

Abstract
Objectives
AICAR, an adenosine analog, has been shown to exhibit vascular protective effects through activation of AMP-activated protein kinase (AMPK). However, it remains unclear as to whether adenosine kinase-mediated ZMP formation or adenosine receptor activation contributes to AICAR-mediated AMPK activation and/or vasorelaxant response in vascular smooth muscle.
Methods and Results
In the present study using endothelium-denuded rat aortic ring preparations, isometric tension measurements revealed that exposure to 1 mM AICAR for 30 min resulted in inhibition of phenylephrine (1 μM)-induced smooth muscle contractility by ∼35%. Importantly, this vasorelaxant response by AICAR was prevented after pretreatment of aortic rings with an AMPK inhibitor (compound C, 40 µM) and adenosine kinase inhibitor (5-iodotubercidin, 1 µM), but not with an adenosine receptor blocker (8-sulfophenyltheophylline, 100 µM). Immunoblot analysis of respective aortic tissues showed that AMPK activation seen during vasorelaxant response by AICAR was abolished by compound C and 5-iodotubercidin, but not by 8-sulfophenyltheophylline, suggesting ZMP involvement in AMPK activation. Furthermore, LC–MS/MS MRM analysis revealed that exposure of aortic smooth muscle cells to 1 mM AICAR for 30 min enhanced ZMP level to 2014.9 ± 179.4 picomoles/mg protein (vs. control value of 8.5 ± 0.6; p<0.01), which was accompanied by a significant decrease in ATP/ADP ratio (1.08 ± 0.02 vs. 2.08 ± 0.06; p<0.01).
Conclusions
Together, the present findings demonstrate that AICAR-mediated ZMP elevation and the resultant AMPK activation in vascular smooth muscle contribute to vasorelaxation.
Keywords: AICAR, AMPK, vascular smooth muscle, vasorelaxation, ZMP
Introduction
5-aminoimidazole-4-carboxamide-1-β-D-ribofuranosyl-5′-monophosphate (ZMP) is a key metabolic intermediate in the de novo pathway of purine synthesis in vascular smooth muscle and striated muscles such as cardiac muscle and skeletal muscle [1], [2], [3]. In addition to its role as a purine precursor [4], ZMP can function as an adenosine 5′-monophosphate (AMP) mimetic to activate AMP-activated protein kinase (AMPK), a cellular energy sensor [5, 6]. Studies by several investigators including our previous study have shown that 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR), an adenosine analog and a pro-drug for ZMP, activates AMPK in the vessel wall to promote vascular protective effects [7], [8], [9]. However, the extent of alterations in ZMP and cellular energy state remain unclear especially during AICAR-mediated AMPK activation and vasorelaxation.
Several lines of evidence suggest that AICAR has blood pressure-lowering effects in different animal models. For instance, in insulin-resistant obese Zucker rats that exhibit hypertension, long-term administration of AICAR for 6 weeks results in a marked lowering of systolic blood pressure [10]. In a rat model of preeclampsia with placental ischemia-induced hypertension, AICAR administration for 2–3 weeks mitigates the increase in mean arterial pressure [11]. In apoE knockout mice on a high fat diet for 6–12 weeks, AICAR administration for 2 weeks or 45 min leads to significant decreases in mean arterial pressure [12]. Notably, in spontaneously hypertensive rats, AICAR administration for 30 min leads to a profound decrease in mean arterial pressure [8]. Together, the blood pressure-lowering effects of AICAR can be attributed to its ability to promote vasodilation via AMPK activation in vascular endothelium [13], [14], [15] and vascular smooth muscle [7, 9, 15]. In normotensive rats, acute infusion of AICAR for 60 min enhances microvascular perfusion in skeletal muscle through AMPK activation and the associated endothelial nitric oxide-mediated vasodilation [14]. In addition, we and several other investigators have shown that in endothelium-denuded rodent aorta, acute exposure to AICAR activates AMPK and promotes vasorelaxant response [7, 9, 15]. Although it is known that AICAR is an adenosine analog, it remains unclear as to whether activation of adenosine kinase and/or adenosine receptor is involved in AICAR-mediated AMPK activation and vasorelaxation. In addition, the likely alterations in ZMP and ATP/ADP ratio in AICAR-treated vascular smooth muscle cells are not fully understood.
The present study is therefore aimed at examining the effects of AICAR on phenylephrine-induced smooth muscle contraction and AMPK activation using endothelium-denuded rat aorta after exposure to: (i) compound C, an AMPK inhibitor; (ii) 5-iodotubercidin, an adenosine kinase inhibitor; or (iii) 8-sulfophenyltheophylline (8-SPT), an adenosine receptor blocker. In addition, we examined the effects of AICAR on the changes in ATP, ADP, AMP, and ZMP in aortic smooth muscle cells.
Materials and methods
Materials
Phenylephrine hydrochloride, acetylcholine chloride, 5-iodotubercidin, and 8-SPT [8-(p-sulfophenyl) theophylline hydrate] were purchased from Sigma-Aldrich (St. Louis, MO). AICAR (5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside) and compound C (6-[4-(2-piperidin-1-yl-ethoxy)-phenyl]-3-pyridin-4-yl-pyrrazolo[1,5-a]-pyrimidine or dorsomorphin dihydrochloride) were purchased from Tocris Bioscience (Minneapolis, MN). Adenosine 5′-triphosphate disodium (ATP), adenosine 5′-diphosphate monosodium (ADP), adenosine 5′-monophosphate disodium (AMP), and 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranosyl-5′-monophosphate (ZMP or AICAR 5′-monophosphate) were purchased from EMD Millipore Chemicals (Billerica, MA). The primary antibodies for AMPKα (2,532) and phospho-AMPKα(Thr172) (2,535) were purchased from Cell Signaling Technology (Danvers, MA). The primary antibody for β-actin was purchased from Abcam (Cambridge, MA). HRP-conjugated goat anti-rabbit secondary antibody was purchased from Bio-Rad (Hercules, CA). All other chemicals were from Fisher Scientific (Fair Lawn, NJ) or Sigma-Aldrich (St. Louis, MO).
