

SUMMARY
Natural killer T (NKT) cells operate distinctly different metabolic programming from CD4 T cells, including a strict requirement for glutamine to regulate cell homeostasis. However, the underlying mechanisms remain unknown. Here, we report that at a steady state, NKT cells have higher glutamine levels than CD4 T cells and that NKT cells increase glutaminolysis on activation. Activated NKT cells use glutamine to fuel the tricarboxylic acid cycle and glutathione synthesis. In addition, glutamine-derived nitrogen enables protein glycosylation via the hexosamine biosynthesis pathway (HBP). Each of these branches of glutamine metabolism seems to be critical for NKT cell homeostasis and mitochondrial functions. Glutaminolysis and HBP differentially regulate interleukin-4 (IL-4) and interferon γ (IFNγ) production. Glutamine metabolism appears to be controlled by AMP-activated protein kinase (AMPK)-mammalian target of rapamycin complex 1 (mTORC1) signaling. These findings highlight a distinct metabolic requirement of NKT cells compared with CD4 T cells, which may have therapeutic implications in the treatment of certain nutrient-restricted diseases.
In brief
Kumar et al. report that NKT cells rely on glutamine for their homeostasis and functions. Glutamine does this by supporting mitochondrial functions, maintaining redox balance, and supporting glycosylation processes in NKT cells.
Graphical Abstract

INTRODUCTION
Cellular metabolism plays a significant role in modulating T cell functions. Activated T cells undergo metabolic rewiring to fulfill the demands of clonal expansion, as well as cytokine synthesis and secretion. A recent body of evidence has highlighted the role of cellular metabolism in regulating T cell plasticity. T cells shift glucose metabolism from a more glycolytic phenotype to a more oxidative phenotype after activation, a process known as metabolic reprogramming (Geiger et al., 2016; Pearce and Pearce, 2013; Voss et al., 2021). This metabolic reprogramming is orchestrated by a series of signaling pathways and transcriptional networks (Gubser et al., 2013; Johnson et al., 2018; Shyer et al., 2020). Additionally, the various T cell subsets operate distinct metabolic profiles that are critical for their specific effector functions (Johnson et al., 2018; Klysz et al., 2015).
Invariant natural killer T (NKT) cells are innate-like lymphocytes that recognize glycolipid antigens in the context of the non-classical MHC molecule CD1d, which is present on antigen-presenting cells. NKT cells are selected by cortical thymocytes expressing CD1d and mature through a series of stages (Kovalovsky et al., 2008; Savage et al., 2008). Thymic NKT cells are capable of producing the cytokines interferon γ (IFNγ), interleukin-4 (IL-4), and IL-17 and are thus termed NKT1, NKT2, and NKT17, respectively (Wang and Hogquist, 2018). NKT cells are a vital part of the defense system against infectious diseases (Baron et al., 2002; Crosby and Kronenberg, 2016; Durante-Mangoni et al., 2004) and also play a role in the development of autoimmunity (Beaudoin et al., 2002; Illes et al., 2000) and asthma (Lisbonne et al., 2003). Additionally, NKT cells mediate potent antitumor immune responses and have been utilized in immunotherapy for cancer patients using various immunomodulatory approaches (Cui et al., 1997; Dhodapkar et al., 2003; Metelitsa et al., 2004; Viale et al., 2012).
NKT cells express promyelocytic leukemia zinc finger (PLZF; encoded by Zbtb16), a transcription factor required for NKT cell development and function (Kovalovsky et al., 2008; Kreslavsky et al., 2009; Savage et al., 2011). Several studies have shown that metabolic signals are critical for NKT cell development and function. Mammalian target of rapamycin (mTOR) complex 1 (mTORC1) and complex 2 (mTORC2) integrate various environmental cues to regulate cellular growth, proliferation, and metabolism (Roy et al., 2018; Salmond, 2018). Deletion of either mTORC1 or mTORC2 leads to a block in NKT cell development during which NKT cells accumulate in the early developmental stages (Prevot et al., 2015; Shin et al., 2014; Zhang et al., 2014). Additionally, mTORC1 is a critical regulator of glycolysis and amino acid transport in T cells (Ho et al., 2015; Sena et al., 2013). mTORC1 has been shown to be negatively regulated by AMP-activated protein kinase (AMPK) in T cells (Blagih et al., 2015). AMPK senses cellular energy levels and in turn activates pathways necessary to maintain cellular energy balance. Additionally, loss of the AMPK-interacting adaptor protein folliculin-interacting protein 1 (Fnip1) results in defective NKT cell development (Park et al., 2014).
As NKT cells develop and mature in the thymus, they become more quiescent and display lower metabolic activity in the peripheral organs compared with conventional T cells (Salio et al., 2014). We have shown that resting NKT cells have lower glucose uptake and mitochondrial function compared with conventional T cells, which are regulated by PLZF (Kumar et al., 2019). Furthermore, high environmental levels of lactate are detrimental for NKT cell homeostasis and cytokine production, suggesting that reduced glycolysis is essential for NKT cell maintenance (Kumar et al., 2019). Interestingly, NKT cells preferentially partition glucose into the pentose phosphate pathway (PPP) and contribute less carbon into the glycolysis than CD4 T cells. Recently, lipid synthesis has also emerged as a critical regulator of NKT cell responses (Fu et al., 2020).
In addition to glucose, rapidly proliferating cells require the amino acid glutamine to produce ATP, biosynthetic precursors, and reducing agents (Carr et al., 2010; Johnson et al., 2018). Glutaminolysis refers to the breakdown of glutamine to fuel metabolism. In some proliferating cell types, glutaminolysis can take place in the mitochondria, where glutamine is converted to glutamate by the glutaminase (GLS) enzyme. From here, glutamate can undergo several metabolic fates. For one, glutamate can be deaminated into the tricarboxylic acid (TCA) cycle intermediate α-ketoglutarate (αKG) by either glutamate dehydrogenase (GDH) or aminotransferases. Glutamate can also be transported back into the cytosol and produce glutathione (GSH), a critical mediator of cellular redox balance (Mak et al., 2017). Additionally, glutamine-derived nitrogen can be used to fuel de novo glycosylation precursor biogenesis in the hexosamine biosynthesis pathway (HBP) (Araujo et al., 2017; Swamy et al., 2016).
A growing body of work has recently begun to highlight the importance of glutamine metabolism in modulating T cell-mediated immunity. Activated T cells not only upregulate amino acid transport but also increase the expression of enzymes involved in glutamine metabolism (Carr et al., 2010; Johnson et al., 2018). In addition, glutamine deprivation suppresses tumor growth and induces cell death in several cancer types (Chen and Cui, 2015; Qing et al., 2012). The glutamine dependency displayed by cancerous cells has been referred to as glutamine addiction (Abu Aboud et al., 2017; Yoo et al., 2020). Similarly, we have previously shown that NKT cells rely on glutamine for their survival and proliferation (Kumar et al., 2019). Despite this, the precise metabolic pathways and outputs of glutamine metabolism in NKT cells remain unknown.
In the current study, we report that NKT cells have higher glutamine metabolism than CD4 T cells, and that NKT cells increase glutamine metabolism after activation. NKT cells use glutamine-derived carbon to fuel the TCA cycle and glutamine-derived nitrogen to fuel the HBP while simultaneously supporting GSH generation via glutamine-derived glutamate. More importantly, these processes are critical for NKT cell survival and proliferation. NKT cells require glutaminolysis for IL-4 production, but they use the HBP to support IFNγ production. Furthermore, we demonstrate that NKT cells are glutamine addicted, because glucose is not sufficient to fuel mitochondrial function in the absence of glutamate oxidation. Lastly, AMPK-mTORC1 signaling is involved in the regulation of glutamine metabolism in NKT cells.
RESULTS
NKT cells upregulate glutamine metabolism on activation
We previously reported that resting NKT cells are less glycolytic than CD4 T cells and rely on glutamine for their survival and proliferation (Kumar et al., 2019). To gain a better understanding of glutamine metabolism in NKT cells, we assessed metabolite levels in freshly sorted NKT and CD4 T cells using liquid chromatography-coupled tandem mass spectrometry (LC-MS/MS)-based metabolomics. Metabolomic analysis showed that NKT cells have lower levels of metabolites related to glycolysis but higher levels of metabolites related to glutaminolysis compared with CD4 T cells (Figure 1A). Pathway enrichment analysis revealed increased amino acid metabolism in NKT cells, which includes glutamine metabolism (Figure S1A). In addition to glutamine, other metabolites, such as glutamate, arginine, and asparagine, were relatively high in NKT cells compared with CD4 T cells (Figure 1B). To investigate whether NKT cells upregulate glutaminolysis on activation, we measured intracellular metabolite levels after 3 days of stimulation using LC-MS/MS. Metabolites from the culture media were measured simultaneously. Metabolites downstream of glutamine metabolism were increased in cells and decreased in the culture media on activation (Figures 1C and 1D), suggesting that NKT cells enhance both glutamine import and utilization during activation. Indeed, the expression of the amino acid transporter (CD98), which aids in the glutamine transport (Nicklin et al., 2009), was increased on activated NKT cells (Figure S1B). Moreover, the levels of metabolites derived from glutamine, such as glutamate, αKG, and GSH, were increased after activation (Figures 1C and S1C–S1E). We also observed that the expression of genes encoding key enzymes involved in glutamine metabolism was elevated after activation (Figure S1F). Overall, NKT cells upregulate glutamine metabolism on activation.
Figure 1. NKT cells increase glutaminolysis on activation.

(A and B) Freshly sorted splenic NKT and CD4 T cells from C57BL/6 mice were subjected to metabolomic analysis through LC-MS/MS. (A) The volcano graph depicts upregulated and downregulated metabolites shown in red in resting NKT cells compared with CD4 T cells (n = 3). (B) Graph shows relative levels of the indicated metabolites in resting NKT cells and CD4 T cells (n = 3).
(C and D) NKT cells were stimulated with α-galactosylceramide (αGalCer; 100 ng/mL) for 3 days. The cell lysate was prepared from unstimulated (day [D] 0) and stimulated (D3) NKT cells. Media were also collected on D3 of activation. Cell lysate and media samples were subjected to metabolomic analysis through LC-MS/MS (n = 3). (C) Heatmap represents relative levels of metabolites in unstimulated and stimulated NKT cells (n = 3). (D) Heatmap shows relative levels of metabolites in fresh media and the media collected from stimulated NKT cells (n = 3).
Data are shown as mean ± SEM. *p < 0.05, **p < 0.01 were considered significant.
