

Abstract
Background/Aims:
Despite the published evidence implicating phosphoinositide 3-kinase (PI3-kinase) in the regulation of islet function, limited information is available on the putative contributory roles of its downstream signaling steps, including the phosphatidylinositol-3,4,5-trisphosphate-dependent Rac exchange factor 1 (P-Rex1) signaling pathway in the islet β-cell. Therefore, we investigated potential roles for P-Rex1 in glucose-stimulated Rac1 activation and insulin secretion in insulin-secreting (INS-1 832/13) β-cells.
Methods:
Glucose-stimulated Insulin secretion (GSIS) was quantified by ELISA. Expression of endogenous P-Rex1 and RhoG was suppressed by siRNA transfection using the DharmaFect1 reagent. Total membrane and cytosolic fractions were isolated using the Mem-PER Plus Membrane Extraction Kit. The degree of activation of Rac1 was determined by the pull-down assay.
Results:
P-Rex1 is expressed in INS-1 832/13 cells, normal rat islets and human islets. siRNA-mediated knockdown of P-Rex1 attenuated glucose-induced Rac1 activation, membrane association and insulin secretion. RhoG, which has been implicated in PI3-kinase-mediated Rac1 activation in other cell types, appears not to contribute to GSIS since the siRNA-mediated knockdown of RhoG failed to exert significant effects on GSIS. LY294002, a known inhibitor of PI3-kinase, potentiated GSIS without affecting glucose-induced Rac1 activation.
Conclusion:
Based on these findings, we conclude that P-Rex1 plays a novel regulatory role in glucose-induced Rac1 activation and insulin secretion.
Keywords: Rac1, P-Rex1, Insulin secretion, G proteins, Guanine nucleotide exchange factors, Pancreatic β-cell
Introduction
Insulin secretion from the pancreatic β-cell is regulated precisely by the ambient concentration of glucose [1, 2]. However, the molecular and cellular mechanisms underlying the stimulus-secretion coupling of glucose-stimulated insulin secretion (GSIS) remain only partially understood. Available evidence along these lines suggests that GSIS is mediated largely via the generation of hydrophobic (e.g., diacylglycerol, phosphatidylinositol and lysophospholipids) as well as hydrophilic (e.g., inositol triphosphates, cyclic nucleotides) second messengers. In addition, alterations in cationic events (e.g., intracellular calcium levels) have also been shown to play a critical regulatory role in the cascade of events leading to GSIS [3–7].
It is noteworthy that, in addition to adenine nucleotides (ATP), several earlier studies have documented evidence in support of critical regulatory roles for GTP in physiological insulin secretion [8, 9]. For example, using specific inhibitors for inosine monophosphate dehydrogenase (e.g., mycophenolic acid), Metz and coworkers have demonstrated regulatory roles for GTP in GSIS [8, 10, 11]. Furthermore, GTP has been shown to be necessary for the regulation of islet β-cell functions, including proliferation and GSIS via activation of a variety of heterotrimeric and small molecular mass GTP-binding proteins (G proteins) [12]. In this context, numerous studies have established that small G proteins (e.g., Arf6, Cdc42, Rac1, RhoA and Rap1) play significant roles in cytoskeletal remodeling thereby favoring mobilization of secretory granules to the plasma membrane for fusion and release of their cargo into circulation in a glucose-stimulated β-cell [reviewed in [13–15]]. Furthermore, small G proteins belonging to the Rab G protein family (e.g., Rab3, Rab27A) have been shown to regulate insulin secretion by promoting insulin-granule translocation and fusion with the plasma membrane [13–15]. Lastly, in addition to small G proteins, glucose has been shown to mediate activation of heterotrimeric G proteins either via classical (canonical) G protein-coupled receptor (GPCR)-mediated mechanisms or via non-canonical signaling pathways involving activation of novel histidine kinase-derived signaling mechanisms [15, 16].
It is well established that G proteins undergo an activation-deactivation cycle between their inactive (GDP-bound) and active (GTP-bound) conformations, which are tightly controlled by specific regulatory proteins/factors [13–15]. At least three major types of regulatory proteins/factors have been described for activation-deactivation of small G proteins (e.g., Rac1; the focus of the current investigation). The first group is comprised of guanine nucleotide exchange factors (GEFs), which facilitate the conversion of the GDP-bound (inactive) forms to their corresponding GTP-bound (active) forms. The second group that regulates small G proteins is the GDP-dissociation inhibitors (GDIs), which prevent the dissociation of GDP from G proteins, and hence are considered negative modulators of the G protein activation cascade. Lastly, the third group represents the GTPase-activating proteins (GAPs); these proteins promote the conversion of the GTP-bound G proteins to their GDP-bound (inactive) conformation to complete the GTP hydrolytic cycle. Recent studies from several laboratories including our own have identified GEFs, GDIs and GAPs for small G proteins, including Rac1 in the islet β-cell. These aspects of beta cell biology have been reviewed in [13–15].
Earlier evidence suggests that P-Rex1, a GEF for the Rho subfamily of G proteins, specifically Rac1, plays critical regulatory roles in cell function [17, 18]. This predominantly cytosolic GEF is activated by phosphatidylinositol-3,4,5-trisphosphate (PIP3), which is generated via the activation of PI3-kinase, and the βγ subunits of heterotrimeric G proteins in a GPCR-dependent fashion [17–20]. Along these lines recent studies have shown regulatory roles for PI3-kinase in cell motility in islet morphogenesis [21] and insulin secretion from the islet β-cell [22–24]. Using two structurally distinct inhibitors of PI3-kinase (Wortmannin and LY294002), Kolic and associates have demonstrated modulatory roles for PI3-kinase in insulin secretion, which appear to involve cellular homeostasis of cAMP. They also presented evidence for PI3-Kγ in insulin secretion induced by glucose-dependent insulinotropic polypeptide [25, 26].
Despite the above evidence implicating PI3-kinase in the regulation of islet function, including mitogenesis and insulin secretion, very little is known about potential regulatory roles of its downstream signaling steps, including the P-Rex1-Rac1 signaling pathway in the islet β-cell. Therefore, we undertook the current investigation to examine the P-Rex1-Rac1 signaling module in GSIS in insulin-secreting INS-1 832/13 cells. We present evidence to support the hypothesis that P-Rex1 serves as a GEF for Rac1 in the cascade of events leading to GSIS.
Materials and Methods
Materials
P-Rex1 antibody was from R&D Systems (Minneapolis, MN, USA). Antisera directed against RhoG was from Santa Cruz Biotechnology (CA, USA). E-Cadherin, GAPDH and HRP-conjugated secondary antibodies were from Cell Signaling (Danvers, MA, USA). Rac1 antibody was from EMD Millipore (Burlington, MA, USA). Rat high range insulin ELISA was from ALPCO (Salem, NH, USA). Target sequence for the ON-TARGETplus Non-targeting siRNA#1 (Catalog Item- D-001810–01-20) is: UGGUUUACAUGUCGACUAA. The target sequences for the ON-TARGETplus Rat Prex1(311647) siRNA-SMARTpool (Catalog Item L-093719–02-0010 that has 4 sequences that it targets) are: GCAUGGAGCGCGACGCAUA, CAACAACAACGGCGAGUAU, UCCUGAAAGUCAACGGCAA and CCAUCAACGCCCUGGACGA. The above on-target P-Rex1siRNA SMARTpool and non-targeting control siRNA (Con-si), as well as DharmaFect1, were from Dharmacon (Lafayette, CO, USA). Antibody for β-actin and all other reagents used in the current studies were from Sigma Aldrich (St. Louis, MO, USA). Mem-PER Plus Membrane Extraction Kit was from Thermo Fisher Scientific (Waltham, MA, USA). The pull down assay kit used for the Rac1 activation was from Cytoskeleton (Denver, CO, USA).
