

ABSTRACT
Malaria is a mosquito-borne fatal infectious disease that affects humans and is caused by Plasmodium parasites, primarily Plasmodium falciparum. Widespread drug resistance compels us to discover novel compounds and alternative drug discovery targets. The coenzyme A (CoA) biosynthesis pathway is essential for the malaria parasite P. falciparum. The last enzyme in CoA biosynthesis, dephospho-CoA kinase (DPCK), is essential to the major life cycle development stages but has not yet been exploited as a drug target in antimalarial drug discovery. We performed a high-throughput screen of a 210,000-compound library using recombinant P. falciparum DPCK (PfDPCK). A high-throughput enzymatic assay using a 1,536-well platform was developed to identify potential PfDPCK inhibitors. PfDPCK inhibitors also inhibited parasite growth in a P. falciparum whole-cell asexual blood-stage assay in both drug-sensitive and drug-resistant strains. Hit compounds were selected based on their potency in cell-free (PfDPCK) and whole-cell (Pf3D7 and PfDd2) assays, selectivity over the human orthologue (HsCOASY) and no cytotoxicity (HepG2). The compounds were ranked using a multiparameter optimization (MPO) scoring model, and the specific binding and the mechanism of inhibition were investigated for the most promising compounds.
KEYWORDS: malaria, coenzyme A, high-throughput screening, dephospho-CoA kinase, Plasmodium falciparum, inhibitor, antimalarial agents
INTRODUCTION
Malaria is a protozoan infection caused by Plasmodium species and transmitted by mosquito bites. Malaria causes approximately an estimated 241 million cases and kills an estimated 627,000 people in 2020, mainly in tropical and subtropical regions worldwide, representing more cases and deaths than in 2019 (1). Plasmodium falciparum is the most dangerous and deadliest among five human-infecting species. Despite significant global health investments in recent years, which decreased malaria mortality by more than 50% between 2000 and 2016, malaria remains one of the major infectious killers. This is largely attributable to the lack of an effective vaccine and the emergence of resistance to virtually all available antimalarial chemotherapeutics, including the standard chemotherapeutic regimens, which include artemisinin and its partner drugs in artemisinin combination therapies (ACT). Thus, new antimalarial agents that act on different targets, preferably in multiple life cycle stages, and have novel mechanisms of action, are urgently needed to overcome the lack or shortage of effective therapeutic options (2–5).
One of the validated and unexplored drug targets for the development of antimalarials is the coenzyme A (CoA) biosynthetic pathway (6). CoA is an essential cofactor that acts as an acyl group carrier and involved in approximately 9% of all (3,500) cellular activities (https://www.brenda-enzymes.info/). CoA is synthesized by 4 to 5 enzymatic steps (Fig. 1), and the pathway is initiated by a conversion of pantothenic acid (vitamin B5) to 4-phosphopantothenate, followed by later steps in which l-cysteine and pyrimidine/purine nucleotides are used as the substrates (7). Some bacteria, archaea, mammals, and plants are able to produce pantothenate de novo (8–10), while the others can scavenge it from the environment, including the hosts or prey (8, 11). The first enzyme in the pathway, pantothenate kinase (PanK), has been well characterized and considered to be a rational drug target against P. falciparum (8, 11, 12). The last enzyme in this pathway, dephospho-CoA kinase (DPCK; EC 2.7.1.24), has also been proven to be essential, and it has been demonstrated that DPCK is rate limiting and allosterically regulated in P. falciparum (13, 14). Similarly, DPCK has been proven to be indispensable in the enteric protozoan Entamoeba histolytica and thus underpinned as a potential drug target (15). However, no specific inhibitors against P. falciparum dephospho-CoA kinase (PfDPCK), which can be further developed for antimalarial drug discovery, have been documented.
FIG 1.

Coenzyme A biosynthetic pathway in P. falciparum. ID numbers of individual enzymes in PlasmoDB are also shown. PfPanK, P. falciparum pantothenate kinase; PfPPCS, P. falciparum phosphopantothenoylcysteine synthetase; PfPPCDC P. falciparum phosphopantothenoylcysteine decarboxylase; PfPPAT, P. falciparum phosphopantetheine adenylyltransferase; PfDPCK, P. falciparum dephosphocoenzyme A kinase.
In this study, we achieved expression and purification of PfDPCK using an Escherichia coli expression system. We enzymatically characterized PfDPCK and developed a high-throughput screening protocol to screen approximately 210,000 structurally diversified compounds from the Drug Discovery Initiative (DDI) library to discover potential inhibitors. Among these PfDPCK inhibitors, 99 compounds (80%) showed growth inhibition toward both drug-sensitive and -resistant P. falciparum strains. The modes of inhibition of the representative inhibitors were determined. The specific binding and inhibition of PfDPCK, but not its human counterpart, by the representative inhibitor was confirmed by in silico modeling of the two enzymes and the inhibitor.
RESULTS
Identification of DPCK from P. falciparum.
Since CoA plays an indispensable role in P. falciparum (16) and DPCK catalyzes the final committed step in its biosynthesis, we were prompted to identify and characterize the enzyme from Plasmodium falciparum. We found a single 825-bp-long protein coding sequence (PF3D7_1443700) encoding 274-amino-acid PfDPCK with the calculated molecular mass of 31.9 kDa, from the genome database of the P. falciparum 3D7 strain (https://plasmodb.org/plasmo/).
DPCK is highly conserved among Plasmodium species. PfDPCK exhibits 88 to 93% amino acid identity to Plasmodium vivax, Plasmodium malariae, Plasmodium ovale, Plasmodium knowlesi, and Plasmodium yoelii. PfDPCK also shows the highest identity to, other than orthologs from Plasmodium, bacterial DPCK from Haemophilus influenzae and Aquifex aeolicus (33 and 30%, respectively). In contrast, similarity of PfDPCK to the human counterpart (bifunctional coenzyme A synthase [HsCOASY]) is limited (24%). Phylogenetic analysis of 43 DPCK protein sequences based on 190 aligned positions inferred by maximum-likelihood analysis (see Fig. S1 in the supplemental material) also suggests that Plasmodium DPCKs are highly diverged from HsCOASY.
Determination of kinetic properties of PfDPCK.
We successfully produced and purified recombinant PfDPCK for enzymological characterization by using the E. coli expression system. The purity of the protein was estimated to be 90 to 95% by densitometric scanning of the Coomassie brilliant blue (CBB)-stained gels after SDS-PAGE (Fig. 2A). The apparent molecular mass of the recombinant PfDPCK was consistent with the predicted mass of approximately 32 kDa plus 2.6 kDa corresponding to the histidine tag. The kinetic parameters of PfDPCK were determined by measuring the initial rates obtained with different concentrations of ATP and dephospho-CoA (Fig. 2B and C). Both ATP and dephospho-CoA exhibited hyperbolic saturation kinetics. With the saturating concentrations of both substrates, PfDPCK showed the apparent maximum rate of metabolism (Vmax) of 5.18 ± 0.29 μmol/min/mg. The Km values of PfDPCK were 88.14 ± 11.03 μM and 105.3 ± 10.2 μM for ATP and dephospho-CoA, respectively. PfDPCK was unable to utilize pantothenate, the precursor of CoA biosynthesis, as a substrate (the specific activity, <0.01 μmol/min/mg).
FIG 2.

