

Universal multi-gene testing in hepatobiliary cancers was associated with heritable mutations in over 15% of patients, most of whom would not have been tested using current guidelines. 55% were potentially eligible for approved precision therapy and/or clinical treatment trials. Germline testing should be considered in all patients with hepatobiliary cancers.
Abstract
Data from germline testing in unselected patients with hepatobiliary cancers are limited. Identification of germline predisposition can have important implications on cancer treatment and family counseling. To determine prevalence of pathogenic germline variants (PGV) in patients with hepatobiliary cancer, we undertook a prospective multi-site study of germline sequencing using a >80-gene next-generation sequencing platform among patients with hepatobiliary cancers receiving care at Mayo Clinic Cancer Centers between April 1, 2018 and March 31, 2020. Patients were not selected on the basis of stage, family cancer history, ethnicity, or age. Family cascade testing was offered at no cost. Of 205 patients, the median age was 65 years, 58.5% were male, 81% were White, and 64.4% had cholangiocarcinoma, 21.5% hepatocellular carcinoma, 7.8% gallbladder cancer, and 4.3% carcinoma of ampulla of Vater. PGV were found in 15.6% (n = 32) of patients, including 23 (71%) in moderate and high penetrance cancer susceptibility genes. A total of 75% of patients with a positive result would not have been detected using guidelines for genetic evaluation. Prevalence of PGV was 15.7% in intrahepatic cholangiocarcinoma, 17% in extrahepatic cholangiocarcinoma, 15.9% in hepatocellular cancer, and 33% in carcinoma of ampulla of Vater. On the basis of these genetic findings, 55% were potentially eligible for approved precision therapy and/or clinical treatment trials. Universal multi-gene panel testing in hepatobiliary cancers was associated with detection of heritable mutations in over 15% of patients most of whom would not have been tested using current guidelines. Germline testing should be considered in all patients with hepatobiliary cancers.
Prevention Relevance:
Universal multi-gene testing in hepatobiliary cancers was associated with heritable mutations in over 15% of patients, most of whom would not have been tested using current guidelines. 55% were potentially eligible for approved precision therapy and/or clinical treatment trials. Germline testing should be considered in all patients with hepatobiliary cancers.
Introduction
Hepatobiliary cancers are a set of malignant tumors that arises from different regions including the liver and biliary tract (1). This group comprises several different tumors, including hepatocellular carcinoma (HCC), intrahepatic cholangiocarcinoma (IHC), extrahepatic cholangiocarcinoma (EHC), gallbladder cancer, and cancer of the ampulla of Vater (1). The incidence of HCC and IHC is increasing in the last decades in the United States, with an estimated incidence of 42,810 new cases diagnosed and 30,160 deaths in 2020 (2–4).
Limited data are available about a hereditary component in the development of hepatobiliary cancers (5). A retrospective analysis of 267 patients with hepatobiliary cancer referred for germline testing found 41 patients (15%) were carriers of pathogenic or likely pathogenic germline variants (P/LP GV; ref. 6). In this study, 32% of these PGV detected could have clinical utility for eligibility of the patients in ongoing treatment trials (6). Another group evaluated germline testing in 131 patients with biliary tract cancers (IHC, EHC, and gallbladder cancer) and found 21 patients (16%) with PGV, with a third of mutations being present in the high-penetrance cancer susceptibility genes BRCA1 and BRCA2 (7). Germline mutations particularly in genes related to DNA damage response could have implications for treatment selection and response, such as platinum-based regimens and PARP inhibitors (8, 9).
Currently, germline testing is not recommended as standard of care for all HBCs. The recommendation for germline testing or referral to for genetic evaluation is based on a family history of Lynch- or BRCA-associated cancers (1). Comprehensive studies are still necessary to address the prevalence and characteristics of germline susceptibility in this heterogeneous group of cancers. In this article, we report the clinical characteristic and outcomes of a multi-center prospective cohort of patients with hepatobiliary cancer who underwent germline testing with next-generation sequencing (NGS) using a >80 genes platform. Patients were unselected for stage of disease, family history of cancer, ethnicity or age. We also include in this report cases of carcinoma of ampulla of Vater, a rare tumor that arises from the ampulla of Vater at the duodenal confluence of the distal common bile duct (10).