Animals
All animal experiments were performed in accordance with the Charlie Norwood Veterans Affairs Medical Center Institutional Animal Care and Use Committee guidelines and were approved by the committee. Adult male Wistar rats (280 to 350 g) were purchased from Charles River Laboratories, Inc. (Wilmington, MA). The rats were maintained in a room at a controlled temperature of 23 °C with a 12:12 hr dark-light cycle. The rats had free access to water and standard rodent chow diet.
Preparation of aortic rings and isometric tension measurements
After sacrificing the rats, thoracic aorta was isolated and immediately placed in a petri-dish containing ice-cold oxygenated Krebs–Henseleit bicarbonate (KHB) buffer (118 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4, 1.2 mM KH2PO4, 2.5 mM CaCl2, 25 mM NaHCO3, and 11 mM glucose; pH 7.4), as described [9]. The aorta was carefully cleaned free of adherent fat and connective tissue. Endothelium was removed by gently rubbing the luminal surface using a polyethylene tube. Endothelium-denuded aorta was then cut into 2 mm rings. Subsequently, aortic rings were mounted in the organ bath system (8 chambers, Radnoti Glass Technology, Monrovia, CA). After passing the stainless-steel wires through the lumen, aortic rings were suspended from the isometric force displacement transducers (Model FT03; Grass Technologies, West Warwick, RI) and kept immersed in 10 mL of KHB buffer in the organ bath system. The KHB buffer was bubbled continuously with a gas mixture of 95% O2 and 5% CO2 and maintained at 37 °C. Isometric tension was measured as changes in millinewtons (mN) of force using Octal Bridge Amplifier and PowerLab 8/35 data-acquisition system and recorded using LabChart Pro V7 software (ADInstruments, Colorado Springs, CO). Each aortic ring was gradually stretched to a basal tension of 19.6 mN (2 g) over a period of ∼1 h and then equilibrated for additional 1.5 to 2 h. During this equilibration period, KHB buffer was changed every 30 min. After equilibration, the maximal contractile response to 80 mM KCl was determined followed by washes with KHB buffer until the passive basal tension is restored. To verify endothelial denudation, aortic rings were pre-contracted with phenylephrine (an α-adrenergic agonist) followed by addition of acetylcholine (a muscarinic agonist). The absence of relaxant response to acetylcholine confirmed the denudation of aortic rings. Subsequently, aortic rings were washed with KHB buffer until the passive basal tension is restored. As described in the respective figure legends, aortic rings were then exposed to vehicle (control) or treated with AMPK inhibitor, adenosine kinase inhibitor, adenosine receptor blocker, and/or AICAR followed by stimulation with phenylephrine to determine the changes in contractile response.
Extraction and quantification of proteins in aortic tissues
After contractility studies, aortic rings were immediately rinsed in ice-cold fresh phosphate-buffered saline, blotted to dryness, snap-frozen in liquid nitrogen, and stored at −80 °C until analysis. Aortic ring tissues were then thawed and homogenized in 100 µl RIPA lysis buffer containing protease and phosphatase inhibitors (Thermo Scientific, Rockford, IL) using TissueLyser LT (Qiagen, Valencia, CA) at a setting of 50 Hz for 5 min with samples being placed on ice intermittently. The homogenates were incubated at 4 °C for 1 h on a rotator and centrifuged at 1,000×g for 10 min at 4 °C to remove tissue debris. The supernatants were mixed with 2× laemmli sample buffer at a ratio of 1:1 followed by heating at 67.5 °C for 10 min. Proteins were quantified using Bio-Rad DC assay kit (Bio-Rad, Hercules, CA), as described [9].
Immunoblot analysis
Aortic tissue samples (20 µg protein each) were then electrophoresed using pre-cast 4–12% NuPage mini-gels (Life Technologies, Carlsbad, CA), and the resolved proteins were transferred to nitrocellulose membranes (Hybond C, GE Healthcare Life Sciences, Piscataway, NJ). The membranes were blocked in 5% bovine serum albumin, and probed with the primary antibodies specific for phospho-AMPKα (Thr172), AMPKα, or β-actin. After extensive washes, the immunoreactivity was detected using specific HRP-conjugated secondary antibodies followed by enhanced chemiluminescence (GE Healthcare Life Sciences). The protein bands were quantified by densitometric analysis using Image J [9].
Quantification of nucleotides (ATP, ADP, AMP, and ZMP) in aortic smooth muscle cells using LC-MS/MS MRM
Human aortic smooth muscle cells, vascular cell basal medium (Cat # PCS-100-030), and vascular smooth muscle cell growth kit (Cat # PCS-100-042) were purchased from ATCC (Manassas, VA). Aortic smooth muscle cells (passages 3 to 6) were maintained in culture with vascular cell basal medium containing vascular smooth muscle cell growth kit and antibiotic/antimycotic solution in a humidified atmosphere of 95% air and 5% CO2 at 37 °C. After the attainment of confluence (∼6–7 days), smooth muscle cells were trypsinized, centrifuged, and 3 million cells were seeded on to 150 mm petri dishes each, as described previously [16]. Subconfluent smooth muscle cells were deprived of growth supplements for 2 days followed by incubation with Krebs–Henseleit bicarbonate (KHB) buffer for 90 min. Subsequently, smooth muscle cells were exposed to vehicle control or 1 mM AICAR for 30 min. Smooth muscle cells were then washed twice with ice-cold PBS and collected in 1 mL ice-cold PBS. The cell suspension was centrifuged at 1,000×g for 5 min at 4 °C to pellet the cells. After discarding the supernatant, the cell pellet was snap-frozen using liquid N2 and stored at −80 °C until analysis. On the day of nucleotide extraction, 200 µl of 5% ice-cold perchloric acid was added to the cell pellet to lyse the cells and sample was further centrifuged at 10,000×g for 3 min at 4 °C to remove the acid-insoluble material. Perchloric acid in the collected supernatant was extracted by three washes with 10% excess volume of a 1:1 mixture of tri-n-octylamine and 1,1,2-trichlorotrifluroethane. The nucleotides remaining in the aqueous phase were then analyzed using liquid chromatography/tandem mass spectrometry (LC-MS/MS) with multiple reaction monitoring (MRM) on a 4000 QTRAP LC/MS/MS system (Applied Biosystems, Carlsbad, CA) [9]. The concentrations of nucleotides (ATP, ADP, AMP, and ZMP) in the samples were calculated using the standard curve prepared from serial dilutions of the respective nucleotide stock solution (1 mM). In brief, the samples and standards were run on an Amide XBridge HPLC column (Cat # 186,004,860; 3.5 µM particle size; 2.1 mm inner diameter×100 mm length, Waters, Milford, MA) using buffer A (20 mM ammonium hydroxide and 20 mM ammonium acetate in 5% acetonitrile, pH 9.0) and buffer B (100% acetonitrile) at a flow rate of 0.3 mL/min for 10 min. The mobile phase consisted of isocratic elusion with 20% buffer B. The detailed specifications of MRM transitions for nucleotides are described in Table 1.
