

Abstract
Vaccine platforms enable fast development, testing, and manufacture of more affordable vaccines. Here, we evaluated Generalized Modules for Membrane Antigens (GMMA), outer membrane vesicles (OMVs) generated by genetically modified Gram‐negative bacteria, as a vaccine platform for viral pathogens. Influenza A virus hemagglutinin (HA), either physically mixed with GMMA (HA+STmGMMA mix), or covalently linked to GMMA surface (HA‐STmGMMA conjugate), significantly increased antigen‐specific humoral and cellular responses, with HA‐STmGMMA conjugate inducing further enhancement than HA+STmGMMA mix. HA‐STmGMMA conjugate protected mice from lethal challenge. The versatility for this platform was confirmed by conjugation of rabies glycoprotein (RABVG) onto GMMA through the same method. RABVG+STmGMMA mix and RABVG‐STmGMMA conjugate exhibited similar humoral and cellular response patterns and protection efficacy as the HA formulations, indicating relatively consistent responses for different vaccines based on the GMMA platform. Comparing to soluble protein, GMMA was more efficiently taken up in vivo and exhibited a B‐cell preferential uptake in the draining lymph nodes (LNs). Together, GMMA enhances immunity against viral antigens, and the platform works well with different antigens while retaining similar immunomodulatory patterns. The findings of our study imply the great potential of GMMA‐based vaccine platform also against viral infectious diseases.
Keywords: immune response, influenza, outer membrane vesicle, rabies, vaccine platform
1. INTRODUCTION
Vaccines need to confer efficacious and long‐lasting protective immunity while minimizing adverse effects. Commercialized vaccines against viruses are mostly developed based on the principles of Pasteur's “isolating, inactivating and injecting the causative microorganisms” (Rappuoli, 2004). Many of these vaccines, albeit effective, often lead to adverse effects. In addition to efficacy and safety, time required to develop a vaccine is also an important factor. A fast response to emerging infectious diseases is crucial to bring outbreaks under control (Noad et al., 2019). Traditional vaccines are usually developed by a “one bug, one vaccine” strategy, which takes both time and resources to develop, manufacture and deploy (Adalja et al., 2020).
A vaccine platform is to use certain carriers for different antigens to develop vaccines against specific pathogens. The establishment of vaccine platforms is advantageous in several ways. A well‐defined vaccine platform can standardize the induced immune responses and adverse effects (Gerritzen et al., 2017). Core components remain the same when a vaccine platform is used to develop different vaccines, therefore, uncertainties regarding the recipient responses, both immune responses and side effects, will be reduced. In addition, a vaccine platform allows easy switching of antigens, enabling faster and cheaper development, testing and manufacture of new vaccines (Gerritzen et al., 2017; Kis et al., 2019; Kushnir et al., 2012; Lurie et al., 2020).
Gram‐negative bacteria naturally release blebs of the outer membrane as extracellular vesicles (EVs), also called outer membrane vesicles (OMVs). OMVs contain a lot of excellent intrinsic immunostimulatory components including lipopolysaccharides (LPS), lipoproteins and peptidoglycans, which make these vesicles attractive for the design of vaccines (Gerritzen et al., 2017; Mancini et al., 2020; Micoli et al., 2020; Tan et al., 2018; van der Pol et al., 2015). Bacteria have also been engineered to produce high levels of OMVs with reduced reactogenicity to make OMVs more suitable as a vaccine platform (Balhuizen et al., 2021; De Benedetto et al., 2017; Gerke et al., 2015; Micoli et al., 2018; Rossi et al., 2014, 2016; van der Pol et al., 2015). We call the OMVs produced by these genetically modified bacteria as Generalized Modules for Membrane Antigens (GMMA), which can be produced at very high yields with simple manufacture process, leading to affordable vaccines (Kis et al., 2019; Li & Liu, 2020). GMMA have been shown to effectively promote immunogenicity of homologous and heterologous bacteria‐derived antigens (Launay et al., 2019; Mancini et al., 2020; Micoli et al., 2020; Obiero et al., 2017).
In the current study, GMMA were evaluated as an OMV‐based vaccine platform for viral pathogens using the influenza A virus hemagglutinin (HA) and rabies virus glycoprotein (RABVG) as model antigens. Our data suggest that GMMA enhance antigen‐specific humoral and cell‐mediated responses, and the display of antigens on the GMMA surface by chemical conjugation leads to further enhancement in immune responses than physical mix. Moreover, the GMMA platform not only shows better efficacy over traditional inactivated vaccines, but also is compatible with different antigens while retaining similar patterns of immune enhancement. The findings in this study imply the great potential of GMMA as an OMV‐based vaccine platform targeting infectious diseases.
2. RESULTS
2.1. Preparation and characterization of HA‐STmGMMA conjugate
HA was chemically linked to GMMA surface proteins through bissulfosuccinimidyl suberate (BS3) chemistry, which linked the lysine residues of HA to GMMA proteins previously activated with this amine‐reactive homobifunctional linker (Figure 1A).
FIGURE 1.

Conjugation and characterization of HA‐STmGMMA conjugate. (A) Reaction scheme: Salmonella Typhimurium OAg‐negative GMMA (STmGMMA) were derivatized with BS3 linker and chemically conjugated to HA. (B) Anti‐HA flow cytometry, (C) SDS‐PAGE analysis and (D) anti‐HA western blot analysis and (E) anti‐STmGMMA western blot analysis of the conjugate compared to STmGMMA and HA alone and their physical mixture. (F) Summary table with conjugate characteristics: HA percentage on HA‐STmGMMA conjugate (estimated by amino acid analysis [AAA]) and particles size distribution (Z‐average diameter and polydispersivity index (PdI)). (G) DLS profiles of HA‐STmGMMA conjugate and unconjugated STmGMMA.
OMV flow cytometry analysis and immuno‐TEM showed that HA was successfully detected on HA‐STmGMMA surface (Figures 1B and S1). SDS‐PAGE analysis (Figure 1C) further proved that conjugation reaction worked efficiently, with many STmGMMA protein bands (visible in lane 2 and 5 of STmGMMA alone or physically mixed with HA) disappearing and moving to molecular weights similar or higher than HA antigen alone (lane 3) in HA‐STmGMMA conjugate. With the BS3 chemistry, HA could be conjugated to any protein on the STmGMMA with exposed NH2 groups. Depending on the protein that HA was conjugated to, the size of the conjugated complex could vary significantly (Figure 1C). Anti‐HA western blot analysis (Figure 1D) confirmed presence of the viral antigen in the HA‐STmGMMA conjugate, showing a main band similar to unconjugated HA with some smear above the band. The successful conjugation was also evidenced by the anti‐STmGMMA western blot analysis. As shown in Figure 1E, the band profile showed an apparent shift in the HA‐STmGMMA comparing to STmGMMA alone or HA+STmGMMA physical mix. Absence of free viral antigen after conjugate purification was confirmed by HPLC‐SEC analysis (Figure S2). The amount of HA antigen linked to STmGMMA, as estimated by amino acid analysis, was 19.80% by weight of total conjugate protein content (Figure 1F). Moreover, dynamic light scattering (DLS) revealed that the size distribution profile of HA‐STmGMMA conjugate was comparable to that of unconjugated STmGMMA, indicating the absence of aggregate formation in the conjugates (Figure 1G).
2.2. STmGMMA enhance HA‐specific antibody and cell‐mediated responses
To test whether STmGMMA formulated HA vaccines could induce better antibody and cell‐mediated responses over HA alone, BALB/c mice were injected intramuscularly (i.m.) with STmGMMA alone, HA alone, HA+STmGMMA mix and HA‐STmGMMA conjugate twice in a “prime‐boost” strategy (Figure 2A,B). Current commercially available influenza vaccines are usually inactivated or live‐attenuated virus propagated in chicken eggs (C. f. D. Control, 2020). To compare the effectiveness of our GMMA vaccine platform with the traditional influenza vaccine, egg‐propagated formalin‐inactivated X31 at the same dose was also included in our study (Figure 2A). Four weeks after the prime and 2 weeks after the boost, mice sera were collected and HA‐specific IgG response was quantified by direct ELISA. As shown in Figure 2C and Table S1, HA alone induced detectable level of IgG response after the prime, and the response was further enhanced by the boost. HA+STmGMMA mix significantly increased the HA‐specific IgG level comparing to HA alone (45 folds at week 4 and 21 folds at week 6), and the formulation with conjugated HA showed even further enhancement (762 folds at week 4 and 78 folds at week 6). Of note, both STmGMMA formulations seemed to be more efficient in inducing HA‐specific IgG response in mice than inactivated X31. After the prime, the IgG level in the HA+STmGMMA mix group was comparable to that in the inactivated X31 group, while a significantly higher response was observed in the HA‐STmGMMA conjugate group. After the boost, both HA+STmGMMA mix and HA‐STmGMMA conjugate induced significantly higher IgG response than inactivated X31 (Figure 2C and Table S1).
FIGURE 2.
