

SUMMARY
Three-dimensional (3D) chromatin organization maps help dissect cell type-specific gene regulatory programs. Furthermore, 3D chromatin maps contribute to elucidating the pathogenesis of complex genetic diseases by connecting distal regulatory regions and genetic risk variants to their respective target genes. To understand the cell type-specific regulatory architecture of diabetes risk, we generated transcriptomic and 3D epigenomic profiles of human pancreatic acinar, alpha, and beta cells using single-cell RNA-seq, single-cell ATAC-seq, and high-resolution Hi-C of sorted cells. Comparisons of these profiles revealed differential A/B (open/closed) chromatin compartmentalization, chromatin looping, and transcriptional factor mediated control of cell type-specific gene regulatory programs. We identified a total of 4,750 putative causal-variant-to-target-gene pairs at 194 type 2 diabetes GWAS signals using pancreatic 3D chromatin maps. We found that the connections between candidate causal variants and their putative target effector genes are cell-type stratified and emphasize previously underappreciated roles for alpha and acinar cells in diabetes pathogenesis.
eTOC
Su et al. use 3D chromatin organization maps of human pancreatic alpha, beta, and acinar cells to establish that connections between candidate causal variants and their putative target effector genes are cell-type stratified and emphasize previously underappreciated roles for alpha and acinar cells in diabetes pathogenesis.
INTRODUCTION
Type 2 diabetes (T2D) is a serious and costly disease that impacted an estimated 463 million people worldwide in 2019 (Saeedi et al., 2019). Disease onset is typically late (>40 years old), with incidence rising due to population aging and increasing rates of obesity. Of concern, more children and young adults are now developing T2D (Zimmet et al., 2001). Chronic complications include accelerated development of cardiovascular and microvascular disease; furthermore, diabetes is a major cause of blindness. While much of the increased prevalence of T2D can be attributed to the rising prevalence of obesity and a sedentary lifestyle, increasing evidence suggests that genetic risk factors contribute to the pathogenesis of this common disease. As the genetic contribution to T2D is relatively weak and multifactorial when contrasted with that of type 1 diabetes (T1D)(Clayton, 2009; Rich, 1990), it is more challenging to identify risk loci for T2D, earning the disease the nickname of ‘a geneticist’s nightmare’ prior to the advent of genome-wide association studies (GWAS).
GWAS have revolutionized the field of complex disease genetics and yielded compelling evidence for over 500 genetic loci associated with T2D (Vujkovic et al., 2020). The role of large consortia and international collaborations for achieving the scale and power necessary for larger GWAS analyses through the collection of material, combining genotype scores, and releasing data to the scientific community has greatly increased the number of risk loci uncovered for this disease. Unfortunately, GWAS only report genomic signals associated with a given trait and rarely the precise identity of the effector transcript and corresponding affected cell-types. As such, over the past 15 years GWAS has defined a period of signal discovery, rather than an era of gene target discovery.
Because gene expression in mammals is frequently controlled via long range interactions over large genomic distances, many regulatory elements do not control the nearest genes and can reside tens or hundreds of kilobases (kb) away. Therefore, attention is required when considering the biology underpinning an established GWAS signal, as frequently the gene nearest the lead SNP does not encode the effector transcript that impacts the phenotype. In addition, for complex diseases such as T2D, multiple organ systems contribute to the disease phenotype; in the case of diabetes, at a minimum this includes skeletal muscle, adipose tissue, liver, brain, and pancreas. Due to the predominant role of pancreatic islet regulatory sites for T2D risk (Torres et al., 2020), three-dimensional chromatin maps of intact human pancreatic islets and the β cell cellular model EndoC-βH1 have been used to annotate T2D risk signals to effector genes (Greenwald et al., 2019; Lawlor et al., 2019; Miguel-Escalada et al., 2019). However, because islets consist of multiple cell types with distinct functions and because different cell types can employ alternative cis-regulatory elements to control the same gene, it is necessary to further dissect variant-to-gene connections at pancreatic cell-type level to fully understand the molecular basics of T2D risk.
In this study, we combined 3D chromatin maps from the major pancreatic cell types with matched single cell transcriptome and chromatin accessibility data to investigate cell-type-specific gene regulation program in the human pancreas. This approach enabled mapping of more than 100 T2D GWAS signals to their likely effector transcript(s) and corresponding cell type(s) of action. Remarkably, in addition to the expected beta-cell acting variants, we also discovered variant-to-gene combinations that likely function in pancreatic alpha and acinar cells, suggesting an important contribution for these underappreciated cell types in T2D pathogenesis.
RESULTS
Single-cell transcriptomic and chromatin accessibility profiles of the human pancreas
To explore the cell type-specific impact of chromatin architecture and accessibility on gene expression in the human pancreas, we performed high-resolution Hi-C on three distinct primary pancreatic cell subsets (acinar, alpha, and beta cells) FACS-sorted from multiple human organ donors without diabetes. All islet preparations were obtained from organ donors without diabetes and assessed for beta cell function using islet perifusion assays, demonstrating glucose-responsive insulin secretion (data not shown, available on PANC-DB; https://hpap.pmacs.upenn.edu). We coupled our Hi-C approach with scATAC-seq and scRNA-seq on whole pancreatic islets from three matched donors (Figure 1A and Supplemental Table 1) and performed integrative data analysis.
Figure 1. General validation of transcriptomic, chromatin accessibility and 3D architecture profiles in human pancreas cell subsets.
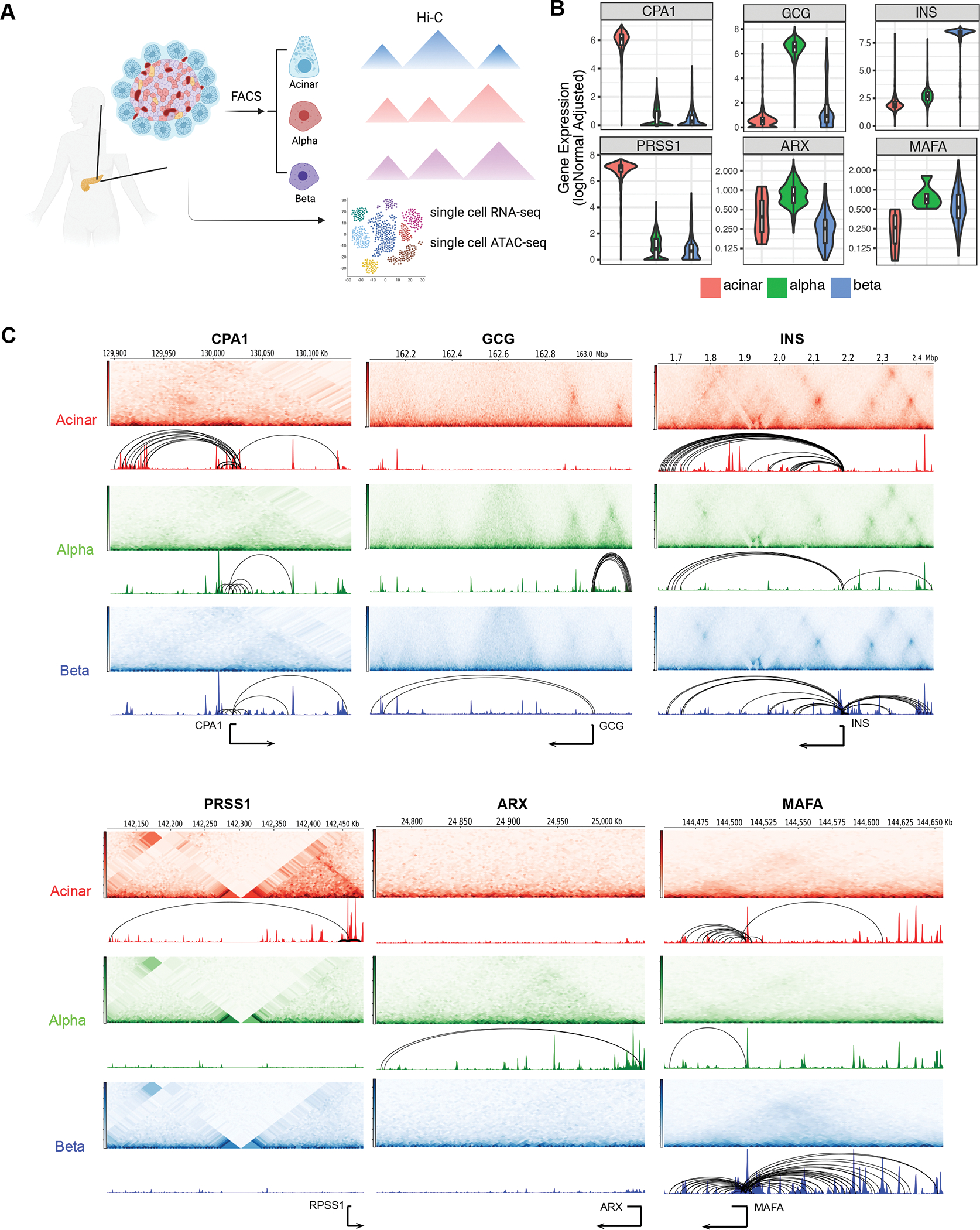
A, Experimental design used in this study. Pancreatic islets obtained from 3 human donors were used for single cell experiments. Acinar, alpha and beta cells from 5 human donors were purified using FACS and followed by Arima Hi-C assays. See also Supplemental Table S1B, Single-cell expression profiles for selected marker genes. Acinar: CPA1 and PRSS1; alpha: GCG and ARX; beta: INS and MAFA. C, Aggregate single-cell chromatin accessibility, Hi-C contact frequency matrix and chromatin loops for selected marker genes. Hi-C contact heatmaps represent the interaction frequency between regions along chromosome following along the diagonal from a given point on the track (darker color indicates higher interaction frequency). Genomic tracks represent chromatin accessibility and significant loops.
For the transcriptomes, we first clustered 5,288 quality-controlled single cells across the three donors from scRNA-seq into nine clusters (Supplemental Figure 1A) using cell-type-specific markers (Supplemental Figure 1B). Donors were evenly represented across each of the identified cell types (Supplemental Figure 1B) with highly reproducible transcriptomic profiles (Supplemental Figure 1C). Acinar (19.8%), alpha (52.2%) and beta cells (16.1%) contributed to the three largest cell clusters from human pancreatic islets with distinctly expressed cell markers (Figure 1B, Supplemental Figure 1B).
Next, we performed snATAC-seq on islet cells from the same set of donors and clustered accessible chromatin profiles from 12,473 high-quality single cells into nine cell type groups, similar to that observed with scRNA-seq (Supplemental Figure 2A). The promoter regions of marker genes showed high chromatin accessibility in the corresponding cell types, and each cell type was represented by all three donors (Supplemental Figure 2B). We further compared aggregated reads from acinar, alpha and beta cells in scATAC-seq to previously published bulk ATAC-seq (Ackermann et al., 2016), and observed enriched accessibility signals at both gene bodies and promoter regions of the marker genes CPA1, GCG and IGF2 in acinar, alpha, and beta cells, respectively, as expected (Supplemental Figure 2C,D). Further comparison of scATAC-seq cell clusters to the independently identified transcriptomic clusters revealed highly specific correlations between scRNA-seq and scATAC-seq (Supplemental Figure 2E), confirming the high quality and reliable cell-type clustering of single cell chromatin accessibility profiles.
The 3D chromatin architecture of the major pancreatic cell types
To define target genes of regulatory chromatin in a lineage-specific manner, we generated 3D chromatin maps of the three major pancreatic cell types using high-resolution Hi-C chromosome conformation capture on FACS-sorted cell subsets from two biological replicates pooled from 5 healthy donors. To confirm the purity of FACS-sorted cell populations, we performed bulk RNA-seq on sorted subsets of cells that were used for Hi-C library preparation and deconvoluted them based on the single cell transcriptomic profile. On average, 95.5% of the sorted alpha gene program and 76% of the sorted beta cell program matched to the corresponding cell type cluster from scRNA-seq analysis (Supplemental Figure 1E). Each Hi-C library replicate was sequenced to over 2.2 billion reads, and 1.8 billion reads passing quality control metrics per library were used to construct 3D chromatin maps (Supplemental Table 2). Contact matrices were strongly correlated between replicates (stratum-adjusted correlation coefficient >0.9; Supplemental Figure 3A). The endocrine cell types (alpha and beta) were more similar to each other than to acinar cells in terms of contact frequency, as expected from their developmental origin and functional properties (Supplemental Figure 3A). We observed approximately 5,000 topologically associating domains (TADs) with a median size of 360kb for each cell type (Supplemental Figure 3B,C). We further called chromatin contacts at 1kb, 2kb and 4kb resolution independently in each cell type using Mustache (Roayaei Ardakany et al., 2020) and Fit-Hi-C2 (Kaul et al., 2020) from pooled reads for both replicates (Supplemental Table 3). We observed higher than ~4-fold enrichment of Hi-C contacts for the loop calls with respect to their local background (Supplemental Figure 3D), confirming that the samples allowed for high-fidelity chromatin loop identification.
