

ABSTRACT
Imprinted genes – critical for growth, metabolism, and neuronal function – are expressed from one parental allele. Parent-of-origin-dependent CpG methylation regulates this expression at imprint control regions (ICRs). Since ICRs are established before tissue specification, these methylation marks are similar across cell types. Thus, they are attractive for investigating the developmental origins of adult diseases using accessible tissues, but remain unknown. We determined genome-wide candidate ICRs in humans by performing whole-genome bisulphite sequencing (WGBS) of DNA derived from the three germ layers and from gametes. We identified 1,488 hemi-methylated candidate ICRs, including 19 of 25 previously characterized ICRs (https://humanicr.org/). Gamete methylation approached 0% or 100% in 332 ICRs (178 paternally and 154 maternally methylated), supporting parent-of-origin-specific methylation, and 65% were in well-described CTCF-binding or DNaseI hypersensitive regions. This draft of the human imprintome will allow for the systematic determination of the role of early-acquired imprinting dysregulation in the pathogenesis of human diseases and developmental and behavioural disorders.
KEYWORDS: Epigenetics, genomic imprinting, foetal origins, whole genome, methylation, imprint control regions
Introduction
Data from model systems and humans demonstrate that environmentally induced epigenetic modifications occurring early in development can cause long-term gene expression changes in important mechanistic pathways involved in disease pathogenesis. Affected diseases include neurological disorders [1,2], cardio- and cerebrovascular diseases [3], and cancers [4–6], and their major risk factors, such as obesity and the attendant dysfunction in metabolism, nutrient acquisition, fat deposition, appetite, and satiety [7,8].
Covalent DNA methylation of cytosines in CpG dinucleotide sites is the most studied epigenetic modification and is hypothesized to link environmental exposures to these diseases. Nevertheless, data generated from case-control or cross-sectional studies, the most cost-efficient epidemiologic study designs, have been difficult to interpret. This is because methylation marks measured in easily accessible peripheral cell types from otherwise healthy individuals do not always reflect methylation in inaccessible cell types, tissues, and organs involved in the formation or progression of a chronic disease. Moreover, DNA methylation at susceptible loci can adaptively change throughout life in response to environmental exposures or disease. Indeed, methylation levels can also naturally diverge with normal cell differentiation, ageing, and environmental influences [9–12].
Known exceptions are CpG methylation marks that are stochastically established before specification that control metastable epiallele expression [13] and imprinting control regions (ICRs) that regulate the monoallelic expression of imprinted genes [6,14]. CpG methylation of metastable epialleles and ICRs is established before gastrulation and is mitotically heritable. Thus, these epigenetic marks are normally similar across tissues and cell types throughout an individual’s life. Unlike metastable epialleles however, ICRs are defined by parent-of-origin specific methylation marks that are important gene dosage regulators based on the allele’s parental origin. Consequently, in contrast to epigenetic marks controlling metastable epiallele expression, methylation marks regulating imprinted genes are similar across individuals [15,16]. Importantly, changes in the methylation patterns in ICRs are implicated in adult-onset diseases suspected to have foetal origins, including neurological disorders, cancers, and metabolic diseases stemming from abnormal growth and nutrient acquisition disorders [17,18].
Together, these features make ICRs attractive targets for dissecting disease aetiology, particularly since imprinted genes comprise an estimated 1–6% of the human genome, are over-selected for growth regulators, and are critical in early embryonic development [3,19]. The stability of these methylation marks with age also makes them long-term ‘records’ of early exposures that are difficult to obtain through questionnaires or other exposure assessment assays [6]. Yet, despite their biological and clinical relevance, only 25 of the ICRs regulating the 100–150 identified human imprinted genes, and of the 300–1,000 genes predicted to be imprinted [19], are currently known [14].
Leveraging existing specimens, recent advances in genome sequencing, and computational capabilities, we sought to comprehensively characterize human ICRs using DNA methyl-sequencing of tissues representing the three germ layers as well as the gametes, to create a catalogue of the set of imprint regulatory DNA methylation marks in humans, the ‘human imprintome’ [14]. This characterization of human ICRs should enable a more detailed understanding of the epigenetic basis of numerous pathophysiologies with foetal origins, and with that, advance the ability to diagnose, prevent, and treat a number of developmental disorders and diseases.
Materials and methods
Materials and subjects
Sperm from three adults and tissues from twelve 65 to 95-day-old embryos of both sexes (confirmed by sex-linked marker genotyping) and of African and European descent were selected for bisulphite DNA sequencing. These tissues were obtained from the National Institutes of Health funded Laboratory of Human Embryology at the University of Washington, Seattle, WA; they were snap frozen to preserve DNA/RNA integrity (NCSU Institutional Review Board #3565). Embryonic tissues were used for identifying ICRs because the gametic and somatic imprint marks are intact, and because monoallelic gene expression of imprinted genes occurs primarily during embryonic development [20–23]. Sperm DNA was from the TIEGER study at Duke [23].
Whole-genome bisulphite sequencing (WGBS)
Libraries for NextSeq sequencing using HiSeq 2500 were prepared from bisulphite converted DNA derived from tissues representing the three germ layers using previously described methods [21]. Thirty (three from sperm obtained as previously described [23,24] and 27 from somatic tissues) of the 36 samples passed quality control standards for sequencing by Illumina NextSeq with 12–15X coverage. The sequenced somatic tissue libraries were 8 kidney (mesoderm), 8 liver (endoderm), and 11 brain (ectoderm). Libraries were index-tagged for separating reads after multiplex sequencing and pooled into groups of nine, with each group split for sequencing into three separate lanes. Splitting samples across lanes ensured that no single sample was disproportionally affected by technical variability specific to an individual sequencing lane (e.g., low read numbers or low read quality). If a problem persisted, samples exhibiting consistently low quality across lanes were rerun or removed from analysis. These sequence data were supplemented with publicly available oocyte sequence data (JGAS00000000006) [25] and part of control sperm data from PRJNA754049 (control males in this study were otherwise healthy individuals who did not use cannabis) [26]. Details are in the Supplementary Materials and Methods.
Bioinformatic approaches to identify ICRs
Samples were separated by index sequences and aligned to a reference in silico bisulphite-converted genome. Reads without unique alignment to the reference sequence due to either repetitive sequence or loss of information because of cytosine conversion were eliminated, as were duplicate reads, indicative of clonally amplified original random DNA fragments. From these reads, methylation fractions and read counts were calculated for all CpG sites in the genome. We developed a candidate ICR identification pipeline (putICR) application using a Ruffus framework in Python. The workflow is described in detail in Supplementary Materials and Methods and outlined in Supplementary Figure S7.
This application was designed to scan the genome and identify regions of allelic differential methylation based on four criteria that define genomic imprinting: 1) ≥5 consecutive CpG sites, consistent with known cis-acting imprinted control regions [14]; 2) methylation levels of 50% ±15%, supportive of monoallelic methylation (i.e., approximately 100% methylation on one parental allele and 0% on the other); 3) similarity of methylation levels in tissues from the three germ layers (i.e., brain, liver, and kidney), as expected for methylation marks established before tissue specification; and 4) similarity of methylation across individuals, indicative of sequence regions playing critical roles in regulating imprinted gene dosage, which should not generally vary by sex, ethnicity, developmental age, or from person-to-person. Based on the methylation levels, we developed an online tool, the imprintome browser, which is linked to standard genome browsers to visualize the methylation level for each CG site in the 1,488 candidate ICR regions (https://humanicr.org/).
To assess the reproducibility of the putICR pipeline, the raw sequence data and the same four criteria were provided to an independent bioinformatics group (Sciome, Inc, Research Triangle Park, NC) (Supplementary Materials and Methods). Additionally, fully methylated or unmethylated regions from gametic sequences were compared, as these are the original inherited parent-of-origin specific regions.
We used pyrosequencing (sequencing primers in Supplementary Table S3) to determine if methylation patterns at previously identified ICRs in the three germ layers in embryos were similar to those obtained from DNA from accessible adult tissues frequently used in epidemiologic studies, such as peripheral blood components and human umbilical vein endothelial cells (HUVECs) (Figure 3).
Figure 3.
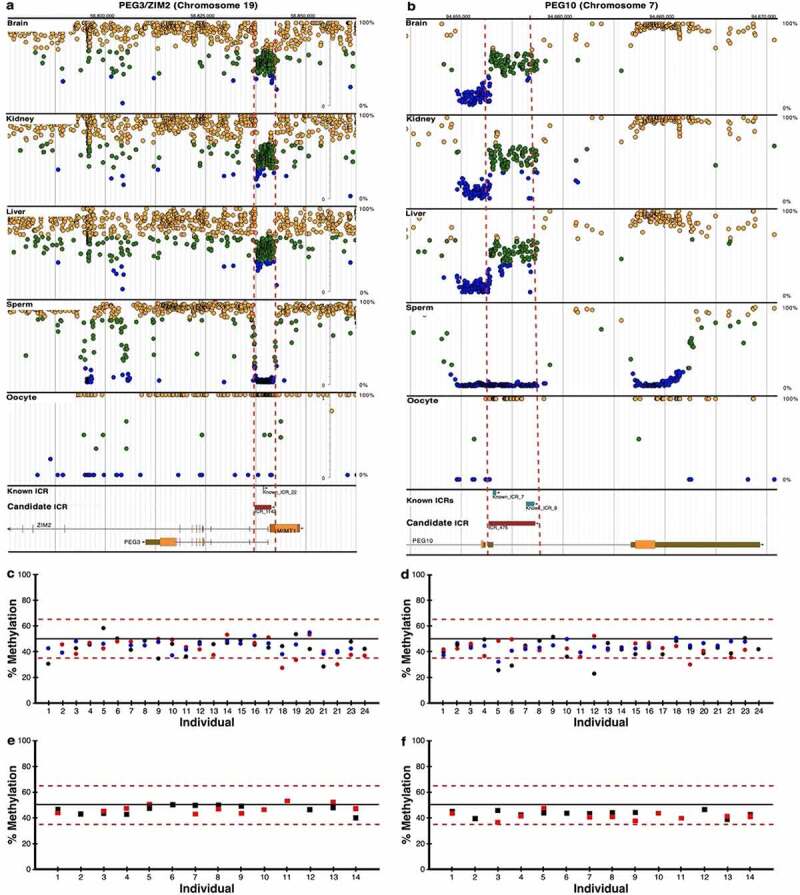
Pyrosequencing results for previously known ICRs. DNA methylation profiles determined by whole genome bisulphite sequencing (WGBS) are shown for imprinted genes (a) PEG3/ZIM2 (ICR_1142) and (b) PEG10 (ICR_475) with previously known (blue boxes) and candidate ICRs (red boxes); visualize at https://humanicr.org/. DNA methylation levels determined by WGBS in sperm and oocytes are also provided. These sequence data were supplemented with publicly available gametic sequence (accession number JGAS00000000006) and part of control sperm data from PRJNA754049 [25,26]. Dots indicate hemi-methylation (green), hypomethylation (blue) and hypermethylation (yellow) based on (WGBS). The vertical dashed red lines delineate the candidate ICR regions. Confirmation of 50 ± 15% DNA methylation of the ICRs determined by WGBS is shown for (c) PEG3/ZIM2 and (d) PEG10 using pyrosequencing of kidney (black circles), liver (red circles), and brain (blue circles) tissues from 24 embryos. Confirmation of 50 ± 15% DNA methylation determined by WGBS is shown for (e) PEG3/ZIM2 and (f) PEG10 using pyrosequencing for CD14 monocytes (red squares) and HUVECs (black squares) from 14 adult individuals.
