

Abstract
Background
Lysosomal acid lipase deficiency (LAL-D) is a phenotypic continuum between the severe Wolman disease and the attenuated cholesteryl ester storage disease (CESD).
Objective
To study if the amount of residual LAL enzymatic activity in dried blood spots (DBS) correlates with the LAL-D disease severity.
Methods
DBS from Wolman and CESD patients, LAL-D carriers, and presumably unaffected random newborns were acquired. LAL enzymatic activity in DBS were measured using a novel, highly specific LAL substrate.
Results
Patients with Wolman disease displayed significantly lower LAL enzymatic activity compared to CESD patients. This was not observed with the traditional assay in which a non-specific substrate was used together with an LAL-specific inhibitor.
Conclusion
The new LAL enzymatic activity assay using the specific substrate offers an improved biochemical genetics method for the diagnosis of LAL-D in symptomatic patients and more importantly, for the prognosis of asymptomatic patients who test positive in population-wide LAL-D newborn screening.
Keywords: Lysosomal acid lipase deficiency (LAL-D), Enzymatic activity, Dried blood spot (DBS), Newborn screening
1. Introduction
Lysosomal acid lipase (LAL, EC 3.1.1.13) deficiency (LAL-D, OMIM 278000) is a rare autosomal recessive lysosomal storage disorder (LSD) caused by biallelic pathogenic variants in LIPA. Under acidic condition, LAL hydrolases cholesterol esters and triglycerides into free cholesterol, glycerol, and fatty acids, and its deficiency leads to accumulation of esterified lipids, especially in the lysosome of hepatocytes and cells of mononuclear phagocyte system [1,2].
LAL-D is a phenotypic continuum between the severe Wolman disease and the attenuated cholesteryl ester storage disease (CESD), with a combined prevalence estimated to be 1:40,000 to 1:300,000 [2,3]. Patients with Wolman disease typically present in the first months of life with failure to thrive, malabsorption, hepatosplenomegaly, liver failure and bilateral adrenal calcifications [1]. Without treatment, the children die from multi-organ failure within the first year of life [3]. On the contrary, CESD patients have a more variable disease course and typically present later in life with dyslipidemia, hepatosplenomegaly, and/or elevated liver enzymes [1].
LAL-D can be treated, with substantial improvements to quality of life, by enzyme replacement therapy (ERT) using recombinant human LAL (sebelipase alfa), which has obtained regulatory approval in United States, European Union, and Japan [4,5]. Since early diagnosis and treatment initiation is crucial for optimal outcomes, newborn screening for LAL-D may be considered in the near future [3,5].
The diagnosis of LAL-D can be established by demonstration of reduced LAL activity in dried blood spots (DBS) or leukocytes and/or identification of biallelic pathogenic variants in LIPA. Traditionally, LAL activity is measured in DBS with a non-specific fluorogenic substrate in the presence and absence of lalistat 2, a specific LAL inhibitor [6]. This necessitates two incubations per sample, and LAL activity is defined as lalistat 2-sensitive lipase activity, calculated by subtracting the lipase activity in the presence of lalistat 2 from that in the absence of lalistat 2 [6]. Parallel incubation can be cumbersome and limits the assay throughput. In addition, the measurement of LAL activity is not as accurate and precise, especially at the lower end, since its calculation is based on subtraction of two substantial activity values and the error and imprecision of the two measurements propagates. Whilst being excellent for diagnostic purposes, this poses a challenge in using DBS LAL activity to predict disease severity. Indeed, patients with Wolman disease and CESD demonstrate unequivocally substantially reduced LAL activity in DBS, regardless of their phenotype [[7], [8], [9]]. On the contrary, correlation between residual LAL activity in fibroblast or leukocytes with phenotype has been reported [7,8,10].
More recently, a highly specific LAL substrate, 4-propyl-8-methyl-7-hydroxycoumarin (P-PMHC), was developed in our laboratory and the assay can be analyzed by either ultra-performance liquid chromatography tandem mass spectrometry (UPLC-MS/MS) or fluorometry [11]. P-PMHC was found to be >98% selective for LAL in DBS, therefore one incubation without lalistat 2 is sufficient. Furthermore, the UPLC-MS/MS platform offers additional advantages to its fluorometry counterpart, including better sensitivity and specificity, along with the multiplexing ability [11]. The new LAL assay was further consolidated into a high throughput 18-plex UPLC-MS/MS assay, setting the stage for large-scale screening of LAL-D [12]. A prospective pilot study using the new LAL assay is being conducted in New York, US to assess the analytical and clinical validity of LAL-D screening [13].
