

Abstract
Background
Cardiovascular calcifications are prevented by matrix Gla protein (MGP), a vitamin K–dependent protein. Haemodialysis patients exhibit marked vitamin K deficiency. The randomized, prospective, open-label, multicentre VitaVasK trial analysed whether vitamin K1 supplementation reduces progression of coronary artery calcifications (CACs) and thoracic aortic calcifications (TACs).
Methods
Patients with pre-existing CACs were randomized to continue on standard care or to additionally receive 5 mg of vitamin K1 orally thrice weekly. Hierarchically ordered primary endpoints were progression of TAC and CAC in computed tomography scans at 18 months. Linear mixed effects models with repeated measures at baseline and 12 and 18 months assessed treatment effects after adjusting for study site.
Results
Of 60 randomized patients, 20 dropped out for reasons unrelated to vitamin K1, resulting in 23 control and 17 vitamin K1 patients. The trial was stopped early due to slow recruitment. At 18 months, the average TAC progression was 56% lower in the vitamin K1 compared with the control group (p = .039). CAC significantly progressed within the control group, but not within the vitamin K1 group. Average progression at 18 months was 68% lower in the vitamin K1 compared to the control group (P = .072). Vitamin K1 reduced plasma levels of pro-calcific uncarboxylated MGP by 69% at 18 months. No treatment-related adverse events were noted.
Conclusion
Vitamin K1 intervention is a potent, safe and cost-effective approach to correct vitamin K deficiency and to potentially reduce cardiovascular calcification in this high-risk population.
Keywords: matrix Gla protein, valvular calcification, vascular calcification, vitamin K
Graphical Abstract
Graphical Abstract.

INTRODUCTION
Patients on maintenance haemodialysis (HD) exhibit a greatly increased cardiovascular mortality associated with cardiovascular calcifications [1]. Cardiovascular calcifications result from a positive phosphate and calcium balance, as well as reduced activity of inhibitors of calcification [e.g. matrix Gla protein (MGP)] [2, 3]. MGP is an arterial wall- and cardiac valve-based inhibitor of calcification [4]. MGP requires post-translational modification by vitamin K–dependent gamma-glutamyl carboxylation to be fully active. In vitamin K deficiency or antagonism, uncarboxylated, inactive MGP (ucMGP) accumulates. Therapeutic vitamin K antagonism accelerates the development of cardiovascular calcifications and the risk of calciphylaxis in HD patients [5–7].
Vitamin K encompasses two forms, namely vitamin K1 (phylloquinone) and a number of K2 species [menaquinones (MK) 4–13] [5]. In humans, the major forms of vitamin K2 that have been investigated are MK-4 and MK-7, as well as menaquinones derived from intestinal bacterial synthesis. MK-4 can also be generated enzymatically from vitamin K (all forms) by UbiA prenyltransferase domain-containing protein 1 (UBIAD1) [8].
Hepatic vitamin K status is adequate in most populations, as indicated by normal coagulation parameters. However, most healthy individuals have detectable levels of circulating dephosphorylated (dp)-ucMGP, suggesting a suboptimal vascular supply of vitamin K [5]. In chronic dialysis patients, circulating levels of dp-ucMGP are markedly higher than in the normal population in about two-thirds of patients and predict both calcification and mortality [5, 7, 9]. This subclinical vitamin K deficiency results from dietary restrictions [10], possibly impaired vitamin K recycling in uraemia [11] and uraemia-associated alterations of vitamin K uptake and transport in lipoproteins [12].
The above observations, combined with the lack of any known human toxicity of dietary vitamin K supplementation, have laid the basis for a number of intervention trials targeting cardiovascular calcification in HD patients. Supplementation of vitamin K2, specifically MK-7, at doses of up to 860 µg/day (recommended daily vitamin K intake ranges from 75 to 120 µg/day) rapidly lowered serum levels of dp-ucMGP by up to 50% [13–18]. Despite this biochemical benefit of MK-7 supplementation, several intervention trials in advanced chronic kidney disease (CKD) or HD patients consistently failed to note any benefit for the cardiovascular system, such as calcification progression, pulse wave velocity, blood pressure or cardiovascular events [13, 15–19].
In 2010, a pathway was discovered whereby vitamin K1 was converted into K2 (MK-4, but not MK-7) [8], which allowed us to use high-dose, drug-grade vitamin K1 for an intervention. To show that vitamin K1 therapy at 15 mg/week added to standard care reduces the progression of cardiovascular calcification compared with standard care alone, we conducted the randomized multicentre VitaVasK trial [20]. The primary endpoints are the progression of thoracic aortic and coronary artery calcification [CAC; calculated as absolute changes in the volume scores at the 18-month multislice computed tomography (MSCT) versus the baseline MSCT]. Secondary endpoints comprise changes in the Agatston score, mitral and aortic valve calcification as well as major adverse cardiovascular events (MACEs) and all-cause mortality.
MATERIALS AND METHODS
Trial design, population and intervention
VitaVasK is an investigator-initiated, randomized (1:1 in two parallel groups), controlled, prospective, fixed sample, open-label but assessor-blinded clinical trial conducted in four sites in Germany, two sites in Belgium and one site in Sweden (see Acknowledgements). The study protocol was approved by the ethics committees of all participating sites. The study was registered on ClinicalTrials.gov (NCT01742273) and EudraCT (2010-021264-14). Written informed consent was obtained from all patients. There were no changes in the protocol during the conduct of the trial.
The rationale and design of VitaVasK has been described previously [20]. We calculated a total sample size of 348 patients in order for the trial to have 80% power at a two-sided type 1 error of 0.05, based on the assumptions that the mean difference in the increase in the thoracic aortic calcification (TAC) score will be 30% lower in the vitamin K1 group {absolute difference ∼90 [standard deviation (SD) 235]} after 18 months and the dropout rate will be 37.5% due to comorbid conditions in dialysis patients [20].
Adults on chronic maintenance HD for at least 6 months with a baseline CAC volume score >100 mm3 were eligible for inclusion. Key exclusion criteria included intake of vitamin K, history of thrombosis [except arteriovenous (AV) shunt occlusion], intake of vitamin K antagonists at baseline or in the 3 months prior to baseline, inflammatory bowel disease, short-bowel syndrome, significant liver dysfunction, any condition likely to impair vitamin K absorption (i.e. chronic pancreatitis), malignancy and more than one stent in one coronary artery plus one or more stents in an additional artery (Supplementary Table S1). Randomization lists were generated by the Institute of Medical Statistics using the randomization tool RITA (version 1.5.0; Evidat, Germany). The randomization procedure utilized, stratified by centre, was Efron's biased coin, whereby the probability was chosen as two-thirds. Participants were randomly assigned following simple randomization procedures (computerized random numbers) to one of two treatment groups. Whereas patients and physicians allocated to the intervention group were aware of the allocated arm, outcome assessors and data analysts were kept blinded to the allocation.