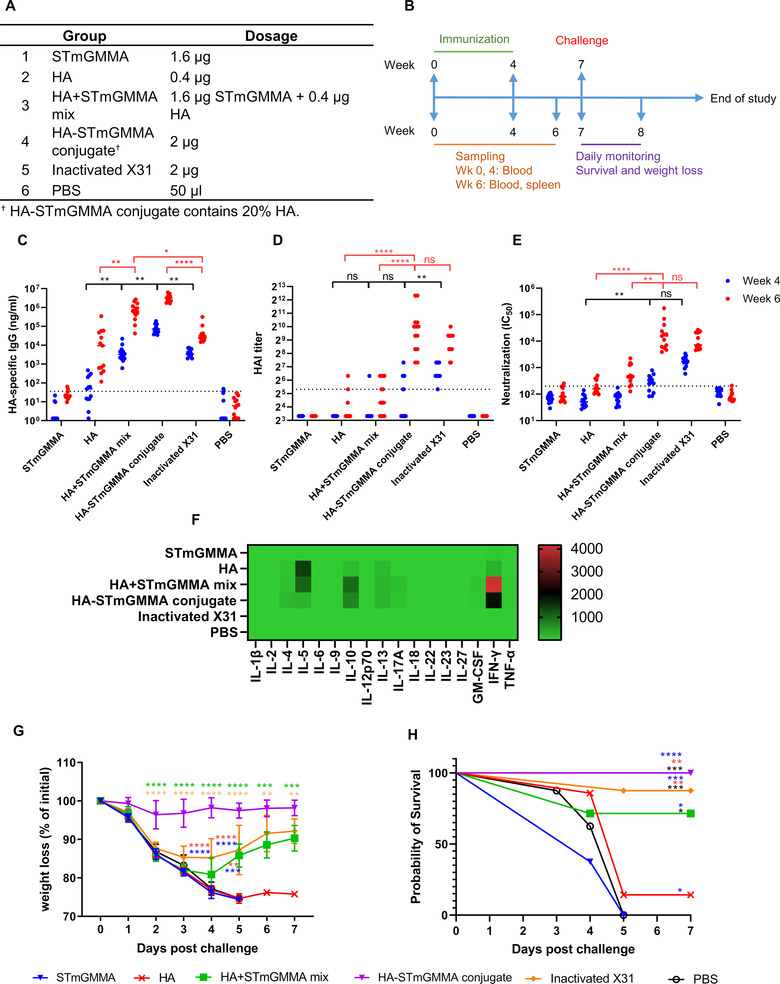
HA‐STmGMMA conjugate induces protective immune responses against lethal challenges. (A) Animal group and dosage information. (B) Immunization and challenge schedule. (C) HA‐specific IgG response, (D) HAI assay and (E) microneutralization in sera from vaccinated animals (n = 13) at week 4 (blue) and week 6 (red). ns, statistically not significant; * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001. (F) Th1/2/9/17/22/Treg‐related cytokines production from HA‐restimulated splenocytes isolated from vaccinated animals (n = 5) at week 6. Heat map shows the fold change of individual cytokine expression relating to PBS group. (G) Weight loss and (H) survival of animals (n = 7 for HA and HA+STmGMMA mix groups and n = 8 for other groups) challenged with lethal dose X31 Influenza virus at week 7. ns, statistically not significant; * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001.
Sera neutralization activity was then assessed by HAI and microneutralization assay. Although HA+STmGMMA mix and HA‐STmGMMA conjugate induced significantly higher binding antibodies than HA and inactivated X31, their ability to induce neutralization seemed to be not as profound (Figure 2D,E). In detail, HA‐STmGMMA conjugate and inactivated X31 induced similar levels of neutralization, which was significantly higher than HA+STmGMMA mix and HA alone. The neutralization induced by HA+STmGMMA mix was also comparable to that by HA alone. Although other factors may also be involved, it is possible that the difference in conformation between our recombinant soluble HA and the native HA on the virus particles resulted in the discrepancy between binding IgG levels and neutralization titers. The recombinant soluble HA might have exposed some highly immunogenic non‐neutralizing epitopes that were conceived in the native HA. These exposed non‐neutralizing epitopes on HA may be a result of suboptimal design of soluble HA and/or the BS3‐based conjugation process.
The impact of STmGMMA on the cell‐mediated immune responses was also determined by quantification of Th1/Th2/Th9/Th17/Th22/Treg‐related cytokines secreted by antigen‐restimulated mice splenocytes in vitro. Similar to what was seen in the humoral response, both STmGMMA formulations showed much stronger enhancement in both the breadth and magnitude of antigen‐specific cytokines than HA alone (Figures 2F and S3). In detail, in mice injected with HA alone, an elevation of IL‐4, IL‐5, IL‐10, IL‐13, and IFN‐γ was detected. In mice injected with either HA+STmGMMA mix or HA‐STmGMMA conjugate, the level of IL4, IL‐5, IL‐10, IL‐13, IL‐17A, GM‐CSF,IFN‐γ, and TNF‐α was considerably increased. In addition, a higher elevation of cytokines was observed in mice injected with HA+STmGMMA mix and HA‐STmGMMA conjugate than HA alone, except IL‐5, which was higher in HA than in the two STmGMMA formulations. Of note, unlike the antibody response, HA‐STmGMMA conjugate did not show better enhancement in the secretion of Th‐associated cytokines than HA+STmGMMA mix. Unexpectedly, almost all the measured cytokines in the inactivated X31 group were close to the PBS control group, indicating that inactivated X31 is poor in induction of cell‐mediated immune responses.
2.3. STmGMMA‐formulated HA vaccine protects animals from lethal viral challenge
To determine the protective effect of the different formulations, mice at 3 weeks post the boost were challenged intranasally with a lethal dose of X31 and weight loss and survival rate were recorded daily. As shown in Figure 2G,H, the negative control groups (PBS group and STmGMMA groups) showed continuously sharp weight loss since Day 1 and 100% death was observed on Day 5. The HA group also demonstrated the same weight loss profile as the negative groups, but the survival rate was slightly higher, with one of eight mice that survived the challenge. The HA+STmGMMA mix and inactivated X31 groups offered similar levels of protection, which were both significantly better than HA alone. These two groups also showed rapid weight loss over the first 4 days after challenge, but weight was slowly regained since. At the end of the challenge study, five out of seven in HA+STmGMMA mix group and seven out of eight in the inactivated X31 group survived. A full protection was only observed in the HA‐STmGMMA conjugate group. Weight in the HA‐STmGMMA conjugate group only slightly dropped at first 2 days after challenge and then it remained stable throughout the challenge. Of note, the inactivated X31 group with good antibody response but less efficient cell‐mediated responses showed good survival rate but with significant weight loss after lethal challenge (Figure 2G,H). These data suggested that good antibody response was sufficient to provide protection against mortality, but cell‐mediated responses were required to further prevent symptoms.
2.4. The GMMA platform is easily adaptable to other viral antigens
To test whether the GMMA platform could be easily adapted to other viral antigens, we next investigated this platform with rabies glycoprotein (RABVG). RABVG was chemically conjugated to STmGMMA through the exact same method as HA (Figure S4A). Conjugate formation was confirmed by SDS‐PAGE and western blot (Figure S4B,C). As RABVG antigen is not characterized by a single defined band in SDS‐PAGE/WB, it is difficult from this analysis confirming absence of unconjugated protein. Also, in HPLC‐SEC analysis, the peak of the conjugate overlapped with that of RABVG (Figure S5). However, removal of free antigen was followed during conjugate purification by ultracentrifugation by analyzing the supernatants in HPLC‐SEC, verifying presence of 2.4% only of free RABVG in the last supernatant respect to its total amount put in reaction. The conjugation resulted in 25% by weight of RABVG within RABVG‐STmGMMA conjugate (Figure S4D). Absence of aggregates after conjugation was confirmed by DLS analysis (Figure S4D,E).
To test whether STmGMMA formulated RABVG vaccines could induce better antibody and cell‐mediated responses over RABVG alone, BALB/c mice were injected intramuscularly (i.m.) with RABVG alone, GMMA alone, RABVG+STmGMMA mix and RABVG‐STmGMMA conjugate by the same “prime‐boost” strategy tested with HA antigen (Figure 3A,B). Four weeks after the prime and 2 weeks after the boost, mice sera were collected and RABVG‐specific IgG response was quantified by direct ELISA. As shown in Figure 2C and Table S2, low dose RABVG did not induce detectable IgG response and high dose RABVG only induced marginal IgG response in serum after the boost. Similar to the results from the HA study, both RABVG+STmGMMA mix and RABVG‐STmGMMA conjugate formulations (low and high doses) induced significantly higher IgG titres than RABVG alone, indicating the potent adjuvant effect of GMMA. Compared to RABVG+STmGMMA mix, RABVG‐STmGMMA conjugate showed further enhancement in antibody induction (>2 log higher at the low dose and >1 log higher at the high dose). Noticeably, GMMA‐based vaccines, both the physical mix and the conjugate, induced decent antigen‐specific immune response after the first injection, implying the possibility of single‐dose vaccines for the GMMA platform (Figure 3C).
FIGURE 3.
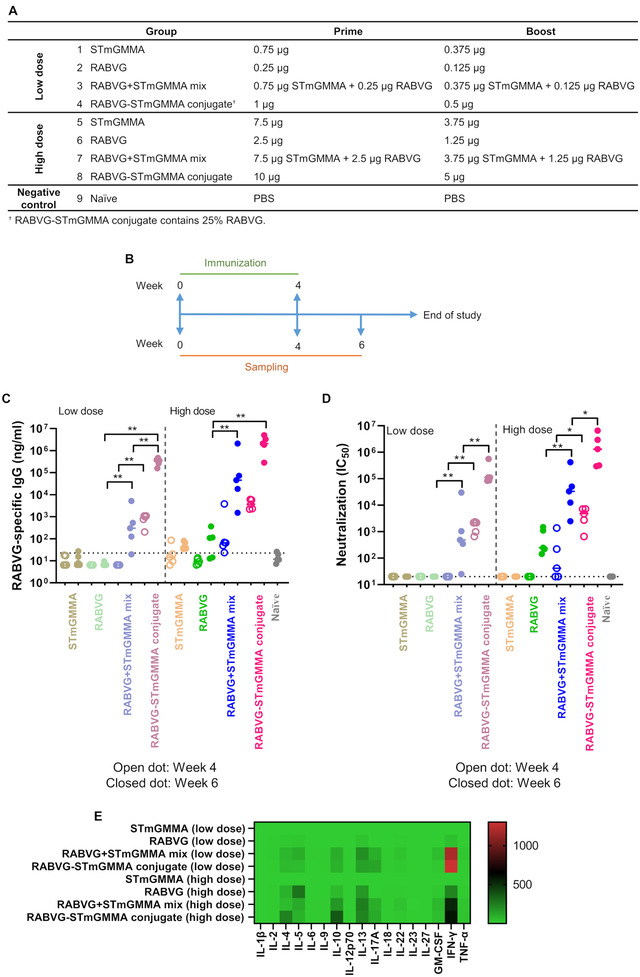
STmGMMA enhances RABVG‐specific humoral and cell‐mediated responses. (A) Animal group and dosage information. (B) Immunization schedule. (C) RABVG‐specific IgG response in vaccinated animals (n = 5) at week 4 (open dots) and week 6 (closed dots). * p < 0.05; ** p < 0.01; dotted line on the y‐axis represents assay detection limit. (D) Pseudotyped virus neutralization of sera from vaccinated animals (n = 5) at week 4 (open dots) and week 6 (closed dots). * p < 0.05; ** p < 0.01; dotted line on the y‐axis represents assay detection limit. (E) Th1/2/9/17/22/Treg‐related cytokines production from RABVG‐restimulated splenocytes isolated from vaccinated animals (n = 5) at week 6. Heat map shows the fold change of individual cytokine expression relating to GMMA (low dose) group.