Biochemical characterization of recombinant PfDPCK and HsCOASY. (A) Expression and purification of recombinant PfDPCK. Samples at each step (lanes 1 to 4) of purification were subjected to 15% SDS-PAGE under reducing conditions and then stained with Coomassie brilliant blue R250. (B) Validation of the purity of recombinant HsCOASY. CBB staining (left) and immunoblot using anti-His antibody (right) are shown. (C to F) Kinetic analysis of PfDPCK (C, D) and HsCOASY (E, F) to determine the Km values of ATP (C, E) and dephospho-CoA (D, F).
Identification of potent PfDPCK inhibitors by a high-throughput screening of the structurally diverse chemical library of 210,000 compounds.
In order to identify inhibitors against recombinant PfDPCK, we successfully established the high-throughput screening protocol using a small reaction volume (2 μL) on a 1,536-well plate platform in a one-step enzyme-coupled fluorescence assay (17). A chemical library from the Drug Discovery Initiative, The University of Tokyo, Japan, composed of 210,000 structurally diverse compounds, was tested for PfDPCK inhibition at a single final concentration of 10 μM. The screening was robust, with the mean Z′ factor (18) being 0.80 (Fig. 3A). With the selection criteria of >40% inhibition, 1,241 primary hits were identified with an overall hit rate of 0.6% (Fig. 3B; Fig. 4). In the second screening, the primary hits that showed >40% inhibition of PfDPCK activity at 10 μM were tested in quadruplicate to exclude false positives that inhibited the coupled assay, but not PfDPCK, which yielded 359 secondary hits (Fig. 3C). In the tertiary screening, the remaining hits were further tested against their human counterpart (HsCOASY) at a final concentration of 10 μM, and the hits that showed >10% inhibition toward HsCOASY were excluded to yield 127 tertiary hits, which were 0.06% from total compounds tested, which corresponds to 10.2% of the 1,241 primary hit compounds (Fig. 3D). The 50% inhibitory concentration (IC50) values against PfDPCK and the 50% effective concentration (EC50) values against P. falciparum drug-sensitive 3D7, the chloroquine-resistant Dd2 strain, and human hepatoma HepG2 cells (19) of the 127 tertiary hits were determined using a five-dose titration assay in quadruplicate (Table S1).
FIG 3.

(A) Performance metrics of high-throughput screening of DDI library against PfDPCK. Quality control values of all 150 1,536-well assay plates in the primary screening of the whole DDI library (210,000 compounds). The values of the S/B ratio (SB), Z′, coefficient of variation (CV) of positive control (CV_HC), and CV of negative control (CV_LC) are shown. (B) Plot showing percentage inhibition of PfDPCK activity by all DDI compounds in the primary assay. (C) Scatterplot showing percentage inhibition of PfDPCK activity (average of quadruplicate, y axis) and that of the ADP-coupled assay (x axis) of 825 secondary confirmed hits. (D) Scatterplot of percentage inhibition of PfDPCK activity (the average of quadruplicate, y axis) and that of HsCOASY activity (x axis) of 359 tertiary hits. Arrowhead indicates compounds selected for titration assay.
FIG 4.

Screening cascade of the high-throughput screening of DDI library. The cascade is divided into two legs, screening based on the selectivity toward PfDPCK over HsCOASY (left leg) and screening based on anti-erythrocytic-stage growth inhibition (right leg). The number of technical replicate assays is indicated (n). Individual steps of the cascade are described in the text.
We manually screened the 127 quaternary hits (Table S1) to further exclude undesirable hits, and we selected 21 compounds (Table 1). Our criteria of filtering were based on the previous experience of scientists at Medicines for Malaria Venture (MMV) and included the following: (i) being capable of inhibiting both PfDPCK and P. falciparum erythrocytic-stage parasites of both drug-sensitive 3D7 and resistant Dd2 strains, (ii) not being frequent hits from other screening campaigns, (iii) relatively high selectivity against P. falciparum 3D7 and Dd2 strains compared to HepG2 human cell line (index > 30; either HepG2 to 3D7 or Dd2 or HepG2 to both strains), and (iv) the IC50 values being <10 μM.
TABLE 1.
IC50 values of hit compounds against PfDPCK enzyme, P. falciparum cell drug-sensitive (3D7) and drug-resistant (Dd2) strains, and liver human cell line (HepG2)a
| Compound | Chemical structure | IC50 (μM)b of: |
Selectivity index |
||||
|---|---|---|---|---|---|---|---|
| PfDPCK | Pf cell 3D7 | Pf cell Dd2 | HepG2 | IC50 (HepG2/3D7) | IC50 (HepG2/Dd2) | ||
| A-2 |

|
0.40 ± 0.07 | 4.43 ± 0.04 | 0.98 ± 0.50 | >100 | >23 | >102 |
| A-4 |

|
0.52 ± 0.09 | 4.45 ± 0.05 | 1.59 ± 0.23 | ND | NA | NA |
| A-7 |

|
1.09 ± 0.03 | 1.10 ± 0.11 | 0.13 ± 0.01 | 9.04 ± 0.01 | 8.2 | 69.5 |
| A-13 |

|
1.69 ± 0.05 | 1.30 ± 0.06 | 3.40 ± 0.35 | ND | NA | NA |
| A-16 |
|
2.09 ± 0.04 | 2.21 ± 0.07 | 0.23 ± 0.01 | 39.64 ± 2.30 | 17.9 | 171.7 |
| A-19 |

|
2.55 ± 0.03 | 3.64 ± 0.04 | 2.65 ± 0.13 | 81.50 | 22.4 | 30.8 |
| A-22 |

|
3.25 ± 0.02 | 30.61 ± 1.50 | 6.32 ± 0.35 | ND | NA | NA |
| A-23 |

|
3.31 ± 0.02 | 4.47 ± 0.12 | 7.42 ± 0.46 | ND | NA | NA |
| A-25 |

|
3.45 ± 0.07 | 1.64 ± 0.03 | >50 | ND | NA | NA |
| A-26 |

|
3.50 ± 0.04 | 14.45 ± 0.08 | 3.41 ± 1.22 | ND | NA | NA |
| A-30 |

|
3.83 ± 0.03 | 0.08 ± 0.01 | 0.09 ± 0.06 | 3.75 ± 3.55 | 45.1 | 43.5 |
| A-31 |

|
3.83 ± 0.04 | >50 | 0.67 ± 0.02 | ND | NA | NA |
| A-38 |
|
4.87 ± 0.05 | 29.06 ± 0.20 | 0.70 ± 0.10 | >100 | >3.4 | >142.8 |
| A-44 |

|
6.33 ± 0.11 | 3.96 ± 0.04 | 1.75 ± 0.05 | ND | NA | NA |
| A-45 |
|
6.68 ± 0.24 | 15.63 ± 0.61 | 1.29 ± 0.02 | ND | NA | NA |
| A-50 |
|
6.99 ± 0.13 | >50 | 6.62 ± 1.12 | ND | NA | NA |
| A-51 |

|
7.05 ± 0.08 | 15.76 ± 0.12 | 0.62 ± 0.23 | >100 | >6.3 | >166.6 |
| A-60 |

|
8.35 ± 0.05 | 34.14 ± 0.04 | 5.53 ± 0.95 | ND | NA | NA |
| A-63 |

|
8.69 ± 0.01 | 6.04 ± 0.05 | 0.70 ± 0.22 | >100 | >16.6 | >142.8 |
| A-65 |

|
9.21 ± 0.12 | 4.40 ± 0.05 | 0.36 ± 0.10 | ND | NA | NA |
| Atovaquone |

|
ND | 0.22 ± 0.03 nM | 0.16 ± 0.028 nM | 79.90 ± 1.79 | 363,181 | 499,375 |
| Mefloquine |