Table 1:
MRM transitions for nucleotides (ATP, ADP, AMP, and ZMP).
| Compound | Q1 (Da) | Q3 (Da) | Dwell Time (ms) | CE (V) | DP (V) | EP (V) | CXP (V) |
|---|---|---|---|---|---|---|---|
| ATP | 506 | 159 | 50 | −50 | −93 | −10 | −5 |
| 506 | 79 | 50 | −40 | −93 | −10 | −5 | |
| ADP | 426 | 134 | 50 | −35 | −93 | −10 | −5 |
| 426 | 79 | 50 | −65 | −93 | −10 | −5 | |
| AMP | 346 | 97 | 50 | −36 | −93 | −10 | −5 |
| 346 | 79 | 50 | −60 | −93 | −10 | −5 | |
| ZMP | 337 | 97 | 50 | −30 | −93 | −10 | −5 |
| 337 | 79 | 50 | −55 | −93 | −10 | −5 |
CE, collision energy; DP, declustering potential; EP, entrance potential; CXP, collision cell exit potential.
Statistical analysis
Results are expressed as means ± SEM values. The n value represents the number of animals. To determine E max and EC50 values (−log EC50 or pEC50), nonlinear regression analysis was performed using GraphPad Prism software (version 6.01, GraphPad Software, Inc., La Jolla, CA). Statistical analyses of the data among groups were performed by paired t-test (parametric) or one-way repeated measures ANOVA followed by Bonferroni t-test. Values of p<0.05 were considered statistically significant.
Results
Denudation of endothelium prevents acetylcholine-induced relaxation in aortic rings
Prior to determining the effects of AICAR on vascular smooth muscle contraction, initial studies included the use of endothelium-intact vs. endothelium-denuded aorta to confirm denudation of endothelium. As shown in Figure 1, in endothelium-intact aortic rings pre-contracted with phenylephrine (PE; 1 µM), exposure to increasing concentrations of acetylcholine (10−10 to 10−4 M) led to a progressive increase in vasorelaxant response. The E max and pEC50 values for acetylcholine-induced vasorelaxation were 69.8 ± 4.2% and 6.9 ± 0.06 M, respectively. In parallel, in endothelium-denuded aortic rings pre-contracted with PE (1 µM), exposure to increasing concentrations of acetylcholine (10−10 to 10−4 M) did not show any vasorelaxant response.
Figure 1:

Effects of acetylcholine on rat aortic rings precontracted with phenylephrine. Endothelium-intact aortic rings and endothelium-denuded aortic rings were precontracted with 1 µM phenylephrine for 10 min, followed by exposure to cumulative concentrations of acetylcholine (ACh, 10−10 to 10−4 M). The percent changes in ACh-induced relaxant response were then determined as described in ‘Materials and methods’. The data shown are the means ± SEM values obtained with aortic rings from 4 rats. ##p<0.01, compared with endothelium-intact aortic rings (∆).
AICAR-induced vasorelaxation in aortic smooth muscle is prevented by inhibitors of AMPK and adenosine kinase, but not by adenosine receptor blocker
We initially examined the effects of compound C, an AMPK inhibitor, on AICAR-mediated vasorelaxation. To this end, we pretreated the aortic rings with 40 µM compound C for 30 min prior to AICAR exposure, as described. As shown in Figure 2A, in the control group, stimulation with cumulative concentrations of phenylephrine (PE; 10−10 to 10−4 M) led to a progressive increase in contractile response. The E max and pEC50 values for phenylephrine-induced contractility were 7.09 ± 0.12 mN/mm and 8.11 ± 0.05 M, respectively. In AICAR-treated aortic rings, the E max and pEC50 values for PE-induced contractility decreased to 5.28 ± 0.21 mN/mm (p<0.05) and 7.28 ± 0.06 M (p<0.001), respectively, compared with control. Pretreatment with compound C (40 µM) alone did not significantly affect PE-induced contractility. However, after pretreatment with compound C followed by AICAR treatment, the E max and pEC50 values for PE-induced contractility were 6.49 ± 0.17 mN/mm (p<0.05) and 7.26 ± 0.04 M (ns), respectively, compared with AICAR treatment. Thus, compound C prevented the inhibitory effects of AICAR on PE-induced contractility.
Figure 2:

Effects of AMPK inhibitor, adenosine kinase inhibitor, and adenosine receptor blocker on AICAR-induced vasorelaxation in aortic smooth muscle. Endothelium-denuded aortic rings were exposed to vehicle control or pretreated with 40 µM compound C (AMPK inhibitor) or 1 µM 5-iodotubercidin (Iodo; adenosine kinase inhibitor) or 100 µM 8-SPT (adenosine receptor blocker) for 30 min. Subsequently, a parallel set of these control and pretreated aortic rings were subjected to treatments with 1 mM AICAR for 30 min. The aortic rings were then challenged with cumulative concentrations of phenylephrine (PE; 10−10 to 10−4 M) to determine the changes in contractile response. The E max and pEC50 values for PE-induced contractility were then calculated as described under ‘Materials and methods’. The data shown are the means ± SEM values obtained with aortic rings from 3 to 4 rats. #p<0.05; compared with control (+PE); $p<0.05; compared with AICAR treatment (+PE).
