

The genetic determinants of the somatic mutational processes in cancer elucidate the biology underlying somatic selection and evolution of cancers and demonstrate complementary predictive power across cancer types.
Abstract
The somatic landscape of the cancer genome results from different mutational processes represented by distinct “mutational signatures.” Although several mutagenic mechanisms are known to cause specific mutational signatures in cell lines, the variation of somatic mutational activities in patients, which is mostly attributed to somatic selection, is still poorly explained. Here, we introduce a quantitative trait, mutational propensity (MP), and describe an integrated method to infer genetic determinants of variations in the mutational processes in 3,566 cancers with specific underlying mechanisms. As a result, we report 2,314 candidate determinants with both significant germline and somatic effects on somatic selection of mutational processes, of which, 485 act via cancer gene expression and 1,427 act through the tumor–immune microenvironment. These data demonstrate that the genetic determinants of MPs provide complementary information to known cancer driver genes, clonal evolution, and clinical biomarkers.
Significance:
The genetic determinants of the somatic mutational processes in cancer elucidate the biology underlying somatic selection and evolution of cancers and demonstrate complementary predictive power across cancer types.
Introduction
Cancer acquires malignant phenotypes through various somatic mutations in the genome, which result in functional gains or losses contributing to the tumor fitness (1). Somatic driver mutations are critical for cancer initiation and evolution and cause the genetic heterogeneity, which determines the clonal architecture in cancers (2). The mutations occur through different mutational processes, which are evidenced in distinct mutational signatures represented by the frequencies of mutations within corresponding nucleotide contexts (3). The mutational signatures surrogate for the mutational processes during tumorigenesis, which drive the clonal evolution (4), and in turn, impact the complex clinical phenotypes of the disease (5).
So far, there are 67 signatures of single-base substitution (SBS) identified, of which, 49 were considered likely to be of biological origin (6). The mutagenic mechanisms of some mutational signatures have been elucidated in cell lines or mouse models (7, 8). External mutagens, such as tobacco smoking and UV, result in specific mutational signatures; then alterations of certain biological functions, such as deficiencies in double-strand breaks (DSB) repair mechanism and APOBEC enzymatic activities give rise to specific mutational signatures (3, 6). Finally, the random mutations can change the fitness of the tumor cells; hence, they are subject to extrinsic selective pressures such as immune responses, chemotherapy regimen, and targeted therapies (9–11). For instance, APOBEC mutational signature is a predictive marker for immunotherapy response in non–small cell lung cancer.
The activities of the mutational signature vary substantially among individual cancers, suggesting complex biological mechanisms underlying somatic selection of mutational processes (3, 12). The mutagenic processes only partially account for the activities of the mutational signatures in patients with cancer. For example, APOBEC3B activity contributes to SBS2 and SBS13; however, APOBEC3B expression only explains 20% to 30% of the total variance of the APOBEC mutational signature (Spearman r = 0.3), whereas the causes of the rest of the variation are still unknown (13). Likewise, SBS4 is caused by exposure to tobacco smoking (9). However, no more than 20% of the SBS4 activities are explained by the tobacco exposure level in lung squamous cell carcinomas and lung adenocarcinomas (14). It appears that, although the causal mutagenetic factors of the mutational processes are known, the majority of the intertumor variations in the activities of the mutational processes in real patients are still largely unexplained.
However, there still lacks reliable methodology for identification of the genetic determinants of somatic selection of mutational processes (15). The major challenges are, first, the measure of the activities of the mutational signatures, which are regularly confounded by contamination, sequencing errors, and mapping biases among the cancer samples (16). Moreover, the distribution of the signature activities in cancers are usually heavily skewed, to which most of the linear models do not fit and fail to reveal the statistical associations. Therefore, in this study, we introduced the quantitative trait of mutational propensity (MP), which is tethered to the relative activity between a given signature and a reference signature. The MP is quasi-normal distributed and more explainable by linear models. Then, we described a regression model integrating evidences from both germline and somatic levels to identify the genetic determinants of somatic selection of mutational processes. In addition, we inferred the causal biological mechanisms for the candidate determinants of the mutational processes via cancer gene expression and tumor–immune microenvironment (TIME).
Previous studies identify cancer driver genes (or driver mutations), of which, the mutational statuses are significantly associated with specific mutational processes (17, 18). However, the genetic determinants that impact the mutational processes are rarely reported until recently (19). We hypothesized that the intra- and intertumor variations of the activities of the mutational signatures largely result from the selection processes during somatic evolution, which is caused by the heterogeneity in the genetic background, the somatic landscape, and the microenvironment. Recent studies reported several cancer genes with functional and clinical significance based on the effects on the mutational processes (20, 21).
Our analyses provide a systematic view of the genetic determination of somatic selection of the major mutational processes in 3,566 cancers from The Cancer Genome Atlas (TCGA) and suggest highly relevant biological processes with clinical and therapeutic implications. In addition, the study described an alternative approach to identify cancer genes based on the process of clonal evolution, which further broadens our understanding of the formation of intratumor heterogeneity and informs the future precision medicine for cancer.
Materials and Methods
Data collection
We analyzed multiomics data on 13 cancer types including breast Cancer (BRCA, N = 690), colorectal cancer (COAD, N = 344), esophageal adenocarcinomas (EAC, N = 82), squamous-cell carcinomas (ESCC, N = 31), glioblastoma multiforme (GBM, N = 326), kidney renal clear cell carcinoma (KIRC, N = 299), liver hepatocellular carcinoma (LIHC, N = 121), lung adenocarcinoma (LUAD, N = 427), ovarian cancer (OV, N = 345), prostate adenocarcinoma (PRAD, N = 231), stomach adenocarcinoma (STAD, N = 213), thyroid carcinoma (THCA, N = 82), and uterine corpus endometrial carcinoma (UCEC, N = 375; Supplementary Table S1).
The genotype data, germline, and somatic variants and mRNA expressions were downloaded from Genomic Data Commons Data Portal. The gene-based somatic copy number alterations (SCNA) and DNA methylation were downloaded from UCSC Xena TCGA hub (https://tcga.xenahubs.net). The gene fusions were downloaded from Tumor Fusion Gene Data Portal (https://tumorfusions.org). The immune characteristics were downloaded from the published study (22, 23), including intratumor heterogeneity (ITH), immune cell fractions, subtypes, and clone number (Supplementary Methods).
To avoid biases from the populational background, it is a general practice to control for the ethnicity of the population. To infer the ancestry, we performed principal component analysis using five populations of the 1000 Genomes Project Phase 3 (1KGP) as references. For a certain population, the genotypes were prephased using SHAPEIT2 and imputed using IMPUTE2 with the 1KGP reference panel for further analyses (Supplementary Methods; refs. 24, 25).
We calculated the gene-wise genetic burden for each class of germline variants (missense, truncated, and structural variant) and encoded the gene-wise statuses of somatic nonsynonymous mutations (somatic nsy-mutations), SCNAs, methylation aberrations in the promoter regions (TSS-methylation), and fusions for integrated regression analyses (Supplementary Methods).
The matched multilevel data of 1,631 cancer cell lines were downloaded from the Cancer Cell Line Encyclopedia project (CCLE; https://portals.broadinstitute.org/ccle), including the somatic mutations, SCNAs, DNA methylations, and mRNA expressions. The profiling protocols were consistent with the preference rules of TCGA (Supplementary Methods). The 563 cell lines' CERES scores were downloaded from The Cancer Dependency Map Project at Broad Institute (DepMap, v19q2; https://depmap.org). CERES score is a computational method to estimate gene-dependency levels from CRISPR-Cas9 essentiality screens while accounting for the copy number–specific effect (26). The drug sensitivity data (IC50) of 305 drugs in 988 cell lines were downloaded from Genomics of Drug Sensitivity in Cancer V8 (GDSC; https://www.cancerrxgene.org).