We hypothesized that patients with Wolman disease and CESD can be stratified based on residual LAL activity in DBS using P-PMHC as the substrate, due to its high specificity and the use of UPLC-MS/MS instead of fluorometry. To test the hypothesis, we measured LAL activity in DBS from confirmed LAL-D patients and carriers using P-PMHC. CESD patients were found to have higher residual DBS LAL activity when compared to Wolman patients, indicating LAL-D patients can be stratified based on residual activity using the UPLC-MS/MS-based LAL assay.
2. Materials and methods
2.1. Materials
This study using DBS from de-identified newborns was approved by the Washington State Institutional Review Board. DBS were shared by the Washington State Department of Health (WA DOH) after being stored for 30–60 days at room temperature.
De-identified DBS from genetically and/or biochemically confirmed LAL-D patients were obtained from the Queen Elizabeth University Hospital, United Kingdom. According to The Human Tissue Acts of England, Wales and Northern Ireland (2004) and Scotland (2006), de-identified, remaining materials can be used for method validation and/or performance assessment without the need for ethical approval. The DBS was stored at −20 °C with desiccant for <2 years prior to the current study.
P-PMHC was synthesized in the Gelb laboratory (University of Washington, US) as previously described [11]. It is also available at GelbChem, LLC (Seattle, US).
3. Methods
The LAL activity in DBS was measured with P-PMHC using a previously described protocol [12]. In brief, DBS extract was incubated with an assay cocktail containing P-PMHC and an isotope-labeled internal standard at 37 °C for 3 h. The mixture was then quenched, cleaned up by liquid-liquid extraction, and analyzed by UPLC-MS/MS. The enzymatic activity was calculated based on the ratio between the enzymatic product and the internal standard [12].
Statistical analysis was carried out using GraphPad Prism 8.
4. Results
LAL activity from LAL-D patients and carriers, along with the presumably unaffected random newborns is summarized in Fig. 1. The mean LAL activity in the 10 Wolman patients was 0.89 μM/h (range: undetectable-4.01 μM/h). The mean LAL activity in the 11 CESD patients was 3.98 μM/h (range: 2.21–8.13 μM/h). The mean LAL activity in the 5 LAL-D carriers was 154 μM/h (range: 104–232 μM/h). The mean LAL activity in the 9 random newborns was 154 μM/h (range: 120–197 μM/h).
Fig. 1.
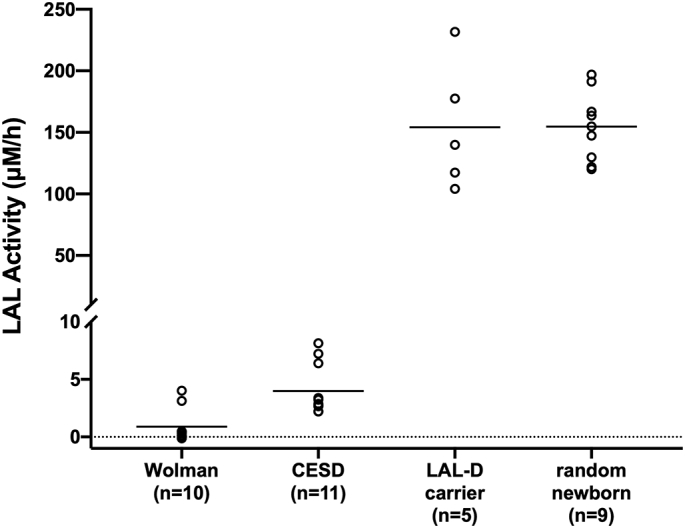
LAL activity in 10 Wolman patients, 11 CESD patients, 5 LAL-D carriers, and 9 random newborns. The horizontal line within each group indicates the mean activity.
Despite the overlap, the LAL activity from the Wolman patients was statistically lower (p < 0.001) than that from the CESD patients (Fig. 1). The mean LAL activity from the 21 LAL-D patients (Wolman and CESD) was 2.51 μM/h (range: undetectable-8.13 μM/h), which is 60-fold lower than the mean activity from the 5 LAL-D carriers. Therefore, the assay can detect LAL-D patients with 100% sensitivity. No statistical difference was observed between the LAL-D carriers and the random newborns, which may be ascribed to the non-ideal storage condition of the random newborn DBS (see Discussion).