Materials and Methods
Patient selection
From April 1, 2018 through March 31, 2020, a total of 2,984 unselected adult patients with a new or active diagnosis of cancer were recruited from multi-disciplinary clinics at any of the Mayo Clinic destination Cancer Centers in Rochester, MN; Jacksonville, FL or Phoenix, AZ, and a community oncology practice in Eau Claire, WI—Mayo Clinic Interrogating Cancer Etiology using Proactive Genetic Testing (INTERCEPT) study (11). Patients undergoing surveillance post curative cancer or with hematologic malignances were excluded. Research coordinators of each site recruited patients using central lists of daily oncology clinic visits. Germline sequencing using a NGS panel of 83 genes (84 genes as of July 2019) was offered at no cost for all participants and had disclosure of results (11). All cancer-predisposing genes identified in the American College of Medical Genetics and Genomics guidelines were included in the panel. Patients in this study were not selected on the basis of clinical characteristics, including family or personal history of cancer, cancer type, stage of disease, ethnicity, or age at diagnosis. This cohort included 205 patients with a diagnosis of HBC and ampullary carcinoma and comprises the patients analyzed in this study. Patients with a previously established molecular diagnosis of a cancer genetic syndrome were excluded from the INTERCEPT study; however, none of the patients in the HBC cohort had a prior genetic diagnosis.
All patients viewed a standard pretest education video before undergoing genetic testing and additional pretest genetic counseling was offered. The test results were reviewed by physicians with expertise in cancer genetics or certified genetic professional. Genetic counseling and family variant cascade testing was offered to all individuals with pathogenic or likely pathogenic variants at no cost.
Clinical outcomes information was collected in this study either from medical records or self-administered electronic questionnaires for family pedigree information. Mayo Clinic Institutional Review Board (IRB 18-00326) approved this study. Written informed consent is provided from all the patients. Data were deidentified with the exception of two investigators (N. Jewel Samadder and K.L. Kunze).
Sequencing, variant calling, and result reporting
All patients underwent NGS germline genetic testing with a multi-gene cancer panel of 83 genes (84 genes as of July 2019) on the Invitae Multi-cancer panel (Supplementary Table S1). Invitae performed the full gene sequencing and variant interpretation. Independent review of the test results by a medical geneticist confirmed the variant findings. The classification of the genes were based on disease risk and prior modeling, classified as high [relative risk (RR) > 4], intermediate (RR, 2–4) or low (RR < 2) penetrant, recessive or of uncertain clinical actionability.
Statistical analysis
Descriptive statistics for demographic, clinical, and treatment-related characteristics of the cohort were examined. Rates of detection of clinically actionable findings using 2018 and 2020 National Comprehensive Cancer Network (NCCN) guidelines were calculated. Rates of uptake of family variant testing (FVT) and findings in tested family members were examined.
Results
Cohort characteristics
From April 1, 2018 through March 31, 2020, 3,095 patients were enrolled into the INTERCEPT study, 111 patients were ultimately excluded because of: (i) no blood sample was obtained for genetic testing (n = 12), (ii) consent withdrawn by patient (n = 96), (iii) failure of genetic testing at Invitae (n = 3), leaving 2,984 of whom 205 were patients with a diagnosis of hepatobiliary cancer (Supplementary Fig. S1). The distribution of sex, age, comorbidities, and stage stratified by primary tumor location are shown in Table 1; Supplementary Table S2. The most common tumor type was 64.4% cholangiocarcinoma (64.4%), followed by HCC (21.5%), gallbladder cancer (7.8%), and carcinoma of ampulla of Vater (4.3%). The median age at diagnosis was 65 years and 58.5% were male. Overall, 54.6% of patients were smokers, 17.1% had a body mass index over 30, 20% had type 2 diabetes, and 30.7% hypertension. The proportions of patients with early stage (I and II) disease were 42% and late stage (III and IV) were 58%. Race and ethnicity distributions included 10.2% Hispanic/Latino, 4.4% Black/African American, and 81.5% White. Eighteen patients with biliary cancers had history of primary sclerosing cholangitis and 59% (26/44) of patients with HCC had hepatitis B or C. Detailed family history information was available on 91 patients (44.4%), of whom 61 patients (29.8%) had a family history of any cancer in a first-degree relative. Of the 205 patients with HBC, 31% (n = 64) were new/incident diagnosis and 69% (n = 151) were active or prevalent cases in continued oncology care. Clinical outcomes of the entire cohort are shown in Table 1; Supplementary Table S3.