A pseudo‐typed rabies neutralization assay was performed with the immunized mice sera to check whether STmGMMA‐induced enhancement of RABVG‐specific antibody production would lead to enhanced neutralization. Consistent with the ELISA data, without STmGMMA, RABVG at low dose did not show much neutralization even after boost and RABVG at high dose only induced moderate neutralization after boost, whereas both RABVG+STmGMMA mix and RABVG‐STmGMMA conjugate led to considerably higher neutralization titres in the sera (Figure 3D). In particular, a single dose of 10 μg RABVG+STmGMMA mix and RABVG‐STmGMMA conjugate induced neutralization similar to or even higher than two doses of RABVG alone (Figure 3D). When comparing the physical mix and the conjugate formulations, the conjugated version (RABVG‐STmGMMA conjugate) offered further enhancement in neutralization (Figure 3D).
A cell‐mediated response similar to the HA‐STmGMMA conjugate study was also observed in this RABVG‐STmGMMA conjugate study, as determined by quantification of Th1/Th2/Th9/Th17/Th22/Treg‐related cytokines produced from antigen restimulated splenocytes (Figures 3E and S6). Of note, unlike the antibody response, RABVG‐STmGMMA conjugate did not show better enhancement in the secretion of Th‐associated cytokines than RABVG+STmGMMA mix. Besides, the low dose RABVG+STmGMMA mix and RABVG‐STmGMMA conjugate formulations seemed to induce better IFN‐γ production than the high dose.
A challenge study was also performed on BALB/c mice to determine the protective effect of the RABVG‐STmGMMA conjugate vaccine. As shown in Figure 4A,B, mice were first immunized twice in 4‐week intervals, either with a low or high dose of RABVG‐STmGMMA conjugate, GMMA alone, or a commercial rabies vaccine (VeroRab), and then were challenged intracerebrally with a lethal dose of rabies virus (CVS‐11) 2 weeks after the second vaccination. Our data showed that mice receiving GMMA alone started to show signs of rabies infection on Day 6 and 100% of mice (n = 12) displayed clinical signs on Day 12 (Figure 4C). One mouse out of 11 in the low dose RABVG‐STmGMMA conjugate group also showed signs on Day 10, but the rest of the group survived until the end of the study. All mice from the high‐dose RABVG‐STmGMMA conjugate and VeroRab groups were fully protected (Figure 4C). Rabies N protein was assayed in the mouse brains, all of those that survived to the end of the experiment were negative for rabies N protein, and all those from the GMMA group were positive for rabies N protein except for one mouse euthanized on Day 12 that was negative for rabies N protein (data not shown). The body weights of mice were also monitored weekly, and no apparent weight differences were observed between different groups until after the challenge when the weight losses were seen in the GMMA group (Figure 4D).
FIGURE 4.
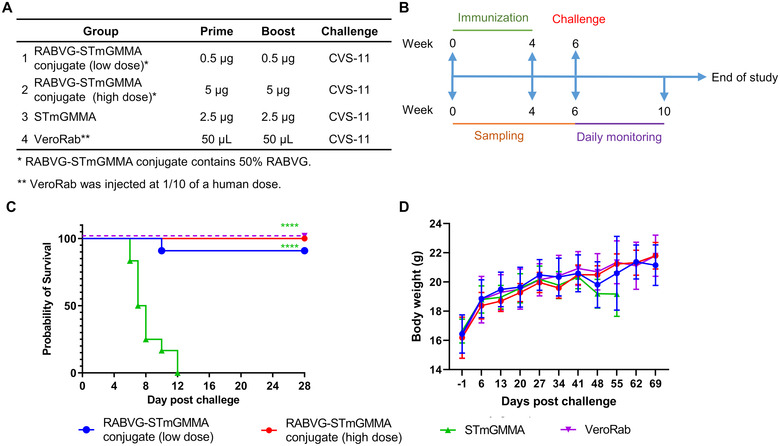
RABVG‐STmGMMA conjugate protects mice from lethal challenge. (A) Animal group and dosage information. (B) Immunization and challenge schedule. (C) Survival and (D) weekly weight changes of animals (n = 11 for RABV‐STmGMMA [low dose] and VeroRab groups and n = 12 for other two groups) challenged with a lethal dose of rabies CVS‐11 at week 6. **** p < 0.0001.
Taken together, our data suggest that the GMMA platform is versatile and can be easily adapted to another antigen for quick development of vaccines for another viral pathogen, and the GMMA‐formulated vaccines induce similar humoral and cell‐mediated immune responses profiles between different viral antigens. In addition, GMMA formulations are more efficient than traditional inactivated virus formulation in immune induction.
2.5. STmGMMA is more efficiently taken up in vivo by B cells than soluble protein BSA
We next tested if STmGMMA was taken up by cells in vivo in a pattern distinctive to soluble proteins. Soluble protein BSA and STmGMMA were first fluorescently labelled with AF488, and then injected in mice intramuscularly. One hour later, muscle at the injection site and draining lymph nodes (LNs) were isolated and AF488+ cells were assessed. Our data showed that significantly more AF488+ cells were detected in both muscle and draining LNs in STmGMMA‐AF488 mice than in BSA‐AF488 mice, indicating STmGMMA was more efficiently taken up in vivo (Figures 5A and S7). Cell subsets in the muscle and draining LNs taking up BSA‐AF488 and STmGMMA‐AF488 were also analyzed (Figure S8). In the muscle, both BSA‐AF488 and STmGMMA‐AF488 were mainly taken up by myofibril cells, endothelial cells and FAPs (Figure 5B and Table S3). Of note, although AF488+ satellite cells did not constitute a major ratio of total AF488+ cells, there were significantly more AF488+ satellite cells in the STmGMMA‐AF488 muscles than in the BSA‐AF488 ones (Table S3). In the draining LNs, BSA‐AF488 was mainly taken up by DC, B cells and macrophage, whereas STmGMMA‐AF488 showed a B cell‐preference uptake (Figure 5B and Table S3). Taken together, our data here indicate that STmGMMA is more efficiently taken up in vivo than soluble protein BSA and the uptake of these two antigen forms shows different cell type preferences.
FIGURE 5.
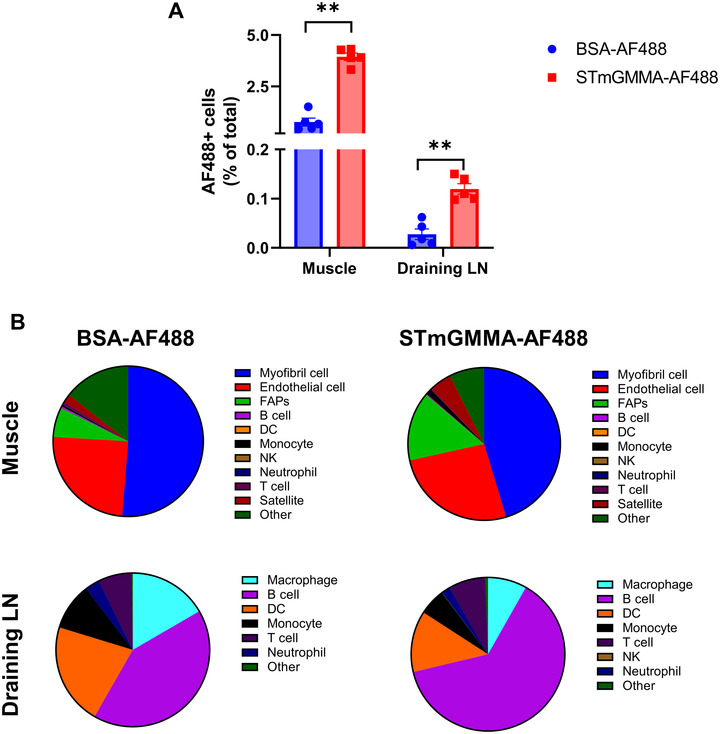
STmGMMA is more efficiently taken up in vivo. (A) The percentage of AF488+ cells in the injection site (muscle) and draining LNs 1 h after injection. (B) The distribution of AF488+ cell subsets in the muscle and draining LNs (n = 5).
3. DISCUSSION
OMV‐based vaccines have been investigated as a powerful platform for the development of bacterial vaccines (Mancini et al., 2021, 2020). In this study, we investigated GMMA as an OMV‐based vaccine platform for viral pathogens. Our data demonstrated that decoration of viral antigens onto GMMA can be easily achieved through chemical conjugation and the antigen‐STmGMMA conjugate induced significantly enhanced humoral and cellular responses. The induced responses offered protective immunity against lethal viral challenges in mice. Our findings suggest that the GMMA‐based vaccine platform initially designed for bacterial vaccines is a promising approach also for vaccine design against viral pathogens.
GMMA decoration with heterologous antigens is a key step in the production of GMMA‐based vaccines. There are currently two popular approaches to load foreign antigens onto OMVs. One is bioengineering of bacteria to express foreign antigens on the surface of OMVs, the other is chemical conjugation of purified foreign antigen onto OMVs. Bioengineering to express foreign proteins has been successful for various proteins in several bacteria strains (Bartolini et al., 2013; Cheng et al., 2021; Kuipers et al., 2015; Rappazzo et al., 2016). In particular, the ectodomain of influenza A M2 protein (M2e) was successfully loaded in Escherichia coli‐derived OMVs by transformation of plasmid encoding transmembrane protein ClyA‐M2e fusion protein. The M2e‐OMVs protected immunized mice from lethal challenge (Rappazzo et al., 2016). The advantage of this approach is simple production and purification process once the bacteria bioengineering is complete. However, this approach requires bacteria engineering for each vaccine antigen. Viral antigens are usually glycoproteins and glycans are important for these proteins to retain their structure, biological function and immunogenicity (Solá & Griebenow, 2010; Varki, 2017). Bacteria, in general, do not naturally have a fully developed glycosylation system, and engineering bacteria to produce glycoproteins with the correct glycosylation patterns is complicated and challenging (Keys & Aebi, 2017). Although M2e was successful through this method, other glycosylated antigens loading via the same method may require more optimization and be less efficient in protective immune induction. In addition to the extensive modifications required, GMMA produced by these engineered bacteria may have distinctive features, which could impact immune induction. Chemical conjugation, on the other hand, can circumvent the above problems, although more purification steps are introduced (Micoli et al., 2020). With chemical conjugation, all GMMA particles can be produced from the same bacteria and no antigen‐specific bioengineering is required. Antigens, despite the origin, size and glycosylation status can be conjugated onto GMMA through the same chemistry reaction, enabling easy antigen switch for different vaccines. Therefore, chemical conjugation‐based GMMA vaccines can enable a more standardized production and should have more consistent features and immune induction profiles. Our study has shown that an easy swap of RABVG to HA through the same BS3 chemistry is possible and both RABVG‐STmGMMA conjugate and HA‐STmGMMA conjugate have induced similar humoral and cellular immunity profiles. Although not being investigated in this study, multivalent vaccines are also possible through the chemical conjugation process (Micoli et al., 2020).