Next, maintaining the highest resolution, we merged the resultant contact calls from all three cell types to create a combined set of 860,565 consensus chromatin contacts (Supplemental Figure 3E). The median distance between loop anchors was 318kb, with 15.6% of called contacts connecting chromatin regions more than 1Mb apart. We annotated these chromatin loops to genes and regulatory chromatin by overlapping loop anchors with gene promoters (−1,500bp to +500bp relative to the transcriptional start site [TSS]) and the open chromatin regions we had identified via scATAC-seq in the corresponding cell types. On average, 58% of the chromatin contacts had at least one anchor annotated to open chromatin regions, while 46% of the chromatin contacts mapped to promoters of protein-coding or long noncoding RNA genes (Supplemental Figure 3F). Approximately 20% of the chromatin contacts were annotated to gene-to-open-chromatin-region pairs, in which one anchor overlapped with a gene promoter while the other anchor overlapped open chromatin regions from the corresponding cell type. Compared to random gene-to-open-chromatin-region pairs, those connected by chromatin contacts demonstrated significantly higher co-accessibility of gene promoter and open chromatin region (Supplemental Figure 3G, two-sided Wilcoxon rank sum test P-value < 2.2e–16). Consistent with previous chromatin conformation capture assays (Javierre et al., 2016; Su et al., 2021), we also observed a larger number of contacts between genes and distal chromatin regions in the cell types in which these genes were specifically expressed (Figure 1C), suggesting a quantitative regulatory effect of chromatin contacts on gene expression.
To further explore the putative regulatory impact of chromatin contacts, we mapped loop anchors to previously characterized chromHMM chromatin states, i.e. active promoters, poised enhancers, ‘active enhancers’ (as annotated by chromatin marks, but not functionally tested as such), etc., in endocrine and acinar cells (Arda et al., 2018). Compared to distance-matched non-significant Hi-C contacts, significant chromatin contacts were enriched for enhancers (acinar: active enhancer 1, odds ratio (OR) =1.68, active enhancer 2, OR=1.54; alpha: active enhancer 1, OR=1.60, active enhancer 2, OR=1.60; beta: active enhancer 1, OR=1.44, active enhancer 2, OR=1.43) and active TSS (acinar: active TSS OR=2.08; alpha: active TSS OR=1.87; beta active TSS OR=1.68), with ~68% of chromatin contacts mapping to active enhancers and 8% of contacts mapping to active TSS in the corresponding cell types (Supplemental Figure 3H,I). Collectively, these results demonstrate that our robust cell purification approach coupled with high-resolution Hi-C analysis successfully generated comprehensive, high-quality 3D regulatory maps that connect regulatory elements to their target genes in the major human pancreatic cell types.
Cell type-specific A/B compartmentalization distinguishes pancreatic endocrine and exocrine cells
The genome is divided on the megabase scale into two compartments, termed “A” and “B”, with the former associated with open and the latter with closed chromatin (Dixon et al., 2015; Fortin and Hansen, 2015). To investigate if A/B compartmentalization contributes to pancreatic endocrine/exocrine differentiation, we estimated A/B compartments in acinar, alpha and beta cells by eigenvector analysis of the genome contact matrix at 40kb resolution after observed/expected frequency normalization (Figure 2A). Prior studies (Dixon et al., 2015; Fortin and Hansen, 2015) show that A/B compartments exhibit cell-type specificity, and we likewise detected variation between the pancreatic cell types that substantially exceeded technical variation (Figure 2B). Endocrine cell types (alpha and beta) were more similar to each other (jaccard coefficient of A-B assignment = 0.62) than to acinar cells (jaccard coefficient of A-B assignment = 0.51, Figure 2B). We observed that 16.9% of the genome was differentially compartmentalized between alpha and beta cells, which was significantly lower than the variant compartmentalization between either endocrine cell type and acinar cells (Figure 2C). Further examination of differentially compartmentalized regions between cell types revealed that, compared to invariant compartmentalized regions, the genome was significantly less accessible at regions that were annotated to compartment B in one cell type and compartment A in the other cell types, whereas the genome was more open in differential compartments annotated as A (Figure 2D, Wilcoxon Rank Sum Test of all pairwise comparisons, P-value < 2.1e–277). Similarly, genes that reside in a compartment A in one cell type and in compartment B in another showed reduced expression in the latter cell type, while genes located in the A compartment showed increased expression as expected (Figure 2E, Wilcoxon Rank Sum Test all pairwise comparison p-value < 3.8e–67).
Figure 2. Cell type-specific A/B compartments and their relations to gene expression and regulatory chromatin.
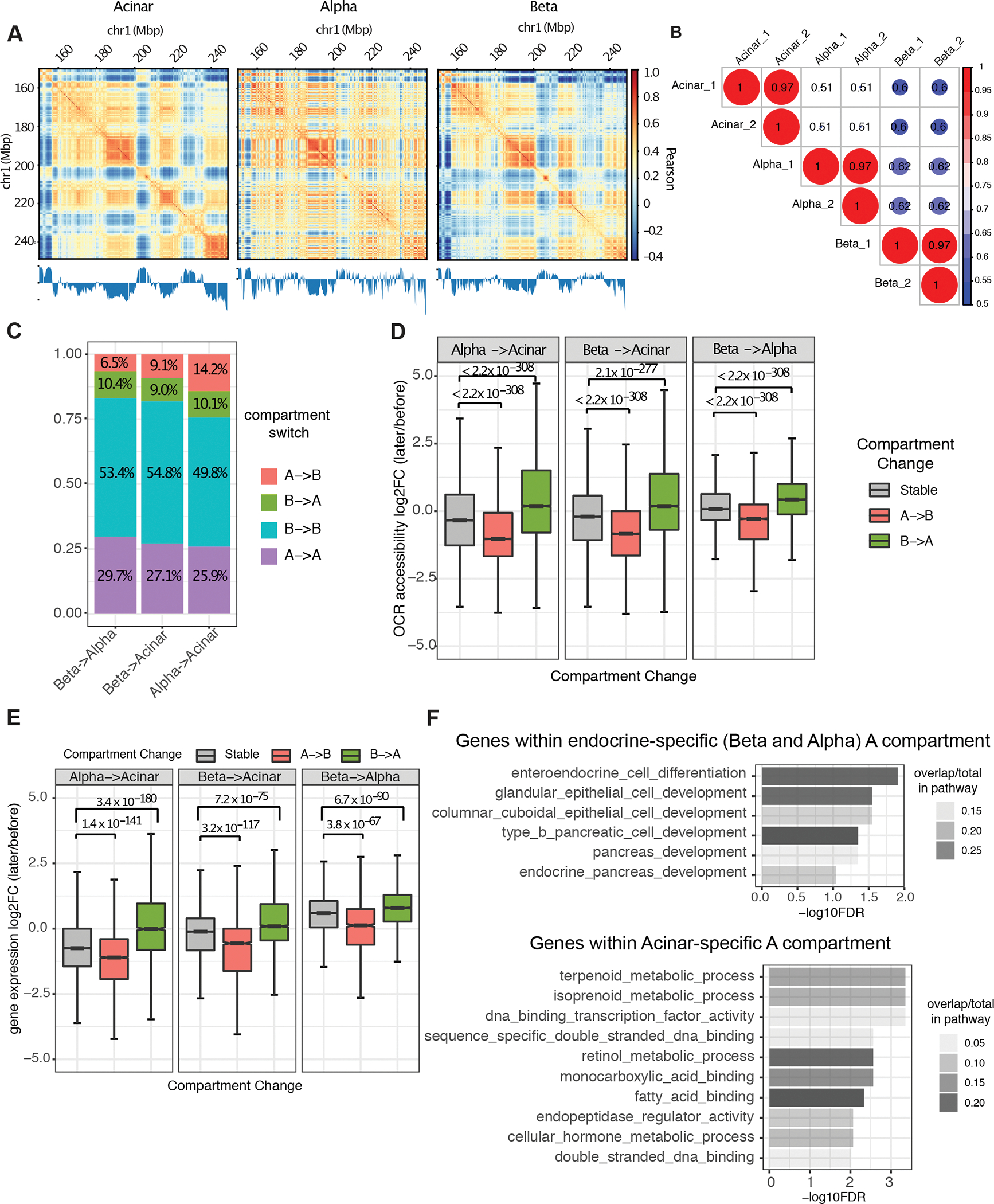
A, Example of A/B compartment identification using Pearson correlation matrix at 40kb resolution on chr1:150,000,000–250,000,000. The plaid pattern suggests that chromatin spatially segregates into two compartments. The first eigenvector of the correlation matrix is shown below to derive compartment type, with genomic GC content as reference. B, Jaccard coefficient of A/B compartment assignment in pairwise comparison of each replicate sample derived from acinar, alpha, and beta cells. A/B compartment assignment was defined by the sign of first eigenvector for each 40kb bin and compared between Hi-C samples. C, Composition of genomic regions that change compartment status or remain the same in pairwise comparisons between cell-types. D and E, Distribution of fold-change in OCR (open chromatin region) accessibility (D) and gene expression (E) at dynamic (A to B or B to A) or stable (A to A or B to B) compartmentalized regions. P value was calculated by two-sided Wilcoxon sum rank test; whiskers correspond to interquartile range. F, Pathway enrichment for genes located at endocrine and acinar-specific A compartment. P-values are calculated by hypergeometric test with FDR correction.
When considering acinar cells, we found 763 genes residing in the A compartment that were not present in the A compartment in endocrine cells. These gene sets were significantly enriched for functions associated with the exocrine pancreas such as isoprenoid, terpenoid, and retinol metabolism and endopeptidase regulator activity (Figure 2F, Supplemental Table 4). On the other hand, 998 genes that were unambiguously located at A compartments of both alpha and beta cells but not in acinar cells were enriched for endocrine cell differentiation (Figure 2F, Supplemental Table 4). Taken together, these results suggest that the differential organization of A and B compartments contribute to shaping the gene expression patterns that drive differentiation of exocrine and endocrine cells in the human pancreas.
The strength and pattern of chromatin contacts recapitulates cell type-specific gene expression in the human pancreas
To determine how the chromatin contacts that we identified compare between cell types, we performed a quantitative comparison of contact frequency matrices using multiCompareHiC v1.8.0 (Stansfield et al., 2018) to identify statistically significant differential looping (FDR < 0.05). Using this approach, we identified 11,052 contacts that were differentially enriched in at least one pairwise comparison (Supplemental Table 5). These ‘cell type enriched loops’ (CTELs) were further grouped hierarchically into four clusters based on normalized interaction frequencies, with cluster 1 (2,786 loops) representing acinar-specific CTELs, cluster 2 (4,348 loops) representing beta-specific CTELs, cluster 3 (2,378 loops) representing alpha-specific CTELs and cluster 4 (1,540 loops) representing endocrine-shared CTELs (Figure 3A).
Figure 3. Cell-type differential loops (CTDLs) and their effect on gene regulation.