Cell culture and treatment conditions
INS-1 832/13 cells were cultured in RPMI-1640 medium containing 10% FBS supplemented with 100 IU/ ml penicillin and 100 IU/ml streptomycin, 1 mM sodium pyruvate, 50 μM 2-mercapto-ethanol, and 10 mM HEPES (pH 7.4). The cultured cells were sub-cloned twice weekly. Cells were starved overnight in a low glucose /low serum growth medium prior to the treatment with different concentrations of glucose (2.5 or 20 mM) for various time points, as indicated in the text. For inhibitor studies, cells were pretreated with either Wortmannin (100 nM) or LY294002 (10 μM) and then exposed to LG (2.5 mM) and HG (20 mM) for indicated time points. DMSO was used as a vehicle control.
Islets isolation
All protocols involving animal care and use were reviewed and approved by Wayne State University and John D. Dingell VA Medical Center Institutional Animal Care and Use Committees. Islets from 8 to 10-week-old male Sprague–Dawley rats were isolated by the collagenase digestion method [27–29]. Human islets were from Prodo Labs (Aliso Viejo, CA). Studies involving human islets were approved by the Biosafety Committee at the John D. Dingell VA Medical Center.
siRNA-mediated knockdown of expression of P-Rex1 and RhoG
Expression of endogenous P-Rex1 or RhoG was suppressed by siRNA transfection as per manufacturer’s protocol. Cells were transfected with siRNA at a final concentration of 100 nM using the DharmaFect1 reagent. To assess the specificity of RNA interference, cells were transfected (as above) with non-targeting siRNA (i.e., control siRNA) duplexes specific for rat genome. Transfected cells were maintained in complete growth medium for 48 hrs. Cells were washed with ice-cold PBS and collected in RIPA lysis buffer containing protease inhibitors. Efficiency of the knockdown was determined by immunoblotting of lysates derived from control siRNA and P-Rex1 siRNA transfected cells. Actin was used as a loading control.
Insulin secretion assay
Following a 60 min pre-incubation at 37°C in the presence or absence of inhibitors, the cells were exposed to LG (2.5 mM) or HG (20 mM) for 45min. Insulin released into the medium was quantified by ELISA per the manufacturer’s instructions. Data were expressed as ng/ml insulin secreted in the medium.
Isolation of membrane and cytosolic fractions
Total membrane and cytosolic fractions were isolated using Mem-PER Plus Membrane Extraction Kit, as per manufacturer instructions, and used for the determination of relative abundance of P-Rex1 in these fractions by Western blotting. The purity of cytosolic and membrane fractions was assessed by enrichment of these fractions with specific protein markers, namely GAPDH and E-Cadherin, respectfully [29].
Rac1 activation assay
The degree of activation of Rac1 was determined using a pull-down assay kit [28–30]. In brief, INS-1 cells were exposed to basal (2.5 mM) or high (20 mM) glucose concentrations for 15min in Krebs-Ringer buffer. Lysates were clarified by centrifugation and p21-binding domain of p21-activated kinase beads were added to the supernatant. The mixture was rotated for 1 h at 4°C and pelleted by centrifuging at 4,000g for 3 min. The pellets were washed once with wash buffer then reconstituted in Laemmli buffer and boiled for 5min. Proteins were resolved by SDS-PAGE, transferred to a nitrocellulose membrane, and the relative abundance of Rac1 was determined by Western blotting.
Statistical analysis
Data are presented as mean ± SD of at least three independent experiments. Statistical analysis for differences between groups was done using the Student t-test. A p value of < 0.05 was considered statistically significant.
Results
P-Rex1 is expressed in INS-1 832/13 cells, normal rat and human islets, and siRNA-mediated knockdown of P-Rex1 attenuates GSIS in INS-1 832/13 cells
At the outset, we determined, by Western blotting, the expression of P-Rex1 in INS-1 832/13 cells, normal rat islets and human islets. Data in Fig. 1 (Panel A) indicate that P-Rex1 is expressed in all three insulin-secreting cells studied. We next investigated the role of P-Rex1 in GSIS. To address this, we employed siRNA-P-Rex1 to deplete the expression of endogenous P-Rex1 in INS-1 832/13 cells. Data depicted in Fig. 1 (Panel B) indicate a significant reduction in the expression of P-Rex1 following the transfection of siRNA-P-Rex1. Transfection of these cells with control siRNA did not exert any effect on the expression of P-Rex1 under these experimental conditions. Data from GSIS studies (Fig. 1; Panel C) indicated significant reduction in GSIS in INS-1 832/13 cells transfected with siRNA-P-Rex1, but not control siRNA. No significant effects of control siRNA or siRNA-P-Rex1 were seen on basal insulin secretion. Together, these data demonstrate that P-Rex1 plays a contributory role in the stimulus-secretion coupling of GSIS.
Fig. 1.
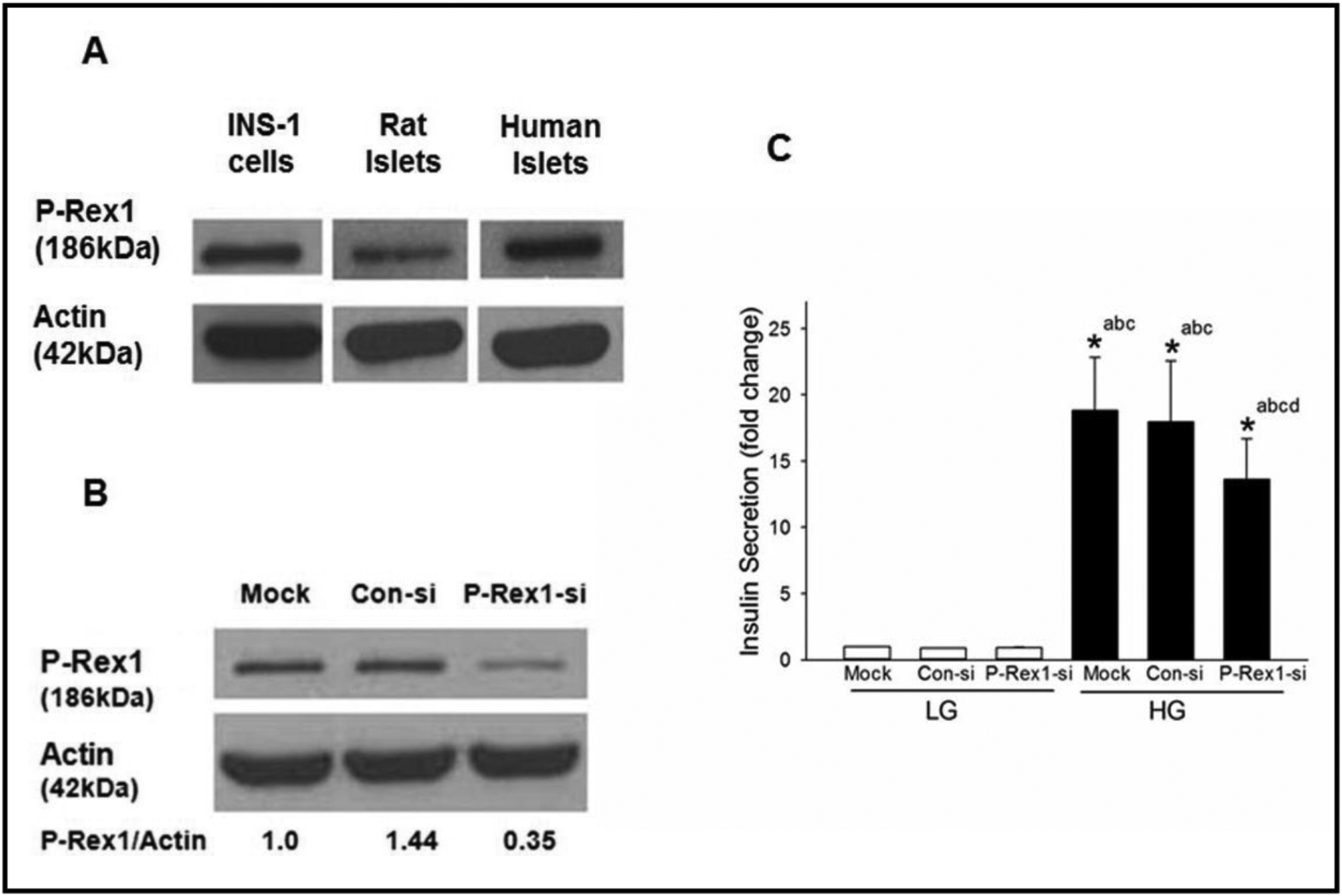
Expression of P-Rex1 in clonal INS-1 832/13 cells, rodent islets and human islets cells and siRNA mediated knockdown of P-Rex1 significantly attenuates GSIS in INS-1 832/13 cells. Panel A: Lysates from INS-1 832/13 cells, rat and human islets were analyzed for P-Rex1 protein expression by Western blot analysis. Actin was used as loading control. Panel B: INS-1 832/13 cells were transfected with Con-siRNA or siRNA targeted to P-Rex1 (P-Rex1-si). Cell lyates were analyzed by Western blotting for the expression of P-Rex1. Actin was used as loading control. A representative blot from three independent experiments is shown here. Panel C: GSIS was quantified in mock, Con-si and P-Rex1-si transfected INS-1 832/13 cells (see Methods for additional details). Data are mean ± SD from three experiments. The data are expressed as fold change relative to LG-Mock. (* p< 0.05) Comparisons shown: a -significant compared with LG treated mock; b - significant compared with LG-treated Con-si; c- significant compared with LG-treated P-Rex1-si; d: significant compared with HG-treated mock.