Subsequently, we examined whether AICAR promotes vasorelaxation via adenosine kinase activation in vascular smooth muscle. To this end, we used 5-iodotubercidin, an inhibitor of adenosine kinase. The aortic rings were pretreated with 1 µM 5-iodotubercidin for 30 min prior to AICAR exposure, as described. As shown in Figure 2B, in control aortic rings, stimulation with cumulative concentrations of phenylephrine (PE; 10−10 to 10−4 M) led to a progressive increase in contractile response. The E max and pEC50 values for phenylephrine-induced contractility were 7.09 ± 0.13 mN/mm and 8.09 ± 0.05 M, respectively. In AICAR-treated aortic rings, the E max and pEC50 values for PE-induced contractility decreased to 5.57 ± 0.10 mN/mm (p<0.05) and 7.16 ± 0.11 M (p<0.001), respectively, compared with control. In aortic rings pretreated with 5-iodotubercidin (1 µM) alone, there were no significant changes in PE-induced contractility. However, pretreatment with 5-iodotubercidin followed by AICAR exposure restored the E max and pEC50 values for PE-induced contractility to 7.17 ± 0.16 mN/mm (p<0.05) and 8.09 ± 0.06 M (p<0.001), respectively, compared with AICAR treatment alone.
To determine whether adenosine receptor activation is involved in AICAR-mediated effects in vascular smooth muscle, we used 8-sulfophenyltheophylline (8-SPT), an adenosine receptor blocker [17, 18]. As shown in Figure 2C, pretreatment of aortic rings with 100 µM 8-SPT for 30 min prior to vehicle or AICAR exposure did not affect phenylephrine-induced contractility or AICAR inhibition of phenylephrine-induced contractility.
AICAR-induced vasorelaxation in aortic smooth muscle is mediated by AMPK activation
To determine whether AICAR-induced vasorelaxation is dependent on AMPK activation, we pretreated the aortic rings with vehicle control or with 40 µM compound C for 30 min prior to AICAR exposure, as described. Subsequently, the aortic rings were challenged with a fixed concentration of phenylephrine (PE; 1 µM) for 6 min.
As shown in Figure 3A (upper panel), AICAR treatment resulted in a significant decrease in PE-induced contractility by 32.5% (p<0.05), compared with control (+PE). Compound C alone did not induce significant changes in PE-induced contractility. However, compound C pretreatment significantly prevented AICAR inhibition of PE-induced contractility (p<0.05), compared with AICAR treatment (+PE). Neither AICAR nor compound C produced significant changes in passive basal tension, compared with control (lower panel).
Figure 3:

Effects of compound C on AICAR-induced vasorelaxation and AMPK activation in aortic smooth muscle.
(A) Endothelium-denuded aortic rings were exposed to vehicle control or pretreated with 40 µM compound C (AMPK inhibitor) for 30 min. Subsequently, a parallel set of these control and pretreated aortic rings were subjected to treatments with 1 mM AICAR for 30 min. The aortic rings were then challenged with a fixed concentration of phenylephrine (PE; 1 µM) for 30 min to determine the changes in contractile response. (B) After contractility studies, the respective aortic rings were subjected to immunoblot analysis using primary antibodies specific for phospho-AMPKα (Thr172) and total AMPKα. β-actin was used as an internal control. The extent of changes in AMPK phosphorylation is illustrated by the representative immunoblots (upper panel) and pAMPK/AMPK ratio (lower panel). The data shown in the bar graphs are the means ± SEM values obtained with aortic rings from 3 rats. #p<0.05; ##p<0.01, ###p<0.001 compared with control (+PE); *p<0.05; **p<0.01; ***p<0.001 compared with control (−PE); $p<0.05; $$p<0.01 compared with AICAR treatment (+PE); ¢¢ p<0.01 compared with AICAR treatment (−PE).
To determine the extent of changes in AMPK phosphorylation in vascular smooth muscle under the above-mentioned treatment conditions, the respective aortic rings were snap-frozen in liquid nitrogen immediately after contractility studies. Using these aortic tissues, we performed immunoblot analysis with the primary antibodies specific for phospho-AMPKα (Thr172) and total AMPKα. As shown in Figure 3A and B, AICAR-induced vasorelaxation was accompanied by enhanced AMPK phosphorylation. In particular, AMPK phosphorylation by AICAR was further increased upon stimulation with PE (lanes 3 and 4). Importantly, the ability of compound C to prevent AICAR-induced vasorelaxation was reflected by its inhibitory effect on AMPK phosphorylation (lanes 7 and 8). Together, these findings demonstrate that AMPK activation contributes to AICAR-induced vasorelaxation in vascular smooth muscle.
AICAR-induced vasorelaxation and AMPK activation in aortic smooth muscle are mediated by activation of adenosine kinase, but not by adenosine receptor
To determine whether AICAR-induced vasorelaxation involves the intermediary activation of AMPK via adenosine kinase or adenosine receptor, we pretreated the aortic rings with vehicle control, 1 µM 5-iodotubercidin (adenosine kinase inhibitor) or 100 µM 8-SPT (adenosine receptor blocker) for 30 min prior to AICAR exposure, as described. Subsequently, the aortic rings were challenged with a fixed concentration of phenylephrine (PE; 1 µM) for 6 min.
As shown in Figure 4A (upper panel), in control aortic rings, AICAR treatment led to a significant diminution in PE-induced contractility by 38.1% (p<0.05), compared with vehicle (+PE). Importantly, 5-iodotubercidin pretreatment, but not 8-SPT, prevented AICAR inhibition of PE-induced smooth muscle contractility (p<0.05), compared with AICAR treatment (+PE). Pretreatment with 5-iodotubercidin or 8-SPT alone did not induce significant changes in PE-induced contractility, compared with control (+PE). In addition, 5-iodotubercidin, or 8-SPT did not produce significant changes in the passive basal tension, compared with control (lower panel).
Figure 4:

Effects of 5-iodotubercidin vs. 8-SPT on AICAR-induced vasorelaxation and AMPK activation in aortic smooth muscle.
