of blood capillaries located within Bowman’s capsule. Blood enters the capillaries through the afferent arteriole and leaves through the efferent arteriole and moves from there into various peritubular capillaries. Glomeruli contain 4 different cell types: podocytes, mesangial cells (MCs), endothelial cells, and parietal epithelial cells of Bowman’s capsule. B: scanning electron micrograph showing a mouse glomerulus with several capillary loops, a capillary lumen (asterisk), podocytes with their cell body (marked P), and the primary processes that emanate from the cell body (marked with an arrowhead). Bowman’s capsule can also be seen (arrow). Image adapted from Ref. 25 with permission. C: intravital imaging of the superficial glomerulus in the rat kidney. Texas Red rat-labeled serum albumin shows the organization of peritubular capillaries (arrowheads) and glomerular capillaries (asterisk). Nuclei are stained with a blue dye (Hoechst). See glossary for abbreviations.
Most of the MCs are located on the interior of the GBM and play an essential role in maintaining the structural integrity of the glomerular capillary (25, 48, 49). MCs comprise approximately one-third of the cells within Bowman’s capsule. Most MCs are in sufficiently close contact with endothelial cells and podocytes to allow for reciprocal paracrine interactions (48). However, a subset of MCs, known as extraglomerular MCs, form part of the juxtaglomerular apparatus (JGA) and are in close contact with cells of the macula densa, which is a region of specialized epithelia in the distal tubule that plays an important role in sensing tubular Na+ content and communicating that status to other cells. The extraglomerular MCs are also in close contact with the afferent and efferent arterioles (50–52). Loss or deletion of MCs initially results in extreme dilation of capillary loops (53, 54), and MCs are thought to sustain glomerular structure in the face of substantial changes in hydrostatic pressure, in part by contractions that occur through mechanisms similar to those of vascular smooth muscle (55). MC contraction may also fine-tune the surface area for glomerular filtration and thereby contribute to the regulation of the glomerular filtration rate (GFR) (56, 57). MCs also secrete a complex extracellular matrix (ECM), known as the mesangial matrix, that connects MCs to each other and to the GBM (48). The composition of this matrix is distinctly different from that of the GBM (58). MCs also secrete a host of soluble growth factors and chemokines that are thought to regulate the functions of other cells within the glomerulus and to orchestrate responses to injury and immunological insults (25, 48, 59, 60). In addition, MCs take up proteins by endocytosis (59, 61), and they receive signals from cells in the macula densa (51, 61, 62). MCs respond to a number of different endocrine signaling factors, such as insulin, endothelin (ET), and angiotensin II (ANG II) (55, 56, 63–65), as well as to paracrine factors such as adenosine triphosphate (ATP) (66, 67). Importantly, endothelial cells and MCs are capable of regenerating after injury (60, 68).
Podocytes, sometimes referred to as visceral epithelial cells, are highly specialized polarized cells located on the external face of the GBM (26, 30, 34). Although they are considered to be epithelial cells, they share many structural similarities with neurons and astrocytes. A number of primary processes emanate from a large cell body and wrap around the GBM (FIGURES 1 and 2). Normally, a series of small foot processes, also known as pedicels, extend at regular spatial intervals from the major processes of podocytes. The basement domains of these foot processes adhere to the GBM (30). In addition, the individual foot processes form junctions with each other through the specialized adhesion molecules nephrin and Neph1, as well as through other junctional proteins. These junctions form a highly porous structure known as the slit diaphragm that is 30–40 nM in width (30). The water and solutes that pass through the slit diaphragm will then move into Bowman’s space and from there into the proximal tubule, a process driven by Starling’s forces. Historically, the glomerular filter has been modeled as a series of three sequential hydrostatic barriers (25). However, a portion of the filtrate that passes through the slit diaphragms will enter the so-called subpodocyte space and will enter Bowman’s space only after traversing a structure known as the podocyte pore complex (71). FIGURE 2 illustrates some of these structural features.
FIGURE 2.

Higher-resolution images of glomeruli seen from different locations. A: helium ion microscopic imaging of glomerular endothelial cells. Two adjacent endothelial cells from a glomerular capillary as seen from the luminal side. Adapted from Ref. 69 with permission. B: scanning ion-conductance microscopy (SICM) 3-dimensional image of a region of denuded glomerular basement membrane (GBM) in a glomerulus isolated from a rat with an advanced stage of diabetic nephropathy. The corresponding high-magnification inset shows adjacent endothelial cells imaged from the GBM side (color corresponds to the z-axis profile scale shown on the side). Adapted from Ref. 70 with permission. C: helium ion microscopy imaging shows the interior of Bowman’s capsule containing a layer of squamous epithelial parietal cells. Each parietal epithelial cell displays a single, long central cilium. Adapted from Ref. 69. D: the glomerular capillary loop shows complex interdigitations of podocytes and their foot processes. Adapted from Ref. 69). E: 3-dimensional topographic SICM image of a glomerular capillary isolated from a normotensive rat. The corresponding high-magnification inset reveals interdigitated foot processes that wrap around the glomerular capillaries (color corresponds to the z-axis profile scale shown on the side). Adapted from Ref. 70 with permission.
In contrast to glomerular endothelial cells and MCs, podocytes are terminally differentiated cells (30, 72, 73), and there is only a limited capacity to regenerate podocytes that die or that detach from the GBM surface (74–76). There is evidence that some of the parietal cells that normally comprise Bowman’s capsule can differentiate into podocytes after an injury (77–79), but this is not sufficient to restore glomerular function in the face of chronic disease, and glomerulosclerosis proceeds once a threshold number of podocytes have been lost (80, 81). Podocytes can be lost through processes of apoptotic or necrotic cell death, or they can detach from the glomerular surface (30, 82, 83). Indeed, it is possible to culture viable podocytes isolated from the urine of humans and animals with kidney disease (84, 85). Podocytes undergo a distinctive and substantial change in morphology when stressed through certain mechanical or metabolic stimuli. This process, referred to as foot process effacement, is characterized by major rearrangements of the foot process cytoskeleton as the individual foot processes become wider and shorter, in some cases being resorbed all the way back into the major process (30, 86–88). It has been suggested that foot process effacement is a structural adaptation to prevent the detachment of podocytes and their subsequent loss in urine (86, 87, 89), and it is notable that foot process effacement is a reversible process in certain acute animal models (90). During conditions in which foot process effacement occurs, some portions of the outer face of GBM will be uncovered, and foot process effacement is almost always accompanied by some degree of albuminuria.
2.2. Regulation of Glomerular Filtration via Tubuloglomerular Feedback
GFR is defined as the volume of plasma that flows from the glomerulus into Bowman’s space over a period of time. It is one of the most important indexes of overall renal function, as a loss of functional nephrons inevitably results in a decline in GFR. The GFR is regulated through several different mechanisms, and, as a result, substantial changes in blood pressure over a wide range produce only small changes in GFR (91). Myogenic autoregulation is a process in certain arteries and arterioles in which an increase in pressure within the vessel is sufficient to trigger a contraction of the vessel smooth muscle through a mechanotransduction cascade within the smooth muscle itself. Myogenic autoregulation within the renal vasculature, especially in the afferent arterioles, plays a crucial role in protecting glomeruli from barotrauma (92, 93). This intrinsic myogenic effect is relatively rapid in onset (it occurs in 1–2 s) and is mediated by a mechanotransduction pathway that is thought to include TRPC channels in the smooth muscle cells of the afferent arteriole (93, 94). GFR is also regulated by the sympathetic nervous system, which projects to afferent and efferent arterioles as well as to the juxtaglomerular apparatus (95, 96). Morphologically distinct sympathetic systems appear to independently regulate the afferent and efferent arterioles, resulting in complex effects on GFR and the filtration fraction (97). Finally, there are important intrarenal feedback mechanisms whereby portions of the distal nephron communicate directly with the afferent arterioles, thereby triggering a signal that ultimately propagates to all cells within the glomeruli (67) and to nearby nephrons (23, 98). These processes include tubuloglomerular feedback (TGF) (22, 23, 93) and connecting tubule-glomerular feedback (CTGF) (24, 99). Both of these processes are slower than myogenic autoregulation and occur over a timescale of 20–40 s.
TGF is a negative feedback loop by which the preglomerular vascular resistance, and especially the tone of the afferent arteriole, is controlled by signals that originate in the macula densa of the distal tubule (93, 100). The specialized epithelial cells of the macula densa absorb Na+ and Cl− in proportion to the amount delivered to that portion of the distal tubule. NaCl delivery is influenced by the filtered load of Na+ and Cl−, which is usually directly proportional to the GFR and the amount of Na+ that is reabsorbed in more proximal portions of the nephron. Macula densa cells absorb Na+ and Cl− primarily through apical Na-K-2Cl (NKCC) transporters and, to a lesser extent, by the Na+/H+ exchanger (NHE) (101–103). This, in turn, leads to increases in intracellular Na+ and pH (104) and an increase in intracellular free Ca2+ (67). This Ca2+ signal triggers the release of ATP through a basolateral maxi-anion channel (66) into the juxtaglomerular interstitium. A portion of this ATP is converted to adenosine by 5′-nucleotidases (105, 106), and adenosine A1 receptors on the afferent arteriole appear to be essential for TGF (107, 108). ATP can also activate P2X receptors on the afferent arteriole (109). These purinergic signals drive the contraction of the afferent arteriole. However, an important study has shown that activation of macula densa cells evokes an increase in intracellular Ca2+ in extraglomerular MCs and juxtaglomerular granular cells (67). This Ca2+ wave then propagates toward proximal segments of the afferent arteriole and into intraglomerular cells, including MCs and podocytes. Indeed, the Ca2+ wave reaches extraglomerular MCs before reaching the afferent arteriole (67). In other words, TGF signaling is not limited to the afferent arteriole but appears to involve the entire glomerulus. The Ca2+ signal reaches podocytes ∼40 s after initiation of the TGF signal in the macula densa, and propagation of this Ca2+ wave requires a combination of purinergic signaling and gap junctions (67, 110). It has been observed that Ca2+ signaling in podocytes in vivo under certain conditions can be correlated with contraction of the entire glomerular capillary tuft (111). It should also be noted that MCs are necessary for TGF to exert effects on the afferent arteriole (112, 113), further suggesting that the communication between the macula densa and the afferent arteriole is at least partly indirect (51).
In contrast to TGF, CTGF is a positive feedback loop that is triggered by an increase in Na+ delivery to the distal tubule, a portion of which is also in close anatomical contact with the afferent arteriole (24). Epithelial Na+ channels (ENaCs) are the most abundant cation channels in this segment, and Na+ influx through those channels is required for CTGF (114). CTGF leads to dilation of the afferent arteriole, a process that is due in part to the secretion of prostanoids and other arachidonic acid metabolites (24, 115). Importantly, CTGF is thought to play an important role in resetting the sensitivity of the TGF mechanism (116). It is not known whether CTGF evokes signals that propagate through the entire glomerulus, but it is likely that it is able to modify TGF signals that involve all three types of glomerular cells.
2.3. Paracrine Signaling in Glomeruli
The physical proximity of podocytes, MCs, and glomerular endothelial cells allows paracrine signaling to occur between these cells, and this appears to occur even during embryonic development (117–120). Based on precedents in other systems, it is likely that some of these signals entail signaling through ion channels. Podocytes are separated from the endothelial cells and MCs by the highly permeable GBM. However, there are no physical barriers that separate MCs and endothelial cells. The existence of paracrine signaling between the three types of glomerular cells has been implied from observations in animal models and in human diseases and in some cases has been observed directly (66, 67). “Mesangiolysis” refers to the dissolution or attenuation of the mesangial matrix, which may be accompanied by the degeneration of MCs. Notably, this can occur after damage to glomerular endothelial cells induced by reactive oxygen species (ROS), toxins, or antibodies to the endothelium (117, 121, 122). Cellular cross talk between endothelial cells, MCs, and podocytes also occurs in diabetic nephropathy (DN) (123–126), which is characterized by both mesangial expansion and mesangiolysis (127). Mesangiolysis is typically accompanied by proteinuria and changes in podocyte ultrastructure, and it is thought that MCs play a role in the regulation of podocyte structure and function (128). Conversely, injuries to podocytes can drive changes in MCs. For example, MC proliferation is observed in hereditary forms of nephrotic syndrome that are caused by mutations in genes selectively expressed in podocytes (129). In mice, it has been reported that glomerular endothelial cell injury precedes changes in podocytes in doxorubicin-induced nephropathy (130).
Multiple soluble factors have been found to transmit signals between glomerular cells (128, 131). The role of ATP was mentioned in sect. 2.2 in the context of TGF. In addition, all three types of glomerular cells generate and respond to a variety of growth factors and cytokines. For example, vascular endothelial growth factor A (VEGF-A) and platelet-derived growth factor B (PDGF-B) have been implicated in signaling between podocytes and endothelial cells and between endothelial cells and MCs, respectively (48, 128, 132, 133). VEGF-A is the best-characterized member of the VEGF family, which includes VEGF-A, VEGF-B, VEGF-C, VEGF-D, and the placental growth factors. VEGF-A regulates the proliferation, migration, specialization, and survival of endothelial cells by binding to cell surface VEGF receptors 1 and 2 (132). Podocytes are both sources and targets of VEGF-A (134). Glomerular endothelial cells and MCs express VEGF receptors 1 and 2 (118, 135), and podocyte-derived VEGF-A signals to glomerular endothelial cells (136), resulting in changes in endothelial cell migration, differentiation, and survival (119, 137). This pathway also plays a role in regulating the permeability of the glomerular filtration barrier (138, 139). VEGF-A produced by podocytes also plays a role in MC survival and differentiation (137, 140). Glomerular endothelial cells can produce PDGF-B (141), which then acts on MCs through activation of PDGF-B receptors (48, 137). These various paracrine interactions are thought to play a role in glomerular capillary development during embryogenesis, as well as in the pathogenesis of adult glomerular diseases (131, 141). Several other macromolecules are thought to function in signaling between glomerular cells. For instance, nephronectin, an activating ligand of α8β1-integrin, is secreted by podocytes and deposited into the glomerular basement membrane. It then binds to α8β1-integrin expressed by MCs at the sites where they protrude into the base of the capillary loops (142). More recently, there have been reports of paracrine interactions between glomerular cells mediated through secreted microRNAs (143).
A common form of paracrine signaling, especially in vascular tissues, is mediated by small gaseous molecules, including nitric oxide (NO), hydrogen sulfide (H2S), and possibly carbon monoxide (CO). There is now evidence that these substances contribute to cell-cell communication within the glomerulus. For example, MC function is regulated by NO, which is released by glomerular endothelial cells (144) and by podocytes (145). Podocytes, MCs, and endothelial cells also produce and respond to H2S (146). Generally, these secreted gaseous signals are protective to glomerular cells, and deficits or impairment of these pathways has been reported in several glomerular disease models (146–149).
2.4. Glomerular Diseases
Glomeruli are often damaged during disease processes that are systemic and widely disseminated, but disease processes can also originate within the glomeruli. In either case, the progression of glomerular diseases usually causes changes in other renal compartments. Moreover, a number of extrarenal manifestations can be observed as overall renal function declines, such as increases in systemic blood pressure, peripheral edema, reductions in erythropoiesis, changes in bone density, increases in soft tissue calcification, and a marked increase in all-cause mortality, especially as a result of cardiovascular diseases. It is beyond the scope of this review to provide a complete classification of glomerulopathies, their clinical presentation, or their associated histopathological features. There are excellent resources elsewhere that review this vast subject (150, 151).
The most common glomerulopathies are a secondary result of systemic diseases such as diabetes mellitus, chronic uncontrolled hypertension, and systemic autoimmune disorders such as lupus erythematosus. However, some glomerulopathies, such as certain familial forms of FSGS, are classified as primary diseases. In many cases, primary glomerular diseases are idiopathic, and there is no known secondary cause. The term “glomerulonephritis” refers to diseases in which there is a strong inflammatory component within the glomeruli themselves, even at the early stages of the disease. Examples include lupus nephritis and IgA nephritis. It is not unusual to see traces of blood cells in the urine in glomerulonephritis. “Nephrotic syndrome” is a term that refers to a massive urinary excretion of proteins such as albumin, for example, >3 g per day in an adult. Urinary albumin excretion of this magnitude often drives systemic manifestations, including marked dyslipidemias. Although nephrosis sometimes occurs in the context of glomerulonephritis, it is more typically seen in syndromes, such as FSGS or minimal change disease (MCD), in which the inflammatory component is a relatively minor feature of the disease process, at least at the early stages. Precise diagnosis of a glomerular disease in many cases requires a renal biopsy along with measurement of specific biomarkers and assessments of urine protein excretion and estimation of the GFR. Modern analysis of a renal biopsy entails standard histopathology, assessment of other biochemical features such as immunoglobulin and complement deposition, and determination of glomerular ultrastructure. Features of special diagnostic significance include the amount of mesangial matrix, hypercellularity within the glomeruli, and the presence of segmental scars, glomerular crescents, or even fully collapsed glomeruli. In addition, ultrastructure can reveal changes in podocyte foot process morphology as well as alterations in the GBM or the presence of immune deposits on one or both sides of the GBM. In recent years it has become increasingly possible to classify glomerular diseases according to etiology rather than the histological pattern of injury (152). It is again worth emphasizing that podocytes have a special significance in glomerular pathology because there is only a minimal capacity to regenerate those cells. Once a sufficient number of podocytes are lost, glomerulosclerosis and the eventual loss of that nephron will inevitably follow. The cellular details of these processes, including the formation of adhesions or synechiae between Bowman’s capsule and a bare section of the GBM, have been described in detail elsewhere (27, 29, 30, 32).
3. ION CHANNELS OF MESANGIAL CELLS, ENDOTHELIAL CELLS, AND PARIETAL CELLS
MCs are a unique cell type that can behave in a manner that resembles fibroblasts (60) or contractile vascular smooth muscle cells (55). In either case, the function of MCs is tightly coupled to the activity of multiple classes of ion channels. To date, seven different classes of plasma membrane ion channels have been functionally characterized in MCs, including voltage-activated Ca2+ channels (Cav), two types of Cl− channels, large-conductance Ca2+-activated K+ channels (KCa1.1), ATP-sensitive K+ channels, canonical transient receptor potential (TRPC) channels, and store-operated Ca2+ (SOC) channels, as summarized in FIGURE 3. Recent studies have also demonstrated that transient receptor potential ankyrin 1 (TRPA1) channels are expressed in MCs and play a role in IL-1β-induced MC proliferation (153), but additional studies are required to firmly establish the role of these channels in MCs. FIGURE 3 also shows some of the signaling pathways that converge on ion channels in MCs.
FIGURE 3.

Distribution and activation of major ion channels in glomerular mesangial cells (MCs). Cav, voltage-activated Ca2+ channel; ClCa, Ca2+ activated Cl− channel; DAG, diacylglycerol; ER, endoplasmic reticulum; GPCR, G protein-coupled receptor; IP3, inositol 1,4,5-trisphosphate; KCa1.1, large-conductance Ca2+-activated K+ channel (also known as BK or Slo1); PIP2, phosphatidylinositol 4,5-bisphosphate; PLC, phospholipase C; RTK, receptor tyrosine kinase; SOC, store-operated Ca2+ channel; SR, sarcoplasmic reticulum; TRPC, canonical transient receptor potential channel.
3.1. Voltage-Activated Ca2+ Channels
Voltage-activated Ca2+ (Cav) channels were among the first to be characterized in MCs (55, 154). On the basis of biophysical and pharmacological criteria, Cav channels were initially divided into three classes, known as T-type, L-type, and N-type channels (155). T-type channels have a low unitary channel conductance (<10 pS in 100 mM Ba2+) and a low activation threshold, and they rapidly inactivate in response to sustained membrane depolarization. They are also highly sensitive to blockade by Ni2+, whereas more selective inhibition can be achieved with organic molecules such as R(−)-efonidipine, TH177, and mibefradil. L-type channels exhibit a larger single-channel conductance (>20 pS in 100 mM Ba2+) and much slower activation and inactivation kinetics. They are selectively sensitive to various dihydropyridine compounds, such as BAY K8644 (which enhances their activation) and nifedipine (which inhibits the channels). N-type channels are primarily expressed in neurons and have an intermediate conductance and a high activation threshold. R-type, Q-type, and P-type channels were subsequently identified in neurons, primarily based on pharmacological criteria, including inhibition by various venom toxins.
The presence of Cav channels in MCs was first demonstrated in cultured rat cells by fluorescence microscopy (156, 157). Direct electrophysiological evidence for Cav channels was reported later in rat (158) and human (159) MCs. These currents were enhanced by BAY K8644 and blocked by nifedipine and diltiazem and were therefore classified as L-type channels (158, 159). T-type Ca2+ channels have also been detected in human MCs on the basis of electrophysiological criteria and the presence of transcripts encoding Cav3.2 (160).
In excitable cells, activation of Cav channels by a small initial depolarization can drive a much larger regenerative depolarization. This initial depolarizing stimulus typically occurs through the activation of other types of channels. For example, TRPC channels are also expressed in MCs and are discussed in detail in sect. 3.5. Here we note that TRPC activation will result in membrane depolarization sufficient to cause activation of Cav channels in MCs. Other types of channels could play a similar role upstream of Cav in MCs. Regardless of the initial stimulus, the resulting increases in cytosolic free Ca2+ can initiate a wide variety of cellular responses including cell contraction, changes in metabolic status, cell hypertrophy, and changes in the cell cycle. In this regard, Cav channels have been suggested to play a role in both MC contraction and proliferation. For example, vasoactive peptides derived from the systemic circulation or produced locally, such as ANG II, vasopressin, and ET-1, can depolarize MCs, thereby driving Cav-dependent MC contraction (156–161). L-type and T-type Cav channels also play a role in MC growth and proliferation. Thus, in cultured rat and human MCs, dihydropyridine inhibitors of L-type channels have been shown to suppress MC proliferation and the production of ECM (162, 163). Similarly, T-type Cav channel blockers, including R(−)-efonidipine, TH177, and mibefradil, suppressed the proliferation of primary cultures of human and rat MCs (160, 164) and of rat MCs in situ after subtotal nephrectomy and during Thy1 nephritis (164–166).
3.2. KCa1.1 Channels
KCa1.1 is the preferred name for a group of large-conductance Ca2+-activated K+ channels found in a wide variety of cell types (167), including podocytes and MCs, as well as in the preglomerular vasculature. Within the literature, they are also referred to as BK channels, maxi-K channels, BKCa channels, and Slo1 channels. The primary pore-forming α-subunit of KCa1.1 is encoded by a single gene (KCNMA1) that encodes a subunit with six membrane-spanning domains (168, 169) and that assembles as a tetramer to form a functional channel (170). The KCNMA1 gene is expressed in a large number of different splice variants (>20) that differ with respect to gating (168), posttranslational regulation (171), and trafficking to the cell surface (172, 173). The native KCa1.1 channels in mammalian cells typically assemble with auxiliary subunits, which modify the properties of the channel. These include the β-subunits (β1–β4) (174), which are integral membrane proteins with two membrane-spanning domains linked by an extracellular loop domain (174). Functional KCa1.1 channels can have as many as four associated β-subunits (i.e., in a 1:1 relationship with pore-forming α-subunits) but may have fewer (175–177). The γ-subunits (γ1–4) are structurally unrelated to β-subunits (178). They have a single transmembrane domain with a large extracellular domain containing six leucine-rich repeats (179). The stoichiometry of γ-subunits with respect to the pore-forming α-subunits in native channels is not known. In addition, it has been shown that auxiliary subunits previously thought to associate primarily with other types of ion channels and transporters (such as the β1-subunits of certain Cav channels and the β1-subunits of Na+-K+-ATPase) can also interact directly with KCa1.1 channels and modify their gating or trafficking (180, 181).
KCa1.1 channels have several highly characteristic features, including a very large unitary conductance (182, 183), activation by binding of Ca2+ to cytoplasmic domains of the channel, and voltage-dependent activation (184, 185). The measured unitary conductance of KCa1.1 depends on recording conditions but is typically at least 200 pS when recordings from excised patches are made in symmetrical 150 mM KCl solutions (179, 182). With more physiological ionic gradients, the unitary conductance is substantially reduced but is still greater than that of most other K+ channels. For comparison, the renal outer medullary potassium channel (ROMK), also known as Kir1.1 or the SK channel, typically exhibits a unitary conductance of ∼35 pS (186). Application of Ca2+ to the cytoplasmic face of excised membrane patches causes KCa1.1 channels to become active. The Ca2+ sensitivity is a function of the membrane potential (167, 185) and depends on which, if any, auxiliary subunits are present (167, 179). Additional heterogeneity results from formation of heterotetramers composed of different α-subunit splice variants. It is important to note that the pharmacological properties of KCa1.1 channels also can vary depending on their subunit composition (167, 187, 188). For example, KCa1.1 channels that assemble with β4-subunits are relatively resistant to scorpion toxins such as iberiotoxin, slotoxin, and charybdotoxin, which are potent inhibitors of KCa1.1 in the absence of those particular auxiliary subunits (189). The Ca2+ sensitivity of KCa1.1 channels in most cases requires them to be closely colocalized with some Ca2+-permeable channel so that they remain within the “spark” nanodomain that surrounds active Cav channels or other Ca2+ sources (190–193). For example, KCa1.1 channels in various types of native cells can often be activated when cells are loaded with EGTA but not if they are loaded with BAPTA, a Ca2+-buffering agent with similar Ca2+ affinity but much faster binding kinetics (194). The consequences of this for overall cell physiology will depend on how Ca2+ influx varies as a function of membrane potential, as discussed further below.
KCa1.1 channels have been identified in podocytes and glomerular MCs and in several tubule segments, including principal and intercalated cells of connecting tubules and cortical collecting ducts (41, 195). In MCs the properties of KCa1.1 channels were initially described by single-channel recording methods (196), and it was subsequently shown that the MC KCa1.1 channels contain β1-subunits, similar to the KCa1.1 channels in smooth muscle cells (197). KCa1.1 channels are likely to function as a negative feedback mechanism downstream of the activation of Cav channels of MCs (198, 199). Thus, an increase in the intracellular Ca2+ concentration secondary to the opening of Cav channels drives activation of KCa1.1 (198). This in turn causes the membrane potential to return to a more negative membrane potential, resulting in the deactivation of the Cav channels. This negative feedback loop may contribute to physiological regulation of MC tone. However, as discussed in sect. 4.2, KCa1.1 channels may have a distinctly different function in cells such as podocytes that lack Cav channels.
The activity of KCa1.1 channels in MCs is regulated by multiple intracellular intermediates. Their activation appears to be enhanced by cyclic guanosine monophosphate (cGMP), cGMP-dependent protein kinase (PKG), and mitogen-activated protein kinase (MAPK), whereas they tend to be inhibited by protein phosphatase 2A (PP2A) (55, 200, 201). Consequently, KCa1.1 can contribute to responses evoked by several hormones and paracrine factors, including atrial natriuretic peptide (ANP), insulin, NO, and transforming growth factor-β1 (TGF-β1) (200, 202). It is generally thought that MC tone contributes to the regulation of glomerular filtration function by changing the filtration coefficient (Kf) (48, 55). Activation of KCa1.1 is expected to reduce MC tone, which could therefore lead to an increase in the GFR. Activation of KCa1.1 may also play a role in limiting the effects of certain vasoconstrictor substances such as ANG II, while enhancing the effects of certain vasodilator substances. For example, the vasodilator ANP induces increases in GFR and is known to activate KCa1.1 through activation of cGMP-PKG signaling pathways (203, 204). Studies carried out in vivo support a possible role for KCa1.1 in the regulation of glomerular filtration, although it should be noted that this effect is likely to entail effects on many different vascular tissues and cell types. For example, knocking out the KCa1.1 β1-subunit in mice prevented increases in GFR that generally occur after blood volume expansion (205).
In addition to the regulation of MC tone, KCa1.1 channels also promote MC proliferation, migration, and apoptosis (206). Thus, KCa1.1 channels are required for high-glucose-induced proliferation, migration, apoptosis, and production of extracellular matrix proteins in cultured MCs. This effect is mediated in part through the activation of TGF-β1/Smad2/3 signaling pathways (206). Insulin also modulates KCa1.1 channels in MCs by increasing both the activity and the number of available channels (202).
3.3. ATP-Sensitive K+ Channels
ATP-sensitive K+ channels (KATP) are large multisubunit complexes that function to couple membrane potential and excitability to the bioenergetic state of a cell. KATP activity is suppressed by the binding of ATP and enhanced by the binding of ADP to different sites on the cytoplasmic face of the channel complex. Therefore, the KATP gating state will depend on the overall energy charge of the cell. Functional KATP complexes include the pore-forming subunits (Kir6.1, and Kir6.2), which are members of the large KCNJ family of inwardly rectifying K+ channels. They also contain sulfonylurea receptors (SUR1, SUR2A, and SUR2B), which belong to the ATP-binding cassette superfamily.
Immunohistochemical studies have shown that Kir6.1 is expressed in rat glomerular MCs (207), and whole cell recordings have identified functional KATP channels in both mouse and rat MCs (208, 209). The physiological role of KATP channels in MCs is not well understood, but there is evidence that they are inhibited in the presence of elevated external glucose, as is seen in other cell types. Moreover, their activation tends to suppress the proliferation of cultured MCs and inhibit ECM production (209). The role of KATP in MC function in vivo is not known.
3.4. Cl− Channels
Glomerular MCs possess two populations of Cl− channels, including a Ca2+-activated Cl− (ICl.Ca) channel and a volume-sensitive Cl− (ICl.vol) channel (210). These channels have distinct gating mechanisms and regulate different aspects of MC function. Because the Cl− equilibrium potential in MCs is less negative than the resting membrane potential, the opening of Cl− channels leads to depolarization (211). The ICl.Ca channels in MCs become active in response to the application of certain vasoactive agonists or treatment with the Ca2+ ionophore A-23187 (210), and direct electrophysiological evidence for ICl.Ca channels has been obtained in cultured human and immortalized murine MCs (161, 210). In those experimental systems, the conductance mediated by ICl.Ca channels increases linearly with an increase in intracellular free Ca2+ concentration and is blocked by niflumic acid (210). It is likely that the primary physiological role of ICl.Ca in MCs is to contribute to depolarization of the plasma membrane during the actions of vasoconstrictors such as ANG II, arginine vasopressin (AVP), and ET-1, which act in part to stimulate the release of Ca2+ from intracellular stores. This allows activation of ICl.Ca channels, resulting in membrane depolarization and activation of Cav channels in the plasma membrane. ICl.vol has been observed in both human MCs and an SV40-immortalized mouse MC cell line (210). It exhibits marked outward rectification and is modulated by extracellular osmolarity but not by cytosolic Ca2+ (210). ICl.vol channels have been implicated in MC alkalinization induced by hyperosmolality (212) and in MC apoptosis caused by oxidative stress (213).
3.5. TRPC Channels
Transient receptor potential canonical (TRPC) channels belong to the transient receptor potential (TRP) superfamily (214) and are by far the most intensely studied family of channels in glomerular cells. There are seven subtypes of TRPC proteins, designated TRPC1–7, although it should be noted that TRPC2 is encoded by a pseudogene in humans. Based on their primary structures, TRPC1 is most closely related to TRPC4 and TRPC5, whereas TRPC3 is grouped with TRPC6 and TRPC7 (214). TRPCs can function as store-operated channels (215–217), receptor-operated channels (218, 219), redox-sensitive channels (220–222), and channels that can be activated in response to mechanical stimuli applied to cells (223–226). TRPCs are nonselective cation channels and allow for at least some Ca2+ influx under most conditions, although the permselectivity of these channels exhibits complex characteristics (227, 228).
The various TRPC channels each have a distinct tissue distribution, and it is likely that heteromultimerization of different TRPC subunits increases the heterogeneity of this family of channels (229, 230). A systematic evaluation of TRPC interactions in heterologous expression systems found that TRPC channels generally interact only within their own subfamilies. Thus, direct interactions between TRPC1, TRPC4, and TRPC5 are possible. Similarly, TRPC3, TRPC6, and TRPC7 can interact (214, 230). TRPC channels are known to be expressed in MCs and in podocytes. In this section, we review TRPC channels in MCs. TRPC channels of podocytes are discussed in sect. 4.1. As noted in sect. 2.1, glomerular MCs and the ECM that they secrete form the central stalk of the glomerulus (59, 60). Signaling through Ca2+ plays a central role in regulating MC physiology (154), and this process entails effects on multiple TRPC channels.
TRPC1 proteins were initially found in glomerular MC lysates from rat kidneys (231), and a later study detected TRPC1 and TRPC4 in mouse MCs (232). Immunoblot analysis and immunocytochemistry detected TRPC3 and TRPC6 channels in addition to TRPC1 and TRPC4 in cultured human MCs, whereas TRPC5 and TRPC7 were not detectable by either method (233). Coimmunoprecipitation and immunofluorescence double staining have shown biochemical interactions of TRPC1 with TRPC4 and TRPC6 (234), whereas no interactions were detected among other TRPC isoforms (233). Human glomerular MCs are reported to express TRPC1, 3, 4, and 6 proteins (233, 235), and TRPC1 expression in an immortalized human MC line appears to be under the control of MiR-135a (236). The activity or expression of TRPC6 in MCs can be stimulated by ANG II, chronic hypoxia, and phenylephrine (237–241). These various studies have used different assays that are likely to have different sensitivities, including the use of different antibodies. Nevertheless, there appears to be a consensus that multiple TRPC channels are expressed in glomerular MCs, including TRPC1 and TRPC6.
Glomerular MCs have a contractile phenotype similar to that of vascular smooth muscle cells, especially when they are grown in dissociated culture. The contractile properties of MCs may enable them to alter the intraglomerular capillary flow and glomerular ultrafiltration surface area and hence GFR (55, 60, 242). Similar to vascular smooth muscle cells, MC contractile function is controlled by the cytosolic Ca2+ concentration, and especially by Ca2+ influx through various Ca2+-permeable channels in the plasma membrane. TRPC channels are permeable to Ca2+, more so at more negative membrane potentials (227, 228). They can be activated by chemical stimuli such as diacylglycerol (DAG) and by mechanical stimuli (223, 224, 226, 243), both of which are physiologically relevant for MCs.
TRPC1 appears to contribute to Ca2+ entry and thereby modulate the contraction of human MCs in response to ANG II. It has been proposed that inhibition of mesangial TRPC1-mediated Ca2+ influx should result in increases in GFR and should also attenuate the decline in GFR that occurs as a result of MC contraction. This prediction was supported by an in vivo study in which infusion of a TRPC1 antibody directed against an extracellular epitope of TRPC1 resulted in significant attenuation of the ANG II-induced decrease in GFR in rats (244). FIGURE 4 summarizes the effects of ANG II on TRPC1 activity in MCs and on GFR. In this regard, ratiometric Ca2+ imaging has suggested that store-operated Ca2+ entry (SOCE) was significantly reduced by knocking down TRPC1 and enhanced by overexpressing TRPC1 (233). SOCE mechanisms are discussed in more detail in sect. 3.6.
FIGURE 4.
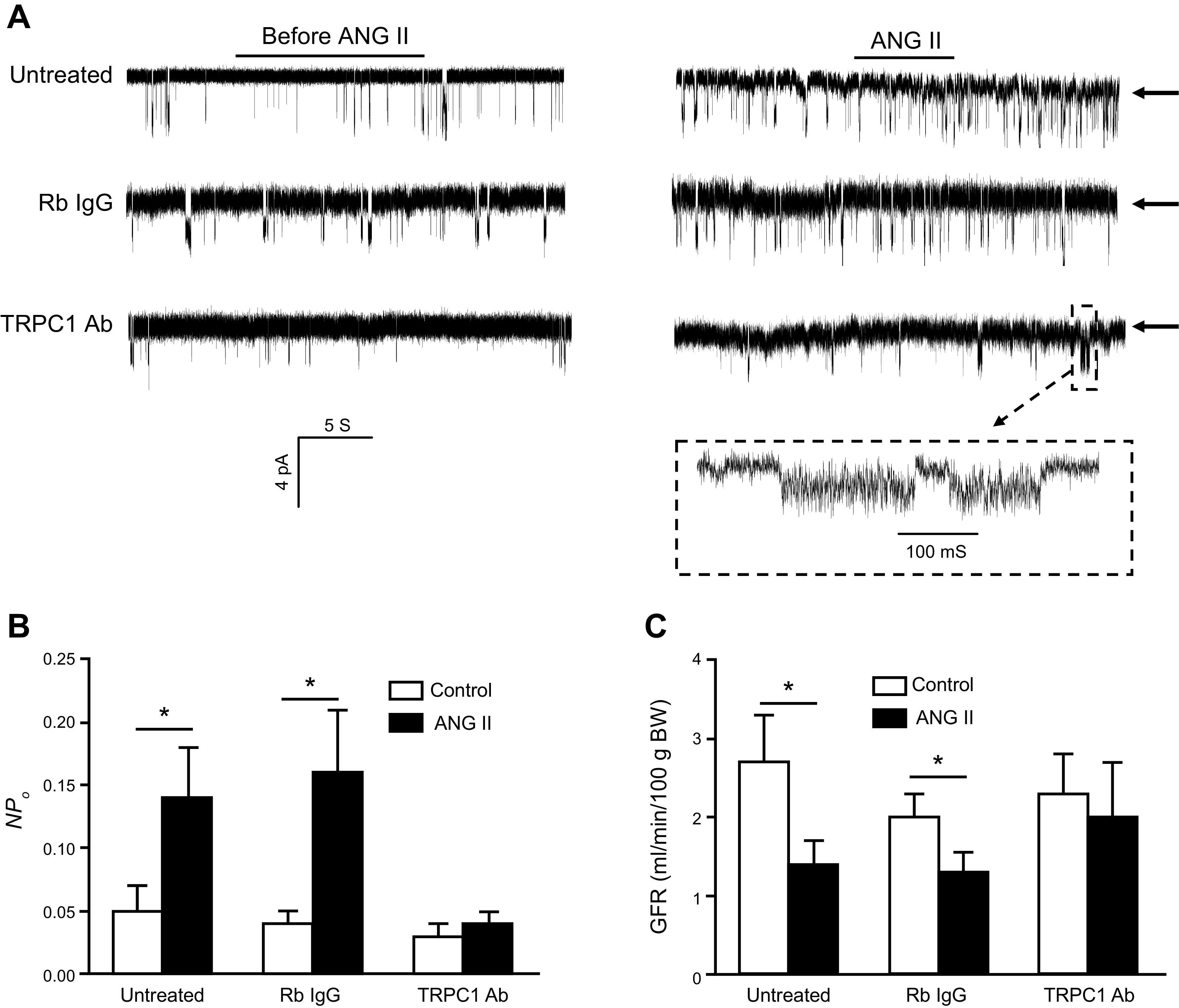
Activation of TRPC1 channels in glomerular mesangial cells (MCs) and its role in the regulation of the glomerular filtration rate (GFR). A: representative cell-attached single-channel currents evoked by application of 1 µM ANG II to cultured MCs in the presence or absence of rabbit (Rb IgG) or TRPC1 antibodies (Abs). Arrows indicate the closed state of the channels. Downward deflections indicate inward currents. The recording electrode was held at −80 mV. The bottom trace (inside the dashed rectangle) is the time-expanded portion of the trace indicated by a small dashed rectangle above. B: single-channel TRPC1 activity (NPO) before and after application of ANG II in untreated, Rb IgG-, and TRPC1 Ab-treated MCs. C: GFR, evaluated by inulin clearance, before and during infusion of ANG II [1.7 ng/min per 100 g body weight (BW)] in the presence or absence of an inactivating TRPC1 or Rb IgG antibodies. *P < 0.05 between the indicated groups. Adapted from Ref. 244 with permission.
TRPC6 also appears to play a role in regulating Ca2+ entry and contraction in MCs. In cultured human MCs overexpression of TRPC6 significantly enhanced, and knockdown of TRPC6 significantly attenuated, ANG II-stimulated cation currents and Ca2+ influx (245) as well as ANG II-stimulated contraction (246). ANG II-stimulated contraction and Ca2+ entry were also reduced in primary MCs isolated from TRPC6-deficient mice (247). Notably, the reduced Ca2+ response in TRPC6-deleted mouse MCs could be rescued by reintroducing TRPC6. These observations have been supported by studies carried out in vivo. Thus, GFR has been analyzed in conscious wild-type and TRPC6 knockout mice and in anesthetized rats with and without in vivo knockdown of TRPC6 in the kidneys. TRPC6-deficient mice exhibited a greater GFR, as indicated by a reduction in serum creatinine. In addition, local knockdown of TRPC6 in kidneys with a TRPC6-specific shRNA significantly attenuated ANG II-induced suppression of GFR in rats (247). It should be noted that deletion or inhibition of a particular TRPC channel (TRPC1 or TRPC6) was not specific for MCs in these in vivo studies. These manipulations may have induced changes in blood pressure, renal blood flow, and the tone of the afferent and/or efferent arterioles and may also reflect contributions from podocytes. Unfortunately, at the present time there is no MC-specific promoter that would allow a TRPC channel knockout to be specific for this population of cells. However, a targeted nanoparticle-siRNA in vivo system has recently been developed that can deliver siRNAs into MCs with high selectivity in mice (248, 249). FIGURE 5 illustrates the principles underlying this approach, which may provide a basis for understanding the role of TRPC and other channels in MCs.
FIGURE 5.