Table 1.
Clinical and demographic characteristics of patients stratified by tumor type.
| HCC | IHC | EHC | Gallbladder | Ampulla of Vater | Total | |
|---|---|---|---|---|---|---|
| (N = 44) | (N = 83) | (N = 53) | (N = 16) | (N = 9) | (N = 205) | |
| Region | ||||||
| Southwest | 21 (47.7%) | 56 (67.5%) | 35 (66.0%) | 8 (50.0%) | 6 (66.7%) | 126 (61.5%) |
| Midwest | 9 (20.5%) | 13 (15.7%) | 6 (11.3%) | 4 (25.0%) | 2 (22.2%) | 34 (16.6%) |
| Southeast | 14 (31.8%) | 14 (16.9%) | 12 (22.6%) | 4 (25.0%) | 1 (11.1%) | 45 (22.0%) |
| Sex | ||||||
| Male participant | 36 (81.8%) | 41 (49.4%) | 35 (66.0%) | 4 (25.0%) | 4 (44.4%) | 120 (58.5%) |
| Female participant | 8 (18.2%) | 42 (50.6%) | 18 (34.0%) | 12 (75.0%) | 5 (55.6%) | 85 (41.5%) |
| Age (years) | ||||||
| Mean (SD) | 64.7 (9.9) | 61.0 (12.2) | 61.3 (12.3) | 61.1 (12.3) | 65.6 (8.2) | 62.1 (11.6) |
| Median | 68.0 | 65.0 | 62.0 | 57.5 | 67.0 | 65.0 |
| Range | 31.0–78.0 | 26.0–79.0 | 28.0–80.0 | 45.0–80.0 | 50.0–74.0 | 26.0–80.0 |
| Race | ||||||
| White | 37 (84.1%) | 69 (83.1%) | 42 (79.2%) | 13 (81.2%) | 6 (66.7%) | 167 (81.5%) |
| Hispanic/Latino | 2 (4.5%) | 7 (8.4%) | 7 (13.2%) | 3 (18.8%) | 2 (22.2%) | 21 (10.2%) |
| Black/African American | 5 (11.4%) | 3 (3.6%) | 1 (1.9%) | 0 (0.0%) | 0 (0.0%) | 9 (4.4%) |
| Asian | 0 (0.0%) | 1 (1.2%) | 1 (1.9%) | 0 (0.0%) | 0 (0.0%) | 2 (1.0%) |
| American Indian/Alaskan Native | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 1 (11.1%) | 1 (0.5%) |
| Other | 0 (0.0%) | 3 (3.6%) | 2 (3.8%) | 0 (0.0%) | 0 (0.0%) | 5 (2.4%) |
Abbreviations: EHC, extrahepatic cholangiocarcinoma; HCC, hepatocellular carcinoma; IHC, intrahepatic cholangiocarcinoma.
Variants detection
Of the 205 patients undergoing germline analysis, 32 patients (15.6%) harbored 34 pathogenic/likely pathogenic variants conferring cancer predisposition, with 23 (71.8%) of the PGV in high and moderate penetrance genes (Fig. 1). The most common pathogenic variants in high and moderate penetrance genes were found in DNA damage repair (DDR) genes including BRCA1 and BRCA2 (11.8%), NBN (11.8%), ATM (8.8%), CHEK2 (2.9%), RAD51C (2.9%), RAD51D (2.9%; Tables 2 and 3). Three patients (8.8%) were detected with Lynch syndrome, 2 of whom had PGV in MLH1 and one in MSH6. Figure 2 shows the distribution of PGV by gene and tumor site. When stratified by tumor type, 15.9% of patients with HCC, 15.7% of IHC, 17% of EHC, and 33% of ampullary cancer were diagnosed as carriers of a PGV, respectively (Supplementary Table S4). No PGV were identified in patients with gallbladder cancer. The prevalence of pathogenic mutations was similar in both incident and prevalent cases of HBC (16.3% and 14.1%). Of the 34 PGV variants, 19 (55%) were potentially eligible for approved precision therapy and/or clinical treatment trials (Supplementary Table S5).
Figure 1.