Prohibited medications during the trial were only vitamin K antagonists and supplements containing vitamin K. Patients were encouraged to maintain their usual diet during the trial.
Eligible patients were randomized to receive 5 mg of vitamin K1, provided in liquid form at a concentration of 10 mg/ml (Ka-Vit, Infectopharm, Heppenheim, Germany), given orally thrice weekly under observation during HD in addition to usual standard care. This dose was chosen based on our prior trial in which 2 mg/day of vitamin K1 reduced valvular progression in patients with an estimated glomerular filtration rate >60 ml/min/1.73 m2 [21]. VitaVasK control patients continued to receive usual standard care. Participants were recruited from October 2013 to January 2019. Clinical, serum-based laboratory and MSCT data were obtained at baseline and at 12 and 18 months. Additional biochemical data were obtained at 4 weeks after randomization. Adverse events were collected by questioning the patients or the treating physicians as well as by monitoring vital signs and routine laboratory tests.
Imaging
To assess cardiovascular calcifications, unenhanced, electrocardiogram-synchronized MSCT of the chest, extending from the aortic arch to the oesophageal hiatus of the diaphragm were performed. To homogenize image quality between study sites, calibration scans with incremental current time products at 120-kV tube voltage were conducted using an anthropomorphic coronary calcification phantom [22]. Two phantom belts simulating different obese conditions were additionally used to enable three study protocols on each scanner for small, medium and obese patients. Image noise was evaluated as the SD of the density value determination of a measurement area (field of view >1 square centimetre) in a water-isodense area of the phantom. For each MSCT scanner and each of the three patient sizes, the protocol with image noise <25 HU that required the lowest X-ray tube current was selected to minimize radiation exposure and to provide consistent image quality.
MSCT scans were performed at baseline and at 12 and 18 months. In all datasets, the Agatston score, the volume score and the mass score [23] were determined for the four locations of calcifications: thoracic aorta, aortic and mitral valves and the coronary arteries. The software Syngo.Via VA20 (Siemens Healthineers, Forchheim, Germany) was used for quantification purposes. The Agatston score was determined using a 3 mm CT slice thickness with an increment of 3 mm (non-overlapped). A detection threshold of ≥130 HU involving ≥1 mm2 area/lesion (3 pixels) was applied. The mass scores at baseline are reported in Supplementary Table S2.
All scans were evaluated independently and in a blinded fashion by two experienced radiologists (SR and RS). Relevant deviations between the two readers were countered by a joint evaluation in which the relevant measured values were determined by mutual agreement and consistently across all measurement time points. In case of coronary intervention during the study period, the affected area of the coronary artery was excluded from the evaluation and the previous or subsequent examinations were consistently evaluated in the same way.
Biochemical measurements
Fasting blood samples were obtained prior to dialysis, centrifuged for 10 min at 3000 g within 30–60 min and immediately stored at −80°C until dry ice shipment for centralized analysis. Serum levels of vitamin K were measured using liquid chromatography tandem mass spectroscopy (LC-MS/MS) consisting of an initial sample purification step prior to tandem MS detection (Magtivio, Nuth, The Netherlands). The assay performance was evaluated through participation in the international Vitamin K External Quality Assurance Scheme. Specific measurement of MK-4 in serum was not considered, since this is only detectable after very high oral MK-4 doses [24].
Circulating dp-ucMGP was quantified in plasma using the commercially available In Vitro Diagnostic CE-marked chemiluminescent InaKtif MGP assay on the IDS-iSYS system (IDS, Boldon, UK). In brief, samples were exposed to magnetic particles coated with monoclonal antibodies against dp-MGP and ucMGP. Trigger reagents were then added, resulting in light emission that was directly proportional to the level of dp-ucMGP in the sample. The within-run and total variations of this assay are 0.8–6.2% and 3.0–8.2%, respectively. The assay quantitation range was between 50 and 12 000 pmol/l and was linear up to 11 651 pmol/l.
Routine blood analyses were performed at locally certified laboratories.
Outcomes
The two hierarchical co-primary endpoints were progression of TAC (i.e. the absolute change in the volume score at the 18-month MSCT versus the baseline MSCT) as well as progression of CAC (i.e. the absolute change in the volume score at the 18-month MSCT versus the baseline MSCT). Secondary endpoints included progression of TAC or CAC assessed via the Agatston score method (see below), progression of aortic and mitral valve calcification and mortality from any cause or MACE within 18 months after the start of treatment (Supplementary Table S1). VitaVasK was stopped early in January 2019 because of slow recruitment.
Statistical methods
Statistical analyses were performed using SAS 9.4 software (SAS Institute, Cary, NC, USA) and graphical presentations were done using R version 4.1.0 (R Foundation for Statistical Computing, Vienna, Austria). For the first primary endpoint analysis of the TAC (volume score), we applied a linear mixed effects model with repeated effect for the time points baseline, 12 months and 18 months (proc MIXED in SAS). We modelled treatment and centre as fixed effects and also included the interaction between treatment and time point. We adjusted for baseline calcification, as repeated mixed effects models were used, and the changes from baseline status were used for interpretation. The spatial power covariance structure was used and residual plots were examined visually to assess the model fit (Supplementary Fig. S1 and S2). Thus extreme outliers (n = 1 per primary endpoint or 1–2 per secondary endpoints) based on the restricted likelihood distance were excluded. Group differences in the changes of TAC from baseline to 18 months were computed with linear contrasts. As the trial was stopped prematurely, the p-values are explorative and were denoted as statistically significant if they fell below the usual significance level of 5%. Applying the significance level to the hierarchical decision rule, as planned in the protocol, the second primary endpoint, CAC (volume score), was analysed using the same statistical model.
For the first and second primary endpoints, two sensitivity analyses were performed. As the randomization was not stratified by gender, we accounted for this confounder in the analysis. For this purpose we first included the confounders gender and age and in the second we included further confounders gender, age, smoking status and diabetes mellitus as fixed effects. For the secondary endpoints TAC (Agatston score), CAC (Agatston score), aortic valve calcification (volume and Agatston scores) and mitral valve calcification (volume and Agatston scores), the statistical models were fitted analogously to the primary endpoints and sensitivity analyses were performed accordingly. For the secondary endpoints all-cause mortality and major cardiovascular events, we performed Fisher exact tests to investigate the occurrence between the two treatment groups. Further analyses were performed for serum vitamin K concentrations and dp-ucMGP levels, applying a linear mixed effects model with repeated effect for the time points baseline, 1 month, 12 months and 18 months and with the same specifications as before.