Results
WGBS library preparation
WGBS libraries were prepared from three human tissues – brain (ectoderm), kidney (mesoderm), and liver (endoderm) – representing the three germ layers from 12 embryos (6 male, 6 female), and three sperm samples, resulting in 39 total libraries. Twenty-seven of the tissue libraries (i.e., 8 kidney (mesoderm), 8 liver (endoderm), and 11 brain (ectoderm)), and the three sperm libraries passed quality checks for Illumina HiSeq2500 (Illumina, San Diego, California, USA), resulting in 30 total libraries sequenced (27 somatic and 3 gametic). Gametic sequence data were augmented with publicly available human oocyte sequence data (accession number JGAS00000000006 [25]) and control sperm sequence data from PRJNA754049 [26]. The average number of per-sample reads was 153 million (range 74–231 million), covering an average of 23.1 billion bases per sample (range 11.2–34.9 billion). Approximately 80% of reads uniquely aligned to the in silico bisulphite-converted human genome (hg38). Of the 29.2 million CpG sites in the human genome [27], an average of 26.6 million (91%, range 86–94%) were covered by aligned reads for the set of 30 samples.
Characteristics of candidate ICRs from tissues representing the three germ layers
Most of the sequences obtained from the brain, kidney, and liver (75%) had a sequence coverage of greater than 20X. The sequence coverage for oocytes (accession number JGAS00000000006) was lower due to the decreased availability of DNA from this source [25] while the sperm coverage, by incorporating data from Schrott et al. [26], was higher (Figure 1 (a)). Using the percent methylation fractions calculated for each CpG site, candidate ICRs were defined based on the following criteria: five or more consecutive CpG sites within a 300 bp region having methylation levels of approximately 50 ± 10–20% in tissues from all three germ layers. These criteria are consistent with an ICR being a genomic series of cis-acting CpG sites that are established in embryonic stem cells, resulting in one parental allele being near fully methylated (~100%) while the other is unmethylated (~0%) for ≥80% of the sites.
Figure 1.
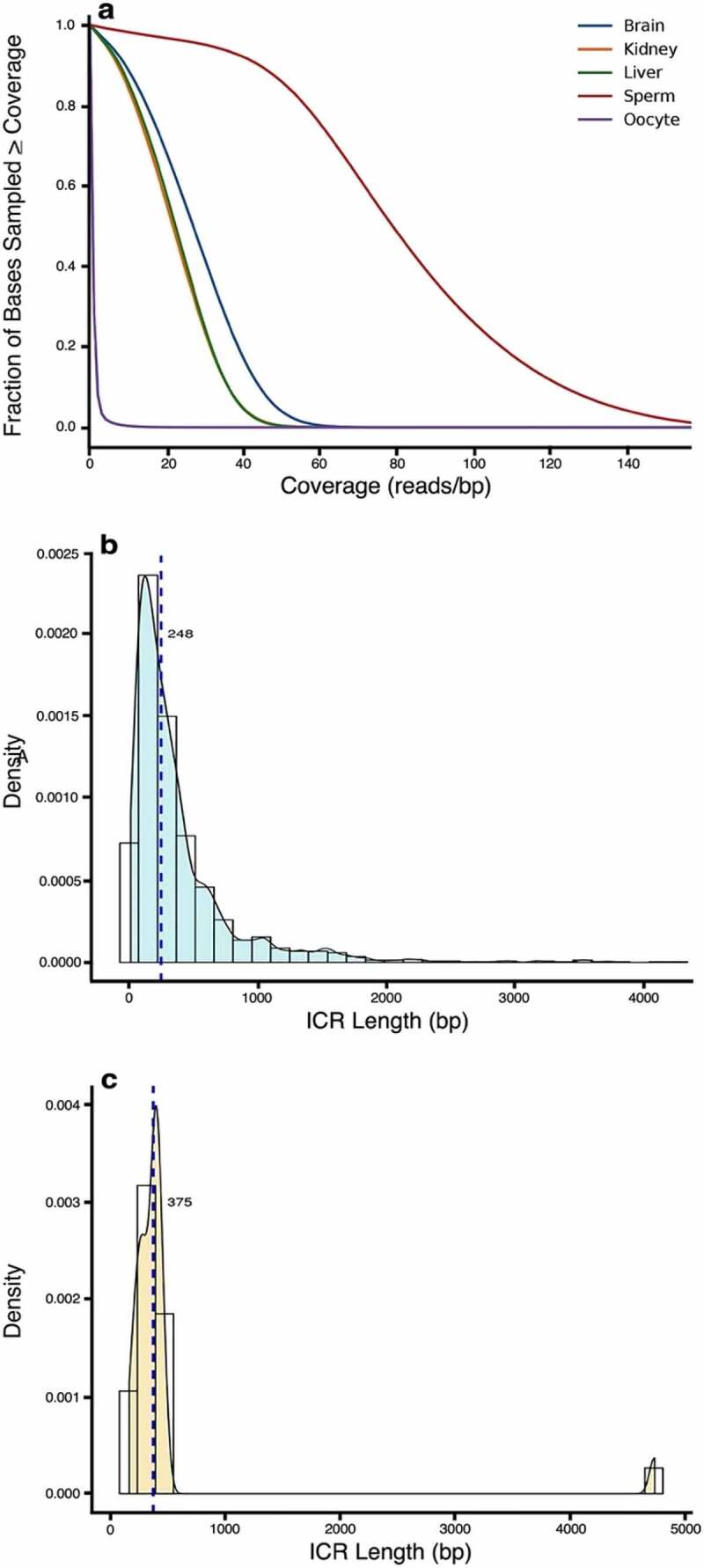
Detection of ICRs genome-wide. A bioinformatics selection algorithm, putICR, was used to identify methylation fractions using whole-genome bisulphite sequencing (WGBS) of DNA from kidney, liver, and brain embryonic tissues and gametes (i.e., sperm and oocyte). (a) Genome coverage was >20X for the somatic tissues in more than 75% of the fraction base sampled. The genome coverage was lower in the available oocyte gametic sequence data (accession number JGAS00000000006), but higher in sperm because our data were supplemented with control sperm data from PRJNA754049 [25,26]. (b) The size range of candidate imprint control regions (ICRs) averaged 248 bp. (c) This is similar to previously identified ICRs (Avg: 375 bp) [14].
Using the most relaxed criteria (50 ± 20%), we identified 7,559 candidate ICRs, including 21 of the 25 known ICRs [14]. A more stringent methylation criterion of 50 ± 15% decreased the number of candidate ICRs to 1,488 while still detecting 19 of 25 known ICRs (Table 1 and Supplementary Table S1). Further, restricting the window to 50% ±10% decreased the number of candidate ICRs to 127, including 15 of the known ICRs.
Table 1.
Candidate ICRs+ with gametic origin of methylation.
| ID | Genomic Coordinates | Parental Origin of Methylation | Nearest Transcript | Distance to Nearest Transcript |
|---|---|---|---|---|
| ICR_2^ | chr1:628959-630792 | P | MTND1P23|MTND2P28 | 0 |
| ICR_3^ | chr1:632183-632834 | P | MTCO1P12|MIR12136|MTCO2P12 | 0 |
| ICR_4^ | chr1:633381-634921 | P | MTCO2P12|MTATP8P1|MTATP6P1|MTCO3P12 | 0 |
| ICR_6* | chr1:1174554-1174597 | P | TTLL10 | 0 |
| ICR_10^ | chr1:2469095-2469433 | P | PLCH2 | 0 |
| ICR_12 | chr1:2661644-2661722 | M | TTC34 | 0 |
| ICR_15 | chr1:6461637-6461737 | M | TNFRSF25 | 0 |
| ICR_16^ | chr1:7199286-7199687 | M | CAMTA1 | 0 |
| ICR_17^ | chr1:8117511-8117827 | P | RPL7AP18 | 59,024 |
| ICR_20^ | chr1:10682902-10683413 | P | CASZ1 | 0 |
| ICR_21 | chr1:10808891-10809149 | P | CASZ1 | 12,241 |
| ICR_22^ | chr1:16164023-16164515 | P | EPHA2 | 7919 |
| ICR_25^ | chr1:22428873-22429209 | M | ZBTB40 | 0 |
| ICR_26 | chr1:24210504-24210603 | P | LINC02800 | 0 |
| ICR_32*^ | chr1:32471040-32471395 | M | ZBTB8B | 0 |
| ICR_35^ | chr1:35699589-35699951 | M | C1orf216 | 13,925 |
| ICR_36^ | chr1:36572484-36573080 | P | FTLP18 | 57,255 |
| ICR_39 | chr1:38210131-38210429 | P | LINC01343 | 0 |
| ICR_40^ | chr1:39559036-39559674 | M | PPIEL | 0 |
| ICR_41*^ | chr1:41423557-41423666 | P | FOXO6 | 39,967 |
| ICR_43^ | chr1:47963613-47963993 | P | TRABD2B | 0 |
| ICR_44^ | chr1:53145868-53146595 | P | SLC1A7 | 3230 |
| ICR_46*^ | chr1:68046822-68047535 | M | DIRAS3 | 0 |
| ICR_47^# | chr1:68049858-68051097 | M | DIRAS3 | 0 |
| ICR_48*^ | chr1:68051239-68051861 | M | DIRAS3 | 0 |
| ICR_52^ | chr1:91171791-91172929 | P | HFM1 | 87,837 |
| ICR_62 | chr1:148011459-148011977 | P | PDZK1P1|LINC02804 | 0 |
| ICR_65 | chr1:156840564-156840978 | P | NTRK1|INSRR | 0 |
| ICR_73 | chr1:168056307-168056468 | M | DCAF6|GCSHP5 | 0 |
| ICR_80*^ | chr1:204829545-204831249 | P | NFASC | 0 |
| ICR_81^ | chr1:208044765-208045294 | P | PLXNA2 | 0 |
| ICR_83^ | chr1:226127234-226127411 | P | ACBD3 | 17,268 |
| ICR_100^ | chr1:234955629-234956306 | P | RN7SL668P | 51141 |
| ICR_101^ | chr1:236120016-236120293 | P | GPR137B | 22236 |
| ICR_107 | chr1:245688553-245688677 | M | KIF26B | 0 |
| ICR_116^ | chr2:28342638-28342806 | P | BABAM2 | 3737 |
| ICR_117 | chr2:31581451-31581676 | M | SRD5A2 | 0 |
| ICR_121^ | chr2:44885640-44886022 | P | LINC01833 | 35,058 |
| ICR_123^ | chr2:49229257-49229842 | P | FSHR | 74730 |
| ICR_125 | chr2:54289850-54290281 | M | ACYP2 | 0 |
| ICR_133^ | chr2:95078802-95079392 | P | MRPS5 | 5977 |
| ICR_144 | chr2:120526146-120526533 | P | LINC01101 | 59,797 |
| ICR_158 | chr2:161340716-161341095 | M | PSMD14|MXRA7P1 | 0 |
| ICR_159^ | chr2:162243085-162244421 | P | FAP | 0 |
| ICR_161^ | chr2:173217299-173217391 | M | MAP3K20 | 0 |
| ICR_167^ | chr2:206257411-206257639 | P | HMGN1P6 | 1989 |
| ICR_168 | chr2:206257801-206258345 | P | HMGN1P6 | 1283 |
| ICR_169* | chr2:206258596-206260336 | P | HMGN1P6|RN7SKP260 | 0 |
| ICR_170* | chr2:206260650-206261209 | P | RN7SKP260 | 436 |
| ICR_177 | chr2:218890039-218890255 | P | WNT10A | 0 |
| ICR_182^ | chr2:234168141-234168738 | P | SPP2 | 91,007 |
| ICR_184*^ | chr2:236642778-236643076 | P | ACKR3 | 60,420 |
| ICR_187*^ | chr2:241901656-241902337 | M | LINC01237 | 0 |
| ICR_188 | chr2:241902453-241902725 | M | LINC01237 | 0 |
| ICR_191 | chr3:147595-148344 | P | CHL1 | 48,244 |
| ICR_194^ | chr3:14366359-14366571 | P | LINC01267 | 13,791 |
| ICR_198 | chr3:28200466-28200710 | M | CMC1 | 40,883 |
| ICR_199 | chr3:30893926-30894319 | M | GADL1 | 0 |
| ICR_202 | chr3:44580918-44580941 | P | ZKSCAN7|MPRIPP1 | 0 |
| ICR_207 | chr3:50481168-50481455 | P | CACNA2D2 | 0 |
| ICR_209 | chr3:72928978-72929098 | M | GXYLT2|FTH1P23 | 0 |
| ICR_227*^ | chr3:147297676-147298367 | P | RPL21P71 | 20,389 |
| ICR_228*^ | chr3:150865529-150865545 | M | MINDY4B | 4831 |
| ICR_229^ | chr3:156120781-156120929 | M | KCNAB1 | 0 |
| ICR_230^ | chr3:184563668-184564356 | P | EPHB3 | 0 |
| ICR_239 | chr4:1053635-1054116 | P | RNF212 | 2132 |
| ICR_243*^ | chr4:2463493-2463856 | M | CFAP99 | 530 |
| ICR_244 | chr4:3702565-3703061 | P | LINC02171 | 24,710 |
| ICR_246^ | chr4:3771074-3771481 | P | ADRA2C | 2548 |
| ICR_249 | chr4:4863227-4864029 | M | MSX1 | 0 |
| ICR_250^ | chr4:6105593-6105924 | M | JAKMIP1 | 0 |
| ICR_251 | chr4:8581258-8581331 | M | GPR78 | 0 |
| ICR_254^ | chr4:39406796-39407320 | P | KLB | 0 |
| ICR_255 | chr4:41876102-41876861 | P | LINC00682 | 2660 |
| ICR_263 | chr4:49270757-49270848 | M | MTND3P22 | 23,842 |