In vivo knockdown of Orai1 with small interfering RNA/cyclodextrin-containing polymer nanoparticles (siRNA/NPs) in mesangial cells (MCs) resulted in an increase in glomerular ECM protein deposition in mice. A: schematic assembly of a siRNA/NP. When mixed in an aqueous solution (5% dextrose), the cationic cyclodextrins (CDP) assemble with the negatively charged siRNA molecules. As a result, 5-kDa polyethylene glycol (PEG) molecules are covalently linked to the small molecule adamantane (AD) and form guest/host interactions with the nanoparticle’s CDP component stabilizing the nanoparticles. In addition, the distal end of the AD-PEG molecules can be covalently linked to targeting ligands (TL) that facilitate cellular internalization of the nanoparticles (adapted from Ref. 248 with permission). B: schematic of nanoparticle deposition in glomerular MCs and in the mesangium. ECM, mesangial extracellular matrix; GBM, glomerular basement membrane; NP, nanoparticle. C: representative images show localization of nanoparticles containing Cy3-tagged Orai1 siRNA (NP-Cy3-siOrai1) (red signals) in glomeruli (indicated by arrows) but not in tubules. D: localization of NP-Cy3-siOrai1 in MCs (left) but not in podocytes (right). MCs and podocytes were stained with integrin-8 (green) and synaptopodin (green), respectively. NP-Cy3-siOrai1 is shown as red signals. E and F: expression of fibronectin (E) and collagen IV (Col IV) (F) in glomeruli of mice treated with NP containing scrambled siRNA (NP-Con) and NP-Cys-siOrai1. Both fibronectin and Col IV are shown as green signals. A bright-field image of the kidney section was captured in NP-Con-treated mice to show the glomerulus. In NP-Cy3-siOrai1, the distribution of NP-Cy3-siOrai1 is indicated by Cy3 signals (red). Arrows indicate glomeruli. Original magnification ×200. C, E, and F are adapted from Ref. 42, and D is adapted from Ref. 250 with permission.
MCs secrete and respond to various growth factors. In this regard, TRPC6 has been shown to be involved in ANG II-induced MC proliferation, which is mediated by AT1 receptors (AT1Rs) acting through extracellular signal-regulated kinases (ERKs) (237). Stimulation of the Ca2+-sensing receptor (CsR) also induces the proliferation of human MCs (235), and this effect appears to be mediated by TRPC3 and TRPC6 acting as receptor-operated channels (235).
It is likely that TRPC channels function as heterotetramers in many types of native cells (230, 234). This raises the question of whether individual TRPC channels in MCs act as homomers or as heteromultimeric complexes with other pore-forming proteins. TRPC1 has multiple binding partners in MCs, and, as noted above, both TRPC1 and TRPC6 are required for ANG II-induced MC contraction (234, 246, 247). Moreover, TRPC1 is reported to physically interact with TRPC6 in human MCs (233), and these isoforms may assemble to form heteromeric channels in MCs. In this regard, single knockdown of TRPC1 or TRPC6, or double knockdown of both proteins, caused comparable reductions in ANG II-stimulated Ca2+ influx. This observation is consistent with the hypothesis that TRPC1 and TRPC6 are components of the same heteromultimeric channels (247).
Finally, it should be mentioned that MCs are reported to express TRPP2 channels (also known as PKD2 channels) (251), which have received considerable attention in the context of polycystic kidney diseases and which are distantly related to TRPC channels. TRPP2 channels are able to coimmunoprecipitate with TRPC1 and TRPC4 and have been shown to contribute to the responses evoked by ANG II in MCs.
3.6. Store-Operated Ca2+ Channels
Store-operated Ca2+ channels (SOCs) are defined as channels that open in response to the depletion of internal Ca2+ stores in ER and related “calciosomes” (252–254). Activation of β isozymes of phospholipase C (PLC-β) by GPCRs, or activation of PLCγ isozymes by receptor tyrosine kinases, results in the hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2), thereby generating inositol 1,4,5-trisphosphate (IP3) and DAG. IP3 stimulates the release of Ca2+ from intracellular stores in the ER, and the depletion of those stores subsequently results in the activation of SOCs in the plasma membrane (255). The influx of Ca2+ via SOCs was initially referred to as “capacitative Ca2+ entry,” and this term is still occasionally encountered, but the term “store-operated Ca2+ entry” (SOCE) is now preferred. Substantial information on the biophysical and pharmacological features of SOC and their function is now available (252, 255–257). SOCs appear to be heterogeneous, and the term “SOC” encompasses both Ca2+-selective and nonselective cationic SOCs (252, 254). The characteristics of SOCs depend on the cell type and tissue examined (254, 256–258). It is important to note that SOC activation is never dependent on cytosolic Ca2+ concentration but is instead triggered by a reduction of Ca2+ within certain internal Ca2+ stores (256, 259). This unique property is in marked contrast to other cation channels, such as Ca2+-activated nonselective cation channels.
There are three major hypotheses regarding the activation mechanisms of SOCs. These include 1) diffusible messengers (260); 2) vesicle fusion/exocytosis (261); and 3) direct coupling between endoplasmic reticulum (ER) IP3 receptor channels and plasma membrane Ca2+ channels (262, 263). Early studies suggested that TRPC proteins are components of SOCs (232, 264–266). However, significant differences in the biophysical and pharmacological properties between TRPC channels and native SOCs raised doubts about TRPC channels as the molecular mediators of SOCs. A major advance came with the discovery of the stromal interaction molecule (STIM) (267, 268) and Orai protein families (269, 270). STIM1 is a single-pass transmembrane protein located primarily in the ER, which functions as a sensor of the ER luminal Ca2+ concentration. Orai1 is a small plasma membrane protein that constitutes a pore-forming unit of SOC. Upon depletion of ER Ca2+, STIM1 aggregates and translocates to the vicinity of ER-plasma membrane junctions, where it physically interacts with Orai1. This interaction results in activation of Orai1, thereby causing Ca2+ influx into the cytosol (270, 271). In addition to STIM1 and Orai1, STIM2 (a closely related mammalian ortholog of STIM1) and Orai2 and Orai3 (additional mammalian orthologs of Orai1) may also contribute to the formation of SOCs, albeit with distinct functional properties (272–274). SOCE pathways are further complicated by the existence of splice variants of both Orai1 (Orai1α and Orai1β) (275, 276) and STIM1 (STIM1 and STIM1L) (277, 278), which results in additional functional heterogeneity. Several TRPC proteins can interact with STIM1 and/or Orai1 (266, 279–281).
Evidence for SOCs in MCs was initially obtained in ratiometric Ca2+ imaging experiments in which depletion of ER Ca2+ stores by ANG II, thapsigargin, or ionomycin was observed to significantly potentiate Ca2+ influx in cultured human MCs (282). These early observations were subsequently confirmed in rat and human MCs (257, 283–285). Direct evidence for SOCs in MCs was also obtained from single-channel recordings. Thus, unitary SOC currents became active in cell-attached patches in response to thapsigargin in cultured human MCs preincubated with BAPTA-AM, a rapidly acting membrane-permeant Ca2+ buffer. These currents had a very low unitary conductance (2.1 pS with Ba2+ in the recording electrode), high Ca2+ selectivity (87/8.2/1 for Ca2+/Ba2+/K+), and a markedly positive reversal potential (63 mV with 90 mM Ba2+ in the pipette solution) and were blocked by low concentrations of La3+ (2 µM) (257). In addition, the open probability of these channels was independent of membrane potential. These properties are similar to those of SOCs described in other cell types (255, 286, 287). Rectifying macroscopic currents through SOCs were also observed in whole cell recordings from MCs in response to thapsigargin or ANG II (288).
SOCs are known to be heterogeneous (289), and this has raised the issue of the molecular identity of SOCs in MCs. As described above, multiple TRPC proteins, including TRPC1 and TRPC4, are expressed in MCs. Moreover, transient knockdown of TRPC4 resulted in reduced Ca2+ influx induced by internal Ca2+ store depletion, suggesting that this channel contributes to SOCE in MCs (232). Orai1 and STIM1 proteins have also been detected in rat (290) and human (234, 291) MCs, and Ca2+ imaging and patch-clamp recordings have provided functional evidence that Orai1 and STIM1 are elements of SOCE in MCs. However, it is important to note that TRPC1 and TRPC4 can form a heteromeric complex that can interact with STIM1 in human MCs. Moreover, siRNA knockdown of STIM1, TRPC1, or TRPC4 significantly reduced thapsigargin-induced membrane currents (234). It is not known whether Orai1 and TRPCs function independently as distinct SOCs, but it is possible that they interact to form a common SOC. The resolution of this question will require a more detailed analysis of the biophysical and pharmacological properties of the Orai1-mediated and TRPC-mediated SOC in MCs.
SOC-mediated Ca2+ signaling plays a role in the regulation of a wide variety of cellular processes, including exocytosis, enzyme activity, gene transcription, cell proliferation, and apoptosis (252, 289). In addition, there are several potential pathways for SOC activation in MCs (253). Certain vasoactive factors, including ANG II and thromboxane A2, can activate SOCs through GPCRs coupled to PLC/DAG/IP3 pathways in MCs (282, 288). In addition, epidermal growth factor (EGF) can stimulate SOC-mediated channel activity and Ca2+ entry in cultured human MCs through effects mediated at least in part on plasma membrane tyrosine kinase receptors (283). Interestingly, in contrast to most other stimuli, EGF does not trigger detectable ER Ca2+ release and seems to activate SOCE in an IP3-independent manner, albeit with PLC as a crucial element (288). Several isoforms of PKC can become active as a result of PLC signaling. It has been suggested that PKC may modulate SOC activity directly, i.e., independently of the state of Ca2+ stores. It has also been reported that calphostin C, a specific PKC inhibitor, diminished thapsigargin-activated SOCE in cultured human MCs (292). Moreover, activation of PKC or application of an active catalytic subunit of PKC directly to excised inside-out patches also activated channels that resembled SOCs (292). A later study provided evidence that PKCα was the specific isoform of PKC responsible for this effect (285). Similar results have been obtained in endothelial cells, in which SOCE activated by thrombin or thapsigargin was significantly diminished by pharmacological or genetic inhibition of PKCα (293). On the other hand, it has been suggested that PKC acts as an inhibitor of SOC in cultured human MCs because phorbol 12-myristate 13-acetate (PMA), an activator of PKC, inhibited ANG II-stimulated Ca2+ influx assessed by microfluorometry (294). The interpretation of that result is open to question because the ANG II-induced Ca2+ influx required an increase in cytosolic free Ca2+, in contrast to what is seen with SOCs. However, it has been shown that phosphorylation of Orai1 on Ser 27 and Ser 30 residues by PKCβ1 results in SOCE suppression in HEK-293 cells (295). It is possible that the effect of any PKC enzyme of SOCE in MCs depends on the pathway whereby it becomes active.
Other pathways may regulate SOCs in MCs. For example, PKG-mediated phosphorylation of vasodilator-stimulated phosphoprotein (VASP), a focal adhesion molecule highly expressed in MCs, causes VASP to associate with TRPC4 and to inhibit the associated SOCE (296). Although the precise mechanism of this inhibition is not clear, phosphorylated VASP may inhibit SOCE by dissociating TRPC4 from the SOC complex.
3.7. Purinergic Signaling and P2X Receptors in Mesangial Cells
Purinergic signaling plays an essential role in renal physiology, and dysregulation of these systems may contribute to renal disease (297). The nature of P1 adenosine receptors, various P2 receptor systems, and the extracellular molecules derived from ATP has been reviewed previously (108). This section focuses on P2X receptor expression and function in MCs. The term “P2,” as opposed to “P1,” is a purely pharmacological designation, based on the agonists that can activate these receptors (e.g., ATP and certain other nucleotides) and is not based on current understanding of the structure of these receptors. For this reason, the various P2 receptors include large families of ligand-gated ionotropic P2X receptors as well as the G protein-coupled P2Y receptors. There is no structural relationship between P2X and P2Y receptors, although both are tied to Ca2+ signaling. Ionotropic P2X channels open rapidly upon binding of ATP or other extracellular purines to accessible ectofacial domains. In fact, P2X receptors are structurally related to epithelial sodium channels (ENaCs) (298, 299) and as such contain three subunits. Each subunit has two membrane-spanning domains with both the NH2 and COOH termini extending into the cytosol (FIGURE 6A). Seven different genes encode P2X subunits. They can form heteromeric complexes, but it is unknown whether heteromerization is a feature of the endogenous receptors in glomerular cells or other portions of the nephron. P2X receptors are nonselective Ca2+-permeable cation channels (300), and the expression of these receptors is dynamic and appears to be modulated by several pathophysiological factors and as a result of aging. The concentration of extracellular P2 receptor ligands such as ATP can change rapidly during pathological conditions (301, 302), which can drive changes in the expression and composition of different types of P2 receptors in the plasma membrane. Chronic exposure to high concentrations of ATP appears to modulate the permselectivity of P2X receptor pores, increasing their permeability to somewhat larger organic molecules (e.g., N-methylglucoasamine) (303, 304).
FIGURE 6.
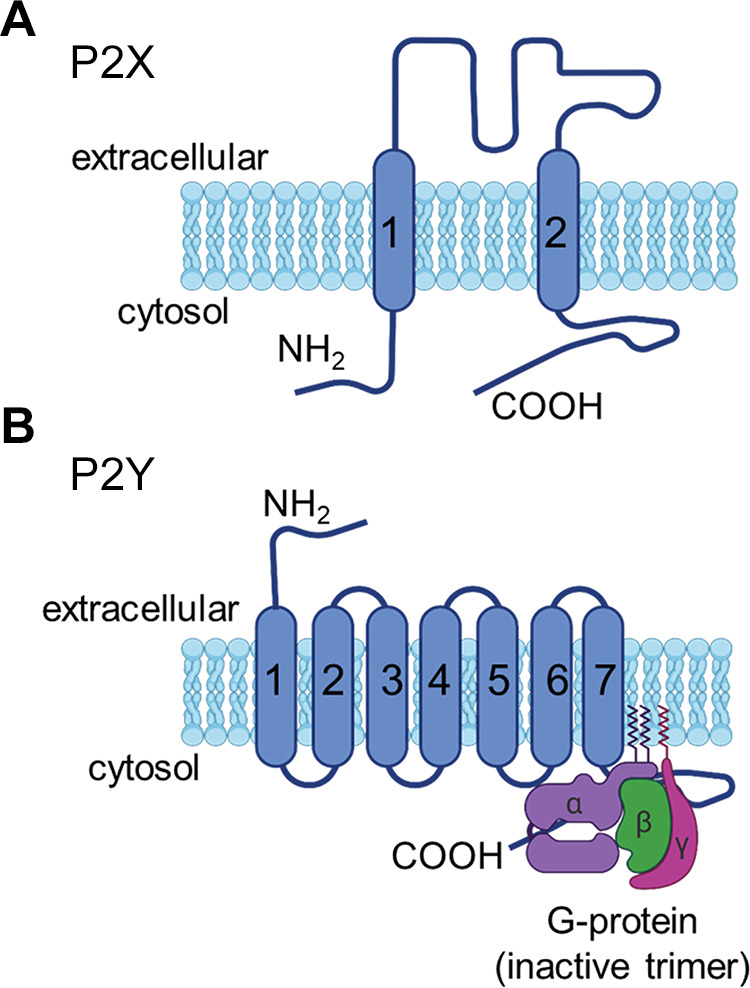
Structure of P2X and P2Y receptors. A: ionotropic P2X receptors are trimeric proteins formed by subunits with 2 membrane-spanning domains. The amino and carboxy termini extend into the cytosol. B: metabotropic P2Y receptors have 7 membrane-spanning domains and are coupled to heterotrimeric G proteins.
By contrast, the metabotropic P2Y receptors are seven-transmembrane GPCRs (FIGURE 6B) that bind extracellular nucleotides including ATP, ADP, UTP, UDP, and UDP-glucose, leading to the generation of a variety of intracellular signals. P2Y receptors are expressed throughout the nephron (305). The expression of P2Y1 and P2Y2 receptors in MCs and glomerular epithelial cells was initially demonstrated by immunohistochemical staining and by measuring Ca2+ signals evoked by specific nucleotide agonists (306, 307). Recent studies suggest that P2Y receptors are normally the dominant pathway for purinergic signaling in glomerular cells (43, 308). By contrast, P2X receptors are generally present at lower levels, but they are markedly increased under specific pathological conditions in which a sustained high concentration of extracellular ATP may trigger the remodeling of purinergic signaling to include a more significant ionotropic component.
Activation of the various P2 receptors in MCs results in a rapid transient response to the application of ATP (309). However, activation of the different types of P2 receptors can lead to opposing effects depending on what output is measured. For example, in rat glomerular MCs, ATP-mediated activation of P2Y2 and/or P2Y4 receptors triggered an increase in DNA synthesis and induced cell proliferation (310). By contrast, P2X7 activation by 3′-O-(4-benzoyl)benzoyl ATP (BzATP) promoted apoptotic cell death (310).
3.8. Ion Channels of Endothelial Cells and Parietal Cells
In contrast to other glomerular cell types, very little is known about ion channels of glomerular endothelial cells or parietal cells. Histochemical studies have indicated that both of these cells express an intrinsically mechanosensitive cation channel known as Piezo1 (311), but to date no functional studies have been carried out to determine whether these channels can become active. Interestingly, Piezo2 expression was also recently reported in mouse MCs, and it was suggested that Piezo2 plays a role in the regulation of glomerular filtration (312). Endothelial cells and parietal cells are likely to express other types of channels, and this is a topic that needs additional investigation, especially given that endothelial cells participate in a variety of signaling pathways described in sect. 2.3.
4. ION CHANNELS OF PODOCYTES
4.1. TRPC Channels of Podocytes
The general features of TRPC family channels are discussed in sect. 3, where we emphasize that this is the most extensively studied family of channels in glomerular cells. We discuss their role in podocyte Ca2+ signaling in sect. 5 and their dysregulation and contributions to glomerular disease, especially podocyte disease, in sect. 6. This section describes some of the gating and permeation properties of TRPC channels in podocytes. TRPC6 channels are found both in the foot processes and in the cell body of podocytes. It is likely that these channels have different functions and are subject to different modes of regulation depending on where they are located within the cells. For example, TRPC6 channels in foot processes may be more relevant to the regulation of the local cytoskeleton, whereas TRPC6 channels in the cell body may be more critical in the regulation of gene expression.
TRP superfamily channels are often multimodal when expressed in cells, meaning they can become active in response to several different stimuli (226). For example, TRPC6 channels are required for pressure-induced myogenic autoregulation in isolated cerebral resistance vessels (313). Multimodal activation has been extensively studied in the case of the TRPC6 channels of podocytes. TRPC6 can be activated through several GPCRs that are coupled to Gq and PLCβ. For example, podocyte TRPC6 channels are activated by ANG II acting through AT1 receptors and by ATP acting through various P2Y receptors (FIGURE 7A). The pathway for TRPC6 activation by GPCRs in podocytes has certain unique features compared to other cell types, which we describe further below. Here we note that in every case studied to date, GPCR-mediated TRPC6 activation in podocytes requires the localized generation of ROS (314–316). TRPC6 channels in podocytes also become active when various mechanical stimuli are applied to the cells (226). However, the mechanisms whereby mechanical stimuli can activate TRPC6 channels in cells are not well understood. A recent study has shown that TRP channels such as TRPC6 are not mechanosensitive when they are the only proteins present in lipid bilayers (317), and it has been argued that the pathways that lead to mechanical activation of TRPC6 require activation of GPCRs and/or production of DAG (317). Mechanosensitivity of a channel can arise from interactions with cytoskeletal elements that are not present in artificial bilayers (318). That could in turn depend on how the cytoskeleton is arranged and also on whether or not the channel is present in a lipid raft (319), which is the membrane environment of TRPC6 channels in podocyte foot processes. Whatever the biophysical details may be, the mechanisms underlying stretch activation of TRPC6 in podocytes are biochemically and pharmacologically distinct from those used for activation by GPCRs (226) (FIGURE 7B). For example, knockdown of podocin, a raft-domain protein unique to podocytes, enhances TRPC6 activation by mechanical stimuli, whereas this manipulation suppresses activation by GPCRs (226, 320). In addition, mechanical activation of TRPC6 persists when all G protein signaling is inhibited or in the presence of ROS quenchers such as TEMPOL, whereas TRPC6 responses to ANG II and ATP are completely blocked by these procedures (226, 314). Mechanical activation of TRPC6 in podocytes is greatly enhanced by depolymerization of the actin cytoskeleton with cytochalasin D (226). Mechanical activation of TRPC6 is also blocked by GsMTx4, a small spider toxin that inhibits the activity of several types of mechanosensitive channels (226), probably by altering the interactions of transmembrane proteins with the lipid bilayer. On the other hand, both mechanical and GPCR activation of podocyte TRPC6 channels are inhibited by agents such as SAR-7334 and La3+ that act directly on the channels (226, 321, 322).
FIGURE 7.

Polymodal gating of TRPC6 channels in podocytes. A: activation of podocyte GPCRs leads to activation of PLC, generation of DAG, and Rac1-dependent activation of the NADPH oxidase NOX2, leading to a localized increase in ROS generation in the immediate vicinity of TRPC6. The catalytic subunits of NOX2 form a complex with podocin and TRPC6. These interactions with podocin are essential for TRPC6 activation by GPCRs. B: activation of podocyte TRPC6 channels by mechanical stimuli, which does not require activation of any cellular GTPases or generation of ROS. Podocin functions to suppress activation of podocyte TRPC6 by mechanical stimuli, possibly by regulating membrane stiffness or by modulating interactions of the channel complex with cytoskeletal elements. Podocin has a cholesterol-binding domain and functions as a scaffold to hold its binding partners within lipid raft domains in foot processes. See glossary for abbreviations.
There are at least two other TRPC channels expressed in podocytes. TRPC3 channels are easily detected in cultured podocytes and acutely isolated glomeruli, and it is likely that at least some of the TRPC6 channels in podocytes form a complex with TRPC3. Inactivation of TRPC6 channels in rats in vivo leads to an increase in the abundance of glomerular TRPC3 protein, but although they are readily detected by immunoblot, the TRPC3 channels cannot be activated by application of ATP in the absence of TRPC6 (39). It is possible that TRPC3 subunits in podocytes are modified in such a way that they cannot form functional channels by themselves or they do not traffic to the cell surface. There is also evidence that TRPC5 channels are expressed in podocytes, certainly in mice and rats. The factors that cause TRPC5 channels to become active in normal podocytes are not known. Most of the conditions in which they can be detected by electrophysiological methods appear to resemble disease conditions, and we therefore defer a detailed discussion of these channels until sect. 6, where we discuss the contributions of ion channels to glomerular disease in more depth, as well as the potential of those channels as therapeutic targets.
4.2. KCa1.1 Channels and Associated Subunits
The general properties of KCa1.1 channels are discussed in sect. 3.2. The topological features of KCa1.1 α-, β-, and γ-subunits are shown in FIGURE 8A. KCa1.1 channels are expressed in rodent glomeruli, and a portion of these channels closely colocalize with synaptopodin (FIGURE 8B), indicating that they are expressed in podocyte foot processes, although they are also present in the podocyte cell body and in MCs (323). The KCa1.1 channels in human glomeruli colocalize with nephrin, again indicating endogenous expression in podocytes (324). Functional KCa1.1 channels have been characterized in immortalized podocytes derived from humans (324) and mice (173, 323, 325–328). The KCa1.1 channels of immortalized human podocytes become active during membrane stretch (324), a phenomenon observed in other cell types including vascular smooth muscle cells (329, 330). Stretch activation of KCa1.1 in human podocytes was observed at membrane potentials 100 mV positive to the resting membrane potential. The available data have not yet established whether this response can occur at physiological membrane potentials. In addition, stretch activation of KCa1.1 may be occurring secondary to mechanical activation of Ca2+-permeable TRPC6 channels (226).
FIGURE 8.

Membrane topology of KCa1.1 pore-forming subunits (α-subunits) and auxiliary subunits. A: endogenous channels contain 4 pore-forming KCa1.1 subunits, which can assemble with various auxiliary β- and γ-subunits. The cytosolic domain of KCa1.1 is composed of 2 high-affinity Ca2+ binding sites known as RCK1 and RCK2 (regulator of Ca2+ conductance). The β-subunits have 2 membrane-spanning domains with the amino- and carboxy-terminal domains extending into the cytosol. The γ-subunits have a single membrane-spanning domain. B: immunofluorescence showing the distribution of KCa1.1 α-subunits in a mouse glomerulus. Note extensive colocalization with synaptopodin, a marker for podocyte cell bodies and foot processes. Adapted from Ref. 323 with permission.
In human and mouse podocytes, it is possible to detect KCa1.1 and several auxiliary subunits, including β3 and β4 (324, 325, 331), as well as γ3, also known as LRRC55 (178, 332). β4 and γ3 are often considered to be “brain-type” subunits for KCa1.1 channels (167), and for this reason some of the properties of the podocyte channels share certain characteristics with those found in the CNS (as opposed to those found in, e.g., smooth muscle). For example, the KCa1.1 channels of human and mouse podocytes are relatively resistant to inhibition by iberiotoxin and charybdotoxin, as the blockade is incomplete even at high concentrations. However, they are completely blocked by small-molecule fungal toxins such as penitrem A and paxilline (324, 325). Auxiliary subunits are known to produce large effects on the gating of podocyte KCa1.1 channels (187), and β4-subunits, in particular, are known to markedly slow the activation kinetics of BK channels (333–335). In this regard, the native KCa1.1 channels of mouse podocytes have unusually slow activation kinetics as assessed by whole cell recordings (325, 326) even when they are compared to neuronal KCa1.1 channels examined under identical conditions (326). In those studies, Ca2+ was supplied through the recording pipette (325, 326), and therefore the slow activation of podocyte KCa1.1 cannot be attributed to the kinetics of the Ca2+ source.
At least some of the endogenous subunits of native KCa1.1 channels of mouse podocytes end in a VEDEC motif at the carboxy terminus, which is also found in neurons (325). This pentapeptide motif has profound effects on the trafficking of KCa1.1 channels to the cell surface. Channels with this motif tend to be retained within intracellular compartments but can move to the cell surface if specific signaling pathways are activated or if the channels form a complex with certain other proteins. By contrast, KCa1.1 variants with the motifs EMVYR or QEERL at the carboxy terminus tend to exhibit high levels of constitutive trafficking to the cell surface (172, 173, 336). Notably, subunits with the VEDEC motif can have a dominant-negative effect on the surface expression of other KCa1.1 splice variants (173). In this regard, a number of podocyte proteins have been shown to interact directly with native KCa1.1 channels and appear to be necessary for normal surface expression of KCa1.1. These include the slit diaphragm adhesion molecules nephrin (325) and Neph1 (326), as well as TRPC6 channels (328). It is interesting to note that TRPC3 channels, also present in podocytes, can interact with the VEDEC forms of KCa1.1 but by themselves are unable to stimulate their trafficking to the cell surface (328). Trafficking of KCa1.1 channels to the podocyte cell surface also seems to require interactions with synaptopodin and actin (323, 337). It is possible that proteins such as nephrin and TRPC6 function in a steric manner to block access of some inhibitory systems to the KCa1.1 VEDEC motifs. The scaffolding molecule MAGI-1 reduces the trafficking of native KCa1.1 channels to the surface in podocytes as well as in neurons (327). It is therefore likely that protein complexes containing KCa1.1 in podocytes are large and dynamic.
The available data suggest that the trafficking of endogenous KCa1.1 channels to the podocyte cell surface is a highly regulated process. In neurons, this process can be regulated by multiple growth and differentiation factors (338), including factors that require Akt signaling (339). Through an analogous process, exposure of mouse podocytes to physiological concentrations of insulin markedly increases the steady-state surface expression of functional KCa1.1 channels within minutes (331, 340). Insulin also induces a rapid increase in the surface expression of TRPC6 channels in podocytes (341, 342), and it is tempting to hypothesize that some or all of the KCa1.1 channels in podocytes traffic to the cell surface as a complex with TRPC6 (328). On the other hand, there are conditions in which TRPC6 and KCa1.1 trafficking is uncoupled. For example, exposure of podocytes to high glucose or H2O2 results in a rapid increase in the steady-state surface expression of TRPC6 (341, 343, 344), and the effects of many physiological signals on podocyte TRPC6 are mediated by localized production of ROS through activation of NADPH oxidases (314–316, 345, 346). By contrast, exposure to high glucose or H2O2 inhibits surface expression of KCa1.1 in podocytes and can even abrogate the stimulatory effects of insulin (340). The effects of insulin on KCa1.1 in podocytes appear to follow a standard transduction pathway that entails activation of Akt, Erk, and phosphatidylinositol 3-kinase (PI3-kinase) (340) along with the activation and possible dimerization of PKG type 1 subunit (331).
The physiological roles of KCa1.1 channels in podocytes are not entirely understood. In general, the role of KCa1.1 channels depends on the sources of Ca2+ that stimulate their activation. For example, the KCa.1 channels in excitable cells often occur in a complex with various types of voltage-activated Cav channels. Because Cav channels are activated by depolarization, the coordinated coactivation of KCa1.1 will tend to oppose the further activation of Cav. Depending on the kinetics of the KCa1.1, this could lead to repolarization of a neuronal action potential, generation of a hyperpolarizing afterpotential, and/or modulation of the frequency of action potential discharge (347). In smooth muscle, the activation of KCa1.1 secondary to Cav channels opposes depolarization-induced contraction (348). However, podocytes do not express functional Cav channels. Instead, at least some KCa1.1 channels occur in a complex with TRPC6 channels (328), which can function as a Ca2+ source for their activation (FIGURE 9). The gating of podocyte TRPC6 channels is not appreciably voltage dependent. However, the permeation properties of TRPC6 are highly voltage dependent (227, 228), as depolarization causes the channels to function primarily as monovalent cation channels and to exclude Ca2+ (228). This property arises from the fact that Ca2+ can block as well as permeate TRPC6 (228). Because TRPC6 activation will markedly depolarize cells, including podocytes, this permeation property effectively results in Ca2+ influx with a voltage dependence that is the opposite of what would occur through Cav channels. Therefore, for TRPC6 to be an efficient pathway for Ca2+ influx into nonexcitable cells that lack Cav channels, there needs to be a mechanism to limit depolarization that would occur as a result of TRPC6 activation (227, 228, 349). Coordinated activation of KCa1.1 could serve that function and is likely to arise from the physical interactions between KCa1.1 and TRPC6 that have been documented in podocytes (328). This hypothesis predicts that KCa1.1 activation would enhance Ca2+ influx through TRPC6 activation in podocytes, whereas inhibition of KCa1.1 gating or expression on the cell surface should reduce Ca2+ influx (227, 228, 349).
FIGURE 9.

Interaction of KCa1.1 and TRPC6 channels and its functional significance. A: interaction of native podocyte KCa1.1 and TRPC6 subunits as revealed by reciprocal coimmunoprecipitation (IP). Adapted from Ref. 328 with permission. B: whole cell recordings of K+ currents in cultured mouse podocytes evoked by a series of depolarizing voltage steps made from a holding potential of −60 mV. The 2 sets of current traces at top were made from the same cell with recording electrodes containing the Ca2+ buffer EGTA. Note that the currents get larger after the application of OAG, a membrane-permeable analog of DAG that activates TRPC6. Traces on the bottom were recorded from a different cell with electrodes containing the Ca2+ buffer BAPTA, which has much faster Ca2+ binding kinetics than EGTA but has a similar Ca2+-binding affinity. Under those conditions application of OAG was no longer able to induce an increase in macroscopic K+ currents. The bar graph on right shows the summary analysis of these experiments. Error bars represent SE using immunoblotting (IB). Im, membrane current; n.s., non significant; *P < 0.05. C: this diagram shows 1 interpretation of this result, namely that TRPC6 provides a Ca2+ source for activation of KCa1.1 but that this requires colocalization of the channels within the nanodomain of elevated Ca2+ surrounding an active TRPC6 channel for KCa1.1 to become active. In the presence of BAPTA, this nanodomain of elevated Ca2+ will be reduced in size because of its more rapid quenching of free Ca2+, and the coupling between the channels will be less efficient. See glossary for abbreviations.
A recent study has confirmed this prediction in immortalized human podocytes in vitro and mouse podocytes examined in acutely isolated glomeruli. In those experiments, the application of ANG II resulted in activation of KCa1.1 secondary to TRPC6-mediated Ca2+ influx, and the coordinated activation of KCa1.1 resulted in enhanced ANG II-mediated Ca2+ influx (332). The effects of the γ3-subunit probably reflect its ability to enhance the Ca2+ sensitivity of KCa1.1 channels (350), although it is also possible that the γ3-subunit could increase steady-state surface expression of the pore-forming α-subunits. The consequence of this would be similar to those that occur after enhanced TRPC6 activation. It has also been suggested that K+ depletion from podocytes could enhance the loss of podocytes (350).
4.3. SOCs in Podocytes
It has been reported that the application of palmitic acid resulted in STIM1 oligomerization in the ER membrane of mouse podocytes, an effect that may be mediated through PLC signaling (351). As noted in sect. 3.6, oligomerization of STIM1 is the initial step of SOC activation, and palmitate treatment significantly increased SOCE as expected. It was subsequently observed that the mRNA expression levels of both STIM1 and Orai1 were significantly greater in the renal cortex of mice with nephropathy induced by doxorubicin (352). In addition, overexpression of STIM1 and Orai1 in conditionally immortalized mouse podocytes significantly decreased the protein abundance of podocin and CD2-associated protein (CD2AP), and increased α-actinin-4 protein content. These proteins are localized at slit diaphragm domains of foot processes and are essential for maintaining foot process ultrastructure. Consistent with this, overexpression of STIM1 and Orai1 induces an increase in the protein permeability of cultured podocyte monolayers (352). Although an increase in Ca2+ entry through SOCs was not verified in these experiments, it suggests that SOC signaling proteins are associated with podocyte physiology and biology. Moreover, overexpression of STIM1 and Orai1 has been observed to increase Ca2+ influx in podocytes in vivo and in vitro (352). The importance of SOCs in podocyte structural and functional integrity was also recently reported in vivo and in vitro. It was shown that insulin-stimulated plasma membrane trafficking of Orai1 resulted in an increase in SOCE in cultured podocytes. The insulin-activated SOCE triggered actin remodeling and transepithelial albumin leakage via the Ca2+-calcineurin pathway in podocytes. Genetic manipulations with Orai1 in mice further revealed the role of this channel in podocyte injury and albuminuria (353). Another recent study in cultured podocytes has shown that SOCE signaling can regulate the abundance of nephrin, one of the key elements of the slit diaphragm, and that Orai1-mediated SOCE contributed to the cytoskeletal organization in these cells (354). Enhancement of Ca2+ signaling through this pathway resulted in cytoskeletal remodeling, which might correlate with injury responses (e.g., foot process effacement) if it were to occur in vivo (30). In addition, pharmacological inhibition of Orai1 by BTP2 and genetic deletion of Orai1 by CRISPR-Cas9 gene editing blunted high-glucose-induced podocyte injury, suggesting that enhanced SOCE in podocytes could play a role in diabetic nephropathy (354).
4.4. Ionotropic NMDA and P2X Receptors in Podocytes
Receptors for the dicarboxylic amino acids l-glutamate and d-aspartate are best known for their role in excitatory synaptic transmission throughout the CNS (355). l-glutamate and related compounds can activate several types of heteromultimeric ionotropic receptors. Signaling through ionotropic glutamate receptors can induce a host of responses within the CNS over a variety of timescales, ranging from milliseconds to weeks. These include regulation of the initial development and refinement of neuronal circuits (356) as well as long-term changes in synaptic strength, neuronal ultrastructure, and gene expression in fully formed circuits (357, 358). Dysregulation of CNS ionotropic glutamate receptors can cause a form of neurodegeneration known as excitotoxicity (359). A subset of ionotropic glutamate receptors, referred to as N-methyl-d-aspartic acid (NMDA) receptors, have been implicated in the pathogenesis of multiple neurodegenerative disorders and other diseases of cognition and behavior (360). These receptors received their name from NMDA, the prototypical agonist of these receptors. NMDA is a pharmacological tool and is not endogenously expressed in animals.
For many years it has been known that NMDA receptors are expressed in nonneuronal peripheral tissues, including the kidney (361–364). Thus, expression of NMDA receptors has been shown in the proximal tubule (365–367), principal cells of the cortical collecting duct (368), and glomeruli (362, 369–371). They may also be expressed within the renal vasculature but at locations that appear to be independent of any vasculature innervation (364, 372). Renal NMDA receptors have been proposed to play roles in the regulation of reabsorption from proximal tubules, GFR (365), and the regulation of urine osmolarity (368). It is likely that NMDA receptors in multiple renal compartments play significant roles in physiology and disease. However, NMDA receptors have only been characterized functionally in podocytes, where several properties of the receptors have been examined directly with whole cell recordings (369). It is possible that NMDA receptors in other renal cell types may have different properties.
The NMDA receptors in immortalized podocytes and podocytes in primary culture have several unusual features compared to most NMDA receptors in neurons. As with neuronal receptors, these are extracellular ligand-gated cation channels. Podocyte NMDA receptors are robustly activated by the prototype agonist NMDA, with an ED50 of ∼100 μM and a Hill coefficient of 1.98, similar to what is seen in neurons. However, in marked and surprising contrast to neurons, they are resistant to activation by either l-glutamate or l-aspartate (369), which are considered the usual endogenous ligands for the vast majority of synaptic NMDA receptors. As with neuronal forms, the podocyte NMDA receptors can be activated by d-aspartate and l-homocysteic acid, both of which can occur endogenously. These effects are potentiated by d-serine, which acts at glycine-binding sites on the NR1 subunits of NMDA receptors (369). Those sites do not appear to be very glycine sensitive in podocyte NMDA receptors. In addition, podocyte NMDA receptors exhibit almost no inactivation during sustained continuous application of NMDA, and although voltage-dependent inhibition by Mg2+ can occur, this is seen at somewhat higher concentrations than are observed with neuronal NMDA receptors (369). Podocyte NMDA receptors have a significant permeability to Ca2+ as well as to Na+ and K+. Thus, the Ca2+ permeability of these receptors relative to Na+ (PCa/PNa) is ∼2.1, which is less than the value of 4.0 typically seen in neurons monitored under similar conditions (369, 373). Collectively, these features suggest that podocyte NMDA receptors are well suited to function at the physiological membrane potentials of podocytes and to respond to metabolites whose concentrations fluctuate slowly, as opposed to very fast synaptic signaling processes. Finally, podocyte NMDA receptors can be inhibited by many of the standard pan-NMDA receptor antagonists, such as MK-801 (also known as dizocilpine), d-2-aminophosphonovaleric acid, and memantine, which act on NR2 subunits. They are also inhibited by L689560, which acts on the glycine- and d-serine-binding sites on NR1 subunits (369, 374).
The molecular basis for the distinctive pharmacological and biophysical features of podocyte NMDA receptors is not known. Functional NMDA receptors are heterotetramers assembled from various subunits (NR1, NR2A, NR2B, NR2C, NR2D, NR3A, and NR3B) that are encoded by seven different genes. A functional receptor requires two NR1 subunits, which occur in various splice variants transcribed from a single gene, and either two NR2 subunits or one NR2 subunit and one NR3 subunit (FIGURE 10) (361, 375, 376). The NR2 subunits contain the binding sites for dicarboxylic acid agonists (such as l-glutamate and NMDA), whereas NR1 subunits contain the allosteric sites that are activated by d-serine or glycine (375). NR3 subunits also contain glycine-binding sites, and it is possible to create excitatory glycine receptors from combinations of NR1 and NR3 subunits (377). Transcripts encoding NR1, NR2A, NR2C, and NR2D subunits can be detected in immortalized mouse podocytes, and immunoblot analysis has confirmed the presence of each of these proteins (44). Several of these NMDA receptor subunits are also detectable in cultured MCs (44) and by immunohistochemistry within mouse (44) and rat (370) glomeruli. None of these molecular features is sufficient to explain the unusual biophysical and pharmacological characteristics of the podocyte receptors, and they may arise from an interaction with some other protein. Many such interactions are possible. For example, podocyte NR1 subunits interact directly with synaptopodin (369). These properties might also emerge from posttranslational modifications, presumably occurring on NR2 subunits (378, 379).
FIGURE 10.

Structure and agonist binding sites of NMDA receptor subunits and responses of podocytes to NMDA and other agonists. A: NMDA receptors are formed from NR1 and NR2 subunits. NMDA and other diacidic agonists bind to NR2 subunits, whereas glycine and d-serine bind to NR1 subunits. Both types of subunits must be occupied by their respective agonists for receptor activation. Each subunit has 4 semiautonomous domains, including an amino-terminal domain (ATD), an agonist-binding domain (ABD), a series of 3 helical membrane-spanning domains (M1, M2, and M4), a membrane reentrant loop (M2), and the intracellular COOH-terminal domains. The ABD is formed by 2 polypeptide segments that fold into a bilobed structure with an upper and a lower lobe. B: schematic showing the experimental design to analyze whole cell responses to NMDA and other agonists in podocytes. C: summary of prototypical agonists (left) and antagonists (right) that act on NR2 and NR1 subunits of NMDA receptors. Note that the NMDA receptor pore exhibits voltage-dependent blockade by Mg2+, which is relieved by membrane depolarization. D-APV, D-2-amino-5-phosphonovalerate; HCA, L-homocysteic acid; Quin, quinolinate. D: currents evoked by NMDA application in a cultured podocyte at a holding potential of −60 mV. NMDA-mediated currents in podocytes differ from most neuronal responses in that they can be evoked repeatedly and show no tendency to inactivate, even with a sustained application of NMDA lasting for 1 min. E: responses to NMDA are potentiated after bath application of d-serine. Adapted from Ref. 361 with permission.
The endogenous ligands that activate podocyte NMDA receptors are not known with certainty. It has been suggested that l-glutamate can be secreted from immortalized podocytes by an exocytotic process (362, 371). However, because the podocyte receptors are so glutamate resistant, secreted l-glutamate is unlikely to exert any effect on podocyte NMDA receptors (369), although it could conceivably exert effects on metabotropic glutamate receptors (380) or on NMDA receptors on other renal cells (381). Plasma concentrations of l-glutamate and l-aspartate occur in the range of 25–35 and 20–30 µM, respectively (382, 383). Therefore, the lowest range of these concentrations would saturate most neuronal NMDA receptors, especially given that both of those dicarboxylic amino acids will always be present (384). By contrast, free extracellular amino acid concentrations in the extracellular space in the brain and the spinal cord are much lower because of the blood-brain barrier and a host of local amino acid transport systems, especially in glial cells, that control the neuronal microenvironment (385). Therefore, other ligands are likely to be endogenous agonists for podocyte NMDA receptors. These could include l-homocysteic acid, a spontaneous oxidation product of l-homocysteine, which is produced during metabolism of l-methionine in the liver and elsewhere (386, 387). In this regard, increased activation of NMDA receptors has been demonstrated in glomeruli of hyperhomocysteinemic rats (370). Quinolinic acid, a normal product of tryptophan metabolism (388), is a potent agonist of NMDA receptors (384), and it is markedly elevated in the plasma of patients with diabetes and kidney disease (389, 390). Circulating d-amino acids such as d-aspartate and d-serine could also lead to increased activation of podocyte NMDA receptors, and it is worth noting that both of these are produced by intestinal microbiota (391, 392) as well as by various mammalian enzymes with intrinsic racemase activity (393, 394). Indeed, d-serine is thought to be a physiological modulator of NMDA receptors in the brain (395), and infusing d-serine produces a number of nephrotoxic effects (380, 396). Recent studies have presented evidence that plasma d-serine concentrations can be used as a biomarker for kidney disease (397, 398). Finally, guanidinosuccinate, produced during the metabolism of arginine, is another potential endogenous agonist of peripheral NMDA receptors, such as those on podocytes (399, 400).
Activation of NMDA receptors produces a number of downstream effects, several of which are likely to be secondary to Ca2+ influx. These are summarized in FIGURE 11. Thus, NMDA is able to induce activation of Erk/MAPK, Akt, and RhoA in immortalized mouse podocytes (369, 401). NMDA treatment also causes an increase in ROS production, which occurs secondary to activation of NOX2 (401). A similar response is evoked by l-homocysteine (402). Increased ROS production following stimulation of NMDA receptors also occurs in MCs (370). The increase in ROS production evoked by NMDA was sufficient to stimulate an increase in surface abundance of TRPC6 channels in podocytes that could be detected by biochemical and electrophysiological procedures (401). NMDA treatment also induced activation of calcineurin-NFATc1 signaling cascades, which increased NFATc1 localization in nuclei (401). More sustained exposure to NMDA (24 h) resulted in internalization and loss of podocin and nephrin but no loss of cells. An even longer exposure to NMDA or l-homocysteic acid, for 72 h, resulted in apoptotic cell death. Importantly, all of these effects were inhibited by the NMDA receptor antagonist MK-801. It is likely that several of these effects occurred downstream of TRPC6 mobilization, as they were inhibited by the pan-TRPC inhibitor SKF-96365 (401).
FIGURE 11.

A: schematic of pathways to cell death in podocytes through sustained activation of NMDA receptors. Exposure of cultured podocytes to NMDA or other NMDA agonists triggers apoptotic cell death after 72 h. Certain products of tryptophan and methionine metabolism, such as L-homocysteic acid and quinolinic acid, are NMDA receptor agonists and can potentially trigger podocyte apoptosis. Activation of NFAT transcription factors in kidney podocytes is driven by Ca2+-calcineurin and could drive the loss of podocytes. B: sustained exposure to NMDA receptors can result in elevated cell surface expression of TRPC6 channels via increased production of ROS as well as by activation of NFAT signaling. See glossary for abbreviations.
As with MCs, podocytes respond to the extracellular application of ATP, and both ionotropic P2X and metabotropic P2Y receptors can be detected by biochemical methods in podocytes (43, 108, 297, 403). The metabotropic receptors P2Y1 and P2Y2 are especially important in mediating responses to ATP and related nucleotides in podocytes. Here we note that there are also conditions in which ATP can affect podocyte Ca2+ dynamics by activating ionotropic P2X receptors. As discussed in sect. 5.2, purinergic signaling in glomeruli appears to undergo remodeling in certain disease conditions, such as diabetes (43). Therefore, podocyte P2X receptors in podocytes seem to be most likely to be active in disease states.
5. ION CHANNELS AND CA2+ SIGNALING IN PODOCYTES
Above we emphasized that podocytes are terminally differentiated cells that are highly vulnerable in a variety of disease conditions. There are multiple signaling mechanisms controlling intracellular Ca2+ levels in podocytes that converge on ion channels in podocytes. TRPC6 is by far the most extensively studied channel in the context of podocyte Ca2+ signaling (404–408). FIGURE 12 outlines some of the signaling pathways controlling TRPC6 expression and activity. Several of these factors have been implicated in glomerular disease processes, and in sect. 6 we focus on TRPC6 channels as a driver of diseases affecting podocytes. A common feature in many of the pathways that converge on TRPC6 is an essential role for ROS in activation of trafficking of these channels to the cell surface.
FIGURE 12.

Signaling mechanisms in disease states can lead to excessive signaling through TRPC6 channels in podocytes. Circulating permeability factors such as suPAR can bind to αVβ3-integrin, triggering increased generation of ROS and stimulating the trafficking of TRPC6 channels to the cell surface. This effect occurs within 6−24 h. A similar effect is seen in podocytes exposed to elevated external glucose concentrations. Sustained elevation of a metabotropic pathway through ANG II receptors [AT1 receptor (AT1R)] or P2Y receptors (P2YRs) would also be expected to cause chronic hyperactivation of TRPC6. ROS plays a critical role in TRPC6 activation by GPCRs and stimulates both trafficking and gating, as shown in the inset. See glossary for other abbreviations.
5.1. Podocyte Ion Channel Modulation by ANG II
ANG II exerts its effects through two different GPCRs, known as AT1 and AT2, both of which are expressed in podocytes (409–411). ANG II and the expression of its receptors are often increased in patients with glomerular diseases (412), and retrospective studies of kidney transplant recipients have shown that the presence of AT1 receptor antibodies is associated with posttransplant FSGS and proteinuria (413). Interestingly, although most studies report that AT1 is the dominant receptor in podocytes (414, 415), both AT1 and AT2 can affect Ca2+ signaling and glomerular permeability (322, 409).
Early studies suggested that ANG II depolarizes podocytes in the intact rat glomerulus (416). Several in vitro studies showed that ANG II induced a biphasic increase in cytosolic Ca2+ in podocytes through mobilization from intracellular stores and an influx of Ca2+ through nonselective cation channels (414, 416). These observations have been repeatedly confirmed (315, 322, 345, 416–418). For example, ANG II increased cationic currents in rat podocytes in an isolated glomerulus preparation, and this effect was blocked by the pan-TRP inhibitor SKF-96365, micromolar La3+, and siRNA knockdown of TRPC6, indicating that TRPC6 is the primary channel responsible for ANG II-evoked cation currents in rat podocytes (315). Similarly, no ANG II-evoked channel activity was detected in any of the recordings made from podocytes of TRPC6 knockout mice (FIGURE 13, A–C) (345). This and several subsequent studies showed that this effect of ANG II requires the generation of ROS in podocytes (316, 419). In addition, the ANG II-TRPC6 pathway controls glomerular permeability in a three-dimensional whole glomerulus ex vivo imaging assay (322). Activation of TRPC6 channels with flufenamic acid, an activator of TRPC6, produced effects similar to TRPC6 on glomerular volume dynamics, whereas the specific TRPC6 channel inhibitor SAR7334 (420) restored glomerular volume changes to those seen in the absence of ANG II (FIGURE 13D) (322).
FIGURE 13.

Modulation of Ca2+ signaling in podocytes and glomerular volume by ANG II pathway. A: a representative current trace from the cells transfected with AT1 receptor (AT1R) and TRPC6 before and after application of 1 μM ANG II, during a washout, and after a second ANG II application. This recording has a total length of 30 min. An expanded region shows 10 s of channel activity. B: representative current traces of a TRPC6 channel in a cell-attached patch on a podocyte from a freshly isolated wild-type (WT) mouse glomerulus. A continuous current trace (top) and addition of ANG II (1 μM) to the external bath solution (bottom) are shown. c and oi denote closed and open current levels, respectively. A summary graph for the channel open probability (Po) before and after application of ANG II is shown on right. *P < 0.01 vs. before ANG II. C: a representative recording made from the podocytes in a glomerulus freshly isolated from a TRPC6 knockout mouse. No channel activity was recorded in any of the patches before or after application of ANG II. A–C adapted from Ref. 345 with permission. D: the effects of activation of TRPC6 channels by flufenamic acid (FFA, 100 µM) or TRPC6 inhibition by SAR7334 (1 µM) on ANG II-induced glomerulus volume dynamics (top) and a summary plot for the end point of glomerular volume (bottom). *P < 0.05 vs. control. D adapted from Ref. 322 with permission.
Some other physiological processes in podocytes, including cytoskeletal organization and apoptosis, are affected by ANG II. For example, podocyte-specific knockout of Dynamin 1 and Dynamin 2 suppressed AT1 receptor internalization and accentuated Rac1 activation and membrane ruffling. Podocyte-specific deletion of the AT1 receptor in these mice resulted in improved albuminuria and kidney function (421). Intravital imaging has shown that ANG II enhances the endocytosis and transcytosis of plasma albumin by podocytes, resulting in impaired podocyte function (422), and ANG II signaling is elevated in several animal models in which there is podocyte damage and proteinuria (415). ANG II also contributes to enhanced TRPC6-mediated Ca2+ influx in podocytes during chronic hyperglycemia (423). Overexpression of the human AT1 receptor in rat podocytes leads to early development of proteinuria and glomerulosclerosis (424), and it is associated with increased TRPC6 expression that occurs through the activation of the calcineurin-NFAT pathway (425). In this regard, ANG II-induced albuminuria is reduced in TRPC6 knockout mice (426).
5.2. Modulation by Purinergic Signals
ATP is one of the primary paracrine signals acting within glomeruli (108), especially in the context of TGF (see sect. 2.2) (67, 109). ATP stimulates a rapid increase in Ca2+ influx in podocytes, which occurs both in isolated cells (427) and in vivo during TGF (67). ATP signaling contributes to the rapid Ca2+ responses that occur in podocytes after increases in glomerular filtration pressure (403, 428), and ATP is quickly released in response to changes in renal perfusion pressure (302). The effects of ATP in healthy glomeruli are primarily mediated by metabotropic P2Y receptors, and it should be noted that the responses to ATP in podocytes are greater than those evoked by other GPCR-mediated stimuli, including ANG II (429). Although podocytes clearly respond to ATP, there is evidence that they can also release ATP, for example, in response to mechanical stimulation (428). Podocyte Ca2+ responses are driven by Gq-coupled GPCRs that are highly responsive to nucleotides such as adenosine diphosphate (ADP), an agonist of P2Y1, or uracil triphosphate (UTP), an agonist of P2Y2 (314, 430). P2Y1 receptor signaling appears to be the predominant P2Y purinergic pathway in podocytes studied in situ in glomeruli isolated from healthy rats (403). Several agonists, including MRS-2365 (a specific P2Y1 receptor activator) and 2-meSADP (an agonist for P2Y1 and P2Y12/P2Y13), are able to evoke Ca2+ flux into podocytes. Notably, only the P2Y1-specific antagonist MRS-2500 can block the effects of ATP at concentrations where its actions are specific (403). A physiological role for other P2Y receptors, such as P2Y2, in renal function has been inferred in mice on the basis of gene knockout and immunofluorescence (108, 431).
ATP acting through P2Y receptors induces robust activation of TRPC6 channels in podocytes (314, 432) (FIGURE 14A). TRPC6 activation also occurs to a lesser extent in response to ADP, UTP, and UDP, and these results suggest that ATP should evoke simultaneous activation of more than one subclass of P2Y receptors (314). As with ANG II, ATP activation of TRPC6 requires the generation of ROS and is completely suppressed after podocin knockdown (314), which is also seen with DAG analogs (320). Therefore, it is possible that sustained excessive purinergic signaling could contribute to foot process effacement, detachment of podocytes, and rapid progression of glomerular disease through its effects on TRPC6.
FIGURE 14.