Deriving the consensus mutational signatures from multiple cancer types
The distribution of mutational signatures (MS) activities is heavily skewed; thus, it prevents the development of an optimized model for the discovery of driver genes for these mutagenic processes. To overcome the statistical obstacle, we retrieved the somatic mutational signatures using “pmsignature” based on 5-nucleotide context (27). This algorithm introduced a “background signature” that is designed to capture biases in intrinsic genome sequence composition and is calculated from the composition of consecutive nucleotides of the human genome sequence (27). All the mutational signatures were compared with the results of the SBS mutational signatures in the COSMIC (https://cancer.sanger.ac.uk/cosmic) using cosine similarity (CS) measure. To account for the confounding effects in the future analysis, we set a threshold of
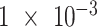 for the present call of a given signature k. Then, we used the background signature as the reference and generated a new statistical term called MP, which is the relative activity of mutational signature as the ratio between the activities of a given signature k and the reference (Eq. A).
for the present call of a given signature k. Then, we used the background signature as the reference and generated a new statistical term called MP, which is the relative activity of mutational signature as the ratio between the activities of a given signature k and the reference (Eq. A).
 |
Here,
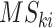 is the activity of the
is the activity of the
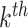 signature of
signature of
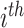 individual;
individual;
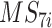 is the reference signature (MS7);
is the reference signature (MS7);
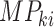 is the
is the
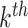 mutational propensity.
mutational propensity.
We used Bioconductor package “deconstructSigs” to determine the activities of the conserved mutational signatures in cancer cell lines (28), and the MPs were computed in the same way as aforementioned. We compared the mean of MPs between the TCGA and CCLE cohorts based on cosine similarity.
Integrated regression analyses suggest driver genes of mutational processes
To estimate the effects of each gene on the mutational processes, we combined seven classes of gene variations: germline variants (missense, truncated, and structural variant), somatic nsy-mutations, SCNAs, TSS-methylation, and gene fusions. We excluded highly polymorphic genes from the analysis, namely the human leukocyte antigen genes and olfactory receptor genes. Then, for pan-Cancer and cancer-specific analyses, we excluded the samples with low present call (N < 30) and genes with very low mutation/variation rate (N < 5). We used the MPs to evaluate the effects of the mutational status of each gene using a linear model for both pan-cancer (Eq. B) and cancer-specific analysis (Eq. C). The regression coefficients of
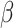 represent the effect sizes of the variations of genes.
represent the effect sizes of the variations of genes.
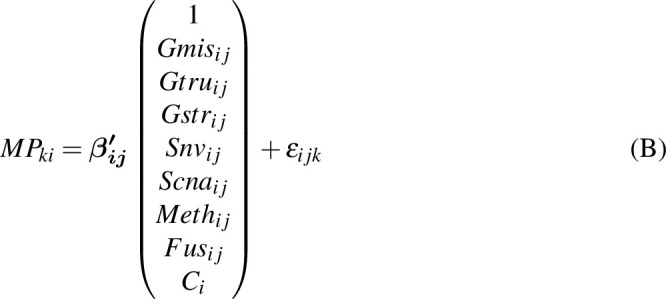 |
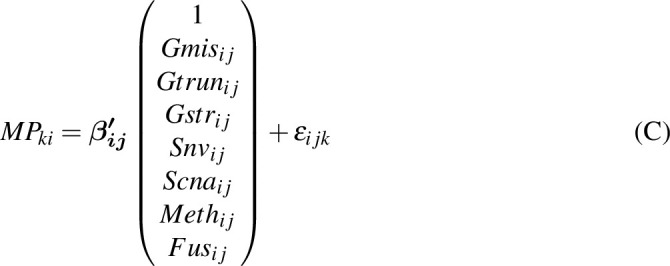 |
Here,
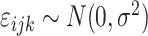 is a Gaussian error term;
is a Gaussian error term;
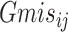 refers to the
refers to the
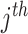 gene's missense genetic burden of
gene's missense genetic burden of
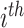 individual; and
individual; and
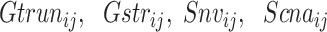 ,
, 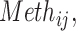 and
and 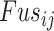 are the truncation genetic burden, structural genetic burden, somatic nsy-mutation status, SCNA status, TSS-methylation levels, and fusion status, respectively;
are the truncation genetic burden, structural genetic burden, somatic nsy-mutation status, SCNA status, TSS-methylation levels, and fusion status, respectively;
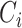 is the cancer type.
is the cancer type.
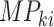 is the
is the
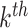 mutational propensity. We define a driver gene, of which, the germline variants and somatic variants are both significantly associated with the MPs.
mutational propensity. We define a driver gene, of which, the germline variants and somatic variants are both significantly associated with the MPs.
Instrumental variable regression analysis
We then aimed to find the genes, of which, the genetic features impact the mutational processes through their expression levels by performing instrumental variable analysis using the Julia (v1.1.1) package of “FixedEffectModels.” Briefly, the dependent variable is the MPs, the independent variable is the expression levels of genes that significantly associated with MPs, and the genetic instruments are the genetic features in Eq. B and Eq. C, which were previously found significant (Eq. D).
Here,
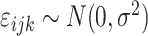 is a Gaussian error term;
is a Gaussian error term;
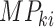 is the
is the
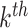 mutational propensity of the
mutational propensity of the
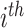 individual;
individual;
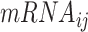 is the
is the
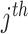 gene expression;
gene expression;
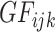 are the genetic features;
are the genetic features;
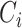 is the cancer type.
is the cancer type.
Models with instrument variables were estimated using Two-Stage least squares (2SLS; Julia package, FixedEffectModels). To determinate the significance of the independent variables, we calculated FDR based on the P values of the regression coefficients using Benjamini–Hochberg procedure. To determinate the significance of the instrumental variables, we used the weak instruments test P values (29) based on the Kleibergen–Paap rank Wald F-statistic and estimated FDRweak. Finally, we chose genes, of which, the expression levels and genetic instruments were both significant to call E-genes (FDR < 0.1 and FDRweak < 0.1).
Similarly, we performed the IV analysis to find the genes, of which the genetic statuses impact the mutational processes through interacting with the TIME. The dependent variable is the MPs, the independent variable is the immune cell fraction (22), the genetic instruments are the genetic features. Finally, we selected a set of genes, of which genetic features associated with immune cell fractions, and in turn, impacted somatic mutational processes (FDR < 0.1 and FDRweak < 0.1), which were called I-genes.
Identify the drug-related candidate genes in cancer cell lines
To evaluate the effects of the candidate genes on the drug sensitivity/resistance to therapy, we used a linear model to calculate the association between the mRNA expression levels of E-genes and I-genes and the IC50 of drugs (Eq. E):
Here,
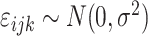 is a Gaussian error term;
is a Gaussian error term;
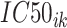 is the IC50 of the
is the IC50 of the
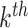 drug of
drug of
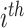 cell line;
cell line;
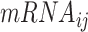 is the mRNA expression of the
is the mRNA expression of the
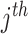 gene;
gene;
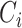 is the cancer type.
is the cancer type.
Identify the genes associated with anti–PD-1 therapy response
To identify the genes associated with anti–PD-1 therapy response, we performed a one-sided Student t test to capture genes that were differentially expressed between the nonresponse groups (progressive/stable disease, PD/SD) and response groups (partial/complete response, PR/CR) in melanoma (N = 55; ref. 30) and metastatic gastric cancer (N = 45; ref. 31). A P < 0.05 was considered significant.
Identify common noncoding variants influencing the mutational processes
To examine the association between noncoding germline variants and the mutational processes in cancers, we took the MP as a quantitative trait and performed a whole-genome quantitative trait loci (QTL) mapping based on linear model in both pan-cancer level (Eq. F) as well as cancer-specific level (Eq. G), which resulted in a set of mpQTLs significantly associated with the MPs (FDR < 0.1).