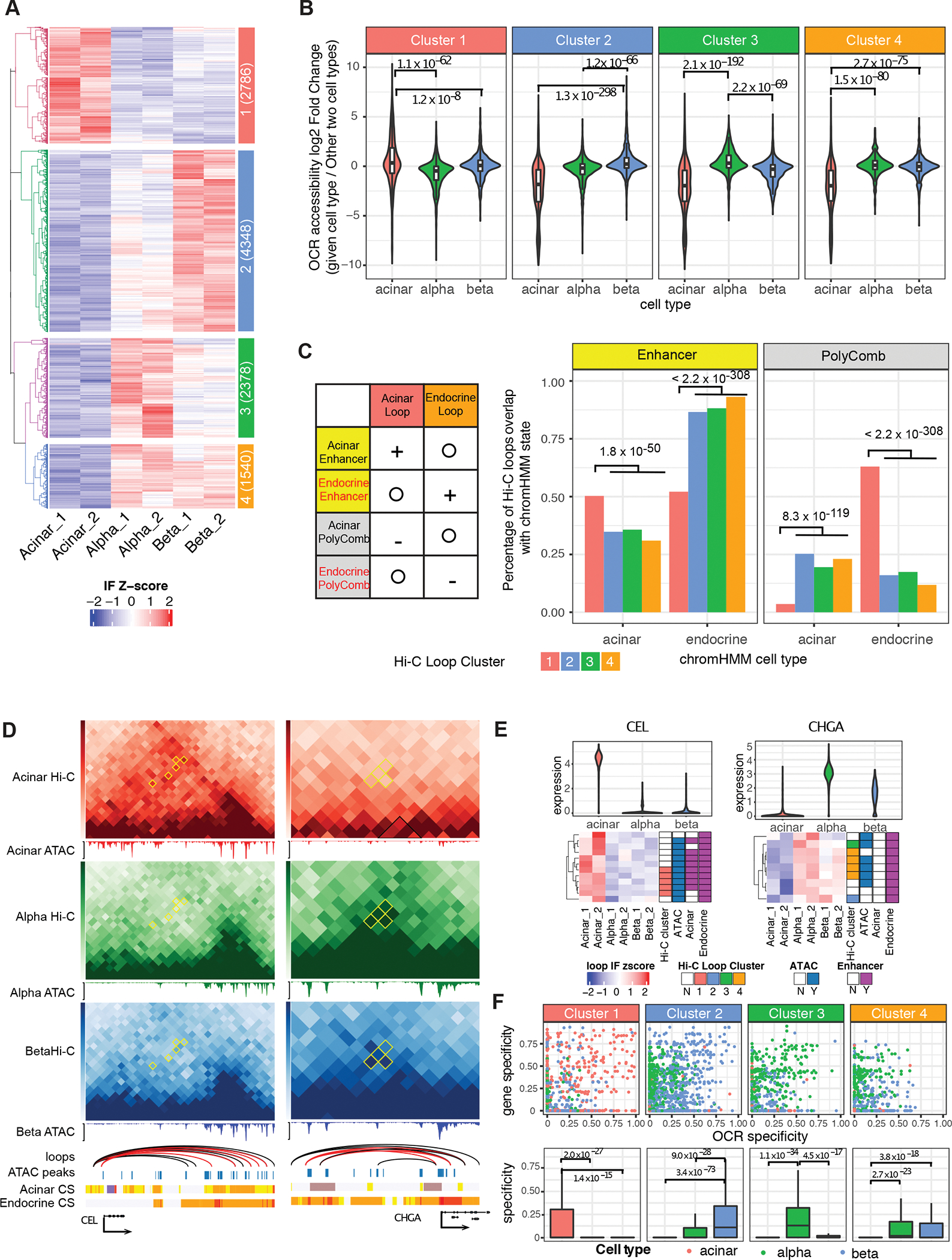
A, The interaction frequency (IF) heatmap shows distinct clustering of cell-type enriched loops (CTELs). Each row represents a unique CTEL, and individual samples are organized in columns; four clusters are defined by hierarchical clustering. Cluster notion and number of differential chromatin loops are indicated in colored annotation box at left of heatmap. B, Distribution of accessibility fold-change of OCRs located at anchors of CTELs. P value is calculated by two-sided Wilcoxon sum rank test; whiskers correspond to interquartile range. C, The ratio of clustered CTELs overlapped with enhancer and polycomb-binding chromatin (PolyComb) from acinar and endocrine cells. Left panel illustrates the expected result based on chromatin enrichment of all chromatin loops in Supplemental Figure 3I. Plus sign means “enrichment”, minus sign means “depletion” and circle means “irrelevant”. Right panel shows the observed result in terms of the percentage of clustered CTELs overlapping with given chromatin state. P value is calculated by two-side two sample proportion test. D, CTELs connecting promoters of CEL and CHGA to distal regulatory elements. The Hi-C contact frequency matrix is shown in heatmap with CTDLs (cell type differential loops) in yellow diamonds. Consensus chromatin loops are represented as arcs, with CTELs labelled in red. scATAC-seq is shown as normalized aggregated fragment abundance in each cell type with consensus peaks highlighted by blue bars. Chromatin states (CS) for both acinar and endocrine cells are represented by the color scheme explained in Supplemental Figure 3H. E, The gene regulation scheme of CEL and CHGA. The interaction frequency (IF) heatmap is shown for all the chromatin loops that connect the gene promoters to distal chromatin, with CTELs annotated with corresponding cluster color scheme below. The chromatin loops are also annotated based on whether distal chromatin anchors are overlapped with consensus ATAC-seq peaks (union of the aggregated ATAC-seq peaks called from acinar, alpha, and beta cells) and cell-type enhancer chromatin. F, Cell-specificity of gene and OCRs connected by CTELs. The specificity of genes and OCRs in each cell type are calculated using scRNA-seq and scATAC-seq data. The overall specificity of gene-OCR pair is represented by the geometric mean of specificity values of each gene and plotted in the boxplots. P-value is calculated by two-sided Wilcoxon sum rank test; whiskers correspond to interquartile range. Red:Acinar cell, Green:Alpha cell, Blue:Beta cell, Orange: Alpha-Beta shared.
To determine if cell type-specific chromatin contacts are likewise associated with cell type-specific enrichment of regulatory chromatin, we first measured the accessibility of open chromatin regions that overlapped CTEL anchors. This analysis demonstrated that genomic regions with higher interaction frequencies showed increased chromatin accessibility in a given cell type. For example, acinar-specific CTELs connected open chromatin regions that were more open in acinar than in alpha or beta cells (Figure 3B). In addition, a comparison with chromHMM-identified chromatin states revealed that acinar-specific CTEL anchors were preferentially enriched for acinar enhancers and, conversely, depleted of acinar polycomb-repressed chromatin, while endocrine-specific CTEL anchors (clusters 2 to 4) were enriched for endocrine enhancers and depleted of endocrine polycomb-repressed chromatin (Figure 3C, two-sided two sample proportion test, all P-values < 1.8e–50). These results indicate that cell type-enriched chromatin contacts preferentially connect active regulatory regions and recapitulate cell type-specific regulatory signatures.
To explore how cell type-specific chromatin architecture affects gene expression, we focused on signature genes whose promoters were linked to distal regulatory genomic regions by CTELs. For example, CEL encodes a lipase secreted from the pancreas into the digestive tract (Johansson et al., 2018); it was both highly expressed in acinar cells and linked by five acinar-enriched CTELs to distal genomic regions enriched for regulatory chromatin signatures (Figure 3D, E). Although these distal regulatory regions were annotated as enhancers in both acinar and endocrine cell types, the enhancer-gene contacts were formed more frequently in acinar than endocrine cells, suggesting that increased physical contact between the CEL promoter and distal enhancers drives the gene expression difference between the cell types. Another example is CHGA, which encodes chromogranin A, a critical component of secretory vesicles of endocrine cells (Beumer et al., 2020). Here, six out of nine CHGA promoter-connected contacts were found to be specific to endocrine cells, and all distal interacting regions corresponded to endocrine-specific enhancers (Figure 3D, E). These findings establish that quantitatively identified CTELs are enriched for cell type-specific enhancers regions.
To globally evaluate the relationship between gene expression, distal cis-regulatory element activity, and physical gene-to-cis-regulatory-element contacts, we focused on the 2,910 (26.3%) CTELs that we could annotate to gene-open chromatin region pairs, and derived the gene expression and open chromatin region accessibility specificity weight matrix from our single cell scRNA-seq and scATAC-seq data using CELLEX (Timshel et al., 2020). We found that gene-to-open chromatin region pairs with high cell type-specificity were preferentially joined to contacts enriched in the same cell type (Figure 3F), i.e. acinar-specific CTELs (cluster 1) connect open chromatin regions and genes with high specificity in acinar cells, while beta-specific CTELs (cluster 2) connect open chromatin regions and genes with high specificity values in beta cells (Figure 3F). Overall, these findings suggest that differential chromatin contacts play an important role in cell type-specific regulation of gene expression in the pancreas.
Cell type-specific transcription factor accessibility and promoter connectivity cooperate to establish lineage-specific gene expression programs
Changes in chromatin structure at genomic regions are generally accompanied by dynamic binding of transcription factors (Reizel et al., 2021; Voss and Hager, 2014). The development and differentiation of the pancreas is governed by the coordinated action of lineage-specific transcription factors that direct cell-selective transcriptional programs (Bramswig and Kaestner, 2011; Gao et al., 2010; Gao et al., 2008; Gao et al., 2007; Kropp et al., 2019; van der Meulen and Huising, 2015). To investigate which transcription factors are enriched in cell type-specific regulatory chromatin organization in the human pancreas, we employed chromVAR (Schep et al., 2017) to assess the global accessibility of transcription factor motifs from the JASPAR 2020 database (Fornes et al., 2020) in the data derived from scATAC-seq. Motif enrichment in each cell type was represented by an accessibility deviation score reflecting the accessibility of peak sets in the selected cell type relative to the basal assumption of equal chromatin accessibility across all cells. Figure 4A summarizes these data, which confirm previously known motif enrichment patterns. For example, the motif occupied by the endocrine transcription factor NEUROD1 was enriched in alpha, beta, delta and epsilon cells, while the motif preferred by NKX6–1, PDX1 and PAX4 was enriched in beta and delta cells. Additionally, the binding sites of the exocrine transcription factors NR5A2, GATA4 and GATA6 were enriched in ductal and acinar cells (Figure 4A, B). Further clustering of cell types based on motif enrichment revealed close relationships between acinar and ductal (Pearson correlation = 0.78), endothelial and mesenchyme (Pearson correlation = 0.77), alpha and epsilon (Pearson correlation = 0.8), and beta and delta cells (Pearson correlation = 0.64, Figure 4C). These findings are consistent with the transcriptome profiles of these cell types measured by scRNA-seq (Supplemental Figure 1D), suggesting that cell lineage-enriched transcription factors together with differential regulatory chromatin accessibility change across cell types to determine cell-specific gene expression.
Figure 4. Cell-type specific transcription factor (TF) motif enrichment and TF-gene network.
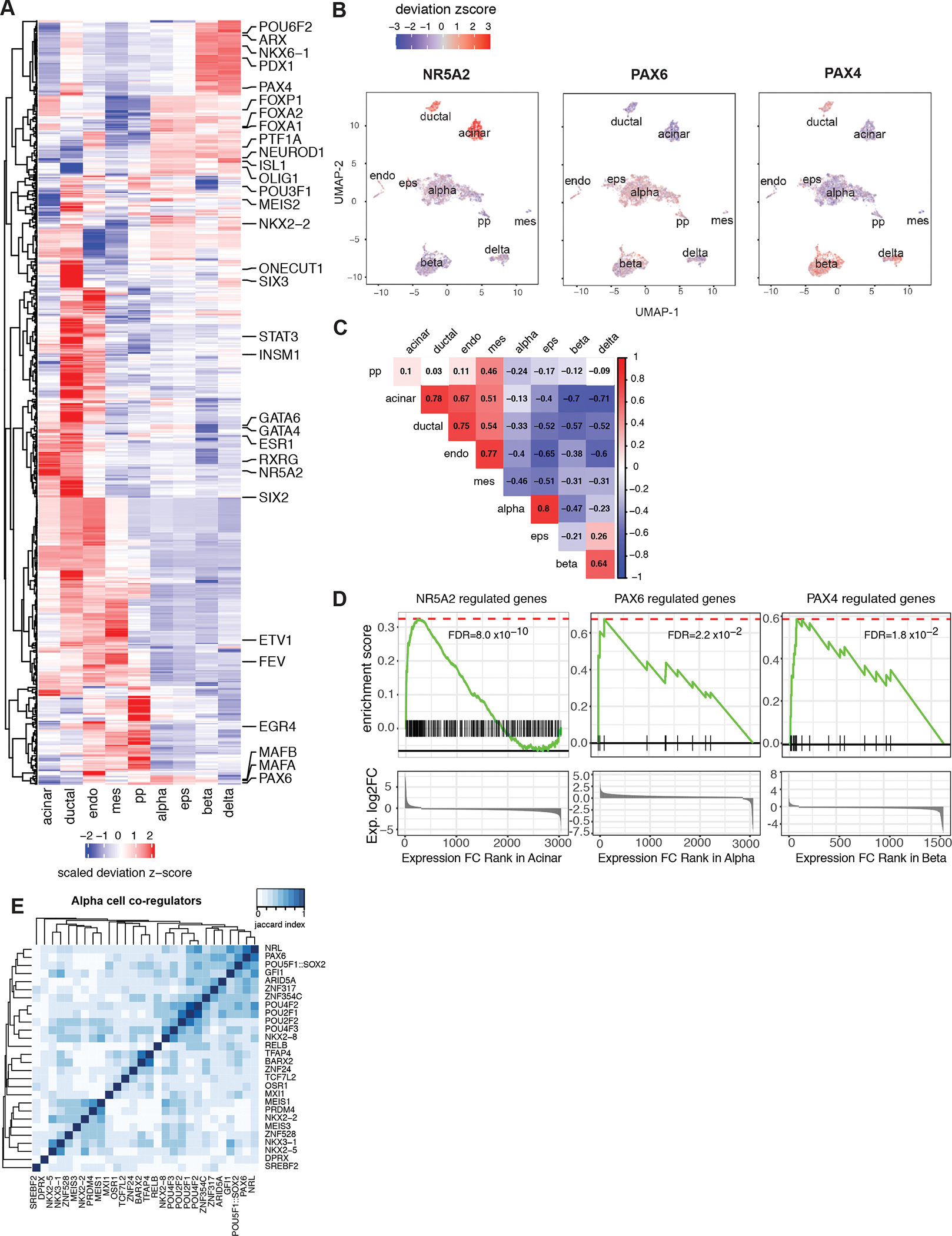
A, chromVAR motif enrichment for 438 cell-type-specific TFs. The identification of cell-type-specific TFs is explained in Methods, for full list see Supplemental Table 6. The enrichment was measured by deviation z-score. B, The enrichment for NR5A2, PAX6 and PAX4 motifs for each cell projected onto UMAP. C, Pearson correlation of TF enrichment between cell types. The deviation z-score was used for correlation. D, GSEA pre-rank enrichment of cell type-specific expressed genes identified as NR5A2, PAX6 and PAX4 downstream genes by motif analysis. Genes are ranked by gene expression log2 fold change in the given cell type from highest to the lowest. Gene sets are defined by TF downstream genes of which either promoters or distal cis-regulatory elements contain the given TF motif. Details of enrichment are given in Supplemental Table 7. E, Association index analysis reveals alpha-specific TF modules. TF sets are defined cell-specific TFs with significant GSEA enrichment (Supplemental Table 7). Matrices show the Jaccard similarity between TF pairs based on occurrence of co-regulated downstream gene.