Effects of ATP on podocytes and changes in purinergic signaling in diabetes. A: representative images of a glomerulus freshly isolated from a 12-wk-old rat with type 2 diabetes. Measurements of free Ca2+ with fluo-4 (green pseudocolor) and fura red AM (red pseudocolor) before and after application of ATP. Scale bar = 25 μm. B: examples of ATP-induced intracellular Ca2+ concentration ([Ca2+]i) transients simultaneously recorded from several podocytes in glomeruli isolated from age-matched Wistar or type 2 diabetic nephropathy (T2DN) rats. Note the much slower decay of [Ca2+]i transient observed in podocytes of diabetic stain. a.u., Arbitrary units. C: expression of P2 receptors in renal cortex of nondiabetic (Wistar), diabetic without DN (Goto-Kakizaki, GK), and T2DN rats. Note significant shift from metabotropic (P2Y1) to ionotropic (P2X4, P2X7) signaling in diabetes. A–C adapted from Ref. 43 with permission. D: schematic of ATP-mediated purinergic pathways in podocytes under normal and pathophysiological conditions.
There are also conditions in which ATP can affect podocyte Ca2+ dynamics by activating ionotropic P2X receptors. Purinergic signaling in glomeruli appears to change substantially in certain disease conditions, such as in diabetes (FIGURE 14, B and C) (43). In podocytes examined from diabetic animals, there is a marked increase in ATP signaling due to the upregulation of functional ionotropic P2X4 and P2X7 receptors (43) that are not readily detected in podocytes from healthy animals (43, 403). Interestingly, remodeling of the P2 receptor profiles in glomeruli from metabotropic P2Y to ionotropic P2X receptors was reported for both type 1 and type 2 diabetes (43). Podocyte-specific changes in P2 receptor expression have also been seen with single-cell transcriptomics from early human DN; specifically, P2RX7 expression increases while P2RY1 expression decreases (433). The upregulation of P2X receptors is likely to contribute to Ca2+ overload, loss of podocytes, and glomerulosclerosis (FIGURE 14D) (43). Increased expression of P2X receptors also occurs in immortalized podocyte cell lines that have not differentiated fully into a podocyte phenotype (434). Furthermore, P2X7 receptor protein glomerular expression was shown in renal biopsy tissue of patients with autoimmune-related glomerulonephritis, and the experimental rat model of proliferative glomerulonephritis coincided with the onset of proteinuria (435). It bears noting that the remodeling of P2 receptors is not unique to podocytes, since previous studies have shown that increases in the expression of P2X, and specifically P2X7 receptor, are linked to polycystic kidney disease, glomerulonephritis, lupus nephritis, and hypertension-associated renal injury (108, 436, 437).
5.3. Modulation by Insulin
Podocytes are one of several renal targets for insulin signaling (438, 439). Here we note that insulin increases the functional expression of TRPC6 channels in podocytes (341). Thus, exposure of cultured podocytes to physiological concentrations of insulin results in an increase in the abundance of TRPC6 channels on the cell surface that can be observed within minutes. This is accompanied by an increase in functional TRPC6 channels that can be detected by whole cell recordings (341). This effect is mediated by an increase in intracellular ROS generation. However, it is interesting to note that insulin modulates podocyte TRPC6 through assembly and activation of NADPH oxidase 4 (NOX4) on the cell surface (341). The functional significance of insulin modulation of podocyte ion channels is not fully understood, and it needs to be considered along with concurrent insulin effects on other renal cells. Insulin effects on podocytes, along with its effects in MCs (202), could provide a mechanism to allow the glomerular filtration barrier to adjust to acute changes in the glomerular capillary that occur in response to an acute glucose load (440, 441). There is evidence that insulin increases glomerular protein permeability through TRPC6-dependent activation of PKGIα signaling (442) and cytoskeleton reorganization (443). Insulin also increases mobilization of KCa1.1 channels in podocytes, although in part through different transduction mechanisms than are used for mobilization of TRPC6 (331, 340). An enhancement of KCa1.1 could serve to enhance Ca2+ efflux triggered by activation of TRPC6 by helping to maintain an adequate driving force to allow for efficient Ca2+ permeation (227).
5.4. Other Signaling Pathways That Modulate Ion Channels in Podocytes
20-Hydroxyeicosatetraenoic acid (20-HETE) is an ω-hydroxylated metabolite of arachidonic acid. It has been proposed to play a variety of roles in the normal function and dysfunction of the glomerular filtration barrier (444), including stimulation of Ca2+ influx and contraction of preglomerular vessels (445). 20-HETE also causes activation of TRPC6 channels in cultured podocytes (446). Thus, acute exposure of podocytes to 20-HETE results in increased current through TRPC6 that occurs within minutes. The effect of 20-HETE is additive with that of membrane-permeable DAG analogs and, importantly, does not occlude the effects of DAG (446). It is possible that this effect of 20-HETE is mediated by a G protein-coupled receptor, as it is blocked by agents that inhibit G protein signaling, requires the presence of podocin, and is inhibited by agents that quench ROS (446). In these aspects, the effects of 20-HETE in podocytes resemble those of ANG II and ATP.
It is well established that NO regulates glomerular function. Podocytes can release NO, and this may be involved in paracrine and autocrine regulation of podocytes and surrounding cells (145). It was recently shown that NO and a soluble guanylate cyclase (sGC) activator increase cGMP synthesis in podocytes and decrease doxorubicin-induced TRPC6 expression, thereby attenuating TRPC6-mediated Ca2+ influx and reducing podocyte injury (447).
Activation of certain opioid receptors (ORs) also contributes to the dysregulation of intracellular Ca2+ homeostasis in podocytes (448). Stimulation of κ-ORs, but not μ-ORs or δ-ORs, evoked a transient Ca2+ influx in podocytes, which was blocked by prior treatment with the TRPC6 inhibitor SAR7334. Hypertensive Dahl SS rats chronically treated with the κ-OR agonist BRL52537 exhibited signs of Ca2+ overload in podocytes, accompanied by nephrinuria, albuminuria, changes in electrolyte balance, and augmented blood pressure (448). Other pathways and drugs, such as nonsteroidal anti-inflammatory drugs (449), vitamin D (450, 451), and others, may also modulate the activity of TRPC6 channels in podocytes.
6. ION CHANNEL DYSREGULATION AND GLOMERULAR DISEASES
6.1. Mutations in TRPC6 Associated with FSGS
A role for TRPC6 channels in driving glomerular disease was initially reported in a 2005 study that described a gain-of-function mutation in TRPC6, the gene that encodes TRPC6 channels (36). This mutation was identified by analysis of a large extended family with a severe familial form of FSGS in which the disease exhibited an autosomal dominant pattern of inheritance (452). The disease caused by this mutation presented with an adult onset, and ∼60% of the affected individuals progressed to ESKF, usually within 10 yr after the initial presentation of the disease. The nephrosis did not recur after a kidney transplant (36, 453). This mutation, TRPC6-P112Q, is localized within the first ankyrin-repeat domain, which is highly conserved among all TRPC family members. Functional studies of TRPC6-P112Q and wild-type TRPC6 channels were carried out by heterologous expression in HEK-293 cells using two different methods to activate the channels. Transfected cells (cotransfected with AT1 receptors or M1 muscarinic receptors) were activated by exposure to the membrane-permeable DAG analog 1-oleoyl-2-acetyl-sn-glycerol (OAG) or by application of ANG II or carbachol (36). Responses were also seen when cells were treated with UTP, an agonist of P2Y receptors that are endogenously expressed in HEK-293 cells. TRPC6 activation was monitored by examining Ca2+ influx with Ca2+ imaging or measuring macroscopic cationic currents. These analyses showed that currents through TRPC6-P112Q were two- to threefold greater than through wild-type channels, even though the proteins were expressed at comparable levels. Cell surface biotinylation assays suggested that this effect was due at least in part to increased abundance of the mutant TRPC6 channel on the cell surface, presumably through effects on trafficking into or out of the plasma membrane (36).
Shortly after the appearance of this initial report, analyses of several other families with inherited autosomal dominant FSGS led to the identification of five additional TRPC6 mutations: TRPC6-N143S, TRPC6-S270T, TRPC6-K874*, TRPC6-R895C, and TRPC6-E897K. As with TRPC6-P112Q, these mutations occurred at residues conserved among TRPC channels (37). Individuals with these mutations also exhibited severe adult-onset FSGS, with the first presentation occurring at 17–52 yr of age. Most of the people with these mutations progressed to ESKF. The functional properties of these mutations were characterized in HEK293-M1 cells stably transfected with the M1 muscarinic receptor, and channels were activated by the application of carbachol. Two of the mutations, TRPC6-R895C and TRPC6-E897K, resulted in currents that were consistently larger than those seen with wild-type TRPC6 channels across a wide range of membrane potentials (−100 to +100 mV). The other three mutations did not produce abnormal currents in response to carbachol (37). A later study confirmed that TRPC6-N143S yields currents similar to wild-type TRPC6 in response to heterotrimeric G protein signaling (through purinergic receptors) in CHO-K1 cells (432). The gain of function of the TRPC6-P112Q, TRPC6-R895C, and TRPC6-R895C mutations was subsequently confirmed by demonstrating that their activation in HEK-293 cells resulted in increased NFAT-dependent transcription and ERK activation compared to wild-type channels (454, 455).
Since these initial reports, several other TRPC6 mutations have been identified in patients with both familial and sporadic nephrotic syndromes. These include TRPC6-M132T (456); TRPC6-P15S (457); TRPC6-N110H (458); TRPC6-G109S, TRPC6-N125S, and TRPC6-L780P (459); TRPC6-R175Q (460); TRPC6-R360H (461); TRPC6-L395A and TRPC6-A404V (462); TRPC6-Q889K (463); TRPC6-G757D (464); and TRPC6-H218L and TRPC6-R895L (465). The majority of these mutations show a gain of function when the channels are expressed in heterologous expression systems and activated by GPCRs (460, 463, 466). These mutations occur within cytosolic domains of the TRPC6 channels, either in one of the ankyrin-repeat domains near the amino terminus or in domains near the carboxy terminal (349), some of which may affect the formation of the channel tetramer (466). There is evidence that several of these gain-of-function mutations affect the structure of domains that can bind small TRPC6-activating molecules. In addition, several of these mutations may destabilize the closed state of the channel, i.e., they enhance gating (467). FIGURE 15 summarizes a number of known mutations, including an indication of the terminus on which they are located.
FIGURE 15.

TRPC6 mutations associated with familial focal segmental glomerulosclerosis (FSGS). Domain structure of the TRPC6 channel. Human mutations associated with FSGS are shown in boxes. All of the known disease-causing mutations are located in the amino- and carboxy-terminal portions of TRPC6 that extend into the cytosol. Adapted from Ref. 349 with permission.
Although the initial reports described TRPC6 mutations that caused adult-onset disease, it is now known that at least some TRPC6 mutations can cause FSGS in infants and children. The most extreme example described in the literature is TRPC6-G757D, which was first identified in a girl whose nephrosis presented within the first year of life (466). In addition, TRPC6-M132T was identified in a 9-yr-old girl (456), TRPC6-H218L occurred in a boy whose nephrosis first appeared at 8 yr of age (465), and TRPC6-R895L was identified in a girl with a severe collapsing glomerulopathy that occurred at 2 yr of age (465). TRPC6-L780P was identified in a 9-yr-old girl with severe steroid-resistant nephrotic syndrome with hematuria (459).
This raises the question of whether there is something functionally different about TRPC6 mutations associated with FSGS of very early onset. In the case of TRPC6-M132T, expression in HEK-293 cells resulted in channels with a very large gain of function compared to wild-type TRPC6 when the channels were activated through M1 muscarinic receptors (456). Moreover, podocyte-specific overexpression of TRPC6-M131T (the mouse ortholog to human TRPC6-M132T) resulted in severe albuminuria and glomerulosclerosis by 8 wk of age (468). The resulting phenotype was more severe than was seen following overexpression of TRPC6-P11Q (orthologous to human P112Q) and TRPC6-E896K in podocytes (468, 469) or by constitutive global overexpression of TRPC6-E896K (470).
By contrast, at least five TRPC6 mutations have been associated with very early disease onset (TRPC6-N125S, TRPC6-L395A, TRPC6-G757D, TRPC6-L780P, and TRPC6-R895L) and result in a loss of channel function when these variants are activated with either a DAG analog or carbachol (466). Indeed, TRPC6-G757D is able to suppress the formation of active channels when it is heterologously coexpressed with wild-type TRPC6 channels, suggesting that it produces some type of dominant-negative effect on wild-type channels or other channels with which it can heteromerize (466). The mechanism of this effect is not known but could occur, for example, if TRPC6-G757D blocks the trafficking of channels to the cell surface or if it targets channels for degradation. Alternatively, it could also act to suppress gating of any surface channel complex in which it is present. An additional complication is that whether there is a gain or loss of function of TRPC6 can depend on how the channels are activated. For example, in contrast to wild-type TRPC6 channels, TRPC6-N143S channels expressed in CHO cells do not become active in response to membrane stretch, although they exhibit responses similar to or greater than wild type when they are activated by GPCR signaling pathways (432). Along with other studies mentioned above, this shows that the mechanisms whereby TRPC6 channels become active in response to membrane stretch are fundamentally different from activation driven through GPCR signaling (226).
It is certainly possible that a basal level of TRPC6 function is required for normal glomerular function in humans at perinatal stages of development but that sustained hyperactivation of TRPC6 will eventually cause glomerular disease. In this regard, podocyte-specific expression of a constitutively active Gq also facilitates glomerular disease in mice through a mechanism that requires TRPC6 (471). With respect to basal function, TRPC6 knockout mice do not exhibit apparent deficits in renal function, although it has been reported that they eventually become insulin resistant (472). A possible protective effect of TRPC6 has been observed in podocytes exposed to activated complement (473). A basal level of TRPC6 activity may also be necessary for normal glomerular responses during TGF (67), in response to changes in intraglomerular pressure (428), and during insulin signaling (341).
6.2. Effects of Mutations in Other Genes on the Function of TRPC6 in Podocytes
The most direct mechanism for an inherited alteration in TRPC6 is through a mutation in the TRPC6 gene itself. However, it is possible that TRPC6 function in podocytes is altered in other genetic glomerular diseases. Within podocytes, some of the TRPC6 channels are located at slit diaphragms in a complex with other proteins, including nephrin and podocin (37, 474). Podocin is a cholesterol-binding protein, encoded by the NPHS2 gene (475), that forms large aggregates (476) that organize cholesterol-rich lipid rafts at the slit diaphragm domains of podocyte foot processes (477, 478). Podocin binds directly to TRPC6 channels (37, 226, 474) through interactions between the carboxy-terminal domains of the two proteins (226). These observations raised the question as to how podocin might affect TRPC6 channels in podocytes.
A homologous protein has been identified in Caenorhabditis elegans, where it was shown to mediate mechanical activation of degenerin-family cation channels involved in cutaneous sensation (474), and on this basis it was predicted that it would mediate mechanical activation of TRPC6 channels in podocytes (479). Podocin modulates the mechanical activation of TRPC6 channels in podocytes but, surprisingly, not in the way that was originally predicted. Specifically, knockdown of podocin results in a marked increase in activation of podocyte TRPC6 channels evoked by membrane stretch (226). This could occur, for example, if TRPC6 is held longer within cholesterol-rich membrane domains or if interactions with cytoskeletal elements are disrupted. Podocin has a scaffolding function that among other things allows TRPC6 channels to form a complex with NOX2 catalytic subunits (320). In this way, podocin may function to retain TRPC6 within a zone where ROS production is locally elevated. Because of this, knockdown of podocin reduces or eliminates responses to modulators such as DAG (226, 320) and ATP (314). The effects of podocin in the regulation of podocyte TRPC6 are summarized in FIGURE 16, A and B. It is worth noting that podocin expression is reduced in certain disease models, such as chronic PAN nephrosis (408) and exposure to FSGS permeability factors (321, 346), and this results in changes in TRPC6 gating modes similar to those seen following podocin knockdown. Many different mutations within NPHS2 have been identified, several of which result in severe autosomal recessive forms of nephrotic syndrome that can occur at 0–3 yr of age (475, 480, 481). Therefore one would expect that loss-of-function NPHS2 mutations would result in the sustained activation of TRPC6 at foot processes as a result of mechanical forces that are always present, while preventing activation via neurohumoral and paracrine pathways acting on GPCRs. It is reasonable to suspect that this would lead to sustained hyperactivation of TRPC6.
FIGURE 16.

Modulation of TRPC6 function by podocin and alpha-actinin (ACTN4). A: whole cell currents activated by membrane stretch in cultured podocytes treated with a control siRNA or an siRNA targeting TRPC6, as indicated. Knockdown of TRPC6 nearly eliminates stretch-evoked cationic currents in fully differentiated podocytes. VM, membrane potential. B: knockdown of podocin in cultured podocytes resulted in a large increase in stretch-evoked cation currents. A and B adapted from Ref. 226 with permission. C: effect of ACTN4 K255E mutant on the organization of the actin cytoskeleton. CHO cells were transiently transfected with plasmids encoding wild-type (WT) or mutant K255E ACTN4. Scale bars, 50 μm. a: Rhodamine-phalloidin emission (red). Merged image of rhodamine-phalloidin (red), GFP-labeled α-actinin-4 (green), and Hoechst-33342 (nuclei, blue) emissions. d and e are zoomed areas from c (marked by white square) for rhodamine-phalloidine and GFP-labeled α-actinin-4, respectively. D: summary graphs of the channel activity (NPo) of the TRPC6 channels recorded in CHO cells cotransfected with TRPC6 and ACTN4 or ACTN4 K255E before and after OAG (100 µM) stimulation; *P < 0.05. C and D adapted from Ref. 485 with permission.
Mutations in other genes expressed in podocytes also result in familial forms of FSGS. In this regard, a recent study has shown that a mutation in ACTN4 that has been previously implicated in FSGS (ACTN4-K255E) (482–484) leads to decreased activity of single TRPC6 channels when these proteins are coexpressed in CHO cells (FIGURE 16, C and D) (485). This effect is associated with altered cytoskeletal dynamics, and it could be associated with increased stretch activation of TRPC6 in podocytes (226). An effect of cytochalasin D to activate previously silent endogenously expressed podocyte TRPC6 channels (485) may also reflect the effects of cytoskeletal elements that include ACTN4 on the stiffness and deformability of plasma membrane domains containing TRPC6 channels (483). A possible interaction of TRPC6 with other proteins critical to the actin cytoskeleton, including those associated with FSGS (such as ARHGAP24 and ARHGDIA) (89, 486, 487), requires further investigation.
6.3. TRPC6 Dysregulation Caused by Oxidative Stress
TRPC6 are redox-regulated channels that are targeted in various ways by ROS, such as and H2O2 (220, 488). The detailed mechanisms whereby ROS modulate TRPC6 are unknown but probably entail actions on several proteins and depend to some extent on what cell type is considered. By analogy to other TRP family channels, it is possible that ROS act in part on cysteine residues in the ankyrin-repeat domains to modulate gating and trafficking (489). This is a normal physiological process, and the generation of ROS is essential for the activation of podocyte TRPC6 channels through GPCR-mediated signaling by ANG II (315, 322, 345) and ATP (314), as well as by lipids such as DAG (320) and 20-HETE (446). One would therefore expect this kind of modulation to be an important process in TGF, for example. Activation of NOX4 is required for insulin-induced mobilization and activation of podocyte TRPC6 (341) and may also be important in certain chronic disease settings (316, 425). These modulatory effects of ROS are due at least in part to the increased steady-state abundance of TRPC6 on the cell surface (408), probably through exocytotic processes that, in some cases, occur rapidly (220, 490–492). These processes contribute to the coupling of oxidative stress and altered Ca2+ dynamics (493) and could be especially important in hyperglycemia or hypoxia (488).
Evidence for ROS effects on glomerular TRPC6 channels in vivo was initially obtained with the chronic puromycin aminonucleoside nephrosis (PAN) model in rats, in which increases in glomerular TRPC6 expression were blocked by treatment with the NADPH oxidase inhibitor apocynin (494). Because this is a chronic disease model, it is important to note that TRPC6 activation leads to an increase in its own transcription (425). More recent evidence has verified the increase in TRPC6 abundance and has directly implicated TRPC6 as a causal factor driving albuminuria and glomerulosclerosis in the chronic PAN model (39). Similarly, exposure of podocytes to high glucose produces a marked increase in the activity of TRPC6 channels (343, 344, 495), which over time can lead to apoptotic cell death (343, 496, 497). The effect of high glucose in podocytes is mimicked by exposing cells to H2O2 (316, 341, 343, 495) or peroxynitrite (495) and is blocked by quenching ROS with agents such as TEMPOL (343, 495). In addition, increased generation of ROS in podocytes drives increased TRPC6 expression on the cell surface (341). A contribution of a NOX4/TRPC6 pathway in podocyte Ca2+ regulation has also been supported by experiments in STZ-treated Dahl SS rats in which NOX4 was knocked out compared to STZ-treated Dahl SS rats with normal NOX4 (316). In these experiments, the NOX4 knockout diabetic rats exhibited significantly lower basal intracellular Ca2+ levels, reduced ANG II-evoked Ca2+ influx in podocytes, and reduced nephropathy following STZ treatment (316, 322, 429).
However, it is important to note that the effects of high glucose on TRPC6 appear to be cell type dependent. Thus, exposing MCs to high glucose for 2–6 h causes a marked decrease in the abundance of TRPC6 (the opposite of what occurs in podocytes), and this effect is mimicked by H2O2 and blocked by knockdown of NOX4 or by application of membrane-permeable preparations of superoxide dismutase or catalase (246). The pathway whereby external glucose suppresses TRPC6 expression in MCs includes the generation of ROS by NOX4 and subsequent activation of PKC and nuclear factor-κB (NF-κB) (246, 498). In this regard, the triterpenoid saponin astragaloside IV, which can function as a potent antioxidant, blocks the high-glucose-induced decrease in TRPC6 protein content by inhibiting the NOX4/Akt/NF-κB pathway (499). This pathway is summarized in FIGURE 17.
FIGURE 17.

Schematic illustration of a signaling pathway for downregulation of TRPC6 expression in the presence of elevated external glucose in MCs. High glucose stimulates the production of ROS via activation of the NADPH oxidase NOX4. Activation of PKCα by H2O2 causes activation of NF-κB (p50 and p65 subunits), which binds to the NF-κB binding site within the TRPC6 promoter resulting in repression of its transcription. A decrease in TRPC6 channel proteins in the plasma membrane reduces TRPC6-mediated Ca2+ entry and impairs the contractile function of MCs. See glossary for abbreviations.
Although the effects of high glucose in podocytes and MCs may appear contradictory at first glance, they may be part of an integrated response that protects glomeruli in the face of sustained hyperglycemia. It has long been known that increases in circulating glucose produce an increase in GFR (500) due to increased proximal tubule Na+ reabsorption that leads to TGF at the macula densa and afferent arteriole (501, 502). Moreover, whereas MCs can be regenerated, podocytes are highly vulnerable to shear force-induced detachment from the glomerular capillary surface (87) and the capacity to regenerate podocytes is extremely limited (503). We have already noted that MCs are contractile through pathways that entail activation of TRPC6 (246, 247). Therefore, sustained hyperglycemia through suppression of TRPC6 in MCs might induce a relaxation of the glomerular capillary that prevents damaging increases in intraglomerular pressure. At the same time, hyperglycemia-induced TRPC6 mobilization and activation in podocytes might, at least over short periods of time, induce cytoskeletal changes that work to resist detachment of those cells (473). Glucose-induced hyperfiltration could also provide a physiological context for increases in podocyte TRPC6 channels evoked by insulin (340, 341). Other Ca2+-permeable channels, such as SOCs, may also contribute to a short-term protective effect on podocytes during hyperglycemia (353). However, if the oxidative stress in podocytes is sustained for a long enough time one would expect to see decompensation as a result of Ca2+ overload and other factors that eventually lead to glomerular disease.
6.4. TRPC6 Dysregulation in Acquired Forms of FSGS and in Glomerulonephritis
FSGS is a histopathological lesion with multiple etiologies, including primary, genetic, and adaptive forms of the disease (504, 505). The final common pathway, regardless of etiology, appears to be an injury, loss, or dedifferentiation of podocytes (27, 504, 505). In sect. 6.1, we discuss some of the genetic forms of FSGS, for example, FSGS due to mutations in TRPC6 or NPHS2. Mutations in other genes, such as ACTN4 and INF2, can also cause severe forms of FSGS (506, 507), and patients with certain allelic variants of APOL1 have a markedly increased probability of renal diseases, including FSGS (508–510). However, the majority of FSGS cases are not genetic or familial in nature and can be considered to be acquired forms.
Acquired FSGS also has multiple etiologies. A subset of FSGS patients have a primary or idiopathic form of the disease that is driven by one or more circulating factors thought to be associated in some way with innate immunity (511, 512). A substantial number of primary FSGS patients do not respond to current therapies and will progress to ESKF. Moreover, as many as 40% of patients with primary FSGS will experience recurrence of their disease after receiving a kidney transplant (513, 514). Therapeutic modalities such as plasmapheresis or plasma exchange can sometimes bring primary FSGS patients into remission and may slow the progression to ESKF (514, 515). Identifying the circulating factors that drive primary FSGS has been an active area of research, and it is possible that multiple circulating factors may drive the pathology (516). Other patients have an adaptive form of FSGS that occurs as a consequence of chronic hypertension, a reduction in renal mass and the number of functional nephrons, sickle cell disease, or other factors but that does not appear to be mediated by soluble factors in the systemic circulation (517). A collapsing form of FSGS can be seen in patients with viral infections such as HIV (518). There are also drug-induced forms of FSGS, for example, due to treatment with calcineurin inhibitors, certain antiviral agents, interferons, or anabolic steroids (517). There is now evidence that some of the pathological mechanisms in acquired forms of FSGS converge on TRPC6 channels (38, 321, 408).
Patients with certain acquired glomerular diseases, such as FSGS and membranous glomerulonephritis, have an increased abundance of TRPC6 transcripts and proteins within glomeruli (38). This may be sufficient to drive glomerular pathology, as overexpression of wild-type TRPC6 in mouse podocytes has been shown to induce proteinuria and glomerulosclerosis (468, 469). In the acute (38, 494) and chronic (39) PAN models of FSGS in rats, there is also an increase in glomerular TRPC6 abundance. Moreover, podocytes in glomeruli isolated acutely from rats with chronic PAN-induced FSGS exhibit a marked increase in TRPC6-mediated currents evoked by membrane stretch (FIGURE 18) (408). TRPC6 inactivation using CRISPR-Cas9 methods in Sprague-Dawley rats markedly reduced albuminuria, azotemia, uremia, glomerulosclerosis, and tubulointerstitial fibrosis in the chronic PAN model of FSGS (39), which is considered an animal model for adaptive forms of FSGS (519). By contrast, TRPC6 inactivation had no protective effect with respect to urine albumin excretion during the acute phase of the PAN model (39), a stage at which no glomerular damage is discernible at the light microscopic level (519). Doxorubicin-induced nephropathy, which is similar to the chronic PAN model, also produces an increase in glomerular TRPC6 expression in rats (520).
FIGURE 18.

Dysregulation of TRPC6 in the chronic PAN nephrosis model of adaptive FSGS in rats. The experiment in this diagram used 2 injections of PAN given at a 30-day interval. A: at 60 days after an initial injection of PAN, marked glomerulosclerosis can be observed with periodic acid-Schiff staining in rats treated with PAN but not in saline-treated control rats. Arrow indicates segmental sclerotic lesions. B: TRPC6 abundance is markedly increased in glomeruli isolated from PAN-treated rats compared with saline-treated control rats. However, podocin expression is markedly decreased, whereas nephrin is not changed. C: cationic currents recorded from podocytes in acutely isolated glomeruli showing responses to membrane-stretch. Stretch-evoked currents are much more prominent in podocytes from PAN-treated rats. However, responses to a membrane-permeable DAG analog (OAG) are decreased. Vm, membrane voltage. D: summary of responses to stretch and OAG in podocytes from saline- or PAN-treated animals. See glossary for abbreviations. C and D adapted from Ref. 408 with permission.
There are very few animal models of primary FSGS. However, there is evidence that soluble circulating factors implicated in primary FSGS can cause dysregulation of podocyte TRPC6 channels in vitro. Thus, exposing immortalized podocytes to serum or plasma samples collected from patients with recurrent forms of primary FSGS has been shown to activate integrin signaling, alter cytoskeletal dynamics and cell motility, and profoundly alter the trafficking of slit diaphragm proteins (521–526). Exposing cultured podocytes to recurrent FSGS plasma or serum samples also causes marked increases in the steady-state abundance of TRPC6 channel on the cell surface (321). In the majority of the patient samples tested, this was accompanied by a loss of podocin (321), which is initially internalized and then degraded under these conditions (521). The loss of podocin and the increase in surface TRPC6 occur over a period of 6–24 h. However, a subset of the patient samples had no effect on podocin over that time period, although they still increased surface TRPC6 (321, 526). The more common pattern (i.e., an increase in cell surface TRPC6 accompanied by a loss of podocin) was closely mimicked by treating podocytes with recombinant soluble urokinase plasminogen activator receptor (suPAR) (321, 346, 527). suPAR is the name of a class of circulating glycoproteins that are shed from several different cell types (528, 529) and that are elevated in the blood (530) and urine (531, 532) of a substantial subset of FSGS patients. Elevated suPAR is not a selective biomarker for FSGS. Instead, it predicts poor future patient outcomes in a number of forms of chronic and acute kidney disease, and it is probably a significant contributor to FSGS pathology in some patients. Overexpression of suPAR in mouse adipocytes is sufficient to drive albuminuria and glomerulosclerosis (533). Podocytes exposed to recurrent FSGS serum samples or recombinant suPAR also showed marked increases in TRPC6 activation evoked by membrane stretch, which likely reflects the loss of podocin that also occurred in response to these treatments (226, 321).
There is also evidence that tumor necrosis factor (TNF) can drive glomerulosclerosis and recurrent FSGS in some patients, especially in children (534, 535), and it also produces cytoskeletal changes in podocytes (522, 536). TNF induces a marked increase in cell surface expression of TRPC6 in cultured podocytes but, in contrast to suPAR, does not cause internalization or degradation of podocin. Because of this, TNF does not alter the dominant mode of TRPC6 gating in podocytes (321). The effects of suPAR and TNF on podocyte TRPC6 channels are highly synergistic, and complete inhibition of the responses to some recurrent FSGS samples requires the simultaneous immunoneutralization of both factors (321). This observation is consistent with a model in which primary FSGS emerges from an abnormal extracellular milieu in which multiple factors affect podocytes.
The effects of suPAR on podocyte TRPC6 channels have been studied in some detail. This factor initially acts on a cell surface complex that contains αvβ3-integrin and the receptor for advanced glycation end-products (RAGE) (321, 346, 527). Activation of one or more components in this large complex leads to Rac1-dependent activation of NOX2 and an increase in ROS generation that is required for TRPC6 mobilization to the cell surface (346). The increase in ROS generation stimulates activation of Src-family tyrosine kinases, which is likely to produce a host of effects in podocytes, including direct phosphorylation of TRPC6 (346, 537).
There are other potential circulating factors that could play a role in FSGS and other glomerular diseases, at least in part through actions on TRPC6. Thus, exposing isolated podocytes to TGF-β1 also evokes an increase in TRPC6 expression, and, in addition, TRPC6 knockdown reduces podocyte apoptosis induced by TGF-β1 in vitro (520). Soluble ectodomains of syndecan-4 also modulate podocyte TRPC6 channels and are detected in the urine of rats during acute PAN nephrosis (538). Moreover, syndecan-4 knockdown can reduce albuminuria induced by albumin overload in mice (539).
TRPC6 channels have also been examined in inflammatory glomerular disease models and in glomerulonephritis. In an anti-GBM model of rapidly progressing glomerulonephritis in Sprague-Dawley rats, a global knockout of TRPC6 resulted in a significant reduction in glomerular disease as assessed by histopathology (540). However, there was no effect of TRPC6 knockout on tubulointerstitial disease in this model, and overall renal function, as assessed from serum creatinine levels, was not improved. A similar pattern was observed in aging Sprague-Dawley rats in which TRPC6 knockout resulted in a reduction in glomerulosclerosis but had no protective effect in other renal compartments and no improvement in overall renal function (540).
By contrast, TRPC6 knockout mice show a marked reduction in tubulointerstitial fibrosis evoked by a 24-h unilateral ureteral obstruction (541–543). This is not a glomerular disease model, as the initial injury occurs to distal tubular epithelia. The fairly robust protective effects in this model included reduced renal expression of numerous fibrosis markers (collagen-1, α-smooth muscle actin, vimentin, matrix metalloproteinases 2 and 9, and TGF-β1) (541), which may reflect suppression of epithelial-mesenchymal transition in tubule cells (543). Protective effects may also reflect a role for TRPC6 in leukocyte migration (544) and/or in the transdifferentiation of myofibroblasts (545). It is interesting to note that administration of soluble klotho reduced fibrosis evoked by ureteral obstruction in wild-type but not TRPC6 knockout mice and that klotho causes a decrease in TRPC6 expression throughout the kidney (541), including in podocytes (546, 547). On the other hand, TRPC6 inactivation had no effect on interstitial fibrosis or proximal tubule damage in Sprague-Dawley rats subjected to sustained (2–4 wk) albumin overload (548). Thus, whether or not TRPC6 inactivation produces antifibrotic effects in the kidney may depend on the species investigated, the nature of the initial insult that triggers the fibrosis, and possibly the time course of the disease process. In the future, it will be important to determine the extent to which species and disease time course affect responses to knockout of TRPC6 (and other channels) and the responses to TRPC6 inhibitors.
6.5. Role of TRPC Channel Dysregulation in Diabetes and Hyperglycemia
Deleterious changes in renal function occur in as many as 40% of patients with diabetes mellitus, and DN is the leading cause of ESKF throughout the world (404, 549). The cardinal feature of diabetes mellitus is hyperglycemia, and the effects of elevated glucose depend on which glomerular cell type one considers. In sect. 6.3 we note that elevated extracellular glucose increases the activity of podocyte TRPC6 channels through increased generation of ROS (316, 341, 343, 495, 496) but also causes a fall in the expression of TRPC6 channels in MCs (245). Part of the effect of hyperglycemia on glomerular TRPC6 expression in vivo is driven by ANG II (423), which causes ROS-dependent activation of TRPC6 channels in podocytes, and this, in turn, enhances TRPC6 expression through calcineurin-NFAT cascades (315, 316, 345, 425). Given that sustained TRPC6 hyperactivation is generally considered to be pathogenic for podocytes, it is quite surprising that TRPC6 knockout or inactivation has not produced impressive renoprotection in rodent models of diabetes. For example, in the Akita mouse model of type 1 diabetes, knockout of TRPC6 reduced albuminuria in young animals (12–16 wk of age). However, this protective effect disappeared as animals got older (20 wk of age), and TRPC6 knockout promoted a state of insulin resistance in glomeruli and in isolated podocytes, possibly due to reduced expression of insulin receptor substrate 2 and an increase in the expression of cyclooxygenase 2 (472, 550). This transient protective effect is reminiscent of the effect of TRPC6 knockout on glomerular pathology induced by chronic infusions of ANG II (426). In another study, combined global knockout of TRPC3, TRPC6, and TRPC7 produced protection with respect to albuminuria and histological kidney damage in STZ-induced diabetes in mice for 12 wk (551), but it is not known whether the protection would have been more sustained.
The role of TRPC6 in driving renal complications has also been studied in diabetic rats. Thus, STZ treatment induced type 1 diabetes that was associated with increased TRPC6 expression in glomeruli of Dahl SS rats. However, TRPC6 inactivation using CRISPR-Cas9 gene editing did not ameliorate elevated blood glucose or urine albumin excretion. However, there was a reduction in urine nephrin excretion, suggesting a decrease in podocyte detachment and some reduction in basal Ca2+ levels (552). A similar result was obtained in Sprague-Dawley rats, in which there was no protective effect of TRPC6 inactivation on hyperglycemia, albuminuria, histological kidney injury, azotemia, plasma creatinine, or urine nephrin excretion (553). In that study, in contrast to what was observed in mice (472), there was never a stage in the disease process at which TRPC6 inactivation was protective. The differences in these various studies may reflect both species and genetic background, which can have significant effects on the severity of renal complications in diabetes (554). On the other hand, NOX4 knockout is protective in STZ-induced diabetes and is associated with markedly reduced Ca2+ influx in podocytes from diabetic Dahl SS rats (316). Recall that NOX4 can modulate TRPC6 expression and trafficking in podocytes (341), but it is certainly going to affect other proteins as well. It has also been reported that glucagon-like peptide-1 (GLP-1) agonists such as liraglutide reduce the expression of glomerular TRPC6 in STZ-induced diabetes in Sprague-Dawley rats and may act to reduce interactions between TRPC6 and NADPH oxidases (555).
One aspect that complicates interpretation of many in vivo experiments is that multiple types of TRPC channels, including TRPC6, are also expressed in MCs and may play a distinctly different role there, especially in the context of DN. Glomerular MCs often acquire a myofibroblast phenotype when challenged by barotrauma, inflammation, or localized injuries. This transition is characterized in part by the production of α-smooth muscle actin (556). An important early change in the diabetic kidney is the development of glomerular hyperfiltration (557), which is thought to drive at least some of the later pathological features of DN (558). Diabetic hyperfiltration occurs in part as a result of a decreased responsiveness of the afferent arterioles and glomerular MCs to vasoconstrictors (284, 559, 560). Mesangial contractility is impaired in diabetes, and reduced Ca2+ influx into MCs is believed to be a major contributing factor. Since TRPC6 mediates at least some of the Ca2+ influx into MCs, it has been proposed that dysfunction of that channel may play a role in diabetic hyperfiltration. The protein abundance of TRPC6, but not of TRPC1 or TRPC3, was reduced in cultured MCs treated for a sustained period of time with elevated external glucose (245). It should be noted that the decrease in abundance of MC TRPC6 protein appears to be a quite early change in the development of DN. High-glucose treatment and knockdown of TRPC6 attenuated ANG II-stimulated membrane currents and Ca2+ influx in MCs. It bears noting that the decrease in TRPC6 protein expression caused by high glucose or diabetes is a somewhat unique feature of MCs, was not observed in the aorta and the heart tissue isolated from the same animals (246), and does not occur in cultured podocytes (343).
TRPC6 also plays a role in MC proliferation, which is a cardinal pathological feature in many glomerular diseases, including DN. Activation of CsRs and AT1 receptors can induce proliferation in various cell types, including MCs. In addition, it has been observed that ANG II stimulates MC proliferation by increasing the expression level of TRPC6 protein (239). Chronic hypoxia can contribute to both the initiation and progression of chronic renal diseases and can impact all glomerular cells. Chronic hypoxia alters MC proliferation (561) and induces actin reorganization and an upregulation of TRPC6 protein (238). Although it should be noted that these results suggest that inhibition of TRPC6 could represent a therapeutic strategy for kidney diseases in which there is prominent MC proliferation, it is possible that these effects could be countered by maladaptive effects on MC contractility. These effects of TRPC6 on the physiology of MCs might explain why global TRPC6 inactivation in rats does not produce large protective effects (552, 553).
Changes in TRPC1 may also contribute to DN. TRPC1 expression in MCs is reduced in Zucker diabetic fatty rats (an animal model for type 2 diabetes mellitus) and in STZ-treated rats, as well as in kidney biopsies from patients with DN (562). In the db/db mouse model of type 2 diabetes, there was a slight reduction in TRPC1 transcripts at 12 wk of age and a large reduction at 26 wk compared to wild-type control mice (563). In addition, MiR-135a is markedly upregulated in serum and renal tissue in patients with DN, as well as in db/db mice, and this change is correlated with the development of microalbuminuria and renal fibrosis (236). TRPC1 appears to be a target of miR-135a during renal injury, and overexpression of TRPC1 reversed the pathological effects of miR-135a on MC proliferation and ECM secretion. In addition, knockdown of miR-135a in diabetic kidneys restored levels of TRPC1 and reduced the synthesis of fibronectin and collagen 1 in vivo (236). Therefore, in marked contrast to TRPC6, TRPC1 functions as a suppressor of MC proliferation.
6.6. Podocyte Pathways Downstream of TRPC6
The loss of a threshold number of podocytes is a key event in any renal disease process because there is a very limited capacity to replace those cells (503). Excessive activation of TRPC6 channels appears to trigger podocyte pathology through multiple downstream pathways in different cell compartments, most of which are likely to occur secondary to increased Ca2+ influx. Note that the pathways downstream of TRPC6 in foot processes will be different from those occurring in the cell body. The most extensively studied pathway entails the activation of calcineurin, which in turn can cause activation of NFAT transcription factors and Erk MAP kinases in the podocyte cell body (454, 455, 473). This will lead to changes in gene expression, including changes in the expression of TRPC6 itself (425). Calcineurin activation downstream of TRPC6 activation can also lead to modulation of the actin cytoskeleton through effects on synaptopodin, which leads to modulation of Rho GTPases (564–566). Those effects would be most important in foot processes. TRPC6 activation has also been linked to activation of the protease calpain in podocytes (567, 568), and there is evidence that this is due at least in part to the conformational coupling of TRPC6 and calpain rather than Ca2+ influx (568). TRPC6-mediated calpain activation has been linked to podocyte pathology in vivo (567). However, it should also be noted that irreversible cysteine cathepsin inhibitor E-64, chronically infused in Dahl SS rats, had no effect on basal calcium levels in podocytes (569). If conformation-coupled activation of calpain represents a dominant mechanism driving podocyte pathology, it might limit the types of TRPC6 inhibitors that would be effective, as standard pore blockers would not be expected to alter gating conformations of these channels.
It has been reported that TRPC6 activation leads to changes in podocyte motility in vitro, which has often been correlated with altered behavior of foot processes in vivo. However, the results published to date are difficult to reconcile. Thus activation of TRPC6 has been reported to cause a decrease in podocyte motility due to activation of Rho (418). On the other hand, podocyte cell lines derived from TRPC6−/− mice are less motile compared with podocyte cell lines from wild-type mice, owing to greater adhesion to the substrate (568). Differences in the cell lines and culture conditions may explain these differences.
6.7. Pharmacological Strategies for Targeting TRPC6
The evidence reviewed so far suggests that inhibition of various TRPC channels might be a useful therapeutic strategy for at least some glomerular diseases in which there is a currently unmet need, especially in familial and acquired forms of FSGS. A conceptually straightforward approach is to develop agents that block or inhibit TRPC6 channels directly. The structures of several TRPC inhibitors are shown in FIGURE 19. There are a number of small molecules that can inhibit TRPC6 with varying degrees of specificity. Among the most specific are SAR7334 (420), laryxil acetate (571), BI 749327 (570), and SH045 (574). All are active in the nanomolar range and have more than an order of magnitude of selectivity for TRPC6 over the closely related TRPC3 and TRPC7 channels. They do not block more distantly related TRP family channels, including TRPC4 and TRPC5. Importantly, laryxil acetate and BI 749327 are able to inhibit mutant forms of TRPC6 implicated in familial FSGS (570, 575). BI 749327 is especially interesting because it also blocks renal fibrosis induced by unilateral ureteral obstruction in mice, improves left heart function, and reduces cardiac hypertrophy and interstitial fibrosis in mice subjected to chronic pressure overload (570). These TRPC6 inhibitors are orally active and represent useful lead compounds. Several other small molecules can inhibit TRPC6 channels, albeit with somewhat less specificity. Some of these will block TRPC3 and TRPC6 with approximately equal potency, for example, certain analino-thiazoles (576) and benzothiazole amides (577). SH045 and BI 749327 were recently used to evaluate the contribution of TRPC6 in the response of the kidney to acute ischemia (578). In vivo inhibition of TRPC6 by SH045 also was reported to attenuate renal fibrosis in a mouse model of metabolic syndrome (579). It is likely that more TRPC6 inhibitors will be developed, as the structure of TRPC6 has recently been established by cryo-electron microscopy (cryo-EM) (467, 580), which should allow for more rational development of TRPC6 inhibitors and modulators. The utility of this approach is illustrated by the development of M085 and GSK1702934A, two small molecules that directly activate TRPC6 by acting on extracellular sites formed by the pore helix and transmembrane helix S6 (581). Although there is value in developing inhibitors with high specificity, it is likely that at least some of the TRPC6 channels in podocytes are heteromers containing TRPC3 subunits (328, 408). In this regard, TRPC3/6 heteromers are reported to be more stretch sensitive than either of the homomeric channels, at least in certain cell lines (582). In any case, it is not clear that high specificity for TRPC6 over TRPC3 is necessary to develop a useful therapeutic agent for kidney disease (572), a point we return to in sect. 6.8.
FIGURE 19.
Structures of TRPC inhibitors. A: TRPC6 [BI 749327 (570), SAR7334 (420), larixyl acetate (571), and GSK 2833503A (572)] inhibitors. B: TRPC5 [AC-1903 (40) and GFB-8438 (573)] inhibitors.
There are a number of drugs that can affect TRPC6 function or expression indirectly, at least in podocytes, and that have been examined in animal models of kidney diseases, including adaptive forms of FSGS. One of the most notable is sildenafil, an inhibitor of phosphodiesterase-5 (PDE-5), which reduces the expression of TRPC6 channels in podocytes and robustly reduced proteinuria in rats treated with doxorubicin (583). The effect of sildenafil is mediated by a pathway that entails cGMP and PKG (583, 584), leading to phosphorylation of peroxisome proliferator-activated receptor γ (PPARγ) (585), which binds to the Trpc6 promoter and thereby suppresses TRPC6 expression (583). A similar pathway occurs in pulmonary artery smooth muscle (586). In this regard, there is evidence that agents that increase cGMP accumulation can reduce renal fibrosis in animal models (587). Through the same pathway, PPARγ agonists such as pioglitazone can also reduce proteinuria in the doxorubicin model (583) and may be useful clinically in CKD (585). The importance of these studies lies in the fact that PDE-5 inhibitors and PPARγ agonists are already in widespread clinical use and are well tolerated. In addition, supplementation with 1,25-dihydroxyvitamin D3 has been shown to reduce TRPC6 expression in podocytes and may also have renoprotective effects (451). Vitamin D treatment also reduces renal urokinase receptor (uPAR) expression (588), which could result in reduced local shedding of suPAR within glomeruli (408), and a reduction of TRPC6 expression on the cell surface (321). It is also possible that agents that inhibit the podocyte surface receptor for uPAR may prove to be effective agents for primary FSGS, in part through inhibition of TRPC6 signaling. Agents in this category include RGD peptides such as cilengitide, which act by inhibiting αV-containing integrins (321, 527, 530), and RAGE antagonists such as azeliragon (527). Obviously, those agents will have a host of effects on other systems as well. It has also been reported that certain plant-derived natural products can also reduce TRPC6 expression or activity in podocytes (589–591). These agents warrant additional study in animal models of FSGS, but it will be important to identify the active components of these preparations and to work with purified molecules. One limitation of TRPC6 as a drug target is its unusually wide pattern of expression, including in smooth muscle, cardiac muscle, immune cells, and even platelets. TRPC6 knockout rats and mice appear healthy and readily reproduce, but it is still possible that adverse effects of pharmacological TRPC6 inhibition could ultimately limit their utility, even if they prove to be efficacious in FSGS or other nephrotic syndromes.
6.8. TRPC5 Dysregulation in Glomerular Disease Models and Its Potential as a Therapeutic Target
TRPC5 channels can be detected by biochemical methods in cultured podocytes (341, 418) and in rodent glomeruli (540, 553, 592), where they colocalize with synaptopodin, indicating expression in podocytes (592). The physiological factors that normally cause TRPC5 channels to become active are not well understood. There is evidence that TRPC5 can be activated by DAG when the channels are part of a complex containing other proteins, such as Na+/H+ exchange-regulatory factors (593). Depletion of PIP2 secondary to PLC activation may allow this activation to be sustained during signaling through Gq (594, 595). TRPC5 channels can be activated by mechanical stimuli applied to at least some types of cells (596, 597). They are also redox sensitive (598) and can be activated by certain oxidized phospholipids (599).
An early study detected active TRPC5 and TRPC6 channels in a line of immortalized mouse podocytes exposed to ANG II (418). Notably, the cell line used in that study stably overexpressed AT1 receptors for ANG II (418, 600). TRPC5 channels in those cells were enhanced by La3+ (a unique and well-established feature of TRPC5) and had a unitary conductance of ∼33 pS in an excised outside-out patch. By contrast, the TRPC6 channels were blocked by La3+ (418). TRPC5 can also be detected in recordings from podocytes in glomeruli isolated from rats that overexpress AT1 receptors (40). (−)-Englerin A is a sesquiterpene compound that enhances the activation of TRPC5 and TRPC4 channels (601, 602), although it appears to have several other activities as well (603–605). Active TRPC5 channels were detected when human induced pluripotent stem cell (iPSC)-derived podocytes were exposed to (−)-Englerin A (606). On the other hand, the application of ANG II to other podocyte cell lines primarily activates TRPC6 channels (426), and this pattern is also seen in podocytes in glomeruli isolated acutely from mice (345) and rats (315, 322). It is possible that podocyte TRPC5 channels primarily become active in disease conditions in a manner similar to P2X receptors (43), but as yet this has not been confirmed.
A role for TRPC5 has been studied in several glomerular disease models, although to date no data on TRPC5 expression have been published from human renal biopsies to suggest that there are changes in its abundance comparable to those that occur with TRPC6. TRPC5 knockout or inhibition of TRPC5 using the small-molecule inhibitor ML-204 reduced albuminuria and cytoskeletal rearrangements in mice treated with protamine sulfate (PS) or lipopolysaccharide (LPS) (592). PS is a polycation that evokes an acute injury in rodent glomeruli due to the neutralization of anionic charges in the GBM and in glomerular glycocalyces (607, 608). Consequently, infusion of PS produces a rapid and reversible effacement of podocyte foot processes, but this is an acute protocol that is not considered a model for any particular human disease (608). LPS is the name of a class of glycolipids from gram-negative bacteria that are more typically used as a model for sepsis owing to their activation of Toll-like receptor 4 (TLR-4). However, LPS can produce transient albuminuria and cytoskeletal changes in mouse podocytes (609). This is also an acute model, as the changes are seen over a period of ∼48 h. Foot process effacement evoked by PS requires activation of the GTPase Rac1 (610). In cultured podocytes, Rac1 activation may occur downstream of the activation of TRPC5 (418), but it is also required for activation of NOX2, which in turn drives activation of TRPC6 (314–316, 320, 345, 446). It should be noted that ML-204 is not a selective inhibitor of TRPC5 (and indeed can inhibit TRPC6) and was used at a very high dose in this study (2 injections at 20 mg/kg) (592). Moreover, ML-204 does not affect ANG II-induced Ca2+ flux in podocytes from acutely isolated rat glomeruli (322). TRPC5 knockout is a specific manipulation, and in those animals there was a partial reduction in PS-evoked foot process effacement (∼30%) compared to wild-type control animals. Given this, it is notable that TRPC5 knockout resulted in the complete elimination of albuminuria evoked by PS and also blocked Ca2+ influx evoked by PS (592). However, the interpretation of this experiment is not as simple as it might seem because TRPC5 knockdown affects the trafficking of TRPC6 channels to the cell surface and vice versa (408, 611, 612).
A novel TRPC5 inhibitor known as AC1903 has recently been developed (40). Chronic administration of this compound reduced albuminuria and glomerulosclerosis in a strain of transgenic rats that selectively overexpress AT1 receptors (AT1Rs) in podocytes (40). Progression of kidney disease in these animals is characterized by the appearance of pseudocysts within podocytes, followed by foot process effacement and ultimately glomerulosclerosis (424). When these rats were treated with AC1903 twice per day for 7 days there was a reduction in podocyte pseudocysts and albuminuria in AT1R rats. Notably, this treatment protocol was reported to be effective even when it was initiated in older AT1R rats (40) that already had advanced glomerular disease (424). This is a remarkable observation since it would seem to suggest that TRPC5 plays some role in podocyte regeneration, as well as in the reabsorption of ECM. This is because glomerulosclerosis is a consequence of a loss of podocytes and is characterized by dense deposition of ECM (27, 504, 613). AC1903 has also been reported to reduce albuminuria in Dahl SS rats given high NaCl intake to induce systemic hypertension. AC1903 has more recently been shown to reduce albuminuria and podocyte foot process effacement in the acute PAN nephrosis model in Sprague-Dawley rats, in this case measured 7 days after administration of a single dose of PAN (606). Note that previous studies have shown that TRPC6 inactivation is not protective during the acute phase of PAN nephrosis in rats (39). In addition, a larger number of functional riluzole-activated TRPC5 channels could be detected by patch-clamp recordings from podocytes in glomeruli from PAN-treated rats. Those channels were blocked by AC1903 (606).
A recent study has questioned the selectivity of AC1903 and presented evidence that it can inhibit multiple TRPC channels, including TRPC3, TRPC4, TRPC5, TRPC6, TRPC4-C1, and TRPC5-C1 (614). It is interesting to note that the effect of AC1903 was mimicked by nonselective TRPC inhibitors but not by selective inhibitors of TRPC1/4/5 (Pico145, GFB-8438) or TRPC3/6/7 (SAR7334). Given these observations, it is possible that drug-development efforts might benefit by considering oral bioavailability and useful pharmacokinetics at least as much as attempting to develop inhibitors selective for a single type of TRPC channel.
Another family of TRPC5 inhibitors has recently been discovered from high-throughput screening, the most notable of which are GFB-8438 and GFB-887 (573). These molecules appear to have superior pharmacokinetic properties compared with other TRPC5 inhibitors described to date. Treatment with GFB-8438 over a period of 3 wk reduced proteinuria and albuminuria in the deoxycorticosterone acetate (DOCA) rat model of hypertension-induced FSGS (573). Notably, this occurred without any reductions in mean arterial pressure. A Phase 2a clinical trial is currently being planned to test the effects of GFB-887 in patients with FSGS, treatment-resistant minimal change disease, or DN (615).
However, it is important to note that a role for TRPC5 in driving kidney disease has not been seen in every animal model. Overexpression of either a wild-type TRPC5 or a dominant-negative TRPC5 pore mutant (TRPC5-DN) in mice at 2 or 8 mo of age did not result in detectable albuminuria, foot process effacement, or glomerulosclerosis (616), in contrast to what is seen with overexpression of at least some TRPC6 variants (468, 469). An increase in TRPC5 was detected in the podocytes of these animals compared with wild-type control animals. In addition, animals overexpressing TRPC5 did not exhibit increased albuminuria in response to treatment with either (−)-Englerin A or LPS, and LPS-induced albuminuria was not affected by ML-204 applied at two doses of 2 mg/kg (616), a dose shown previously to produce robust effects on TRPC5-mediated processes in vivo (617). The reasons for the difference between this study and the earlier results are not known. Finally, we note that sustained albumin overload in Sprague-Dawley rats, a model that produces albuminuria, proximal tubule damage, and interstitial fibrosis, causes a loss of TRPC5 expression, which is accompanied by increases in TRPC6 and TRPC3 (548).
6.9. KCa1.1 Channels in Glomerular Diseases
It is well established that sustained hyperactivation of TRPC6 can drive podocyte dysfunction in humans and in animal models (36, 37, 321, 469, 471). A recent study has shown that coordinated activation of KCa1.1 and TRPC6 in podocytes can result in increased Ca2+ influx through TRPC6 (332), a consequence of the permeation properties of TRPC6 (227) and its direct interaction with KCa1.1 (328). This raises the question of whether KCa1.1 can also contribute to the progression of glomerular diseases. A recent study addressed this issue directly. It was shown that KCa1.1 γ3-subunit transcripts and protein are markedly elevated in podocytes of patients with FSGS, DN, and membranous nephropathy but not in patients with interstitial nephritis or minimal change disease (332). As with the KCa1.1 pore-forming subunits (323), the γ3-subunits colocalize with synaptopodin, indicating expression in podocyte foot processes. Increased γ3 expression was also observed in mice with albuminuria induced by chronic infusions of ANG II. Importantly, knockout of Kcnma1 (the gene encoding pore-forming subunits of KCa1.1 in mice) reduced podocyte injury induced by ANG II infusion (332). The γ3-subunits were increased by ANG II by a pathway that requires the increased activity of the transcription factor NFATc3, and suppression of NFATc3 blocked ANG II-evoked increases in the γ3-subunit (332). Because KCa1.1 channels are expressed in multiple cell types in the glomeruli, including within arterioles and in MCs, it is not entirely certain that the protective effect of Kcnma1 knockout can be attributed to changes in podocytes. However, the fact that γ3-subunits are elevated in human glomerular diseases and mouse disease models is strongly suggestive, because this subunit is not generally found in vascular smooth muscle.
6.10. NMDA and P2X Receptors in Glomerular Diseases
There is considerable evidence from rodent models and isolated cells that renal NMDA receptors can contribute to kidney pathology. As noted above, activation of renal NMDA receptors results in substantial Ca2+ influx accompanied by marked increases in the generation of ROS, which induce numerous downstream effects (369, 370, 401, 402). The effect of NMDA on Ca2+ influx is likely to be amplified by ROS-dependent increases in functional cell surface TRPC6 channels in podocytes (401). The combined effects of these cation channels will activate calcineurin-NFAT signaling systems (401), which can cause marked changes in gene expression, including increases in the abundance of TRPC6 (342) as well as increased expression of γ3-subunits of KCa1.1 (332). Note that Ca2+-dependent NFAT activation is considered an essential aspect of glomerular disease caused by dysregulation of TRPC6 (425, 454, 468, 618). The resulting Ca2+ influx may also increase calpain activation (567), which has long been known to be a downstream target of pathological NMDA receptor signaling in neurons. Sustained NMDA receptor activation produces several other potentially pathological effects in podocytes, including inhibition of Cdc42, with accompanying changes in cell cytoskeleton and morphology (44), internalization and eventual loss of the key slit diaphragm proteins nephrin and podocin, and ultimately cell death associated with increased expression of caspase-3, caspase-6, and Bax (401).
l-homocysteic acid is an effective agonist of podocyte NMDA receptors (369) and is capable of inducing apoptosis in podocytes with sustained exposure (401). l-homocysteic acid is a spontaneous oxidation product of l-homocysteine, a sulfur-containing amino acid produced during the metabolism of methionine. Therefore it is important to note that rats with markedly elevated concentrations of l-homocysteine develop marked glomerulosclerosis (370). Hyperhomocysteinemia in rats can be induced by maintaining animals on a folate-free diet or by adding l-methionine to their drinking water (370). After 6 wk, the rats exhibited marked glomerulosclerosis and increased extracellular matrix deposition that could be blocked by treating rats with the NMDA antagonist MK-801 (370). The treatment with MK-801 had no effect on the hyperhomocysteinemia but protected against the renal consequences of this metabolic dysregulation. Glomeruli from these animals had markedly increased ROS production due to increased activation of NOX2, as is seen in cultured podocytes exposed to NMDA agonists (369).
NMDA receptors also appear to contribute to the progression of DN (44, 619, 620). Thus, expression of NR1, NR2A, and NR2C subunits is increased in podocytes cultured in high glucose (619) and in Akita mice, a model for type 1 diabetes (44, 620). Importantly, treating diabetic mice continuously with MK-801 for 28 days resulted in marked reductions in glomerulosclerosis and podocyte foot process effacement (FIGURE 20) (44). This was also seen in mice made diabetic by treatment with STZ (44, 619). A similar protective effect was observed after treating mice with memantine, an orally bioavailable NMDA antagonist currently used for the treatment of neurodegenerative diseases in humans (44). Treatment with the NMDA antagonists had no effect on blood glucose or blood pressure, and it is likely that the protective effects were exerted directly on glomeruli (44, 619). Treating Akita mice with MK-801 also normalized the expression of connexins in the diabetic kidney (620). In addition, reducing NMDA receptor NR1 subunit expression by infusion of a targeted shRNA reduced glomerulosclerosis in db/db mice, a model for type 2 diabetes (619). This treatment also reduced mesangial expansion and preserved expression of WT1 and synaptopodin, indicating a protective effect on podocytes (619). In this regard, NMDA antagonists are reported to induce a reduction in GFR. The mechanism of that effect is not known, but it could provide an additional basis for its glomeruloprotective effect in diabetes (621).
FIGURE 20.