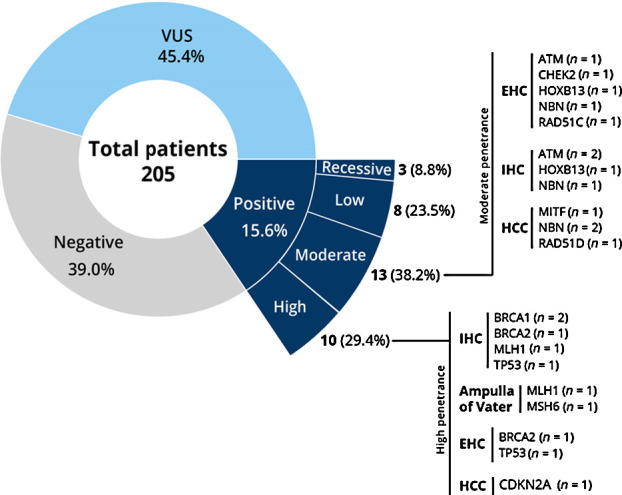
Germline testing results.
Table 2.
Distribution of the 34 PGVs by penetrance status.
| PGV | Total (n = 34) | |
|---|---|---|
| High penetrance | BRCA1 | 2 (5.9%) |
| BRCA2 | 2 (5.9%) | |
| CDKN2A | 1 (2.9%) | |
| MLH1 | 2 (5.9%) | |
| MSH6 | 1 (2.9%) | |
| TP53 | 2 (5.9%) | |
| Moderate penetrance | ATM | 3 (8.8%) |
| CHEK2 | 1 (2.9%) | |
| HOXB13 | 2 (5.9%) | |
| MITF | 1 (2.9%) | |
| NBN | 4 (11.8%) | |
| RAD51C | 1 (2.9%) | |
| RAD51D | 1 (2.9%) | |
| Low penetrance | BARD1 | 1 (2.9%) |
| MUTYH (monoallelic) | 6 (17.6%) | |
| RAD50 | 1 (2.9%) | |
| Recessive alleles | BLM (monoallelic) | 1 (2.9%) |
| RECQL4 | 1 (2.9%) | |
| WRN | 1 (2.9%) |
Table 3.
Pathogenic/likely pathogenic variant types.
| Overall (N = 34) | |
|---|---|
| Type of variant | |
| Deletion | 9 (26.5%) |
| Duplication | 4 (11.8%) |
| Missense | 21 (61.8%) |
Figure 2.

PGVs and tumor primary site.
Application of clinical genetic referral criteria
After application of clinical genetic referral criteria, 75% of the patients found as carriers of a PGV would not be detected using NCCN, National Society of Genetic Counselors, or American College of Medical Genetics and Genomics (ACMG) testing guidelines. Only 34% of PGV carriers met guidelines based on family history regardless of personal history (Supplementary Table S5). Even when this analysis was restricted to patients with complete pedigree information, 73.7% of PGV carriers would not meet current clinical practice criteria for genetic evaluation.
Family variant cascade testing
No cost FVT was offered to all blood relatives of affected participants. Only 3 (1.5%) patients with PGV had family members undergo FVT within a 3-month window of their test result.
Clinical implications of PGVs
Of the 34 patients found to have a PGV, 82% (n = 28) had PGVs that are qualifiers for potential clinically actionable management and treatment changes (Supplementary Table S6). These can be categorized into precision therapy options (8%, contingent on patients meeting other clinical indications), clinical treatment trials (47%, contingent on patients meeting other clinical inclusion criteria) or published clinical guideline management recommendations (76%, such as NCCN and ACMG).
Discussion
Germline testing results in hepatobiliary cancer are limited, with most data based on retrospective analysis of samples (5, 6, 12). In this prospective study, universal multi-gene panel testing in unselected patients with HBC was able to identify 15.6% as carriers of PGV, equating to nearly 1 in 6 patients with HBC harboring a germline genetic predisposition to cancer. The majority of these patients (75%) with a PGV would not have been detected applying currently genetic referral criteria, and over two-thirds of PGV were in high and moderate penetrance genes with established guidelines for management and/or treatment implications. Of the 34 patients with PGVs, 19 (55%) had PGVs in genes that would be qualifiers for approved precision therapy and/or clinical treatment trials, contingent upon the patients meeting appropriate clinical criteria (e.g., disease stage, Eastern Cooperative Oncology Group status, prior treatment, etc.). Overall, 28 (82%) of these patients had PGV in genes with available precision therapies, clinical trial and/or published management implications.