siRNA-mediated knockdown of P-Rex1 inhibits glucose-induced Rac1 activation in INS-1 832/13 cells
Next series of studies were aimed at understanding potential roles for P-Rex1 as a GEF in the cascade of events leading to glucose-induced activation of Rac1, which has been shown to be a requisite for GSIS to occur [13, 28, 31]. To address this, we quantified activation of glucose-induced activation of Rac1 in INS-1 832/13 cells under mock, control siRNA, or P-Rex siRNA transfection conditions. Data shown in Fig. 2 (Panel A) indicate a modest, but insignificant increase in Rac1 activation under basal conditions in cells transfected with either control siRNA or P-Rex1 siRNA. In line with previously published evidence [31], exposure of these cells to stimulatory glucose (20 mM for 15 min) markedly increased Rac1 activation under mock and control siRNA conditions. Depletion of P-Rex1 expression in these cells significantly suppressed glucose-induced Rac1 activation. Taken together, these findings suggest that P-Rex1 mediates glucose-induced Rac1 activation. Pooled data from multiple experiments are shown in Fig. 2 (Panel B).
Fig. 2.
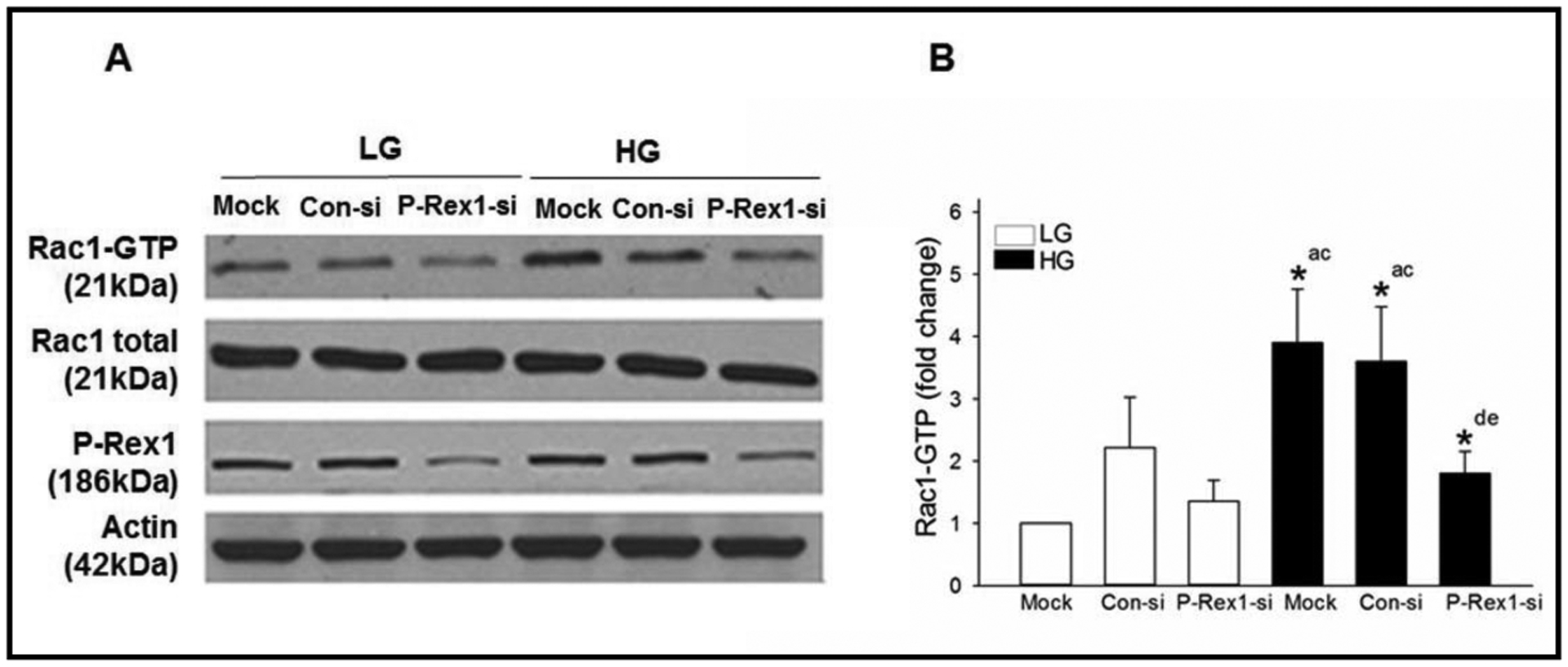
Knockdown of P-Rex1 expression inhibits glucose-stimulated Rac1 activation in INS-1 832/13 cells. Panel A: INS-1 832/13 cells were transfected with Con-si or P-Rex1-si as described in the Methods section. After 48 hours of transfection, cells were subjected to overnight starvation and then were treated with LG (2.5 mM) or HG (20 mM) for 15 mins. Rac1 activation was quantified by Rac1 pull down assay. Expression of total Rac1, P-Rex1 and actin in respective cell lysates is also provided. Representative blots from three independent studies are provided. Panel B: Densitometric quantitation of activated Rac1 in Panel A is shown here. The results from three independent experiments are presented as means ± SD. The data are expressed as fold change relative to LG-mock. (* p< 0.05) Comparisons shown: a - significant compared with LG-treated mock; b - significant compared with LG treated Con-si; c - significant compared with LG treated P-Rex1-si; d - significant compared with HG treated mock.
siRNA-mediated depletion of P-Rex1 inhibits glucose-induced membrane association of Rac1 in INS-1 832/13 cells
Previous studies in pancreatic beta cells have shown membrane targeting of Rac1 following exposure to stimulatory glucose conditions [29, 32]. Therefore, given our findings of glucose-induced P-Rex1 mediated activation of Rac1 (Fig. 2), we asked if P-Rex1 mediates targeting of Rac1 to the membrane fraction in the glucose-stimulated beta cell. To address this, relative abundance of Rac1 was determined, by Western blotting, in the cytosol and membrane fractions in INS-1 832/13 cells transfected with either control-siRNA or P-Rex1-siRNA under basal or glucose-stimulated conditions. The purity of cytosol and membrane fractions was assessed by determining the abundance of GAPDH and E-cadherin, respectively. Data in Fig. 3 (panel A) show that both cytosolic and membrane fractions isolated for these studies were pure. Furthermore, we noted that P-Rex1 is predominantly (~ 80% of total; n=5 experiments) cytosolic in distribution. We also observed a significant reduction in P-Rex1 expression in both cytosolic and membrane fractions following P-Rex1-siRNA transfection. In addition, we observed a modest, but significant, increase in membrane-associated Rac1 in glucose-stimulated INS-1 832/13 cells transfected with control-siRNA compared to basal conditions. More importantly, significant inhibition of membrane-associated Rac1 was seen in cells exposed to stimulatory, but not basal glucose (Fig. 3; Panel A). Pooled data from multiple experiments are shown in Fig. 3; Panel B. Taken together, data represented in Fig. 1–3 suggest that P-Rex1 might play contributory roles in glucose-induced membrane association and activation of Rac1 culminating in insulin secretion. In the next series of investigations, we explored potential mechanisms downstream to P-Rex1-mediated activation of Rac1 and stimulation of insulin secretion.