Glutaminolysis is essential for NKT cell survival and proliferation
Glutamine is a major source of energy and carbon molecules in rapidly proliferating cells, such as immune cells and cancerous cells (Yoo et al., 2020). We have previously shown that NKT cells rely on glutamine for their survival and proliferation (Kumar et al., 2019). We first determined the lowest amount of glutamine required for optimal NKT cell homeostasis and proliferation. To do this, we cultured NKT cells in media with decreasing amounts of glutamine. A 200 μM concentration of glutamine was observed to be required for optimal NKT cell survival and proliferation, because lower concentrations decreased these phenotypes (Figure 2A). A similar pattern was seen for mitochondrial function as measured by mitochondrial mass and membrane potential (Figure S2A). These observations prompted us to investigate whether this dependency on glutamine is due to glutaminolysis. We used a variety of pharmacological inhibitors to examine the importance of each branch of glutamine catabolism for NKT cell responses (Figure 2B). To understand how NKT cells are different in glutamine metabolism from CD4 T cells, we activated CD4 T cells in the presence and absence of these inhibitors. To begin, we measured glutamate in NKT cells activated under glutamine deprivation conditions. We found that glutamate levels were decreased in the absence of glutamine (Figure 2C). Next, to confirm whether the oxidation of glutamine into glutamate is functionally important for NKT cell survival and proliferation, we activated cells in the presence or absence of the GLS inhibitor CB839. GLS inhibition impaired NKT cell survival, proliferation, and activation (Figures 2D and S2B), which seems to be associated with lower mitochondrial function (Figure S2C). NKT cells seemed to be more sensitive to GLS inhibition compared with CD4 T cells because CB839 treatment mildly affected the CD4 T cell proliferation (Figure S2D). To confirm whether glutamine contributes to mitochondrial energy production in NKT cells, we measured ATP. ATP levels decreased significantly after GLS inhibition, suggesting that glutamate supports mitochondrial ATP production (Figure S2E).
Figure 2. Glutamine metabolism is essential for NKT cell survival and proliferation.
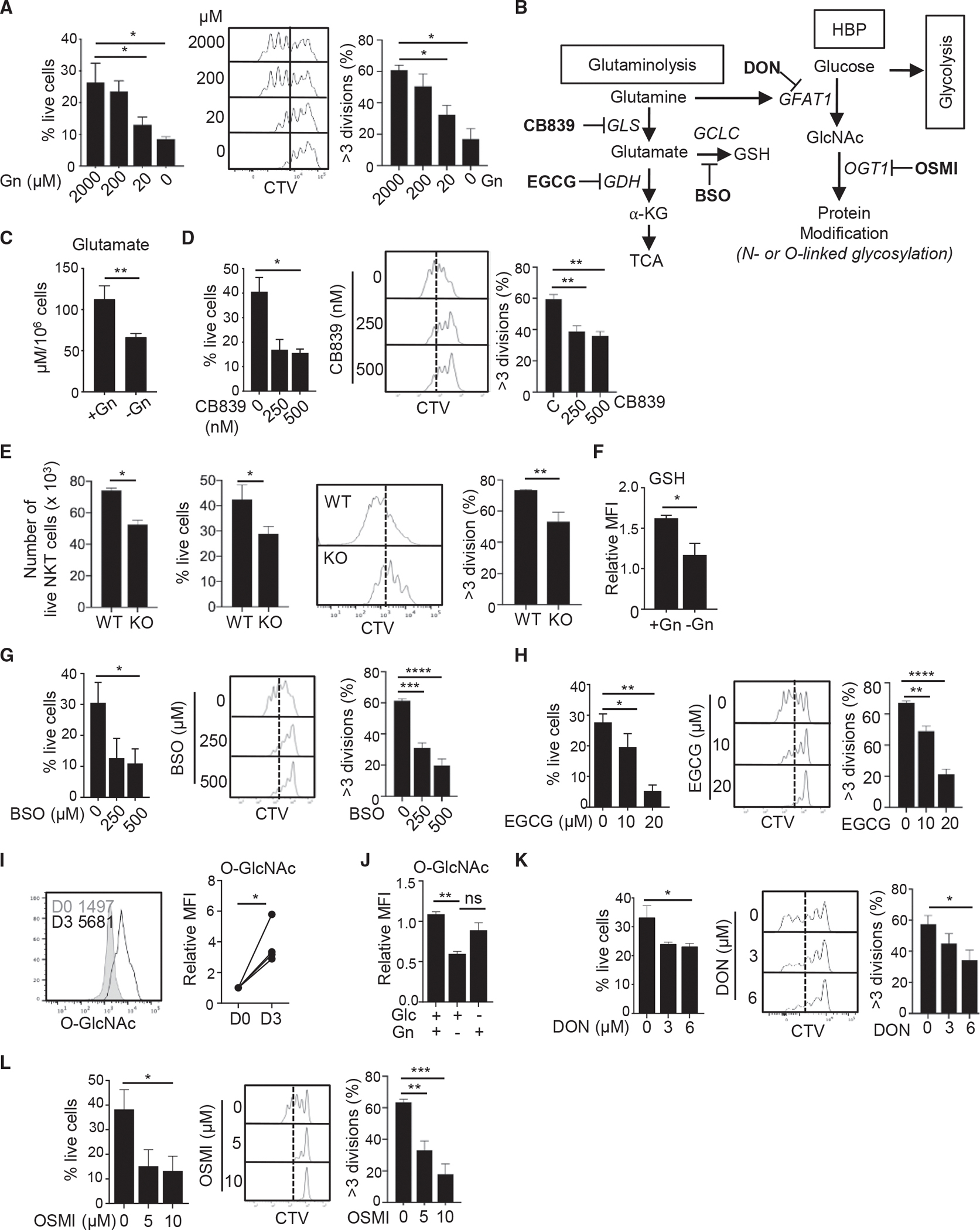
(A–E) Sorted splenic NKT cells from C57BL/6 mice were labeled with 5 μM CellTrace Violet (CTV) and stimulated for 3 days in the indicated culture conditions. (A) The data show percentages of cell survival as measured by live/dead marker staining (left panel) and cell proliferation (right panel) in media containing the indicated amounts of glutamine (n = 3). (B) The schematic depicts key branches of glutamine metabolism producing αKG and GSH, as well as utilization of glutamine in the HBP to synthesize O-GlcNAc. Pathway-specific inhibitors are shown in bold, and the names of target enzymes are italicized. (C) The graph shows glutamate levels in NKT cells activated in the presence or absence of glutamine (n = 3). (D) Cell survival and proliferation of NKT cells activated in the presence or absence of CB839 (n = 4). (E) Total live cell numbers (left panel), the percentages of live cells (middle panel), and cell proliferation from WT and GLS1 KO mice are shown after 3 days of activation (n = 3).
(F) Sorted splenic NKT cells were activated in the presence or absence of glutamine. GSH levels on day 3 of activation are shown (n = 3).
(G and H) NKT cells were activated for 3 days in the presence or absence of BSO (G) or EGCG (H). Cell survival and proliferation are shown (n = 3).
(I) The levels of O-GlcNAc in NKT cells with and without activation were compared (n = 3).
(J) Sorted NKT cells were stimulated for 3 days in the presence or absence of either glutamine or glucose as indicated. The graph shows the relative mean fluorescent intensity (MFI) of O-GlcNAc on day 3 of activation (n = 3).
(K and L) NKT cells were activated for 3 days in the presence or absence of DON (K) or OSMI (L). Cell survival and proliferation are shown (n = 3). All relative levels were calculated using the average of the control values as a reference point. All data are representative of or combined from at least three independent experiments.
Data are shown as mean ± SEM. *p < 0.05, **p < 0.01. ns, not significant.
Next, we used mice having a T cell-specific deletion of GLS1 (GLS1fl/fl CD4-Cre, referred to as GLS1 knockout [KO]) (Johnson et al., 2018) to validate the responses caused by the pharmacological inhibitor. GLS1 deficiency did not affect NKT cell development in the thymus (Figure S2F); however, the frequency of the NKT17 subset was significantly decreased in GLS1 KO spleens (Figure S2G). Additionally, NKT cell numbers were also slightly reduced in the spleens of these mice (Figure S2H), suggesting a role for glutamine in peripheral NKT cell maintenance. Next, we measured cell survival and proliferation in activated wild-type (WT) and GLS1 KO NKT cells. Similar to what was seen with CB839, GLS1-deficient cells not only died more than WT cells but also proliferated worse than WT cells (Figure 2E).
Because glutamine contributes to cellular redox regulation through GSH synthesis, we investigated whether glutamate is converted to GSH in the absence of glutamine. As expected, GSH levels were decreased in NKT cells grown without glutamine (Figure 2F). GSH was found to be critical for NKT cell homeostasis because cell survival and proliferation were impaired when GSH synthesis was inhibited by adding buthionine sulfoximine (BSO) to the culture media (Figure 2G). In contrast, CD4 T cells were resistant to BSO treatment evidenced by little difference in survival and a small decrease in proliferation, suggesting that CD4 T cells are more tolerant to the low levels of antioxidants than NKT cells (Figure S2I).
In addition to GSH, glutamate can be converted into αKG, which then can enter the TCA cycle. The elevated levels of αKG in activated NKT cells (Figure S1D) prompted us to examine whether the conversion of glutamate to αKG is critical for NKT cells. Like GLS inhibition, GDH inhibition using the pan dehydrogenase inhibitor epigallocatechin-3-gallate (EGCG) reduced cell survival and proliferation (Figure 2H). Furthermore, GDH inhibition decreased mitochondrial mass and membrane potential in NKT cells (Figure S2J). Therefore, glutaminolysis is necessary not only for mitochondrial anaplerosis but also for GSH synthesis and ATP production in NKT cells. In contrast with this, we observed that CD4 T cells were relying to a lesser extent on GDH activity for their survival and proliferation (Figure S2K) despite a slight reduction in mitochondrial membrane potential (Figure S2L). Collectively, glutaminolysis is critical for NKT cell survival and optimal proliferation, potentially by supporting mitochondrial function.
NKT cell homeostasis depends on the contribution of glutamine-derived nitrogen to the HBP
Glucose and glutamine contribute carbon and nitrogen, respectively, via the HBP to generate uridine diphosphate N-acetylglucosamine (UDP-GlcNAc), the primary donor for cellular glycosylation (Figure 2B) (Swamy et al., 2016). This step is catalyzed by the enzyme glutamine-fructose-6-phosphate transaminase 1 (GFAT1). From UDP-GlcNAc, O- and N-linked glycosylation marks are deposited on proteins by O-GlcNAc transferase (OGT) necessary for protein stability and function. To test the role of de novo glycosylation biosynthesis via the HBP in NKT cells, we examined total protein glycosylation in the absence of glutamine by measuring O-GlcNAc-ylation of the proteome. Activated NKT cells showed increased total protein glycosylation (Figure 2I), as well as higher mRNA expression of both the Gfat1 and Ogt genes (Figure S3A). Next, to understand how nutrient limitation impacts the HBP, we measured O-GlcNAc levels in cells stimulated in the presence of glucose only, glutamine only, or both. We found that glutamine limitation reduced de novo O-GlcNAc synthesis significantly more than glucose limitation did in activated NKT cells (Figure 2J), suggesting that NKT cells are capable of increasing their usage of the salvage pathway for HBP synthesis under glucose restriction, but not glutamine restriction.