(A) Endothelium-denuded aortic rings were exposed to vehicle control or pretreated with 1 µM 5-iodotubercidin or 100 µM 8-SPT for 30 min. Subsequently, a parallel set of these control and pretreated aortic rings were subjected to treatments with 1 mM AICAR for 30 min. The aortic rings were then challenged with a fixed concentration of phenylephrine (PE; 1 µM) for 30 min to determine the changes in contractile response. (B) After contractility studies, the respective aortic rings were subjected to immunoblot analysis using primary antibodies specific for phospho-AMPKα (Thr172) and total AMPKα. β-actin was used as an internal control. The data shown in the bar graphs are the means ± SEM values obtained with aortic rings from 3 rats. #p<0.05; ###p<0.001 compared with control (+PE); $p<0.05; $$$p<0.001 compared with AICAR treatment.
To determine the extent of changes in AMPK phosphorylation in vascular smooth muscle under the above-mentioned treatment conditions, the respective aortic rings were snap-frozen in liquid nitrogen immediately after contractility studies. The aortic tissues were then subjected to immunoblot analysis using primary antibodies specific for phospho-AMPKα (Thr172) and total AMPKα. As shown in Figure 4A and B, AICAR-induced vasorelaxation was accompanied by enhanced AMPK phosphorylation (lane 2 vs. lane 1). Importantly, the ability of 5-iodotubercidin to prevent AICAR-induced vasorelaxation was reflected by its inhibitory effect on AMPK phosphorylation (lanes 7 and 8). In contrast, 8-SPT did not affect AICAR-induced vasorelaxation or AMPK phosphorylation. Together, these findings suggest that adenosine kinase-mediated ZMP formation but not adenosine receptor activation is involved in AICAR-induced AMPK activation and the resultant vasorelaxation.
AICAR enhances ZMP level and decreases ATP/ADP ratio in aortic smooth muscle cells
To further determine the extent of changes in ZMP (an AMP mimetic) upon AICAR exposure, aortic smooth muscle cells were treated with 1 mM AICAR for 30 min, as described in ‘Materials and methods’. The lysates were then subjected to LC-MS/MS MRM analysis to quantify ZMP level. Using the same lysates, we also quantified the levels of AMP, ADP, and ATP to determine alterations in cellular energy state under these conditions. Figure 5A shows that under control conditions, ATP, ADP, AMP, and ZMP levels were 2,751.6 ± 126.7, 1,335.6 ± 55.2, 264.7 ± 20.7, and 8.5 ± 0.6 picomoles/mg protein, respectively. Importantly, AICAR treatment resulted in significant changes in the nucleotide levels. The respective changes in ATP, ADP, AMP, and ZMP levels were 1,230.3 ± 81.3 (p<0.01), 1,141.4 ± 96.7 (n.s.), 166.1 ± 16.4 (p<0.01), and 2014.9 ± 179.4 (p<0.01) picomoles/mg protein, compared with control treatment. Thus, AICAR treatment led to a robust increase in ZMP level with an associated decrease in ATP level. As shown in Figure 5B, data analysis of nucleotides revealed a significant decrease in ATP/ADP ratio to 1.08 ± 0.02 (p<0.01) upon AICAR treatment, compared with ATP/ADP ratio of 2.08 ± 0.06 for control. Table 1 shows the parameters used for the quantification of all nucleotides using LC-MS/MS MRM analysis.
Figure 5:
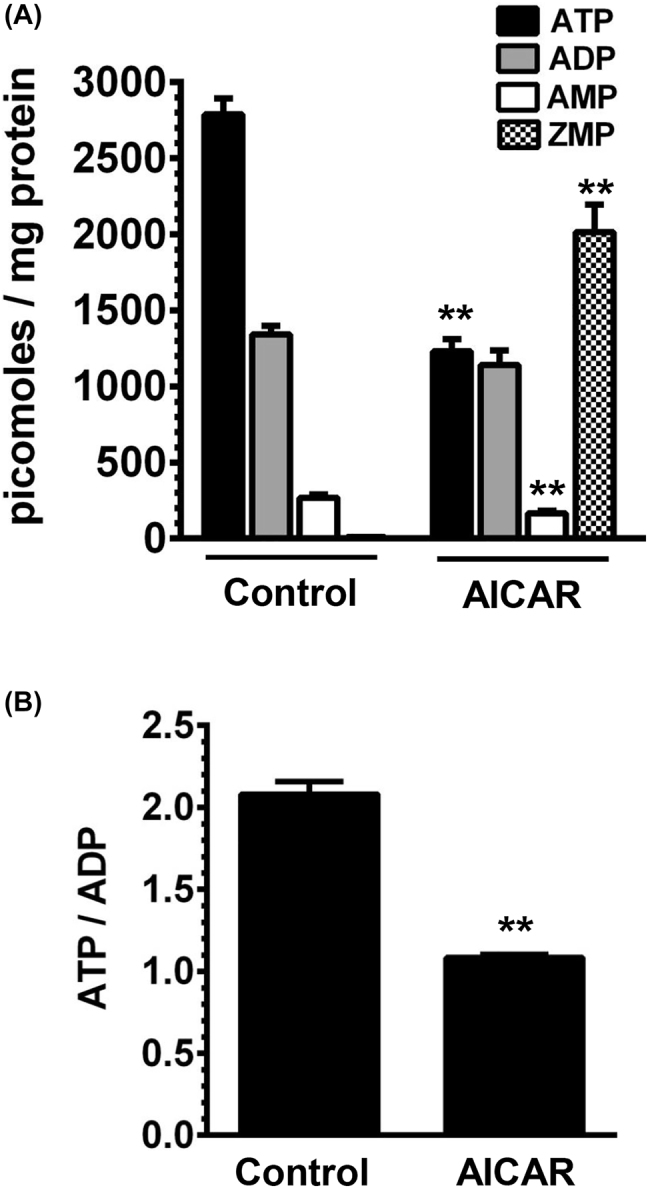
Effects of AICAR on ZMP and adenosine nucleotides (AMP, ADP, and ATP) in aortic smooth muscle cells. Aortic smooth muscle cells were exposed to vehicle control or 1 mM AICAR for 30 min. The lysates were then subjected to LC-MS/MS MRM analysis for the quantification of ATP, ADP, AMP, and ZMP levels, as described in ‘Materials and methods’. The respective nucleotide levels (A) and ATP/ATDP ratio (B) shown in the bar graphs are the means ± SEM values. **p<0.01 compared with control; n = 3.