Fig. 3.
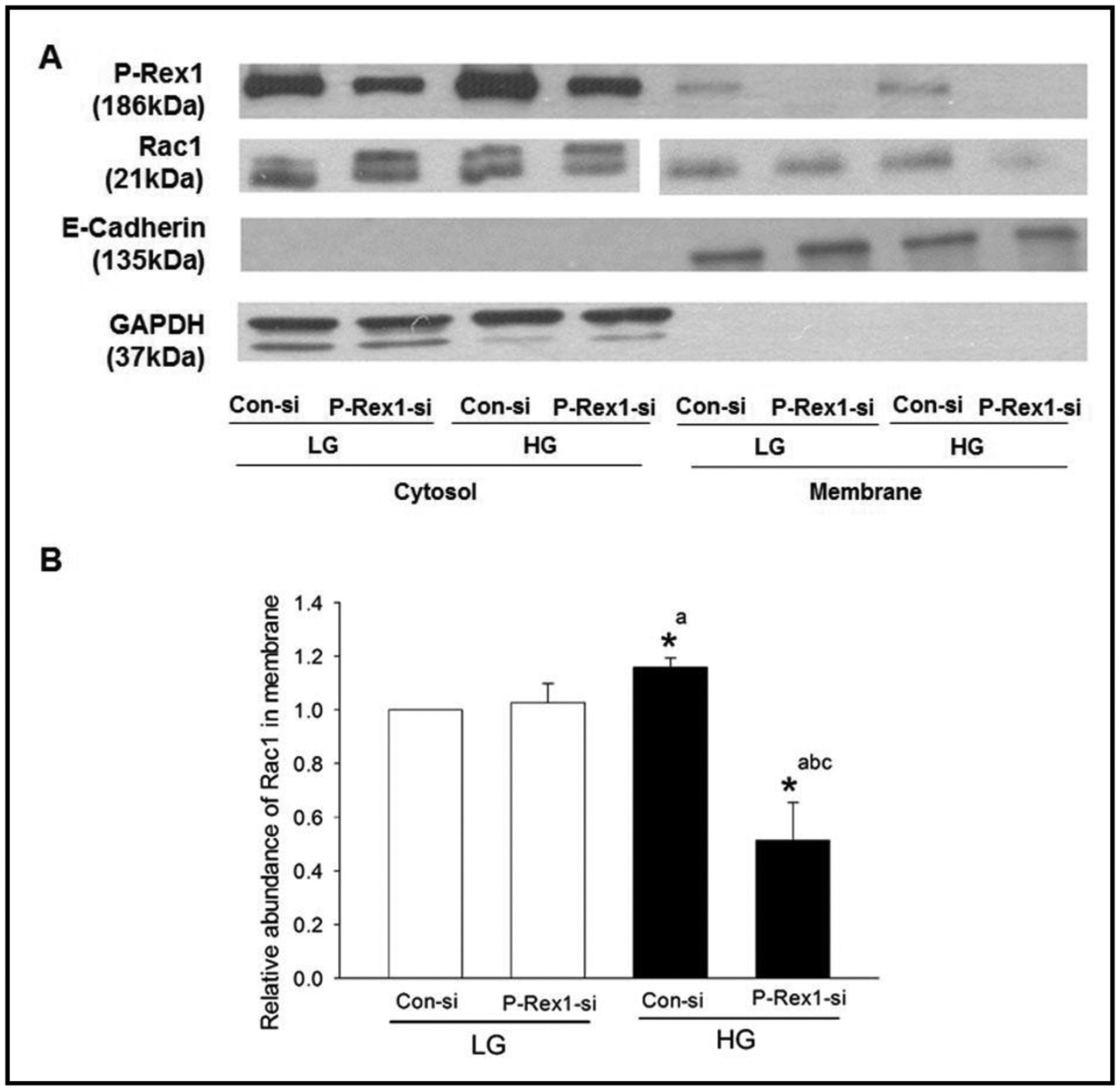
Depletion of P-Rex1 suppresses glucose-induced membrane targeting of Rac1 in INS-1 832/13 cells. Panel A: INS-1 832/13 cells were transfected with con-si or P-Rex1-si and exposed to either low glucose (LG, 2.5 mM) or high glucose (HG, 20 mM) for 15 minutes. Following incubation, total membrane and cytosolic fractions were isolated using a commercially available kit (see Methods), and relative abundance of P-Rex1 and Rac1 was determined by Western blotting. Purity of the cytosol and membrane fractions was assessed by expression of GAPDH and E-Cadherin in those fractions, respectively. A representative blot from five independent studies is shown. Panel B: Densitometric analysis of relative abundance of Rac1 in the membrane fraction obtained from studies described in Panel A. Data are expressed as mean ± SD from three experiments. (* p< 0.05) Comparisons shown: a- significant compared with LG Con-si; b- significant compared with LG P-Rex1-si; c- significant compared with HG Con-si.
RhoG, a known regulator of P-Rex1-Rac signaling axis, is expressed in INS-1 832/13 cells, normal rat and human islets, but exerts minimal effects on GSIS in INS-1 832/13 cells
Earlier studies by Damoulakis and coworkers have implicated activation of RhoG, a small G protein, in P-Rex1-mediated G protein coupled receptor-driven activation of Rac1 and actin cytoskeletal polarity in neutrophils [17]. Data from these investigations have suggested that P-Rex1 plays the role of a GEF for RhoG activation thereby regulating downstream signaling steps including Rac1 activation and cytoskeletal functions. Therefore, we next explored the roles of RhoG signaling pathway in GSIS. Data in Fig. 4 (Panel A) provide the first evidence for the expression of RhoG in INS-1 832/13 cells, normal rat and human islets. Transfection of INS-1 832/13 cells with siRNA-RhoG, but not control siRNA, markedly suppressed the endogenous expression of RhoG in these cells (Fig. 4; Panel B). Interestingly, however, no significant effects of RhoG knockdown were seen on either basal insulin secretion or GSIS (Fig. 4; Panel C). These findings rule out potential regulatory roles for RhoG in GSIS. They also suggest that RhoG is not downstream to glucose-induced P-Rex1-sensitive signaling mechanisms involved in GSIS.
Fig. 4.