The analysis population was the full analysis set following International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use E9 guidelines [25].
RESULTS
Trial participants and baseline parameters
Of the 60 patients randomized into the trial, 20 patients dropped out over the subsequent 18 months, 12 of 29 patients in the K1 group and 8 of 31 in the standard care group [odds ratio 2.0 (95% confidence interval 0.68–6.05)]. This is expected in a multimorbid population [20], and there were no obvious differences in causes for dropping out dominating in either group (Fig. 1).
Figure 1:

Consolidated Standards of Reporting Trials (CONSORT) flow diagram.
The final analysis population consisted of 40 patients, mostly men, mostly non-diabetic and on chronic HD for an average of 75 months (Table 1). There were numerically more smokers or ex-smokers in the vitamin K1 group and more diabetic patients in the control group, with no differences in current haemoglobin A1c levels.
Table 1:
Baseline characteristics of all participants included in the analysis of the VitaVasK study.
| Characteristics | Controls (n = 23) | Vitamin K1 (n = 17) |
|---|---|---|
| Female, n (%) | 6 (26) | 6 (38) |
| Age (years) | 64.4 ± 13.3 | 62.5 ± 12.3 |
| Dialysis vintage (months) | 82.1 ± 65.6 | 65.2 ± 56.6 |
| Body mass index (kg/m2) | 27.4 ± 6 | 26.7 ± 4.9 |
| Smoking status,an (%)Current or ex-smokerNon-smoker | 12 (55)10 (46) | 11 (69)5 (31) |
| Systolic blood pressureb (mmHg) | 141.9 ± 18.4 | 138.9 ± 19.7 |
| Diastolic blood pressureb (mmHg) | 69.6 ± 13.6 | 75.0 ± 16.7 |
| Diabetes mellitus, n (%) | 8 (34.8) | 2 (11.8) |
| Haemoglobin A1c (%) | 5.5 ± 0.8 | 5.5 ± 1 |
| International normalized ratio | 0.98 ± 0.09 | 0.96 ± 0.07 |
| Haemoglobin (g/l) | 111.5 ± 8.6 | 109.9 ± 9.9 |
| Serum ionized calcium (mmol/l) | 1.1 ± 0.1 | 1.1 ± 0.2 |
| Serum phosphate (mmol/l) | 1.8 ± 0.5 | 1.9 ± 1.2 |
| Serum intact parathyroid hormone (ng/l) | 265.3 ± 306.7 | 536.1 ± 426.8 |
| Serum alkaline phosphatase (U/I) | 93 ± 52 | 93 ± 48 |
| Bone-specific alkaline phosphatase (U/I) | 27 ± 23 | 36 ± 33 |
| 25-hydroxyvitamin D3 (μg/l) | 45.2 ± 20.2 | 31.7 ± 19.7 |
| Serum total magnesium (mmol/l) | 0.9 ± 0.1 | 1 ± 0.2 |
| Total cholesterol (mg/dl) | 169.4 ± 41.3 | 166.7 ± 34.3 |
| C-reactive protein (mg/l) | 5.5 ± 8.6 | 4.7 ± 4.3 |
| Statin therapy, n (%) | 11 (47.8) | 8 (47.0) |
| Native vitamin D therapy, n (%) | 18 (85.7) | 14 (93.3) |
| Vitamin D receptor agonist therapy, n (%) | 15 (71.4) | 8 (53.3) |
| Calcium-free phosphate binder therapy, n (%) | 12 (65.2) | 13 (82.4) |
| Calcium-containing phosphate binder therapy, n (%) | 15 (71.4) | 9 (60.0) |
| Calcimimetic therapy, n (%) | 9 (42.9) | 7 (46.7) |
| Months from baseline CT to planned 12-month CT | 12.6 ± 1.3 | 12.7 ± 1.4 |
| Months from baseline CT to planned 18-month CT | 19.6 ± 1.4 | 18.3 ± 1.1 |
| Coronary artery disease, n (%) | 11 (34.8) | 8 (41.8) |
| TAC (mm3) | 4864.6 ± 4792.0 | 4830.3 ± 10 621.3 |
| Aortic valve calcification (mm3) | 148.2 ± 246.3 | 133.5 ± 171.0 |
| CAC (mm3) | 1886.9 ± 1143.1 | 1550.5 ± 1419.7 |
| Mitral valve calcification (mm3) | 1063.1 ± 2237.1 | 936.6 ± 1764.8 |
Values are presented as mean ± SD unless stated otherwise. All calcification values are volume scores.
aInformation on smoking status was missing for one person in each group.
bPre-dialysis blood pressure after long interdialytic interval.
Times from baseline MSCT to the 12-month MSCT were similar between the two groups, whereas MSCTs for the primary endpoint at 18 months were performed on average 1.3 months later in the controls compared with the vitamin K1 group. The mean baseline calcification volume scores of the thoracic aorta were very similar in the two groups, whereas mean coronary calcification scores at baseline were ∼20% higher in the control group compared with the vitamin K1 group (Table 1).
Primary endpoint
As shown in Fig. 2A and Table 2, TAC progressed significantly between baseline and 12 and 18 months in both study populations. However, the progression of TAC was significantly reduced by an average of 56% {892.1 mm3 [standard error (SE) 423.0], model 1}) in the vitamin K1 group compared with the control group at 18 months (P =.039). Progression at 12 months was reduced by a mean of 41% [325.8 mm3 (SE 334.0), model 1] in the vitamin K1 group compared with the control group, but this difference failed to reach statistical significance (P = .333). Of the various parameters potentially affecting calcification progression, only age at baseline exerted a significant effect on the endpoint (Supplementary Table S3). Consistent with this, multivariate adjustment in the different models did not lead to any qualitative change in the above statements (Table 2).
