

Keywords: antagonists, disease, homeostasis, proteases, receptors
Abstract
Proteases are signaling molecules that specifically control cellular functions by cleaving protease-activated receptors (PARs). The four known PARs are members of the large family of G protein-coupled receptors. These transmembrane receptors control most physiological and pathological processes and are the target of a large proportion of therapeutic drugs. Signaling proteases include enzymes from the circulation; from immune, inflammatory epithelial, and cancer cells; as well as from commensal and pathogenic bacteria. Advances in our understanding of the structure and function of PARs provide insights into how diverse proteases activate these receptors to regulate physiological and pathological processes in most tissues and organ systems. The realization that proteases and PARs are key mediators of disease, coupled with advances in understanding the atomic level structure of PARs and their mechanisms of signaling in subcellular microdomains, has spurred the development of antagonists, some of which have advanced to the clinic. Herein we review the discovery, structure, and function of this receptor system, highlight the contribution of PARs to homeostatic control, and discuss the potential of PAR antagonists for the treatment of major diseases.
CLINICAL HIGHLIGHTS
-
1)
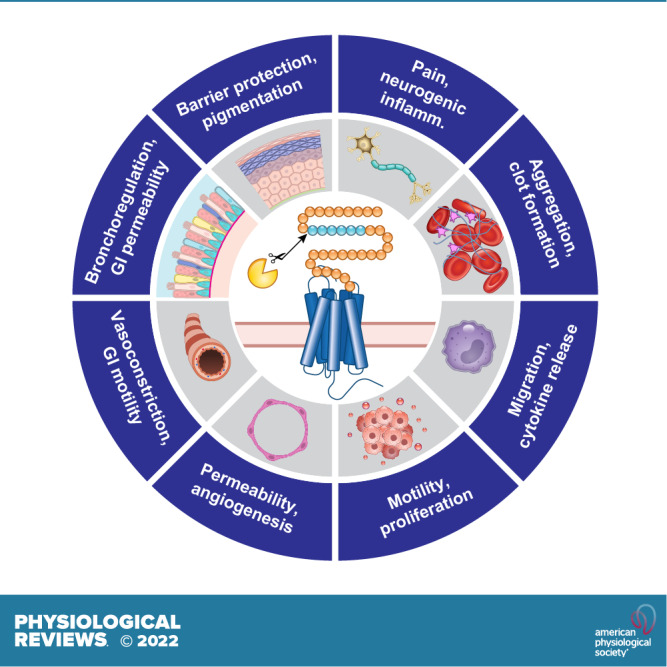
PARs are widely expressed on platelets, epithelial cells, endothelial cells, smooth muscle cells, innate and adaptive immune cells, neurons, and cancer cells and contribute to the physiology and pathology of every organ system.
-
2)
PAR activation by proteases, including coagulation factors, position the receptor as a key target for major causes of death including coronary artery disease, cerebrovascular disease, cancer, and, more recently, SARS-CoV-2 infection.
-
3)
Drugs, including peptidic, small molecules, and antibodies, have been developed to target PAR1, PAR2, and PAR4. Vorapaxar, a PAR1 antagonist, has been tested for the treatment of acute coronary syndrome and the prevention of atherothrombotic events; vorapaxar has been approved for specific indications.
-
4)
Given their central role in highly prevalent diseases including coronary artery disease and cerebrovascular disease, PARs will continue to be attractive targets for drug development; however, much of our knowledge regarding PAR physiology is based on genetically engineered mouse models. There are species differences with PAR function, therefore the translation of laboratory findings to clinical management will require an increase in the number of studies involving humans.
1. INTRODUCTION
Proteases are critically important regulators of homeostasis and mediators of disease. The coordinated cascade of proteolytic events that lead to the formation of a blood clot illustrates the importance of proteases in homeostatic control. The requirement of proteases for the entry of viruses into cells exemplifies their contributions to diseases of global relevance. In line with their important regulatory roles, the activity of proteases is tightly controlled; most proteases are formed as inactive zymogens, and activity is further regulated by endogenous inhibitors or by intracellular and extracellular acidity. Herein, we review another aspect of the regulatory role of proteases: their ability to function as hormones or paracrine substances that regulate cells by cleaving and activating protease-activated receptors (PARs). PARs are a subfamily of four G protein-coupled receptors (GPCRs), the largest family of cell surface receptors and the target of one-third of therapeutic drugs. All PARs share the main features of GPCRs, with seven transmembrane (TM) domains, three extracellular loop (ECL) and three intracellular loop (ICL) domains, an extracellular NH2-terminal domain, and an intracellular COOH-terminal domain. However, proteases activate PARs by a unique mechanism. Although some proteases can dock with their receptors, binding per se does not lead to signaling. Instead, proteolysis either reveals a tethered ligand domain that binds to and activates the cleaved PAR or results in a conformational change that initiates signaling (FIGURE 1). Synthetic peptides that mimic the tethered ligand sequence, referred to herein as activating peptides, can directly activate some PARs without the need for proteolysis. Activating peptides are useful tools for studying PAR activation and are a starting point for the development of selective ligands.
FIGURE 1.
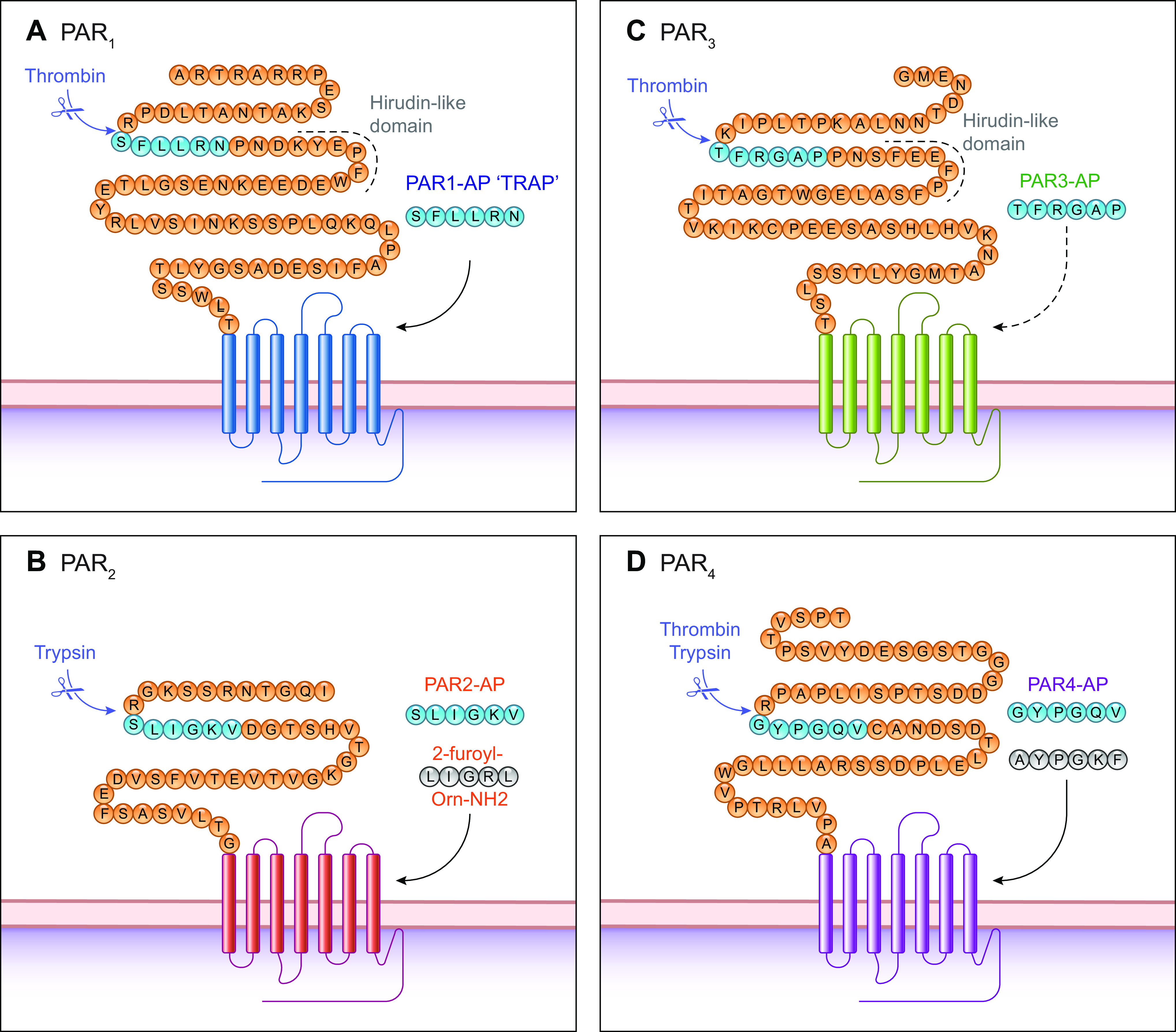
Canonical protease-activated receptor (PAR) cleavage sites and activation mechanisms. A–D: NH2-terminal domains of PAR1-4 highlighting the canonical cleavage site for thrombin or trypsin in the human receptors. Cleavage can reveal a tethered ligand (blue) from which soluble activating peptides (APs) were derived; PAR3-AP is unable to activate PAR3. APs have been modified to generate selective and potent activating peptides (e.g., for PAR2 and PAR4; gray). TRAP, thrombin receptor-activating peptide.
The prototypical PAR, PAR1, was discovered as a mediator of thrombin-induced aggregation of platelets and revealed a major role for PARs in hemostasis in the circulatory system. PARs are now known to mediate the hormone-like actions of diverse proteases, including enzymes from the circulation and those secreted by inflammatory, immune, epithelial, and cancer cells. Proteases from commensal and pathogenic microorganisms can also activate PARs. Proteases and PARs are known to control most tissues and organ systems and to regulate important homeostatic and disease processes. This review summarizes the discovery, structure, and function of PARs in the major cell types and organ systems. The review then highlights the mechanisms by which activated PARs engage the intracellular signaling machinery to control physiological and pathological processes and discusses how new knowledge about the structure and function of PARs has informed the development of selective antagonists, some of which have advanced to the clinic.
2. PAR DISCOVERY AND GENE AND PROTEIN STRUCTURE
The key features of the gene and protein structure of PARs are summarized in TABLE 1.
Table 1.
Summary of the key properties of PARs, highlighting the gene and protein structure, mechanisms of activation, and sites of expression of human receptors
| PAR1 | PAR2 | PAR3 | PAR4 | |
|---|---|---|---|---|
| Amino acids* | 425 | 397 | 374 | 385 |
| Gene name | F2r | F2rl1 | F2rl2 | F2rl3 |
| Gene structure | 2 exons | 2 exons | 2 exons | 2 exons |
| Chromosome | 5q13.3 | 5q13.3 | 5q13.3 | 19p13.11 |
| Highest transcript expression | Spleen, gall bladder, skin, endometrium, and lung | Colon, duodenum, stomach, small intestine, and gall bladder | Fat, stomach, thyroid, colon, and thyroid | Fat, stomach, thyroid, colon, and thyroid |
| Canonical cleavage site | Arg41/Ser42 | Arg36/Ser37 | Lys38/Thr39 | Arg47/Gly48 |
| Exposed canonical NH2-terminal domain | S42FLLRN | S37LIGKV | T39FRGAP | G48YPGQV |
| Canonical activating | ||||
| Peptides | SFLLRN (hPAR1) | SLIGKV (hPAR2) | TFRGAP (hPAR3) inactive | GYPGQV (hPAR4) |
| Analogs | 2-furoyl-LIGRLO-NH2 | AYPGKF | ||
| Activating proteases(canonical and biased) | Thrombin APC Plasmin MMP1 Proteinase-3 Elastase FVIIa FXa |
Trypsin Tryptase Elastase Proteinase-3 Cathepsin G Cathepsin S FXa KLK-4 Chymase Legumain |
Thrombin APC |
Thrombin Trypsin Plasmin Cathepsin G Cathepsin S KLK-14 |
APC, activated protein C; MMP, matrix metalloprotease; PAR, protease-activated receptor. *Including the signal sequence, obtained from UniProt.
2.1. Discovery and Gene Structure
2.1.1. PAR1.
The concept that proteases can act as hormonal regulators of cellular functions is illustrated by the ability of thrombin to aggregate human platelets by cleaving PAR1. The search for the platelet thrombin receptor was motivated by a desire to develop treatments for vascular diseases and coagulopathies, in which thrombin was implicated. Schmidt hypothesized in 1872 that a protease would mediate the conversion of fibrinogen to fibrin; thrombin was ultimately identified as the responsible protease (1). Thrombin also promotes platelet activation and aggregation. In 1990, two groups reported that microinjection of poly(A)+ mRNA from two thrombin-responsive cell lines, hamster lung fibroblasts (2) and human umbilical vein endothelial cells (HUVECs; Ref. 3), into Xenopus oocytes led to thrombin responsiveness. Platelets were not used as the source of mRNA because they contain little nucleic acid. An expression cloning approach in which cDNA from human erythroleukemic cell lines (HEL; Dami) was injected into Xenopus oocytes led to cloning of the thrombin receptor (4). F2r, the official HUGO Gene Nomenclature Committee gene symbol for PAR1, contains two exons separated by a large intron. The second exon contains most of the coding sequence, including the critical thrombin cleavage site. F2r exhibits ubiquitous expression with the highest expression levels in the spleen, gall bladder, skin, endometrium, and lung.
2.1.2. PAR2.
Different PAR variants were hypothesized given the expansive role of serine proteases in physiological processes. In 1994, low-stringency screening of a mouse genomic library using probes corresponding to TM2 and TM6 of the bovine substance K receptor revealed a gene for which the predicted GPCR was homologous to PAR1 and thus designated PAR2. PAR2 is not a second thrombin receptor; α-thrombin did not activate PAR2 in an oocyte system, but trypsin did. The trypsin cleavage site on PAR2 was comparable to the thrombin cleavage site on PAR1. The HUGO symbol for the PAR2 gene is F2rl1. F2rl1 encodes a 397 amino acid protein. PAR2 and PAR1 share ∼30% identity, but some key differences explain different mechanisms of activation and signaling. The extracellular NH2 terminus of PAR2 is similar to that of PAR1 at the cleavage site but shorter and lacking the acidic residues responsible for thrombin affinity. The intracellular COOH terminus of PAR1 and PAR2 is also dissimilar, which likely accounts for different signaling properties (5). The genes for PAR1 and PAR2 share a common locus, leading to the proposal that they arose from a gene-duplication event (6). The PAR2 gene spans over 13 kb. The two exons are separated by an intron of ∼10 kb. F2rl1 is expressed widely in the colon, duodenum, stomach, small intestine, and gall bladder.
2.1.3. PAR3.
To explain the cellular effects of thrombin not mediated by PAR1, investigators sought to discover other thrombin receptors. The second thrombin receptor, PAR3, was discovered in a study that characterized the phenotype of mice in which the gene for PAR1 was deleted (6). Differential genomic blotting using cDNA probes that encompassed the PAR1 and PAR2 genes was used to identify the human PAR3 gene. The query did not reveal an associated gene with PAR1-specific probes; however, the PAR2-specific probes induced a band that was identified as PAR3. Confirmation came when the protein that represented the band produced phosphatidylinositol 4,5-bisphosphate (PIP2) hydrolysis when activated by thrombin (6). The PAR3 gene F2rl2 is almost identical to PAR1 and PAR2. F2rl2 contains two exons. Like PAR1 and PAR2, the second exon contains the protease cleavage site. PAR3 transcripts are present in both platelets and endothelial cells. PAR3 shows biased expression in the fat, stomach, thyroid, colon, and thyroid.
2.1.4. PAR4.
Human PAR4 was cloned in 1998 in an expressed sequence tag (EST) database. The full-length cDNA was then isolated from a lymphoma cell cDNA library (7). The amino acid sequences for PAR1, PAR2, and PAR3 were used to query available databases. An EST sequence matched the TM4 domain of the PARs. Translation of the identified sequence identified PAR4 as a GPCR of 385 amino acids with 33% identity with PAR1, PAR2, and PAR3. However, the extracellular NH2 terminus and intracellular COOH terminus of PAR4 do not resemble the homologous regions of the remaining PARs. The PAR4 gene F2rl3 does not exhibit the same characteristics common to the genes of the other PAR types. The gene is not on the same chromosome as the other PARs, and the intron is smaller. PAR4 likely resulted from a combination of gene duplication and translocation events (8). The PAR4 gene is expressed in most tissues, with the highest expression level in fat, stomach, thyroid, colon, and thyroid.
2.1.5. Interspecies gene homology.
Like other GPCRs, the PAR genes are clustered (9, 10). The PAR1, PAR2, and PAR3 genes are on chromosome 5q13.3 for humans and chromosome 13 for mice. The PAR4 gene is on chromosome 19p13.11 in humans and chromosome 8 in mice. PAR gene evolution has been characterized in the context of phylogenetics, chromosomal location, selective pressure, and functional divergence (11). Phylogenetic tree analysis suggests that PARs1-4 originate from four invertebrates. A maximum likelihood tree of the PAR family shows that PAR1 and PAR2 cluster into one subfamily while PAR3 and PAR4 cluster into another. The genes for PAR1 show evidence of environmental adaptation, while PAR2, PAR3, and PAR4 are highly conserved in vertebrates.
2.2. Protein Structure
PARs are classified within the family of class A rhodopsin-like GPCRs. A brief summary of the structures shared by all PARs will expedite structural comparisons between PAR types. All four PARs comprise seven TM helices, an extracellular NH2-terminal domain, three ICL and three ECL domains, and an intracellular COOH terminus (FIGURES 1 and 2). Posttranslational modifications of PARs include phosphorylation, ubiquitination, palmitoylation, and N-glycosylation. GPCR stability is bolstered by a disulfide bond across cysteines between TM3 and ECL3. N-glycosylation occurs at the following sites: the mature NH2 termini of all four PARs, ECL2 of PAR1 and PAR2, and the ECL3 of PAR3.
FIGURE 2.
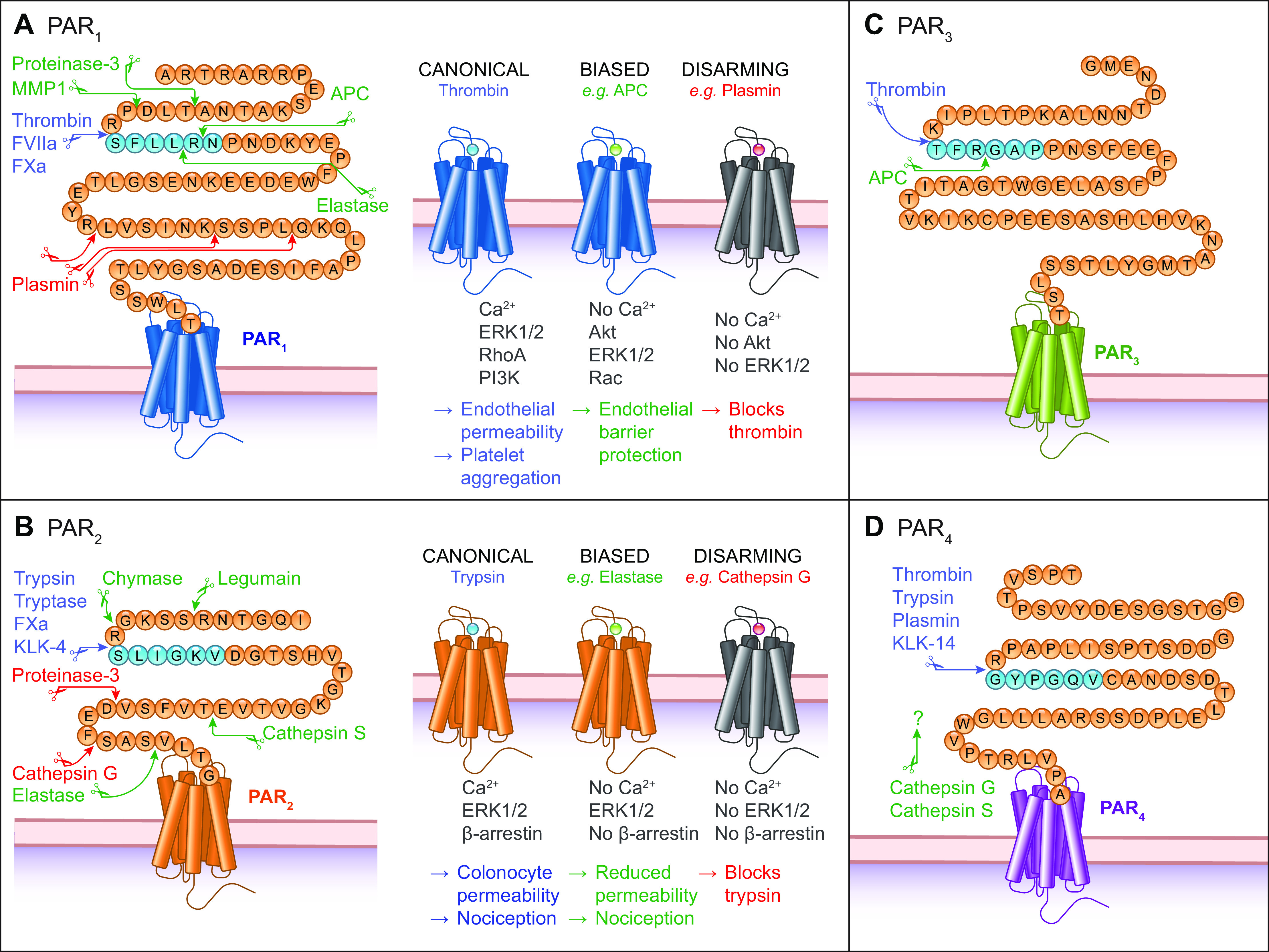
Cleavage by noncanonical proteases. A–D: NH2-terminal domains of protease-activated receptors 1 to 4 (PAR1-4) showing cleavage sites for proteases that activate PARs by canonical (blue) or biased (green) mechanisms or those that disarm the receptor (red). Residues are shown for the human receptors without the predicted signal sequence. APC, activated protein C; MMP, matrix metalloproteases.
2.2.1. PAR1.
Human and murine PAR1 possess a canonical thrombin cleavage site at Arg41/Ser42 (4). Cleavage reveals the tethered ligand SFLLRN as the new NH2 terminus of the human receptor (4). Adjacent to this canonical cleavage site, PAR1 contains a hirudin-like domain that mediates high-affinity thrombin binding (7). Mutation of this domain reduces the potency of thrombin to induce Ca2+ signaling (12). Other proteases that cleave the canonical site include factor Xa (FXa; Ref. 13) and FVIIa (14; FIGURE 2A). Advances in protein engineering and crystallography enabled elucidation of a 2.2 Å resolution structure of PAR1 bound to its antagonist vorapaxar (sect. 6.1). PAR1 was stabilized to permit crystallization by insertion of T4 lysozyme into ICL3, mutation of N-linked glycosylation sites in ECL2, and removal of the NH2-terminal exodomain. Key structural features included a conserved disulphide bond between helix III and ECL2 and two antiparallel β-strands, which are formed by residues situated toward the NH2 terminus of Cys254 loop outward toward Phe6.44 (numbered according to the Ballesteros-Weinstein scheme). Phe6.44 is a position that is conserved across class A (i.e., rhodopsin-like) GPCRs (15). PAR1 differs from other class A GPCRs with regard to its transmission of the signal from the extracellular side to the cytoplasmic domains that interact with G proteins, as well as associated structural rearrangements (16, 17). Interactions between TM5, TM6, and TM7, and conserved motifs in the former two TMs, facilitate structural rearrangement upon receptor activation in class A GPCRs. However, the interactions between TMs 5, 6, and 7 and the conserved motifs in TM6 and TM7 are different in the PARs. In this regard, the β2 adrenergic receptor (β2-AR) serves as a good comparator to the PARs. The TM6 motif is FxxCWxP, and the TM7 motif is NPxxY, whereby the amino acid identity of X is not critical. Based on differences in the active and inactive states of β2-AR, Phe6.44, along with Ile3.40 and Pro5.50, is important for G-protein binding and undergo rearrangement. PAR1 also contains Phe6.44. However, the interaction between Phe3226.44, Ile1903.40, and Pro2825.50 in PAR1 during the switch between the active and inactive state is different from the interaction between the associated amino acid residues within β2-AR. For many GPCRs, the Trp residue within the motif FxxCW6.48xP operates as a molecular switch following receptor activation; however, all PARs are characterized by Phe6.48. For PAR1, the DP7.50xxY in the above-referenced TM7 motif (which is NP7.50xxY in other class A GPCRs) mediates sodium sensitivity.
The crystal structure of PAR1 was consistent with the hypothesis that thrombin cleavage exposes a tethered ligand that interacts superficially with the heptahelical bundle, rather than deep within the TM core (18). Support for superficial activation was provided by mutagenesis experiments. The substitution of the tethered ligand binding region of ECL2 (Asn259 to Ala268) on human PAR1 with the associated Xenopus ECL2 sequences increased basal activity by 10-fold (19).
2.2.2. PAR2.
PAR2 was discovered as a receptor for trypsin (5, 20, 21). Trypsin cleaves human PAR2 at Arg36/Ser37 to reveal the tethered ligand SLIGKV. Trypsin cleaves mouse PAR2 at Arg38/Ser39, exposing the tethered ligand SLIGRL (5). The PAR2 canonical site (FIGURE 2B) is also cleaved by tryptase (22, 23), FXa (24), KLK-14 (25), KLK-4 (26), and KLK-5 (27).
Crystal structures of PAR2 in complex with two antagonists and a blocking antibody were reported in 2017 (28). For thermostabilization, nine mutations were introduced into PAR2. Crystallization was optimized by replacing residues 1–54 in the NH2 terminus with T4 lysozyme, replacing residues 270–275 on ICL3 with cytochrome b562RIL, mutating Asn222 to Gln, and truncating the COOH terminus after Lys377. The crystal structure of PAR2 revealed similarities and differences with PAR1. The ECL2 of PAR2 fills the top half of the binding pocket, similar to the PAR1 structure (29). On PAR2, His227ECL2 and Tyr1563.33 form a hydrogen bond; this intermolecular interaction is not present on PAR1.
The crystal structures for PAR1 and PAR2 provide a possible explanation for the selectivity of the antagonist vorapaxar for PAR1 and the antagonist AZ8838 for PAR2. Vorapaxar does not bind PAR2 because of the interaction between TM5, TM6, and TM7. Inward movement of TM5 and TM6 leads to steric clashes between vorapaxar and Tyr2425.38, Phe2435.39, His3106.58, and Tyr3116.59; therefore, the vorapaxar binding pocket on PAR2 is eliminated. With regard to the selectivity of AZ8838 for PAR2, the hydroxyl group of AZ8838 makes a hydrogen bond to a side chain on His1352.64. While His1352.64 is present on PAR3 and PAR4, it is not present on PAR1, and the corresponding amino acid is Tyr1622.64. If His1352.64 is replaced with Tyr, AZ8838 no longer inhibits PAR2. The binding site for AZ8838 is buried in the crystal structure, which leads to slow binding kinetics. AZ8838 inhibition, as measured with Ca2+ conductance and β-arrestin recruitment, requires 1 hour for complete inhibition. The crystal structure of PAR2 also provided information regarding receptor activation. For all four PARs, the residue equivalent to Asp228ECL2 in PAR2 is critical to activation.
The final crystal structure of PAR2 to be characterized was PAR2 bound to a monoclonal antibody, MAB3949. The antibody binds the following extracellular PAR2 residues: 59–63 from the NH2 terminus, 220 and 232–234 from ECL2, and 315–319 from TM6 and ECL3. Residues 57–62 on the NH2 terminus are important to the PAR2-binding epitope. MAB3949 antagonizes PAR2 activation by trypsin or SLIGRL as measured by an increase in intracellular Ca2+.
2.2.3. PAR3.
The canonical thrombin cleavage site of human PAR3 is Lys38/Thr39 (30). Similarly to PAR1, this is followed by a hirudin-like domain. There are questions regarding the signaling capacity of PAR3. In oocytes expressing PAR3, thrombin, FXa, trypsin, elastase, and chymotrypsin evoke Ca2+ signaling (30). Thrombin cleavage reveals the NH2-terminal sequence of TFRGAP (FIGURE 2C). However, peptides that mimic this NH2-terminal sequence fail to initiate any signaling response via PAR3.
2.2.4. PAR4.
PAR4 is the most structurally divergent of the PARs. PAR4 is considered the “low-affinity” thrombin receptor. Thrombin and trypsin cleave human PAR4 at Arg47/Gly48 to reveal the tethered ligand GYPGQV (7, 8) (FIGURE 2D). PAR4 lacks a hirudin-like thrombin-binding domain, instead it contains an anionic cluster (DDED) that interacts with thrombin to facilitate cleavage (31). The lack of the hirudin site explains the low potency for thrombin activation of PAR4 (4, 7, 30). PAR1 and PAR4 act together to mediate platelet responses to a range of thrombin concentrations. Whereas low-thrombin concentrations lead to PAR1-mediated platelet activation, high-thrombin concentrations maximally activate platelets through PAR4 (32). The NH2 terminus and COOH terminus share little with the corresponding regions of the other PARs. PAR4 shares only three amino acids (CHD) with the common sequence (ITTCHDV) present in PAR1, PAR2, and PAR3 in ECL2, the region that binds the tethered ligand (7). ECL2 is key to the activity of PAR4. A series of Asp residues within ECL2 mediate the interaction and activation of platelets. Following cleavage, the tethered ligand that consists of the sequence GYPGQV binds ECL2. PAR4 does not have the high-affinity binding domain for thrombin that is present in PAR1 and PAR3. PAR4 lacks the hirudin-like binding domain present in PAR1 and PAR3; however, a series of anionic residues including Asp57, Asp59, Glu62, and Asp65 increase the Km of α-thrombin fourfold and impede thrombin dissociation (33).
Less is known regarding the structure of PAR3 and PAR4. The crystal structures of fragments of murine PAR3 and PAR4 reveal information regarding cleavage by thrombin (34). Thrombin is characterized by an active site and two exosites. Binding at exosite I orients the substrate toward the active site. Thrombin Trp60d acts like a swinging gate and permits diffusion of the substrate to the active site. Upon binding of PAR3 to exosite I, the 60-loop on thrombin rotates 3.8 Å upward, a movement that allows Trp60d to rotate 180º. This open gate makes the active site accessible.
A further key finding from the crystal structures of PAR3 and PAR4 was that cleaved PAR3 and intact PAR4 do not overlap when bound to thrombin, a finding that suggests that a ternary complex is structurally feasible. Thus cleaved PAR3 can be bound to the thrombin exosite I and still allow for thrombin/PAR4 binding. In this manner, PAR3 can act as an allosteric modulator to promote cleavage of PAR4 (34). Exosite I on thrombin plays a critical role in the allosteric regulation of the active site. This role is best exemplified by hirudin, a potent natural anticoagulant found in leech saliva. When the COOH-terminal tail of hirudin is bound to exosite I, the active site of thrombin is accessible (35). Hirudin binds thrombin at both exosite I and the active site and prevents activation of fibrinogen. PAR1 has a hirudin-like region that is rich in aromatic and acidic residues (DKYEPF). PAR1 residues Tyr52, Glu53, and Phe55, which are similar to Phe56, Glu57, and Ile59 of hirudin, facilitate the interaction with thrombin and bind its anion-binding exosite. In the case of cleaved PAR1, the hirudin-like domain remains bound to exosite I of thrombin. In humans, PAR1 and PAR4 can form heterodimers. Cleaved PAR1 facilitates the association between platelets and thrombin while leaving the active site of thrombin free to bind PAR4. In this manner, PAR1 enhances the cleavage of PAR4 by thrombin. The allosteric role of PAR1 in mouse platelets, which lack PAR1, is taken up by PAR3. Cleaved PAR3 binds exosite I of thrombin, similarly to hirudin (36). Mouse PAR3 enhances cleavage of PAR4 by opening the active site with the shift in 60-loop and flip of Trp60d described above (8, 37). With PAR3 bound to the exosite I of thrombin, diffusion of PAR4 to the active site is enhanced; this configuration thus favors hydrolysis. PAR3 has a short cytoplasmic domain, which could also limit its involvement in signaling.
2.3. Summary
PARs are a family of G protein-coupled receptors. Common elements of all four PARs include seven TM helices, an extracellular NH2-terminal domain, three ICL and three ECL domains, and an intracellular COOH terminus. The location of NH2-terminal domain cleavage and the remaining peptide fragment determines subsequent cellular signaling and physiological activity. PAR2 differs from the other three PARs by lacking an extracellular NH2-terminal domain that is cleaved by thrombin. The crystal structures of PAR1 and PAR2 have been revealed with the use of antagonists, monoclonal antibodies, mutagenesis, and domain removal. Less is known about the structure of PAR3 and PAR4. PAR4 is an outlier with regard to the structure of the NH2 terminus and COOH terminus; it also lacks the hirudin-like binding domain of PAR1 and PAR3. PAR1 and PAR3 enhance binding and cleavage of PAR4.
3. CELL TYPE EXPRESSION OF PARS
One challenge in reporting the cellular expression of PARs is concern over the specificity of GPCR antibodies (38). Evidence of expression of PARs should thus include analysis of transcript, as well as functional responses (e.g., Ca2+ imaging) to selective agonists and antagonists. Another approach to localize PARs with high specificity has been to study knockin mice expressing PARs fused to a fluorescent protein. With the use of this approach, PAR2-GFP has been detected in intestinal epithelial cells and a subpopulation of primary sensory neurons of dorsal root ganglia with high specificity and sensitivity (39). TABLE 1 and FIGURE 3 summarize the distribution of PARs in different cells, tissues, and organs.
FIGURE 3.

Patterns of protease-activated receptors (PAR) expression. Sites of PAR1-4 expression across different organ systems (arrows) and cell types (listed).
3.1. Epithelial Cells
Epithelial cells of the skin, airway, gastrointestinal (GI) tract, urinary tract, and exocrine and endocrine glands differentially express PARs, which contribute to the physiology and pathology of these tissues (sect. 5).
3.1.1. Cutaneous epithelia.
The presence of proteases, protease inhibitors, and PARs in the skin suggests a major role for this system in cutaneous biology (sect. 5.2). PARs regulate the function of keratinocytes, which interact with nerves, melanocytes, Langerhans cells, and immune cells of the skin. PARs also contribute to keratinocyte maturation, replication, wound repair, migration, inflammation, and immune response. PAR1 and PAR2 activation have opposing effects on keratinocyte growth but similar effects on differentiation. PAR1 activation enhances keratinocyte growth, whereas PAR2 activation inhibits cell growth (40).
PAR2 activity in keratinocytes was discovered based on functional assays and localization of immunoreactive receptors (41, 42). PAR2 was detected in keratinocytes but not fibroblasts, while PAR1 was detected in both cell types. PAR2 on keratinocytes is activated by tissue factor (TF)/coagulation factor VIIa (FVIIa), as well as FXa is generated by TF/FVIIa (43). In keratinocytes, PAR2 activation evokes release of Ca2+ from store-operated calcium entry (SOCE), which makes use of Ca2+ release-activated channels in the plasma membrane, such as ORAI1, also known as calcium release-activated calcium (CRAC) channel protein 1. Keratinocytes regulate the local immune response in the skin, and PARs contribute to immune regulation. In differentiated human primary keratinocytes, PAR2 upregulates the release of inflammatory mediators, which involves the transient receptor potential vanilloid 1 (TRPV1) channel (44). Activation of PAR2 on human primary keratinocytes depletes Ca2+ stores. However, the effect requires both TRPV1 and the inositol 1,4,5-trisphosphate (IP3) receptor on the endoplasmic membrane, rather than SOCE.
3.1.2. Respiratory epithelia.
All four PARs are expressed in the human lung (45) (sect. 5.3). In the human lung, immunoreactive PAR2 is localized to bronchial smooth muscle and epithelium (46).
3.1.3. Gastrointestinal epithelia.
Across species, PARs are expressed in GI epithelia (sect. 5.4). PAR2 is highly expressed in the intestinal mucosa, including at the apical membrane of enterocytes where it can be cleaved by luminal trypsin (47). In situ hybridization in the mouse reveals PAR2 transcript in epithelial cells of the small intestine, where it is primarily localized to the upper two-thirds of the intestinal villi and less so in the crypts.
3.1.4. Genitourinary epithelia.
PAR1 and PAR2 are expressed in the kidney. PAR1 expression in the kidney epithelium regulates inflammation (48). PAR2 is strongly expressed in the epithelium of the ureter (49). Activation of PAR2 inhibits rhythmic beating of the ureter. PAR2 is expressed in renal cortical collecting duct cells and in cultured M-1 mouse cortical collecting duct cells (50).
3.2. Endothelial Cells
Our understanding of PAR physiology has largely been driven by the study of thrombin-mediated PAR signaling in endothelial cells. For example, the observation that the tethered ligand alone could act as a receptor agonist was demonstrated in human platelets and endothelial cells (4). PAR endothelial signaling affects homeostasis and contributes to cerebrovascular disease, cardiovascular disease, and carcinogenesis (sect. 5.2). The effects of activation on platelets and endothelial cells exemplify the role of PAR activation in hemostasis. PAR1, PAR3, and PAR4 mediate the effects of thrombin following blood vessel trauma. Thrombin activation of PARs leads to cytokine production by endothelial cells (51–53). Vitamin K-dependent coagulation proteases (e.g., FVIIa, FXa) except for FIXa, as well as thrombin, activate PARs on the surface of endothelial cells. PAR1 and PAR4 expression by endothelial cells in mice serve redundant roles; endothelial cells that do not express PAR1 or PAR4 show no response to thrombin (54). PAR1 (4), PAR2 (55, 56), PAR3 (57), and PAR4 (58) are expressed on human endothelial cells. Thrombin activates PAR1, PAR3, and PAR4 but not PAR2. PAR1 on endothelial cells is a clinically important target due to its potential role in thrombosis. Quantitative phosphoproteomics have been used to study the effects of thrombin activation PAR1 and of PAR1 antagonism on endothelial cells, including the finding that the antagonist properties of vorapaxar and parmodulin-2 are distinct (59).
Human dermal microvascular endothelial cells (HMEC-1) have revealed the effects of PAR1 activation and downstream trafficking (60). PAR1 activation on endothelial cells leads to the secretion of inflammatory cytokines including interleukin (IL)-1, IL-6, tumor necrosis factor (TNF)-α, and cell adhesion molecules including selectins, intracellular adhesion molecule 1 (ICAM-1), and vascular cell adhesion molecule 1 (VCAM-1). PAR1 is the effector receptor for endothelial protein C receptor (EPCR), which is responsible for the cytoprotective effects of activated protein C (APC). This includes anti-inflammation, antiapoptotic effects, and protection of endothelial barrier functions (61–63). PAR1 exists in the native and cleaved form within intracellular pools of endothelial cells. When endothelial cells are exposed to an agonist, both native and cleaved PAR1 will translocate to the cell surface (64, 65).
PAR1 on endothelial cells exhibits biased agonism (sect. 4). APC is a serine protease that proteolytically inactivates FVa and FVIIIa; it therefore opposes coagulation and is also involved in inflammation. APC leads to endothelial barrier protection, as measured by zymogen-affected permeability of an endothelial cell monolayer. Based on its effects on systemic inflammation, recombinant human APC was approved for treatment of sepsis in patients (66). APC cleaves PAR1; however, PAR1 remains on the endothelial cell surface, as opposed to PAR1 cleavage by thrombin (66, 67) (sect. 4.8). When thrombin cleaves PAR1, the receptor internalizes for lysosomal degradation. While the two activators of PAR1 have differential effects, thrombin and APC can cleave the same scissile bond on PAR1 (68). APC can, however, also cleave downstream from the canonical site at Arg46/Asn47 (FIGURE 2).
3.3. Smooth Muscle Cells
Before the discovery and characterization of PARs, thrombin was well-known to cause vasoconstriction and induce biogenesis in vascular smooth cells (69, 70). Activation of “the thrombin receptor” on smooth muscle cells (SMCs) by thrombin led to our early understanding of the selectivity of activating peptides for PARs (71). PARs are also expressed on SMCs in the respiratory system (72), GI tract (73, 74), and vasculature (75, 76). PAR activation on SMCs leads to protein phosphorylation, gene expression, contraction, relaxation, and mitogenesis. PAR2 is expressed by human vascular SMCs and contributes to proliferation and migration (77). Vascular SMCs express PAR1, PAR3, and PAR4, which mediate thrombin-induced proliferation and migration (78).
3.4. Platelets
Thrombin is the most potent activator of platelets. In 1967, Davey and Luscher (79) showed that thrombin’s catalytic activity was responsible for its action at the thrombin receptor on platelets. In the late 1970s and early 1980s, evidence was building that thrombin activation of platelets produced downstream signaling that involved GTP-binding proteins and second messengers, signaling processes that would later be characterized as a consequence of PAR activation (80). In 1990, a receptor for serine protease was identified in leukocytes (81). The authors proposed the name “effector cell protease receptor-1” for this receptor. The authors made the seminal finding that activation involved release of the NH2 terminus of the receptor and rearrangement of the tethered ligand into a pocket within the receptor. Soon after the thrombin receptor was cloned, it was discovered that thrombin-induced cleavage of the NH2 terminus and receptor activation by the tethered ligand led to granule secretion, shape change, and platelet activation (82). PAR1 activation leads to the secretion of mediators that alter platelet function, including ATP and ADP. The search for a second thrombin-responsive receptor was motivated by data that showed thrombin-mediated effects following activation of platelets from PAR1 knockout mice. Further investigation of effects not mediated by PAR1 led to cloning and characterization of PAR3 (30).
PAR expression on platelets is species specific. In spite of this, thrombin receptor subtypes work in concert in mice and humans. Human platelets do not express PAR2. Rat platelets do not express PAR1 or PAR2. Similarly, mouse platelets express PAR3 and PAR4; in mice, PAR3 is the high-affinity receptor. Knockout of either PAR subtype affects platelet activation by thrombin (83). However, mice deficient in PAR3 do not exhibit spontaneous bleeding or have abnormally long bleeding times. Human platelets express PAR1 and PAR4; in the case of humans, PAR1 acts as the high-affinity receptor. The role of the dual thrombin receptor system is not known. Coughlin and colleagues (83) hypothesized that the role of two PARs might allow for ligands other than thrombin to activate the receptors, or for thrombin to signal through different pathways with distinct kinetics. An example is the expression of PAR3 on Dami cells (derived from a patient with megaloblastic leukemia) but not on human platelets. This suggests that PAR3 is expressed by hematopoietic cells in the megakaryocyte lineage; however, expression is lost on megakaryocytes as the cells mature into platelets (84).
Although thrombin activates both PAR1 and PAR4, the affinity for PAR1 is higher and the cellular consequences are different. PAR4 lacks the hirudin-like binding site. Glycoprotein Ib and PAR3 mediate binding of thrombin to PAR1 and PAR4, respectively (85, 86). Activation of PAR1 or PAR4 leads to secretion and aggregation of human platelets, and if both PARs are inhibited, platelets do not respond to even high doses of thrombin. This suggested that PAR1 and PAR4 were responsible for the action of thrombin on platelets (84), while PAR3 does not participate in human platelet activation.
Mouse knockout models and heterologous expression systems have been used to elucidate the downstream mechanisms following activation of the PARs on platelets; however, species differences in expression of PARs limit the inference of the result to effects expected in human platelets. Activation of PAR1 and PAR4 results in human platelet aggregation, secretion, and increased intracellular Ca2+ (84). The differences between PAR1 and PAR4 activation remain a question. However, work has shown that PAR1 has a dominant role in fibrinolysis, whereas PAR4 mediates clot elasticity (87).
Proteases besides thrombin can activate PARs on platelets. For example, cathepsin G from neutrophils activates PAR4, whereas cathepsin G does not activate PAR1. Cathepsin G/PAR4 activation leads to secretion from platelets and platelet aggregation (see Supplemental Table S4; all Supplemental material is available at https://doi.org/10.6084/m9.figshare.19739830). The effect mediated by PAR4 can be blocked with an antibody against the thrombin cleavage site and desensitization with an activating peptide (88). Human kallikreins (KLKs) are another family of proteases that activate PARs and induce platelet aggregation. Activating peptides for both PAR1 and PAR4 generate a strong Ca2+ signal (25). The advantage of a Ca2+ assay over platelet aggregation is that the latter allows for a sequential and concurrent evaluation of activation of PAR1 and PAR4 on platelets.
3.5. Neurons and Glial Cells
In 1978, it was demonstrated that a glial-derived protease inhibitor induced neurite outgrowth in neuroblastoma cells (89). Neurite outgrowth is a cytoarchitectural change consistent with differentiation in neurons and glial cells. In contrast, thrombin was shown to reduce neurite outgrowth a decade later (90). Hirudin, the thrombin inhibitor, led to neurite outgrowth of neuroblastoma cells (91). Thrombin was known to bind to a receptor on neuroblastoma cells, and the binding resulted in an increase in cGMP (92). Collectively, these findings pointed to PAR1 expression on neurons. Subsequently, data supported PAR expression in the central nervous system (CNS) on neurons and astrocytes; PAR1 is highly expressed in the former and moderately expressed in the latter (93). PAR1 on astrocytes responds to activation by thrombin in the same way that PAR1 responds to activation on human platelets (94). On astrocytes, the strongest expression of PAR1 is on the cell body and the distal neuronal endplate that innervates capillaries. Human astrocytes in culture respond to PAR1 activation by increasing intracellular Ca2+.
Of the four PARs, the role of PAR2 on sensory neurons and its role in neurogenic inflammation and pain is best characterized (95, 96) (sect. 5.5). Activation of PAR2 induces neurogenic inflammation and pain and sensitizes ion channels including TRPV1 (97). PAR2 is expressed on peripheral nerve endings and the cell body of dorsal root ganglia (DRG); however, PAR2 does not appear to be expressed in the central terminals in the spinal cord (98). While most of the work on PAR2 expression in sensory ganglia has been done in DRG, PAR2 has also been shown to be strongly expressed in trigeminal ganglia (99). Trigeminal neurons innervating the nasal mucosa in rats were retrograde labeled, and then labeled ganglia were stained for PAR2. Over 40% of trigeminal neurons were positive for PAR2. Functional evidence also showed PAR2 was expressed in trigeminal ganglia (100). Administration of the PAR2-activating peptide SLIGRL-NH2 produced c-Fos expression in the trigeminal nucleus caudalis of the caudal brainstem and superior cervical spinal cord. Further evidence for PAR2 expression in the trigeminal ganglia was provided by experiments showing that activation of PAR2 on rat trigeminal neurons leads to functional competence of δ-opioid receptor, which is inactive under basal conditions (101).
The question regarding expression of PAR2/F2rl1 on neurons has been controversial. Despite functional in vitro and in vivo data that supported PAR2 expression on neurons, confirmation of expression was complicated by the nonspecific nature of antibodies for PAR2. A publication by Price and colleagues in 2020 (102) clarified the conflicting functional and anatomic results; the publication also resolved some of the controversial ideas with regard to neuronal PAR2 and pain. The first demonstration of the role of PAR2 in pain was published by Vergnolle and colleagues in 2001 (96). This publication was one of the first to use a genetic knockout model to confirm the role of a receptor in mediating nociception. The authors showed that activation of PAR2 with SLIGRL produced mechanical allodynia and thermal hyperalgesia. Subsequently, there were publications over two decades that confirmed the role of PAR2 in somatic and visceral hyperalgesia; however, RNA sequencing data also showed extremely low levels of expression of F2rl1 (103, 104). Price and colleagues (102) demonstrated that F2rl1 is expressed on a small subset of neurons using available RNA sequencing data. Approximately 4% of DRG neurons express F2rl1. Using RNAScope, the authors showed that F2rl1 is expressed on nonpeptidergic neurons that express P2rx3. These F2rl1-expressing neurons also coexpress IL31ra and nppb (naturietic precursor peptide b), genes that mediate itch. The authors used a conditional knockout with the Pirt promoter, where F2rlflox mice were crossed with PirtCre mice. The promoter for Pirt is expressed in primary sensory neurons in the DRG and TG; however, it is not expressed in glia or skin (106). Using the conditional knockout mice, the authors showed that activation of F2rl1 on neurons by three different agonists produces mechanical allodynia, agonists including at-LIGRL, neutrophil elastase, and compound 48/80, which leads to mast cell degranulation of tryptase. The dose of at-LIGRL (<10 µM) is specific for PAR2 (107). Thermal hyperalgesia was induced by at-LIGRL through PAR2. In contrast, compound 48/80 and neutrophil elastase produced thermal hyperalgesia; however, neuronal PAR2 was not required. This key result shows that activation of nonneuronal PAR2 by endogenous proteases mediates the thermal effect. Before this study, most studies that reported on the role of neuronal PAR2 in pain have used reflexive assays. Price and colleagues (102) used the facial grimace assay that measures the affective component of nociception (108). They showed that at-LIGRL injected into the paw produces nociception measured by facial grimace. The nociception is dependent on neuronal F2rl1. The investigators also looked at the role of PAR2 in itch. Using the at-LIGRL dose specific for PAR2 as well as F2rl1-/-, they showed that the low at-LIGRL dose induces pain but not itch. This finding is consistent with the work of Dong and colleagues (109) who showed that SLIGRL mediates itch through activation of murine MrgprC11. Homolog to the human mas-related GPCR member X1 (MRGPRX1), this pruritogenic receptor is an orphan receptor expressed by sensory neurons. The authors showed that the pentapeptide SLIGR specifically activated PAR2 and not MrgprC11; this induced nociception but not pain.
PAR4 was shown to be expressed on sensory neurons in 2002 (110). While PAR4 was later shown to be strongly expressed in the gut, it was first shown to be expressed on visceral primary afferent neurons in 2009 (111).
Following early studies looking at PAR expression on sensory neurons, motor neurons, and the brain, investigators started to look at PAR expression on neurons that innervate the GI tract. Myenteric neurons strongly (>60%) express PAR1 and PAR2, at the transcript and protein level (23). PAR1 and PAR2 expression was studied in neurons isolated from the guinea pig small intestine. These neurons responded to mast cell tryptase and trypsin, proteases that have a central role in trauma and inflammation, as measured by Ca2+ imaging. The neurons also responded to agonist peptides selective for PAR1 [AF(pF)RchaCitY-NH2 and TFLLRN-NH2] and PAR2 (SFLLRN-NH2). The response of these myenteric neurons to PAR2 agonists was further characterized in the guinea pig ileum (112). SLIGRL-NH2 or trypsin activation of the myenteric neurons produced prolonged depolarization in tetrodotoxin-resistant manner, which suggested direct activation of PAR2 on myenteric neurons. Myenteric neurons have been classified into two types based on electrophysiologic parameters: AH/type 2 (less excitable, one to two spikes following depolarizing current pulses) and S/type 1 (more excitable, repetitive spikes following depolarizing current pulses) (113, 114).
3.6. Immune and Inflammatory Cells
The interpretation of studies that report PAR expression on leukocytes should consider the cellular treatment such as centrifugation, purification, or even repeated pipetting; each step can lead to PAR upregulation (115). Early evidence suggested that leukocytes express PARs (116) (sect. 5.6). Activation of PAR1 on mast cells, either from trypsin or the activating peptide, produced a change in mast cell activity and secretion of IL-6 (117, 118). PAR1 is expressed on memory CD4+ and CD8+ T cells (119).
Work with neutrophils and thrombin led to the speculation that there were additional PARs beyond PAR1. Neutrophils were known to respond to thrombin and that response was mediated by a proteolytically activated receptor. It was also known that a peptide analog of the NH2-terminal region following its cleavage could activate neutrophils in the absence of thrombin (4). Work with thrombin and its activating peptide, thrombin receptor-activating peptide (TRAP), led to the suggestion of additional PARs, including PAR2 (120). The peptide sequence for TRAP (SFLLRN) and the specific PAR2-activating peptide (SLIGRL) share homology (20).
Cutaneous human primary skin mast cells and the human mast cell line 1 (HMC-1) express all four PARs; however, PAR2 mediates the action of mast cells. PAR2 agonists cause the release of histamine by mast cells. Tryptase and PAR2 colocalize on the surface of mast cells (121). Human mast cells express PAR2, which, when activated, leads to mast cell degranulation. Tryptase, which is released by degranulation, cleaves PAR2 (121–123).