In a prior retrospective analysis of 267 patients with HBC referred for germline counseling, 15% were found to have a PGV (there was no overlap of patients with the current study; ref. 6). In addition, in another retrospective analysis of 146 Japanese patients diagnosed with biliary tract cancers (BTC), 11% of the patients were identified as PGV carriers (13). Around half of the cases were in patients with IHC, just one case in a patient with gallbladder cancer (13). In a smaller prospective cohort of 131 patients with BTC (63.4% patients with IHC), they reported a PGV prevalence of 16% (7). The prevalence of PGV in patients with HBC in these studies is nearly identical to our finding of 15.6%. The distribution of PGV in IHC and EHC was similar in this prospective cohort of 131 BTC, with a slightly higher rate in EHC similar to our findings (7). With the data provided in this study and corroboration by prior literature, the overall risk of a patient with cholangiocarcinoma harboring a PGV is comparable with other solid tumors including colorectal, breast, and pancreatic cancers (11, 14).
In 44 patients with HCC, 7 (15.9%) patients were identified as carriers of a PGV. Prior liver disease and known risks factors including non-alcoholic fatty liver disease and viral hepatitis are causally related to the development of HCC (15–17). Prior studies in HCC have identified somatic pathogenic variants related to development of HCC including variants in LZTR1, EEF1A1, SF3B1, and SMARCA4 (18, 19). The impact of germline testing in unselected patients with HCC without known risk factors to carry PGVs has not been well characterized. Incorporation of multi-gene panels to identify PGV in patients with HCC could be helpful to delineate relationship of genetic predisposition and environmental risk factors in the landscape of this disease.
Though data in ampulla of Vater cancers are not widely available, few studies suggest appreciable rates of PGV in small case series. In our cohort, 3 in 9 patients with ampullary cancer had a PGV detected, 2 of them with Lynch syndrome. As part of the MSK-IMPACT study, 44 patients with this rare gastrointestinal cancer underwent germline sequencing with a multi-gene panel (76–88 genes) and 18% were found to have a PGV (10). These results suggest that ampullary cancers are associated with PGV and incorporation of routine germline testing in these patients can have therapeutic implications and improve family counseling and cancer prevention.
The overall survival associated with BTC is low and precision guided therapy is still evolving. In our study, over 50% of the PGV were detected in DDR-related genes, including BRCA1, BRCA2, ATM, CHEK2, NBN, RAD 50, RAD 51C, RAD 51D, BARD1, BLM, and WRN. Pathogenic variants in genes related to homologous recombination in patients with BTC can identify subgroups of patients with diverse patterns of disease and possible response to targeted therapies (8, 9). Interestingly, DDR genes with PGV were detected in 5 patients with HCC (11%), 6 IHC (7.2%), 6 EHC (11%), and 1 ampullary carcinoma, suggesting prevalence in all subgroups analyzed. Similar results were observed in MSK-IMPACT study (7). Monoallelic MUTYH mutations have a prevalence around 2% in overall population and thus their finding in this series may be associated with the disease or could be incidental findings expected on the basis of population prevalence. It's worth noting that the PGVs in these patients do not appear enriched within a particular gene or subset of genes tested. This may in part be related to the size of the cohort and underscores the need for further research to elucidate whether PGVs in particular genes confer a predisposition to HBC.
Referral for genetic testing is traditionally based on clinical guidelines that utilize tumor type, patient age, and family cancer history as predictors of a PGV. Utilizing the 2020 NCCN guidelines, 75% of patients with BTC in our study detected with a PGV would have been missed. Furthermore, the PGV prevalence of 15.6% in HBC is comparable with that observed in pancreatic and ovarian cancers (15% and 20%, respectively), guidelines for both of which recommend universal testing of patients with these cancers. These results reinforce the need to incorporate germline testing for all patients with HBC regardless of guideline-based criteria.