To determine the role of the HBP in NKT cell responses, we treated NKT cells with 6-diazo-5-oxo-L-nor-leucine (DON), a pan glutamine-deamidase inhibitor (Anderson et al., 2017), during activation. We observed that DON treatment reduced O-GlcNAc levels (Figure S3B) leading to impaired NKT cell survival accompanied by reduced cell proliferation and activation (Figures 2K and S3C). Similarly, inhibition of OGT by (αR)-α-[{1,2-Dihydro-2-oxo-6-quinolinyl)sulfonyl}amino]-N-(2-furanylmethyl)-2-methoxy-N-(2-thienylmethyl)-benzeneacetamide (OSMI-1) resulted in more cell death and less cell proliferation than in untreated cells (Figure 2L). Similarly, CD4 T cells were also sensitive to impaired HBP for their survival and proliferation (Figures S3D and S3E). These data suggest that HBP is essential for the homeostasis of both NKT and CD4 T cells.
NKT cell homeostasis requires GSH-mediated redox balance
NKT cells rely on glutamine to produce GSH, which is vital for the effective management of the reactive oxygen species (ROS) (Cetinbas et al., 2016). We have previously shown that NKT cells are highly susceptible to oxidative stress (Kim et al., 2017). Therefore, NKT cells may be susceptible to cell death in the absence of GSH. To investigate this, we examined total ROS production in NKT cells treated with the GSH inhibitor BSO. The presence of the inhibitor resulted in greater total ROS levels than the control (Figure 3A). In contrast, GSH inhibition reduced mitochondrial ROS, mitochondrial mass, and membrane potential (Figures 3B and 3C), suggesting that GSH is critical for mitochondrial functions.
Figure 3. GSH-mediated redox balance is essential for NKT cells homeostasis.

Sorted splenic NKT cells from C57BL/6 mice were stimulated for 3 days in the presence or absence of BSO.
(A–C) Histograms and graphs show total ROS levels measured using 2′,7′-dichlorodihydrofluorescein diacetate (DCFDA) (A), mitochondrial ROS measured by MitoSOX (B), mitochondrial mass by MitoTracker, and mitochondrial membrane potential by tetramethylrhodamine methyl ester perchlorate (TMRM) (C) in activated NKT cells (n = 3).
(D and E) NKT cells were activated for 3 days in the presence or absence of BSO (250 μM) and NAC (10 μM). Histograms and graphs show total ROS levels (D), cell survival, and cell proliferation (E) (n = 3). All relative levels were calculated using the average of control value as a reference point. All data are representative of or combined from at least three different experiments.
Data are shown as mean ± SEM. *p < 0.05, **p < 0.01.
These observations suggest that the high levels of cell death in NKT cells after inhibition of GSH synthesis could be because of increased ROS. To test this, we treated cells with the ROS scavenger N-acetyl-cysteine (NAC) to reduce ROS in GSH-inhibited cells by BSO. We found that NAC lowered ROS levels back to the control levels in GSH-inhibited cells (Figure 3D). Interestingly, NKT cell survival was rescued by NAC treatment, whereas cell proliferation was not rescued (Figure 3E). Together, these data suggest that NKT cell survival is supported by GSH-mediated redox balance.
Mitochondrial anaplerosis fueled by glutamine is necessary for NKT cell homeostasis and effector function
Glucose can be metabolized through glycolysis and the PPP. Previously, we have shown that the expression of PPP genes was significantly higher in NKT cells compared with CD4 T cells (Kumar et al., 2019). Consequently, the levels of glycolytic metabolites were lower in NKT cells than CD4 T cells (Figure 1A). The absence of glucose did not affect NKT cell survival or proliferation, raising the possibility that NKT cells rely primarily on glutamine (Kumar et al., 2019). To determine whether NKT cells are addicted to glutamine, we first measured the expression of hexokinase 2 (HK2), which converts glucose into glucose 6-phosphate during the first step of glycolysis. Consistent with our previous finding that CD4 T cells take up more glucose than NKT cells on activation (Kumar et al., 2019), HK2 expression was also higher in activated CD4 T cells compared with NKT cells (Figure 4A). Next, we measured PPP metabolites in NKT and CD4 T cells by LC-MS/MS. Compared with CD4 T cells, NKT cells have notably higher levels of PPP metabolites before activation (Figure 4B). Additionally, PPP metabolite levels were further increased on activation of NKT cells (Figure 4C). Considering the lower level of lactate produced by NKT cells (Kumar et al., 2019), these data suggest that NKT cells have a higher rate of diversion of glucose into the PPP compared with CD4 T cells.
Figure 4. NKT cells exhibit a glutamine-addicted phenotype.

(A) The graph shows hexokinase 2 (HK2) expression in NKT and CD4 T cells with and without stimulation (n = 3).
(B) Heatmap shows relative levels of the indicated PPP metabolites in resting NKT cells compared with resting CD4 T cells analyzed after LC-MS/MS analysis (n = 3).
(C) Heatmap shows PPP metabolites in NKT cells with and without stimulation as analyzed by LC-MS/MS (n = 3).
(D and E) Cell survival and proliferation of sorted NKT cells stimulated in the presence or absence of EGCG (20 μM) with or without sodium pyruvate (1 mM) or DMαKG (1.5 mM) (n = 3).
(F and G) WT and GLS1 KO mice were injected with αGalCer (5 μg/mouse), and splenic NKT cells were sorted on day 3 of activation. The representative graphs show ECAR using the glycolytic stress test (F) and OCR using the Mito Stress test (G) in Seahorse assay (n = 6 replicates per group pooled from two mice). The graph in (G) shows basal respiration (BR), maximum respiration capacity (MRC), and reserve capacity (RC) (n = 3).
(H and I) NKT from WT and GLS1 KO mice were stimulated in the presence or absence of DMαKG (1.5 mM). (H) Cell survival and proliferation of NKT cells are shown (n = 3). (I) Representative histograms and summary graphs show mitochondrial mass and membrane potential (n = 3). All relative levels were calculated using the average control value as a reference point. All data are representative of or combined from at least two or three different experiments. Seahorse data are shown as mean ± SD. All other data are shown as mean ± SEM. *p < 0.05, **p < 0.01, ****p < 0.0001.
Elevated PPP gene expression suggests that glucose is metabolized mainly via the PPP in NKT cells (Kumar et al., 2019); therefore, glucose-derived pyruvate would not be sufficient to supplement mitochondrial anaplerosis in NKT cells. We tested this hypothesis by adding sodium pyruvate to cells treated with the GDH inhibitor EGCG, which blocks the conversion of glutamate to αKG. We then determined whether the reduced cell survival and proliferation observed after GDH inhibition can be rescued by providing pyruvate. Indeed, we observed that adding pyruvate enhanced cell survival, but not proliferation, in the control group, suggesting that insufficient glycolysis affects NKT cell survival (Figure 4D). Furthermore, cell survival and proliferation were restored to control levels when sodium pyruvate was added to GDH-inhibited cells (Figure 4D), further supporting inefficient glycolysis in NKT cells.
To further investigate the role of glutaminolysis in supporting αKG production, we provided dimethyl αKG (DMαKG), a cell-permeable αKG analog, to NKT cells grown in either the absence of glutamine or the presence of CB839 or EGCG. Cell survival was not rescued by DMαKG in glutamine deprivation conditions (Figure S4A), and cell proliferation was partially rescued by DMαKG when GLS activity was inhibited (Figure S4B). However, providing DMαKG restored cell survival to normal levels and cell proliferation partially in GDH-inhibited NKT cells (Figure 4E).
Because mitochondrial function was affected by glutaminolysis inhibition, we investigated whether glucose metabolism was also affected. To do this, we measured glucose uptake in NKT cells grown under glutamine deprivation conditions. We also measured glycolysis using a glycolytic stress test in GLS1 KO NKT cells. The results showed that despite lower glucose uptake capacity under glutamine deprivation conditions (Figure S4C), GLS1 deficiency did not affect the glycolytic rate (Figure 4F). Because glutamine is critical for optimal mitochondrial functions, we next measured ATP production under the conditions without glucose or glutamine. Indeed, NKT cells grown under both conditions have reduced ATP production (Figure S4D). However, a reduction in ATP during glutamine deprivation could be an additive effect of both reduced glutaminolysis and glycolysis. To test the idea that glutaminolysis is responsible for mitochondrial function, we assessed the effect of GLS1 deficiency on cellular oxygen consumption rate (OCR) between WT and GLS1 KO NKT cells. NKT cells lacking GLS1 displayed lower levels of mitochondrial activity, as reflected by the lower levels of basal respiration (BR), maximum respiration capacity (MRC), and reserve capacity (RC) in these cells compared with WT NKT cells (Figure 4G). DMαKG supplementation also partially rescued cell survival while fully recovering cell proliferation in GLS1 KO NKT cells (Figure 4H). However, mitochondrial function was partially rescued by DMαKG in GLS1 KO NKT cells (Figure 4I) but completely rescued during GDH inhibition (Figure S4E). These observations suggest that branching of glutamine into different utilization pathways is essential to maintain optimal NKT cell homeostasis and mitochondrial function.
In all, these data demonstrate that NKT cells exhibit lower levels of glycolysis, resulting in insufficient levels of glucose-derived metabolites to fuel the TCA cycle. As a result, NKT cells primarily rely on glutamine metabolism to support mitochondrial functions for their survival and proliferation.
Distinct glutamine oxidation pathways regulate NKT cell effector functions
We have previously shown that glucose availability is critical for NKT cell cytokine production (Kumar et al., 2019). Because glutamine fuels both glutaminolysis and the HBP (Figure 2B), we were interested in investigating the role of glutamine in cytokine expression. Glutamine deprivation reduced the expression of IFNγ and IL-4 by NKT cells (Figure 5A), suggesting a distinct role for glutamine metabolic pathways in cytokine expression in NKT cells. To investigate whether glutaminolysis has any role in cytokine production, we activated NKT cells with and without the GLS inhibitor CB839. Additionally, we activated WT and GLS1 KO NKT cells. GLS activity was observed to be critical for IL-4 production in NKT cells, because IL-4+ cells were significantly reduced on CB839 treatment (Figure S5A). Similarly, both intracellular and secreted levels of IL-4 were lower in GLS1 KO than WT NKT cells (Figures 5B and S5B). However, GDH inhibition reduced percentages of both IL-4+ and IFNγ+ NKT cells (Figure S5C).