|
ND | 0.013 ± 0.006 | 0.19 ± 0.07 | 1.73 ± 0.7 | 133 | 8.8 |
Atovacuone and mefloquine were included as anti-Plasmodium drug control. ND, no inhibition detected in our maximum concentration tested and considered not toxic; NA, not applicable. The assays were carried out three times independently, and the results are shown as means ± SEMs of triplicates.
Values represent micromolar unless otherwise specified.
One hundred twenty-seven potential PfDPCK inhibitors were manually categorized into eight groups based on structural similarity (i.e., common scaffolds) by manual inspection. The eight groups are composed of 2 large groups, Gr1 and Gr2, with 13 and 9 hit members, respectively; 6 small groups (Gr3 to Gr8) with 2 to 6 hit members; and 87 singletons (Fig. S5). We selected Gr4, Gr5, and Gr8 as the potent PfDPCK-inhibitory scaffolds based on their overall PfDPCK and cell growth-inhibitory activities. Table 2 shows representative structures of these 3 groups. Especially Gr4 may represent a novel PfDPCK inhibitory scaffold because it inhibited both PfDPCK and P. falciparum cells with comparable IC50 values, and it did not show toxicity toward HepG2. However, further structure-activity relationship (SAR) studies are needed to validate the scaffold as a reasonable initial PfDPCK inhibitor candidate. Regarding Gr2, it showed reasonable potency against PfDPCK and P. falciparum cells, but it was excluded in the downstream analysis because of the poor physicochemical properties (e.g., high molecular weight, complex structure, and low solubility).
TABLE 2.
Structure-activity relationship of PfDPCK potent inhibitor of hit compounds
| Scaffold | Compound ID | Structure | R1 | R2 | R3 | IC50 (μM) |
|---|---|---|---|---|---|---|
Gr4 
|
A-4 |
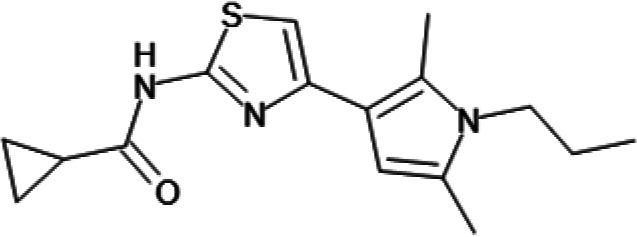
|

|

|
0.52 ± 0.09 | |
| A-19 |

|

|
|
2.55 ± 0.03 | ||
| A-44 |
|

|

|
6.33 ± 0.11 | ||
| A-96 |

|

|

|
14.7 ± 0.28 | ||
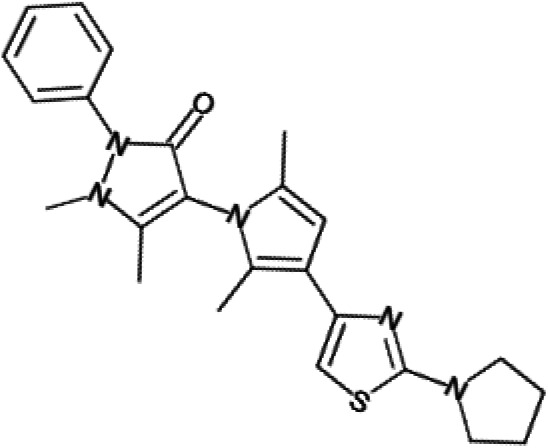
Gr5 
|
A-17 |

|

|
|

|
2.15 ± 0.07 |
| A-42 |
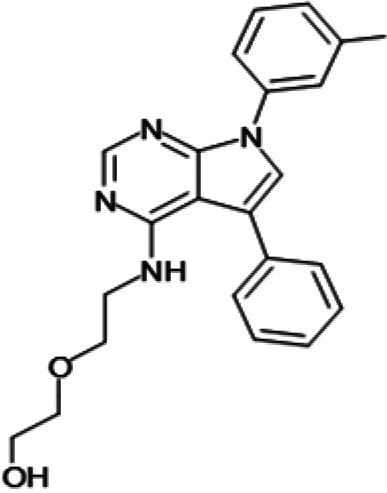
|

|

|
|
6.14 ± 0.28 | |
Gr8 
|
A-15 |
|

|
1.93 ± 0.04 | ||
| A-30 |

|

|
3.83 ± 0.03 |
Identification and prioritization of malaria-specific DPCK inhibitors and elucidation of their mode of inhibition.
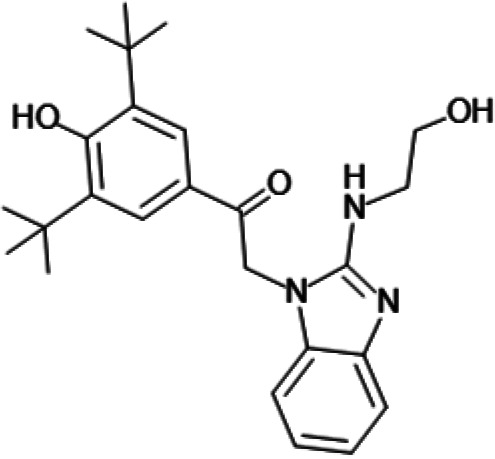
In order to understand the mechanism of PfDPCK inhibition of the identified compounds, we independently selected compounds from 359 PfDPCK-specific inhibitor candidates (showing >40% inhibition at 10 μM against PfDPCK), based on the more stringent criteria of >80% inhibition at 10 μM against PfDPCK and <10% inhibition against HsCOASY, to yield 30 hits. The hits were prioritized using a multiparameter optimization (MPO) scoring model, developed by MMV (https://www.optibrium.com/downloads/scoring-profiles/mmv-antimalarial-scoring-profile/) based on calculated physicochemical properties, including molecular weight, logP, hydrogen bond donors and acceptors, rotatable bonds, and structural alerts (20, 21). We finally chose six representative compounds (A-15, A-33, and A-69, shown in Table S1, and other three compounds; A-126, A-127, and A-128) by manual inspection with “chemist eye” for chemical attractiveness (druglikeness). All six compounds except A-127 showed no growth inhibition to 3D7 at 2 μM (Table S1). Compound A-127, which is a quinoline-containing compound, showed 80% inhibition against 3D7 at 2 μM. We found that two compounds, A-126 and A-128, also inhibit many other targets than kinase, based on the previous experimental database provided by DDI, suggesting that they are nonspecific inhibitors against numerous biological targets. Furthermore, two compounds, A-33 and A-69, are considered to be pan-assay interference compounds (PAINS) (phenolic Mannich base) (22). We consequently focused on two commercially available compounds (A-15 and A-127) (Fig. 6A) and investigated their mechanism of inhibition. We differentiated the type of inhibition by Lineweaver-Burk plot with various concentrations of one substrate (dephospho-CoA or ATP), while the other substrate was given in saturating concentrations in kinetic studies (Fig. 5). We found that compound A-15 competitively inhibits PfDPCK in respect of both dephospho-CoA and ATP, with Ki values of 0.47 and 10.42 μM, respectively (Fig. 5A and B). On the other hand, compound A-127 showed uncompetitive or noncompetitive inhibition for dephospho-CoA and ATP substrate, with the Ki values of 14.89 and 8.99 μM, respectively (Fig. 5C and D).
FIG 6.