Here,
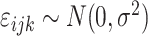 is a Gaussian error term;
is a Gaussian error term;
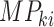 is the
is the
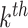 mutational propensity of the
mutational propensity of the
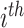 individual;
individual;
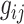 is the
is the
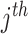 SNP's genotype;
SNP's genotype;
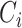 is the cancer type.
is the cancer type.
Then, we used the significant variants as genetic instruments to examine the association between the nearby genes of variants (< 1 Mb) and the corresponding MPs in each cancer type using Julia (v1.1.1) package FixedEffectModels (Eq. H):
Here,
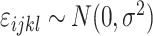 is a Gaussian error term;
is a Gaussian error term;
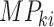 is the
is the
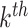 mutational propensity of the
mutational propensity of the
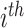 individual;
individual;
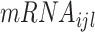 is the
is the
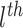 gene expression nearby the
gene expression nearby the
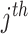 SNP (<1 Mb);
SNP (<1 Mb);
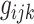 is the genotype of the
is the genotype of the
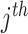 SNP, which is significantly associated with the
SNP, which is significantly associated with the
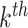 MP;
MP;
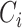 is the cancer type.
is the cancer type.
Annotation of biological processes determine the mutational processes
The above E-genes and I-genes were then subjected to gene set enrichment analysis using gene sets including Reactome (MSigDB v6.1) and Hallmark (MSigDB v6.1), Kyoto Encyclopedia of Genes and Genomes (MSigDB v6.1).
Gene sets enrichment test
We performed Fisher exact test to evaluate the enrichment of E-genes and I-genes enriched for known cancer driver gene sets using R packages GeneOverlap.
Clinical analysis
To assess the effects of E-genes and I-genes on the treatment outcome, we collected a cohort of 60 patients with LUAD from The First Affiliated Hospital of Xiamen University (FHXU, Xiamen, China). Informed written consent was obtained from each subject or each subject's guardian. The usage of patient data is approved by the ethics committee/institutional review board of FHXU (Xiamen, China). We used a logistic regression to evaluate the interaction between somatic mutational burden of E-genes or I-genes and the targeted therapies on the binary clinical outcomes (PR and PD/SD), adjusting for clinical covariates including stage and smoking. A P < 0.05 was considered significant.
The Kaplan–Meier method was utilized to estimate overall survival, and difference between groups was assessed using the log-rank test. A P < 0.05 was considered significant.
Data availability statement
The Code Ocean capsule containing the necessary data and codes to replicate the results of this study can be found at https://codeocean.com/capsule/6181333/tree/v1 (DOI: 10.24433/CO.2000361.v1).
Results
Mutational propensity in thirteen cancer types
We obtained a dataset of 13 cancer types from TCGA with matched germline variants, somatic mutations, SCNAs, DNA methylation, and expression of mRNA (32). After filtering for populational background and removal of unmatched individuals, we identified a population of 3,566 Utah residents of northern and western European ancestry (CEU; Fig. 1; Supplementary Fig. S1A and S1B; Supplementary Table S1) for the following analysis.
Figure 1.

The schematic view of this study. In this study, we introduced the quantitative trait of MP, which is tethered to the relative activity between a given signature and a reference signature. Then, we described a regression model integrating evidences from both germline and somatic levels to identify the genetic determinants of somatic selection of mutational processes and also performed mpQTL analysis for MP-associated loci. In addition, we inferred the causal biological mechanisms for the candidate determinants of the mutational processes via cancer gene expression and TIME.
To derive a quantitative trait for intertumor variations in the mutational processes, we first retrieved seven highly conserved somatic mutational signatures (MS) from the mutational profiles based on a 5-nucleotide context (Fig. 2A; Supplementary Fig. S2A–S2C). These signatures are highly comparable with the known SBS signatures in the COSMIC according to the cosine similarity (CS; Supplementary Fig. S2D; ref. 6). For example, MS1 is highly similar to SBS1 (the deamination of 5-methylcytosine, characterized by C>T at NpCpG trinucleotide, CS = 0.97). MS2 associates with SBS2 (CS = 0.79), which is attributed to the activity of the AID/APOBEC family of cytidine deaminases. MS3 correlates with SBS6 (CS = 0.75), which is associated with defective DNA mismatch repair (dMMR). MS4 correlates to SBS4 (CS = 0.87) caused by tobacco smoking. MS5 and MS6 correlate with SBS10a (CS = 0.96) and SBS10b (CS = 0.91), respectively, both of which are caused by polymerase epsilon exonuclease (POLE) domain mutations (3, 6). Finally, the MS7 universally correlates with the multiple mutational signatures in the COSMIC databases (Supplementary Fig. S2D). Among all MSs, MS7 is abundant in all nucleotide contexts, representing the most recurrent and conserved mutations in the genome, and is universally present in all cancer types; hence, it is a suitable reference mutational signature in the following analysis.
Figure 2.

The mutational propensities in thirteen cancer types. A, The conserved MSs based on 5-nucleotide context are correlated with COMISC single-base substitution signatures. B, The quantile–quantile plot shows the abnormal distribution of the MSs at pan-cancer level. C, The quantile–quantile plot shows the quasi-normal distribution of the MPs at pan-cancer level. D, The MPs of each individual in different TCGA cancer types. Each bar represents an individual. The cancer types are ranked from left to right in the order of increased median TMB. In each cancer type, the individuals are ranked from left to the right in the order of decreased activities of the MS7. Different colors represent different MPs. E, The MP and the MS activities show consistent effects (Spearman correlation) on TMB. The effect sizes are calculated as Spearman rho between MP or MS and TMB. F, The MP and the MS activities show consistent effects (Spearman correlation) on ITH. The effect sizes are calculated as Spearman rho between MP or MS and TMB. G, The MPs are highly consistent between TCGA cancer tissues and CCLE cancer cell lines representing the same cancer types.
To control for the latent confounders of between-sample variation and improve the skewed distribution of MSs, we introduced MP as a quantitative trait for somatic selection pressure over the MSs. MP is defined as the natural log ratio of the activities between a signature of interest and the reference signature (Eq. A). Hence, a positive MP indicates that the corresponding mutational process is positively selected in the given cancer. After removal of the extreme values, the MPs in the cancer population follow approximately normal distribution (Fig. 2B and C).
The MPs retain the important biological properties of the original MSs (Fig. 2D). Of note, in LUAD, COAD, and UECE, tumor with high tumor mutation burden (TMB) inclined to specific MPs, suggesting the corresponding mutational processes are dominant in high TMB tumors. Among the 13 cancer types, the effect sizes (Spearman rho) of MPs on TMB are strongly correlated with those of the original MSs (R = 0.822, P < 2.20 × 10–16; Fig. 2E). We calculated the Shannon index based on the activities of MS as a measure of diversity of the mutational processes. Our data suggested that tumors with more diverse mutational processes showed significantly increased number of clones (P = 7.00 × 10–4; Supplementary Fig. S3A–S3C) and the effect sizes (Spearman rho) of MPs on ITH are strongly correlated with those of MSs (R = 0.795, P < 2.20 × 10–16; Fig. 2F). We also noticed that the MPs are significantly associated with tumor immune subtypes (Supplementary Fig. S4).
To further validate the MPs in cell lines, we retrieved the six MPs in 544 cancer cell lines of CCLE representing 13 cancer types. The MPs are highly consistent between cancer tissues and the cell lines of the same cancer type with an overall correlation of 0.957 (P = 1.00 × 10–6; Fig. 2G). Within each cancer type, the cosine similarity between TCGA sample and cell lines ranges from 0.938 (ESCC) to 0.996 (EAC). The results suggested that the MPs are highly conserved in both cancer tissues and cell lines; hence, they are a robust surrogate for the mutational processes in cancer (33).