Pharmacological inhibition of NMDA receptors reduces DN in the Akita mouse model of type 1 diabetes. A: diabetic mice exhibit marked increases in renal cortical NMDA receptor subunit expression compared with wild-type normoglycemic control mice (DBA/2J). B: treatment with the NMDA antagonist MK-801 for 28 days resulted in a decrease in urine albumin excretion accompanied by a decrease in podocyte foot process effacement. *P < 0.05. C: representative electron microscopy (EM) images of foot process effacement (arrowheads) and GBM thickening (asterisk) in Akita mice treated with saline. The ultrastructure was markedly improved in Akita mice treated with MK-801. See glossary for abbreviations. Adapted from Ref. 44 with permission.
NMDA receptor blockade has protective effects in several other kidney disease models, such as in ischemia-reperfusion injuries (363, 622–625), renal fibrosis induced by unilateral ureteral obstruction (626), sepsis-induced acute kidney injury (AKI) (627), and kidney insufficiency induced by infusion of lipopolysaccharide (628). NMDA receptors may also play a role in vitamin D metabolism in the kidney (629). Although glomerular pathology is not a cardinal feature of these disease models, it underscores the potential therapeutic efficacy of NMDA antagonists in kidney disease.
There is evidence that P2X receptors are altered in glomerular disease conditions and may play a role in driving pathologies. There is a marked upregulation of functional ionotropic P2X4 and P2X7 receptors in podocytes examined from diabetic animals compared to podocytes from healthy animals (43, 403). The upregulation of functional P2X receptors may contribute to Ca2+ overload, loss of podocytes, and glomerulosclerosis in DN and possibly other glomerular diseases (FIGURE 14) (43). These studies are consistent with the reports demonstrating that activation of P2X7 receptors contributes to the high prevalence of kidney disease found in people with diabetes. Renal P2X7 receptor expression was associated with severe mesangial expansion, impaired glomerular filtration, and increased interstitial fibrosis in diabetic patients (630).
P2X receptors of MCs may also contribute to glomerular pathology (345). Thus activation of P2X4 receptors in human MCs can drive nucleotide-induced apoptosis (631). Moreover, P2X receptors contribute to mesangial matrix expansion that occurs in diabetes and ANG II-dependent hypertension. The combination of hyperglycemia and high concentrations of extracellular ATP promote mesangial matrix expansion and increase TGF-β production in rat MCs (308). In the rat model of hypertension and kidney disease mediated by chronic ANG II infusion, MC transformation was prevented by blockade of P2X signaling (632).
6.11. SOCs and Glomerular Disease
SOC dysregulation has been most extensively studied in the context of DN, especially in MCs. Mesangial expansion and deposition of ECM proteins occur early in the development of DN, before the development of glomerular fibrosis and glomerulosclerosis (633). SOCE regulates ECM synthesis and deposition by glomerular MCs and becomes active in response to neuroendocrine and paracrine signaling. However, certain aspects of this literature are controversial.
It is a common experimental design (albeit having some limitations) to mimic the diabetic milieu by exposing cultured cells to elevated external glucose for various periods of time. Early studies that used this approach reported contradictory results. In one such study, exposing cultured rat MCs to high glucose for 5 days inhibited resting and vasopressin-induced Ca2+ influx, and the authors argued that this was due to reductions in SOCE secondary to inactivation of PKC (634). However, a different study reported that exposure of cultured rat MCs to high glucose for up to 7 days did not affect SOCE, although it did reduce receptor-mediated Ca2+ influx evoked by ET-1 (284). More recent studies have examined high glucose effects on SOCs with experimental approaches that went beyond ratiometric Ca2+ imaging and that examined multiple time points. Thus, elevated glucose for 8–24 h decreased Orai1 protein content in rat and human MCs (635), whereas a 7-day exposure to high glucose increased the abundance of STIM1 and Orai1 proteins and enhanced SOCE induced by thapsigargin (291). The enhanced Ca2+ entry seen with more sustained exposure to high glucose was inhibited by GSK-7975A, an inhibitor of SOC, and also by Orai1 knockdown, verifying that Ca2+ entry in those experiments was true SOC. In addition, the more sustained high-glucose treatment markedly augmented SOC currents measured by whole cell recording (291). Finally, diabetic rats showed increased STIM1 and Orai1 protein abundance in the renal cortex and in glomeruli (291). The current consensus is that the effects of high glucose on SOC and SOCE are time dependent. Longer-duration exposures to high glucose are likely to be a better model for diabetic dysregulation because the hyperglycemia that occurs in diabetes is a chronic condition.
SOCs regulate ECM dynamics in MCs. Thus, activation of SOCE by thapsigargin treatment suppressed the synthesis of the ECM proteins fibronectin and collagen IV by cultured human MCs, whereas inhibition of SOCs with 2-aminoethyl diphenylborinate had the opposite effect (42). Orai1 knockdown also increased ECM protein abundance in cultured human MCs (42), and a similar effect was seen in vivo when a siRNA targeting Orai1 was delivered into mice with a nanoparticle system (42) or with an adenoviral vector (636) (see FIGURE 5). Orai1 knockdown also caused marked reductions in mesangial expansion and renal fibrosis (41, 319). This effect of SOCs in MCs is mediated at least in part by suppression of profibrotic Smad1 and Smad3 activation (250, 637). It should be noted that in vivo studies using a nanoparticle-siRNA delivery system have shown that knocking down Orai1 in MCs significantly increased the expression of both Smad1 and Smad3 in mouse glomeruli (250, 637). FIGURE 21 shows a schematic diagram of the signaling pathway for suppression of ECM production by Orai1-mediated SOCE in glomerular MCs. The mechanisms whereby SOC inhibits Smad1 and Smad3 activation are not known. It is possible that SOC inhibits the kinase activity of a type II TGF-β1 receptor dimer that phosphorylates the type I dimer. Alternatively, SOCE could drive activation of a Ca2+-dependent phosphatase or inhibit a protein kinase leading to increased phosphorylation of Smad1/3. In either case, it is known that SOCE elevates pro-/anti-inflammatory signaling that contributes to ECM protein production by MCs (638). In sect 3.6 we note that TRPC4 contributes to SOCE in MCs, and it has been reported that activation of TRPC4-mediated SOCE by urotensin II promoted MC proliferation and ECM protein accumulation in the presence of high glucose (639).
FIGURE 21.