OMVs contain a wide range of pathogen‐associated molecular patterns (PAMPs) and therefore are considered good adjuvants with intrinsic immunostimulatory properties (Gnopo et al., 2017; Kaparakis‐Liaskos & Ferrero, 2015). In addition, we have also observed that mixing GMMA with RABVG or HA enhanced both humoral and cellular immune responses comparing to antigen alone. In particular, the cell‐based response induced by physical mix of GMMA with RABVG and HA was comparable to that by RABVG‐STmGMMA and HA‐STmGMMA conjugates. These data imply that GMMA has the potential to be used as adjuvant for vaccine development, and the physical mix of GMMA with antigens will further simplify the production of GMMA‐based vaccines. In our current study, our main interest was to check whether the decoration of Ag on GMMA surface could provide any benefits on immune induction, while the investigation on GMMA as an adjuvant was not within the scope of the study. However, it would be interesting to compare the adjuvant effect of GMMA with other OMVs and classical adjuvants, like MF59 and alum etc.
We have also observed that conjugation of antigen onto GMMA provided further immune enhancement comparing physical mix of GMMA with antigen. In the HA study, HA‐STmGMMA conjugate has even induced better protective immunity against lethal viral challenge comparing to inactivated virus, a commercially used formulation. Using fluorescently labelled soluble protein (BSA) and GMMA, we have discovered that GMMA is more efficiently taken up in vivo and exhibit a B cell‐preferential uptake in the draining LN. Although further investigation is required, conjugation of antigens onto GMMA may provide more efficient antigen uptake as cells are activated by GMMA's immunomodulatory properties. In addition, the B‐cell preferential targeting may increase antigen processing efficiency, B cell differentiation and the formation germinal centres, thereby enhancing immune response (Suan et al., 2017; Yuseff et al., 2013).
As a proof‐of‐concept study, antigen‐STmGMMA conjugates were generated using the BS3 chemistry without extensive optimization. Although the conjugation was successful every time, the conjugation efficiency varied considerably (Figures 2, 3, and 4A). Further condition optimization will be required to generate conjugates with more consistent conjugation efficiency. In addition, the quality of the purified antigen for GMMA conjugation may represent a notable factor for the efficacy of the conjugated vaccine. In the HA study, HA‐STmGMMA conjugate induced a HA‐specific IgG titre of about 60 folds higher than inactivated X31, however, the neutralization titers between the two groups were comparable (Figure 4C–E). This indicates that HA on the GMMA surface is somehow different from its native form on the viral particles, which has probably exposed more non‐neutralizing epitopes. Such structure alteration may be caused by strategy to make HA soluble and/or the BS3 chemistry‐based conjugation process. Although the suboptimal HA used in our study still provided protective immunity, the antigen quality for other viruses may be a factor of concern. In particular, non‐neutralizing antibodies induced by suboptimal antigens may trigger antibody‐dependent enhancement (ADE) effect, which may pose a significant impact on vaccine efficacy (Dutry et al., 2011; Winarski et al., 2019). Although no ADE effect was detected in our current HA‐STmGMMA conjugate formulation, it is important to further confirm that GMMA‐based vaccines like HA‐STmGMMA conjugate do not induce ADE effect in preclinical and clinical studies (Figure S9). The BS3 chemistry can be used to conjugate antigens onto GMMA via any orientation, which may slightly impair protein conformation and immunogenicity. Ad hoc conjugation strategies can be selected to directionally conjugate antigen onto GMMA surface in a way that resembles that on viral particles and reduce the induction of non‐neutralizing antibodies. A previous study successfully loaded SARS‐CoV2 spike protein by attaching a cathelicidin‐related antimicrobial peptide (mCRAMP), a strong LPS binding motif, to the C terminal, which could bind to the LPS on the OMV surface (van der Ley et al., 2021). Comparing to the BS3 chemical conjugation, the binding of OMV through affinity sequences like mCRAMP may introduce less interference on protein structure and also can assure unidirectional binding of proteins onto OMVs. Although beyond the scope of our current study, it would be beneficial to investigate whether non‐neutralizing antibody level could be reduced by adopting mCRAMP to our HA‐STmGMMA system.
The adaptive immunity consists of two interconnected arms, humoral response and T‐cell‐based response. The former can be measured by antigen‐specific antibody level while the latter is usually characterized by specific cytokine profiles (Araujo‐Pires et al., 2014; Kudlay et al., 2022; Ye et al., 2018). In our current study, we determined the humoral response by antigen‐specific IgG level and sera neutralization activity and investigated cell‐based responses by measurement of Th1/Th2/Th9/Th17/Th22/Treg‐related cytokines. For humoral response, our data showed that a simple physical mix of STmGMMA with antigen could significantly enhance antigen‐specific antibody response while the conjugation of antigen onto STmGMMA could further increase such enhancement, indicating the adjuvant properties of STmGMMA for viral antigens. Of note, one dose of Ag‐STmGMMA conjugate could induce an antibody response comparable to that of two doses of Ag+STmGMMA mix, implying the potential of single‐dose vaccines with the GMMA platform (Figures 2C–E and 3C,D). For cell‐mediated response, antigen alone (RABVG or HA) induced a Th1(IFN‐γ and IL‐10) and Th2(IL‐4, IL‐5, and IL‐13) response while the inclusion of STmGMMA, either in physical mix or in conjugation, could further increase the Th1/2 responses. In addition, formulations with STmGMMA also enhanced Th17(IL‐17A) response, which is considered important for mucosal immunity (Figures 2F and 3E) (Kolls & Khader, 2010). Interestingly, a higher dose did not seem to induce better cell‐mediated response in the STmGMMA formulations. Our data in the RABVG study showed that the low dose RABVG+STmGMMA mix and the RABVG‐STmGMMA conjugate induced higher levels of Th‐related cytokines than the high dose vaccines, implying inappropriately overdosed STmGMMA may present an inhibitory effect on cell‐mediated response (Figure 3E). In addition, immune responses‐induced by STmGMMA‐based formulations, either physical mix or conjugation, are quite different from the responses induced by inactivated viral particles. In our HA study, both HA‐STmGMMA conjugate and inactivated X31 virus‐induced very good neutralizing antibody responses at comparable levels (Figure 2D,E). However, the inactivated X31 did not seem to induce any detectable cell‐mediated responses while a very significant enhancement of Th1/2/17 responses were observed in HA‐STmGMMA conjugate group (Figure 2F). While the mechanism regarding the difference in immune induction between HA‐STmGMMA conjugate inactivated X31 remains to be further explored, the challenge data highlighted the importance of cell‐mediated response to offer better protection against lethal viral challenge (Figure 2G).
Our study has shown that the GMMA has a great potential as a vaccine platform for the development of vaccines against viral pathogens too. This versatile platform can generate well‐defined vaccines at low cost and enable easy and fast adaption to other antigens to generate vaccines against new pathogens in response to outbreaks.
4. MATERIALS AND METHODS
4.1. Ethical statement
The rabies challenge study was undertaken at the Animal & Plant Health Agency (APHA), under the terms of a project license (PCA17EA73) and personal licenses. All project licenses and personal licenses were granted by the UK Home Office under the Animals (Scientific Procedures) Act 1986. All protocols and procedures including the statistical power of the study design were reviewed and approved by the Animal Welfare and Ethical Review Body (AWERB) at APHA.
All other animal experiments in the current study were performed at Imperial College London, in accordance with the terms of a project license (P63FE629C) and the authority of personal licenses. All protocols and procedures including the statistical power of the study design were reviewed and approved by the Animal Welfare and Ethical Review Body (AWERB) at Imperial College London.
4.2. Cells, virus, bacteria, and plasmids
HEK293T/17, MDCK, and BHK‐21 cell lines were purchased from LGC standards and cultured in DMEM supplemented with 10 % FBS and antibiotics. FreeStyle™ 293‐F cells were purchased from Thermo Fisher Scientific and cultured in FreeStyle™ 293 Expression Medium (Gibco; Thermo Fisher Scientific).
Influenza virus H3N2 (X31 strain) was kindly provided by Prof. Wendy S. Barclay (Imperial College London) and propagated in MDCK cells. Inactivated X31 was purchased from Charles River (Cat no: 10100784) and used for animal injections. The inactivated X31 was propagated in specific‐pathogen‐free (SPF) eggs in the allantoic cavity and purified by a sucrose gradient and inactivated by 0.2% buffered formalin.
Salmonella Typhimurium isolate 1418 (LT2 collection, University of Calgary) was chosen as parent strain and engineered to obtain the hypervesiculating phenotype (removal of the tolR gene) and the loss of LPS O‐antigen chains (removal of the rfbP‐U genes in the biosynthetic locus). The null mutations were obtained by sequentially replacing the genes of interest with the kanamycin resistance cassette aph by homologous recombination using lambda red recombineering system (Datsenko & Wanner, 2000). In the case of the tolR mutation, the antibiotic cassette was removed using the pCP20 plasmid, as previously described (Datsenko & Wanner, 2000). STmGMMA were produced at lab scale from Salmonella typhimurium mutant strain as follows. After over day growth in HTMC medium, bacteria were pelleted through centrifugation at 5000× g for 45 min and culture supernatant, containing STmGMMA, was collected and filtered through 0.22 μm Stericup filters (Millipore). Filtered supernatant was ultracentrifuged at 175,000× g for 2 h at 4°C and the resulting pellet washed with phosphate‐buffered saline (PBS), further ultra‐centrifuged at 175,000× g for 2 h and finally resuspended in PBS. STmGMMA were then characterized as previously described (Gasperini et al., 2021).