To further explore this concept, we mapped regions harboring accessible transcription factor motifs to the genes they likely regulate via physical contact in acinar, alpha and beta cells using our genome-scale chromatin connectivity datasets. Focusing on transcription factor binding sites in cell type-specific open chromatin, we identified between 23 and 2,179 (mean 580) likely downstream genes per transcription factor motif. Among all 438 TF motifs exhibiting cell type-specific accessibility (Supplemental Table 6), 65% (283) were physically connected to genes with significantly higher expression in that same cell type compared to other cell types (Supplemental Table 7). For example, NR5A2, whose motif was specifically enriched in the open chromatin landscape of acinar and ductal cells (Figure 4A, B), was predicted to regulate 138 downstream genes through its binding to promoters or promoter-connected distal regions. These 138 genes, including CPA1, CTRB2, CTRB1 and PLA2G1B, were enriched for high expression in acinar cells compared to other cell types (GSEA pre-rank permutation test FDR = 8.0e–10, Figure 4D, Supplemental Table 7). Similarly, PAX6 and PAX4, whose motifs were specifically enriched in endocrine cells, were localized to downstream genes with elevated expression in alpha and beta cells, respectively (GSEA pre-rank permutation test PAX6: FDR = 0.02; PAX4: FDR=0.018, Figure 4D, Supplemental Table 7).
Next, we calculated the binding similarities between transcription factors to define jointly acting regulators in a cell-specific manner, based on their shared downstream target genes (Supplemental Table 7). As expected, TFs with similar motif positional weight matrices clustered together (e.g. POU class 2 and class 4 TF motifs in alpha co-regulators; Figure 4E). However, we also found TFs with divergent motifs that grouped together because they target similar sets of downstream genes. For example, NRL, PAX6 and POU5F1/SOX2 motifs are highly colocalized with alpha cell open chromatin, all having motifs in the alpha-specific genes GCG, IRX2 and C5orf38 (Figure 4E, Supplemental Table 7). Similarly, NKX6–1 and POU6F1 motifs, which are enriched in beta cell open chromatin, are each connected to the MAFA, HADH, ADCYAP1, SYNE2, SORL1, GNAS, SAMD11 and SCD5 genes that are likewise highly expressed in beta cells (Supplemental Table 7). In addition, we were able to identify cell-specific “hub” regulators based on the number of targeted downstream genes and their involvement in the cohesiveness of TF-target gene transcription regulatory network. In alpha cells, POU2F1 and RELB motifs targeted 8 alpha-specific genes that showed elevated expression in alpha cells (Supplemental Table 6 and 7). In beta cells, NHLH2 and ARID3A motifs targeted 14 and 25 downstream genes, respectively, that were specifically expressed in beta cells, and NHLH2 itself showed elevated expression in beta cells (Supplemental Table 6 and 7). This integrated analysis of the transcriptome, open chromatin landscape, and nuclear architecture of the major cell types of the human pancreas refines our understanding of lineage-specific regulators that operate to establish pancreatic gene regulatory networks.
Cell type-specific assignment of diabetes-associated variants to their target effector genes
Genetic variants associated with diabetes and diabetes-relevant traits are enriched at chromatin accessible sites in various pancreatic islet cell types (Chiou et al., 2021). However, our current 3D epigenomic analyses of purified human pancreatic alpha, beta, and acinar cells allows a previously unattainable, direct view of specific genes that these T2D variants target in the major cell types of the pancreas and adds physical contact maps as evidence for variant to gene interactions. To achieve this goal, we focused on accessible chromatin regions that were connected to at least one target gene through either long-range chromatin conformation or promoter proximity, and performed partitioned heritability linkage disequilibrium (LD) score regression (LDSC) (Finucane et al., 2015) on type 1 and 2 diabetes (Chiou et al., 2021; Mahajan et al., 2018), glycemic traits (Chen et al., 2021) and other complex traits (Dashti et al., 2019; Jones et al., 2019; Rashkin et al., 2020; Roselli et al., 2018) independently in acinar, alpha and beta cells. Overall, we found significant enrichment of fasting glucose (alpha: enrichment=23.8, p-value=1.1e–6; beta: enrichment=35.0, P-value=2.3e–5), T2D (alpha: enrichment=13.8, p-value=3.3e–3; beta: enrichment=17.1, P-value=2.4e–4), sleep duration (beta: enrichment=7.6, p-value=6.9e–5) and chronotype (alpha: enrichment=8.6, P-value=3.9e–6; beta: enrichment=10.0, P-value=2.7e–7) in beta and alpha cells, and enrichment of pancreatic cancer (acinar: enrichment=5379, P-value=1.6e–3) in acinar cells (FDR < 0.1, Figure 5A, Supplemental Table 8). We also observed additional significant enrichment of fasting glucose (enrichment=17.3, P-value=3.0e–3) and a more nominal enrichment of T2D (enrichment=14.4, P-value=7.5e–3) in acinar cells, which implicates epigenetic chromatin-structure-based connections between pancreatic exocrine cells and T2D. The link of sleep-related traits to genes expressed in pancreatic islets was recently explored further (Lasconi et al., 2022).
Figure 5. Chromatin contacts link T2D risk variants to target genes with cell-type specific stratification.
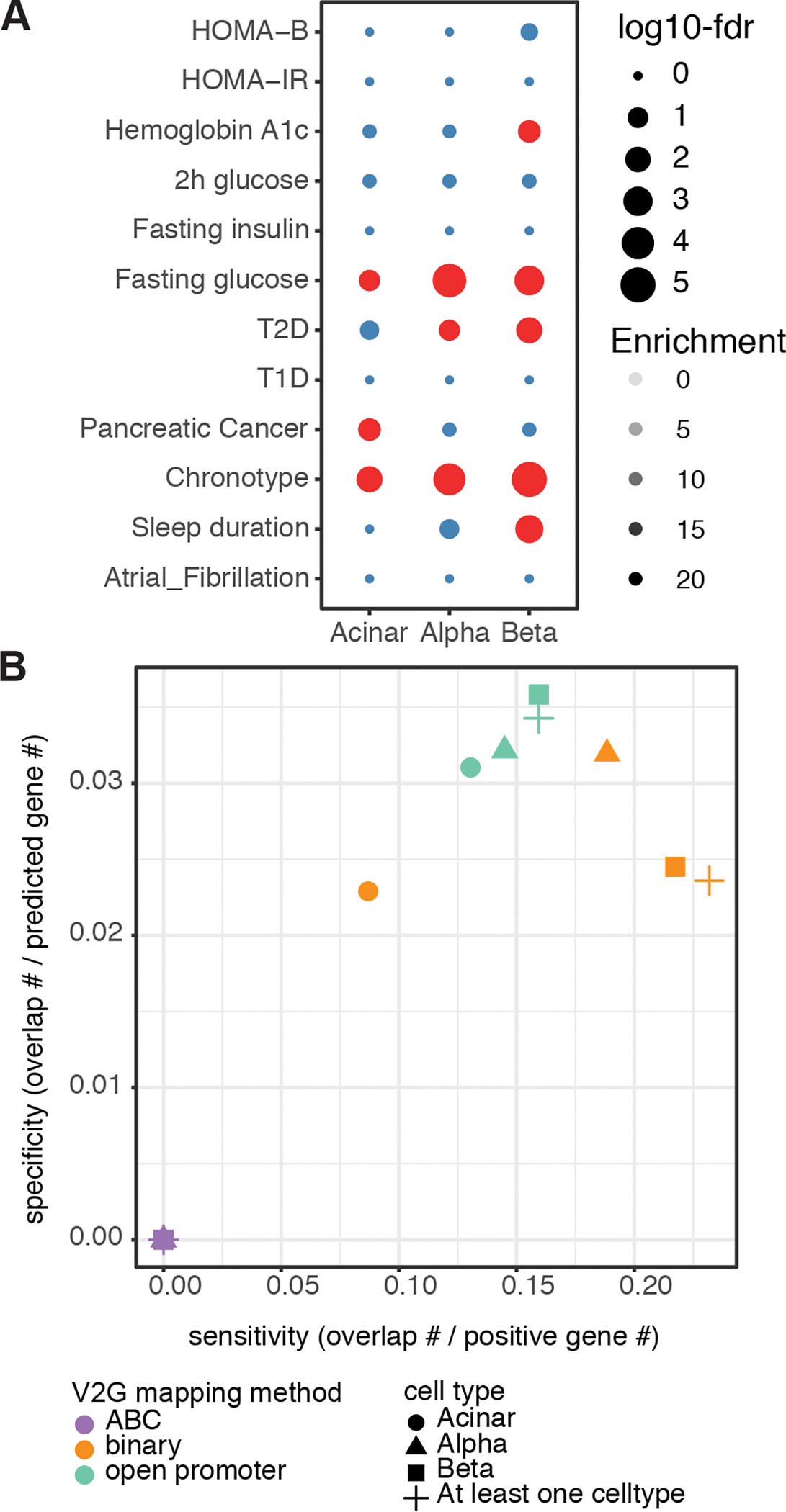
A, Partitioned LD score regression enrichment of gene-connected regulatory chromatin for T2D and related relevant traits. Significant enrichment FDR < 0.1 is indicated with red. B, Specificity and sensitivity of variant-to-gene mapping approaches using chromatin maps from pancreatic cells to detect positive control genes previously defined for T2D. The approaches “ABC” and “binary” are detailed in Methods. The “open promoter” approach defined candidate genes as the proximal genes whose promoter regions (−1,500bp to +500bp of TSS) overlapped with proxies located in open chromatin regions (Supplemental Figure 4, right branch), while the “open nearest genes” were defined by proximal genes closest to open proxies regardless of distance.
A number of approaches have been developed to assign variants to genes by leveraging regulatory chromatin landscape and/or nuclear architectural data. Here we compared two general approaches: (1) The Activity-by-Contact (ABC) model (Fulco et al., 2019), which computes a score that predicts gene-enhancer connections by combining the read counts of enhancer accessibility (ATAC-seq or DNAse-seq) and presumed activity (H3K27ac ChIP-seq) with the normalized contact frequency (HiC) between putative enhancers and gene promoters, or (2) binary calls using the intersection of significant cell-specific loops called from our Hi-C data and cis-regulatory elements defined by ATAC-seq with corresponding gene promoters (Supplemental Figure 4, see Methods). Notably, it was previously reported that an averaged HiC matrix of several cell lines performed similarly to cell type matched HiC data in the ABC model (Fulco et al., 2019; Nasser et al., 2021). Using our cell-type-specific Hi-C contact frequencies in acinar, alpha and beta, the ABC model linked 12,114 putative enhancers to 9,062 target genes (31,103 enhancer-gene pairs) in all three cell types (ABC score >= 0.02, Gene Cis-element Interactome in Pancreas Hi-C shinyApp). In contrast, our chromatin loop binary calls identified four times as many gene-distal element connections, with 146,472 cis-regulatory-element-gene pairs associating 55,003 open chromatin regions to 20,029 genes in at least one of the three cell types (Gene Cis-element Interactome in Pancreas Hi-C shinyApp).
Even though our chromatin loop binary calls did not constrain the cis-regulatory-elements to open chromatin regions marked by the H3K27ac signal like the ABC model does, we observed that on average fewer than 10% of the gene to putative enhancer pairs identified by the ABC model were also present in the binary calls. In order to evaluate how the two different approaches affect variant-to-gene mapping, we applied the two gene to distal-element maps to T2D GWAS signals (Mahajan et al., 2018). The currently known 403 sentinel SNPs for T2D (Supplemental Table 9) correspond to 8,859 proxy SNPs that are within close linkage disequlibrium (LD, r2 > 0.8) using the 1,000 genome European population as the reference and 38,758 fine-mapped variants of which 5,768 variants are shared between proxies (R2 > 0.8) and the 95% credible set. The chromatin loops we mapped above in the major pancreatic cell types revealed contacts related to 165 sentinels, for which we could map 1,162 open SNPs to 922 distally located gene promoters. Limiting to just the 165 sentinel SNPs for which our pancreas-specific Hi-C maps and ATAC-seq data were informative, our experimental approach narrowed down the list of possible causal SNPs from 34,993 to 1,162, i.e. a >96% reduction of the search space for causal variants.
In contrast, the ABC model linked only 301 putative causal variants (corresponding to 96 sentinel SNPs) to 411 gene promoters in our three cell types (Supplemental Table 10). Although 56.8% of the proxy SNPs identified by the ABC model were also implicated by our chromatin loop binary approach, the two methods linked SNPs to very different sets of genes, with only 42% of the genes (8.4% proxy-gene pairs) from the ABC model recapitulated by the chromatin loop binary approach.