5. Discussion
In previous studies, both the traditional fluorometry-based and the new UPLC-MS/MS-based DBS LAL assay demonstrated clear distinction between LAL-D patients and carriers [6,7,11]. In the current study, we demonstrated that most of the Wolman patients can be distinguished from the CESD patients using the new UPLC-MS/MS-based DBS LAL assay. All but 2 of the 10 Wolman DBS displayed LAL activity below the range observed in the 11 CESD DBS (Fig. 1). In marked contrast, all the 21 LAL-D samples displayed undetectable LAL activity using the traditional fluorometry assay (data not shown).
Stratification of LAL-D patients has great implications for patient counseling and management. Given that enzyme replacement therapy is available for LAL-D, newborn screening for this disorder may be warranted in the future. The new UPLC-MS/MS-based DBS LAL assay may be useful for stratification of the initially asymptomatic patients identified by newborn screening. Genotyping may be informative, but annotation of all the LIPA variants is incomplete, and biochemical evaluation will provide complementary information [14].
Shen et al. reported a patient who was clinically consistent with LAL-D and responded well to ERT. The diagnosis was complicated by inconclusive genetic testing due to the second LIPA variant being a variant of unknown significance (VUS), and the LAL activity measured by the traditional fluorometry assay did not support a diagnosis of LAL-D. However, the LAL activity was found to be reduced to the affected range when determined by the specific UPLC-MS/MS-based LAL assay reported here [15]. This case underlies the importance of using clinical, biochemical, and genetic evidence when evaluating patients with rare diseases, and also the importance of using a highly specific and sensitive enzyme assay.
The enzymatic activities in LAL-D patients were significant lower (60-fold) compared to LAL-D carriers (Fig. 1). Such difference is substantially larger than that in other LSDs (e.g. 5-fold in Krabbe disease, < 2-fold in Pompe disease) [16,17]. Therefore, we envision that the false positive rate caused by a carrier status in LAL-D screening will be lower than with other LSDs screening. To the best of our knowledge, pseudodeficiency has not been reported for LAL-D and therefore should not cause concerns for false positives in large-scale screening. However, we note that this statement may change when population screening starts. In addition, the ongoing pilot study for the newborn screening of LAL-D will provide more information on the analytical and clinical validity of the test [13]. The cutoff value needs to be carefully validated with larger cohorts of patient and control samples. Furthermore, the false positive rate should be examined in large-scale screening studies to determine whether additional second-tier screening is needed.
We did not observe any statistical difference between the LAL-D carriers and the random newborns (Fig. 1), as opposed to the previous studies demonstrating clear separation [6,7]. In addition, the LAL activity in random newborns reported in the current study is lower compared to a previous study using the same LAL-specific substrate [11]. This may be ascribed to two limitations of the study. First, storage condition of the random newborn DBS (30–60 days at room temperature) was non-ideal. A 50% loss of LAL activity was reported when the DBS is stored at room temperature for 2 months [6]. At −20 °C, an average of 5% of LAL activity is lost per year (personal communication, Hamilton). Unfortunately, DBS are stored under room temperature at the WA DOH and those stored <30 days cannot be shared for research purpose according to the policy. Secondly, our DBS samples from the LAL-D patients were not age-matched with the newborn controls, which could bias our results since hematocrit and leukocyte counts are age dependent. Therefore, the results reported in this study cannot be used as reference ranges.
Currently, we do not have detailed clinical information for the LAL-D patients whose samples were analyzed in this study. In the future, LAL activity can be measured in a larger CESD cohort to examine if the level of residual activity correlates with the severity of the highly variable CESD. In addition to DBS, leukocytes or fibroblast may be considered for such purpose.
6. Conclusion
LAL activity was measured using a novel, highly specific LAL substrate, P-PMHC, by UPLC-MS/MS. The LAL-D patients (Wolman and CESD) displayed 60-fold lower activity compared to the carriers, allowing 100% sensitivity for LAL-D detection. More importantly, the Wolman patients displayed significantly lower activity compared to the CESD patients, demonstrating a correlation between biochemical and clinical phenotype. This has not been observed when the LAL activity is measured using the nonspecific substrate. Such correlation has implications when evaluating asymptomatic at-risk patients, such as those identified by newborn screening.