The signaling pathway for suppressing extracellular matrix protein production by Orai1-mediated store-operated Ca2+ entry (SOCE) in glomerular mesangial cells (MCs). ECM, extracellular matrix; P-Smad1, phosphorylated Smad1; P-Smad2/3, phosphorylated Smad2/3; TGF-β1: transforming growth factor β.
There are several other neuroendocrine and paracrine signaling pathways that can activate SOCE in MCs and thereby alter matrix production. Thus, liraglutide, an agonist of the G protein-coupled glucagon-like peptide-1 receptor (GLP-1R), causes activation of SOCs in MCs, thereby reducing high-glucose-induced ECM production (640). This can also occur through Wnt/β-catenin signaling pathways (641). Of note, GLP-1, the endogenous agonist of GLP-1R, is used clinically to promote insulin secretion in the treatment of type 2 diabetes (642). Cross talk between the SOCE and Wnt/β-catenin pathways in MCs has not been studied. However, it has been shown that micro-RNAs such as miR-29a promote Wnt/β-catenin signaling in MCs (643). An important unresolved question is whether SOC signaling activates miR-29a to augment the Wnt/β-catenin signaling machinery to downregulate ECM proteins.
Nitric oxide (NO) and its metabolites are mediators of inflammation in renal diseases (644), and, as described above, TRPC1 and TRPC4 are necessary for SOCE in MCs. It has been reported that NO inhibits TRPC4-associated SOCE via the PKG/VASP pathway in human MCs (296). Therefore, a pathway comprised of NO/PKG/VASP/TRPC4/SOCE in MCs may be involved in glomerular inflammation. Podocytes are capable of releasing NO, and this could represent a paracrine signaling pathway to modify function of MCs (145). There is also evidence supporting a role for TRPC1 channels. Thus, TRPC1-mediated SOCE reduced the synthesis of ECM proteins in human MCs exposed to high glucose (236). Thus, the effect of TRPC1 is similar to that of Orai (234). As mentioned in sect. 3.6, TRPC1 and Orai may function together as part of a larger complex. In this regard, miR-135a attenuated SOCE in human MCs by reducing TRPC1 abundance and, consequently, augmented fibronectin and collagen I synthesis (236).
Although the available data suggest that targeting SOC channels could be a useful therapeutic strategy in certain glomerular diseases, several physiological factors may limit this approach. First, SOCs are ubiquitously expressed in both excitable and nonexcitable cells (289). Therefore, the systemic application of SOC regulators would be expected to have a wide range of effects, possibly including adverse effects on multiple tissues. Another issue is that although a variety of SOC inhibitors have been used experimentally for several decades, none is highly selective for SOC (645). Indeed, inhibition of SOCs could result in counteracting effects even within a single organ such as the kidney. Thus, activation of SOC in glomerular MCs inhibits ECM production, which might be beneficial in DN (42, 250, 637), although at least one report suggests that SOC stimulates MC proliferation and ECM production (639). At the same time, SOC activation in proximal tubular cells may exacerbate renal fibrosis (636), and inhibition of SOC is reported to exacerbate albuminuria in mice with DN by impairing albumin uptake by proximal tubular cells (646).
7. CONCLUSIONS
This review has summarized the properties and functions of ion channels in glomerular cells, especially MCs and podocytes. Both of these cell types express multiple populations of plasma membrane ion channels, and nearly all of the channels studied to date play some role in Ca2+ signaling that occurs during neural, endocrine, or paracrine signaling, in response to the external environment (e.g., mechanosensitivity), and/or as a result of the metabolic status of the cells. It is interesting to note that the most substantial evidence relates to the contributions of some of these channels to cellular processes that are closely related to disease states, for example, secretion of matrix by MCs, or changes in the ultrastructure of podocytes. Indeed, the strongest argument for the pathophysiological relevance of glomerular ion channels remains the observation that mutations in TRPC6 channels result in devastating glomerular diseases in humans. Further evidence comes from animal models in which various channels have been manipulated by genetic and pharmacological procedures, as well as in cell culture systems. Based on these studies, several ion channels can be considered as promising targets for drug discovery in glomerular disease, and drug development efforts targeting TRPC5 and TRPC6 are already well underway. Some of the other channels discussed in this review, such as NMDA receptors, P2X receptors, and SOCs, probably deserve more attention in this context, and it is possible that a bias in favor of agents with a high degree of selectivity for a single type of channel may actually cause potentially effective agents to be overlooked and discarded.
It is somewhat paradoxical that much less is known about the role of ion channels in the normal function of glomerular cells. It is now well established that Ca2+ signaling occurs in all glomerular cells on an ongoing basis during processes such as TGF, but the effect that these signals have on glomerular cells in situ is not well understood. Indeed, surprisingly little is known about dynamic processes in MCs and podocytes in the regulation of glomerular filtration. Although one could get the impression that the glomerular capillary is simply a sieve and that all of the critical regulation of glomerular filtration occurs at the level of the afferent and efferent arterioles, it remains clear that glomerular cells are not static. In addition to Ca2+ signals, these cells can secrete a wide range of substances, can rearrange their cytoskeleton, and can generate force. They can also reenter the cell cycle. Indeed, it is likely that glomerular cells play a normal role in fine-tuning the glomerular filtration barrier on a moment-to-moment basis and in compensating when other physiological processes (e.g., blood pressure or glucose concentration) are outside of the normal range. Indeed, certain processes, such as mesangial matrix expansion and podocyte foot process effacement, are probably protective, at least for some period of time. On the other hand, at a certain point, for example, with severe and sustained Ca2+ overload, these processes become maladaptive and the system decompensates. It is likely that the development of new tools to allow the in vivo manipulation of ion channels and other signaling processes in glomerular cells, and in particular to do this in a cell type-specific manner, will lead to a greater understanding of renal physiology as a whole, especially if these experimental approaches are combined with sophisticated imaging of glomerular dynamics and electrophysiology.
GLOSSARY
- 20-HETE
20-Hydroxyeicosatetraenoic acid
- ADP
Adenosine diphosphate
- AKI
Acute kidney injury
- ANG II
Angiotensin II
- ANP
Atrial natriuretic peptide
- ATP
Adenosine triphosphate
- AVP
Arginine vasopressin
- BK (KCa1.1)
Large-conductance calcium-activated potassium channel
- BzATP
3′-O-(4-benzoyl)benzoyl ATP
- Cav
Voltage-activated Ca2+ channel
- CD2AP
CD2-associated protein
- cGMP
Cyclic guanosine monophosphate
- CKD
Chronic kidney disease
- CNS
Central nervous system
- CO
Carbon monoxide
- CsR
Ca2+-sensing receptor
- CTGF
Connecting tubule-glomerular feedback
- DAG
Diacylglycerol
- DN
Diabetic nephropathy
- DOCA
Deoxycorticosterone acetate
- ECF
Extracellular fluid
- ECM
Extracellular matrix
- EGF
Epidermal growth factor
- ENaC
Epithelial Na+ channel
- ERK
Extracellular signal-regulated kinase
- ESKF
End-stage kidney failure
- ET
Endothelin
- FSGS
Focal segmental glomerulosclerosis
- GBM
Glomerular basement membrane
- GFR
Glomerular filtration rate
- H2S
Hydrogen sulfide
- I Cl.Ca
Ca2+-activated Cl− channel
- I Cl.vol
Volume-sensitive Cl− channel
- IP3
Inositol 1,4,5-trisphosphate
- JGA
Juxtaglomerular apparatus
- KATP
ATP-sensitive K+ channels
- K f
Filtration coefficient
- Kir
Inwardly rectifying K+ channel
- LPS
Lipopolysaccharide
- MAPK
Mitogen-activated protein kinase
- MC
Mesangial cell
- MCD
Minimal change disease
- NFAT
Nuclear factor of activated T cell
- NF-κB
Nuclear factor-κB
- NHE
Na+/H+ exchanger
- NKCC
Na-K-2Cl cotransporter
- NMDA
N-methyl-d-aspartate
- NO
Nitric oxide
- NOX
NADPH oxidase
- OAG
1-Oleoyl-2-acetyl-sn-glycerol
- PAN
Puromycin aminonucleoside nephrosis
- PDE-5
Phosphodiesterase-5
- PDGF-B
Platelet-derived growth factor B
- PIP2
Phosphatidylinositol 4,5-bisphosphate
- PKG
cGMP-dependent protein kinase
- PLC
Phospholipase C
- PMA
Phorbol 12-myristate 13-acetate
- PP2A
Protein phosphatase 2A
- RAGE
Receptor for advanced glycation end-products
- ROMK
Renal outer medullary potassium channel
- ROS
Reactive oxygen species
- sGC
Soluble guanylate cyclase
- SOC
Store-operated channel
- SOCE
Store-operated Ca2+ entry
- STIM
Stromal interaction molecule
- STZ
Streptozotocin
- suPAR
Soluble urokinase plasminogen activator receptor
- SUR
Sulfonylurea receptor
- TGF
Tubuloglomerular feedback
- TGF-β1
Transforming growth factor-β1
- TLR-4
Toll-like receptor 4
- TNF
Tumor necrosis factor
- TRP
Transient receptor potential
- TRPA1
Transient receptor potential ankyrin 1
- TRPC
Transient receptor potential canonical
- UTP
Uridine-5′-triphosphate
- VASP
Vasodilator-stimulated phosphoprotein
- VEGF
Vascular endothelial growth factor
GRANTS
Research in the authors’ laboratories was supported by National Institutes of Health Grants R35 HL135749 and R21 DK129882 (to A.S.), R01 DK129227 (to A.S. and O.P.), R01 DK126720 (to O.P.), R01 DK104708 (to S.E.D.), and R01 DK115424 (to R.M.) and Department of Veterans Affairs Grant I01 BX004024 (to A.S.).
DISCLOSURES
S.E.D. has received research contracts from Pfizer Inc. and Walden Biosciences Inc. He has received speakers honoraria from Amgen Inc., Walden Bioscience Inc., and Aredelyx Inc., and serves on the scientific advisory board of Actio Biosciences Inc. No other authors have any conflicts of interest, financial or otherwise, to disclose.
AUTHOR CONTRIBUTIONS
A.S., R.M., O.P., and S.E.D. prepared figures; A.S., R.M., O.P., and S.E.D. drafted manuscript; A.S., R.M., O.P., and S.E.D. edited and revised manuscript; A.S., R.M., O.P., and S.E.D. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors apologize to the investigators of glomerular ion channels whose relevant publications were inadvertently not discussed directly. We also thank all past and current members of the authors’ laboratories. Graphical figures were created with BioRender.com.
REFERENCES
- 1. Vallon V. Glucose transporters in the kidney in health and disease. Pflugers Arch 472: 1345–1370, 2020. doi: 10.1007/s00424-020-02361-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hoorn EJ, Gritter M, Cuevas CA, Fenton RA. Regulation of the renal NaCl cotransporter and its role in potassium homeostasis. Physiol Rev 100: 321–356, 2020. doi: 10.1152/physrev.00044.2018. [DOI] [PubMed] [Google Scholar]
- 3. Bröer S. Amino acid transport across mammalian intestinal and renal epithelia. Physiol Rev 88: 249–286, 2008. doi: 10.1152/physrev.00018.2006. [DOI] [PubMed] [Google Scholar]
- 4. Ellison DH, Welling P. Insights into salt handling and blood pressure. N Engl J Med 385: 1981–1993, 2021. doi: 10.1056/NEJMra2030212. [DOI] [PubMed] [Google Scholar]
- 5. Rossier BC, Baker ME, Studer RA. Epithelial sodium transport and its control by aldosterone: the story of our internal environment revisited. Physiol Rev 95: 297–340, 2015. doi: 10.1152/physrev.00011.2014. [DOI] [PubMed] [Google Scholar]
- 6. Mutchler SM, Kirabo A, Kleyman TR. Epithelial sodium channel and salt-sensitive hypertension. Hypertension 77: 759–767, 2021. doi: 10.1161/HYPERTENSIONAHA.120.14481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Clase CM, Carrero JJ, Ellison DH, Grams ME, Hemmelgarn BR, Jardine MJ, Kovesdy CP, Kline GA, Lindner G, Obrador GT, Palmer BF, Cheung M, Wheeler DC, Winkelmayer WC, Pecoits-Filho R, Conference Participants. Potassium homeostasis and management of dyskalemia in kidney diseases: conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int 97: 42–61, 2020. doi: 10.1016/j.kint.2019.09.018. [DOI] [PubMed] [Google Scholar]
- 8. Staruschenko A. Beneficial effects of high potassium: contribution of renal basolateral K+ channels. Hypertension 71: 1015–1022, 2018. doi: 10.1161/HYPERTENSIONAHA.118.10267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Matikainen N, Pekkarinen T, Ryhänen EM, Schalin-Jäntti C. Physiology of calcium homeostasis: an overview. Endocrinol Metab Clin North Am 50: 575–590, 2021. doi: 10.1016/j.ecl.2021.07.005. [DOI] [PubMed] [Google Scholar]
- 10. Figueres L, Beck-Cormier S, Beck L, Marks J. The complexities of organ crosstalk in phosphate homeostasis: time to put phosphate sensing back in the limelight. Int J Mol Sci 22: 5701, 2021. doi: 10.3390/ijms22115701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hernando N, Gagnon K, Lederer E. Phosphate transport in epithelial and nonepithelial tissue. Physiol Rev 101: 1–35, 2021. doi: 10.1152/physrev.00008.2019. [DOI] [PubMed] [Google Scholar]
- 12. Wagner CA, Imenez Silva PH, Bourgeois S. Molecular pathophysiology of acid-base disorders. Semin Nephrol 39: 340–352, 2019. doi: 10.1016/j.semnephrol.2019.04.004. [DOI] [PubMed] [Google Scholar]
- 13. Lobel L, Cao YG, Fenn K, Glickman JN, Garrett WS. Diet posttranslationally modifies the mouse gut microbial proteome to modulate renal function. Science 369: 1518–1524, 2020. doi: 10.1126/science.abb3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pluznick JL. The gut microbiota in kidney disease. Science 369: 1426–1427, 2020. doi: 10.1126/science.abd8344. [DOI] [PubMed] [Google Scholar]
- 15. Vanholder R, De Smet R, Glorieux G, Argilés A, Baurmeister U, Brunet P, Clark W, Cohen G, De Deyn PP, Deppisch R, Descamps-Latscha B, Henle T, Jörres A, Lemke HD, Massy ZA, Passlick-Deetjen J, Rodriguez M, Stegmayr B, Stenvinkel P, Tetta C, Wanner C, Zidek W; the European Uremic Toxin Work Group (EUTox). Review on uremic toxins: classification, concentration, and interindividual variability. Kidney Int 63: 1934–1943, 2003. doi: 10.1046/j.1523-1755.2003.00924.x. [DOI] [PubMed] [Google Scholar]
- 16. Lim YJ, Sidor NA, Tonial NC, Che A, Urquhart BL. Uremic toxins in the progression of chronic kidney disease and cardiovascular disease: mechanisms and therapeutic targets. Toxins 13: 142, 2021. doi: 10.3390/toxins13020142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sands JM, Layton HE. Advances in understanding the urine-concentrating mechanism. Annu Rev Physiol 76: 387–409, 2014. doi: 10.1146/annurev-physiol-021113-170350. [DOI] [PubMed] [Google Scholar]
- 18. Rinschen MM, Palygin O, El-Meanawy A, Domingo-Almenara X, Palermo A, Dissanayake LV, Golosova D, Schafroth MA, Guijas C, Demir F, Jaegers J, Gliozzi ML, Xue J, Hoehne M, Benzing T, Kok BP, Saez E, Bleich M, Himmerkus N, Weisz OA, Cravatt BF, Krüger M, Benton HP, Siuzdak G, Staruschenko A. Accelerated lysine metabolism conveys kidney protection in salt-sensitive hypertension. Nat Commun 13: 4099, 2022. doi: 10.1038/s41467-022-31670-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rinschen MM, Palygin O, Guijas C, Palermo A, Palacio-Escat N, Domingo-Almenara X, Montenegro-Burke R, Saez-Rodriguez J, Staruschenko A, Siuzdak G. Metabolic rewiring of the hypertensive kidney. Sci Signal 12: eaax9760, 2019. doi: 10.1126/scisignal.aax9760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cherney DZ, Kanbay M, Lovshin JA. Renal physiology of glucose handling and therapeutic implications. Nephrol Dial Transplant 35: i3–i12, 2020. doi: 10.1093/ndt/gfz230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang X, Garrett MR. Nephron number, hypertension, and CKD: physiological and genetic insight from humans and animal models. Physiol Genomics 49: 180–192, 2017. doi: 10.1152/physiolgenomics.00098.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Marsh DJ, Postnov DD, Sosnovtseva OV, Holstein-Rathlou NH. The nephron-arterial network and its interactions. Am J Physiol Renal Physiol 316: F769–F784, 2019. doi: 10.1152/ajprenal.00484.2018. [DOI] [PubMed] [Google Scholar]
- 23. Zehra T, Cupples WA, Braam B. Tubuloglomerular feedback synchronization in nephrovascular networks. J Am Soc Nephrol 32: 1293–1304, 2021. doi: 10.1681/ASN.2020040423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Romero CA, Carretero OA. Tubule-vascular feedback in renal autoregulation. Am J Physiol Renal Physiol 316: F1218–F1226, 2019. doi: 10.1152/ajprenal.00381.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Haraldsson B, Nyström J, Deen WM. Properties of the glomerular barrier and mechanisms of proteinuria. Physiol Rev 88: 451–487, 2008. doi: 10.1152/physrev.00055.2006. [DOI] [PubMed] [Google Scholar]
- 26. Daehn IS, Duffield JS. The glomerular filtration barrier: a structural target for novel kidney therapies. Nat Rev Drug Discov 20: 770–788, 2021. doi: 10.1038/s41573-021-00242-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kriz W, LeHir M. Pathways to nephron loss starting from glomerular diseases—insights from animal models. Kidney Int 67: 404–419, 2005. doi: 10.1111/j.1523-1755.2005.67097.x. [DOI] [PubMed] [Google Scholar]
- 28. Fan L, Gao W, Nguyen BV, Jefferson JR, Liu Y, Fan F, Roman RJ. Impaired renal hemodynamics and glomerular hyperfiltration contribute to hypertension-induced renal injury. Am J Physiol Renal Physiol 319: F624–F635, 2020. doi: 10.1152/ajprenal.00239.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Löwen J, Gröne EF, Groß-Weißmann ML, Bestvater F, Gröne HJ, Kriz W. Pathomorphological sequence of nephron loss in diabetic nephropathy. Am J Physiol Renal Physiol 321: F600–F616, 2021. doi: 10.1152/ajprenal.00669.2020. [DOI] [PubMed] [Google Scholar]
- 30. Pavenstädt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev 83: 253–307, 2003. doi: 10.1152/physrev.00020.2002. [DOI] [PubMed] [Google Scholar]
- 31. Wrede C, Hegermann J, Mühlfeld C. Novel cell contact between podocyte microprojections and parietal epithelial cells analyzed by volume electron microscopy. Am J Physiol Renal Physiol 318: F1246–F1251, 2020. doi: 10.1152/ajprenal.00097.2020. [DOI] [PubMed] [Google Scholar]
- 32. Lemley KV. Glomerular pathology and the progression of chronic kidney disease. Am J Physiol Renal Physiol 310: F1385–F1388, 2016. doi: 10.1152/ajprenal.00099.2016. [DOI] [PubMed] [Google Scholar]
- 33. Rovin BH, Adler SG, Barratt J, Bridoux F, Burdge KA, Chan TM, et al. Executive summary of the KDIGO 2021 Guideline for the Management of Glomerular Diseases. Kidney Int 100: 753–779, 2021. doi: 10.1016/j.kint.2021.05.015. [DOI] [PubMed] [Google Scholar]
- 34. Benzing T, Salant D. Insights into glomerular filtration and albuminuria. N Engl J Med 384: 1437–1446, 2021. doi: 10.1056/NEJMra1808786. [DOI] [PubMed] [Google Scholar]
- 35. Levey AS, Eckardt KU, Dorman NM, Christiansen SL, Hoorn EJ, Ingelfinger JR, et al. Nomenclature for kidney function and disease: report of a Kidney Disease: Improving Global Outcomes (KDIGO) Consensus Conference. Kidney Int 97: 1117–1129, 2020. doi: 10.1016/j.kint.2020.02.010. [DOI] [PubMed] [Google Scholar]
- 36. Winn MP, Conlon PJ, Lynn KL, Farrington MK, Creazzo T, Hawkins AF, Daskalakis N, Kwan SY, Ebersviller S, Burchette JL, Pericak-Vance MA, Howell DN, Vance JM, Rosenberg PB. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science 308: 1801–1804, 2005. doi: 10.1126/science.1106215. [DOI] [PubMed] [Google Scholar]
- 37. Reiser J, Polu KR, Möller CC, Kenlan P, Altintas MM, Wei C, Faul C, Herbert S, Villegas I, Vila-Casado C, McGee M, Sugimoto H, Brown D, Kalluri R, Mundel P, Smith PL, Clapham DE, Pollak MR. TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat Genet 37: 739–744, 2005. doi: 10.1038/ng1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Möller CC, Wei C, Altintas MM, Li J, Greka A, Ohse T, Pippin JW, Rastaldi MP, Wawersik S, Schiavi S, Henger A, Kretzler M, Shankland SJ, Reiser J. Induction of TRPC6 channel in acquired forms of proteinuric kidney disease. J Am Soc Nephrol 18: 29–36, 2007. doi: 10.1681/ASN.2006091010. [DOI] [PubMed] [Google Scholar]
- 39. Kim EY, Yazdizadeh Shotorbani P, Dryer SE. Trpc6 inactivation confers protection in a model of severe nephrosis in rats. J Mol Med (Berl) 96: 631–644, 2018. doi: 10.1007/s00109-018-1648-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhou Y, Castonguay P, Sidhom EH, Clark AR, Dvela-Levitt M, Kim S, Sieber J, Wieder N, Jung JY, Andreeva S, Reichardt J, Dubois F, Hoffmann SC, Basgen JM, Montesinos MS, Weins A, Johnson AC, Lander ES, Garrett MR, Hopkins CR, Greka A. A small-molecule inhibitor of TRPC5 ion channels suppresses progressive kidney disease in animal models. Science 358: 1332–1336, 2017. doi: 10.1126/science.aal4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tao J, Lan Z, Wang Y, Hei H, Tian L, Pan W, Zhang X, Peng W. Large-conductance calcium-activated potassium channels in glomerulus: from cell signal integration to disease. Front Physiol 7: 248, 2016. doi: 10.3389/fphys.2016.00248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wu P, Wang Y, Davis ME, Zuckerman JE, Chaudhari S, Begg M, Ma R. Store-operated Ca2+ channels in mesangial cells inhibit matrix protein expression. J Am Soc Nephrol 26: 2691–2702, 2015. doi: 10.1681/ASN.2014090853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Palygin O, Klemens CA, Isaeva E, Levchenko V, Spires DR, Dissanayake LV, Nikolaienko O, Ilatovskaya DV, Staruschenko A. Characterization of purinergic receptor 2 signaling in podocytes from diabetic kidneys. iScience 24: 102528, 2021. doi: 10.1016/j.isci.2021.102528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Roshanravan H, Kim EY, Dryer SE. NMDA receptors as potential therapeutic targets in diabetic nephropathy: increased renal NMDA receptor subunit expression in Akita mice and reduced nephropathy following sustained treatment with memantine or MK-801. Diabetes 65: 3139–3150, 2016. doi: 10.2337/db16-0209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Naylor RW, Morais M, Lennon R. Complexities of the glomerular basement membrane. Nat Rev Nephrol 17: 112–127, 2021. doi: 10.1038/s41581-020-0329-y. [DOI] [PubMed] [Google Scholar]
- 46. Deen WM, Lazzara MJ, Myers BD. Structural determinants of glomerular permeability. Am J Physiol Renal Physiol 281: F579–F596, 2001. doi: 10.1152/ajprenal.2001.281.4.F579. [DOI] [PubMed] [Google Scholar]
- 47. Jeansson M, Haraldsson B. Morphological and functional evidence for an important role of the endothelial cell glycocalyx in the glomerular barrier. Am J Physiol Renal Physiol 290: F111–F116, 2006. doi: 10.1152/ajprenal.00173.2005. [DOI] [PubMed] [Google Scholar]
- 48. Schlöndorff D, Banas B. The mesangial cell revisited: no cell is an island. J Am Soc Nephrol 20: 1179–1187, 2009. doi: 10.1681/ASN.2008050549. [DOI] [PubMed] [Google Scholar]
- 49. Schlöndorff D, Wyatt CM, Campbell KN. Revisiting the determinants of the glomerular filtration barrier: what goes round must come round. Kidney Int 92: 533–536, 2017. doi: 10.1016/j.kint.2017.06.003. [DOI] [PubMed] [Google Scholar]
- 50. Shroff UN, Gyarmati G, Izuhara A, Deepak S, Peti-Peterdi J. A new view of macula densa cell protein synthesis. Am J Physiol Renal Physiol 321: F689–F704, 2021. doi: 10.1152/ajprenal.00222.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Goligorsky MS, Iijima K, Krivenko Y, Tsukahara H, Hu Y, Moore LC. Role of mesangial cells in macula densa to afferent arteriole information transfer. Clin Exp Pharmacol Physiol 24: 527–531, 1997. doi: 10.1111/j.1440-1681.1997.tb01240.x. [DOI] [PubMed] [Google Scholar]
- 52. Briggs JP, Skøtt O, Schnermann J. Cellular mechanisms within the juxtaglomerular apparatus. Am J Hypertens 3: 76–80, 1990. doi: 10.1093/ajh/3.1.76. [DOI] [PubMed] [Google Scholar]
- 53. Bradfield JW, Cattell V, Smith J. The mesangial cell in glomerulonephritis. II. Mesangial proliferation caused by Habu snake venom in the rat. Lab Invest 36: 487–492, 1977. [PubMed] [Google Scholar]
- 54. Ziegler V, Fremter K, Helmchen J, Witzgall R, Castrop H. Mesangial cells regulate the single nephron GFR and preserve the integrity of the glomerular filtration barrier: an intravital multiphoton microscopy study. Acta Physiol (Oxf) 231: e13592, 2021. doi: 10.1111/apha.13592. [DOI] [PubMed] [Google Scholar]
- 55. Stockand JD, Sansom SC. Glomerular mesangial cells: electrophysiology and regulation of contraction. Physiol Rev 78: 723–744, 1998. doi: 10.1152/physrev.1998.78.3.723. [DOI] [PubMed] [Google Scholar]
- 56. Blantz RC, Gabbai FB, Tucker BJ, Yamamoto T, Wilson CB. Role of mesangial cell in glomerular response to volume and angiotensin II. Am J Physiol 264: F158–F165, 1993. doi: 10.1152/ajprenal.1993.264.1.F158. [DOI] [PubMed] [Google Scholar]
- 57. Butt L, Unnersjö-Jess D, Höhne M, Edwards A, Binz-Lotter J, Reilly D, Hahnfeldt R, Ziegler V, Fremter K, Rinschen MM, Helmstädter M, Ebert LK, Castrop H, Hackl MJ, Walz G, Brinkkoetter PT, Liebau MC, Tory K, Hoyer PF, Beck BB, Brismar H, Blom H, Schermer B, Benzing T. A molecular mechanism explaining albuminuria in kidney disease. Nat Metab 2: 461–474, 2020. doi: 10.1038/s42255-020-0204-y. [DOI] [PubMed] [Google Scholar]
- 58. Couchman JR, Beavan LA, McCarthy KJ. Glomerular matrix: synthesis, turnover and role in mesangial expansion. Kidney Int 45: 328–335, 1994. doi: 10.1038/ki.1994.42. [DOI] [PubMed] [Google Scholar]
- 59. Schlöndorff D. Roles of the mesangium in glomerular function. Kidney Int 49: 1583–1585, 1996. doi: 10.1038/ki.1996.229. [DOI] [PubMed] [Google Scholar]
- 60. Avraham S, Korin B, Chung JJ, Oxburgh L, Shaw AS. The mesangial cell—the glomerular stromal cell. Nat Rev Nephrol 17: 855–864, 2021. doi: 10.1038/s41581-021-00474-8. [DOI] [PubMed] [Google Scholar]
- 61. Bryniarski MA, Yee BM, Chaves LD, Stahura CM, Yacoub R, Morris ME. Megalin-mediated albumin endocytosis in cultured murine mesangial cells. Biochem Biophys Res Commun 529: 740–746, 2020. doi: 10.1016/j.bbrc.2020.05.166. [DOI] [PubMed] [Google Scholar]
- 62. Cowley AW. Salt intake and the dance of the macula densa cells. Am J Physiol Renal Physiol 320: F375–F377, 2021. doi: 10.1152/ajprenal.00051.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ling BN, Seal EE, Eaton DC. Regulation of mesangial cell ion channels by insulin and angiotensin II. Possible role in diabetic glomerular hyperfiltration. J Clin Invest 92: 2141–2151, 1993. doi: 10.1172/JCI116815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Barrera-Chimal J, Lima-Posada I, Bakris GL, Jaisser F. Mineralocorticoid receptor antagonists in diabetic kidney disease—mechanistic and therapeutic effects. Nat Rev Nephrol 18: 56–70, 2022. doi: 10.1038/s41581-021-00490-8. doi: 10.1038/s41581-021-00490-8. [DOI] [PubMed] [Google Scholar]
- 65. Barton M, Sorokin A. Endothelin and the glomerulus in chronic kidney disease. Semin Nephrol 35: 156–167, 2015. doi: 10.1016/j.semnephrol.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bell PD, Lapointe JY, Sabirov R, Hayashi S, Peti-Peterdi J, Manabe K, Kovacs G, Okada Y. Macula densa cell signaling involves ATP release through a maxi anion channel. Proc Natl Acad Sci USA 100: 4322–4327, 2003. doi: 10.1073/pnas.0736323100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Peti-Peterdi J. Calcium wave of tubuloglomerular feedback. Am J Physiol Renal Physiol 291: F473–F480, 2006. doi: 10.1152/ajprenal.00425.2005. [DOI] [PubMed] [Google Scholar]
- 68. Hugo C, Shankland SJ, Bowen-Pope DF, Couser WG, Johnson RJ. Extraglomerular origin of the mesangial cell after injury. A new role of the juxtaglomerular apparatus. J Clin Invest 100: 786–794, 1997. doi: 10.1172/JCI119592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Rice WL, Van Hoek AN, Păunescu TG, Huynh C, Goetze B, Singh B, Scipioni L, Stern LA, Brown D. High resolution helium ion scanning microscopy of the rat kidney. PLoS One 8: e57051, 2013. doi: 10.1371/journal.pone.0057051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Palygin O, Spires D, Levchenko V, Bohovyk R, Fedoriuk M, Klemens CA, Sykes O, Bukowy JD, Cowley AW Jr, Lazar J, Ilatovskaya DV, Staruschenko A. Progression of diabetic kidney disease in T2DN rats. Am J Physiol Renal Physiol 317: F1450–F1461, 2019. doi: 10.1152/ajprenal.00246.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Neal CR, Crook H, Bell E, Harper SJ, Bates DO. Three-dimensional reconstruction of glomeruli by electron microscopy reveals a distinct restrictive urinary subpodocyte space. J Am Soc Nephrol 16: 1223–1235, 2005. doi: 10.1681/ASN.2004100822. [DOI] [PubMed] [Google Scholar]
- 72. Marshall CB, Shankland SJ. Cell cycle regulatory proteins in podocyte health and disease. Nephron Exp Nephrol 106: e51–e59, 2007. doi: 10.1159/000101793. [DOI] [PubMed] [Google Scholar]
- 73. Shankland SJ, Wang Y, Shaw AS, Vaughan JC, Pippin JW, Wessely O. Podocyte aging: why and how getting old matters. J Am Soc Nephrol 32: 2697–2713, 2021. doi: 10.1681/ASN.2021050614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Suzuki T, Eng DG, McClelland AD, Pippin JW, Shankland SJ. Cells of NG2 lineage increase in glomeruli of mice following podocyte depletion. Am J Physiol Renal Physiol 315: F1449–F1464, 2018. doi: 10.1152/ajprenal.00118.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Shankland SJ, Freedman BS, Pippin JW. Can podocytes be regenerated in adults? Curr Opin Nephrol Hypertens 26: 154–164, 2017. doi: 10.1097/MNH.0000000000000311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wang Y, Eng DG, Kaverina NV, Loretz CJ, Koirala A, Akilesh S, Pippin JW, Shankland SJ. Global transcriptomic changes occur in aged mouse podocytes. Kidney Int 98: 1160–1173, 2020. doi: 10.1016/j.kint.2020.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zhang J, Pippin JW, Vaughan MR, Krofft RD, Taniguchi Y, Romagnani P, Nelson PJ, Liu ZH, Shankland SJ. Retinoids augment the expression of podocyte proteins by glomerular parietal epithelial cells in experimental glomerular disease. Nephron Exp Nephrol 121: e23–e37, 2012. doi: 10.1159/000342808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Hackl MJ, Burford JL, Villanueva K, Lam L, Suszták K, Schermer B, Benzing T, Peti-Peterdi J. Tracking the fate of glomerular epithelial cells in vivo using serial multiphoton imaging in new mouse models with fluorescent lineage tags. Nat Med 19: 1661–1666, 2013. doi: 10.1038/nm.3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Chan GC, Eng DG, Miner JH, Alpers CE, Hudkins K, Chang A, Pippin JW, Shankland SJ. Differential expression of parietal epithelial cell and podocyte extracellular matrix proteins in focal segmental glomerulosclerosis and diabetic nephropathy. Am J Physiol Renal Physiol 317: F1680–F1694, 2019. doi: 10.1152/ajprenal.00266.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Wharram BL, Goyal M, Wiggins JE, Sanden SK, Hussain S, Filipiak WE, Saunders TL, Dysko RC, Kohno K, Holzman LB, Wiggins RC. Podocyte depletion causes glomerulosclerosis: diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J Am Soc Nephrol 16: 2941–2952, 2005. doi: 10.1681/ASN.2005010055. [DOI] [PubMed] [Google Scholar]
- 81. Okabe M, Yamamoto K, Miyazaki Y, Motojima M, Ohtsuka M, Pastan I, Yokoo T, Matsusaka T. Indirect podocyte injury manifested in a partial podocytectomy mouse model. Am J Physiol Renal Physiol 320: F922–F933, 2021. doi: 10.1152/ajprenal.00602.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Chen A, Feng Y, Lai H, Ju W, Li Z, Li Y, Wang A, Hong Q, Zhong F, Wei C, Fu J, Guan T, Liu B, Kretzler M, Lee K, He JC. Soluble RARRES1 induces podocyte apoptosis to promote glomerular disease progression. J Clin Invest 130: 5523–5535, 2020. doi: 10.1172/JCI140155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Yin L, Yu L, He JC, Chen A. Controversies in podocyte loss: death or detachment? Front Cell Dev Biol 9: 771931, 2021. doi: 10.3389/fcell.2021.771931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Vogelmann SU, Nelson WJ, Myers BD, Lemley KV. Urinary excretion of viable podocytes in health and renal disease. Am J Physiol Renal Physiol 285: F40–F48, 2003. doi: 10.1152/ajprenal.00404.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Bondue T, Arcolino FO, Veys KR, Adebayo OC, Levtchenko E, van den Heuvel LP, Elmonem MA. Urine-derived epithelial cells as models for genetic kidney diseases. Cells 10: 1413, 2021. doi: 10.3390/cells10061413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kriz W, Shirato I, Nagata M, LeHir M, Lemley KV. The podocyte’s response to stress: the enigma of foot process effacement. Am J Physiol Renal Physiol 304: F333–F347, 2013. doi: 10.1152/ajprenal.00478.2012. [DOI] [PubMed] [Google Scholar]
- 87. Kriz W, Lemley KV. Mechanical challenges to the glomerular filtration barrier: adaptations and pathway to sclerosis. Pediatr Nephrol 32: 405–417, 2017. doi: 10.1007/s00467-016-3358-9. [DOI] [PubMed] [Google Scholar]
- 88. Ning L, Suleiman HY, Miner JH. Synaptopodin deficiency exacerbates kidney disease in a mouse model of Alport syndrome. Am J Physiol Renal Physiol 321: F12–F25, 2021. doi: 10.1152/ajprenal.00035.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Sever S. Role of actin cytoskeleton in podocytes. Pediatr Nephrol 36: 2607–2614, 2021. doi: 10.1007/s00467-020-04812-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Asanuma K, Kim K, Oh J, Giardino L, Chabanis S, Faul C, Reiser J, Mundel P. Synaptopodin regulates the actin-bundling activity of alpha-actinin in an isoform-specific manner. J Clin Invest 115: 1188–1198, 2005. doi: 10.1172/JCI200523371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Steinhausen M, Endlich K. Controversies on glomerular filtration from Ludwig to the present. Pflugers Arch 432: R73–R81, 1996. [PubMed] [Google Scholar]
- 92. Gilmore JP, Cornish KG, Rogers SD, Joyner WL. Direct evidence for myogenic autoregulation of the renal microcirculation in the hamster. Circ Res 47: 226–230, 1980. doi: 10.1161/01.res.47.2.226. [DOI] [PubMed] [Google Scholar]
- 93. Carlström M, Wilcox CS, Arendshorst WJ. Renal autoregulation in health and disease. Physiol Rev 95: 405–511, 2015. doi: 10.1152/physrev.00042.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Fellner SK, Arendshorst WJ. Angiotensin II-stimulated Ca2+ entry mechanisms in afferent arterioles: role of transient receptor potential canonical channels and reverse Na+/Ca2+ exchange. Am J Physiol Renal Physiol 294: F212–F219, 2008. doi: 10.1152/ajprenal.00244.2007. [DOI] [PubMed] [Google Scholar]
- 95. Osborn JW, Tyshynsky R, Vulchanova L. Function of renal nerves in kidney physiology and pathophysiology. Annu Rev Physiol 83: 429–450, 2021. doi: 10.1146/annurev-physiol-031620-091656. [DOI] [PubMed] [Google Scholar]
- 96. van de Borne P. The kidney and the sympathetic system: a short review. Curr Clin Pharmacol 8: 175–181, 2013. doi: 10.2174/15748847113089990049. [DOI] [PubMed] [Google Scholar]
- 97. Denton KM, Luff SE, Shweta A, Anderson WP. Differential neural control of glomerular ultrafiltration. Clin Exp Pharmacol Physiol 31: 380–386, 2004. doi: 10.1111/j.1440-1681.2004.04002.x. [DOI] [PubMed] [Google Scholar]
- 98. Marsh DJ, Toma I, Sosnovtseva OV, Peti-Peterdi J, Holstein-Rathlou NH. Electrotonic vascular signal conduction and nephron synchronization. Am J Physiol Renal Physiol 296: F751–F761, 2009. doi: 10.1152/ajprenal.90669.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Ren Y, Garvin JL, Liu R, Carretero OA. Crosstalk between the connecting tubule and the afferent arteriole regulates renal microcirculation. Kidney Int 71: 1116–1121, 2007. doi: 10.1038/sj.ki.5002190. [DOI] [PubMed] [Google Scholar]
- 100. Gyarmati G, Shroff UN, Riquier-Brison A, Kriz W, Kaissling B, Neal CR, Arkill KP, Ahmadi N, Gill IS, Moon JY, Desposito D, Peti-Peterdi J. A new view of macula densa cell microanatomy. Am J Physiol Renal Physiol 320: F492–F504, 2021. doi: 10.1152/ajprenal.00546.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Orlov SN, Mongin AA. Salt-sensing mechanisms in blood pressure regulation and hypertension. Am J Physiol Heart Circ Physiol 293: H2039–H2053, 2007. doi: 10.1152/ajpheart.00325.2007. [DOI] [PubMed] [Google Scholar]
- 102. Fowler BC, Chang YS, Laamarti A, Higdon M, Lapointe JY, Bell PD. Evidence for apical sodium proton exchange in macula densa cells. Kidney Int 47: 746–751, 1995. doi: 10.1038/ki.1995.114. [DOI] [PubMed] [Google Scholar]
- 103. Peti-Peterdi J, Chambrey R, Bebok Z, Biemesderfer D, St John PL, Abrahamson DR, Warnock DG, Bell PD. Macula densa Na+/H+ exchange activities mediated by apical NHE2 and basolateral NHE4 isoforms. Am J Physiol Renal Physiol 278: F452–F463, 2000. doi: 10.1152/ajprenal.2000.278.3.F452. [DOI] [PubMed] [Google Scholar]
- 104. Bell PD, Lapointe JY, Peti-Peterdi J. Macula densa cell signaling. Annu Rev Physiol 65: 481–500, 2003. doi: 10.1146/annurev.physiol.65.050102.085730. [DOI] [PubMed] [Google Scholar]
- 105. Castrop H, Huang Y, Hashimoto S, Mizel D, Hansen P, Theilig F, Bachmann S, Deng C, Briggs J, Schnermann J. Impairment of tubuloglomerular feedback regulation of GFR in ecto-5'-nucleotidase/CD73-deficient mice. J Clin Invest 114: 634–642, 2004. doi: 10.1172/JCI21851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Ren Y, Garvin JL, Liu R, Carretero OA. Role of macula densa adenosine triphosphate (ATP) in tubuloglomerular feedback. Kidney Int 66: 1479–1485, 2004. doi: 10.1111/j.1523-1755.2004.00911.x. [DOI] [PubMed] [Google Scholar]
- 107. Sun D, Samuelson LC, Yang T, Huang Y, Paliege A, Saunders T, Briggs J, Schnermann J. Mediation of tubuloglomerular feedback by adenosine: evidence from mice lacking adenosine 1 receptors. Proc Natl Acad Sci USA 98: 9983–9988, 2001. doi: 10.1073/pnas.171317998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Vallon V, Unwin R, Inscho EW, Leipziger J, Kishore BK. Extracellular nucleotides and P2 receptors in renal function. Physiol Rev 100: 211–269, 2020. doi: 10.1152/physrev.00038.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Inscho EW, Cook AK, Imig JD, Vial C, Evans RJ. Physiological role for P2X1 receptors in renal microvascular autoregulatory behavior. J Clin Invest 112: 1895–1905, 2003. doi: 10.1172/JCI18499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Hanner F, von Maltzahn J, Maxeiner S, Toma I, Sipos A, Krüger O, Willecke K, Peti-Peterdi J. Connexin45 is expressed in the juxtaglomerular apparatus and is involved in the regulation of renin secretion and blood pressure. Am J Physiol Regul Integr Comp Physiol 295: R371–R380, 2008. doi: 10.1152/ajpregu.00468.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Burford JL, Villanueva K, Lam L, Riquier-Brison A, Hackl MJ, Pippin J, Shankland SJ, Peti-Peterdi J. Intravital imaging of podocyte calcium in glomerular injury and disease. J Clin Invest 124: 2050–2058, 2014. doi: 10.1172/JCI71702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Ren Y, Carretero OA, Garvin JL. Role of mesangial cells and gap junctions in tubuloglomerular feedback. Kidney Int 62: 525–531, 2002. doi: 10.1046/j.1523-1755.2002.00454.x. [DOI] [PubMed] [Google Scholar]
- 113. Iversen BM, Kvam FI, Matre K, Mørkrid L, Horvei G, Bagchus W, Grond J, Ofstad J. Effect of mesangiolysis on autoregulation of renal blood flow and glomerular filtration rate in rats. Am J Physiol Renal Physiol 262: F361–F366, 1992. doi: 10.1152/ajprenal.1992.262.3.F361. [DOI] [PubMed] [Google Scholar]
- 114. Wang H, D’Ambrosio MA, Ren Y, Monu SR, Leung P, Kutskill K, Garvin JL, Janic B, Peterson EL, Carretero OA. Tubuloglomerular and connecting tubuloglomerular feedback during inhibition of various Na transporters in the nephron. Am J Physiol Renal Physiol 308: F1026–F1031, 2015. doi: 10.1152/ajprenal.00605.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Ren Y, D’Ambrosio MA, Garvin JL, Wang H, Carretero OA. Possible mediators of connecting tubule glomerular feedback. Hypertension 53: 319–323, 2009. doi: 10.1161/HYPERTENSIONAHA.108.124545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Wang H, D’Ambrosio MA, Garvin JL, Ren Y, Carretero OA. Connecting tubule glomerular feedback mediates acute tubuloglomerular feedback resetting. Am J Physiol Renal Physiol 302: F1300–F1304, 2012. doi: 10.1152/ajprenal.00673.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Eremina V, Jefferson JA, Kowalewska J, Hochster H, Haas M, Weisstuch J, Richardson C, Kopp JB, Kabir MG, Backx PH, Gerber HP, Ferrara N, Barisoni L, Alpers CE, Quaggin SE. VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med 358: 1129–1136, 2008. doi: 10.1056/NEJMoa0707330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Simon M, Gröne HJ, Jöhren O, Kullmer J, Plate KH, Risau W, Fuchs E. Expression of vascular endothelial growth factor and its receptors in human renal ontogenesis and in adult kidney. Am J Physiol Renal Physiol 268: F240–F250, 1995. doi: 10.1152/ajprenal.1995.268.2.F240. [DOI] [PubMed] [Google Scholar]
- 119. Eremina V, Sood M, Haigh J, Nagy A, Lajoie G, Ferrara N, Gerber HP, Kikkawa Y, Miner JH, Quaggin SE. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J Clin Invest 111: 707–716, 2003. doi: 10.1172/JCI17423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Dimke H, Maezawa Y, Quaggin SE. Crosstalk in glomerular injury and repair. Curr Opin Nephrol Hypertens 24: 231–238, 2015. doi: 10.1097/MNH.0000000000000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Mizuno M, Nozaki M, Morine N, Suzuki N, Nishikawa K, Morgan BP, Matsuo S. A protein toxin from the sea anemone Phyllodiscus semoni targets the kidney and causes a severe renal injury with predominant glomerular endothelial damage. Am J Pathol 171: 402–414, 2007. doi: 10.2353/ajpath.2007.060984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Morita T, Yamamoto T, Churg J. Mesangiolysis: an update. Am J Kidney Dis 31: 559–573, 1998. doi: 10.1053/ajkd.1998.v31.pm9531171. [DOI] [PubMed] [Google Scholar]
- 123. Jiang S, Luo M, Bai X, Nie P, Zhu Y, Cai H, Li B, Luo P. Cellular crosstalk of glomerular endothelial cells and podocytes in diabetic kidney disease. J Cell Commun Signal 16: 313–331, 2022. doi: 10.1007/s12079-021-00664-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Gil CL, Hooker E, Larrivée B. diabetic kidney disease, endothelial damage, and podocyte-endothelial crosstalk. Kidney Med 3: 105–115, 2021. doi: 10.1016/j.xkme.2020.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Fu J, Lee K, Chuang PY, Liu Z, He JC. Glomerular endothelial cell injury and cross talk in diabetic kidney disease. Am J Physiol Renal Physiol 308: F287–F297, 2015. doi: 10.1152/ajprenal.00533.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Wei Y, Gao X, Li A, Liang M, Jiang Z. Single-nucleus transcriptomic analysis reveals important cell cross-talk in diabetic kidney disease. Front Med (Lausanne) 8: 657956, 2021. doi: 10.3389/fmed.2021.657956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Thomas MC. Targeting the pathobiology of diabetic kidney disease. Adv Chronic Kidney Dis 28: 282–289, 2021. doi: 10.1053/j.ackd.2021.07.001. [DOI] [PubMed] [Google Scholar]
- 128. Ebefors K, Bergwall L, Nyström J. The glomerulus according to the mesangium. Front Med 8: 740527, 2021. doi: 10.3389/fmed.2021.740527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Li Q, Gulati A, Lemaire M, Nottoli T, Bale A, Tufro A. Rho-GTPase activating protein myosin MYO9A identified as a novel candidate gene for monogenic focal segmental glomerulosclerosis. Kidney Int 99: 1102–1117, 2021. doi: 10.1016/j.kint.2020.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Sun YB, Qu X, Zhang X, Caruana G, Bertram JF, Li J. Glomerular endothelial cell injury and damage precedes that of podocytes in adriamycin-induced nephropathy. PLoS One 8: e55027, 2013. doi: 10.1371/journal.pone.0055027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Banas B, Wörnle M, Berger T, Nelson PJ, Cohen CD, Kretzler M, Pfirstinger J, Mack M, Lipp M, Gröne HJ, Schlöndorff D. Roles of SLC/CCL21 and CCR7 in human kidney for mesangial proliferation, migration, apoptosis, and tissue homeostasis. J Immunol 168: 4301–4307, 2002. doi: 10.4049/jimmunol.168.9.4301. [DOI] [PubMed] [Google Scholar]
- 132. Bartlett CS, Jeansson M, Quaggin SE. Vascular growth factors and glomerular disease. Annu Rev Physiol 78: 437–461, 2016. doi: 10.1146/annurev-physiol-021115-105412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Harvey TW, Engel JE, Chade AR. Vascular endothelial growth factor and podocyte protection in chronic hypoxia: effects of endothelin-A receptor antagonism. Am J Nephrol 43: 74–84, 2016. doi: 10.1159/000444719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Veron D, Aggarwal PK, Li Q, Moeckel G, Kashgarian M, Tufro A. Podocyte VEGF-A knockdown induces diffuse glomerulosclerosis in diabetic and in eNOS knockout mice. Front Pharmacol 12: 788886, 2021. doi: 10.3389/fphar.2021.788886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Thomas S, Vanuystel J, Gruden G, Rodríguez V, Burt D, Gnudi L, Hartley B, Viberti G. Vascular endothelial growth factor receptors in human mesangium in vitro and in glomerular disease. J Am Soc Nephrol 11: 1236–1243, 2000. doi: 10.1681/ASN.V1171236. [DOI] [PubMed] [Google Scholar]
- 136. Sison K, Eremina V, Baelde H, Min W, Hirashima M, Fantus IG, Quaggin SE. Glomerular structure and function require paracrine, not autocrine, VEGF-VEGFR-2 signaling. J Am Soc Nephrol 21: 1691–1701, 2010. doi: 10.1681/ASN.2010030295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Suyama M, Miyazaki Y, Matsusaka T, Sugano N, Ueda H, Kawamura T, Ogura M, Yokoo T. Forced expression of vascular endothelial growth factor-A in podocytes decreases mesangial cell numbers and attenuates endothelial cell differentiation in the mouse glomerulus. Clin Exp Nephrol 22: 266–274, 2018. doi: 10.1007/s10157-017-1450-5. [DOI] [PubMed] [Google Scholar]
- 138. Salmon AH, Neal CR, Bates DO, Harper SJ. Vascular endothelial growth factor increases the ultrafiltration coefficient in isolated intact Wistar rat glomeruli. J Physiol 570: 141–156, 2006. doi: 10.1113/jphysiol.2005.099184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Qiu Y, Ferguson J, Oltean S, Neal CR, Kaura A, Bevan H, Wood E, Sage LM, Lanati S, Nowak DG, Salmon AH, Bates D, Harper SJ. Overexpression of VEGF165b in podocytes reduces glomerular permeability. J Am Soc Nephrol 21: 1498–1509, 2010. doi: 10.1681/ASN.2009060617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Eremina V, Cui S, Gerber H, Ferrara N, Haigh J, Nagy A, Ema M, Rossant J, Jothy S, Miner JH, Quaggin SE. Vascular endothelial growth factor a signaling in the podocyte-endothelial compartment is required for mesangial cell migration and survival. J Am Soc Nephrol 17: 724–735, 2006. doi: 10.1681/ASN.2005080810. [DOI] [PubMed] [Google Scholar]
- 141. Eng E, Holgren C, Hubchak S, Naaz P, Schnaper HW. Hypoxia regulates PDGF-B interactions between glomerular capillary endothelial and mesangial cells. Kidney Int 68: 695–703, 2005. doi: 10.1111/j.1523-1755.2005.00448.x. [DOI] [PubMed] [Google Scholar]