In addition, the MPs are significantly associated with IC50 of 116 drugs (FDR < 0.1). For example, MP3 is associated with the IC50 of BRAF inhibitor (dabrafenib, FDR = 0.0781 and PLX-4720, FDR = 0.0240); MP4 is associated with the IC50 of HSP90 inhibitors (CCT-018159; FDR = 0.0568; Supplementary Fig. S5), which is consistent with the previous reports that treatment influences the mutability of cancer (10, 34).
The genetic determinants of somatic selection of mutational propensities in cancers
Next, we sought to identify genes that influence somatic selection of MPs. We defined a regression model integrating the effects of the germline statuses, the somatic statuses, and the epigenetic statuses (TSS methylation levels). To determine the germline statuses of the genes, we classified the 524,912 rare variants into three subtypes: 9,584 structural variants (>50 bp), 467,709 missense variants, and 47,619 truncated variants. Then, we computed the burden of each class of variations separately for 17,810 genes. For the somatic mutational statuses, we obtained a total of 820,907 single-nucleotide variants (SNV), 163,544 deletions, and 54,084 insertions from TCGA for 3,566 cancers. We, then, retrieved 19,105 genes' somatic mutational statuses based on 797,814 nonsynonymous variants. The samples with high TMB (>20) occur mostly in UCEC (17.6%), COAD (17.4%), and STAD (16.0%), which is consistent with prior studies (35).
We evaluated 17,027 genes in 3,566 cancers and found 87.5% (N = 14,903) genes, of which at least one type of genetic status was significantly associated with the MPs (FDR < 0.1), suggesting a wide influence of the mutational processes. Among the genes associated with the MPs, 14,373 (84.4%) act at the pan-cancer level and 13 (0.0763%, ESCC) to 9,596 (56.4%, UCEC) are cancer specific. As for the effect sizes, the somatic nsy-mutations were significant in 12,836 genes and accounted for most of the variations of MPs, especially for MP5 (1.77% to 54.1%), MP6 (0.0864% to 27.0%), and MP3 (0.798% to 29.9%; Fig. 3A); followed by the SCNAs, which were significant in 11,721 genes and accounted for 0.764% to 48.8% of the variance of MP1 and MP3, respectively. Of note, there are a small subset of genes, of which, SCNA statuses accounted for more than 40% of the variance of MP1 (adjusted R2 > 0.4), including PIK3CA (FDR = 6.68 × 10–4, adjusted R2 = 0.477) and MAP3K1 (FDR = 1.25 × 10–4, adjusted R2 = 0.478; Fig. 3A). In addition, the TSS-methylation of 8,199 genes accounted for over 10% of the variance of MP2 and 20% of the variance of MP6. Genes in this category include CDH1 (MP2, FDR = 2.75 × 10–5, adjusted R2 = 0.162) and ERCC3 (MP6, FDR = 0.0318, adjusted R2 = 0.227; Fig. 3A). The effect sizes of the germline variants are much smaller than those of the somatic mutations (Fig. 3A). The germline missense mutations were significant in 3,304 genes, followed by the truncated mutations, which were significant in 737 genes, and the structural mutations, which were significant in 107 genes. Gene fusions were significant in only 48 genes. Nevertheless, the fusion statuses of 12 genes showed very strong effects on MP1 (R2 > 0.4).
Figure 3.

Genes' genetic statuses influence the mutational propensities. A, The fraction of variance (adjusted R2) of 6 MPs explained by different statuses of genes (FDR < 0.1), including three types of germline variations (missense, truncated, structural), four types of somatic alterations (somatic nsy-mutation, SCNA, fusion), and epigenetic stats (TSS-methylation). The benchmark genes with different colors are the known cancer driver genes, of which, germline or somatic mutations are associated with the certain MSs. B, The Upset plot shows the overlaps among the gene sets, each of which is significantly associated with the MPs by a distinct genetic status. The black bar shows the total 2,314 unique genes, which are considered as candidate genes, of which, germline genetic burden and somatic or epigenetic status are simultaneously significantly associated with the same MP (FDR < 0.1). C, The number of significant genes, of which, genetic statuses impact the MPs at pan-cancer level or cancer-specific level. D, The number of significant genes, of which, genetic statuses associate with one or multiple MPs.
To further validate the methodology, we chose a set of cancer genes with known germline pathogenic effects for an internal validation, including MBD4, BRCA1, BRCA2, and several dMMR genes (MLH1, MSH2, MSH6, and PMS2). As a result, we found that MP1 (the deamination of 5-methylcytosine) is influenced by the germline truncation of MBD4 (P = 9.17 × 10–3, adjusted R2 = 0.467) and BRCA2 (P = 3.79 × 10–5, adjusted R2 = 0.468) at pan-cancer levels, which is consistent with the previous report (19). MP3 (dMMR-related) is influenced by germline missense variants of PMS2 in LUAD (P = 0.0431, adjusted R2 = 0.0122) and MSH6 in BRCA (P = 3.93 ×10–4, adjusted R2 = 0.0232). And MP2 (APOBEC-related) is influenced by the missense variant of APOBEC3H (P = 3.61 ×10–3, adjusted R2 = 0.156; Fig. 3A).
Our data indicated that many genes are associated with the MPs, but such associations do not necessarily imply any causal effects. To ascertain the causal effect of a given gene, we mandated the significance of both germline effect and nongermline effect on the same MP (Supplementary Methods). Thus, we identified 2,314 candidate driver genes of MPs in cancer based on FDR of 0.1 (Fig. 3B). Among the candidate genes, 1,268 (54.8%) genes influence the MPs at pan-cancer level, 935 (40.4%) in specific cancer types, and 111 (4.80%) are significant at both pan-cancer and cancer-specific level, such as BRCA1/2, FAT3, and SETDB1 (Fig. 3C). The majority of the cancer-specific candidate genes of MP are identified in UCEC (38.4%, N = 888), COAD (5.49%, N = 127), and BRCA (1.17%, N = 27). On the other hand, 87.9% of the 2,314 candidate driver genes are associated with unique MPs (Fisher exact test P < 0.01), suggesting the biological mechanisms underlying somatic selection of mutational processes are highly specific; 10.46% (N = 242) are associated with two and only 1.64% (N = 38) are associated with more than two MPs (Fig. 3D). MP2 (N = 661), MP5 (N = 491), MP6 (N = 452), MP1 (N = 362), and MP3 (N = 352; Supplementary Table S2A) are the mutational processes with the most genetic determinants (90.3% total), whereas MP4, which is caused by tobacco smoking has the least (N = 310).
We also validated the candidate genes in the population of African Ancestry in Southwest United States (ASW, N = 564) and Han Chinese in Beijing, China (CHB, N = 363). Among the 2,314 candidate genes, the germline genetic burdens of 9 genes in ASW and 4 in CHB are significantly associated with the same MPs as in CEU, and the somatic statuses of 223 genes in ASW and 18 in CHB are significant (Supplementary Table S2A).
To infer the biological processes mediating the effects of the candidate determinant genes and the selection of mutational processes, we performed an instrumental variable (IV) regression for the 2,314 candidate driver genes based on two possible mechanisms of somatic selection: first, the genetic and/or epigenetic statuses of a candidate gene directly alter its expression level in cancers and influence the fitness of the cell. As a result, we identified 485 unique “expression-associated determinant genes” (E-genes; FDR < 0.1 and FDRweak < 0.1; Fig. 3B; Supplementary Table S2B). In another scenario, the genetic and/or epigenetic statuses influence the selection of mutational process through interacting with the TIME. Thus, we identified another set of 1,427 “Immune-interactive-genes” (I-genes; FDR < 0.1 and FDRweak < 0.1; Fig. 3B; Supplementary Table S2C).