There is functional and anatomic evidence for PAR2 expression in human eosinophils. PAR2 and eosinophils mediate airway disease. While there is a transcript for PAR2 and PAR3 in human eosinophils, there is no transcript for PAR1 or PAR4. Human eosinophils respond to trypsin and the activating peptide for PAR2, but not thrombin or the activating peptides for PAR1, PAR3, and PAR4 (124). In a subsequent study, eosinophils were isolated from human blood. Tryptase caused release of interleukin-6 (IL-6) and IL-8 from eosinophils (125). A later study used RT-PCR, Western blotting, and flow cytometry to analyze expression of the PARs in human eosinophils, neutrophils, and mononuclear cells (126). In contrast to the earlier study that did not detect PAR1 in eosinophils, this study, which used complementary techniques, showed expression of PAR1 and PAR2. Similarly, mononuclear cells showed expression of PAR1 and PAR2. Neutrophils expressed PAR2, which had been shown in an earlier study in which a PAR2-activating peptide led to an increase in intracellular Ca2+ and a change in cellular morphology (127).
PAR2 on T lymphocytes is involved with leukocyte rolling and adhesion (128, 129). PAR2 is also expressed in the Jurkat T-cell leukemia line (130). PAR2 is not expressed on B cells (131). With the use of a preparation that involves exteriorization of the midjejunum and in vivo microscopy, it was demonstrated that activation of PAR2 contributes to leukocyte rolling, adhesion, and extravasation (132). Trypsin-mediated activation of neutrophils (133), eosinophils (124, 126), and lymphocytes (134) leads to the release of reactive oxygen species.
Differential expression of PARs on human monocytes was studied using RT-PCR and flow cytometric analysis to measure expression of PARs; Ca2+ imaging then demonstrated functional expression (135). PAR3, and a lower level of PAR1, are expressed by human monocytes. When monocytes were treated with granulocyte-macrophage colony-stimulating factor (GM-CSF) to differentiate into macrophages, or with GM-CSF and IL-4 to differentiate into dendritic cells, both macrophages and dendritic cells expressed PAR1, PAR2, and PAR3.
PAR4 is present on the surface of rat neutrophils (110). In a separate in vivo study measuring neutrophil function, they demonstrated that PAR4 on neutrophils participated in the features of inflammation including edema and granulocyte recruitment. Carrageenan-induced inflammation was reduced with a PAR4 antagonist. Conversely, injection of the PAR4 agonist produced paw edema and granulocyte recruitment (136).
3.7. Cancer Cells
Soon after their discovery, PARs were detected in cancer cells (sect. 5.7), including expression of PAR2 in lung and colon adenocarcinoma cells (21) and expression of PAR1 in pancreatic tumor cells (137). The role of thrombin in metastasis was demonstrated in metastatic breast cancers (138). PAR1 was expressed in metastatic breast carcinoma cell lines but not in nonmetastatic breast carcinoma cell lines. For metastatic cell lines, PAR1 correlated with metastatic potential. A key study recently used data from The Cancer Genome Atlas and the Genotype-Tissue Expression projects to highlight PARs overexpressed by human cancer cells (139).
PAR1 and PAR2 are not only expressed by cancer cells but also by nonmalignant cells in the tumor microenvironment that support carcinogenesis (140). The key finding of this study was that PAR1 and PAR2 are upregulated and showed moderate to strong staining by stromal fibroblasts that express smooth muscle actin. Myofibroblasts are known to be critical for carcinogenesis (141).
3.8. Summary
PARs are widely expressed on epithelial cells (skin, respiratory system, GI tract, urinary tract, and exocrine and endocrine glands), endothelial cells, platelets, smooth muscle cells, neurons, glial cells, innate and adaptive immune and inflammatory cells, and cancer cells. PAR expression on the different cell type positions PARs to contribute to homeostasis and pathology. Our understanding of PAR physiology has been driven by the study of the effect of PAR activation on different cell types. PAR expression and function differ across species.
4. ACTIVATION AND SIGNALING OF PARS IN HEALTH AND DISEASE
4.1. Canonical and Biased Mechanisms of PAR Activation
PAR activation entails hydrolysis of peptide bonds in the extracellular NH2-terminal domain, revealing a sequence of amino acids (tethered ligand) that can interact with the orthosteric site within the TM domains of the cleaved receptor. While a growing variety of proteases are known to cleave PARs, their canonical sites are defined by the protease linked to their discovery. Biased signaling is a phenomenon whereby different ligands stabilize conformations of the same receptor that favor distinct signaling pathways. Biased signaling is of particular interest for proteases that cleave PARs at different sites and reveal distinct tethered ligands or stabilize unique receptor conformations (FIGURE 2). Biased mechanisms of PAR activation trigger different downstream signaling pathways and physiological outcomes. For example, APC cleaves PAR1 at both the canonical Arg41/Ser42 site and distal at Arg46/Asn47 (142) (see Supplemental Table S1). Whereas thrombin leads to endothelial permeability, APC stabilizes the endothelial barrier with anti-inflammatory and cytoprotective effects (sect. 5.1; see FIGURE 6).
FIGURE 6.

Role of protease-activated receptors (PARs) in the circulatory system. A: PARs are expressed in various cell types in the vasculature and circulation. B: human platelets expressing PAR1 and PAR4 respond to low- or high-thrombin concentrations to aggregate platelets. C: endothelial PAR1 has distinct signaling outcomes in response to thrombin or activated protein C (APC) bound to endothelial protein C receptor (EPCR). NO, nitric oxide.
Noncanonical cleavage sites can occur proximal and distal to the canonical cleavage site. In some cases, cleavage reveals a distinct peptide that retains some residues of the canonical tethered ligand but includes a sequence that can independently activate the receptor. Matrix metalloprotease 1 (MMP1) is a matrix-degrading enzyme released by platelets that modulates platelet survival and hemostatic function. MMP1 cleaves PAR1 at Asp39/Pro40, two residues proximal to the canonical thrombin cleavage site, to reveal PR-SFLLRN (143) (FIGURE 2; see Supplemental Table S1). This activates signaling pathways that regulate platelet motility and aggregation. Proteinase-3 cleaves PAR1 at Ala36/Thr37, revealing TLDPR-SFLLRN (144). Proteinase-3 cleavage causes biased signaling, leading to ERK1/2 phosphorylation but lacking Ca2+ mobilization (FIGURE 2; see Supplemental Table S1). Activating peptides mimicking the PAR1-tethered ligand revealed by MMP1 or proteinase-3 reproduce signaling pathways observed by protease exposure. Similarly, cathepsin S cleaves PAR2 at Glu56/Thr57, distal to the canonical site, which reveals a distinct tethered ligand (145) (FIGURE 2; see Supplemental Table S2). Cathepsin S and its activating peptide are biased for Gαs signaling, while lacking Ca2+ signaling or β-arrestin recruitment.
Some proteases can activate PARs without exposing a tethered ligand, presumably by stabilizing unique active conformations. Elastase cleaves PAR1 and PAR2 distal to their canonical site at Leu45/Arg46 and Ser67/Val68, respectively (144, 146, 147) (FIGURE 2). While elastase does not initiate PAR2-mediated Ca2+ signaling, it activates ERK1/2 (146) and protein kinase D (PKD; Refs. 146, 148) (see Supplemental Table S2). The activating peptide for elastase initiates PAR1 signaling as a tethered ligand (144). In contrast, the elastase/PAR2-activating peptide does not signal in its own right (146). Legumain, an asparaginyl endopeptidase resident in late endosomes, also leads to biased PAR2 signaling. Cleavage at Asn30/Arg31 reveals RSSKGR-SLIGKV (149), leading to ERK, Ca2+, and cyclic adenosine monophosphate (cAMP) signaling without β-arrestin recruitment (FIGURE 2; see Supplemental Table S2). Likewise, the activating peptide for legumain/PAR2 cleavage does not initiate signaling (149).
4.2. Proteolytic Disarming of PARs
Cleavage distal to the canonical site of activation can “disarm” PARs by permanently removing the canonical cleavage site and destroying the tethered ligand sequence. If these cleavage events do not themselves activate PARs, they can render the cleaved receptor unresponsive to proteases that activate PARs by canonical mechanisms. Mass spectrometry studies show plasmin, calpain, elastase, cathepsin G, and proteinase-3 cleave PAR1 at multiple sites distal of the canonical site (150). Plasmin, for example, can cleave at both the canonical site and distal at Arg70/Leu71, Lys76/Ser77, and Lys82/Gln83 (68). Plasmin desensitizes PAR1 Ca2+ signaling in response to thrombin (see Supplemental Table S1). Cathepsin G cleaves PAR2 at Phe64/Ser65 and renders the receptor unresponsive to trypsin; it does not, however, prevent signaling in response to the activating peptide SLIGKV (151). Nonmammalian proteases also disarm PARs. Streptococcal pyrogenic exotoxin B (SpeB), a virulence factor secreted from Group A streptococcus, cleaves PAR1 at Leu44/Leu45 within the SFLLRN tethered ligand (152). SpeB inhibits thrombin-induced ERK1/2 signaling in endothelial cells and prevents thrombin-evoked platelet aggregation. Bacterial proteases can also disarm PAR2. Elastolytic metalloproteinase (Epa) from Pseudomonas aeruginosa cleaves PAR2 at Ser37/Leu38 to reveal LIGKV and disrupt the canonical tethered ligand (153).
4.3. Tethered Ligands and Soluble Activating Peptides
Structure/function studies of the interaction between the tethered ligand domain and the cleaved receptor provide insights into the mechanism of intramolecular activation of PARs. The PAR1 tethered ligand interacts with ECL2 Glu160, as identified by mutagenesis (154). This was corroborated by the PAR1 crystal structure in complex with an orthosteric antagonist (29) (sect. 2.2). A synthetic peptide mimicking the PAR1 tethered ligand (TRAP) can directly activate PAR1 without requiring receptor cleavage (4). Modified versions of SFLLRN have a varying capacity to activate PAR1 and aggregate platelets (155). The potency depends on peptide length, whereby reduction from 14 to 5 residues enables platelet aggregation while reducing potency fivefold (156, 157). At least five amino acids are required to activate PAR1 (155, 156) and PAR2 (158). In contrast, PAR3 does not itself respond to activating peptides TFRGAP-NH2 (human) or SFNGGP-NH2 (murine) (159). The PAR4-activating peptide was modified to AYPGKF to enhance its potency (160). Other modifications of activating peptides can enhance biological activity. Modification of the PAR2-activating peptide to 2-furoyl-LIGKV-OH enhances bioavailability by protection from endogenous aminopeptidases (161), leading to development of 2-furoyl-LIGRLO-NH2, a potent and selective PAR2 agonist (162).
Activating peptides have been widely used to probe PAR functions. While they usually recapitulate the actions of proteases, there are some differences. Thrombin aggregates platelets and stimulates Ca2+ in all species, whereas SFLLRN only activates PARs in guinea pigs, monkeys, and humans (40). Signaling kinetics also differ; thrombin-induced Ca2+ signaling in platelets is more sustained than SFLLRN or GYPGQV (163). Activating peptides exhibit varying degrees of subtype selectivity, which can complicate the interpretation of their effects. PAR4-activating peptides cannot activate PAR1 and PAR2 (164). In contrast, the PAR1-activating peptide activates both PAR1 and PAR2 (165). This lack of selectivity raises the possibility that tethered ligands from different PAR subtypes could activate another via transactivation and coactivation (FIGURE 4A). For example, PAR1-PAR2 heterodimers have been directly observed using biophysical techniques (166). PAR1-PAR4 heterodimers also occur via TM4 interactions induced by thrombin (167). Activating peptides can also activate non-PARs, such as the activation of MrgprC11 by the PAR2-activating peptide (109).
FIGURE 4.

Modulation of protease-activated receptors (PAR) signaling. A: heteromers can form between different PAR subtypes, such as PAR1 and PAR2. Tethered ligands from one subtype can transactivate another. B: heteromers can form with other G protein-coupled receptors (GPCRs), such as between the chemokine receptor 4 (CXCR4; shown) or with β-adrenoceptors in cardiac fibroblasts. C: GPCRs transactivate receptor tyrosine kinases (RTKs), such as the epidermal growth factor receptor (EGFR). PAR1-mediated Src activation can phosphorylate (Ph) EGFR (1). PAR1 also activates matrix metalloproteases (MMPs) that liberate membrane-tethered EGFR ligands, such as amphiregulin (2). D: PAR signaling can modulate ion channel signaling, such as PAR2-mediated sensitization of transient receptor potential vanilloid 1 (TRPV1) in sensory neurons. TRPV1 sensitization is mediated by PKC activation downstream of PAR2, causing phosphorylation (Ph) of TRPV1 (Ser residues 502 and 800 in human TRPV1).
PAR3 does not respond to synthetic peptides that mimic the putative tethered ligand. PAR3 signaling remains an enigma. Autonomous PAR3-mediated signaling was shown in response to thrombin using HEK293 cells lacking PAR1 (168). However, thrombin can have additional functions, such as integrin-induced chemotaxis (169). Thus observed ERK phosphorylation and IL-8 release could be PAR3 independent. PAR3-activating peptides can also signal through other PAR subtypes (as in FIGURE 4A). PAR3 acts as a cofactor in platelets, signaling via PAR4 (86). PAR3 activation could also act through PAR1, as the PAR3-activating peptide initiates ERK1/2 signaling that was abolished by a PAR1 antagonist or using PAR1 knockout fibroblasts (170). PAR3 modulation of thrombin/PAR1 signaling causes biased signaling in endothelial cells, whereby siRNA against PAR3 corresponds with increased endothelial barrier permeability independently of Ca2+ signaling (171).
4.4. Accessory Proteins for PAR Activation
Interactions with other membrane proteins can bias the outcomes of PAR activation. This concept is illustrated by the role of EPCR in APC-mediated activation of PAR1 (63, 172) (sect. 3.2). APC activates PAR1, yet it leads to divergent phenotypic outcomes compared to thrombin. APC also cleaves PAR3 distal from its canonical site in an EPCR-dependent manner with a barrier-protective phenotype (173). EPCR binds the other PAR1-cleaving proteases FXa (174) and FVIIa (14, 175). FXa/EPCR interactions reduce endothelial cell permeability in a manner sensitive to the EPCR inhibitor RCR252 (174).
Other membrane proteins can modulate the protease activity or PAR-signaling pathways. For example, integrins are receptors linking the extracellular matrix (ECM) with the cytoskeleton. Plasmin interacts with α9β1-integrin, leading to enhanced migration blocked by a PAR1 inhibitor (e.g., RWJ 58259) (176). Coexpression of another integrin subtype, αIIbβ3-integrin, enhances MMP2-mediated cleavage of PAR1 (177). Thrombin interacts with αVβ5-integrin, a membrane protein localized in the same region as PAR1 in lung carcinoma cells (178).
Other GPCRs can also modulate PAR signaling (reviewed in Ref. 179; FIGURE 4B). In addition to heteromers between PAR subtypes, PAR1 can interact with β-adrenoceptors expressed by cardiac fibroblasts and cardiomyocytes (180). Considering PAR1 is the highest GPCR expressed in numerous fibroblast subtypes (181), modulation could have important implications in cardiac dysfunction with β-adrenoceptor overstimulation. PAR4 interacts with Gαi-coupled P2Y1 and P2Y12 purinergic receptors to induce platelet activation (182). There is also evidence that PAR1 forms a heteromer with chemokine receptor 4 (CXCR4) (183).
4.5. Pathways of Canonical and Biased PAR Signaling
PARs activate multiple pathways of intracellular signaling, which depend on the activating proteases and the receptor in question (see Supplemental Tables S1–S4). PARs couple to multiple subtypes of heterotrimeric guanosine triphosphate-binding proteins (G proteins) that are composed of Gα and Gβγ subunits. GPCR activation causes the Gα subunit to exchange GDP for GTP, leading to dissociation of “active” GTP-bound Gα protein from the Gβγ subunits. Subsequent signaling cascades are typically determined by the Gα protein subtype, where PAR1 and PAR2 interact with Gαq/11, Gαi/o, and Gα12/13 (184).
4.5.1. Calcium.
Gαq/11 activates phospholipase C (PLC) to hydrolyze PIP2 into IP3 and diacylglycerol. IP3 initiates Ca2+ release from the endoplasmic reticulum. Ca2+ mobilization was monitored upon first characterization of thrombin/PAR1 (4), trypsin/PAR2 (5, 21), thrombin/PAR3 (30), and thrombin/PAR4 (8). This leads to protein kinase C (PKC) activation; e.g., PKCε recruitment to the plasma membrane of DRG neurons in response to thrombin (185). Multiple regions of PAR receptors are involved in Gαq/Ca2+ signaling, including PAR1 helix 8 (186) or a PAR2 palmitoylation site (187). PAR1-mediated Ca2+ signaling is sensitive to thapsigargin, a Ca2+/ATPase transporter inhibitor (188). It can also be modulated by store-operated Ca2+ influx via TRPC (189). In endothelial cells, thrombin and activating peptides for PAR1 and PAR2 induce Gαq-mediated Ca2+, with no role of Gαi (190). While many proteases induce Ca2+ signaling [e.g., legumain-activated PAR2 (149)], some proteases do not mobilize Ca2+ [e.g., elastase/PAR2 (146, 148)].
Ca2+ signaling has distinct consequences depending on cell type. For example, PAR1-mediated Ca2+ signaling in astrocytic end feet is important for protease influx upon disturbance of the blood-brain barrier (93). With respect to platelets, Ca2+ signaling is linked to platelet aggregation. Two rare patients with deficiencies in Gαq or PLC-β2 had a mild bleeding disorder with mucosal bleeding and bruising. This was accompanied by reduced Ca2+ signaling via PAR1 and PAR4 in platelets (191). A racially dimorphic mutation in platelet PAR4 that is prevalent in Black populations enhances Ca2+ signaling, with elevated platelet aggregation in response to AYPGKF (192). This Thr120Ala mutation (rs773902) in TM2 implicated responses to therapeutics, with resistance to inhibition by selective PAR4 antagonist YD-3 (193).
Ca2+-signaling kinetics differ between PAR subtypes. PAR4 has a slower, sustained Ca2+-signaling profile, compared to rapid, transient Ca2+ mobilization in response to PAR1 (84, 194, 195) or PAR2 (196). Akin to Ca2+ kinetics, platelet aggregation induced by the PAR4-activating peptide is slower than thrombin/PAR1 (197). There are also distinctions in kinetics between proteases, whereby FXa Ca2+ mobilization in endothelial cells is more delayed than thrombin (13, 198, 199). Other factors can influence Ca2+-signaling kinetics, such as TRPV4 coexpression resulting in more sustained Ca2+ in response to PAR2 (200).
4.5.2. MAPK kinases.
PAR activation leads to the phosphorylation of mitogen-activated protein kinases (MAPK), including ERK1/2 or p38 MAPK. Canonical ERK1/2 signaling is downstream of Gαq/Ca2+ for PAR2 (201) and PAR4 (197). Trypsin causes PKC-dependent ERK1/2 activation inhibited by a nonselective PKC inhibitor and a PKC-β inhibitor (201). Trypsin-mediated PAR2 ERK1/2 signaling is also reduced by inhibitors of Src, Gαi, and Rho kinase (202). Activation of PAR2-mediated nuclear or cytosolic ERK1/2 is sensitive to Gαq and Gβγ inhibitors (203). ERK1/2 is linked to chemotactic pathways, as PAR2 is expressed in motile cells (e.g., macrophages, tumor cells). PAR2-activating peptide induces cytoskeletal rearrangements and extended polarized pseudopodia in a manner sensitive to an inhibitor of MEK1/2, a kinase upstream of ERK1/2 (204). siRNA against PAR2 demonstrated its role in cancer cell migration, where ERK1/2 activation peaks within 15 minutes and remains elevated in response to FXa or activating peptides (140). Other physiological consequences are linked to ERK1/2, such as PAR2-mediated hypersensitivity in DRG neurons. Likewise, incubation with a MEK1/2 inhibitor inhibited the development of mechanical hypersensitivity (205). Considering PAR1 is an oncogene in metastatic breast cancer, cell proliferation is also a consequence of ERK1/2 signaling. Janus kinase 2 (JAK2) is another kinase upstream of Ras/Raf/MEK/ERK in thrombin-stimulated vascular SMC growth (206). Thrombin/PAR1 also activates p38 MAPK in vascular SMCs (207). PAR1 p38 MAPK activation leads to increased endothelial permeability, downstream of Src-mediated activation of E3 ubiquitin ligase (208).
Despite lacking Ca2+ signaling, elastase and proteinase-3 activate ERK1/2 via PAR1 and Gαi/o signaling (144). Similarly, elastase triggers Ca2+-independent PAR2 ERK1/2 phosphorylation in a Rho kinase-dependent manner (146). ERK1/2 signaling can be modulated by other receptors. PAR1 heteromerization with CXCR4 (FIGURE 4B) modulates thrombin-induced Ca2+ and ERK1/2 signaling in endothelial cells (183). Via TM2 PAR1/CXCR4 interactions, ERK1/2 phosphorylation kinetics are modulated where CXCR4 siRNA reduces ERK1/2 phosphorylation.
4.5.3. PI3K/Akt.
Gαq-mediated PI3K activation phosphorylates Akt (also known as protein kinase B), a Ser/Thr-specific kinase that regulates cell survival and proliferation. For PAR2, this leads to chemotaxis downstream of Gαq/Ca2+-activated PI3K (209). Trypsin-mediated Akt activation can enhance motility in tumor metastasis via actin polymerization promoting microvesicle generation in MDA-MB-231 cancer cells (210). PAR2/Akt also maintains intestinal epithelium homeostasis by inhibiting cytokine-induced apoptosis in colonic epithelial cells, in a manner sensitive to MEK1/2 and PI3K inhibitors (211). In response to various proteases, PI3K/Akt signaling also regulates platelet aggregation downstream of PAR1 (212) and PAR4 (213). MMP2 potentiates thrombin-induced platelet aggregation in a PI3K-dependent manner (214). Later identified to reveal “DPR-TRAP,” MMP2 triggers Gαq and Gα12/13 signaling that lead to Akt activation in platelets, as well as p38 MAPK and Ca2+ (177). Key phenotypic differences between APC- and thrombin-induced PAR1 signaling in endothelial cells have been attributed to Akt signaling. APC or its activating peptide “TR47” cause Akt phosphorylation in endothelial cells, whereas thrombin or “TRAP” did not (142). Likewise, mice expressing PAR1 Arg46Asn exhibit reduced Akt phosphorylation, whereas mutating the canonical site have reduced Ca2+ signaling (215).
4.5.4. Adenylyl cyclase and cAMP.
Whereas Gαs increases cAMP via activation of adenylyl cyclase, Gαi/o inhibits local cAMP production. PAR1 activation leads to reduced forskolin-induced cAMP in platelets (155). PAR2 also reduces cAMP formation in SMCs, such as in response to tryptase (216) or its activating peptide (217). There are disagreements regarding PAR2-induced cAMP changes across cell types. Unlike PAR1, the same study suggests PAR2 does not modulate cAMP (184). In contrast, there is evidence of PAR2/Gαs signaling in response to trypsin in a manner sensitive to PAR2 antagonist I-343 (203). Cathepsin S is biased for Gαs signaling, while lacking Ca2+ signaling or β-arrestin recruitment (145). Legumain also leads to cAMP accumulation (149). In addition to monitoring cAMP, studies demonstrate the concentration dependence of PAR1- or PAR2-Gαi coupling, with rapid Gαi recruitment to PAR1 and evidence for preassembled complexes (218). PAR4 can induce Gαi-mediated reduction in cAMP. Activating peptides for PAR1 (SFLLRN) and PAR4 (AYPGKF) inhibit adenylyl cyclase in platelets in an ADP-dependent manner (219). This is reduced by an antagonist of the P2Y12 receptor, a receptor for ADP. Platelets from a patient lacking P2Y12 no longer respond to thrombin with respect to cAMP inhibition, resulting in reduced platelet aggregation (219). There is evidence that PAR4/P2Y12 interactions occur via TM4 (214).
4.5.5. Cytoskeletal changes via Rho GTPase.
Gα12/13 activation modulates the cytoskeleton via RhoGEFs, RhoA, and phosphorylation of myosin light chain (MLC). Early studies demonstrated that thrombin activates Gα12/13 [e.g., platelets (220), endothelial cells (221)]. Thrombin-mediated RhoGEF activation regulates the cytoskeleton (222). In endothelial cells, thrombin-mediated activation of RhoA increases endothelial permeability important for vascular inflammation (223) via rearrangement of the cytoskeleton (224). Changes in endothelial permeability induced by thrombin or PAR1-activating peptide are sensitive to inhibitors of Gα12/13-mediated Rho kinase (190). Cytoskeletal changes can also involve cofilin, an actin filament-severing protein that leads to cytoskeletal reorganization and chemotaxis. PAR2 activates cofilin by dephosphorylation independently of Gαq/Ca2+; this is reduced in the absence of β-arrestin (225). PAR1 and PAR2 activate RhoA via both Gαq/11 and Gα12/13 (185). PAR4 also leads to changes in cell shape in a Gα12/13-dependent manner in response to AYPGKF, sensitive to a ROCK inhibitor or CRISPR/Cas9-mediated knockout of RhoA (226). In contrast to the rapid, transient recruitment of Gαi to PAR1 and PAR2, Gα12/13 coupling is delayed and sustained (218, 227). Gα subtypes interact with different interfaces on the cytoplasmic face of GPCRs. While inhibition of PAR1 helix 8 reduces Gαq signaling, there is no effect on Gα12/13-mediated changes in platelet shape or transepithelial resistance (228).
There are contrasting effects of thrombin and APC on endothelial barrier permeability. Whereas thrombin increases permeability by a RhoA pathway, APC/EPCR stabilizes the endothelial barrier by a Rac1 pathway (229). Phosphoproteomic studies comparing thrombin and APC in endothelial cells demonstrate differences in numerous phosphorylation targets that modulate adherens junctions, such as afadin and adducin-1 (230). Other proteases exhibit bias with respect to cytoskeletal changes. Despite cleaving noncanonical sites, elastase and proteinase-3 induce MAPK signaling and actin stress fiber formation in endothelial cells (144). MMP1/PAR1 also demonstrate bias, causing migration through cytoskeletal changes via Rho activation and MAPK (143). This was shown in platelets (143), endothelial cells (231), and SMCs (232), in addition to triggering cancer cell invasiveness (233). Whereas thrombin induces a contractile phenotype, MMP1 triggers migration and proliferation (232). MMP1 migration is PAR1 dependent, blocked by a cell-penetrating pepducin (sect. 6.2) or inhibitor RWJ-56110 (231). Other factors can modulate cytoskeletal responses, such as protease concentration. Whereas high thrombin concentrations disrupt endothelial barriers, picomolar concentrations are protective in a PAR1-dependent manner (234). Posttranslational modifications can also alter Gα coupling, whereby lacking PAR1 ECL2 glycosylation reduces Gα12/13-dependent RhoA activation and stress fiber formation in contrast to Gαq signaling (235). Heteromer PAR1/PAR3 formation can also increase Gα12/13 signaling without affecting Ca2+ pathways, causing increased endothelial permeability and PAR1/Gα12/13 coupling (171).
4.5.6. β-Arrestin recruitment.
β-Arrestins mediate desensitization and endocytosis of many GPCRs. Whereas PAR2 endocytosis requires β-arrestin recruitment (FIGURE 5; sect. 4.8) (201), β-arrestin is not required for endocytosis of PAR1 (236) or PAR4 (237). With respect to PAR1, β-arrestin interactions are phosphorylation independent (236) with slow, sustained kinetics (238). Rapid β-arrestin recruitment to PAR2, on the other hand, peaks within minutes (227). PAR1/PAR2 heteromer formation results in increased β-arrestin recruitment (166). Some proteases that activate PAR2 are unable to recruit β-arrestin, such as elastase (146, 147), cathepsin S (145), and legumain (149).
FIGURE 5.

Trafficking of protease-activated receptors (PARs). A: PAR2 activation leads to Gα/βγ recruitment, phosphorylation (Ph; red) by G protein-coupled receptor (GPCR)-regulated kinase (GRK), and β-arrestin (βARR) recruitment (1). This triggers clathrin-mediated endocytosis (CME). Internalized PAR2 continues to signal from endosomes (2). Ubiquitination (Ub; blue) of the COOH terminus triggers trafficking to lysosomes for degradation (3). To replace the cleaved receptor, PAR2 signaling induces Gβγ-mediated activation of PKD in the Golgi apparatus, which mobilizes newly synthesized PAR2 to repopulate the plasma membrane (4). B: PAR1 is subject to constitutive CME upon ubiquitination and AP-2 recruitment (1). In contrast to PAR2, it is not subject to βARR-dependent endocytosis. PAR1 can signal from endosomes in a ubiquitin-driven manner with adaptor proteins TABs to drive p38 MAPK signaling (2). Uncleaved PAR1 is returned to the plasma membrane via recycling endosomes. Cleaved PAR1 signals with Gα/βγ. Similarly, PAR1 is then degraded in the lysosome (3).
4.6. Sustained PAR Signaling in Subcellular Compartments
GPCRs were conventionally considered to signal predominantly from the plasma membrane, where GPCRs interact with extracellular ligands and couple to intracellular G proteins and β-arrestins. Plasma membrane signaling is often transient, where β-arrestin-mediated desensitization and endocytosis were viewed to terminate cell surface GPCR signaling. However, it is now clear that GPCRs, including PARs, can continue to signal from within the cell (239). Moreover, different signaling outcomes can originate from receptors in distinct subcellular regions.
When activated by canonical mechanisms, PAR1 (240), PAR2 (122, 201, 241, 242), and PAR4 (237) all undergo clathrin-mediated endocytosis (sect. 4.8). While it is unknown whether extracellular proteases internalize alongside PARs, fluorescent PAR2-activating peptides rapidly internalize (243). The capacity of PAR2 to signal from endosomes has been extensively studied. Trypsin and activating peptides induce the assembly of PAR2, G protein, and β-arrestin signalosomes in model cell lines (HEK, KNRK), colonocyte cell lines, and DRG neurons (201, 203). Endosomal PAR2 signaling leads to activation of ERK1/2 in the cytosol and nucleus, mediating the effects of trypsin and tryptase on paracellular permeability of colonocytes (244) and excitability of nociceptors (203). PAR1 and PAR4 also signal from endosomes. Ubiquitinated PAR1 in endosomes interacts with adaptor proteins to mediate p38 MAPK signaling (208). Although β-arrestin knockdown has no effect on PAR4 endocytosis, siRNA targeting a subunit of adaptor protein-2 (AP-2) or mutations in PAR4 ICL3 inhibit internalization. Inhibition of PAR4 endocytosis enhances ERK1/2 phosphorylation, whereas internalization is required for Akt signaling (237). Some proteases do not cause PAR2 internalization, including elastase (146, 147), cathepsin S (145), and legumain (149). As such, inhibiting endocytosis has no effect on signaling or nociceptive responses initiated by these proteases (203).
PAR signaling can also be modulated by receptor localization in membrane microdomains, including lipid rafts. PAR1 and EPCR colocalize in cholesterol-rich lipid rafts in endothelial cells (245). Lipid rafts are vital for the protective barrier effects of APC since methyl-β-cyclodextrin, a cholesterol-chelating agent that disrupts lipid rafts, blocks the cytoprotective effects of APC (246). Filipin, which also disrupts lipid rafts, inhibits thrombin and MLC kinase-mediated cytoskeletal changes in endothelial cells (247). Cavaeloe (“little caves”) are specialized lipid rafts. Disruption of caveolae using siRNA against caveolin-1 has no effect on thrombin-mediated cytoskeletal changes (247). In contrast, the cytoprotective effects of APC via Rac1 activation and protection from endothelial barrier permeability are abolished in cells lacking caveolin-1 (229).
4.7. Downstream Targets of PAR Signaling
4.7.1. Transactivation with receptor tyrosine kinases.
GPCRs transactivate receptor tyrosine kinases (RTKs) and vice versa. A seminal study of GPCR/RTK transactivation investigated PAR-mediated upregulation of epidermal growth factor receptor (EGFR) signaling (248; FIGURE 4C). Rapid RTK phosphorylation is observed in response to thrombin and PAR1-activating peptide (248). Considering these agonists could act through multiple PAR subtypes, it was later shown to be PAR1 dependent (249). PAR/EGFR transactivation can occur through multiple mechanisms. PAR activation can stimulate the release of EGFR agonists following MMP activation (250), cleaving amphiregulin (heparin-binding EGF) to enable EGFR activation (FIGURE 4C). Additionally, Src kinase can mediate thrombin- or TRAP-induced EGFR transactivation, as demonstrated using an Src kinase inhibitor or immunoprecipitation (249, 251). PAR2/EGFR transactivation is also observed in HT-29 colon cancer cells, where PAR2 activation by trypsin or PAR2-activating peptide leads to Src kinase activation and MMP-dependent release of an EGFR ligand (252). In lung-derived A-549 epithelial cells, SLIGRL-mediated activation of PAR2 leads to EGFR phosphorylation, as well as delayed, persistent p38 MAPK activation blocked by the EGFR inhibitor PD153035 (253). PAR2-mediated EGFR transactivation is also seen in epithelial cells via an Src-dependent mechanism (254). Src-mediated PAR2/EGFR transactivation is observed in response to trypsin or activating peptide in ovarian cancer cells that endogenously overexpress PAR2 with an enhanced migratory and invasive phenotype. PAR2 transactivates EGFR through Gαq/11, Gα12/13, and β-arrestin but not Gαs or Gαi (255). In lung cancer cells, coinhibition with the EGFR inhibitor gefitinib and 2pal-18S leads to synergistic inhibition of EGFR phosphorylation, ERK1/2 phosphorylation, and tumor growth (255).
PARs can also transactivate platelet-derived growth factor receptors (PDGFRs). PAR1 and PDGFRs are coexpressed in the spinal cord, where thrombin-induced hyperalgesia is reduced in the presence of recombinant mouse PDGFR/Fc chimera (256). PAR2-induced Src signaling mediates PDGFR transactivation, leading to chemotaxis in monocytes, fibroblasts, and endothelial cells (257). PAR2 also transactivates the hepatocyte growth factor (HGF) receptor Met involved in liver cancer metastasis (258). PAR2 activation causes Met transactivation in hepatocarcinoma cells independently of HGF release. PAR2-mediated cell invasion via ERK1/2 is sensitive to Met inhibition following siRNA knockdown or pharmacological inhibition with SU 11274 or PHA 665752. In endothelial cells, thrombin/PAR1 signaling also transactivates the vascular endothelial growth factor (VEGF) receptor 2 via MAPK phosphatase and ERK1/2 signaling (259).
4.7.2. TRP ion channels.
Numerous studies have shown PAR activation sensitizes TRP channels, which is relevant to protease-evoked neurogenic inflammation and pain (sect. 5.5; FIGURE 4D). PAR2 activation sensitizes TRPV1 in DRG neurons, which results in thermal hyperalgesia (97, 260, 261). The TRPV1 antagonist capsazepine blocks thermal hyperalgesia induced by selective PAR2 agonists (262). Mechanistically, PAR2-mediated TRPV1 sensitization is mediated by PKCε and protein kinase A (PKA) (97, 260, 261). TRPV1 sensitization in response to activating peptides for PAR1 or PAR4 is also mediated by PKCε signaling (185). PAR2-mediated TRPV1 potentiation in response to SLIGRL is absent in PAR2 knockout mice (109). PAR2-mediated sensitization of TRP channels is responsible for chemotherapy-induced neuropathy following paclitaxel exposure (263). Pain responses are inhibited by a selective PAR2 antagonist, as well as inhibitors of PKCε and PKA. Paclitaxel-induced hyperalgesia is reduced by inhibitors of TRPV4 (RN1734), TRPV1 (capsazepine, SB366791), and TRPA1 (HC030031).
PAR2 also sensitizes TRPV4 signaling, with relevance to pain (147, 200, 264). PAR2-induced mechanical allodynia is absent in TRPV4 knockout mice (264). PAR2-activating peptide leads to enhanced firing of colonic DRG neurons in response to TRPV4 agonists (265). PAR2 activation induces a sustained TRPV4-mediated Ca2+ influx from extracellular ions (200, 266). Dependent on the TRPV4 Tyr110 residue, this is sensitive to inhibitors of PLA2 and arachidonic-derived lipid mediators, the latter suggesting links to the release of TRPV4 agonists such as epoxyeicosatrienoic acid (200). Trypsin and PAR2 agonist-mediated sensitization of mouse aorta contractions via TRPV4 is reduced in the presence of PKC inhibitor Go6983 (267). In the hippocampus, the PAR2 agonist AC55541 also leads to long-term depression following PKA-dependent TRPV4 phosphorylation (268). TRPV4 sensitization has been shown for different proteases cleaving PAR2, including cathepsin S (145). Cathepsin S/PAR2-evoked sensitization of TRPV4 is dependent on adenylyl cyclase and PKA. Elastase evokes PAR2-dependent sensitization of TRPV4 by a similar mechanism (147). TRPV4 potentiation by PAR2 is inhibited in the presence of the EGFR kinase inhibitor AG1478 in the mouse aorta (267). TRPV4 sensitization is also observed in response to PAR1 activation in endothelial cells, whereby TRPV4 contributes to sustained Ca2+ signaling and endothelial junction remodeling (269). There are also links between PAR4 and TRP channel sensitization. DRG neurons do not signal in response to PAR4-selective AYPGKF. Instead, AYPGKF reduces capsaicin-induced Ca2+ signaling and reverts inflammatory hyperalgesia in a dose-dependent manner (270). In response to PAR4 agonist peptide AYPGKF-NH2, Ca2+ mobilization is reduced when stimulated with pronociceptive PAR2 agonists or TRPV4 agonist 4αPDD in colonic sensory neurons (111). This endogenously acts to reduce nociception.
PAR2 also potentiates responses to TRPA1 (271). TRPA1 is expressed on sensory neurons, including those that innervate the intestine, where it mediates mechanosensation (272). TRPA1 is activated by allyl isothiocyanate (AITC), the spicy component of mustard that also activates TRPV1. Covalent modification of cytosolic NH2-terminal TRPA1 residues (Cys621, Cys641, Cys665) leads to channel activation (273). TRPA1 sensitization is partly mediated by increased channel insertion into the membrane (274). PKA phosphorylates four serine residues (Ser86, Ser317, Ser428, Ser972) proposed to contribute to TRPA1 sensitization. Potentiated currents are sensitive to inhibitors or PIP2 or PLC, showing that PAR2 sensitizes TRPA1 via membrane hydrolysis of PIP2. In comparison to TRPV1 sensitization, the mechanism of PAR2-mediated sensitization of TRPA1 does not involve PKC. Studies have also shown that PAR2 sensitizes responses to acid-sensing ion channel 3 (ASIC3). Potentiation by PAR2-activating peptide causes a dose-dependent increase in acetic acid-induced hyperalgesia in rats in a PAR2-specific manner (275).
4.7.3. Genes.
It is well-established that PAR signaling leads to transcriptional changes that alter gene expression. Early studies showed thrombin or FXa stimulation activates NF-κB, a known regulator of gene transcription (13). Gene expression microarrays have since demonstrated hundreds of genes are regulated by PAR1 (276) or PAR2 (277) signaling. Considering their central role in the immune system, PAR activation leads to the upregulation of proinflammatory cytokines and chemokines across cell types. For example, IL-6 and IL-8 are released from HUVECs in response to FXa and thrombin (199). IL-8 secretion induced by thrombin has also been attributed to PAR3 activation in lung epithelial cells and human astrocytoma cells (168). Activation of both PAR1 and PAR2 leads to IL-8 expression in epithelial cells through NF-κB- and ERK1/2-dependent pathways (278). In human lung A-549 epithelial cells, PAR2 stimulation leads to prostaglandin E2 (PGE2) formation (253, 279). PAR2-mediated IL-8 upregulation in A-549 cells is sensitive to MEK, JNK, and EGFR inhibitors (280). Treating mice with a PAR2 agonist leads to the release of IL-4, IL-6, IL-13, and TNF-α, many of which are reduced in β-arrestin knockout mice (281). Studies have demonstrated PAR2-dependent release, as CXCL8, IL-8, IL-6, CTGF, and CSF2 are released in response to the PAR2 agonist and inhibited by I-191 (282). SLIGRL-mediated activation of PAR2 in A-549 epithelial cells upregulates cyclooxygenases (COX-1, COX-2) (253). PAR2 activation in A-549 cells upregulates prostaglandin E synthetase isoform 1 (mPGE-1) and COX-2, dependent on prostanoids formed via MEK/ERK/cPLA2/COX-1 (279).
Noncanonical proteases can alter gene expression, such as downstream of APC/PAR1/EPCR in HUVECs (63). This alters expression of genes encoding the nuclear hormone receptor TR3, the immunomodulatory monocyte chemoattractant MCP-1, and a negative regulator of calcineurin (63). Granzyme K also causes a concentration-dependent release of IL-6, IL-8, and MCP-1 from human lung fibroblasts, sensitive to PAR1 or ERK1/2 inhibitors (283). KLK-5 leads to PAR2/NF-κB signaling and changes in microRNA expression in oral cells (284). In addition to distinctions between proteases, gene expression is modulated by other membrane proteins. For example, PAR2 is expressed alongside Toll-like receptor 4 (TLR4) in macrophages and colonic epithelial cells. Functional cross talk between PAR2 and TLR4 can induce NF-κB signaling (285). Growth factors are also released in response to PAR activation, such as the release of PDGF in response to FXa stimulation of vascular smooth muscle (286). PAR1-activating peptide induces PDGF secretion from epithelial cells derived from the lung (287). PAR2-activating peptide and FXa stimulate the expression of connective tissue growth factor involved in its profibrotic events in endothelial cells (174).
Genes involving the ECM network of proteoglycans and glycosaminoglycan chains are also modulated by PAR signaling. FXa and thrombin activation increases expression of ECM-associated proteins that modulate adhesion, Cyr61, and CTGF (13). PAR1 signaling also induces ECM production, whereby FXa and thrombin both stimulate procollagen gene expression in mouse fibroblasts, absent in the PAR1-deficient mouse fibroblasts (288). Thrombin also increases expression of a glycosaminoglycan biosynthesizing enzyme via PAR1 RTK transactivation (289).
PARs therefore act as the first step that triggers “feed-forward signaling” through a multitude of downstream outcomes mediated by other receptors. These signaling events can have different kinetics and degrees of signal amplification, which enable both a rapid and prolonged response to proteases. For example, TRP channel sensitization and RTK transactivation are rapid (seconds to minutes), whereas transcriptional regulation is more sustained (minutes to hours). The release of agonists acting through other receptors can also have a variable timeframe, such as prostanoids secreted from epithelial cells. Considering there is context dependence across even different cell lines, we have a limited understanding of feed-forward signaling in endogenous human systems.
4.8. Termination and Recovery of PAR Signaling
4.8.1. PAR desensitization.
Desensitization rapidly terminates signaling of PARs. There are distinct mechanisms through which PAR signaling is terminated that differ in their kinetics and downstream outcomes. These mechanisms include receptor desensitization due to irreversible proteolysis and G-protein uncoupling, endocytosis-mediated trafficking and subsequent receptor degradation. Once cleaved, PARs can no longer respond to proteases due to removal of the cleavage site. This is a rapid process occurring on a timescale of seconds to minutes. For example, exposure of DRG neurons to tryptase prevents subsequent Ca2+ responses to trypsin (95). Similarly, exposure to trypsin prevents subsequent thrombin/PAR1 activation in Jurkat cells (159). Protease-cleaved PARs can, however, still respond to activating peptide, as shown for thrombin-activated PAR1 (290).
Another method of PAR desensitization involves endocytosis. Relative to desensitization, endocytosis and degradation are slower. The trafficking of many GPCRs is regulated by phosphorylation of the COOH-terminal domain by GPCR kinases (GRKs). The COOH terminus of PAR1 (291, 292) and PAR2 (242) are rapidly phosphorylated at Ser/Thr residues in response to activation. GPCR phosphorylation is often required for β-arrestin recruitment and receptor endocytosis. However, PAR1 recruitment of β-arrestin is phosphorylation independent (236). Despite this, mutagenesis of PAR1 Ser/Thr residues causes reduced endocytosis, slower desensitization, and more robust signaling (291, 293, 294). Accordingly, enhancing PAR1 phosphorylation by GRK overexpression reduces thrombin-induced signaling (291, 295). There are also differences between GRK isoforms, whereby the termination of PAR1 signaling involves GRK5 (295). Another mechanism of PAR desensitization involves regulators of G-protein signaling (RGS proteins). RGS proteins are cytoplasmic GTPase-activating proteins that accelerate GTP hydrolysis, thereby reducing GPCR signaling. These directly interact with PAR1 at ICL3, reducing Ca2+-activated chloride currents and ERK1/2 phosphorylation (296, 297). This is also observed for PAR2 (298) and PAR4 (299).
Some studies have solely attributed β-arrestin to receptor desensitization. Although not required for PAR1 endocytosis, β-arrestin overexpression reduces thrombin-induced IP3 production (236). Similarly, phosphatidylinositol hydrolysis is reduced upon β-arrestin overexpression in cells expressing PAR1 (300). On the other hand, studies have linked β-arrestin knockdown to signaling. In a pathway-specific manner, thrombin-induced PI3K and Akt signaling is reduced by dominant-negative β-arrestin without affecting ERK1/2 (212). PAR1/β-arrestin coupling is linked to APC-mediated cytoprotective signaling, in contrast to thrombin-mediated ERK1/2 and Rho GTPase endothelial disruption (301). FVIIa/PAR1-mediated Akt phosphorylation is also reduced in endothelial cells with β-arrestin siRNA (14). Contradicting studies have also been shown for PAR2. Implicating its role in desensitization, numerous PAR2-expressing cell types lacking β-arrestin have increased phosphatidylinositol hydrolysis (302). Likewise, mutant PAR2 unable to interact with β-arrestin (Ser636Ala/Thr366Ala) has sustained Ca2+ and ERK1/2 signaling kinetics (201). β-Arrestin knockout cells expressing PAR2 have prolonged Ca2+ signaling in response to SLIGRL or trypsin (187). β-Arrestin knockdown also causes distinct kinetics in ERK1/2 phosphorylation with a sustained signaling profile in response to trypsin in the absence of desensitization (302). In contrast, other studies have shown that ERK1/2 phosphorylation is reduced in the absence of β-arrestin (146). This is also shown for PAR2-mediated inflammation, as β-arrestin knockout mice have reduced proinflammatory cytokine production or leukocyte recruitment in response to a PAR2 agonist (281). There is evidence for direct interactions between PAR2, β-arrestin, and ERK1/2 using coimmunoprecipitation (201), as well as between β-arrestin and PI3K, albeit with an inhibitory outcome on Akt signaling (209).
While there are distinctions between PAR2 and PAR1/PAR4 trafficking, each subtype undergoes clathrin-mediated endocytosis (FIGURE 5). Fluorophore-labeled PAR2 shows β-arrestin recruitment to the plasma membrane within 5 minutes of trypsin stimulation, followed by receptor/β-arrestin internalization within 10 minutes in KNRK cells (241). PAR2 is internalized into Rab5+ endosomes (303). There has since been evidence using BRET between luciferase-tagged PAR2 and fluorescent subcellular markers to demonstrate trafficking in response to trypsin within 5–10 minutes sensitive to dominant-negative dynamin (203). This was demonstrated in colonocytes using BRET, as well as in vivo using mice expressing PAR2 fused to green fluorescent protein (39).
Unlike PAR2 (FIGURE 5A), the trafficking of PAR1 and PAR4 away from the plasma membrane occurs through distinct mechanisms. PAR1 endocytosis is phosphorylation and β-arrestin independent (236, 300, 304, 305; FIGURE 5B). PAR1 is subject to dynamin-mediated endocytosis into clathrin-coated pits (240) and rapidly internalizes in response to SFLLRN (306) or thrombin (307). Unlike PAR2, PAR1 is also subject to constitutive endocytosis (293). PAR1 constitutive endocytosis is regulated by ubiquitylation, whereby mutating ubiquitinated Lys residues causes enhanced constitutive endocytosis (304). This is mediated by interactions of AP-2 with a COOH-terminal tyrosine motif in PAR1 that can be mutated to AKKAA (305, 306). However, cultured cells may secrete proteases that endogenously activate PARs by an autocrine mechanism. These proteases could activate PAR1, evoke endocytosis, and thus account for spontaneous PAR turnover. PAR4 is internalized with slower kinetics than PAR1 in response to stimulation, albeit similarly independent of phosphorylation or β-arrestin (194, 237). PAR4 endocytosis is, however, clathrin mediated with inhibition by dominant-negative dynamin (237). PAR4 is also internalized from the plasma membrane via AP-2 binding, instead of through a highly conserved tyrosine motif in the ICL3 (237).
4.8.2. PAR lysosomal targeting and downregulation.
PARs are activated in an irreversible manner and are ultimately degraded rather than recycled. There are differences between PAR1 and PAR2 with respect to pathways that lead to receptor degradation. PAR2 is ubiquitinated and then targeted to lysosomes for degradation (308–310). PAR2 signaling leads to phosphorylation and activation of c-Cbl, an E3 ligase, that directly ubiquitinates PAR2 Lys residues (310). In the absence of Lys residues or in cells expressing dominant-negative c-Cbl, PAR2 is retained in endosomes without degradation. Lysosomal PAR2 degradation then occurs via a component of ESCRT-0, the HGF-regulated tyrosine kinase substrate (HRS) (309). As observed for endocytosis, not all proteases cause lysosomal degradation. Trypsin and its activating peptide lead to colocalization with a lysosomal marker within 2 hours, whereas elastase does not (311).
In contrast, PAR1 is targeted to lysosomes in an ubiquitin-independent manner through an ESCRT-III–dependent pathway (312). PAR1 is directed to lysosomes via interactions with a Tyr-based motif in ICL2, interacting with adaptor protein ALG-2-interacting protein X (ALIX) in multivesicular bodies. Direct interactions with an ESCRT complex bypass the requirement for ubiquitination. siRNA against ALIX, or PAR1 mutagenesis to Tyr206Ala, highlights the importance of this interaction for rapid lysosomal sorting. PAR1 lysosomal sorting is also regulated by palmitoylation at Cys residues in the COOH terminus (313). In the absence of this palmitoylation site, there is increased constitutive internalization and degradation mediated by AP-3. PAR1 lysosomal degradation is modulated by tumor suppressor α-arrestin domain-containing protein-3 (ARRDC3), a protein that acts via ALIX to mediate PAR1 degradation (314). Invasive breast cancers exhibit a loss of ARRDC3, thus reducing lysosomal degradation for sustained, aberrant PAR1 signaling.
In terms of the other PAR subtypes, initial studies have identified PAR4 in lysosomes after agonist stimulation (237). Although in a similar localization to PAR1, this is independent of the COOH terminus. PAR4 degradation is mediated through a conserved tyrosine motif in ICL3 involved in AP-2 interactions.
4.8.3. Recovery of PAR signaling.
Many GPCRs for hormones and neurotransmitters recycle from endosomes to the plasma membrane, typically involving Rab11 GTPases that control vesicle formation; this recycling mediates resensitization. Constitutive PAR1 endocytosis is considered to enable rapid recovery of signaling by returning uncleaved PAR1 to the plasma membrane (292, 315). Uncleaved PAR1 is directed to the plasma membrane via Rab11b-dependent sorting, in opposition with Rab11a-mediated targeting for lysosomal degradation (316). The requirement of constitutive PAR1 internalization for resensitization to signaling is demonstrated in endothelial cells. Mutating the tyrosine motif responsible for AP-2 interactions, or using siRNA against AP-2, prevents endothelial cells from gaining responsiveness to thrombin (306).
Following protease stimulation, PARs are not recycled to return to the plasma membrane given their nature of irreversible cleavage. This was investigated with respect to PAR1 using a “P/S” chimera with the COOH terminus of neurokinin 1 receptor (NK1R, substance P receptor) (317, 318). Whereas wild-type PAR1 was destined for lysosomal degradation, the protease-stimulated chimera recycled to the plasma membrane, allowing persistent signaling to thrombin and activating peptide.
Recovery of PAR2 signaling at the cell surface occurs by a different mechanism that requires mobilization of intact receptors from Golgi stores in a Rab11-dependent manner (303, 308) (FIGURE 5A). Dominant-negative Rab11 Ser25Asn causes receptor retention in the Golgi apparatus, whereas Rab11a overexpression accelerates trafficking from the Golgi to the plasma membrane (303). PAR2 translocation from the Golgi is Gβγ and PKD dependent and is responsible for recovery of signaling at the cell surface (148, 319). This mechanism of recovery is similar to when PAR2 is activated by canonical (trypsin) or biased (cathepsin S, elastase) mechanisms (148, 319). This process underlies sustained protease- and PAR2-evoked pain since inhibitors of Gβγ and PKD attenuate mechanical allodynia in response to proteases in mice.