Figure 5. IFNγ and IL-4 production in NKT cells rely on distinct branches of glutamine metabolism.

(A) Sorted splenic NKT cells from C57BL/6 mice were stimulated for 3 days with or without glutamine (Gn), and cytokine expression was compared after PMA/Ionomycin re-stimulation.
(B) Representative dot plots show cytokine expression in sorted NKT cells from WT and GLS1 KO mice stimulated for 3 days. Graphs show cumulative data from three independent experiments.
(C and D) Sorted splenic NKT cells from C57BL/6 mice were stimulated for 3 days in the presence or absence of DON (6 μM). Intracellular cytokine expression (C) and the levels of cytokine secreted into the media by ELISA (D) are shown (n = 3).
(E) The graph shows cytokine expression in NKT cells stimulated in the presence or absence of BSO (n = 3).
(F) NKT cells were stimulated for 3 days in the presence or absence of glutamine (2 mM) in combination with DMαKG (1.5 mM). Graphs show relative percentages of cytokine-positive NKT cells (n = 3). Data are shown as mean ± SEM. All data are representative of or combined from at least three independent experiments.
*p < 0.05, **p < 0.01.
We next asked whether the HBP regulates cytokine production in NKT cells. In contrast with GLS inhibition, inhibition of GFAT1 via DON treatment decreased IFNγ, but not IL-4, production (Figures 5C and 5D). However, inhibition of OGT significantly reduced IFNγ expression but only moderately affected IL-4 expression (Figure S5D). Overall, our data suggest that de novo HBP activity is critical for cytokine production by NKT cells.
ROS seem to be important for NKT cell effector functions at a steady state, but they decrease on activation (Kim et al., 2017). To examine whether GSH-mediated redox balance modulates NKT cell effector function, we activated NKT cells in the presence or absence of BSO and measured cytokine expression. Interestingly, cytokine production was not affected by GSH inhibition (Figures 5E and S5E), even though GSH-inhibited cells have higher total ROS levels (Figure 3A).
As expected, DMαKG supplementation partially restored cytokine production under either glutamine-deficient culture conditions (Figures 5F and S5F) or GLS inhibition (Figure S5G). Similarly, DMαKG corrected IL-4 cytokine production by GDH-inhibited cells (Figure S5H). Overall, glutamine metabolism distinctly regulates IL-4 and IFNγ production in NKT cells.
GLS1 is crucial for proper NKT cell responses to Listeria monocytogenes infection
To investigate the role of glutamine metabolism in NKT cell-mediated immune responses in vivo, we used the Listeria infection model. We injected Listeria monocytogenes expressing ovalbumin intraperitoneally to WT and GLS1 KO mice. Bacterial load and NKT cell-specific functions were analyzed after 2 days of infection. This time point allows us to study NKT cell-mediated effects on bacterial infection, because CD4 and CD8 T cells are not able to mount an immune response in this short time frame. To examine whether NKT cell metabolic responses are changed after Listeria infection, we compared GSH and CD98 expression in WT mice. Both CD98 expression (Figure 6A) and GSH levels (Figure 6B) were greatly increased in splenic and hepatic NKT cells in infected mice compared with PBS-injected controls. When bacterial loads were compared, GLS1 KO mice had higher bacterial loads than WT mice in the spleens and livers (Figure 6C). Higher bacterial burden correlated with impaired activation of NKT cells from GLS1 KO mice (Figure 6D). We then asked whether the high bacterial load in GLS1 KO mice was correlated with slower NKT cell expansion or diminished cytokine expression. We observed that cell proliferation was impaired in NKT cells from GLS1 KO mice in response to bacterial challenge (Figure 6E), supporting the important role for glutamine metabolism in NKT cell responses. However, like our in vitro observations, Listeria infection did not significantly affect IFNγ production in GLS1 KO NKT cells (Figure 6F). Overall, GLS-mediated glutaminolysis is essential for NKT cells to mediate protective immune responses against Listeria infection.
Figure 6. Glutaminolysis is important for NKT cell responses to Listeria infection.

WT and GLS1 KO mice were injected with either 105 CFUs/mouse of LM-Ova (Lm) or PBS intraperitoneally. Two days after infection, spleens and livers were harvested and analyzed for bacterial load, NKT cell proliferation, and IFNγ expression.
(A and B) Graphs show levels of CD98 expression and GSH in NKT cells from the spleen (left panel) and the liver (right panel) of PBS- and Lm-injected WT mice (n = 6).
(C) Graphs show bacterial loads in the spleens and livers of infected WT and GLS1 KO mice (n = 6).
(D) Graphs show CD69 expression in splenic and hepatic NKT cells from WT and GLS1 KO mice (n = 6).
(E) Representative dot plots and graphs show cell proliferation as measured by Ki-67 expression in NKT cells from the spleens and livers of WT and GLS1 KO mice (n = 6).
(F) To assess IFNγ expression, we incubated total splenocytes in the presence of Monensin for 2 h to prevent cytokine secretion followed by comparing intracellular expression of IFNγ in splenic (top panel) and hepatic (bottom panel) NKT cells (n = 6). The data are pooled from two independent experiments.
Data are shown as mean ± SEM. *p < 0.05, **p < 0.01.
The AMPK- mTORC1 axis regulates NKT cell glutamine metabolism
Studies have linked mTORC1 activation to glutamine addiction in some types of cancer cell (Choo et al., 2010). Moreover, mTOR signaling is critical for the development and function of NKT cells (Prevot et al., 2015; Shin et al., 2014; Sklarz et al., 2017). We have previously shown that mTORC1 activity is enhanced on NKT cell activation (Kumar et al., 2019). Furthermore, NKT cells stimulated in the presence of high lactate showed reduced mTORC1 activity accompanied by poor proliferation (Kumar et al., 2019). As such, we reasoned that mTORC1 signaling might affect glutamine metabolism in NKT cells. To test this, we used the pharmacological reagent rapamycin to inhibit mTORC1 activity because mTORC1 deficiency compromises NKT cell development (Shin et al., 2014). We stimulated NKT cells in the presence of rapamycin and examined the indicators of glycolysis and amino acid transport by comparing the expression of HK2 and CD98, respectively. We also measured glutamine, glutamate, and GSH to study glutaminolysis. mTORC1 inhibition by rapamycin resulted in reduced HK2 expression, CD98 expression, and GSH levels (Figures 7A–7C). Interestingly, mTORC1 inhibition also reduced proteome O-GlcNAc levels (Figure 7D), suggesting that mTORC1 is a key regulator of glucose and glutamine metabolism and glycosylation in NKT cells.
Figure 7. mTORC1-AMPK signaling regulates both glucose and glutamine metabolism in NKT cells.

(A–D) Sorted splenic NKT cells were stimulated for 3 days in the presence or absence of rapamycin (2 nM). Representative histograms and summary graphs show relative HK2 expression (A), CD98 expression (B), GSH production (C), and O-GlcNAc levels (D) in stimulated NKT cells (n = 3).
(E and F) NKT cells were activated for 3 days under the indicated conditions, and relative expression levels of pS6Ser235/236 (E) and c-Myc (F) were compared (n = 3).
(G) Relative expression of pAMPK before (day [D] 0) and after activation for 3 days (D3) is shown (n = 3).
(H) Cell survival and proliferation of WT and AMPK KO splenic NKT cells after 3 days of activation are shown (n = 3).
(I–K) WT and AMPK KO NKT cells were sorted and stimulated for 3 days. Glutamate and αKG (I), GlcNAc (J), and L-PHA (K) expression levels were compared (n = 3).
(L) Graphs show cytokine expression in WT and AMPK KO NKT cells after 3 days of activation (n = 3).
(M) Histograms show cell proliferation in WT and AMPK KO NKT cells activated with and without CB839 (250 nM) (n = 3). All relative levels were calculated using the average control value as a reference point. All data are combined from at least three independent experiments.
Data are shown as mean ± SEM. *p < 0.05, **p < 0.01.
mTORC1 signaling integrates growth factors and nutrient signals to regulate cell growth. mTORC1 is unresponsive to these signals under amino acid deprivation (Jewell et al., 2015). In particular, glutamine and glutamate are essential for maintaining mTORC1 activity in T cells (Duran et al., 2012; Klysz et al., 2015). Therefore, we asked whether glutamine or glutamate availability was necessary for mTORC1 activity in NKT cells. Indeed, both glutamine deprivation and GLS inhibition reduced the phosphorylation of ribosomal protein S6 (pS6), a substrate of mTORC1 (Figure 7E). Similarly, inhibition of GSH production and OGT activity also decreased mTORC1 activity (Figure 7E). mTORC1 is known to enhance c-Myc expression (Gera et al., 2004). We found that c-Myc levels were reduced in NKT cells grown under glutamine deprivation conditions, as well as after CB839 treatment (Figure 7F), indicating that GLS activity regulates mTORC1 signaling in NKT cells. Together, these data suggest that crosstalk between glutamine metabolism and mTORC1 signaling regulates cell proliferation in NKT cells.
The levels of phosphorylated AMPK (pAMPK) increase during primary T cell responses in vivo (Pearce et al., 2009), and pAMPK is known to negatively regulate mTORC1 activity in T cells (Blagih et al., 2015). Having observed that mTORC1 promotes glutamine metabolism in NKT cells, we investigated the role of the AMPK. We found that pAMPK levels were greatly increased in stimulated NKT cells compared with unstimulated cells (Figure 7G). Next, we used T cell-specific AMPK KO mice to test our hypothesis that AMPK deficiency would elevate glutaminolysis, which would have a beneficial effect on NKT cells. AMPK KO mice have no observed defects in conventional T cell (Zarrouk et al., 2013) or NKT cell development in the thymus (Figure S6A). Additionally, although NKT cell subsets were skewed toward NKT17, peripheral maintenance of NKT cells was normal in these mice (Figures S6A and S6B). We next analyzed activated NKT cell survival and proliferation in WT and AMPK KO mice. AMPK KO NKT cells were more resistant to cell death and proliferated better than WT cells (Figure 7H). As expected, AMPK KO NKT cells have increased levels of glutamate and αKG (Figure 7I), indicators of glutaminolysis. AMPK NKT cells also have elevated HBP as evidenced by high levels of O-GlcNAc (Figure 7J) and lectin phytohemagglutinin-L (L-PHA), which is the marker of protein glycosylation (Figure 7K). Higher glutaminolysis and HBP in AMPK KO NKT cells were correlated with higher mTORC1 activity (Figure S6C). These observations suggest that glutamine metabolism is enhanced in AMPK KO NKT cells. AMPK KO NKT cells expressed more IL-4 but similar levels of IFNγ (Figures 7L and S6D). Furthermore, in contrast with WT cells, AMPK KO NKT cells proliferated efficiently even in the presence of GLS inhibitor (Figure 7M). Together, these data suggest that the AMPK-mTORC1 signaling axis controls glutamine metabolism in NKT cells.