Structure of the representative PfDPCK inhibitors (A) and docking simulation of compound A-15 (B, C). (A) Structure of the representative PfDPCK inhibitors A-15 and A-127 and their calculated physicochemical properties. MW, molecular weight; logP, partition coefficient; HBD, hydrogen bond donor; HBA, hydrogen bond acceptor. (B, C) Docking simulation of compound A-15 to the catalytic pocket of PfDPCK (B) and HsCOASY (C). The three-dimensional (3D) docking poses of compound A-15 (white sticks; oxygens are shown in red, and nitrogens are shown in dark blue) in the active sites of PfDPCK (B) and HsCOASY (C) are shown. Key amino acid residues were determined in the 3D position in active sites. (B) The binding pose of compound A-15 with the best docking score to PfDPCK is shown. (C) The structure of HsCOASY is superimposed with Arg514 depicted with cyan sticks, showing structural hindrance with compound A-15.
FIG 5.

Lineweaver-Burk plots showing inhibition of two compounds, compound A-15 (A, B) and compound A-127 (C, D) on the PfDPCK activity. The enzymatic activities were determined with various concentrations of dephospho-CoA and 200 μM ATP (A, C) or various concentrations of ATP and 200 μM dephospho-CoA (B, D) in the presence of a range of concentrations of inhibitors. Data are shown in means ± SEMs of triplicate.
Binding mode of PfDPCK by compound A-15.
To better understand the mechanism of inhibition by compound A-15 at the structural level, we performed in silico docking of compound A-15 as a representative PfDPCK competitive inhibitor. The binding pose of compound A-15 with the best docking score is shown in Fig. 6A. The tert-butyl group of compound A-15 was found to be in contact with the hydrophobic side chain of PfDPCK Ile230. In contrast, when the structure of HsCOASY was superimposed, the hydrophilic side chain of HsCOASY Arg514 collided with the position of the tert-butyl group (in Fig. 6B). This difference is consistent with the experimental results with the reciprocal plot analysis showing that compound A-15 inhibits PfDPCK, but not HsCOASY.
DISCUSSION
Enzymes on the CoA biosynthetic pathway have been validated as antimalarial drug targets. Among the multiple enzymes involved in the pathway, PfDPCK, which catalyzes the last committed step of the pathway, is a promising target for drug development because it is encoded by a single gene, expressed in all life cycle stages, including the liver and erythrocytic stages in humans and mosquitos (23, 24), proven to be essential (13) and largely different from their human counterparts. This enzyme has an N-terminal bipartite apicoplast-trafficking peptide (25) and is confirmed as an essential apicoplast-localized protein (16).
In the present study, we identified potential hits for malaria-specific inhibitors targeting the CoA biosynthetic pathway. Although inhibitors against PanK, which catalyzes the first step of the pathway, were previously identified, our current study has provided the first case in which PfDPCK was exploited as the drug target, leading to the discovery of potential leads from the structurally elucidated compound library. Although the identified PfDPCK inhibitors show only moderate antiproliferative activities against the erythrocytic-stage parasites at submicro- to micromolar concentrations, our discovery of the compounds that showed comparable IC50 and EC50 values against PfDPCK and the parasite have provided a proof of concept for the enzyme and the pathway are exploitable for drug discovery, despite some skepticism due to the lack of correlation between antitarget (enzymatic) and antiproliferative (cellular) activity (26) in the current trends of antimalarial discovery using phenotypic cell-based screening for the primary screening in recent years (27–29). It is worth noting that among 127 PfDPCK-specific (i.e., not inhibiting HsCOASY) inhibitors we tested against P. falciparum blood-stage parasites, only 10 compounds (8%) show inhibition against both P. falciparum drug-sensitive 3D7 and drug-resistant Dd2 strains (with the criteria of IC50 of <5 μM for both enzymatic and cellular inhibition), suggesting poor permeability or instability of most of the identified PfDPCK inhibitors. Thus, further chemical modifications of the identified PfDPCK inhibitors have the potential to be further optimized to yield the derivatives with improved anti-erythrocytic-stage activity.
Our second-stage cell-based screening of the hits obtained by PfDPCK-selective (not inhibiting HsCOASY) screening yielded six candidates which have the IC50 values of 0.40 to 4.87 μM against PfDPCK and those of 0.08 to 6.04 μM against 3D7 and/or Dd2. The most promising compounds show reasonable agreement of the IC50 values against PfDPCK and cells (i.e., the differences in the IC50 values are < 5-fold, and the compounds are more potent against the enzyme than the cells); however, some compounds show marked disparity for reasons that are not yet understood. For example, compound A-30 shows 4- to 10-fold lower IC50 values in the cell-based assay than the enzyme-based assay. These data are consistent with the hypothesis that the target of the compounds for growth inhibition is not solely via inhibition of CoA synthesis. In addition, several compounds, including A-16, A-38, A-51, and A-63, show a marked decrease in the IC50 values against the drug-resistant Dd2 strain compared to the drug-sensitive 3D7 (7- to 42-fold), suggesting that the resistance to one agent may lead to increased susceptibility to another. It was previously shown (30) that the development of resistance to one line of antimalarials is accompanied by the loss of resistance to other drugs. Our observation on the increased sensitivity against A-16, A-38, A-51, and A-63 in chloroquine-resistant Dd2 may also indicate that a similar interconnection of drug action may occur. In contrast, in a reverse trend, an increase in resistance was seen in Dd2 for compound A-25, suggestive of shared mechanisms of resistance (Table 1).
We also selected 30 inhibitors exclusively by virtue of selective inhibition toward PfDPCK over HsCOASY. Among the 30 compounds, only 16 are included in the 127 compounds selected as described above. A multiparameter optimization (MPO) scoring profile was used to prioritize six compounds. The MPO was developed by MMV and allows us to rank compounds based on physicochemical properties, including molecular weight, logP, hydrogen bond acceptors and donors, and rotatable bonds, as well as structural alert and malaria drug fragments (21). Two representative PfDPCK-specific inhibitors identified from our high-throughput screening (HTS) demonstrated a distinct mechanism of inhibition as suggested by Lineweaver-Burk plot analysis: compound A-15 displayed competitive inhibition with both of two substrates, ATP and dephospho-CoA. The selectivity for PfDPCK over the human orthologue, HsCOASY, was rationalized by in silico molecular docking showing that the tert-butyl group of compound A-15 collides with the hydrophilic side chain of HsCOASY Arg514, which substitutes the important residue for strong substrate binding for PfDPCK (the hydrophobic side chain of PfDPCK Ile230). Thus, our docking simulation provides structural basis of the specificity of compound A-15 toward PfDPCK, but not its human counterpart.