A human codon‐optimized fragment encoding a soluble rabies glycoprotein (Pasteur strain; GenBank accession number: NP_056796.1) was synthesized by GeneArt (Thermo Fisher Scientific) and assembled into pcDNA3.1(+) with NEB HiFi assembly (New England BioLabs) and designated as p‐sRABVG. This soluble rabies glycoprotein ends at Asparagine N455 of the Pasteur strain glycoprotein, followed by a GSGSGS linker, a GCN4 trimerization motif, a further GSGSGS linker, a thrombin site, a Myc tag, another GSGSGS linker and finally an 8×His tag to enable protein purification. A plasmid (designated as p‐sHA) encoding soluble influenza (X31 strain) HA was also constructed in the same way. This plasmid contains the first 514 amino acids of X31 HA, followed by a (G4S)2 linker, a T4 fibritin (foldon) trimerization motif, a Myc tag, a GSGSGS linker and finally a 8×His tag. The following three plasmids were gifts from Prof. Julian Ma, St George's University of London and used in the current study for pseudo‐rabies virus packaging: a HIV‐1 gag‐pol plasmid (pCMV‐Δ8.91), A firefly luciferase reporter plasmid (pCSFLW) and a plasmid encoding the wild type rabies glycoprotein (pCVS‐G; CVS‐11 strain; GenBank Sequence Accession: EU352767).
4.3. Recombinant soluble glycoprotein production and purification
Recombinant soluble rabies glycoprotein was produced and purified as previously described with modifications (Fu et al., 2019; McKay et al., 2020). In brief, HEK293T/17 cells were first transfected with p‐sRABVG using PEI MAX (Polysciences) for 4 h, and then the transfection medium was removed and cells were cultured in FreeStyle 293 Expression medium (Thermo Fisher Scientific) for 3 days. Conditioned medium was then collected and sequentially purified with a HisTrap HP column (Cytiva) and an ENrich™ SEC 650 size exclusion chromatography column (Bio‐Rad). Purified protein was first analyzed by Native‐PAGE and Western blot, and then sterilized by 0.22 μm filtration, aliquoted and store at ‐80°C.
Recombinant soluble X31 HA was produced with the FreeStyle™ 293 Expression System (Gibco; Thermo Fisher Scientific), according to the manufacturer's instructions. In brief, FreeStyle™ 293‐F cells were transfected with p‐sHA using 293fectin™ Transfection Reagent (Gibco; Thermo Fisher Scientific) and cultured for 7 days. After culture, conditioned medium was harvested, filtrated to remove cell debris. Protein was sequentially purified by a HisTrap HP column (Cytiva) and a HiPrep™ 16/60 Sephacryl™ S‐300 HR SEC column (Cytiva). Purified protein was analyzed and stored the same as the recombinant rabies glycoprotein.
4.4. Ag‐STmGMMA conjugation
Both RABVG and HA proteins were conjugated to STmGMMA via BS3 chemistry. BS3 linker was added to STmGMMA suspension in 100 mM borate buffer, pH 9 to have final linker and STmGMMA concentrations of 50 mg/ml and 4.2 mg/ml, respectively, in the reaction mixture. The mixture was incubated at 25°C for 30 min, then activated STmGMMA were purified by two rounds of ultracentrifugation (110,000 rpm, 16 min 4°C). Purified activated STmGMMA were then added to viral antigens, RABVG or HA, in PBS buffer with a w/w ratio of STmGMMA to viral antigen of 1:1 and a STmGMMA concentration close to 450 μg/ml. After gently mixing overnight on a rotator at room temperature, RABVG‐conjugate was purified by two rounds of ultracentrifugation (110,000 rpm, 4°C, 1 h) and then resuspended in PBS. Alternatively, HA‐STmGMMA conjugate was purified through ultrafiltration with Amicon Ultra‐15 100K using PBS with 1 M NaCl for the first two washes and PBS for the remaining nine ones.
4.5. Ag‐STmGMMA conjugates characterization
Conjugates were characterized by microBCA for total protein recovery and SDS‐PAGE and Western blot analyses to confirm conjugate formation. In the case of HA antigen, TEM and FACS analyses were performed on STmGMMA and corresponding conjugate to confirm linkage of the viral antigen to GMMA surface. Absence of unconjugated viral antigen was verified by analyzing supernatants from conjugates ultracentrifugation by HPLC‐SEC (TSK gel 6000‐4000 PW columns in series). To quantify the amount of linked protein antigen, a method based on amino acid analysis (AAA) was used, as previously described (Micoli et al., 2020). In brief, dried STmGMMA, protein antigen and conjugate samples, 20 μg each, were hydrolyzed in triplicate through gas‐phase acid hydrolysis at 110°C for 24 h. Hydrolysates were then resuspended in 100 μl of 100 mM HCl, vortexed, then 10 μl were derivatized with 6‐aminoquinolyl‐N‐hydroxysuccinimidyl carbamate (AQC) and analyzed by Ultra‐Performance Liquid Chromatography (UPLC) following Waters AccQTag Ultra kit instructions. Each amino acid was quantified respect to a calibration curve (built using the Amino Acid Hydrolysate Standard from the kit), detecting 260 nm absorbance of the derivatized amino acids through Photometric Diode Array (PDA) detector. The proportion of amino acids in the conjugate coming from the antigen (βa ) and from the STmGMMA (βG ) was determined by finding the values of βa and βG that minimized the sum of errors squared (Equation (1)).
| (1) |
where αci is the proportion of the ith amino acid in the conjugate hydrolysate, αai is the proportion of the ith amino acid in the antigen hydrolysate and αGi is the proportion of the ith amino acid in the STmGMMA hydrolysate, and this is summed over 15 amino acids (20 natural amino acids excluding Asn and Gln that are converted to Asp and Glu, respectively, and Trp, Cys and Met, that are totally or partially destroyed during hydrolysis). Note that βa + βG = 1. Eventual aggregates formation was assessed through DLS (Zetasizer, Malvern) (De Benedetto, Cescutti et al., 2017).
4.6. SDS‐PAGE and western blot
SDS‐PAGE and western blot analyses were performed as previously described with modifications (Fu et al., 2019; Hu et al., 2017, 2020). In brief, samples were first mixed with Bolt LDS Sample Buffer supplemented with reducing reagent (Thermo Fisher Scientific) and heated at 70°C for 10 min. Heated samples were then resolved by a 4%–12% or 10% Bis‐Tris SDS‐PAGE gel (Thermo Fisher Scientific). For SDS‐PAGE gel staining, the gel was stained in PageBlue Staining Solution (Thermo Fisher Scientific) for 1 h at room temperature with agitation, followed by three washes with deionized water to remove excess dye. The stained gel was then imaged by a CCD camera under white light. For Western blot, the resolved proteins were transferred from the SDS‐PAGE gel onto a PVDF membrane using the Trans‐Blot Turbo Transfer System (Bio‐Rad) according to the manufacturer's instructions. The membrane was then blocked in 5% non‐fat milk for 1 h at room temperature and sequentially incubated with primary antibodies overnight at 4°C and HRP‐conjugated secondary antibodies for 1 h at room temperature, respectively. For HRP‐conjugated primary antibodies, incubation overnight at 4°C was performed. Following extensive washes with PBST, the membrane was incubated with Immobilon Crescendo Western HRP substrate (Merck Millipore) and the immuno‐bands were captured by a CCD camera. The following primary antibodies were used: anti‐His‐HRP (1:20,000, R931‐25, Thermo Fisher Scientific), rabbit anti‐influenza H3N2 HA antibody (1:1000; PA5‐34930; Thermo Fisher Scientific), mouse anti‐STmGMMA (1:1000; in‐house produced anti‐STmGMMA murine sera). The following HRP‐conjugated secondary antibodies were used: goat anti‐human IgG‐HRP (ab6858; Abcam), mouse anti‐rabbit IgG‐HRP (sc‐2357; Santa Cruz) and m‐IgGκ BP‐HRP (sc‐516102; Santa Cruz). All secondary antibodies were used at 1:20,000 dilution.
4.7. OMV flow cytometry analysis
Unconjugated STmGMMA and HA‐STmGMMA conjugate were diluted at 40 μg/ml in PBS with 1% BSA and incubated with mouse anti‐His primary antibody (1:1000 in PBS with 1% BSA) for 1 h at 0°C. Samples were then ultracentrifuged (110,000 rpm, 1 h, 4°C) to remove primary antibody excess. After pellets resuspension in PBS with 1% BSA, samples were incubated with goat anti‐mouse AlexaFluor647 secondary antibody (1:1000 in PBS with 1% BSA) for 1 h at 0°C. A second round of ultracentrifugation was performed to remove secondary antibody excess; pellets were finally resuspended in 200 μl of 0.02 μm‐filtered PBS and analyzed with BD Accuri C6 flow cytometer.
4.8. Immunogold electron microscopy
Five microlitres of HA‐STmGMMA conjugate, diluted at 80 ng/μl in PBS, were adsorbed to 300‐mesh nickel grids, blocked in PBS with 0.5% BSA and incubated with primary anti‐X31‐HA antibody purified from immunized mice sera (1:5000 in PBS with 1% BSA) for 1 h. Grids were washed several times in PBS with 1% BSA and incubated with gold‐labelled anti‐mouse secondary antibody (1:20 in PBS with 1% BSA) for 1 h. After several washes with distilled water, grids were negatively stained and analyzed using a FEI TECNAI G2 Spirit transmission microscope operating at 120 kV, equipped with Tvips TemCam‐F216.
4.9. Fluorochrome labelling of BSA and STmGMMA
Both STmGMMA and BSA soluble protein were fluorescently labelled targeting lysine residues with Alexa Fluor 488(Alexa Fluor 488 carboxylic acid, succinimidyl ester, Invitrogen, A20000). STmGMMA and BSA were diluted to final concentration of 10 mg/ml in PBS pH 7.2, following addition of AF488 dye in a 1:20 w/w ratio.
After 1 h at room temperature in the dark, samples were purified through ultrafiltration with Amicon Ultra‐15 30K using PBS with 1 M NaCl for the first two washes and PBS for the remaining eight ones.
Labelled STmGMMA and BSA were characterized by HPLC‐SEC (SK gel 6000‐4000 PW columns in series). Fluorescence intensities were measured at 494/517 nm Ex/Em, where an increased signal intensity was observed confirming the successful labelling, while absence of unconjugated dye was also verified.
4.10. Animal immunization and sampling
Six‐to‐eight‐week‐old BALB/c mice (Charles River) were used in this study. Mice were injected intramuscularly (i.m.) with 50 μl of different vaccine formulations, according to Figures 2A,B and 4A,B, twice in 4‐week intervals. For negative control (naïve) group, mice were housed in the same condition without injections. Blood samples were taken at weeks 0, 4, and 6, and clarified serum samples were aliquoted and store at ‐80°C. At week 6, mice were sacrificed and spleens were harvested and processed to single cell suspension for in vitro antigen stimulation.