Authors' statement
This study using DBS from de-identified newborns was approved by the Washington State Institutional Review Board. DBS were shared by the Washington State Department of Health (WA DOH).
De-identified DBS from genetically and/or biochemically confirmed LAL-D patients were obtained from the Queen Elizabeth University Hospital, United Kingdom. According to The Human Tissue Acts of England, Wales and Northern Ireland (2004) and Scotland (2006), de-identified, remanent materials can be used for method validation and/or performance assessment without the need for ethical approval.
This work was supported by the funding from the National Institutes of Health (R01 DK067859).
M.H. Gelb is a consultant for PerkinElmer and a co-founder for GelbChem, LLC. Awarded and filed patents filed by M.H. Gelb and co-workers at the University of Washington include US20140249054A1, US20160298166A1, US8802833B2, EP2191006B1, and EP2385950B1. The other authors do not have conflicts of interest to disclose.
Declaration of Competing Interest
M.H. Gelb is a co-founder for GelbChem, LLC. Awarded and patents filed by M.H. Gelb and co-workers at the University of Washington include US20140249054A1, US20160298166A1, US8802833B2, EP2191006B1, and EP2385950B1. The other authors do not have conflicts of interest to disclose.
Acknowledgments
We thank Dr. John Hamilton from the Queen Elizabeth University Hospital for providing DBS samples from LAL-D patients and carriers, and for his valuable input during the sample analysis and data interpretation.
This work was supported by the funding from the National Institutes of Health (R01 DK067859).
Contributor Information
Xinying Hong, Email: hongx@chop.edu.
Michael H. Gelb, Email: gelb@uw.edu.
Data availability
No data was used for the research described in the article.
References
- 1.Reiner Z., Guardamagna O., Nair D., Soran H., Hovingh K., Bertolini S., Jones S., Coric M., Calandra S., Hamilton J., Eagleton T., Ros E. Lysosomal acid lipase deficiency--an under-recognized cause of dyslipidaemia and liver dysfunction. Atherosclerosis. 2014;235:21–30. doi: 10.1016/j.atherosclerosis.2014.04.003. [DOI] [PubMed] [Google Scholar]
- 2.Pericleous M., Kelly C., Wang T., Livingstone C., Ala A. Wolman's disease and cholesteryl ester storage disorder: the phenotypic spectrum of lysosomal acid lipase deficiency. Lancet Gastroenterol. Hepatol. 2017;2:670–679. doi: 10.1016/S2468-1253(17)30052-3. [DOI] [PubMed] [Google Scholar]
- 3.Demaret T., Lacaille F., Wicker C., Arnoux J.B., Bouchereau J., Belloche C., Gitiaux C., Grevent D., Broissand C., Adjaoud D., Abi Warde M.T., Plantaz D., Bekri S., de Lonlay P., Brassier A. Sebelipase alfa enzyme replacement therapy in Wolman disease: a nationwide cohort with up to ten years of follow-up. Orphanet. J. Rare. Dis. 2021;16:507. doi: 10.1186/s13023-021-02134-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burton B.K., Balwani M., Feillet F., Baric I., Burrow T.A., Camarena Grande C., Coker M., Consuelo-Sanchez A., Deegan P., Di Rocco M., Enns G.M., Erbe R., Ezgu F., Ficicioglu C., Furuya K.N., Kane J., Laukaitis C., Mengel E., Neilan E.G., Nightingale S., Peters H., Scarpa M., Schwab K.O., Smolka V., Valayannopoulos V., Wood M., Goodman Z., Yang Y., Eckert S., Rojas-Caro S., Quinn A.G. A phase 3 trial of sebelipase alfa in lysosomal acid lipase deficiency. N. Engl. J. Med. 2015;373:1010–1020. doi: 10.1056/NEJMoa1501365. [DOI] [PubMed] [Google Scholar]