DISCUSSION
Glucose and glutamine are the two primary nutrients utilized by highly proliferative cells, including T cells (Vander Heiden et al., 2009). Unlike glucose, glutamine can provide both carbon and nitrogen for anabolic reactions (Hensley et al., 2013). Indeed, glutamine-derived nitrogen is critical for the synthesis of nitrogenous compounds, such as nucleic acids, glycosoamino glycans, and non-essential amino acids (Ma et al., 2013). Here, we demonstrate that glutamine is metabolized via glutaminolysis and the HBP in activated NKT cells to support their survival, proliferation, and effector functions. We also show that NKT cells are glutamine addicted because their low glycolytic rate cannot spare enough glucose to support the TCA cycle. Moreover, glutamine metabolism seems to be regulated by AMPK-mTORC1 signaling in NKT cells.
Glutamine metabolism is differentially regulated in the various T cell subsets (Johnson et al., 2018; Klysz et al., 2015). In addition to synthesizing glutamine de novo, proliferating cells can acquire glutamine from the extracellular environment to meet their energetic requirements. Resting NKT cells have higher glutamine levels than CD4 T cells, which may explain why they rely on glutamine on activation for their survival and proliferation. This idea is supported by the fact that GLS1 KO mice exhibit lower NKT cell frequencies in the spleen compared with WT. In addition to glutamine, the levels of other amino acids were also higher in activated NKT cells compared with CD4 T cells. Whether NKT cells have enhanced uptake or increased synthesis of these amino acids from glutamine warrants further investigation. The high rate of glutamine consumption in NKT cells suggests that these cells use glutamine for multiple roles beyond protein synthesis. We found that NKT cells use glutamine in the HBP to modulate protein modification processes such as glycosylation. It is important to note that TCA cycle intermediates can regulate epigenetic signatures in activated T cells (Johnson et al., 2018; Klysz et al., 2015). These metabolites are critical in regulating T helper cell subsets and their cytokine production (Ichiyama et al., 2015). In corroboration with these facts, inhibiting glutamate oxidation reduced cytokine production by NKT cells, which was rescued by αKG supplementation. Interestingly, NKT cells were observed to rely on glutaminolysis primarily for IL-4 production but depend on glutamine oxidation from the HBP for IFNγ production. We also showed that GLS1 does not control IFNγ production in response to Listeria infection, indicating that GLS1 largely controls NKT cell homeostasis. Extending these findings to their in vivo relevance, we observed slower NKT cell proliferation and higher bacterial burden after Listeria infection in GLS1 KO mice.
Glutamate can replenish TCA cycle metabolites either by GDH or by using transaminases to produce non-essential amino acids. IFNγ expression was more severely affected by GDH inhibition than by GLS1 deletion, indicating that a compensatory mechanism involving blockage of transaminases may rescue IFNγ in GLS1 KO cells. Additionally, the off-target effects of EGCG, such as alteration of the cell cycle (Gupta et al., 2004), suppression of the mitogen-activated protein kinase (MAPK) pathway (Shimizu et al., 2011), and epigenetic changes in gene expression, have been previously reported (Lee et al., 2005; Lin et al., 2012). However, studying the role of transaminases and these off-target effects in NKT cells is beyond the scope of this study.
Mitochondrial homeostasis is critical for NKT cell development (Prevot et al., 2015; Sklarz et al., 2017). T cell-specific deletion of Rieske iron-sulfur protein (RISP) (T-Uqcr−/−), a nuclear-encoded protein subunit of mitochondrial complex III, has recently been shown to block NKT cell development (Weng et al., 2021). Additionally, conventional T cells use glucose to produce lactate and fuel mitochondrial metabolism for their homeostasis and effector function (Chang et al., 2013). NKT cells have low glucose uptake but high PPP enzyme expression (Kumar et al., 2019) and metabolite abundance. Together, these results suggest that less glucose-derived carbon is oxidized through glycolysis, and therefore less is available to support TCA cycling in NKT cells. Based on the reduced availability of glucose intermediates to fuel respiration, NKT cells might depend on glutamine to support mitochondrial function. Similar to T cells, we observed that NKT cells also undergo activation-induced cell death, and the survival of activated NKT cells is typically lower than CD4 T cells.
ROS can act as signaling messengers and positively modify protein structure; however, high concentrations of ROS can lead to cell death (Gorrini et al., 2013; Mak et al., 2017). Antioxidation via GSH supports activation-induced metabolic reprogramming in T cells (Mak et al., 2017). Additionally, NKT cells reduce intracellular ROS levels on activation (Kim et al., 2017), suggesting that high levels of ROS may be detrimental for activated NKT cells. Our data suggest that glutamine also contributes to GSH synthesis in NKT cells, which is critical for maintaining the redox balance necessary for cell survival. GSH also supports mitochondrial function in NKT cells, and this phenomenon might be because of mTORC1 activation. Further investigation is warranted to shed light on this mechanism.
Lymphocytes must balance a wide range of metabolic pathways to maintain homeostasis after activation. T cells use glutamine-dependent oxidative phosphorylation to produce ATP and remain viable in low-glucose environments. Because NKT cells have low glycolytic capacity, AMPK is triggered on activation to regulate glutamine metabolism. AMPK has been reported to regulate mTORC1 in T cells (Blagih et al., 2015). mTORC1 supports glycolysis by directly regulating pathway-specific gene expression in T cells (Duvel et al., 2010). Previously, we have shown that mTORC1 inhibition by rapamycin not only compromised NKT cell survival and proliferation but also reduced glucose uptake (Kumar et al., 2019). Here, we showed that rapamycin treatment negatively affected various steps of glutamine metabolism, including glutamine transporter expression, HBP pathway activity, and GSH synthesis. Unlike GLS1 KO NKT cells, AMPK-deficient NKT cells have not been reported to show any difference in bacterial burden after Listeria infection (Blagih et al., 2015), suggesting that WT NKT cells are capable of clearing bacteria, and the enhanced response shown by AMPK KO NKT cells does not add additional effect.
We report in this study that glutamine is essential for NKT cell homeostasis and effector function, shedding light on a potential mechanism for NKT cell survival in the tumor microenvironment (TME). Low availability of glucose in the TME (Anderson et al., 2017; Ho et al., 2015) reduces conventional T cell proliferation and cytokine production (Ota et al., 2016; Renner et al., 2015). However, glucose restriction likely does not affect NKT cell homeostasis, because these cells are more dependent on glutamine (Kumar et al., 2019). Our findings led us to propose that NKT cells can be used as an effective immunotherapeutic agent against glucose-reliant tumors.
In conclusion, glutamine oxidation is pivotal for NKT cell survival and proliferation. Because NKT cells display inefficient glycolysis, we predict that they cannot effectively use glucose to fuel mitochondrial metabolism. Glutamine-derived GSH is critical in maintaining redox balance in NKT cells, which is essential for their survival. This study also reveals that NKT cells use different glutamine oxidation pathways for IL-4 and IFNγ production. Moreover, AMPK-mTORC1 signaling regulates glutamine metabolism in NKT cells. Taken together, NKT cells have distinct metabolic requirements from CD4 T cells, and a better understanding of these requirements may contribute to the development of new therapeutic targets to improve T cell-based therapies in the future.
Limitations of the study
Although we comprehensively investigated glutamine metabolism in NKT cells using pathway-specific inhibitors and T cell-specific GLS1 KO mice, our study has limitations. First, genetically modified mice lacking metabolic genes of interest in T cells are not available. Knockdown of a gene using specific siRNAs in primary NKT cells is also difficult. Therefore, we mostly relied on inhibitors. Second, because of the low number of NKT cells in mice, it is highly challenging to investigate metabolic requirements in depth, such as glutamine tracing. Using NKT cell lines is not ideal because metabolism is different in immortalized cells. Lastly, the current study is focused on the role of glutamine in the total NKT cell pool. However, functional subsets of NKT cells exist (Wang and Hogquist, 2018), and each subset may require a different metabolism. Moreover, this study has been carried out in C57BL/6 mice, in which the NKT1 subset is the dominant cell population. Studying the metabolic regulation in NKT2 or NKT17 is warranted, which requires mouse models that are enriched with each subset.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Cheong-Hee Chang (heechang@umich.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Unprocessed data underlying the display items in the manuscript, related to figures are available from the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Male and female C57BL/6 mice ranging from 8–12 weeks of age were either bred in-house or purchased from The Jackson Laboratory. T cell-specific GLS1 deficient mice (referred to as GLS1 KO) and AMPK deficient mice (referred to as AMPK KO) were generated by crossing GLS1fl/fl and AMPKfl/fl mice with CD4-Cre expressing mice purchased from The Jackson Laboratory. In all experiments, WT littermates were used as controls. All mice were bred and maintained under specific pathogen free conditions. All animal experiments were performed in accordance with the Institutional Animal Care and Use Committee of the University of Michigan.
METHOD DETAILS
Cell isolation and activation
Primary cell suspensions were prepared from spleens as per standard protocol (Kim et al., 2017). To sort NKT and CD4 T cells, B cells were excluded from whole splenocytes by incubating with anti-CD19 beads (Miltenyi Biotec) or by using the EasySep™ mouse CD19 positive selection kit (STEMCELL Technologies) as per the manufacturers’ protocols. NKT and CD4 T cells were sorted based on TCR-β and PBS-57 loaded CD1d tetramer expression using a FACS Aria III (BD Biosciences). To study activated NKT cells, cells were stimulated with α-Galactosylceramide (αGalCer; 100 ng/mL) in RPMI 1640 medium supplemented with 10% FBS, 2 mM glutamine, and penicillin/streptomycin at 37°C. In the similar culture condition CD4 T cells were activated with αCD3 and αCD28. For glucose and glutamine deprivation assays, sorted NKT cells were stimulated in glucose- and glutamine-free RPMI 1640 media supplemented with 10% dialyzed FBS (Sigma Aldrich). To inhibit GLS1 activity, CB839 (Sigma Aldrich) was used at 250 nM, or 500 nM. To inhibit GDH activity, epigallocatechin-3-gallate (EGCG) (Sigma Aldrich) was used at 10 μM or 20 μM. To inhibit GSH synthesis, L-buthionine-sulfoximine (BSO) (Sigma Aldrich) was used at 250 μM or 500 μM. Rapamycin (Sigma Aldrich) was used to inhibit mTORC1 activity at a concentration of 2nM. 6-diazo-5-oxo-L-norleucine (DON) was used to inhibit O-GlcNAc production at a concentration of 3 μM, 6 μM, or 12 μM. To inhibit OGT1 activity, OSMI-1 (Sigma Aldrich), an OGT-1 inhibitor was used at 10 μM or 20 μM concentrations. For αKG supplementation assays, dimethyl-2-oxoglutarate (DMαKG) (Sigma Aldrich) was used at a 1mM concentration. N-acetyl cysteine (NAC) (Sigma Aldrich) was used at a 1mM concentration as an antioxidant.