In addition, not only the universal testing can improve the discovery of a PGV in the patient, but it can also improve the guidance for their relatives. In our study, the traditional barrier of cost was removed for the first 3 months following a positive test result. Family variant testing was pursued in less than 2% of families of probands with a PGV which was a disheartening realization though not completely surprising. Low adherence to cascade family testing is consistent in multiple studies. The uptake of free cascade testing was around 20% in a study conducted in Singapore (20). Other groups evaluated family testing in hereditary syndromes including Lynch and gynecologic cancers and observed similar findings (21, 22). In another approach including an online initiative to cascade testing, 47.5% of invited first-degree relatives underwent genetic testing and only 12% continued the cascade (23). Multiple factors can be associated with the low uptake including communication barriers, poor understanding of the test, fear of discrimination or eventual procedures related to the findings, outside the financial barriers. Education material as websites, videos, letters, and brochures can help to support disclosure of results (21). An annotated copy of the family tree indicating which members should receive genetic testing may help ensure that the information is shared with patients and relatives (22). Although contrary to U.S. privacy laws, empowerment of the clinician or testing laboratory to directly reach out relatives may be fruitful (23).
As has been reported previously, concerns have been raised about high rates of variant of uncertain significance (VUS) identified in multi-gene panel testing. Consistent with prior studies (11) we report a VUS rate of 45%. Several studies (24–26) have described the limited confidence that oncologists have with the interpretation and correct management of VUS results. A related concern is when genes with unknown or unclear clinical relevance may prompt invasive procedures or morbid prophylactic operations. Referral of patients with VUS results to a genetic counselor or clinical geneticist is an effective approach to help mitigate these concerns. Although one might argue that smaller, less comprehensive gene panel should be used to reduce VUS rates, decreasing costs of testing allow broader application of comprehensive panels, which enables identification of clinically relevant PGVs that might otherwise be missed because of limited family history or a nonclassical phenotype. These issues will be important to address as broader genetic testing is incorporated into practice.
Strengths of this study include the prospective, multi-center design, with a broad disease stage distribution and use of a large NGS gene panel. Some limitations of our results include demographic inclusivity with 81% of patients being white. A study with long-term follow-up is necessary to address implications of PGV status on treatment selection and survival outcomes. Family pedigree information was not available on all patients, which is reflective of real-world practice however limits the ability to fully apply clinical practice guidelines which rely heavily on this factor. Finally, integrated tumor analysis was not performed in this cohort, yet all the PGV found are possible genetic drivers related to the development of cancer.
Conclusion
To our knowledge, this study is the largest prospective, multi-center study evaluating germline sequencing in unselected patients with hepatobiliary cancer. Our findings show that nearly 1 in 6 patients with hepatobiliary cancer carry a germline PGV. This is similar to other malignant tumors including colorectal, breast, and pancreatic cancers that are more commonly associated with germline predisposition. Incorporation of germline sequencing for all patients with HBC in clinical practice could improve understanding of the disease, application of precision therapies, and the development of clinical trials with personalized medicine and strategies for family counseling and cancer prevention.
Authors' Disclosures
D. Ahn reports other support from Genentech, Eisai, Exelixis, and Advanced Accelerator Applications outside the submitted work. E.D. Esplin reports other support from Invitae during the conduct of the study. R.L. Nussbaum reports personal fees from Invitae during the conduct of the study; personal fees from Pfizer; personal fees and other support from Genome Medical and Maze Therapeutics outside the submitted work. A. Keith Stewart reports other support from Genomics England LLC, Helix Inc., and personal fees from Tempus Inc. outside the submitted work. T. Bekaii-Saab reports research funding (to institution): Agios, Arys, Arcus, Atreca, Boston Biomedical, Bayer, Amgen, Merck, Celgene, Lilly, Ipsen, Clovis, Seattle Genetics, Genentech, Novartis, Mirati, Merus, Abgenomics, Incyte, Pfizer, BMS; consulting (to institution): Ipsen, Arcus, Array Biopharma, Pfizer, Seattle Genetics, Bayer, Genentech, Incyte, and Merck; Consulting (to self): Stemline, AbbVie, Boehringer Ingelheim, Janssen, Eisai, Daichii Sankyo, Natera, TreosBio, Celularity, Exact Science, Sobi, Beigene, Kanaph, Xilis, AstraZeneca, and Foundation Medicine; IDMC/DSMB (to self): Suzhou Kintor, Astra Zeneca, Exelixis, Lilly, PanCan, and 1Globe; is scientific advisory board member with Imugene, Immuneering, and Sun Biopharma; Inventions/Patents: WO/2018/183488 and WO/2019/055687. No disclosures were reported by the other authors.