We further analyzed candidate genes in detail and found that the 485 E-genes and 1,427 I-genes are both significantly enriched for known cancer driver genes, such as the COSMIC cancer gene consensus (CGCs; 2.39-fold, P = 1.30 × 10–5; 1.74-fold, P = 2.80 × 10–5) and three other cancer driver gene sets (P < 0.05, OR = 1.78–4.05; Fig. 4A; refs. 15, 36, 37), suggesting the genetic determinants of somatic selection of mutational processes we found are highly consistent to the known cancer driver genes.
Figure 4.

The E-genes and I-genes influence the mutational propensities. A, The Upset plot shows the overlap between the E-genes, I-genes, COSMIC CGCs, and another three published benchmark cancer gene sets. B, The E-genes and I-genes are enriched for determinants of different MPs at the pan-cancer level based on the background of 2,314 candidate genes. The dot color represents the −log10 (FDR). The dot size represents the OR of enrichment based on the background of 2,314 candidate genes. C, The pathways enriched in E-genes and I-genes. The dot color represents the −log10 (FDR). The dot size represents the OR of enrichment. D, The I-genes impact on the MPs through different immune cell activities in TIME.
Cancer gene expression intermediates the genetic determinants of mutational propensity
Of the 485 “E-genes,” 210 influence the MPs at pan-cancer level, which mainly affect the mutational processes of MP1 (N = 73, OR = 15.5, P = 3.41 × 10–43) and MP3 (N = 23, OR = 5.91, P = 7.55 × 10–10; Fig. 4B). There are also 283 “E-genes” acting in specific cancer types, most of which are identified in UCEC (N = 244), COAD (N = 42), and BRCA (N = 10, Supplementary Table S2B).
The 485 E-genes are significantly enriched in pathways of cancer cell growth and proliferation pathways, such as metabolism of RNA (FDR = 2.26 × 10–6), RNA polymerase III transcription (FDR = 3.72 × 10–3), cell cycle (FDR = 3.86 × 10–3), and metabolism of lipids (FDR = 3.21 × 10–5; Fig. 4C). Many E-genes are well-known cancer driver genes, such as BRCA1 (MP2, FDR = 7.20 × 10–3), PIK3R1 (MP6, FDR = 1.89 × 10–5), while others are newly reported, such as PPIP5K2 (MP1, FDR = 0.00250) and SNX24 (MP6, FDR = 2.51 × 10–6).
Tumor–immune microenvironment intermediates the genetic determinants of mutational propensity
Of the 1,427 “I-genes,” 797 significantly influence MPs at pan-cancer level, which are significantly enriched for determinants of the mutational processes corresponding to MP2 (N = 360; OR = 4.09; P = 1.70 × 10–48), MP3 (N = 58; OR = 8.42; P = 4.45 × 10–10), MP5 (N = 180; OR = 3.98; P = 3.89 × 10–28), and MP6 (N = 138; OR = 3.82; P = 4.20 × 10–22; Fig. 4B). There are another 680 “I-genes” acting in specific cancer types, most of which are identified in UCEC (N = 599), COAD (N = 54), and BRCA (N = 23, Supplementary Table S2C).
The pathways significantly enriched in the I-genes are consistent with the functions in cancer immunity. The I-genes are enriched in cytokine signaling in immune system pathways (FDR = 3.16 × 10–7) and innate immune system pathway (FDR = 1.29 × 10–5, Fig. 4C); hence, they have an effect on the activities of tumor microenvironment. Then, the I-genes are also enriched in cancer invasion and metastasis pathways, such as epithelial mesenchymal transition (EMT; FDR = 2.59 ×10–5) and adhesion (FDR = 1.66 ×10–22), suggesting that intercellular communications are involved in somatic selection and influence the clonal evolution (38). Finally, certain metabolism pathways, such as the lipid metabolism (FDR = 5.41 × 10–3) are also enriched in the I-genes.
Of note, 350 of the 485 E-genes are also I-genes, which suggest the genetic determinants can influence the mutational processes in both ways (Supplementary Table S2D). Many of these 350 genes are known cancer genes, such as MSH2, MSH6, BRCA1, ALK, ABL2, MAPK6, and NF1.
In addition, the impact of the somatic statuses on the MPs is related to the activities of specific immune cells. For example, the TSS-methylations are strongly associated with MP2 through neutrophil activity (25.2%, N = 360, Fig. 4D), and the somatic nsy-mutations influence MP5 through the activities of T follicular helper cells (19.1%, N = 273) and M1 macrophages (9.95%, N = 142). These findings suggest specific immune responses underlie somatic selection of mutational processes, which are activated in response to different types of somatic alterations in I-genes.
The genetic determinants of somatic mutational propensity impact carcinogenesis and cancer therapy
As the genetic determinants of MPs influence many cancer-related biological processes, we asked how these genes impact carcinogenesis and the consequent biological–clinical characteristics of cancer.
Recent advances in gene-editing techniques enable highly specific evaluation of the genetic dependency of cancer proliferation. We first evaluated the enrichment of genes annotated for cancer dependency by CRISPR-Cas9 screening in 233 cell lines (26). We categorized 10,343 genes of cancer dependency according to the median CERES score across different cell lines. Then we compared the fold enrichment of benchmark gene sets (15, 36, 37), such as COSMIC CGCs and susceptibility cancer driver gene sets with those of our E-genes and I-genes. As a result, both E-genes and I-genes are significantly enriched in the oncogenes, of which, the CERES score is less than zero (Fig. 5A). For example, E-genes are significantly enriched in the gene set (CERES = −1.6 ∼ −1.2; P = 0.024, fold enrichment = 9.18) and I-genes are significantly enriched in the gene set (CERES = −2.2 ∼ −2; P = 0.0137, fold enrichment = 17.7). Of note, the tendency of enrichment of the E-genes and I-genes remains stable after removal of known cancer genes.
Figure 5.

The carcinogenesis and cancer therapy of E-genes and I-genes. A, The genetic determinants of the MPs enrich for genes to which cancer cells manifest strong genetic dependency. B, Comparison fractions of genes significantly associated with drug IC50 in cancer cell lines among different gene sets. C, The E-genes of different MPs are predictive of drug IC50 targeting at different pathways. The dot size represents the fraction of drugs in each category, which are predicted. Different colors represent the average absolute effect sizes. D, The fractions of genes significantly associated with anti–PD-1 therapy response in melanoma among different gene sets. E, The fractions of genes significantly associated with anti–PD-1 therapy response in metastatic gastric cancer among different gene sets.
We compared the CERES scores of E-genes and I-genes. Consistent with the corresponding biological basis, the median CERES score of the E-genes is significantly lower than those of the I-genes (P = 0.0075; Supplementary Fig. S6), suggesting that I-genes overall show weaker cancer dependency than E-genes in vitro.
We assessed the therapeutic implications of the E-genes in 300 cancer cell lines treated with 320 drugs with specific targets. Among the six benchmark gene sets (15, 36, 37), E-genes showed the highest fraction of genes that are significantly predictive of the IC50 in all cell lines (70.5%, N = 148, FDR < 0.1), whereas the fraction of predictive I-genes is the lowest (63.9%, Fig. 5B). Each of the 320 cancer drugs targets a specific signaling pathway in cancer. Our results suggest that E-genes extensively influence the efficacies of targeted therapies in cell lines, especially on hormone-related pathway and cytoskeleton pathway (MP2) and chromatin histone methylation pathway (MP3 and MP6; Fig. 5C). Moreover, in a cohort of 60 LUAD that received chemotherapy and targeted therapies (Supplementary Table S3), the somatic mutation burdens of 17 E-genes showed significant interactive effect on the clinical benefit from EGFR-targeted therapies (PR vs. PD/SD; P = 0.0361; OR = 2.26); whereas the mutation burdens of I-genes showed no such effect.