- 142. Zimmerman SE, Hiremath C, Tsunezumi J, Yang Z, Finney B, Marciano DK. Nephronectin regulates mesangial cell adhesion and behavior in glomeruli. J Am Soc Nephrol 29: 1128–1140, 2018. doi: 10.1681/ASN.2017070752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Müller-Deile J, Sopel N, Ohs A, Rose V, Gröner M, Wrede C, Hegermann J, Daniel C, Amann K, Zahner G, Schiffer M. Glomerular endothelial cell-derived microRNA-192 regulates nephronectin expression in idiopathic membranous glomerulonephritis. J Am Soc Nephrol 32: 2777–2794, 2021. doi: 10.1681/ASN.2020121699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Datta PK, Lianos EA. Nitric oxide induces metallothionein-I gene expression in mesangial cells. Transl Res 148: 180–187, 2006. doi: 10.1016/j.trsl.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 145. Palygin O, Ilatovskaya DV, Levchenko V, Endres BT, Geurts AM, Staruschenko A. Nitric oxide production by glomerular podocytes. Nitric Oxide 72: 24–31, 2018. doi: 10.1016/j.niox.2017.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Beck KF, Pfeilschifter J. The pathophysiology of H2S in renal glomerular diseases. Biomolecules 12: 207, 2022. doi: 10.3390/biom12020207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Beck KF, Pfeilschifter J. Gasotransmitter synthesis and signalling in the renal glomerulus. Implications for glomerular diseases. Cell Signal 77: 109823, 2021. doi: 10.1016/j.cellsig.2020.109823. [DOI] [PubMed] [Google Scholar]
- 148. Ma Y, Li W, Shotorbani PY, Dubansky BH, Huang L, Chaudhari S, Wu P, Wang LA, Ryou MG, Zhou Z, and Ma R. Comparison of diabetic nephropathy between male and female eNOS−/− db/db mice. Am J Physiol Renal Physiol 316: F889–F897, 2019. doi: 10.1152/ajprenal.00023.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Fu J, Akat KM, Sun Z, Zhang W, Schlondorff D, Liu Z, Tuschl T, Lee K, He JC. Single-cell RNA profiling of glomerular cells shows dynamic changes in experimental diabetic kidney disease. J Am Soc Nephrol 30: 533–545, 2019. doi: 10.1681/ASN.2018090896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Bargman JM, Skorecki K. Chronic kidney disease. In: Harrison's Principles of Internal Medicine, edited by Kasper D, Fauci A, Hauser S, Longo D, Jameson JL, Loscalzo J.. New York: McGraw-Hill Education, 2014. [Google Scholar]
- 151. Kopp JB, Anders HJ, Susztak K, Podestà MA, Remuzzi G, Hildebrandt F, Romagnani P. Podocytopathies. Nat Rev Dis Primers 6: 68, 2020. doi: 10.1038/s41572-020-0196-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Sethi S, Fervenza FC. Standardized classification and reporting of glomerulonephritis. Nephrol Dial Transplant 34: 193–199, 2019. doi: 10.1093/ndt/gfy220. [DOI] [PubMed] [Google Scholar]
- 153. Soni H, Kumar R, Kanthakumar P, Adebiyi A. Interleukin 1 beta-induced calcium signaling via TRPA1 channels promotes mitogen-activated protein kinase-dependent mesangial cell proliferation. FASEB J 35: e21729, 2021. doi: 10.1096/fj.202100367R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Ma R, Pluznick JL, Sansom SC. Ion channels in mesangial cells: function, malfunction, or fiction. Physiology (Bethesda) 20: 102–111, 2005. doi: 10.1152/physiol.00050.2004. [DOI] [PubMed] [Google Scholar]
- 155. Zamponi GW, Striessnig J, Koschak A, Dolphin AC. The physiology, pathology, and pharmacology of voltage-gated calcium channels and their future therapeutic potential. Pharmacol Rev 67: 821–870, 2015. doi: 10.1124/pr.114.009654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Takeda K, Meyer-Lehnert H, Kim JK, Schrier RW. Effect of angiotensin II on Ca2+ kinetics and contraction in cultured rat glomerular mesangial cells. Am J Physiol Renal Physiol 254: F254–F266, 1988. doi: 10.1152/ajprenal.1988.254.2.F254. [DOI] [PubMed] [Google Scholar]
- 157. Yu YM, Lermioglu F, Hassid A. Modulation of Ca by agents affecting voltage-sensitive Ca channels in mesangial cells. Am J Physiol Renal Physiol 257: F1094–F1099, 1989. doi: 10.1152/ajprenal.1989.257.6.F1094. [DOI] [PubMed] [Google Scholar]
- 158. Nishio M, Tsukahara H, Hiraoka M, Sudo M, Kigoshi S, Muramatsu I. Calcium channel current in cultured rat mesangial cells. Mol Pharmacol 43: 96–99, 1993. [PubMed] [Google Scholar]
- 159. Hall DA, Carmines PK, Sansom SC. Dihydropyridine-sensitive Ca2+ channels in human glomerular mesangial cells. Am J Physiol Renal Physiol 278: F97–F103, 2000. doi: 10.1152/ajprenal.2000.278.1.F97. [DOI] [PubMed] [Google Scholar]
- 160. Mulgrew CJ, Cove-Smith A, McLatchie LM, Brooks G, Shattock MJ, Hendry BM. Inhibition of human mesangial cell proliferation by targeting T-type calcium channels. Nephron Exp Nephrol 113: e77–e88, 2009. doi: 10.1159/000232590. [DOI] [PubMed] [Google Scholar]
- 161. Ling BN. Regulation of mesangial chloride channels by insulin and glucose: role in diabetic nephropathy. Clin Exp Pharmacol Physiol 23: 89–94, 1996. doi: 10.1111/j.1440-1681.1996.tb03068.x. [DOI] [PubMed] [Google Scholar]
- 162. Ono T, Liu N, Kusano H, Nogaki F, Makino T, Muso E, Sasayama S. Broad antiproliferative effects of benidipine on cultured human mesangial cells in cell cycle phases. Am J Nephrol 22: 581–586, 2002. doi: 10.1159/000065266. [DOI] [PubMed] [Google Scholar]
- 163. Sugiura T, Imai E, Moriyama T, Horio M, Hori M. Calcium channel blockers inhibit proliferation and matrix production in rat mesangial cells: possible mechanism of suppression of AP-1 and CREB activities. Nephron 85: 71–80, 2000. doi: 10.1159/000045633. [DOI] [PubMed] [Google Scholar]
- 164. Sugano N, Wakino S, Kanda T, Tatematsu S, Homma K, Yoshioka K, Hasegawa K, Hara Y, Suetsugu Y, Yoshizawa T, Hara Y, Utsunomiya Y, Tokudome G, Hosoya T, Saruta T, Hayashi K. T-type calcium channel blockade as a therapeutic strategy against renal injury in rats with subtotal nephrectomy. Kidney Int 73: 826–834, 2008. doi: 10.1038/sj.ki.5002793. [DOI] [PubMed] [Google Scholar]
- 165. Cove-Smith A, Mulgrew CJ, Rudyk O, Dutt N, McLatchie LM, Shattock MJ, Hendry BM. Anti-proliferative actions of T-type calcium channel inhibition in Thy1 nephritis. Am J Pathol 183: 391–401, 2013. doi: 10.1016/j.ajpath.2013.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Cove-Smith A, Sharpe CC, Shattock MJ, Hendry BM. Ion-Channel modulator TH1177 reduces glomerular injury and serum creatinine in chronic mesangial proliferative disease in rats. BMC Nephrol 21: 187, 2020. doi: 10.1186/s12882-020-01842-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Latorre R, Castillo K, Carrasquel-Ursulaez W, Sepulveda RV, Gonzalez-Nilo F, Gonzalez C, Alvarez O. Molecular determinants of BK channel functional diversity and functioning. Physiol Rev 97: 39–87, 2017. doi: 10.1152/physrev.00001.2016. [DOI] [PubMed] [Google Scholar]
- 168. Tseng-Crank J, Foster CD, Krause JD, Mertz R, Godinot N, DiChiara TJ, Reinhart PH. Cloning, expression, and distribution of functionally distinct Ca2+-activated K+ channel isoforms from human brain. Neuron 13: 1315–1330, 1994. doi: 10.1016/0896-6273(94)90418-9. [DOI] [PubMed] [Google Scholar]
- 169. McCobb DP, Fowler NL, Featherstone T, Lingle CJ, Saito M, Krause JE, Salkoff L. A human calcium-activated potassium channel gene expressed in vascular smooth muscle. Am J Physiol Heart Circ Physiol 269: H767–H777, 1995. doi: 10.1152/ajpheart.1995.269.3.H767. [DOI] [PubMed] [Google Scholar]
- 170. Tao X, Hite RK, MacKinnon R. Cryo-EM structure of the open high-conductance Ca2+-activated K+) channel. Nature 541: 46–51, 2017. doi: 10.1038/nature20608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Tian L, Duncan RR, Hammond MS, Coghill LS, Wen H, Rusinova R, Clark AG, Levitan IB, Shipston MJ. Alternative splicing switches potassium channel sensitivity to protein phosphorylation. J Biol Chem 276: 7717–7720, 2001. doi: 10.1074/jbc.C000741200. [DOI] [PubMed] [Google Scholar]
- 172. Kim EY, Ridgway LD, Zou S, Chiu YH, Dryer SE. Alternatively spliced C-terminal domains regulate the surface expression of large conductance calcium-activated potassium channels. Neuroscience 146: 1652–1661, 2007. doi: 10.1016/j.neuroscience.2007.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Chiu YH, Alvarez-Baron C, Kim EY, Dryer SE. Dominant-negative regulation of cell surface expression by a pentapeptide motif at the extreme COOH terminus of an Slo1 calcium-activated potassium channel splice variant. Mol Pharmacol 77: 497–507, 2010. doi: 10.1124/mol.109.061929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Brenner R, Peréz GJ, Bonev AD, Eckman DM, Kosek JC, Wiler SW, Patterson AJ, Nelson MT, Aldrich RW. Vasoregulation by the beta1 subunit of the calcium-activated potassium channel. Nature 407: 870–876, 2000. doi: 10.1038/35038011. [DOI] [PubMed] [Google Scholar]
- 175. Wang YW, Ding JP, Xia XM, Lingle CJ. Consequences of the stoichiometry of Slo1 alpha and auxiliary beta subunits on functional properties of large-conductance Ca2+-activated K+ channels. J Neurosci 22: 1550–1561, 2002. doi: 10.1523/JNEUROSCI.22-05-01550.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Tao X, MacKinnon R. Molecular structures of the human Slo1 K+ channel in complex with β4. Elife 8: e51409, 2019. doi: 10.7554/eLife.51409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Grimm PR, Foutz RM, Brenner R, Sansom SC. Identification and localization of BK-beta subunits in the distal nephron of the mouse kidney. Am J Physiol Renal Physiol 293: F350–F359, 2007. doi: 10.1152/ajprenal.00018.2007. [DOI] [PubMed] [Google Scholar]
- 178. Li Q, Fan F, Kwak HR, Yan J. Molecular basis for differential modulation of BK channel voltage-dependent gating by auxiliary γ subunits. J Gen Physiol 145: 543–554, 2015. doi: 10.1085/jgp.201511356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Gonzalez-Perez V, Lingle CJ. Regulation of BK channels by beta and gamma subunits. Annu Rev Physiol 81: 113–137, 2019. doi: 10.1146/annurev-physiol-022516-034038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Zou S, Jha S, Kim EY, Dryer SE. The beta 1 subunit of L-type voltage-gated Ca2+ channels independently binds to and inhibits the gating of large-conductance Ca2+-activated K+ channels. Mol Pharmacol 73: 369–378, 2008. doi: 10.1124/mol.107.040733. [DOI] [PubMed] [Google Scholar]
- 181. Jha S, Dryer SE. The beta1 subunit of Na+/K+-ATPase interacts with BKCa channels and affects their steady-state expression on the cell surface. FEBS Lett 583: 3109–3114, 2009. doi: 10.1016/j.febslet.2009.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Blatz AL, Magleby KL. Ion conductance and selectivity of single calcium-activated potassium channels in cultured rat muscle. J Gen Physiol 84: 1–23, 1984. doi: 10.1085/jgp.84.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Yellen G. Ionic permeation and blockade in Ca2+-activated K+ channels of bovine chromaffin cells. J Gen Physiol 84: 157–186, 1984. doi: 10.1085/jgp.84.2.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Miranda P, Holmgren M, Giraldez T. Voltage-dependent dynamics of the BK channel cytosolic gating ring are coupled to the membrane-embedded voltage sensor. Elife 7: e40664, 2018. doi: 10.7554/eLife.40664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Horrigan FT, Aldrich RW. Coupling between voltage sensor activation, Ca2+ binding and channel opening in large conductance (BK) potassium channels. J Gen Physiol 120: 267–305, 2002. doi: 10.1085/jgp.20028605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Welling PA, Ho K. A comprehensive guide to the ROMK potassium channel: form and function in health and disease. Am J Physiol Renal Physiol 297: F849–F863, 2009. doi: 10.1152/ajprenal.00181.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Torres YP, Granados ST, Latorre R. Pharmacological consequences of the coexpression of BK channel α and auxiliary β subunits. Front Physiol 5: 383, 2014. doi: 10.3389/fphys.2014.00383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Abbott GW. Control of biophysical and pharmacological properties of potassium channels by ancillary subunits. Handb Exp Pharmacol 267: 445–480, 2021. doi: 10.1007/164_2021_512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Meera P, Wallner M, Toro L. A neuronal beta subunit (KCNMB4) makes the large conductance, voltage- and Ca2+-activated K+ channel resistant to charybdotoxin and iberiotoxin. Proc Natl Acad Sci USA 97: 5562–5567, 2000. doi: 10.1073/pnas.100118597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Irie T, Trussell LO. Double-nanodomain coupling of calcium channels, ryanodine receptors, and BK Channels controls the generation of burst firing. Neuron 96: 856–870.e4, 2017. doi: 10.1016/j.neuron.2017.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Vivas O, Moreno CM, Santana LF, Hille B. Proximal clustering between BK and CaV1.3 channels promotes functional coupling and BK channel activation at low voltage. Elife 6: e28029, 2017. doi: 10.7554/eLife.28029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Robitaille R, Garcia ML, Kaczorowski GJ, Charlton MP. Functional colocalization of calcium and calcium-gated potassium channels in control of transmitter release. Neuron 11: 645–655, 1993. doi: 10.1016/0896-6273(93)90076-4. [DOI] [PubMed] [Google Scholar]
- 193. Pérez GJ, Bonev AD, Nelson MT. Micromolar Ca2+ from sparks activates Ca2+-sensitive K+ channels in rat cerebral artery smooth muscle. Am J Physiol Cell Physiol 281: C1769–C1775, 2001. doi: 10.1152/ajpcell.2001.281.6.C1769. [DOI] [PubMed] [Google Scholar]
- 194. Prakriya M, Lingle CJ. Activation of BK channels in rat chromaffin cells requires summation of Ca2+ influx from multiple Ca2+ channels. J Neurophysiol 84: 1123–1135, 2000. doi: 10.1152/jn.2000.84.3.1123. [DOI] [PubMed] [Google Scholar]
- 195. Grimm PR, Sansom SC. BK channels in the kidney. Curr Opin Nephrol Hypertens 16: 430–436, 2007. doi: 10.1097/MNH.0b013e32826fbc7d. [DOI] [PubMed] [Google Scholar]
- 196. Stockand JD, Sansom SC. Large Ca2+-activated K+ channels responsive to angiotensin II in cultured human mesangial cells. Am J Physiol Cell Physiol 267: C1080–C1086, 1994. doi: 10.1152/ajpcell.1994.267.4.C1080. [DOI] [PubMed] [Google Scholar]
- 197. Kudlacek PE, Pluznick JL, Ma R, Padanilam B, Sansom SC. Role of hbeta1 in activation of human mesangial BK channels by cGMP kinase. Am J Physiol Renal Physiol 285: F289–F294, 2003. doi: 10.1152/ajprenal.00046.2003. [DOI] [PubMed] [Google Scholar]
- 198. Sansom SC, Stockand JD. Physiological role of large, Ca2+-activated K+ channels in human glomerular mesangial cells. Clin Exp Pharmacol Physiol 23: 76–82, 1996. doi: 10.1111/j.1440-1681.1996.tb03066.x. [DOI] [PubMed] [Google Scholar]
- 199. Stockand JD, Sansom SC. Role of large Ca2+-activated K+ channels in regulation of mesangial contraction by nitroprusside and ANP. Am J Physiol Cell Physiol 270: C1773–C1779, 1996. doi: 10.1152/ajpcell.1996.270.6.C1773. [DOI] [PubMed] [Google Scholar]
- 200. Stockand JD, Sansom SC. Mechanism of activation by cGMP-dependent protein kinase of large Ca2+-activated K+ channels in mesangial cells. Am J Physiol Cell Physiol 271: C1669–C1677, 1996. doi: 10.1152/ajpcell.1996.271.5.C1669. [DOI] [PubMed] [Google Scholar]
- 201. Sansom SC, Stockand JD, Hall D, Williams B. Regulation of large calcium-activated potassium channels by protein phosphatase 2A. J Biol Chem 272: 9902–9906, 1997. doi: 10.1074/jbc.272.15.9902. [DOI] [PubMed] [Google Scholar]
- 202. Foutz RM, Grimm PR, Sansom SC. Insulin increases the activity of mesangial BK channels through MAPK signaling. Am J Physiol Renal Physiol 294: F1465–F1472, 2008. doi: 10.1152/ajprenal.00012.2008. [DOI] [PubMed] [Google Scholar]
- 203. Alioua A, Tanaka Y, Wallner M, Hofmann F, Ruth P, Meera P, Toro L. The large conductance, voltage-dependent, and calcium-sensitive K+ channel, Hslo, is a target of cGMP-dependent protein kinase phosphorylation in vivo. J Biol Chem 273: 32950–32956, 1998. doi: 10.1074/jbc.273.49.32950. [DOI] [PubMed] [Google Scholar]
- 204. Nara M, Dhulipala PD, Ji GJ, Kamasani UR, Wang YX, Matalon S, Kotlikoff MI. Guanylyl cyclase stimulatory coupling to KCa channels. Am J Physiol Cell Physiol 279: C1938–C1945, 2000. doi: 10.1152/ajpcell.2000.279.6.C1938. [DOI] [PubMed] [Google Scholar]
- 205. Pluznick JL, Wei P, Carmines PK, Sansom SC. Renal fluid and electrolyte handling in BKCa-beta1-/- mice. Am J Physiol Renal Physiol 284: F1274–F1279, 2003. doi: 10.1152/ajprenal.00010.2003. [DOI] [PubMed] [Google Scholar]
- 206. Wu Z, Yin W, Sun M, Si Y, Wu X, Chen M. BKCa mediates dysfunction in high glucose induced mesangial cell injury via TGF-β1/Smad2/3 signaling pathways. Int J Endocrinol 2020: 3260728, 2020. doi: 10.1155/2020/3260728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Zhou M, He HJ, Suzuki R, Tanaka O, Sekiguchi M, Yasuoka Y, Kawahara K, Itoh H, Abe H. Expression of ATP sensitive K+ channel subunit Kir6.1 in rat kidney. Eur J Histochem 51: 43–51, 2007. [PubMed] [Google Scholar]
- 208. Barber RD, Woolf AS, Henderson RM. Potassium conductances and proliferation in conditionally immortalized renal glomerular mesangial cells from the H-2Kb-tsA58 transgenic mouse. Biochim Biophys Acta 1355: 191–203, 1997. doi: 10.1016/s0167-4889(96)00124-3. [DOI] [PubMed] [Google Scholar]
- 209. Zhang B, Shi YQ, Zou JJ, Chen XF, Tang W, Ye F, Liu ZM. High glucose stimulates cell proliferation and Collagen IV production in rat mesangial cells through inhibiting AMPK-KATP signaling. Int Urol Nephrol 49: 2079–2086, 2017. doi: 10.1007/s11255-017-1654-3. [DOI] [PubMed] [Google Scholar]
- 210. Cipleu CD, Palant CE, Sanders KM, Dick GM. Separation of two Cl- currents in cultured human and murine mesangial cells: biophysical and pharmacological characteristics of ICl.vol and ICl.Ca. J Vasc Res 39: 426–436, 2002. doi: 10.1159/000064516. [DOI] [PubMed] [Google Scholar]
- 211. Mallis L, Guber H, Adler SG, Palant CE. Intracellular chloride activity in cultured mesangial cells. Ren Physiol Biochem 14: 12–18, 1991. doi: 10.1159/000173383. [DOI] [PubMed] [Google Scholar]
- 212. Miyata Y, Muto S, Yanagiba S, Asano Y. Extracellular Cl- modulates shrinkage-induced activation of Na+/H+ exchanger in rat mesangial cells. Am J Physiol Cell Physiol 278: C1218–C1229, 2000. doi: 10.1152/ajpcell.2000.278.6.C1218. [DOI] [PubMed] [Google Scholar]
- 213. Jiao JD, Xu CQ, Yue P, Dong DL, Li Z, Du ZM, Yang BF. Volume-sensitive outwardly rectifying chloride channels are involved in oxidative stress-induced apoptosis of mesangial cells. Biochem Biophys Res Commun 340: 277–285, 2006. doi: 10.1016/j.bbrc.2005.11.175. [DOI] [PubMed] [Google Scholar]
- 214. Chen X, Sooch G, Demaree IS, White FA, Obukhov AG. Transient receptor potential canonical (TRPC) channels: then and now. Cells 9: 1983, 2020. doi: 10.3390/cells9091983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Bodnar D, Chung WY, Yang D, Hong JH, Jha A, Muallem S. STIM-TRP pathways and microdomain organization: Ca2+ influx channels: the Orai-STIM1-TRPC complexes. Adv Exp Med Biol 993: 139–157, 2017. doi: 10.1007/978-3-319-57732-6_8. [DOI] [PubMed] [Google Scholar]
- 216. Yuan JP, Zeng W, Huang GN, Worley PF, Muallem S. STIM1 heteromultimerizes TRPC channels to determine their function as store-operated channels. Nat Cell Biol 9: 636–645, 2007. doi: 10.1038/ncb1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Shalygin A, Kolesnikov D, Glushankova L, Gusev K, Skopin A, Skobeleva K, Kaznacheyeva EV. Role of STIM2 and Orai proteins in regulating TRPC1 channel activity upon calcium store depletion. Cell Calcium 97: 102432, 2021. doi: 10.1016/j.ceca.2021.102432. [DOI] [PubMed] [Google Scholar]
- 218. Thakur DP, Wang Q, Jeon J, Tian JB, Zhu MX. Intracellular acidification facilitates receptor-operated TRPC4 activation through PLCδ1 in a Ca2+-dependent manner. J Physiol 598: 2651–2667, 2020. doi: 10.1113/JP279658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Gudermann T, Mederos y Schnitzler M, Dietrich A. Receptor-operated cation entry–more than esoteric terminology? Sci STKE 2004: pe35, 2004. doi: 10.1126/stke.2432004pe35. [DOI] [PubMed] [Google Scholar]
- 220. Graham S, Ding M, Ding Y, Sours-Brothers S, Luchowski R, Gryczynski Z, Yorio T, Ma H, Ma R. Canonical transient receptor potential 6 (TRPC6), a redox-regulated cation channel. J Biol Chem 285: 23466–23476, 2010. doi: 10.1074/jbc.M109.093500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Xu SZ, Sukumar P, Zeng F, Li J, Jairaman A, English A, Naylor J, Ciurtin C, Majeed Y, Milligan CJ, Bahnasi YM, Al-Shawaf E, Porter KE, Jiang LH, Emery P, Sivaprasadarao A, Beech DJ. TRPC channel activation by extracellular thioredoxin. Nature 451: 69–72, 2008. doi: 10.1038/nature06414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Poteser M, Graziani A, Rosker C, Eder P, Derler I, Kahr H, Zhu MX, Romanin C, Groschner K. TRPC3 and TRPC4 associate to form a redox-sensitive cation channel. Evidence for expression of native TRPC3-TRPC4 heteromeric channels in endothelial cells. J Biol Chem 281: 13588–13595, 2006. doi: 10.1074/jbc.M512205200. [DOI] [PubMed] [Google Scholar]
- 223. Spassova MA, Hewavitharana T, Xu W, Soboloff J, Gill DL. A common mechanism underlies stretch activation and receptor activation of TRPC6 channels. Proc Natl Acad Sci USA 103: 16586–16591, 2006. doi: 10.1073/pnas.0606894103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Inoue R, Jensen LJ, Jian Z, Shi J, Hai L, Lurie AI, Henriksen FH, Salomonsson M, Morita H, Kawarabayashi Y, Mori M, Mori Y, Ito Y. Synergistic activation of vascular TRPC6 channel by receptor and mechanical stimulation via phospholipase C/diacylglycerol and phospholipase A2/omega-hydroxylase/20-HETE pathways. Circ Res 104: 1399–1409, 2009. doi: 10.1161/CIRCRESAHA.108.193227. [DOI] [PubMed] [Google Scholar]
- 225. Khayyat NH, Tomilin VN, Zaika O, Pochynyuk O. Polymodal roles of TRPC3 channel in the kidney. Channels (Austin) 14: 257–267, 2020. doi: 10.1080/19336950.2020.1804153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Anderson M, Kim EY, Hagmann H, Benzing T, Dryer SE. Opposing effects of podocin on the gating of podocyte TRPC6 channels evoked by membrane stretch or diacylglycerol. Am J Physiol Cell Physiol 305: C276–C289, 2013. doi: 10.1152/ajpcell.00095.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Dryer SE, Kim EY. Permeation and rectification in canonical transient receptor potential-6 (TRPC6) channels. Front Physiol 9: 1055, 2018. doi: 10.3389/fphys.2018.01055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Estacion M, Sinkins WG, Jones SW, Applegate MA, Schilling WP. Human TRPC6 expressed in HEK 293 cells forms non-selective cation channels with limited Ca2+ permeability. J Physiol 572: 359–377, 2006. doi: 10.1113/jphysiol.2005.103143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Gao YY, Tian W, Zhang HN, Sun Y, Meng JR, Cao W, Li XQ. Canonical transient receptor potential channels and their modulators: biology, pharmacology and therapeutic potentials. Arch Pharm Res 44: 354–377, 2021. doi: 10.1007/s12272-021-01319-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Hofmann T, Schaefer M, Schultz G, Gudermann T. Subunit composition of mammalian transient receptor potential channels in living cells. Proc Natl Acad Sci USA 99: 7461–7466, 2002. doi: 10.1073/pnas.102596199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Goel M, Sinkins WG, Zuo CD, Estacion M, Schilling WP. Identification and localization of TRPC channels in the rat kidney. Am J Physiol Renal Physiol 290: F1241–F1252, 2006. doi: 10.1152/ajprenal.00376.2005. [DOI] [PubMed] [Google Scholar]
- 232. Wang X, Pluznick JL, Wei P, Padanilam BJ, Sansom SC. TRPC4 forms store-operated Ca2+ channels in mouse mesangial cells. Am J Physiol Cell Physiol 287: C357–C364, 2004. doi: 10.1152/ajpcell.00068.2004. [DOI] [PubMed] [Google Scholar]
- 233. Sours S, Du J, Chu S, Ding M, Zhou XJ, Ma R. Expression of canonical transient receptor potential (TRPC) proteins in human glomerular mesangial cells. Am J Physiol Renal Physiol 290: F1507–F1515, 2006. doi: 10.1152/ajprenal.00268.2005. [DOI] [PubMed] [Google Scholar]
- 234. Sours-Brothers S, Ding M, Graham S, Ma R. Interaction between TRPC1/TRPC4 assembly and STIM1 contributes to store-operated Ca2+ entry in mesangial cells. Exp Biol Med (Maywood) 234: 673–682, 2009. doi: 10.3181/0809-RM-279. [DOI] [PubMed] [Google Scholar]
- 235. Meng K, Xu J, Zhang C, Zhang R, Yang H, Liao C, Jiao J. Calcium sensing receptor modulates extracellular calcium entry and proliferation via TRPC3/6 channels in cultured human mesangial cells. PLoS One 9: e98777, 2014. doi: 10.1371/journal.pone.0098777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. He F, Peng F, Xia X, Zhao C, Luo Q, Guan W, Li Z, Yu X, Huang F. MiR-135a promotes renal fibrosis in diabetic nephropathy by regulating TRPC1. Diabetologia 57: 1726–1736, 2014. doi: 10.1007/s00125-014-3282-0. [DOI] [PubMed] [Google Scholar]
- 237. Zhang N, Ji Z. Effects of caveolin-1 and P-ERK1/2 on Ang II-induced glomerular mesangial cell proliferation. Ren Fail 35: 971–977, 2013. doi: 10.3109/0886022X.2013.808956. [DOI] [PubMed] [Google Scholar]
- 238. Liao C, Yang H, Zhang R, Sun H, Zhao B, Gao C, Zhu F, Jiao J. The upregulation of TRPC6 contributes to Ca2+ signaling and actin assembly in human mesangial cells after chronic hypoxia. Biochem Biophys Res Commun 421: 750–756, 2012. doi: 10.1016/j.bbrc.2012.04.075. [DOI] [PubMed] [Google Scholar]
- 239. Qiu G, Ji Z. AngII-induced glomerular mesangial cell proliferation inhibited by losartan via changes in intracellular calcium ion concentration. Clin Exp Med 14: 169–176, 2014. doi: 10.1007/s10238-013-0232-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Kong F, Ma L, Zou L, Meng K, Ji T, Zhang L, Zhang R, Jiao J. Alpha1-adrenergic receptor activation stimulates calcium entry and proliferation via TRPC6 channels in cultured human mesangial cells. Cell Physiol Biochem 36: 1928–1938, 2015. doi: 10.1159/000430161. [DOI] [PubMed] [Google Scholar]
- 241. Soni H, Adebiyi A. TRPC6 channel activation promotes neonatal glomerular mesangial cell apoptosis via calcineurin/NFAT and FasL/Fas signaling pathways. Sci Rep 6: 29041, 2016. doi: 10.1038/srep29041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Mené P, Simonson MS, Dunn MJ. Physiology of the mesangial cell. Physiol Rev 69: 1347–1424, 1989. doi: 10.1152/physrev.1989.69.4.1347. [DOI] [PubMed] [Google Scholar]
- 243. Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 397: 259–263, 1999. doi: 10.1038/16711. [DOI] [PubMed] [Google Scholar]
- 244. Du J, Sours-Brothers S, Coleman R, Ding M, Graham S, Kong DH, Ma R. Canonical transient receptor potential 1 channel is involved in contractile function of glomerular mesangial cells. J Am Soc Nephrol 18: 1437–1445, 2007. doi: 10.1681/ASN.2006091067. [DOI] [PubMed] [Google Scholar]
- 245. Graham S, Ding M, Sours-Brothers S, Yorio T, Ma JX, Ma R. Downregulation of TRPC6 protein expression by high glucose, a possible mechanism for the impaired Ca2+ signaling in glomerular mesangial cells in diabetes. Am J Physiol Renal Physiol 293: F1381–F1390, 2007. doi: 10.1152/ajprenal.00185.2007. [DOI] [PubMed] [Google Scholar]
- 246. Graham S, Gorin Y, Abboud HE, Ding M, Lee DY, Shi H, Ding Y, Ma R. Abundance of TRPC6 protein in glomerular mesangial cells is decreased by ROS and PKC in diabetes. Am J Physiol Cell Physiol 301: C304–C315, 2011. doi: 10.1152/ajpcell.00014.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Li W, Ding Y, Smedley C, Wang Y, Chaudhari S, Birnbaumer L, Ma R. Increased glomerular filtration rate and impaired contractile function of mesangial cells in TRPC6 knockout mice. Sci Rep 7: 4145, 2017. doi: 10.1038/s41598-017-04067-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Zuckerman JE, Gale A, Wu P, Ma R, Davis ME. siRNA delivery to the glomerular mesangium using polycationic cyclodextrin nanoparticles containing siRNA. Nucleic Acid Ther 25: 53–64, 2015. doi: 10.1089/nat.2014.0505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Zuckerman JE, Davis ME. Targeting therapeutics to the glomerulus with nanoparticles. Adv Chronic Kidney Dis 20: 500–507, 2013. doi: 10.1053/j.ackd.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 250. Chaudhari S, Li W, Wang Y, Jiang H, Ma Y, Davis ME, Zuckerman JE, and Ma R. Store-operated calcium entry suppressed the TGF-β1/Smad3 signaling pathway in glomerular mesangial cells. Am J Physiol Renal Physiol 313: F729–F739, 2017. doi: 10.1152/ajprenal.00483.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Du J, Ding M, Sours-Brothers S, Graham S, Ma R. Mediation of angiotensin II-induced Ca2+ signaling by polycystin 2 in glomerular mesangial cells. Am J Physiol Renal Physiol 294: F909–F918, 2008. doi: 10.1152/ajprenal.00606.2007. [DOI] [PubMed] [Google Scholar]
- 252. Emrich SM, Yoast RE, Trebak M. Physiological functions of CRAC channels. Annu Rev Physiol 84: 355–379, 2022. doi: 10.1146/annurev-physiol-052521-013426. [DOI] [PubMed] [Google Scholar]
- 253. Chaudhari S, Mallet RT, Shotorbani PY, Tao Y, Ma R. Store-operated calcium entry: pivotal roles in renal physiology and pathophysiology. Exp Biol Med (Maywood) 246: 305–316, 2021. doi: 10.1177/1535370220975207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Lewis RS. Store-operated calcium channels: from function to structure and back again. Cold Spring Harb Perspect Biol 12: a035055, 2020. doi: 10.1101/cshperspect.a035055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Hoth M, Penner R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature 355: 353–356, 1992. doi: 10.1038/355353a0. [DOI] [PubMed] [Google Scholar]
- 256. Trepakova ES, Gericke M, Hirakawa Y, Weisbrod RM, Cohen RA, Bolotina VM. Properties of a native cation channel activated by Ca2+ store depletion in vascular smooth muscle cells. J Biol Chem 276: 7782–7790, 2001. doi: 10.1074/jbc.M010104200. [DOI] [PubMed] [Google Scholar]
- 257. Ma R, Smith S, Child A, Carmines PK, Sansom SC. Store-operated Ca2+ channels in human glomerular mesangial cells. Am J Physiol Renal Physiol 278: F954–F961, 2000. doi: 10.1152/ajprenal.2000.278.6.F954. [DOI] [PubMed] [Google Scholar]
- 258. Bugaj V, Alexeenko V, Zubov A, Glushankova L, Nikolaev A, Wang Z, Kaznacheyeva E, Bezprozvanny I, Mozhayeva GN. Functional properties of endogenous receptor- and store-operated calcium influx channels in HEK293 cells. J Biol Chem 280: 16790–16797, 2005. doi: 10.1074/jbc.M500192200. [DOI] [PubMed] [Google Scholar]
- 259. Hofer AM, Fasolato C, Pozzan T. Capacitative Ca2+ entry is closely linked to the filling state of internal Ca2+ stores: a study using simultaneous measurements of ICRAC and intraluminal [Ca2+]. J Cell Biol 140: 325–334, 1998. doi: 10.1083/jcb.140.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Randriamampita C, Tsien RY. Emptying of intracellular Ca2+ stores releases a novel small messenger that stimulates Ca2+ influx. Nature 364: 809–814, 1993. doi: 10.1038/364809a0. [DOI] [PubMed] [Google Scholar]
- 261. Fasolato C, Hoth M, Penner R. A GTP-dependent step in the activation mechanism of capacitative calcium influx. J Biol Chem 268: 20737–20740, 1993. doi: 10.1016/S0021-9258(19)36843-7. [DOI] [PubMed] [Google Scholar]
- 262. Irvine RF. ‘Quantal’ Ca2+ release and the control of Ca2+ entry by inositol phosphates—a possible mechanism. FEBS Lett 263: 5–9, 1990. doi: 10.1016/0014-5793(90)80692-C. [DOI] [PubMed] [Google Scholar]
- 263. Rossi AM, Riley AM, Dupont G, Rahman T, Potter BV, Taylor CW. Quantal Ca2+ release mediated by very few IP3 receptors that rapidly inactivate allows graded responses to IP3. Cell Rep 37: 109932, 2021. doi: 10.1016/j.celrep.2021.109932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Liu X, Bandyopadhyay BC, Singh BB, Groschner K, Ambudkar IS. Molecular analysis of a store-operated and 2-acetyl-sn-glycerol-sensitive non-selective cation channel. Heteromeric assembly of TRPC1-TRPC3. J Biol Chem 280: 21600–21606, 2005. doi: 10.1074/jbc.C400492200. [DOI] [PubMed] [Google Scholar]
- 265. Ma R, Rundle D, Jacks J, Koch M, Downs T, Tsiokas L. Inhibitor of myogenic family, a novel suppressor of store-operated currents through an interaction with TRPC1. J Biol Chem 278: 52763–52772, 2003. doi: 10.1074/jbc.M309610200. [DOI] [PubMed] [Google Scholar]
- 266. Lopez JJ, Jardin I, Sanchez-Collado J, Salido GM, Smani T, Rosado JA. TRPC channels in the SOCE scenario. Cells 9: 126, 2020. doi: 10.3390/cells9010126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, Kozak JA, Wagner SL, Cahalan MD, Veliçelebi G, Stauderman KA. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol 169: 435–445, 2005. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, Stauderman KA, Cahalan MD. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 437: 902–905, 2005. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Prakriya M, Feske S, Gwack Y, Srikanth S, Rao A, Hogan PG. Orai1 is an essential pore subunit of the CRAC channel. Nature 443: 230–233, 2006. doi: 10.1038/nature05122. [DOI] [PubMed] [Google Scholar]
- 270. Li Y, Yang X, Shen Y. Structural insights into Ca2+ permeation through Orai channels. Cells 10: 3062, 2021. doi: 10.3390/cells10113062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Wang Y, Deng X, Gill DL. Calcium signaling by STIM and Orai: intimate coupling details revealed. Sci Signal 3: pe42, 2010. doi: 10.1126/scisignal.3148pe42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. Soboloff J, Spassova MA, Hewavitharana T, He LP, Xu W, Johnstone LS, Dziadek MA, Gill DL. STIM2 is an inhibitor of STIM1-mediated store-operated Ca2+ entry. Curr Biol 16: 1465–1470, 2006. doi: 10.1016/j.cub.2006.05.051. [DOI] [PubMed] [Google Scholar]
- 273. DeHaven WI, Smyth JT, Boyles RR, Putney JW Jr.. Calcium inhibition and calcium potentiation of Orai1, Orai2, and Orai3 calcium release-activated calcium channels. J Biol Chem 282: 17548–17556, 2007. doi: 10.1074/jbc.M611374200. [DOI] [PubMed] [Google Scholar]
- 274. Yoast RE, Emrich SM, Zhang X, Xin P, Johnson MT, Fike AJ, Walter V, Hempel N, Yule DI, Sneyd J, Gill DL, Trebak M. The native ORAI channel trio underlies the diversity of Ca2+ signaling events. Nat Commun 11: 2444, 2020. doi: 10.1038/s41467-020-16232-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Fukushima M, Tomita T, Janoshazi A, Putney JW. Alternative translation initiation gives rise to two isoforms of Orai1 with distinct plasma membrane mobilities. J Cell Sci 125: 4354–4361, 2012. doi: 10.1242/jcs.104919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Desai PN, Zhang X, Wu S, Janoshazi A, Bolimuntha S, Putney JW, Trebak M. Multiple types of calcium channels arising from alternative translation initiation of the Orai1 message. Sci Signal 8: ra74, 2015. doi: 10.1126/scisignal.aaa8323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277. Saüc S, Bulla M, Nunes P, Orci L, Marchetti A, Antigny F, Bernheim L, Cosson P, Frieden M, Demaurex N. STIM1L traps and gates Orai1 channels without remodeling the cortical ER. J Cell Sci 128: 1568–1579, 2015. doi: 10.1242/jcs.164228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278. Darbellay B, Arnaudeau S, Bader CR, Konig S, Bernheim L. STIM1L is a new actin-binding splice variant involved in fast repetitive Ca2+ release. J Cell Biol 194: 335–346, 2011. doi: 10.1083/jcb.201012157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279. Kim MS, Zeng W, Yuan JP, Shin DM, Worley PF, Muallem S. Native store-operated Ca2+ influx requires the channel function of Orai1 and TRPC1. J Biol Chem 284: 9733–9741, 2009. doi: 10.1074/jbc.M808097200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Ambudkar IS, de Souza LB, Ong HL. TRPC1, Orai1, and STIM1 in SOCE: Friends in tight spaces. Cell Calcium 63: 33–39, 2017. doi: 10.1016/j.ceca.2016.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281. Liao Y, Erxleben C, Abramowitz J, Flockerzi V, Zhu MX, Armstrong DL, Birnbaumer L. Functional interactions among Orai1, TRPCs, and STIM1 suggest a STIM-regulated heteromeric Orai/TRPC model for SOCE/Icrac channels. Proc Natl Acad Sci USA 105: 2895–2900, 2008. doi: 10.1073/pnas.0712288105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Menè P, Teti A, Pugliese F, Cinotti GA. Calcium release-activated calcium influx in cultured human mesangial cells. Kidney Int 46: 122–128, 1994. doi: 10.1038/ki.1994.251. [DOI] [PubMed] [Google Scholar]
- 283. Ma R, Sansom SC. Epidermal growth factor activates store-operated calcium channels in human glomerular mesangial cells. J Am Soc Nephrol 12: 47–53, 2001. doi: 10.1681/ASN.V12147. [DOI] [PubMed] [Google Scholar]
- 284. Nutt LK, O’Neil RG. Effect of elevated glucose on endothelin-induced store-operated and non-store-operated calcium influx in renal mesangial cells. J Am Soc Nephrol 11: 1225–1235, 2000. doi: 10.1681/ASN.V1171225. [DOI] [PubMed] [Google Scholar]
- 285. Ma R, Kudlacek PE, Sansom SC. Protein kinase Calpha participates in activation of store-operated Ca2+ channels in human glomerular mesangial cells. Am J Physiol Cell Physiol 283: C1390–C1398, 2002. doi: 10.1152/ajpcell.00141.2002. [DOI] [PubMed] [Google Scholar]
- 286. Lückhoff A, Clapham DE. Calcium channels activated by depletion of internal calcium stores in A431 cells. Biophys J 67: 177–182, 1994. doi: 10.1016/S0006-3495(94)80467-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287. Kaznacheyeva E, Zubov A, Gusev K, Bezprozvanny I, Mozhayeva GN. Activation of calcium entry in human carcinoma A431 cells by store depletion and phospholipase C-dependent mechanisms converge on ICRAC-like calcium channels. Proc Natl Acad Sci USA 98: 148–153, 2001. doi: 10.1073/pnas.98.1.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288. Li WP, Tsiokas L, Sansom SC, Ma R. Epidermal growth factor activates store-operated Ca2+ channels through an inositol 1,4,5-trisphosphate-independent pathway in human glomerular mesangial cells. J Biol Chem 279: 4570–4577, 2004. doi: 10.1074/jbc.M304334200. [DOI] [PubMed] [Google Scholar]
- 289. Parekh AB, Putney JW Jr.. Store-operated calcium channels. Physiol Rev 85: 757–810, 2005. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- 290. Shen B, Zhu J, Zhang J, Jiang F, Wang Z, Zhang Y, Li J, Huang D, Ke D, Ma R, Du J. Attenuated mesangial cell proliferation related to store-operated Ca2+ entry in aged rat: the role of STIM 1 and Orai 1. Age (Dordr) 35: 2193–2202, 2013. doi: 10.1007/s11357-013-9511-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Chaudhari S, Wu P, Wang Y, Ding Y, Yuan J, Begg M, Ma R. High glucose and diabetes enhanced store-operated Ca2+ entry and increased expression of its signaling proteins in mesangial cells. Am J Physiol Renal Physiol 306: F1069–F1080, 2014. doi: 10.1152/ajprenal.00463.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292. Ma R, Pluznick J, Kudlacek P, Sansom SC. Protein kinase C activates store-operated Ca2+ channels in human glomerular mesangial cells. J Biol Chem 276: 25759–25765, 2001. doi: 10.1074/jbc.M011241200. [DOI] [PubMed] [Google Scholar]
- 293. Ahmmed GU, Mehta D, Vogel S, Holinstat M, Paria BC, Tiruppathi C, Malik AB. Protein kinase Calpha phosphorylates the TRPC1 channel and regulates store-operated Ca2+ entry in endothelial cells. J Biol Chem 279: 20941–20949, 2004. doi: 10.1074/jbc.M313975200. [DOI] [PubMed] [Google Scholar]
- 294. Menè P, Pugliese F, Cinotti GA. Regulation of capacitative calcium influx in cultured human mesangial cells: roles of protein kinase C and calmodulin. J Am Soc Nephrol 7: 983–990, 1996. doi: 10.1681/ASN.V77983. [DOI] [PubMed] [Google Scholar]
- 295. Kawasaki T, Ueyama T, Lange I, Feske S, Saito N. Protein kinase C-induced phosphorylation of Orai1 regulates the intracellular Ca2+ level via the store-operated Ca2+ channel. J Biol Chem 285: 25720–25730, 2010. doi: 10.1074/jbc.M109.022996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296. Wang X, Pluznick JL, Settles DC, Sansom SC. Association of VASP with TRPC4 in PKG-mediated inhibition of the store-operated calcium response in mesangial cells. Am J Physiol Renal Physiol 293: F1768–F1776, 2007. doi: 10.1152/ajprenal.00365.2007. [DOI] [PubMed] [Google Scholar]
- 297. Burnstock G, Evans LC, Bailey MA. Purinergic signalling in the kidney in health and disease. Purinergic Signal 10: 71–101, 2014. doi: 10.1007/s11302-013-9400-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298. Staruschenko A, Adams E, Booth RE, Stockand JD. Epithelial Na+ channel subunit stoichiometry. Biophys J 88: 3966–3975, 2005. doi: 10.1529/biophysj.104.056804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299. Noreng S, Bharadwaj A, Posert R, Yoshioka C, Baconguis I. Structure of the human epithelial sodium channel by cryo-electron microscopy. Elife 7: e39340, 2018. doi: 10.7554/eLife.39340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300. North RA. Molecular physiology of P2X receptors. Physiol Rev 82: 1013–1067, 2002. doi: 10.1152/physrev.00015.2002. [DOI] [PubMed] [Google Scholar]
- 301. Palygin O, Ilatovskaya DV, Levchenko V, Klemens CA, Dissanayake L, Williams AM, Pavlov TS, Staruschenko A. Characterization of purinergic receptor expression in ARPKD cystic epithelia. Purinergic Signal 14: 485–497, 2018. doi: 10.1007/s11302-018-9632-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302. Palygin O, Evans LC, Cowley AW Jr, Staruschenko A. Acute in vivo analysis of ATP release in rat kidneys in response to changes of renal perfusion pressure. J Am Heart Assoc 6: e006658, 2017. doi: 10.1161/JAHA.117.006658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303. Harkat M, Peverini L, Cerdan AH, Dunning K, Beudez J, Martz A, Calimet N, Specht A, Cecchini M, Chataigneau T, Grutter T. On the permeation of large organic cations through the pore of ATP-gated P2X receptors. Proc Natl Acad Sci USA 114: E3786–E3795, 2017. doi: 10.1073/pnas.1701379114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304. Li Q, Luo X, Muallem S. Regulation of the P2X7 receptor permeability to large molecules by extracellular Cl- and Na+. J Biol Chem 280: 26922–26927, 2005. doi: 10.1074/jbc.M504966200. [DOI] [PubMed] [Google Scholar]
- 305. Vallon V, Stockand J, Rieg T. P2Y receptors and kidney function. Wiley Interdiscip Rev Membr Transp Signal 1: 731–742, 2012. doi: 10.1002/wmts.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306. Bailey MA, Turner CM, Hus-Citharel A, Marchetti J, Imbert-Teboul M, Milner P, Burnstock G, Unwin RJ. P2Y receptors present in the native and isolated rat glomerulus. Nephron Physiol 96: 79–90, 2004. doi: 10.1159/000076753. [DOI] [PubMed] [Google Scholar]
- 307. Gutierrez AM, Lou X, Erik A, Persson G, Ring A. Ca2+ response of rat mesangial cells to ATP analogues. Eur J Pharmacol 369: 107–112, 1999. doi: 10.1016/s0014-2999(99)00032-1. [DOI] [PubMed] [Google Scholar]
- 308. Solini A, Iacobini C, Ricci C, Chiozzi P, Amadio L, Pricci F, Di Mario U, Di Virgilio F, Pugliese G. Purinergic modulation of mesangial extracellular matrix production: role in diabetic and other glomerular diseases. Kidney Int 67: 875–885, 2005. doi: 10.1111/j.1523-1755.2005.00152.x. [DOI] [PubMed] [Google Scholar]
- 309. Rivera I, Zhang S, Fuller BS, Edwards B, Seki T, Wang MH, Marrero MB, Inscho EW. P2 receptor regulation of [Ca2+]i in cultured mouse mesangial cells. Am J Physiol Renal Physiol 292: F1380–F1389, 2007. doi: 10.1152/ajprenal.00349.2006. [DOI] [PubMed] [Google Scholar]
- 310. Harada H, Chan CM, Loesch A, Unwin R, Burnstock G. Induction of proliferation and apoptotic cell death via P2Y and P2X receptors, respectively, in rat glomerular mesangial cells. Kidney Int 57: 949–958, 2000. doi: 10.1046/j.1523-1755.2000.00911.x. [DOI] [PubMed] [Google Scholar]
- 311. Dalghi MG, Clayton DR, Ruiz WG, Al-Bataineh MM, Satlin LM, Kleyman TR, Ricke WA, Carattino MD, Apodaca G. Expression and distribution of PIEZO1 in the mouse urinary tract. Am J Physiol Renal Physiol 317: F303–F321, 2019. doi: 10.1152/ajprenal.00214.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312. Mochida Y, Ochiai K, Nagase T, Nonomura K, Akimoto Y, Fukuhara H, Sakai T, Matsumura G, Yamaguchi Y, Nagase M. Piezo2 expression and its alteration by mechanical forces in mouse mesangial cells and renin-producing cells. Sci Rep 12: 4197, 2022. doi: 10.1038/s41598-022-07987-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313. Welsh DG, Morielli AD, Nelson MT, Brayden JE. Transient receptor potential channels regulate myogenic tone of resistance arteries. Circ Res 90: 248–250, 2002. doi: 10.1161/hh0302.105662. [DOI] [PubMed] [Google Scholar]
- 314. Roshanravan H, Dryer SE. ATP acting through P2Y receptors causes activation of podocyte TRPC6 channels: role of podocin and reactive oxygen species. Am J Physiol Renal Physiol 306: F1088–F1097, 2014. doi: 10.1152/ajprenal.00661.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315. Anderson M, Roshanravan H, Khine J, Dryer SE. Angiotensin II activation of TRPC6 channels in rat podocytes requires generation of reactive oxygen species. J Cell Physiol 229: 434–442, 2014. doi: 10.1002/jcp.24461. [DOI] [PubMed] [Google Scholar]
- 316. Ilatovskaya DV, Blass G, Palygin O, Levchenko V, Pavlov TS, Grzybowski MN, Winsor K, Shuyskiy LS, Geurts AM, Cowley AW Jr, Birnbaumer L, Staruschenko A. A NOX4/TRPC6 pathway in podocyte calcium regulation and renal damage in diabetic kidney disease. J Am Soc Nephrol 29: 1917–1927, 2018. doi: 10.1681/ASN.2018030280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317. Nikolaev YA, Cox CD, Ridone P, Rohde PR, Cordero-Morales JF, Vásquez V, Laver DR, Martinac B. Mammalian TRP ion channels are insensitive to membrane stretch. J Cell Sci 132: jcs238360, 2019. doi: 10.1242/jcs.238360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318. Martinac B. The ion channels to cytoskeleton connection as potential mechanism of mechanosensitivity. Biochim Biophys Acta 1838: 682–691, 2014. doi: 10.1016/j.bbamem.2013.07.015. [DOI] [PubMed] [Google Scholar]
- 319. Lei L, Lu S, Wang Y, Kim T, Mehta D, Wang Y. The role of mechanical tension on lipid raft dependent PDGF-induced TRPC6 activation. Biomaterials 35: 2868–2877, 2014. doi: 10.1016/j.biomaterials.2013.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320. Kim EY, Anderson M, Wilson C, Hagmann H, Benzing T, Dryer SE. NOX2 interacts with podocyte TRPC6 channels and contributes to their activation by diacylglycerol: essential role of podocin in formation of this complex. Am J Physiol Cell Physiol 305: C960–C971, 2013. doi: 10.1152/ajpcell.00191.2013. [DOI] [PubMed] [Google Scholar]
- 321. Kim EY, Roshanravan H, Dryer SE. Changes in podocyte TRPC channels evoked by plasma and sera from patients with recurrent FSGS and by putative glomerular permeability factors. Biochim Biophys Acta Mol Basis Dis 1863: 2342–2354, 2017. doi: 10.1016/j.bbadis.2017.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322. Ilatovskaya DV, Palygin O, Levchenko V, Endres BT, Staruschenko A. The role of angiotensin II in glomerular volume dynamics and podocyte calcium handling. Sci Rep 7: 299, 2017. doi: 10.1038/s41598-017-00406-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323. Kim EY, Suh JM, Chiu YH, Dryer SE. Regulation of podocyte BKCa channels by synaptopodin, Rho, and actin microfilaments. Am J Physiol Renal Physiol 299: F594–F604, 2010. doi: 10.1152/ajprenal.00206.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324. Morton MJ, Hutchinson K, Mathieson PW, Witherden IR, Saleem MA, Hunter M. Human podocytes possess a stretch-sensitive, Ca2+-activated K+ channel: potential implications for the control of glomerular filtration. J Am Soc Nephrol 15: 2981–2987, 2004. doi: 10.1097/01.ASN.0000145046.24268.0D. [DOI] [PubMed] [Google Scholar]