4.9. Summary
Proteases with varying selectivity can cleave PARs at distinct sites, leading to divergent pathways of receptor signaling and trafficking. Biased signaling can arise from exposure of different tethered ligands or stabilization of unique receptor conformations. Alternatively, proteases can disarm PARs by removal of cleavage sites or tethered ligand domains. Synthetic peptides that mimic tethered ligand domains (activating peptides) have been instrumental in delineating PAR signaling pathways and the complex modulation of PARs in heteromeric complexes with other GPCRs. While less is known for PAR3, the other PAR subtypes activate signaling pathways linked to Ca2+, cAMP, MAPK/ERK, PI3K/Akt, and Rho GTPases. PARs can also induce RTK transactivation, ion channel sensitization, and changes in gene expression. PARs also undergo endocytosis to regulate their localization and turnover with distinctions between mechanisms governing PAR1 and PAR2.
5. PARS IN PHYSIOLOGY AND PATHOLOGY
There are several challenges to delineating the function of PARs in health and disease states. Multiple proteases are capable of activating PARs, and, in some instances, the relevant proteases are not known. Although thrombin is an established agonist of PAR1 in the circulatory system, the proteases that activate PAR2 in different healthy and diseased tissues are not well understood. Since proteases are regulated through control of their activity, rather than expression, insight into the proteases that activate PARs requires profiling activated proteases. Activity-based probes that irreversibly bind to activated proteases are valuable tools for profiling proteases that are activated in diseased tissues (320). Pharmacological studies of PARs are hampered by the inadequate potency and selectivity of some PAR antagonists and the promiscuity of certain activating peptides that signal through multiple PAR subtypes. Genetic deletion studies, particularly cell-specific deletion of PARs, have provided valuable insights into functions, although compensatory changes can complicate the interpretation of results. Section 5 will discuss the role of PARs by the organ system in health and disease states.
5.1. PARs in the Circulatory System
5.1.1. Physiology.
The role of PARs in hemodynamic maintenance was proposed soon after their discovery, by the finding that intravenous administration of trypsin or thrombin produced endothelium-dependent acute hypotension in dogs (321). However, the effects of PAR activation on the vascular system (FIGURE 6A) depend on species, age, whether endothelial cells or vascular SMCs are studied, and the proteases under investigation. For example, canonical activation of PAR1 by thrombin produces a contractile, dedifferentiated phenotype, whereas biased activation of PAR1 by MMP1 leads to dedifferentiation and hyperplasia of SMCs (232).
PAR1 and PAR4 mediate thrombin signaling in platelets and thrombosis (322; FIGURE 6B). PAR1-mediated contraction is endothelium dependent and mediated by prostacyclin and nitric oxide (NO), an effect observed in humans (323, 324). PAR1 activation contributes to vascular permeability and edema, which involves substance P activation of NK1R on endothelial cells, the first evidence that thrombin mediated neurogenic inflammation (325). PAR2 activation leads to vasodilation by release of NO and endothelin from endothelial cells (326, 327). At a systems level, PAR2 activation induces hypotension by the l-arginine/NO pathway. The vasodilatory effects of PAR2 activation are greater than for PAR1 and PAR4 (164). PAR2-mediated vasodilation has been proposed to protect against heart ischemia, disseminated intravascular coagulation, and endotoxemia (328).
Proteases that participate in coagulation that can cleave PARs include thrombin, APC, TF-FVIIa, and TF-FVIIa-FXa. TF-FVIIa generates FXa, which can cleave PAR1 and PAR2. The complex of TF-FVIIa-Xa and EPCR can also cleave PAR1 or PAR2 (329). The APC-EPCR complex, as part of the protein C pathway, can cleave PAR1 (63; FIGURE 6C). FVIIa-EPCR can cleave PAR1 on endothelial cells (330). TF-FVIIa cleaves and signals through PAR2, which contributes to several pathologic conditions, especially cancer. PAR2 signaling following cleavage by TF-FVIIa affects integrin function and angiogenesis (331). The ternary complex TF-FVIIA-FXa cleaves PAR2. The TF pathway inhibitor inhibits the ternary complex; therefore, PAR signaling is also reduced (332). Thrombin activation of platelets leads to the secretion of endostatin, which inhibits angiogenesis. This effect is mediated by PAR4 (333).
The processes of thrombosis and inflammation are interlinked and reciprocal (334). Coagulation mediates both processes, where the factors and associated proteases activate PARs. For example, mannose-binding lectin-associated serine protease-1 (MASP-1) can activate PAR1 and PAR4 on platelets and endothelial cells, while MASP-2 activates the complement system starting with C4 and C2 (335, 336). Thrombin activates PAR1, PAR3, and PAR4, and plasmin activates PAR1 and PAR4. PAR2 is cleaved by trypsin, tryptase, TF, FVIIa, and FXa (43). Thrombin activation of PAR1 and PAR4 in humans leads to procoagulant activity through the release of α-granules and dense granules from platelets, which facilitates inflammation.
Following activation by the proteases associated with coagulation, PARs mediate a myriad of cellular effects on leukocytes that contribute to their role in the innate immune system. Activation of PAR1 on murine mast cells increases adhesion to fibronectin and laminin (117). Thrombin/PAR1 signaling mediates inflammation by endothelial cell activation, which leads to monocyte activation and increases the permeability of endothelial cells. Activation of PAR2 on eosinophils leads to the generation of superoxide anion and degranulation (124, 126). Similarly, activation of PAR2 either by trypsin or the activating peptide SLIGKV leads to degranulation of histamine from mast cells (121, 337). Activation of PARs leads to leukocyte recruitment, an effect that was demonstrated by injection of proteases and activating peptides into the airways of mice (specifically, thrombin activation of PAR4), which leads to the recruitment of polymorphonuclear cells (338). Catalytic activity of thrombin is not required for activation and chemotaxis of innate immune cells; the mechanism likely involves integrin activation (169, 339, 340). PARs also contribute to the adaptive immune response that follows coagulation. Activation of PAR2 of TH2 lymphocytes in mice leads to secretion of IL-31, which activates neurons and enhances neuroimmune signaling (341). The activation of PAR1 on T lymphocytes leads to the secretion of IL-6 and IL-8 (342).
In addition to differences in PAR subtype expressed by platelets (rat/mouse: PAR3, PAR4; human: PAR1, PAR4; sect. 3.4), there are species differences in the signaling pathways and platelet response to PAR activation. This must be considered as drugs are developed for the treatment of diseases that are a consequence of dysregulation of the coagulation system. Both thrombin and SFLLRN, which activate PAR1, have the following effect on platelets: activation of PLC, PI3K, and PKC, inhibition of adenylyl cyclase, and tyrosine phosphorylation of platelet proteins including a 47-kDa protein known as P47 or pleckstrin (343). While thrombin produces platelet aggregation and an increase in cytosolic Ca2+ in all species, SFLLRN activates PAR in guinea pigs, monkeys, and human platelets (40).
The activation of PAR1 and PAR4 on human platelets by their selective activating peptides has divergent effects with angiogenesis (333). Activation of both PARs leads to platelet aggregation; activation of PAR4 alone leads to platelet retraction (344). Activation of PAR1 induces VEGF release and suppresses endostatin release. In contrast, activation of PAR4 results in endostatin release, as occurs in the rat, and suppresses VEGF secretion. PAR1 is not expressed by rat or mouse platelets; however, rat platelets demonstrate a response to PAR1 agonists (345, 346). Moreover, in vivo effects (gastric ulcer healing) are observed in rats in response to a PAR1 antagonist (333). As such, studies in rodent platelets should be extrapolated with caution to humans.
5.1.2. Pathology.
Cells including monocytes, macrophages, lymphocytes, vascular SMCs, and platelets contribute to circulatory system pathology. PARs expressed on vascular SMCs, macrophages, and platelets contribute to diseases of the circulatory system, especially atherosclerosis and thrombosis.
In addition to its contribution to atherosclerosis, PAR2 on monocytes, macrophages, and endothelial cells contributes to vessel stability and health. PAR2 on monocytes mediates the effects of ischemia on vessel walls (347). Moreover, inflammation upregulates PAR2 on the endothelial cells (324, 348). Accordingly, PAR2 is upregulated in human coronary atheromas. Similarly, atherosclerotic aortas from mice that are ApoE-/- exhibit upregulation of PAR2 (349). Plaques from PAR2-/- mice are characterized by reduced levels of MMP9 and smooth muscle actin, along with increased collagen content in the plaques. Activation of PAR2 induces expression of inflammatory cytokines in macrophages (350). On the other hand, PAR2 stabilizes atherosclerotic plaques through the following: plaque progression is halted and stabilized, vascular inflammation and the associated macrophage response are reduced, and the adhesion between macrophages and vascular SMCs is lowered. Decreased adhesion between macrophages and vascular SMCs leads to reciprocal effects of the two cell types; namely, decreased expression of VCAM-1 and ICAM-1 by vascular SMCs and reduced TNF-α and MCP-1 expression by macrophages. PAR2 further affects plaque stability through new blood vessel formation and revascularization (351, 352).
PAR1 and PAR4 on platelets facilitate aggregation and subsequent thrombosis. When thrombin cleaves PAR1, PLCγ leads to increased intracellular Ca2+, in addition to the activation of integrin αIIbβ3 that leads to platelet aggregation. Antiplatelet therapy has been investigated extensively to reduce the generation of thrombi. Vorapaxar is a PAR1 antagonist approved for the treatment of patients who have suffered a myocardial infarction (sect. 6.1). Despite the central role of thrombin activation of PARs in atherosclerosis, platelet activation by mediators other than thrombin is sufficient to produce atheromas (353). This finding was demonstrated using ApoE-/- mice, which are prone to develop atherosclerotic plaques. The role of PAR4 was demonstrated using PAR4-/- mice. Mouse platelets require PAR4 for activation by thrombin, and platelets from PAR4-/- mice do not respond to thrombin, as measured by Ca2+ mobilization (322, 354). The investigators showed that atheromas on the aorta of the mice did not differ in size, extent, or location between PAR4-/- and WT mice.
Due to the differential expression and signaling of PAR1 and PAR4 on platelets and their role in forming a clot (FIGURE 6B), the two PARs are an attractive target for the development of antagonists to treat cardiovascular disease and cerebrovascular disease. Studies related to PAR1 led to the development of vorapaxar, which is selective for PAR1 but not PAR4. PAR1 mediates strong blood clotting, while the effect of PAR4 activation leads only to platelet aggregation, a response similar to that seen with weaker agonists such as epinephrine and ADP (163).
Most publications on the role of PARs on vascular SMCs report effects on these cells harvested from the aorta, and few studies have focused on cerebral arterioles. Misaki et al. (355) studied the response of SMCs from rat cerebral arterioles. Thrombin and PAR1-activating peptide induce increased Ca2+ mobilization from intracellular sources and contraction. On the other hand, PAR2 induced a decrease in Ca2+ thought to be mediated by endothelium-derived NO and Ca2+ sequestration. A rabbit double subarachnoid hemorrhage model was studied to understand the role of thrombin and PAR1 activation. PAR1 was found to have an autoregulatory role because thrombin led to upregulation of PAR1, which enhanced the contractile response to thrombin. The aforementioned effects could be prevented with intracisternal PAR1 antagonist. In contrast, activating peptides for PAR2 and PAR4 did not induce a contractile response in the subarachnoid hemorrhage model. Previous reports have confirmed thrombin-induced upregulation of PAR1 in isolated rings of the basilar artery (356).
In addition to vessel narrowing and coagulation, PARs contribute to systolic dysfunction, overload, and fibrosis. Ischemia increases expression of PAR1 and PAR2, while cardiac overload leads to overexpression of PAR2 (357, 358). Investigators used cardiomyocyte-specific PAR1 overexpression in a mouse model to show that PAR1 is responsible for systolic dysfunction and enlargement of the left ventricle (359). Genetic deletion or pharmacological antagonism of PAR1 abrogates cardiac systolic dysfunction and fibrosis (360). More recently, investigators have used more refined models and measures of cardiac dysfunction to confirm the role of PAR1 in congestive heart failure (361). Renin-overexpressing hypertensive (Ren-Tg) mice, in which the liver secretes high levels of renin that yield elevated levels of renin and angiotensin II in the plasma, exhibit elevated blood pressure and congestive heart failure. Experiments with the selective PAR1 antagonist SCH79797 implicated PAR1 in the resultant circulatory pathology. In this pathological Ren-Tg mouse model, PAR1 antagonism had several effects: decreased systolic blood pressure, reduced cardiac hypertrophy, and fibrosis. Additional effects included fewer monocytes and macrophages, along with proinflammatory and profibrotic cytokines. Thrombin or FXa produces cardiac inflammation and remodeling through PAR1 (362). Thrombin or FXa activates PAR1 in cardiac fibroblasts to phosphorylate ERK1/2. Given the finding that PAR1 antagonism does not affect systolic blood pressure or vascular tone, the role of PAR1 inhibition in reducing cardiac hypertrophy and fibrosis is independent of blood pressure mechanisms.
PAR activation contributes to the related processes of inflammation and thromboembolic events that characterize pathologic conditions affecting the circulatory system. For example, SARS-CoV-2 is associated with a pathologic increase in cytokines (i.e., cytokine storm), which include interferon-γ, TNF-α, IL-1, IL-6, and IL-2, and coagulation factors (363, 364). Following infection by SARS-CoV-2, the increase in cytokines leads to the upregulation of coagulation factors including TF (365). Moreover, the high level of cytokines can lead to massive engagement of macrophages, resulting in macrophage activation syndrome. This perpetuates activation of coagulation factors, disinhibition of coagulation through the loss of inhibitors, venous thrombosis, pulmonary embolism, and multiple organ failure, a process that leads to death in persons with severe SARS-CoV-2 infections (366, 367). Because of its role in coagulation and its expression on endothelial cells and the lung, PAR1 has been thought to play a prominent role in the hypercoagulable state and lung pathology associated with SARS-CoV-2 infection (368). Due to PAR2 expression on lung epithelial cells, neutrophils, and fibroblasts (sect. 3), PAR2 is likely to contribute directly or indirectly to the cytokine storm, innate immune response, organ inflammation, and ultimate organ failure that characterize SARS-CoV-2 infection.
5.2. PARs in the Integumentary System
5.2.1. Physiology.
Three layers comprise the skin: epidermis, dermis, and subcutis. The outer layer or the epidermis consists of five strata: stratum corneum, stratum lucidum, stratum granulosum, stratum spinosum, and stratum basale. PAR expression occurs early in development. As an example, PAR2 shows up in the epidermis at embryonic day 17 in mice. For context, PAR2 is first expressed in the CNS and cardiac muscle at embryonic day 12 (369).
Shortly after its discovery, PAR2 was shown to be expressed by keratinocytes (sect. 3.1); expression was confirmed with Ca2+ imaging. PAR2 is highly expressed by keratinocytes that comprise the granular layer; however, there is expression in basal and spinous layers (42). PAR2 localizes to lipid rafts in human and mouse keratinocytes (370). Within the stratum corneum, there are three families of proteases produced: human tissue KLKs, cysteine proteases, and aspartate proteases. There are eight KLKs that are expressed in the stratum corneum; all KLKs are tryptic proteases, except KLK-7, which is chymotryptic (371). Activation of PAR2 on keratinocytes by KLK-5 and KLK-14 mediates homeostatic functions in the skin including barrier protection, immune response, inflammation, pigmentation, and tumor surveillance (372, 373). PAR activation by KLKs on keratinocytes also leads to pigmentation, differentiation, and cornification. KLK-5 and KLK-14 cleave PAR2 and favor keratinocyte maturation, rather than keratinocyte proliferation (373, 374).
5.2.1.1. differentiation.
Stem cells generate keratinocytes that progress from the immature to the mature stage, followed by cell death and cornification. PAR2 activation participates in keratinocyte progression through these different steps (370). The role of PAR2 in differentiation of keratinocytes is equivocal; some publications support the concept that PAR2 activation leads to differentiation, while others show that PAR2 activation opposes differentiation. Based on the evidence that Ca2+ mediates differentiation, investigators expected that PAR2 activation would promote differentiation given its mobilization of intracellular Ca2+. One of the earliest papers that looked at the roles of PAR1 and PAR2 activation on differentiation found that the PARs exhibited divergent effects (40). Thrombin activation of PAR1 increased cell growth and decreased differentiation. PAR2 activation inhibited both cell growth and differentiation. Opposing work has shown that PAR2 activation promotes differentiation (375).
5.2.1.2. barrier function.
Before the description of the role of PAR2 in cutaneous barrier function, PAR2 activation was shown to disrupt the barrier in the cornea, vagina, respiratory system, kidney, and GI tract. Skin barrier protection involves keratinocyte cornification, lamellar body section, and formation of intercellular junctions. PAR2 activation delays barrier recovery. The reverse, that is barrier recovery following PAR2 antagonism, has been shown. In PAR2-/- mice, barrier recovery is accelerated (370). PAR2 regulates lamellar body secretion. PAR2 activation suppressed lamellar body secretion and barrier recovery; PAR2-mediated alterations of actin mediated the decrease in lamellar body secretion (376).
5.2.1.3. inflammation and immune responses.
Immune cells within the skin contribute to protection mediated by PARs. Activation of PAR1 on monocytes and T lymphocytes leads to the production of IL-6 and IL-8. PAR2, expressed by eosinophils, mast cells, monocytes, and neutrophils, leads to the secretion of IL-6, IL-8, IL-13, IL-1β, ICAM-1, thymic stromal lymphopoietin, and TNF-α. PAR1- and PAR2-mediated production of inflammatory mediators cause inflammation within the deeper layers of the skin (342).
PAR2 activation induces an inflammatory response, which is observed at the tissue level and is manifested as edema, vascular permeability, and mobilization of granulocytes. PAR2, which mediates neurogenic inflammation in the skin, contributes to smooth muscle contraction, vasodilation, and plasma extravasation (377). The PAR2-mediated inflammatory effect is balanced by the anti-inflammatory effect of PAR1 activation. As in FIGURE 6C, EPCR acts as a coreceptor for the anticoagulant APC. APC cleaves PAR1 and is antiapoptotic and neuroprotective (61). When APC binds to EPCR and activates PAR1, keratinocyte proliferation and barrier protection are reinforced, along with reduced inflammation (378, 379).
PAR2 activation on keratinocytes mediates phagocytosis. Activation by trypsin, SLIGRL, or SLIGKV mediates ingestion of microspheres and bioparticles (380). The mechanisms by which PAR2 activation increases phagocytosis involve actin polymerization, α-actinin reorganization, cellular morphological changes, and protease activity. PAR2 activation of keratinocytes, in this case the simian virus-transformed human keratinocyte SVK14 cell line, also leads to the secretion of cytokines, including IL-8 (381).
5.2.1.4. pigmentation.
Evidence supporting the role of PAR2 in skin color includes the finding that pigmented human skin exhibits higher expression of PAR2 and its canonical activator, trypsin, relative to lighter human skin; moreover, dark skin exhibits increased PAR2-mediated phagocytosis (382). PAR2 activation by trypsin and SLIGRL on keratinocytes upregulates melanosome transfer and phagocytosis (383).
5.2.2. Pathology.
5.2.2.1. inflammatory skin disorders.
Of the PAR subtypes, PAR2 features prominently in inflammatory skin disorders. These disorders are common: acne affects 50% of teenagers, atopic dermatitis (AD) affects ∼20% of the population, rosacea affects 10% of adults, and psoriasis affects ∼3% of the population (384).
PAR2 has a clear role in AD. AD is characterized by increased expression of PAR2 in keratinocytes and associated nerve fibers (377). Skin affected by AD contains a higher concentration of tryptase, which induces itch through PAR2. KLK-5 and KLK-14, which cleave PAR2, are upregulated in skin samples from patients with AD. Not all KLKs are capable of cleaving PAR2, for example, KLK-7 or KLK-8 (385). Komatsu et al. (385) generated their data from lesional and nonlesional skin samples harvested from the same six patients, with patients serving as their own control. In a separate study, investigators found that activated PAR2 in diseased skin from patients with AD leads to upregulation of ICAM-1 through the NF-κB pathway.
PAR2 activation resulting from proteases secreted by the house dust mite also contributes to AD. Investigators used a PAR2-overexpressing mouse in which the gene for PAR2 was inserted into the start codon of the grainyhead-like-3 gene Grhl3Par2/+ to study the role of house dust mite in AD (386, 387). The AD model was generated by repeated application of an extract of house dust mite to the nape of the neck. The AD mouse model exhibited spontaneous eczema and an intense pruritic-like behavioral response and impaired skin barrier. DRG harvested from the overexpressing mice exhibited a greater response to SLIGRL, which can activate PAR2 and MrgprC11. The authors propose that the PAR2-overexpressing mice are a more accurate model of pediatric AD compared to flaky tail mice, or the SPINK5-/-, and flaggrin-/- mouse models (388–390). PAR2 activation of TH2 cells in the PAR2-overexpressing mice leads to secretion of IL-31, which activates neurons and enhances neuroimmune signaling.
Netherton syndrome is a rare autosomal recessive skin disease associated with skin inflammation, scaling, and itch as well as with dehydration and growth abnormalities. Patients exhibit a mutation in the gene for serine protease inhibitor Kazal-type 5 (SPINK5), also known as lympho-epithelial Kazal-type-related inhibitor (LEKTI). LEKTI regulates desquamation and inhibits three KLK-related peptidases: KLK-5, KLK-7, and KLK-14. With the loss of the serine protease inhibitors, there is increased activation of PAR2 by the KLKs. PAR2 then signals through NF-κB (388). The KLK-5/PAR2 cascade leads to an increase in thymic stromal lymphopoietin production, which generates a proallergic microenvironment. This leads to the formation of eczematous skin lesions and a systemic TH2 response; an unweighted TH2 response contributes to allergy (391). Thymic stromal lymphopoietin released by keratinocytes induces Langerhans cells to accelerate sensitization to allergens from sources such as cockroaches and dust mites that activate PAR2 and contribute to a disruption of the skin barrier. Allergens disrupt the skin barrier through several mechanisms including increased protease activity, delayed barrier recovery, and delayed lamellar body secretion. These PAR2-mediated effects explain why PAR2 antagonists normalize the time required for barrier recovery (392).
5.2.2.2. itch.
PAR2 activation produces itch (393). Injection of PAR2 agonist into humans induces pain, followed by itch (377). Most patients suffering from chronic itch have AD or psoriasis (394). There are differences in the role of PAR2 in itch associated with AD versus psoriasis. Nattkemper and colleagues (395) performed RNA sequencing on skin from 25 patients with AD and 25 patients with psoriasis; 30 patients without skin lesions provided site-matched skin biopsies, which served as controls. For the patients with psoriasis or AD, the pruritic, lesional skin was compared to nonpruritic, nonlesional skin. Pruritic skin from patients with AD overexpressed F2rl1 (the gene for PAR2); however, pruritic psoriatic skin did not.
PAR2 contributes to itch associated with several disorders besides AD and psoriasis, including uremia, dermatophyte infection, and liver failure (396, 397). Uremic pruritis is associated with end-stage renal disease. A recent study evaluated differences in serine protease activity and PAR2 expression in patients with end-stage renal disease without uremic pruritis, patients with end-stage renal disease and uremic pruritis, and healthy controls. They found a significant positive correlation between elevated PAR2 expression and uremic pruritis, as measured with a visual analog scale (397).
Dermatophytes are fungi that infect the skin. These organisms secrete itch- and pain-inducing proteases. For example, PAR2 is activated by Der p1 (cysteine proteases), p3 (trypsins), and p9 (serine proteases) secreted by the house dust mites Dermatophagoides pteronyssinus and Dermatophagoides farina (398–400).
Cholestatic liver disease, characterized by reduced or blocked flow of bile from the liver, is associated with itch. A rat model was developed in which the bile duct was ligated; the rats ultimately exhibit cirrhosis and hepatic encephalopathy (396). In this model, the roles of proteases and PAR2 were investigated by administering gabexate mesylates, lima bean trypsin inhibitor, and the PAR2 antagonist FSLLRY-NH2. The protease inhibitors and PAR2 antagonist attenuated the scratching behavior. DRG removed from the rat bile duct ligation model demonstrated PAR2 upregulation and greater TRPV1 activity. Furthermore, there were more peptidergic neurons that expressed PAR2 and TRPV1; medium-sized neurons exhibited expression of TRPV1. PAR2 activation leads to TRPV1 phosphorylation and subsequent sensitization with a rapid decrease in activation threshold through PKCε and PKA (260).
5.3. PARs in the Respiratory System
5.3.1. Physiology.
All four PARs are expressed in the lungs (45, 53). PAR2 is expressed on the apical surface of ciliated columnar epithelial cells in the trachea, main bronchi, and first-order bronchi. PAR2 staining is less commonly seen on airway smooth muscle. PAR1 immunoreactivity is faint on epithelium and SMCs.
The differential actions of PAR1 and PAR2 in the lung have been studied with activating peptides. SFLLRN-NH2 activates PAR1 and PAR2, while SLIGRL-NH2 is specific to PAR2. In preparations of mouse bronchi from which the epithelium has been denuded, SLIGRL-NH2 does not cause contraction; however, SFLLRN-NH2 does cause contraction through activation of PAR1. One of the early studies that reported on the role of PAR2 in the bronchi, that is bronchodilation, was by Cocks et al. in 1999 (74). The authors proposed that PAR2 activation was protective for the bronchi, which is PGE2 dependent. In a bronchial ring preparation, constriction can be induced with the muscarinic agonist carbachol. Subsequent relaxation in the preparation is induced by the mouse-activating peptide SLIGRL-NH2 and trypsin; PAR2-mediated bronchorelaxation is rapid and concentration dependent. PAR2 activation inhibited bronchoconstriction. This protective action in the bronchi points to the role of PAR2 has in reactive airway disease.
In addition to regulation of bronchoconstriction and bronchodilation, PAR2 regulates ciliary beating, which facilitates mucociliary clearance. Mucociliary clearance is a mechanism of defense against inhaled pathogens. Expression and subsequent activation of PAR2 on different regions of the cell (apical versus basolateral) determine sinonasal epithelial functions such as ciliary beating frequency (401).
5.3.2. Pathology.
The four PAR variants are expressed in the lung by different cell types including cells in the epithelium and blood vessels. PARs contribute to pathologic conditions in the lungs that usually involve a combination of fibrosis, proliferation, edema, and inflammation.
PAR1 is expressed on lung epithelium and lung fibroblasts (402–404; sect. 3.1). PAR1 activation on epithelium leads to inflammation through the release of cytokines. Activation of PAR1 on fibroblasts drives differentiation to myofibroblasts and subsequent generation of proteins that form the ECM (231, 405). A common cause of iatrogenic lung injury involves the administration of high-volume ventilation, which induces pulmonary edema. This outcome involves PAR1, transforming growth factor-β (TGFβ), and the αVβ6-integrin (406). Thrombin is a PAR1 agonist active during lung injury (407, 408). TGFβ is normally latent and activated by αVβ6. PAR1 regulates TGFβ activity through RhoA and Rho kinase signaling (sect. 4.5). The PAR1/αVβ6 /TGFβ axis mediates a distinct form of lung injury that is induced by bleomycin, a chemotherapeutic agent that induces DNA double-strand breaks. Bleomycin leads to increased lung permeability and edema. In PAR1-/- mice, TGFβ activation and the lung permeability induced by bleomycin are reduced (406, 409). Exposure of airway SMCs to thrombin leads to proliferation through ERK2 activation, which is inhibited by activation of cAMP-dependent protein kinases by forskolin. PAR1 has a role in pulmonary fibrosis; however, PAR2 is required. Simultaneous antagonism of PAR1 and PAR2 is not more effective than antagonism of either PAR alone (410).
Much of the pathology of the respiratory system involves bronchoconstriction and bronchodilation. Investigating the role of PARs in constriction and dilation in lung pathology is challenging. The effect of PAR activation on airway smooth muscle, i.e., contraction versus dilation, as well as the effect of proteases versus activating peptides, is species dependent. Moreover, airway preparation variability affects the results. In rat trachea-derived smooth muscle cells, the activating peptides for PAR1, PAR2, PAR3, and PAR4 were tested, along with thrombin and trypsin (411). To test whether PARs induce dilation, precontraction was induced by carbachol. PAR1-, PAR2-, and PAR4-activating peptides, along with trypsin and thrombin, produced dilation, which was indomethacin dependent. PAR1- and PAR4-activating peptides induced dilation that was preceded by a rapid and transient contraction. The contractile effect was not induced by thrombin or trypsin.
Fresh human lung tissue is difficult to obtain for translational experiments. However, bronchial rings can be obtained from patients who require pneumonectomy or lobectomy for carcinoma. Intact human bronchial rings obtained from such patients were used to test the effect of α-thrombin on bronchial tone. α-Thrombin increased bronchial tone, which did not depend on the respiratory epithelium. Transcripts for PAR1, PAR2, and PAR3 were present, while protein for only PAR1 and PAR2 could be detected (412). Experiments on isolated rat airways, including the trachea and main bronchi and first-order bronchi, revealed that epithelium must be present to induce smooth muscle contraction and relaxation (413). Trypsin and thrombin did not have an effect on the airway. This study also revealed differences in the actions of proteases or activating peptide. PAR1-activating peptide caused contraction of the airway, while PAR2- and PAR4-activating peptides caused airway dilation; the effect of the PAR2-activating peptide was greater than the effect of the PAR4-activating peptide. The effect of PAR2 activation was inhibited by redox states of hemoglobin, which was independent of hemoglobin’s NO- and CO-scavenging properties.
PAR2 expressed by circulating immune cells also contributes to asthma. In fact, PAR2 on CD14++CD16+ monocytes in the blood of asthma patients has been proposed as a biomarker for asthma severity (414). Monocytes are classified according to expression of CD14 (the lipopolysaccharide coreceptor) and CD16 (the Fcγ receptor). CD14++CD16+ are intermediate monocytes. Concentration of these monocytes is increased in patients with type 2 diabetes and sarcoidosis (415, 416).
The prominent role of PARs in inflammation contributes to scar formation and fibrosis in the lungs (417, 418). PAR3 (along with PAR1) on alveolar epithelium plays a role in the epithelial-to-mesenchymal transition (45). PAR3 is overexpressed in the lungs of patients with idiopathic pulmonary fibrosis and in mice with bleomycin-induced lung fibrosis. PAR1 and PAR3 colocalize within alveolar type II cells, which comprise 60% of alveolar epithelial cells. PAR3 is overexpressed primarily on alveolar type II cells and is thought to contribute to lung fibrosis. Activation of PAR3 on alveolar type II cells leads to secretion of IL-8 (168) (sect. 4.7). In the lung, the PAR2/EGFR axis also leads to secretion of IL-8 (280). PAR4 contributes to the role that EGFR plays in mediating the transition from epithelial cells to mesenchyme. This transition contributes to lung fibrosis (53).
5.4. PARs in the Gastrointestinal System
5.4.1. Physiology.
The site of highest PAR expression is in the GI system (sect. 3). The GI system contains and is subject to more proteases (from exocrine pancreas, inflammatory cells, microorganisms) than any other location or organ in the body; therefore, PARs are anatomically set up to contribute to GI homeostasis, including enterocyte renewal, permeability, barrier function, motility, immune response, cytokine function, neurogenic inflammation, and sensation. Within the GI system, PARs are most highly expressed in the small intestine, colon, liver, and pancreas; however, other sites within the GI system that exhibit PAR expression include the oral cavity, salivary glands, exocrine pancreas, and liver (419). At a cellular level, PARs are expressed on enterocytes (basolateral and apical membranes), endothelial cells, SMCs, immune cells, and neurons. Most reports on PAR function address the roles of PAR1, PAR2, and PAR4; we know little about the expression and function of PAR3 in the GI system.
Based on the role of PAR1 in electrolyte transport on epithelial cells, such as renal proximal tubule cells, PAR1 was proposed to contribute to intestinal epithelial cell function. The role of PAR1 in intestinal epithelial cells was demonstrated in 2001 using a nontransformed, chloride-secreting human epithelial crypt cell line (SCBN) (420). PAR1 activation on the basolateral surface of the SCBN cells led to chloride secretion that is directed toward the apical portion of the cell; a mechanism that is mediated by the release of intracellular stores of Ca2+.
5.4.1.1. epithelial renewal.
Similar to their role in organ development, PARs contribute to epithelial cell renewal and turnover. PAR2 is involved in human colonocyte homeostasis and maintenance (211). Intestinal cell homeostasis involves cellular proliferation and differentiation from stem cells within the base of mucosal crypts; the cells undergo differentiation, maturation, and ultimately senescence as they migrate to the mucosal surface. Trypsin activation of PAR2 on enterocytes stabilizes yes-associated protein 1 (YAP), a transcriptional regulator and member of the Hippo signaling pathway. YAP activation contributes to cellular turnover (421). PAR2 contributes to colon carcinogenesis, which involves YAP1, an oncogene. PAR2 also regulates proliferation of intestinal stem cells through activation of glycogen synthase kinase-3β (422).
5.4.1.2. barrier function.
The critical function of the GI system is the movement of nutrients and water from the lumen into the vasculature and lymphatic system, while also serving as a barrier to bacteria; accordingly, GI permeability, transcellular and paracellular, is central to homeostasis. The protease/PAR axis regulates transcellular and paracellular permeability (FIGURE 7A).
FIGURE 7.

Protease-activated receptor (PAR) signaling in the gastrointestinal system. A: colonocytes express PAR1 and PAR2. The apical membrane facing the gut lumen is exposed to proteases, including digestive enzymes and proteases from commensal and pathogenic microorganisms. The basolateral membrane is exposed to proteases released by immune cells, epithelial cells, blood vessels, and other cell types. B: activation of PAR2 at the basolateral region of cells leads to ERK1/2 signaling that causes paracellular permeability via disruption of tight junctions. C: PAR2 activation on sensory neurons, including those that innervate the gut, leads to Ca2+ mobilization. Subsequent PKC activation sensitizes transient receptor potential (TRP) channels, leading to ionic influx and activation of voltage-gated channels to trigger action potentials. This also triggers release of neuropeptides, such as substance P (SP) or calcitonin gene-related peptide (CGRP), which mediate neurogenic inflammation and pain. ZO-1, zona occludens.
PAR1 and PAR2 contribute to intestinal epithelial barrier function and ionic transport (423). PAR1 agonists disrupt tight junctional zonula occludens (ZO). PAR1 activation disrupts ZO-1 and apoptotic nuclear condensation, mechanisms that involve caspase-3, tyrosine kinase, and MLC kinase (423). Most of the work on PARs and enterocyte paracellular permeability has focused on PAR2. PAR2 mediates intestinal epithelial inflammation across multiple species (424–426). Activation of PAR2 on enterocytes by a low dose of SLIGRL leads to increased paracellular permeability through calmodulin activation, which leads to MLC phosphorylation and ZO-1 disruption (427). Paracellular permeability is mediated through activation of PAR2 on capsaicin-sensitive sensory neurons and subsequent neurogenic inflammation (428). Intraperitoneal PAR2 activation (that is, activation of PAR2 on the basolateral surface) leads to paracellular permeability (FIGURE 7B), which involves ERK1/2 and β-arrestin (sect. 4.5).
If homeostasis of the tight junctions is lost, toxins and bacteria migrate between enterocytes, which then generates an inflammatory response. The inflammatory response, characterized by increased epithelial permeability and bacterial translocation, follows PAR2 activation in the colon (425). Indirect evidence also confirms that PAR2 mediates colonic permeability. Ampicillin, a broad-spectrum antibiotic that inhibits growth of Gram-positive and Gram-negative bacteria, was given to animals in the drinking water (429). The antibiotic reduced serine protease activity and PAR2 expression. PAR2 activity was restored with trypsin administration. The results of this study show the relationship between bacteria, luminal proteases, PAR2 expression, and colonocyte paracellular permeability. Moreover, apical expression of PAR2 changes with antibiotics (429).
5.4.1.3. motility.
Interpretation of the effect of PAR activation on GI motility needs to be considered in the context of the cell, tissue (e.g., gastric versus colon), or whole animal that is studied. Species differences should also be considered, along with the assay. An in vitro assay quantifies circular muscle activity by measuring endoluminal pressure and uses isometric tension as an index of longitudinal muscle activity. As an example of the importance of tissue specificity in considering GI motility, activation of PAR2 reduces the amplitude of rhythmic contractions of the circular and longitudinal muscle of the rat colon. PAR2 is expressed (shown by immunofluorescence) in the muscularis externa of the rat colon as well as the mucosa; therefore, activation of PAR2 at either site could impact GI motility. The investigators ruled out the effect of PAR2 on the mucosa by removing this tissue (122). The literature contains equivocal results with regard to PAR activation and motility. For example, activation of PAR2 in the mouse and rat stomach, by trypsin and activating peptides, leads to stimulation of gastric motility, as demonstrated with a gastric longitudinal muscle preparation (327). In another study, PAR2 activation, along with PAR1 activation, in the mouse gastric fundus produces relaxation followed by contraction (72). Relaxation requires small-conductance Ca2+-activated potassium channels (SK or small potassium). Ryanodine also inhibited the effect. In a separate study, PAR2 activation in the rat colon inhibits the amplitude of rhythmic contractions (122). Colonic myocytes respond, as measured by an increase in Ca2+, to numerous PAR2 agonists including trypsin, SLIGRL-NH2, mast cell tryptase, and filtrate collected from degranulated mast cells. The PAR2-mediated effect on gastric motility is Ca2+ dependent.
PAR1 and PAR2 activity, induced by either selective activating peptides or trypsin for the latter PAR, cause contraction in the guinea pig and human gall bladder (430). Contraction of the gall bladder strips was measured with an isometric transducer. RT-PCR confirmed expression of PAR1 and PAR2 in the gall bladder. Contraction did not depend on tetrodotoxin or atropine; therefore, the effects of PAR1 and PAR2 activation were direct. PAR4 activation had no effect on gall bladder contraction.
5.4.1.4. secretion.
PAR2 activation has effects on secretion in the stomach; PAR2 activation stimulates mucus secretion that involves TRPV1+ neurons. PAR2 activation also increases gastric mucosal blood flow and has an inhibitory effect on gastric acid secretion. Activation of PAR2 in the stomach, but not in the duodenum, leads to secretion of mucus. The mechanism involves the release of calcitonin gene-related peptide (CGRP) and tachykinins on sensory neurons (431) (FIGURE 7C). PAR2 is expressed on gastric chief cells and leads to secretion of pepsinogen. PAR2 on pancreatic and parotid gland acinar cells leads to secretion of amylase (432). Ablation of TRPV1+ neurons reversed the gastric mucosal secretion induced by PAR2 activation. PAR2 activation leads to exocrine secretion by the salivary glands and the pancreas.
5.4.1.5. sensation.
Pain homeostasis is a balance of nociceptive and antinociceptive mechanisms, including visceral sensitivity in the GI system. PAR4 activation is antinociceptive in a number of visceral hypersensitivity models. PAR4 has also been shown to reverse nociception in the somatic system (270). Activation of PAR4 by the activating peptide AYPGKF-NH2 inhibits the Ca2+ current induced by capsaicin in rat DRG sensory neurons. At a behavioral level, the PAR4-activating peptide heightened mechanical and thermal nociceptive thresholds. The PAR4-mediated antinociceptive effect that was demonstrated in the somatic system is also seen in DRG that innervate the colon (433). Activation of PAR4 was shown to counterbalance the excitatory effects of PAR2-induced excitability in dissociated neurons, as measured by whole cell patch-clamp recording. Antagonism of PAR2-mediated nociception by PAR4 activation was also shown at a systems level (111). PAR2 sensitizes TRPV4, which contributes to visceral hypersensitivity. When the PAR4-activating peptide was injected into the colon, there was reduced visceral hypersensitivity. A combination of PAR2 and TRPV4 agonists induced allodynia and hyperalgesia, as measured by visceromotor responses that occurs following colorectal distension. PAR4 agonism reversed the allodynia and hyperalgesia.
5.4.2. Pathology.
5.4.2.1. oral cavity.
Soon after the discovery of PARs, these receptors were found to contribute to the physiology and pathology of the GI system, especially the intestines. More recently, the role of PARs in pathologic conditions of the oral cavity, including teeth, has emerged. Pulpitis, inflammation of the dental pulp, causes odontogenic (i.e., tooth) pain. This process is mediated by PAR1 (434). In the setting of pulpitis, PAR1 can be activated by KLK, a serine protease that cleaves kininogen to generate bradykinin and a well-established algogen (i.e., a mediator that is responsible for pain). KLK produces pulpal inflammation by generating a Ca2+ flux that upregulates COX-2 and production of PGE2. SCH79797, the PAR1 antagonist, reverses KLK-induced pulpitis. PAR1 in periodontal tissues (i.e., the tissues supporting teeth) mediates inflammatory and reparative processes. Activation of PAR1 on oral epithelial cells by Arg-gingipain leads to an increase in IL-1α, IL-1β, IL-6, TNF-α, and CXCL5. Chronic periodontitis causes downregulation of PAR1 expression, which leads to an increase in IL-6, IL-8, TNF-α, IFN-γ, and MMP2 in crevicular fluid (435). On the other hand, periodontal therapy leads to an increase in PAR1 expression by epithelial cells. Thrombin activates PAR1 on oral epithelial cells, which attracts granulocytes through chemokine induction and contributes to the inflammation that characterizes periodontal disease. Trypsin-like cysteine proteases in the gingipains family are secreted by Porphyromonas gingivalis. These proteases cleave PAR1 on monocytes, which generates secretion of cytokines (436) and contributes to periodontal disease (437).
5.4.2.2. inflammatory bowel disease.
Inflammatory bowel disease (IBD), which includes Crohn’s disease and ulcerative colitis, is characterized by dysmotility, a process that contributes to overgrowth of bacteria, increased intestinal permeability, bacterial translocation, inflammation (which reciprocally worsens dysmotility), diarrhea, and constipation. Inflammation affects sensory and motor neuronal function, which further impacts motility. Inflammation is responsible for sensory and motor neuronal hypertrophy, hyperplasia, axonal degeneration, and necrosis.
GI dysmotility results in constipation and diarrhea. Intestinal inflammation suppresses spontaneous phasic contractions and contributes to ultrarapid propulsion (438). Peristalsis involves ascending contraction and descending relaxation. PARs mediate contraction, relaxation and/or both (i.e., biphasic; 419, 439, 440). In a dextran sulfate sodium (DSS)-induced colitis model in rats, both trypsin and the PAR2-activating peptide SLIGRL-NH2 pathologically altered relaxation (441).
Permeability of the gut contributes to GI pathology, including Crohn’s disease and ulcerative colitis. Of all the PARs, PAR2 is the most prominent subtype that contributes to permeability (FIGURE 8B). The role of PAR2 in GI pathology, including permeability, has been demonstrated in preclinical models and patients. PAR2 is overexpressed in patients with IBD (442). Trypsin administered into the colon produces an inflammatory reaction that contributes to permeability (425). GI inflammation is characterized by high levels of trypsinogen I-II, which activate PAR2. Trypsinogen IV, a human trypsin that is resistant to inhibitors and proteolytic breakdown, activates PAR2 and PAR4 (443). In a pathologic context, activation of PAR2 leads to fluid secretion and inflammation through neurogenic inflammation mediated by CGRP and substance P (444, 445). Along with endogenous proteases, microbial proteases contribute to increased intestinal permeability (446). In IBD patients, Enterococcus faecalis produces gelatinase, which breaks down E-cadherin through PAR2 activation and leads to increased permeability (447). Serratia marcescens produces serralysin that activates PAR2.
FIGURE 8.

Protease-activated receptor (PAR) signaling in pain. A: primary (1º) sensory neurons with cell bodies in dorsal root ganglia (DRG) project fibers to peripheral tissues and the dorsal horn of the spinal cord. Second-order (2º) neurons in the dorsal horn transmit painful signals centrally. B: proteases that are released from immune cells cleave PAR2 expressed on sensory nerve fibers in the periphery. Some proteases (e.g., mast cell tryptase) trigger PAR2 endocytosis and trafficking to signaling endosomes that generate signals leading to sensitization or activation of ion channels. Other proteases (e.g., neutrophil elastase, macrophage cathepsin S, and legumain) activate PAR2 by biased mechanisms that do not evoke endocytosis; PAR2 signals from the plasma membrane to sensitize or activate ion channels. Neurons can release substance P (SP) and calcitonin gene-related peptide (CGRP) locally, causing neurogenic inflammation, and centrally, leading to pain transmission. TRP, transient receptor potential.
As PAR2 is so widely expressed in the GI system (epithelium, SMCs, endothelium, enteric neurons, fibroblasts, and infiltrating immune cells; sect. 3), the cellular source and associated PAR2 activity that contributes to IBD is in question. The role of PAR2 on some of the cell types has been addressed using a variety of models. The role of PAR2 on bone marrow-derived blood cells was shown to contribute to a trinitrobenzenesulfonic acid (TNBS)-induced colitis model, which recapitulates human IBD (448). PAR2-/- recipient mice did not develop colitis induced by DSS or TNBS. PAR2 on bone marrow-derived cells plays an important role in the latter model but not the former. In patients with IBD-associated colitis and patients with Hirschsprung’s-associated enterocolitis, PAR1 and PAR2 are overexpressed and thought to contribute to their pathology (449).
5.4.2.3. irritable bowel syndrome.
Irritable bowel syndrome (IBS) affects over 10% of the population and consists of symptoms that include abdominal pain, abdominal bloating, and dysregulated bowel movements. The latter is used to distinguish different types of IBS: IBS-D (IBS with diarrhea); IBS-C (IBS with constipation); IBS-M (IBS with diarrhea and constipation); and IBS-U (IBS with neither diarrhea nor constipation). Because of the role of proteases in visceral pain, the role of proteases in IBS is expected. Sources of proteases in IBS include tryptase from mast cells and trypsin from the pancreas as well as epithelial and endothelial cells (443).
Investigators have used biopsies from patients with IBS to understand the role of proteases and PARs in the etiology of IBS and associated symptoms, especially pain. Studies by several groups have demonstrated the following: 1) colonic biopsy supernatant collected from patients diagnosed with IBS contains proteases and shows proteolytic activity at an Arg site; and 2) proteases in this supernatant produce hyperalgesia through PAR2 (450). Evaluation of the proteases in the supernatant reveals increased levels of trypsin and tryptase, without an increase in the quantity of mast cells. Investigators applied the supernatant to dissociated DRG neurons and measured the neuronal response with Ca2+ imaging. Supernatants collected from the four subgroups of IBS patients led to an increase in Ca2+ signaling in DRG neurons. There was no distinction between tissue removed from the ascending colon or rectum. The general serine protease inhibitor FUT-175 reversed the effect. The level of expression of α-1-antiproteinase in the supernatants was unaltered; it is inferred that the increased proteolytic activity was not due to decreased inhibition. DRG neurons from PAR2-/- did not exhibit a Ca2+ response. This finding implicates PAR2 on neurons. In addition to the in vitro results, the investigators demonstrated that the supernatant had an in vivo effect. Supernatants induced visceral hypersensitivity, which recapitulates the human condition. Moreover, the supernatant produced both somatic thermal hyperalgesia and mechanical allodynia. These nociceptive effects were PAR2 dependent. IBS is characterized by decreased expression of PAR4, based on its expression in healthy control patients relative to IBS patients. Soon after the discovery that PAR2 contributes to somatic nociception, it was shown that activation of PAR2 by either trypsin or its activating peptide produces abdominal contractions (a measure of visceral nociception), as well as c-Fos expression in the relevant spinal cord level (451, 452). Human colonic biopsies from IBS patients were kept in oxygenated media for 24 hours, and the supernatant was subsequently collected. The supernatant produced hypersensitivity through PAR2 (450).
IBS pain is typically chronic and maintained by continued PAR2 signaling in endosomes (203; sect. 4.6). Supernatant from colonic mucosal biopsies contained proteases that led to the activation and endocytosis of PAR2. Activation of PAR2 from endosomes sustained nociceptor activation. A PAR2 antagonist was conjugated with cholestanol to allow the passage of the antagonist through the plasma membrane and into the endosomes (sect. 6.2), with enhanced inhibition of nociception. The investigators studied trypsin, elastase, and cathepsin S and their effect on PAR2 endocytosis, nociceptor sensitivity, and allodynia.
Supernatants generated from colonic biopsies from patients with IBS were demonstrated to act through PAR1 (453). Proteases at high concentrations included elastase 3a, cathepsin L, and proteasome α-subunit-4. The supernatants generated from colonic samples were then tested on submucosal plexus preparations from humans and guinea pigs and neuronal responses were assessed with Ca2+ imaging. The role of PAR1 in IBS was shown with human thoracic DRG neurons, which were also tested with supernatant from human colonic biopsies. The thoracic DRG neurons responded to a PAR1 agonist but not to PAR2 or PAR4 activation. The results were confirmed with a PAR1 antagonist (454).
5.4.2.4. visceral pain.
Visceral pain is a symptom associated with many common GI disorders including IBS, gastroenteritis, and IBD. Similar to somatic pain, visceral pain is mediated primarily by peptidergic (i.e., those fibers that express and secrete substance P and CGRP) unmyelinated C-fibers and thinly myelinated Aδ-fibers. Anatomically relevant sensory nerve fibers innervating the colon identified by retrograde tracing express TRPA1 and PAR2 (455).
TRPV4 is also required for PAR2-induced visceral mechanical hyperalgesia (265; sect. 4.7). TRPV4, like TRPA1, responds to mechanical stimulation (456). In 2007, it was shown that PAR2 sensitizes the response of TRPV4 to mechanical stimulation in the somatic system; however, it was not clear whether this effect occurred with visceral hyperalgesia (264). Somatic nociception and visceral nociception differ in their mechanisms (457). Anatomic and functional studies reveal that TRPV4 and PAR2 are coexpressed and colocalized on spinal afferent neurons that innervate the colon. Administration of a PAR2 antagonist potentiates this hyperalgesia. The effect of PAR2-induced visceral hyperalgesia was not observed in TRPV4-/- mice. Whole cell patch-clamp electrophysiology recording of Fast Blue-labeled DRG neurons showed that pretreatment of neurons with a PAR2 agonist led to increased current density following application of the TRPV4 agonist 4αPDD.
Cathepsins have several roles including antigen presentation and pain modulation through spinal microglial cells (458). In addition, cathepsins contribute to visceral pain. Activity-based probes were used to demonstrate the role of cathepsin S in colitis-related pain. Cathepsin S cleaves the transmembrane chemokine fractalkine, which mediates allodynia alongside cathepsin S itself (459). Stimulation of primary afferent neurons leads to secretion of fractalkine. This process requires cathepsin S from activated microglial cells. Microglial cathepsin S liberates fractalkine, leading to p38 MAPK phosphorylation in microglia, which, in turn, causes release of pronociceptive mediators from neurons. The cathepsin S inhibitor morpholinurea-leucine-homophenylalanine-vinyl phenyl sulfone (LHVS) reverses hyperalgesia and allodynia in neuropathic rats. The authors conclude that cathepsin S in microglial cells, but not cathepsin B and cathepsin L, maintains rather than initiates mechanical hypersensitivity. Activity-based probes were also used to demonstrate that cathepsin S is responsible for cleaving PAR2 and contributes to visceral hyperalgesia (460). A series of probes were used to label cathepsin B, cathepsin L, and cathepsin S. Cathepsin S was localized to macrophages in the colon, spinal cells, and microglia in mouse models of colitis. Two models of colitis were studied: piroxicam-induced in IL-10-/- mice and TNBS-induced colitis. The former recapitulates Crohn’s disease, while the latter allows for the study of genetic knockout mice.
5.4.2.5. extraintestinal pathology.
PARs contribute to pathology related to the hepatobiliary system. Thrombin activation of PAR1 contributes to hepatocellular injury through lipopolysaccharide, an endotoxin in the cell wall of gram-negative bacteria (461–463). Hepatocellular damage secondary to lipopolysaccharide involves neutrophils. Lipopolysaccharide is increased in the setting of sepsis. Sepsis leads to activation of coagulation factors, including thrombin, which contribute to hepatocellular injury. Thrombin also leads to an increase in neutrophil degranulation that further contributes to lipopolysaccharide-induced injury. Activation of PAR1 by TRAP recapitulated the effect of thrombin following hepatocellular injury induced by lipopolysaccharide (464).
PAR2 has a dual role, protective and injurious, in pancreatitis. Acute pancreatitis leads to multiple organ failure that affects the lungs (pulmonary dysfunction is the most common cause of death with pancreatitis), kidneys, heart, nervous system, and coagulation system. PAR2 is upregulated in acute pancreatitis, as shown in a rat pancreatitis model induced by taurocholate in rats (465). Ion exchange, especially Cl−/ exchange, is critical for pancreatic function. Pancreatic epithelial duct cells express PAR2 and mediate ion channel function following activation by trypsin (466).