We then compared the I-genes with genes associated with responses to immune checkpoint inhibitors (ICI, anti–PD-1) in melanoma (N = 55; ref. 30) and metastatic gastric cancer (N = 45; ref. 31). As a result, I-genes showed the strongest overlapping with genes predictive of anti–PD-1 therapy response in both cohorts (9.91%, N = 79 and 4.52%, N = 36), compared with the other five benchmark gene sets (Fig. 5D and E). Moreover, the 79 I-genes that overlapped with the anti–PD-1 therapy response genes are enriched in pathways, such as fatty acid metabolism (FDR = 0.0113) and adipogenesis (FDR = 0.0113; Supplementary Fig. S7). As for the long-term outcomes, the somatic nsy-mutation statuses of 6 I-genes, such as CREBBP (MP3; HR = 0.415; P = 5.66 × 10–4) and PARP1 (MP6; HR = 0.169; P = 0.0123) were significantly associated with better overall survival in 1,661 patients who received ICI treatment. (Supplementary Fig. S8; ref. 39).
In summary, our results suggest that E-genes and I-genes influence specific mutational processes, respectively. E-genes represent the proliferation capacity; hence, they have an impact on the age-related mutational process, such as deamination of methylated cytosines, which occurs throughout the patient's life time (9). The I-genes, which influence somatic selection of mutational processes via TIME, are more effective in the mutational processes associated with cancer immunity, such as AID/APOBEC processes (40).
Quantitative trait loci of the mutational propensities
Finally, we assessed the effects of 6,103,818 noncoding common germline variants on six MPs. We found 127 unique noncoding germline variants, which mapped to two MPs (mpQTLs; P < 5 × 10–8, Fig. 6A; Supplementary Table S4A). Of note, the effect sizes of the noncoding common germline variations on the mutational processes are comparable with those of the coding variants. We found that the MP2 (APOBEC-related) is the most influenced by mpQTLs (20q11.21, 15q21.2, and 16q12.2; 77.2%, N = 98), followed by MP6 (mut-POLE–related; 10q24.1, 9.45%, N = 12) and MP1 (NpCpG-related; 5q12.3, 5.51%, N = 7; Fig. 6A). Especially, we found that 6 SNPs in 22q13.1 are significantly associated with MP2 (APOBEC), such as rs112045173 (P < 8.21 × 10–6) and rs17824310 (P < 8.49 × 10–6), which are consistent with the previous study (19). Among the germline variants associated with the MPs, 99 loci (78.0%) act at the pan-cancer level while the others are reported mainly in three cancer types, BRCA (N = 14, 0.110%), THCA (N = 7, 5.51%), and KIRC (N = 4, 3.15%; Supplementary Table S4A).
Figure 6.

The quantitative loci of the mutational propensities. A, The genomic distribution of the SNP loci. Each box represents a chromosome and each dot a SNP locus. The different colors represent the different MPs significantly associated. The colored dots represent the mpQTLs that reached genome-wide significance (P < 5.0 × 10–8), and the rest of SNPs are labeled in gray. B and C, The associations among the rs6060924 genotypes with the MP2 and the COX4I2 expression. D and E, The associations among the rs35413356 genotypes with the MP2 and the ALDH5A1 expression. The P values are based on Student t test. *, P < 0.05; **, P < 0.01; ***, P < 1.0 × 10–3; ****, P < 1.0 × 10–4; *****, P < 1.0 × 10–5.
We noticed that all of the unique 127 mpQTLs are known eQTL loci in cancer (41), suggesting the impacts on mutational process are related to cancer gene expression. To reveal the underlying mechanisms through which the mpQTLs exert their functions, we performed IV regression based on gene expression in cis of the mpQTLs (Supplementary Methods). The results suggested that 13 mpQTLs significantly impact the MP2 (APOBEC-related) via acting on mRNA transcript levels in cis (Supplementary Table S4B). For example, rs6060924 acts through mitochondrial cytochrome c oxidase subunit 4 isotype 2 (COX4I2) to impact MP2 (Fig. 6B and C); another SNP in linkage disequilibrium is reported as an eQTL of COX4I2 in a variety of cancer (41). COX4I2 is a component of Warburg effect and related to tumor progression and metastasis (42). The other mpQTL (rs35413356) acts through aldehyde dehydrogenase 5 family member A1 (ALDH5A1), which also impacts MP2 (Fig. 6D and E).
Discussion
In our study, we used the MPs as a measure of the propensity of mutational processes in cancer. Compared with the raw activities of MSs, the MPs show better normality, robustness to sample purity and heterogeneity, and stronger clinical relevance. MPs work as a robust linkage to somatic selection of mutational processes, which facilitate the discovery of relevant cancer genes. Future study with larger sample size can improve the statistical power to identify more cancer-evolution–related genes.
Although the germline variants that directly cause MSs are mainly reported for homologous recombination (HR; ref. 43) and dMMR (44)-related genes. Recent studies demonstrated that germline variants other than HR and dMMR also influence the MSs (45). The germline determinants of MPs include both causal mutagenic processes and intrinsic or extrinsic biological processes contributing to somatic selection and clonality of cancer. In this study, we hypothesized that many more germline variants can influence the “fitness” of cancer. Therefore, the candidate genes in our study included both genes from the causal mutagenic processes and intrinsic or extrinsic biological processes contributing to somatic selection and clonality of cancer.
Most of the studies trying to identify “cancer drivers” are based on the significantly high recurrence of somatic mutations in the cancer populations. Such methods have yielded many meaningful driver genes and mutations, which are successfully used in cancer diagnosis, prediction, and treatment. On the other hand, the clinical benefit of treatments still vary substantially among patients, which is largely attributed to the clonal evolution of cancer. This study is designed to identify genes and variants that potentially influence the processes of somatic selection and thereby suggest new candidate for cancer driver genes.
ITH is extensively studied for its association to clonal evolution and somatic selection, and is a major cause of resistance to treatments (46). However, most of the ITH studies are still based on limited sample size, whereas the clinical phenotypic heterogeneity of cancer is observed and measured in large population of cancer. Alternatively, our approach is based on the intertumor variations of the evolution processes in a reasonably large population, which offers the capability to assess the effects of genetic heterogeneity, especially rare mutational events, on the somatic evolution, which cannot be addressed in small sample.
Here, we report two gene sets (E-genes and I-genes), which exhibit additional predictive power in the treatment responses and innate immune status in TIME. Our findings confirm the importance of somatic evolution in the development of cancer and provide an alternative way to identify genes, which further explains the heterogenous clinical phenotypes beyond the known driver genes.
Our data suggest that I-genes are enriched in lipid metabolism. For example, HSPH1 (heat shock protein family H member 1) can induces macrophage differentiation (47). HSPH1 also interacts with BCL6, which specifies and promotes T follicular helper cell program (48). Therefore, we deduce that some of the I-genes modulate T follicular helper cell and M1 macrophage activities via lipid metabolism and thereby impact MP5. Consistently, recent studies show that tumors with elevated lipid metabolism have increased antigen presentation and are associated with response to anti–PD-1 or TIL-based immunotherapy (49).
Some of the I-genes can also be functionally pleiotropic in cancer. For example, EEF2 (eukaryotic elongation factor 2, CERES score = −2.03), elicits both humoral and cellular immune responses; it also promotes tumor cell proliferation, angiogenesis, metastasis, and invasion (50). TYRO3 is involved in both cell proliferation and survival pathways and immune response regulation (51). CXCR4 promotes cancer cell proliferation (52) and also plays a role in the recruitment of immunosuppressive cells such as regulatory T cells (Treg), M2, and N2 neutrophils to limit the effectiveness of immune responses (53, 54). Altogether, our results showed that the I-genes and E-genes have multiple roles in both cancer cell survival and immune response.