Figure 2:
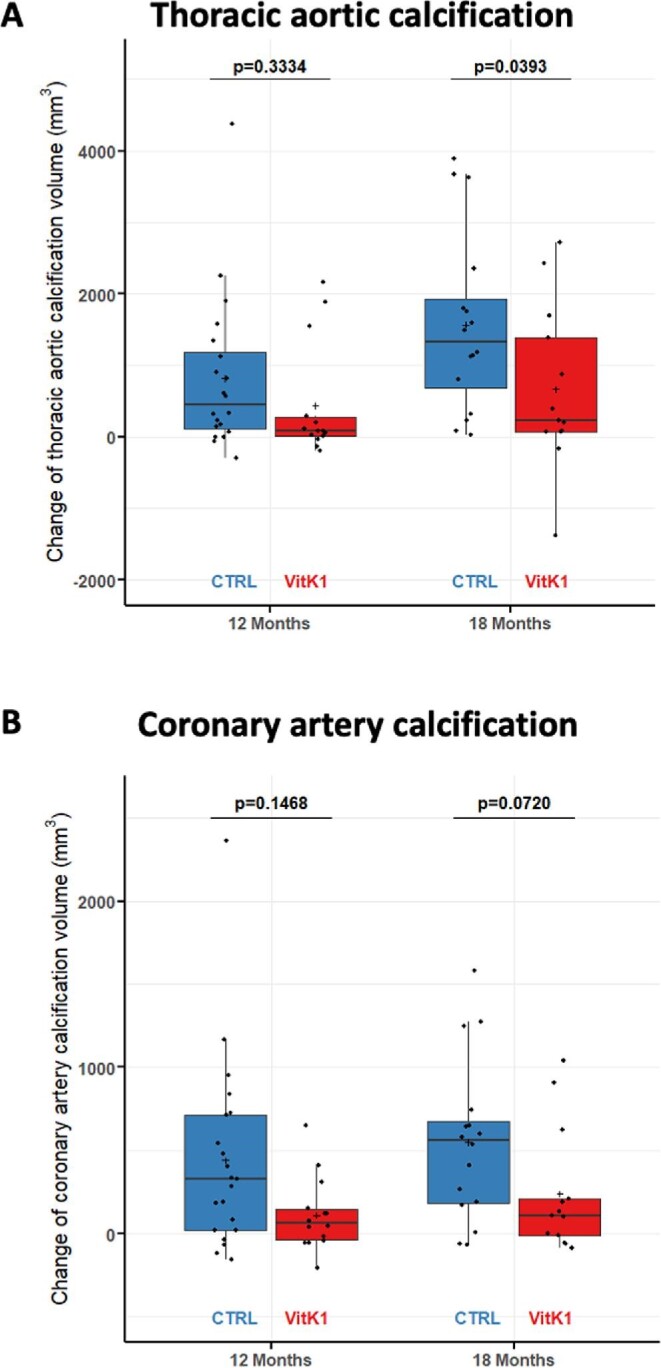
Primary endpoints of the VitaVasK trial: (A) progression of TAC (i.e. the absolute change of the volume score at the 12- and 18-month MSCT versus the baseline MSCT) as well as (B) progression of CAC (i.e. the absolute change in the volume score at the 12- and 18-month MSCT versus the baseline MSCT). Individual data, means and SDs are shown.
Table 2:
Primary endpoint TAC score: changes in volume scores (mm3) between baseline and 12 and 18 months in participants of the VitaVasK study.
| Model 1 | Model 2 | Model 3 | ||
|---|---|---|---|---|
| TAC volume estimates (SE) | ||||
| Vitamin K | Baseline | 3273.6 (1328.6) | 4729.3 (1313.4) | 1409.6 (1402.5) |
| 12 months | 3752.5 (1329.9) | 5200.7 (1314.1) | 1880.8 (1403.2) | |
| 18 months | 3983.1 (1332.3) | 5430.6 (1318.2) | 2115.4 (1406.2) | |
| Control | Baseline | 5231.7 (1066.3) | 5971.6 (1026.3) | 3794.7 (1075.9) |
| 12 months | 6036.4 (1067.3) | 6775.8 (1027.3) | 4615.4 (1077.2) | |
| 18 months | 6833.3 (1071.0) | 7578.3 (1032.9) | 5413.0 (1080.1) | |
| Changes in volume score versus baseline within groups | ||||
| Vitamin K | Baseline versus 12 months | 478.9 (256.4), 0.0668 | 471.4 (263.6), 0.0791 | 471.2 (265.9), 0.0820 |
| Baseline versus 18 months | 709.5 (322.6), 0.0318 | 701.3 (336.8), 0.0418 | 705.7 (339.6), 0.0424 | |
| Control | Baseline versus 12 months | 804.7 (214.1), 0.0004 | 804.1 (215.9), 0.0004 | 820.7 (228.4), 0.0007 |
| Baseline versus 18 months | 1601.6 (273.6), <0.0001 | 1606.7 (275.5), <0.0001 | 1618.3 (285.0), <0.0001 | |
| Changes in volume score versus baseline between groups | ||||
| Vitamin K versus control | Baseline versus 12 months | 325.8 (334.0), 0.3334 | 332.8 (340.7), 0.3329 | 349.5 (350.6), 0.3231 |
| Baseline versus 18 months | 892.1 (423.0), 0.0393 | 905.4 (435.1), 0.0420 | 912.6 (443.4), 0.0443 | |
Data are linear mixed model estimates (SE) and P-value.
Model 1: adjusted for centre; model 2: adjusted for centre, gender and age; model 3: adjusted for centre, gender, age, smoking status and diabetes mellitus. All models automatically adjust for baseline calcification as repeated mixed effects models were used.
The extent of CAC also increased significantly in both groups between baseline and 18 months (Fig. 2B, Table 3). Progression at 18 months was lower by an average of 68% [412.7 mm3 (SE 225.4), model 1] in the vitamin K1 group compared with the control group (P = .072). Progression at 12 months was reduced by a mean of 65% [265.7 mm3 (SE 180.8), model 1] in the vitamin K1 group compared with the control group (P = .147). Sensitivity analyses yielded qualitatively similar data (Supplementary Table S3).
Table 3:
Primary endpoint CAC score: changes in volume scores (mm3) between baseline and 12 and 18 months in participants of the VitaVasK study.
| Model 1 | Model 2 | Model 3 | ||
|---|---|---|---|---|
| CAC volume estimates (SE) | ||||
| Vitamin K | Baseline | 1045.6 (347.8) | 1237.0 (400.4) | 1381.2 (400.4) |
| 12 months | 1187.4 (349.9) | 1378.1 (401.7) | 1521.8 (401.7) | |
| 18 months | 1242.2 (351.5) | 1433.1 (404.5) | 1580.0 (403.9) | |
| Control | Baseline | 1388.4 (286.5) | 1422.3 (322.5) | 1517.8 (313.0) |
| 12 months | 1796.0 (287.4) | 1829.8 (323.2) | 1904.2 (313.9) | |
| 18 months | 1997.8 (291.8) | 2032.6 (328.6) | 2109.8 (317.4) | |
| Changes in volume score versus baseline within groups | ||||
| Vitamin K | Baseline versus 12 months | 141.8 (138.3), 0.3092 | 141.1 (141.9), 0.3241 | 140.7 (143.4), 0.3308 |
| Baseline versus 18 months | 196.6 (169.5), 0.2505 | 196.2 (176.8), 0.2716 | 198.8 (178.1), 0.2690 | |
| Control | Baseline versus 12 months | 407.5 (116.4), 0.0009 | 407.5 (117.4), 0.0010 | 386.4 (124.3), 0.0029 |
| Baseline versus 18 months | 609.3 (148.6), 0.0001 | 610.3 (149.9), 0.0001 | 592.0 (154.9), 0.0003 | |
| Changes in volume score versus baseline between groups | ||||
| Vitamin K versus control | Baseline versus 12 months | 265.7 (180.8), 0.1468 | 266.4 (184.2), 0.1534 | 245.7 (189.8), 0.2006 |
| Baseline versus 18 months | 412.7 (225.4), 0.0720 | 414.2 (231.7), 0.0790 | 393.2 (236.0), 0.1012 | |
Data are linear mixed model estimates (SE) and P-value.