Our understanding of pancreatitis has become more refined with the use of fluorescently quenched activity-based probes. Legumain is a cysteine protease found in lysosomes and is capable of cleaving PAR2 (149) (sect. 4.1). Legumain was implicated in pancreatitis when increased legumain expression was identified in patients with this disease (467). The use of an activity-based probe was used to show that legumain contributed to acute (up to 12 hours) and chronic (5 weeks) pancreatitis, in a model that was induced with caerulein (468). The source of legumain was localized to CD68+ macrophages, rather than pancreatic acinar cells. Increased legumain activity was confirmed in tissues from patients with pancreatitis. The authors propose that macrophages that express legumain might be the source of inflammation, which ultimately could lead to pancreatic cancer.
5.4.2.6. pathologic infection.
In 2011, Clostridium difficile infection occurred in ∼500,000 Americans and caused nearly 29,000 deaths; it is the leading cause of death due to enteritis (469, 470). C. difficile infection is often iatrogenic following antibiotic administration. C. difficile secretes multiple toxins including an enterotoxin [toxin A (TxA)] and a cytotoxin (toxin B), which produce diarrhea. TxA is responsible for other effects including fluid secretion, enterocyte damage, edema, and myeloperoxidase (MPO) activity. MPO activity is used as an index of colonic inflammation (471). PAR2 is pivotal for translating the toxic effects of TxA into signs and symptoms of C. difficile infection (472). C. difficile colitis can be induced by administration of TxA into the small intestine of mice, which recapitulates infection in humans. TxA administration leads to the following: fluid secretion, infiltration of neutrophils, edema, and cellular damage. In PAR2-deficient mice, these effects are significantly reduced. Tryptase, which must be activated by dipeptidyl peptidase I (DPPI), is secreted by mast cells and activates PAR2 (FIGURE 2). Mast cell tryptase contributes to long-term hyperexcitability on submucosal neurons (473). Similar to PAR2 deletion, DPPI deletion reverses fluid secretion, granulocyte infiltration, and the pathologic changes (e.g., edema and necrosis) induced by TxA. Antagonists of trypsin and tryptase can mimic the effects of deletion of PAR2 and DPPI. While the studies were done in mice, the mechanisms are likely similar in humans. TxA leads to increased expression and activity of PAR2 in human colonocytes; trypsin IV expression is also increased by TxA. PAR2-mediated TxA-induced colitis involves neurogenic inflammation through the NK1R. Trypsinogen IV or brain trypsin can activate PAR1, PAR2, and PAR4. This human trypsin is resistant to inhibitors (475). Trypsinogen IV was identified in the brain and is a splice variant of mesotrypsinogen (476). Trypsinogen IV is also expressed in normal human colonic mucosa (443).
5.5. PARs in the Nervous System
5.5.1. Physiology.
The early evidence to suggest that PARs mediated nervous system physiology included the finding that thrombin induced a change in neuron morphology and neurite retraction; moreover, continued activation by thrombin led to neuronal apoptosis (90). Within a few years of demonstrating thrombin’s effects on neurons, PAR1 was shown to be responsible (477–479). Additional studies confirmed that continuous exposure of neurons to thrombin led to apoptosis from PAR1 activation (480–483). In 1994 it was demonstrated that granzyme A, a serine protease released by T lymphocytes, activates PAR1 on neurons and astrocytes (484). Activation of PAR1 by thrombin or granzyme A results in neurite retraction, an action that can be inhibited by function-inhibiting antibodies to PAR1. Thrombin also produced a change in the morphology, proliferation, and differentiation of neuroblastoma cells (485) and astrocytes (90, 483, 486).
Following the discovery of PAR2 expression on neurons (sect. 3.5), the role of PAR2 in neurogenic inflammation would soon follow (FIGURE 7). Neurogenic inflammation is characterized by arteriolar vasodilation, opening of gaps in postcapillary venules, plasma extravasation, and neutrophil infiltration into tissues (73, 487–489). It was known that neurogenic inflammation was mediated by neuropeptides. Also, arteriolar vasodilation is mediated by CGRP through activation of the CGRP1 receptor, now known as CLR/RAMP1 (490, 491). Gap formation between endothelial cells was mediated by substance P, which activates NK1R and leads to plasma extravasation and neutrophil mobilization (492, 493). In 2000, the link between PAR2 and neurogenic inflammation was demonstrated (95). The authors showed that PAR2 was coexpressed with neuropeptides on DRG, providing anatomic evidence for the role of PAR2 in neurogenic inflammation. Functional evidence was provided by data confirming that PAR2 stimulation led to secretion of CGRP and substance P and tissue edema. The PAR2-activating peptide SLIGRL-NH2 led to paw edema and infiltration of neutrophils, effects that could be inhibited by antagonists of CLR/RAMP1 and NK1R. In the same year, it was shown independently that the same PAR2-activating peptide SLIGRL-NH2 produced another feature of neurogenic inflammation, nociception (431; FIGURE 7). PAR2 agonism produced thermal, but not mechanical, hyperalgesia, as measured by paw withdrawal. Moreover, the animals exhibited nociceptive behavior including biting and licking. The hyperalgesic effect had a rapid onset and was not lost following mast cell degranulation, suggesting that activation of PAR2 on sensory afferents directly led to neuronal activation and hyperalgesia. Hyperalgesia induced by PAR2 agonism was independently demonstrated (96).
The roles of PAR1, PAR3, and PAR4 in nociception are poorly characterized compared to PAR2. PAR1 is not expressed by TRPV1+ neurons (260). PAR1 and PAR4, at the appropriate protease concentration, mediate antinociception (494, 495). McNaughton and colleagues (185) have characterized the expression and function of PARs besides PAR2 on neurons that mediate nociception. PAR1 and PAR4 are expressed on small peptidergic neurons and larger myelinated neurons; the former location is proposed to mediate nociception, and the latter site is proposed to mediate antinociception.
Mesotrypsin, which is encoded by PRSS3 and also known as brain trypsin, is a serine protease that is resistant to inhibitors. Mesotrypsin is isolated from the brain. PAR1 on astrocytes in rats is activated by mesotrypsin, which has been proposed to contribute to physiologic and pathologic conditions in the brain (496). Wang et al. (496) did not find that PAR2 is activated by mesotrypsin; however, other investigators have shown that mesotrypsin activates PAR2 to induce inflammation and pain (475).
A looming question in the GPCR field has been how ligand binding and/or cleavage of the receptor leads to membrane depolarization. A recent paper addressed this question by studying how PAR1 activation on nodose ganglia neurons mediated a change in current (497). PARs on pulmonary sensory neurons are involved with neuronal excitability, similar to their role on peripheral sensory neurons. C-fiber activation in the lungs contributes to inflammatory disease. TRP channels have been proposed to possibly mediate membrane depolarization. Undem and colleagues (497) showed that PAR1 activation leads to an increase in intracellular Ca2+ that involves TRPA1 but not TRPV1. PAR1 is expressed by mouse nodose C-fibers but not jugular ganglion neurons. In some nodose C-fibers, TRPA1 contributes to the intracellular Ca2+ increase following selective activation of PAR1 by TFLLR but not membrane depolarization. The PAR1-mediated action potential discharge depended on the PLCβ3 isozyme of PLC. The investigators showed that neither TRPA1 nor TRPV1 are required for PLC-dependent activation. C-fiber neurons from wild-type and TRPA1/TRPV1 double knockout mice exhibited the same action potential number and peak discharge frequency when activated by TFLLR. Another example of regulation of TRP channel activity by PARs is reflected by the increase in vascular permeability following activation of PAR1, which is mediated through TRPV4 (269; sect. 4.7).
The analysis of pulmonary jugular and nodose ganglia in response to PAR2 activation also sheds light on the question of how activation of PAR2 leads to neuronal activation. PAR2 activation leads to hyperexcitability of pulmonary jugular and nodose ganglia neurons through regulation of large-conductance Ca2+-activated potassium (BK or big potassium; also known as KCa1.1) channels in the rat (498). BK channels respond to an increase in Ca2+ conductance, which leads to opening of potassium channels and neuronal repolarization. The second messengers PLC, PKC, PKA, and MEK/ERK were not involved in PAR2-mediated regulation of BK channels. Like sensitization of TRP channels by PAR2 activation on sensory neurons (261) (sect. 4.7), PAR2 is activated by serine proteases secreted by house dust mites on vagal pulmonary sensory neurons. These are anatomically relevant pulmonary sensory neurons identified by DiI labeling. These proteases secreted from house dust mites sensitize the response to capsaicin, as measured by Ca2+ fluorimetry, in a PLC- and PKC-mediated manner (499). The action of PAR2 on nociceptors has also pointed to a role in inhibition of potassium channels, which mediate neuronal activation and nociception (500). Inhibition of the M current was mediated by PLC, Ca2+, and PIP2.
PAR1 agonists have neuroprotective effects on CNS neurons in conditions of hypoglycemia, as well as oxidative and environmental stress (480). Brain astrocytes undergo morphological changes, proliferate and secrete endothelin-1 and nerve growth factor upon activation of PAR1 (479, 501, 502). Activation of PAR1 and PAR3 by APC has neuroprotective effects. APC is a serine protease that along with its neuroprotective effects, has anticoagulant properties (sects. 3.1 and 4.1). Mutated forms of APC have been generated to disambiguate APC’s anticoagulant and neuroprotective properties, as well as whether effects are observed in rodents are observed in human cells. 3K3A-APC is a mutated form of human APC (503–505). 3K3A-APC has less than 10% of APC’s anticoagulant properties, but it has APC’s cell signaling and neuroprotective effects in models of amyotrophic lateral sclerosis (506), traumatic brain injury (507), and stroke (508). Both APC and 3K3A-APC exert their neuroprotective effects through PAR1 and PAR3 (508–513). APC induces proliferation of neural progenitor cells (NPCs) in ischemic (514) and traumatic injury (515) mouse models; the proliferative effect is not observed in PAR1 knockout mice. Fetal human neural stem and progenitor cells divide and differentiate when treated with human 3K3A-APC, similar to the cellular effects observed in response to fibroblast growth factor and brain-derived growth factor (516). At the same time, 3K3A-APC inhibits differentiation of astroglial cells (517). This response, neuronal differentiation and astrogliosis, is the opposite of that found with CNS injury and pathology; therefore, mutated forms of APC might hold potential for therapy. The neuroprotective effects of APC require both PAR1 and PAR3 in most cell types; however, only PAR1 is required in other cell types (509–511, 518). The neurogenic effects of 3K3A-APC in human neural progenitor cells is mediated through PAR1- and/or PAR3-sphingosine-1-phosphate receptor-1 (S1PR1)-mediated Akt signaling (519). Activation of S1PR1 is involved in the proliferation and migration of neural progenitors, likely through PI3K/Akt signaling (520).
5.5.2. Pathology.
Thrombin and PARs contribute to a number of neurologic diseases, including, but not limited to, ischemia, hemorrhage, HIV-associated dementia, Alzheimer’s disease, and traumatic head injury. As tissue plasminogen activator is effective with occlusive stroke, the role of serine proteases on CNS neuronal function following ischemic injury is of interest. When thrombin is infused into the caudate putamen, the resulting cellular features are consistent with those seen following trauma, such as the formation of a glial scar (521, 522). PAR1 mediates glial proliferation, retraction of neurites, and cell death (90). Thrombin also precipitates seizures when it is injected into the basal ganglia of rats. PAR1 and PAR2 activation induces neuronal inflammation and cytotoxicity (523, 524).
Neurologic conditions affect PAR1 expression. Trauma, such as nerve transection, impacts PAR1 expression; for example, facial nerve transection produces a decrease in PAR1 mRNA expression in facial motoneurons (525). Ischemia, on the other hand, leads to an increase in the expression of PAR1 in hippocampal neurons and glia (526). In the CNS, thrombin activation of PAR1 on glial cells produces neuroinflammation (527).
PAR1 is upregulated in HIV encephalitis and in the brains of patients with HIV-associated neurocognitive disorders (528). HIV-infected macrophages are a source of proteases that can activate PAR2, including cathepsin B and cathepsin G (529). Trypsin, the canonical activator of PAR2, is absent from the CNS. CNS mast cells secrete tryptase that cleave PAR2 in the nervous system. Furin, a PAR2 protease that cleaves at the canonical site, is neuroprotective in the setting of HIV (530). Activation of PAR2 in the brain prevents neuronal death and prevents behavioral changes associated with HIV (523).
5.6. PARs in the Immune System
5.6.1. Physiology.
PARs are an ideal immune system regulator, based on cellular expression and the change in local concentrations of proteases that accompany tissue injury, inflammation, and barrier compromise. PAR expression on epithelial surfaces that line the skin and internal organs (sect. 3.1), as well as on nerves that innervate these tissues (sect. 3.5), allows these GPCRs to serve as sensors for proteases secreted by microorganisms such as fungi, mites, and bacteria. Expression of PARs on immune cells allows these cells to alter their activity and development based on protease activation.
The link between coagulation and inflammation has long been appreciated. A thrombin-like receptor, rather than the thrombin receptor itself, produced immune cell activation and chemotaxis. Evidence for a thrombin-like receptor came from neutrophils, as inactivated thrombin could produce neutrophil chemotaxis (531–533). This finding led to the hypothesis that proteases at sites of inflammation could generate fragments of thrombin, which did not have proteolytic activity but could activate processes associated with inflammation, such as leukocyte chemotaxis. Supporting evidence showed that if the cleavage site of the thrombin receptor was inactivated with antibodies, thrombin could still induce neutrophil chemotaxis. While it was recognized that proteolytic activity was not required for neutrophil chemotaxis, it was also known that the actions of thrombin could occur through a receptor that was not the thrombin receptor. The PAR1-activating peptide TRAP was shown to increase intracellular Ca2+ but did not require a proteolytically activated thrombin receptor (i.e., PAR1), which had been described 4 years earlier (4). Experiments showed that there was a receptor that caused neutrophil chemotaxis that did not require proteolysis. Evidence from neutrophils in 1995 suggested that neutrophils have a receptor that was activated by a protease that was not thrombin (120). It was known that neutrophils did not stain for an antibody to the thrombin receptor (127). An inactive mutant of thrombin (Ser195Ala) produced neutrophil chemotaxis of the same amplitude as that produced by thrombin. Neutrophil chemotaxis was not impacted by antibodies to the cleavage site of PAR1. Moreover, TRAP did not stimulate neutrophil chemotaxis (120). Hirudin, or the COOH-terminal fragments of hirudin, inhibited thrombin-induced neutrophil chemotaxis (532, 533). Translational relevance is seen with increased expression of PAR2 on neutrophils in patients with sepsis (534). Early experimental results showed that TRAP produced an increase in Ca2+, while thrombin did not, in certain cells. When alanine was substituted on positions 1, 2, 3, and 5, the response of neutrophils to TRAP was reduced. This finding was a surprise based on what was anticipated for thrombin receptor agonist activity. There were further distinctions between the response to TRAP and thrombin. While serine in position 1 is not essential for thrombin activation, there was diminished activity if leucine in position 4 is replaced by alanine (156). The solution to this question came 3 years later when it was shown that TRAP could activate PAR2 (41, 165) (sect. 4.3). PAR2 was cloned from endothelial cells and shown to be thrombin insensitive. PAR2 was also upregulated when exposed to inflammatory mediators (348). The effect of IL-1α and TNF-α on the expression of PAR2 on endothelial cells further supported the role of PAR2 in the inflammatory response (165).
Activation of PAR2 mediates the migration of leukocytes through lymph nodes. Thrombin activates dendritic cells, which affects their migration through lymph nodes in models of endotoxemia. PAR2 signaling mediates the uptake of antigens and T-cell trafficking by dendritic cells (535). Following PAR2 activation of CD11chigh dendritic cells, these cells showed increased expression of major histocompatibility complex class II and CD86 and migration toward draining lymph nodes. Furthermore, activated T cells, which included CD4+ and CD8+ T cells, also migrated to draining lymph nodes. The role of PAR2 was confirmed using global knockout mice and a PAR2-specific-activating peptide (536). As mentioned previously, there are also distinctions between in vivo rodent models with respect to inflammation.
5.6.2. Pathology.
PARs contribute to the pathologic effects of systemic inflammation, including platelet activation, endothelial cell damage, leukocytosis, organ damage, and death. The effect of PARs on inflammatory disease is not straightforward, as PAR activation can improve or worsen inflammatory disease. Infusion of lipopolysaccharide induces a state of endotoxemia, and this model is commonly used to study the contribution of PARs to systemic inflammation; however, this model may not accurately recapitulate systemic inflammation in conditions such as bacterial sepsis in humans. The roles of PAR1, PAR2, and PAR4 have been studied in an endotoxemia model generated in global knockout mice. Mice deficient in one of these PARs were studied, along with double knockouts for PAR1/PAR2 and PAR2/PAR4 (537).
PAR2 has a central role in the development of inflammatory diseases. PAR2 is expressed by lymphocytes, neutrophils, eosinophils, and monocytes; endogenous and exogenous inflammatory mediators upregulate PAR2. Activation of PAR2 leads to erythema, edema, and pain. The interaction of PAR2 with the cytoplasmic domain of TF contributes to its role in inflammation. Sustained activation of the innate immune system contributes to disseminated intravascular coagulation that is associated with sepsis. Disseminated intravascular coagulation is defined by uncontrolled generation of blood clots, which leads to occlusion of smaller blood vessels, and is characterized with thrombin-antithrombin and antithrombin III levels, along with microvascular fibrin deposition. The condition can occur in the setting of sepsis, trauma, pregnancy, and cancer. In a lipopolysaccharide infusion model, deletion of PAR2 did not impact the outcome or measurements of glucose, alanine transaminase, creatinine, or lipase in the serum of a mouse model; however, fever was reduced (538).
The role of PAR2 in neurogenic inflammation is well characterized (FIGURE 7). Its presence on sensory afferent neurons that innervate cutaneous and mucosal epithelia enables PAR2 to mediate the clinical manifestations of inflammation including edema, redness, warmth, and pain. Sensory neurons extend from the periphery to the spinal cord, where activation of PAR2 at both locations leads to the secretion of the substance P and CGRP (95, 539). Edema is mediated by plasma extravasation, which involves substance P release and activation of the NK1R on endothelial cells in postcapillary venules. Plasma extravasation is accompanied by granulocyte infiltration. Increased blood flow leads to an elevated temperature, redness, and edema, clinical features attributed to substance P. Several clinical features of neurogenic inflammation can be reduced or blocked by depletion of PAR2 or through the use of a PAR2 antagonist. For example, injection of carrageenan leads to paw edema; this is not present in F2rl1-/- mice or mice treated with a cell-penetrating PAR2 pepducin antagonist (540). PAR2-mediated neurogenic inflammation is regulated by insulin; insulin reduces the PAR2-mediated inflammatory effect in an insulin-deficient murine type I diabetes model (541).
5.7. PARs in Cancer
The hallmarks of cancer, as described by Hanahan and Weinberg (542), include sustained proliferative signaling, evasion of growth suppression, resisting cell death, replicative immortality, angiogenesis, and invasion and metastasis. PARs, especially PAR1 and PAR2, play a role in these hallmarks (FIGURE 9). This was first described for thrombin-mediated cancer cell invasion via PAR1 (reviewed in Ref. 543).
FIGURE 9.

Protease-activated receptor (PAR) signaling in cancer. Proteases and PARs contribute to numerous pathways involved in the “hallmarks of cancer” (542). The acidic tumor microenvironment can enhance protease activity. PARs activate downstream signaling pathways and transactivate other receptors that promote cancer. EGFR, epidermal growth factor receptor; TGFα, transforming growth factor-α.
5.7.1. Proliferation.
PAR1 agonists induce cell proliferation and motility in cancers. Tumor-associated macrophages also play a role in the contribution of PAR1 to carcinogenesis. PAR1 activation on macrophages leads to the secretion of growth factors and thrombin (544). Trypsin, tryptase, and thrombin within the cancer microenvironment activate PAR1, as well as PAR2, to drive the differentiation of monocytes into macrophages.
PAR2 and its activating proteases (e.g., trypsin, mast cell tryptase, type II transmembrane serine protease (TMPRSS2), uPA, FXa, FVIIa, tissue factor or factor III, matriptase, and KLKs) are upregulated in human cancers including colorectal cancer, lung adenocarcinoma, glioma, cutaneous squamous cell carcinoma (SCC), and oral SCC (545–550).
PAR2, acting through direct and indirect mechanisms, is a mitogenic factor in cancers. Direct mechanisms involve Ca2+ release, activation of PKCα, and activation of ERK1/2 (sect. 4.5). One indirect mechanism involves transactivation of EGFR (sect. 4.7), which then activates ERK1/2 and NF-κB. Trypsin activation of PAR2 leads to engagement of integrin α5β1 to bind fibronectin, which stimulates proliferation of human gastric carcinoma cells (551, 552). Downregulation of the microRNA miR-34a is an indirect mechanism through which PAR2 contributes to cancer cell proliferation. The G1/S checkpoint regulator cyclin D1 is negatively regulated by miR-34a. PAR2 activation suppresses miR-34a through TGFβ, which leads to disinhibition of cyclin D1, activation of E2F transcription factors, and induction of cellular proliferation in colon cancer cells. Knockdown of PAR2 in cells used to generate a xenograft model yielded tumors of decreased weight (553). PAR1 can also indirectly contribute to cellular proliferation. PAR1 activation by an activating peptide leads to downregulation of p21Cip1/Waf1 and upregulation of cyclin D1. p21Cip1/Waf1, also known as cyclin-dependent kinase inhibitor 1, mediates cell cycle arrest following DNA damage through p53 (554).
EGFR is transactivated by GPCRs, including PAR2, which contributes to proliferation in several types of cancers (252, 555). EGFR internalization, which depends on dynamin fission in clathrin-independent pathways and nuclear translocation, leads to tumor progression (556–558). PAR2-EGFR transactivation affects survival in patients with colon, cervical, and gastric cancer (252, 555, 559). A 2020 study made use of data from The Cancer Genome Atlas and found that PAR2 is overexpressed in ovarian cancer relative to normal tissue (255). Transactivation of PAR2 induces expression of transcription factors including FOS, MYC, and STAT3, along with PTSGS2 through MEK-ERK1/2 signaling, which increases PGE2. Transactivation of EGFR and the increase in PGE2 lead to increased ovarian cancer cell migration and invasion.
5.7.2. Motility, invasion, and metastasis.
Cancers express PARs (sect. 3.7), as well as their respective activating proteases, therefore there is a molecular basis for autocrine regulation that contributes to the hallmarks of cancer. The finding that activation of PAR1 and PAR2 by their activating peptides across different prostate cancer cell lines leads to increased expression and activity of proteases provides further evidence for an autocrine function between PARs and proteases in cancer (560). The same effect, secretion and increased activity of proteases, is seen with activation of PAR1 by thrombin or PAR1-activating peptide in oral SCC (561).
PAR1 mediates both invasion and metastasis. Expression of PAR1 is low or absent in normal breast tissue, benign breast lesions, and noninvasive breast carcinoma; however, PAR1 is overexpressed in breast cancers (138, 562, 563). A causative role for PAR1 in breast cancer invasion and metastasis has been demonstrated. In an experiment using PAR1 antisense treatment of a breast cancer cell line that exhibits aggressive metastatic behavior, the antisense treatment reduced invasion in a Matrigel migration assay (564).
Thrombin activation of PAR1 mediates cancer cell attachment to platelets and endothelial cells in melanoma (565–568). While PAR2 expression in human melanomas is comparable to levels in benign melanocytic nevi, PAR1 expression is increased in atypical nevi and melanoma compared to common melanocytic nevi (569). The effect of thrombin on cancer cell motility is thought to be mediated by PAR1; however, a potentially intriguing mechanism that drives cancer cell motility involves thrombin activation of PAR1 and transactivation of PAR2. This finding was revealed by desensitizing PAR2 using an activating peptide; when PAR2 was desensitized, the migratory effect of thrombin was reduced. PAR2 is transactivated following thrombin activation of PAR1, exposure of the PAR1 tethered ligand, and formation of a PAR1-PAR2 heterodimer (FIGURE 4A), a process that contributed to cancer cell metastasis in melanoma and prostate cancer. Once TF generates and activates FVIIa and FXa, both factors can activate PAR1 and PAR2. The roles of PAR1 and PAR2 in cancer cell migration were demonstrated with interfering RNAs to deplete breast cancer cells of the two PARs. The studies demonstrated that coagulant proteases FVIIa and FXa signal through PAR2, and not PAR1, to mediate breast cancer cell migration and invasion. PAR2 and TF show increased expression within the invasive component of human breast cancer tissues (549).
PAR2 activation produces several processes associated with carcinogenesis including migration (140), angiogenesis (351, 570), and signaling; however, the responsible activating proteases are less clearly defined. TF is the primary activator of the coagulation cascade. TF is responsible for generating coagulation factors (i.e., proteases) on the plasma membrane of cancer cells. Proteases degrade proteins, adhesion molecules, and the ECM; proteases also activate kinases and growth factors. Proteases are regulated by endogenous inhibitors. Proteases are commonly overexpressed in cancer. The lysosomal cysteine protease cathepsin S is unique because it is active at the pH consistent of the extracellular environment. Accordingly, cathepsin S can activate PAR2 in the setting of cancer. Cathepsin S is overexpressed in several cancers including cancer of the GI system [hepatocellular carcinoma (571), pancreatic cancer (572), colon cancer (573), prostate cancer (574, 575), uveal melanoma (576), astrocytoma (577), and glioblastoma (578)]. Elevated cathepsin S expression within breast cancer, at the primary site, correlates with reduced metastasis-free survival in breast cancer patients (579). The role of cathepsin S and PAR2 in mediating the processes of cancer has been investigated with hepatocellular carcinoma. When cathepsin S expression is inhibited with siRNA, there was inhibition of cancer proliferation, invasion, and angiogenesis as measured by in vitro assays and tumor volume in a mouse model (571). Silencing cathepsin S also induces apoptosis in cells; this process is mediated by PAR2 regulation of NF-κB signaling. Serum levels of cathepsin S correlate with cancer behavior (tumor, size, venous invasion) in humans (580). Cathepsin S expression was also found to correlate with poor survival in patients with glioblastoma; expression was higher in grade IV tumors relative to grade I–III tumors. In the normal human brain, cathepsin S expression is low or absent (578).
5.7.3. Angiogenesis.
PAR1 activation leads to an increase in VEGF expression at the transcript and protein level, a key mediator of angiogenesis (581). The role of PAR2 in regulating VEGF in cancers has been demonstrated with solid cancers, including breast, stomach, and pancreas (582–584). A series of in vitro studies have supported the role of PAR2 in mediating angiogenesis in pancreatic cancer. PAR2 activation regulates angiogenesis in pancreatic cancer through induced TGF-α and VEGF-A expression; the former is mediated by integrin-linked kinase/hypoxia-inducible factor-1α and the latter by MEK (582). TGF-α activates EGFR. Activated EGFR contributes to proliferation, differentiation, and invasion (585). PAR2 also directly activates EGFR (552). As mentioned, thrombosis is a well-established feature of cancer. Factors that mediate thrombosis, including TF and FVII, activate PAR2. Blocking TF/PAR2 signaling on cancer cells decreases vessel density (586). PAR2 deficiency, but not PAR1 deficiency, decreases the transformation rate of adenoma to adenocarcinoma in a spontaneous breast cancer development model based on the oncogenic middle T-antigen protein, a model that recapitulates human breast cancer development (587). PAR2 regulates angiogenesis in models besides cancer, including a mouse model of hindlimb ischemia (588).
Because of the role of PARs in several stages of carcinogenesis, PARs are attractive targets for the treatment of cancer. Liposomal delivery of PAR1 siRNA blocked melanoma proliferation and metastasis in a xenograft model. The most immediate advantage of liposomes over viruses for the delivery of siRNA is that the former does not generate the cytokine storm associated with viruses (589). PAR antagonists have been combined with chemotherapeutics to produce a combined or possibly synergistic effect. A PAR1 inhibitor in the form of a pepducin (sect. 6.2) has been combined with the chemotherapeutic agent taxotere. The pepducin/taxotere combination reduces melanoma growth and metastasis (590). PAR1 is an ideal cancer target because it is expressed by breast carcinoma (sect. 3.7) but not by benign tissues. MMP1 contributes to progression of several cancers including breast, colon, and esophageal. The MMP1-PAR1-Akt axis participates in breast cancer progression. A pepducin, P1pal-7, was developed to inhibit PAR1 and disrupt MMP1-PAR1-Akt signaling. The advantage of pepducin is its selective cytotoxicity in breast cancer cells; benign breast cells were not affected. When the pepducin was combined with taxotere, as it often is for breast cancer, the combination of drugs was synergistic and highly effective in a breast cancer xenograft model. The mechanism of the two drugs involved inhibition of tumor growth and induction of apoptosis through blockade of Akt, which is required for cell survival.
5.8. Summary
Due to their extensive tissue expression and their activation by proteases, PARs are central to the normal function and pathological states affecting every organ system. Moreover, in the case of the nervous system, PARs serve a protective role. The related and dependent processes of coagulation and inflammation, which are both regulated by PARs, lead to thromboembolic events that characterize diseases affecting the vascular system in the periphery, brain, lungs, and heart. Accordingly, PARs are considered high-value targets for drug development to reduce major causes of morbidity and death including cardiovascular and cerebrovascular disease.
6. DRUG DEVELOPMENT TARGETING PARS
6.1. Existing PAR Agonists and Antagonists: Peptides, Small Molecules, and Antibodies
Preclinical studies that implicated PARs in disease spurred drug discovery efforts to develop PAR antagonists from the late 1990s. The development of PAR antagonists was initially met with skepticism in light of the intramolecular mechanisms of proteolytic activation by a tethered ligand, as this was deemed challenging to inhibit. However, PARs have since been successfully antagonized by peptidic compounds based on activating peptide sequences, as well as small molecules and PAR-directed antibodies (FIGURE 10). Antagonists can disrupt PAR signaling by several mechanisms. Orthosteric PAR antagonists block the intermolecular interaction between the tethered ligand domain and the cleaved receptor. Allosteric PAR antagonists interact with a site different from that occupied by the tethered ligand and either suppress or amplify PAR signaling (negative or positive allosteric modulators, respectively). Antagonists can also interact with the cleavage site to suppress proteolytic activation or with protease binding sites (if present) to disrupt binding and efficient proteolysis.
FIGURE 10.

Therapeutic targeting of protease-activated receptors (PARs). A: multiple drug classes have been developed to target PAR signaling, including peptidic and small molecule antagonists and monoclonal antibodies (mAbs). B: receptor signaling can be modified by allosteric modulators that either enhance agonist activity [positive allosteric modulator (PAM)] or decrease signaling [negative allosteric modulator (NAM)]. C: examples of PAR inhibitors, including mAbs (gray), peptides (yellow), small molecules (red), NAMs (green), or pepducins targeting intracellular loops (blue). These can target allosteric sites or the orthosteric binding site of the tethered ligand (dashed).
6.1.1. PAR1.
PAR1 antagonists were developed as antithrombotic drugs to prevent protease-mediated platelet aggregation. PAR1 antagonists include peptidic (e.g., BMS-200261, RWJ-58259) and nonpeptidic molecules (e.g., FR-171113, SCH 530348/Vorapaxar, E555/atopaxar, F 16618; Refs. 591–596). Antagonistic antibodies can also target various PAR1 domains, including the NH2 terminus immediately distal to the canonical cleavage site (597) and the hirudin-like thrombin-binding domain (598).
Peptidomimetic antagonists are analogs of activating peptides designed to compete with the orthosteric site of the intramolecular interaction. BMS-200261 was developed as a peptidic antagonist arising from NH2-terminal acylation of tetrapeptides mimicking SFLLR binding (591). BMS-200261 had a nanomolar IC50 with the ability to reduce platelet aggregation induced by SFLLRN-NH2. RWJ-56110 was discovered as a potent, selective PAR1 antagonist that interfered with Ca2+ signaling, platelet aggregation, and cell proliferation (599).
Small molecule antagonists were pursued to overcome the limitations of peptidomimetics, such as their limited bioavailability. Vorapaxar (SCH 530348) was the first small molecule inhibitor of PAR1 (600). Vorapaxar occupies the orthosteric binding site (Ref. 29; sect. 2.2). Atopaxar (E5555) is a small molecule inhibitor that interacts with ECL2 of PAR1 (601). Vorapaxar was Food and Drug Administration-approved in 2014 as a first-in-class oral PAR1 antagonist. Vorapaxar, however, led to increased bleeding in Phase II trials (602). This was attributed to its slow kinetic dissociation (603). This increased bleeding risk led to a general “black box warning” against serious bleeding and its withdrawal from European Union recommendations. On-target side effects are a liability of PAR1 agonists considering its widespread distribution. For example, PAR1 expression on endothelial cells was attributed to the intracranial bleeding that occurs with PAR1 antagonism. PAR1 activation maintains endothelial cell integrity via APC; thus antagonism also impacts platelet aggregation and vessel wall integrity (604). While PAR4 has a central role in hemostasis, it is not as widely expressed. Accordingly, PAR4 has the potential as a more tractable target for inhibitors for cardiovascular indications. Additionally, APC-like agonists were developed as an alternative due to their neuroprotective functions (605, 606). 3K3A-APC, an analog of APC (607), has since demonstrated promising outcomes with successful data emerging from clinical trials (608, 609).
Negative allosteric modulators have been developed to inhibit PAR1 signaling. For example, ML 161 is a small molecular negative allosteric modulator with potent inhibition (IC50: ∼0.3 µM) of SFLLRN-induced P-selectin expression in platelets (610). Parmodulins are a class of small molecule inhibitors developed as allosteric PAR1 antagonists that interfere with Gα-protein binding (611). This key study demonstrated bias, with preferential inhibition of Gαq signaling compared to Gα12/13 in response to PAR1. Unlike vorapaxar, parmodulins can maintain cytoprotective APC signaling while inhibiting thrombin-mediated signaling. Whereas an antagonist inhibits any signaling response, a partial agonist leads to a submaximal response relative to the full receptor agonist (612). Partial agonism can be a useful pharmacological property for drugs by preventing the endogenous full agonist from signaling, while producing a weak signal that can maintain basal activity. Having been investigated as antagonists, parmodulins were also shown to be partial agonists. Parmodulins themselves can trigger cytoprotective APC-like signaling in the endothelium and activate Gβγ, PI3K, and NF-κB (613).
6.1.2. PAR2.
The contributions of PAR2 to neurogenic inflammation, pain, and other disease processes catalyzed the search for PAR2 antagonists (reviewed in Ref. 614). The first PAR2 antagonists, FSLLRY-NH2 and LSIGRL-NH2, were peptidic and based on modifications of the canonical tethered ligand domain (196). Other peptide antagonists included K-12940 (615) and K-14585, which showed evidence of partial agonism (616). These compounds favored inhibition against activating peptides rather than trypsin, which limited further development. Peptidic C-391 showed micromolar potency and antagonism of hyperalgesia in vivo (617).
The poor bioavailability and potency of peptidic antagonists led to the development of nonpeptidic ligands, including full agonist AC-55541 and partial agonist AC-98170 (262, 618). Modifications were introduced to make molecules more drug-like by developing GB110 and GB83 with selectivity for PAR2 over PAR1 (619). This led to the discovery of the biased antagonist GB88 (620). GB88 was an antagonist in Gαq-mediated Ca2+ signaling, but a partial agonist with respect to cAMP inhibition, ERK1/2 phosphorylation, and Rho kinase signaling (621).
Small molecular PAR2 antagonists have been developed by Heptares Therapeutics and AstraZeneca, where structural studies have mapped the interaction of these antagonists with PAR2 (28; sect. 2.2). AZ3541 is a negative allosteric modulator that binds outside the helix domains. AZ8838 binds in a pocket near the extracellular surface and exhibits slow binding kinetics. Vertex Pharmaceuticals also developed a series of imidazopyridazine compounds that inhibit PAR2 signaling (622). In subsequent studies, antagonist I-191 shows improved potency relative to GB88 (282). This also included I-287, a negative allosteric modulator that inhibits Gαq and Gα12/13 pathways without affecting Gαi/o or β-arrestin2 signaling (623). Crucially, this led to reduced inflammation in vivo. The value of small molecule PAR2 inhibitors with oral bioavailability, subtype selectivity, and high potency is evident from the variety of pathologies that implicate PAR2 signaling (sect. 5).
There have been successful examples of antibodies developed against PAR2. Boehringer Ingelheim developed a humanized blocking antibody against the PAR2 protease cleavage site with picomolar affinity (624). AstraZeneca developed a monoclonal antibody against PAR2, MEDI0618, that is undergoing Phase I clinical trials as the first in human safety trial for chronic pain (NCT04198558; Ref. 614).
6.1.3. PAR1 and PAR2.
Bivalent compounds simultaneously target more than one receptor. A bivalent antagonist of PAR1 and PAR2 was developed by appending PAR1-selective RWJ-58259 to PAR2-selective antagonists based on the imidazopyridine scaffold and joined by a polyethylene glycol linker (625). The optimal bivalent antagonist compound 13d successfully reduced Ca2+ signaling in endothelial cells and cancer cells.
6.1.4. PAR4.
Given the liabilities of PAR1-targeted antagonists, recent drug discovery programs have investigated PAR4 antagonists as improved antiplatelet therapeutics (reviewed in Ref. 626). Bristol-Myers Squibb developed BMS-986120, a potent, selective, first-in-class small molecule PAR4 antagonist (627). This study crucially demonstrated reduced bleeding risk in monkeys compared to typical platelet drugs. BMS-986120 has since completed numerous Phase I clinical trials, including positive results on human thrombin formation ex vivo (628).
6.2. Subcellular Targeting of PAR Antagonists
The realization that PARs can signal at the plasma membrane and from intracellular domains prompted the development of antagonists targeted at subcellular microdomains.
6.2.1. Plasma membrane targeting.
Cell-penetrating antagonists were developed to penetrate cells and inhibit PARs from an intracellular site. Pepducins are peptidic compounds derived from an intracellular region of PARs. These have a palmitoyl modification as a lipid chain to aid association with the plasma membrane. Pepducins were developed against PAR1 using peptide sequences based on the ICL3 of PAR1 and named according to their length (629). Whereas P1pal-13 (palmitate-AVANRSKKSRALF) is an agonist, P1pal-12S (palmitate-RSLSSSAVANRS) is a full antagonist for PAR1. Interactions with ICL3 interfere with Gα protein binding. This PAR1 pepducin antagonist, later designated PZ-128, was successful in Phase II clinical trials in patients with coronary artery disease (630).
Pepducins have also been developed against PAR4. Based on a COOH-terminal motif of PAR4, “RAG8” (RAGLFQRS) was developed. This is coupled to a palmitoylated region as a cell-penetrating peptide (213). This reduces PAR4-mediated Ca2+ signaling, Akt phosphorylation, and binding to β-arrestin but not MAPK signaling. Given the structural differences between PAR1 and PAR4 in this region, RAG8 is PAR4 selective. Importantly, PAR1-mediated activation is not affected. When the COOH-terminal sequence of PAR4 is coupled to RAG8, platelet activation is blocked in vitro and clot consolidation blocked in vivo.
6.2.2. Endosomal membrane targeting.
The discovery that PAR2 can continue to signal from endosomes and that sustained endosomal signaling mediates long-term nociceptor hyperexcitability and pain prompted the development of endosomally targeted PAR2 antagonists (203). Tripartite antagonists comprising cholestanol, a PEG linker, and a cargo of I-343 PAR2 antagonist accumulate in endosomes. These abrogate endosomal signaling and protease-evoked hyperexcitability of nociceptors (sect. 5.5). This antagonist also blocks the effects of proteases released from mucosal biopsies on nociceptor hyperexcitability, raising the possibility of using endosome-targeted PAR2 antagonists for the treatment of IBS pain.
6.3. Future of Drugs Targeting PARs
Vorapaxar is the only PAR-directed drug approved for clinical use. Given the roles of other PARs in pathology in many organ systems (sect. 5), there remain ample opportunities to target PARs for the treatment of diverse diseases. However, the therapeutic targeting of PARs faces several challenges. The widespread expression of PARs (sect. 3) and their roles in essential physiological processes (sect. 5) increase the risks of severe on-target side effects (i.e., mediated by the same receptor). For example, vorapaxar (PAR1-selective antagonist) effectively inhibits platelet aggregation and can be used to treat thrombotic events, but a small number of patients experienced intracranial bleeds due to the impact on endothelial cell function (602). One approach to limiting on-target side effects is to administer drugs to particular tissues using biomaterials, such as nanoparticles. The incorporation of vorapaxar into cardiac stents would be expected to provide beneficial local protection against inappropriate coagulation without detrimental systemic actions. Similarly, inhaled drugs that preferentially target the airway epithelium, or oral drugs that target the intestinal epithelium, might be useful approaches for treatment of inflammatory diseases of the airway and intestine, minimizing side effects in other tissues.
Another approach that has been used to minimize on-target side effects has been to design ligands that preferentially target receptors in diseased tissue. This approach has been exploited to develop agonists of the µ-opioid receptor that preferentially activate receptors in the acidified microenvironment of diseased tissues (e.g., sites of inflammation and cancer). An analog of fentanyl designed to preferentially interact with µ-opioid receptors in acidic environments provides analgesia without the well-known on-target side effects of respiratory depression and constipation mediated by µ-opioid receptors in normal tissues (631). Given the established roles of PARs in inflamed tissues and cancer, which are usually associated with extracellular acidification, pH-dependent PAR ligands could provide another layer of selectivity.
The concept of biased signaling has attracted considerable attention for GPCR drug development. For example, biased ligands (antagonists or agonists) targeting GPCR signaling pathways that mediate detrimental events but sparing those that mediate beneficial outcomes could provide effective therapy while minimizing side effects. However, a prerequisite for the successful development of biased ligands is that the signaling pathways that mediate the detrimental versus beneficial outcomes of GPCR signaling are known and sufficiently distinct, which is not always the case. The complexities of PAR signaling complicate whether an antagonist or agonist would benefit. For example, a PAR1 agonist with respect to APC-induced PAR1 signaling, but an antagonist for thrombin-induced signaling, would be ideal for circulatory pathologies (sect. 5.1). To better target PARs in pathology, it therefore remains vital to understand how PARs function with respect to in vivo physiology and in vitro molecular pharmacology, progressing studies from isolated models to represent the human condition.
6.4. Summary
A range of peptidic, small molecules, and antibody-based therapeutics have been developed against PAR1, PAR2, and more recently PAR4. Vorapaxar is the only PAR-directed drug approved for use in patients, as a first-in-class antiplatelet drug acting through PAR1. They have since been drugs in clinical trials, including a small molecule inhibitor of PAR4 as an alternative antiplatelet drug to reduce bleeding risk (BMS-986120), the recombinant APC-like agonist (3K3A-APC), as well as an antibody against PAR2 for the treatment of pain (MEDI0618). Considering the range of pathologies that PAR signaling implicates, increased understanding of PARs at a molecular level in the human setting will be instrumental in developing better drugs across disease states.
7. CONCLUSIONS
The discovery that thrombin causes the aggregation of platelets by cleaving PAR1 provided the impetus for intensive investigations of the receptor-mediated actions of proteases. They resulted in the identification of the family of four widely expressed PARs. These GPCRs are now known to regulate mechanisms of homeostasis and disease in most tissues and organs. Antagonists of PAR1 have advanced to the clinic for the treatment of coagulation disorders, and antagonists of PAR2 and PAR4 are in clinical development. Structural studies of PAR1 and PAR2 complexed to antagonists have provided an understanding of the mechanisms of activation and antagonism. Future structural studies of PARs in their activated state and complexed with signaling partners and regulatory proteins (e.g., G proteins, β-arrestins) will further enhance the development of selective ligands that may advance to approved drugs. The original concept that proteases activate PARs by cleavage at a single site and that cleavage at different sites merely disarms the receptor has been superseded by the realization that proteases can activate PARs by diverse noncanonical mechanisms. Thus proteases that cleave PARs at distinct sites can activate PARs by biased mechanisms leading to unique cellular responses. The four PARs can, therefore, be considered as molecular integrators of the biological actions of a very large number of proteases, many of which likely derive from microorganisms. Proteases from the microbiome could regulate host cell functions by cleaving PARs. This might be of particular importance in the colon, where proteases from commensal and pathogenic bacteria are likely to regulate colonocyte function in health and disease states by cleaving PARs. It is evident that there are major differences between signaling cascades of PAR subtypes, despite structural and genetic similarities. In particular, PAR3 signaling in its own right remains a major question. The realization that proteases and PARs can signal from subcellular microdomains, which differ depending on the mechanism of PAR activation, adds additional complexity. This also provides an opportunity of developing antagonists that preferentially target PARs in subcellular microdomains with enhanced selectivity and efficacy. The rapidly evolving field of protease and PAR signaling is likely to provide new insights into physiological control and mechanisms of disease, with multiple opportunities for therapeutic intervention.
SUPPLEMENTAL DATA
Supplemental Tables S1–S5 are available at https://doi.org/10.6084/m9.figshare.19739830.
GRANTS
Work in the authors’ laboratories is supported by grants from the National Institutes of Health (NS102722, DE026806, DK118971, and DE029951 to N.W.B. and B.L.S.), Department of Defense (W81XWH1810431 and W81XWH2210239 to N.W.B. and B.L.S.), the Leon Levy Foundation for the Leon Levy Fellowship in Neuroscience (to C.J.P.), the Grimwade Fellowship funded by the Russell and Mab Grimwade Miegunyah Fund at the University of Melbourne (to L.E.E.), and a DECRA Fellowship from the Australian Research Council (ARC; DE180100418 to L.E.M.).
DISCLOSURES
N. W. Bunnett is a founding scientist of Endosome Therapeutics Inc. None of the other authors has any conflicts of interest, financial or otherwise, to disclose.
AUTHOR CONTRIBUTIONS
N.W.B. conceived and designed research; C.J.P., L.E.E., N.W.B., and B.L.S. prepared figures; C.J.P., L.E.E., N.W.B., and B.L.S. drafted manuscript; C.J.P., L.E.E., N.W.B., and B.L.S. edited and revised manuscript; C.J.P., L.E.E., N.W.B., and B.L.S. approved final version of manuscript.
ACKNOWLEDGMENTS
Parts of the figures were created with BioRender.com, with permission.