| ICR_274 | chr4:82758740-82759068 | P | SCD5 | 0 |
| ICR_275^ | chr4:88697244-88698085 | M | HERC3|NAP1L5 | 0 |
| ICR_278 | chr4:121932565-121932847 | M | TRPC3 | 0 |
| ICR_281 | chr4:152009424-152009915 | P | RNA5SP169 | 37,811 |
| ICR_282 | chr4:154781727-154782004 | M | RBM46 | 0 |
| ICR_284^ | chr4:173608770-173608942 | P | MORF4 | 6794 |
| ICR_295 | chr4:190089486-190089578 | M | DUX4L3 | 497 |
| ICR_297^ | chr5:346459-346789 | M | AHRR | 0 |
| ICR_300 | chr5:580390-580767 | P | CEP72 | 31,573 |
| ICR_319^ | chr5:80650716-80650931 | P | DHFR|MTRNR2L2 | 0 |
| ICR_323 | chr5:101783627-101784015 | M | OR7H2P | 31868 |
| ICR_324 | chr5:110894251-110894443 | M | BCLAF1P1 | 51,830 |
| ICR_325^ | chr5:134927018-134927085 | P | PCBD2|MTND4P12 | 0 |
| ICR_326*^ | chr5:136079156-136079563 | M | TGFBI | 15338 |
| ICR_332 | chr5:140829825-140830061 | M | PCDHA1-6 | 0 |
| ICR_340 | chr5:141345713-141345776 | M | PCDHGA1-3 | 0 |
| ICR_349 | chr5:158737352-158737526 | M | EBF1 | 0 |
| ICR_352 | chr5:171319025-171319892 | P | TLX3 | 6886 |
| ICR_353 | chr5:172602845-172603197 | P | NEURL1B | 38066 |
| ICR_358 | chr5:176785238-176785536 | P | UNC5A | 25023 |
| ICR_360^ | chr5:179167525-179167654 | M | ADAMTS2 | 0 |
| ICR_366*^# | chr6:3848794-3850307 | M | FAM50B | 0 |
| ICR_376 | chr6:27827550-27827724 | M | H4C11 | 3070 |
| ICR_380 | chr6:28742326-28742467 | M | RPSAP2 | 9352 |
| ICR_385^ | chr6:30781799-30782146 | P | HCG20 | 0 |
| ICR_393^ | chr6:39367315-39368095 | P | KIF6 | 0 |
| ICR_394^ | chr6:41627093-41627387 | P | MDFI | 9631 |
| ICR_404^# | chr6:144006941-144008825 | M | PLAGL1|HYMAI | 0 |
| ICR_407 | chr6:149986298-149986661 | P | BTF3P10 | 7609 |
| ICR_409 | chr6:160005401-160005610 | M | IGF2R|AIRN | 0 |
| ICR_410^ | chr6:160006184-160006584 | M | IGF2R|AIRN | 0 |
| ICR_414^ | chr6:167341434-167342866 | P | TTLL2 | 0 |
| ICR_416^ | chr6:167667943-167668201 | P | LINC02538 | 0 |
| ICR_418* | chr6:168417326-168417653 | P | SMOC2 | 23,500 |
| ICR_420*^ | chr6:169421049-169421128 | P | WDR27 | 5292 |
| ICR_426^ | chr7:343522-343641 | P | FOXL3 | 52,034 |
| ICR_427^ | chr7:360518-360693 | P | FOXL3 | 69,030 |
| ICR_432 | chr7:1663342-1663514 | M | ELFN1 | 2561 |
| ICR_436*^ | chr7:3089399-3089604 | P | CARD11 | 45,532 |
| ICR_439*^ | chr7:5144439-5144757 | M | ZNF890P | 0 |
| ICR_443^ | chr7:23490490-23491453 | M | RPS2P32 | 0 |
| ICR_450 | chr7:39834493-39834768 | M | SSBP3P1 | 0 |
| ICR_451^ | chr7:41990814-41991507 | P | GLI3 | 0 |
| ICR_452^ | chr7:45564015-45564354 | P | ADCY1 | 9786 |
| ICR_453^ | chr7:47576623-47576932 | P | TNS3 | 0 |
| ICR_454*^# | chr7:50781638-50783354 | M | GRB10 | 0 |
| ICR_468^ | chr7:67772905-67773321 | P | MTCO3P41 | 144,556 |
| ICR_469 | chr7:69233760-69233973 | P | MTCO2P25 | 96,725 |
| ICR_474*^ | chr7:76499727-76499956 | M | DTX2 | 0 |
| ICR_475*^ | chr7:94656360-94658647 | M | PEG10 | 0 |
| ICR_481*^# | chr7:130490640-130494200 | M | MEST|MESTIT1 | 0 |
| ICR_487^ | chr7:150111840-150112171 | M | ACTR3C | 0 |
| ICR_489 | chr7:152910856-152911190 | P | ACTR3B | 0 |
| ICR_490*^ | chr7:154892532-154892702 | M | DPP6 | 0 |
| ICR_491 | chr7:155071148-155071376 | M | HTR5A | 0 |
| ICR_492 | chr7:155383638-155384080 | P | BLACE | 15705 |
| ICR_500 | chr7:159112595-159112828 | M | VIPR2 | 0 |
| ICR_503 | chr8:882678-883012 | M | DLGAP2 | 0 |
| ICR_505^ | chr8:1373020-1373561 | M | DLGAP2 | 0 |
| ICR_506*^ | chr8:1548800-1548981 | M | DLGAP2 | 0 |
| ICR_508^ | chr8:2727637-2727760 | M | CSMD1 | 207,593 |
| ICR_510 | chr8:8703090-8703190 | M | CLDN23 | 0 |
| ICR_515 | chr8:22118860-22119045 | P | HR | 0 |
| ICR_516 | chr8:22696910-22697886 | P | EGR3 | 3430 |
| ICR_518*^ | chr8:27472461-27472768 | P | CHRNA2 | 0 |
| ICR_522^ | chr8:37699369-37699633 | M | ZNF703 | 0 |
| ICR_523^ | chr8:37747687-37748265 | M | ERLIN2 | 0 |
| ICR_534*^ | chr8:60713942-60714366 | M | CHD7 | 0 |
| ICR_539*^ | chr8:94119578-94120523 | M | CDH17 | 6639 |
| ICR_541*^ | chr8:101277219-101278202 | P | LINC02844 | 14,243 |
| ICR_542*^ | chr8:102528555-102528786 | P | ODF1 | 22,803 |
| ICR_548*^ | chr8:140098048-140100981 | M | TRAPPC9|PEG13 | 0 |
| ICR_549 | chr8:140349279-140349564 | M | TRAPPC9 | 0 |
| ICR_550 | chr8:141723822-141724064 | P | MROH5 | 216,592 |
| ICR_553 | chr8:143399766-143399977 | P | RHPN1 | 15,545 |
| ICR_555^ | chr8:143728031-143728348 | M | FAM83H | 0 |
| ICR_561^ | chr9:21997429-22000964 | P | CDKN2B | 1939 |
| ICR_562*^ | chr9:35036051-35036291 | M | C9orf131 | 4804 |
| ICR_563 | chr9:37800197-37800386 | M | DCAF10 | 168 |
| ICR_564 | chr9:38487573-38487989 | M | TCEA1P3 | 8029 |
| ICR_570 | chr9:41267302-41267410 | M | PTGER4P1 | 12,049 |
| ICR_578 | chr9:62844696-62844981 | M | CDK2AP2P2 | 0 |
| ICR_598^ | chr9:84272060-84272363 | M | SLC28A3 | 760 |
| ICR_600 | chr9:87944657-87944766 | M | SPATA31C1 | 21,000 |
| ICR_601^ | chr9:89282235-89282503 | M | CKS2 | 28,692 |
| ICR_603^ | chr9:95313103-95313511 | M | FANCC | 0 |
| ICR_604*^ | chr9:96177087-96177730 | P | EIF4BP3 | 29,122 |
| ICR_609*^ | chr9:117642714-117642890 | M | RPL35AP22 | 42,368 |
| ICR_616^ | chr9:128415922-128416668 | P | CERCAM | 0 |
| ICR_617 | chr9:130327055-130327332 | P | HMCN2 | 0 |
| ICR_620^ | chr9:134626633-134626781 | P | COL5A1 | 15,008 |
| ICR_623 | chr9:135605044-135605311 | P | PAEPP1 | 13,151 |
| ICR_625^ | chr9:136656981-136657090 | P | HSPC324|EGFL7 | 0 |
| ICR_626 | chr9:137145779-137145957 | P | GRIN1 | 0 |
| ICR_627^ | chr9:137306698-137307023 | P | EXD3 | 0 |
| ICR_630*^ | chr10:789268-789465 | P | LARP4B | 17449 |
| ICR_633^ | chr10:5645451-5645631 | P | ASB13 | 0 |
| ICR_640^ | chr10:18261294-18261435 | M | CACNB2 | 0 |
| ICR_641^ | chr10:24519852-24520359 | M | KIAA1217 | 0 |
| ICR_643*^ | chr10:27413523-27414477 | M | PTCHD3 | 0 |
| ICR_644^ | chr10:28326170-28327001 | P | ZNF101P1 | 12,276 |
| ICR_664^ | chr10:71266448-71266685 | P | UNC5B | 0 |
| ICR_665^ | chr10:71279400-71279528 | P | UNC5B | 0 |
| ICR_666^ | chr10:78356394-78357287 | P | LINC00595 | 76,181 |
| ICR_667^ | chr10:80136121-80136538 | P | PLAC9 | 0 |
| ICR_674^ | chr10:97974158-97974573 | M | CRTAC1 | 0 |
| ICR_675 | chr10:100669022-100669173 | P | PAX2 | 66,223 |
| ICR_679 | chr10:104275410-104275792 | M | GSTO2 | 0 |
| ICR_681*^ | chr10:119817943-119819030 | M | INPP5F | 0 |
| ICR_686*^ | chr10:126882020-126882047 | P | DOCK1 | 23,362 |
| ICR_687^ | chr10:127911380-127911781 | P | PTPRE | 0 |
| ICR_689^ | chr10:130301062-130301272 | M | LINC02646 | 0 |
| ICR_693 | chr10:133099321-133099567 | P | ADGRA1 | 0 |
| ICR_694 | chr10:133246176-133246423 | P | MIR202HG | 56 |
| ICR_695 | chr10:133569810-133570081 | M | SPRNP1 | 0 |
| ICR_703 | chr10:133740759-133740834 | M | DUX4L29 | 0 |
| ICR_708*^ | chr11:396655-396870 | P | PKP3 | 0 |
| ICR_709*^ | chr11:397385-397461 | P | PKP3 | 0 |
| ICR_711 | chr11:420604-420662 | M | ANO9 | 0 |
| ICR_716*^# | chr11:1997886-1999417 | P | MRPL23|H19 | 0 |
| ICR_717*^# | chr11:1999793-2000383 | P | MRPL23|H19 | 0 |
| ICR_718*^# | chr11:2000487-2001247 | P | MRPL23|H19 | 0 |
| ICR_719*^# | chr11:2001655-2003118 | P | MRPL23 | 0 |
| ICR_720*^# | chr11:2698157-2699485 | M | KCNQ1|KCNQ1OT1 | 0 |
| ICR_721*^# | chr11:2699814-2701210 | M | KCNQ1|KCNQ1OT1 | 0 |
| ICR_724*^ | chr11:7088963-7089048 | M | NLRP14|RBMXL2 | 0 |
| ICR_726^ | chr11:14259048-14259382 | M | SPON1 | 0 |
| ICR_730 | chr11:44328419-44329478 | P | ALX4 | 18,280 |
| ICR_737*^ | chr11:62371153-62371400 | M | ASRGL1 | 0 |
| ICR_751*^ | chr11:119942219-119942412 | P | LINC02744 | 45,533 |
| ICR_752*^ | chr11:119982974-119983610 | P | LINC02744 | 4335 |
| ICR_755^ | chr11:132792676-132793068 | M | OPCML | 0 |
| ICR_756 | chr11:133081840-133082221 | M | OPCML | 0 |
| ICR_757 | chr11:133222318-133222823 | P | OPCML | 0 |
| ICR_767 | chr12:31120459-31120874 | M | OVOS2 | 0 |
| ICR_779 | chr12:49543394-49543481 | P | KCNH3 | 0 |