Next, we compared the implicated genes to a set of independently curated T2D positive control genes, which include (1) genes whose perturbation causes a Mendelian form of diabetes, (2) genes whose encoded protein acts as a drug target for the common disease, or (3) coding variants discovered in case-control exome studies of T2D (Forgettta et al., 2020). None of the genes identified by the ABC model overlapped with these T2D positive control genes, while the chromatin loop binary calls detected 23.2% of positive control gene set with various sensitivity and specificity among the different cell types (Figure 5B). Given these observations, we elected to use the gene to distal-element map identified by our chromatin loop binary approach for variant-to-gene mapping and complimented the list of putative causal variants with open proxies located at gene promoters we had identified (Figure 5B, Supplemental Table 10).
Finally, we found that 194 of the 403 T2D GWAS sentinels (32%) were in LD with at least one variant situated in an open gene promoter (147 sentinels) or a distal OCR found in direct contact with a gene promoter (165 sentinels) in the three human pancreatic cell types analyzed. This represents 1,388 putative casual variants mapping to a total of 1,091 genes across acinar, alpha and beta cells at these loci (Supplemental Table 10). Out of 4,750 casual-variant-gene pairs identified by our method, only 369 (7.8%) were reproduced by previously islet promoter-enhancer promoter capture Hi-C (Miguel-Escalada et al., 2019). Conversely, of the prior whole islet variant-gene pairs reproduced here, 98.4% (363) are physically connected by Hi-C loops in alpha and beta cells, as expected. Further evaluation of a variant’s impact on the immediate surrounding genomic location found that 869 proxies (57.5%) show a strong disruptive effect on at least one TF binding site (Supplemental Table 11). We also compared the genes implicated by our method to a recent study that used single cell ATAC-seq to identify cell type specific gene assignments for T2D GWAS signals using co-accessibility of ATAC-seq peaks. Seven genes (SPRED2, INPP5E, WASF3, THOC7, IGF2BP3, PDE8B, POLR3K) were found to be implicated specifically in beta cells in both studies.
Importantly, in addition to dramatically reducing the number of likely putative causal variants for T2D, our analyses mapped each of the 194 T2D risk loci containing at least one putative causal-variant-gene pair to their likely effector transcripts. In total, we provide variant-to gene-to cell type mapping for 900 genes, many of which previously were either not supported, or only scantly supported, as T2D relevant (Supplemental Table 12), facilitating functional interrogation of these gene in the future. To further dissect the cell-specificity of T2D variant-to-gene connections, we examined cell type-specific gene expression, variant-containing open chromatin region accessibility, and gene-open chromatin region contact frequency for each putative causal-variant-gene pair. We discovered 16 putative effector genes that, together with their interacting variant-containing open chromatin regions, demonstrated high cell-type specificity (specificity weight >=0.5, Figure 6A,B). As expected, most of cell-type specific putative effector genes, e.g., INS, DGKB, and P2RY1, were specific to insulin-producing beta cells (Figure 6B). However, we also implicated 5 putative effector genes likely to function in non-beta cells; the alpha cell-enriched WFS1 encoding Wolframin, a transmembrane protein regulating cellular Ca2+ homeostasis, and the acinar-specific gene KCNQ1, SLC39A8, CTRB1, and CPA4 (Figure 6B; Supplemental Figure 5B). Importantly, with the exception of TRPM5, all chromatin interactions of these causal-variant-gene pairs demonstrated higher contact frequency in their specific cell type of action (Figure 6B), suggesting the importance of chromatin looping in the cell-type specific regulation of T2D pathogenesis. For example, at the TH locus (rs4929965), a beta-cell specific open chromatin region harboring three putative causal variants (rs7482891 A>G r2=0.98, rs4929964 T>G r2=0.98 and rs4929965 A>G r2=1), physically contacted the INS promoter over 12kb away preferentially in beta cells (Figure 6C). The same open chromatin region also interacted with the TRPM5 promoter over 250kbp downstream in both alpha and beta cells, with a higher contact frequency in alpha cells (Figure 6C, Supplemental Table 10). Further examination of the three putative causal variants revealed that all of them resided at transcriptional factor binding sites (rs4929965 A>G: KLF4, ZNF148, MAZ and PLAGL2; rs4929964 T>G: NR2C2; rs7482891 A>G: KLF12 and GLIS1) and were predicted to strongly disrupt factor occupancy (Supplemental Figure 5C, Supplemental Table 11). Similarly, at the WFS1 locus (rs10937721), a putative endocrine-specific causal proxy variant (rs4234731 A>G r2=0.88) in intron 7 of WFS1 physically contacted the WFS1 promoter in both alpha and beta cells, with an additional alpha-cell specific contact further upstream (Figure 6D). Importantly, this variant is predicted to strongly disrupt the binding of the endocrine transcription factor NEUROD2 (Supplemental Figure 5D). Numerous publications have associated with WFS1 locus with Mendelian and common forms of diabetes (Sandhu et al., 2007). A mouse study found that disruption of WFS1 gene causes progressive beta cell loss and impairs insulin exocytosis (Ishihara et al., 2004). However, our chromatin looping approach found the connection between the putative causal variant rs4234731 and WFS1 promoter is more specific to alpha cells; elevated gene expression, increased variant accessibility, and higher contact frequency are all observed in in alpha cells compared to beta cells (Figure 6B), necessitating the future investigation of WFS1 function in the alpha cell context.
Figure 6. Chromatin contacts link T2D risk variants to target genes with cell-type specific stratification.
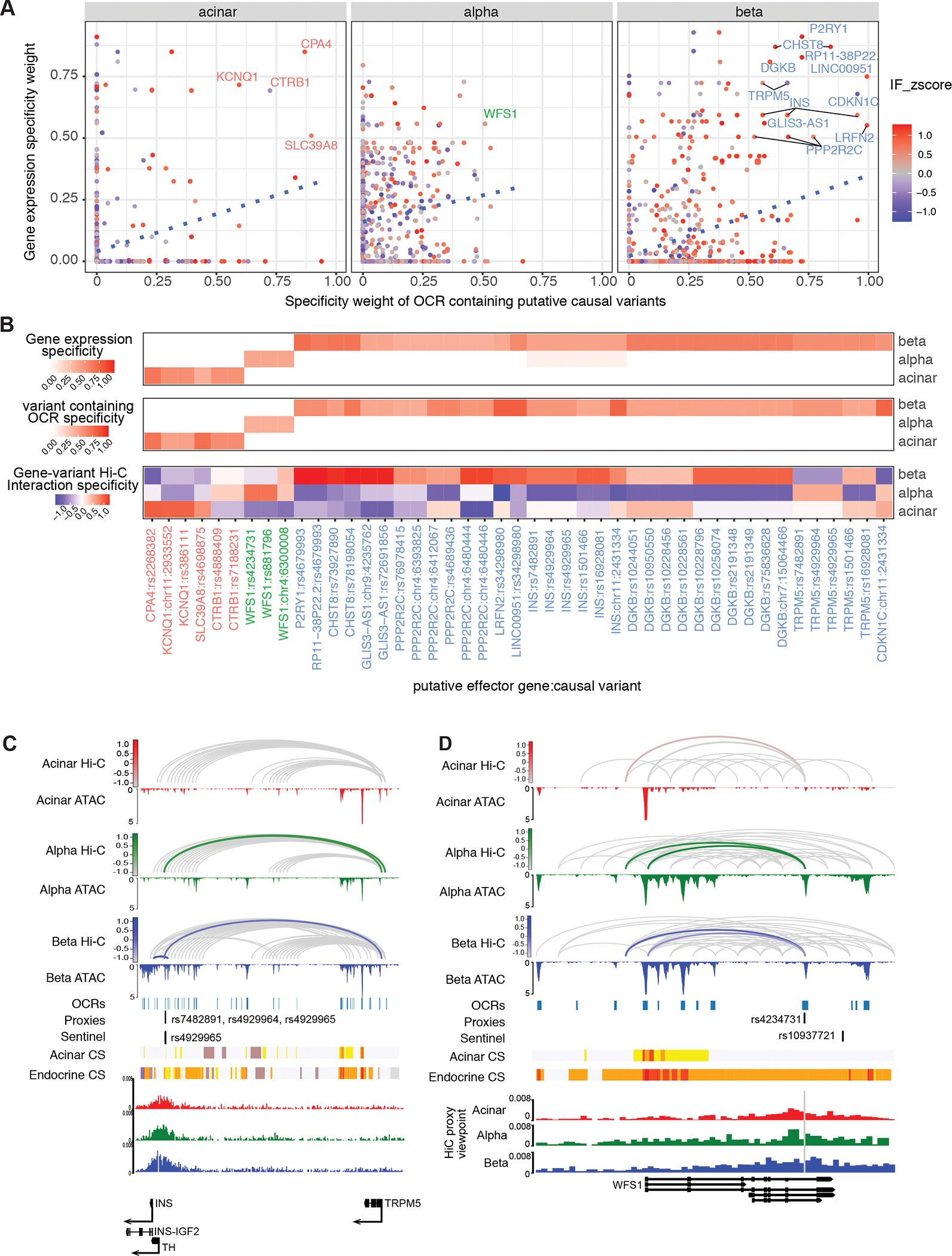
A, Cell type-specificity of putative causal variants and their target gene. The variant-containing gene-OCR pairs with high cell-specificity (specificity >= 0.5) were labelled with target gene name for each cell type B, The gene-causal-variant pairs were clustered based on cell-type specificity C, Cell type-specific chromatin looping between causal variants (rs7482891, rs4929964 and rs4929965) and target genes INS and TRPM5 and the T2D TH (rs4929965) locus. The chromatin loops that connect causal variants and target genes are highlighted with cell-type specific color (acinar: red, alpha: green and beta: blue), with hue scale indicating Hi-C interaction frequency. D, Cell-specific chromatin looping between variant rs4234731 and the target gene WFS1 at the T2D rs10937721 locus. The chromatin loops that connect putative causal variants and target genes are highlighted with cell-type specific color (acinar: red, alpha: green and beta: blue) with the hue scale indicating the Hi-C interaction frequency.
To further prioritize causal genes and elucidate mechanisms involved in T2D, we compared the variant-gene pairs predicted for T2D to our analyses of other relevant traits (Supplemental Table 13), including fasting glucose (FG), Hemoglobin A1C (HbA1c), fasting insulin (FI), 2h glucose (2hGluc) and T1D. Among 4,750 putative causal gene-variant pairs in T2D, 241 pairs (5.1%) were identified within one or more other relevant trait (Supplemental Figure 6A) and correspond to 23 T2D sentinel signals. Although T1D and T2D shared most gene-variant pairs, 15 out of 16 signals that show statistically significant multi-trait genetic etiology with T2D were only colocalized with glycemic traits (Supplemental Table 14), suggesting pleiotropic effects of genetic variants mainly between T2D and glycemic traits. Next, we employed colocalization with glycemic traits to infer potential mechanisms for causal variants in T2D. For example, fasting glucose and hemoglobin A1C were colocalized with T2D at sentinel signal rs10228066 with a posterior probability of 0.819 (Supplemental Figure 6B). Variant rs10228796, which connects specifically in beta cells to the promoter of diacylglycerol kinase (DGKB), (Figure 6B, Supplemental Figure 5E), explained 33.6% of this colocalization by variant to gene-mapping (Supplemental Figure 6C), implicating the beta-cell specific chromatin connection rs10228796-DGKB in genetically contributing to the elevated levels of fasting glucose and hemoglobin A1C observed in people with T2D. Mechanistically, the inhibition of diacylglycerol kinase reduces beta cell insulin secretion and leads to disruption of peripheral glucose metabolism (Kurohane Kaneko et al., 2013). In addition, defective glucose homeostasis increases synthesis of diacylglycerols which leads to lipotoxicity of the beta cells, resulting in beta cell dysfunction in T2D (Briaud et al., 2001). This example of prioritizing the rs10228796-DGKB interaction by both multi-trait variant-to-gene-mapping and genetic colocalization data demonstrates that our pancreatic cell chromatin architecture-guided variant-to-gene mapping not only sheds light on the molecular mechanistic effect of causal variants on associations among genetic-relevant traits, but also infers the specific cell-type context for further functional study on the relationship between these traits.
DISCUSSION
T2D is a major health concern and an immense economic burden on our health care system. Due to the sheer prevalence of type 2 diabetes, this disease has received widespread attention by human geneticists and has often led the way in how to best tackle the discovery of loci, and has thus been the focus of more GWAS analyses than any other disorder studied to date. The GWAS approach has identified hundreds of loci that are associated with T2D disease risk with high statistical significance that have been reproduced over multiple patient cohorts. Unfortunately, given the complexity of gene regulation in humans, in many cases the effector transcript and cell type of action of a given locus are uncertain. Of note, the GWAS approach was never designed with the explicit intention of interrogating the actual underlying causal variant conferring increased risk for disease, but only to ‘tag’ the approximate location of a disease variant typically down to a few hundreds of kilobases (Altshuler et al., 2008; Manolio et al., 2008; McCarthy et al., 2008). Thus, for almost all signals, the effector transcript and the cell type(s) of action of the relevant gene are unknown. However, the identification of the effector transcript and site of action is needed in order for us to understand better the etiology of T2D and to identify novel drug targets.