REFERENCES
- 1. Schmidt A. Neue Untersuchungen ueber die Fasserstoffesgerinnung. Pflüger Arch 6: 413–538, 1872. doi: 10.1007/BF01612263. [DOI] [Google Scholar]
- 2. Van Obberghen-Schilling E, Chambard JC, Lory P, Nargeot J, Pouyssegur J. Functional expression of Ca2(+)-mobilizing alpha-thrombin receptors in mRNA-injected Xenopus oocytes. FEBS Lett 262: 330–334, 1990. doi: 10.1016/0014-5793(90)80221-4. [DOI] [PubMed] [Google Scholar]
- 3. Pipili-Synetos E, Gershengorn MC, Jaffe EA. Expression of functional thrombin receptors in xenopus oocytes injected with human endothelial cell mRNA. Biochem Biophys Res Commun 171: 913–919, 1990. doi: 10.1016/0006-291X(90)90770-N. [DOI] [PubMed] [Google Scholar]
- 4. Vu TK, Hung DT, Wheaton VI, Coughlin SR. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell 64: 1057–1068, 1991. doi: 10.1016/0092-8674(91)90261-V. [DOI] [PubMed] [Google Scholar]
- 5. Nystedt S, Emilsson K, Wahlestedt C, Sundelin J. Molecular cloning of a potential proteinase activated receptor. Proc Natl Acad Sci U S A 91: 9208–9212, 1994. doi: 10.1073/pnas.91.20.9208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Connolly AJ, Ishihara H, Kahn ML, Farese RV Jr, Coughlin SR. Role of the thrombin receptor in development and evidence for a second receptor. Nature 381: 516–519, 1996. doi: 10.1038/381516a0. [DOI] [PubMed] [Google Scholar]
- 7. Xu WF, Andersen H, Whitmore TE, Presnell SR, Yee DP, Ching A, Gilbert T, Davie EW, Foster DC. Cloning and characterization of human protease-activated receptor 4. Proc Natl Acad Sci U S A 95: 6642–6646, 1998. doi: 10.1073/pnas.95.12.6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kahn ML, Zheng YW, Huang W, Bigornia V, Zeng D, Moff S, Farese RV Jr, Tam C, Coughlin SR. A dual thrombin receptor system for platelet activation. Nature 394: 690–694, 1998. doi: 10.1038/29325. [DOI] [PubMed] [Google Scholar]
- 9. Somers GR, Hammet F, Woollatt E, Richards RI, Southey MC, Venter DJ. Chromosomal localization of the human P2y6 purinoceptor gene and phylogenetic analysis of the P2y purinoceptor family. Genomics 44: 127–130, 1997. doi: 10.1006/geno.1997.4841. [DOI] [PubMed] [Google Scholar]
- 10. Yang-Feng TL, Xue FY, Zhong WW, Cotecchia S, Frielle T, Caron MG, Lefkowitz RJ, Francke U. Chromosomal organization of adrenergic receptor genes. Proc Natl Acad Sci U S A 87: 1516–1520, 1990. doi: 10.1073/pnas.87.4.1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jin M, Yang HW, Tao AL, Wei JF. Evolution of the protease-activated receptor family in vertebrates. Int J Mol Med 37: 593–602, 2016. doi: 10.3892/ijmm.2016.2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ishii K, Gerszten R, Zheng YW, Welsh JB, Turck CW, Coughlin SR. Determinants of thrombin receptor cleavage. Receptor domains involved, specificity, and role of the P3 aspartate. J Biol Chem 270: 16435–16440, 1995. doi: 10.1074/jbc.270.27.16435. [DOI] [PubMed] [Google Scholar]
- 13. Riewald M, Kravchenko VV, Petrovan RJ, O’Brien PJ, Brass LF, Ulevitch RJ, Ruf W. Gene induction by coagulation factor Xa is mediated by activation of protease-activated receptor 1. Blood 97: 3109–3116, 2001. doi: 10.1182/blood.V97.10.3109. [DOI] [PubMed] [Google Scholar]
- 14. Kondreddy V, Pendurthi UR, Xu X, Griffin JH, Rao LV. FVIIa (Factor VIIa) induces biased cytoprotective signaling in mice through the cleavage of PAR (protease-activated receptor)-1 at xanonical Arg41 (Arginine41) site. Arterioscler Thromb Vasc Biol 40: 1275–1288, 2020. doi: 10.1161/ATVBAHA.120.314244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Isberg V, de Graaf C, Bortolato A, Cherezov V, Katritch V, Marshall FH, Mordalski S, Pin JP, Stevens RC, Vriend G, Gloriam DE. Generic GPCR residue numbers–aligning topology maps while minding the gaps. Trends Pharmacol Sci 36: 22–31, 2015. doi: 10.1016/j.tips.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rasmussen SG, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, Devree BT, Rosenbaum DM, Thian FS, Kobilka TS, Schnapp A, Konetzki I, Sunahara RK, Gellman SH, Pautsch A, Steyaert J, Weis WI, Kobilka BK. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature 469: 175–180, 2011. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, Mathiesen JM, Shah ST, Lyons JA, Caffrey M, Gellman SH, Steyaert J, Skiniotis G, Weis WI, Sunahara RK, Kobilka BK. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 477: 549–555, 2011. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gerszten RE, Chen J, Ishii M, Ishii K, Wang L, Nanevicz T, Turck CW, Vu TK, Coughlin SR. Specificity of the thrombin receptor for agonist peptide is defined by its extracellular surface. Nature 368: 648–651, 1994. doi: 10.1038/368648a0. [DOI] [PubMed] [Google Scholar]
- 19. Nanevicz T, Wang L, Chen M, Ishii M, Coughlin SR. Thrombin receptor activating mutations. Alteration of an extracellular agonist recognition domain causes constitutive signaling. J Biol Chem 271: 702–706, 1996. doi: 10.1074/jbc.271.2.702. [DOI] [PubMed] [Google Scholar]
- 20. Nystedt S, Emilsson K, Larsson AK, Strombeck B, Sundelin J. Molecular cloning and functional expression of the gene encoding the human proteinase-activated receptor 2. Eur J Biochem 232: 84–89, 1995. doi: 10.1111/j.1432-1033.1995.tb20784.x. [DOI] [PubMed] [Google Scholar]
- 21. Böhm SK, Kong W, Brömme D, Smeekens SP, Anderson DC, Connolly A, Kahn M, Nelken NA, Coughlin SR, Payan DG, Bunnett NW. Molecular cloning, expression and potential functions of the human proteinase-activated receptor-2. Biochem J 314: 1009–1016, 1996. doi: 10.1042/bj3141009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Molino M, Barnathan ES, Numerof R, Clark J, Dreyer M, Cumashi A, Hoxie JA, Schechter N, Woolkalis M, Brass LF. Interactions of mast cell tryptase with thrombin receptors and PAR-2. J Biol Chem 272: 4043–4049, 1997. doi: 10.1074/jbc.272.7.4043. [DOI] [PubMed] [Google Scholar]
- 23. Corvera CU, Dery O, McConalogue K, Gamp P, Thoma M, Al-Ani B, Caughey GH, Hollenberg MD, Bunnett NW. Thrombin and mast cell tryptase regulate guinea-pig myenteric neurons through proteinase-activated receptors-1 and -2. J Physiol 517: 741–756, 1999. doi: 10.1111/j.1469-7793.1999.0741s.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rana S, Yang L, Hassanian SM, Rezaie AR. Determinants of the specificity of protease-activated receptors 1 and 2 signaling by factor Xa and thrombin. J Cell Biochem 113: 977–984, 2012. doi: 10.1002/jcb.23427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Oikonomopoulou K, Hansen KK, Saifeddine M, Tea I, Blaber M, Blaber SI, Scarisbrick I, Andrade-Gordon P, Cottrell GS, Bunnett NW, Diamandis EP, Hollenberg MD. Proteinase-activated receptors, targets for kallikrein signaling. J Biol Chem 281: 32095–32112, 2006. doi: 10.1074/jbc.M513138200. [DOI] [PubMed] [Google Scholar]
- 26. Ramsay AJ, Dong Y, Hunt ML, Linn M, Samaratunga H, Samaratunga H, Clements J, Hooper JD, Hooper JD. Kallikrein-related peptidase 4 (KLK4) initiates intracellular signaling via protease-activated receptors (PARs). KLK4 and PAR-2 are co-expressed during prostate cancer progression. J Biol Chem 283: 12293–12304, 2008. doi: 10.1074/jbc.M709493200. [DOI] [PubMed] [Google Scholar]
- 27. Stefansson K, Brattsand M, Roosterman D, Kempkes C, Bocheva G, Steinhoff M, Egelrud T. Activation of proteinase-activated receptor-2 by human kallikrein-related peptidases. J Invest Dermatol 128: 18–25, 2008. doi: 10.1038/sj.jid.5700965. [DOI] [PubMed] [Google Scholar]
- 28. Cheng RK, Fiez-Vandal C, Schlenker O, Edman K, Aggeler B, Brown DG, Brown GA, Cooke RM, Dumelin CE, Dore AS, Geschwindner S, Grebner C, Hermansson NO, Jazayeri A, Johansson P, Leong L, Prihandoko R, Rappas M, Soutter H, Snijder A, Sundstrom L, Tehan B, Thornton P, Troast D, Wiggin G, Zhukov A, Marshall FH, Dekker N. Structural insight into allosteric modulation of protease-activated receptor 2. Nature 545: 112–115, 2017. doi: 10.1038/nature22309. [DOI] [PubMed] [Google Scholar]
- 29. Zhang C, Srinivasan Y, Arlow DH, Fung JJ, Palmer D, Zheng Y, Green HF, Pandey A, Dror RO, Shaw DE, Weis WI, Coughlin SR, Kobilka BK. High-resolution crystal structure of human protease-activated receptor 1. Nature 492: 387–392, 2012. doi: 10.1038/nature11701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ishihara H, Connolly AJ, Zeng D, Kahn ML, Zheng YW, Timmons C, Tram T, Coughlin SR. Protease-activated receptor 3 is a second thrombin receptor in humans. Nature 386: 502–506, 1997. doi: 10.1038/386502a0. [DOI] [PubMed] [Google Scholar]
- 31. Nieman MT. Protease-activated receptor 4 uses anionic residues to interact with α-thrombin in the absence or presence of protease-activated receptor 1. Biochemistry 47: 13279–13286, 2008. doi: 10.1021/bi801334s. [DOI] [PubMed] [Google Scholar]
- 32. Duvernay MT, Temple KJ, Maeng JG, Blobaum AL, Stauffer SR, Lindsley CW, Hamm HE. Contributions of protease-activated receptors PAR1 and PAR4 to thrombin-induced GPIIbIIIa activation in human platelets. Mol Pharmacol 91: 39–47, 2017. doi: 10.1124/mol.116.106666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jacques SL, Kuliopulos A. Protease-activated receptor-4 uses dual prolines and an anionic retention motif for thrombin recognition and cleavage. Biochem J 376: 733–740, 2003. doi: 10.1042/bj20030954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bah A, Chen Z, Bush-Pelc LA, Mathews FS, Di Cera E. Crystal structures of murine thrombin in complex with the extracellular fragments of murine protease-activated receptors PAR3 and PAR4. Proc Natl Acad Sci U S A 104: 11603–11608, 2007. doi: 10.1073/pnas.0704409104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vindigni A, White CE, Komives EA, Di Cera E. Energetics of thrombin-thrombomodulin interaction. Biochemistry 36: 6674–6681, 1997. doi: 10.1021/bi962766a. [DOI] [PubMed] [Google Scholar]
- 36. Vijayalakshmi J, Padmanabhan KP, Mann KG, Tulinsky A. The isomorphous structures of prethrombin2, hirugen-, and PPACK-thrombin: changes accompanying activation and exosite binding to thrombin. Protein Sci 3: 2254–2271, 1994. doi: 10.1002/pro.5560031211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Scarborough RM, Naughton MA, Teng W, Hung DT, Rose J, Vu TK, Wheaton VI, Turck CW, Coughlin SR. Tethered ligand agonist peptides. Structural requirements for thrombin receptor activation reveal mechanism of proteolytic unmasking of agonist function. J Biol Chem 267: 13146–13149, 1992. doi: 10.1016/S0021-9258(18)42184-9. [DOI] [PubMed] [Google Scholar]
- 38. Adams MN, Pagel CN, Mackie EJ, Hooper JD. Evaluation of antibodies directed against human protease-activated receptor-2. Naunyn Schmiedebergs Arch Pharmacol 385: 861–873, 2012. doi: 10.1007/s00210-012-0783-6. [DOI] [PubMed] [Google Scholar]
- 39. Latorre RA, Hegron A, Peach CJ, Teng SA, Tonello RA, Retamal JA, Klein-Cloud RA, Bok D, Jensen DA, Gottesman-Katz L, Rientjes J, Veldhuis NA, Poole DA, Schmidt BL, Pothoulakis CH, Rankin CO, Xie Y, Koon HA, Bunnett NA. Mice expressing fluorescent PAR(2) reveal that endocytosis mediates colonic inflammation and pain. Proc Natl Acad Sci U S A 119: e2112059119, 2022. doi: 10.1073/pnas.2112059119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Derian CK, Eckardt AJ, Andrade-Gordon P. Differential regulation of human keratinocyte growth and differentiation by a novel family of protease-activated receptors. Cell Growth Differ 8: 743–749, 1997. [PubMed] [Google Scholar]
- 41. Santulli RJ, Derian CK, Darrow AL, Tomko KA, Eckardt AJ, Seiberg M, Scarborough RM, Andrade-Gordon P. Evidence for the presence of a protease-activated receptor distinct from the thrombin receptor in human keratinocytes. Proc Natl Acad Sci U S A 92: 9151–9155, 1995. doi: 10.1073/pnas.92.20.9151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Steinhoff M, Corvera CU, Thoma MS, Kong W, McAlpine BE, Caughey GH, Ansel JC, Bunnett NW. Proteinase-activated receptor-2 in human skin: tissue distribution and activation of keratinocytes by mast cell tryptase. Exp Dermatol 8: 282–294, 1999. [DOI] [PubMed] [Google Scholar]
- 43. Camerer E, Huang W, Coughlin SR. Tissue factor- and factor X-dependent activation of protease-activated receptor 2 by factor VIIa. Proc Natl Acad Sci U S A 97: 5255–5260, 2000. doi: 10.1073/pnas.97.10.5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gouin O, L’Herondelle K, Buscaglia P, Le Gall-Ianotto C, Philippe R, Legoux N, Mignen O, Buhe V, Leschiera R, Sakka M, Kerfant N, Carre JL, Le Garrec R, Lefeuvre L, Lebonvallet N, Misery L. Major role for TRPV1 and InsP3R in PAR2-elicited inflammatory mediator production in differentiated human keratinocytes. J Invest Dermatol 138: 1564–1572, 2018. doi: 10.1016/j.jid.2018.01.034. [DOI] [PubMed] [Google Scholar]
- 45. Wygrecka M, Didiasova M, Berscheid S, Piskulak K, Taborski B, Zakrzewicz D, Kwapiszewska G, Preissner KT, Markart P. Protease-activated receptors (PAR)-1 and -3 drive epithelial-mesenchymal transition of alveolar epithelial cells–potential role in lung fibrosis. Thromb Haemost 110: 295–307, 2013. doi: 10.1160/TH12-11-0854. [DOI] [PubMed] [Google Scholar]
- 46. D’Andrea MR, Derian CK, Leturcq D, Baker SM, Brunmark A, Ling P, Darrow AL, Santulli RJ, Brass LF, Andrade-Gordon P. Characterization of protease-activated receptor-2 immunoreactivity in normal human tissues. J Histochem Cytochem 46: 157–164, 1998. doi: 10.1177/002215549804600204. [DOI] [PubMed] [Google Scholar]
- 47. Kong W, McConalogue K, Khitin LM, Hollenberg MD, Payan DG, Bohm SK, Bunnett NW. Luminal trypsin may regulate enterocytes through proteinase-activated receptor 2. Proc Natl Acad Sci U S A 94: 8884–8889, 1997. doi: 10.1073/pnas.94.16.8884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cunningham MA, Rondeau E, Chen X, Coughlin SR, Holdsworth SR, Tipping PG. Protease-activated receptor 1 mediates thrombin-dependent, cell-mediated renal inflammation in crescentic glomerulonephritis. J Exp Med 191: 455–462, 2000. doi: 10.1084/jem.191.3.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Moffatt JD, Cocks TM. The role of protease-activated receptor-2 (PAR2) in the modulation of beating of the mouse isolated ureter: lack of involvement of mast cells or sensory nerves. Br J Pharmacol 128: 860–864, 1999. doi: 10.1038/sj.bjp.0702871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bertog M, Letz B, Kong W, Steinhoff M, Higgins MA, Bielfeld-Ackermann A, Fromter E, Bunnett NW, Korbmacher C. Basolateral proteinase-activated receptor (PAR-2) induces chloride secretion in M-1 mouse renal cortical collecting duct cells. J Physiol 521: 3–17, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chi L, Li Y, Stehno-Bittel L, Gao J, Morrison DC, Stechschulte DJ, Dileepan KN. Interleukin-6 production by endothelial cells via stimulation of protease-activated receptors is amplified by endotoxin and tumor necrosis factor-alpha. J Interferon Cytokine Res 21: 231–240, 2001. doi: 10.1089/107999001750169871. [DOI] [PubMed] [Google Scholar]
- 52. Fujiwara M, Jin E, Ghazizadeh M, Kawanami O. Differential expression of protease-activated receptors 1, 2, and 4 on human endothelial cells from different vascular sites. Pathobiology 71: 52–58, 2004. doi: 10.1159/000072962. [DOI] [PubMed] [Google Scholar]
- 53. Jesmin S, Gando S, Zaedi S, Sakuraya F. Differential expression, time course and distribution of four PARs in rats with endotoxin-induced acute lung injury. Inflammation 30: 14–27, 2007. doi: 10.1007/s10753-006-9017-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kataoka H, Hamilton JR, McKemy DD, Camerer E, Zheng YW, Cheng A, Griffin C, Coughlin SR. Protease-activated receptors 1 and 4 mediate thrombin signaling in endothelial cells. Blood 102: 3224–3231, 2003. doi: 10.1182/blood-2003-04-1130. [DOI] [PubMed] [Google Scholar]
- 55. Hwa JJ, Ghibaudi L, Williams P, Chintala M, Zhang R, Chatterjee M, Sybertz E. Evidence for the presence of a proteinase-activated receptor distinct from the thrombin receptor in vascular endothelial cells. Circ Res 78: 581–588, 1996. doi: 10.1161/01.RES.78.4.581. [DOI] [PubMed] [Google Scholar]
- 56. Mirza H, Yatsula V, Bahou WF. The proteinase activated receptor-2 (PAR-2) mediates mitogenic responses in human vascular endothelial cells. J Clin Invest 97: 1705–1714, 1996. doi: 10.1172/JCI118597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Schmidt VA, Nierman WC, Maglott DR, Cupit LD, Moskowitz KA, Wainer JA, Bahou WF. The human proteinase-activated receptor-3 (PAR-3) gene. Identification within a Par gene cluster and characterization in vascular endothelial cells and platelets. J Biol Chem 273: 15061–15068, 1998. doi: 10.1074/jbc.273.24.15061. [DOI] [PubMed] [Google Scholar]
- 58. Fujiwara M, Jin E, Ghazizadeh M, Kawanami O. Activation of PAR4 induces a distinct actin fiber formation via p38 MAPK in human lung endothelial cells. J Histochem Cytochem 53: 1121–1129, 2005. doi: 10.1369/jhc.4A6592.2005. [DOI] [PubMed] [Google Scholar]
- 59. Van Den Eshof BL, Hoogendijk AJ, Simpson PJ, Van Alphen FP, Zanivan S, Mertens K, Meijer AB, Van Den Biggelaar M. Paradigm of biased PAR1 (protease-activated receptor-1) activation and inhibition in endothelial cells dissected by phosphoproteomics. Arterioscler Thromb Vasc Biol 37: 1891–1902, 2017. doi: 10.1161/ATVBAHA.117.309926. [DOI] [PubMed] [Google Scholar]
- 60. Asteriti S, Daniele S, Porchia F, Dell’Anno MT, Fazzini A, Pugliesi I, Trincavelli ML, Taliani S, Martini C, Mazzoni MR, Gilchrist A. Modulation of PAR(1) signalling by benzimidazole compounds. Br J Pharmacol 167: 80–94, 2012. doi: 10.1111/j.1476-5381.2012.01974.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Domotor E, Benzakour O, Griffin JH, Yule D, Fukudome K, Zlokovic BV. Activated protein C alters cytosolic calcium flux in human brain endothelium via binding to endothelial protein C receptor and activation of protease activated receptor-1. Blood 101: 4797–4801, 2003. doi: 10.1182/blood-2002-12-3680. [DOI] [PubMed] [Google Scholar]
- 62. Mosnier LO, Griffin JH. Inhibition of staurosporine-induced apoptosis of endothelial cells by activated protein C requires protease-activated receptor-1 and endothelial cell protein C receptor. Biochem J 373: 65–70, 2003. doi: 10.1042/bj20030341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Riewald M, Petrovan RJ, Donner A, Mueller BM, Ruf W. Activation of endothelial cell protease activated receptor 1 by the protein C pathway. Science 296: 1880–1882, 2002. doi: 10.1126/science.1071699. [DOI] [PubMed] [Google Scholar]
- 64. Hoxie JA, Ahuja M, Belmonte E, Pizarro S, Parton R, Brass LF. Internalization and recycling of activated thrombin receptors. J Biol Chem 268: 13756–13763, 1993. doi: 10.1016/S0021-9258(18)86921-6. [DOI] [PubMed] [Google Scholar]
- 65. Woolkalis MJ, DeMelfi TM Jr, Blanchard N, Hoxie JA, Brass LF. Regulation of thrombin receptors on human umbilical vein endothelial cells. J Biol Chem 270: 9868–9875, 1995. doi: 10.1074/jbc.270.17.9868. [DOI] [PubMed] [Google Scholar]
- 66. Schuepbach RA, Feistritzer C, Brass LF, Riewald M. Activated protein C-cleaved protease activated receptor-1 is retained on the endothelial cell surface even in the presence of thrombin. Blood 111: 2667–2673, 2008. doi: 10.1182/blood-2007-09-113076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Shapiro MJ, Coughlin SR. Separate signals for agonist-independent and agonist-triggered trafficking of protease-activated receptor 1. J Biol Chem 273: 29009–29014, 1998. doi: 10.1074/jbc.273.44.29009. [DOI] [PubMed] [Google Scholar]
- 68. Kuliopulos A, Covic L, Seeley SK, Sheridan PJ, Helin J, Costello CE. Plasmin desensitization of the PAR1 thrombin receptor: kinetics, sites of truncation, and implications for thrombolytic therapy. Biochemistry 38: 4572–4585, 1999. doi: 10.1021/bi9824792. [DOI] [PubMed] [Google Scholar]
- 69. Walz DA, Anderson GF, Ciaglowski RE, Aiken M, Fenton JW 2nd.. Thrombin-elicited contractile responses of aortic smooth muscle. Proc Soc Exp Biol Med 180: 518–526, 1985. doi: 10.3181/00379727-180-42211. [DOI] [PubMed] [Google Scholar]
- 70. Huang CL, Ives HE. Growth inhibition by protein kinase C late in mitogenesis. Nature 329: 849–850, 1987. doi: 10.1038/329849a0. [DOI] [PubMed] [Google Scholar]
- 71. Hollenberg MD, Yang SG, Laniyonu AA, Moore GJ, Saifeddine M. Action of thrombin receptor polypeptide in gastric smooth muscle: identification of a core pentapeptide retaining full thrombin-mimetic intrinsic activity. Mol Pharmacol 42: 186–191, 1992. [PubMed] [Google Scholar]
- 72. Cocks TM, Sozzi V, Moffatt JD, Selemidis S. Protease-activated receptors mediate apamin-sensitive relaxation of mouse and guinea pig gastrointestinal smooth muscle. Gastroenterology 116: 586–592, 1999. doi: 10.1016/S0016-5085(99)70180-0. [DOI] [PubMed] [Google Scholar]
- 73. Hollenberg MD, Saifeddine M, Al-Ani B. Proteinase-activated receptor-2 in rat aorta: structural requirements for agonist activity of receptor-activating peptides. Mol Pharmacol 49: 229–233, 1996. [PubMed] [Google Scholar]
- 74. Cocks TM, Fong B, Chow JM, Anderson GP, Frauman AG, Goldie RG, Henry PJ, Carr MJ, Hamilton JR, Moffatt JD. A protective role for protease-activated receptors in the airways. Nature 398: 156–160, 1999. doi: 10.1038/18223. [DOI] [PubMed] [Google Scholar]
- 75. Magazine HI, King JM, Srivastava KD. Protease activated receptors modulate aortic vascular tone. Int J Cardiol 53, Suppl: S75–S80, 1996. doi: 10.1016/0167-5273(96)02569-7. [DOI] [PubMed] [Google Scholar]
- 76. Hamilton JR, Nguyen PB, Cocks TM. Atypical protease-activated receptor mediates endothelium-dependent relaxation of human coronary arteries. Circ Res 82: 1306–1311, 1998. doi: 10.1161/01.RES.82.12.1306. [DOI] [PubMed] [Google Scholar]
- 77. Wei M, Liu Y, Zheng M, Wang L, Ma F, Qi Y, Liu G. Upregulation of protease-activated receptor 2 promotes proliferation and migration of human vascular smooth muscle cells (VSMCs). Med Sci Monit 25: 8854–8862, 2019. doi: 10.12659/MSM.917865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Schror K, Bretschneider E, Fischer K, Fischer JW, Pape R, Rauch BH, Rosenkranz AC, Weber AA. Thrombin receptors in vascular smooth muscle cells–function and regulation by vasodilatory prostaglandins. Thromb Haemost 103: 884–890, 2010. doi: 10.1160/TH09-09-0627. [DOI] [PubMed] [Google Scholar]
- 79. Davey MG, Luscher EF. Actions of thrombin and other coagulant and proteolytic enzymes on blood platelets. Nature 216: 857–858, 1967. doi: 10.1038/216857a0. [DOI] [PubMed] [Google Scholar]
- 80. Haslam RJ, Davidson MM. Potentiation by thrombin of the secretion of serotonin from permeabilized platelets equilibrated with Ca2+ buffers. Relationship to protein phosphorylation and diacylglycerol formation. Biochem J 222: 351–361, 1984. doi: 10.1042/bj2220351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Altieri DC, Edgington TS. Identification of effector cell protease receptor-1. A leukocyte-distributed receptor for the serine protease factor Xa. J Immunol 145: 246–253, 1990. [PubMed] [Google Scholar]
- 82. Norton KJ, Scarborough RM, Kutok JL, Escobedo MA, Nannizzi L, Coller BS. Immunologic analysis of the cloned platelet thrombin receptor activation mechanism: evidence supporting receptor cleavage, release of the N-terminal peptide, and insertion of the tethered ligand into a protected environment. Blood 82: 2125–2136, 1993. [PubMed] [Google Scholar]
- 83. Hamilton JR, Cornelissen I, Coughlin SR. Impaired hemostasis and protection against thrombosis in protease-activated receptor 4-deficient mice is due to lack of thrombin signaling in platelets. J Thromb Haemost 2: 1429–1435, 2004. doi: 10.1111/j.1538-7836.2004.00783.x. [DOI] [PubMed] [Google Scholar]
- 84. Kahn ML, Nakanishi-Matsui M, Shapiro MJ, Ishihara H, Coughlin SR. Protease-activated receptors 1 and 4 mediate activation of human platelets by thrombin. J Clin Invest 103: 879–887, 1999. doi: 10.1172/JCI6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. De Candia E, Hall SW, Rutella S, Landolfi R, Andrews RK, De Cristofaro R. Binding of thrombin to glycoprotein Ib accelerates the hydrolysis of Par-1 on intact platelets. J Biol Chem 276: 4692–4698, 2001. doi: 10.1074/jbc.M008160200. [DOI] [PubMed] [Google Scholar]
- 86. Nakanishi-Matsui M, Zheng YW, Sulciner DJ, Weiss EJ, Ludeman MJ, Coughlin SR. PAR3 is a cofactor for PAR4 activation by thrombin. Nature 404: 609–613, 2000. doi: 10.1038/35007085. [DOI] [PubMed] [Google Scholar]
- 87. Vretenbrant K, Ramstrom S, Bjerke M, Lindahl TL. Platelet activation via PAR4 is involved in the initiation of thrombin generation and in clot elasticity development. Thromb Haemost 97: 417–424, 2007. doi: 10.1160/TH06-07-0397. [DOI] [PubMed] [Google Scholar]
- 88. Sambrano GR, Huang W, Faruqi T, Mahrus S, Craik C, Coughlin SR. Cathepsin G activates protease-activated receptor-4 in human platelets. J Biol Chem 275: 6819–6823, 2000. doi: 10.1074/jbc.275.10.6819. [DOI] [PubMed] [Google Scholar]
- 89. Schurch-Rathgeb Y, Mongard D. Brain development influences the appearance of glial factor-like activity in rat brain primary cultures. Nature 273: 308–309, 1978. doi: 10.1038/273308a0. [DOI] [PubMed] [Google Scholar]
- 90. Gurwitz D, Cunningham DD. Thrombin modulates and reverses neuroblastoma neurite outgrowth. Proc Natl Acad Sci U S A 85: 3440–3444, 1988. doi: 10.1073/pnas.85.10.3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Monard D, Niday E, Limat A, Solomon F. Inhibition of protease activity can lead to neurite extension in neuroblastoma cells. Prog Brain Res 58: 359–364, 1983. doi: 10.1016/S0079-6123(08)60037-0. [DOI] [PubMed] [Google Scholar]
- 92. Snider RM, McKinney M, Fenton JW 2nd, Richelson E. Activation of cyclic nucleotide formation in murine neuroblastoma N1E-115 cells by modified human thrombins. J Biol Chem 259: 9078–9081, 1984. doi: 10.1016/S0021-9258(17)47267-X. [DOI] [PubMed] [Google Scholar]
- 93. Junge CE, Lee CJ, Hubbard KB, Zhang Z, Olson JJ, Hepler JR, Brat DJ, Traynelis SF. Protease-activated receptor-1 in human brain: localization and functional expression in astrocytes. Exp Neurol 188: 94–103, 2004. doi: 10.1016/j.expneurol.2004.02.018. [DOI] [PubMed] [Google Scholar]
- 94. Debeir T, Vige X, Benavides J. Pharmacological characterization of protease-activated receptor (PAR-1) in rat astrocytes. Eur J Pharmacol 323: 111–117, 1997. doi: 10.1016/S0014-2999(97)00018-6. [DOI] [PubMed] [Google Scholar]
- 95. Steinhoff M, Vergnolle N, Young SH, Tognetto M, Amadesi S, Ennes HS, Trevisani M, Hollenberg MD, Wallace JL, Caughey GH, Mitchell SE, Williams LM, Geppetti P, Mayer EA, Bunnett NW. Agonists of proteinase-activated receptor 2 induce inflammation by a neurogenic mechanism. Nat Med 6: 151–158, 2000. doi: 10.1038/72247. [DOI] [PubMed] [Google Scholar]
- 96. Vergnolle N, Bunnett NW, Sharkey KA, Brussee V, Compton SJ, Grady EF, Cirino G, Gerard N, Basbaum AI, Andrade-Gordon P, Hollenberg MD, Wallace JL. Proteinase-activated receptor-2 and hyperalgesia: a novel pain pathway. Nat Med 7: 821–826, 2001. doi: 10.1038/89945. [DOI] [PubMed] [Google Scholar]
- 97. Amadesi S, Nie J, Vergnolle N, Cottrell GS, Grady EF, Trevisani M, Manni C, Geppetti P, McRoberts JA, Ennes H, Davis JB, Mayer EA, Bunnett NW. Protease-activated receptor 2 sensitizes the capsaicin receptor transient receptor potential vanilloid receptor 1 to induce hyperalgesia. J Neurosci 24: 4300–4312, 2004. doi: 10.1523/JNEUROSCI.5679-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Alier KA, Endicott JA, Stemkowski PL, Cenac N, Cellars L, Chapman K, Andrade-Gordon P, Vergnolle N, Smith PA. Intrathecal administration of proteinase-activated receptor-2 agonists produces hyperalgesia by exciting the cell bodies of primary sensory neurons. J Pharmacol Exp Ther 324: 224–233, 2008. doi: 10.1124/jpet.107.129171. [DOI] [PubMed] [Google Scholar]
- 99. Dinh QT, Cryer A, Dinh S, Trevisani M, Georgiewa P, Chung F, Geppetti P, Heppt W, Klapp BF, Fischer A. Protease-activated receptor 2 expression in trigeminal neurons innervating the rat nasal mucosa. Neuropeptides 39: 461–466, 2005. doi: 10.1016/j.npep.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 100. Kawabata A, Itoh H, Kawao N, Kuroda R, Sekiguchi F, Masuko T, Iwata K, Ogawa A. Activation of trigeminal nociceptive neurons by parotid PAR-2 activation in rats. Neuroreport 15: 1617–1621, 2004. doi: 10.1097/01.wnr.0000134991.97051.6b. [DOI] [PubMed] [Google Scholar]
- 101. Patwardhan AM, Diogenes A, Berg KA, Fehrenbacher JC, Clarke WP, Akopian AN, Hargreaves KM. PAR-2 agonists activate trigeminal nociceptors and induce functional competence in the delta opioid receptor. Pain 125: 114–124, 2006. doi: 10.1016/j.pain.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 102. Hassler SN, Kume M, Mwirigi J, Ahmad A, Shiers S, Wangzhou A, Ray P, Belugin SN, Naik DK, Burton MD, Vagner J, Boitano S, Akopian AN, Dussor G, Price TJ. The cellular basis of protease activated receptor type 2 (PAR2) evoked mechanical and affective pain. JCI Insight 5: e137393, 2020. doi: 10.1172/jci.insight.137393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Ray P, Torck A, Quigley L, Wangzhou A, Neiman M, Rao C, Lam T, Kim JY, Kim TH, Zhang MQ, Dussor G, Price TJ. Comparative transcriptome profiling of the human and mouse dorsal root ganglia: an RNA-seq-based resource for pain and sensory neuroscience research. Pain 159: 1325–1345, 2018. doi: 10.1097/j.pain.0000000000001217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Usoskin D, Furlan A, Islam S, Abdo H, Lonnerberg P, Lou D, Hjerling-Leffler J, Haeggstrom J, Kharchenko O, Kharchenko PV, Linnarsson S, Ernfors P. Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat Neurosci 18: 145–153, 2015. doi: 10.1038/nn.3881. [DOI] [PubMed] [Google Scholar]
- 105. Li CL, Li KC, Wu D, Chen Y, Luo H, Zhao JR, Wang SS, Sun MM, Lu YJ, Zhong YQ, Hu XY, Hou R, Zhou BB, Bao L, Xiao HS, Zhang X. Somatosensory neuron types identified by high-coverage single-cell RNA-sequencing and functional heterogeneity. Cell Res 26: 967, 2016. doi: 10.1038/cr.2016.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Kim YS, Chu Y, Han L, Li M, Li Z, LaVinka PC, Sun S, Tang Z, Park K, Caterina MJ, Ren K, Dubner R, Wei F, Dong X. Central terminal sensitization of TRPV1 by descending serotonergic facilitation modulates chronic pain. Neuron 81: 873–887, 2014. doi: 10.1016/j.neuron.2013.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Boitano S, Hoffman J, Tillu DV, Asiedu MN, Zhang Z, Sherwood CL, Wang Y, Dong X, Price TJ, Vagner J. Development and evaluation of small peptidomimetic ligands to protease-activated receptor-2 (PAR2) through the use of lipid tethering. PLoS One 9: e99140, 2014. doi: 10.1371/journal.pone.0099140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Langford DJ, Bailey AL, Chanda ML, Clarke SE, Drummond TE, Echols S, Glick S, Ingrao J, Klassen-Ross T, Lacroix-Fralish ML, Matsumiya L, Sorge RE, Sotocinal SG, Tabaka JM, Wong D, van den Maagdenberg AM, Ferrari MD, Craig KD, Mogil JS. Coding of facial expressions of pain in the laboratory mouse. Nat Methods 7: 447–449, 2010. doi: 10.1038/nmeth.1455. [DOI] [PubMed] [Google Scholar]
- 109. Liu Q, Weng HJ, Patel KN, Tang Z, Bai H, Steinhoff M, Dong X. The distinct roles of two GPCRs, MrgprC11 and PAR2, in itch and hyperalgesia. Sci Signal 4: ra45, 2011. doi: 10.1126/scisignal.2001925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Vergnolle N, Derian CK, D’Andrea MR, Steinhoff M, Andrade-Gordon P. Characterization of thrombin-induced leukocyte rolling and adherence: a potential proinflammatory role for proteinase-activated receptor-4. J Immunol 169: 1467–1473, 2002. doi: 10.4049/jimmunol.169.3.1467. [DOI] [PubMed] [Google Scholar]
- 111. Augé C, Balz-Hara D, Steinhoff M, Vergnolle N, Cenac N. Protease-activated receptor-4 (PAR4): a role as inhibitor of visceral pain and hypersensitivity. Neurogastroenterol Motil 21: 1189–e1107, 2009. doi: 10.1111/j.1365-2982.2009.01310.x. [DOI] [PubMed] [Google Scholar]
- 112. Linden DR, Manning BP, Bunnett NW, Mawe GM. Agonists of proteinase-activated receptor 2 excite guinea pig ileal myenteric neurons. Eur J Pharmacol 431: 311–314, 2001. doi: 10.1016/S0014-2999(01)01447-9. [DOI] [PubMed] [Google Scholar]
- 113. Bornstein JC, Furness JB, Kunze WA. Electrophysiological characterization of myenteric neurons: how do classification schemes relate? J Auton Nerv Syst 48: 1–15, 1994. doi: 10.1016/0165-1838(94)90155-4. [DOI] [PubMed] [Google Scholar]
- 114. Wood JD. Application of classification schemes to the enteric nervous system. J Auton Nerv Syst 48: 17–29, 1994. doi: 10.1016/0165-1838(94)90156-2. [DOI] [PubMed] [Google Scholar]
- 115. Johansson U, Lawson C, Dabare M, Syndercombe-Court D, Newland AC, Howells GL, Macey MG. Human peripheral blood monocytes express protease receptor-2 and respond to receptor activation by production of IL-6, IL-8, and IL-1{beta}. J Leukoc Biol 78: 967–975, 2005. doi: 10.1189/jlb.0704422. [DOI] [PubMed] [Google Scholar]
- 116. Mari B, Imbert V, Belhacene N, Far DF, Peyron JF, Pouyssegur J, Van Obberghen-Schilling E, Rossi B, Auberger P. Thrombin and thrombin receptor agonist peptide induce early events of T cell activation and synergize with TCR cross-linking for CD69 expression and interleukin 2 production. J Biol Chem 269: 8517–8523, 1994. doi: 10.1016/S0021-9258(17)37225-3. [DOI] [PubMed] [Google Scholar]
- 117. Gordon JR, Zhang X, Stevenson K, Cosford K. Thrombin induces IL-6 but not TNFalpha secretion by mouse mast cells: threshold-level thrombin receptor and very low level FcepsilonRI signaling synergistically enhance IL-6 secretion. Cell Immunol 205: 128–135, 2000. doi: 10.1006/cimm.2000.1714. [DOI] [PubMed] [Google Scholar]
- 118. Strukova SM, Chistov IV, Umarova BA, Dugina TN, Storozhevykh TP, Pinelis VG, Glusa E. Modulation of mast cell activity by a peptide agonist of the thrombin receptor: role of nitric oxide. Biochemistry (Mosc) 64: 658–664, 1999. [PubMed] [Google Scholar]
- 119. Hurley A, Smith M, Karpova T, Hasley RB, Belkina N, Shaw S, Balenga N, Druey KM, Nickel E, Packard B, Imamichi H, Hu Z, Follmann D, McNally J, Higgins J, Sneller M, Lane HC, Catalfamo M. Enhanced effector function of CD8(+) T cells from healthy controls and HIV-infected patients occurs through thrombin activation of protease-activated receptor 1. J Infect Dis 207: 638–650, 2013. doi: 10.1093/infdis/jis730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Jenkins AL, Howells GL, Scott E, Le Bonniec BF, Curtis MA, Stone SR. The response to thrombin of human neutrophils: evidence for two novel receptors. J Cell Sci 108: 3059–3066, 1995. doi: 10.1242/jcs.108.9.3059. [DOI] [PubMed] [Google Scholar]
- 121. Moormann C, Artuc M, Pohl E, Varga G, Buddenkotte J, Vergnolle N, Brehler R, Henz BM, Schneider SW, Luger TA, Steinhoff M. Functional characterization and expression analysis of the proteinase-activated receptor-2 in human cutaneous mast cells. J Invest Dermatol 126: 746–755, 2006. doi: 10.1038/sj.jid.5700169. [DOI] [PubMed] [Google Scholar]
- 122. Corvera CU, Déry O, Mcconalogue K, Böhm SK, Khitin LM, Caughey GH, Payan DG, Bunnett NW. Mast cell tryptase regulates rat colonic myocytes through proteinase-activated receptor 2. J Clin Invest 100: 1383–1393, 1997. doi: 10.1172/JCI119658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. D’Andrea MR, Rogahn CJ, Andrade-Gordon P. Localization of protease-activated receptors-1 and -2 in human mast cells: indications for an amplified mast cell degranulation cascade. Biotech Histochem 75: 85–90, 2000. doi: 10.3109/10520290009064152. [DOI] [PubMed] [Google Scholar]
- 124. Miike S, Mcwilliam AS, Kita H. Trypsin induces activation and inflammatory mediator release from human eosinophils through protease-activated receptor-2. J Immunol 167: 6615–6622, 2001. doi: 10.4049/jimmunol.167.11.6615. [DOI] [PubMed] [Google Scholar]
- 125. Temkin V, Kantor B, Weg V, Hartman ML, Levi-Schaffer F. Tryptase activates the mitogen-activated protein kinase/activator protein-1 pathway in human peripheral blood eosinophils, causing cytokine production and release. J Immunol 169: 2662–2669, 2002. doi: 10.4049/jimmunol.169.5.2662. [DOI] [PubMed] [Google Scholar]
- 126. Bolton SJ, McNulty CA, Thomas RJ, Hewitt CR, Wardlaw AJ. Expression of and functional responses to protease-activated receptors on human eosinophils. J Leukoc Biol 74: 60–68, 2003. doi: 10.1189/jlb.0702351. [DOI] [PubMed] [Google Scholar]
- 127. Howells GL, Macey MG, Chinni C, Hou L, Fox MT, Harriott P, Stone SR. Proteinase-activated receptor-2: expression by human neutrophils. J Cell Sci 110: 881–887, 1997. doi: 10.1242/jcs.110.7.881. [DOI] [PubMed] [Google Scholar]
- 128. Lopez ML, Soriano-Sarabia N, Bruges G, Marquez ME, Preissner KT, Schmitz ML, Hackstein H. Expression pattern of protease activated receptors in lymphoid cells. Cell Immunol 288: 47–52, 2014. doi: 10.1016/j.cellimm.2014.02.004. [DOI] [PubMed] [Google Scholar]
- 129. Mari B, Guerin S, Far DF, Breitmayer JP, Belhacene N, Peyron JF, Rossi B, Auberger P. Thrombin and trypsin-induced Ca(2+) mobilization in human T cell lines through interaction with different protease-activated receptors. FASEB J 10: 309–316, 1996. doi: 10.1096/fasebj.10.2.8641564. [DOI] [PubMed] [Google Scholar]
- 130. Joyce DE, Chen Y, Erger RA, Koretzky GA, Lentz SR. Functional interactions between the thrombin receptor and the T-cell antigen receptor in human T-cell lines. Blood 90: 1893–1901, 1997. doi: 10.1182/blood.V90.5.1893. [DOI] [PubMed] [Google Scholar]
- 131. Bushell TJ, Cunningham MR, McIntosh KA, Moudio S, Plevin R. Protease-activated receptor 2: are common functions in glial and immune cells linked to inflammation-related CNS disorders? Curr Drug Targets 17: 1861–1870, 2016. doi: 10.2174/1389450117666151209115232. [DOI] [PubMed] [Google Scholar]
- 132. Vergnolle N, Hollenberg MD, Sharkey KA, Wallace JL. Characterization of the inflammatory response to proteinase-activated receptor-2 (PAR2)-activating peptides in the rat paw. Br J Pharmacol 127: 1083–1090, 1999. doi: 10.1038/sj.bjp.0702634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Williams JD, Topley N, Alobaidi HM, Harber MJ. Activation of human polymorphonuclear leucocytes by particulate zymosan is related to both its major carbohydrate components: glucan and mannan. Immunology 58: 117–124, 1986. [PMC free article] [PubMed] [Google Scholar]
- 134. Lim SY, Tennant GM, Kennedy S, Wainwright CL, Kane KA. Activation of mouse protease-activated receptor-2 induces lymphocyte adhesion and generation of reactive oxygen species. Br J Pharmacol 149: 591–599, 2006. doi: 10.1038/sj.bjp.0706905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Colognato R, Slupsky JR, Jendrach M, Burysek L, Syrovets T, Simmet T. Differential expression and regulation of protease-activated receptors in human peripheral monocytes and monocyte-derived antigen-presenting cells. Blood 102: 2645–2652, 2003. doi: 10.1182/blood-2002-08-2497. [DOI] [PubMed] [Google Scholar]
- 136. Houle S, Papez MD, Ferazzini M, Hollenberg MD, Vergnolle N. Neutrophils and the kallikrein-kinin system in proteinase-activated receptor 4-mediated inflammation in rodents. Br J Pharmacol 146: 670–678, 2005. doi: 10.1038/sj.bjp.0706371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Kaufmann R, Schafberg H, Nowak G. Proteinase-activated receptor-2-mediated signaling and inhibition of DNA synthesis in human pancreatic cancer cells. Int J Pancreatol 24: 97–102, 1998. doi: 10.1007/BF02788566. [DOI] [PubMed] [Google Scholar]
- 138. Even-Ram S, Uziely B, Cohen P, Grisaru-Granovsky S, Maoz M, Ginzburg Y, Reich R, Vlodavsky I, Bar-Shavit R. Thrombin receptor overexpression in malignant and physiological invasion processes. Nat Med 4: 909–914, 1998. doi: 10.1038/nm0898-909. [DOI] [PubMed] [Google Scholar]
- 139. Arakaki AK, Pan WA, Trejo JA. GPCRs in cancer: protease-activated receptors, endocytic adaptors and signaling. Int J Mol Sci 19: 1886–1824, 2018. doi: 10.3390/ijms19071886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Morris DR, Ding Y, Ricks TK, Gullapalli A, Wolfe BL, Trejo J. Protease-activated receptor-2 is essential for factor VIIa and Xa-induced signaling, migration, and invasion of breast cancer cells. Cancer Res 66: 307–314, 2006. doi: 10.1158/0008-5472.CAN-05-1735. [DOI] [PubMed] [Google Scholar]
- 141. Schmitt-Graff A, Desmouliere A, Gabbiani G. Heterogeneity of myofibroblast phenotypic features: an example of fibroblastic cell plasticity. Virchows Arch 425: 3–24, 1994. [DOI] [PubMed] [Google Scholar]
- 142. Mosnier LO, Sinha R, Burnier L, Bouwens E, Griffin JH. Biased agonism of protease-activated receptor 1 by activated protein C caused by noncanonical cleavage at Arg46. Blood 120: 5237–5246, 2012. doi: 10.1182/blood-2012-08-452169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Trivedi V, Boire A, Tchernychev B, Kaneider NC, Leger AJ, O’Callaghan K, Covic L, Kuliopulos A. platelet matrix metalloprotease-1 mediates thrombogenesis by activating PAR1 at a cryptic ligand site. Cell 137: 332–343, 2009. doi: 10.1016/j.cell.2009.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Mihara K, Ramachandran R, Renaux B, Saifeddine M, Hollenberg MD. Neutrophil elastase and proteinase-3 trigger G protein-biased signaling through proteinase-activated receptor-1 (PAR1). J Biol Chem 288: 32979–32990, 2013. doi: 10.1074/jbc.M113.483123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Zhao P, Lieu T, Barlow N, Metcalf M, Veldhuis NA, Jensen DD, Kocan M, Sostegni S, Haerteis S, Baraznenok V, Henderson I, Lindström E, Guerrero-Alba R, Valdez-Morales EE, Liedtke W, McIntyre P, Vanner SJ, Korbmacher C, Bunnett NW. Cathepsin S causes inflammatory pain via biased agonism of PAR2 and TRPV4. J Biol Chem 289: 27215–27234, 2014. doi: 10.1074/jbc.M114.599712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Ramachandran R, Mihara K, Chung H, Renaux B, Lau CS, Muruve DA, Defea KA, Bouvier M, Hollenberg MD. Neutrophil elastase acts as a biased agonist for proteinase-activated receptor-2 (PAR2). J Biol Chem 286: 24638–24648, 2011. doi: 10.1074/jbc.M110.201988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Zhao P, Lieu T, Barlow N, Sostegni S, Haerteis S, Korbmacher C, Liedtke W, Jimenez-Vargas NN, Vanner SJ, Bunnett NW. Neutrophil elastase activates protease-activated receptor-2 (PAR2) and transient receptor potential vanilloid 4 (TRPV4) to cause inflammation and pain. J Biol Chem 290: 13875–13887, 2015. doi: 10.1074/jbc.M115.642736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Zhao P, Pattison LA, Jensen DD, Jimenez-Vargas NN, Latorre R, Lieu T, Jaramillo JO, Lopez-Lopez C, Poole DP, Vanner SJ, Schmidt BL, Bunnett NW. Protein kinase D and Gβγ mediate sustained nociceptive signaling by biased agonists of protease-activated receptor-2. J Biol Chem 294: 10649–10662, 2019. doi: 10.1074/jbc.RA118.006935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Tu NH, Jensen DD, Anderson BM, Chen E, Jimenez-Vargas NN, Scheff NN, Inoue K, Tran HD, Dolan JC, Meek TA, Hollenberg MD, Liu CZ, Vanner SJ, Janal MN, Bunnett NW, Edgington-Mitchell LE, Schmidt BL. Legumain induces oral cancer pain by biased agonism of protease-activated receptor-2. J Neurosci 41: 193–210, 2021. doi: 10.1523/JNEUROSCI.1211-20.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Loew D, Perrault C, Morales M, Moog S, Ravanat C, Schuhler S, Arcone R, Pietropaolo C, Cazenave JP, Van Dorsselaer A, Lanza F. Proteolysis of the exodomain of recombinant protease-activated receptors: prediction of receptor activation or inactivation by MALDI mass spectrometry. Biochemistry 39: 10812–10822, 2000. doi: 10.1021/bi0003341. [DOI] [PubMed] [Google Scholar]
- 151. Dulon S, Candé C, Bunnett NW, Hollenberg MD, Chignard M, Pidard D. Proteinase-activated receptor-2 and human lung epithelial cells. Am J Respir Cell Mol Biol 28: 339–346, 2003. doi: 10.1165/rcmb.4908. [DOI] [PubMed] [Google Scholar]
- 152. Ender M, Andreoni F, Zinkernagel AS, Schuepbach RA. Streptococcal SpeB cleaved PAR-1 suppresses ERK phosphorylation and blunts thrombin-induced platelet aggregation. PLoS One 8: e81298, 2013. doi: 10.1371/journal.pone.0081298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Dulon S, Leduc D, Cottrell GS, D’Alayer J, Hansen KK, Bunnett NW, Hollenberg MD, Pidard D, Chignard M. Pseudomonas aeruginosa elastase disables proteinase-activated receptor 2 in respiratory epithelial cells. Am J Respir Cell Mol Biol 32: 411–419, 2005. doi: 10.1165/rcmb.2004-0274OC. [DOI] [PubMed] [Google Scholar]
- 154. Nanevicz T, Ishii M, Wang L, Chen M, Chen J, Turck CW, Cohen FE, Coughlin SR. Mechanisms of thrombin receptor agonist specificity. J Biol Chem 270: 21619–21625, 1995. doi: 10.1074/jbc.270.37.21619. [DOI] [PubMed] [Google Scholar]
- 155. Vassallo RR, Kieber-Emmons T, Cichowski K, Brass LF. Structure-function relationships in the activation of platelet thrombin receptors by receptor-derived peptides. J Biol Chem 267: 6081–6085, 1992. doi: 10.1016/S0021-9258(18)42664-6. [DOI] [PubMed] [Google Scholar]
- 156. Chao BH, Kalkunte S, Maraganore JM, Stone SR. Essential groups in synthetic agonist peptides for activation of the platelet thrombin receptor. Biochemistry 31: 6175–6178, 1992. doi: 10.1021/bi00142a001. [DOI] [PubMed] [Google Scholar]
- 157. Hui KY, Jakubowski JA, Wyss VL, Angleton EL. Minimal sequence requirement of thrombin receptor agonist peptide. Biochem Biophys Res Commun 184: 790–796, 1992. doi: 10.1016/0006-291X(92)90659-9. [DOI] [PubMed] [Google Scholar]
- 158. Maryanoff BE, Santulli RJ, Mccomsey DF, Hoekstra WJ, Hoey K, Smith CE, Addo M, Darrow AL, Andrade-Gordon P. Protease-activated receptor-2 (PAR-2): structure-function study of receptor activation by diverse peptides related to tethered-ligand epitopes. Arch Biochem Biophys 386: 195–204, 2001. doi: 10.1006/abbi.2000.2207. [DOI] [PubMed] [Google Scholar]
- 159. Hansen KK, Saifeddine M, Hollenberg MD. Tethered ligand-derived peptides of proteinase-activated receptor 3 (PAR3) activate PAR1 and PAR2 in Jurkat T cells. Immunology 112: 183–190, 2004. doi: 10.1111/j.1365-2567.2004.01870.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Faruqi TR, Weiss EJ, Shapiro MJ, Huang W, Coughlin SR. Structure-function analysis of protease-activated receptor 4 tethered ligand peptides. Determinants of specificity and utility in assays of receptor function. J Biol Chem 275: 19728–19734, 2000. doi: 10.1074/jbc.M909960199. [DOI] [PubMed] [Google Scholar]
- 161. Ferrell WR, Lockhart JC, Kelso EB, Dunning L, Plevin R, Meek SE, Smith AJ, Hunter GD, McLean JS, McGarry F, Ramage R, Jiang L, Kanke T, Kawagoe J. Essential role for proteinase-activated receptor-2 in arthritis. J Clin Invest 111: 35–41, 2003. doi: 10.1172/JCI16913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Mcguire JJ, Saifeddine M, Triggle CR, Sun K, Hollenberg MD. 2-Furoyl-LIGRLO-amide: a potent and selective proteinase-activated receptor 2 agonist. J Pharmacol Exp Ther 309: 1124–1131, 2004. doi: 10.1124/jpet.103.064584. [DOI] [PubMed] [Google Scholar]