| ICR_782 | chr12:52321080-52321266 | M | KRT83 | 0 |
| ICR_784*^ | chr12:55996580-55996638 | M | RAB5B|SUOX | 0 |
| ICR_789^ | chr12:92503489-92504056 | P | LINC02397 | 19,542 |
| ICR_795 | chr12:110501049-110501164 | M | VPS29 | 0 |
| ICR_799^ | chr12:124259431-124259804 | P | RFLNA | 29360 |
| ICR_802^ | chr12:124759333-124759536 | P | SCARB1 | 17,320 |
| ICR_807* | chr12:132436944-132437228 | P | MUC8 | 34,343 |
| ICR_809 | chr12:132603392-132603498 | M | LRCOL1 | 0 |
| ICR_814 | chr13:20142811-20142911 | M | GJA3 | 0 |
| ICR_818*^ | chr13:21242471-21243094 | P | LINC01046 | 7267 |
| ICR_825 | chr13:48317373-48317679 | M | RB1|PPP1R26P1 | 0 |
| ICR_826# | chr13:48317894-48321417 | M | RB1|PPP1R26P1 | 0 |
| ICR_827*^ | chr13:60267612-60268519 | M | LINC00434 | 0 |
| ICR_829^ | chr13:80654682-80655272 | M | PWWP2AP1 | 125 |
| ICR_835 | chr13:110813578-110813640 | M | LINC00567 | 494 |
| ICR_836 | chr13:111411595-111411773 | M | TEX29 | 67,347 |
| ICR_838^ | chr13:113729641-113730537 | P | GRK1 | 0 |
| ICR_853 | chr14:33799786-33799948 | M | NPAS3 | 0 |
| ICR_854 | chr14:35825391-35825732 | M | BRMS1L | 0 |
| ICR_859^ | chr14:74265554-74265792 | P | VSX2 | 2816 |
| ICR_866^ | chr14:96588928-96589205 | P | PAPOLA | 21812 |
| ICR_869^ | chr14:100714137-100714444 | P | DLK1 | 12,421 |
| ICR_871^ | chr14:100810929-100811087 | P | MIR2392 | 3404 |
| ICR_873*^# | chr14:100824556-100828242 | S | MEG3 | 0 |
| ICR_875*^ | chr14:104171069-104171501 | M | KIF26A | 0 |
| ICR_878^ | chr14:105060530-105060974 | P | GPR132 | 0 |
| ICR_879 | chr14:105628650-105629100 | P | IGH | 0 |
| ICR_887^# | chr15:23647239-23648622 | S | MAGEL2 | 0 |
| ICR_888# | chr15:23686523-23686574 | S | NDN | 0 |
| ICR_893*^ | chr15:24954592-24956828 | M | SNHG14|SNRPN|SNURF | 0 |
| ICR_898*^ | chr15:40769605-40770075 | P | DNAJC17|C15orf62 | 0 |
| ICR_907^ | chr15:66877196-66878321 | P | LINC02206 | 53,216 |
| ICR_909^ | chr15:70605464-70606129 | P | SALRNA3 | 9418 |
| ICR_912^ | chr15:95287058-95287471 | P | LINC01197 | 0 |
| ICR_913^ | chr15:98865375-98866277 | M | IGF1R | 0 |
| ICR_914^ | chr15:99476322-99476786 | P | LINC02244 | 73,869 |
| ICR_917 | chr16:159416-159850 | P | HBZP1 | 3217 |
| ICR_919 | chr16:801561-801834 | P | GNG13 | 827 |
| ICR_920 | chr16:817208-818000 | M | PRR25 | 3347 |
| ICR_921^ | chr16:1043980-1044213 | P | SSTR5 | 28,543 |
| ICR_926^ | chr16:3431819-3432009 | M | ZNF597 | 405 |
| ICR_928 | chr16:5490312-5490585 | M | RBFOX1 | 0 |
| ICR_930* | chr16:14990248-14990306 | M | PDXDC1 | 0 |
| ICR_946 | chr16:33808459-33808588 | P | ENPP7P13 | 24,184 |
| ICR_956* | chr16:35271909-35272955 | P | C2orf69P5 | 7713 |
| ICR_957 | chr16:35397156-35398017 | P | LINC01566 | 5443 |
| ICR_962*^ | chr16:46757358-46757691 | P | MYLK3 | 0 |
| ICR_964^ | chr16:57889831-57890669 | P | CNGB1 | 0 |
| ICR_982 | chr17:1961067-1961305 | P | RTN4RL1 | 0 |
| ICR_990 | chr17:16856437-16856709 | P | COTL1P1 | 316 |
| ICR_999 | chr17:21685888-21686090 | M | KCNJ18 | 6433 |
| ICR_1006 | chr17:34040220-34040257 | M | ASIC2 | 0 |
| ICR_1010^ | chr17:40014561-40014758 | P | CSF3 | 682 |
| ICR_1020^ | chr17:50639135-50639654 | P | ABCC3 | 0 |
| ICR_1024*^ | chr17:76079666-76079946 | P | ZACN | 0 |
| ICR_1025^ | chr17:76542972-76543727 | P | CYGB|PRCD | 0 |
| ICR_1027 | chr17:79517963-79518428 | P | RBFOX3 | 0 |
| ICR_1028^ | chr17:80191857-80192326 | P | CARD14 | 0 |
| ICR_1036^ | chr18:13379767-13380531 | P | LDLRAD4 | 0 |
| ICR_1038 | chr18:37258900-37259288 | P | CELF4 | 0 |
| ICR_1039 | chr18:47526284-47526407 | M | MIR4527HG | 0 |
| ICR_1048^ | chr18:79616752-79617334 | M | CTDP1 | 62,469 |
| ICR_1051 | chr18:79638645-79638833 | M | CTDP1 | 40,970 |
| ICR_1052^ | chr18:80147168-80147833 | M | ADNP2 | 6822 |
| ICR_1053^ | chr18:80147992-80149033 | M | ADNP2 | 7646 |
| ICR_1054*^ | chr19:386289-386791 | P | THEG | 9596 |
| ICR_1062 | chr19:1779885-1780102 | P | ONECUT3 | 0 |
| ICR_1068^ | chr19:3006289-3006627 | M | TLE2 | 0 |
| ICR_1079 | chr19:6509209-6509630 | P | TUBB4A | 6361 |
| ICR_1083 | chr19:7863802-7864121 | M | EVI5L | 0 |
| ICR_1085 | chr19:9912721-9912951 | M | OLFM2 | 0 |
| ICR_1086*^ | chr19:9953808-9954321 | P | COL5A3 | 5240 |
| ICR_1091*^ | chr19:14061704-14062308 | P | PALM3 | 0 |
| ICR_1093 | chr19:15777135-15778369 | P | CYP4F24P | 0 |
| ICR_1095 | chr19:15940417-15941089 | P | CYP4F11 | 5551 |
| ICR_1099* | chr19:19120808-19121015 | M | TMEM161A | 0 |
| ICR_1101*^ | chr19:19997705-19997967 | P | BNIP3P12 | 0 |
| ICR_1107^ | chr19:29662034-29662704 | P | PLEKHF1 | 2718 |
| ICR_1109^ | chr19:33298571-33298842 | P | CEBPA | 1092 |
| ICR_1120 | chr19:43406341-43406626 | M | TEX101 | 0 |
| ICR_1129 | chr19:50489534-50489611 | M | EMC10 | 0 |
| ICR_1135*^ | chr19:53537445-53538957 | M | ZNF331 | 0 |
| ICR_1136^ | chr19:53553906-53554999 | M | ZNF331 | 0 |
| ICR_1141^ | chr19:56478788-56479078 | M | ZNF667 | 723 |
| ICR_1142*^# | chr19:56837320-56841439 | M | ZIM2|PEG3|MIMT1 | 0 |
| ICR_1149^ | chr20:890832-892113 | P | ANGPT4 | 0 |
| ICR_1155^ | chr20:23164173-23164463 | P | RNA5SP478 | 3202 |
| ICR_1161 | chr20:29325493-29325868 | M | DUX4L33 | 444 |
| ICR_1190^ | chr20:31496438-31497008 | P | LINC00028 | 8864 |
| ICR_1191*^ | chr20:31547027-31548129 | M | HM13|MCTS2P | 0 |
| ICR_1192^# | chr20:37520202-37521842 | M | BLCAP|NNAT | 0 |
| ICR_1193^# | chr20:37522341-37522993 | M | BLCAP|NNAT | 0 |
| ICR_1194*^# | chr20:43513725-43515256 | M | L3MBTL1 | 0 |
| ICR_1205*^# | chr20:58839107-58842875 | M | GNAS | 0 |
| ICR_1206*^# | chr20:58850158-58852357 | M | GNAS | 0 |
| ICR_1207*^ | chr20:58853850-58856828 | M | GNAS | 0 |
| ICR_1209*^ | chr20:59220788-59221858 | M | ZNF831 | 0 |
| ICR_1211 | chr20:62581580-62581898 | P | RPL7P3 | 8195 |
| ICR_1213 | chr20:63003240-63003541 | P | BHLHE23 | 2386 |
| ICR_1216 | chr20:63474459-63475096 | P | KCNQ2 | 1804 |
| ICR_1315*^ | chr21:29155405-29155931 | M | MAP3K7CL | 0 |
| ICR_1321*^ | chr21:42063975-42064308 | P | UMODL1 | 0 |
| ICR_1327^ | chr21:45359946-45360583 | P | MTCO1P3 | 15,608 |
| ICR_1355*^ | chr22:18967907-18968273 | P | DGCR5 | 2225 |
| ICR_1356^ | chr22:19749148-19749533 | P | TBX1 | 7170 |
| ICR_1366 | chr22:31104622-31104758 | M | SMTN | 0 |
| ICR_1372 | chr22:39664168-39664319 | M | CACNA1I | 0 |
| ICR_1374^ | chr22:40040395-40041632 | P | FAM83F | 0 |
| ICR_1375^ | chr22:41681861-41682938 | M | SNU13 | 0 |
| ICR_1377*^ | chr22:42532792-42533280 | P | RRP7A | 12996 |
| ICR_1378 | chr22:42547749-42547909 | P | SERHL2 | 5941 |
| ICR_1384 | chr22:45877926-45878087 | P | ATXN10 | 32,619 |
| ICR_1385* | chr22:48238911-48239473 | P | MIR3201 | 34,891 |
| ICR_1394 | chrX:2325208-2325958 | M | DHRSX | 0 |
| ICR_1395^ | chrX:2962476-2962843 | P | ARSL | 0 |
| ICR_1398 | chrX:11138801-11139297 | M | ARHGAP6 | 0 |
| ICR_1399*^ | chrX:14244065-14244239 | M | UBE2E4P | 26 |
| ICR_1400* | chrX:17655055-17655175 | M | NHS | 0 |
| ICR_1408* | chrX:38474430-38475389 | P | FTLP16 | 9372 |
| ICR_1413^ | chrX:40243438-40244131 | P | BCOR | 66048 |
| ICR_1417*^ | chrX:47637837-47638168 | M | ELK1 | 0 |
| ICR_1441^ | chrX:99939597-99940400 | M | B3GNT2P1 | 51,491 |
| ICR_1468^ | chrX:133437318-133438444 | P | GPC4 | 21,829 |
| ICR_1484^ | chrX:154408925-154409317 | P | DNASE1L1 | 0 |
Regions of differential methylation meeting criteria reported herein for ICRs that overlap with regions of gamete-specific methylation. Parental allele methylated indicates which gamete source showed hypermethylation – Paternal (P – sperm) or Maternal (M – oocyte) based upon both sperm and oocyte methylation data. Previously reported ICRs that do not have gamete specific methylation are also included and labelled as having Somatic (S) methylation. ICRs overlapping ENCODE annotated regions of CTCF binding and DNase I hypersensitivity are indicated by * and ^, respectively. ICRs overlapping previously published ICRs of imprinted genes are indicated by #.