4.11. Viral challenge and monitoring
For the rabies challenge study, BALB/c mice (female, 3–4 weeks old) were first vaccinated according to the schedule detailed in Figure 3A,B. Two weeks after boost, mice were anaesthetized and challenged intracerebrally with a lethal dose of rabies virus (CVS‐11 strain, 50 ffu/30 μl). Mice were closely monitored at least twice daily. Mice were euthanized when they reached a humane endpoint, that is, ruffled fur, hunched back and affected gait (slow movement or losing coordination). The presence of rabies antigens in the mouse brains was tested using the fluorescent antibody test (FAT) through detecting the rabies N protein (Chapter, 2018).
Influenza challenge study was performed as previously described with modifications (Blakney et al., 2020). In brief, mice at 3 weeks after the second injection were anaesthetized with isoflurane and challenged intranasally with 4×105 PFU of H3N2×31. Mouse weight was recorded daily for 7 days and weight loss and survival rate were calculated. Mice with weight loss of 20% for three continuous days or 25% at any time were considered to have reached the experimental endpoint and were humanely culled.
4.12. ELISA
The antigen‐specific IgG titres in mouse serum samples were measured by a semi‐quantitative ELISA, as previously described with modifications (McKay et al., 2020). In brief, Nunc MaxiSorp ELISA plates (Thermo Fisher Scientific) were coated with 100 μl 1 μg/ml purified recombinant rabies glycoprotein or X31 HA overnight at 4°C. For the standard IgG curve generation, each plate contained wells coated with goat anti‐mouse Kappa (Cat No. 1050‐01, Southern Biotech) and goat anti‐mouse Lamda light chains (Cat No. 1060‐01, Southern Biotech). After coating, plates were washed, blocked with 1% BSA, and then incubated with diluted serum samples or 5‐fold serially diluted standard mouse IgG (standard curve) for 1 h at 37°C. Plates were then washed again and incubated with HRP‐conjugated goat anti‐mouse IgG (Cat No. 1030‐50) for another 1 h at 37°C. Following incubation and washes, plates were added with SureBlue TMB substrate (Insight Biotechnologies) for 5 min and then the reaction was stopped with stop solution (Insight Biotechnologies). Immediately after the addition of stop solution, plates were read at a testing wavelength of 450 nm and a reference wavelength of 800 nm on a Versamax Spectrophotometer (BioTek Industries). Antibody titres were calculated from the standard curve generated on each plate.
4.13. Pseudo‐rabies neutralization assay
The rabies pseudo‐neutralization assay was performed as previously described with modifications (Fu et al., 2019; Wright et al., 2008). In brief, pseudo‐rabies virus was produced by cotransfection of HEK293T/17 cells with pCMV‐Δ8.91, pCSFLW and pCVS‐G at a ratio of 1: 1.5: 1. Seventy‐two hours after transfection, virus‐containing medium was harvested and filtrated through a 0.45 μm membrane and store at ‐80°C. For sera neutralization assay, heat‐inactivated serum samples were serially diluted and incubated with virus for 1 h at 37°C. After incubation, 10,000 BHK‐21 cells/well were added to the virus‐serum mixture and cultured for 48 h at 37°C. Following culture, cells were lysed, and luciferase activity was measured by the addition of Bright‐Glo luciferase substrate (Promega). Sera neutralization was calculated as IC50, using GraphPad Prism 8.4(GraphPad).
4.14. Hemagglutination inhibition (HAI) assay
The HAI assay was performed as previously described with modifications (Blakney et al., 2020; Organization., 2013). In brief, mice sera were first treated with receptor‐destroying enzyme (RDE II) (Denka Seiken Co.) for 18 h and then heat‐inactivated for 30 min at 56°C. Following that, sera were two‐fold serially diluted with PBS in the final volume of 25 μl and incubated with four hemagglutination units (HAU)/25 μl of X31 virus for 30 min at room temperature (RT). After incubation, 50 μl 0.5% chicken red blood cells (Envigo) were added and the HAI titres were recorded 30 min later. The HAI titre was defined as the highest serum dilution that causes complete inhibition of hemagglutination.
4.15. Influenza virus microneutralization assay
Influenza microneutralization assay was performed as previously described with modifications (Grund et al., 2011). In brief, sera samples were first treated with RDE II and heated inactivated, as described in the HAI assay above. Then, sera were serially diluted with serum‐free DMEM (Merck) supplemented with penicillin/streptomycin, L‐glutamine and 1 μg/ml TPCK‐Trypsin, and incubated with X31 virus for 1 h at 37°C. Following incubation, the sera‐virus mixture was transferred to preseeded MDCK cell monolayer which was washed with serum‐free DMEM, and cultured at 37°C. Twenty‐four hours later, cells were washed with PBS, fixed with 80% ice‐cold acetone and the infection was quantified by an influenza nucleoprotein ELISA. Plates were first blocked with 5% non‐fat milk in PBST, and then sequentially incubated with rabbit anti‐NP antibody (PA5‐32242; Thermo Fisher Scientific) and mouse anti‐rabbit IgG‐HRP (sc2357; Santa Cruz). After extensive washes, plates were developed with TMB solution (Sera Care) for 5 min at RT and stopped with TMB stop solution (Sera Care). Plates were finally read at a testing wavelength of 450 nm and a reference wavelength of 800 nm, and the IC50 was calculated for each sample.
4.16. Luminex
Two weeks after the final immunization, mice were sacrificed and spleens were harvested. Splenocytes were prepared by pressing the spleens through a 70 μm cell strainer, followed by density gradient centrifugation using Lymphoprep™ and the SepMate™‐15 tubes (both from Stem Cell). After washes with complete DMEM, 1×106 splenocytes in 100 μl were restimulated with 10 μg/ml antigen (RABVG or X31 HA) in equal volume at 37°C for 7 days. Con A (5 μg/ml) and complete DMEM were served as positive and negative stimulation control, respectively. After stimulation, cell culture supernatants were harvested and antigen‐stimulated cytokines were measured by Th1/Th2/Th9/Th17/Th22/Treg Cytokine 17‐Plex Mouse ProcartaPlex™ Panel (Invitrogen Thermo Fisher Scientific) on a BioPlex 200(Bio‐Rad) platform, according to the manufacturers’ instructions.
4.17. In vivo uptake of fluorochrome‐labelled GMMA and BSA
Six‐to‐eight weeks old female BALB/c mice were first injected with 10 μg of AF488‐BSA or AF488‐STmGMMA in 50 μl PBS intramuscularly. One hour later, mice were sacrificed and muscle at the inject site and draining LNs were harvested. Cell isolation from muscle was performed as previously described with modifications (Blakney et al., 2020). In brief, muscle samples were first digested in serum‐free DMEM supplemented with 1 mg/ml collagenase P (Sigma‐Aldrich, Merck) and 5 mg/ml Dispase II (Sigma‐Aldrich, Merck) for 30 min at 37°C in an orbital shaker. After digestion, the samples were then filtered through a 70 μm cell strainer and isolated cells were pelleted by centrifugation. Draining LNs were harvested in serum‐free DMEM and cells were isolated by pressing the LNs through a 70 μm cell strainer. Isolated cells were then washed and stained with LIVE/DEAD™ Fixable Aqua Dead Cell Stain Kit (Thermo Fisher Scientific) for 30 min at room temperature. After washes with FACS staining buffer, cells were first incubated with Mouse BD Fc Block (BD Biosciences) and cell surface staining antibodies for 5 and 30 min at 4°C, respectively. Cells were then washed twice with FACS staining buffer and fixed and permeabilized with BD Cytofix/Cytoperm™ Kit (BD Biosciences) for 20 min at 4°C. After permeabilization, cells were washed twice with BD Perm/Wash buffer (BD Biosciences) and then stained with intracellular antibodies for another 30 min at 4°C. After final washes with BD Perm/Wash buffer, cells were fixed with BD Fixation buffer (BD Biosciences) and evaluated on a BD LSRFortessa flow cytometer. Total AF488+ cells and cell subsets were analyzed with Flowjo (v10.7.1). Gating strategy was illustrated in Figure S8. The following antibodies were used in the current study: muscle cells were stained with BV421‐CD34 (fibroadipogenic progenitor, FAP; 1.25 μl/reaction; 562608; BD Biosciences), PE/CY7‐CD146 (endothelial cell; 1.25 μl/reaction; 134714), BV605‐CD19 (B cell; 1.25 μl/reaction; 115540), BV711‐CD3e (T cell; 1.25 μl/reaction; 100349), BV650‐Ly‐6G (neutrophil; 5 μl/reaction; 127641), PE‐CD11b (monocyte; 1.25 μl/reaction; 101207), BV785‐CD11c (dendritic cell, DC; 1.25 μl/reaction; 117336), APC/CY7‐CD49b (NK; 5 μl/reaction; 108920), AF594‐PAX7(satellite cell; 2.5 μl/reaction; sc‐81648 AF594; Santa Cruz) and AF647‐DESMIN (myofibril; 5 μl/reaction; NBP2‐54503AF647; Novus Biologicals), while LN cells were stained with BV605‐CD19 (B cell; 1.25 μl/reaction), BV711‐CD3e (T cell; 1.25 μl/reaction;), BV650‐Ly‐6G (neutrophil; 5 μl/reaction;), PE‐CD11b (monocyte; 1.25 μl/reaction), BV785‐CD11c (DC; 1.25 μl/reaction), APC/CY7‐CD49b (NK; 5 μl/reaction) and BV421‐F4/80 (macrophage; 5 μl/reaction; 123131). Unless otherwise specified, all antibodies used for flow cytometry were purchased from BioLegend.
4.18. Statistical analyses
All data were expressed as mean ± standard deviation (SD) and all statistical analyses were performed with GraphPad Prism (version 8.4.3). For comparisons between two groups, Mann‐Whitney test was used. For comparisons among three and more groups, Kruskal‐Wallis test plus Dunn's multiple comparisons test was used. Animal survival analyses were performed with Kaplan‐Meier survival analysis. A p‐value <0.05 was considered statistically significant.