Finally, our discovery of PfDPCK-specific inhibitors targeting the CoA biosynthetic pathway should provide a new validated and exploitable metabolic target that has a potential to be further pursued in the antimalarial drug development pipeline. DPCK-targeting drugs can be used in combination with existing antimalarials or new drug candidates in preclinical development or clinical trials, such as a variety of drug candidates targeting dihydroorotate dehydrogenase (DHODH); ATP4; phosphatidylinositol-4 kinase; elongation factor; acetyl-CoA synthetase; lysine-, proline-, tyrosine, and phenylalanine tRNA synthetases; proteasome; phosphodiesterase (PDE); and plasmepsin (5, 31–33).
Conclusion.
We have provided a proof of concept that inhibitors of PfDPCK can be identified from a target-based high-throughput screen. In addition, potent PfDPCK inhibitors that also inhibit parasite growth in a phenotypic whole-cell asexual blood-stage assay were identified. Furthermore, the structural basis of PfDPCK-specific inhibition was elucidated by a docking simulation. Although further optimization of the hits that were identified is necessary to improve efficacy, drug metabolism and pharmacokinetics (DMPK), and safety required for development as potential antimalarials, the inhibitors discovered in this study can be used as pharmacological tools for further target validation.
MATERIALS AND METHODS
Chemicals, microplates, organisms, and cultivation.
All chemicals of analytical grade were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise stated. ADP-hexokinase from Thermococcus litoralis (52.6 U/mg solid) was purchased from Asahi Kasei Pharma (Tokyo, Japan). Diaphorase I from Bacillus stearothermophilus (1.8 kU/mg protein) was purchased from Nipro (Osaka, Japan). Recombinant glucose-6-phosphate dehydrogenase from Leuconostoc sp. (G6PDH; 754 U/mg protein) and NADP+ were obtained from Oriental Yeast Co. (Tokyo, Japan). Triton X-100 and Tween 20 were purchased from Alfa Aesar (Lancashire, UK) and Tokyo Chemical Industry (Tokyo, Japan). N-ethylmaleimide (NEM), dithiothreitol (DTT), and bovine serum albumin (BSA) were purchased from Fujifilm Wako Pure Chemical (Osaka, Japan). The 1,536-well polypropylene microplates used for the kinase assay and 384-well microplates for the malaria growth inhibition assay were purchased from Greiner Bio-One (Frickenhausen, Germany).
The Plasmodium falciparum drug-sensitive 3D7 strain (GL Clone) MRA-1001 (BEI Resources, NIAID, NIH) and drug-resistant Dd2 strain were used for asexual blood-stage phenotypic assay. The cytotoxicity assay was conducted using the human hepatocarcinoma HepG2 cell line. AlbuMax II, RPMI 1640 medium, and gentamicin were purchased from Gibco (Life Technologies, Carlsbad, CA, USA); hypoxanthine was purchased from Sigma, sodium l-lactate and nitro-tetrazolium blue chloride (NBT) were purchased from Fujifilm (Wako), and 3-acetylpyridine adenine dinucleotide (APAD) was from Oriental Yeast (Japan). Dulbecco’s modified Eagle’s medium, low glucose (d-MED; with 1,000 mg/L glucose), and fetal bovine serum were purchased from Sigma-Aldrich. Trypsin-EDTA was purchased from Gibco. The overexpression of protein recombinant harboring Escherichia coli BL21(DE3) was purchased from Invitrogen (Carlsbad, CA, USA). Purification of recombinant protein used Ni2+-nitrilotriacetic acid (NTA) agarose, purchased from Novagen (Darmstadt, Germany). All other chemicals were analytical grade, purchased from Sigma-Aldrich (Tokyo, Japan) unless otherwise stated.
Phylogenetic analyses of P. falciparum DPCK.
We collected 43 DPCK protein sequences from representative taxa by BLASTp search using the PfDPCK protein sequence (PlasmoDB ID PF3D7_1443700; GenPept accession no. XP_001348589) as a query and nonredundant (nr) protein sequences database of National Center for Biotechnology Information (NCBI; http://www.ncbi.nlm.nih.gov/). Only protein sequences with an E value of 1 × 10−10 or less were selected. Sequences were aligned using the MUSCLE program (34) in SeaView package version 4.6.1 (35). The data matrices for phylogeny were subjected to the IQ-TREE program (36). The maximum-likelihood (ML) analysis implemented in the RAxML program version 7.2.6 (37) was used to infer ML tree. Trees were constructed using FigTree program version 1.4.2 (http://tree.bio.ed.ac.uk/software/figtree/). Bootstrap values higher than 50 are indicated on the corresponding internal branches of the ML tree.
Expression and purification of recombinant PfDPCK and HsCOASY.
The protein-coding sequences of the codon-optimized PfDPCK and HsCOASY genes were inserted into BamHI and SalI sites of the plasmid pCold1 His tag vector (TaKaRa) to produce pCold-PfDPCK and pCold-HsCOASY. Escherichia coli BL21Star (DE3) chemically competent cells (Thermo Fisher Scientific, Waltham, MA, USA) were transformed with these plasmids and cultured at 37°C in 500 mL of Luria-Bertani medium (LB, Invitrogen) in the presence of 100 μg/mL ampicillin (Nacalai Tesque). The overnight culture was used to inoculate 1 L of fresh LB medium. The culture was continued at 37°C with shaking at 180 rpm until the A600 reached 0.8. After isopropyl β-d-thiogalactopyranoside (IPTG) was added at a final concentration of 0.5 mM, cultivation was continued for another 24 h at 15°C. Cultured E. coli cells were harvested by centrifugation at 5,000 × g for 20 min at 4°C. The cell pellet was collected and resuspended in 40 mL of the lysis buffer (50 mM Tris-HCl, pH 8.0, 300 mM NaCl, and 10 mM imidazole) containing 0.1% Triton X-100 (vol/vol), 0.7 M trehalose, 100 μg/mL lysozyme, and 1 mM phenylmethylsulfonyl fluoride (PMSF) and incubated at room temperature for 30 min. The cells were then passed through a French press (Ohtake, Tokyo) with a pressure of 800 kg/cm2, and the lysate was centrifuged at 25,000 × g for 30 min at 4°C to remove debris. The supernatant obtained was mixed with 2 mL of 50% Ni2+-NTA His-bind slurry (Qiagen, Germany) and then incubated at 4°C with mild shaking for 1 h. The resin bound to recombinant enzymes was washed three times with washing buffer (50 mM Tris-HCl, pH 8.0, and 300 mM NaCl, containing 20 mM imidazole and 0.1% [vol/vol] Triton X-100). Bound enzymes were eluted with washing buffer containing stepwise gradient concentrations of imidazole (20 to 300 mM). The purity of recombinant protein was confirmed by 12% SDS-PAGE analysis, followed by Coomassie brilliant blue (CBB) staining. Purified proteins were dialyzed against a 300-fold volume of dialysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, containing 10% glycerol [vol/vol] supplemented with Complete Mini protease inhibitor cocktail [Roche, Mannheim, Germany]) at 4°C for 18 h to remove residual imidazole. Enzymes were stored at −80°C with a final 20% glycerol in small aliquots until use.
Enzyme assays.
PfDPCK and HsCOASY activity was measured by quantifying ADP produced in the reaction by a coupling assay using the ADP Hunter Plus assay kit (DiscoverX, USA) according to the manufacturer's instructions. Briefly, enzymatic reactions were carried out in a 20-μL mixture of assay buffer containing 15 mM HEPES, 20 mM NaCl, 1 mM EGTA, 0.02% Tween 20, 10 mM MgCl2, 0.1% bovine gamma globulin, 50 ng of recombinant PfDPCK or HsCOASY, 4 to 256 μM dephospho-CoA, and 5 to 300 μM ATP on a black microplate. All reactions were performed for 2 h, reagents A and B were added, and they were then reincubated for 60 min at 30°C. After reactions were terminated by addition of ADP Hunter Stop solution, the fluorescent signal was measured using SpectraMax Paradigm (Molecular Devices, CA, USA) at excitation and emission wavelengths of 530 and 590 nm, respectively. The kinetic parameters were calculated using the nonlinear regression function using the single saturating concentrations of the substrates by the GraphPad Prism software (GraphPad Software Inc., San Diego, CA). The experiments were repeated three times in triplicate using proteins purified from independent cultures. Kinetic values of enzymes are presented as the means ± standard errors (SEs).