+Criteria for an ICR: 1) ≥5 consecutive CpG sites, 2) methylation levels of 50% ±15%, 3) similarity of methylation levels in tissues from the three germ layers (i.e., brain, liver, and kidney), and 4) similarity of methylation across individuals.
The 1,488 novel ICRs methylated at 50% ±15% ranged from 10 to ~4,000 bp long, with a median length of 248 bp (Figure 1(b)). This is similar to the median size of the known ICRs (375 bp) (Figure 1(c)), but with a tailing distribution of much longer candidates. As expected for ICRs, differentiated tissues derived from the three embryonic germ layers exhibited similar methylation levels, consistent with the establishment of these methylation marks in the stem cells before tissue specification. Importantly, these regions also showed similar methylation fractions across individuals, consistent with ICRs controlling gene dosage. We have developed a corresponding imprintome browser depicting methylation fractions for each CpG site in the 1,488 candidate ICRs identified for each embryonic germ layer (https://humanicr.org/).
As a sensitivity analysis, Sciome, Inc. (Research Triangle Park, NC, USA) used an independent calling pipeline with similar procedures for adapter trimming and alignment to hg38 and the four ICR call criteria, but without pooling aligned reads. This generated 1,225 ICRs, including 19 of the 25 characterized ICRs. Of the 1,225 ICRs discovered by Sciome, Inc. (Research Triangle Park, NC, USA), 900 (62%) were found in our initial analysis (Supplementary Table S1 and Supplementary Table S2).
Parent-of-origin methylation patterns in gametes
Parent-of-origin methylation patterns are a property of authentic inherited ICRs and can be discerned from the methylation patterns of gametes. We examined the parent-of-origin methylation patterns of the novel ICRs using WGBS results from sperm and oocyte sequence data. Oocyte data was obtained from public databases (accession number JGAS00000000006) [25]. Our sperm sequence data was supplemented with partial control sperm samples from PRJNA754049 [26]. We compared the embryonic and gametic methylation patterns of the 1,488 candidate ICRs seeking to identify those either fully unmethylated (<10%) or fully methylated (>90%) in gametes. This methylation pattern indicates control of genomic imprinting, which once established in the gametes and inherited persists throughout development. As proof of concept, we first examined whether this strategy could successfully identify known ICRs, and 19 of the 25 known ICRs [14] were captured using this strategy (Table 1 and Supplementary Table S1). Despite the low read-depth of the oocyte sequence data, we found that 332 (Table 1) of the 1,488 candidate ICRs (Supplementary Table 1) had methylation patterns consistent with originating in either the sperm or egg (visualize in https://humanicr.org/).
Our data show that two previously characterized ICRs [14], PLAGL1 (ICR_404, Figure 2(a), Table 1 and Supplementary Table S1) and MEG3 (ICR_872, 873, Figure 2(b), Table 1 and Supplementary Table S1) have somatic DNA methylation approximating the expected 50% level, but they appear significantly longer than the regions currently defined for these ICRs. Additional examples of this are shown for imprinted genes L3MBTL1 (ICR_1194, Supplementary Figure S1A, Table 1 and Supplementary Table S1) [28] and BLCAP/NNAT (ICR_1192, 1193, Supplementary Figure S1B, Table 1 and Supplementary Table S1) [29,30].
Figure 2.
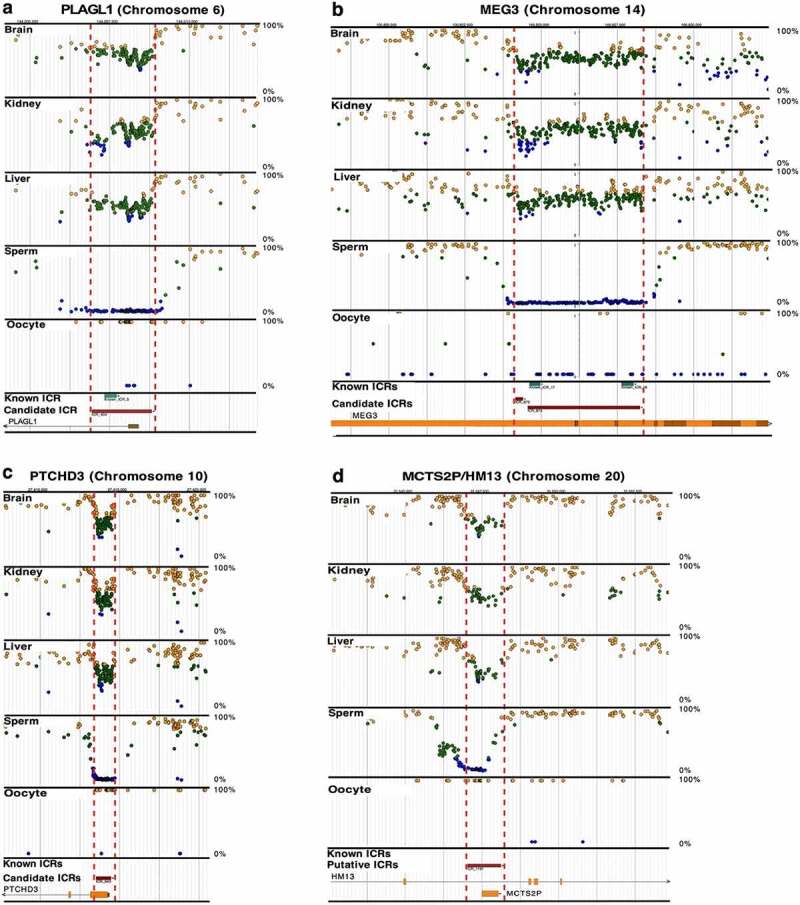
Previously known and candidate ICRs. (a) PLAGL1 (ICR_404) and (b) MEG3 (ICR_872,873) imprinted genes contain previously known ICRs (blue boxes) [14]. They were also identified in our whole-genome bisulphite sequencing (WGBS) screen for candidate ICRs (red boxes); visualize at https://humanicr.org/. The ICRs defined herein overlap and extend beyond the currently known ICRs. (c) PTCHD3 (ICR_643) and (d) MCTS2P/HM13 (ICR_1191) contain only candidate ICRs (red boxes) determined in this study, and potentially control novel imprinted genes. The methylation levels for ICRs is 50 ± 15% for the candidate ICRs. DNA methylation levels determined by WGBS in sperm and oocytes are also provided. These sequence data were supplemented with publicly available gametic sequence (accession number JGAS00000000006) and part of the control sperm data from PRJNA754049 [25,26]. Dots indicate hemi-methylation (green), hypomethylation (blue), and hypermethylation (yellow) based on WGBS. The vertical dashed red lines delineate the candidate ICR regions.
The ICRs for most known imprinted human genes (https://www.geneimprint.com/site/genes-by-species) are unknown, but most are now defined, at least in part, by the present human imprintome. For example, the ICRs for ZDBF2 (ICR_165-176) are shown in Supplementary Figure S2A. An ICR for the tumour suppressor gene IGF2R [31] (ICR_409, 410) is found only in intron 2 in humans (Supplementary Figure S2B). This is similar to what has been observed in the dog [32], but different from that observed in the mouse, which also has an ICR in the promoter region [33].
We also identified novel candidate ICRs mapping near PTCHD3 (ICR_643, Figure 2(c), Table 1) and MCTS2P/HM13 (ICR_1191, (Figure 2(d)), (Table 1). Other novel candidate ICRs include those mapping near WNT10A (ICR_177, Supplementary Figure S3A, Table 1) and ADNP2/PARD6G (ICR_1052, 1053, Supplementary Figure S3B, Table 1). Such findings indicate the presence of additional imprinted genes in the human genome.
Similarity of methylation marks across accessible tissues
For diagnostics and public health screening, it is critically important that DNA methylation marks be reproducible using other sequencing technologies and be similar to that in otherwise healthy human’s accessible tissues (e.g., peripheral blood, maternal or foetal tissues discarded at birth (e.g., placenta), or HUVECs). These tissues also often serve as controls in epidemiologic studies, and act as surrogates for affected inaccessible tissues.
To assess reproducibility, we selected two well-characterized ICRs regulating the imprinted expression of PEG3/ZIM2 (ICR_1142, (Figure 3(a)), (Table 1) and Supplementary Table S1) and PEG10 (ICR_475, Figure 3(b), Table 1 and Supplementary Table S1) for which we were also able to developed pyrosequencing assays using the PCR primer sets provided in Supplementary Table S3 to determine DNA methylation levels. We used pyrosequencing to test whether CpG methylation averaged 50% in these canonical ICRs samples. The DNA methylation patterns determined with pyrosequencing of embryonic brain, kidney, and liver fell between 35% and 65% and averaged approximately 50%. This was comparable to that found with WGBS in these well-characterized ICRs (Figure 3(c,d)).