Acknowledgments
The study was funded by Mayo Transform the Practice Grant, Mayo Clinic Center for Individualized Medicine, Desert Mountain Members' CARE Foundation, David and Twila Woods Foundation. The funding sources did not play a role in the design, conduct or reporting of the study or in the decision to submit the article for publication.
Support for this project was provided by Mayo Transform the Practice Grant, Mayo Clinic Center for Individualized Medicine, Desert Mountain Members' CARE Foundation, David and Twila Woods Foundation and a Faculty Career Development Award from the Gerstner Foundation (N. Jewel Samadder).
We would like to thank the following persons for their assistance with this project—Sydney Welp, Jessie Fox, Plush Gutierrez, Sara Hernandez, Sharon Levy, Eric Nelson, Rachel Colburn, Anne Bofferding, Arta Palaj, Lorelei Bandel, Megan Mulcahy, and David Upjohn.
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Cancer Prevention Research Online (http://cancerprevres.aacrjournals.org/).
Authors' Contributions
P. LS Uson Junior: Conceptualization, writing–original draft, writing–review and editing. K.L. Kunze: Data curation, formal analysis, supervision. M.A. Golafshar: Data curation, formal analysis, writing–review and editing. D. Riegert-Johnson: Conceptualization, investigation, writing–review and editing. L. Boardman: Conceptualization, investigation, writing–review and editing. M.J. Borad: Conceptualization, investigation, writing–review and editing. D. Ahn: Conceptualization, investigation, writing–review and editing. M.B. Sonbol: Conceptualization, investigation, writing–review and editing. D.O. Faigel: Investigation, writing–review and editing. N. Fukami: Investigation, writing–review and editing. R. Pannala: Investigation, writing–review and editing. K. Barrus: Investigation, writing–review and editing. L. Mountjoy: Investigation, writing–review and editing. E.D. Esplin: Conceptualization, investigation, writing–review and editing. R. Nussbaum: Conceptualization, investigation, writing–review and editing. A. Keith Stewart: Conceptualization, resources, supervision, funding acquisition, writing–review and editing. T. Bekaii-Saab: Conceptualization, investigation, writing–review and editing. N. Jewel Samadder: Conceptualization, resources, supervision, funding acquisition, investigation, writing–original draft, writing–review and editing.
References
- 1. Benson AB, D'Angelica MI, Abbott DE, Abrams TA, Alberts SR, Anaya DA, et al. Guidelines Insights: Hepatobiliary Cancers, Version 2.2019. J Natl Compr Canc Netw 2019;17:302–10. [DOI] [PubMed] [Google Scholar]
- 2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin 2020;70:7–30. [DOI] [PubMed] [Google Scholar]
- 3. Saha SK, Zhu AX, Fuchs CS, Brooks GA. Forty-year trends in cholangiocarcinoma incidence in the U.S.: intrahepatic disease on the rise. Oncologist 2016;21:594–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bertuccio P, Malvezzi M, Carioli G, Hashim D, Boffetta P, El-Serag HB, et al. Global trends in mortality from intrahepatic and extrahepatic cholangiocarcinoma. J Hepatol 2019;71:104–14. [DOI] [PubMed] [Google Scholar]
- 5. Germline testing in biliary tract cancers. Available from: http://dailynews.ascopubs.org/do/10.1200/ADN.19.190399/full/.