Model 1: adjusted for centre; model 2: adjusted for centre, gender and age; model 3: adjusted for centre, gender, age, smoking status and diabetes mellitus. All models automatically adjust for baseline calcification as repeated mixed effects models were used.
Secondary endpoints
When progression of TAC was assessed using the Agatston score instead of the volume score, qualitatively similar results were obtained: progression was significantly reduced in the vitamin K1 group at 18 months but not at 12 months (Fig. 3A, Supplementary Table S4). Progression at 18 months was reduced by an average of 57% [1166.1 (SE 551.4), model 1; P = .039; Supplementary Table S4). With respect to progression of CAC, Agatston score progression was significantly decreased in the vitamin K1 group at 18 months [average reduction 72%; 565.8 (SE 252.0), model 1; P = .028] but not at 12 months (Fig. 3B, Supplementary Table S5). Multivariate adjustment with the different models did not lead to any qualitative change in the above statements (Supplementary Tables S4 and S5).
Figure 3:

Secondary endpoints of the VitaVasK trial: (A) progression of TAC (i.e. the absolute change in the Agatston score at the 12- and 18-month MSCT versus the baseline MSCT), (B) progression of CAC (i.e. the absolute change in the Agatston score at the 12- and 18-month MSCT versus the baseline MSCT), (C) progression of aortic valve calcification (i.e. the absolute change in the volume score at the 12- and 18-month MSCT versus the baseline MSCT) and (D) progression of mitral valve calcification (i.e. the absolute change in the volume score at the 12- and 18-month MSCT versus the baseline MSCT). Individual data, means and SDs are shown.
Progression of aortic valve calcification measured by volume scores was on average 76% lower at 18 months in the vitamin K1 group, but this result failed to reach statistical significance (Fig. 3C, Supplementary Table S6). Similar findings were obtained using the Agatston score (data not shown). Calcification progression of the mitral valve at 18 months measured by the volume score was reduced by a mean of 38% in the vitamin K1 group; again, this was not statistically significant (Fig. 3D, Supplementary Table S7). Assessment via the Agatston score yielded similar findings (data not shown).
All-cause mortality at 18 months did not differ between the two groups (i.e. all 60 randomized patients) (P = .703) (Table 4). Five major cardiovascular events (myocardial infarction, stroke, acute coronary syndrome, embolism, symptom-driven revascularization, death from cardiovascular cause) were observed in the control group and three in the vitamin K1 group (Table 4), a non-significant difference (P = .751).
Table 4:
Number of participants with adverse events according to the Medical Dictionary for Regulatory Activities System Order Class version 23.1 and number of deaths in the VitaVasK study.
| Event | Controls (n = 32) | Vitamin K1 (n = 30) |
|---|---|---|
| Any adverse event | 91 | 55 |
| Any serious adverse event | 64 | 53 |
| DeathsDeath from cardiovascular cause within 18 months after start of treatment | 31 | 40 |
| Blood and lymphatic | 2 | 1 |
| Myocardial infarctionAcute coronary syndromeOther cardiaca | 3213 | 124 |
| Congenital and genetic | 0 | 0 |
| Ear and labyrinth | 0 | 0 |
| Endocrine | 1 | 1 |
| Eye | 2 | 2 |
| Gastrointestinal | 10 | 14 |
| General | 14 | 7 |
| Hepatobiliary | 1 | 1 |
| Immune system | 0 | 0 |
| Infections and infestations | 10 | 8 |
| Injury, poisoning and procedural complications | 8 | 2 |
| Investigations | 8 | 5 |
| Metabolism and nutrition | 0 | 0 |
| Musculoskeletal | 9 | 6 |
| Neoplasms | 0 | 3 |
| Nervous systemStroke | 61 | 21 |
| Pregnancy | 0 | 0 |
| Product issues | 0 | 0 |
| Psychiatric | 3 | 1 |
| Renal and urinary | 0 | 0 |
| Reproductive and breast | 0 | 0 |
| Respiratory | 11 | 14 |
| Skin | 2 | 1 |
| Social | 0 | 0 |
| Surgical/medical procedures | 17 | 18 |
| EmbolismSymptoms-driven revascularizationAV fistula thrombosisOther vascular | 002010 | 0163 |
Arrhythmia, hypertensive crisis, hypotension, stable angina pectoris.
Biochemical measurements
Fig. 4A and Supplementary Table S8 show that serum vitamin K concentrations progressively increased in the vitamin K1 cohort (+590% at 18 months), but not in the control group (−4% at 18 months). Circulating plasma dp-ucMGP showed a rapid, pronounced reduction of the baseline level to 36.5 ± 2.0% at the week 4 visit, 33.9 ± 35.2% at the 12-month visit and 30.9 ± 64.2% at the 18-month visit in the vitamin K1 group as opposed to 109.1 ± 5.6% (week 4), 115.6 ± 4.7% (12 months) and 101.7 ± 20.9% (18 months), respectively, in the control group (Fig. 4B and Supplementary Table S9).
Figure 4:

Biochemical endpoints of the VitaVasK trial: (A) change in serum vitamin K1 concentration versus baseline at follow-up visits of 4 weeks, 12 months and 18 months and (B) change in plasma ucMGP concentration versus baseline at follow-up visits of 4 weeks, 12 months and 18 months. Individual data, means and SDs are shown.
Vitamin K uptake is potentially affected by sevelamer via intestinal binding [26]. However, when comparing patients receiving sevelamer versus non-sevelamer phosphate binders, there were no significant differences in vitamin K1 or dp-ucMGP levels or progression of calcification over 18 months (data not shown). Serum concentrations of phosphate, intact parathyroid hormone and bone-specific alkaline phosphatase remained relatively constant during the study period with no obvious trends in either group (Supplementary Table S9).