Discussion
AICAR, an AMPK activator, has previously been shown to exhibit blood pressure-lowering effects in different animal models of hypertension [8, 10], [11], [12]. The present study shows that AICAR activates adenosine kinase in vascular smooth muscle to enhance the formation of ZMP, which in turn activates AMPK to promote vasorelaxation. Together, these findings suggest that AICAR-mediated ZMP elevation and the resultant AMPK activation in vascular smooth muscle may in part contribute to its vasodilatory and antihypertensive effects.
Several lines of evidence suggest that AICAR exhibits cardioprotective effects through adenosine release and subsequent adenosine receptor activation [17, 18]. Although adenosine receptor is expressed in vascular smooth muscle [19], it is not involved in AICAR-mediated vasorelaxant response in the present study using endothelium-denuded rat aorta. This is supported by our findings that showed a lack of inhibitory effect of 8-SPT, an adenosine receptor blocker, on AICAR-mediated AMPK activation and vasorelaxant response in aortic smooth muscle. Our results are consistent with a previous study using mouse aortic smooth muscle, where AICAR-mediated vasorelaxation occurs independent of adenosine receptors [7]. Together, these findings suggest that, although adenosine-mediated adenosine receptor activation in ventricular myocytes may have cardioprotective effects [20], adenosine receptor activation in vascular smooth muscle cells does not play an intermediary role in AICAR-mediated vasorelaxation regardless of species.
Because AICAR is a nucleoside analog, its uptake via adenosine transporters can result in its phosphorylation to ZMP via adenosine kinase, as evidenced in different cell types [21, 22]. Importantly, previous studies have demonstrated the expression of high-affinity adenosine transporter in porcine coronary vascular smooth muscle [23]. This suggests that AICAR uptake via adenosine transporter in vascular smooth muscle is a necessary step for its vasorelaxant effect. It would in turn allow AICAR to be acted upon by intracellular adenosine kinase to form ZMP, an AMP mimetic. Accordingly, AICAR-mediated AMPK activation and vasorelaxant response in aortic smooth muscle were completely abolished after pretreatment with 5-iodotubercidin, an inhibitor of adenosine kinase. The present findings indicate that adenosine kinase-mediated formation of ZMP is an obligatory intermediary event in the pharmacological activation of AMPK by AICAR and the resultant vasorelaxant response in aortic smooth muscle.
Furthermore, the present study demonstrates a marked elevation of ZMP concentration in AICAR-treated aortic smooth muscle cells. This increase in ZMP is accompanied by significant decreases in ATP concentration and ATP/ADP ratio, suggesting ATP utilization during adenosine kinase-mediated phosphorylation of AICAR to ZMP. Because ZMP is an AMP mimetic, it can regulate the activity of heterotrimeric AMPK that consists of a catalytic α subunit and β and γ regulatory subunits. In particular, ZMP has been shown to interact directly with the regulatory γ subunit of AMPK to induce a conformational change to facilitate phosphorylation of Thr172 residue of the catalytic α subunit of AMPK [6, 24]. Thus, AICAR-mediated ZMP elevation contributes to AMPK-mediated vasorelaxation as evidenced in the present study using aortic smooth muscle.
Previous studies have shown that compound C, a cell-permeable pyrrazolopyrimidine derivative, is an ATP-competitive inhibitor of AMPK [25]. This is achieved by the ability of compound C to bind to the highly conserved active site of AMPK [26]. To further support the intermediary role of AMPK in its vasorelaxant response, aortic smooth muscle was therefore pretreated with compound C prior to AICAR exposure. The present findings reveal that pharmacological inhibition of AMPKα phosphorylation/activation by compound C prevents the vasorelaxant response to AICAR, thus supporting an essential link between ZMP-mediated AMPKα activation and vasorelaxation. Using AMPKα isoform-specific knockout mouse models, a previous study has shown that AICAR promotes vasorelaxation of precontracted aortic rings in AMPKα2-deficient condition but not with AMPKα1-deficiency, suggesting the critical intermediary role of AMPKα1 toward vasorelaxation in endothelium-denuded aortic smooth muscle [7].
Although AICAR enhances ZMP to promote AMPK-mediated vasorelaxation and lower blood pressure as evidenced in ex vivo studies (present observations) and in vivo studies [8, 10], [11], [12], respectively, the clinical utility of AICAR as a therapeutic agent is largely limited by its pharmacokinetic properties. For instance, AICAR has poor bioavailability when administered orally, has short half-life when administered intravenously, and thereby can elevate the circulating concentrations of lactic acid and uric acid in human subjects [27], [28], [29]. It is noteworthy that ZMP elevation, as observed in the present study with AICAR-treated vascular smooth muscle cells, can also occur upon exposure to chemotherapeutic drugs such as pemetrexed and methotrexate [30]. As noted in the Introduction, ZMP is a key metabolic intermediate in the de novo pathway of purine synthesis [4, 31]. In this pathway, endogenously formed ZMP undergoes intermediary conversion to form inosine monophosphate (IMP) via activation of two successive enzymes that include AICAR-Transformylase and IMP-Cyclohydrolase (together called ATIC). Importantly, ATIC is subject to inhibition by pemetrexed and methotrexate, thereby accounting for endogenous ZMP elevation by these chemotherapeutic drugs [30, 32, 33]. Thus, methotrexate has the potential to promote ZMP-mediated AMPK as evidenced in different tissues including skeletal muscle [33]. Intriguingly, in patients with rheumatoid arthritis, treatment with methotrexate has been shown to lower blood pressure [34]. It is likely that methotrexate-mediated ZMP accumulation and the resultant AMPK activation in the vessel wall contribute to the lowering of blood pressure. Recently, methotrexate has been shown to improve perivascular adipose tissue/endothelial dysfunction by activating the signaling pathway involving activation of AMPK and endothelial nitric oxide synthase [35]. Future studies should therefore utilize animal models of hypertension to determine the extent to which methotrexate treatment results in ZMP accumulation and AMPK activation in arterial smooth muscle vs. endothelium.