- 6. Samadder J, Rupp M, Yang S, Michalski ST, Lincoln SE, Nussbaum RL, et al. Landscape of germline mutations in hepatobiliary carcinoma: unrealized risk, untapped clinical trial opportunities. J Clin Oncol 37:4s, 2019. (suppl; abstr 236). [Google Scholar]
- 7. Maynard H, Stadler ZK, Berger MF, Solit DB, Ly M, Lowery MA, et al. Germline alterations in patients with biliary tract cancers: a spectrum of significant and previously underappreciated findings. Cancer 2020;126:1995–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ahn DH, Bekaii-Saab T. Biliary tract cancer and genomic alterations in homologous recombinant deficiency: exploiting synthetic lethality with PARP inhibitors. Chin Clin Oncol 2020;9:6. [DOI] [PubMed] [Google Scholar]
- 9. Golan T, Raitses-Gurevich M, Kelley RK, Bocobo AG, Borgida A, Shroff RT, et al. Overall survival and clinical characteristics of BRCA-associated cholangiocarcinoma: a multicenter retrospective study. Oncologist 2017;22:804–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wong W, Lowery MA, Berger MF, Kemel Y, Taylor B, Zehir A, et al. Ampullary cancer: evaluation of somatic and germline genetic alterations and association with clinical outcomes. Cancer 2019;125:1441–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Samadder NJ, Riegert-Johnson D, Boardman L, Rhodes D, Wick M, Okuno S, et al. Comparison of universal genetic testing vs guideline-directed targeted testing for patients with hereditary cancer syndrome. JAMA Oncol 2021;7:230–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Terashima T, Umemoto K, Takahashi H, Hosoi H, Takai E, Kondo S, et al. Germline mutations in cancer-predisposition genes in patients with biliary tract cancer. Oncotarget 2019;10:5949–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wardell CP, Fujita M, Yamada T, Simbolo M, Fassan M, Karlic R, et al. Genomic characterization of biliary tract cancers identifies driver genes and predisposing mutations. J Hepatol 2018;68:959–69. [DOI] [PubMed] [Google Scholar]
- 14. Lowery MA, Wong W, Jordan EJ, Lee JW, Kemel Y, Vijai J, et al. Prospective evaluation of germline alterations in patients with exocrine pancreatic neoplasms. J Natl Cancer Inst 2018;110:1067–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. de Martel C, Maucort-Boulch D, Plummer M, Franceschi S. World-wide relative contribution of hepatitis B and C viruses in hepatocellular carcinoma. Hepatology 2015;62:1190–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Thiele M, Gluud LL, Fialla AD, Dahl EK, Krag A. Large variations in risk of hepatocellular carcinoma and mortality in treatment naïve hepatitis B patients: systematic review with meta-analyses. PLoS One 2014;9:e107177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kanwal F, Kramer JR, Mapakshi S, Natarajan Y, Chayanupatkul M, Richardson PA, et al. Risk of hepatocellular cancer in patients with non-alcoholic fatty liver disease. Gastroenterology 2018;155:1828–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ally A, Balasundaram M, Carlsen R, Chuah E, Clarke A, Dhalla N, et al. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell. 2017;169:1327–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zucman-Rossi J, Villanueva A, Nault JC, Llovet JM. Genetic landscape and biomarkers of hepatocellular carcinoma. Gastroenterology 2015;149:1226–39. [DOI] [PubMed] [Google Scholar]
- 20. Courtney E, Chok AK-L, Ting Ang ZL, Shaw T, Li S-T, Yuen J, et al. Impact of free cancer predisposition cascade genetic testing on uptake in Singapore. NPJ Genom Med 2019;4:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Griffin NE, Buchanan TR, Smith SH, Leon AA, Meyer MF, Liu J, et al. Low rates of cascade genetic testing among families with hereditary gynecologic cancer: an opportunity to improve cancer prevention. Gynecol Oncol 2020;156:140–6. [DOI] [PubMed] [Google Scholar]
- 22. Stoffel EM, Ford B, Mercado RC, Punglia D, Kohlmann W, Conrad P, et al. Sharing genetic test results in Lynch syndrome: communication with close and distant relatives. Clin Gastroenterol Hepatol 2008;6:333–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Caswell-Jin JL, Zimmer AD, Stedden W, Kingham KE, Zhou AY, Kurian AW. Cascade genetic testing of relatives for hereditary cancer risk: results of an online initiative. J Natl Cancer Inst 2019;111:95–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pal T, Cragun D, Lewis C, Doty A, Rodriguez M, Radford C, et al. A statewide survey of practitioners to assess knowledge and clinical practices regarding hereditary breast and ovarian cancer. Genet Test Mol Biomarkers 2013;17:367–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kurian AW, Friese CR, Bondarenko I, Jagsi R, Li Y, Hamilton AS, et al. Second opinions from medical oncologists for early-stage breast cancer: prevalence, correlates, and consequences. JAMA Oncol 2017;3:391–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kurian AW, Li Y, Hamilton AS, Ward KC, Hawley ST, Morrow M, et al. Gaps in incorporating germline genetic testing into treatment decision-making for early-stage breast cancer. J Clin Oncol 2017;35:2232–9. [DOI] [PMC free article] [PubMed] [Google Scholar]