Adverse events
In the 60 patients allocated to the treatment groups, no new safety signal evolved in the group receiving vitamin K1 (Table 4). In particular, we noted no increase in thromboembolic events in this group, but rather a trend towards lower event rates of AV fistula thrombosis (20 cases in the control group versus 6 cases in the vitamin K1 group; P = .185).
DISCUSSION
Our multicentre, randomized controlled VitaVasK trial is the first to document a marked attenuation of aortic calcification progression in chronic HD patients with vitamin K1 therapy. We noted a similar trend towards less rapid progression of CAC in the group of patients receiving vitamin K1, which reached statistical significance if evaluated using the Agatston score.
A number of recent randomized controlled trials have failed to observe any cardiovascular benefit from vitamin K supplementation in HD patients or patients with advanced CKD [13, 15–18]. The central difference between these earlier trials and our VitaVasK trial is that we administered 15 mg of vitamin K1 per week, which is ∼29 times higher than the daily recommended vitamin K intake of 75 µg. Previous trials in dialysis patients used 200–400 µg of MK-7 [13, 16, 17], i.e. 2.7–5.3-fold higher than the recommended intake, or 90–400 µg daily in non-dialysis CKD patients [15, 18]. Second, all other trials focused on vitamin K2, specifically MK-7. Long-chain menaquinones such as MK-7 are believed to act in non-hepatic tissues, exhibit a long half-life in healthy subjects [24, 27] and, among the vitamin K2 species, MK-7 is available in synthetic form. While this renders MK-7 attractive in cardiovascular intervention trials, we recently reported that in chronic dialysis patients, the uraemic high-density lipoprotein particles, i.e. important transport vehicles of MK-7 in healthy individuals, hardly incorporate any MK-7 [12]. Thus our very high vitamin K1 dosage, the thrice weekly administration under observation and the altered biodistribution of MK-7 in uraemia can explain why we found a rapid and sustained ∼75% reduction in ucMPG levels with vitamin K1, which can convert into the vitamin K2 species MK-4. In the earlier trials, correction of vitamin K deficiency was less potent and dp-ucMGP plasma levels decreased either slowly and maximally by 47% [17], transiently by 39% [16] or to a small extent only in warfarin-naïve patients [13] by 11% [15] or by 17% [18].
Other aspects may also account for the discrepant observations of benefit versus no benefit from vitamin K therapy. Thus several of the above-mentioned trials lasted only 9–12 months [15, 17, 18], which would not have yielded a significant benefit in VitaVasK in any case, even though a trend towards lesser calcification progression with vitamin K was apparent at this time point. In addition, whereas in VitaVasK, 40 of 60 randomized patients were available for analysis at the 18-month study endpoint, other trials had dropout rates of ∼50% at 12 months [17] or 56% over 24 months [16]. Important differences also relate to the Valkyrie trial, which, like VitaVasK, lasted 18 months. Valkyrie included a very particular HD population, notable for an average age of ∼80 years, non-valvular atrial fibrillation in all patients and prior vitamin K antagonist therapy in the majority of patients [13]. Not unexpectedly, the extent of baseline calcification, e.g. in the thoracic aorta, was therefore ∼1.5–2-fold higher in Valkyrie compared with VitaVasK. Nevertheless, a trend towards less calcification progression in the thoracic aorta with MK-7 supplementation was present in Valkyrie as well [13].
The key finding of the multivariate analysis in VitaVasK, namely slower progression of calcification in vitamin K1–treated patients, is consistent with a number of studies in patients without or with only modest CKD. Healthy individuals with CACs experienced slower progression if they received 500 µg vitamin K1 per day for 36 months [28]. In our prior trial in patients with calcific aortic valve stenosis and CKD stages 0–3, daily administration of 2 mg of vitamin K1 for 12 months led to less progression of radiographic calcification and better haemodynamic outcomes than in controls [21]. However, in non-CKD populations there are also clinical trials in which vitamin K1 or MK-7 failed to affect the progression of CAC or haemodynamic changes such as pulse wave velocity [29, 30].
While experimentally the combination of any phosphate binder with vitamin K2 was more potent than either measure alone to retard progression of calcification [31], there are also data indicating that sevelamer in particular may interfere with vitamin K absorption in dialysis patients [26, 32]. However, within the limitations of our trial, we found no evidence for any significant effect of sevelamer on the efficacy of our vitamin K1 therapy.
A major limitation of our trial is that it had to be stopped early, given the slow recruitment, most likely mainly due to the lack of financial incentives and competing trials with better financial rewards. However, our final analysed population is in a similar range compared with previous vitamin K studies in the dialysis population [15–17]. Our power calculation was largely based on the study by Mazzaferro et al. [33] and the ADVANCE trial [34], which focused on progression of calcification in dialysis patients, as these were the only available studies when we designed VitaVasK. However, the cohorts in both studies are different from that studied in ViKaVasK, in particular younger and ∼40–50% less calcified at baseline. Both of these are likely to result in much slower progression of calcification. In contrast, VitaVasK may have a much higher power to detect changes in calcification. The open-label nature of the trial does not appear to be a major limitation, given that all radiographic and biochemical endpoints were obtained and analysed in a blinded fashion. In our partially unblinded design, patients might have changed their dietary habits (Hawthorne effect), but stable dp-ucMGP levels in control patients argue against major changes in dietary vitamin K intake. Further, as the RITA software was used for randomization, which was organized centrally, predictability, and thus allocation bias, was mitigated. An imbalance among the covariates of both groups was addressed by the adjusted statistical models and analyses chosen, as adjustment for these covariates was implemented. Missing data are another problem that we dealt with in the statistical analysis model. A central strength of VitaVasK is the administration of vitamin K1 under observation during dialysis, which allows for very high patient adherence compared with daily oral regimens, and thus mitigates compliance bias [16–18].
In summary, despite early termination, this trial identifies vitamin K1 as a promising drug for correcting vitamin K deficiency and decreasing the progression of vascular calcification in HD patients. Vitamin K1 therapy is a potent, safe and cost-effective approach to reducing cardiovascular calcification in this high-risk population.