- 163. Andersen H, Greenberg DL, Fujikawa K, Xu W, Chung DW, Davie EW. Protease-activated receptor 1 is the primary mediator of thrombin-stimulated platelet procoagulant activity. Proc Natl Acad Sci U S A 96: 11189–11193, 1999. doi: 10.1073/pnas.96.20.11189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Hollenberg MD, Saifeddine M, Al-Ani B, Gui Y. Proteinase-activated receptor 4 (PAR4): action of PAR4-activating peptides in vascular and gastric tissue and lack of cross-reactivity with PAR1 and PAR2. Can J Physiol Pharmacol 77: 458–464, 1999. doi: 10.1139/cjpp-77-6-458, . [DOI] [PubMed] [Google Scholar]
- 165. Blackhart BD, Emilsson K, Nguyen D, Teng W, Martelli AJ, Nystedt S, Sundelin J, Scarborough RM. Ligand cross-reactivity within the protease-activated receptor family. J Biol Chem 271: 16466–16471, 1996. doi: 10.1074/jbc.271.28.16466. [DOI] [PubMed] [Google Scholar]
- 166. Lin H, Trejo J. Transactivation of the PAR1-PAR2 heterodimer by thrombin elicits β-arrestin-mediated endosomal signaling. J Biol Chem 288: 11203–11215, 2013. doi: 10.1074/jbc.M112.439950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Arachiche A, Mumaw MM, De La Fuente M, Nieman MT. Protease-activated receptor 1 (PAR1) and PAR4 heterodimers are required for PAR1-enhanced cleavage of PAR4 by α-thrombin. J Biol Chem 288: 32553–32562, 2013. doi: 10.1074/jbc.M113.472373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Ostrowska E, Reiser G. The protease-activated receptor-3 (PAR-3) can signal autonomously to induce interleukin-8 release. Cell Mol Life Sci 65: 970–981, 2008. doi: 10.1007/s00018-008-7555-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Bar-Shavit R, Sabbah V, Lampugnani M, Marchisio P, Fenton J 2nd, Vlodavsky I, Dejana E. An Arg-Gly-Asp sequence within thrombin promotes endothelial cell adhesion. J Cell Biol 112: 335–344, 1991. doi: 10.1083/jcb.112.2.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Kaufmann R, Schulze B, Krause G, Mayr L, Settmacher U, Henklein P. Proteinase-activated receptors (PARs)–the PAR3 Neo-N-terminal peptide TFRGAP interacts with PAR1. Regul Pept 125: 61–66, 2005. doi: 10.1016/j.regpep.2004.07.032. [DOI] [PubMed] [Google Scholar]
- 171. Mclaughlin JN, Patterson MM, Malik AB. Protease-activated receptor-3 (PAR3) regulates PAR1 signaling by receptor dimerization. Proc Natl Acad Sci U S A 104: 5662–5667, 2007. doi: 10.1073/pnas.0700763104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Bae JS, Yang L, Manithody C, Rezaie AR. The ligand occupancy of endothelial protein C receptor switches the protease-activated receptor 1-dependent signaling specificity of thrombin from a permeability-enhancing to a barrier-protective response in endothelial cells. Blood 110: 3909–3916, 2007. doi: 10.1182/blood-2007-06-096651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Burnier L, Mosnier LO. Novel mechanisms for activated protein C cytoprotective activities involving noncanonical activation of protease-activated receptor 3. Blood 122: 807–816, 2013. doi: 10.1182/blood-2013-03-488957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Schuepbach RA, Riewald M. Coagulation factor Xa cleaves protease-activated receptor-1 and mediates signaling dependent on binding to the endothelial protein C receptor. J Thromb Haemost 8: 379–388, 2010. doi: 10.1111/j.1538-7836.2009.03682.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Kondreddy V, Wang J, Keshava S, Esmon CT, Rao LV, Pendurthi UR. Factor VIIa induces anti-inflammatory signaling via EPCR and PAR1. Blood 131: 2379–2392, 2018. doi: 10.1182/blood-2017-10-813527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Majumdar M, Tarui T, Shi B, Akakura N, Ruf W, Takada Y. Plasmin-induced migration requires signaling through protease-activated receptor 1 and integrin α9β1. J Biol Chem 279: 37528–37534, 2004. doi: 10.1074/jbc.M401372200. [DOI] [PubMed] [Google Scholar]
- 177. Sebastiano M, Momi S, Falcinelli E, Bury L, Hoylaerts MF, Gresele P. A novel mechanism regulating human platelet activation by MMP-2–mediated PAR1 biased signaling. Blood 129: 883–895, 2017. doi: 10.1182/blood-2016-06-724245. [DOI] [PubMed] [Google Scholar]
- 178. Zhu L, Wang X, Wu J, Mao D, Xu Z, He Z, Yu A. Cooperation of protease-activated receptor 1 and integrin ανβ5 in thrombin-mediated lung cancer cell invasion. Oncol Rep 28: 553–560, 2012. doi: 10.3892/or.2012.1851. [DOI] [PubMed] [Google Scholar]
- 179. Lin H, Liu AP, Smith TH, Trejo J. Cofactoring and dimerization of proteinase-activated receptors. Pharmacol Rev 65: 1198–1213, 2013. doi: 10.1124/pr.111.004747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Jaffré F, Friedman AE, Hu Z, Mackman N, Blaxall BC. β-Adrenergic receptor stimulation transactivates protease-activated receptor 1 via matrix metalloproteinase 13 in cardiac cells. Circulation 125: 2993–3003, 2012. doi: 10.1161/CIRCULATIONAHA.111.066787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Snead AN, Insel PA. Defining the cellular repertoire of GPCRs identifies a profibrotic role for the most highly expressed receptor, protease-activated receptor 1, in cardiac fibroblasts. FASEB J 26: 4540–4547, 2012. doi: 10.1096/fj.12-213496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Smith TH, Li JG, Dores MR, Trejo J. Protease-activated receptor-4 and purinergic receptor P2Y12 dimerize, co-internalize, and activate Akt signaling via endosomal recruitment of β-arrestin. J Biol Chem 292: 13867–13878, 2017. doi: 10.1074/jbc.M117.782359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Gao X, Cheng YH, Enten GA, DeSantis AJ, Gaponenko V, Majetschak M. Regulation of the thrombin/protease-activated receptor 1 axis by chemokine (CXC motif) receptor 4. J Biol Chem 295: 14893–14905, 2020. doi: 10.1074/jbc.RA120.015355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. McCoy KL, Traynelis SF, Hepler JR. PAR1 and PAR2 couple to overlapping and distinct sets of G proteins and linked signaling pathways to differentially regulate cell physiology. Mol Pharmacol 77: 1005–1015, 2010. doi: 10.1124/mol.109.062018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Vellani V, Kinsey AM, Prandini M, Hechtfischer SC, Reeh P, Magherini PC, Giacomoni C, McNaughton PA. Protease activated receptors 1 and 4 sensitize TRPV1 in nociceptive neurones. Mol Pain 6: 61, 2010. doi: 10.1186/1744-8069-6-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Swift S, Leger AJ, Talavera J, Zhang L, Bohm A, Kuliopulos A. Role of the PAR1 receptor 8th helix in signaling. J Biol Chem 281: 4109–4116, 2006. doi: 10.1074/jbc.M509525200. [DOI] [PubMed] [Google Scholar]
- 187. Thibeault PE, Ramachandran R. Role of the helix-8 and C-terminal tail in regulating proteinase activated receptor 2 signaling. ACS Pharmacol Transl Sci 3: 868–882, 2020. doi: 10.1021/acsptsci.0c00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Kim YV, Di Cello F, Hillaire C, Kim KS. Differential Ca2+ signaling by thrombin and protease-activated receptor-1-activating peptide in human brain microvascular endothelial cells. Am J Physiol Cell Physiol 286: C31–C42, 2004. doi: 10.1152/ajpcell.00157.2003. [DOI] [PubMed] [Google Scholar]
- 189. Tiruppathi C, Freichel M, Vogel SM, Paria BC, Mehta D, Flockerzi V, Malik AB. Impairment of store-operated Ca2+ entry in TRPC4-/- mice interferes with increase in lung microvascular permeability. Circ Res 91: 70–76, 2002. doi: 10.1161/01.RES.0000023391.40106.A8. [DOI] [PubMed] [Google Scholar]
- 190. Mclaughlin JN, Shen L, Holinstat M, Brooks JD, Dibenedetto E, Hamm HE. Functional selectivity of G protein signaling by agonist peptides and thrombin for the protease-activated receptor-1. J Biol Chem 280: 25048–25059, 2005. doi: 10.1074/jbc.M414090200. [DOI] [PubMed] [Google Scholar]
- 191. Vaidyula VR, Rao AK. Role of Galphaq and phospholipase C-beta2 in human platelets activation by thrombin receptors PAR1 and PAR4: studies in human platelets deficient in Galphaq and phospholipase C-beta2. Br J Haematol 121: 491–496, 2003. doi: 10.1046/j.1365-2141.2003.04296.x. [DOI] [PubMed] [Google Scholar]
- 192. Tourdot BE, Conaway S, Niisuke K, Edelstein LC, Bray PF, Holinstat M. Mechanism of race-dependent platelet activation through the protease-activated receptor-4 and Gq signaling axis. Arterioscler Thromb Vasc Biol 34: 2644–2650, 2014. doi: 10.1161/ATVBAHA.114.304249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Edelstein LC, Simon LM, Lindsay CR, Kong X, Teruel-Montoya R, Tourdot BE, Chen ES, Ma L, Coughlin S, Nieman M, Holinstat M, Shaw CA, Bray PF. Common variants in the human platelet PAR4 thrombin receptor alter platelet function and differ by race. Blood 124: 3450–3458, 2014. doi: 10.1182/blood-2014-04-572479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Shapiro MJ, Weiss EJ, Faruqi TR, Coughlin SR. Protease-activated receptors 1 and 4 are shut off with distinct kinetics after activation by thrombin. J Biol Chem 275: 25216–25221, 2000. doi: 10.1074/jbc.M004589200. [DOI] [PubMed] [Google Scholar]
- 195. Covic L, Gresser AL, Kuliopulos A. Biphasic kinetics of activation and signaling for PAR1 and PAR4 thrombin receptors in platelets. Biochemistry 39: 5458–5467, 2000. doi: 10.1021/bi9927078. [DOI] [PubMed] [Google Scholar]
- 196. Al-Ani B, Wijesuriya SJ, Hollenberg MD. Proteinase-activated receptor 2: differential activation of the receptor by tethered ligand and soluble peptide analogs. J Pharmacol Exp Ther 302: 1046–1054, 2002. doi: 10.1124/jpet.302.3.1046. [DOI] [PubMed] [Google Scholar]
- 197. Thibeault PE, LeSarge JC, Arends D, Fernandes M, Chidiac P, Stathopulos PB, Luyt LG, Ramachandran R. Molecular basis for activation and biased signaling at the thrombin-activated GPCR proteinase activated receptor-4 (PAR4). J Biol Chem 295: 2520–2540, 2020. doi: 10.1074/jbc.RA119.011461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Bono F, Schaeffer P, Hérault JP, Michaux C, Nestor AL, Guillemot JC, Herbert JM. Factor Xa activates endothelial cells by a receptor cascade between EPR-1 and PAR-2. Arterioscler Thromb Vasc Biol 20: e107–e112, 2000. doi: 10.1161/01.atv.20.11.e107. [DOI] [PubMed] [Google Scholar]
- 199. Daubie V, Cauwenberghs S, Senden NH, Pochet R, Lindhout T, Buurman WA, Heemskerk JW. Factor Xa and thrombin evoke additive calcium and proinflammatory responses in endothelial cells subjected to coagulation. Biochim Biophys Acta 1763: 860–869, 2006. doi: 10.1016/j.bbamcr.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 200. Poole DP, Amadesi S, Veldhuis NA, Abogadie FC, Lieu T, Darby W, Liedtke W, Lew MJ, Mcintyre P, Bunnett NW. Protease-activated receptor 2 (PAR2) protein and transient receptor potential vanilloid 4 (TRPV4) protein coupling is required for sustained inflammatory signaling. J Biol Chem 288: 5790–5802, 2013. doi: 10.1074/jbc.M112.438184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Defea KA, Zalevsky J, Thoma MS, Déry O, Mullins RD, Bunnett NW. βArrestin–dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated Erk1/2. J Cell Biol 148: 1267–811281, 2000. doi: 10.1083/jcb.148.6.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Ramachandran R, Mihara K, Mathur M, Rochdi MD, Bouvier M, Defea K, Hollenberg MD. Agonist-biased signaling via proteinase activated receptor-2: differential activation of calcium and mitogen-activated protein kinase pathways. Mol Pharmacol 76: 791–801, 2009. doi: 10.1124/mol.109.055509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Jimenez-Vargas NN, Pattison LA, Zhao P, Lieu T, Latorre R, Jensen DD, Castro J, Aurelio L, Le GT, Flynn B, Herenbrink CK, Yeatman HR, Edgington-Mitchell L, Porter CJ, Halls ML, Canals M, Veldhuis NA, Poole DP, McLean P, Hicks GA, Scheff N, Chen E, Bhattacharya A, Schmidt BL, Brierley SM, Vanner SJ, Bunnett NW. Protease-activated receptor-2 in endosomes signals persistent pain of irritable bowel syndrome. Proc Natl Acad Sci U S A 115: E7438–E7447, 2018. doi: 10.1073/pnas.1721891115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Ge L, Ly Y, Hollenberg M, DeFea K. A beta-arrestin-dependent scaffold is associated with prolonged MAPK activation in pseudopodia during protease-activated receptor-2-induced chemotaxis. J Biol Chem 278: 34418–34426, 2003. doi: 10.1074/jbc.m300573200. [DOI] [PubMed] [Google Scholar]
- 205. Tillu DV, Hassler SN, Burgos-Vega CC, Quinn TL, Sorge RE, Dussor G, Boitano S, Vagner J, Price TJ. Protease-activated receptor 2 activation is sufficient to induce the transition to a chronic pain state. Pain 156: 859–867, 2015. doi: 10.1097/j.pain.0000000000000125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Madamanchi NR, Li S, Patterson C, Runge MS. Thrombin regulates vascular smooth muscle cell growth and heat shock proteins via the JAK-STAT pathway. J Biol Chem 276: 18915–18924, 2001. doi: 10.1074/jbc.M008802200. [DOI] [PubMed] [Google Scholar]
- 207. Kanda Y, Mizuno K, Kuroki Y, Watanabe Y. Thrombin-induced p38 mitogen-activated protein kinase activation is mediated by epidermal growth factor receptor transactivation pathway. Br J Pharmacol 132: 1657–1664, 2001. doi: 10.1038/sj.bjp.0703952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Grimsey NJ, Aguilar B, Smith TH, Le P, Soohoo AL, Puthenveedu MA, Nizet V, Trejo J. Ubiquitin plays an atypical role in GPCR-induced p38 MAP kinase activation on endosomes. J Cell Biol 210: 1117–1131, 2015. doi: 10.1083/jcb.201504007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Wang P, Defea KA. Protease-activated receptor-2 simultaneously directs β-arrestin-1-dependent inhibition and Gαq-dependent activation of phosphatidylinositol 3-kinase. Biochemistry 45: 9374–9385, 2006. doi: 10.1021/bi0602617. [DOI] [PubMed] [Google Scholar]
- 210. Das K, Prasad R, Roy S, Mukherjee A, Sen P. The protease activated receptor 2 promotes Rab5a mediated generation of pro-metastatic microvesicles. Sci Rep 8: 1886, 2018. doi: 10.1038/s41598-018-20099-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Iablokov V, Hirota CL, Peplowski MA, Ramachandran R, Mihara K, Hollenberg MD, Macnaughton WK. Proteinase-activated receptor 2 (PAR2) decreases apoptosis in colonic epithelial cells. J Biol Chem 289: 34366–34377, 2014. doi: 10.1074/jbc.M114.610485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Goel R, Phillips-Mason PJ, Raben DM, Baldassare JJ. α-Thrombin induces rapid and sustained Akt phosphorylation by β-arrestin1-dependent and -independent mechanisms, and only the sustained Akt phosphorylation is essential for G1 phase progression. J Biol Chem 277: 18640–18648, 2002. doi: 10.1074/jbc.M108995200. [DOI] [PubMed] [Google Scholar]
- 213. Ramachandran R, Mihara K, Thibeault P, Vanderboor CM, Petri B, Saifeddine M, Bouvier M, Hollenberg MD. Targeting a proteinase-activated receptor 4 (PAR4) carboxyl terminal motif to regulate platelet function. Mol Pharmacol 91: 287–295, 2017. doi: 10.1124/mol.116.106526. [DOI] [PubMed] [Google Scholar]
- 214. Falcinelli E, Guglielmini G, Torti M, Gresele P. Intraplatelet signaling mechanisms of the priming effect of matrix metalloproteinase-2 on platelet aggregation. J Thromb Haemost 3: 2526–2535, 2005. doi: 10.1111/j.1538-7836.2005.01614.x. [DOI] [PubMed] [Google Scholar]
- 215. Sinha RK, Wang Y, Zhao Z, Xu X, Burnier L, Gupta N, Fernández JA, Martin G, Kupriyanov S, Mosnier LO, Zlokovic BV, Griffin JH. PAR1 biased signaling is required for activated protein C in vivo benefits in sepsis and stroke. Blood 131: 1163–1171, 2018. doi: 10.1182/blood-2017-10-810895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Berger P, Perng DW, Thabrew H, Compton SJ, Cairns JA, McEuen AR, Marthan R, Tunon De Lara JM, Walls AF. Tryptase and agonists of PAR-2 induce the proliferation of human airway smooth muscle cells. J Appl Physiol (1985) 91: 1372–1379, 2001. doi: 10.1152/jappl.2001.91.3.1372. [DOI] [PubMed] [Google Scholar]
- 217. Sriwai W, Mahavadi S, Al-Shboul O, Grider JR, Murthy KS. Distinctive G protein-dependent signaling by protease-activated receptor 2 (PAR2) in smooth muscle: feedback inhibition of RhoA by cAMP-independent PKA. PLoS One 8: e66743, 2013. doi: 10.1371/journal.pone.0066743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Ayoub MA, Trinquet E, Pfleger KD, Pin JP. Differential association modes of the thrombin receptor PAR1 with Galphai1, Galpha12, and beta-arrestin 1. FASEB J 24: 3522–3535, 2010. doi: 10.1096/fj.10-154997. [DOI] [PubMed] [Google Scholar]
- 219. Kim S, Foster C, Lecchi A, Quinton TM, Prosser DM, Jin J, Cattaneo M, Kunapuli SP. Protease-activated receptors 1 and 4 do not stimulate Gi signaling pathways in the absence of secreted ADP and cause human platelet aggregation independently of Gisignaling. Blood 99: 3629–3636, 2002. doi: 10.1182/blood.V99.10.3629. [DOI] [PubMed] [Google Scholar]
- 220. Offermanns S, Laugwitz KL, Spicher K, Schultz G. G proteins of the G12 family are activated via thromboxane A2 and thrombin receptors in human platelets. Proc Natl Acad Sci U S A 91: 504–508, 1994. doi: 10.1073/pnas.91.2.504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Garcia JGN, Davis HW, Patterson CE. Regulation of endothelial cell gap formation and barrier dysfunction: role of myosin light chain phosphorylation. J Cell Physiol 163: 510–522, 1995. doi: 10.1002/jcp.1041630311. [DOI] [PubMed] [Google Scholar]
- 222. Majumdar M, Seasholtz TM, Buckmaster C, Toksoz D, Brown JH. A Rho exchange factor mediates thrombin and Gα12-induced cytoskeletal responses. J Biol Chem 274: 26815–26821, 1999. doi: 10.1074/jbc.274.38.26815. [DOI] [PubMed] [Google Scholar]
- 223. Mehta D, Rahman A, Malik AB. Protein kinase C-α signals Rho-guanine nucleotide dissociation inhibitor phosphorylation and Rho activation and regulates the endothelial cell barrier function. J Biol Chem 276: 22614–22620, 2001. doi: 10.1074/jbc.M101927200. [DOI] [PubMed] [Google Scholar]
- 224. Holinstat M, Mehta D, Kozasa T, Minshall R, Malik AB. Protein kinase Calpha-induced p115RhoGEF phosphorylation signals endothelial cytoskeletal rearrangement. J Biol Chem 278: 28793–28798, 2003. doi: 10.1074/jbc.M303900200. [DOI] [PubMed] [Google Scholar]
- 225. Zoudilova M, Kumar P, Ge L, Wang P, Bokoch GM, DeFea KA. Beta-arrestin-dependent regulation of the cofilin pathway downstream of protease-activated receptor-2. J Biol Chem 282: 20634–20646, 2007. doi: 10.1074/jbc.M701391200. [DOI] [PubMed] [Google Scholar]
- 226. Vanderboor CM, Thibeault PE, Nixon KC, Gros R, Kramer J, Ramachandran R. Proteinase-activated receptor 4 activation triggers cell membrane blebbing through RhoA and β-arrestin. Mol Pharmacol 97: 365–376, 2020. doi: 10.1124/mol.119.118232. [DOI] [PubMed] [Google Scholar]
- 227. Ayoub MA, Pin JP. Interaction of protease-activated receptor 2 with G proteins and β-arrestin 1 studied by bioluminescence resonance energy transfer. Front Endocrinol 4: 196, 2013. doi: 10.3389/fendo.2013.00196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Dowal L, Sim DS, Dilks JR, Blair P, Beaudry S, Denker BM, Koukos G, Kuliopulos A, Flaumenhaft R. Identification of an antithrombotic allosteric modulator that acts through helix 8 of PAR1. Proc Natl Acad Sci U S A 108: 2951–2956, 2011. doi: 10.1073/pnas.1014863108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Russo A, Soh UJ, Paing MM, Arora P, Trejo J. Caveolae are required for protease-selective signaling by protease-activated receptor-1. Proc Natl Acad Sci U S A 106: 6393–6397, 2009. doi: 10.1073/pnas.0810687106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Lin Y, Wozniak JA, Grimsey NJ, Girada S, Patwardhan A, Molinar-Inglis O, Smith TH, Lapek JD, Gonzalez DJ, Trejo JA. Phosphoproteomic analysis of protease-activated receptor-1 biased signaling reveals unique modulators of endothelial barrier function. Proc Natl Acad Sci USA 117: 5039–5048, 2020. doi: 10.1073/pnas.1917295117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Tressel SL, Kaneider NC, Kasuda S, Foley C, Koukos G, Austin K, Agarwal A, Covic L, Opal SM, Kuliopulos A. A matrix metalloprotease-PAR1 system regulates vascular integrity, systemic inflammation and death in sepsis. EMBO Mol Med 3: 370–384, 2011. doi: 10.1002/emmm.201100145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Austin KM, Nguyen N, Javid G, Covic L, Kuliopulos A. Noncanonical matrix metalloprotease-1-protease-activated receptor-1 signaling triggers vascular smooth muscle cell dedifferentiation and arterial stenosis. J Biol Chem 288: 23105–23115, 2013. doi: 10.1074/jbc.M113.467019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Boire A, Covic L, Agarwal A, Jacques S, Sherifi S, Kuliopulos A. PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell 120: 303–313, 2005. doi: 10.1016/j.cell.2004.12.018. [DOI] [PubMed] [Google Scholar]
- 234. Bae JS, Kim YU, Park MK, Rezaie AR. Concentration dependent dual effect of thrombin in endothelial cells via Par-1 and Pi3 kinase. J Cell Physiol 219: 744–751, 2009. doi: 10.1002/jcp.21718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Soto AG, Smith TH, Chen B, Bhattacharya S, Cordova IC, Kenakin T, Vaidehi N, Trejo J. N-linked glycosylation of protease-activated receptor-1 at extracellular loop 2 regulates G-protein signaling bias. Proc Natl Acad Sci U S A 112: E3600–E3608, 2015. doi: 10.1073/pnas.1508838112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Chen CH, Paing MM, Trejo J. Termination of protease-activated receptor-1 signaling by β-arrestins is independent of receptor phosphorylation. J Biol Chem 279: 10020–10031, 2004. doi: 10.1074/jbc.M310590200. [DOI] [PubMed] [Google Scholar]
- 237. Smith TH, Coronel LJ, Li JG, Dores MR, Nieman MT, Trejo J. Protease-activated receptor-4 signaling and trafficking is regulated by the clathrin adaptor protein complex-2 independent of β-arrestins. J Biol Chem 291: 18453–18464, 2016. doi: 10.1074/jbc.M116.729285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Ayoub MA, Maurel D, Binet V, Fink M, Prezeau L, Ansanay H, Pin JP. Real-time analysis of agonist-induced activation of protease-activated receptor 1/Galphai1 protein complex measured by bioluminescence resonance energy transfer in living cells. Mol Pharmacol 71: 1329–1340, 2007. doi: 10.1124/mol.106.030304. [DOI] [PubMed] [Google Scholar]
- 239. Thomsen AR, Jensen DD, Hicks GA, Bunnett NW. Therapeutic targeting of endosomal G-protein-coupled receptors. Trends Pharmacol Sci 39: 879–891, 2018. doi: 10.1016/j.tips.2018.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Trejo J, Altschuler Y, Fu HW, Mostov KE, Coughlin SR. Protease-activated receptor-1 down-regulation. J Biol Chem 275: 31255–31265, 2000. doi: 10.1074/jbc.M003770200. [DOI] [PubMed] [Google Scholar]
- 241. Déry O, Thoma MS, Wong H, Grady EF, Bunnett NW. Trafficking of proteinase-activated receptor-2 and β-arrestin-1 tagged with green fluorescent protein. J Biol Chem 274: 18524–18535, 1999. doi: 10.1074/jbc.274.26.18524. [DOI] [PubMed] [Google Scholar]
- 242. Ricks TK, Trejo J. Phosphorylation of protease-activated receptor-2 differentially regulates desensitization and internalization. J Biol Chem 284: 34444–34457, 2009. doi: 10.1074/jbc.M109.048942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Hollenberg MD, Renaux B, Hyun E, Houle S, Vergnolle N, Saifeddine M, Ramachandran R. Derivatized 2-furoyl-LIGRLO-amide, a versatile and selective probe for proteinase-activated receptor 2: binding and visualization. J Pharmacol Exp Ther 326: 453–462, 2008. doi: 10.1124/jpet.108.136432. [DOI] [PubMed] [Google Scholar]
- 244. Jacob C, Yang PC, Darmoul D, Amadesi S, Saito T, Cottrell GS, Coelho AM, Singh P, Grady EF, Perdue M, Bunnett NW. Mast cell tryptase controls paracellular permeability of the intestine. Role of protease-activated receptor 2 and beta-arrestins. J Biol Chem 280: 31936–31948, 2005. doi: 10.1074/jbc.M506338200. [DOI] [PubMed] [Google Scholar]
- 245. Bae JS, Yang L, Rezaie AR. Receptors of the protein C activation and activated protein C signaling pathways are colocalized in lipid rafts of endothelial cells. Proc Natl Acad Sci U S A 104: 2867–2872, 2007. doi: 10.1073/pnas.0611493104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Bae JS, Rezaie AR. Protease activated receptor 1 (PAR-1) activation by thrombin is protective in human pulmonary artery endothelial cells if endothelial protein C receptor is occupied by its natural ligand. Thromb Haemost 100: 101–109, 2008. doi: 10.1160/TH08-02-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Carlile-Klusacek M, Rizzo V. Endothelial cytoskeletal reorganization in response to PAR1 stimulation is mediated by membrane rafts but not caveolae. Am J Physiol Heart Circ Physiol 293: H366–H375, 2007. doi: 10.1152/ajpheart.01044.2006. [DOI] [PubMed] [Google Scholar]
- 248. Daub H, Weiss FU, Wallasch C, Ullrich A. Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature 379: 557–560, 1996. doi: 10.1038/379557a0. [DOI] [PubMed] [Google Scholar]
- 249. Sabri A, Short J, Guo J, Steinberg SF. Protease-activated receptor-1–mediated DNA synthesis in cardiac fibroblast is via epidermal growth factor receptor transactivation. Circ Res 91: 532–539, 2002. doi: 10.1161/01.RES.0000035242.96310.45. [DOI] [PubMed] [Google Scholar]
- 250. Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, Wallasch C, Ullrich A. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature 402: 884–888, 1999. doi: 10.1038/47260. [DOI] [PubMed] [Google Scholar]
- 251. Schauwienold D, Sastre AP, Genzel N, Schaefer M, Reusch HP. The transactivated epidermal growth factor receptor recruits Pyk2 to regulate Src kinase activity. J Biol Chem 283: 27748–27756, 2008. doi: 10.1074/jbc.M801431200. [DOI] [PubMed] [Google Scholar]
- 252. Darmoul D, Gratio V, Devaud H, Laburthe M. Protease-activated receptor 2 in colon cancer: trypsin-induced MAPK phosphorylation and cell proliferation are mediated by epidermal growth factor receptor transactivation. J Biol Chem 279: 20927–20934, 2004. doi: 10.1074/jbc.M401430200. [DOI] [PubMed] [Google Scholar]
- 253. Kawao N, Nagataki M, Nagasawa K, Kubo S, Cushing K, Wada T, Sekiguchi F, Ichida S, Hollenberg MD, Macnaughton WK, Nishikawa H, Kawabata A. Signal transduction for proteinase-activated receptor-2-triggered prostaglandin E2 formation in human lung epithelial cells. J Pharmacol Exp Ther 315: 576–589, 2005. doi: 10.1124/jpet.105.089490. [DOI] [PubMed] [Google Scholar]
- 254. van der Merwe JQ, Hollenberg M, MacNaughton WK. EGF receptor transactivation and MAP kinase mediate proteinase-activated receptor-2-induced chloride secretion in intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol 294: G441–G451, 2008. doi: 10.1152/ajpgi.00303.2007. [DOI] [PubMed] [Google Scholar]
- 255. Jiang Y, Zhuo X, Fu X, Wu Y, Mao C. Targeting PAR2 overcomes gefitinib resistance in non-small-cell lung cancer cells through inhibition of EGFR transactivation. Front Pharmacol 12: 625289, 2021. doi: 10.3389/fphar.2021.625289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Narita M, Usui A, Narita M, Niikura K, Nozaki H, Khotib J, Nagumo Y, Yajima Y, Suzuki T. Protease-activated receptor-1 and platelet-derived growth factor in spinal cord neurons are implicated in neuropathic pain after nerve injury. J Neurosci 2510000–10009, 2005. doi: 10.1523/JNEUROSCI.2507-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Siegbahn A, Johnell M, Nordin A, Aberg M, Velling T. TF/FVIIa transactivate PDGFRβ to regulate PDGF-BB–induced chemotaxis in different cell types. Arterioscler Thromb Vasc Biol 28: 135–141, 2008. doi: 10.1161/ATVBAHA.107.155754. [DOI] [PubMed] [Google Scholar]
- 258. Kaufmann R, Oettel C, Horn A, Halbhuber KJ, Eitner A, Krieg R, Katenkamp K, Henklein P, Westermann M, Bohmer FD, Ramachandran R, Saifeddine M, Hollenberg MD, Settmacher U. Met receptor tyrosine kinase transactivation is involved in proteinase-activated receptor-2-mediated hepatocellular carcinoma cell invasion. Carcinogenesis 30: 1487–1496, 2009. doi: 10.1093/carcin/bgp153. [DOI] [PubMed] [Google Scholar]
- 259. Chandrasekharan UM, Waitkus M, Kinney CM, Walters-Stewart A, Dicorleto PE. Synergistic induction of mitogen-activated protein kinase phosphatase-1 by thrombin and epidermal growth factor requires vascular endothelial growth factor receptor-2. Arterioscler Thromb Vasc Biol 30: 1983–1989, 2010. doi: 10.1161/ATVBAHA.110.212399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Dai Y, Moriyama T, Higashi T, Togashi K, Kobayashi K, Yamanaka H, Tominaga M, Noguchi K. Proteinase-activated receptor 2-mediated potentiation of transient receptor potential vanilloid subfamily 1 activity reveals a mechanism for proteinase-induced inflammatory pain. J Neurosci 24: 4293–4299, 2004. doi: 10.1523/JNEUROSCI.0454-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Amadesi S, Cottrell GS, Divino L, Chapman K, Grady EF, Bautista F, Karanjia R, Barajas-Lopez C, Vanner S, Vergnolle N, Bunnett NW. Protease-activated receptor 2 sensitizes TRPV1 by protein kinase Cepsilon- and A-dependent mechanisms in rats and mice. J Physiol 575: 555–571, 2006. doi: 10.1113/jphysiol.2006.111534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Gardell LR, Ma JN, Seitzberg JG, Knapp AE, Schiffer HH, Tabatabaei A, Davis CN, Owens M, Clemons B, Wong KK, Lund B, Nash NR, Gao Y, Lameh J, Schmelzer K, Olsson R, Burstein ES. Identification and characterization of novel small-molecule protease-activated receptor 2 agonists. J Pharmacol Exp Ther 327: 799–808, 2008. doi: 10.1124/jpet.108.142570. [DOI] [PubMed] [Google Scholar]
- 263. Chen Y, Yang C, Wang ZJ. Proteinase-activated receptor 2 sensitizes transient receptor potential vanilloid 1, transient receptor potential vanilloid 4, and transient receptor potential ankyrin 1 in paclitaxel-induced neuropathic pain. Neuroscience 193: 440–451, 2011. doi: 10.1016/j.neuroscience.2011.06.085. [DOI] [PubMed] [Google Scholar]
- 264. Grant AD, Cottrell GS, Amadesi S, Trevisani M, Nicoletti P, Materazzi S, Altier C, Cenac N, Zamponi GW, Bautista-Cruz F, Lopez CB, Joseph EK, Levine JD, Liedtke W, Vanner S, Vergnolle N, Geppetti P, Bunnett NW. Protease-activated receptor 2 sensitizes the transient receptor potential vanilloid 4 ion channel to cause mechanical hyperalgesia in mice. J Physiol 578: 715–733, 2007. doi: 10.1113/jphysiol.2006.121111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Sipe WE, Brierley SM, Martin CM, Phillis BD, Cruz FB, Grady EF, Liedtke W, Cohen DM, Vanner S, Blackshaw LA, Bunnett NW. Transient receptor potential vanilloid 4 mediates protease activated receptor 2-induced sensitization of colonic afferent nerves and visceral hyperalgesia. Am J Physiol Gastrointest Liver Physiol 294: G1288–G1298, 2008. doi: 10.1152/ajpgi.00002.2008. [DOI] [PubMed] [Google Scholar]
- 266. Grace MS, Lieu T, Darby B, Abogadie FC, Veldhuis N, Bunnett NW, Mcintyre P. The tyrosine kinase inhibitor bafetinib inhibits PAR2-induced activation of TRPV4 channelsin vitroand painin vivo. Br J Pharmacol 171: 3881–3894, 2014. doi: 10.1111/bph.12750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Saifeddine M, El-Daly M, Mihara K, Bunnett NW, Mcintyre P, Altier C, Hollenberg MD, Ramachandran R. GPCR-mediated EGF receptor transactivation regulates TRPV4 action in the vasculature. Br J Pharmacol 172: 2493–2506, 2015. doi: 10.1111/bph.13072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Shavit-Stein E, Artan-Furman A, Feingold E, Ben Shimon M, Itzekson-Hayosh Z, Chapman J, Vlachos A, Maggio N. Protease activated receptor 2 (PAR2) induces long-term depression in the hippocampus through transient receptor potential vanilloid 4 (TRPV4). Front Mol Neurosci 10: 42, 2017. doi: 10.3389/fnmol.2017.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Peng S, Grace MS, Gondin AB, Retamal JS, Dill L, Darby W, Bunnett NW, Abogadie FC, Carbone SE, Tigani T, Davis TP, Poole DP, Veldhuis NA, Mcintyre P. The transient receptor potential vanilloid 4 (TRPV4) ion channel mediates protease activated receptor 1 (PAR1)-induced vascular hyperpermeability. Lab Invest 100: 1057–1067, 2020. doi: 10.1038/s41374-020-0430-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Asfaha S, Cenac N, Houle S, Altier C, Papez MD, Nguyen C, Steinhoff M, Chapman K, Zamponi GW, Vergnolle N. Protease-activated receptor-4: a novel mechanism of inflammatory pain modulation. Br J Pharmacol 150: 176–185, 2007. doi: 10.1038/sj.bjp.0706975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Dai Y, Wang S, Tominaga M, Yamamoto S, Fukuoka T, Higashi T, Kobayashi K, Obata K, Yamanaka H, Noguchi K. Sensitization of TRPA1 by PAR2 contributes to the sensation of inflammatory pain. J Clin Invest 117: 1979–1987, 2007. doi: 10.1172/JCI30951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. Brierley SM, Hughes PA, Page AJ, Kwan KY, Martin CM, O’Donnell TA, Cooper NJ, Harrington AM, Adam B, Liebregts T, Holtmann G, Corey DP, Rychkov GY, Blackshaw LA. The ion channel TRPA1 is required for normal mechanosensation and is modulated by algesic stimuli. Gastroenterology 137: 2084–2095.e3, 2009. doi: 10.1053/j.gastro.2009.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Hinman A, Chuang HH, Bautista DM, Julius D. TRP channel activation by reversible covalent modification. Proc Natl Acad Sci U S A 103: 19564–19568, 2006. doi: 10.1073/pnas.0609598103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. Schmidt M, Dubin AE, Petrus MJ, Earley TJ, Patapoutian A. Nociceptive signals induce trafficking of TRPA1 to the plasma membrane. Neuron 64: 498–509, 2009. doi: 10.1016/j.neuron.2009.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Wu J, Liu TT, Zhou YM, Qiu CY, Ren P, Jiao M, Hu WP. Sensitization of ASIC3 by proteinase-activated receptor 2 signaling contributes to acidosis-induced nociception. J Neuroinflammation 14: 150, 2017. doi: 10.1186/s12974-017-0916-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. McLaughlin JN, Mazzoni MR, Cleator JH, Earls L, Perdigoto AL, Brooks JD, Muldowney JA, Vaughan DE, Hamm HE. Thrombin modulates the expression of a set of genes including thrombospondin-1 in human microvascular endothelial cells. J Biol Chem 280: 22172–22180, 2005. doi: 10.1074/jbc.M500721200. [DOI] [PubMed] [Google Scholar]
- 277. Suen JY, Gardiner B, Grimmond S, Fairlie DP. Profiling gene expression induced by protease-activated receptor 2 (PAR2) activation in human kidney cells. PLoS One 5: e13809, 2010. doi: 10.1371/journal.pone.0013809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278. Wang H, Moreau F, Hirota CL, Macnaughton WK. Proteinase‐activated receptors induce interleukin‐8 expression by intestinal epithelial cells through ERK/RSK90 activation and histone acetylation. FASEB J 24: 1971–1980, 2010. doi: 10.1096/fj.09-137646. [DOI] [PubMed] [Google Scholar]
- 279. Nagataki M, Moriyuki K, Sekiguchi F, Kawabata A. Evidence that PAR2-triggered prostaglandin E2 (PGE2) formation involves the ERK-cytosolic phospholipase A2-COX-1-microsomal PGE synthase-1 cascade in human lung epithelial cells. Cell Biochem Funct 26: 279–282, 2008. doi: 10.1002/cbf.1434. [DOI] [PubMed] [Google Scholar]
- 280. Moriyuki K, Nagataki M, Sekiguchi F, Nishikawa H, Kawabata A. Signal transduction for formation/release of interleukin-8 caused by a PAR2-activating peptide in human lung epithelial cells. Regul Pept 145: 42–48, 2008. doi: 10.1016/j.regpep.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 281. Nichols HL, Saffeddine M, Theriot BS, Hegde A, Polley D, El-Mays T, Vliagoftis H, Hollenberg MD, Wilson EH, Walker JK, Defea KA. beta-Arrestin-2 mediates the proinflammatory effects of proteinase-activated receptor-2 in the airway. Proc Natl Acad Sci U S A 109: 16660–16665, 2012. doi: 10.1073/pnas.1208881109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Jiang Y, Yau MK, Lim J, Wu KC, Xu W, Suen JY, Fairlie DP. A potent antagonist of protease-activated receptor 2 that inhibits multiple signaling functions in human cancer cells. J Pharmacol Exp Ther 364: 246–257, 2018. doi: 10.1124/jpet.117.245027. [DOI] [PubMed] [Google Scholar]
- 283. Cooper DM, Pechkovsky DV, Hackett TL, Knight DA, Granville DJ. Granzyme K activates protease-activated receptor-1. PLoS One 6: e21484, 2011. doi: 10.1371/journal.pone.0021484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284. Johnson JJ, Miller DL, Jiang R, Liu Y, Shi Z, Tarwater L, Williams R, Balsara R, Sauter ER, Stack MS. Protease-activated receptor-2 (PAR-2)-mediated Nf-κB activation suppresses inflammation-associated tumor suppressor microRNAs in oral squamous cell carcinoma. J Biol Chem 291: 6936–6945, 2016. doi: 10.1074/jbc.M115.692640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285. Rallabhandi P, Nhu QM, Toshchakov VY, Piao W, Medvedev AE, Hollenberg MD, Fasano A, Vogel SN. Analysis of proteinase-activated receptor 2 and TLR4 signal transduction. J Biol Chem 283: 24314–24325, 2008. doi: 10.1074/jbc.M804800200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286. Ko FN, Yang YC, Huang SC, Ou JT. Coagulation factor Xa stimulates platelet-derived growth factor release and mitogenesis in cultured vascular smooth muscle cells of rat. J Clin Invest 98: 1493–1501, 1996. doi: 10.1172/JCI118938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287. Shimizu S, Gabazza EC, Hayashi T, Ido M, Adachi Y, Suzuki K. Thrombin stimulates the expression of PDGF in lung epithelial cells. Am J Physiol Lung Cell Mol Physiol 279: L503–L510, 2000. doi: 10.1152/ajplung.2000.279.3.L503. [DOI] [PubMed] [Google Scholar]
- 288. Blanc-Brude OP, Archer F, Leoni P, Derian C, Bolsover S, Laurent GJ, Chambers RC. Factor Xa stimulates fibroblast procollagen production, proliferation, and calcium signaling via PAR1 activation. Exp Cell Res 304: 16–27, 2005. doi: 10.1016/j.yexcr.2004.10.021. [DOI] [PubMed] [Google Scholar]
- 289. Burch ML, Ballinger ML, Yang SN, Getachew R, Itman C, Loveland K, Osman N, Little PJ. Thrombin stimulation of proteoglycan synthesis in vascular smooth muscle is mediated by protease-activated receptor-1 transactivation of the transforming growth factor β type I receptor. J Biol Chem 285: 26798–26805, 2010. doi: 10.1074/jbc.M109.092767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290. Hammes SR, Coughlin SR. Protease-activated receptor-1 can mediate responses to SFLLRN in thrombin-desensitized cells: evidence for a novel mechanism for preventing or terminating signaling by PAR1’s tethered ligand. Biochemistry 38: 2486–2493, 1999. doi: 10.1021/bi982527i. [DOI] [PubMed] [Google Scholar]
- 291. Ishii K, Chen J, Ishii M, Koch WJ, Freedman NJ, Lefkowitz RJ, Coughlin SR. Inhibition of thrombin receptor signaling by a G-protein coupled receptor kinase. Functional specificity among G-protein coupled receptor kinases. J Biol Chem 269: 1125–1130, 1994. doi: 10.1016/S0021-9258(17)42230-7. [DOI] [PubMed] [Google Scholar]
- 292. Hein L, Ishii K, Coughlin SR, Kobilka BK. Intracellular targeting and trafficking of thrombin receptors. A novel mechanism for resensitization of a G protein-coupled receptor. J Biol Chem 269: 27719–27726, 1994. doi: 10.1016/S0021-9258(18)47045-7. [DOI] [PubMed] [Google Scholar]
- 293. Shapiro MJ, Trejo J, Zeng D, Coughlin SR. Role of the thrombin receptor’s cytoplasmic tail in intracellular trafficking. J Biol Chem 271: 32874–32880, 1996. doi: 10.1074/jbc.271.51.32874. [DOI] [PubMed] [Google Scholar]
- 294. Hammes SR, Shapiro MJ, Coughlin SR. Shutoff and agonist-triggered internalization of protease-activated receptor 1 can be separated by mutation of putative phosphorylation sites in the cytoplasmic tail. Biochemistry 38: 9308–9316, 1999. doi: 10.1021/bi9902236. [DOI] [PubMed] [Google Scholar]
- 295. Tiruppathi C, Yan W, Sandoval R, Naqvi T, Pronin AN, Benovic JL, Malik AB. G protein-coupled receptor kinase-5 regulates thrombin-activated signaling in endothelial cells. Proc Natl Acad Sci U S A 97: 7440–7445, 2000. doi: 10.1073/pnas.97.13.7440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296. Ghil S, Mccoy KL, Hepler JR. Regulator of G protein signaling 2 (RGS2) and RGS4 form distinct G protein-dependent complexes with protease activated-receptor 1 (PAR1) in live cells. PLoS One 9: e95355, 2014. doi: 10.1371/journal.pone.0095355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297. Lee J, Ghil S. Regulator of G protein signaling 8 inhibits protease-activated receptor 1/G i/o signaling by forming a distinct G protein-dependent complex in live cells. Cell Signal 28: 391–400, 2016. doi: 10.1016/j.cellsig.2016.01.015. [DOI] [PubMed] [Google Scholar]
- 298. Kim K, Lee J, Ghil S. The regulators of G protein signaling RGS16 and RGG18 nhibit protease‐activated receptor 2/Gi/o signaling through distinct interactions with Gα in live cells. FEBS Lett 592: 3126–3138, 2018. doi: 10.1002/1873-3468.13220. [DOI] [PubMed] [Google Scholar]
- 299. Kim Y, Ghil S. Regulators of G-protein signaling, RGS2 and RGS4, inhibit protease-activated receptor 4-mediated signaling by forming a complex with the receptor and Gα in live cells. Cell Commun Signal 18: 86. 2020. doi: 10.1186/s12964-020-00552-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300. Paing MM, Stutts AB, Kohout TA, Lefkowitz RJ, Trejo J. β-Arrestins regulate protease-activated receptor-1 desensitization but not internalization or down-regulation. J Biol Chem 277: 1292–1300, 2002. doi: 10.1074/jbc.M109160200. [DOI] [PubMed] [Google Scholar]
- 301. Soh UJ, Trejo J. Activated protein C promotes protease-activated receptor-1 cytoprotective signaling through -arrestin and dishevelled-2 scaffolds. Proc Natl Acad Sci U S A 108: E1372–E1380, 2011. doi: 10.1073/pnas.1112482108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302. Stalheim L, Ding Y, Gullapalli A, Paing MM, Wolfe BL, Morris DR, Trejo J. Multiple independent functions of arrestins in the regulation of protease-activated receptor-2 signaling and trafficking. Mol Pharmacol 67: 78–87, 2005. doi: 10.1124/mol.104.006072. [DOI] [PubMed] [Google Scholar]
- 303. Roosterman D, Schmidlin F, Bunnett NW. Rab5a and rab11a mediate agonist-induced trafficking of protease-activated receptor 2. Am J Physiol Cell Physiol 284: C1319–C1329, 2003. doi: 10.1152/ajpcell.00540.2002. [DOI] [PubMed] [Google Scholar]
- 304. Wolfe BL, Marchese A, Trejo J. Ubiquitination differentially regulates clathrin-dependent internalization of protease-activated receptor-1. J Cell Biol 77: 905–916, 2007. doi: 10.1083/jcb.200610154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305. Chen B, R DM, Grimsey N, Canto I, Barker BL, Trejo J. Adaptor protein complex-2 (AP-2) and epsin-1 mediate protease-activated receptor-1 internalization via phosphorylation- and ubiquitination-dependent sorting signals. J Biol Chem 286: 40760–40770, 2011. doi: 10.1074/jbc.M111.299776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306. Paing MM, Johnston CA, Siderovski DP, Trejo J. Clathrin adaptor AP2 regulates thrombin receptor constitutive internalization and endothelial cell resensitization. Mol Cell Biol 26: 3231–3242, 2006. doi: 10.1128/MCB.26.8.3231-3242.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307. Alvarez-Arce A, Lee-Rivera I, López E, Hernández-Cruz A, López-Colomé AM. Thrombin-induced calpain activation promotes protease-activated receptor 1 internalization. Intl J Cell Biol 2017: 1908310, 2017. doi: 10.1155/2017/1908310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308. Böhm SK, Khitin LM, Grady EF, Aponte G, Payan DG, Bunnett NW. Mechanisms of desensitization and resensitization of proteinase-activated receptor-2. J Biol Chem 271: 22003–22016, 1996. doi: 10.1074/jbc.271.36.22003. [DOI] [PubMed] [Google Scholar]
- 309. Hasdemir B, Bunnett N, Cottrell GS. Hepatocyte growth factor-regulated tyrosine kinase substrate (HRS) mediates post-endocytic trafficking of protease-activated receptor 2 and calcitonin receptor-like receptor. J Biol Chem 282: 29646–29657, 2007. doi: 10.1074/jbc.M702974200. [DOI] [PubMed] [Google Scholar]
- 310. Jacob C, Cottrell GS, Gehringer D, Schmidlin F, Grady EF, Bunnett NW. c-Cbl mediates ubiquitination, degradation, and down-regulation of human protease-activated receptor 2. J Biol Chem 280: 16076–16087, 2005. doi: 10.1074/jbc.M500109200. [DOI] [PubMed] [Google Scholar]
- 311. Hasdemir B, Murphy JE, Cottrell GS, Bunnett NW. Endosomal deubiquitinating enzymes control ubiquitination and down-regulation of protease-activated receptor 2. J Biol Chem 284: 28453–28466, 2009. doi: 10.1074/jbc.M109.025692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312. Dores MR, Chen B, Lin H, Soh UJ, Paing MM, Montagne WA, Meerloo T, Trejo J. ALIX binds a YPX3L motif of the GPCR PAR1 and mediates ubiquitin-independent ESCRT-III/MVB sorting. J Cell Biol 197: 407–419, 2012. doi: 10.1083/jcb.201110031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313. Canto I, Trejo J. Palmitoylation of protease-activated receptor-1 regulates adaptor protein complex-2 and -3 interaction with tyrosine-based motifs and endocytic sorting. J Biol Chem 288: 15900–15912, 2013. doi: 10.1074/jbc.M113.469866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314. Arakaki AK, Pan WA, Lin H, Trejo J. The α-arrestin ARRDC3 suppresses breast carcinoma invasion by regulating G protein–coupled receptor lysosomal sorting and signaling. J Biol Chem 293: 3350–3362, 2018. doi: 10.1074/jbc.RA117.001516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315. Ishii K, Hein L, Kobilka B, Coughlin SR. Kinetics of thrombin receptor cleavage on intact cells. Relation to signaling. J Biol Chem 268: 9780–9786, 1993. doi: 10.1016/S0021-9258(18)98415-2. [DOI] [PubMed] [Google Scholar]
- 316. Grimsey NJ, Coronel LJ, Cordova IC, Trejo J. Recycling and endosomal sorting of protease-activated receptor-1 is distinctly regulated by Rab11A and Rab11B proteins. J Biol Chem 291: 2223–2236, 2016. doi: 10.1074/jbc.M115.702993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317. Trejo J, Hammes SR, Coughlin SR. Termination of signaling by protease-activated receptor-1 is linked to lysosomal sorting. Proc Natl Acad Sci U S A 95: 13698–13702, 1998. doi: 10.1073/pnas.95.23.13698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318. Trejo J, Coughlin SR. The cytoplasmic tails of protease-activated receptor-1 and substance p receptor specify sorting to lysosomes versus recycling. J Biol Chem 274: 2216–2224, 1999. doi: 10.1074/jbc.274.4.2216. [DOI] [PubMed] [Google Scholar]
- 319. Jensen DD, Zhao P, Jimenez-Vargas NN, Lieu T, Gerges M, Yeatman HR, Canals M, Vanner SJ, Poole DP, Bunnett NW. Protein kinase D and Gβγ subunits mediate agonist-evoked translocation of protease-activated receptor-2 from the golgi apparatus to the plasma membrane. J Biol Chem 291: 11285–11299, 2016. doi: 10.1074/jbc.M115.710681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320. Edgington LE, Verdoes M, Bogyo M. Functional imaging of proteases: recent advances in the design and application of substrate-based and activity-based probes. Curr Opin Chem Biol 15: 798–805, 2011. doi: 10.1016/j.cbpa.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321. Ku DD. Mechanism of thrombin-induced endothelium-dependent coronary vasodilation in dogs: role of its proteolytic enzymatic activity. J Cardiovasc Pharmacol 8: 29–36, 1986. doi: 10.1097/00005344-198601000-00005. [DOI] [PubMed] [Google Scholar]