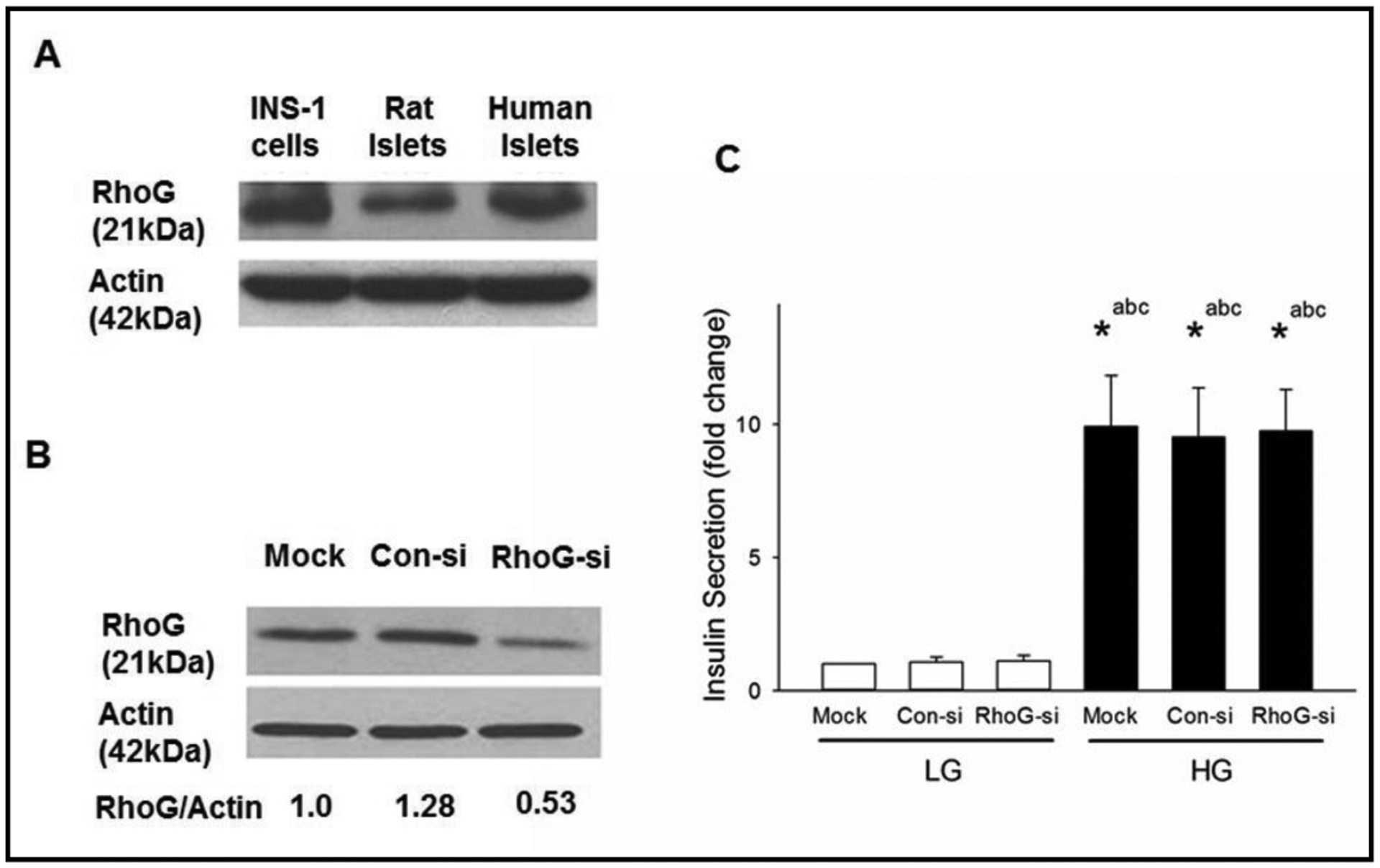
Lack of effects of Rho G, a known regulator of P-Rex1-Rac1 module, on GSIS in INS-1 832/13 cells. Panel A: Lysates from INS-1 832/13 cells, rats and human islets were analyzed for RhoG protein expression by Western blot analysis. Actin was used as loading control. Panel B: INS-1 832/13 cells were transfected with Con-si or RhoG-si as described in Methods. Cell lysates were analyzed by Western blotting for the expression of RhoG. Actin was used as loading control. A representative blot from three independent studies is shown here. Panel C: Following 48 hours of transfection, cells were subjected to overnight starvation and then were treated with LG (2.5 mM) or HG (20 mM) for 45 mins. Insulin secretion in the media was determined as described in the Methods section. Data are mean ± SD from three experiments. The data are expressed as fold change relative to LG-Mock. (* p< 0.05). Comparisons shown: a - significant compared with LG-treated mock; b – significant compared with LG-treated Con-si; c - significant compared with LG-treated RhoG-si.
PI3-kinase inhibitors exert differential effects on GSIS in INS-1 832/13 cells without significantly affecting glucose-induced Rac1 activation in INS-1 832/13 cells
It is well established that functional activation of P-Rex1 is downstream to PI3-kinase activation [18–20]. Despite compelling evidence on roles of PI3-kinase in GSIS [23, 24, 26], putative roles of PI3-kinase-Rac1 signaling axis in GSIS remain unknown. Therefore, we undertook a series of investigations to determine the roles of PI3-kinase in glucose-induced Rac1 activation and insulin secretion via two different experimental approaches. In the first, we quantified GSIS in INS-1 832/13 cells in the absence and presence of two structurally distinct inhibitors of PI3- kinase, namely Wortmannin [33] and LY294002 [34]. In the second set of experiments, we determined the effects of these two inhibitors on glucose-induced Rac1 activation in INS-1 832/13 cells.
Data in Fig. 5 (Panel A) demonstrate no significant effects of either of these inhibitors (following one-hour pre-incubation) on basal insulin secretion. A robust stimulation of insulin secretion was seen in the presence of stimulatory glucose. Wortmannin failed to exert any effect on GSIS under our current experimental conditions. However, we observed a significant potentiation of GSIS in cells exposed to the LY294002 inhibitor (Panel A). Similar potentiating effect, by LY294002, of GSIS was noted in cells following overnight pre-incubation of INS-1 832/13 cells to the inhibitor (data not shown). A modest, but significant increase in basal insulin secretion was seen in cells incubated with Wortmannin following one hour pre-exposure conditions (Panel A). However, in a manner akin to findings in Panel A, we observed no effects of Wortmannin on GSIS even after exposure of the cells overnight with the inhibitor (not shown). Together, these findings demonstrate differential effects of two structurally distinct inhibitors of PI3-kinase on insulin secretion from INS-1 832/13 cells.
Fig. 5.