Samples of internal tissues are generally unavailable for research, diagnostics or public health screening purposes. To determine if methylation marks at known ICRs are similar in accessible tissues from healthy individuals, we again characterized ICRs regulating the imprinted expression of PEG3/ZIM2 (ICR_1142, (Figure 3(a)), (Table 1) and Supplementary Table S1) and PEG10 (ICR_475, Figure 3(b), Table 1 and Supplementary Table S1) in CD14− monocytes from newborn cord blood and HUVECs. The methylation patterns determined with pyrosequencing were comparable to those found with WGBS in these well-characterized ICRs in the CD14− monocytes and HUVECs (Figure 3(e,f)), falling between 35% and 65% methylation and averaging 50% in these accessible tissues as expected for bona fide ICRs. These results indicate that accessible peripheral tissues can be used to assess the effects on the human imprintome from early life exposures to chemicals, physical agents (e.g., radiation, blunt force trauma, heat stress, etc.), and other adverse physiological conditions.
Novel ICR methylation and human pathology
Many of the novel ICRs identified are in regions previously implicated in the pathogenesis of human diseases. For example, four candidate differentially methylated ICRs are located within the Down syndrome (DS) critical region at chromosome 21q22 [34]. ICR_1317 resides in the promoter of HLCS (Supplementary Figure S4A, Supplementary Table S1); ICR_1318 is in intron 1 of RIPPLY3 (Supplementary Figure S4A, Supplementary Table S1); ICR_1319 is close to the 3’ UTR of KCNJ6 (Supplementary Figure S4B, Supplementary Table S1); and ICR_1320 is in intron 1 of GET1 (Supplementary Figure S4C, Supplementary Table S1).
The functions of these genes have developmental implications, playing roles in the metabolism required for infant growth and development (HLCS); transcriptional regulation controlling pharyngeal development (RIPPLY3); cell membrane potential regulating G-protein coupled receptors in both cardiac and neuronal signalling, with mutations connected to developmental delay with facial abnormalities and intellectual phenotypes (KCNJ6); and intracellular transport and positioning of proteins involved in signal transduction pathways connected to retinal deterioration and nystagmus (GET1).
Interestingly, ICR_1319 and the highly restricted Down syndrome critical region (HR-DSCR) of only 34 kb [35] flank KCNJ6 on distal 21q22.13, suggesting that not only gene duplication but also imprinting dysregulation may be involved in DS. Moreover, the first intron of the GET1 gene harbours a single nucleotide polymorphism (rs2244352, chr21:39,386,047) (Supplementary Figure S4C) implicated in comitant esotropia, a condition resulting in poor binocular vision that affects ~20% of Down syndrome cases. The SNP rs2244352 is associated with risk in a parent-of-origin dependent manner, with significant association between incidence and paternal inheritance of the minor ‘T’ allele [36,37]. The major allele for rs2244352 is G. Thus, carrying the minor T allele would eliminate the formation of a CG dinucleotide able to be methylated; however, loss of methylation on the paternal chromosome is likely not the causative mechanism.
It has been shown that this region is differentially methylated, but that it is the maternal allele that is methylated [36]. Our gametic data is also consistent with maternal methylation, showing hypomethylation in sperm and hypermethylation in oocytes (Supplementary Figure S4C). With no loss of paternal methylation possible to cause dysregulation of this putative ICR, we hypothesize an alternate mechanism, in which repressor binding occurs on unmethylated CG sites in this region. This would be comparable to regulation at the H19/Igf2 ICR, by CTCF binding of the unmethylated allele [14]. Thus, if ‘CG’ to ‘TG’ substitution alters the recognition sequence for repressor binding, paternal GET1 expression could be activated, adding to the expression from the maternal copy protected from repression by methylation of the putative ICR.
Similarly, four candidate ICRs are located within the DiGeorge syndrome critical region at chromosome location 22q11.2 (Supplementary Figure S5 A, C) [38,39]. Hemizygous microdeletions in this crucial region result in developmental disorders, such as velopharyngeal insufficiency, variable conotruncal heart defects, and cognitive and behavioural disorders (e.g., schizophrenia, bipolar disorder, and autism) [38]. Moreover, differential brain effects have been reported with the maternal or paternal deletion of 22q11.2, suggesting a role of imprinted genes in the aetiology of these psychiatric abnormalities [38]. The ICRs identified are ICR_1355 in the promoter of DGCR5 (Supplementary Figure S5A, Supplementary Table S1); ICR_1356 is in the promoter region of TBX1 and ICR_1357 is intronic (Supplementary Figure S5B, Table 1 and Supplementary Table S1). ICR_1358, which overlaps pseudogene ABHD17AP4 (Supplementary Figure S5C, Supplementary Table S1), is upstream of SERPIND1 and SNAP29 (Supplementary Table S1). Functionally, DGCR5 is a long non-coding RNA that acts as a regulator of apoptosis and proliferation; TBX1 is a transcription factor involved in the regulation of developmental processes, with its deletion directly linked to physical malformations seen in DiGeorge syndrome; SERPIND1 is a protease inhibitor with critical functions in blood clotting; and SNAP29 is a member of a family of synaptic vesicle trafficking regulators associated with developmental disorders of the CNS.
We previously used a computer algorithm to predict the genome-wide imprint status of human genes from sequence features [19]. DLGAP2 was predicted to be imprinted and demonstrated to be paternally expressed. It is a membrane-associated protein that plays a role in synapse organization and signalling in neuronal cells. It was subsequently implicated in autism based upon CNV analyses [40]. In this study, we identified an ICR_502 in intron 1 (Supplementary Figure S6A, Supplementary Table S1); this gene also contains ICR_503-506 (Table 1 and Supplementary Table S1). Thus, the function of DLGAP2 could potentially be altered both genetically and epigenetically in the formation of autism. Interestingly, of the 102 annotated genes we predicted with the use of computer algorithms to be imprinted [19], we have now identified ICRs for 35 of them (34%) (Supplementary Table S1 and Supplementary Table S4), providing additional supporting evidence that they are imprinted.
PRDM16 functions as a transcription coregulator in the development of brown adipocytes and increased expression may protect against obesity [41]. We previously predicted it to be imprinted and paternally expressed in both mice [42] and humans [19]. Interestingly, ICR_11 and ICR_12 (Supplementary Table S1), while overlapping TTC34, are also within 500kb of PRDM16. Although the oocyte methylation data are sparse, it appears that the paternal allele at these ICR regions is sparsely or entirely unmethylated while the maternal allele is methylated, suggesting that PRDM16 is paternally expressed as predicted by Luedi et al. [19]. Using less stringent criteria (i.e., 50 + 20% DNA methylation), there is also an ICR in the promoter region of PRDM16 (ICR_3070_54) and three additional intronic ICRs (i.e., ICR_3070_55-57; Supplementary Figure S6B). Should this gene be experimentally confirmed to be imprinted, genomic imprinting would be involved in the maturation of both white [43] and brown fat cells, making the role of imprinted gene expression in metabolism and obesity more extensive than previously appreciated.
Imprinted genes have been identified on the X-chromosome in mice [44], but not in humans. Nevertheless, we have identified 98 putative ICRs on the X-chromosome (ICR_1391-1488, Supplementary Table S1), indicating that imprinted genes are also present on the human X chromosome. Gamete DNA methylation was adequate to determine parent-of-origin methylation for 11 of these ICRs (6 maternally methylated and 5 paternally methylated, Table 1). Only DHRSX (ICR_1394), a secretory protein associated with starvation-induced autophagy [45], is located in a pseudoautosomal region – PAR1 (Figure 4). The role of these candidate imprinted genes in the genesis of parental-dependent behavioural disorders, such as those observed in Turner syndrome [46] needs further investigation.
Figure 4.
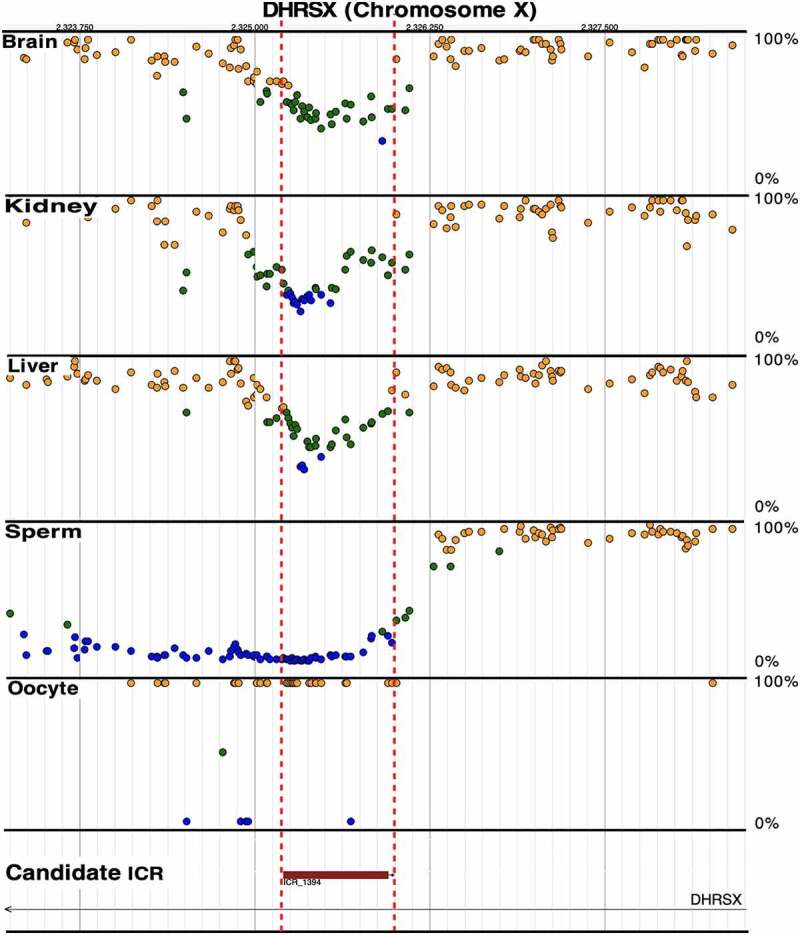
Candidate ICR for DHRSX (ICR_1394) in the pseudo autosomal region, PAR1, on the X chromosome; visualize at https://humanicr.org/. Whole genome bisulphite sequencing (WGBS) identified a candidate ICR (red box) in embryonic brain, kidney, and liver tissue for a candidate imprinted gene associated with starvation induced autophagy [45]. DNA methylation determined by WGBS in sperm and oocytes is also shown; these sequence data were supplemented with publicly available gametic sequence (accession number JGAS00000000006) and part of control sperm data from PRJNA754049 [25,26]. Dots indicate hemi-methylation (green), hypomethylation (blue) and hypermethylation (yellow) based on WGBS. The vertical dashed red lines delineate the candidate ICR regions.
Functional significance of ICR methylation
Bona fide ICRs typically regulate gene expression by controlling access to transcriptional regulatory sites within the imprinted genes. We examined the overlap of our 1,488 candidate ICRs with DNase I hypersensitive sites, regulatory motifs, and transcription factor-binding sites for nearby genes. Of the 332 ICRs that were either unmethylated or fully methylated in gametes, 200 (60%) overlapped with DNase I hypersensitive regions. Of those 332 ICRs, 178 were hypermethylated in sperm DNA sequences, and 154 were hypermethylated in oocytes.
Moreover, for each ICR, we looked for regulatory motifs within ±5,000 bp using Analysis of Motif Enrichment (AME) [47] against the Homo Sapiens Comprehensive Model Collection (HOCOMOCO). This consists of motifs for 680 known transcription factors. Approximately one-third of the 680 known transcription factor-binding sites are within or near candidate ICRs, supporting the functional significance of these ICRs (Supplementary Table S5).