AUTHOR CONTRIBUTIONS
Conceptualization: Kai Hu, Elena Palmieri, Paul F. McKay, Robin J. Shattock, Francesca Micoli. Methodology: Kai Hu, Elena Palmieri, Karnyart Samnuan, Beatrice Ricchetti, Davide Oldrini, Paul F. McKay, Guanghui Wu, Robin J. Shattock, Francesca Micoli. Investigation: Kai Hu, Elena Palmieri, Karnyart Samnuan, Beatrice Ricchetti, Davide Oldrini, Paul F. McKay, Guanghui Wu, Leigh Thorne, Anthony R. Fooks, Lorraine M. McElhinney, Hooman Goharriz, Megan Golding. Visualization: Kai Hu, Elena Palmieri, Guanghui Wu. Supervision: Paul F. McKay, Robin J. Shattock, Francesca Micoli, Lorraine M. McElhinney. Writing—original draft: Kai Hu, Elena Palmieri, Francesca Micoli. Writing—review & editing: Kai Hu, Elena Palmieri, Guanghui Wu, Leigh Thorne, Robin J. Shattock, Francesca Micoli, Lorraine M. McElhinney, Anthony R. Fooks.
CONFLICT OF INTEREST
Elena Palmieri, Davide Oldrini, Francesca Micoli are employees of the GSK group of companies. Beatrice Ricchetti was also an employee of the GSK group of companies at the time of the experimental work.
Supporting information
Table S1. HA‐specific IgG response in vaccinated animals at week 4 and 6
Table S2. RABV‐specific IgG response in vaccinated animals at week 4 and 6
Table S3. In vivo uptake of BSA‐AF488 and STmGMMA‐AF488 (n = 5)
Fig. S1. Detection of HA on HA‐STmGMMA by immuno‐TEM. HA‐STmGMMA.
Fig. S2. HPLC‐SEC analysis to verify absence of free protein in HA‐STmGMMA conjugates.
Fig. S3. GMMA enhances HA‐specific cell‐mediated responses.
Fig. S4. Conjugation and characterization of RABVG‐STmGMMA conjugate.
Fig. S5. HPLC‐SEC analysis to verify absence of free protein in RABVG‐GMMA conjugate.
Fig. S6. STmGMMA enhances RABVG‐specific cell‐mediated responses.
Fig. S7. Fluorescent Intensity of BSA‐AF488 and STmGMMA‐AF488 in vitro.
Fig. S8. Gating strategy for STmGMMA‐AF488 in vivo uptake assay.
Fig. S9. HA‐GMMA conjugate and inactivated X31 vaccines do not induce antibody‐dependent enhancement (ADE) effect.
ACKNOWLEDGEMENTS
We thank Christopher L Pinder for his assistance in the design of the antibody panel for flow cytometry, and Dr. Benjamin F Pierce for the administrative support for the project, and Animal Service Unit of APHA for their support on rabies challenge study, and Gianmarco Gasperini for OMV FACS analysis and Fabiola Giusti for TEM imaging. This study was funded by the Department of Health and Social Care using UK Aid funding and is managed by the Engineering and Physical Sciences Research Council (EPSRC, grant number: EP/R013764/1, note: the views expressed in this publication are those of the author(s) and not necessarily those of the Department of Health and Social Care).
Hu, K. , Palmieri, E. , Samnuan, K. , Ricchetti, B. , Oldrini, D. , McKay, P. F. , Wu, G. , Thorne, L. , Fooks, A. R. , McElhinney, L. M. , Goharriz, H. , Golding, M. , Shattock, R. J. , & Micoli, F. (2022). Generalized Modules for Membrane Antigens (GMMA), an outer membrane vesicle‐based vaccine platform, for efficient viral antigen delivery. Journal of Extracellular Vesicles, 11, e12247. 10.1002/jev2.12247
Contributor Information
Robin J. Shattock, Email: r.shattock@imperial.ac.uk.
Francesca Micoli, Email: francesca.x.micoli@gsk.com.
DATA AVAILABILITY STATEMENT
All data are available in the main text or the supplementary materials.
REFERENCES
- Adalja, A. A. , Watson, M. , Cicero, A. , & Inglesby, T. (2020). Vaccine platform technologies: a potent tool for emerging infectious disease vaccine development. Health security, 18(1), 59–60. [DOI] [PubMed] [Google Scholar]
- Araujo‐Pires, A. C. , Francisconi, C. F. , Biguetti, C. C. , Cavalla, F. , Aranha, A. M. F. , Letra, A. , Trombone, A. P. F. , Faveri, M. , Silva, R. M. , & Garlet, G. P. (2014). Simultaneous analysis of T helper subsets (Th1, Th2, Th9, Th17, Th22, Tfh, Tr1 and Tregs) markers expression in periapical lesions reveals multiple cytokine clusters accountable for lesions activity and inactivity status. Journal of Applied Oral Science, 22, 336–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balhuizen, M. D. , Veldhuizen, E. J. A. , & Haagsman, H. P. (2021). Outer membrane vesicle induction and isolation for vaccine development. Frontiers in Microbiology, 12, 629090–629090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartolini, E. , Ianni, E. , Frigimelica, E. , Petracca, R. , Galli, G. , Berlanda Scorza, F. , Norais, N. , Laera, D. , Giusti, F. , Pierleoni, A. , & Grifantini, R. (2013). Recombinant outer membrane vesicles carrying Chlamydia muridarum HtrA induce antibodies that neutralize chlamydial infection in vitro. Journal of extracellular vesicles, 2(1), 20181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakney, A. K. , Zhu, Y. , McKay, P. F. , Bouton, C. R. , Yeow, J. , Tang, J. , Hu, K. , Samnuan, K. , Grigsby, C. L. , Shattock, R. J. , & Stevens, M. M. (2020). Big is beautiful: enhanced saRNA delivery and immunogenicity by a higher molecular weight, bioreducible, cationic polymer. ACS nano, 14(5), 5711–5727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention, Flu Vaccine and People with Egg Allergies . (2020). https://www.cdc.gov/flu/prevent/egg-allergies.htm#:~:text=Changes%3A,egg%20should%20receive%20flu%20vaccine
- Chapter, O. (2018), in World Organization for Animal Health, Rabies (infection with rabies virus) and other lyssaviruses. https://www.woah.org/fileadmin/Home/eng/Health_standards/tahm/2.01.17_RABIES.pdf [Google Scholar]
- Cheng, K. , Zhao, R. , Li, Y. , Qi, Y. , Wang, Y. , Zhang, Y. , Qin, H. , Qin, Y. , Chen, L. , Li, C. , & Nie, G. (2021). Bioengineered bacteria‐derived outer membrane vesicles as a versatile antigen display platform for tumor vaccination via Plug‐and‐Display technology. Nature communications, 12(1), 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko, K. A. , & Wanner, B. L. (2000). One‐step inactivation of chromosomal genes in Escherichia coli K‐12 using PCR products. Proceedings of the National Academy of Sciences of the United States of America, 97, 6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Benedetto, G. , Alfini, R. , Cescutti, P. , Caboni, M. , Lanzilao, L. , Necchi, F. , Saul, A. , MacLennan, C. A. , Rondini, S. , & Micoli, F. (2017). Characterization of O‐antigen delivered by Generalized Modules for Membrane Antigens (GMMA) vaccine candidates against nontyphoidal Salmonella. Vaccine, 35(3), 419–426. [DOI] [PubMed] [Google Scholar]
- De Benedetto, G. , Cescutti, P. , Giannelli, C. , Rizzo, R. , & Micoli, F. (2017). Multiple techniques for size determination of generalized modules for membrane antigens from Salmonella typhimurium and Salmonella enteritidis . ACS Omega, 2, 8282–8289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutry, I. , Yen, H.‐l. , Lee, H. , Peiris, M. , & Jaume, M. (2011). Antibody‐Dependent Enhancement (ADE) of infection and its possible role in the pathogenesis of influenza. BMC Proceedings, 5, P62.22373307 [Google Scholar]
- Fu, M. , Hu, K. , Hu, H. , Ni, F. , Du, T. , Shattock, R. J. , & Hu, Q. (2019). Antigenicity and immunogenicity of HIV‐1 gp140 with different combinations of glycan mutation and V1/V2 region or V3 crown deletion. Vaccine, 37(51), 7501–7508. [DOI] [PubMed] [Google Scholar]
- Gasperini, G. , Raso, M. M. , Arato, V. , Aruta, M. G. , Cescutti, P. , Necchi, F. , & Micoli, F. (2021). Effect of O‐antigen chain length regulation on the immunogenicity of Shigella and Salmonella generalized modules for membrane antigens (GMMA). International journal of molecular sciences, 22(3), 1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerke, C. , Colucci, A. M. , Giannelli, C. , Sanzone, S. , Vitali, C. G. , Sollai, L. , Rossi, O. , Martin, L. B. , Auerbach, J. , Di Cioccio, V. , & Saul, A. (2015). Production of a Shigella sonnei vaccine based on generalized modules for membrane antigens (GMMA), 1790GAHB. PLoS one, 10(8), e0134478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerritzen, M. J. H. , Martens, D. E. , Wijffels, R. H. , van der Pol, L. , & Stork, M. (2017). Bioengineering bacterial outer membrane vesicles as vaccine platform. Biotechnology Advances, 35, 565–574. [DOI] [PubMed] [Google Scholar]
- Gnopo, Y. M. D. , Watkins, H. C. , Stevenson, T. C. , DeLisa, M. P. , & Putnam, D. (2017). Designer outer membrane vesicles as immunomodulatory systems—Reprogramming bacteria for vaccine delivery. Advanced Drug Delivery Reviews, 114, 132–142. [DOI] [PubMed] [Google Scholar]
- Grund, S. , Adams, O. , Wählisch, S. , & Schweiger, B. (2011). Comparison of hemagglutination inhibition assay, an ELISA‐based micro‐neutralization assay and colorimetric microneutralization assay to detect antibody responses to vaccination against influenza A H1N1 2009 virus. Journal of Virological Methods, 171, 369–373. [DOI] [PubMed] [Google Scholar]
- Hu, K. , He, S. , Xiao, J. , Li, M. , Luo, S. , Zhang, M. , & Hu, Q. (2017). Interaction between herpesvirus entry mediator and HSV‐2 glycoproteins mediates HIV‐1 entry of HSV‐2‐infected epithelial cells. Journal of General Virology, 98(9), 2351–2361. [DOI] [PubMed] [Google Scholar]
- Hu, K. , Fu, M. , Wang, J. , Luo, S. , Barreto, M. , Singh, R. , Chowdhury, T. , Li, M. , Zhang, M. , Guan, X. , & Hu, Q. (2020). HSV‐2 infection of human genital epithelial cells upregulates TLR9 expression through the SP1/JNK signaling pathway. Frontiers in immunology, 11, 356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaparakis‐Liaskos, M. , & Ferrero, R. L. (2015). Immune modulation by bacterial outer membrane vesicles. Nature Reviews Immunology, 15, 375–387. [DOI] [PubMed] [Google Scholar]