- 325. Kim EY, Choi KJ, Dryer SE. Nephrin binds to the COOH terminus of a large-conductance Ca2+-activated K+ channel isoform and regulates its expression on the cell surface. Am J Physiol Renal Physiol 295: F235–F246, 2008. doi: 10.1152/ajprenal.00140.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326. Kim EY, Chiu YH, Dryer SE. Neph1 regulates steady-state surface expression of Slo1 Ca2+-activated K+ channels: different effects in embryonic neurons and podocytes. Am J Physiol Cell Physiol 297: C1379–C1388, 2009. doi: 10.1152/ajpcell.00354.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327. Ridgway LD, Kim EY, Dryer SE. MAGI-1 interacts with Slo1 channel proteins and suppresses Slo1 expression on the cell surface. Am J Physiol Cell Physiol 297: C55–C65, 2009. doi: 10.1152/ajpcell.00073.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328. Kim EY, Alvarez-Baron CP, Dryer SE. Canonical transient receptor potential channel (TRPC)3 and TRPC6 associate with large-conductance Ca2+-activated K+ (BKCa) channels: role in BKCa trafficking to the surface of cultured podocytes. Mol Pharmacol 75: 466–477, 2009. doi: 10.1124/mol.108.051912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329. Piao L, Ho WK, Earm YE. Actin filaments regulate the stretch sensitivity of large-conductance, Ca2+-activated K+ channels in coronary artery smooth muscle cells. Pflugers Arch 446: 523–528, 2003. doi: 10.1007/s00424-003-1079-y. [DOI] [PubMed] [Google Scholar]
- 330. Dopico AM, Bukiya AN, Jaggar JH. Calcium- and voltage-gated BK channels in vascular smooth muscle. Pflugers Arch 470: 1271–1289, 2018. doi: 10.1007/s00424-018-2151-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331. Piwkowska A, Rogacka D, Audzeyenka I, Kasztan M, Angielski S, Jankowski M. Insulin increases glomerular filtration barrier permeability through PKGIα-dependent mobilization of BKCa channels in cultured rat podocytes. Biochim Biophys Acta 1852: 1599–1609, 2015. doi: 10.1016/j.bbadis.2015.04.024. [DOI] [PubMed] [Google Scholar]
- 332. Hu S, Han R, Chen L, Qin W, Xu X, Shi J, Zhu X, Zhang M, Zeng C, Tang Z, Bao H, Liu Z. Upregulated LRRC55 promotes BK channel activation and aggravates cell injury in podocytes. J Exp Med 218: e20192373, 2021. doi: 10.1084/jem.20192373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333. Behrens R, Nolting A, Reimann F, Schwarz M, Waldschütz R, Pongs O. hKCNMB3 and hKCNMB4, cloning and characterization of two members of the large-conductance calcium-activated potassium channel beta subunit family. FEBS Lett 474: 99–106, 2000. doi: 10.1016/s0014-5793(00)01584-2. [DOI] [PubMed] [Google Scholar]
- 334. Brenner R, Jegla TJ, Wickenden A, Liu Y, Aldrich RW. Cloning and functional characterization of novel large conductance calcium-activated potassium channel beta subunits, hKCNMB3 and hKCNMB4. J Biol Chem 275: 6453–6461, 2000. doi: 10.1074/jbc.275.9.6453. [DOI] [PubMed] [Google Scholar]
- 335. Weiger TM, Holmqvist MH, Levitan IB, Clark FT, Sprague S, Huang WJ, Ge P, Wang C, Lawson D, Jurman ME, Glucksmann MA, Silos-Santiago I, DiStefano PS, Curtis R. A novel nervous system beta subunit that downregulates human large conductance calcium-dependent potassium channels. J Neurosci 20: 3563–3570, 2000. doi: 10.1523/JNEUROSCI.20-10-03563.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336. Ma D, Nakata T, Zhang G, Hoshi T, Li M, Shikano S. Differential trafficking of carboxyl isoforms of Ca2+-gated (Slo1) potassium channels. FEBS Lett 581: 1000–1008, 2007. doi: 10.1016/j.febslet.2007.01.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337. Zou S, Jha S, Kim EY, Dryer SE. A novel actin-binding domain on Slo1 calcium-activated potassium channels is necessary for their expression in the plasma membrane. Mol Pharmacol 73: 359–368, 2008. doi: 10.1124/mol.107.039743. [DOI] [PubMed] [Google Scholar]
- 338. Dryer SE, Lhuillier L, Cameron JS, Martin-Caraballo M. Expression of KCa channels in identified populations of developing vertebrate neurons: role of neurotrophic factors and activity. J Physiol Paris 97: 49–58, 2003. doi: 10.1016/j.jphysparis.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 339. Chae KS, Martin-Caraballo M, Anderson M, Dryer SE. Akt activation is necessary for growth factor-induced trafficking of functional KCa channels in developing parasympathetic neurons. J Neurophysiol 93: 1174–1182, 2005. doi: 10.1152/jn.00796.2004. [DOI] [PubMed] [Google Scholar]
- 340. Kim EY, Dryer SE. Effects of insulin and high glucose on mobilization of slo1 BKCa channels in podocytes. J Cell Physiol 226: 2307–2315, 2011. doi: 10.1002/jcp.22567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341. Kim EY, Anderson M, Dryer SE. Insulin increases surface expression of TRPC6 channels in podocytes: Role of NADPH oxidases and reactive oxygen species. Am J Physiol Renal Physiol 302: F298–F307, 2012. doi: 10.1152/ajprenal.00423.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342. Xia S, Liu Y, Li X, Thilo F, Tepel M. Insulin increases expression of TRPC6 channels in podocytes by a calcineurin-dependent pathway. Cell Physiol Biochem 38: 659–669, 2016. doi: 10.1159/000438658. [DOI] [PubMed] [Google Scholar]
- 343. Liu BC, Song X, Lu XY, Li DT, Eaton DC, Shen BZ, Li XQ, Ma HP. High glucose induces podocyte apoptosis by stimulating TRPC6 via elevation of reactive oxygen species. Biochim Biophys Acta 1833: 1434–1442, 2013. doi: 10.1016/j.bbamcr.2013.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344. Lu XY, Liu BC, Cao YZ, Song C, Su H, Chen G, Klein JD, Zhang HX, Wang LH, Ma HP. High glucose reduces expression of podocin in cultured human podocytes by stimulating TRPC6. Am J Physiol Renal Physiol 317: F1605–F1611, 2019. doi: 10.1152/ajprenal.00215.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345. Ilatovskaya DV, Palygin O, Chubinskiy-Nadezhdin V, Negulyaev YA, Ma R, Birnbaumer L, Staruschenko A. Angiotensin II has acute effects on TRPC6 channels in podocytes of freshly isolated glomeruli. Kidney Int 86: 506–514, 2014. doi: 10.1038/ki.2014.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346. Kim EY, Hassanzadeh Khayyat N, Dryer SE. Mechanisms underlying modulation of podocyte TRPC6 channels by suPAR: Role of NADPH oxidases and Src family tyrosine kinases. Biochim Biophys Acta Mol Basis Dis 1864: 3527–3536, 2018. doi: 10.1016/j.bbadis.2018.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347. Sah P. Ca2+ activated K+ currents in neurones: types, physiological roles and modulation. Trends Neurosci 19: 150–154, 1996. doi: 10.1016/S0166-2236(96)80026-9. [DOI] [PubMed] [Google Scholar]
- 348. Wu RS, Marx SO. The BK potassium channel in the vascular smooth muscle and kidney: α- and β-subunits. Kidney Int 78: 963–974, 2010. doi: 10.1038/ki.2010.325. [DOI] [PubMed] [Google Scholar]
- 349. Dryer SE, Reiser J. TRPC6 channels and their binding partners in podocytes: role in glomerular filtration and pathophysiology. Am J Physiol Renal Physiol 299: F689–F701, 2010. doi: 10.1152/ajprenal.00298.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350. Yan J, Aldrich RW. BK potassium channel modulation by leucine-rich repeat-containing proteins. Proc Natl Acad Sci USA 109: 7917–7922, 2012. doi: 10.1073/pnas.1205435109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351. Xu S, Nam SM, Kim JH, Das R, Choi SK, Nguyen TT, Quan X, Choi SJ, Chung CH, Lee EY, Lee IK, Wiederkehr A, Wollheim CB, Cha SK, Park KS. Palmitate induces ER calcium depletion and apoptosis in mouse podocytes subsequent to mitochondrial oxidative stress. Cell Death Dis 6: e1976, 2015. doi: 10.1038/cddis.2015.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352. Miao L, Wei D, Zhang Y, Liu J, Lu S, Zhang A, Huang S. Effects of stromal interaction molecule 1 or Orai1 overexpression on the associated proteins and permeability of podocytes. Nephrology (Carlton) 21: 959–967, 2016. doi: 10.1111/nep.12691. [DOI] [PubMed] [Google Scholar]
- 353. Kim JH, Hwang KH, Dang BT, Eom M, Kong ID, Gwack Y, Yu S, Gee HY, Birnbaumer L, Park KS, Cha SK. Insulin-activated store-operated Ca2+ entry via Orai1 induces podocyte actin remodeling and causes proteinuria. Nat Commun 12: 6537, 2021. doi: 10.1038/s41467-021-26900-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354. Tao Y, Chaudhari S, Shotorbani PY, Ding Y, Chen Z, Kasetti R, Zode G, Ma R. Enhanced Orai1-mediated store-operated Ca2+ channel/calpain signaling contributes to high glucose-induced podocyte injury. J Biol Chem 298: 101990, 2022. doi: 10.1016/j.jbc.2022.101990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355. Watkins JC. l-glutamate as a central neurotransmitter: looking back. Biochem Soc Trans 28: 297–309, 2000. doi: 10.1042/bst0280297. [DOI] [PubMed] [Google Scholar]
- 356. Constantine-Paton M. NMDA receptor as a mediator of activity-dependent synaptogenesis in the developing brain. Cold Spring Harb Symp Quant Biol 55: 431–443, 1990. doi: 10.1101/sqb.1990.055.01.043. [DOI] [PubMed] [Google Scholar]
- 357. Booker SA, Sumera A, Kind PC, Wyllie DJ. Contribution of NMDA receptors to synaptic function in rat hippocampal interneurons. eNeuro 8: ENEURO.0552-20.2021, 2021. doi: 10.1523/ENEURO.0552-20.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358. Murphy KP, Reid GP, Trentham DR, Bliss TV. Activation of NMDA receptors is necessary for the induction of associative long-term potentiation in area CA1 of the rat hippocampal slice. J Physiol 504: 379–385, 1997. doi: 10.1111/j.1469-7793.1997.379be.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359. Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci 11: 682–696, 2010. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360. Liu J, Chang L, Song Y, Li H, Wu Y. The role of NMDA receptors in Alzheimer’s disease. Front Neurosci 13: 43, 2019. doi: 10.3389/fnins.2019.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361. Dryer SE. Glutamate receptors in the kidney. Nephrol Dial Transplant 30: 1630–1638, 2015. doi: 10.1093/ndt/gfv028. [DOI] [PubMed] [Google Scholar]
- 362. Rastaldi MP, Armelloni S, Berra S, Calvaresi N, Corbelli A, Giardino LA, Li M, Wang GQ, Fornasieri A, Villa A, Heikkila E, Soliymani R, Boucherot A, Cohen CD, Kretzler M, Nitsche A, Ripamonti M, Malgaroli A, Pesaresi M, Forloni GL, Schlöndorff D, Holthofer H, D’Amico G. Glomerular podocytes contain neuron-like functional synaptic vesicles. FASEB J 20: 976–978, 2006. doi: 10.1096/fj.05-4962fje. [DOI] [PubMed] [Google Scholar]
- 363. Valdivielso JM, Eritja À, Caus M, Bozic M. Glutamate-gated NMDA receptors: insights into the function and signaling in the kidney. Biomolecules 10: 1051, 2020. doi: 10.3390/biom10071051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364. Deng A, Valdivielso JM, Munger KA, Blantz RC, Thomson SC. Vasodilatory N-methyl-D-aspartate receptors are constitutively expressed in rat kidney. J Am Soc Nephrol 13: 1381–1384, 2002. doi: 10.1097/01.asn.0000013293.11876.4e. [DOI] [PubMed] [Google Scholar]
- 365. Deng A, Thomson SC. Renal NMDA receptors independently stimulate proximal reabsorption and glomerular filtration. Am J Physiol Renal Physiol 296: F976–F982, 2009. doi: 10.1152/ajprenal.90391.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366. Yang CC, Chien CT, Wu MH, Ma MC, Chen CF. NMDA receptor blocker ameliorates ischemia-reperfusion-induced renal dysfunction in rat kidneys. Am J Physiol Renal Physiol 294: F1433–F1440, 2008. doi: 10.1152/ajprenal.00481.2007. [DOI] [PubMed] [Google Scholar]
- 367. Bozic M, de Rooij J, Parisi E, Ortega MR, Fernandez E, Valdivielso JM. Glutamatergic signaling maintains the epithelial phenotype of proximal tubular cells. J Am Soc Nephrol 22: 1099–1111, 2011. doi: 10.1681/ASN.2010070701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368. Sproul A, Steele SL, Thai TL, Yu S, Klein JD, Sands JM, Bell PD. N-methyl-D-aspartate receptor subunit NR3a expression and function in principal cells of the collecting duct. Am J Physiol Renal Physiol 301: F44–F54, 2011. doi: 10.1152/ajprenal.00666.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369. Anderson M, Suh JM, Kim EY, Dryer SE. Functional NMDA receptors with atypical properties are expressed in podocytes. Am J Physiol Cell Physiol 300: C22–C32, 2011. doi: 10.1152/ajpcell.00268.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370. Zhang C, Yi F, Xia M, Boini KM, Zhu Q, Laperle LA, Abais JM, Brimson CA, Li PL. NMDA receptor-mediated activation of NADPH oxidase and glomerulosclerosis in hyperhomocysteinemic rats. Antioxid Redox Signal 13: 975–986, 2010. doi: 10.1089/ars.2010.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371. Giardino L, Armelloni S, Corbelli A, Mattinzoli D, Zennaro C, Guerrot D, Tourrel F, Ikehata M, Li M, Berra S, Carraro M, Messa P, Rastaldi MP. Podocyte glutamatergic signaling contributes to the function of the glomerular filtration barrier. J Am Soc Nephrol 20: 1929–1940, 2009. doi: 10.1681/ASN.2008121286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372. Bądzyńska B, Zakrocka I, Sadowski J, Turski WA, Kompanowska-Jezierska E. Effects of systemic administration of kynurenic acid and glycine on renal haemodynamics and excretion in normotensive and spontaneously hypertensive rats. Eur J Pharmacol 743: 37–41, 2014. doi: 10.1016/j.ejphar.2014.09.020. [DOI] [PubMed] [Google Scholar]
- 373. Mayer ML, Westbrook GL. Permeation and block of N-methyl-D-aspartic acid receptor channels by divalent cations in mouse cultured central neurones. J Physiol 394: 501–527, 1987. doi: 10.1113/jphysiol.1987.sp016883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374. Grimwood S, Moseley AM, Carling RW, Leeson PD, Foster AC. Characterization of the binding of [3H]L-689,560, an antagonist for the glycine site on the N-methyl-D-aspartate receptor, to rat brain membranes. Mol Pharmacol 41: 923–930, 1992. [PubMed] [Google Scholar]
- 375. Paoletti P, Bellone C, Zhou Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci 14: 383–400, 2013. doi: 10.1038/nrn3504. [DOI] [PubMed] [Google Scholar]
- 376. Hansen KB, Wollmuth LP, Bowie D, Furukawa H, Menniti FS, Sobolevsky AI, Swanson GT, Swanger SA, Greger IH, Nakagawa T, McBain CJ, Jayaraman V, Low CM, Dell’Acqua ML, Diamond JS, Camp CR, Perszyk RE, Yuan H, Traynelis SF. Structure, function, and pharmacology of glutamate receptor ion channels. Pharmacol Rev 73: 298–487, 2021. doi: 10.1124/pharmrev.120.000131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377. Chatterton JE, Awobuluyi M, Premkumar LS, Takahashi H, Talantova M, Shin Y, Cui J, Tu S, Sevarino KA, Nakanishi N, Tong G, Lipton SA, Zhang D. Excitatory glycine receptors containing the NR3 family of NMDA receptor subunits. Nature 415: 793–798, 2002. doi: 10.1038/nature715. [DOI] [PubMed] [Google Scholar]
- 378. Lussier MP, Sanz-Clemente A, Roche KW. Dynamic regulation of N-methyl-d-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors by posttranslational modifications. J Biol Chem 290: 28596–28603, 2015. doi: 10.1074/jbc.R115.652750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379. Hubalkova P, Ladislav M, Vyklicky V, Smejkalova T, Hrcka Krausova B, Kysilov B, Krusek J, Naimová Z, Korinek M, Chodounska H, Kudova E, Cerny J, Vyklicky L Jr.. Palmitoylation controls NMDA receptor function and steroid sensitivity. J Neurosci 41: 2119–2134, 2021. doi: 10.1523/JNEUROSCI.2654-20.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380. Krug AW, Völker K, Dantzler WH, Silbernagl S. Why is D-serine nephrotoxic and alpha-aminoisobutyric acid protective? Am J Physiol Renal Physiol 293: F382–F390, 2007. doi: 10.1152/ajprenal.00441.2006. [DOI] [PubMed] [Google Scholar]
- 381. Puliti A, Rossi PI, Caridi G, Corbelli A, Ikehata M, Armelloni S, Li M, Zennaro C, Conti V, Vaccari CM, Cassanello M, Calevo MG, Emionite L, Ravazzolo R, Rastaldi MP. Albuminuria and glomerular damage in mice lacking the metabotropic glutamate receptor 1. Am J Pathol 178: 1257–1269, 2011. doi: 10.1016/j.ajpath.2010.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382. Stegink LD, Filer LJ Jr, Baker GL. Plasma glutamate concentrations in adult subjects ingesting monosodium L-glutamate in consomme. Am J Clin Nutr 42: 220–225, 1985. doi: 10.1093/ajcn/42.2.220. [DOI] [PubMed] [Google Scholar]
- 383. Zhou Y, Qiu L, Xiao Q, Wang Y, Meng X, Xu R, Wang S, Na R. Obesity and diabetes related plasma amino acid alterations. Clin Biochem 46: 1447–1452, 2013. doi: 10.1016/j.clinbiochem.2013.05.045. [DOI] [PubMed] [Google Scholar]
- 384. Patneau DK, Mayer ML. Structure-activity relationships for amino acid transmitter candidates acting at N-methyl-D-aspartate and quisqualate receptors. J Neurosci 10: 2385–2399, 1990. doi: 10.1523/JNEUROSCI.10-07-02385.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385. Hawkins RA. The blood-brain barrier and glutamate. Am J Clin Nutr 90: 867s–874s, 2009. doi: 10.3945/ajcn.2009.27462BB. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386. Finkelstein JD. Pathways and regulation of homocysteine metabolism in mammals. Semin Thromb Hemost 26: 219–225, 2000. doi: 10.1055/s-2000-8466. [DOI] [PubMed] [Google Scholar]
- 387. Werge MP, McCann A, Galsgaard ED, Holst D, Bugge A, Albrechtsen NJ, Gluud LL. The role of the transsulfuration pathway in non-alcoholic fatty liver disease. J Clin Med 10: 1081, 2021. doi: 10.3390/jcm10051081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388. Pires AS, Sundaram G, Heng B, Krishnamurthy S, Brew BJ, Guillemin GJ. Recent advances in clinical trials targeting the kynurenine pathway. Pharmacol Ther 236: 108055, 2022. doi: 10.1016/j.pharmthera.2021.108055. [DOI] [PubMed] [Google Scholar]
- 389. Debnath S, Velagapudi C, Redus L, Thameem F, Kasinath B, Hura CE, Lorenzo C, Abboud HE, O’Connor JC. Tryptophan metabolism in patients with chronic kidney disease secondary to type 2 diabetes: relationship to inflammatory markers. Int J Tryptophan Res 10: 117864691769460, 2017. doi: 10.1177/1178646917694600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390. Zakrocka I, Załuska W. Kynurenine pathway in kidney diseases. Pharmacol Rep 74: 27–39, 2022. doi: 10.1007/s43440-021-00329-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391. Matsumoto M, Kunisawa A, Hattori T, Kawana S, Kitada Y, Tamada H, Kawano S, Hayakawa Y, Iida J, Fukusaki E. Free D-amino acids produced by commensal bacteria in the colonic lumen. Sci Rep 8: 17915, 2018. doi: 10.1038/s41598-018-36244-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392. Nakade Y, Iwata Y, Furuichi K, Mita M, Hamase K, Konno R, Miyake T, Sakai N, Kitajima S, Toyama T, Shinozaki Y, Sagara A, Miyagawa T, Hara A, Shimizu M, Kamikawa Y, Sato K, Oshima M, Yoneda-Nakagawa S, Yamamura Y, Kaneko S, Miyamoto T, Katane M, Homma H, Morita H, Suda W, Hattori M, Wada T. Gut microbiota-derived D-serine protects against acute kidney injury. JCI Insight 3: e97957, 2018. doi: 10.1172/jci.insight.97957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393. Kim PM, Duan X, Huang AS, Liu CY, Ming GL, Song H, Snyder SH. Aspartate racemase, generating neuronal D-aspartate, regulates adult neurogenesis. Proc Natl Acad Sci USA 107: 3175–3179, 2010. doi: 10.1073/pnas.0914706107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394. Wolosker H, Radzishevsky I. Promiscuous enzymes generating d-amino acids in mammals: why they may still surprise us? Biochem J 478: 1175–1178, 2021. doi: 10.1042/BCJ20200988. [DOI] [PubMed] [Google Scholar]
- 395. Stroebel D, Mony L, Paoletti P. Glycine agonism in ionotropic glutamate receptors. Neuropharmacology 193: 108631, 2021. doi: 10.1016/j.neuropharm.2021.108631. [DOI] [PubMed] [Google Scholar]
- 396. Kimura T, Hesaka A, Isaka Y. D-amino acids and kidney diseases. Clin Exp Nephrol 24: 404–410, 2020. doi: 10.1007/s10157-020-01862-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397. Spasova K, Fähling M. D-serine—a useful biomarker for renal injury? Acta Physiol (Oxf) 230: e13531, 2020. doi: 10.1111/apha.13531. [DOI] [PubMed] [Google Scholar]
- 398. Okushima H, Iwata Y, Hesaka A, Sugimori E, Ikeda T, Nakane M, Mita M, Hayashi T, Isaka Y, Kimura T. Intra-body dynamics of D-serine reflects the origin of kidney diseases. Clin Exp Nephrol 25: 893–901, 2021. doi: 10.1007/s10157-021-02052-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399. D’Hooge R, Raes A, Lebrun P, Diltoer M, Van Bogaert PP, Manil J, Colin F, De Deyn PP. N-methyl-D-aspartate receptor activation by guanidinosuccinate but not by methylguanidine: behavioural and electrophysiological evidence. Neuropharmacology 35: 433–440, 1996. doi: 10.1016/0028-3908(96)00011-1. [DOI] [PubMed] [Google Scholar]
- 400. De Deyn PP, Vanholder R, Eloot S, Glorieux G. Guanidino compounds as uremic (neuro)toxins. Semin Dial 22: 340–345, 2009. doi: 10.1111/j.1525-139X.2009.00577.x. [DOI] [PubMed] [Google Scholar]
- 401. Kim EY, Anderson M, Dryer SE. Sustained activation of N-methyl-D-aspartate receptors in podocytes leads to oxidative stress, mobilization of transient receptor potential canonical 6 channels, nuclear factor of activated T cells activation, and apoptotic cell death. Mol Pharmacol 82: 728–737, 2012. doi: 10.1124/mol.112.079376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402. Zhang Q, Conley SM, Li G, Yuan X, Li PL. Rac1 GTPase inhibition blocked podocyte injury and glomerular sclerosis during hyperhomocysteinemia via suppression of nucleotide-binding oligomerization domain-like receptor containing pyrin domain 3 inflammasome activation. Kidney Blood Press Res 44: 513–532, 2019. doi: 10.1159/000500457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403. Ilatovskaya DV, Palygin O, Levchenko V, Staruschenko A. Pharmacological characterization of the P2 receptors profile in the podocytes of the freshly isolated rat glomeruli. Am J Physiol Cell Physiol 305: C1050–C1059, 2013. doi: 10.1152/ajpcell.00138.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404. Staruschenko A, Spires D, Palygin O. Role of TRPC6 in progression of diabetic kidney disease. Curr Hypertens Rep 21: 48, 2019. doi: 10.1007/s11906-019-0960-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405. Ilatovskaya DV, Staruschenko A. TRPC6 channel as an emerging determinant of the podocyte injury susceptibility in kidney diseases. Am J Physiol Renal Physiol 309: F393–F397, 2015. doi: 10.1152/ajprenal.00186.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406. Hall G, Wang L, Spurney RF. TRPC channels in proteinuric kidney diseases. Cells 9: 44, 2019. doi: 10.3390/cells9010044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407. Pablo JL, Greka A. Charting a TRP to novel therapeutic destinations for kidney diseases. Trends Pharmacol Sci 40: 911–918, 2019. doi: 10.1016/j.tips.2019.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408. Dryer SE, Roshanravan H, Kim EY. TRPC channels: regulation, dysregulation and contributions to chronic kidney disease. Biochim Biophys Acta Mol Basis Dis 1865: 1041–1066, 2019. doi: 10.1016/j.bbadis.2019.04.001. [DOI] [PubMed] [Google Scholar]
- 409. Sharma R, Sharma M, Vamos S, Savin VJ, Wiegmann TB. Both subtype 1 and 2 receptors of angiotensin II participate in regulation of intracellular calcium in glomerular epithelial cells. J Lab Clin Med 138: 40–49, 2001. doi: 10.1067/mlc.2001.115493. [DOI] [PubMed] [Google Scholar]
- 410. Yanofsky SM, Dugas CM, Katsurada A, Liu J, Saifudeen Z, El-Dahr SS, Satou R. Angiotensin II biphasically regulates cell differentiation in human iPSC-derived kidney organoids. Am J Physiol Renal Physiol 321: F559–F571, 2021. doi: 10.1152/ajprenal.00134.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411. Sharma M, Sharma R, Greene AS, McCarthy ET, Savin VJ. Documentation of angiotensin II receptors in glomerular epithelial cells. Am J Physiol Renal Physiol 274: F623–F627, 1998. doi: 10.1152/ajprenal.1998.274.3.f623. [DOI] [PubMed] [Google Scholar]
- 412. Wennmann DO, Hsu HH, Pavenstädt H. The renin-angiotensin-aldosterone system in podocytes. Semin Nephrol 32: 377–384, 2012. doi: 10.1016/j.semnephrol.2012.06.009. [DOI] [PubMed] [Google Scholar]
- 413. Abuzeineh M, Aala A, Alasfar S, Alachkar N. Angiotensin II receptor 1 antibodies associate with post-transplant focal segmental glomerulosclerosis and proteinuria. BMC Nephrol 21: 253, 2020. doi: 10.1186/s12882-020-01910-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414. Nitschke R, Henger A, Ricken S, Gloy J, Müller V, Greger R, Pavenstadt H. Angiotensin II increases the intracellular calcium activity in podocytes of the intact glomerulus. Kidney Int 57: 41–49, 2000. doi: 10.1046/j.1523-1755.2000.00810.x. [DOI] [PubMed] [Google Scholar]
- 415. Binz-Lotter J, Jüngst C, Rinschen MM, Koehler S, Zentis P, Schauss A, Schermer B, Benzing T, Hackl MJ. Injured podocytes are sensitized to angiotensin II-induced calcium signaling. J Am Soc Nephrol 31: 532–542, 2020. doi: 10.1681/ASN.2019020109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416. Gloy J, Henger A, Fischer KG, Nitschke R, Mundel P, Bleich M, Schollmeyer P, Greger R, Pavenstädt H. Angiotensin II depolarizes podocytes in the intact glomerulus of the rat. J Clin Invest 99: 2772–2781, 1997. doi: 10.1172/JCI119467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417. Henger A, Huber T, Fischer KG, Nitschke R, Mundel P, Schollmeyer P, Greger R, Pavenstädt H. Angiotensin II increases the cytosolic calcium activity in rat podocytes in culture. Kidney Int 52: 687–693, 1997. doi: 10.1038/ki.1997.383. [DOI] [PubMed] [Google Scholar]
- 418. Tian D, Jacobo SM, Billing D, Rozkalne A, Gage SD, Anagnostou T, Pavenstädt H, Pavenstaedt H, Hsu HH, Schlondorff J, Ramos A, Greka A. Antagonistic regulation of actin dynamics and cell motility by TRPC5 and TRPC6 channels. Sci Signal 3: ra77, 2010. doi: 10.1126/scisignal.2001200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419. Che G, Gao H, Hu Q, Xie H, Zhang Y. Angiotensin II promotes podocyte injury by activating Arf6-Erk1/2-Nox4 signaling pathway. PLoS One 15: e0229747, 2020. doi: 10.1371/journal.pone.0229747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420. Maier T, Follmann M, Hessler G, Kleemann HW, Hachtel S, Fuchs B, Weissmann N, Linz W, Schmidt T, Löhn M, Schroeter K, Wang L, Rütten H, Strübing C. Discovery and pharmacological characterization of a novel potent inhibitor of diacylglycerol-sensitive TRPC cation channels. Br J Pharmacol 172: 3650–3660, 2015. doi: 10.1111/bph.13151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421. Inoue K, Tian X, Velazquez H, Soda K, Wang Z, Pedigo CE, Wang Y, Cross E, Groener M, Shin JW, Li W, Hassan H, Yamamoto K, Mundel P, Ishibe S. Inhibition of endocytosis of clathrin-mediated angiotensin II receptor type 1 in podocytes augments glomerular injury. J Am Soc Nephrol 30: 2307–2320, 2019. doi: 10.1681/ASN.2019010053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422. Schießl IM, Hammer A, Kattler V, Gess B, Theilig F, Witzgall R, Castrop H. Intravital imaging reveals angiotensin II-induced transcytosis of albumin by podocytes. J Am Soc Nephrol 27: 731–744, 2016. doi: 10.1681/ASN.2014111125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423. Sonneveld R, van der Vlag J, Baltissen MP, Verkaart SA, Wetzels JF, Berden JH, Hoenderop JG, Nijenhuis T. Glucose specifically regulates TRPC6 expression in the podocyte in an AngII-dependent manner. Am J Pathol 184: 1715–1726, 2014. doi: 10.1016/j.ajpath.2014.02.008. [DOI] [PubMed] [Google Scholar]
- 424. Hoffmann S, Podlich D, Hähnel B, Kriz W, Gretz N. Angiotensin II type 1 receptor overexpression in podocytes induces glomerulosclerosis in transgenic rats. J Am Soc Nephrol 15: 1475–1487, 2004. doi: 10.1097/01.asn.0000127988.42710.a7. [DOI] [PubMed] [Google Scholar]
- 425. Nijenhuis T, Sloan AJ, Hoenderop JG, Flesche J, van Goor H, Kistler AD, Bakker M, Bindels RJ, de Boer RA, Möller CC, Hamming I, Navis G, Wetzels JF, Berden JH, Reiser J, Faul C, van der Vlag J. Angiotensin II contributes to podocyte injury by increasing TRPC6 expression via an NFAT-mediated positive feedback signaling pathway. Am J Pathol 179: 1719–1732, 2011. doi: 10.1016/j.ajpath.2011.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426. Eckel J, Lavin PJ, Finch EA, Mukerji N, Burch J, Gbadegesin R, Wu G, Bowling B, Byrd A, Hall G, Sparks M, Zhang ZS, Homstad A, Barisoni L, Birbaumer L, Rosenberg P, Winn MP. TRPC6 enhances angiotensin II-induced albuminuria. J Am Soc Nephrol 22: 526–535, 2011. doi: 10.1681/ASN.2010050522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427. Fischer KG, Saueressig U, Jacobshagen C, Wichelmann A, Pavenstädt H. Extracellular nucleotides regulate cellular functions of podocytes in culture. Am J Physiol Renal Physiol 281: F1075–F1081, 2001. doi: 10.1152/ajprenal.2001.281.6.F1075. [DOI] [PubMed] [Google Scholar]
- 428. Gyarmati G, Toma I, Izuhara A, Burford JL, Shroff UN, Papadouri S, Deepak S, Peti-Peterdi J. . The role of TRPC6 calcium channels and P2 purinergic receptors in podocyte mechanical and metabolic sensing. Physiol Int 2021: 2021.00205, 2021. doi: 10.1556/2060.2021.00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429. Ilatovskaya DV, Levchenko V, Lowing A, Shuyskiy LS, Palygin O, Staruschenko A. Podocyte injury in diabetic nephropathy: implications of angiotensin II-dependent activation of TRPC channels. Sci Rep 5: 17637, 2015. doi: 10.1038/srep17637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430. Jacobson KA, Ivanov AA, de Castro S, Harden TK, Ko H. Development of selective agonists and antagonists of P2Y receptors. Purinergic Signal 5: 75–89, 2009. doi: 10.1007/s11302-008-9106-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431. Vallon V, Rieg T. Regulation of renal NaCl and water transport by the ATP/UTP/P2Y2 receptor system. Am J Physiol Renal Physiol 301: F463–F475, 2011. doi: 10.1152/ajprenal.00236.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432. Wilson C, Dryer SE. A mutation in TRPC6 channels abolishes their activation by hypoosmotic stretch but does not affect activation by diacylglycerol or G protein signaling cascades. Am J Physiol Renal Physiol 306: F1018–F1025, 2014. doi: 10.1152/ajprenal.00662.2013. [DOI] [PubMed] [Google Scholar]
- 433. Wilson PC, Wu H, Kirita Y, Uchimura K, Ledru N, Rennke HG, Welling PA, Waikar SS, Humphreys BD. The single-cell transcriptomic landscape of early human diabetic nephropathy. Proc Natl Acad Sci USA 116: 19619–19625, 2019. doi: 10.1073/pnas.1908706116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434. Forst AL, Olteanu VS, Mollet G, Wlodkowski T, Schaefer F, Dietrich A, Reiser J, Gudermann T, Mederos y Schnitzler M, Storch U. Podocyte purinergic P2X4 channels are mechanotransducers that mediate cytoskeletal disorganization. J Am Soc Nephrol 27: 848–862, 2016. doi: 10.1681/ASN.2014111144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435. Turner CM, Tam FW, Lai PC, Tarzi RM, Burnstock G, Pusey CD, Cook HT, Unwin RJ. Increased expression of the pro-apoptotic ATP-sensitive P2X7 receptor in experimental and human glomerulonephritis. Nephrol Dial Transplant 22: 386–395, 2007. doi: 10.1093/ndt/gfl589. [DOI] [PubMed] [Google Scholar]
- 436. Arkhipov SN, Potter DL, Geurts AM, Pavlov TS. Knockout of P2rx7 purinergic receptor attenuates cyst growth in a rat model of ARPKD. Am J Physiol Renal Physiol 317: F1649–F1655, 2019. doi: 10.1152/ajprenal.00395.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437. Shokoples BG, Paradis P, Schiffrin EL. P2X7 receptors: an untapped target for the management of cardiovascular disease. Arterioscler Thromb Vasc Biol 41: 186–199, 2021. doi: 10.1161/ATVBAHA.120.315116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438. Rogacka D. Insulin resistance in glomerular podocytes: potential mechanisms of induction. Arch Biochem Biophys 710: 109005, 2021. doi: 10.1016/j.abb.2021.109005. [DOI] [PubMed] [Google Scholar]
- 439. Welsh GI, Hale LJ, Eremina V, Jeansson M, Maezawa Y, Lennon R, Pons DA, Owen RJ, Satchell SC, Miles MJ, Caunt CJ, McArdle CA, Pavenstädt H, Tavaré JM, Herzenberg AM, Kahn CR, Mathieson PW, Quaggin SE, Saleem MA, Coward RJ. Insulin signaling to the glomerular podocyte is critical for normal kidney function. Cell Metab 12: 329–340, 2010. doi: 10.1016/j.cmet.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440. Vallon V, Thomson SC. Renal function in diabetic disease models: the tubular system in the pathophysiology of the diabetic kidney. Annu Rev Physiol 74: 351–375, 2012. doi: 10.1146/annurev-physiol-020911-153333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441. Persson P, Hansell P, Palm F. Tubular reabsorption and diabetes-induced glomerular hyperfiltration. Acta Physiol (Oxf) 200: 3–10, 2010. doi: 10.1111/j.1748-1716.2010.02147.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442. Rogacka D, Audzeyenka I, Rachubik P, Rychłowski M, Kasztan M, Jankowski M, Angielski S, Piwkowska A. Insulin increases filtration barrier permeability via TRPC6-dependent activation of PKGIα signaling pathways. Biochim Biophys Acta Mol Basis Dis 1863: 1312–1325, 2017. doi: 10.1016/j.bbadis.2017.03.002. [DOI] [PubMed] [Google Scholar]
- 443. Rachubik P, Szrejder M, Rogacka D, Audzeyenka I, Rychłowski M, Angielski S, Piwkowska A. The TRPC6-AMPK pathway is involved in insulin-dependent cytoskeleton reorganization and glucose uptake in cultured rat podocytes. Cell Physiol Biochem 51: 393–410, 2018. doi: 10.1159/000495236. [DOI] [PubMed] [Google Scholar]
- 444. Roman RJ. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev 82: 131–185, 2002. doi: 10.1152/physrev.00021.2001. [DOI] [PubMed] [Google Scholar]
- 445. Zhao X, Falck JR, Gopal VR, Inscho EW, Imig JD. P2X receptor-stimulated calcium responses in preglomerular vascular smooth muscle cells involves 20-hydroxyeicosatetraenoic acid. J Pharmacol Exp Ther 311: 1211–1217, 2004. doi: 10.1124/jpet.104.070797. [DOI] [PubMed] [Google Scholar]
- 446. Roshanravan H, Kim EY, Dryer SE. 20-Hydroxyeicosatetraenoic acid (20-HETE) modulates canonical transient receptor potential-6 (TRPC6) channels in podocytes. Front Physiol 7: 351, 2016. doi: 10.3389/fphys.2016.00351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447. ’t Hart D, Li J, van der Vlag J, Nijenhuis T. Repurposing riociguat to target a novel paracrine nitric oxide-TRPC6 pathway to prevent podocyte injury. Int J Mol Sci 22: 12485, 2021. doi: 10.3390/ijms222212485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448. Golosova D, Palygin O, Bohovyk R, Klemens CA, Levchenko V, Spires DR, Isaeva E, El-Meanawy A, Staruschenko A. Role of opioid signaling in kidney damage during the development of salt-induced hypertension. Life Sci Alliance 3: e202000853, 2020. doi: 10.26508/lsa.202000853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449. Ilatovskaya DV, Levchenko V, Ryan RP, Cowley AW Jr, Staruschenko A. NSAIDs acutely inhibit TRPC channels in freshly isolated rat glomeruli. Biochem Biophys Res Commun 408: 242–247, 2011. doi: 10.1016/j.bbrc.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450. Zhang X, Song Z, Guo Y, Zhou M. The novel role of TRPC6 in vitamin D ameliorating podocyte injury in STZ-induced diabetic rats. Mol Cell Biochem 399: 155–165, 2015. doi: 10.1007/s11010-014-2242-9. [DOI] [PubMed] [Google Scholar]
- 451. Sonneveld R, Ferrè S, Hoenderop JG, Dijkman HB, Berden JH, Bindels RJ, Wetzels JF, van der Vlag J, Nijenhuis T. Vitamin D down-regulates TRPC6 expression in podocyte injury and proteinuric glomerular disease. Am J Pathol 182: 1196–1204, 2013. doi: 10.1016/j.ajpath.2012.12.011. [DOI] [PubMed] [Google Scholar]
- 452. Winn MP, Conlon PJ, Lynn KL, Howell DN, Slotterbeck BD, Smith AH, Graham FL, Bembe M, Quarles LD, Pericak-Vance MA, Vance JM. Linkage of a gene causing familial focal segmental glomerulosclerosis to chromosome 11 and further evidence of genetic heterogeneity. Genomics 58: 113–120, 1999. doi: 10.1006/geno.1999.5828. [DOI] [PubMed] [Google Scholar]
- 453. Conlon PJ, Lynn K, Winn MP, Quarles LD, Bembe ML, Pericak-Vance M, Speer M, Howell DN. Spectrum of disease in familial focal and segmental glomerulosclerosis. Kidney Int 56: 1863–1871, 1999. doi: 10.1046/j.1523-1755.1999.00727.x. [DOI] [PubMed] [Google Scholar]
- 454. Schlöndorff J, Del Camino D, Carrasquillo R, Lacey V, Pollak MR. TRPC6 mutations associated with focal segmental glomerulosclerosis cause constitutive activation of NFAT-dependent transcription. Am J Physiol Cell Physiol 296: C558–C569, 2009. doi: 10.1152/ajpcell.00077.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 455. Chiluiza D, Krishna S, Schumacher VA, Schlöndorff J. Gain-of-function mutations in transient receptor potential C6 (TRPC6) activate extracellular signal-regulated kinases 1/2 (ERK1/2). J Biol Chem 288: 18407–18420, 2013. doi: 10.1074/jbc.M113.463059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456. Heeringa SF, Möller CC, Du J, Yue L, Hinkes B, Chernin G, Vlangos CN, Hoyer PF, Reiser J, Hildebrandt F. A novel TRPC6 mutation that causes childhood FSGS. PLoS ONE 4: e7771, 2009. doi: 10.1371/journal.pone.0007771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457. Obeidová L, Reiterová J, Lněnička P, Štekrová J, Šafránková H, Kohoutová M, Tesař V. TRPC6 gene variants in Czech adult patients with focal segmental glomerulosclerosis and minimal change disease. Folia Biol (Praha) 58: 173–176, 2012. [PubMed] [Google Scholar]
- 458. Barua M, Brown EJ, Charoonratana VT, Genovese G, Sun H, Pollak MR. Mutations in the INF2 gene account for a significant proportion of familial but not sporadic focal and segmental glomerulosclerosis. Kidney Int 83: 316–322, 2013. doi: 10.1038/ki.2012.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459. Santín S, Ars E, Rossetti S, Salido E, Silva I, García-Maset R, et al. TRPC6 mutational analysis in a large cohort of patients with focal segmental glomerulosclerosis. Nephrol Dial Transplant 24: 3089–3096, 2009. doi: 10.1093/ndt/gfp229. [DOI] [PubMed] [Google Scholar]
- 460. Hofstra JM, Lainez S, van Kuijk WH, Schoots J, Baltissen MP, Hoefsloot LH, Knoers NV, Berden JH, Bindels RJ, van der Vlag J, Hoenderop JG, Wetzels JF, Nijenhuis T. New TRPC6 gain-of-function mutation in a non-consanguineous Dutch family with late-onset focal segmental glomerulosclerosis. Nephrol Dial Transplant 28: 1830–1838, 2013. doi: 10.1093/ndt/gfs572. [DOI] [PubMed] [Google Scholar]
- 461. Büscher AK, Konrad M, Nagel M, Witzke O, Kribben A, Hoyer PF, Weber S. Mutations in podocyte genes are a rare cause of primary FSGS associated with ESRD in adult patients. Clin Nephrol 78: 47–53, 2012. doi: 10.5414/cn107320. [DOI] [PubMed] [Google Scholar]
- 462. Mir S, Yavascan O, Berdeli A, Sozeri B. TRPC6 gene variants in Turkish children with steroid-resistant nephrotic syndrome. Nephrol Dial Transplant 27: 205–209, 2012. doi: 10.1093/ndt/gfr202. [DOI] [PubMed] [Google Scholar]
- 463. Zhu B, Chen N, Wang ZH, Pan XX, Ren H, Zhang W, Wang WM. Identification and functional analysis of a novel TRPC6 mutation associated with late onset familial focal segmental glomerulosclerosis in Chinese patients. Mutat Res 664: 84–90, 2009. doi: 10.1016/j.mrfmmm.2008.11.021. [DOI] [PubMed] [Google Scholar]
- 464. Büscher AK, Kranz B, Büscher R, Hildebrandt F, Dworniczak B, Pennekamp P, Kuwertz-Bröking E, Wingen AM, John U, Kemper M, Monnens L, Hoyer PF, Weber S, Konrad M. Immunosuppression and renal outcome in congenital and pediatric steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol 5: 2075–2084, 2010. doi: 10.2215/CJN.01190210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465. Gigante M, Caridi G, Montemurno E, Soccio M, d’Apolito M, Cerullo G, Aucella F, Schirinzi A, Emma F, Massella L, Messina G, De Palo T, Ranieri E, Ghiggeri GM, Gesualdo L. TRPC6 mutations in children with steroid-resistant nephrotic syndrome and atypical phenotype. Clin J Am Soc Nephrol 6: 1626–1634, 2011. doi: 10.2215/CJN.07830910. [DOI] [PubMed] [Google Scholar]
- 466. Riehle M, Büscher AK, Gohlke BO, Kaßmann M, Kolatsi-Joannou M, Bräsen JH, Nagel M, Becker JU, Winyard P, Hoyer PF, Preissner R, Krautwurst D, Gollasch M, Weber S, Harteneck C. TRPC6 G757D loss-of-function mutation associates with FSGS. J Am Soc Nephrol 27: 2771–2783, 2016. doi: 10.1681/ASN.2015030318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467. Bai Y, Yu X, Chen H, Horne D, White R, Wu X, Lee P, Gu Y, Ghimire-Rijal S, Lin DC, Huang X. Structural basis for pharmacological modulation of the TRPC6 channel. Elife 9: e53311, 2020. doi: 10.7554/eLife.53311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468. Canales CP, Krall P, Kairath P, Perez IC, Fragoso MA, Carmona-Mora P, Ruiz P, Reiser J, Young JI, Walz K. Characterization of a Trpc6 transgenic mouse associated with early onset FSGS. Br J Med Res 5: 1198–2012, 2015. doi: 10.9734/bjmmr/2015/12493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469. Krall P, Canales CP, Kairath P, Carmona-Mora P, Molina J, Carpio JD, Ruiz P, Mezzano SA, Li J, Wei C, Reiser J, Young JI, Walz K. Podocyte-specific overexpression of wild type or mutant trpc6 in mice is sufficient to cause glomerular disease. PLoS One 5: e12859, 2010. doi: 10.1371/journal.pone.0012859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470. Brown BJ, Boekell KL, Stotter BR, Talbot BE, Schlondorff JS. Gain-of-function, focal segmental glomerulosclerosis Trpc6 mutation minimally affects susceptibility to renal injury in several mouse models. PLoS One 17: e0272313, 2022. doi: 10.1371/journal.pone.0272313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471. Wang L, Jirka G, Rosenberg PB, Buckley AF, Gomez JA, Fields TA, Winn MP, Spurney RF. Gq signaling causes glomerular injury by activating TRPC6. J Clin Invest 125: 1913–1926, 2015. doi: 10.1172/JCI76767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 472. Wang L, Chang JH, Buckley AF, Spurney RF. Knockout of TRPC6 promotes insulin resistance and exacerbates glomerular injury in Akita mice. Kidney Int 95: 321–332, 2019. doi: 10.1016/j.kint.2018.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473. Kistler AD, Singh G, Altintas MM, Yu H, Fernandez IC, Gu C, Wilson C, Srivastava SK, Dietrich A, Walz K, Kerjaschki D, Ruiz P, Dryer S, Sever S, Dinda AK, Faul C, Reiser J. Transient receptor potential channel 6 (TRPC6) protects podocytes during complement-mediated glomerular disease. J Biol Chem 288: 36598–36609, 2013. doi: 10.1074/jbc.M113.488122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474. Huber TB, Schermer B, Müller RU, Höhne M, Bartram M, Calixto A, Hagmann H, Reinhardt C, Koos F, Kunzelmann K, Shirokova E, Krautwurst D, Harteneck C, Simons M, Pavenstädt H, Kerjaschki D, Thiele C, Walz G, Chalfie M, Benzing T. Podocin and MEC-2 bind cholesterol to regulate the activity of associated ion channels. Proc Natl Acad Sci USA 103: 17079–17086, 2006. doi: 10.1073/pnas.0607465103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475. Boute N, Gribouval O, Roselli S, Benessy F, Lee H, Fuchshuber A, Dahan K, Gubler MC, Niaudet P, Antignac C. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet 24: 349–354, 2000. doi: 10.1038/74166. [DOI] [PubMed] [Google Scholar]
- 476. Mulukala SKN, Irukuvajjula SS, Kumar K, Garai K, Venkatesu P, Vadrevu R, Pasupulati AK. Structural features and oligomeric nature of human podocin domain. Biochem Biophys Rep 23: 100774, 2020. doi: 10.1016/j.bbrep.2020.100774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477. Schwarz K, Simons M, Reiser J, Saleem MA, Faul C, Kriz W, Shaw AS, Holzman LB, Mundel P. Podocin, a raft-associated component of the glomerular slit diaphragm, interacts with CD2AP and nephrin. J Clin Invest 108: 1621–1629, 2001. doi: 10.1172/JCI12849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 478. Huber TB, Simons M, Hartleben B, Sernetz L, Schmidts M, Gundlach E, Saleem MA, Walz G, Benzing T. Molecular basis of the functional podocin-nephrin complex: mutations in the NPHS2 gene disrupt nephrin targeting to lipid raft microdomains. Hum Mol Genet 12: 3397–3405, 2003. doi: 10.1093/hmg/ddg360. [DOI] [PubMed] [Google Scholar]
- 479. Huber TB, Schermer B, Benzing T. Podocin organizes ion channel-lipid supercomplexes: implications for mechanosensation at the slit diaphragm. Nephron Exp Nephrol 106: e27–e31, 2007. doi: 10.1159/000101789. [DOI] [PubMed] [Google Scholar]
- 480. Bouchireb K, Boyer O, Gribouval O, Nevo F, Huynh-Cong E, Morinière V, Campait R, Ars E, Brackman D, Dantal J, Eckart P, Gigante M, Lipska BS, Liutkus A, Megarbane A, Mohsin N, Ozaltin F, Saleem MA, Schaefer F, Soulami K, Torra R, Garcelon N, Mollet G, Dahan K, Antignac C. NPHS2 mutations in steroid-resistant nephrotic syndrome: a mutation update and the associated phenotypic spectrum. Hum Mutat 35: 178–186, 2014. doi: 10.1002/humu.22485. [DOI] [PubMed] [Google Scholar]
- 481. Zenker M, Machuca E, Antignac C. Genetics of nephrotic syndrome: new insights into molecules acting at the glomerular filtration barrier. J Mol Med (Berl) 87: 849–857, 2009. doi: 10.1007/s00109-009-0505-9. [DOI] [PubMed] [Google Scholar]
- 482. Feng D, Steinke JM, Krishnan R, Birrane G, Pollak MR. Functional validation of an alpha-actinin-4 mutation as a potential cause of an aggressive presentation of adolescent focal segmental glomerulosclerosis: implications for genetic testing. PLoS One 11: e0167467, 2016. doi: 10.1371/journal.pone.0167467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 483. Feng D, Notbohm J, Benjamin A, He S, Wang M, Ang LH, Bantawa M, Bouzid M, Del Gado E, Krishnan R, Pollak MR. Disease-causing mutation in α-actinin-4 promotes podocyte detachment through maladaptation to periodic stretch. Proc Natl Acad Sci USA 115: 1517–1522, 2018. doi: 10.1073/pnas.1717870115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 484. Feng D, Kumar M, Muntel J, Gurley SB, Birrane G, Stillman IE, Ding L, Wang M, Ahmed S, Schlondorff J, Alper SL, Ferrante T, Marquez SL, Ng CF, Novak R, Ingber DE, Steen H, Pollak MR. Phosphorylation of ACTN4 leads to podocyte vulnerability and proteinuric glomerulosclerosis. J Am Soc Nephrol 31: 1479–1495, 2020. doi: 10.1681/ASN.2019101032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 485. Shalygin A, Shuyskiy LS, Bohovyk R, Palygin O, Staruschenko A, Kaznacheyeva E. Cytoskeleton rearrangements modulate TRPC6 channel activity in podocytes. Int J Mol Sci 22: 4396, 2021. doi: 10.3390/ijms22094396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 486. Akilesh S, Suleiman H, Yu H, Stander MC, Lavin P, Gbadegesin R, Antignac C, Pollak M, Kopp JB, Winn MP, Shaw AS. Arhgap24 inactivates Rac1 in mouse podocytes, and a mutant form is associated with familial focal segmental glomerulosclerosis. J Clin Invest 121: 4127–4137, 2011. doi: 10.1172/JCI46458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487. Gee HY, Saisawat P, Ashraf S, Hurd TW, Vega-Warner V, Fang H, Beck BB, Gribouval O, Zhou W, Diaz KA, Natarajan S, Wiggins RC, Lovric S, Chernin G, Schoeb DS, Ovunc B, Frishberg Y, Soliman NA, Fathy HM, Goebel H, Hoefele J, Weber LT, Innis JW, Faul C, Han Z, Washburn J, Antignac C, Levy S, Otto EA, Hildebrandt F. ARHGDIA mutations cause nephrotic syndrome via defective RHO GTPase signaling. J Clin Invest 123: 3243–3253, 2013. doi: 10.1172/JCI69134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488. Ma R, Chaudhari S, Li W. Canonical transient receptor potential 6 channel: a new target of reactive oxygen species in renal physiology and pathology. Antioxid Redox Signal 25: 732–748, 2016. doi: 10.1089/ars.2016.6661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489. Martinez GQ, Gordon SE. Multimerization of Homo sapiens TRPA1 ion channel cytoplasmic domains. PLoS One 14: e0207835, 2019. doi: 10.1371/journal.pone.0207835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 490. Cayouette S, Lussier MP, Mathieu EL, Bousquet SM, Boulay G. Exocytotic insertion of TRPC6 channel into the plasma membrane upon Gq protein-coupled receptor activation. J Biol Chem 279: 7241–7246, 2004. doi: 10.1074/jbc.M312042200. [DOI] [PubMed] [Google Scholar]