In this study, we addressed the key issues of causality at key T2D GWAS-implicated loci in order to identify ‘culprit’ genes. We tackled this challenge through the integration of GWAS findings with scATAC-seq, scRNA-seq and high-resolution Hi-C analyses from sorted key cell types of the human pancreas, allowing us to map variant-to-gene-by-cell type relationships for 900 genes (Supplemental Table 12), thus delivering critical information required for testing and evaluating gene function in individual pancreatic cell types. This complements the previously variant-to-gene mapping work by combining these state-of-the-art techniques to drill down to cell-type specific causal effector genes within pancreas for the key loci established in T2D GWAS efforts. This in turn offers the potential to uncover corresponding diagnostically actionable variants harbored within these genes to better serve precision medicine in the T2D field going forward.
Compared to the discoveries reported by Chiou and colleagues (Chiou et al., 2021), there are discrepancies in the annotation of risk variants. A likely reason for the divergent results is the different approaches employed: Chiou and colleagues utilized co-accessibility analyses of single-cell ATAC-seq data, while we employed physical contact maps obtained by HiC analysis.
With the advantage of chromatin conformation maps established in sorted pancreatic cell types, our study also revealed that not all T2D loci are likely to act within the insulin-producing beta cell. For example, we discovered that the Wolfram syndrome 1 (WSF1) gene, previously shown to be critical for beta cell survival in mice (Ishihara et al., 2004; Riggs et al., 2005), is likely to function additionally in glucagon-producing alpha cells in humans (Figure 6). While further experimental validation is required, this finding suggests that caution is warranted when inferring phenotypes of null alleles in mice to those of weak enhancer mutations that most T2D risk variants represent. Perhaps even more surprising is our discovery of that the T2D risk locus tagged by rs2268382 connects to CPA4, encoding Carboxypeptidase 4 (CPA4) specifically in acinar cells, suggesting the exocrine pancreas as the likely tissue of action for this T2D risk locus. In support of this finding, exocrine pancreas insufficiency is frequently associated with diabetes and has been suggested as a critical factor in the development of the disease (Hardt et al., 2008). In sum, our integration of GWAS findings with cell type-specific high-resolution Hi-C, single cell ATAC-seq and transcriptome data has allowed for the identification of hundreds of likely variant-to-gene pairs and will serve as a crucial resource for the diabetes research community.
Limitations of Study:
While we were able to determine the 3D chromatin architecture of three major pancreatic cell types, that is alpha, beta, and acinar cells, it is of course possible and even likely that additional T2D GWAS effector transcripts are active in other pancreatic cell types such as ductal cells or endothelial cells that could not be profiled here due to the large numbers of cells required for the Hi-C analysis. Thus, for instance, the GWAS T2D risk gene HHEX was shown to function in the somatostatin-producing delta cells which we could not profile (Zhang et al., 2014). In addition, we analyzed only organs from donors without diabetes, and it would be of interest to determine if 3D chromatin architecture is altered in people with T2D. In addition, it is possible that chromatin organization differs with developmental time and that certain variants affect gene function during fetal or neonatal life. To address this possibility, it will be desirable to analyze chromatin state by cell type during human development. Finally, it would be desirable if the chromatin contacts identified here by Hi-C could be validated in the future using imaging-based methodologies.
STAR METHODS
RESOURCE AVILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Klaus H. Kaestner (kaestner@pennmedicine.upenn.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Single cell RNAseq and scATACseq data can be obtained from PancDB (https://hpap.pmacs.upenn.edu). HiC data have been deposited GEO under GSE188311. Raw sequencing data will be made available upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human pancreatic islets
Pancreatic islets were procured by the HPAP consortium under the Human Islet Research Network (https://hirnetwork.org/) with approval from the University of Florida Institutional Review Board (IRB # 201600029) and the United Network for Organ Sharing (UNOS). A legal representative for each donor provided informed consent prior to organ retrieval. Organs were recovered and processed as previously described (Campbell-Thompson et al., 2012). Supplemental Table 1 summarizes donor information. Pancreatic islets were cultured for 24 to 48 hours before dissociation into single cells as described (Ackermann et al., 2016). Total dissociated cells were subjected to FACS sorting as reported previously (Dorrell et al., 2011). Pancreatic islets were assessed for function by the Human Islet Phenotyping Program for HPAP (https://iidp.coh.org/hpap/entry/HPAP-XXX) as described (Brissova et al., 2019).
METHOD DETAILS
Hi-C library preparation
Hi-C library preparation on FACS-sorted pancreatic cells was performed using the Arima-HiC kit (Arima Genomics Inc; cat# A410030), according to the manufacturer’s protocols. Briefly, cells were crosslinked using formaldehyde. Crosslinked cells were then subject to the Arima-HiC protocol, which utilizes multiple restriction enzymes to digest chromatin. Arima-HiC sequencing libraries were prepared by first shearing purified proximally-ligated DNA and then size-selecting 200–600 bp DNA fragments using AmpureXP beads (Beckman Coulter; cat# A63882). The size-selected fragments were then enriched using Enrichment Beads (provided in the Arima-HiC kit), and then converted into Illumina-compatible sequencing libraries with the Swift Accel-NGS 2S Plus DNA Library Kit (Swift, 21024) and Swift 2S Indexing Kit (Swift, 26148). The purified, PCR-amplified DNA underwent standard QC (qPCR, Bioanalyzer, and KAPA Library Quantification [Roche, KK4824]) and was sequenced with unique single indexes on the Illumina NovaSeq 6000 Sequencing System at 2×101 base pairs.
scRNA-seq data generation and analysis
The Single Cell 3’ Reagent Kit v2 or v3 was used for generating scRNA-seq data. 3,000 cells were targeted for recovery per donor. All libraries were validated for quality and size distribution using a BioAnalyzer 2100 (Agilent) and quantified using Kapa (Illumina). For samples prepared using ‘The Single Cell 3’ Reagent Kit v2’, the following chemistry was performed on an Illumina HiSeq4000: Read 1: 26 cycles, i7 Index: 8 cycles, i5 index: 0 cycles, and Read 2: 98 cycles. For samples prepared using ‘The Single Cell 3’ Reagent Kit v3’, the following chemistry was performed on an Illumina HiSeq 4000: Read 1: 28 cycles, i7 Index: 8 cycles, i5 index: 0 cycles, and Read 2: 91 cycles. Raw sequencing reads were aligned to the hg19 reference genome and quantified using CellRanger v3.1.0 (http://10xgenomics.com). Seurat R package v3.0 (Stuart et al., 2019) was used for data quality control, preprocessing and dimensional reduction analysis. After gene-cell data matrix generation for different donors, matrices were merged and poor-quality cells with < 200 expressed genes and mitochondrial gene percentages > 10% were excluded. Expression values depicted were generated using Seurat NormalizeData with the default parameters (normalization.factor = logNormalize and scale factor=10,000). These values correspond to log(10,000 × feature readcount/cell total readcount) + 1) for each cell.
scATAC-seq data generation and analysis
Handpicked islets (approximately 5,000 IEQs) were transferred into a 15 mL conical tube and 10 mL of 1xPBS w/o Ca2+, Mg2+ (Rockland, MB-008) added. Islets were centrifuged for 2 min at RT, 180 × g and the supernatant removed. Islets were then incubated with 1 mL of warm (37 °C) 0.05% Trypsin (Invitrogen, 25300054) at 37 °C for 9 min with tituration with a 1 ml pipette at 0, 2, 4 and 7 min. The digestion was stopped by addition of 1 mL of 100% FBS (Hyclone, SH3091003) and dissociated islets passed through a BD FACs tube with strainer top (Corning 352235). After rinsing with 1 mL of 100% FBS and transfer to a 15 mL conical cells were centrifuged for 4 min at 400 × g. Cell pellets were resuspended in 1 ml PBS with 10% FBS and centrifuged for 4 min, 400 × g two times before the cells were counted after resuspension in 0.04% BSA in PBS. 5,000 nuclei were processed for scATACseq using Chromium Single Cell ATAC Reagent Kits and sequenced on an Illumina Novaseq 6000 instrument.
Raw fastq files were aligned to the hg19 reference genome and quantified using CellRanger atac v1.2.0 (http://10xgenomics.com). The resulting sorted BAM files were hierarchically structured into hdf5 snapfiles using snaptools v 1.0.0. Quality control was conducted using SnapATAC v.1.0.0 (Fang et al., 2021) by only keeping the cells with less than 3~6 logUMI, 10% ~ 60%promoter ratio. The cells with more than 10% mitochondrial ratio and less than 1000 mapped fragments were also removed. Then, we binarized fragments into 1-kb bins and removed bins on mitochondria, chromosome X, chromosome Y and ambiguous chromosomes, overlapping with the ENCODE blacklist as well as top 5% bins that overlap with invariant features such as the housekeeping gene promoters. Cells with a bin coverage less than 1,000 were excluded.
‘Diffusion map’ was applied as a dimension reduction method using function runDiffusionMaps on 50 eigenvectors and the first 14 eigenvectors were then selected for further batch effect correction using Harmony v.1.0.0 (Korsunsky et al., 2019) and a k-nearest neighbor graph generation using function runKNN with k = 15. Clustering was performed using the igraph algorithm and UMAP embeddings were generated to visualize the data. We manually assigned each identified cluster to pancreatic cell types based on promoter accessibility of known marker genes.
Fragments originating from cells belonging to the same clusters were pooled using snaptools, and pseudo bulk open chromatin regions were called for each cell type using MACS2 v2.1.2 (Zhang et al., 2008) with the parameters --nomodel --shift 100 --ext 200 --qval 5e–2 -B --SPMR. Differential accessible regions (DARs) across different cell types were next identified using the findDAR function in the SnapATAC package with exactTest and the parameter cluster.neg.method as”knn” and bcv as 0.4. DARs were determined by an FDR < 0.05 and absolute log fold change > 1. Cell-type specific open chromatin regions were DARs that are only significant in that given cell type but not others.
Hi-C data analysis
Paired-end reads from two replicates were pre-processed using the HICUP pipeline v0.7.4 (Wingett et al., 2015), with bowtie as aligner and hg19 as the reference genome. The alignment .bam file were parsed to .pairs format using pairtools v 0.3.0 and pairix v0.3.7, and eventually converted to pre-binned Hi-C matrix in .cool format by cooler v 0.8.10 with multiple resolutions (500bp, 1kbp, 2kbp, 2.5kbp, 4kbp, 5kbp, 10kbp, 25kbp, 40kbp, 50kbp, 100kbp, 250kbp, 500kbp, 1Mbp and 2.5Mbp) and normalized with ICE method (Imakaev et al., 2012). Replicate similarity was determined by HiCRep v1.12.2 (Yang et al., 2017) at 10K resolution.
For each sample, eigenvectors were determined from an ICE balanced Hi-C matrix with 40kb resolution using cooltools v0.3.2 and first principal components were used to determine A/B compartments with GC% of genome region as reference track to determine the sign. Hierarchical TADs were determined by directional index using software HiTAD v.0.4.2 (Wang et al., 2017) on bi-replicated 10kb binned matrix. Finally, for each cell type, significant intra-chromosomal interaction loops were determined under multiple resolutions (1kb, 2kb and 4kb) using the Hi-C loop callers mustache v1.0.1 (q-value < 0.1) (Roayaei Ardakany et al., 2020) and Fit-Hi-C2 (Kaul et al., 2020) v2.0.7 (FDR < 1e–6) on merged replicates matrix. The called loops were merged between both callers to represent cell type loops at each resolution. The consensus chromatin loops within resolution were identified by combining all three cell types. These sets of loops were used as consensus for quantitative differential analysis explained below. The final consensus interaction loops for visualization were collected by merging loops from all the resolutions with preference to keep the highest resolution.
Quantitative loop differential analysis across cell types was performed on fast lasso normalized interaction frequency (IF) by multiCompareHiC v1.8.0 (Stansfield et al., 2018) for each chromosome at resolution 1kb, 2kb and 4kb independently. The contacts with zero interaction frequency (IF) among more than 80% of the samples and average IF less than 5 were excluded from differential analysis. The QLF test based on a generalized linear model was performed in cell type-pairwise comparisons, and p-values were corrected with FDR. The final differential loops were identified by overlapping differential IF contacts with consensus interaction loops.
Cell-specificity score
We derived the cell type specificity weight of scRNA-seq and scATAC-seq data using CELLEX (CELL-type EXpression-specificity) (Timshel et al., 2020) on UMI counts for each gene and consensus pseudo-bulk open chromatin regions across acinar, alpha and beta cells. A gene or open chromatin region with a specificity estimate of 0 would indicate no expression in that cell type while a specificity estimate of 1 would indicate exclusive cell type-specific expression in that given cell type. The loop interaction frequency specificity was estimated by z-score across all Hi-C samples and further averaged among replicates within the same cell type. Thus, a loop with a negative z-score would indicate that it is less likely to be formed, while a positive z-score indicates loops that are more likely to formed in the given cell type.