Nevertheless, this study is limited in certain aspects. Although we consider the effects of both germline and somatic statuses, the variants can cause either gain or loss of the function, which lead to opposite effects on the signature activity; and the current analysis cannot control for the consistency of the effects as the functional consequence of the variants are unknown. Unlike the germline variants, there was no significant difference in selection pressure among these somatic variants (missense, nonsense, and splice site; ref. 55). Therefore, we combined all somatic mutations into a binary status to take the advantage of sample size. Eventually, our validation based on separated mutational statuses indicates that the determinant genes of MPs are highly conserved (Supplementary Table S2A).
Theoretically, the method described is applicable to all MSs. But for many rarer mutational processes that occur only in certain cancer subtypes, analyzing such MSs together with common ones will cause imbalance in the data and introduce unnecessary biases.
In addition, there are other important biological mechanisms, which influence the somatic evolution, such as age and individual mutation order (56, 57). However, the current analysis based on TCGA samples cannot address the effects of such factors due to the lack of information. Other factors, such as irradiation (58) or chemotherapy (10) also influence somatic selection of mutational processes. However, as TCGA samples receive different treatments, this study cannot directly assess the effects caused by a specific therapy.
Our findings can be further validated in different levels. For example, the correlation between the deterministic genes and the MPs can be validated by CRISPR-Cas9–mediated knockout or knockin in cell lines or mouse models. Moreover, the MPs can also be derived from targeted tumor-sequencing tests, such as MSK-IMPACT (59) and Praxis Extended RAS Panel (60). Thus, the correlation between MPs and clinical phenotypes, such as imaging, pathology, and treatment outcomes can be evaluated in much larger cohorts.
In summary, we provide a systematic view of the landscape of genetic determination of somatic selection of mutational processes in cancers. Our findings can inform the identification of cancer genes with highly potential clinical and therapeutic values.
Authors' Disclosures
R. Yu reports being a shareholder of Aginome Scientific. Q. Li reports grants from the Fundamental Research Funds for the Chinese Central Universities during the conduct of the study. No disclosures were reported by the other authors.
Supplementary Material
The mutational propensities and PCA results of each patients.
The genetic determinants of the mutational propensities. A) The integrated regression results of 2,314 unique candidate genes. B) The instrumental variable regression results of 485 unique E-genes. C) The instrumental variable regression results of 1,427 unique I-genes. D) The overlapped genes between E-genes and I-genes.
The clinical information of 60 LUAD patients which received chemotherapy and target therapies.
The quantitative trait loci of the mutational propensities. A) The results of 127 unique mpQTLs. B) The instrumental variable regression results of 13 mpQTLs.
Supplementary Methods, Figures S1-S8, Table S1-S4
Acknowledgments
The authors thank Dr. Jiahuai Han, Dr. Bing Xu, Dr. Dan Du, and Dr. Tianhai Ji for critical reading of and feedback to our work. The results published here are, in whole or part, based upon data generated by the TCGA Research Network. https://www.cancer.gov/tcgaThis work was supported by the Fundamental Research Funds for the Chinese Central Universities (20720190101 to Q. Li); the National Natural Science Foundation of China (81802823 to Y. Zhou); the Natural Science Foundation of Fujian Province of China (2018J01054 to Y. Zhou); National Natural Science Foundation of China (grant no. 81702414 to W. Tang); Natural Science Foundation of Fujian Province of China (grant no. 2020J05306 to W. Tang); and Xiamen Science and Technology Planning Project (grant no. 3502Z20194003 to W. Tang).
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Authors' Contributions
J. Guo: Data curation, software, formal analysis, investigation, visualization, methodology, writing–original draft, writing–review and editing. Y. Zhou: Methodology, writing–original draft, writing–review and editing. Chaoqun Xu: Data curation. Q. Chen: Data curation. Z. Sztupinszki: Resources, writing–review and editing. J. Börcsök: Resources. Canqiang Xu: Resources. F. Ye: Resources. W. Tang: Resources. J. Kang: Resources. L. Yang: Resources. J. Zhong: Data curation, software. T. Zhong: Data curation, software. T. Hu: Writing–review and editing. R. Yu: Resources, writing–review and editing. Z. Szallasi: Writing–review and editing. X. Deng: Writing–review and editing. Q. Li: Conceptualization, resources, supervision, methodology, writing–original draft, project administration, writing–review and editing.
References
- 1. Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell 2017;171:934–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nam AS, Chaligne R, Landau DA. Integrating genetic and non-genetic determinants of cancer evolution by single-cell multi-omics. Nat Rev Genet 2020;22:3–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature 2013;500:415–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. McGranahan N, Favero F, de Bruin EC, Birkbak NJ, Szallasi Z, Swanton C. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci Transl Med 2015;7:283ra54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. PCAWG Evolution & Heterogeneity Working Group, PCAWG Consortium, Gerstung M, Jolly C, Leshchiner I, Dentro SC, et al. The evolutionary history of 2,658 cancers. Nature 2020;578:122–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. PCAWG Mutational Signatures Working Group, PCAWG Consortium, Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, et al. The repertoire of mutational signatures in human cancer. Nature 2020;578:94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Huang MN, Yu W, Teoh WW, Ardin M, Jusakul A, Ng AWT, et al. Genome-scale mutational signatures of aflatoxin in cells, mice, and human tumors. Genome Res 2017;27:1475–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zou X, Owusu M, Harris R, Jackson SP, Loizou JI, Nik-Zainal S. Validating the concept of mutational signatures with isogenic cell models. Nat Commun 2018;9:1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Helleday T, Eshtad S, Nik-Zainal S. Mechanisms underlying mutational signatures in human cancers. Nat Rev Genet 2014;15:585–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Russo M, Crisafulli G, Sogari A, Reilly NM, Arena S, Lamba S, et al. Adaptive mutability of colorectal cancers in response to targeted therapies. Science 2019;366:1473–80. [DOI] [PubMed] [Google Scholar]
- 11. Wang S, Jia M, He Z, Liu X-S. APOBEC3B and APOBEC mutational signature as potential predictive markers for immunotherapy response in non-small cell lung cancer. Oncogene 2018;37:3924–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Turajlic S, Sottoriva A, Graham T, Swanton C. Resolving genetic heterogeneity in cancer. Nat Rev Genet 2019;20:404–16. [DOI] [PubMed] [Google Scholar]
- 13. Roberts SA, Lawrence MS, Klimczak LJ, Grimm SA, Fargo D, Stojanov P, et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat Genet 2013;45:970–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Alexandrov LB, Ju YS, Haase K, Van Loo P, Martincorena I, Nik-Zainal S, et al. Mutational signatures associated with tobacco smoking in human cancer. Science 2016;354:618–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dietlein F, Weghorn D, Taylor-Weiner A, Richters A, Reardon B, Liu D, et al. Identification of cancer driver genes based on nucleotide context. Nat Genet 2020;52:208–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Baez-Ortega A, Gori K. Computational approaches for discovery of mutational signatures in cancer. Brief Bioinform 2017;20:77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Letouzé E, Shinde J, Renault V, Couchy G, Blanc J-F, Tubacher E, et al. Mutational signatures reveal the dynamic interplay of risk factors and cellular processes during liver tumorigenesis. Nat Commun 2017;8:1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Temko D, Tomlinson IPM, Severini S, Schuster-Böckler B, Graham TA. The effects of mutational processes and selection on driver mutations across cancer types. Nat Commun 2018;9:1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature 2020;578:82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Middlebrooks CD, Banday AR, Matsuda K, Udquim K-I, Onabajo OO, Paquin A, et al. Association of germline variants in the APOBEC3 region with cancer risk and enrichment with APOBEC-signature mutations in tumors. Nat Genet 2016;48:1330–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim J, Mouw KW, Polak P, Braunstein LZ, Kamburov A, Tiao G, et al. Somatic ERCC2 mutations are associated with a distinct genomic signature in urothelial tumors. Nat Genet 2016;48:600–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Yang T-HO, et al. The immune landscape of cancer. Immunity. 2018;48:812–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Raynaud F, Mina M, Tavernari D, Ciriello G. Pan-cancer inference of intra-tumor heterogeneity reveals associations with different forms of genomic instability. PLoS Genet 2018;14:e1007669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Delaneau O, Zagury J-F, Marchini J. Improved whole-chromosome phasing for disease and population genetic studies. Nat Methods 2013;10:5–6. [DOI] [PubMed] [Google Scholar]