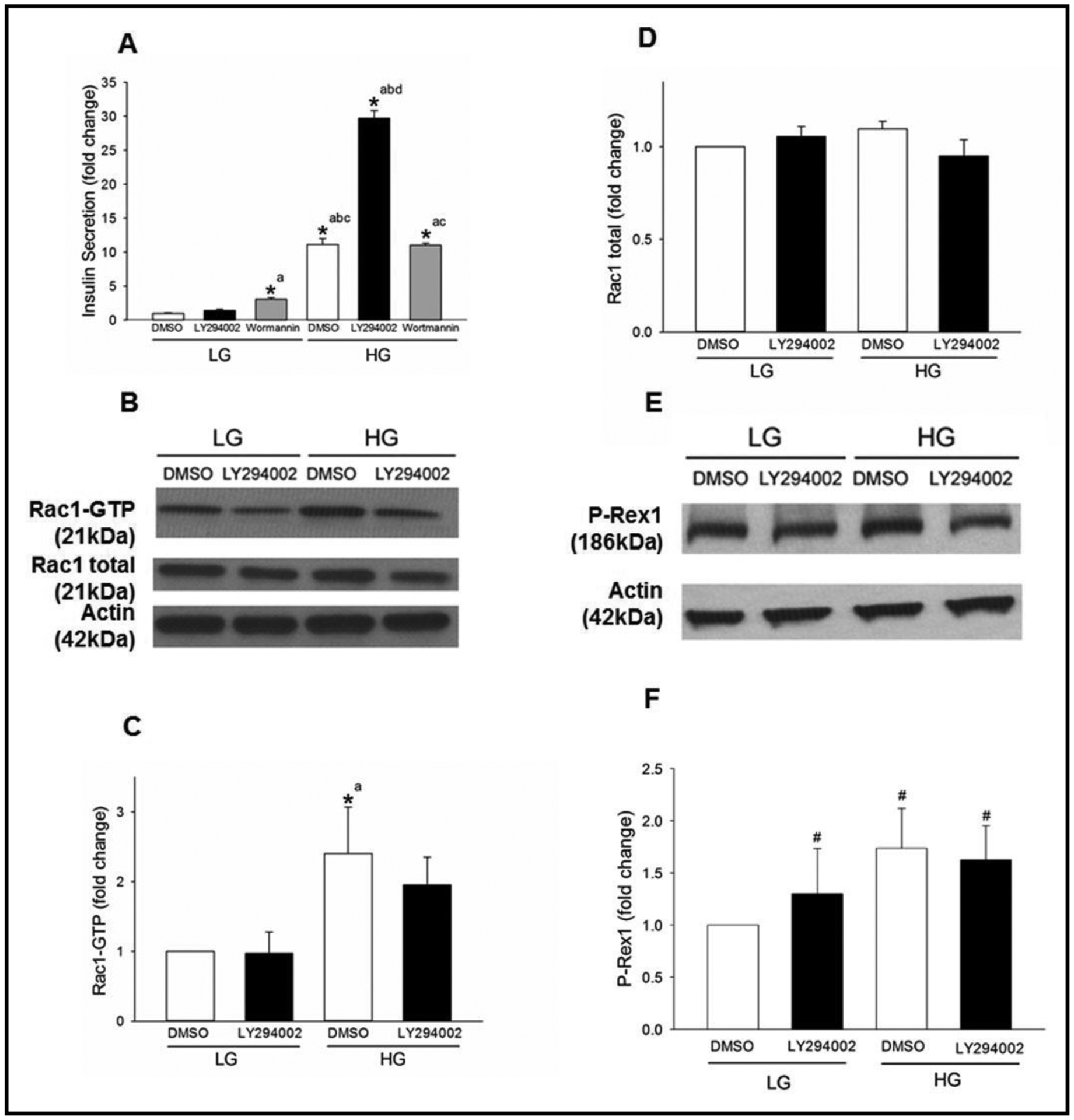
LY294002, but not Wortmannin, potentiates GSIS but elicits no significant effects on glucose- induced Rac1 activation in INS-1 832/13 cells. Panel A: INS-1 832/13 cells were treated 1hr with LY294002 (10 μM) or Wortmannin (100nM) and subjected to LG (2.5mM) or HG (20mM) treatment for 45 mins. DMSO was used as a vehicle control. Amounts of insulin secreted into the media was quantified as described under Methods. The graph is a representative experiment performed in a single run. Data are mean ± SD from four replicates and expressed as fold change relative to LG DMSO control. (* p< 0.05) Comparisons shown: a - significant compared with LG DMSO control; b - significant compared with LG LY294002; c- significant compared with LG Wortmannin; d- significant compared with HG DMSO control. Panel B: INS-1 832/13 cells were treated with LY294002 (10 μM) for 1hr and subjected to LG (2.5 mM) or HG (20 mM) treatment for 15 mins. Rac1 activation was quantified by Rac1 pull down assay. Representative blots from four independent studies are provided. Panel C: Densitometry quantitation of activated Rac1 (shown in Panel B) is depicted. The results from four independent experiments presented as fold change relative to LG DMSO and as means ± SD. (* p< 0.05) Comparisons shown: a - significant compared with LG-treated DMSO control. Panel D: Densitometry quantitation of total Rac1 in Panel B is shown here. Panel E: INS-1 832/13 cells were treated with LY294002 (10 μM) for 1hr and subjected to LG (2.5mM) or HG (20mM) treatment for 15 mins. Expression of P-Rex1 in the cell lysates was detected by Western blotting. A representative blot from three independent studies is provided. Panel F: Densitometry quantitation of P-Rex1 in studies described in Panel E is shown here. # denotes not significant from LG with diluent (means ± SD; n=4 experiments).
Next, we wanted to know if LY290042-mediated potentiation of GSIS (Fig. 5; panel A) involves Rac1 activation. To address this, the degree of glucose-induced Rac1 activation was quantified in INS-1 832/13 cells incubated (60 min pre-incubation) in the absence and presence of LY290042. Data in Fig. 5 (panel B) indicate significant activation of Rac1 in these cells exposed to stimulatory glucose. However, glucose-induced Rac1 activation was not affected by the inhibitor (Panel B). Under these conditions, we detected no significant changes in total Rac1 (Fig. 5; panels C and D). Furthermore, no significant differences in the expression of P-Rex1 were noticed in INS-1 832/13 cells under basal or glucose-stimulated conditions in the absence or presence of LY290042 (Fig. 5; panels E and F). Together, these data suggest that the potentiating effect, by LY290042, on GSIS might not involve activation of Rac1.
Discussion
A growing body of evidence supports the overall hypothesis that small G proteins (e.g., Rac1) play regulatory roles in islet β-cell function, including insulin secretion [13–15]. Extant studies have suggested regulatory roles for Rac1 in islet function including the recruitment of secretory granules through actin cytoskeletal reorganization for GSIS and islet morphogenesis [21, 31, 35]. For example, studies of Asahara et al. have shown that Rac1-null [βRac1−/−] mice exhibited impaired glucose tolerance and hypoinsulinemia. Glucose-, but not KCl-induced insulin secretion, was markedly attenuated in islets from the Rac1 null mice. The β-cell mass or islet density remained unaltered in these mice. Based on these findings, it was concluded that Rac1 plays a key regulatory role in insulin secretion primarily through regulating cytoskeletal reorganization [35]. Altogether, the above studies provide significant support to the viewpoint that Rac1 plays a positive modulatory role in islet function including GSIS.
One of the goals of the current studies was to determine the roles of P-Rex1 as a GEF for Rac1 in the cascade of events leading to insulin secretion. Our findings indicate that P-Rex1 is expressed in INS-1 832/13 cells and normal rodent and human islets. siRNA-mediated knockdown of expression of endogenous P-Rex1 attenuated GSIS in INS-1 832/13 cells. Our findings also suggest that depletion of P-Rex1 expression results in significant inhibition of glucose induced activation of Rac1 in these cells. It is noteworthy that, we observed a significant reduction in the abundance of membrane-associated Rac1 in INS-1 832/13 cells exposed to stimulatory glucose, suggesting potential roles for P-Rex1 in membrane association of Rac1. Together, these data affirm that P-Rex1 might sub-serve the function of a GEF for Rac1 in the cascade of events leading to GSIS.
Damoulakis et al. have reported novel roles for RhoG, a small G protein, in P-Rex1-mediated regulation of cellular functions. They demonstrated that P-Rex1 serves as a GEF for RhoG in vitro as well as in fMPLP-stimulated (i.e., GPCR-mediated) primary mouse neutrophils. Interestingly, inhibition of either P-Rex1 or RhoG functions resulted in a marked reduction in GPCR-mediated Rac1-NADPH oxidase signaling axis suggesting that P-Rex1-RhoG module plays regulatory roles upstream to Rac1 activation [17]. Based on these observations we asked if RhoG plays a contributory role(s) in the events leading to GSIS. This has not been addressed before. In this context, we provide the first evidence for the expression of RhoG in normal rodent and human islets and INS-1 832/13 cells. Interestingly, however, the siRNA-mediated knockdown of RhoG expression exerted no effects on GSIS, thus ruling out potential contributory roles of RhoG at least under acute regulatory conditions.
In an effort to determine potential roles of PI3-kinase in glucose-induced Rac1 activation and insulin secretion, we utilized two structurally distinct inhibitors of PI3-kinase for their effects on GSIS and glucose-induced activation of Rac1. Compatible with observations of Collier and coworkers, we observed significant amplification of GSIS in cells exposed to LY294002, but not Wortmannin [24]. Interestingly, however, LY294002 failed to exert significant regulatory effects on glucose-induced activation of Rac1, thereby suggesting that the amplification of GSIS consequential to PI3-kinase inhibition does not involve (or require) activation of Rac1. Instead, the amplification process might involve an increase in intracellular cAMP (due to inhibition of specific isoforms of phosphodiesterase) and activation of protein kinase A signaling steps [36, 37].