Transcription factor motif enrichment and cell-specific transcription factor identification
ChromVAR v1.5.0 (Schep et al., 2017) was applied to scATAC-seq to estimate transcription factor motif enrichment across pancreatic cell types. First, we constructed consensus open chromatin regions by merging pseudo-bulk open chromatin regions called for each cell type and built a cell sparse binary matrix on consensus open chromatin regions as the input for chromVAR. GC bias was corrected on BSgenome.Hsapiens.UCSC.hg19 using the addGCBias function, and the cells were filtered with a minimum fragment count of 1,500 per library and a minimum fraction of samples within peaks of 0.15. Next, we calculated the bias-corrected deviation z-scores for 746 transcription factor motifs from the nonredundant JASPAR 2020 CORE vertebrate set (Fornes et al., 2020). Motifs with differential accessibility among acinar, alpha and beta cells were identified for a given cell type compared to the other two cell types with the differentialDeviations function using a one-side t-test. Cell-specific TF motifs were determined by applying an FDR < 0.01 to the target cell type while FDR > 0.01 to the control cell types. 438 cell-specific TFs were identified by further filtering with gene expression > 0 from scRNA-seq in at least one cell type among acinar, alpha, and beta cells. To determine cell-specific TF-regulated genes, we mapped TF binding sites to the differentially accessible regions that either contact gene promoters through Hi-C loops or reside at gene promoters (−1,500bp ~ 500bp around TSS) in at least one cell type among acinar, alpha and beta cells. We performed Prerank gene set enrichment analysis with the R package fgsea (Korotkevich et al., 2021) using the gene expression log2 fold change the between target cell type and the other two cell types as rank.
Bulk RNA-seq generation and analysis
Bulk RNA-seq libraries were constructed as described (Ackermann et al., 2016) and sequenced on an Illumina Novaseq 6000 instrument. The pair-end fastq files were mapped to the genome assembly hg19 by STAR (v2.6.0c) independently for each replicate. GencodeV19 annotation was used for gene feature annotation and the raw read count for gene feature was calculated by htseq-count (v0.6.1) with parameter settings -f bam -r pos -s yes -t exon -m union. The gene features localized on chrM or annotated as rRNAs, small coding RNA, or pseudo genes were removed from the final sample-by-gene read count matrix. Bulk RNA-seq data were compared to scRNA-seq to perform cell deconvolution on each donor and each cell type independently. The function music_prop from R package MuSiC v0.1.1(Wang et al., 2019) was used to estimate the proportion of cell types from scRNA-seq in bulk RNA-seq on sorted cells. The set of selected marker features that was used for the deconvolution can be found in Supplemental Figure 2B.
Variant to gene mapping by Hi-C and single cell data
Sentinel SNPs were obtained from the original GWAS publications (Dashti et al., 2019; Jones et al., 2019; Mahajan et al., 2018; Rashkin et al., 2020; Roselli et al., 2018) (Supplemental Table 9). Proxies within high LD (r2 >=0.8) with sentinel SNPs were determined using the LDlink (Machiela and Chanock, 2015) API R package LDlinkR with the European population genotype from the 1000 Genome Project (1000G) phase 3 as reference. The proxies were filtered by overlapping with the open chromatin regions from our pseudo-bulk scATAC-seq and annotated to genes either by contacting with gene promoters through Hi-C loops or by residing at gene promoters (−1,500bp to 500bp around TSS) in the corresponding cell type.
Variant to gene mapping was also performed using the Activity-by-Contact (ABC) model (Fulco et al., 2019) to link SNP-containing putative enhancers to target genes with the cell type-specific Hi-C matrix. ATAC-seq signals were obtained by pooling fragment mapping of scATAC-seq in acinar, alpha and beta cells, respectively, while the H3K27ac ChIP-seq signals were derived from publicly available resources for acinar (GEO:GSE79468) (Arda et al., 2018), alpha and beta cells (GEO:GSE140500) (Alvarez-Dominguez et al., 2020), and pre-processed using the Encode ChIP-seq pipeline (https://github.com/ENCODE-DCC/chip-seq-pipeline2). The candidate enhancer regions were identified and quantified by mapping scATAC-seq and H3K27ac ChIP-seq signals to pseudo bulk open chromatin regions merged from acinar, alpha and beta cells, and quantile normalized against the K562 cell line epigenetic signal provided by the software. Protein-coding and long non-coding RNA gene promoters were defined by Gencode V19 annotation with ~500bp around TSS. The ABC scores were calculated for each candidate region-to-transcript pair by integrating cell type-specific Hi-C contact frequencies at 4kb resolution from our acinar, alpha and beta cell data. Any putative enhancer-to-transcript pair with an ABC score above 0.02 was identified as a significant interaction and used to overlap with T2D risk proxies as identified above.
Disruptive effect of T2D putative causal variants on JASPAR 2020 transcription factor binding sites was estimated using motifbreakR (Coetzee et al., 2015) with relative entropy algorithm (method = “ic”). The results were filtered with PWM match p-value smaller than 1e–3 and TF gene expression > 0 in corresponding cell types.
Partitioned heritability LD score regression enrichment analysis
Partitioned heritability LD Score Regression (v1.0.0) (Finucane et al., 2015) was used to identify heritability enrichment with GWAS summary statistics and open chromatin regions annotated to genes. The baseline analysis was performed using LDSCORE data (https://data.broadinstitute.org/alkesgroup/LDSCORE) with LD scores, regression weights, and allele frequencies from the 1000G phase 1 data. The summary statistics were obtained from GWAS publications (Chiou et al., 2021; Dashti et al., 2019; Mahajan et al., 2018; Rashkin et al., 2020; Roselli et al., 2018) and harmonized. The Partitioned LD score regression annotations were generated using the coordinates of open chromatin regions which either contact gene promoters through Hi-C loops or reside at gene promoters for each cell type. Finally, partitioned LD scores were compared to baseline LD scores to measure enrichment fold change and enrichment p-values, which were adjusted with FDR across all comparisons.
Multi-trait colocalization analysis
Sentinel signals in T2D GWAS were obtained from putative causal variant to gene pairs shared between T2D and other relevant diabetes traits and used as input SNPs to identify multi-trait colocalized association signals. Summary statistics on different traits were harmonized to the 1000 genome reference data set and the missing variants were imputed using scripts from https://github.com/hakyimlab/summary-gwas-imputation. The effect size and standard deviation for imputed variants were calculated from estimated z-scores, reference allele frequencies and effective sample sizes (Foley et al., 2021). The colocalization analysis was performed for 1Mbp regions around each input sentinel signal using HyPrColoc (Zhu et al., 2016), and the posterior probability of colocalization at putative causal variants identified by variant-to-gene mapping was approximated by setting snpscores as TRUE in the hyprcoloc function.
QUANTIFICATION AND STATISTICAL ANALYSIS
Details on quantification and statistical analysis are contained in the methods outlines above and the figure legends.
Supplementary Material
Table S12: Effector genes implicated by our variant to gene to cell type mapping approach. Related to Figure 6.
Table S13: Putative proxy variants linked to additional diabetes related traits. Related to Figures 5 and 6.
Table S9: Sentinel and proxy SNPs for T2D and other traits analyzed and the source publications. Related to Figure 5.
Table S7: Cell Type Specific Transcription Factor Motifs. Related to Figure 4.
Table S6: Transcription Factor Motifs with Differential Accessibility among Human Pancreas Cell Types. Related to Figure 4.
Table S5: Location of consensus loops and interaction strength by cell type. Related to Figure 3.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Organ Donor | ||
| HPAP040 | Human Pancreas Analysis Program | RRID:SAMN19776471 |
| HPAP045 | Human Pancreas Analysis Program | RRID:SAMN19776476 |
| HPAP054 | Human Pancreas Analysis Program | RRID:SAMN19776484 |
| HPAP066 | Human Pancreas Analysis Program | RRID:SAMN19842595 |
| HPAP072 | Human Pancreas Analysis Program | RRID:SAMN19842601 |
| Arima HiC Kit | Arima Genomics Inc. | Cat# A410030 |
| AmpureXP beads | Beckman Coulter | Cat# A63882 |
| Kapa Library Quantification | Roche Inc. | Cat# KK4824 |
| Trypsin | Invitrogen Inc. | Cat# 25300054 |
| Fetal Bovine Serum | Hyclone | Cat# SH3091003 |
| FACS tube with strainer top | Corning | Cat# 352235 |
HIGHLIGHTS.
Established 3D chromatin maps for human pancreatic α, β, and acinar cells
Mapped causal-variant-to-target-gene pairs at 194 type 2 diabetes GWAS loci
Identified pancreatic α and acinar cells as likely site of action for T2D effectors
ACKNOWLEDGEMENTS
We acknowledge and appreciate the efforts of nPOD (https://www.jdrfnpod.org/) for its role in obtaining donor organs utilized in these studies, and Dr. Marcela Brissova and her team at Vanderbilt University for assessment of islet function, supported through NIH grant U01- DK123716. We acknowledge the help of the Functional Genomics Core of the Penn Diabetes Research Center (P30-DK19525). This work was supported by NIH grant 5U01-DK123594 and 5UM-1DK126194 to KHK.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES:
- Ackermann AM, Wang Z, Schug J, Naji A, and Kaestner KH (2016). Integration of ATAC-seq and RNA-seq identifies human alpha cell and beta cell signature genes. Mol Metab 5, 233–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altshuler D, Daly MJ, and Lander ES (2008). Genetic mapping in human disease. Science 322, 881–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Dominguez JR, Donaghey J, Rasouli N, Kenty JHR, Helman A, Charlton J, Straubhaar JR, Meissner A, and Melton DA (2020). Circadian Entrainment Triggers Maturation of Human In Vitro Islets. Cell Stem Cell 26, 108–122 e110. [DOI] [PubMed] [Google Scholar]
- Arda HE, Tsai J, Rosli YR, Giresi P, Bottino R, Greenleaf WJ, Chang HY, and Kim SK (2018). A Chromatin Basis for Cell Lineage and Disease Risk in the Human Pancreas. Cell Syst 7, 310–322 e314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beumer J, Puschhof J, Bauza-Martinez J, Martinez-Silgado A, Elmentaite R, James KR, Ross A, Hendriks D, Artegiani B, Busslinger GA, et al. (2020). High-Resolution mRNA and Secretome Atlas of Human Enteroendocrine Cells. Cell 182, 1062–1064. [DOI] [PubMed] [Google Scholar]
- Bramswig NC, and Kaestner KH (2011). Transcriptional regulation of alpha-cell differentiation. Diabetes Obes Metab 13 Suppl 1, 13–20. [DOI] [PubMed] [Google Scholar]
- Briaud I, Harmon JS, Kelpe CL, Segu VB, and Poitout V (2001). Lipotoxicity of the pancreatic beta-cell is associated with glucose-dependent esterification of fatty acids into neutral lipids. Diabetes 50, 315–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brissova M, Niland JC, Cravens J, Olack B, Sowinski J, and Evans-Molina C (2019). The Integrated Islet Distribution Program Answers the Call for Improved Human Islet Phenotyping and Reporting of Human Islet Characteristics in Research Articles. Diabetes 68, 1363–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell-Thompson M, Wasserfall C, Kaddis J, Albanese-O’Neill A, Staeva T, Nierras C, Moraski J, Rowe P, Gianani R, Eisenbarth G, et al. (2012). Network for Pancreatic Organ Donors with Diabetes (nPOD): developing a tissue biobank for type 1 diabetes. Diabetes Metab Res Rev 28, 608–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Spracklen CN, Marenne G, Varshney A, Corbin LJ, Luan J, Willems SM, Wu Y, Zhang X, Horikoshi M, et al. (2021). The trans-ancestral genomic architecture of glycemic traits. Nat Genet 53, 840–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiou J, Geusz RJ, Okino ML, Han JY, Miller M, Melton R, Beebe E, Benaglio P, Huang S, Korgaonkar K, et al. (2021). Interpreting type 1 diabetes risk with genetics and single-cell epigenomics. Nature 594, 398–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton DG (2009). Prediction and interaction in complex disease genetics: experience in type 1 diabetes. PLoS Genet 5, e1000540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coetzee SG, Coetzee GA, and Hazelett DJ (2015). motifbreakR: an R/Bioconductor package for predicting variant effects at transcription factor binding sites. Bioinformatics 31, 3847–3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dashti HS, Jones SE, Wood AR, Lane JM, van Hees VT, Wang H, Rhodes JA, Song Y, Patel K, Anderson SG, et al. (2019). Genome-wide association study identifies genetic loci for self-reported habitual sleep duration supported by accelerometer-derived estimates. Nat Commun 10, 1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Jung I, Selvaraj S, Shen Y, Antosiewicz-Bourget JE, Lee AY, Ye Z, Kim A, Rajagopal N, Xie W, et al. (2015). Chromatin architecture reorganization during stem cell differentiation. Nature 518, 331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorrell C, Grompe MT, Pan FC, Zhong Y, Canaday PS, Shultz LD, Greiner DL, Wright CV, Streeter PR, and Grompe M (2011). Isolation of mouse pancreatic alpha, beta, duct and acinar populations with cell surface markers. Mol Cell Endocrinol 339, 144–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang R, Preissl S, Li Y, Hou X, Lucero J, Wang X, Motamedi A, Shiau AK, Zhou X, Xie F, et al. (2021). Comprehensive analysis of single cell ATAC-seq data with SnapATAC. Nat Commun 12, 1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finucane HK, Bulik-Sullivan B, Gusev A, Trynka G, Reshef Y, Loh PR, Anttila V, Xu H, Zang C, Farh K, et al. (2015). Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat Genet 47, 1228–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley CN, Staley JR, Breen PG, Sun BB, Kirk PDW, Burgess S, and Howson JMM (2021). A fast and efficient colocalization algorithm for identifying shared genetic risk factors across multiple traits. Nat Commun 12, 764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forgettta V, Jiang L, Vulpescu NA, Hogan MS, Chen S, Morris JA, Grinek S, Brenner C, Jang D-K, Hoang Q, et al. (2020). An Effector Index to Predict Causal Genes at GWAS Loci. BioRxiv. [DOI] [PubMed] [Google Scholar]