In conclusion, administration of exogenous AICAR has the potential to elevate ZMP concentration in arterial smooth muscle. This is achieved by adenosine kinase-mediated conversion of AICAR to ZMP. In addition, ZMP-mediated AMPK activation can result in vasorelaxation in an endothelium-independent manner. As noted above, strategies to enhance endogenous ZMP concentration in the vessel wall would promote AMPK-mediated vasodilation to lower blood pressure, and thereby may provide new therapeutic options for the management of hypertension.
Acknowledgments
This work was supported by the National Heart, Lung, and Blood Institute/National Institutes of Health Grant (R01-HL-097090). This material is the result of work supported with resources and the use of facilities at the Charlie Norwood Veterans Affairs Medical Center, Augusta, Georgia, USA. The contents of this article do not represent the views of the U.S. Department of Veterans Affairs or the United States Government.
Footnotes
Research funding: This work was supported by the National Heart, Lung, and Blood Institute/National Institutes of Health Grant (R01-HL-097090).
Author contribution: All authors have accepted responsibility for the entire content of this manuscript and approved its submission.
Competing interests: Authors state no conflict of interest.
Informed consent: Not applicable.
Ethical approval: All animal experiments were performed in accordance with the Charlie Norwood Veterans Affairs Medical Center Institutional Animal Care and Use Committee guidelines and were approved by the committee.
References
- 1.Sabina RL, Kernstine KH, Boyd RL, Holmes EW, Swain JL. Metabolism of 5-amino-4-imidazolecarboxamide riboside in cardiac and skeletal muscle. Effects on purine nucleotide synthesis. J Biol Chem. 1982;257:10178–83. doi: 10.1016/s0021-9258(18)34001-8. [DOI] [PubMed] [Google Scholar]
- 2.Barany M, Barron JT, Gu L, Barany K. Exchange of the actin-bound nucleotide in intact arterial smooth muscle. J Biol Chem. 2001;276:48398–403. doi: 10.1074/jbc.m106227200. [DOI] [PubMed] [Google Scholar]
- 3.James SG, Appleby GJ, Miller KA, Steen JT, Colquhoun EQ, Clark MG. Purine and pyrimidine nucleotide metabolism of vascular smooth muscle cells in culture. Gen Pharmacol. 1996;27:837–44. doi: 10.1016/0306-3623(95)02087-x. [DOI] [PubMed] [Google Scholar]
- 4.Ni L, Guan K, Zalkin H, Dixon JE. De novo purine nucleotide biosynthesis: cloning, sequencing and expression of a chicken PurH cDNA encoding 5-aminoimidazole-4-carboxamide-ribonucleotide transformylase-IMP cyclohydrolase. Gene. 1991;106:197–205. doi: 10.1016/0378-1119(91)90199-l. [DOI] [PubMed] [Google Scholar]
- 5.Corton JM, Gillespie JG, Hawley SA, Hardie DG. 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur J Biochem. 1995;229:558–65. doi: 10.1111/j.1432-1033.1995.0558k.x. [DOI] [PubMed] [Google Scholar]
- 6.Day P, Sharff A, Parra L, Cleasby A, Williams M, Horer S, et al. Structure of a CBS-domain pair from the regulatory gamma1 subunit of human AMPK in complex with AMP and ZMP. Acta Crystallogr D Biol Crystallogr. 2007;63:587–96. doi: 10.1107/s0907444907009110. [DOI] [PubMed] [Google Scholar]
- 7.Goirand F, Solar M, Athea Y, Viollet B, Mateo P, Fortin D, et al. Activation of AMP kinase alpha1 subunit induces aortic vasorelaxation in mice. J Physiol. 2007;581:1163–71. doi: 10.1113/jphysiol.2007.132589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ford RJ, Teschke SR, Reid EB, Durham KK, Kroetsch JT, Rush JW. AMP-activated protein kinase activator AICAR acutely lowers blood pressure and relaxes isolated resistance arteries of hypertensive rats. J Hypertens. 2012;30:725–33. doi: 10.1097/hjh.0b013e32835050ca. [DOI] [PubMed] [Google Scholar]
- 9.Pyla R, Osman I, Pichavaram P, Hansen P, Segar L. Metformin exaggerates phenylephrine-induced AMPK phosphorylation independent of CaMKKbeta and attenuates contractile response in endothelium-denuded rat aorta. Biochem Pharmacol. 2014;92:266–79. doi: 10.1016/j.bcp.2014.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buhl ES, Jessen N, Pold R, Ledet T, Flyvbjerg A, Pedersen SB, et al. Long-term AICAR administration reduces metabolic disturbances and lowers blood pressure in rats displaying features of the insulin resistance syndrome. Diabetes. 2002;51:2199–206. doi: 10.2337/diabetes.51.7.2199. [DOI] [PubMed] [Google Scholar]
- 11.Banek CT, Bauer AJ, Needham KM, Dreyer HC, Gilbert JS. AICAR administration ameliorates hypertension and angiogenic imbalance in a model of preeclampsia in the rat. Am J Physiol Heart Circ Physiol. 2013;304:H1159–65. doi: 10.1152/ajpheart.00903.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Greig FH, Ewart MA, McNaughton E, Cooney J, Spickett CM, Kennedy S. The hypotensive effect of acute and chronic AMP-activated protein kinase activation in normal and hyperlipidemic mice. Vasc Pharmacol. 2015;74:93–102. doi: 10.1016/j.vph.2015.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bosselaar M, Boon H, van Loon LJ, van den Broek PH, Smits P, Tack CJ. Intra-arterial AICA-riboside administration induces NO-dependent vasodilation in vivo in human skeletal muscle. Am J Physiol Endocrinol Metab. 2009;297:E759–66. doi: 10.1152/ajpendo.00141.2009. [DOI] [PubMed] [Google Scholar]