- 5.Jones S.A., Rojas-Caro S., Quinn A.G., Friedman M., Marulkar S., Ezgu F., Zaki O., Gargus J.J., Hughes J., Plantaz D., Vara R., Eckert S., Arnoux J.B., Brassier A., Le Quan Sang K.H., Valayannopoulos V. Survival in infants treated with sebelipase alfa for lysosomal acid lipase deficiency: an open-label, multicenter, dose-escalation study. Orphanet. J. Rare. Dis. 2017;12:25. doi: 10.1186/s13023-017-0587-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hamilton J., Jones I., Srivastava R., Galloway P. A new method for the measurement of lysosomal acid lipase in dried blood spots using the inhibitor Lalistat 2. Clin. Chim. Acta. 2012;413:1207–1210. doi: 10.1016/j.cca.2012.03.019. [DOI] [PubMed] [Google Scholar]
- 7.Lukacs Z., Barr M., Hamilton J. Best practice in the measurement and interpretation of lysosomal acid lipase in dried blood spots using the inhibitor Lalistat 2. Clin. Chim. Acta. 2017;471:201–205. doi: 10.1016/j.cca.2017.05.027. [DOI] [PubMed] [Google Scholar]
- 8.Civallero G., De Mari J., Bittar C., Burin M., Giugliani R. Extended use of a selective inhibitor of acid lipase for the diagnosis of Wolman disease and cholesteryl ester storage disease. Gene. 2014;539:154–156. doi: 10.1016/j.gene.2014.02.003. [DOI] [PubMed] [Google Scholar]
- 9.Fasano T., Pisciotta L., Bocchi L., Guardamagna O., Assandro P., Rabacchi C., Zanoni P., Filocamo M., Bertolini S., Calandra S. Lysosomal lipase deficiency: molecular characterization of eleven patients with Wolman or cholesteryl ester storage disease. Mol. Genet. Metab. 2012;105:450–456. doi: 10.1016/j.ymgme.2011.12.008. [DOI] [PubMed] [Google Scholar]
- 10.Aslanidis C., Ries S., Fehringer P., Büchler C., Klima H., Schmitz G. Genetic and biochemical evidence that CESD and Wolman disease are distinguished by residual lysosomal acid lipase activity. Genomics. 1996;33:85–93. doi: 10.1006/geno.1996.0162. [DOI] [PubMed] [Google Scholar]
- 11.Masi S., Chennamaneni N., Turecek F., Scott C.R., Gelb M.H. Specific substrate for the assay of lysosomal acid lipase. Clin. Chem. 2018;64:690–696. doi: 10.1373/clinchem.2017.282251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hong X., Sadilek M., Gelb M.H. A highly multiplexed biochemical assay for analytes in dried blood spots: application to newborn screening and diagnosis of lysosomal storage disorders and other inborn errors of metabolism. Genet. Med. 2020;22:1262–1268. doi: 10.1038/s41436-020-0790-9. [DOI] [PubMed] [Google Scholar]
- 13.Kelly N., Boychuk N., Wasserstein M. OP056: ScreenPlus pilot newborn screening: recruitment and engagement findings from the first 300 consented infants. Genet. Med. 2022;24:S383. [Google Scholar]
- 14.Bernstein D.L., Hulkova H., Bialer M.G., Desnick R.J. Cholesteryl ester storage disease: review of the findings in 135 reported patients with an underdiagnosed disease. J. Hepatol. 2013;58:1230–1243. doi: 10.1016/j.jhep.2013.02.014. [DOI] [PubMed] [Google Scholar]
- 15.Shen J.J., Davis J.L., Hong X., Laningham F.H., Gelb M.H., Kim G.E. A case of lysosomal acid lipase deficiency confirmed by response to sebelipase alfa therapy. J. Pediatr. Gastroenterol. Nutr. 2020;71:726–730. doi: 10.1097/MPG.0000000000002870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Orsini J.J., Morrissey M.A., Slavin L.N., Wojcik M., Biski C., Martin M., Keutzer J., Zhang X.K., Chuang W.L., Elbin C., Caggana M. Implementation of newborn screening for Krabbe disease: population study and cutoff determination. Clin. Biochem. 2009;42:877–884. doi: 10.1016/j.clinbiochem.2009.01.022. [DOI] [PubMed] [Google Scholar]
- 17.Ficicioglu C., Ahrens-Nicklas R.C., Barch J., Cuddapah S.R., DiBoscio B.S., DiPerna J.C., Gordon P.L., Henderson N., Menello C., Luongo N., Ortiz D., Xiao R. Newborn screening for pompe disease: Pennsylvania experience. Int. J. Neonatal. Screen. 2020;6 doi: 10.3390/ijns6040089. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No data was used for the research described in the article.