Supplementary Material
APPENDIX
VitaVask Investigators
Jürgen Floege, Turgay Saritas, Sebastian Reinartz, Robert Siepmann, Stephanie Wied, Ralf-Dieter Hilgers, Christoph Kuppe, Roland Böhm, Maria Ritzerfeld, Jörg Saupe, Luigi Villa, Yvonne Grafen, Beate Schneider, Bernhard Holschbach and Leon Schurgers (For Aachen, Germany); Johannes Stegbauer, Ralf Westenfeld, Claudia Schmidt, Georg Schlieper, Gerd R. Hetzel, Frank Dellana, Stephanie Taub and Thilo Krüger (Düsseldorf, Germany); Christoph Kopp, Michael Leidig, Yvonne Thoß and Michaela Streubert (Erlangen, Germany); Orfeas Liangos, Patrick Biggar, Monika Wittig and Markus Ketteler (Coburg, Germany); Pieter Evenepoel, Helga Wielandt, Joanna De Vis, Veerle Verbeek and Steven Dymarkowski (Leuven, Belgium); Michel Jadoul, Laura Labriola, Mercedes Vignioble, Vinciane De Backer, Marie-Agnes Ronsyn and Perrine Triqueneaux (Brussels, Belgium); Peter Stenvinkel, Olof Heimburger and Ulrika Jensen Durgé (Stockholm, Sweden).
Contributor Information
Turgay Saritas, Division of Nephrology, RWTH Aachen University Hospital, Aachen, Germany; Institute of Experimental Medicine and Systems Biology, RWTH Aachen University, Aachen, Germany.
Sebastian Reinartz, Department of Radiology, RWTH Aachen University Hospital, Aachen, Germany.
Thilo Krüger, MVZ DaVita Geilenkirchen, Geilenkirchen, Germany.
Markus Ketteler, General Internal Medicine and Nephrology, Robert-Bosch-Hospital, Stuttgart, Germany.
Orfeas Liangos, KfH Kidney Center Lichtenfels, Lichtenfels, Germany; Division of Nephrology, Faculty of Medicine, University of Würzburg, Würzburg, Germany.
Laura Labriola, Division of Nephrology, Cliniques Universitaires Saint Luc, Université Catholique de Louvain, Brussels, Belgium.
Peter Stenvinkel, Division of Renal Medicine, Department of Clinical Science, Technology and Intervention, Karolinska University Hospital, Stockholm, Sweden.
Christoph Kopp, Department of Nephrology and Hypertension, Friedrich-Alexander-University Erlangen-Nürnberg, Erlangen, Germany.
Ralf Westenfeld, Division of Cardiology, Pulmonology, and Vascular Medicine, Medical Faculty, Heinrich Heine University, Düsseldorf, Germany.
Pieter Evenepoel, Department of Nephrology, KU Leuven University Hospitals Leuven, Leuven, Belgium.
Robert Siepmann, Department of Radiology, RWTH Aachen University Hospital, Aachen, Germany.
Stephanie Wied, Department of Medical Statistics, RWTH Aachen University Hospital, Aachen, Germany.
Ralf-Dieter Hilgers, Department of Medical Statistics, RWTH Aachen University Hospital, Aachen, Germany.
Leon Schurgers, Department of Biochemistry and Cardiovascular Research Institute Maastricht, School for Cardiovascular Diseases, Maastricht University, Maastricht, The Netherlands.
Jürgen Floege, Division of Nephrology, RWTH Aachen University Hospital, Aachen, Germany.
VitaVasK Investigators:
Jürgen Floege, Turgay Saritas, Sebastian Reinartz, Robert Siepmann, Stephanie Wied, Ralf-Dieter Hilgers, Christoph Kuppe, Roland Böhm, Maria Ritzerfeld, Jörg Saupe, Luigi Villa, Yvonne Grafen, Beate Schneider, Bernhard Holschbach, Leon Schurgers, Johannes Stegbauer, Ralf Westenfeld, Claudia Schmidt, Georg Schlieper, Gerd R Hetzel, Frank Dellana, Stephanie Taub, Thilo Krüger, Christoph Kopp, Michael Leidig, Yvonne Thoß, Michaela Streubert, Johannes, Orfeas Liangos, Patrick Biggar, Monika Wittig, Markus Ketteler, Pieter Evenepoel, Helga Wielandt, Joanna De Vis, Veerle Verbeek, Steven Dymarkowski, Michel Jadoul, Laura Labriola, Mercedes Vignioble, Vinciane De Backer, Marie-Agnes Ronsyn, Perrine Triqueneaux, Peter Stenvinkel, Olof Heimburger, and Ulrika Jensen Durgé
FUNDING
We gratefully acknowledge the funding by the European Renal Association as well as the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) (TRR 219, Project ID 322900939). The vitamin K1 used for the study was donated by InfectoPharm, Heppenheim, Germany.
AUTHORS’ CONTRIBUTIONS
J.F., L.S., R.D.H., T.K. and S.R. were responsible for the research idea and study design. T.S., S.R., T.K., M.K., O.L., L.L., P.S., C.K., R.W., P.E., R.S. and J.F. were responsible for data acquisition. R.S. and S.R. were responsible for the quantification of cardiovascular calcifications. L.S. measured vitamin K and uncarboxylated MGP. S.W. and R.D.H. were responsible for the statistical analysis. T.S. and J.F. were responsible for original draft preparation and editing. Each author contributed important intellectual content during manuscript drafting or revision and accepts accountability for the overall work by ensuring that questions pertaining to the accuracy or integrity of any portion of the work are appropriately investigated and resolved.
DATA AVAILABILITY STATEMENT
Public posting of individual-level participant data is not covered by the informed patient consent form.
CONFLICT OF INTEREST STATEMENT
The authors have no financial conflicts of interest to disclose.