The limited number of ICRs previously characterized [14] are over-selected for growth effectors, with dysregulation associated with a wide range of conditions, including metabolic disorders, cancers, neurological diseases, language development deficits, schizophrenia, and bipolar affective disorders [48,49]. Thus, we examined the predicted functions of the 914 genes associated with the 332 candidate ICRs with gamete-specific methylation (Supplementary Table S6) by performing gene ontology (GO) analysis using Gorilla (http://cbl-gorilla.cs.technion.ac.il/) (Supplementary Table S7). The most significant high-level biological processes identified by Gorilla were cAMP biosynthetic processes, cellular response to glucagon, and adenylate cyclase-activating G protein-coupled receptor signalling pathway (Supplementary Table S7). Functional analysis of candidate regions using the Comparative Toxicogenomics Database (CTD) identified protein-binding and DNA-binding as enriched GO terms in the functional annotations (Supplementary Table S8). Of the top five pathways identified by Ingenuity Pathway Analysis (IPA) (QIAGEN Inc., https://www.qiagenbioinformatics.com/products/ingenuity- pathway-analysis), three were important neurological pathways: gonadotropin-releasing hormone (GnRH) signalling, endocannabinoid neuronal synapse pathway, and g-aminobutyric acid receptor signalling (Supplementary Table S9). Five hundred eight genes were associated with neurological disease, while 168 were related to nervous system development and function. Of the top networks identified by IPA, RNA post-transcriptional modification, cell cycle and DNA replication, recombination, and repair had the highest score, with auditory disease, cellular compromise, and neurological disease coming in second. Results of KEGG and REACTOME analysis of enriched pathways included neuronal system, transmission across chemical synapses, signal transduction, pathways in cancers, circadian entrainment, axon guidance, cholinergic synapse, glutamatergic synapse, and calcium signalling pathways (Supplementary Table S10) [50].
We also explored the role of the candidate imprinted genes in human disease using the Mouse Genome Informatics (MGI) database and the CTD and their curation of OMIM disease categories and inferred diseases via chemical-gene chemical-disease associations [51]. Consistent with the preceding analyses, several neurological disorders correlated with a subset of the genes in MGI, including Alzheimer’s disease (n = 2), autism spectrum disorder (n = 6), various ataxias, and cranial developmental disorders. When inferred disease associations were factored into the CTD analysis, the number of gene-disease associations restricted to neurological disorders expanded greatly, e.g., 242 genes were associated with Alzheimer’s disease and 243 with Parkinson’s disease. For Alzheimer’s disease, 16 of the 242 genes were unique to the disorder (ABHD17AP4, BRE, C2ORF27A, CCDC144B, CERS3, CUTALP, DHRSX, DUX4L1, FAM155B, FRG1CP, FRG2B, GPR78, HERC2P4, JRKL, KBTBD13, KCNAB1); whereas, for Parkinson’s disease, 17 were unique (ADARB2, AFF2, CDH24, CMC4, CPAMD8, CRTC1, DBH-AS1, DIRAS3, DNAJC17, DNM1P46, DTX2, DUX4L9, EPHA10, ERAS, EXD3, HTR5A-AS1, HYMAI); the remaining genes were shared by both disorders, indicating overlapping chemical mechanisms at the molecular level in the aetiology of these diseases.
Discussion
Epigenetic mechanisms, such as DNA methylation, are believed to link adverse intrauterine exposures to adult disease susceptibility; however, supporting empirical evidence from humans has remained scarce because of a lack of recognizable and archivable patterns of early epigenetic effects that can be detected and quantified in the epigenome [52–54]. This is especially important for human studies that rely on DNA from sample types accessible in otherwise healthy human populations (e.g., saliva or peripheral blood). While sequence regions controlling the monoallelic expression of imprinted genes have been previously proposed as targets for such studies [6], until now, only 25 of these regions have been described [14], with potentially hundreds more unknown.
Herein, we determined that the human imprintome is comprised of 1,488 regions with characteristics typically observed in known ICRs. Interestingly, the overlap between the 850,000 CpG sites in the Illumina Infinium Methylation EPIC microarray (Illumina, Inc., San Diego, CA, USA) and the 22,279 CpG sites in the human imprintome is only 7%. Using genomic DNA obtained from three tissue types isolated from embryos of both sexes and at least two ethnicities, these regions include most of the previously identified ICRs [14]. Of the 1,488 regions, 332 also exhibited parent-of-origin specific methylation – the hallmark of an ICR. Of those ICRs, 209 overlapped with DNase I hypersensitive regions. These novel ICRs have a median CpG dinucleotide content of 248, similar to the 25 previously characterized ICRs with a size range of 10 to ~4,000 bp. When an overlap was identified between a novel and a known ICR, the novel sequence typically extended beyond the boundaries of the prior reported sequence. Chromosomes frequently contain clusters of genes that are controlled by a single ICR (i.e., imprinted domains). Thus, our 200 strongest candidate ICRs, with overlapping DNase I hypersensitive sites and gamete-specific methylation, could regulate as many as 400 imprinted genes if each ICR controlled only two genes, which is commonly observed. Expanding this to include all 332 candidate ICRs with gamete-specific methylation increases the number of potentially imprinted genes to a little more than 500 or ~3% of the genome.
Many known imprinted genes have developmental functions involving the regulation of cell function and growth such as pruning of synapses and adipocyte accrual affecting the life course. Thus, ICRs are of particular interest in studying the early origins of a wide range of common chronic diseases, including neurological disorders and metabolic diseases such as obesity, type 2 diabetes, and cancers. The majority (90%) of our candidate ICRs fall within 5,000 bp of genes involved in fundamental processes, including cAMP biosynthetic processes, cellular responses to glucagon and adenylate cyclase-activating G protein-coupled receptor signalling. Notably, a large proportion of the candidate ICRs are near genes involved in metabolic and neurological diseases, consistent with the observations of previously characterized ICRs. Characterizing and experimentally confirming the complete repertoire of ICRs could lead to the development of dietary interventions and pharmacological targeting of specific regions to advance the precision of nutritional and chemical therapies.
For example, if the candidate ICR proximal to the zinc finger transcription factor gene PRDM16 is experimentally confirmed to be imprinted, altered methylation can be detected in any tissue as early as birth. As PRDM16 controls the bidirectional fate decision between brown adipocytes and myoblasts [55], early detection of alteration in an imprinted ICR could provide opportunities for pharmacological or dietary manipulation to enhance the expression of PRDM16, preventing metabolic dysfunction, including obesity. Similarly, disposition to hypoplasia of the thymus and parathyroid glands and conotruncal cardiac malformations and schizophrenia that potentially result from imprinting disorders in the DiGeorge syndrome critical region could be identified earlier, and potentially even during gestation when interventions may be more effective.
ICRs have the unique features of the early establishment of DNA methylation marks, similarity across cell types and tissues, and stability over the lifespan. These characteristics facilitate their broad use as stable archives of early developmental exposures that may alter metabolic and other developmental and behavioural processes in adulthood [56–58]. Thus, ICRs are logical targets for evaluating the early origins of disease using accessible cell types obtained at variable ages [59–62]. A predetermined reference panel of ICRs could be vital in identifying early exposures. Their effects on DNA methylation have a wide range of potential uses, particularly in chronic disease epidemiology where relevant past exposures may be difficult to quantify. Such a predetermined reference panel could also be key in unravelling epigenetic responses to early life chemical and non-chemical stressors that result in molecular ‘wear and tear’ as reflected in methylation changes in accessible tissue, and may improve the precision and usefulness of epigenetic clocks [63,64]. The expectation that the baseline methylation fraction of ICRs is approximately 50% and is stable over time, with little epigenetic drift, also supports the use of ICR methylation as biomarkers of adult disease susceptibility at any time during the life course. Therefore, having the complete repertoire of bona fide ICRs – the imprintome – should improve our understanding of the early origins of adult diseases.
While the use of methylation marks as exposure proxies has long been advocated, and methylation patterns of ICRs provide a rare ‘epigenetic responsive’ window to early exposures, our results should be interpreted in the context of the study limitations. Firstly, our algorithm used to identify candidate genome-wide ICRs was tested at three methylation thresholds: relaxed 30–70% (50 ± 20%), moderate 35–65% (50 ± 15%), and stringent 45-55% (50 ± 5%). We selected the moderate set for further analysis. While this approach is pragmatic, it does not accommodate the possibility that methylation patterns defining ICR boundaries may be more fluid, requiring additional statistical and experimental interrogation of ICRs to refine the boundaries and, with that, the imprinted genes they regulate. Bioinformatic approaches such as change-point modelling that have been used in copy number variant analyses [65,66], coupled with experimental validation, could also be deployed in the future. Secondly, we have not performed clonal-allele analysis of candidate regions to definitively prove that they are bona fide ICRs, with methylation patterns consistent in cis for the parental alleles. Thus, we do not expect all candidates to overlap true ICRs; however, based on the characteristics of known ICRs, we are confident that a majority of human ICRs are captured within the 1,488 candidate ICRs.
Despite these limitations, we have used a combination of bioinformatic and sequencing approaches to provide the first draft of the complete repertoire of human ICRs – the human imprintome. To facilitate its use, we have also developed an online tool, the imprintome browser, which is linked to the UCSC Genome Browser to visualize the data at: https://humanicr.org/. Further refinement is needed to identify bona fide ICRs and their exact boundaries. This will be bioinformatically iterative and will require experimental validation. To our knowledge, however, this is the first study to create a human ICR compendium. As these sequence regions are also present in accessible peripheral blood DNA, we anticipate our data will greatly facilitate the ability to quantify the contribution of exposures in early development to a wide range of adult-onset chronic diseases; pave the way for novel early-detection tools; and eventually reveal the molecular underpinnings of vulnerability in disease processes, especially in early life.
Supplementary Material
Funding Statement
This work was supported in part by National Institutes of Health grants R01HD098857 (D.A.S., C.H., R.L.J.), R01MD011746 (C.H., D.A.S, R.L.J.), R01MD011746-S1 (C.H., D.A.S., R.L.J.), and Office of Extramural Research, National Institutes of Health. R01HD098857, R01MD011746NICHD, NIMHD, NIEHS [R01HD098857, R01MD011746-S1, P30ES025128].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Author contributions:
C.H. and R.L.J. conceived the idea and obtained funding, D.D.J., A.M.R, F.W. and J.H. contributed bioinformatics expertise, D.A.S., A.P., A.L., S.E.C, S.S.P. and M.C. contributed mechanistic expertise to advancing the thesis. All authors contributed to drafting and editing the manuscript.
Data availability
Data will be available from the authors upon request, and at the website https://humanicr.org/.