- 25. Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 2009;5:e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, et al. Computational correction of copy number effect improves specificity of CRISPR–Cas9 essentiality screens in cancer cells. Nat Genet 2017;49:1779–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shiraishi Y, Tremmel G, Miyano S, Stephens M. A simple model-based approach to inferring and visualizing cancer mutation signatures. PLos Genet 2015;11:1005657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rosenthal R, McGranahan N, Herrero J, Taylor BS, Swanton C. DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol 2016;17:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pflueger CE, Wang S. A robust test for weak instruments in stata. Stata J 2015;15:216–25. [Google Scholar]
- 30. Cui C, Xu C, Yang W, Chi Z, Sheng X, Si L, et al. Ratio of the interferon-γ signature to the immunosuppression signature predicts anti-PD-1 therapy response in melanoma. NPJ Genom Med 2021;6:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kim ST, Cristescu R, Bass AJ, Kim K-M, Odegaard JI, Kim K, et al. Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat Med 2018;24:1449–58. [DOI] [PubMed] [Google Scholar]
- 32. Weinstein JN, Collisson EA, Mills GB, Shaw KRM, Ozenberger BA, Ellrott K, et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet 2013;45:1113–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ghandi M, Huang FW, Jané-Valbuena J, Kryukov GV, Lo CC, McDonald ER, et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019;569:503–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu K, Guo J, Liu K, Fan P, Zeng Y, Xu C, et al. Integrative analysis reveals distinct subtypes with therapeutic implications in KRAS-mutant lung adenocarcinoma. EBioMedicine 2018;36:196–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med 2017;9:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Martínez-Jiménez F, Muiños F, Sentís I, Deu-Pons J, Reyes-Salazar I, Arnedo-Pac C, et al. A compendium of mutational cancer driver genes. Nat Rev Cancer 2020;20:555–72. [DOI] [PubMed] [Google Scholar]
- 37. Huang K, Mashl RJ, Wu Y, Ritter DI, Wang J, Oh C, et al. Pathogenic germline variants in 10,389 adult cancers. Cell 2018;173:355–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Walens A, Lin J, Damrauer JS, McKinney B, Lupo R, Newcomb R, et al. Adaptation and selection shape clonal evolution of tumors during residual disease and recurrence. Nat Commun 2020;11:5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hsiehchen D, Hsieh A, Samstein RM, Lu T, Beg MS, Gerber DE, et al. DNA repair gene mutations as predictors of immune checkpoint inhibitor response beyond tumor mutation burden. Cell Rep Med 2020;1:100034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Driscoll CB. APOBEC3B-mediated corruption of the tumor cell immunopeptidome induces heteroclitic neoepitopes for cancer immunotherapy. Nat Commun 2020;11:790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zheng Z, Huang D, Wang J, Zhao K, Zhou Y, Guo Z, et al. QTLbase: an integrative resource for quantitative trait loci across multiple human molecular phenotypes. Nucleic Acids Res 2020;48:D983–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mazzio EA, Boukli N, Rivera N, Soliman KFA. Pericellular pH homeostasis is a primary function of the Warburg effect: inversion of metabolic systems to control lactate steady state in tumor cells. Cancer Sci 2012;103:422–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Polak P, Kim J, Braunstein LZ, Karlic R, Haradhavala NJ, Tiao G, et al. A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat Genet 2017;49:1476–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Grolleman JE, Díaz-Gay M, Franch-Expósito S, Castellví-Bel S, de Voer RM. Somatic mutational signatures in polyposis and colorectal cancer. Mol Aspects Med 2019;69:62–72. [DOI] [PubMed] [Google Scholar]
- 45. Ramroop JR, Gerber MM, Toland AE. Germline variants impact somatic events during tumorigenesis. Trends Genet 2019;35:515–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ramón y Cajal S, Sesé M, Capdevila C, Aasen T, De Mattos-Arruda L, Diaz-Cano SJ, et al. Clinical implications of intratumor heterogeneity: challenges and opportunities. J Mol Med 2020;98:161–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Berthenet K, Boudesco C, Collura A, Svrcek M, Richaud S, Hammann A, et al. Extracellular HSP110 skews macrophage polarization in colorectal cancer. Oncoimmunology 2016;5:e1170264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liu X, Yan X, Zhong B, Nurieva RI, Wang A, Wang X, et al. Bcl6 expression specifies the T follicular helper cell program in vivo. J Exp Med 2012;209:1841–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lim AR, Rathmell WK, Rathmell JC. The tumor microenvironment as a metabolic barrier to effector T cells and immunotherapy. eLife 2020;9:e55185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Oji Y, Tatsumi N, Fukuda M, Nakatsuka S-I, Aoyagi S, Hirata E, et al. The translation elongation factor eEF2 is a novel tumor-associated antigen overexpressed in various types of cancers. Int J Oncol 2014;44:1461–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Smart S, Vasileiadi E, Wang X, DeRyckere D, Graham D. The emerging role of TYRO3 as a therapeutic target in cancer. Cancers 2018;10:474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bianchi ME, Mezzapelle R. The chemokine receptor CXCR4 in cell proliferation and tissue regeneration. Front Immunol 2020;11:2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Li Z, Wang Y, Shen Y, Qian C, Oupicky D, Sun M. Targeting pulmonary tumor microenvironment with CXCR4-inhibiting nanocomplex to enhance anti–PD-L1 immunotherapy. Sci Adv 2020;6:eaaz9240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Scala S. Molecular pathways: targeting the CXCR4–CXCL12 axis—untapped potential in the tumor microenvironment. Clin Cancer Res 2015;21:4278–85. [DOI] [PubMed] [Google Scholar]
- 55. Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, et al. Patterns of somatic mutation in human cancer genomes. Nature 2007;446:153–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rozhok AI, DeGregori J. Toward an evolutionary model of cancer: Considering the mechanisms that govern the fate of somatic mutations. Proc Natl Acad Sci U S A 2015;112:8914–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kent DG, Green AR. Order matters: the order of somatic mutations influences cancer evolution. Cold Spring Harb Perspect Med 2017;7:a027060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Marusyk A, Casás-Selves M, Henry CJ, Zaberezhnyy V, Klawitter J, Christians U, et al. Irradiation alters selection for oncogenic mutations in hematopoietic progenitors. Cancer Res 2009;69:7262–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial sloan kettering-integrated mutation profiling of actionable cancer targets (MSK-IMPACT). J Mol Diagn 2015;17:251–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Udar N, Lofton-Day C, Dong J, Vavrek D, Jung AS, Meier K, et al. Clinical validation of the next-generation sequencing-based Extended RAS Panel assay using metastatic colorectal cancer patient samples from the phase 3 PRIME study. J Cancer Res Clin Oncol 2018;144:2001–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The mutational propensities and PCA results of each patients.
The genetic determinants of the mutational propensities. A) The integrated regression results of 2,314 unique candidate genes. B) The instrumental variable regression results of 485 unique E-genes. C) The instrumental variable regression results of 1,427 unique I-genes. D) The overlapped genes between E-genes and I-genes.
The clinical information of 60 LUAD patients which received chemotherapy and target therapies.
The quantitative trait loci of the mutational propensities. A) The results of 127 unique mpQTLs. B) The instrumental variable regression results of 13 mpQTLs.
Supplementary Methods, Figures S1-S8, Table S1-S4
Data Availability Statement
The Code Ocean capsule containing the necessary data and codes to replicate the results of this study can be found at https://codeocean.com/capsule/6181333/tree/v1 (DOI: 10.24433/CO.2000361.v1).