- 322. Sambrano GR, Weiss EJ, Zheng YW, Huang W, Coughlin SR. Role of thrombin signalling in platelets in haemostasis and thrombosis. Nature 413: 74–78, 2001. doi: 10.1038/35092573. [DOI] [PubMed] [Google Scholar]
- 323. Hollenberg MD, Laniyonu AA, Saifeddine M, Moore GJ. Role of the amino- and carboxyl-terminal domains of thrombin receptor-derived polypeptides in biological activity in vascular endothelium and gastric smooth muscle: evidence for receptor subtypes. Mol Pharmacol 43: 921–930, 1993. [PubMed] [Google Scholar]
- 324. Hamilton JR, Moffatt JD, Frauman AG, Cocks TM. Protease-activated receptor (PAR) 1 but not PAR2 or PAR4 mediates endothelium-dependent relaxation to thrombin and trypsin in human pulmonary arteries. J Cardiovasc Pharmacol 38: 108–119, 2001. doi: 10.1097/00005344-200107000-00012. [DOI] [PubMed] [Google Scholar]
- 325. Vergnolle N, Hollenberg MD, Wallace JL. Pro- and anti-inflammatory actions of thrombin: a distinct role for proteinase-activated receptor-1 (PAR1). Br J Pharmacol 126: 1262–1268, 1999. doi: 10.1038/sj.bjp.0702408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326. Al-Ani B, Saifeddine M, Hollenberg MD. Detection of functional receptors for the proteinase-activated-receptor-2-activating polypeptide, SLIGRL-NH2, in rat vascular and gastric smooth muscle. Can J Physiol Pharmacol 73: 1203–1207, 1995. doi: 10.1139/y95-172. [DOI] [PubMed] [Google Scholar]
- 327. Saifeddine M, Al-Ani B, Cheng CH, Wang L, Hollenberg MD. Rat proteinase-activated receptor-2 (PAR-2): cDNA sequence and activity of receptor-derived peptides in gastric and vascular tissue. Br J Pharmacol 118: 521–530, 1996. doi: 10.1111/j.1476-5381.1996.tb15433.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328. Cicala C. Protease activated receptor 2 and the cardiovascular system. Br J Pharmacol 135: 14–20, 2002. doi: 10.1038/sj.bjp.0704438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329. Disse J, Petersen HH, Larsen KS, Persson E, Esmon N, Esmon CT, Teyton L, Petersen LC, Ruf W. The endothelial protein C receptor supports tissue factor ternary coagulation initiation complex signaling through protease-activated receptors. J Biol Chem 286: 5756–5767, 2011. doi: 10.1074/jbc.M110.201228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330. Mohan Rao LV, Esmon CT, Pendurthi UR. Endothelial cell protein C receptor: a multiliganded and multifunctional receptor. Blood 124: 1553–1562, 2014. doi: 10.1182/blood-2014-05-578328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331. Rothmeier AS, Liu E, Chakrabarty S, Disse J, Mueller BM, Ostergaard H, Ruf W. Identification of the integrin-binding site on coagulation factor VIIa required for proangiogenic PAR2 signaling. Blood 131: 674–685, 2018. doi: 10.1182/blood-2017-02-768218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332. Ahamed J, Belting M, Ruf W. Regulation of tissue factor-induced signaling by endogenous and recombinant tissue factor pathway inhibitor 1. Blood 105: 2384–2391, 2005. doi: 10.1182/blood-2004-09-3422. [DOI] [PubMed] [Google Scholar]
- 333. Ma L, Perini R, McKnight W, Dicay M, Klein A, Hollenberg M, Wallace JL. Proteinase-activated receptors 1 and 4 counter-regulate endostatin and VEGF release from human platelets. Proc Natl Acad Sci U S A 102: 216–220, 2005. doi: 10.1073/pnas.0406682102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334. Jackson SP, Darbousset R, Schoenwaelder SM. Thromboinflammation: challenges of therapeutically targeting coagulation and other host defense mechanisms. Blood 133: 906–918, 2019. doi: 10.1182/blood-2018-11-882993. [DOI] [PubMed] [Google Scholar]
- 335. Ekdahl KN, Teramura Y, Hamad OA, Asif S, Duehrkop C, Fromell K, Gustafson E, Hong J, Kozarcanin H, Magnusson PU, Huber-Lang M, Garred P, Nilsson B. Dangerous liaisons: complement, coagulation, and kallikrein/kinin cross-talk act as a linchpin in the events leading to thromboinflammation. Immunol Rev 271: 245–269, 2016. [DOI] [PubMed] [Google Scholar]
- 336. Megyeri M, Makó V, Beinrohr L, Doleschall Z, Prohászka Z, Cervenak L, Závodszky P, Gál P. Complement protease MASP-1 activates human endothelial cells: PAR4 activation is a link between complement and endothelial function. J Immunol 183: 3409–3416, 2009. doi: 10.4049/jimmunol.0900879. [DOI] [PubMed] [Google Scholar]
- 337. He SH, Xie H, Fu YL. Activation of human tonsil and skin mast cells by agonists of proteinase activated receptor-2. Acta Pharmacol Sin 26: 568–574, 2005. doi: 10.1111/j.1745-7254.2005.00079.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338. Moffatt JD, Lever R, Page CP. Effects of inhaled thrombin receptor agonists in mice. Br J Pharmacol 143: 269–275, 2004. doi: 10.1038/sj.bjp.0705926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339. Bar-Shavit R, Kahn A, Mudd M, Wilner G, Mann K, Fenton JW 2nd.. Localization of a chemotactic domain in human thrombin. Biochemistry 23: 397–400, 1984. doi: 10.1021/bi00298a001. [DOI] [PubMed] [Google Scholar]
- 340. Bar-Shavit R, Kahn A, Mann K, Wilner GD. Identification of a thrombin sequence with growth factor activity on macrophages. Proc Natl Acad Sci U S A 83: 976–980, 1986. doi: 10.1073/pnas.83.4.976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341. Rabenhorst A, Hartmann K. Interleukin-31: a novel diagnostic marker of allergic diseases. Curr Allergy Asthma Rep 14: 423, 2014. doi: 10.1007/s11882-014-0423-y. [DOI] [PubMed] [Google Scholar]
- 342. Shpacovitch V, Feld M, Hollenberg MD, Luger TA, Steinhoff M. Role of protease-activated receptors in inflammatory responses, innate and adaptive immunity. J Leukoc Biol 83: 1309–1322, 2008. doi: 10.1189/jlb.0108001. [DOI] [PubMed] [Google Scholar]
- 343. Rasmussen UB, Gachet C, Schlesinger Y, Hanau D, Ohlmann P, Van Obberghen-Schilling E, Pouyssegur J, Cazenave JP, Pavirani A. A peptide ligand of the human thrombin receptor antagonizes alpha-thrombin and partially activates platelets. J Biol Chem 268: 14322–14328, 1993. doi: 10.1016/S0021-9258(19)85244-4. [DOI] [PubMed] [Google Scholar]
- 344. Kuriri FA, Burchall G, Alanazi F, Antonipillai J, Dobie G, Beachemin N, Jackson DE. mice lacking PECAM-1 and Ceacam1 have enhanced platelet secretion and thrombus growth: novel link with PAR4. Thromb Haemost 122: 961–973, 2022. doi: 10.1055/a-1663-8108. [DOI] [PubMed] [Google Scholar]
- 345. Wallace JL, McKnight W, Del Soldato P, Baydoun AR, Cirino G. Anti-thrombotic effects of a nitric oxide-releasing, gastric-sparing aspirin derivative. J Clin Invest 96: 2711–2718, 1995. doi: 10.1172/JCI118338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346. Ruef J, Kacharava A, Pohl J, Bode C, Runge MS. Indications for the presence of an atypical protease-activated receptor on rat platelets. Ann Hematol 79: 604–611, 2000. doi: 10.1007/s002770000211. [DOI] [PubMed] [Google Scholar]
- 347. van den Hengel LG, Hellingman AA, Nossent AY, van Oeveren-Rietdijk AM, de Vries MR, Spek CA, van Zonneveld AJ, Reitsma PH, Hamming JF, de Boer HC, Versteeg HH, Quax PH. Protease-activated receptor (PAR)2, but not PAR1, is involved in collateral formation and anti-inflammatory monocyte polarization in a mouse hind limb ischemia model. PLoS One 8: e61923, 2013. doi: 10.1371/journal.pone.0061923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348. Nystedt S, Ramakrishnan V, Sundelin J. The proteinase-activated receptor 2 is induced by inflammatory mediators in human endothelial cells. Comparison with the thrombin receptor. J Biol Chem 271: 14910–14915, 1996. doi: 10.1074/jbc.271.25.14910. [DOI] [PubMed] [Google Scholar]
- 349. Hara T, Fukuda D, Tanaka K, Higashikuni Y, Hirata Y, Nishimoto S, Yagi S, Yamada H, Soeki T, Wakatsuki T, Shimabukuro M, Sata M. Rivaroxaban, a novel oral anticoagulant, attenuates atherosclerotic plaque progression and destabilization in ApoE-deficient mice. Atherosclerosis 242: 639–646, 2015. doi: 10.1016/j.atherosclerosis.2015.03.023. [DOI] [PubMed] [Google Scholar]
- 350. Zuo P, Zuo Z, Wang X, Chen L, Zheng Y, Ma G, Zhou Q. Factor Xa induces pro-inflammatory cytokine expression in RAW 264.7 macrophages via protease-activated receptor-2 activation. Am J Transl Res 7: 2326–2334, 2015. [PMC free article] [PubMed] [Google Scholar]
- 351. Uusitalo-Jarvinen H, Kurokawa T, Mueller BM, Andrade-Gordon P, Friedlander M, Ruf W. Role of protease activated receptor 1 and 2 signaling in hypoxia-induced angiogenesis. Arterioscler Thromb Vasc Biol 27: 1456–1462, 2007. doi: 10.1161/ATVBAHA.107.142539. [DOI] [PubMed] [Google Scholar]
- 352. Sitaras N, Rivera JC, Noueihed B, Bien-Aime M, Zaniolo K, Omri S, Hamel D, Zhu T, Hardy P, Sapieha P, Joyal JS, Chemtob S. Retinal neurons curb inflammation and enhance revascularization in ischemic retinopathies via proteinase-activated receptor-2. Am J Pathol 185: 581–595, 2015. doi: 10.1016/j.ajpath.2014.10.020. [DOI] [PubMed] [Google Scholar]
- 353. Hamilton JR, Cornelissen I, Mountford JK, Coughlin SR. Atherosclerosis proceeds independently of thrombin-induced platelet activation in ApoE-/- mice. Atherosclerosis 205: 427–432, 2009. doi: 10.1016/j.atherosclerosis.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354. Furman MI, Benoit SE, Barnard MR, Valeri CR, Borbone ML, Becker RC, Hechtman HB, Michelson AD. Increased platelet reactivity and circulating monocyte-platelet aggregates in patients with stable coronary artery disease. J Am Coll Cardiol 31: 352–358, 1998. doi: 10.1016/s0735-1097(97)00510-x. [DOI] [PubMed] [Google Scholar]
- 355. Misaki T, Satoh Y, Saino T, Ogawa A. The role of protease-activated receptors on the intracellular calcium ion dynamics of vascular smooth muscles, with special reference to cerebral arterioles. Arch Histol Cytol 69: 49–60, 2006. doi: 10.1679/aohc.69.49. [DOI] [PubMed] [Google Scholar]
- 356. Kai Y, Hirano K, Maeda Y, Nishimura J, Sasaki T, Kanaide H. Prevention of the hypercontractile response to thrombin by proteinase-activated receptor-1 antagonist in subarachnoid hemorrhage. Stroke 38: 3259–3265, 2007. doi: 10.1161/STROKEAHA.107.487769. [DOI] [PubMed] [Google Scholar]
- 357. Liu J, Nishida M, Inui H, Chang J, Zhu Y, Kanno K, Matsuda H, Sairyo M, Okada T, Nakaoka H, Ohama T, Masuda D, Koseki M, Yamashita S, Sakata Y. Rivaroxaban suppresses the progression of ischemic cardiomyopathy in a murine model of diet-induced myocardial infarction. J Atheroscler Thromb 26: 915–930, 2019. doi: 10.5551/jat.48405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358. Kondo H, Abe I, Fukui A, Saito S, Miyoshi M, Aoki K, Shinohara T, Teshima Y, Yufu K, Takahashi N. Possible role of rivaroxaban in attenuating pressure-overload-induced atrial fibrosis and fibrillation. J Cardiol 71: 310–319, 2018. doi: 10.1016/j.jjcc.2017.08.007. [DOI] [PubMed] [Google Scholar]
- 359. Pawlinski R, Tencati M, Hampton CR, Shishido T, Bullard TA, Casey LM, Andrade-Gordon P, Kotzsch M, Spring D, Luther T, Abe J, Pohlman TH, Verrier ED, Blaxall BC, Mackman N. Protease-activated receptor-1 contributes to cardiac remodeling and hypertrophy. Circulation 116: 2298–2306, 2007. doi: 10.1161/CIRCULATIONAHA.107.692764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360. Sonin DL, Wakatsuki T, Routhu KV, Harmann LM, Petersen M, Meyer J, Strande JL. Protease-activated receptor 1 inhibition by SCH79797 attenuates left ventricular remodeling and profibrotic activities of cardiac fibroblasts. J Cardiovasc Pharmacol Ther 18: 460–475, 2013. doi: 10.1177/1074248413485434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361. Yokono Y, Hanada K, Narita M, Tatara Y, Kawamura Y, Miura N, Kitayama K, Nakata M, Nozaka M, Kato T, Kudo N, Tsushima M, Toyama Y, Itoh K, Tomita H. Blockade of PAR-1 signaling attenuates cardiac hypertrophy and fibrosis in renin-overexpressing hypertensive mice. J Am Heart Assoc 9: e015616, 2020. doi: 10.1161/JAHA.119.015616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362. Darrow AL, Fung-Leung WP, Ye RD, Santulli RJ, Cheung WM, Derian CK, Burns CL, Damiano BP, Zhou L, Keenan CM, Peterson PA, Andrade-Gordon P. Biological consequences of thrombin receptor deficiency in mice. Thromb Haemost 76: 860–866, 1996. doi: 10.1055/s-0038-1650676. [DOI] [PubMed] [Google Scholar]
- 363. Bindoli S, Felicetti M, Sfriso P, Doria A. The amount of cytokine-release defines different shades of Sars-Cov2 infection. Exp Biol Med (Maywood) 245: 970–976, 2020. doi: 10.1177/1535370220928964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364. Sriram K, Insel PA. Inflammation and thrombosis in COVID-19 pathophysiology: proteinase-activated and purinergic receptors as drivers and candidate therapeutic targets. Physiol Rev 101: 545–567, 2021. doi: 10.1152/physrev.00035.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365. Schouten M, Wiersinga WJ, Levi M, van der Poll T. Inflammation, endothelium, and coagulation in sepsis. J Leukoc Biol 83: 536–545, 2008. doi: 10.1189/jlb.0607373. [DOI] [PubMed] [Google Scholar]
- 366. McGonagle D, Sharif K, O’Regan A, Bridgewood C. The role of cytokines including interleukin-6 in COVID-19 induced pneumonia and macrophage activation syndrome-like disease. Autoimmun Rev 19: 102537, 2020. doi: 10.1016/j.autrev.2020.102537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367. Helms J, Tacquard C, Severac F, Leonard-Lorant I, Ohana M, Delabranche X, Merdji H, Clere-Jehl R, Schenck M, Fagot Gandet F, Fafi-Kremer S, Castelain V, Schneider F, Grunebaum L, Angles-Cano E, Sattler L, Mertes PM, Meziani F, CRICS TRIGGERSEP Group (Clinical Research in Intensive Care and Sepsis Trial Group for Global Evaluation and Research in Sepsis). High risk of thrombosis in patients with severe SARS-CoV-2 infection: a multicenter prospective cohort study. Intensive Care Med 46: 1089–1098, 2020. doi: 10.1007/s00134-020-06062-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368. Rovai ES, Alves T, Holzhausen M. Protease-activated receptor 1 as a potential therapeutic target for COVID-19. Exp Biol Med (Maywood) 246: 688–694, 2021. doi: 10.1177/1535370220978372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369. Jenkins AL, Chinni C, De Niese MR, Blackhart B, Mackie EJ. Expression of protease-activated receptor-2 during embryonic development. Dev Dyn 218: 465–471, 2000. doi:. [DOI] [PubMed] [Google Scholar]
- 370. Hachem JP, Houben E, Crumrine D, Man MQ, Schurer N, Roelandt T, Choi EH, Uchida Y, Brown BE, Feingold KR, Elias PM. Serine protease signaling of epidermal permeability barrier homeostasis. J Invest Dermatol 126: 2074–2086, 2006. doi: 10.1038/sj.jid.5700351. [DOI] [PubMed] [Google Scholar]
- 371. Lundwall A, Brattsand M. Kallikrein-related peptidases. Cell Mol Life Sci 65: 2019–2038, 2008. doi: 10.1007/s00018-008-8024-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372. Rattenholl A, Steinhoff M. Proteinase-activated receptor-2 in the skin: receptor expression, activation and function during health and disease. Drug News Perspect 21: 369–381, 2008. doi: 10.1358/dnp.2008.21.7.1255294. [DOI] [PubMed] [Google Scholar]
- 373. Oikonomopoulou K, Hansen KK, Saifeddine M, Vergnolle N, Tea I, Blaber M, Blaber SI, Scarisbrick I, Diamandis EP, Hollenberg MD. Kallikrein-mediated cell signalling: targeting proteinase-activated receptors (PARs). Biol Chem 387: 817–824, 2006. doi: 10.1515/bc.2006.104. [DOI] [PubMed] [Google Scholar]
- 374. Oikonomopoulou K, Hansen KK, Saifeddine M, Vergnolle N, Tea I, Diamandis EP, Hollenberg MD. Proteinase-mediated cell signalling: targeting proteinase-activated receptors (PARs) by kallikreins and more. Biol Chem 387: 677–685, 2006. doi: 10.1515/BC.2006.086. [DOI] [PubMed] [Google Scholar]
- 375. Roelandt T, Heughebaert C, Hachem JP. Proteolytically active allergens cause barrier breakdown. J Invest Dermatol 128: 1878–1880, 2008. doi: 10.1038/jid.2008.168. [DOI] [PubMed] [Google Scholar]
- 376. Roelandt T, Heughebaert C, Verween G, Giddelo C, Verbeken G, Pirnay JP, Devos D, Crumrine D, Roseeuw D, Elias PM, Hachem JP. Actin dynamics regulate immediate PAR-2-dependent responses to acute epidermal permeability barrier abrogation. J Dermatol Sci 61: 101–109, 2011. doi: 10.1016/j.jdermsci.2010.11.016. [DOI] [PubMed] [Google Scholar]
- 377. Steinhoff M, Neisius U, Ikoma A, Fartasch M, Heyer G, Skov PS, Luger TA, Schmelz M. Proteinase-activated receptor-2 mediates itch: a novel pathway for pruritus in human skin. J Neurosci 23: 6176–6180, 2003. doi: 10.1523/JNEUROSCI.23-15-06176.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378. Julovi SM, Xue M, Dervish S, Sambrook PN, March L, Jackson CJ. Protease activated receptor-2 mediates activated protein C-induced cutaneous wound healing via inhibition of p38. Am J Pathol 179: 2233–2242, 2011. doi: 10.1016/j.ajpath.2011.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379. Xue M, Campbell D, Sambrook PN, Fukudome K, Jackson CJ. Endothelial protein C receptor and protease-activated receptor-1 mediate induction of a wound-healing phenotype in human keratinocytes by activated protein C. J Invest Dermatol 125: 1279–1285, 2005. doi: 10.1111/j.0022-202X.2005.23952.x. [DOI] [PubMed] [Google Scholar]
- 380. Sharlow ER, Paine CS, Babiarz L, Eisinger M, Shapiro S, Seiberg M. The protease-activated receptor-2 upregulates keratinocyte phagocytosis. J Cell Sci 113: 3093–3101, 2000. doi: 10.1242/jcs.113.17.3093. [DOI] [PubMed] [Google Scholar]
- 381. Hou L, Kapas S, Cruchley AT, Macey MG, Harriott P, Chinni C, Stone SR, Howells GL. Immunolocalization of protease-activated receptor-2 in skin: receptor activation stimulates interleukin-8 secretion by keratinocytes in vitro. Immunology 94: 356–362, 1998. doi: 10.1046/j.1365-2567.1998.00528.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382. Babiarz-Magee L, Chen N, Seiberg M, Lin CB. The expression and activation of protease-activated receptor-2 correlate with skin color. Pigment Cell Res 17: 241–251, 2004. doi: 10.1111/j.1600-0749.2004.00133.x. [DOI] [PubMed] [Google Scholar]
- 383. Seiberg M, Paine C, Sharlow E, Andrade-Gordon P, Costanzo M, Eisinger M, Shapiro SS. The protease-activated receptor 2 regulates pigmentation via keratinocyte-melanocyte interactions. Exp Cell Res 254: 25–32, 2000. doi: 10.1006/excr.1999.4692. [DOI] [PubMed] [Google Scholar]
- 384. Bangert C, Brunner PM, Stingl G. Immune functions of the skin. Clin Dermatol 29: 360–376, 2011. doi: 10.1016/j.clindermatol.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 385. Komatsu N, Saijoh K, Kuk C, Shirasaki F, Takehara K, Diamandis EP. Aberrant human tissue kallikrein levels in the stratum corneum and serum of patients with psoriasis: dependence on phenotype, severity and therapy. Br J Dermatol 156: 875–883, 2007. doi: 10.1111/j.1365-2133.2006.07743.x. [DOI] [PubMed] [Google Scholar]
- 386. Buhl T, Ikoma A, Kempkes C, Cevikbas F, Sulk M, Buddenkotte J, Akiyama T, Crumrine D, Camerer E, Carstens E, Schon MP, Elias P, Coughlin SR, Steinhoff M. Protease-activated receptor-2 regulates neuro-epidermal communication in atopic dermatitis. Front Immunol 11: 1740, 2020. doi: 10.3389/fimmu.2020.01740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387. Smith L, Gatault S, Casals-Diaz L, Kelly PA, Camerer E, Metais C, Knaus UG, Eissner G, Steinhoff M. House dust mite-treated PAR2 over-expressor mouse: a novel model of atopic dermatitis. Exp Dermatol 28: 1298–1308, 2019. doi: 10.1111/exd.14030. [DOI] [PubMed] [Google Scholar]
- 388. Briot A, Deraison C, Lacroix M, Bonnart C, Robin A, Besson C, Dubus P, Hovnanian A. Kallikrein 5 induces atopic dermatitis-like lesions through PAR2-mediated thymic stromal lymphopoietin expression in Netherton syndrome. J Exp Med 206: 1135–1147, 2009. doi: 10.1084/jem.20082242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389. Saunders DP, Epstein JB, Elad S, Allemano J, Bossi P, van de Wetering MD, Rao NG, Potting C, Cheng KK, Freidank A, Brennan MT, Bowen J, Dennis K, Lalla RV, Mucositis Study Group of the Multinational Association of Supportive Care in Cancer/International Society of Oral Oncology (MASCC/ISOO). Systematic review of antimicrobials, mucosal coating agents, anesthetics, and analgesics for the management of oral mucositis in cancer patients. Support Care Cancer 21: 3191–3207, 2013. doi: 10.1007/s00520-013-1871-y. [DOI] [PubMed] [Google Scholar]
- 390. Kawasaki H, Nagao K, Kubo A, Hata T, Shimizu A, Mizuno H, Yamada T, Amagai M. Altered stratum corneum barrier and enhanced percutaneous immune responses in filaggrin-null mice. J Allergy Clin Immunol 129: 1538–1546.e6e1536, 2012. doi: 10.1016/j.jaci.2012.01.068. [DOI] [PubMed] [Google Scholar]
- 391. Yoo J, Omori M, Gyarmati D, Zhou B, Aye T, Brewer A, Comeau MR, Campbell DJ, Ziegler SF. Spontaneous atopic dermatitis in mice expressing an inducible thymic stromal lymphopoietin transgene specifically in the skin. J Exp Med 202: 541–549, 2005. doi: 10.1084/jem.20041503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392. Jeong SK, Kim HJ, Youm JK, Ahn SK, Choi EH, Sohn MH, Kim KE, Hong JH, Shin DM, Lee SH. Mite and cockroach allergens activate protease-activated receptor 2 and delay epidermal permeability barrier recovery. J Invest Dermatol 128: 1930–1939, 2008. doi: 10.1038/jid.2008.13. [DOI] [PubMed] [Google Scholar]
- 393. Chung K, Pitcher T, Grant AD, Hewitt E, Lindstrom E, Malcangio M. Cathepsin S acts via protease-activated receptor 2 to activate sensory neurons and induce itch-like behaviour. Neurobiol Pain 6: 100032, 2019. doi: 10.1016/j.ynpai.2019.100032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394. Feramisco JD, Berger TG, Steinhoff M. Innovative management of pruritus. Dermatol Clin 28: 467–478, 2010. doi: 10.1016/j.det.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 395. Nattkemper LA, Tey HL, Valdes-Rodriguez R, Lee H, Mollanazar NK, Albornoz C, Sanders KM, Yosipovitch G. The genetics of chronic itch: gene expression in the skin of patients with atopic dermatitis and psoriasis with severe itch. J Invest Dermatol 138: 1311–1317, 2018. doi: 10.1016/j.jid.2017.12.029. [DOI] [PubMed] [Google Scholar]
- 396. Belghiti M, Estevez-Herrera J, Gimenez-Garzo C, Gonzalez-Usano A, Montoliu C, Ferrer-Montiel A, Felipo V, Planells-Cases R. Potentiation of the transient receptor potential vanilloid 1 channel contributes to pruritogenesis in a rat model of liver disease. J Biol Chem 288: 9675–9685, 2013. doi: 10.1074/jbc.M113.455162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397. Moon SJ, Kim HJ, Cho SB, Lee SH, Choi HY, Park HC, Ha SK. Epidermal proteinase-activated receptor-2 expression is increased in end-stage renal disease patients with pruritus: a pilot study. Electrolyte Blood Press 12: 74–79, 2014. doi: 10.5049/EBP.2014.12.2.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398. Chua KY, Stewart GA, Thomas WR, Simpson RJ, Dilworth RJ, Plozza TM, Turner KJ. Sequence analysis of cDNA coding for a major house dust mite allergen, Der p 1. Homology with cysteine proteases. J Exp Med 167: 175–182, 1988. doi: 10.1084/jem.167.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399. Stewart GA, Ward LD, Simpson RJ, Thompson PJ. The group III allergen from the house dust mite Dermatophagoides pteronyssinus is a trypsin-like enzyme. Immunology 75: 29–35, 1992. [PMC free article] [PubMed] [Google Scholar]
- 400. King C, Simpson RJ, Moritz RL, Reed GE, Thompson PJ, Stewart GA. The isolation and characterization of a novel collagenolytic serine protease allergen (Der p 9) from the dust mite Dermatophagoides pteronyssinus. J Allergy Clin Immunol 98: 739–747, 1996. doi: 10.1016/S0091-6749(96)70121-5. [DOI] [PubMed] [Google Scholar]
- 401. McMahon DB, Workman AD, Kohanski MA, Carey RM, Freund JR, Hariri BM, Chen B, Doghramji LJ, Adappa ND, Palmer JN, Kennedy DW, Lee RJ. Protease-activated receptor 2 activates airway apical membrane chloride permeability and increases ciliary beating. FASEB J 32: 155–167, 2018. doi: 10.1096/fj.201700114RRR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402. Trejo J, Connolly AJ, Coughlin SR. The cloned thrombin receptor is necessary and sufficient for activation of mitogen-activated protein kinase and mitogenesis in mouse lung fibroblasts. Loss of responses in fibroblasts from receptor knockout mice. J Biol Chem 271: 21536–21541, 1996. doi: 10.1074/jbc.271.35.21536. [DOI] [PubMed] [Google Scholar]
- 403. Camerer E, Kataoka H, Kahn M, Lease K, Coughlin SR. Genetic evidence that protease-activated receptors mediate factor Xa signaling in endothelial cells. J Biol Chem 277: 16081–16087, 2002. doi: 10.1074/jbc.M108555200. [DOI] [PubMed] [Google Scholar]
- 404. Howell DC, Goldsack NR, Marshall RP, McAnulty RJ, Starke R, Purdy G, Laurent GJ, Chambers RC. Direct thrombin inhibition reduces lung collagen, accumulation, and connective tissue growth factor mRNA levels in bleomycin-induced pulmonary fibrosis. Am J Pathol 159: 1383–1395, 2001. doi: 10.1016/S0002-9440(10)62525-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405. Bogatkevich GS, Tourkina E, Silver RM, Ludwicka-Bradley A. Thrombin differentiates normal lung fibroblasts to a myofibroblast phenotype via the proteolytically activated receptor-1 and a protein kinase C-dependent pathway. J Biol Chem 276: 45184–45192, 2001. doi: 10.1074/jbc.M106441200. [DOI] [PubMed] [Google Scholar]
- 406. Jenkins RG, Su X, Su G, Scotton CJ, Camerer E, Laurent GJ, Davis GE, Chambers RC, Matthay MA, Sheppard D. Ligation of protease-activated receptor 1 enhances alpha(v)beta6 integrin-dependent TGF-beta activation and promotes acute lung injury. J Clin Invest 116: 1606–1614, 2006. doi: 10.1172/JCI27183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407. Fuchs-Buder T, de Moerloose P, Ricou B, Reber G, Vifian C, Nicod L, Romand JA, Suter PM. Time course of procoagulant activity and D dimer in bronchoalveolar fluid of patients at risk for or with acute respiratory distress syndrome. Am J Respir Crit Care Med 153: 163–167, 1996. doi: 10.1164/ajrccm.153.1.8542111. [DOI] [PubMed] [Google Scholar]
- 408. Kipnis E, Guery BP, Tournoys A, Leroy X, Robriquet L, Fialdes P, Neviere R, Fourrier F. Massive alveolar thrombin activation in Pseudomonas aeruginosa-induced acute lung injury. Shock 21: 444–451, 2004. doi: 10.1097/00024382-200405000-00008. [DOI] [PubMed] [Google Scholar]
- 409. Pittet JF, Griffiths MJ, Geiser T, Kaminski N, Dalton SL, Huang X, Brown LA, Gotwals PJ, Koteliansky VE, Matthay MA, Sheppard D. TGF-beta is a critical mediator of acute lung injury. J Clin Invest 107: 1537–1544, 2001. doi: 10.1172/JCI11963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410. Lin C, von der Thusen J, Daalhuisen J, ten Brink M, Crestani B, van der Poll T, Borensztajn K, Spek CA. Protease-activated receptor (PAR)-2 is required for PAR-1 signalling in pulmonary fibrosis. J Cell Mol Med 19: 1346–1356, 2015. doi: 10.1111/jcmm.12520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411. Shapiro PS, Evans JN, Davis RJ, Posada JA. The seven-transmembrane-spanning receptors for endothelin and thrombin cause proliferation of airway smooth muscle cells and activation of the extracellular regulated kinase and c-Jun NH2-terminal kinase groups of mitogen-activated protein kinases. J Biol Chem 271: 5750–5754, 1996. doi: 10.1074/jbc.271.10.5750. [DOI] [PubMed] [Google Scholar]
- 412. Hauck RW, Schulz C, Schomig A, Hoffman RK, Panettieri RA Jr.. alpha-Thrombin stimulates contraction of human bronchial rings by activation of protease-activated receptors. Am J Physiol Lung Cell Mol Physiol 277: L22–L29, 1999. doi: 10.1152/ajplung.1999.277.1.l22. [DOI] [PubMed] [Google Scholar]
- 413. Chow JM, Moffatt JD, Cocks TM. Effect of protease-activated receptor (PAR)-1, -2 and -4-activating peptides, thrombin and trypsin in rat isolated airways. Br J Pharmacol 131: 1584–1591, 2000. doi: 10.1038/sj.bjp.0703738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414. Shrestha Palikhe N, Nahirney D, Laratta C, Gandhi VD, Vethanayagam D, Bhutani M, Mayers I, Cameron L, Vliagoftis H. Increased protease-activated receptor-2 (PAR-2) expression on CD14++CD16+ peripheral blood monocytes of patients with severe asthma. PLoS One 10: e0144500, 2015. doi: 10.1371/journal.pone.0144500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415. Terasawa T, Aso Y, Omori K, Fukushima M, Momobayashi A, Inukai T. Bezafibrate, a peroxisome proliferator-activated receptor alpha agonist, decreases circulating CD14(+)CD16(+) monocytes in patients with type 2 diabetes. Transl Res 165: 336–345, 2015. doi: 10.1016/j.trsl.2014.07.008. [DOI] [PubMed] [Google Scholar]
- 416. Okamoto H, Mizuno K, Horio T. Circulating CD14+ CD16+ monocytes are expanded in sarcoidosis patients. J Dermatol 30: 503–509, 2003. doi: 10.1111/j.1346-8138.2003.tb00424.x. [DOI] [PubMed] [Google Scholar]
- 417. Chambers RC. Procoagulant signalling mechanisms in lung inflammation and fibrosis: novel opportunities for pharmacological intervention? Br J Pharmacol 153: 367–378, 2008. doi: 10.1038/sj.bjp.0707506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418. Mercer PF, Chambers RC. Coagulation and coagulation signalling in fibrosis. Biochim Biophys Acta 1832: 1018–1027, 2013. doi: 10.1016/j.bbadis.2012.12.013. [DOI] [PubMed] [Google Scholar]
- 419. Kawabata A, Kuroda R, Kuroki N, Nishikawa H, Kawai K. Dual modulation by thrombin of the motility of rat oesophageal muscularis mucosae via two distinct protease-activated receptors (PARs): a novel role for PAR-4 as opposed to PAR-1. Br J Pharmacol 131: 578–584, 2000. doi: 10.1038/sj.bjp.0703590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420. Buresi MC, Schleihauf E, Vergnolle N, Buret A, Wallace JL, Hollenberg MD, MacNaughton WK. Protease-activated receptor-1 stimulates Ca2+-dependent Cl- secretion in human intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol 281: G323–G332, 2001. doi: 10.1152/ajpgi.2001.281.2.G323. [DOI] [PubMed] [Google Scholar]
- 421. He L, Ma Y, Li W, Han W, Zhao X, Wang H. Protease-activated receptor 2 signaling modulates susceptibility of colonic epithelium to injury through stabilization of YAP in vivo. Cell Death Dis 9: 949, 2018. doi: 10.1038/s41419-018-0995-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422. Nasri I, Bonnet D, Zwarycz B, d’Aldebert E, Khou S, Mezghani-Jarraya R, Quaranta M, Rolland C, Bonnart C, Mas E, Ferrand A, Cenac N, Magness S, Van Landeghem L, Vergnolle N, Racaud-Sultan C. PAR2-dependent activation of GSK3beta regulates the survival of colon stem/progenitor cells. Am J Physiol Gastrointest Liver Physiol 311: G221–G236, 2016. doi: 10.1152/ajpgi.00328.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423. Chin AC, Vergnolle N, MacNaughton WK, Wallace JL, Hollenberg MD, Buret AG. Proteinase-activated receptor 1 activation induces epithelial apoptosis and increases intestinal permeability. Proc Natl Acad Sci U S A 100: 11104–11109, 2003. doi: 10.1073/pnas.1831452100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424. Hyun E, Andrade-Gordon P, Steinhoff M, Vergnolle N. Protease-activated receptor-2 activation: a major actor in intestinal inflammation. Gut 57: 1222–1229, 2008. doi: 10.1136/gut.2008.150722. [DOI] [PubMed] [Google Scholar]
- 425. Cenac N, Coelho AM, Nguyen C, Compton S, Andrade-Gordon P, MacNaughton WK, Wallace JL, Hollenberg MD, Bunnett NW, Garcia-Villar R, Bueno L, Vergnolle N. Induction of intestinal inflammation in mouse by activation of proteinase-activated receptor-2. Am J Pathol 161: 1903–1915, 2002. doi: 10.1016/S0002-9440(10)64466-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426. Gobbetti T, Cenac N, Motta JP, Rolland C, Martin L, Andrade-Gordon P, Steinhoff M, Barocelli E, Vergnolle N. Serine protease inhibition reduces post-ischemic granulocyte recruitment in mouse intestine. Am J Pathol 180: 141–152, 2012. doi: 10.1016/j.ajpath.2011.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427. Cenac N, Chin AC, Garcia-Villar R, Salvador-Cartier C, Ferrier L, Vergnolle N, Buret AG, Fioramonti J, Bueno L. PAR2 activation alters colonic paracellular permeability in mice via IFN-gamma-dependent and -independent pathways. J Physiol 558: 913–925, 2004. doi: 10.1113/jphysiol.2004.061721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428. Cenac N, Garcia-Villar R, Ferrier L, Larauche M, Vergnolle N, Bunnett NW, Coelho AM, Fioramonti J, Bueno L. Proteinase-activated receptor-2-induced colonic inflammation in mice: possible involvement of afferent neurons, nitric oxide, and paracellular permeability. J Immunol 170: 4296–4300, 2003. doi: 10.4049/jimmunol.170.8.4296. [DOI] [PubMed] [Google Scholar]
- 429. Roka R, Demaude J, Cenac N, Ferrier L, Salvador-Cartier C, Garcia-Villar R, Fioramonti J, Bueno L. Colonic luminal proteases activate colonocyte proteinase-activated receptor-2 and regulate paracellular permeability in mice. Neurogastroenterol Motil 19: 57–65, 2007. doi: 10.1111/j.1365-2982.2006.00851.x. [DOI] [PubMed] [Google Scholar]
- 430. Lee MC, Huang SC. Proteinase-activated receptor-1 (PAR(1)) and PAR(2) but not PAR(4) mediate contraction in human and guinea-pig gallbladders. Neurogastroenterol Motil 20: 385–391, 2008. doi: 10.1111/j.1365-2982.2007.01041.x. [DOI] [PubMed] [Google Scholar]
- 431. Kawabata A, Kawao N, Kuroda R, Tanaka A, Itoh H, Nishikawa H. Peripheral PAR-2 triggers thermal hyperalgesia and nociceptive responses in rats. Neuroreport 12: 715–719, 2001. doi: 10.1097/00001756-200103260-00020. [DOI] [PubMed] [Google Scholar]
- 432. Kawabata A, Kuroda R, Nishida M, Nagata N, Sakaguchi Y, Kawao N, Nishikawa H, Arizono N, Kawai K. Protease-activated receptor-2 (PAR-2) in the pancreas and parotid gland: Immunolocalization and involvement of nitric oxide in the evoked amylase secretion. Life Sci 71: 2435–2446, 2002. doi: 10.1016/S0024-3205(02)02044-1. [DOI] [PubMed] [Google Scholar]
- 433. Karanjia R, Spreadbury I, Bautista-Cruz F, Tsang ME, Vanner S. Activation of protease-activated receptor-4 inhibits the intrinsic excitability of colonic dorsal root ganglia neurons. Neurogastroenterol Motil 21: 1218–1221, 2009. doi: 10.1111/j.1365-2982.2009.01353.x. [DOI] [PubMed] [Google Scholar]
- 434. Hayama T, Kamio N, Okabe T, Muromachi K, Matsushima K. Kallikrein promotes inflammation in human dental pulp cells via protease-activated receptor-1. J Cell Biochem 117: 1522–1528, 2016. doi: 10.1002/jcb.25437. [DOI] [PubMed] [Google Scholar]
- 435. da Silva HA, Euzebio Alves VT, Spolidorio LC, Cesar Neto JB, Eichler RS, de Carvalho MH, Holzhausen M. Expression of protease activated receptor-1 in chronic periodontitis. J Periodontol 85: 1763–1769, 2014. doi: 10.1902/jop.2014.140172. [DOI] [PubMed] [Google Scholar]
- 436. Uehara A, Imamura T, Potempa J, Travis J, Takada H. Gingipains from Porphyromonas gingivalis synergistically induce the production of proinflammatory cytokines through protease-activated receptors with Toll-like receptor and NOD1/2 ligands in human monocytic cells. Cell Microbiol 10: 1181–1189, 2008. doi: 10.1111/j.1462-5822.2008.01119.x. [DOI] [PubMed] [Google Scholar]
- 437. Curtis MA, Kuramitsu HK, Lantz M, Macrina FL, Nakayama K, Potempa J, Reynolds EC, Aduse-Opoku J. Molecular genetics and nomenclature of proteases of Porphyromonas gingivalis. J Periodontal Res 34: 464–472, 1999. doi: 10.1111/j.1600-0765.1999.tb02282.x. [DOI] [PubMed] [Google Scholar]
- 438. Jouet P, Sarna SK, Singaram C, Ryan RP, Hillard CJ, Telford GL, Fink J, Henderson JD. Immunocytes and abnormal gastrointestinal motor activity during ileitis in dogs. Am J Physiol Gastrointest Liver Physiol 269: G913–G924, 1995. doi: 10.1152/ajpgi.1995.269.6.g913. [DOI] [PubMed] [Google Scholar]
- 439. Kawabata A, Kuroda R, Kuroki N, Nishikawa H, Kawai K, Araki H. Characterization of the protease-activated receptor-1-mediated contraction and relaxation in the rat duodenal smooth muscle. Life Sci 67: 2521–2530, 2000. doi: 10.1016/S0024-3205(00)00835-3. [DOI] [PubMed] [Google Scholar]
- 440. Mule F, Baffi MC, Cerra MC. Dual effect mediated by protease-activated receptors on the mechanical activity of rat colon. Br J Pharmacol 136: 367–374, 2002. doi: 10.1038/sj.bjp.0704746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441. Sato K, Ninomiya H, Ohkura S, Ozaki H, Nasu T. Impairment of PAR-2-mediated relaxation system in colonic smooth muscle after intestinal inflammation. Br J Pharmacol 148: 200–207, 2006. doi: 10.1038/sj.bjp.0706717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442. Kim JA, Choi SC, Yun KJ, Kim DK, Han MK, Seo GS, Yeom JJ, Kim TH, Nah YH, Lee YM. Expression of protease-activated receptor 2 in ulcerative colitis. Inflamm Bowel Dis 9: 224–229, 2003. doi: 10.1097/00054725-200307000-00002. [DOI] [PubMed] [Google Scholar]
- 443. Cottrell GS, Amadesi S, Grady EF, Bunnett NW. Trypsin IV, a novel agonist of protease-activated receptors 2 and 4. J Biol Chem 279: 13532–13539, 2004. doi: 10.1074/jbc.M312090200. [DOI] [PubMed] [Google Scholar]
- 444. Vergnolle N. Modulation of visceral pain and inflammation by protease-activated receptors. Br J Pharmacol 141: 1264–1274, 2004. doi: 10.1038/sj.bjp.0705750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445. Nguyen C, Coelho AM, Grady E, Compton SJ, Wallace JL, Hollenberg MD, Cenac N, Garcia-Villar R, Bueno L, Steinhoff M, Bunnett NW, Vergnolle N. Colitis induced by proteinase-activated receptor-2 agonists is mediated by a neurogenic mechanism. Can J Physiol Pharmacol 81: 920–927, 2003. doi: 10.1139/y03-080. [DOI] [PubMed] [Google Scholar]
- 446. Van Spaendonk H, Ceuleers H, Witters L, Patteet E, Joossens J, Augustyns K, Lambeir AM, De Meester I, De Man JG, Winter BY. Regulation of intestinal permeability: the role of proteases. World J Gastroenterol 23: 2106–2123, 2017. doi: 10.3748/wjg.v23.i12.2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447. Maharshak N, Huh EY, Paiboonrungruang C, Shanahan M, Thurlow L, Herzog J, Djukic Z, Orlando R, Pawlinski R, Ellermann M, Borst L, Patel S, Dotan I, Sartor RB, Carroll IM. Enterococcus faecalis gelatinase mediates intestinal permeability via protease-activated receptor 2. Infect Immun 83: 2762–2770, 2015. doi: 10.1128/IAI.00425-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448. Hyun E, Andrade-Gordon P, Steinhoff M, Beck PL, Vergnolle N. Contribution of bone marrow-derived cells to the pro-inflammatory effects of protease-activated receptor-2 in colitis. Inflamm Res 59: 699–709, 2010. doi: 10.1007/s00011-010-0181-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449. Tomuschat C, O’Donnell AM, Coyle D, Puri P. Increased protease activated receptors in the colon of patients with Hirschsprung’s disease. J Pediatr Surg 55: 1488–1494, 2020. doi: 10.1016/j.jpedsurg.2019.11.009. [DOI] [PubMed] [Google Scholar]
- 450. Cenac N, Andrews CN, Holzhausen M, Chapman K, Cottrell G, Andrade-Gordon P, Steinhoff M, Barbara G, Beck P, Bunnett NW, Sharkey KA, Ferraz JG, Shaffer E, Vergnolle N. Role for protease activity in visceral pain in irritable bowel syndrome. J Clin Invest 117: 636–647, 2007. doi: 10.1172/JCI29255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 451. Vergnolle N, Wallace JL, Bunnett NW, Hollenberg MD. Protease-activated receptors in inflammation, neuronal signaling and pain. Trends Pharmacol Sci 22: 146–152, 2001. doi: 10.1016/S0165-6147(00)01634-5. [DOI] [PubMed] [Google Scholar]
- 452. Coelho AM, Vergnolle N, Guiard B, Fioramonti J, Bueno L. Proteinases and proteinase-activated receptor 2: a possible role to promote visceral hyperalgesia in rats. Gastroenterology 122: 1035–1047, 2002. doi: 10.1053/gast.2002.32387. [DOI] [PubMed] [Google Scholar]
- 453. Buhner S, Hahne H, Hartwig K, Li Q, Vignali S, Ostertag D, Meng C, Hormannsperger G, Braak B, Pehl C, Frieling T, Barbara G, De Giorgio R, Demir IE, Ceyhan GO, Zeller F, Boeckxstaens G, Haller D, Kuster B, Schemann M. Protease signaling through protease activated receptor 1 mediate nerve activation by mucosal supernatants from irritable bowel syndrome but not from ulcerative colitis patients. PLoS One 13: e0193943, 2018. doi: 10.1371/journal.pone.0193943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454. Desormeaux C, Bautzova T, Garcia-Caraballo S, Rolland C, Barbaro MR, Brierley SM, Barbara G, Vergnolle N, Cenac N. Protease-activated receptor 1 is implicated in irritable bowel syndrome mediators-induced signaling to thoracic human sensory neurons. Pain 159: 1257–1267, 2018. doi: 10.1097/j.pain.0000000000001208. [DOI] [PubMed] [Google Scholar]
- 455. Cattaruzza F, Spreadbury I, Miranda-Morales M, Grady EF, Vanner S, Bunnett NW. Transient receptor potential ankyrin-1 has a major role in mediating visceral pain in mice. Am J Physiol Gastrointest Liver Physiol 298: G81–G91, 2010. doi: 10.1152/ajpgi.00221.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456. Suzuki M, Mizuno A, Kodaira K, Imai M. Impaired pressure sensation in mice lacking TRPV4. J Biol Chem 278: 22664–22668, 2003. doi: 10.1074/jbc.M302561200. [DOI] [PubMed] [Google Scholar]
- 457. Cervero F, Laird JM. Visceral pain. Lancet 353: 2145–2148, 1999. doi: 10.1016/S0140-6736(99)01306-9. [DOI] [PubMed] [Google Scholar]
- 458. Clark AK, Gentry C, Bradbury EJ, McMahon SB, Malcangio M. Role of spinal microglia in rat models of peripheral nerve injury and inflammation. Eur J Pain 11: 223–230, 2007. doi: 10.1016/j.ejpain.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 459. Clark AK, Yip PK, Malcangio M. The liberation of fractalkine in the dorsal horn requires microglial cathepsin S. J Neurosci 29: 6945–6954, 2009. doi: 10.1523/JNEUROSCI.0828-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460. Cattaruzza F, Lyo V, Jones E, Pham D, Hawkins J, Kirkwood K, Valdez-Morales E, Ibeakanma C, Vanner SJ, Bogyo M, Bunnett NW. Cathepsin S is activated during colitis and causes visceral hyperalgesia by a PAR2-dependent mechanism in mice. Gastroenterology 141: 1864–1874, 2011. doi: 10.1053/j.gastro.2011.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461. Marra F, DeFranco R, Grappone C, Milani S, Pinzani M, Pellegrini G, Laffi G, Gentilini P. Expression of the thrombin receptor in human liver: up-regulation during acute and chronic injury. Hepatology 27: 462–471, 1998. doi: 10.1002/hep.510270221. [DOI] [PubMed] [Google Scholar]
- 462. Margaretten W, McKay DG, Phillips LL. The effect of heparin on endotoxin shock in the rat. Am J Pathol 51: 61–68, 1967. [PMC free article] [PubMed] [Google Scholar]
- 463. Moulin F, Copple BL, Ganey PE, Roth RA. Hepatic and extrahepatic factors critical for liver injury during lipopolysaccharide exposure. Am J Physiol Gastrointest Liver Physiol 281: G1423–G1431, 2001. doi: 10.1152/ajpgi.2001.281.6.G1423. [DOI] [PubMed] [Google Scholar]
- 464. Copple BL, Moulin F, Hanumegowda UM, Ganey PE, Roth RA. Thrombin and protease-activated receptor-1 agonists promote lipopolysaccharide-induced hepatocellular injury in perfused livers. J Pharmacol Exp Ther 305: 417–425, 2003. doi: 10.1124/jpet.102.046391. [DOI] [PubMed] [Google Scholar]
- 465. Olejar T, Matej R, Zadinova M, Pouckova P. Expression of proteinase-activated receptor 2 during taurocholate-induced acute pancreatic lesion development in Wistar rats. Int J Gastrointest Cancer 30: 113–121, 2001. doi: 10.1385/IJGC:30:3:113. [DOI] [PubMed] [Google Scholar]
- 466. Nguyen TD, Moody MW, Steinhoff M, Okolo C, Koh DS, Bunnett NW. Trypsin activates pancreatic duct epithelial cell ion channels through proteinase-activated receptor-2. J Clin Invest 103: 261–269, 1999. doi: 10.1172/JCI2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467. Chen R, Brentnall TA, Pan S, Cooke K, Moyes KW, Lane Z, Crispin DA, Goodlett DR, Aebersold R, Bronner MP. Quantitative proteomics analysis reveals that proteins differentially expressed in chronic pancreatitis are also frequently involved in pancreatic cancer. Mol Cell Proteomics 6: 1331–1342, 2007. doi: 10.1074/mcp.M700072-MCP200. [DOI] [PubMed] [Google Scholar]
- 468. Edgington-Mitchell LE, Wartmann T, Fleming AK, Gocheva V, van der Linden WA, Withana NP, Verdoes M, Aurelio L, Edgington-Mitchell D, Lieu T, Parker BS, Graham B, Reinheckel T, Furness JB, Joyce JA, Storz P, Halangk W, Bogyo M, Bunnett NW. Legumain is activated in macrophages during pancreatitis. Am J Physiol Gastrointest Liver Physiol 311: G548–G560, 2016. doi: 10.1152/ajpgi.00047.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469. Lessa FC, Mu Y, Bamberg WM, Beldavs ZG, Dumyati GK, Dunn JR, Farley MM, Holzbauer SM, Meek JI, Phipps EC, Wilson LE, Winston LG, Cohen JA, Limbago BM, Fridkin SK, Gerding DN, McDonald LC. Burden of Clostridium difficile infection in the United States. N Engl J Med 372: 825–834, 2015. doi: 10.1056/NEJMoa1408913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470. Hall AJ, Curns AT, McDonald LC, Parashar UD, Lopman BA. The roles of Clostridium difficile and norovirus among gastroenteritis-associated deaths in the United States, 1999–2007. Clin Infect Dis 55: 216–223, 2012. doi: 10.1093/cid/cis386. [DOI] [PubMed] [Google Scholar]
- 471. Bradley PP, Priebat DA, Christensen RD, Rothstein G. Measurement of cutaneous inflammation: estimation of neutrophil content with an enzyme marker. J Invest Dermatol 78: 206–209, 1982. doi: 10.1111/1523-1747.ep12506462. [DOI] [PubMed] [Google Scholar]
- 472. Cottrell GS, Amadesi S, Pikios S, Camerer E, Willardsen JA, Murphy BR, Caughey GH, Wolters PJ, Coughlin SR, Peterson A, Knecht W, Pothoulakis C, Bunnett NW, Grady EF. Protease-activated receptor 2, dipeptidyl peptidase I, and proteases mediate Clostridium difficile toxin A enteritis. Gastroenterology 132: 2422–2437, 2007. doi: 10.1053/j.gastro.2007.03.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473. Reed DE, Barajas-Lopez C, Cottrell G, Velazquez-Rocha S, Dery O, Grady EF, Bunnett NW, Vanner SJ. Mast cell tryptase and proteinase-activated receptor 2 induce hyperexcitability of guinea-pig submucosal neurons. J Physiol 547: 531–542, 2003. doi: 10.1113/jphysiol.2002.032011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474. Roosterman D, Cottrell GS, Schmidlin F, Steinhoff M, Bunnett NW. Recycling and resensitization of the neurokinin 1 receptor. Influence of agonist concentration and Rab GTPases. J Biol Chem 279: 30670–30679, 2004. doi: 10.1074/jbc.M402479200. [DOI] [PubMed] [Google Scholar]
- 475. Knecht W, Cottrell GS, Amadesi S, Mohlin J, Skaregarde A, Gedda K, Peterson A, Chapman K, Hollenberg MD, Vergnolle N, Bunnett NW. Trypsin IV or mesotrypsin and p23 cleave protease-activated receptors 1 and 2 to induce inflammation and hyperalgesia. J Biol Chem 282: 26089–26100, 2007. doi: 10.1074/jbc.M703840200. [DOI] [PubMed] [Google Scholar]
- 476. Wiegand U, Corbach S, Minn A, Kang J, Muller-Hill B. Cloning of the cDNA encoding human brain trypsinogen and characterization of its product. Gene 136: 167–175, 1993. doi: 10.1016/0378-1119(93)90460-K. [DOI] [PubMed] [Google Scholar]
- 477. Jalink K, Moolenaar WH. Thrombin receptor activation causes rapid neural cell rounding and neurite retraction independent of classic second messengers. J Cell Biol 118: 411–419, 1992. doi: 10.1083/jcb.118.2.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 478. Suidan HS, Stone SR, Hemmings BA, Monard D. Thrombin causes neurite retraction in neuronal cells through activation of cell surface receptors. Neuron 8: 363–375, 1992. doi: 10.1016/0896-6273(92)90302-T. [DOI] [PubMed] [Google Scholar]