In the light of current observations, it may be germane to point out that, previous studies have identified Tiam1 and Vav2 as GEFs for Rac1 in clonal β-cells and normal rodent and human islets [15, 38]. These studies have provided compelling evidence for Tiam1-Rac1 and Vav2-Rac1 signaling modules not only in physiological insulin secretion, but also in the constitutive activation of Rac1 by increasing the intracellular oxidative stress and pathogenesis of islet β-cell dysfunction under conditions of metabolic stress [30, 39]. Our current study identifies P-Rex1 as yet another GEF for Rac1 in signaling events leading to GSIS. This raises an important question of why does the β-cell need more than one GEF to activate Rac1? It is likely that these regulatory proteins/factors are involved in spatiotemporal regulation of candidate G proteins (e.g., Rac1) for optimal cell function. This has been reported in platelets recently [40]. Interestingly, recent proteomic analysis of the regulation of Rac1 signaling by GEFs (e.g., Tiam1 and P-Rex1) by Marei and associates have identified distinct sets of interacting partners for Rac1 following its activation by each of these GEFs; such a difference in Rac1 interactome could explain for its differential regulatory roles in eliciting its anti-or pro-migratory effects [41, 42]. It has also been suggested that both of these GEFs sub-serve the functions of not only activating Rac1, but also in dictating Rac1-driven biological outcomes that govern cell migration and invasion [42]. In a manner akin to this, studies of Omelchenko and coworkers have demonstrated regulatory control of spatial localization of Rac1 by β-Pix, a known GEF for Rac1, in facilitating the migration of anterior visceral endoderm cells [43]. Therefore, such non-GEF-dependent functions of GEFs could include the association of Rac1 with its intracellular target/effector proteins in the membrane. Along these lines, using quantitative proteomics approach we have identified novel interaction partners for Rac1 in INS-1 832/13 cells under basal and glucotoxic conditions [44]. Future proteomics investigations along the lines of identification of Rac1 interactome in pancreatic β-cells under conditions of its activation by each of these GEFs (Tiam1, Vav2 and P-Rex-1) should yield valuable insights into these pathways in β-cells in health and metabolic stress.
Conclusion
Based on our observations, we conclude that glucose-induced Rac1 activation and insulin secretion are mediated, in part, via P-Rex1, and such a signaling step(s) might not require the intermediacy of RhoG. Although not tested in our current studies, these signaling steps might involve recently reported non-canonical activation of P-Rex1, which is mediated via binding of the regulatory subunit of protein kinase A (i.e., PKA-Riα) to P-Rex1 thereby promoting its catalytic activation culminating in the activation of Rac1 [37]. This postulation needs to be tested experimentally in the context of GSIS in the pancreatic β-cell. Further, additional signals (i.e., non-canonical) might underlie glucose-induced P-Rex1/Rac1 activation in the stimulus-secretion coupling of GSIS. These might comprise of non-receptor-mediated functional regulation of individual subunits of trimeric G proteins [recently reviewed in [15]], including the post-translational carboxylmethylation of the γ-subunits [45] and histidine phosphorylation of the β-subunits [46] leading to the activation of the putative trimeric G protein(s) that couples PI3-kinase for the activation of P-Rex-1-Rac1 signaling pathway culminating in GSIS. These questions are being addressed in our laboratory currently.
Acknowledgements
Funding
This research is supported by a Merit Review Award (BX004663) from the US Department of Veterans Affairs and the National Institutes of Health (to AK). AK is the recipient of a Senior Research Career Scientist Award (K6BX005383) from the US Department of Veterans Affairs.
Abbreviations
- Arf6
ADP ribosylation factor6
- Cdc42
Cell division control protein 42
- GAP
GTPase-activating protein
- GDI
GDP dissociation inhibitor
- GEF
Guanine nucleotide exchange factor
- GPCR
G-protein-coupled receptor
- GSIS
Glucose-stimulated insulin secretion
- PI3-kinase
Phosphatidylinositol 3-Kinase
- P-Rex1
Phosphatidylinositol-3,4,5-trisphosphate-dependent Rac exchange factor 1
- Rac1
Ras-related C3 botulinum toxin substrate 1
Footnotes
Statement of Ethics
All protocols involving animal care and use were reviewed and approved by Wayne State University and John D. Dingell VA Medical Center Institutional Animal Care and Use Committees. Studies involving human islets were approved by the Biosafety Committee at the John D. Dingell VA Medical Center.
Disclosure Statement
The authors have no conflicts of interest to declare.
References
- 1.Malaisse WJ: Insulin secretion: multifactorial regulation for a single process of release. The Minkowski award lecture delivered on September 7, 1972 before the European Association for the study of Diabetes at Madrid, Spain. Diabetologia 1973;9:167–173. [DOI] [PubMed] [Google Scholar]
- 2.Mayer J: Glucostatic mechanism of regulation of food intake. N Engl J Med 1953;249:13–16. [DOI] [PubMed] [Google Scholar]
- 3.MacDonald MJ: Elusive proximal signals of beta-cells for insulin secretion. Diabetes 1990;39:1461–1466. [DOI] [PubMed] [Google Scholar]
- 4.Newgard CB, McGarry JD: Metabolic coupling factors in pancreatic beta-cell signal transduction. Annu Rev Biochem 1995;64:689–719. [DOI] [PubMed] [Google Scholar]
- 5.Prentki M, Matschinsky FM, Madiraju SR: Metabolic signaling in fuel-induced insulin secretion. Cell Metab 2013;18:162–185. [DOI] [PubMed] [Google Scholar]
- 6.Berggren PO, Leibiger IB: Novel aspects on signal-transduction in the pancreatic beta-cell. Nutr Metab Cardiovasc Dis 2006;16:S7–10. [DOI] [PubMed] [Google Scholar]
- 7.Gilon P, Chae HY, Rutter GA, Ravier MA: Calcium signaling in pancreatic β-cells in health and in Type 2 diabetes. Cell Calcium 2014;56:340–361. [DOI] [PubMed] [Google Scholar]
- 8.Metz SA, Rabaglia ME, Pintar TJ: Selective inhibitors of GTP synthesis impede exocytotic insulin release from intact rat islets. J Biol Chem 1992;267:12517–12527. [PubMed] [Google Scholar]
- 9.Komatsu M, Noda M, Sharp GW: Nutrient augmentation of Ca2+-dependent and Ca2+-independent pathways in stimulus-coupling to insulin secretion can be distinguished by their guanosine triphosphate requirements: studies on rat pancreatic islets. Endocrinology 1998;139:1172–1183. [DOI] [PubMed] [Google Scholar]
- 10.Meredith M, Rabaglia ME, Metz SA: Evidence of a role for GTP in the potentiation of Ca(2+)-induced insulin secretion by glucose in intact rat islets. J Clin Invest 1995;96:811–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Metz SA, Meredith M, Rabaglia ME, Kowluru A: Small elevations of glucose concentration redirect and amplify the synthesis of guanosine 5’-triphosphate in rat islets. J Clin Invest 1993;92:872–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kowluru A: Roles of GTP and Rho GTPases in pancreatic islet beta cell function and dysfunction. Small GTPases 2020:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kowluru A: Small G proteins in islet beta-cell function. Endocr Rev 2010;31:52–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Z, Thurmond DC: Mechanisms of biphasic insulin-granule exocytosis - roles of the cytoskeleton, small GTPases and SNARE proteins. J Cell Sci 2009;122:893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kowluru A: GPCRs, G Proteins, and Their Impact on β-cell Function. Compr Physiol 2020;10:453–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kowluru A: Emerging roles for protein histidine phosphorylation in cellular signal transduction: lessons from the islet beta-cell. J Cell Mol Med 2008;12:1885–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Damoulakis G, Gambardella L, Rossman KL, Lawson CD, Anderson KE, Fukui Y, et al. : P-Rex1 directly activates RhoG to regulate GPCR-driven Rac signalling and actin polarity in neutrophils. J Cell Sci 2014;127:2589–2600. [DOI] [PubMed] [Google Scholar]