Ethics statement
These tissues were obtained from the National Institutes of Health funded Laboratory of Human Embryology at the University of Washington, Seattle, WA; they were snap frozen to preserve DNA/RNA integrity (NCSU Institutional Review Board #3565).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15592294.2022.2091815
References
- [1].Badcock C, Crespi B.. Battle of the sexes may set the brain. Nature. 2008;454(7208):1054–1055. [DOI] [PubMed] [Google Scholar]
- [2].Lorgen-Ritchie M, Murray AD, Ferguson-Smith AC, et al. Imprinting methylation in SNRPN and MEST1 in adult blood predicts cognitive ability. PLoS One. 2019;14(2):e0211799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet. 2007;8(4):253–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Jirtle RL. Genomic imprinting and cancer. Exp Cell Res. 1999;248(1):18–24. [DOI] [PubMed] [Google Scholar]
- [5].Jirtle RL. IGF2 loss of imprinting: a potential heritable risk factor for colorectal cancer. Gastroenterology. 2004;126(4):1190–1193. [DOI] [PubMed] [Google Scholar]
- [6].Hoyo C, Murphy SK, Jirtle RL. Imprint regulatory elements as epigenetic biosensors of exposure in epidemiological studies. J Epidemiol Community Health. 2009;63(9):683–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Franks PW, McCarthy MI. Exposing the exposures responsible for type 2 diabetes and obesity. Science. 2016;354(6308):69–73. [DOI] [PubMed] [Google Scholar]
- [8].Pigeyre M, Rousseaux J, Trouiller P, et al. How obesity relates to socio-economic status: identification of eating behavior mediators. Int J Obes (Lond). 2016;40(11):1794–1801. [DOI] [PubMed] [Google Scholar]
- [9].Arpon A, Milagro FI, Ramos-Lopez O, et al. Methylome-wide association study in peripheral white blood cells focusing on central obesity and inflammation. Genes (Basel). 2019;10(6):444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Bysani M, Perfilyev A, de Mello VD, et al. Epigenetic alterations in blood mirror age-associated DNA methylation and gene expression changes in human liver. Epigenomics. 2017;9(2):105–122. [DOI] [PubMed] [Google Scholar]
- [11].Joubert BR, Felix JF, Yousefi P, et al. DNA methylation in newborns and maternal smoking in pregnancy: genome-wide consortium meta-analysis. Am J Hum Genet. 2016;98(4):680–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Meeks KAC, Henneman P, Venema A, et al. An epigenome-wide association study in whole blood of measures of adiposity among Ghanaians: the RODAM study. Clin Epigenetics. 2017;9:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kessler NJ, Waterland RA, Prentice AM, et al. Establishment of environmentally sensitive DNA methylation states in the very early human embryo. Sci Adv. 2018;4(7):eaat2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Skaar DA, Li Y, Bernal AJ, et al. The human imprintome: regulatory mechanisms, methods of ascertainment, and roles in disease susceptibility. ILAR J. 2012;53(3–4):341–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Murphy SK. Targeting the epigenome in ovarian cancer. Future Oncol. 2012;8(2):151–164. [DOI] [PubMed] [Google Scholar]
- [16].Murphy SK, Adigun A, Huang Z, et al. Gender-specific methylation differences in relation to prenatal exposure to cigarette smoke. Gene. 2012;494(1):36–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Cassidy FC, Charalambous M, Suarez RK. Genomic imprinting, growth and maternal-fetal interactions. J Exp Biol. 2018;221(Pt Suppl 1). DOI: 10.1242/jeb.164517 [DOI] [PubMed] [Google Scholar]
- [18].Kitsiou-Tzeli S, Tzetis M. Maternal epigenetics and fetal and neonatal growth. Curr Opin Endocrinol Diabetes Obes. 2017;24(1):43–46. [DOI] [PubMed] [Google Scholar]
- [19].Luedi PP, Dietrich FS, Weidman JR, et al. Computational and experimental identification of novel human imprinted genes. Genome Res. 2007;17(12):1723–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Green BB, Kappil M, Lambertini L, et al. Expression of imprinted genes in placenta is associated with infant neurobehavioral development. Epigenetics. 2015;10(9):834–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ishida M, Moore GE. The role of imprinted genes in humans. Mol Aspects Med. 2013;34(4):826–840. [DOI] [PubMed] [Google Scholar]
- [22].Lambertini L, Marsit CJ, Sharma P, et al. Imprinted gene expression in fetal growth and development. Placenta. 2012;33(6):480–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Soubry A, Hoyo C, Butt CM, et al. Human exposure to flame-retardants is associated with aberrant DNA methylation at imprinted genes in sperm. Environ Epigenet. 2017;3(1):dvx003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Soubry A, Murphy SK, Vansant G, et al. Opposing epigenetic signatures in human sperm by intake of fast food versus healthy food. Front Endocrinol (Lausanne). 2021;12:625204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Okae H, Chiba H, Hiura H, et al. Genome-wide analysis of DNA methylation dynamics during early human development. PLoS Genet. 2014;10(12):e1004868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Schrott R, Murphy SK, Modliszewski JL, et al. Refraining from use diminishes cannabis-associated epigenetic changes in human sperm. Environ Epigenet. 2021;7(1):dvab009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lander ES, Linton LM, Birren B, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921. [DOI] [PubMed] [Google Scholar]
- [28].Li J, Bench AJ, Vassiliou GS, et al. Imprinting of the human L3MBTL gene, a polycomb family member located in a region of chromosome 20 deleted in human myeloid malignancies. Proc Natl Acad Sci USA. 2004;101(19):7341–7346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Evans HK, Wylie AA, Murphy SK, et al. The neuronatin gene resides in a “micro-imprinted” domain on human chromosome 20q11.2. Genomics. 2001;77(1–2):99–104. [DOI] [PubMed] [Google Scholar]
- [30].Schulz R, McCole RB, Woodfine K, et al. Transcript- and tissue-specific imprinting of a tumour suppressor gene. Hum Mol Genet. 2009;18(1):118–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].De Souza AT, Hankins GR, Washington MK, et al. M6P/IGF2R gene is mutated in human hepatocellular carcinomas with loss of heterozygosity. Nat Genet. 1995;11(4):447–449. [DOI] [PubMed] [Google Scholar]
- [32].O’Sullivan FM, Murphy SK, Simel LR, et al. Imprinted expression of the canine IGF2R, in the absence of an anti-sense transcript or promoter methylation. Evol Dev. 2007;9(6):579–589. [DOI] [PubMed] [Google Scholar]
- [33].Stoger R, Kubicka P, Liu CG, et al. Maternal-specific methylation of the imprinted mouse Igf2r locus identifies the expressed locus as carrying the imprinting signal. Cell. 1993;73(1):61–71. [DOI] [PubMed] [Google Scholar]
- [34].Jiang X, Liu C, Yu T, et al. Genetic dissection of the down syndrome critical region. Hum Mol Genet. 2015;24(22):6540–6551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Antonaros F, Pitocco M, Abete D, et al. Structural characterization of the highly restricted down syndrome critical region on 21q22.13: new KCNJ6 and DSCR4 transcript isoforms. Front Genet. 2021;12:770359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Alves da Silva AF, Machado FB, Pavarino EC, et al. Trisomy 21 alters DNA methylation in parent-of-origin-dependent and -independent manners. PLoS One. 2016;11(4):e0154108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Shaaban S, MacKinnon S, Andrews C, et al. Genome-wide association study identifies a susceptibility locus for comitant Esotropia and suggests a parent-of-origin effect. Invest Ophthalmol Vis Sci. 2018;59(10):4054–4064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Das Chakraborty R, Bernal AJ, Schoch K, et al. Dysregulation of DGCR6 and DGCR6L: psychopathological outcomes in chromosome 22q11.2 deletion syndrome. Transl Psychiatry. 2012;2:e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Motahari Z, Moody SA, Maynard TM, et al. In the line-up: deleted genes associated with DiGeorge/22q11.2 deletion syndrome: are they all suspects? J Neurodev Disord. 2019;11(1):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Catusi I, Garzo M, Capra AP, et al. 8p23.2-pter microdeletions: seven new cases narrowing the candidate region and review of the literature. Genes (Basel). 2021;12(5):652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Seale P, Bjork B, Yang W, et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature. 2008;454(7207):961–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Luedi PP, Hartemink AJ, Jirtle RL. Genome-wide prediction of imprinted murine genes. Genome Res. 2005;15(6):875–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Wylie AA, Murphy SK, Orton TC, et al. Novel imprinted DLK1/GTL2 domain on human chromosome 14 contains motifs that mimic those implicated in IGF2/H19 regulation. Genome Res. 2000;10(11):1711–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Davies W, Isles A, Smith R, et al. Xlr3b is a new imprinted candidate for X-linked parent-of-origin effects on cognitive function in mice. Nat Genet. 2005;37(6):625–629. [DOI] [PubMed] [Google Scholar]
- [45].Zhang G, Luo Y, Li G, et al. DHRSX, a novel non-classical secretory protein associated with starvation induced autophagy. Int J Med Sci. 2014;11(9):962–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Skuse DH, James RS, Bishop DV, et al. Evidence from turner’s syndrome of an imprinted X-linked locus affecting cognitive function. Nature. 1997;387(6634):705–708. [DOI] [PubMed] [Google Scholar]
- [47].Bailey TL, Johnson J, Grant CE, et al. The MEME suite. Nucleic Acids Res. 2015;43(W1):W39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Bartolomei MS, Tilghman SM. Genomic imprinting in mammals. Annu Rev Genet. 1997;31:493–525. [DOI] [PubMed] [Google Scholar]
- [49].Murphy SK, Jirtle RL. Imprinting evolution and the price of silence. Bioessays. 2003;25(6):577–588. [DOI] [PubMed] [Google Scholar]
- [50].Jassal B, Matthews L, Viteri G, et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020;48(D1):D498–D503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Davis AP, Grondin CJ, Johnson RJ, et al. The comparative toxicogenomics database: update 2019. Nucleic Acids Res. 2019;47(D1):D948–D954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Gunasekara CJ, Scott CA, Laritsky E, et al. A genomic atlas of systemic interindividual epigenetic variation in humans. Genome Biol. 2019;20(1):105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Dell’Aversana C, Cuomo F, Longobardi S, et al. Age-related miRNome landscape of cumulus oophorus cells during controlled ovarian stimulation protocols in IVF cycles. Hum Reprod. 2021;36(5):1310–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Poduval DB, Ognedal E, Sichmanova Z, et al. Assessment of tumor suppressor promoter methylation in healthy individuals. Clin Epigenetics. 2020;12(1):131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Becerril S, Gomez-Ambrosi J, Martin M, et al. Role of PRDM16 in the activation of brown fat programming. Relevance to the development of obesity. Histol Histopathol. 2013;28(11):1411–1425. [DOI] [PubMed] [Google Scholar]
- [56].Hoyo C, Murtha AP, Schildkraut JM, et al. Folic acid supplementation before and during pregnancy in the Newborn Epigenetics STudy (NEST). BMC Public Health. 2011;11(1):46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].King K, Murphy S, Hoyo C. Epigenetic regulation of Newborns’ imprinted genes related to gestational growth: patterning by parental race/ethnicity and maternal socioeconomic status. J Epidemiol Community Health. 2015;69(7):639–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Vidal AC, Benjamin Neelon SE, Liu Y, et al. Maternal stress, preterm birth, and DNA methylation at imprint regulatory sequences in humans. Genet Epigenet. 2014;6:37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Cui H, Cruz-Correa M, Giardiello FM, et al. Loss of IGF2 imprinting: a potential marker of colorectal cancer risk. Science. 2003;299(5613):1753–1755. [DOI] [PubMed] [Google Scholar]
- [60].Nakagawa H, Chadwick RB, Peltomaki P, et al. Loss of imprinting of the insulin-like growth factor II gene occurs by biallelic methylation in a core region of H19-associated CTCF-binding sites in colorectal cancer. Proc Natl Acad Sci U S A. 2001;98(2):591–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Sullivan MJ, Taniguchi T, Jhee A, et al. Relaxation of IGF2 imprinting in Wilms tumours associated with specific changes in IGF2 methylation. Oncogene. 1999;18(52):7527–7534. [DOI] [PubMed] [Google Scholar]
- [62].Ulaner GA, Vu TH, Li T, et al. Loss of imprinting of IGF2 and H19 in osteosarcoma is accompanied by reciprocal methylation changes of a CTCF-binding site. Hum Mol Genet. 2003;12(5):535–549. [DOI] [PubMed] [Google Scholar]
- [63].Horvath S, Raj K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat Rev Genet. 2018;19(6):371–384. [DOI] [PubMed] [Google Scholar]
- [64].Levine ME, Lu AT, Quach A, et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging (Albany NY). 2018;10(4):573–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Ray S, McEvoy DS, Aaron S, et al. Using statistical anomaly detection models to find clinical decision support malfunctions. J Am Med Inform Assoc. 2018;25(7):862–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Roberts E, Zhao L. A Bayesian mixture model for changepoint estimation using ordinal predictors. Int J Biostat. 2021;18:57–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be available from the authors upon request, and at the website https://humanicr.org/.