- Keys, T. G. , & Aebi, M. (2017). Engineering protein glycosylation in prokaryotes. Current Opinion in Systems Biology, 5, 23–31. [Google Scholar]
- Kis, Z. , Shattock, R. , Shah, N. , & Kontoravdi, C. (2019). Emerging technologies for low‐cost, rapid vaccine manufacture. Biotechnology Journal, 14, e1800376. [DOI] [PubMed] [Google Scholar]
- Kolls, J. K. , & Khader, S. A. (2010). The role of Th17 cytokines in primary mucosal immunity. Cytokine & Growth Factor Reviews, 21, 443–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudlay, D. , Kofiadi, I. , & Khaitov, M. (2022). Peculiarities of the T cell immune response in COVID‐19. Vaccines, 10, 242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuipers, K. , Daleke‐Schermerhorn, M. H. , Jong, W. S. , Corinne, M. , van Opzeeland, F. , Simonetti, E. , Luirink, J. , & de Jonge, M. I. (2015). Salmonella outer membrane vesicles displaying high densities of pneumococcal antigen at the surface offer protection against colonization. Vaccine, 33(17), 2022–2029. [DOI] [PubMed] [Google Scholar]
- Kushnir, N. , Streatfield, S. J. , & Yusibov, V. (2012). Virus‐like particles as a highly efficient vaccine platform: Diversity of targets and production systems and advances in clinical development. Vaccine, 31, 58–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Launay, O. , Ndiaye, A. G. , Conti, V. , Loulergue, P. , Scire, A. S. , Landre, A. M. , Ferruzzi, P. , Nedjaai, N. , Schütte, L. D. , Auerbach, J. , & Podda, A. (2019). Booster vaccination with GVGH Shigella sonnei 1790GAHB GMMA vaccine compared to single vaccination in unvaccinated healthy European adults: results from a phase 1 clinical trial. Frontiers in immunology, 10, 335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, R. , & Liu, Q. (2020). Engineered bacterial outer membrane vesicles as multifunctional delivery platforms. Frontiers in Materials, 7, 202. [Google Scholar]
- Lurie, N. , Saville, M. , Hatchett, R. , & Halton, J. (2020). Developing covid‐19 vaccines at pandemic speed. New England Journal of Medicine, 382, 1969–1973. [DOI] [PubMed] [Google Scholar]
- Mancini, F. , Micoli, F. , Necchi, F. , Pizza, M. , Berlanda Scorza, F. , & Rossi, O. (2021). GMMA‐Based Vaccines: The Known and The Unknown. Frontiers in Immunology, 3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancini, F. , Rossi, O. , Necchi, F. , & Micoli, F. (2020). OMV vaccines and the role of TLR agonists in immune response. International Journal of Molecular Sciences, 21(12), 4416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay, P. F. , Hu, K. , Blakney, A. K. , Samnuan, K. , Brown, J. C. , Penn, R. , Zhou, J. , Bouton, C. R. , Rogers, P. , Polra, K. , Lin, P. J. , & Shattock, R. J. (2020). Self‐amplifying RNA SARS‐CoV‐2 lipid nanoparticle vaccine candidate induces high neutralizing antibody titers in mice. Nature communications, 11(1), 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micoli, F. , Rondini, S. , Alfini, R. , Lanzilao, L. , Necchi, F. , Negrea, A. , Rossi, O. , Brandt, C. , Clare, S. , Mastroeni, P. , & MacLennan, C. A. (2018). Comparative immunogenicity and efficacy of equivalent outer membrane vesicle and glycoconjugate vaccines against nontyphoidal Salmonella. Proceedings of the National Academy of Sciences, 115(41), 10428–10433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micoli, F. , Alfini, R. , Di Benedetto, R. , Necchi, F. , Schiavo, F. , Mancini, F. , Carducci, M. , Palmieri, E. , Balocchi, C. , Gasperini, G. , & Saul, A. (2020). GMMA is a versatile platform to design effective multivalent combination vaccines. Vaccines, 8(3), 540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noad, R. J. (2019). UK vaccines network: Mapping priority pathogens of epidemic potential and vaccine pipeline developments. Vaccine, 37, 6241–6247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obiero, C. W. , Ndiaye, A. G. , Sciré, A. S. , Kaunyangic, B. M. , Marchetti, E. , Gone, A. M. , Schütte, L. D. , Riccucci, D. , Auerbach, J. , Saul, A. , & Podda, A. (2017). A phase 2a randomized study to evaluate the safety and immunogenicity of the 1790GAHB generalized modules for membrane antigen vaccine against Shigella sonnei administered intramuscularly to adults from a shigellosis‐endemic country. Frontiers in immunology, 8, 1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization, Serological detection of avian influenza A(H7N9) virus infections by modified horse red blood cells haemagglutination‐inhibition assay . (2013). https://www.who.int/publications/m/item/serological-detection-of-avian-influenza-a(h7n9)-virus-infections-by-modified-horse-red-blood-cells-haemagglutination-inhibition-assay
- Rappazzo, C. G. , Watkins, H. C. , Guarino, C. M. , Chau, A. , Lopez, J. L. , DeLisa, M. P. , Leifer, C. A. , Whittaker, G. R. , & Putnam, D. (2016). Recombinant M2e outer membrane vesicle vaccines protect against lethal influenza A challenge in BALB/c mice. Vaccine, 34(10), 1252–1258. [DOI] [PubMed] [Google Scholar]
- Rappuoli, R. (2004). From Pasteur to genomics: Progress and challenges in infectious diseases. Nature Medicine, 10, 1177–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi, O. , Pesce, I. , Giannelli, C. , Aprea, S. , Caboni, M. , Citiulo, F. , Valentini, S. , Ferlenghi, I. , MacLennan, C. A. , D'Oro, U. , & Gerke, C. (2014). Modulation of endotoxicity of Shigella generalized modules for membrane antigens (GMMA) by genetic lipid A modifications: relative activation of TLR4 and TLR2 pathways in different mutants. Journal of Biological Chemistry, 289(36), 24922–24935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi, O. , Caboni, M. , Negrea, A. , Necchi, F. , Alfini, R. , Micoli, F. , Saul, A. , MacLennan, C. A. , Rondini, S. , & Gerke, C. (2016). Toll‐like receptor activation by generalized modules for membrane antigens from lipid A mutants of Salmonella enterica serovars Typhimurium and Enteritidis. Clinical and Vaccine Immunology, 23(4), 304–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solá, R. J. , & Griebenow, K. (2010). Glycosylation of therapeutic proteins. BioDrugs, 24, 9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suan, D. , Sundling, C. , & Brink, R. (2017). Plasma cell and memory B cell differentiation from the germinal center. Current Opinion in Immunology, 45, 97–102. [DOI] [PubMed] [Google Scholar]
- Tan, K. , Li, R. , Huang, X. , & Liu, Q. (2018). Outer membrane vesicles: Current status and future direction of these novel vaccine adjuvants. Frontiers in Microbiology, 9, 783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Ley, P. A. , Zariri, A. , van Riet, E. , & Oosterhoff, D. , Kruiswijk, C. P. (2021). An intranasal OMV‐based vaccine induces high mucosal and systemic protecting immunity against a SARS‐CoV‐2 infection. Frontiers in Immunology, 12, 781280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Pol, L. , Stork, M. , & van der Ley, P. (2015). Outer membrane vesicles as platform vaccine technology. Biotechnology Journal, 10, 1689–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki, A. (2017). Biological roles of glycans. Glycobiology, 27, 3–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winarski, K. L. , Tang, J. , Klenow, L. , Lee, J. , Coyle, E. M. , Manischewitz, J. , Turner, H. L. , Takeda, K. , Ward, A. B. , Golding, H. , & Khurana, S. (2019). Antibody‐dependent enhancement of influenza disease promoted by increase in hemagglutinin stem flexibility and virus fusion kinetics. Proceedings of the National Academy of Sciences, 116(30), 15194–15199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, E. , Temperton, N. J. , Marston, D. A. , McElhinney, L. M. , Fooks, A. R. , & Weiss, R. A. (2008). Investigating antibody neutralization of lyssaviruses using lentiviral pseudotypes: a cross‐species comparison. The Journal of general virology, 89(Pt 9), 2204–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye, J. , Wang, Y. , Wang, Z. , Ji, Q. , Huang, Y. , Zeng, T. , Hu, H. , Ye, D. , Wan, J. , & Lin, Y. (2018). Circulating Th1, Th2, Th9, Th17, Th22, and Treg levels in aortic dissection patients. Mediators of Inflammation, 2018, 5697149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuseff, M.‐I. , Pierobon, P. , Reversat, A. , & Lennon‐Duménil, A.‐M. (2013). How B cells capture, process and present antigens: A crucial role for cell polarity. Nature Reviews Immunology, 13, 475–486. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. HA‐specific IgG response in vaccinated animals at week 4 and 6
Table S2. RABV‐specific IgG response in vaccinated animals at week 4 and 6
Table S3. In vivo uptake of BSA‐AF488 and STmGMMA‐AF488 (n = 5)
Fig. S1. Detection of HA on HA‐STmGMMA by immuno‐TEM. HA‐STmGMMA.
Fig. S2. HPLC‐SEC analysis to verify absence of free protein in HA‐STmGMMA conjugates.
Fig. S3. GMMA enhances HA‐specific cell‐mediated responses.
Fig. S4. Conjugation and characterization of RABVG‐STmGMMA conjugate.
Fig. S5. HPLC‐SEC analysis to verify absence of free protein in RABVG‐GMMA conjugate.
Fig. S6. STmGMMA enhances RABVG‐specific cell‐mediated responses.
Fig. S7. Fluorescent Intensity of BSA‐AF488 and STmGMMA‐AF488 in vitro.
Fig. S8. Gating strategy for STmGMMA‐AF488 in vivo uptake assay.
Fig. S9. HA‐GMMA conjugate and inactivated X31 vaccines do not induce antibody‐dependent enhancement (ADE) effect.
Data Availability Statement
All data are available in the main text or the supplementary materials.