High-throughput screening of structurally diversified chemical library.
High-throughput screening against recombinant PfDPCK was performed using the Drug Discovery Initiative (DDI) full library, available from The University of Tokyo, composed of approximately 210,000 compounds. The primary assay was performed on 1,536-well white plate format with a final volume of 2 μL/well, containing 10 μM test compounds using a Multidrop Combi reagent dispenser (Thermo Fisher Scientific, Waltham, MA). The reaction was conducted in the assay buffer described above with 100 μM ATP and 50 μM dephospho-CoA. The assay was carried out at room temperature for 4 h in a closed plastic container with wet tissues. The reaction was stopped, and produced ADP was quantified using an enzyme-coupled fluorescence ADP detection assay, using PHERAstar microplate reader (BMG Labtech, Offenburg, Germany) with excitation at 540 nm and emission at 590 nm, as previously described (17). The inhibition level was calculated as the percentage inhibition of DPCK activity with each compound compared to the activity with only dimethyl sulfoxide (DMSO) as control (no inhibition) and that with no enzyme as 100% inhibition. IC50 values of the hit compounds were determined under the same assay conditions for the screening: Z′ factor, signal-to-background ratio (S/B), signal-to-noise ratio (S/N), and coefficient of variation (CV) were calculated to estimate the quality of screening system as previously described (18, 38).
Reconfirmation, counterassays, and IC50 determination of PfDPCK inhibitors.
Compounds that showed >40% inhibition against PfDPCK at 10 μM in the primary assay were selected for secondary reconfirmation assay. The primary hits were reassayed in quadruplicate (n = 4) as in the primary screening. We next tested the confirmed secondary hits to exclude false positives that inhibit coupled fluorescence assay, but not PfDPCK. After exclusion of false positives, all tertiary hits were tested against human HsCOASY in the counterassay to exclude compounds that inhibit both PfDPCK and HsCOASY. The compounds showing >10% inhibition against HsCOASY were excluded from further studies. The IC50 values of the hit compounds were determined under the same assay conditions described as above for the primary screening, with the serially diluted compounds at 20, 10, 2.5, 0.5, and 0.1 μM at final concentrations. The assay was conducted in quadruplicate for each dilution, and IC50 values were calculated using GraphPad Software (San Diego, California USA).
Estimation of IC50 values against P. falciparum 3D7 erythrocytic-stage parasite growth.
Selected hit compounds were also tested against erythrocytic-stage parasites of P. falciparum drug-sensitive 3D7 and drug-resistant Dd2 strains (39). For the phenotypic assay, parasite cultures were synchronized with 5% (wt/vol) d-sorbitol as previously described (40). A synchronized culture containing ring-stage parasites of 0.3% parasitemia was initiated with a culture volume of 25 μL/well on a 384-well plate. A series of diluted compounds (at final concentrations of 50, 10, 2, 0.4, and 0.08 μM) were used to calculate IC50 values. Mefloquine at 50 μM and atovaquone at 20 μM were used as no-growth control (100% inhibition). After 72 h of incubation, parasite growth was measured by diaphorase-coupled lactate dehydrogenase (LDH) assay as previously described (41). Absorbance at 650 nm was measured using SpectraMax Paradigm multimode microplate reader (Molecular Devices, San Jose, CA, USA). The IC50 values were calculated as described above.
Cytotoxicity assay.
Cytotoxicity against human cells was evaluated using the human liver carcinoma HepG2 cell line. HepG2 cells were cultured in d-MED supplemented with 4 mM l-glutamine and 10% fetal bovine serum (FBS) in a 75-cm2 flask (Violamo) at 37°C. For the cytotoxicity assay, HepG2 cells in semiconfluent state were detached from the flask by incubating the cells with 5 mL d-MEM containing 0.25% trypsin-EDTA. After detachment, HepG2 cells were resuspended in d-MEM-supplemented l-glutamine and FBS, and cell viability was evaluated by incubating cells with 0.4% Trypan blue on a hemocytometer. An approximately 50-μL suspension containing 3,000 cells was dispensed into each well on a 384-well clear-bottom plate using a Multidrop Combi dispenser. Compounds dissolved in DMSO had been dispensed into wells to yield final concentrations of 100, 20, 4, 0.8, and 0.2 μM before cells were added. After cells were added to the plates, they were cultured at 37°C under 5% CO2. After 48 h cultivation, 5 μL of Cell Counting Kit-8 (Dojindo, Japan) was added, and the plates were further incubated for 2 h to assess cell growth and survival. The absorbance wavelengths of 450 nm were measured on the microplate reader, and the IC50 values were calculated as above.
Creation of protein structure models for docking simulation.
Since the crystal structure of PfDPCK is not available in the public database and our repeated attempts to make a crystal were unsuccessful, we in silico predicted the structure of PfDPCK and HsCOASY by AlphaFold2 (42, 43) (see Fig. S2A in the supplemental material). The predicted structure contains three potential membrane-bound helices in the N terminus (Fig. S2B and S2C). The first membrane-associated alpha helixes from the N terminus are predicted to be a part of signal peptide- and apicoplast-targeting sequence (the amino acid sequence MFLKFFLDKCILCFLAL−). The organisms that PfDPCK show the highest similarity to are DPCK from prokaryotes. The superposition of PfDPCK and DPCK from Mycobacterium paratuberculosis, Sulfolobus solfataricus, and Campylobacter jejuni is shown in Fig. S3. The predicted protein structures were obtained from AlphaFold Protein Structure Database (42), with the database entry IDs of Q8IL34 and Q13057 for PfDPCK and HsCOASY, respectively. The predicted structure of HsCOASY shows a closed conformation of the active site that has contact with the substrates, while the predicted structure of PfDPCK shows an open conformation without the substrate (Fig. S4A). To better align the backbone of PfDPCK with that of HsCOASY, linear morphing was applied using PyMOL (version 2.4.1) (Fig. S4B).
Prediction of the binding of PfDPCK and its inhibitor by docking simulation.
The PfDPCK and HsCOASY structures prepared above were used for docking simulation. The three-dimensional (3D) structure of compound A-15 (registered as CID 2852293) was obtained from PubChem https://pubchem.ncbi.nlm.nih.gov/compound/2852293. The docking simulation was performed using Molegro Virtual Docker (version 7.0.0) (44). We defined the search space as a sphere with a radius of 8 Å centered on the middle region of the binding sites of ATP and dephospho-CoA (Fig. S4C), considering the experimental results of the inhibition mode. Compound A-15 was docked with the default settings of the software with the following modifications: scoring function, PLANTS score (GRID); search algorithm, GPU screening with energy minimization.