- 479. Beecher KL, Andersen TT, Fenton JW 2nd, Festoff BW. Thrombin receptor peptides induce shape change in neonatal murine astrocytes in culture. J Neurosci Res 37: 108–115, 1994. doi: 10.1002/jnr.490370115. [DOI] [PubMed] [Google Scholar]
- 480. Smith-Swintosky VL, Zimmer S, Fenton JW 2nd, Mattson MP. Opposing actions of thrombin and protease nexin-1 on amyloid beta-peptide toxicity and on accumulation of peroxides and calcium in hippocampal neurons. J Neurochem 65: 1415–1418, 1995. doi: 10.1046/j.1471-4159.1995.65031415.x. [DOI] [PubMed] [Google Scholar]
- 481. Vaughan PJ, Pike CJ, Cotman CW, Cunningham DD. Thrombin receptor activation protects neurons and astrocytes from cell death produced by environmental insults. J Neurosci 15: 5389–5401, 1995. doi: 10.1523/JNEUROSCI.15-07-05389.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 482. Festoff BW, Smirnova IV, Ma J, Citron BA. Thrombin, its receptor and protease nexin I, its potent serpin, in the nervous system. Semin Thromb Hemost 22: 267–271, 1996. doi: 10.1055/s-2007-999018. [DOI] [PubMed] [Google Scholar]
- 483. Donovan FM, Pike CJ, Cotman CW, Cunningham DD. Thrombin induces apoptosis in cultured neurons and astrocytes via a pathway requiring tyrosine kinase and RhoA activities. J Neurosci 17: 5316–5326, 1997. doi: 10.1523/JNEUROSCI.17-14-05316.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 484. Suidan HS, Bouvier J, Schaerer E, Stone SR, Monard D, Tschopp J. Granzyme A released upon stimulation of cytotoxic T lymphocytes activates the thrombin receptor on neuronal cells and astrocytes. Proc Natl Acad Sci U S A 91: 8112–8116, 1994. doi: 10.1073/pnas.91.17.8112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 485. Cavanaugh KP, Gurwitz D, Cunningham DD, Bradshaw RA. Reciprocal modulation of astrocyte stellation by thrombin and protease nexin-1. J Neurochem 54: 1735–1743, 1990. doi: 10.1111/j.1471-4159.1990.tb01228.x. [DOI] [PubMed] [Google Scholar]
- 486. Grabham P, Cunningham DD. Thrombin receptor activation stimulates astrocyte proliferation and reversal of stellation by distinct pathways: involvement of tyrosine phosphorylation. J Neurochem 64: 583–591, 2002. doi: 10.1046/j.1471-4159.1995.64020583.x. [DOI] [PubMed] [Google Scholar]
- 487. Emilsson K, Wahlestedt C, Sun MK, Nystedt S, Owman C, Sundelin J. Vascular effects of proteinase-activated receptor 2 agonist peptide. J Vasc Res 34: 267–272, 1997. doi: 10.1159/000159233. [DOI] [PubMed] [Google Scholar]
- 488. Kawabata A, Kuroda R, Minami T, Kataoka K, Taneda M. Increased vascular permeability by a specific agonist of protease-activated receptor-2 in rat hindpaw. Br J Pharmacol 125: 419–422, 1998. doi: 10.1038/sj.bjp.0702063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489. Vergnolle N. Proteinase-activated receptor-2-activating peptides induce leukocyte rolling, adhesion, and extravasation in vivo. J Immunol 163: 5064–5069, 1999. [PubMed] [Google Scholar]
- 490. Brain SD, Williams TJ, Tippins JR, Morris HR, MacIntyre I. Calcitonin gene-related peptide is a potent vasodilator. Nature 313: 54–56, 1985. doi: 10.1038/313054a0. [DOI] [PubMed] [Google Scholar]
- 491. Hughes SR, Brain SD. A calcitonin gene-related peptide (CGRP) antagonist (CGRP8-37) inhibits microvascular responses induced by CGRP and capsaicin in skin. Br J Pharmacol 104: 738–742, 1991. doi: 10.1111/j.1476-5381.1991.tb12497.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 492. Bowden JJ, Garland AM, Baluk P, Lefevre P, Grady EF, Vigna SR, Bunnett NW, McDonald DM. Direct observation of substance P-induced internalization of neurokinin 1 (NK1) receptors at sites of inflammation. Proc Natl Acad Sci U S A 91: 8964–8968, 1994. doi: 10.1073/pnas.91.19.8964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 493. Baluk P, Bertrand C, Geppetti P, McDonald DM, Nadel JA. NK1 receptors mediate leukocyte adhesion in neurogenic inflammation in the rat trachea. Am J Physiol Lung Cell Mol Physiol 268: L263–L269, 1995. doi: 10.1152/ajplung.1995.268.2.l263. [DOI] [PubMed] [Google Scholar]
- 494. Kawabata A, Kawao N, Kuroda R, Tanaka A, Shimada C. The PAR-1-activating peptide attenuates carrageenan-induced hyperalgesia in rats. Peptides 23: 1181–1183, 2002. doi: 10.1016/S0196-9781(02)00053-0. [DOI] [PubMed] [Google Scholar]
- 495. Asfaha S, Brussee V, Chapman K, Zochodne DW, Vergnolle N. Proteinase-activated receptor-1 agonists attenuate nociception in response to noxious stimuli. Br J Pharmacol 135: 1101–1106, 2002. doi: 10.1038/sj.bjp.0704568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 496. Wang Y, Luo W, Wartmann T, Halangk W, Sahin-Toth M, Reiser G. Mesotrypsin, a brain trypsin, activates selectively proteinase-activated receptor-1, but not proteinase-activated receptor-2, in rat astrocytes. J Neurochem 99: 759–769, 2006. doi: 10.1111/j.1471-4159.2006.04105.x. [DOI] [PubMed] [Google Scholar]
- 497. Sun H, Meeker S, Undem BJ. Role of TRP channels in Gq-coupled protease-activated receptor 1-mediated activation of mouse nodose pulmonary C-fibers. Am J Physiol Lung Cell Mol Physiol 318: L192–L199, 2020. doi: 10.1152/ajplung.00301.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498. Moss CR 2nd, Gilbert CA, Gabriel SA, Gu Q. Protease-activated receptor-2 inhibits BK channel activity in bronchopulmonary sensory neurons. Neurosci Lett 589: 13–18, 2015. doi: 10.1016/j.neulet.2015.01.020. [DOI] [PubMed] [Google Scholar]
- 499. Gu Q, Lee LY. House dust mite potentiates capsaicin-evoked Ca2+ transients in mouse pulmonary sensory neurons via activation of protease-activated receptor-2. Exp Physiol 97: 534–543, 2012. doi: 10.1113/expphysiol.2011.060764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 500. Linley JE, Rose K, Patil M, Robertson B, Akopian AN, Gamper N. Inhibition of M current in sensory neurons by exogenous proteases: a signaling pathway mediating inflammatory nociception. J Neurosci 28: 11240–11249, 2008. doi: 10.1523/JNEUROSCI.2297-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 501. Perraud F, Besnard F, Sensenbrenner M, Labourdette G. Thrombin is a potent mitogen for rat astroblasts but not for oligodendroblasts and neuroblasts in primary culture. Int J Dev Neurosci 5: 181–188, 1987. doi: 10.1016/0736-5748(87)90028-1. [DOI] [PubMed] [Google Scholar]
- 502. Ehrenreich H, Costa T, Clouse KA, Pluta RM, Ogino Y, Coligan JE, Burd PR. Thrombin is a regulator of astrocytic endothelin-1. Brain Res 600: 201–207, 1993. doi: 10.1016/0006-8993(93)91374-2. [DOI] [PubMed] [Google Scholar]
- 503. Gale AJ, Tsavaler A, Griffin JH. Molecular characterization of an extended binding site for coagulation factor Va in the positive exosite of activated protein C. J Biol Chem 277: 28836–28840, 2002. doi: 10.1074/jbc.M204363200. [DOI] [PubMed] [Google Scholar]
- 504. Mosnier LO, Gale AJ, Yegneswaran S, Griffin JH. Activated protein C variants with normal cytoprotective but reduced anticoagulant activity. Blood 104: 1740–1744, 2004. doi: 10.1182/blood-2004-01-0110, . [DOI] [PubMed] [Google Scholar]
- 505. Mosnier LO, Zlokovic BV, Griffin JH. The cytoprotective protein C pathway. Blood 109: 3161–3172, 2007. doi: 10.1182/blood-2006-09-003004. [DOI] [PubMed] [Google Scholar]
- 506. Zhang P, Huang W, Wang L, Bao L, Jia ZJ, Bauer SM, Goldman EA, Probst GD, Song Y, Su T, Fan J, Wu Y, Li W, Woolfrey J, Sinha U, Wong PW, Edwards ST, Arfsten AE, Clizbe LA, Kanter J, Pandey A, Park G, Hutchaleelaha A, Lambing JL, Hollenbach SJ, Scarborough RM, Zhu BY. Discovery of betrixaban (PRT054021), N-(5-chloropyridin-2-yl)-2-(4-(N,N-dimethylcarbamimidoyl)benzamido)-5-methoxybenzamide, a highly potent, selective, and orally efficacious factor Xa inhibitor. Bioorg Med Chem Lett 19: 2179–2185, 2009. doi: 10.1016/j.bmcl.2009.02.111. [DOI] [PubMed] [Google Scholar]
- 507. Walker CT, Marky AH, Petraglia AL, Ali T, Chow N, Zlokovic BV. Activated protein C analog with reduced anticoagulant activity improves functional recovery and reduces bleeding risk following controlled cortical impact. Brain Res 1347: 125–131, 2010. doi: 10.1016/j.brainres.2010.05.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 508. Guo H, Wang Y, Singh I, Liu D, Fernandez JA, Griffin JH, Chow N, Zlokovic BV. Species-dependent neuroprotection by activated protein C mutants with reduced anticoagulant activity. J Neurochem 109: 116–124, 2009. doi: 10.1111/j.1471-4159.2009.05921.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 509. Guo H, Liu D, Gelbard H, Cheng T, Insalaco R, Fernandez JA, Griffin JH, Zlokovic BV. Activated protein C prevents neuronal apoptosis via protease activated receptors 1 and 3. Neuron 41: 563–572, 2004. doi: 10.1016/S0896-6273(04)00019-4. [DOI] [PubMed] [Google Scholar]
- 510. Guo H, Singh I, Wang Y, Deane R, Barrett T, Fernandez JA, Chow N, Griffin JH, Zlokovic BV. Neuroprotective activities of activated protein C mutant with reduced anticoagulant activity. Eur J Neurosci 29: 1119–1130, 2009. doi: 10.1111/j.1460-9568.2009.06664.x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 511. Liu D, Cheng T, Guo H, Fernandez JA, Griffin JH, Song X, Zlokovic BV. Tissue plasminogen activator neurovascular toxicity is controlled by activated protein C. Nat Med 10: 1379–1383, 2004. doi: 10.1038/nm1122. [DOI] [PubMed] [Google Scholar]
- 512. Cheng T, Petraglia AL, Li Z, Thiyagarajan M, Zhong Z, Wu Z, Liu D, Maggirwar SB, Deane R, Fernandez JA, LaRue B, Griffin JH, Chopp M, Zlokovic BV. Activated protein C inhibits tissue plasminogen activator-induced brain hemorrhage. Nat Med 12: 1278–1285, 2006. doi: 10.1038/nm1498. [DOI] [PubMed] [Google Scholar]
- 513. Wang Y, Thiyagarajan M, Chow N, Singh I, Guo H, Davis TP, Zlokovic BV. Differential neuroprotection and risk for bleeding from activated protein C with varying degrees of anticoagulant activity. Stroke 40: 1864–1869, 2009. doi: 10.1161/STROKEAHA.108.536680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 514. Thiyagarajan M, Fernandez JA, Lane SM, Griffin JH, Zlokovic BV. Activated protein C promotes neovascularization and neurogenesis in postischemic brain via protease-activated receptor 1. J Neurosci 28: 12788–12797, 2008. doi: 10.1523/JNEUROSCI.3485-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 515. Petraglia AL, Marky AH, Walker C, Thiyagarajan M, Zlokovic BV. Activated protein C is neuroprotective and mediates new blood vessel formation and neurogenesis after controlled cortical impact. Neurosurgery 66: 165–171, 2010. doi: 10.1227/01.NEU.0000363148.49779.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 516. Goldman SA, Kirschenbaum B, Harrison-Restelli C, Thaler HT. Neuronal precursors of the adult rat subependymal zone persist into senescence, with no decline in spatial extent or response to BDNF. J Neurobiol 32: 554–566, 1997. doi:. [DOI] [PubMed] [Google Scholar]
- 517. Nakamura M, Tsuji O, Bregman BS, Toyama Y, Okano H. Mimicking the neurotrophic factor profile of embryonic spinal cord controls the differentiation potential of spinal progenitors into neuronal cells. PLoS One 6: e20717, 2011. doi: 10.1371/journal.pone.0020717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 518. Zhong Z, Ilieva H, Hallagan L, Bell R, Singh I, Paquette N, Thiyagarajan M, Deane R, Fernandez JA, Lane S, Zlokovic AB, Liu T, Griffin JH, Chow N, Castellino FJ, Stojanovic K, Cleveland DW, Zlokovic BV. Activated protein C therapy slows ALS-like disease in mice by transcriptionally inhibiting SOD1 in motor neurons and microglia cells. J Clin Invest 119: 3437–3449, 2009. doi: 10.1172/jci38476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 519. Guo D, Zhou H, Wu Y, Zhou F, Xu G, Wen H, Zhang X. Involvement of ERK1/2/NF-kappaB signal transduction pathway in TF/FVIIa/PAR2-induced proliferation and migration of colon cancer cell SW620. Tumour Biol 32: 921–930, 2011. doi: 10.1007/s13277-011-0194-1. [DOI] [PubMed] [Google Scholar]
- 520. Kimura A, Ohmori T, Ohkawa R, Madoiwa S, Mimuro J, Murakami T, Kobayashi E, Hoshino Y, Yatomi Y, Sakata Y. Essential roles of sphingosine 1-phosphate/S1P1 receptor axis in the migration of neural stem cells toward a site of spinal cord injury. Stem Cells 25: 115–124, 2007. doi: 10.1634/stemcells.2006-0223. [DOI] [PubMed] [Google Scholar]
- 521. Nishino A, Suzuki M, Ohtani H, Motohashi O, Umezawa K, Nagura H, Yoshimoto T. Thrombin may contribute to the pathophysiology of central nervous system injury. J Neurotrauma 10: 167–179, 1993. doi: 10.1089/neu.1993.10.167. [DOI] [PubMed] [Google Scholar]
- 522. Motohashi O, Suzuki M, Shida N, Umezawa K, Sugai K, Yoshimoto T. Hirudin suppresses the invasion of inflammatory cells and the appearance of vimentin-positive astrocytes in the rat cerebral ablation model. J Neurotrauma 14: 747–754, 1997. doi: 10.1089/neu.1997.14.747. [DOI] [PubMed] [Google Scholar]
- 523. Noorbakhsh F, Tsutsui S, Vergnolle N, Boven LA, Shariat N, Vodjgani M, Warren KG, Andrade-Gordon P, Hollenberg MD, Power C. Proteinase-activated receptor 2 modulates neuroinflammation in experimental autoimmune encephalomyelitis and multiple sclerosis. J Exp Med 203: 425–435, 2006. doi: 10.1084/jem.20052148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 524. Lee PR, Johnson TP, Gnanapavan S, Giovannoni G, Wang T, Steiner JP, Medynets M, Vaal MJ, Gartner V, Nath A. Protease-activated receptor-1 activation by granzyme B causes neurotoxicity that is augmented by interleukin-1beta. J Neuroinflammation 14: 131, 2017. doi: 10.1186/s12974-017-0901-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 525. Niclou SP, Suidan HS, Pavlik A, Vejsada R, Monard D. Changes in the expression of protease-activated receptor 1 and protease nexin-1 mRNA during rat nervous system development and after nerve lesion. Eur J Neurosci 10: 1590–1607, 1998. doi: 10.1046/j.1460-9568.1998.00183.x. [DOI] [PubMed] [Google Scholar]
- 526. Striggow F, Riek-Burchardt M, Kiesel A, Schmidt W, Henrich-Noack P, Breder J, Krug M, Reymann KG, Reiser G. Four different types of protease-activated receptors are widely expressed in the brain and are up-regulated in hippocampus by severe ischemia. Eur J Neurosci 14: 595–608, 2001. doi: 10.1046/j.0953-816x.2001.01676.x. [DOI] [PubMed] [Google Scholar]
- 527. Moller T, Hanisch UK, Ransom BR. Thrombin-induced activation of cultured rodent microglia. J Neurochem 75: 1539–1547, 2000. doi: 10.1046/j.1471-4159.2000.0751539.x. [DOI] [PubMed] [Google Scholar]
- 528. Boven LA, Vergnolle N, Henry SD, Silva C, Imai Y, Holden J, Warren K, Hollenberg MD, Power C. Up-regulation of proteinase-activated receptor 1 expression in astrocytes during HIV encephalitis. J Immunol 170: 2638–2646, 2003. doi: 10.4049/jimmunol.170.5.2638. [DOI] [PubMed] [Google Scholar]
- 529. Rodriguez-Franco EJ, Cantres-Rosario YM, Plaud-Valentin M, Romeu R, Rodriguez Y, Skolasky R, Melendez V, Cadilla CL, Melendez LM. Dysregulation of macrophage-secreted cathepsin B contributes to HIV-1-linked neuronal apoptosis. PLoS One 7: e36571, 2012. doi: 10.1371/journal.pone.0036571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 530. Sachan V, Lodge R, Mihara K, Hamelin J, Power C, Gelman BB, Hollenberg MD, Cohen EA, Seidah NG. HIV-induced neuroinflammation: impact of PAR1 and PAR2 processing by Furin. Cell Death Differ 26: 1942–1954, 2019. doi: 10.1038/s41418-018-0264-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 531. Bizios R, Lai L, Fenton JW 2nd, Malik AB. Thrombin-induced thromboxane generation by neutrophils and lymphocytes: dependence on enzymic site. J Cell Physiol 132: 359–362, 1987. doi: 10.1002/jcp.1041320224. [DOI] [PubMed] [Google Scholar]
- 532. Morin A, Marchand-Arvier M, Doutremepuich C, Vigneron C. Effect of hirudin on the chemotactic properties of alpha-thrombin. Haemostasis 21, Suppl 1: 32–35, 1991. doi: 10.1159/000216260. [DOI] [PubMed] [Google Scholar]
- 533. Rowand JK, Marucha P, Berliner LJ. Hirudin C-terminal fragments inhibit thrombin induced neutrophil chemotaxis. Thromb Haemost 67: 289–291, 1992. doi: 10.1055/s-0038-1648433. [DOI] [PubMed] [Google Scholar]
- 534. Shpacovitch VM, Seeliger S, Huber-Lang M, Balkow S, Feld M, Hollenberg MD, Sarma VJ, Ward PA, Strey A, Gerke V, Sommerhoff CP, Vergnolle N, Steinhoff M. Agonists of proteinase-activated receptor-2 affect transendothelial migration and apoptosis of human neutrophils. Exp Dermatol 16: 799–806, 2007. doi: 10.1111/j.1600-0625.2007.00605.x. [DOI] [PubMed] [Google Scholar]
- 535. Fields RC, Schoenecker JG, Hart JP, Hoffman MR, Pizzo SV, Lawson JH. Protease-activated receptor-2 signaling triggers dendritic cell development. Am J Pathol 162: 1817–1822, 2003. doi: 10.1016/S0002-9440(10)64316-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 536. Ramelli G, Fuertes S, Narayan S, Busso N, Acha-Orbea H, So A. Protease-activated receptor 2 signalling promotes dendritic cell antigen transport and T-cell activation in vivo. Immunology 129: 20–27, 2010. doi: 10.1111/j.1365-2567.2009.03144.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 537. Camerer E, Cornelissen I, Kataoka H, Duong DN, Zheng YW, Coughlin SR. Roles of protease-activated receptors in a mouse model of endotoxemia. Blood 107: 3912–3921, 2006. doi: 10.1182/blood-2005-08-3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 538. Kazerani HR, Plevin R, Kawagoe J, Kanke T, Furman BL. Lack of effect of proteinase-activated receptor-2 (PAR-2) deletion on the pathophysiological changes produced by lipopolysaccharide in the mouse: comparison with dexamethasone. J Pharm Pharmacol 56: 1015–1020, 2004. doi: 10.1211/0022357043923. [DOI] [PubMed] [Google Scholar]
- 539. Suckow SK, Caudle RM. Identification and immunohistochemical characterization of colospinal afferent neurons in the rat. Neuroscience 153: 803–813, 2008. doi: 10.1016/j.neuroscience.2008.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 540. Sevigny LM, Zhang P, Bohm A, Lazarides K, Perides G, Covic L, Kuliopulos A. Interdicting protease-activated receptor-2-driven inflammation with cell-penetrating pepducins. Proc Natl Acad Sci U S A 108: 8491–8496, 2011. doi: 10.1073/pnas.1017091108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 541. Hyun E, Ramachandran R, Cenac N, Houle S, Rousset P, Saxena A, Liblau RS, Hollenberg MD, Vergnolle N. Insulin modulates protease-activated receptor 2 signaling: implications for the innate immune response. J Immunol 184: 2702–2709, 2010. doi: 10.4049/jimmunol.0902171. [DOI] [PubMed] [Google Scholar]
- 542. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 100: 57–70, 2000. doi: 10.1016/S0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 543. Bar-Shavit R, Maoz M, Kancharla A, Jaber M, Agranovich D, Grisaru-Granovsky S, Uziely B. Protease-activated receptors (PARs) in cancer: novel biased signaling and targets for therapy. Methods Cell Biol 132: 341–358, 2016. doi: 10.1016/bs.mcb.2015.11.006. [DOI] [PubMed] [Google Scholar]
- 544. White MJ, Gomer RH. Trypsin, tryptase, and thrombin polarize macrophages towards a pro-fibrotic M2a phenotype. PLoS One 10: e0138748, 2015. doi: 10.1371/journal.pone.0138748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 545. Ducroc R, Bontemps C, Marazova K, Devaud H, Darmoul D, Laburthe M. Trypsin is produced by and activates protease-activated receptor-2 in human cancer colon cells: evidence for new autocrine loop. Life Sci 70: 1359–1367, 2002. doi: 10.1016/S0024-3205(01)01519-3. [DOI] [PubMed] [Google Scholar]
- 546. Malfettone A, Silvestris N, Saponaro C, Ranieri G, Russo A, Caruso S, Popescu O, Simone G, Paradiso A, Mangia A. High density of tryptase-positive mast cells in human colorectal cancer: a poor prognostic factor related to protease-activated receptor 2 expression. J Cell Mol Med 17: 1025–1037, 2013. doi: 10.1111/jcmm.12073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 547. Jin E, Fujiwara M, Pan X, Ghazizadeh M, Arai S, Ohaki Y, Kajiwara K, Takemura T, Kawanami O. Protease-activated receptor (PAR)-1 and PAR-2 participate in the cell growth of alveolar capillary endothelium in primary lung adenocarcinomas. Cancer 97: 703–713, 2003. doi: 10.1002/cncr.11087. [DOI] [PubMed] [Google Scholar]
- 548. Magnus N, Garnier D, Rak J. Oncogenic epidermal growth factor receptor up-regulates multiple elements of the tissue factor signaling pathway in human glioma cells. Blood 116: 815–818, 2010. doi: 10.1182/blood-2009-10-250639. [DOI] [PubMed] [Google Scholar]
- 549. Ryden L, Grabau D, Schaffner F, Jonsson PE, Ruf W, Belting M. Evidence for tissue factor phosphorylation and its correlation with protease-activated receptor expression and the prognosis of primary breast cancer. Int J Cancer 126: 2330–2340, 2010. doi: 10.1002/ijc.24921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 550. Lam DK, Dang D, Flynn AN, Hardt M, Schmidt BL. TMPRSS2, a novel membrane-anchored mediator in cancer pain. Pain 156: 923–930, 2015. doi: 10.1097/j.pain.0000000000000130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 551. Miyata S, Koshikawa N, Yasumitsu H, Miyazaki K. Trypsin stimulates integrin alpha(5)beta(1)-dependent adhesion to fibronectin and proliferation of human gastric carcinoma cells through activation of proteinase-activated receptor-2. J Biol Chem 275: 4592–4598, 2000. doi: 10.1074/jbc.275.7.4592. [DOI] [PubMed] [Google Scholar]
- 552. Michel N, Heuze-Vourc’h N, Lavergne E, Parent C, Jourdan ML, Vallet A, Iochmann S, Musso O, Reverdiau P, Courty Y. Growth and survival of lung cancer cells: regulation by kallikrein-related peptidase 6 via activation of proteinase-activated receptor 2 and the epidermal growth factor receptor. Biol Chem 395: 1015–1025, 2014. doi: 10.1515/hsz-2014-0124. [DOI] [PubMed] [Google Scholar]
- 553. Ma Y, Bao-Han W, Lv X, Su Y, Zhao X, Yin Y, Zhang X, Zhou Z, MacNaughton WK, Wang H. MicroRNA-34a mediates the autocrine signaling of PAR2-activating proteinase and its role in colonic cancer cell proliferation. PLoS One 8: e72383, 2013. doi: 10.1371/journal.pone.0072383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 554. Naldini A, Carney DH, Pucci A, Carraro F. Human alpha-thrombin stimulates proliferation of interferon-gamma differentiated, growth-arrested U937 cells, overcoming differentiation-related changes in expression of p21CIP1/WAF1 and cyclin D1. J Cell Physiol 191: 290–297, 2002. doi: 10.1002/jcp.10101. [DOI] [PubMed] [Google Scholar]
- 555. Caruso R, Pallone F, Fina D, Gioia V, Peluso I, Caprioli F, Stolfi C, Perfetti A, Spagnoli LG, Palmieri G, Macdonald TT, Monteleone G. Protease-activated receptor-2 activation in gastric cancer cells promotes epidermal growth factor receptor trans-activation and proliferation. Am J Pathol 169: 268–278, 2006. doi: 10.2353/ajpath.2006.050841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 556. Bazzani L, Donnini S, Finetti F, Christofori G, Ziche M. PGE2/EP3/SRC signaling induces EGFR nuclear translocation and growth through EGFR ligands release in lung adenocarcinoma cells. Oncotarget 8: 31270–31287, 2017. doi: 10.18632/oncotarget.16116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 557. Antonny B, Burd C, De Camilli P, Chen E, Daumke O, Faelber K, Ford M, Frolov VA, Frost A, Hinshaw JE, Kirchhausen T, Kozlov MM, Lenz M, Low HH, McMahon H, Merrifield C, Pollard TD, Robinson PJ, Roux A, Schmid S. Membrane fission by dynamin: what we know and what we need to know. EMBO J 35: 2270–2284, 2016. doi: 10.15252/embj.201694613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 558. Bazzani L, Donnini S, Giachetti A, Christofori G, Ziche M. PGE2 mediates EGFR internalization and nuclear translocation via caveolin endocytosis promoting its transcriptional activity and proliferation in human NSCLC cells. Oncotarget 9: 14939–14958, 2018. doi: 10.18632/oncotarget.24499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 559. Hugo de Almeida V, Guimaraes ID, Almendra LR, Rondon AM, Tilli TM, de Melo AC, Sternberg C, Monteiro RQ. Positive crosstalk between EGFR and the TF-PAR2 pathway mediates resistance to cisplatin and poor survival in cervical cancer. Oncotarget 9: 30594–30609, 2018. doi: 10.18632/oncotarget.25748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 560. Wilson SR, Gallagher S, Warpeha K, Hawthorne SJ. Amplification of MMP-2 and MMP-9 production by prostate cancer cell lines via activation of protease-activated receptors. Prostate 60: 168–174, 2004. doi: 10.1002/pros.20047. [DOI] [PubMed] [Google Scholar]
- 561. Liu Y, Gilcrease MZ, Henderson Y, Yuan XH, Clayman GL, Chen Z. Expression of protease-activated receptor 1 in oral squamous cell carcinoma. Cancer Lett 169: 173–180, 2001. doi: 10.1016/S0304-3835(01)00504-3. [DOI] [PubMed] [Google Scholar]
- 562. D’Andrea MR, Derian CK, Santulli RJ, Andrade-Gordon P. Differential expression of protease-activated receptors-1 and -2 in stromal fibroblasts of normal, benign, and malignant human tissues. Am J Pathol 158: 2031–2041, 2001. doi: 10.1016/S0002-9440(10)64675-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 563. Booden MA, Eckert LB, Der CJ, Trejo J. Persistent signaling by dysregulated thrombin receptor trafficking promotes breast carcinoma cell invasion. Mol Cell Biol 24: 1990–1999, 2004. doi: 10.1128/MCB.24.5.1990-1999.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 564. Even-Ram SC, Maoz M, Pokroy E, Reich R, Katz BZ, Gutwein P, Altevogt P, Bar-Shavit R. Tumor cell invasion is promoted by activation of protease activated receptor-1 in cooperation with the alpha vbeta 5 integrin. J Biol Chem 276: 10952–10962, 2001. doi: 10.1074/jbc.M007027200. [DOI] [PubMed] [Google Scholar]
- 565. Villares GJ, Zigler M, Dobroff AS, Wang H, Song R, Melnikova VO, Huang L, Braeuer RR, Bar-Eli M. Protease activated receptor-1 inhibits the Maspin tumor-suppressor gene to determine the melanoma metastatic phenotype. Proc Natl Acad Sci U S A 108: 626–631, 2011. doi: 10.1073/pnas.1006886108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 566. Trikha M, Timar J, Zacharek A, Nemeth JA, Cai Y, Dome B, Somlai B, Raso E, Ladanyi A, Honn KV. Role for beta3 integrins in human melanoma growth and survival. Int J Cancer 101: 156–167, 2002. doi: 10.1002/ijc.10521. [DOI] [PubMed] [Google Scholar]
- 567. Raso E, Tovari J, Toth K, Paku S, Trikha M, Honn KV, Timar J. Ectopic alphaIIbbeta3 integrin signaling involves 12-lipoxygenase- and PKC-mediated serine phosphorylation events in melanoma cells. Thromb Haemost 85: 1037–1042, 2001. doi: 10.1055/s-0037-1615960. [DOI] [PubMed] [Google Scholar]
- 568. Nierodzik ML, Karpatkin S. Thrombin induces tumor growth, metastasis, and angiogenesis: Evidence for a thrombin-regulated dormant tumor phenotype. Cancer Cell 10: 355–362, 2006. doi: 10.1016/j.ccr.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 569. Massi D, Naldini A, Ardinghi C, Carraro F, Franchi A, Paglierani M, Tarantini F, Ketabchi S, Cirino G, Hollenberg MD, Geppetti P, Santucci M. Expression of protease-activated receptors 1 and 2 in melanocytic nevi and malignant melanoma. Hum Pathol 36: 676–685, 2005. doi: 10.1016/j.humpath.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 570. Belting M, Dorrell MI, Sandgren S, Aguilar E, Ahamed J, Dorfleutner A, Carmeliet P, Mueller BM, Friedlander M, Ruf W. Regulation of angiogenesis by tissue factor cytoplasmic domain signaling. Nat Med 10: 502–509, 2004. doi: 10.1038/nm1037. [DOI] [PubMed] [Google Scholar]
- 571. Wang X, Xiong L, Yu G, Li D, Peng T, Luo D, Xu J. Cathepsin S silencing induces apoptosis of human hepatocellular carcinoma cells. Am J Transl Res 7: 100–110, 2015. [PMC free article] [PubMed] [Google Scholar]
- 572. Ogbomo SM, Shi W, Wagh NK, Zhou Z, Brusnahan SK, Garrison JC. 177Lu-labeled HPMA copolymers utilizing cathepsin B and S cleavable linkers: synthesis, characterization and preliminary in vivo investigation in a pancreatic cancer model. Nucl Med Biol 40: 606–617, 2013. doi: 10.1016/j.nucmedbio.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 573. Gormley JA, Hegarty SM, O’Grady A, Stevenson MR, Burden RE, Barrett HL, Scott CJ, Johnston JA, Wilson RH, Kay EW, Johnston PG, Olwill SA. The role of Cathepsin S as a marker of prognosis and predictor of chemotherapy benefit in adjuvant CRC: a pilot study. Br J Cancer 105: 1487–1494, 2011. doi: 10.1038/bjc.2011.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 574. Fernández PL, Farré X, Nadal A, FernáNdez E, Peiró N, Sloane BF, Shi GP, Chapman HA, Campo EA, Cardesa A. Expression of cathepsins B and S in the progression of prostate carcinoma. Int J Cancer 95: 51–55, 2001. doi:. [DOI] [PubMed] [Google Scholar]
- 575. Lindahl C, Simonsson M, Bergh A, Thysell E, Antti H, Sund M, Wikström P. Increased levels of macrophage-secreted cathepsin S during prostate cancer progression in TRAMP mice and patients. Cancer Genomics Proteomics 6: 149–159, 2009. [PubMed] [Google Scholar]
- 576. Paraoan L, Gray D, Hiscott P, Garcia-Finana M, Lane B, Damato B, Grierson I. Cathepsin S and its inhibitor cystatin C: imbalance in uveal melanoma. Front Biosci (Landmark Ed) 14: 2504–2513, 2009. doi: 10.2741/3393. [DOI] [PubMed] [Google Scholar]
- 577. Flannery T, Gibson D, Mirakhur M, McQuaid S, Greenan C, Trimble A, Walker B, McCormick D, Johnston PG. The clinical significance of cathepsin S expression in human astrocytomas. Am J Pathol 163: 175–182, 2003. doi: 10.1016/S0002-9440(10)63641-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 578. Flannery T, McQuaid S, McGoohan C, McConnell RS, McGregor G, Mirakhur M, Hamilton P, Diamond J, Cran G, Walker B, Scott C, Martin L, Ellison D, Patel C, Nicholson C, Mendelow D, McCormick D, Johnston PG. Cathepsin S expression: an independent prognostic factor in glioblastoma tumours–a pilot study. Int J Cancer 119: 854–860, 2006. doi: 10.1002/ijc.21911. [DOI] [PubMed] [Google Scholar]
- 579. Sevenich L, Bowman RL, Mason SD, Quail DF, Rapaport F, Elie BT, Brogi E, Brastianos PK, Hahn WC, Holsinger LJ, Massague J, Leslie CS, Joyce JA. Analysis of tumour- and stroma-supplied proteolytic networks reveals a brain-metastasis-promoting role for cathepsin S. Nat Cell Biol 16: 876–888, 2014. doi: 10.1038/ncb3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 580. Lee TK, Cheung VC, Lu P, Lau EY, Ma S, Tang KH, Tong M, Lo J, Ng IO. Blockade of CD47-mediated cathepsin S/protease-activated receptor 2 signaling provides a therapeutic target for hepatocellular carcinoma. Hepatology 60: 179–191, 2014. doi: 10.1002/hep.27070. [DOI] [PubMed] [Google Scholar]
- 581. Yin YJ, Salah Z, Maoz M, Even Ram SC, Ochayon S, Neufeld G, Katzav S, Bar-Shavit R. Oncogenic transformation induces tumor angiogenesis: a role for PAR1 activation. FASEB J 17: 163–174, 2003. doi: 10.1096/fj.02-0316com. [DOI] [PubMed] [Google Scholar]
- 582. Chang LH, Pan SL, Lai CY, Tsai AC, Teng CM. Activated PAR-2 regulates pancreatic cancer progression through ILK/HIF-alpha-induced TGF-alpha expression and MEK/VEGF-A-mediated angiogenesis. Am J Pathol 183: 566–575, 2013. doi: 10.1016/j.ajpath.2013.04.022. [DOI] [PubMed] [Google Scholar]
- 583. Liu Y, Mueller BM. Protease-activated receptor-2 regulates vascular endothelial growth factor expression in MDA-MB-231 cells via MAPK pathways. Biochem Biophys Res Commun 344: 1263–1270, 2006. doi: 10.1016/j.bbrc.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 584. Zhang C, Gao GR, Lv CG, Zhang BL, Zhang ZL, Zhang XF. Protease-activated receptor-2 induces expression of vascular endothelial growth factor and cyclooxygenase-2 via the mitogen-activated protein kinase pathway in gastric cancer cells. Oncol Rep 28: 1917–1923, 2012. doi: 10.3892/or.2012.1998. [DOI] [PubMed] [Google Scholar]
- 585. Ciardiello F, Tortora G. EGFR antagonists in cancer treatment. N Engl J Med 358: 1160–1174, 2008. doi: 10.1056/NEJMra0707704. [DOI] [PubMed] [Google Scholar]
- 586. Versteeg HH, Schaffner F, Kerver M, Petersen HH, Ahamed J, Felding-Habermann B, Takada Y, Mueller BM, Ruf W. Inhibition of tissue factor signaling suppresses tumor growth. Blood 111: 190–199, 2008. doi: 10.1182/blood-2007-07-101048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 587. Versteeg HH, Schaffner F, Kerver M, Ellies LG, Andrade-Gordon P, Mueller BM, Ruf W. Protease-activated receptor (PAR) 2, but not PAR1, signaling promotes the development of mammary adenocarcinoma in polyoma middle T mice. Cancer Res 68: 7219–7227, 2008. doi: 10.1158/0008-5472.CAN-08-0419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 588. Milia AF, Salis MB, Stacca T, Pinna A, Madeddu P, Trevisani M, Geppetti P, Emanueli C. Protease-activated receptor-2 stimulates angiogenesis and accelerates hemodynamic recovery in a mouse model of hindlimb ischemia. Circ Res 91: 346–352, 2002. doi: 10.1161/01.RES.0000031958.92781.9E. [DOI] [PubMed] [Google Scholar]
- 589. Villares GJ, Zigler M, Wang H, Melnikova VO, Wu H, Friedman R, Leslie MC, Vivas-Mejia PE, Lopez-Berestein G, Sood AK, Bar-Eli M. Targeting melanoma growth and metastasis with systemic delivery of liposome-incorporated protease-activated receptor-1 small interfering RNA. Cancer Res 68: 9078–9086, 2008. doi: 10.1158/0008-5472.CAN-08-2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 590. Yang E, Boire A, Agarwal A, Nguyen N, O’Callaghan K, Tu P, Kuliopulos A, Covic L. Blockade of PAR1 signaling with cell-penetrating pepducins inhibits Akt-survival pathways in breast cancer cells and suppresses tumor survival and metastasis. Cancer Res 69: 6223–6231-42, 2009. doi: 10.1158/0008-5472.CAN-09-0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 591. Bernatowicz MS, Klimas CE, Hartl KS, Peluso M, Allegretto NJ, Seiler SM. Development of potent thrombin receptor antagonist peptides. J Med Chem 39: 4879–4887, 1996. doi: 10.1021/jm960455s. [DOI] [PubMed] [Google Scholar]
- 592. Damiano BP, Derian CK, Maryanoff BE, Zhang HC, Gordon PA. RWJ-58259: a selective antagonist of protease activated receptor-1. Cardiovasc Drug Rev 21: 313–326, 2003. [DOI] [PubMed] [Google Scholar]
- 593. Kato Y, Kita Y, Hirasawa-Taniyama Y, Nishio M, Mihara K, Ito K, Yamanaka T, Seki J, Miyata S, Mutoh S. Inhibition of arterial thrombosis by a protease-activated receptor 1 antagonist, FR171113, in the guinea pig. Eur J Pharmacol 473: 163–169, 2003. doi: 10.1016/S0014-2999(03)01973-3. [DOI] [PubMed] [Google Scholar]
- 594. Oestreich J. SCH-530348, a thrombin receptor (PAR-1) antagonist for the prevention and treatment of atherothrombosis. Curr Opin Investig Drugs 10: 988–996, 2009. [PubMed] [Google Scholar]
- 595. Serebruany VL, Kogushi M, Dastros-Pitei D, Flather M, Bhatt DL. The in-vitro effects of E5555, a protease-activated receptor (PAR)-1 antagonist, on platelet biomarkers in healthy volunteers and patients with coronary artery disease. Thromb Haemost 102: 111–119, 2009. doi: 10.1160/TH08-12-0805. [DOI] [PubMed] [Google Scholar]
- 596. Dumas M, Nadal-Wollbold F, Gaussem P, Perez M, Mirault T, Letienne R, Bourbon T, Grelac F, Le Grand B, Bachelot-Loza C. Antiplatelet and antithrombotic effect of F 16618, a new thrombin proteinase-activated receptor-1 (PAR1) antagonist. Br J Pharmacol 165: 1827–1835, 2012. doi: 10.1111/j.1476-5381.2011.01668.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 597. Brass LF, Vassallo RR, Belmonte E, Ahuja M, Cichowski K, Hoxie JA. Structure and function of the human platelet thrombin receptor. Studies using monoclonal antibodies directed against a defined domain within the receptor N terminus. J Biol Chem 267: 13795–13798, 1992. doi: 10.1016/S0021-9258(19)49635-X. [DOI] [PubMed] [Google Scholar]
- 598. Hung DT, Vu TK, Wheaton VI, Ishii K, Coughlin SR. Cloned platelet thrombin receptor is necessary for thrombin-induced platelet activation. J Clin Invest 89: 1350–1353, 1992. doi: 10.1172/JCI115721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 599. Andrade-Gordon P, Maryanoff BE, Derian CK, Zhang HC, Addo MF, Darrow AL, Eckardt AJ, Hoekstra WJ, Mccomsey DF, Oksenberg D, Reynolds EE, Santulli RJ, Scarborough RM, Smith CE, White KB. Design, synthesis, and biological characterization of a peptide-mimetic antagonist for a tethered-ligand receptor. Proc Natl Acad Sci U S A 96: 12257–12262, 1999. doi: 10.1073/pnas.96.22.12257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 600. Chackalamannil S, Wang Y, Greenlee WJ, Hu Z, Xia Y, Ahn HS, Boykow G, Hsieh Y, Palamanda J, Agans-Fantuzzi J, Kurowski S, Graziano M, Chintala M. Discovery of a novel, orally active himbacine-based thrombin receptor antagonist (SCH 530348) with potent antiplatelet activity. J Med Chem 51: 3061–3064, 2008. doi: 10.1021/jm800180e. [DOI] [PubMed] [Google Scholar]
- 601. Kogushi M, Matsuoka T, Kawata T, Kuramochi H, Kawaguchi S, Murakami K, Hiyoshi H, Suzuki S, Kawahara T, Kajiwara A, Hishinuma I. The novel and orally active thrombin receptor antagonist E5555 (Atopaxar) inhibits arterial thrombosis without affecting bleeding time in guinea pigs. Eur J Pharmacol 657: 131–137, 2011. doi: 10.1016/j.ejphar.2011.01.058. [DOI] [PubMed] [Google Scholar]
- 602. Morrow DA, Braunwald E, Bonaca MP, Ameriso SF, Dalby AJ, Fish MP, Fox KA, Lipka LJ, Liu X, Nicolau JC, Ophuis AJ, Paolasso E, Scirica BM, Spinar J, Theroux P, Wiviott SD, Strony J, Murphy SA. Vorapaxar in the secondary prevention of atherothrombotic events. N Engl J Med 366: 1404–1413, 2012. doi: 10.1056/NEJMoa1200933. [DOI] [PubMed] [Google Scholar]
- 603. Kosoglou T, Reyderman L, Tiessen RG, Van Vliet AA, Fales RR, Keller R, Yang B, Cutler DL. Pharmacodynamics and pharmacokinetics of the novel PAR-1 antagonist vorapaxar (formerly SCH 530348) in healthy subjects. Eur J Clin Pharmacol 68: 249–258, 2012. doi: 10.1007/s00228-011-1120-6. [DOI] [PubMed] [Google Scholar]
- 604. Feistritzer C, Schuepbach RA, Mosnier LO, Bush LA, Di Cera E, Griffin JH, Riewald M. Protective signaling by activated protein C is mechanistically linked to protein C activation on endothelial cells. J Biol Chem 281: 20077–20084, 2006. doi: 10.1074/jbc.M600506200. [DOI] [PubMed] [Google Scholar]
- 605. Zlokovic BV, Griffin JH. Cytoprotective protein C pathways and implications for stroke and neurological disorders. Trends Neurosci 34: 198–209, 2011. doi: 10.1016/j.tins.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 606. Griffin JH, Zlokovic BV, Mosnier LO. Activated protein C, protease activated receptor 1, and neuroprotection. Blood 132: 159–169, 2018. doi: 10.1182/blood-2018-02-769026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 607. Guo H, Zhao Z, Yang Q, Wang M, Bell R, Wang S, Chow N, Davis T, Griffin J, Goldman S, Zlokovic BV. An activated protein C analog stimulates neuronal production by human neural progenitor cells via a PAR1-PAR3-S1PR1-Akt pathway. J Neurosci 33: 6181–6190, 2013. doi: 10.1523/JNEUROSCI.4491-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 608. Lyden P, Pryor KE, Coffey CS, Cudkowicz M, Conwit R, Jadhav A, Sawyer RN Jr, Claassen J, Adeoye O, Song S, Hannon P, Rost NS, Hinduja A, Torbey M, Lee JM, Benesch C, Rippee M, Rymer M, Froehler MT, Clarke Haley E, Johnson M, Yankey J, Magee K, Qidwai J, Levy H, Mark Haacke E, Fawaz M, Davis TP, Toga AW, Griffin JH, Zlokovic BV; NeuroNEXT Clinical Trials Network NN104 Investigators. Final results of the RHAPSODY Trial: a multi-center, phase 2 trial using a continual reassessment method to determine the safety and tolerability of 3K3A-APC, A recombinant variant of human activated protein C, in combination with tissue plasminogen activator, mechanical thrombectomy or both in moderate to severe acute ischemic stroke. Ann Neurol 85: 125–136, 2019. doi: 10.1002/ana.25383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 609. Lyden PD, Pryor KE, Minigh J, Davis TP, Griffin JH, Levy H, Zlokovic BV. Stroke treatment with PAR-1 agents to decrease hemorrhagic transformation. Front Neurol 12: 593582, 2021. doi: 10.3389/fneur.2021.593582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 610. Dockendorff C, Aisiku O, Verplank L, Dilks JR, Smith DA, Gunnink SF, Dowal L, Negri J, Palmer M, Macpherson L, Schreiber SL, Flaumenhaft R. Discovery of 1,3-diaminobenzenes as selective inhibitors of platelet activation at the PAR1 receptor. ACS Med Chem Lett 3: 232–237, 2012. doi: 10.1021/ml2002696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 611. Aisiku O, Peters CG, De Ceunynck K, Ghosh CC, Dilks JR, Fustolo-Gunnink SF, Huang M, Dockendorff C, Parikh SM, Flaumenhaft R. Parmodulins inhibit thrombus formation without inducing endothelial injury caused by vorapaxar. Blood 125: 1976–1985, 2015. doi: 10.1182/blood-2014-09-599910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 612. Kenakin TP. A Pharmacology Primer (5th ed.), edited by Kenakin TP. New York: Academic Press, 2019, p. 1. [Google Scholar]
- 613. De Ceunynck K, Peters CG, Jain A, Higgins SJ, Aisiku O, Fitch-Tewfik JL, Chaudhry SA, Dockendorff C, Parikh SM, Ingber DE, Flaumenhaft R. PAR1 agonists stimulate APC-like endothelial cytoprotection and confer resistance to thromboinflammatory injury. Proc Natl Acad Sci U S A 115: E982–E991, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 614. Mcintosh KA, Cunningham MR, Bushell T, Plevin R. The development of proteinase-activated receptor-2 modulators and the challenges involved. Biochem Soc Trans 48: 2525–2537, 2020. doi: 10.1042/BST20200191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 615. Kanke T, Kabeya M, Kubo S, Kondo S, Yasuoka K, Tagashira J, Ishiwata H, Saka M, Furuyama T, Nishiyama T, Doi T, Hattori Y, Kawabata A, Cunningham MR, Plevin R. Novel antagonists for proteinase-activated receptor 2: inhibition of cellular and vascular responsesin vitroandin vivo. Br J Pharmacol 158: 361–371, 2009. doi: 10.1111/j.1476-5381.2009.00342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 616. Goh FG, Ng PY, Nilsson M, Kanke T, Plevin R. Dual effect of the novel peptide antagonist K-14585 on proteinase-activated receptor-2-mediated signalling. Br J Pharmacol 158: 1695–1704, 2009. doi: 10.1111/j.1476-5381.2009.00415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 617. Boitano S, Hoffman J, Flynn AN, Asiedu MN, Tillu DV, Zhang Z, Sherwood CL, Rivas CM, Defea KA, Vagner J, Price TJ. The novel PAR2 ligand C391 blocks multiple PAR2 signalling pathwaysin vitroandin vivo. Br J Pharmacol 172: 4535–4545, 2015. doi: 10.1111/bph.13238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 618. Seitzberg JG, Knapp AE, Lund BW, Mandrup Bertozzi S, Currier EA, Ma JN, Sherbukhin V, Burstein ES, Olsson R. Discovery of potent and selective small-molecule PAR-2 agonists. J Med Chem 51: 5490–5493, 2008. doi: 10.1021/jm800754r. [DOI] [PubMed] [Google Scholar]
- 619. Barry GD, Suen JY, Le GT, Cotterell A, Reid RC, Fairlie DP. Novel agonists and antagonists for human protease activated receptor 2. J Med Chem 53: 7428–7440, 2010. doi: 10.1021/jm100984y. [DOI] [PubMed] [Google Scholar]
- 620. Suen JY, Barry GD, Lohman RJ, Halili MA, Cotterell AJ, Le GT, Fairlie DP. Modulating human proteinase activated receptor 2 with a novel antagonist (GB88) and agonist (GB110). Br J Pharmacol 165: 1413–1423, 2012. doi: 10.1111/j.1476-5381.2011.01610.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 621. Suen JY, Cotterell A, Lohman RJ, Lim J, Han A, Yau MK, Liu L, Cooper MA, Vesey DA, Fairlie DP. Pathway-selective antagonism of proteinase activated receptor 2. Br J Pharmacol 171: 4112–4124, 2014. doi: 10.1111/bph.12757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 622. Farmer LJ, Fournier PA, Lessard S, Lui B, St-Onge M, Sturino C, Szychowski J, Yannopoulos C. Vertex Pharmaceuticals Incorporated. Imidazopyridazines Useful as Inhibitors of the PAR-2 Signaling Pathway. Patent WO/2015048245 A1. 2013. https://patentimages.storage.googleapis.com/ad/79/74/681fbe11d4c20b/WO2015048245A1.pdf. [Google Scholar]
- 623. Avet C, Sturino C, Grastilleur S, Gouill CL, Semache M, Gross F, Gendron L, Bennani Y, Mancini JA, Sayegh CE, Bouvier M. The PAR2 inhibitor I-287 selectively targets Gαq and Gα12/13 signaling and has anti-inflammatory effects. Commun Biol 3: 1–13, 2020. doi: 10.1038/s42003-019-0734-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 624. Giblin P, Boxhammer R, Desai S, Kroe-Barrett R, Hansen G, Ksiazek J, Panzenbeck M, Ralph K, Schwartz R, Zimmitti C, Pracht C, Miller S, Magram J, Litzenburger T. Fully human antibodies against the protease-activated receptor-2 (PAR-2) with anti-inflammatory activity. Hum Antibodies 20: 83–94, 2011. doi: 10.3233/HAB-2011-0243. [DOI] [PubMed] [Google Scholar]
- 625. Majewski MW, Gandhi DM, Rosas R, Kodali R, Arnold LA, Dockendorff C. Design and evaluation of heterobivalent PAR1–PAR2 ligands as antagonists of calcium mobilization. ACS Med Chem Lett 10: 121–126, 2019. doi: 10.1021/acsmedchemlett.8b00538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 626. Liu S, Li S, Yuan D, Wang E, Xie R, Zhang W, Kong Y, Zhu X. Protease activated receptor 4 (PAR4) antagonists: research progress on small molecules in the field of antiplatelet agents. Eur J Med Chem 209: 112893, 2021. doi: 10.1016/j.ejmech.2020.112893. [DOI] [PubMed] [Google Scholar]
- 627. Wong PC, Seiffert D, Bird JE, Watson CA, Bostwick JS, Giancarli M, Allegretto N, Hua J, Harden D, Guay J, Callejo M, Miller MM, Lawrence RM, Banville J, Guy J, Maxwell BD, Priestley ES, Marinier A, Wexler RR, Bouvier M, Gordon DA, Schumacher WA, Yang J. Blockade of protease-activated receptor-4 (PAR4) provides robust antithrombotic activity with low bleeding. Sci Transl Med 9: eaaf5294, 2017. doi: 10.1126/scitranslmed.aaf5294. [DOI] [PubMed] [Google Scholar]
- 628. Wilson SJ, Ismat FA, Wang Z, Cerra M, Narayan H, Raftis J, Gray TJ, Connell S, Garonzik S, Ma X, Yang J, Newby DE. PAR4 (protease-activated receptor 4) antagonism with BMS-986120 inhibits human ex vivo thrombus formation. Arterioscler Thromb Vasc Biol 38: 448–456, 2018. doi: 10.1161/ATVBAHA.117.310104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 629. Covic L, Misra M, Badar J, Singh C, Kuliopulos A. Pepducin-based intervention of thrombin-receptor signaling and systemic platelet activation. Nat Med 8: 1161–1165, 2002. doi: 10.1038/nm760. [DOI] [PubMed] [Google Scholar]
- 630. Gurbel PA, Bliden KP, Turner SE, Tantry US, Gesheff MG, Barr TP, Covic L, Kuliopulos A. Cell-penetrating pepducin therapy targeting PAR1 in subjects with coronary artery disease. Arterioscler Thromb Vasc Biol 36: 189–197, 2016. doi: 10.1161/ATVBAHA.115.306777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 631. Jimenez-Vargas NN, Yu Y, Jensen DD, Bok DD, Wisdom M, Latorre R, Lopez C, Jaramillo-Polanco JO, Degro C, Guzman-Rodriguez M, Tsang Q, Snow Z, Schmidt BL, Reed DE, Lomax AE, Margolis KG, Stein C, Bunnett NW, Vanner SJ. Agonist that activates the micro-opioid receptor in acidified microenvironments inhibits colitis pain without side effects. Gut 71: 695–704, 2022. doi: 10.1136/gutjnl-2021-324070. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Tables S1–S5 are available at https://doi.org/10.6084/m9.figshare.19739830.