- 14.Bradley EA, Eringa EC, Stehouwer CD, Korstjens I, van Nieuw Amerongen GP, Musters R, et al. Activation of AMP-activated protein kinase by 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside in the muscle microcirculation increases nitric oxide synthesis and microvascular perfusion. Arterioscler Thromb Vasc Biol. 2010;30:1137–42. doi: 10.1161/atvbaha.110.204404. [DOI] [PubMed] [Google Scholar]
- 15.Ford RJ, Rush JW. Endothelium-dependent vasorelaxation to the AMPK activator AICAR is enhanced in aorta from hypertensive rats and is NO and EDCF dependent. Am J Physiol Heart Circ Physiol. 2011;300:H64–75. doi: 10.1152/ajpheart.00597.2010. [DOI] [PubMed] [Google Scholar]
- 16.Pyla R, Poulose N, Jun JY, Segar L. Expression of conventional and novel glucose transporters, GLUT1, -9, -10, and -12, in vascular smooth muscle cells. Am J Physiol Cell Physiol. 2013;304:C574–589. doi: 10.1152/ajpcell.00275.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mullane K. Acadesine: the prototype adenosine regulating agent for reducing myocardial ischaemic injury. Cardiovasc Res. 1993;27:43–7. doi: 10.1093/cvr/27.1.43. [DOI] [PubMed] [Google Scholar]
- 18.Kitakaze M, Takashima S, Minamino T, Node K, Shinozaki Y, Mori H, et al. Improvement by 5-amino-4-imidazole carboxamide riboside of the contractile dysfunction that follows brief periods of ischemia through increases in ecto-5-nucleotidase activity and adenosine release in canine hearts. Jpn Circ J. 1999;63:542–53. doi: 10.1253/jcj.63.542. [DOI] [PubMed] [Google Scholar]
- 19.Banos G, Martinez F, Grimaldo JI, Franco M. Adenosine participates in regulation of smooth muscle relaxation in aortas from rats with experimental hypothyroidism. Can J Physiol Pharmacol. 2002;80:507–14. doi: 10.1139/y02-064. [DOI] [PubMed] [Google Scholar]
- 20.Auchampach JA, Bolli R. Adenosine receptor subtypes in the heart: therapeutic opportunities and challenges. Am J Physiol. 1999;276:H1113–1116. doi: 10.1152/ajpheart.1999.276.3.h1113. [DOI] [PubMed] [Google Scholar]
- 21.Thorn JA, Jarvis SM. Adenosine transporters. Gen Pharmacol. 1996;27:613–20. doi: 10.1016/0306-3623(95)02053-5. [DOI] [PubMed] [Google Scholar]
- 22.Young ME, Radda GK, Leighton B. Activation of glycogen phosphorylase and glycogenolysis in rat skeletal muscle by AICAR--an activator of AMP-activated protein kinase. FEBS Lett. 1996;382:43–7. doi: 10.1016/0014-5793(96)00129-9. [DOI] [PubMed] [Google Scholar]
- 23.Rubin LJ, Johnson LR, Dodam JR, Dhalla AK, Magliola L, Laughlin MH, et al. Selective transport of adenosine into porcine coronary smooth muscle. Am J Physiol Heart Circ Physiol. 2000;279:H1397–410. doi: 10.1152/ajpheart.2000.279.3.h1397. [DOI] [PubMed] [Google Scholar]
- 24.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–62. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–74. doi: 10.1172/jci13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Handa N, Takagi T, Saijo S, Kishishita S, Takaya D, Toyama M, et al. Structural basis for compound C inhibition of the human AMP-activated protein kinase alpha2 subunit kinase domain. Acta Crystallogr D Biol Crystallogr. 2011;67:480–7. doi: 10.1107/s0907444911010201. [DOI] [PubMed] [Google Scholar]
- 27.Dixon R, Gourzis J, McDermott D, Fujitaki J, Dewland P, Gruber H. AICA-riboside: safety, tolerance, and pharmacokinetics of a novel adenosine-regulating agent. J Clin Pharmacol. 1991;31:342–7. doi: 10.1002/j.1552-4604.1991.tb03715.x. [DOI] [PubMed] [Google Scholar]
- 28.Goodyear LJ. The exercise pill--too good to be true? N Engl J Med. 2008;359:1842–4. doi: 10.1056/nejmcibr0806723. [DOI] [PubMed] [Google Scholar]
- 29.Karagounis LG, Hawley JA. The 5’ adenosine monophosphate-activated protein kinase: regulating the ebb and flow of cellular energetics. Int J Biochem Cell Biol. 2009;41:2360–3. doi: 10.1016/j.biocel.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 30.Racanelli AC, Rothbart SB, Heyer CL, Moran RG. Therapeutics by cytotoxic metabolite accumulation: pemetrexed causes ZMP accumulation, AMPK activation, and mammalian target of rapamycin inhibition. Cancer Res. 2009;69:5467–74. doi: 10.1158/0008-5472.can-08-4979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Greasley SE, Horton P, Ramcharan J, Beardsley GP, Benkovic SJ, Wilson IA. Crystal structure of a bifunctional transformylase and cyclohydrolase enzyme in purine biosynthesis. Nat Struct Biol. 2001;8:402–6. doi: 10.1038/87555. [DOI] [PubMed] [Google Scholar]
- 32.Allegra CJ, Drake JC, Jolivet J, Chabner BA. Inhibition of phosphoribosylaminoimidazolecarboxamide transformylase by methotrexate and dihydrofolic acid polyglutamates. Proc Natl Acad Sci USA. 1985;82:4881–5. doi: 10.1073/pnas.82.15.4881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pirkmajer S, Kulkarni SS, Tom RZ, Ross FA, Hawley SA, Hardie DG, et al. Methotrexate promotes glucose uptake and lipid oxidation in skeletal muscle via AMPK activation. Diabetes. 2015;64:360–9. doi: 10.2337/db14-0508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mangoni AA, Baghdadi LR, Shanahan EM, Wiese MD, Tommasi S, Elliot D, et al. Methotrexate, blood pressure and markers of arterial function in patients with rheumatoid arthritis: a repeated cross-sectional study. Ther Adv Musculoskelet Dis. 2017;9:213–29. doi: 10.1177/1759720x17719850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ma Y, Li L, Shao Y, Bai X, Bai T, Huang X. Methotrexate improves perivascular adipose tissue/endothelial dysfunction via activation of AMPK/eNOS pathway. Mol Med Rep. 2017;15:2353–9. doi: 10.3892/mmr.2017.6225. [DOI] [PubMed] [Google Scholar]