Flow cytometry
The following fluorescently conjugated antibodies were used in the presence of anti-FcγR mAb (2.4G2) for surface and intracellular staining (all from eBioscience): anti-mouse TCRβ (H57-597) Pacific Blue/APC, PBS-57 loaded CD1d tetramer APC/PE/Pacific Blue, anti-mouse CD4 (GK1.5) APC-Cy7, anti-mouse NK1.1 (PK-136) PE-Cy7, anti-mouse CD44 (IM7) PerCp-Cy5.5, anti-mouse IFNγ (XMG1.2) PE/FITC, anti-mouse IL-4 (11B11) PE-Cy7, anti-mouse IL-17 (TC11-18H10) PerCP-Cy5.5, anti-PLZF (Mags-21F7) PE and anti-RORγt (AFKJS-9) Pacific Blue. To identify committed cells, transcription factor staining was performed using the Foxp3/transcription factor staining kit (eBioscience) and intranuclear staining for T-bet, RORγt, GATA3, and PLZF. Ki-67 (SolA15) PerCP-Cy5.5 staining was used to measure in vivo cell proliferation after Listeria infection. CD98 expression was measured by flow cytometry using CD98 (RL388) antibody. RL388 detects a disulfide-linked heterodimer complex (130 kDa) composed of a glycosylated heavy (86 kDa) subunit and a non-glycosylated light (39 kDa) subunit. Dead cells were excluded by staining with LIVE/DEAD Fixable Yellow Dead Cell Stain Kit (405 nm excitation) (Invitrogen).
For intracellular cytokine expression, activated cells were re-stimulated with PMA (50 ng/mL, Sigma Aldrich) and Ionomycin (1.5 μM, Sigma Aldrich) in the presence of Monensin (3 μM, Sigma Aldrich) for 4 h. Cells were then stained for surface antigens and intracellular cytokines according to manufacturer’s instructions (BD Biosciences). For intracellular staining of phosphorylated ribosomal protein S6 (pS6Ser235/236) (Cell Signaling), HK2 staining (EPR20839) (Abcam), and O-GlcNAc (BD Biosciences), cells were permeabilized using 90% methanol. Cells were then incubated with pS6 or HK2 antibody for 1 h. To examine HBP, cells were stained with O-GlcNAc antibody and L-PHA (10 μg/mL, Invitrogen)) for 30 min at RT in the dark in cytoplasmic permeabilization buffer (BD Biosciences). Nuclear permeabilization buffer was used for c-Myc staining. For cell proliferation, NKT cells were labeled with 5 μM CellTrace™ Violet (CTV) (Invitrogen) in 1X PBS containing 0.1% BSA for 30 min at 37°C. Cells were stimulated as indicated and analyzed by flow cytometry on day 3 post-stimulation for CTV dilutions. Cells were acquired on a FACS Canto II (BD Biosciences). The data was analyzed using FlowJo (TreeStar software ver. 10.8.1).
Analysis of metabolic parameters
To measure metabolic parameters, activated NKT cells (1 × 105) were incubated with different reagents as indicated in the figure legends. To measure mitochondrial mass and potential, cells were incubated with 30 nM MitoTracker™ Green (Invitrogen) and 60 nM of tetramethylrhodamine methyl ester perchlorate (TMRM) (Invitrogen), respectively, for 30 min at 37°C in RPMI 1640 complete media. Mitochondrial ROS levels were measured by incubating cells in 2.5 μM MitoSOX (Invitrogen) for 30 min at 37°C in RPMI 1640 complete media. To measure total cellular ROS, activated NKT cells were incubated with 1 mM 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) (Invitrogen) in RPMI complete media for 30 min at 37°C. To measure glucose uptake, cells were incubated in 2-(N-(7-nitrobenz-2-oxa-1,3-diaxol-4-yl) amino)-2-deoxyglucose (2-NBDG) (Invitrogen) (20 μM) for 1 h at 37°C in glucose-free RPMI 1640 media containing 10% dialyzed FBS. To measure GSH, cells were stained using an intracellular glutathione detection assay kit (Abcam) for 20 min at 37°C in RPMI 1640 complete media. Cells were stained for surface antigens and acquired on a FACS Canto II (BD Biosciences).
ATP, glutamine/glutamate and αKG assays
CellTiter-Glo® Luminescent Cell Viability reagent (Promega) was used for ATP measurement. Intracellular glutamate levels were measured using Glutamine/Glutamate-Glo™ Assay kit (Promega). αKG was measured using a colorimetric assay kit (Sigma Aldrich). All kits were used according to manufacturer’s instructions.
ELISA
Supernatants were collected from activated NKT after 3 days of stimulation. ELISA was performed in conjunction with the University of Michigan ELISA core.
Metabolite measurements
Cell lysate was prepared from resting NKT and CD4 T cells as well as stimulated NKT cells (5 × 105 cells per replicate) by incubating the cells with 80% methanol and following a series of vigorous mixing steps. Media was mixed with 100% methanol and vigorously vortexed. Cells and media were spun down at maximum speed for 10 min at 4°C to remove membranous debris, and the lysate was collected for drying using a SpeedVac. Following drying, the lysate was reconstituted using 50/50 methanol/water for mass spectrometry-based metabolomics analysis using an Agilent 1290 Infinity II UHPLC combined with an Agilent 6470 QQQ LC/MS.
RT gene PCR assay
Total RNA was isolated from unstimulated and stimulated NKT cells using a RNeasy Plus mini kit (Qiagen). PCR Array was performed according to the manufacturer’s instructions (Qiagen) using Applied Biosystem’s 7900HT Sequence Detection System. Fold changes were calculated from ΔCt values (gene of interest Ct value - an average of all housekeeping gene Ct values) using the ΔΔCt method. Gene expression of target genes was normalized to β-actin.
Listeria monocytogenes infection
Listeria monocytogenes expressing ovalbumin (LM-Ova strain 10403s) was grown in BHI broth media. Bacteria in a mid-log phase were collected for infection. GLS1 KO and WT littermate mice were injected intraperitoneally with either 200 μL of sterile 1X PBS alone or 200 μL of 1X PBS containing 105 CFU/mouse of LM-Ova. On day two post-infection, the bacterial burden was enumerated from homogenized spleen and liver samples by culturing serially diluted samples on LB agar plates and performing CFU determination. Intracellular cytokine expression by NKT cells was measured as described above.
Metabolic seahorse assay
WT and GLS1 KO mice were injected with 5 μg αGalcer per mouse. On day 3 post-activation, NKT cells were sorted. NKT cells were deposited in the XF96 well microplate coated with polylysine at a density of 2 × 105 NKT cells per well in glucose-free Seahorse media (Sigma Aldrich) and the plate was briefly spun to affix the cells to the bottom of the wells. The plate was incubated for 30 min in a non-CO2 incubator. ECAR was measured using glucose (10 mM) (Sigma Aldrich), oligomycin (1 mM, ATP coupler) (Sigma Aldrich), and 2-deoxyglucose (2-DG, 100 mM) (Sigma Aldrich) in seahorse assay medium. OCR was measured using oligomycin (1 mM), FCCP (1.5 mM, Sigma Aldrich), rotenone (1 mM, Sigma Aldrich) and antimycin (1mM, Sigma Aldrich) in Seahorse assay medium. Both assays were run using a Seahorse XFe96 bioanalyzer (Agilent Technologies).