ACKNOWLEDGMENTS
This work was supported in part by a grant for Science and Technology Research Partnership for Sustainable Development (SATREPS) from the Japan Agency for Medical Research and Development (AMED) and Japan International Cooperation Agency (JICA) (JP20jm0110022 to T.N.), a grant for Research on Emerging and Reemerging Infectious Diseases from AMED (JP20fk0108138 to T.N.), Grant-in-Aid for Scientific Research (B) (JP18H0265, JP21H02723 to T.N.), Grants-in-Aid for Challenging Research (Exploratory) (JP21K19372 to T.N) from the Japan Society for the Promotion of Science, Japan, and a grant for Drug Screening Platform and Hit-to-Lead Platform from Global Health Innovative Technology Fund (S2018-111, H2020-201). This research was (partially) supported by the Platform Project for Supporting Drug Discovery and Life Science Research from AMED under grant number JP20am0101086 (support number 0251).
We gratefully thank the Drug Discovery Initiative (DDI) at The University of Tokyo for providing the DDI library compounds.
We declare no conflicts of interest with the contents of this article.
A.N., the main contributor, designed and performed the experiments, analyzed the data, and wrote the manuscript. R.I. and Y.R. designed the experiment and performed the experiments. E.E.P., D.W., and T.A. performed the experiments for compounds screening and in silico study. K.Y., M.S., and Y.N. performed in silico study, docking, and molecular dynamic assay. T.S. and N.S. performed the in silico study. H.K., T.O., and J.D. identified, selected, and narrowed down malaria-specific DPCK inhibitors and analyzed the data. T.N. conceived the project, acquired funding, and wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.World Health Organization. 2021. World malaria report 2021. https://www.who.int/teams/global-malaria-programme/reports/world-malaria-report-2021.
- 2.Grimberg BT, Mehlotra RK. 2011. Expanding the antimalarial drug arsenal—now, but how? Pharmaceuticals (Basel) 4:681–712. 10.3390/ph4050681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fidock DA. 2010. Drug discovery: priming the antimalarial pipeline. Nature 465:297–298. 10.1038/465297a. [DOI] [PubMed] [Google Scholar]
- 4.Reader J, van der Watt ME, Taylor D, Le Manach C, Mittal N, Ottilie S, Theron A, Moyo P, Erlank E, Nardini L, Venter N, Lauterbach S, Bezuidenhout B, Horatscheck A, van Heerden A, Spillman NJ, Cowell AN, Connacher J, Opperman D, Orchard LM, Llinás M, Istvan ES, Goldberg DE, Boyle GA, Calvo D, Mancama D, Coetzer TL, Winzeler EA, Duffy J, Koekemoer LL, Basarab G, Chibale K, Birkholtz LM. 2021. Multistage and transmission-blocking targeted antimalarials discovered from the open-source MMV Pandemic Response Box. Nat Commun 12:269. 10.1038/s41467-020-20629-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Belete TM. 2020. Recent progress in the development of new antimalarial drugs with novel targets. Drug Des Devel Ther 14:3875–3889. 10.2147/DDDT.S265602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fletcher S, Lucantoni L, Sykes ML, Jones AJ, Holleran JP, Saliba KJ, Avery VM. 2016. Biological characterization of chemically diverse compounds targeting the Plasmodium falciparum coenzyme A synthesis pathway. Parasit Vectors 9:589. 10.1186/s13071-016-1860-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Genschel U, Powell C, Abell C, Smith A. 1999. The final step of pantothenate biosynthesis in higher plants: cloning and characterization of pantothenate synthetase from Lotus japonicus and Oryza sativum (rice). Biochem J 341:669–678. 10.1042/bj3410669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spry C, Kirk K, Saliba KJ. 2008. Coenzyme A biosynthesis: an antimicrobial drug target. FEMS Microbiol Rev 32:56–106. 10.1111/j.1574-6976.2007.00093.x. [DOI] [PubMed] [Google Scholar]
- 9.Begley TP, Kinsland C, Strauss E. 2001. The biosynthesis of coenzyme A in bacteria. Vitam Horm 61:157–171. 10.1016/s0083-6729(01)61005-7. [DOI] [PubMed] [Google Scholar]
- 10.Chakauya E, Coxon KM, Wei M, MacDonald MV, Barsby T, Abell C, Smith AG. 2008. Towards engineering increased pantothenate (vitamin B5) levels in plants. Plant Mol Biol 68:493–503. 10.1007/s11103-008-9386-5. [DOI] [PubMed] [Google Scholar]
- 11.Saliba KJ, Horner Ha, Kirk K. 1998. Transport and metabolism of the essential vitamin pantothenic acid in human erythrocytes infected with the malaria parasite Plasmodium falciparum. J Biol Chem 273:10190–10195. 10.1074/jbc.273.17.10190. [DOI] [PubMed] [Google Scholar]
- 12.Nurkanto A, Jeelani G, Santos HJ, Rahmawati Y, Mori M, Nakamura Y, Goto K, Saikawa Y, Annoura T, Tozawa Y, Sakura T, Inaoka DK, Shiomi K, Nozaki T. 2021. Characterization of Plasmodium falciparum pantothenate kinase and identification of its inhibitors from natural products. Front Cell Infect Microbiol 11:639065. 10.3389/fcimb.2021.639065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hart RJ, Abraham A, Aly ASI. 2017. Genetic characterization of coenzyme A biosynthesis reveals essential distinctive functions during malaria parasite development in blood and mosquito. Front Cell Infect Microbiol 7:260. 10.3389/fcimb.2017.00260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leonardi R, Zhang YM, Rock CO, Jackowski S. 2005. Coenzyme A: back in action. Prog Lipid Res 44:125–153. 10.1016/j.plipres.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 15.Nurkanto A, Jeelani G, Yamamoto T, Hishiki T, Naito Y, Suematsu M, Hashimoto T, Nozaki T. 2018. Biochemical, metabolomic, and genetic analyses of dephospho coenzyme a kinase involved in coenzyme a biosynthesis in the human enteric parasite Entamoeba histolytica. Front Microbiol 9:2902. 10.3389/fmicb.2018.02902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Swift RP, Rajaram K, Liu HB, Prigge ST. 2021. Dephospho-CoA kinase, a nuclear-encoded apicoplast protein, remains active and essential after Plasmodium falciparum apicoplast disruption. EMBO J 40:e107247. 10.15252/embj.2020107247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Imamura RM, Kumagai K, Nakano H, Okabe T, Nagano T, Kojima H. 2019. Inexpensive high-throughput screening of kinase inhibitors using one-step enzyme-coupled fluorescence assay for ADP detection. SLAS Discov 24:284–294. 10.1177/2472555218810139. [DOI] [PubMed] [Google Scholar]
- 18.Zhang JH, Chung TDY, Oldenburg KR. 1999. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen 4:67–73. 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 19.O’Brien PJ, Irwin W, Diaz D, Howard-Cofield E, Krejsa CM, Slaughter MR, Gao B, Kaludercic N, Angeline A, Bernardi P, Brain P, Hougham C. 2006. High concordance of drug-induced human hepatotoxicity with in vitro cytotoxicity measured in a novel cell-based model using high content screening. Arch Toxicol 80:580–604. 10.1007/s00204-006-0091-3. [DOI] [PubMed] [Google Scholar]
- 20.D Segall M. 2012. Multi-parameter optimization: identifying high quality compounds with a balance of properties. Curr Drug Metab 18:1292–1310. 10.2174/138920012799362864. [DOI] [PubMed] [Google Scholar]