- Fornes O, Castro-Mondragon JA, Khan A, van der Lee R, Zhang X, Richmond PA, Modi BP, Correard S, Gheorghe M, Baranasic D, et al. (2020). JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res 48, D87–D92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin JP, and Hansen KD (2015). Reconstructing A/B compartments as revealed by Hi-C using long-range correlations in epigenetic data. Genome Biol 16, 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulco CP, Nasser J, Jones TR, Munson G, Bergman DT, Subramanian V, Grossman SR, Anyoha R, Doughty BR, Patwardhan TA, et al. (2019). Activity-by-contact model of enhancer-promoter regulation from thousands of CRISPR perturbations. Nat Genet 51, 1664–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao N, Le Lay J, Qin W, Doliba N, Schug J, Fox AJ, Smirnova O, Matschinsky FM, and Kaestner KH (2010). Foxa1 and Foxa2 maintain the metabolic and secretory features of the mature beta-cell. Mol Endocrinol 24, 1594–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao N, LeLay J, Vatamaniuk MZ, Rieck S, Friedman JR, and Kaestner KH (2008). Dynamic regulation of Pdx1 enhancers by Foxa1 and Foxa2 is essential for pancreas development. Genes Dev 22, 3435–3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao N, White P, Doliba N, Golson ML, Matschinsky FM, and Kaestner KH (2007). Foxa2 controls vesicle docking and insulin secretion in mature Beta cells. Cell Metab 6, 267–279. [DOI] [PubMed] [Google Scholar]
- Greenwald WW, Chiou J, Yan J, Qiu Y, Dai N, Wang A, Nariai N, Aylward A, Han JY, Kadakia N, et al. (2019). Pancreatic islet chromatin accessibility and conformation reveals distal enhancer networks of type 2 diabetes risk. Nat Commun 10, 2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardt PD, Brendel MD, Kloer HU, and Bretzel RG (2008). Is pancreatic diabetes (type 3c diabetes) underdiagnosed and misdiagnosed? Diabetes Care 31 Suppl 2, S165–169. [DOI] [PubMed] [Google Scholar]
- Imakaev M, Fudenberg G, McCord RP, Naumova N, Goloborodko A, Lajoie BR, Dekker J, and Mirny LA (2012). Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat Methods 9, 999–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara H, Takeda S, Tamura A, Takahashi R, Yamaguchi S, Takei D, Yamada T, Inoue H, Soga H, Katagiri H, et al. (2004). Disruption of the WFS1 gene in mice causes progressive beta-cell loss and impaired stimulus-secretion coupling in insulin secretion. Hum Mol Genet 13, 1159–1170. [DOI] [PubMed] [Google Scholar]
- Javierre BM, Burren OS, Wilder SP, Kreuzhuber R, Hill SM, Sewitz S, Cairns J, Wingett SW, Varnai C, Thiecke MJ, et al. (2016). Lineage-Specific Genome Architecture Links Enhancers and Non-coding Disease Variants to Target Gene Promoters. Cell 167, 1369–1384 e1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson BB, Fjeld K, El Jellas K, Gravdal A, Dalva M, Tjora E, Raeder H, Kulkarni RN, Johansson S, Njolstad PR, et al. (2018). The role of the carboxyl ester lipase (CEL) gene in pancreatic disease. Pancreatology 18, 12–19. [DOI] [PubMed] [Google Scholar]
- Jones SE, Lane JM, Wood AR, van Hees VT, Tyrrell J, Beaumont RN, Jeffries AR, Dashti HS, Hillsdon M, Ruth KS, et al. (2019). Genome-wide association analyses of chronotype in 697,828 individuals provides insights into circadian rhythms. Nat Commun 10, 343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul A, Bhattacharyya S, and Ay F (2020). Identifying statistically significant chromatin contacts from Hi-C data with FitHiC2. Nat Protoc 15, 991–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korotkevich G, Sukhov V, Budin N, Shpak B, Artyomov MN, and Sergushichev A (2021). Fast gene set enrichment analysis. BioRxiv. [Google Scholar]
- Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh PR, and Raychaudhuri S (2019). Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods 16, 1289–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kropp PA, Zhu X, and Gannon M (2019). Regulation of the Pancreatic Exocrine Differentiation Program and Morphogenesis by Onecut 1/Hnf6. Cell Mol Gastroenterol Hepatol 7, 841–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurohane Kaneko Y, Kobayashi Y, Motoki K, Nakata K, Miyagawa S, Yamamoto M, Hayashi D, Shirai Y, Sakane F, and Ishikawa T (2013). Depression of type I diacylglycerol kinases in pancreatic beta-cells from male mice results in impaired insulin secretion. Endocrinology 154, 4089–4098. [DOI] [PubMed] [Google Scholar]
- Lasconi C, Pahl MC, Pippin JA, Su C, Johnson ME, Chesi A, Boehm K, Manduchi E, Ou K, Golson ML, et al. (2022). Variant-to-gene-mapping analyses reveal a role for pancreatic islet cells in conferring genetic susceptibility to sleep-related traits. Sleep. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawlor N, Marquez EJ, Orchard P, Narisu N, Shamim MS, Thibodeau A, Varshney A, Kursawe R, Erdos MR, Kanke M, et al. (2019). Multiomic Profiling Identifies cis-Regulatory Networks Underlying Human Pancreatic beta Cell Identity and Function. Cell Rep 26, 788–801 e786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machiela MJ, and Chanock SJ (2015). LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics 31, 3555–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan A, Taliun D, Thurner M, Robertson NR, Torres JM, Rayner NW, Payne AJ, Steinthorsdottir V, Scott RA, Grarup N, et al. (2018). Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat Genet 50, 1505–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio TA, Brooks LD, and Collins FS (2008). A HapMap harvest of insights into the genetics of common disease. J Clin Invest 118, 1590–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy MI, Abecasis GR, Cardon LR, Goldstein DB, Little J, Ioannidis JP, and Hirschhorn JN (2008). Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet 9, 356–369. [DOI] [PubMed] [Google Scholar]
- Miguel-Escalada I, Bonas-Guarch S, Cebola I, Ponsa-Cobas J, Mendieta-Esteban J, Atla G, Javierre BM, Rolando DMY, Farabella I, Morgan CC, et al. (2019). Human pancreatic islet three-dimensional chromatin architecture provides insights into the genetics of type 2 diabetes. Nat Genet 51, 1137–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasser J, Bergman DT, Fulco CP, Guckelberger P, Doughty BR, Patwardhan TA, Jones TR, Nguyen TH, Ulirsch JC, Lekschas F, et al. (2021). Genome-wide enhancer maps link risk variants to disease genes. Nature 593, 238–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashkin SR, Graff RE, Kachuri L, Thai KK, Alexeeff SE, Blatchins MA, Cavazos TB, Corley DA, Emami NC, Hoffman JD, et al. (2020). Pan-cancer study detects genetic risk variants and shared genetic basis in two large cohorts. Nat Commun 11, 4423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reizel Y, Morgan A, Gao L, Schug J, Mukherjee S, Garcia MF, Donahue G, Baur JA, Zaret KS, and Kaestner KH (2021). FoxA-dependent demethylation of DNA initiates epigenetic memory of cellular identity. Dev Cell 56, 602–612 e604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich SS (1990). Mapping genes in diabetes. Genetic epidemiological perspective. Diabetes 39, 1315–1319. [DOI] [PubMed] [Google Scholar]
- Riggs AC, Bernal-Mizrachi E, Ohsugi M, Wasson J, Fatrai S, Welling C, Murray J, Schmidt RE, Herrera PL, and Permutt MA (2005). Mice conditionally lacking the Wolfram gene in pancreatic islet beta cells exhibit diabetes as a result of enhanced endoplasmic reticulum stress and apoptosis. Diabetologia 48, 2313–2321. [DOI] [PubMed] [Google Scholar]
- Roayaei Ardakany A, Gezer HT, Lonardi S, and Ay F (2020). Mustache: multi-scale detection of chromatin loops from Hi-C and Micro-C maps using scale-space representation. Genome Biol 21, 256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roselli C, Chaffin MD, Weng LC, Aeschbacher S, Ahlberg G, Albert CM, Almgren P, Alonso A, Anderson CD, Aragam KG, et al. (2018). Multi-ethnic genome-wide association study for atrial fibrillation. Nat Genet 50, 1225–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeedi P, Petersohn I, Salpea P, Malanda B, Karuranga S, Unwin N, Colagiuri S, Guariguata L, Motala AA, Ogurtsova K, et al. (2019). Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9(th) edition. Diabetes Res Clin Pract 157, 107843. [DOI] [PubMed] [Google Scholar]
- Sandhu MS, Weedon MN, Fawcett KA, Wasson J, Debenham SL, Daly A, Lango H, Frayling TM, Neumann RJ, Sherva R, et al. (2007). Common variants in WFS1 confer risk of type 2 diabetes. Nat Genet 39, 951–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schep AN, Wu B, Buenrostro JD, and Greenleaf WJ (2017). chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat Methods 14, 975–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stansfield JC, Cresswell KG, Vladimirov VI, and Dozmorov MG (2018). HiCcompare: an R-package for joint normalization and comparison of HI-C datasets. BMC Bioinformatics 19, 279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 e1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su C, Argenziano M, Lu S, Pippin JA, Pahl MC, Leonard ME, Cousminer DL, Johnson ME, Lasconi C, Wells AD, et al. (2021). 3D promoter architecture re-organization during iPSC-derived neuronal cell differentiation implicates target genes for neurodevelopmental disorders. Prog Neurobiol 201, 102000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timshel PN, Thompson JJ, and Pers TH (2020). Genetic mapping of etiologic brain cell types for obesity. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres JM, Abdalla M, Payne A, Fernandez-Tajes J, Thurner M, Nylander V, Gloyn AL, Mahajan A, and McCarthy MI (2020). A Multi-omic Integrative Scheme Characterizes Tissues of Action at Loci Associated with Type 2 Diabetes. Am J Hum Genet 107, 1011–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meulen T, and Huising MO (2015). Role of transcription factors in the transdifferentiation of pancreatic islet cells. J Mol Endocrinol 54, R103–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss TC, and Hager GL (2014). Dynamic regulation of transcriptional states by chromatin and transcription factors. Nat Rev Genet 15, 69–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vujkovic M, Keaton JM, Lynch JA, Miller DR, Zhou J, Tcheandjieu C, Huffman JE, Assimes TL, Lorenz K, Zhu X, et al. (2020). Discovery of 318 new risk loci for type 2 diabetes and related vascular outcomes among 1.4 million participants in a multi-ancestry meta-analysis. Nat Genet 52, 680–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Park J, Susztak K, Zhang NR, and Li M (2019). Bulk tissue cell type deconvolution with multi-subject single-cell expression reference. Nat Commun 10, 380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XT, Cui W, and Peng C (2017). HiTAD: detecting the structural and functional hierarchies of topologically associating domains from chromatin interactions. Nucleic Acids Res 45, e163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingett S, Ewels P, Furlan-Magaril M, Nagano T, Schoenfelder S, Fraser P, and Andrews S (2015). HiCUP: pipeline for mapping and processing Hi-C data. F1000Res 4, 1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Zhang F, Yardimci GG, Song F, Hardison RC, Noble WS, Yue F, and Li Q (2017). HiCRep: assessing the reproducibility of Hi-C data using a stratum-adjusted correlation coefficient. Genome Res 27, 1939–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Zhang F, Hu H, Bakshi A, Robinson MR, Powell JE, Montgomery GW, Goddard ME, Wray NR, Visscher PM, et al. (2016). Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet 48, 481–487. [DOI] [PubMed] [Google Scholar]
- Zimmet P, Alberti KG, and Shaw J (2001). Global and societal implications of the diabetes epidemic. Nature 414, 782–787. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S12: Effector genes implicated by our variant to gene to cell type mapping approach. Related to Figure 6.
Table S13: Putative proxy variants linked to additional diabetes related traits. Related to Figures 5 and 6.
Table S9: Sentinel and proxy SNPs for T2D and other traits analyzed and the source publications. Related to Figure 5.
Table S7: Cell Type Specific Transcription Factor Motifs. Related to Figure 4.
Table S6: Transcription Factor Motifs with Differential Accessibility among Human Pancreas Cell Types. Related to Figure 4.
Table S5: Location of consensus loops and interaction strength by cell type. Related to Figure 3.
Data Availability Statement
Single cell RNAseq and scATACseq data can be obtained from PancDB (https://hpap.pmacs.upenn.edu). HiC data have been deposited GEO under GSE188311. Raw sequencing data will be made available upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.