QUANTIFICATION AND STATISTICAL ANALYSIS
All graphs and statistical analyses were prepared using Prism software (Prism version 9; Graphpad Software, San Diego, CA). In figures showing representative experiments, error bars represent the standard deviation (SD). The graphs depicting repeated experiments show standard error of mean (SEM). For comparison among multiple groups, data were analyzed using one-way ANOVA with the multi-comparison post-hoc test. Unpaired and paired Student’s t-tests were used for comparison between two groups. p < 0.05 was considered statistically significant.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Anti-Mouse CD4 (clone RM4-5) | eBioscience | Cat# 45-0042-82 |
| Anti-Mouse CD8a (clone 53-6.7) | BD Biosciences | Cat# 560778 |
| Anti-Mouse TCRβ (clone H57-597) | eBioscience | Cat# 48-5961-82 |
| Anti O-GlcNAc (clone RL2) | Novus Biologicals | NB300524Y |
| Anti-L PHA | Invitrogen | Cat# L11270 |
| Anti-CD25 (clone PC61.5) | eBioscience | Cat# 50-157-81 |
| Recombinant Anti-HK2 | Abcam (clone EPR20839) | Cat# Ab 209847 |
| Anti-Mouse IFN-γ (clone XMG1.2) | eBioscience | Cat# 11-7311-82 |
| Anti-Mouse CD24 (clone (M1/69) | BD Biosciences | Cat# 562477 |
| Anti-Mouse NK1.1 (clone PK136) | eBioscience | Cat# 45-5941-82 |
| Anti-Mouse CD44 (clone IM7) | eBioscience | Cat# 11-0441-85 |
| Anti-Mouse IL-4 (clone 11B11) | eBioscience | Cat# 25-7041-82 |
| Anti-Mouse IL-17 (clone TC11-18H10) | BD Biosciences | Cat# 560821 |
| Anti-Human/Mouse T-bet (clone eBio4B10) | eBioscience | Cat# 45-5825-82 |
| Anti-Human/Mouse PLZF (clone Mags-21F7) | eBioscience | Cat# 53-9320-82 |
| Anti-Mouse Ki-67 (clone SolA15) | eBioscience | Cat# 46-5698-80 |
| Anti-mouse Glut1 antibody (clone EPR3915) | Abcam | Cat# 115730 |
| Anti-Human/Mouse RORγT (clone AFKJS-9) | eBioscience | Cat# 12-6988-80 |
| Anti-mouse Hexokinase 2 antibody (clone EPR20839) | Abcam | Cat# ab209847 |
| Phospho-S6 Ribosomal Protein (Ser235/236) antibody | Cell Signaling | Cat# 2211 |
| Anti-mouse Total S6 Ribosomal Protein (5G10) antibody | Cell Signaling | Cat# 2217 |
| Anti- β-actin mouse monoclonal antibody | Sigma Aldrich | Cat# A1978 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| Listeria monocytogenesis | Mary O’Riordan lab (University of Michigan) | (LM-Ova strain 10403s) |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Anti-mouse CD3 (145-2C11) monoclonal antibody | Invitrogen | Cat# 16-0031-85 |
| PBS57-loaded mouse CD1d tetramers | NIH tetramer facility | |
| Anti-mouse CD28 (37.5) functional grade | Invitrogen | Cat# 50-112-9711 |
| RPMI 1640 | Gibco™ | Cat#11875093 |
| PMA | Sigma-Aldrich | Cat# P8139 |
| Ionomycin | Sigma-Aldrich | Cat# I0634 |
| GolgiPlug | BD Biosciences | Cat# 555029 |
| α-galactosylceramide (α-GalCer) | Diagnocine | Cat# KRN7000 |
| CB839 GLS inhibitor | Sigma Aldrich | Cat# 533717001 |
| L-buthionine-sulfoximine (BSO) | Sigma Aldrich | Cat# B2515 |
| (−)Epigallocatechin gallate (EGCG) | Sigma Aldrich | Cat# E4143 |
| 6-DIAZO-5-OXO-L-NORLEUCINE (DON) | Sigma Aldrich | Cat# D2141 |
| OMSI | Sigma Aldrich | Cat# SML1621 |
| Sodium pyruvate | Gibco | Cat# 11360-070 |
| Glucose | Gibco | Cat# A24940-01 |
| Glutamine | Gibco | Cat# 25030081 |
| 2- Deoxyglucose (2-DG) | Sigma | Cat# D8375 |
| Dimethyl 2-oxoglutarate (DMαKG) | Sigma | Cat# 349631 |
| Oligomycin | Sigma | Cat# O4876-5MG |
| Carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone) (FCCP) | Sigma | Cat# C2920 |
| Rotenone | Sigma | Cat# R8875 |
| Antimycin | Sigma | Cat# A8674 |
| Corning® Cell-Tak™ Cell and Tissue Adhesive | Corning | Cat# 354240 |
| Guinea pig serum | MP Biomedicals | Cat# 642831 |
| CellTrace™ Violet | Invitrogen | Cat# C34557 |
| DCFDA | Invitrogen | Cat# C6827 |
| 2-NBDG | Cayman Chemicals | Cat# 11046 |
| MitoTracker Green | Invitrogen | Cat# M7514 |
| TMRM | Invitrogen | Cat# T668 |
| SYBR green PCR mix | Applied Biosystems | Cat# 4309155 |
| MitoSOX | Invitrogen | Cat# M36008 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Cytofix/Cytoperm Plus | BD Biosciences | Cat# 554714 |
| Transcription factor staining kit | eBiosciences | Cat # 00-5523-00 |
| RNeasy kit | Qiagen | Cat# 74004 |
| RT2 First Strand Kit | Qiagen | Cat# 330404 |
| aKetoglutarate assay kit | Sigma | Cat# MAK054-1KT |
| Glutamine/glutamate assay kit | Promgea | Cat# J8021 |
| GSH Detection | Abcam | Cat# ab112132 |
| Cell Titer-Glo Luminescent Cell Viability | Promega | Cat# G7570 |
| EasySep Mouse Naïve CD4+Tcell Iso Kit | STEM CELL | Cat# 19765 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: GLS1fl/fl(Glstm2.1Sray/j) | The Jackson Laboratory | Stock No 017894 |
| Mouse: C57BL/6J | The Jackson Laboratory | Stock No: 000664 |
| Mouse: AMPKfl/fl (Prkaa1tm1.1sjm/j) | The Jackson Laboratory | Stock No: 014141 |
| Mouse: B6.Cg-Tg(Cd4-cre)1Cwi/BfluJ | The Jackson Laboratory | Stock No: 022071 |
|
| ||
| Oligonucleotides | ||
|
| ||
| qPCR primers for various genes | This paper | Table S3 |
|
| ||
| Software and algorithms | ||
|
| ||
| ImageJ | ImageJ | https://imagej.nih.gov/ij/ |
| Flowjo ver. 10.8.1 | TreeStar software | https://www.flowjo.com/ |
| BioRender | BioRender | www.biorender.com |
| GraphPad Prism 8 | GraphPad Software | www.graphpad.com |
Highlights.
Glutamine-enabled TCA cycle, GSH synthesis, and HBP are critical for NKT cell homeostasis
Glutaminolysis and HBP differentially regulate IL-4 and IFNγ production by NKT cells
Glutamine metabolism in NKT cells appears to be controlled by AMPK-mTORC1 signaling
ACKNOWLEDGMENTS
We would like to thank Chauna Black for maintaining our mouse colony and performing genetic screening of all mice. We thank Dr. Mary O’Riordan (University of Michigan) for providing the 10403s LM-Ova strain of Listeria monocytogenes. Lastly, we acknowledge the National Institutes of Health Tetramer Facility for providing the CD1d tetramers necessary to study NKT cells. This work was supported by National Institutes of Health (NIH) grants R01 AI121156 and R01 AI148289 (to C.-H.C.). C.A.L. was supported by the National Cancer Institute (NCI) (grants R37 CA237421 and R01 CA248160). Metabolomics studies performed at the University of Michigan were supported by NIH grant DK097153, the Charles Woodson Research Fund, and the UM Pediatric Brain Tumor Initiative.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
DECLARATION OF INTERESTS
C.A.L. has received consulting fees from Astellas Pharmaceuticals and Odyssey Therapeutics and is an inventor on patents pertaining to K-Ras-regulated metabolic pathways and redox-control pathways in cancer and targeting the glutamic-oxaloacetic transaminase 1 (GOT1) pathway as a therapeutic approach.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111516.
REFERENCES
- Abu Aboud O, Habib SL, Trott J, Stewart B, Liang S, Chaudhari AJ, Sutcliffe J, and Weiss RH (2017). Glutamine addiction in Kidney cancer suppresses oxidative stress and can Be Exploited for Real-time Imaging. Cancer Res. 77, 6746–6758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KG, Stromnes IM, and Greenberg PD (2017). Obstacles posed by the tumor microenvironment to T cell activity: a Case for Synergistic therapies. Cancer Cell 31, 311–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araujo L, Khim P, Mkhikian H, Mortales CL, and Demetriou M (2017). Glycolysis and glutaminolysis cooperatively control T cell function by limiting metabolite supply to N-glycosylation. Elife 6, e21330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron JL, Gardiner L, Nishimura S, Shinkai K, Locksley R, and Ganem D (2002). Activation of a nonclassical NKT cell subset in a transgenic mouse model of hepatitis B virus infection. Immunity 16, 583–594. [DOI] [PubMed] [Google Scholar]
- Beaudoin L, Laloux V, Novak J, Lucas B, and Lehuen A (2002). NKT cells inhibit the onset of diabetes by impairing the development of pathogenic T cells specific for pancreatic beta cells. Immunity 17, 725–736. [DOI] [PubMed] [Google Scholar]
- Blagih J, Coulombe F, Vincent EE, Dupuy F, Galicia-Vazquez G, Yurchenko E, Raissi TC, van der Windt GJ, Viollet B, Pearce EL, et al. (2015). The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity 42, 41–54. [DOI] [PubMed] [Google Scholar]
- Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, Turay AM, and Frauwirth KA (2010). Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J. Immunol. 185, 1037–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cetinbas NM, Sudderth J, Harris RC, Cebeci A, Negri GL, Yilmaz OH, DeBerardinis RJ, and Sorensen PH (2016). Glucose-dependent anaplerosis in cancer cells is required for cellular redox balance in the absence of glutamine. Sci. Rep. 6, 32606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CH, Curtis JD, Maggi LB Jr., Faubert B, Villarino AV, O’Sullivan D, Huang SC, van der Windt GJ, Blagih J, Qiu J, et al. (2013). Post-transcriptional control of T cell effector function by aerobic glycolysis. Cell 153, 1239–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, and Cui H (2015). Targeting glutamine induces apoptosis: a cancer Therapy approach. Int. J. Mol. Sci. 16, 22830–22855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo AY, Kim SG, Vander Heiden MG, Mahoney SJ, Vu H, Yoon SO, Cantley LC, and Blenis J (2010). Glucose addiction of TSC null cells is caused by failed mTORC1-dependent balancing of metabolic demand with supply. Mol. Cell 38, 487–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosby CM, and Kronenberg M (2016). Invariant natural killer T cells: front line fighters in the war against pathogenic microbes. Immunogenetics 68, 639–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J, Shin T, Kawano T, Sato H, Kondo E, Toura I, Kaneko Y, Koseki H, Kanno M, and Taniguchi M (1997). Requirement for Valpha14 NKT cells in IL-12-mediated rejection of tumors. Science 278, 1623–1626. [DOI] [PubMed] [Google Scholar]
- Dhodapkar MV, Geller MD, Chang DH, Shimizu K, Fujii S, Dhodapkar KM, and Krasovsky J (2003). A reversible defect in natural killer T cell function characterizes the progression of premalignant to malignant multiple myeloma. J. Exp. Med. 197, 1667–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran RV, Oppliger W, Robitaille AM, Heiserich L, Skendaj R, Gottlieb E, and Hall MN (2012). Glutaminolysis activates Rag-mTORC1 signaling. Mol. Cell 47, 349–358. [DOI] [PubMed] [Google Scholar]
- Durante-Mangoni E, Wang R, Shaulov A, He Q, Nasser I, Afdhal N, Koziel MJ, and Exley MA (2004). Hepatic CD1d expression in hepatitis C virus infection and recognition by resident proinflammatory CD1d-reactive T cells. J. Immunol. 173, 2159–2166. [DOI] [PubMed] [Google Scholar]
- Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S, et al. (2010). Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 39, 171–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu S, He K, Tian C, Sun H, Zhu C, Bai S, Liu J, Wu Q, Xie D, Yue T, et al. (2020). Impaired lipid biosynthesis hinders anti-tumor efficacy of intratumoral iNKT cells. Nat. Commun. 11, 438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, Kogadeeva M, Picotti P, Meissner F, Mann M, et al. (2016). L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell 167, 829–842.e813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gera JF, Mellinghoff IK, Shi Y, Rettig MB, Tran C, Hsu JH, Sawyers CL, and Lichtenstein AK (2004). AKT activity determines sensitivity to mammalian target of rapamycin (mTOR) inhibitors by regulating cyclin D1 and c-myc expression. J. Biol. Chem. 279, 2737–2746. [DOI] [PubMed] [Google Scholar]