- 491. Xie J, An SW, Jin X, Gui Y, Huang CL. Munc13 mediates klotho-inhibitable diacylglycerol-stimulated exocytotic insertion of pre-docked TRPC6 vesicles. PLoS One 15: e0229799, 2020. doi: 10.1371/journal.pone.0229799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 492. Talbot BE, Vandorpe DH, Stotter BR, Alper SL, Schlondorff JS. Transmembrane insertases and N-glycosylation critically determine synthesis, trafficking, and activity of the nonselective cation channel TRPC6. J Biol Chem 294: 12655–12669, 2019. doi: 10.1074/jbc.RA119.008299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 493. Hidalgo C, Donoso P. Crosstalk between calcium and redox signaling: from molecular mechanisms to health implications. Antioxid Redox Signal 10: 1275–1312, 2008. doi: 10.1089/ars.2007.1886. [DOI] [PubMed] [Google Scholar]
- 494. Wang Z, Wei X, Zhang Y, Ma X, Li B, Zhang S, Du P, Zhang X, Yi F. NADPH oxidase-derived ROS contributes to upregulation of TRPC6 expression in puromycin aminonucleoside-induced podocyte injury. Cell Physiol Biochem 24: 619–626, 2009. doi: 10.1159/000257517. [DOI] [PubMed] [Google Scholar]
- 495. Thilo F, Lee M, Xia S, Zakrzewicz A, Tepel M. High glucose modifies transient receptor potential canonical type 6 channels via increased oxidative stress and syndecan-4 in human podocytes. Biochem Biophys Res Commun 450: 312–317, 2014. doi: 10.1016/j.bbrc.2014.05.116. [DOI] [PubMed] [Google Scholar]
- 496. Yang H, Zhao B, Liao C, Zhang R, Meng K, Xu J, Jiao J. High glucose-induced apoptosis in cultured podocytes involves TRPC6-dependent calcium entry via the RhoA/ROCK pathway. Biochem Biophys Res Commun 434: 394–400, 2013. doi: 10.1016/j.bbrc.2013.03.087. [DOI] [PubMed] [Google Scholar]
- 497. Ma R, Wang Y, Xu Y, Wang R, Wang X, Yu N, Li M, Zhou Y. Tacrolimus protects podocytes from apoptosis via downregulation of TRPC6 in diabetic nephropathy. J Diabetes Res 2021: 8832114, 2021. doi: 10.1155/2021/8832114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498. Wang Y, Ding M, Chaudhari S, Ding Y, Yuan J, Stankowska D, He S, Krishnamoorthy R, Cunningham JT, Ma R. Nuclear factor κB mediates suppression of canonical transient receptor potential 6 expression by reactive oxygen species and protein kinase C in kidney cells. J Biol Chem 288: 12852–12865, 2013. doi: 10.1074/jbc.M112.410357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 499. Sun L, Li W, Li W, Xiong L, Li G, Ma R. Astragaloside IV prevents damage to human mesangial cells through the inhibition of the NADPH oxidase/ROS/Akt/NF-κB pathway under high glucose conditions. Int J Mol Med 34: 167–176, 2014. doi: 10.3892/ijmm.2014.1741. [DOI] [PubMed] [Google Scholar]
- 500. Christiansen JS, Frandsen M, Parving HH. Effect of intravenous glucose infusion on renal function in normal man and in insulin-dependent diabetics. Diabetologia 21: 368–373, 1981. doi: 10.1007/BF00252683. [DOI] [PubMed] [Google Scholar]
- 501. Vallon V, Verma S. Effects of SGLT2 Inhibitors on kidney and cardiovascular function. Annu Rev Physiol 83: 503–528, 2021. doi: 10.1146/annurev-physiol-031620-095920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 502. Thomson SC, Vallon V. Effects of SGLT2 inhibitor and dietary NaCl on glomerular hemodynamics assessed by micropuncture in diabetic rats. Am J Physiol Renal Physiol 320: F761–F771, 2021. doi: 10.1152/ajprenal.00552.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 503. Kriz W. The inability of podocytes to proliferate: cause, consequences, and origin. Anat Rec (Hoboken) 303: 2588–2596, 2020. doi: 10.1002/ar.24291. [DOI] [PubMed] [Google Scholar]
- 504. Fogo AB. Causes and pathogenesis of focal segmental glomerulosclerosis. Nat Rev Nephrol 11: 76–87, 2015. doi: 10.1038/nrneph.2014.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 505. De Vriese AS, Wetzels JF, Glassock RJ, Sethi S, Fervenza FC. Therapeutic trials in adult FSGS: lessons learned and the road forward. Nat Rev Nephrol 17: 619–630, 2021. doi: 10.1038/s41581-021-00427-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 506. Yao J, Le TC, Kos CH, Henderson JM, Allen PG, Denker BM, Pollak MR. Alpha-actinin-4-mediated FSGS: an inherited kidney disease caused by an aggregated and rapidly degraded cytoskeletal protein. PLoS Biol 2: e167, 2004. doi: 10.1371/journal.pbio.0020167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 507. Brown EJ, Schlöndorff JS, Becker DJ, Tsukaguchi H, Tonna SJ, Uscinski AL, Higgs HN, Henderson JM, Pollak MR. Mutations in the formin gene INF2 cause focal segmental glomerulosclerosis. Nat Genet 42: 72–76, 2010. doi: 10.1038/ng.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 508. Friedman DJ, Pollak MR. Genetics of kidney failure and the evolving story of APOL1. J Clin Invest 121: 3367–3374, 2011. doi: 10.1172/JCI46263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 509. Friedman DJ, Pollak MR. APOL1 Nephropathy: from genetics to clinical applications. Clin J Am Soc Nephrol 16: 294–303, 2021. doi: 10.2215/CJN.15161219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 510. Datta S, Kataria R, Zhang JY, Moore S, Petitpas K, Mohamed A, Zahler N, Pollak MR, Olabisi OA. Kidney disease-associated APOL1 variants have dose-dependent, dominant toxic gain-of-function. J Am Soc Nephrol 31: 2083–2096, 2020. doi: 10.1681/ASN.2020010079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 511. McCarthy ET, Sharma M, Savin VJ. Circulating permeability factors in idiopathic nephrotic syndrome and focal segmental glomerulosclerosis. Clin J Am Soc Nephrol 5: 2115–2121, 2010. doi: 10.2215/CJN.03800609. [DOI] [PubMed] [Google Scholar]
- 512. Jefferson JA, Shankland SJ. Has the circulating permeability factor in primary FSGS been found? Kidney Int 84: 235–238, 2013. doi: 10.1038/ki.2013.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 513. Hoyer JR, Vernier RL, Najarian JS, Raij L, Simmons RL, Michael AF. Recurrence of idiopathic nephrotic syndrome after renal transplantation. Lancet 2: 343–348, 1972. doi: 10.1016/s0140-6736(72)91734-5. [DOI] [PubMed] [Google Scholar]
- 514. Uffing A, Pérez-Sáez MJ, Mazzali M, Manfro RC, Bauer AC, de Sottomaior Drumond F, et al. Recurrence of FSGS after kidney transplantation in adults. Clin J Am Soc Nephrol 15: 247–256, 2020. doi: 10.2215/CJN.08970719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 515. Alasfar S, Matar D, Montgomery RA, Desai N, Lonze B, Vujjini V, Estrella MM, Manllo Dieck J, Khneizer G, Sever S, Reiser J, Alachkar N. Rituximab and therapeutic plasma exchange in recurrent focal segmental glomerulosclerosis postkidney transplantation. Transplantation 102: e115–e120, 2018. doi: 10.1097/TP.0000000000002008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 516. Saleem MA. The phenomenon of focal segmental glomerulosclerosis post-transplantation–a one-hit wonder? Pediatr Nephrol 27: 2163–2166, 2012. doi: 10.1007/s00467-012-2218-5. [DOI] [PubMed] [Google Scholar]
- 517. De Vriese AS, Sethi S, Nath KA, Glassock RJ, Fervenza FC. Differentiating primary, genetic, and secondary FSGS in adults: a clinicopathologic approach. J Am Soc Nephrol 29: 759–774, 2018. doi: 10.1681/ASN.2017090958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 518. Rosenberg AZ, Naicker S, Winkler CA, Kopp JB. HIV-associated nephropathies: epidemiology, pathology, mechanisms and treatment. Nat Rev Nephrol 11: 150–160, 2015. doi: 10.1038/nrneph.2015.9. [DOI] [PubMed] [Google Scholar]
- 519. Kim YH, Goyal M, Kurnit D, Wharram B, Wiggins J, Holzman L, Kershaw D, Wiggins R. Podocyte depletion and glomerulosclerosis have a direct relationship in the PAN-treated rat. Kidney Int 60: 957–968, 2001. doi: 10.1046/j.1523-1755.2001.060003957.x. [DOI] [PubMed] [Google Scholar]
- 520. Huang H, You Y, Lin X, Tang C, Gu X, Huang M, Qin Y, Tan J, Huang F. Inhibition of TRPC6 signal pathway alleviates podocyte injury induced by TGF-beta1. Cell Physiol Biochem 41: 163–172, 2017. doi: 10.1159/000455985. [DOI] [PubMed] [Google Scholar]
- 521. Coward RJ, Foster RR, Patton D, Ni L, Lennon R, Bates DO, Harper SJ, Mathieson PW, Saleem MA. Nephrotic plasma alters slit diaphragm-dependent signaling and translocates nephrin, Podocin, and CD2 associated protein in cultured human podocytes. J Am Soc Nephrol 16: 629–637, 2005. doi: 10.1681/ASN.2004030172. [DOI] [PubMed] [Google Scholar]
- 522. Bitzan M, Babayeva S, Vasudevan A, Goodyer P, Torban E. TNFα pathway blockade ameliorates toxic effects of FSGS plasma on podocyte cytoskeleton and β3 integrin activation. Pediatr Nephrol 27: 2217–2226, 2012. doi: 10.1007/s00467-012-2163-3. [DOI] [PubMed] [Google Scholar]
- 523. Kachurina N, Chung CF, Benderoff E, Babayeva S, Bitzan M, Goodyer P, Kitzler T, Matar D, Cybulsky AV, Alachkar N, Torban E. Novel unbiased assay for circulating podocyte-toxic factors associated with recurrent focal segmental glomerulosclerosis. Am J Physiol Renal Physiol 310: F1148–F1156, 2016. doi: 10.1152/ajprenal.00349.2015. [DOI] [PubMed] [Google Scholar]
- 524. den Braanker DJ, Maas RJ, Deegens JK, Yanginlar C, Wetzels JF, van der Vlag J, Nijenhuis T. Novel in vitro assays to detect circulating permeability factor(s) in idiopathic focal segmental glomerulosclerosis. Nephrol Dial Transplant 36: 247–256, 2021. doi: 10.1093/ndt/gfaa211. [DOI] [PubMed] [Google Scholar]
- 525. Hattori M, Akioka Y, Chikamoto H, Kobayashi N, Tsuchiya K, Shimizu M, Kagami S, Tsukaguchi H. Increase of integrin-linked kinase activity in cultured podocytes upon stimulation with plasma from patients with recurrent FSGS. Am J Transplant 8: 1550–1556, 2008. doi: 10.1111/j.1600-6143.2008.02287.x. [DOI] [PubMed] [Google Scholar]
- 526. Doublier S, Musante L, Lupia E, Candiano G, Spatola T, Caridi G, Zennaro C, Carraro M, Ghiggeri GM, Camussi G. Direct effect of plasma permeability factors from patients with idiopathic FSGS on nephrin and podocin expression in human podocytes. Int J Mol Med 16: 49–58, 2005. [PubMed] [Google Scholar]
- 527. Kim EY, Dryer SE. RAGE and αVβ3-integrin are essential for suPAR signaling in podocytes. Biochim Biophys Acta Mol Basis Dis 1867: 166186, 2021. doi: 10.1016/j.bbadis.2021.166186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 528. Hahm E, Wei C, Fernandez I, Li J, Tardi NJ, Tracy M, Wadhwani S, Cao Y, Peev V, Zloza A, Lusciks J, Hayek SS, O’Connor C, Bitzer M, Gupta V, Sever S, Sykes DB, Scadden DT, Reiser J. Bone marrow-derived immature myeloid cells are a main source of circulating suPAR contributing to proteinuric kidney disease. Nat Med 23: 100–106, 2017. doi: 10.1038/nm.4242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 529. Wei C, Spear R, Hahm E, Reiser J. suPAR, a circulating kidney disease factor. Front Med (Lausanne) 8: 745838, 2021. doi: 10.3389/fmed.2021.745838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 530. Wei C, El Hindi S, Li J, Fornoni A, Goes N, Sageshima J, Maiguel D, Karumanchi SA, Yap HK, Saleem M, Zhang Q, Nikolic B, Chaudhuri A, Daftarian P, Salido E, Torres A, Salifu M, Sarwal MM, Schaefer F, Morath C, Schwenger V, Zeier M, Gupta V, Roth D, Rastaldi MP, Burke G, Ruiz P, Reiser J. Circulating urokinase receptor as a cause of focal segmental glomerulosclerosis. Nat Med 17: 952–960, 2011. doi: 10.1038/nm.2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 531. Huang J, Liu G, Zhang YM, Cui Z, Wang F, Liu XJ, Chu R, Zhao MH. Urinary soluble urokinase receptor levels are elevated and pathogenic in patients with primary focal segmental glomerulosclerosis. BMC Med 12: 81, 2014. doi: 10.1186/1741-7015-12-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 532. Franco Palacios CR, Lieske JC, Wadei HM, Rule AD, Fervenza FC, Voskoboev N, Garovic VD, Zand L, Stegall MD, Cosio FG, Amer H. Urine but not serum soluble urokinase receptor (suPAR) may identify cases of recurrent FSGS in kidney transplant candidates. Transplantation 96: 394–399, 2013. doi: 10.1097/TP.0b013e3182977ab1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 533. Wei C, Li J, Adair BD, Zhu K, Cai J, Merchant M, Samelko B, Liao Z, Koh KH, Tardi NJ, Dande RR, Liu S, Ma J, Dibartolo S, Hägele S, Peev V, Hayek SS, Cimbaluk DJ, Tracy M, Klein J, Sever S, Shattil SJ, Arnaout MA, Reiser J. uPAR isoform 2 forms a dimer and induces severe kidney disease in mice. J Clin Invest 129: 1946–1959, 2019. doi: 10.1172/JCI124793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 534. Braun F, Homeyer I, Alachkar N, Huber TB. Immune-mediated entities of (primary) focal segmental glomerulosclerosis. Cell Tissue Res 385: 423–434, 2021. doi: 10.1007/s00441-021-03454-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 535. Bustos C, González E, Muley R, Alonso JL, Egido J. Increase of tumour necrosis factor alpha synthesis and gene expression in peripheral blood mononuclear cells of children with idiopathic nephrotic syndrome. Eur J Clin Invest 24: 799–805, 1994. doi: 10.1111/j.1365-2362.1994.tb02022.x. [DOI] [PubMed] [Google Scholar]
- 536. McCarthy ET, Sharma R, Sharma M, Li JZ, Ge XL, Dileepan KN, Savin VJ. TNF-alpha increases albumin permeability of isolated rat glomeruli through the generation of superoxide. J Am Soc Nephrol 9: 433–438, 1998. doi: 10.1681/ASN.V93433. [DOI] [PubMed] [Google Scholar]
- 537. Kanda S, Harita Y, Shibagaki Y, Sekine T, Igarashi T, Inoue T, Hattori S. Tyrosine phosphorylation-dependent activation of TRPC6 regulated by PLC-γ1 and nephrin: effect of mutations associated with focal segmental glomerulosclerosis. Mol Biol Cell 22: 1824–1835, 2011. doi: 10.1091/mbc.E10-12-0929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 538. Kim EY, Roshanravan H, Dryer SE. Syndecan-4 ectodomain evokes mobilization of podocyte TRPC6 channels and their associated pathways: an essential role for integrin signaling. Biochim Biophys Acta 1853: 2610–2620, 2015. doi: 10.1016/j.bbamcr.2015.07.011. [DOI] [PubMed] [Google Scholar]
- 539. Liu Y, Echtermeyer F, Thilo F, Theilmeier G, Schmidt A, Schülein R, Jensen BL, Loddenkemper C, Jankowski V, Marcussen N, Gollasch M, Arendshorst WJ, Tepel M. The proteoglycan syndecan 4 regulates transient receptor potential canonical 6 channels via RhoA/Rho-associated protein kinase signaling. Arterioscler Thromb Vasc Biol 32: 378–385, 2012. doi: 10.1161/ATVBAHA.111.241018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 540. Kim EY, Dryer SE. Effects of TRPC6 inactivation on glomerulosclerosis and renal fibrosis in aging rats. Cells 10: 856, 2021. doi: 10.3390/cells10040856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 541. Wu YL, Xie J, An SW, Oliver N, Barrezueta NX, Lin MH, Birnbaumer L, Huang CL. Inhibition of TRPC6 channels ameliorates renal fibrosis and contributes to renal protection by soluble klotho. Kidney Int 91: 830–841, 2017. doi: 10.1016/j.kint.2016.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 542. Kong W, Haschler TN, Nürnberg B, Krämer S, Gollasch M, Markó L. Renal fibrosis, immune cell infiltration and changes of TRPC channel expression after unilateral ureteral obstruction in Trpc6-/- mice. Cell Physiol Biochem 52: 1484–1502, 2019. doi: 10.1161/ATVBAHA.111.241018. [DOI] [PubMed] [Google Scholar]
- 543. Zhang Y, Yin N, Sun A, Wu Q, Hu W, Hou X, Zeng X, Zhu M, Liao Y. Transient receptor potential channel 6 knockout ameliorates kidney fibrosis by inhibition of epithelial-mesenchymal transition. Front Cell Dev Biol 8: 602703, 2021. doi: 10.3389/fcell.2020.602703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 544. Weber EW, Han F, Tauseef M, Birnbaumer L, Mehta D, Muller WA. TRPC6 is the endothelial calcium channel that regulates leukocyte transendothelial migration during the inflammatory response. J Exp Med 212: 1883–1899, 2015. doi: 10.1084/jem.20150353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 545. Davis J, Burr AR, Davis GF, Birnbaumer L, Molkentin JD. A TRPC6-dependent pathway for myofibroblast transdifferentiation and wound healing in vivo. Dev Cell 23: 705–715, 2012. doi: 10.1016/j.devcel.2012.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 546. Kim JH, Xie J, Hwang KH, Wu YL, Oliver N, Eom M, Park KS, Barrezueta N, Kong ID, Fracasso RP, Huang CL, Cha SK. Klotho may ameliorate proteinuria by targeting TRPC6 channels in podocytes. J Am Soc Nephrol 28: 140–151, 2017. doi: 10.1681/ASN.2015080888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 547. Yao X, Guo H, Sun M, Meng S, Zhu B, Fang J, Huang J, Wang H, Xing L. Klotho ameliorates podocyte injury through targeting TRPC6 channel in diabetic nephropathy. J Diabetes Res 2022: 1329380, 2022. doi: 10.1155/2022/1329380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 548. Kim EY, Dryer SE. TRPC6 inactivation reduces albuminuria induced by protein overload in Sprague Dawley rats. Cells 11: 1985, 2022. doi: 10.3390/cells11131985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 549. Spires D, Manis AD, Staruschenko A. Ion channels and transporters in diabetic kidney disease. Curr Top Membr 83: 353–396, 2019. doi: 10.1016/bs.ctm.2019.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 550. Staruschenko A. TRPC6 in diabetic kidney disease: good guy or bad guy? Kidney Int 95: 256–258, 2019. doi: 10.1016/j.kint.2018.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 551. Liu B, He X, Li S, Xu B, Birnbaumer L, Liao Y. Deletion of diacylglycerol-responsive TRPC genes attenuates diabetic nephropathy by inhibiting activation of the TGFbeta1 signaling pathway. Am J Transl Res 9: 5619–5630, 2017. [PMC free article] [PubMed] [Google Scholar]
- 552. Spires D, Ilatovskaya DV, Levchenko V, North PE, Geurts AM, Palygin O, Staruschenko A. Protective role of Trpc6 knockout in the progression of diabetic kidney disease. Am J Physiol Renal Physiol 315: F1091–F1097, 2018. doi: 10.1152/ajprenal.00155.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 553. Hassanzadeh Khayyat N, Kim EY, Dryer SE. TRPC6 inactivation does not protect against diabetic kidney disease in streptozotocin (STZ)-treated Sprague-Dawley rats. FASEB Bioadv 1: 773–782, 2019. doi: 10.1096/fba.2019-00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 554. Brosius FC 3rd, Alpers CE, Bottinger EP, Breyer MD, Coffman TM, Gurley SB, Harris RC, Kakoki M, Kretzler M, Leiter EH, Levi M, McIndoe RA, Sharma K, Smithies O, Susztak K, Takahashi N, Takahashi T; Animal Models of Diabetic Complications Consortium. Mouse models of diabetic nephropathy. J Am Soc Nephrol 20: 2503–2512, 2009. doi: 10.1681/ASN.2009070721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 555. Youssef N, Noureldein M, Njeim R, Ghadieh HE, Harb F, Azar ST, Fares N, Eid AA. Reno-protective effect of GLP-1 receptor agonists in type1 diabetes: dual action on TRPC6 and NADPH oxidases. Biomedicines 9: 1360, 2021. doi: 10.3390/biomedicines9101360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 556. Johnson RJ, Floege J, Yoshimura A, Iida H, Couser WG, Alpers CE. The activated mesangial cell: a glomerular “myofibroblast”? J Am Soc Nephrol 2: S190–S197, 1992. doi: 10.1681/ASN.V210s190. [DOI] [PubMed] [Google Scholar]
- 557. Cortinovis M, Perico N, Ruggenenti P, Remuzzi A, Remuzzi G. Glomerular hyperfiltration. Nat Rev Nephrol 18: 435–451, 2022. doi: 10.1038/s41581-022-00559-y. [DOI] [PubMed] [Google Scholar]
- 558. Wei P, Lane PH, Lane JT, Padanilam BJ, Sansom SC. Glomerular structural and functional changes in a high-fat diet mouse model of early-stage type 2 diabetes. Diabetologia 47: 1541–1549, 2004. doi: 10.1007/s00125-004-1489-1. [DOI] [PubMed] [Google Scholar]
- 559. Whiteside CI, Hurst RD, Stevanovic ZS. Calcium signaling and contractile response of diabetic glomerular mesangial cells. Kidney Int Suppl 51: S28–S33, 1995. [PubMed] [Google Scholar]
- 560. Yang Y, Xu G. Update on pathogenesis of glomerular hyperfiltration in early diabetic kidney disease. Front Endocrinol (Lausanne) 13: 872918, 2022. doi: 10.3389/fendo.2022.872918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 561. Sahai A, Mei C, Pattison TA, Tannen RL. Chronic hypoxia induces proliferation of cultured mesangial cells: role of calcium and protein kinase C. Am J Physiol Renal Physiol 273: F954–F960, 1997. doi: 10.1152/ajprenal.1997.273.6.F954. [DOI] [PubMed] [Google Scholar]
- 562. Niehof M, Borlak J. HNF4 alpha and the Ca-channel TRPC1 are novel disease candidate genes in diabetic nephropathy. Diabetes 57: 1069–1077, 2008. doi: 10.2337/db07-1065. [DOI] [PubMed] [Google Scholar]
- 563. Zhang D, Freedman BI, Flekac M, Santos E, Hicks PJ, Bowden DW, Efendic S, Brismar K, Gu HF. Evaluation of genetic association and expression reduction of TRPC1 in the development of diabetic nephropathy. Am J Nephrol 29: 244–251, 2009. doi: 10.1159/000157627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 564. Faul C, Donnelly M, Merscher-Gomez S, Chang YH, Franz S, Delfgaauw J, Chang JM, Choi HY, Campbell KN, Kim K, Reiser J, Mundel P. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat Med 14: 931–938, 2008. doi: 10.1038/nm.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 565. Yu H, Kistler A, Faridi MH, Meyer JO, Tryniszewska B, Mehta D, Yue L, Dryer S, Reiser J. Synaptopodin limits TRPC6 podocyte surface expression and attenuates proteinuria. J Am Soc Nephrol 27: 3308–3319, 2016. doi: 10.1681/ASN.2015080896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 566. Asano-Matsuda K, Ibrahim S, Takano T, Matsuda J. Role of Rho GTPase interacting proteins in subcellular compartments of podocytes. Int J Mol Sci 22: 3656, 2021. doi: 10.3390/ijms22073656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 567. Verheijden KA, Sonneveld R, Bakker-van Bebber M, Wetzels JF, van der Vlag J, Nijenhuis T. The calcium-dependent protease calpain-1 links TRPC6 activity to podocyte injury. J Am Soc Nephrol 29: 2099–2109, 2018. doi: 10.1681/ASN.2016111248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 568. Farmer LK, Rollason R, Whitcomb DJ, Ni L, Goodliff A, Lay AC, Birnbaumer L, Heesom KJ, Xu SZ, Saleem MA, Welsh GI. TRPC6 binds to and activates calpain, independent of its channel activity, and regulates podocyte cytoskeleton, cell adhesion, and motility. J Am Soc Nephrol 30: 1910–1924, 2019. doi: 10.1681/ASN.2018070729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 569. Blass G, Levchenko V, Ilatovskaya DV, Staruschenko A. Chronic cathepsin inhibition by E-64 in Dahl salt-sensitive rats. Physiol Rep 4: e12950, 2016. doi: 10.14814/phy2.12950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 570. Lin BL, Matera D, Doerner JF, Zheng N, Del Camino D, Mishra S, Bian H, Zeveleva S, Zhen X, Blair NT, Chong JA, Hessler DP, Bedja D, Zhu G, Muller GK, Ranek MJ, Pantages L, McFarland M, Netherton MR, Berry A, Wong D, Rast G, Qian HS, Weldon SM, Kuo JJ, Sauer A, Sarko C, Moran MM, Kass DA, Pullen SS. In vivo selective inhibition of TRPC6 by antagonist BI 749327 ameliorates fibrosis and dysfunction in cardiac and renal disease. Proc Natl Acad Sci USA 116: 10156–10161, 2019. doi: 10.1073/pnas.1815354116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 571. Urban N, Wang L, Kwiek S, Rademann J, Kuebler WM, Schaefer M. Identification and validation of larixyl acetate as a potent TRPC6 inhibitor. Mol Pharmacol 89: 197–213, 2016. doi: 10.1124/mol.115.100792. [DOI] [PubMed] [Google Scholar]
- 572. Seo K, Rainer PP, Shalkey Hahn V, Lee DI, Jo SH, Andersen A, Liu T, Xu X, Willette RN, Lepore JJ, Marino JP Jr, Birnbaumer L, Schnackenberg CG, Kass DA. Combined TRPC3 and TRPC6 blockade by selective small-molecule or genetic deletion inhibits pathological cardiac hypertrophy. Proc Natl Acad Sci USA 111: 1551–1556, 2014. doi: 10.1073/pnas.1308963111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 573. Yu M, Ledeboer MW, Daniels M, Malojcic G, Tibbitts TT, Coeffet-Le Gal M, Pan-Zhou XR, Westerling-Bui A, Beconi M, Reilly JF, Mundel P, Harmange JC. Discovery of a potent and selective TRPC5 inhibitor, efficacious in a focal segmental glomerulosclerosis model. ACS Med Chem Lett 10: 1579–1585, 2019. doi: 10.1021/acsmedchemlett.9b00430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 574. Häfner S, Burg F, Kannler M, Urban N, Mayer P, Dietrich A, Trauner D, Broichhagen J, Schaefer M. A (+)-larixol congener with high affinity and subtype selectivity toward TRPC6. Chem Med Chem 13: 1028–1035, 2018. doi: 10.1002/cmdc.201800021. [DOI] [PubMed] [Google Scholar]
- 575. Urban N, Neuser S, Hentschel A, Köhling S, Rademann J, Schaefer M. Pharmacological inhibition of focal segmental glomerulosclerosis-related, gain of function mutants of TRPC6 channels by semi-synthetic derivatives of larixol. Br J Pharmacol 174: 4099–4122, 2017. doi: 10.1111/bph.13977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 576. Washburn DG, Holt DA, Dodson J, McAtee JJ, Terrell LR, Barton L, Manns S, Waszkiewicz A, Pritchard C, Gillie DJ, Morrow DM, Davenport EA, Lozinskaya IM, Guss J, Basilla JB, Negron LK, Klein M, Willette RN, Fries RE, Jensen TC, Xu X, Schnackenberg CG, Marino JP Jr.. The discovery of potent blockers of the canonical transient receptor channels, TRPC3 and TRPC6, based on an anilino-thiazole pharmacophore. Bioorg Med Chem Lett 23: 4979–4984, 2013. doi: 10.1016/j.bmcl.2013.06.047. [DOI] [PubMed] [Google Scholar]
- 577. Wei Y, Zhang M, Lyu Z, Yang G, Tian T, Ding M, Zeng X, Xu F, Wang P, Li F, Liu Y, Cao Z, Lu J, Hong X, Wang H. Benzothiazole amides as TRPC3/6 inhibitors for gastric cancer treatment. ACS Omega 6: 9196–9203, 2021. doi: 10.1021/acsomega.1c00514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 578. Zheng Z, Tsvetkov D, Bartolomaeus TU, Erdogan C, Krügel U, Schleifenbaum J, Schaefer M, Nürnberg B, Chai X, Ludwig FA, N’Diaye G, Köhler MB, Wu K, Gollasch M, Markó L. Role of TRPC6 in kidney damage after acute ischemic kidney injury. Sci Rep 12: 3038, 2022. doi: 10.1038/s41598-022-06703-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 579. Zheng Z, Xu Y, Krügel U, Schaefer M, Grune T, Nürnberg B, Köhler MB, Gollasch M, Tsvetkov D, Markó L. In vivo inhibition of TRPC6 by SH045 attenuates renal fibrosis in a New Zealand Obese (NZO) mouse model of metabolic syndrome. Int J Mol Sci 23: 6870, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 580. Tang Q, Guo W, Zheng L, Wu JX, Liu M, Zhou X, Zhang X, Chen L. Structure of the receptor-activated human TRPC6 and TRPC3 ion channels. Cell Res 28: 746–755, 2018. doi: 10.1038/s41422-018-0038-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 581. Yang PL, Li XH, Wang J, Ma XF, Zhou BY, Jiao YF, Wang WH, Cao P, Zhu MX, Li PW, Xiao ZH, Li CZ, Guo CR, Lei YT, Yu Y. GSK1702934A and M085 directly activate TRPC6 via a mechanism of stimulating the extracellular cavity formed by the pore helix and transmembrane helix S6. J Biol Chem 297: 101125, 2021. doi: 10.1016/j.jbc.2021.101125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 582. Quick K, Zhao J, Eijkelkamp N, Linley JE, Rugiero F, Cox JJ, Raouf R, Gringhuis M, Sexton JE, Abramowitz J, Taylor R, Forge A, Ashmore J, Kirkwood N, Kros CJ, Richardson GP, Freichel M, Flockerzi V, Birnbaumer L, Wood JN. TRPC3 and TRPC6 are essential for normal mechanotransduction in subsets of sensory neurons and cochlear hair cells. Open Biol 2: 120068, 2012. doi: 10.1098/rsob.120068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 583. Sonneveld R, Hoenderop JG, Isidori AM, Henique C, Dijkman HB, Berden JH, Tharaux PL, van der Vlag J, Nijenhuis T. Sildenafil prevents podocyte injury via PPAR-γ-mediated TRPC6 inhibition. J Am Soc Nephrol 28: 1491–1505, 2017. doi: 10.1681/ASN.2015080885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 584. Hall G, Rowell J, Farinelli F, Gbadegesin RA, Lavin P, Wu G, Homstad A, Malone A, Lindsey T, Jiang R, Spurney R, Tomaselli GF, Kass DA, Winn MP. Phosphodiesterase 5 inhibition ameliorates angiotensin II-induced podocyte dysmotility via the protein kinase G-mediated downregulation of TRPC6 activity. Am J Physiol Renal Physiol 306: F1442–F1450, 2014. doi: 10.1152/ajprenal.00212.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 585. Agrawal S, He JC, Tharaux PL. Nuclear receptors in podocyte biology and glomerular disease. Nat Rev Nephrol 17: 185–204, 2021. doi: 10.1038/s41581-020-00339-6. [DOI] [PubMed] [Google Scholar]
- 586. Wang J, Yang K, Xu L, Zhang Y, Lai N, Jiang H, Zhang Y, Zhong N, Ran P, Lu W. Sildenafil inhibits hypoxia-induced transient receptor potential canonical protein expression in pulmonary arterial smooth muscle via cGMP-PKG-PPARγ axis. Am J Respir Cell Mol Biol 49: 231–240, 2013. doi: 10.1165/rcmb.2012-0185OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 587. Schinner E, Wetzl V, Schlossmann J. Cyclic nucleotide signalling in kidney fibrosis. Int J Mol Sci 16: 2320–2351, 2015. doi: 10.3390/ijms16022320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 588. Ma J, Zhang B, Liu S, Xie S, Yang Y, Ma J, Deng Y, Wang W, Xu L, Li R, Zhang L, Yu C, Shi W. 1,25-dihydroxyvitamin D3 inhibits podocyte uPAR expression and reduces proteinuria. PLoS One 8: e64912, 2013. doi: 10.1371/journal.pone.0064912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 589. An P, Dong S, Li X, Cai Z, Ye B, Zhang A, Shi X, Wu X. Wenyang huazhuo fang exerts transient receptor potential cation channel subfamily C member-dependent nephroprotection in a rat model of doxorubicin-induced nephropathy. J Tradit Chin Med 40: 613–620, 2020. doi: 10.19852/j.cnki.jtcm.2020.04.010. [DOI] [PubMed] [Google Scholar]
- 590. Wang Q, Tian X, Zhou W, Wang Y, Zhao H, Li J, Zhou X, Zhang H, Zhao T, Li P. Protective role of Tangshen formula on the progression of renal damage in db/db mice by TRPC6/Talin1 pathway in podocytes. J Diabetes Res 2020: 3634974, 2020. doi: 10.1155/2020/3634974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 591. Chen L, Yang J, Zhao SJ, Li TS, Jiao RQ, Kong LD. Atractylodis rhizoma water extract attenuates fructose-induced glomerular injury in rats through anti-oxidation to inhibit TRPC6/p-CaMK4 signaling. Phytomedicine 91: 153643, 2021. doi: 10.1016/j.phymed.2021.153643. [DOI] [PubMed] [Google Scholar]
- 592. Schaldecker T, Kim S, Tarabanis C, Tian D, Hakroush S, Castonguay P, Ahn W, Wallentin H, Heid H, Hopkins CR, Lindsley CW, Riccio A, Buvall L, Weins A, Greka A. Inhibition of the TRPC5 ion channel protects the kidney filter. J Clin Invest 123: 5298–5309, 2013. doi: 10.1172/JCI71165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 593. Storch U, Forst AL, Pardatscher F, Erdogmus S, Philipp M, Gregoritza M, Mederos Y Schnitzler M, Gudermann T. Dynamic NHERF interaction with TRPC4/5 proteins is required for channel gating by diacylglycerol. Proc Natl Acad Sci USA 114: E37–E46, 2017. doi: 10.1073/pnas.1612263114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 594. Trebak M, Lemonnier L, DeHaven WI, Wedel BJ, Bird GS, Putney JW Jr.. Complex functions of phosphatidylinositol 4,5-bisphosphate in regulation of TRPC5 cation channels. Pflugers Arch 457: 757–769, 2009. doi: 10.1007/s00424-008-0550-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 595. Ningoo M, Plant LD, Greka A, Logothetis DE. PIP2 regulation of TRPC5 channel activation and desensitization. J Biol Chem 296: 100726, 2021. doi: 10.1016/j.jbc.2021.100726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 596. Shen B, Wong CO, Lau OC, Woo T, Bai S, Huang Y, Yao X. Plasma membrane mechanical stress activates TRPC5 channels. PLoS One 10: e0122227, 2015. doi: 10.1371/journal.pone.0122227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 597. Jemal I, Soriano S, Conte AL, Morenilla C, Gomis A. G protein-coupled receptor signalling potentiates the osmo-mechanical activation of TRPC5 channels. Pflugers Arch 466: 1635–1646, 2014. doi: 10.1007/s00424-013-1392-z. [DOI] [PubMed] [Google Scholar]
- 598. Ogawa N, Kurokawa T, Mori Y. Sensing of redox status by TRP channels. Cell Calcium 60: 115–122, 2016. doi: 10.1016/j.ceca.2016.02.009. [DOI] [PubMed] [Google Scholar]
- 599. Al-Shawaf E, Naylor J, Taylor H, Riches K, Milligan CJ, O’Regan D, Porter KE, Li J, Beech DJ. Short-term stimulation of calcium-permeable transient receptor potential canonical 5-containing channels by oxidized phospholipids. Arterioscler Thromb Vasc Biol 30: 1453–1459, 2010. doi: 10.1161/ATVBAHA.110.205666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 600. Hsu HH, Hoffmann S, Endlich N, Velic A, Schwab A, Weide T, Schlatter E, Pavenstädt H. Mechanisms of angiotensin II signaling on cytoskeleton of podocytes. J Mol Med (Berl) 86: 1379–1394, 2008. doi: 10.1007/s00109-008-0399-y. [DOI] [PubMed] [Google Scholar]
- 601. Akbulut Y, Gaunt HJ, Muraki K, Ludlow MJ, Amer MS, Bruns A, Vasudev NS, Radtke L, Willot M, Hahn S, Seitz T, Ziegler S, Christmann M, Beech DJ, Waldmann H. (-)Englerin A is a potent and selective activator of TRPC4 and TRPC5 calcium channels. Angew Chem Int Ed Engl 54: 3787–3791, 2015. doi: 10.1002/anie.201411511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 602. Naylor J, Minard A, Gaunt HJ, Amer MS, Wilson LA, Migliore M, Cheung SY, Rubaiy HN, Blythe NM, Musialowski KE, Ludlow MJ, Evans WD, Green BL, Yang H, You Y, Li J, Fishwick CW, Muraki K, Beech DJ, Bon RS. Natural and synthetic flavonoid modulation of TRPC5 channels. Br J Pharmacol 173: 562–574, 2016. doi: 10.1111/bph.13387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 603. de Sousa Valente J, Alawi KM, Bharde S, Zarban AA, Kodji X, Thapa D, Argunhan F, Barrett B, Nagy I, Brain SD. (-)Englerin-A has analgesic and anti-inflammatory effects independent of TRPC4 and 5. Int J Mol Sci 22: 6380, 2021. doi: 10.3390/ijms22126380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 604. Batova A, Altomare D, Creek KE, Naviaux RK, Wang L, Li K, Green E, Williams R, Naviaux JC, Diccianni M, Yu AL. Englerin A induces an acute inflammatory response and reveals lipid metabolism and ER stress as targetable vulnerabilities in renal cell carcinoma. PLoS One 12: e0172632, 2017. doi: 10.1371/journal.pone.0172632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 605. Wu Z, Zhao S, Fash DM, Li Z, Chain WJ, Beutler JA. Englerins: a comprehensive review. J Nat Prod 80: 771–781, 2017. doi: 10.1021/acs.jnatprod.6b01167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 606. Zhou Y, Kim C, Pablo JL, Zhang F, Jung JY, Xiao L, Bazua-Valenti S, Emani M, Hopkins CR, Weins A, Greka A. TRPC5 channel inhibition protects podocytes in puromycin-aminonucleoside induced nephrosis models. Front Med (Lausanne) 8: 721865, 2021. doi: 10.3389/fmed.2021.721865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 607. Seiler MW, Rennke HG, Venkatachalam MA, Cotran RS. Pathogenesis of polycation-induced alterations (“fusion”) of glomerular epithelium. Lab Invest 36: 48–61, 1977. [PubMed] [Google Scholar]
- 608. Pippin JW, Brinkkoetter PT, Cormack-Aboud FC, Durvasula RV, Hauser PV, Kowalewska J, Krofft RD, Logar CM, Marshall CB, Ohse T, Shankland SJ. Inducible rodent models of acquired podocyte diseases. Am J Physiol Renal Physiol 296: F213–F229, 2009. doi: 10.1152/ajprenal.90421.2008. [DOI] [PubMed] [Google Scholar]
- 609. Reiser J, von Gersdorff G, Loos M, Oh J, Asanuma K, Giardino L, Rastaldi MP, Calvaresi N, Watanabe H, Schwarz K, Faul C, Kretzler M, Davidson A, Sugimoto H, Kalluri R, Sharpe AH, Kreidberg JA, Mundel P. Induction of B7-1 in podocytes is associated with nephrotic syndrome. J Clin Invest 113: 1390–1397, 2004. doi: 10.1172/JCI20402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 610. Blattner SM, Hodgin JB, Nishio M, Wylie SA, Saha J, Soofi AA, Vining C, Randolph A, Herbach N, Wanke R, Atkins KB, Gyung Kang H, Henger A, Brakebusch C, Holzman LB, and Kretzler M. Divergent functions of the Rho GTPases Rac1 and Cdc42 in podocyte injury. Kidney Int 84: 920–930, 2013. doi: 10.1038/ki.2013.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 611. Chaudhuri P, Rosenbaum MA, Birnbaumer L, Graham LM. Integration of TRPC6 and NADPH oxidase activation in lysophosphatidylcholine-induced TRPC5 externalization. Am J Physiol Cell Physiol 313: C541–C555, 2017. doi: 10.1152/ajpcell.00028.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 612. Chaudhuri P, Colles SM, Bhat M, Van Wagoner DR, Birnbaumer L, Graham LM. Elucidation of a TRPC6-TRPC5 channel cascade that restricts endothelial cell movement. Mol Biol Cell 19: 3203–3211, 2008. doi: 10.1091/mbc.e07-08-0765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 613. Kriz W. The pathogenesis of ‘classic’ focal segmental glomerulosclerosis—lessons from rat models. Nephrol Dial Transplant 18, Suppl 6: vi39–vi44, 2003. doi: 10.1093/ndt/gfg1064. [DOI] [PubMed] [Google Scholar]
- 614. Baradaran-Heravi A, Bauer CC, Pickles IB, Hosseini-Farahabadi S, Balgi AD, Choi K, Linley DM, Beech DJ, Roberge M, Bon RS. Nonselective TRPC channel inhibition and suppression of aminoglycoside-induced premature termination codon readthrough by the small molecule AC1903. J Biol Chem 298: 101546, 2022. doi: 10.1016/j.jbc.2021.101546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 615. Walsh L, Reilly JF, Cornwall C, Gaich GA, Gipson DS, Heerspink HJ, Johnson L, Trachtman H, Tuttle KR, Farag YM, Padmanabhan K, Pan-Zhou XR, Woodworth JR, and Czerwiec FS. Safety and efficacy of GFB-887, a TRPC5 channel inhibitor, in patients with focal segmental glomerulosclerosis, treatment-resistant minimal change disease, or diabetic nephropathy: TRACTION-2 trial design. Kidney Int Rep 6: 2575–2584, 2021. doi: 10.1016/j.ekir.2021.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 616. Wang X, Dande RR, Yu H, Samelko B, Miller RE, Altintas MM, Reiser J. TRPC5 does not cause or aggravate glomerular disease. J Am Soc Nephrol 29: 409–415, 2018. doi: 10.1681/ASN.2017060682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 617. Alawi KM, Russell FA, Aubdool AA, Srivastava S, Riffo-Vasquez Y, Baldissera L Jr, Thakore P, Saleque N, Fernandes ES, Walsh DA, Brain SD. Transient receptor potential canonical 5 (TRPC5) protects against pain and vascular inflammation in arthritis and joint inflammation. Ann Rheum Dis 76: 252–260, 2017. doi: 10.1136/annrheumdis-2015-208886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 618. Wang Y, Jarad G, Tripathi P, Pan M, Cunningham J, Martin DR, Liapis H, Miner JH, Chen F. Activation of NFAT signaling in podocytes causes glomerulosclerosis. J Am Soc Nephrol 21: 1657–1666, 2010. doi: 10.1681/ASN.2009121253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 619. Shen J, Wang R, He Z, Huang H, He X, Zhou J, Yan Y, Shen S, Shao X, Shen X, Weng C, Lin W, Chen J. NMDA receptors participate in the progression of diabetic kidney disease by decreasing Cdc42-GTP activation in podocytes. J Pathol 240: 149–160, 2016. doi: 10.1002/path.4764. [DOI] [PubMed] [Google Scholar]
- 620. Kundu S, Pushpakumar SB, Tyagi A, Coley D, Sen U. Hydrogen sulfide deficiency and diabetic renal remodeling: role of matrix metalloproteinase-9. Am J Physiol Endocrinol Metab 304: E1365–E1378, 2013. doi: 10.1152/ajpendo.00604.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 621. Mahieu S, Klug M, Millen N, Fabro A, Benmelej A, and Contini MD. Monosodium glutamate intake affect the function of the kidney through NMDA receptor. Life Sci 149: 114–119, 2016. doi: 10.1016/j.lfs.2016.02.023. [DOI] [PubMed] [Google Scholar]
- 622. Singh AP, Singh N, Bedi PM. Estradiol mitigates ischemia reperfusion-induced acute renal failure through NMDA receptor antagonism in rats. Mol Cell Biochem 434: 33–40, 2017. doi: 10.1007/s11010-017-3034-9. [DOI] [PubMed] [Google Scholar]
- 623. Kaur A, Kaur T, Singh B, Pathak D, Singh Buttar H, Pal Singh A. Curcumin alleviates ischemia reperfusion-induced acute kidney injury through NMDA receptor antagonism in rats. Ren Fail 38: 1462–1467, 2016. doi: 10.1080/0886022X.2016.1214892. [DOI] [PubMed] [Google Scholar]
- 624. Singh AP, Singh N, Bedi PM. Pioglitazone ameliorates renal ischemia reperfusion injury through NMDA receptor antagonism in rats. Mol Cell Biochem 417: 111–118, 2016. doi: 10.1007/s11010-016-2718-x. [DOI] [PubMed] [Google Scholar]
- 625. Arora S, Kaur T, Kaur A, Singh AP. Glycine aggravates ischemia reperfusion-induced acute kidney injury through N-methyl-D-aspartate receptor activation in rats. Mol Cell Biochem 393: 123–131, 2014. doi: 10.1007/s11010-014-2052-0. [DOI] [PubMed] [Google Scholar]
- 626. Zhou J, Liu S, Guo L, Wang R, Chen J, Shen J. NMDA receptor-mediated CaMKII/ERK activation contributes to renal fibrosis. BMC Nephrol 21: 392, 2020. doi: 10.1186/s12882-020-02050-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 627. Ying J, Wu J, Zhang Y, Han Y, Qian X, Yang Q, Chen Y, Chen Y, Zhu H. Ligustrazine suppresses renal NMDAR1 and caspase-3 expressions in a mouse model of sepsis-associated acute kidney injury. Mol Cell Biochem 464: 73–81, 2020. doi: 10.1007/s11010-019-03650-4. [DOI] [PubMed] [Google Scholar]
- 628. Lin CS, Hung SF, Huang HS, Ma MC. Blockade of the N-methyl-D-aspartate glutamate receptor ameliorates lipopolysaccharide-induced renal insufficiency. PLoS One 10: e0132204, 2015. doi: 10.1371/journal.pone.0132204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 629. Parisi E, Bozic M, Ibarz M, Panizo S, Valcheva P, Coll B, Fernández E, Valdivielso JM. Sustained activation of renal N-methyl-D-aspartate receptors decreases vitamin D synthesis: a possible role for glutamate on the onset of secondary HPT. Am J Physiol Endocrinol Metab 299: E825–E831, 2010. doi: 10.1152/ajpendo.00428.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 630. Menzies RI, Booth JWR, Mullins JJ, Bailey MA, Tam FW, Norman JT, Unwin RJ. Hyperglycemia-induced renal P2X7 receptor activation enhances diabetes-related injury. EBioMedicine 19: 73–83, 2017. doi: 10.1016/j.ebiom.2017.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 631. Solini A, Santini E, Chimenti D, Chiozzi P, Pratesi F, Cuccato S, Falzoni S, Lupi R, Ferrannini E, Pugliese G, Di Virgilio F. Multiple P2X receptors are involved in the modulation of apoptosis in human mesangial cells: evidence for a role of P2X4. Am J Physiol Renal Physiol 292: F1537–F1547, 2007. doi: 10.1152/ajprenal.00440.2006. [DOI] [PubMed] [Google Scholar]
- 632. Graciano ML, Nishiyama A, Jackson K, Seth DM, Ortiz RM, Prieto-Carrasquero MC, Kobori H, Navar LG. Purinergic receptors contribute to early mesangial cell transformation and renal vessel hypertrophy during angiotensin II-induced hypertension. Am J Physiol Renal Physiol 294: F161–F169, 2008. doi: 10.1152/ajprenal.00281.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 633. Bülow RD, and Boor P. Extracellular matrix in kidney fibrosis: more than just a scaffold. J Histochem Cytochem 67: 643–661, 2019. doi: 10.1369/0022155419849388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 634. Menè P, Pugliese G, Pricci F, Di Mario U, Cinotti GA, Pugliese F. High glucose level inhibits capacitative Ca2+ influx in cultured rat mesangial cells by a protein kinase C-dependent mechanism. Diabetologia 40: 521–527, 1997. doi: 10.1007/s001250050710. [DOI] [PubMed] [Google Scholar]
- 635. Jiang H, Zou S, Chaudhari S, Ma R. Short-term high-glucose treatment decreased abundance of Orai1 protein through posttranslational mechanisms in rat mesangial cells. Am J Physiol Renal Physiol 314: F855–F863, 2018. doi: 10.1152/ajprenal.00513.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 636. Mai X, Shang J, Liang S, Yu B, Yuan J, Lin Y, Luo R, Zhang F, Liu Y, Lv X, Li C, Liang X, Wang W, Zhou J. Blockade of Orai1 store-operated calcium entry protects against renal fibrosis. J Am Soc Nephrol 27: 3063–3078, 2016. doi: 10.1681/ASN.2015080889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 637. Wu P, Ren Y, Ma Y, Wang Y, Jiang H, Chaudhari S, Davis ME, Zuckerman JE, Ma R. Negative regulation of Smad1 pathway and collagen IV expression by store-operated Ca2+ entry in glomerular mesangial cells. Am J Physiol Renal Physiol 312: F1090–F1100, 2017. doi: 10.1152/ajprenal.00642.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 638. Chaudhari S, Yazdizadeh Shotorbani P, Tao Y, Davis ME, Mallet RT, Ma R. Inhibition of interleukin-6 on matrix protein production by glomerular mesangial cells and the pathway involved. Am J Physiol Renal Physiol 318: F1478–F1488, 2020. doi: 10.1152/ajprenal.00043.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 639. Soni H, Adebiyi A. Urotensin II-induced store-operated Ca2+ entry contributes to glomerular mesangial cell proliferation and extracellular matrix protein production under high glucose conditions. Sci Rep 7: 18049, 2017. doi: 10.1038/s41598-017-18143-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 640. Huang L, Ma R, Lin T, Chaudhari S, Shotorbani PY, Yang L, Wu P. Glucagon-like peptide-1 receptor pathway inhibits extracellular matrix production by mesangial cells through store-operated Ca2+ channel. Exp Biol Med (Maywood) 244: 1193–1201, 2019. doi: 10.1177/1535370219876531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 641. Huang L, Lin T, Shi M, Chen X, Wu P. Liraglutide suppresses production of extracellular matrix proteins and ameliorates renal injury of diabetic nephropathy by enhancing Wnt/β-catenin signaling. Am J Physiol Renal Physiol 319: F458–F468, 2020. doi: 10.1152/ajprenal.00128.2020. [DOI] [PubMed] [Google Scholar]
- 642. Rangaswami J, Bhalla V, de Boer IH, Staruschenko A, Sharp JA, Singh RR, Lo KB, Tuttle K, Vaduganathan M, Ventura H, McCullough PA; American Heart Association Council on the Kidney in Cardiovascular Disease; Council on Arteriosclerosis, Thrombosis and Vascular Biology; Council on Cardiovascular and Stroke Nursing; Council on Clinical Cardiology; Council on Lifestyle and Cardiometabolic Health. Cardiorenal protection with the newer antidiabetic agents in patients with diabetes and chronic kidney disease: a Scientific Statement From the American Heart Association. Circulation 142: e265–e286, 2020. doi: 10.1161/CIR.0000000000000920. [DOI] [PubMed] [Google Scholar]
- 643. Hsu YC, Chang PJ, Ho C, Huang YT, Shih YH, Wang CJ, Lin CL. Protective effects of miR-29a on diabetic glomerular dysfunction by modulation of DKK1/Wnt/β-catenin signaling. Sci Rep 6: 30575, 2016. doi: 10.1038/srep30575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 644. Carlström M. Nitric oxide signalling in kidney regulation and cardiometabolic health. Nat Rev Nephrol 17: 575–590, 2021. doi: 10.1038/s41581-021-00429-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 645. Jairaman A, Prakriya M. Molecular pharmacology of store-operated CRAC channels. Channels 7: 402–414, 2013. doi: 10.4161/chan.25292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 646. Zeng B, Chen GL, Garcia-Vaz E, Bhandari S, Daskoulidou N, Berglund LM, Jiang H, Hallett T, Zhou LP, Huang L, Xu ZH, Nair V, Nelson RG, Ju W, Kretzler M, Atkin SL, Gomez MF, Xu SZ. ORAI channels are critical for receptor-mediated endocytosis of albumin. Nat Commun 8: 1920, 2017. doi: 10.1038/s41467-017-02094-y. [DOI] [PMC free article] [PubMed] [Google Scholar]