REFERENCES
- 1. Tonelli M, Muntner P, Lloyd Aet al. Risk of coronary events in people with chronic kidney disease compared with those with diabetes: a population-level cohort study. Lancet 2012;380:807–14. [DOI] [PubMed] [Google Scholar]
- 2. Schlieper G, Schurgers L, Brandenburg Vet al. Vascular calcification in chronic kidney disease: an update. Nephrol Dial Transplant 2016;31:31–39. [DOI] [PubMed] [Google Scholar]
- 3. Durham AL, Speer MY, Scatena Met al. Role of smooth muscle cells in vascular calcification: implications in atherosclerosis and arterial stiffness. Cardiovasc Res 2018;114:590–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Luo G, Ducy P, McKee MDet al. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature 1997;386:78–81. [DOI] [PubMed] [Google Scholar]
- 5. Kaesler N, Schurgers LJ, Floege J.. Vitamin K and cardiovascular complications in CKD patients. Kidney Int 2021;100:1023–36. [DOI] [PubMed] [Google Scholar]
- 6. Holden RM, Morton AR, Garland JSet al. Vitamins K and D status in stages 3–5 chronic kidney disease. Clin J Am Soc Nephrol 2010;5:590–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fusaro M, Crepaldi G, Maggi Set al. Vitamin K, bone fractures, and vascular calcifications in chronic kidney disease: an important but poorly studied relationship. J Endocrinol Invest 2011;34:317–23. [DOI] [PubMed] [Google Scholar]
- 8. Nakagawa K, Hirota Y, Sawada Net al. Identification of UBIAD1 as a novel human menaquinone-4 biosynthetic enzyme. Nature 2010;468:117–21. [DOI] [PubMed] [Google Scholar]
- 9. Schlieper G, Westenfeld R, Kruger Tet al. Circulating nonphosphorylated carboxylated matrix gla protein predicts survival in ESRD. J Am Soc Nephrol 2011;22:387–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cranenburg EC, Schurgers LJ, Uiterwijk HHet al. Vitamin K intake and status are low in hemodialysis patients. Kidney Int 2012;82:605–10. [DOI] [PubMed] [Google Scholar]
- 11. Kaesler N, Magdeleyns E, Herfs Met al. Impaired vitamin K recycling in uremia is rescued by vitamin K supplementation. Kidney Int 2014;86:286–93. [DOI] [PubMed] [Google Scholar]
- 12. Kaesler N, Schreibing F, Speer Tet al. Altered vitamin K biodistribution and metabolism in experimental and human chronic kidney disease. Kidney Int 2022;101:338–48. [DOI] [PubMed] [Google Scholar]
- 13. De Vriese AS, Caluwe R, Pyfferoen Let al. Multicenter randomized controlled trial of vitamin K antagonist replacement by rivaroxaban with or without vitamin K2 in hemodialysis patients with atrial fibrillation: the Valkyrie study. J Am Soc Nephrol 2020;31:186–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Westenfeld R, Krueger T, Schlieper Get al. Effect of vitamin K2 supplementation on functional vitamin K deficiency in hemodialysis patients: a randomized trial. Am J Kidney Dis 2012;59:186–95. [DOI] [PubMed] [Google Scholar]
- 15. Kurnatowska I, Grzelak P, Masajtis-Zagajewska Aet al. Effect of vitamin K2 on progression of atherosclerosis and vascular calcification in nondialyzed patients with chronic kidney disease stages 3–5. Pol Arch Med Wewn 2015;125:631–40. [DOI] [PubMed] [Google Scholar]
- 16. Levy-Schousboe K, Frimodt-Moller M, Hansen Det al. Vitamin K supplementation and arterial calcification in dialysis: results of the double-blind, randomized, placebo-controlled Renakvit trial. Clin Kidney J 2021;14:2114–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Oikonomaki T, Papasotiriou M, Ntrinias Tet al. The effect of vitamin K2 supplementation on vascular calcification in haemodialysis patients: a 1-year follow-up randomized trial. Int Urol Nephrol 2019;51:2037–44. [DOI] [PubMed] [Google Scholar]
- 18. Witham MD, Lees JS, White Met al. Vitamin K supplementation to improve vascular stiffness in CKD: the K4Kidneys randomized controlled trial. J Am Soc Nephrol 2020;31:2434–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Caluwe R, Pyfferoen L, De Boeck Ket al. The effects of vitamin K supplementation and vitamin k antagonists on progression of vascular calcification: ongoing randomized controlled trials. Clin Kidney J 2016;9:273–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Krueger T, Schlieper G, Schurgers Let al. Vitamin K1 to slow vascular calcification in haemodialysis patients (VitaVasK trial): a rationale and study protocol. Nephrol Dial Transplant 2014;29:1633–8. [DOI] [PubMed] [Google Scholar]
- 21. Brandenburg VM, Reinartz S, Kaesler Net al. Slower progress of aortic valve calcification with vitamin K supplementation: results from a prospective interventional proof-of-concept study. Circulation 2017;135:2081–3. [DOI] [PubMed] [Google Scholar]
- 22. McCollough CH, Ulzheimer S, Halliburton SSet al. Coronary artery calcium: a multi-institutional, multimanufacturer international standard for quantification at cardiac CT. Radiology 2007;243:527–38. [DOI] [PubMed] [Google Scholar]
- 23. Rumberger JA, Kaufman L.. A Rosetta stone for coronary calcium risk stratification: Agatston, volume, and mass scores in 11,490 individuals. Am J Roentgenol 2003;181:743–8. [DOI] [PubMed] [Google Scholar]
- 24. Sato T, Schurgers LJ, Uenishi K.. Comparison of menaquinone-4 and menaquinone-7 bioavailability in healthy women. Nutr J 2012;11:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use . Statistical principles for clinical trials. https://www.ich.org/page/ich-guidelines (26 August 2022, date last accessed). [Google Scholar]
- 26. Fusaro M, Cozzolino M, Plebani Met al. Sevelamer use, vitamin K levels, vascular calcifications, and vertebral fractures in hemodialysis patients: results from the VIKI study. J Bone Miner Res 2021;36:500–9. [DOI] [PubMed] [Google Scholar]
- 27. Schurgers LJ, Vermeer C.. Determination of phylloquinone and menaquinones in food. Effect of food matrix on circulating vitamin K concentrations. Haemostasis 2000;30:298–307. [DOI] [PubMed] [Google Scholar]
- 28. Shea MK, O'Donnell CJ, Hoffmann Uet al. Vitamin K supplementation and progression of coronary artery calcium in older men and women. Am J Clin Nutr 2009;89:1799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shea MK, O'Donnell CJ, Vermeer Cet al. Circulating uncarboxylated matrix Gla protein is associated with vitamin K nutritional status, but not coronary artery calcium, in older adults. J Nutr 2011;141:1529–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fulton RL, McMurdo ME, Hill Aet al. Effect of vitamin K on vascular health and physical function in older people with vascular disease—a randomised controlled trial. J Nutr Health Aging 2016;20:325–33. [DOI] [PubMed] [Google Scholar]
- 31. Neradova A, Wasilewski G, Prisco Set al. Combining phosphate binder therapy with vitamin K2 inhibits vascular calcification in an experimental animal model of kidney failure. Nephrol Dial Transplant 2022;37:652–62. [DOI] [PubMed] [Google Scholar]
- 32. Dai L, Meijers BK, Bammens Bet al. Sevelamer use in end-stage kidney disease (ESKD) patients associates with poor vitamin K status and high levels of gut-derived uremic toxins: a drug-bug interaction? Toxins (Basel) 2020;12:351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mazzaferro S, Pasquali M, Taggi Fet al. Progression of coronary artery calcification in renal transplantation and the role of secondary hyperparathyroidism and inflammation. Clin J Am Soc Nephrol 2009;4:685–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Raggi P, Chertow GM, Torres PUet al. The ADVANCE study: a randomized study to evaluate the effects of cinacalcet plus low-dose vitamin D on vascular calcification in patients on hemodialysis. Nephrol Dial Transplant 2011;26:1327–39. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Public posting of individual-level participant data is not covered by the informed patient consent form.