- Gorrini C, Harris IS, and Mak TW (2013). Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 12, 931–947. [DOI] [PubMed] [Google Scholar]
- Gubser PM, Bantug GR, Razik L, Fischer M, Dimeloe S, Hoenger G, Durovic B, Jauch A, and Hess C (2013). Rapid effector function of memory CD8+ T cells requires an immediate-early glycolytic switch. Nat. Immunol. 14, 1064–1072. [DOI] [PubMed] [Google Scholar]
- Gupta S, Hastak K, Afaq F, Ahmad N, and Mukhtar H (2004). Essential role of caspases in epigallocatechin-3-gallate-mediated inhibition of nuclear factor kappa B and induction of apoptosis. Oncogene 23, 2507–2522. [DOI] [PubMed] [Google Scholar]
- Hensley CT, Wasti AT, and DeBerardinis RJ (2013). Glutamine and cancer: cell biology, physiology, and clinical opportunities. J. Clin. Invest. 123, 3678–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, Tsui YC, Cui G, Micevic G, Perales JC, et al. (2015). Phosphoenolpyruvate is a metabolic Checkpoint of anti-tumor T cell responses. Cell 162, 1217–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichiyama K, Chen T, Wang X, Yan X, Kim BS, Tanaka S, Ndiaye-Lobry D, Deng Y, Zou Y, Zheng P, et al. (2015). The methylcytosine dioxygenase Tet2 promotes DNA demethylation and activation of cytokine gene expression in T cells. Immunity 42, 613–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illes Z, Kondo T, Newcombe J, Oka N, Tabira T, and Yamamura T (2000). Differential expression of NK T cell V alpha 24J alpha Q invariant TCR chain in the lesions of multiple sclerosis and chronic inflammatory demyelinating polyneuropathy. J. Immunol. 164, 4375–4381. [DOI] [PubMed] [Google Scholar]
- Jewell JL, Kim YC, Russell RC, Yu FX, Park HW, Plouffe SW, Tagliabracci VS, and Guan KL (2015). Metabolism. Differential regulation of mTORC1 by leucine and glutamine. Science 347, 194–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MO, Wolf MM, Madden MZ, Andrejeva G, Sugiura A, Contreras DC, Maseda D, Liberti MV, Paz K, Kishton RJ, et al. (2018). Distinct regulation of Th17 and Th1 cell differentiation by glutaminase-dependent metabolism. Cell 175, 1780–1795.e1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YH, Kumar A, Chang CH, and Pyaram K (2017). Reactive oxygen species regulate the inflammatory function of NKT cells through promyelocytic leukemia zinc finger. J. Immunol. 199, 3478–3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klysz D, Tai X, Robert PA, Craveiro M, Cretenet G, Oburoglu L, Mongellaz C, Floess S, Fritz V, Matias MI, et al. (2015). Glutamine-dependent alpha-ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci. Signal. 8, ra97. [DOI] [PubMed] [Google Scholar]
- Kovalovsky D, Uche OU, Eladad S, Hobbs RM, Yi W, Alonzo E, Chua K, Eidson M, Kim HJ, Im JS, et al. (2008). The BTB-zinc finger transcriptional regulator PLZF controls the development of invariant natural killer T cell effector functions. Nat. Immunol. 9, 1055–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreslavsky T, Savage AK, Hobbs R, Gounari F, Bronson R, Pereira P, Pandolfi PP, Bendelac A, and von Boehmer H (2009). TCR-inducible PLZF transcription factor required for innate phenotype of a subset of gammadelta T cells with restricted TCR diversity. Proc. Natl. Acad. Sci. USA 106, 12453–12458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Pyaram K, Yarosz EL, Hong H, Lyssiotis CA, Giri S, and Chang CH (2019). Enhanced oxidative phosphorylation in NKT cells is essential for their survival and function. Proc. Natl. Acad. Sci. USA 116, 7439–7448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WJ, Shim JY, and Zhu BT (2005). Mechanisms for the inhibition of DNA methyltransferases by tea catechins and bioflavonoids. Mol. Pharmacol. 68, 1018–1030. [DOI] [PubMed] [Google Scholar]
- Lin CH, Shen YA, Hung PH, Yu YB, and Chen YJ (2012). Epigallocathechin gallate, polyphenol present in green tea, inhibits stem-like characteristics and epithelial-mesenchymal transition in nasopharyngeal cancer cell lines. BMC Complement Altern. Med. 12, 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisbonne M, Diem S, de Castro Keller A, Lefort J, Araujo LM, Hachem P, Fourneau JM, Sidobre S, Kronenberg M, Taniguchi M, et al. (2003). Cutting edge: invariant V alpha 14 NKT cells are required for allergen-induced airway inflammation and hyperreactivity in an experimental asthma model. J. Immunol. 171, 1637–1641. [DOI] [PubMed] [Google Scholar]
- Ma Z, Vocadlo DJ, and Vosseller K (2013). Hyper-O-GlcNAcylation is anti-apoptotic and maintains constitutive NF-kappaB activity in pancreatic cancer cells. J. Biol. Chem. 288, 15121–15130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak TW, Grusdat M, Duncan GS, Dostert C, Nonnenmacher Y, Cox M, Binsfeld C, Hao Z, Brustle A, Itsumi M, et al. (2017). Glutathione Primes T cell metabolism for inflammation. Immunity 46, 675–689. [DOI] [PubMed] [Google Scholar]
- Metelitsa LS, Wu HW, Wang H, Yang Y, Warsi Z, Asgharzadeh S, Groshen S, Wilson SB, and Seeger RC (2004). Natural killer T cells infiltrate neuroblastomas expressing the chemokine CCL2. J. Exp. Med. 199, 1213–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, Yang H, Hild M, Kung C, Wilson C, et al. (2009). Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 136, 521–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota Y, Ishihara S, Otani K, Yasuda K, Nishikawa T, Tanaka T, Tanaka J, Kiyomatsu T, Kawai K, Hata K, et al. (2016). Effect of nutrient starvation on proliferation and cytokine secretion of peripheral blood lymphocytes. Mol. Clin. Oncol. 4, 607–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Tsang M, Iritani BM, and Bevan MJ (2014). Metabolic regulator Fnip1 is crucial for iNKT lymphocyte development. Proc. Natl. Acad. Sci. USA 111, 7066–7071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce EL, and Pearce EJ (2013). Metabolic pathways in immune cell activation and quiescence. Immunity 38, 633–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, Jones RG, and Choi Y (2009). Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature 460, 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prevot N, Pyaram K, Bischoff E, Sen JM, Powell JD, and Chang CH (2015). Mammalian target of rapamycin complex 2 regulates invariant NKT cell development and function independent of promyelocytic leukemia zinc-finger. J. Immunol. 194, 223–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qing G, Li B, Vu A, Skuli N, Walton ZE, Liu X, Mayes PA, Wise DR, Thompson CB, Maris JM, et al. (2012). ATF4 regulates MYC-mediated neuroblastoma cell death upon glutamine deprivation. Cancer Cell 22, 631–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renner K, Geiselhoringer AL, Fante M, Bruss C, Farber S, Schonhammer G, Peter K, Singer K, Andreesen R, Hoffmann P, et al. (2015). Metabolic plasticity of human T cells: Preserved cytokine production under glucose deprivation or mitochondrial restriction, but 2-deoxy-glucose affects effector functions. Eur. J. Immunol. 45, 2504–2516. [DOI] [PubMed] [Google Scholar]
- Roy S, Rizvi ZA, and Awasthi A (2018). Metabolic Checkpoints in differentiation of helper T cells in Tissue inflammation. Front. Immunol. 9, 3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salio M, Puleston DJ, Mathan TS, Shepherd D, Stranks AJ, Adamopoulou E, Veerapen N, Besra GS, Hollander GA, Simon AK, et al. (2014). Essential role for autophagy during invariant NKT cell development. Proc. Natl. Acad. Sci. USA 111, E5678–E5687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmond RJ (2018). mTOR regulation of glycolytic metabolism in T cells. Front. Cell Dev. Biol. 6, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage AK, Constantinides MG, and Bendelac A (2011). Promyelocytic leukemia zinc finger turns on the effector T cell program without requirement for agonist TCR signaling. J. Immunol. 186, 5801–5806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage AK, Constantinides MG, Han J, Picard D, Martin E, Li B, Lantz O, and Bendelac A (2008). The transcription factor PLZF directs the effector program of the NKT cell lineage. Immunity 29, 391–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, Wang CR, Schumacker PT, Licht JD, Perlman H, et al. (2013). Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 38, 225–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu M, Adachi S, Masuda M, Kozawa O, and Moriwaki H (2011). Cancer chemoprevention with green tea catechins by targeting receptor tyrosine kinases. Mol. Nutr. Food Res. 55, 832–843. [DOI] [PubMed] [Google Scholar]
- Shin J, Wang S, Deng W, Wu J, Gao J, and Zhong XP (2014). Mechanistic target of rapamycin complex 1 is critical for invariant natural killer T-cell development and effector function. Proc. Natl. Acad. Sci. USA 111, E776–E783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyer JA, Flavell RA, and Bailis W (2020). Metabolic signaling in T cells. Cell Res. 30, 649–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sklarz T, Guan P, Gohil M, Cotton RM, Ge MQ, Haczku A, Das R, and Jordan MS (2017). mTORC2 regulates multiple aspects of NKT-cell development and function. Eur. J. Immunol. 47, 516–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swamy M, Pathak S, Grzes KM, Damerow S, Sinclair LV, van Aalten DM, and Cantrell DA (2016). Glucose and glutamine fuel protein O-GlcNAcylation to control T cell self-renewal and malignancy. Nat. Immunol. 17, 712–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, and Thompson CB (2009). Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viale R, Ware R, Maricic I, Chaturvedi V, and Kumar V (2012). NKT cell subsets can Exert Opposing effects in autoimmunity, tumor Surveillance and inflammation. Curr. Immunol. Rev. 8, 287–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss K, Hong HS, Bader JE, Sugiura A, Lyssiotis CA, and Rathmell JC (2021). A guide to interrogating immunometabolism. Nat. Rev. Immunol. 21, 637–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, and Hogquist KA (2018). How lipid-specific T cells become effectors: the differentiation of iNKT subsets. Front. Immunol. 9, 1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng X, Kumar A, Cao L, He Y, Morgun E, Visvabharathy L, Zhao J, Sena LA, Weinberg SE, Chandel NS, et al. (2021). Mitochondrial metabolism is essential for invariant natural killer T cell development and function. Proc. Natl. Acad. Sci. USA 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo HC, Yu YC, Sung Y, and Han JM (2020). Glutamine reliance in cell metabolism. Exp. Mol. Med. 52, 1496–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarrouk M, Rolf J, and Cantrell DA (2013). LKB1 mediates the development of conventional and innate T cells via AMP-dependent kinase autonomous pathways. PLoS One 8, e16027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Tschumi BO, Corgnac S, Ruegg MA, Hall MN, Mach JP, Romero P, and Donda A (2014). Mammalian target of rapamycin complex 1 orchestrates invariant NKT cell differentiation and effector function. J. Immunol. 193, 1759–1765. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Unprocessed data underlying the display items in the manuscript, related to figures are available from the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.