- 21.Brenk R, Schipani A, James D, Krasowski A, Gilbert IH, Frearson J, Wyatt PG. 2008. Lessons learnt from assembling screening libraries for drug discovery for neglected diseases. ChemMedChem 3:435–444. 10.1002/cmdc.200700139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baell JB, Holloway GA. 2010. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem 53:2719–2740. 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 23.Gómez-Díaz E, Yerbanga RS, Lefèvre T, Cohuet A, Rowley MJ, Ouedraogo JB, Corces VG. 2017. Epigenetic regulation of Plasmodium falciparum clonally variant gene expression during development in Anopheles gambiae. Sci Rep 7:40655. 10.1038/srep40655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cubi R, Vembar SS, Biton A, Franetich JF, Bordessoulles M, Sossau D, Zanghi G, Bosson-Vanga H, Benard M, Moreno A, Dereuddre-Bosquet N, Le Grand R, Scherf A, Mazier D. 2017. Laser capture microdissection enables transcriptomic analysis of dividing and quiescent liver stages of Plasmodium relapsing species. Cell Microbiol 19:e12735. 10.1111/cmi.12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ralph SA, van Dooren GG, Waller RF, Crawford MJ, Fraunholz MJ, Foth BJ, Tonkin CJ, Roos DS, McFadden GI. 2004. Tropical infectious diseases: metabolic maps and functions of the Plasmodium falciparum apicoplast. Nat Rev Microbiol 2:203–216. 10.1038/nrmicro843. [DOI] [PubMed] [Google Scholar]
- 26.Penzo M, de las Heras-Dueña L, Mata-Cantero L, Diaz-Hernandez B, Vazquez-Muñiz MJ, Ghidelli-Disse S, Drewes G, Fernandez-Alvaro E, Baker DA. 2019. High-throughput screening of the Plasmodium falciparum cGMP-dependent protein kinase identified a thiazole scaffold which kills erythrocytic and sexual stage parasites. Sci Rep 9:7005. 10.1038/s41598-019-42801-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Avery VM, Bashyam S, Burrows JN, Duffy S, Papadatos G, Puthukkuti S, Sambandan Y, Singh S, Spangenberg T, Waterson D, Willis P. 2014. Screening and hit evaluation of a chemical library against blood-stage Plasmodium falciparum. Malar J 13:190. 10.1186/1475-2875-13-190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meister S, Plouffe DM, Kuhen KL, Bonamy GMC, Wu T, Barnes SW, Bopp SE, Borboa R, Bright AT, Che J, Cohen S, Dharia NV, Gagaring K, Gettayacamin M, Gordon P, Groessl T, Kato N, Lee MCS, McNamara CW, Fidock DA, Nagle A, Nam TG, Richmond W, Roland J, Rottmann M, Zhou B, Froissard P, Glynne RJ, Mazier D, Sattabongkot J, Schultz PG, Tuntland T, Walker JR, Zhou Y, Chatterjee A, Diagana TT, Winzeler EA. 2011. Imaging of Plasmodium liver stages to drive next-generation antimalarial drug discovery. Science 334:1372–1377. 10.1126/science.1211936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gamo FJ, Sanz LM, Vidal J, De Cozar C, Alvarez E, Lavandera JL, Vanderwall DE, Green DVS, Kumar V, Hasan S, Brown JR, Peishoff CE, Cardon LR, Garcia-Bustos JF. 2010. Thousands of chemical starting points for antimalarial lead identification. Nature 465:305–310. 10.1038/nature09107. [DOI] [PubMed] [Google Scholar]
- 30.Thu AM, Phyo AP, Landier J, Parker DM, Nosten FH. 2017. Combating multidrug-resistant Plasmodium falciparum malaria. FEBS J 284:2569–2578. 10.1111/febs.14127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paquet T, Le Manach C, Cabrera DG, Younis Y, Henrich PP, Abraham TS, Lee MCS, Basak R, Ghidelli-Disse S, Lafuente-Monasterio MJ, Bantscheff M, Ruecker A, Blagborough AM, Zakutansky SE, Zeeman AM, White KL, Shackleford DM, Mannila J, Morizzi J, Scheurer C, Angulo-Barturen I, Santosmartínez M, Ferrer S, Sanz LM, Gamo FJ, Reader J, Botha M, Dechering KJ, Sauerwein RW, Tungtaeng A, Vanachayangkul P, Lim CS, Burrows J, Witty MJ, Marsh KC, Bodenreider C, Rochford R, Solapure SM, Jiménez-Díaz MB, Wittlin S, Charman SA, Donini C, Campo B, Birkholtz LM, Khanson K, Drewes G, Kocken CM, Delves MJ, Leroy D, Fidock DA, et al. 2017. Antimalarial efficacy of MMV390048, an inhibitor of Plasmodium phosphatidylinositol 4-kinase. Sci Transl Med 9:eaad9735. 10.1126/scitranslmed.aad9735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bouwman SA, Zoleko-Manego R, Renner KC, Schmitt EK, Mombo-Ngoma G, Grobusch MP. 2020. The early preclinical and clinical development of cipargamin (KAE609), a novel antimalarial compound. Travel Med Infect Dis 36:101765. 10.1016/j.tmaid.2020.101765. [DOI] [PubMed] [Google Scholar]
- 33.McCarthy JS, Yalkinoglu Ö, Odedra A, Webster R, Oeuvray C, Tappert A, Bezuidenhout D, Giddins MJ, Dhingra SK, Fidock DA, Marquart L, Webb L, Yin X, Khandelwal A, Bagchus WM. 2021. Safety, pharmacokinetics, and antimalarial activity of the novel plasmodium eukaryotic translation elongation factor 2 inhibitor M5717: a first-in-human, randomised, placebo-controlled, double-blind, single ascending dose study and volunteer infection study. Lancet Infect Dis 21:1713–1724. 10.1016/S1473-3099(21)00252-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gouy M, Guindon S, Gascuel O. 2010. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol 27:221–224. 10.1093/molbev/msp259. [DOI] [PubMed] [Google Scholar]
- 36.Nguyen LT, Schmidt HA, Von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 32:268–274. 10.1093/molbev/msu300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690. 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 38.Chai SC, Goktug AN, Chen T. 2015. Assay validation in high throughput screening-from concept to application. Drug Discov Dev Mol Med 10.5772/59765. [DOI] [Google Scholar]
- 39.Duffey M, Blasco B, Burrows JN, Wells TNC, Fidock DA, Leroy D. 2021. Assessing risks of Plasmodium falciparum resistance to select next-generation antimalarials. Trends Parasitol 37:709–721. 10.1016/j.pt.2021.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lambros C, Vanderberg JP. 1979. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J Parasitol 65:418–420. 10.2307/3280287. [DOI] [PubMed] [Google Scholar]
- 41.Hartuti ED, Inaoka DK, Komatsuya K, Miyazaki Y, Miller RJ, Xinying W, Sadikin M, Prabandari EE, Waluyo D, Kuroda M, Amalia E, Matsuo Y, Nugroho NB, Saimoto H, Pramisandi A, Watanabe YI, Mori M, Shiomi K, Balogun EO, Shiba T, Harada S, Nozaki T, Kita K. 2018. Biochemical studies of membrane bound Plasmodium falciparum mitochondrial L-malate:quinone oxidoreductase, a potential drug target. Biochim Biophys Acta Bioenerg 1859:191–200. 10.1016/j.bbabio.2017.12.004. [DOI] [PubMed] [Google Scholar]
- 42.Tunyasuvunakool K, Adler J, Wu Z, Green T, Zielinski M, Žídek A, Bridgland A, Cowie A, Meyer C, Laydon A, Velankar S, Kleywegt GJ, Bateman A, Evans R, Pritzel A, Figurnov M, Ronneberger O, Bates R, Kohl SAA, Potapenko A, Ballard AJ, Romera-Paredes B, Nikolov S, Jain R, Clancy E, Reiman D, Petersen S, Senior AW, Kavukcuoglu K, Birney E, Kohli P, Jumper J, Hassabis D. 2021. Highly accurate protein structure prediction for the human proteome. Nature 596:590–596. 10.1038/s41586-021-03828-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bouatta N, Sorger P, AlQuraishi M. 2021. Protein structure prediction by AlphaFold2: are attention and symmetries all you need? Acta Crystallogr D Struct Biol 77:982–991. 10.1107/S2059798321007531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thomsen R, Christensen MH. 2006. MolDock: a new technique for high-accuracy molecular docking. J Med Chem 49:3315–3321. 10.1021/jm051197e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material. Download aac.00420-22-s0001.pdf, PDF file, 2.5 MB (2.5MB, pdf)