- 18.Welch HC, Coadwell WJ, Ellson CD, Ferguson GJ, Andrews SR, Erdjument-Bromage H, et al. : P-Rex1, a PtdIns(3,4,5)P3- and Gbetagamma-regulated guanine-nucleotide exchange factor for Rac. Cell 2002;108:809–821. [DOI] [PubMed] [Google Scholar]
- 19.Barber MA, Donald S, Thelen S, Anderson KE, Thelen M, Welch HC: Membrane translocation of P-Rex1 is mediated by G protein betagamma subunits and phosphoinositide 3-kinase. J Biol Chem 2007;282:29967–29976. [DOI] [PubMed] [Google Scholar]
- 20.Hill K, Krugmann S, Andrews SR, Coadwell WJ, Finan P, Welch HC, et al. : Regulation of P-Rex1 by phosphatidylinositol (3,4,5)-trisphosphate and Gbetagamma subunits. J Biol Chem 2005;280:4166–4173. [DOI] [PubMed] [Google Scholar]
- 21.Freudenblum J, Iglesias JA, Hermann M, Walsen T, Wilfinger A, Meyer D, et al. : In vivo imaging of emerging endocrine cells reveals a requirement for PI3K-regulated motility in pancreatic islet morphogenesis. Development 2018;145:dev158477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pigeau GM, Kolic J, Ball BJ, Hoppa MB, Wang YW, Rückle T, et al. : Insulin granule recruitment and exocytosis is dependent on p110gamma in insulinoma and human beta-cells. Diabetes 2009;58:2084–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zawalich WS, Zawalich KC: A link between insulin resistance and hyperinsulinemia: inhibitors of phosphatidylinositol 3-kinase augment glucose-induced insulin secretion from islets of lean, but not obese, rats. Endocrinology 2000;141:3287–3295. [DOI] [PubMed] [Google Scholar]
- 24.Collier JJ, White SM, Dick GM, Scott DK: Phosphatidylinositol 3-kinase inhibitors reveal a unique mechanism of enhancing insulin secretion in 832/13 rat insulinoma cells. Biochem Biophys Res Commun 2004;324:1018–1023. [DOI] [PubMed] [Google Scholar]
- 25.Kolic J, Manning Fox JE, Chepurny OG, Spigelman AF, Ferdaoussi M, Schwede F, et al. : PI3 kinases p110α and PI3K-C2β negatively regulate cAMP via PDE3/8 to control insulin secretion in mouse and human islets. Mol Metab 2016;5:459–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kolic J, Spigelman AF, Smith AM, Manning Fox JE, MacDonald PE: Insulin secretion induced by glucose-dependent insulinotropic polypeptide requires phosphatidylinositol 3-kinase gamma in rodent and human beta-cells. J Biol Chem 2014;289:32109–32120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Veluthakal R, Arora DK, Goalstone ML, Kowluru RA, Kowluru A: Metabolic Stress Induces Caspase-3 Mediated Degradation and Inactivation of Farnesyl and Geranylgeranyl Transferase Activities in Pancreatic β-Cells. Cell Physiol Biochem 2016;39:2110–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Syed I, Kyathanahalli CN, Jayaram B, Govind S, Rhodes CJ, Kowluru RA, et al. : Increased phagocyte-like NADPH oxidase and ROS generation in type 2 diabetic ZDF rat and human islets: role of Rac1-JNK1/2 signaling pathway in mitochondrial dysregulation in the diabetic islet. Diabetes 2011;60:2843–2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thamilselvan V, Kowluru A: Paradoxical regulation of glucose-induced Rac1 activation and insulin secretion by RhoGDIβ in pancreatic β-cells. Small GTPases 2019:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sidarala V, Veluthakal R, Syeda K, Vlaar C, Newsholme P, Kowluru A: Phagocyte-like NADPH oxidase (Nox2) promotes activation of p38MAPK in pancreatic β-cells under glucotoxic conditions: Evidence for a requisite role of Ras-related C3 botulinum toxin substrate 1 (Rac1). Biochem Pharmacol 2015;95:301–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kowluru A: Friendly, and not so friendly, roles of Rac1 in islet β-cell function: lessons learnt from pharmacological and molecular biological approaches. Biochem Pharmacol 2011;81:965–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kowluru A, Veluthakal R: Rho guanosine diphosphate-dissociation inhibitor plays a negative modulatory role in glucose-stimulated insulin secretion. Diabetes 2005;54:3523–3529. [DOI] [PubMed] [Google Scholar]
- 33.Arcaro A, Wymann MP: Wortmannin is a potent phosphatidylinositol 3-kinase inhibitor: the role of phosphatidylinositol 3,4,5-trisphosphate in neutrophil responses. Biochem J 1993;296:297–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vlahos CJ, Matter WF, Hui KY, Brown RF: A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J Biol Chem 1994;269:5241–5248. [PubMed] [Google Scholar]
- 35.Asahara S, Shibutani Y, Teruyama K, Inoue HY, Kawada Y, Etoh H, et al. : Ras-related C3 botulinum toxin substrate 1 (RAC1) regulates glucose-stimulated insulin secretion via modulation of F-actin. Diabetologia 2013;56:1088–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nunoi K, Yasuda K, Tanaka H, Kubota A, Okamoto Y, Adachi T, et al. : Wortmannin, a PI3-kinase inhibitor: promoting effect on insulin secretion from pancreatic beta cells through a cAMP-dependent pathway. Biochem Biophys Res Commun 2000;270:798–805. [DOI] [PubMed] [Google Scholar]
- 37.Holz GG, Chepurny OG, Leech CA: “A-kinase” regulator runs amok to provide a paradigm shift in cAMP signaling. J Biol Chem 2019;294:2247–2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kowluru A: Tiam1/Vav2-Rac1 axis: A tug-of-war between islet function and dysfunction. Biochem Pharmacol 2017;132:9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Syed I, Jayaram B, Subasinghe W, Kowluru A: Tiam1/Rac1 signaling pathway mediates palmitate-induced, ceramide-sensitive generation of superoxides and lipid peroxides and the loss of mitochondrial membrane potential in pancreatic beta-cells. Biochem Pharmacol 2010;80:874–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aslan JE: Platelet Rho GTPase regulation in physiology and disease. Platelets 2019;30:17–22. [DOI] [PubMed] [Google Scholar]
- 41.Marei H, Malliri A: GEFs: Dual regulation of Rac1 signaling. Small GTPases 2017;8:90–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marei H, Carpy A, Macek B, Malliri A: Proteomic analysis of Rac1 signaling regulation by guanine nucleotide exchange factors. Cell Cycle 2016;15:1961–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Omelchenko T, Rabadan MA, Hernández-Martínez R, Grego-Bessa J, Anderson KV, Hall A: β-Pix directs collective migration of anterior visceral endoderm cells in the early mouse embryo. Genes Dev 2014;28:2764–2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Damacharla D, Thamilselvan V, Zhang X, Mestareehi A, Yi Z, Kowluru A: Quantitative proteomics reveals novel interaction partners of Rac1 in pancreatic β-cells: Evidence for increased interaction with Rac1 under hyperglycemic conditions. Mol Cell Endocrinol 2019;494:110489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kowluru A, Li G, Metz SA: Glucose activates the carboxyl methylation of gamma subunits of trimeric GTP-binding proteins in pancreatic beta cells. Modulation in vivo by calcium, GTP, and pertussis toxin. J Clin Invest 1997;100:1596–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kowluru A, Seavey SE, Rhodes CJ, Metz SA: A novel regulatory mechanism for trimeric GTP-binding proteins in the membrane and secretory granule fractions of human and rodent beta cells. Biochem J 1996;313:97–107. [DOI] [PMC free article] [PubMed] [Google Scholar]