

Abstract
Multiple sclerosis (MS) is an inflammatory, demyelinating CNS disease believed to be mediated by CD4 T cells specific for CNS self-antigens. CD8 T cells are also implicated in MS but their function is not well understood. MS lesions are heterogeneous and may reflect variation in the contribution of different types of lymphocytes. Understanding how lymphocytes with different effector functions contribute to MS is essential to develop effective therapies. We investigated how T cells expressing an MHC class I–restricted transgenic TCR specific for myelin basic protein (MBP) contribute to CNS autoimmunity using the mouse model of MS, experimental autoimmune encephalomyelitis. Virus infection triggered cytotoxic TCR-transgenic CD8 T cells to initiate acute experimental autoimmune encephalomyelitis in an IFN-γ– and perforin-dependent manner. Unexpectedly, spontaneous CNS autoimmunity developed in the TCR-transgenic mice that was accelerated by IFN-γ-deficiency. Spontaneous disease was associated with CD4 T cells that develop via endogenous TCR rearrangements but retain specificity for the MHC class I–restricted MBP epitope. The CD4 T cells produced TNF-α without other inflammatory cytokines and caused lesions with striking similarity to active MS lesions. Surprisingly, B cells were the predominant cell type that cross-presented MBP, and their depletion halted disease progression. This work provides a new model of spontaneous CNS autoimmunity with unique similarities to MS that is mediated by T cells with a distinct effector phenotype.
Key Points
CD4 T cells cause spontaneous EAE using an MHC class I–restricted MBP-specific TCR.
Spontaneous disease is dependent on TNF-α without IL-17, IFN-γ, and GM-CSF.
B cells cross-present MBP to drive spontaneous disease.
Introduction
Multiple sclerosis (MS) is a chronic, inflammatory, demyelinating autoimmune disease targeting the CNS that is triggered in genetically predisposed individuals by exposure to environmental factors. Several risk factors have been identified, including infection with EBV (1), a nearly ubiquitous pathogen that can persist throughout life in latent form in B lymphocytes. MS incidence is female biased with an age of onset typically between 20 and 40 y. The pathological hallmark of MS is demarcated focal regions of demyelination, most commonly observed in white matter but can also occur in gray matter (2). Lesions can be classified as active, mixed active/inactive, and inactive lesions depending on the cellularity within the lesion center, localization of activated macrophages/microglia with respect to the demyelinated region, and evidence of ongoing myelin degradation (3). More detailed analyses of lesions defined distinct patterns and noted that individuals generally exhibited only one lesion pattern, suggesting that different pathogenic pathways may exist that culminate in MS (4).
CD4 T cells are thought to play a prominent role in MS, as MHC class II alleles exhibit the strongest association with MS susceptibility, and experimental autoimmune encephalomyelitis (EAE) is induced by activating myelin Ag-specific CD4 T cells. Pathogenic CD4 T cell effector functions have been intensely studied in EAE and focused attention on IL-17, GM-CSF, and IFN-γ as key cytokines that confer encephalitogenicity (5). TNF-α is also important for EAE initiation; however, it has not been considered an essential effector function of pathogenic CD4 T cells because it is also produced by microglia and myeloid cells. The role of B cells is also of great interest because anti-CD20–mediated B cell depletion is beneficial in patients with MS. B cells can produce CNS Ag-specific Abs that facilitate tissue injury and also function as APCs for CD4 T cells. Mice that express transgenic MHC class II–restricted TCRs specific for myelin oligodendrocyte glycoprotein (MOG) demonstrated an important role for B cells as APCs in the initiation of spontaneous EAE (SEAE). Some models combined a transgenic MOG-specific TCR with a transgenic IgH from a MOG-specific Ab (6, 7); however, one TCR-transgenic model demonstrated that nontransgenic B cells could acquire enough endogenous MOG to activate TCR-transgenic CD4 T cells to initiate SEAE (8).
CD8 T cells have also been studied in EAE and MS, as they are the most prevalent lymphocyte seen in MS tissues (9). Several studies implicated a regulatory function for CD8 T cells (10, 11); however, with some exceptions (12), CNS Ag-specific CD8 T cells exert pathogenic activity in animal models. The success of B cell depletion therapy in MS challenges the notion that CD8 T cells could act as the sole pathogenic lymphocyte to sustain chronic CNS autoimmune disease because B cells are not considered efficient in cross-presenting exogenous Ag in the MHC class I pathway (13). However, cytotoxic CD8 T cell effector functions would cause different tissue injury compared with CD4 T cells, suggesting that the relative contribution of pathogenic CD8 versus CD4 T cells among individuals might account for some of the heterogeneity seen in MS lesions. To explore the role of CD8 T cells, we and others developed TCR-transgenic mouse models expressing MHC class I–restricted TCRs specific for CNS Ags. We generated TCR-transgenic mice specific for myelin basic protein (MBP)79–87 associated with the MHC class I molecule Kk, referred to as 8.8 mice (14). Infection of 8.8 mice with wild-type vaccinia virus (Vac-WT) induced an acute CNS disease by activating CD8 T cells that coexpressed the MBP-specific TCR with endogenously rearranged TCRs that recognize vaccinia-derived Ags (15). In Rag−/− 8.8 mice that express only the MBP-specific TCR, infection with a recombinant Vac expressing MBP (Vac-MBP) was required to induce disease. Similarly, acute EAE was induced in TCR-transgenic mice specific for an MHC class I–restricted epitope of the astrocyte-specific protein glial fibrillary acidic protein (GFAP) by infection with recombinant Vac expressing GFAP (16). CNS disease was also initiated in mice coexpressing a transgenic MHC class I–restricted proteolipid protein–specific human TCR with the relevant human MHC class I molecule by immunization with the proteolipid protein peptide, although nontransgenic CD4 T cells with a different myelin Ag specificity were required to sustain disease (17). Collectively, these models showed that activated CNS Ag-specific CD8 T cells acquired a cytotoxic phenotype and initiated acute EAE; however, neither the effector mechanisms employed by the CD8 T cells nor the relevant APCs were identified.
In this study, we demonstrate that both IFN-γ and perforin are required for virally activated 8.8 CD8 T cells to induce acute EAE, and identified multiple APCs that cross-present MBP under these conditions. We also describe a new mouse model of MS in which 8.8 mice develop SEAE characterized by lesions that bear many similarities to active lesions seen in MS. Importantly, SEAE is associated with 8.8 CD4 rather than CD8 T cells that produce TNF-α without IL-17, IFN-γ, or appreciable quantities of GM-CSF. Surprisingly, B cells play a driving role in disease as the predominant cell type cross-presenting the MHC class I–restricted MBP ligand.
Materials and Methods
Mice
C3HeB/FeJ, Pfp−/− (C576L/6-Prf1tm1Sdz/J), and β2-microglobulin (β2m)−/− (C3.129P2(B6)-B2mtm1Unc/J) mice were originally purchased from The Jackson Laboratory. 8.8, 8.8 IFN-γ−/−, 8.8 Pfp−/−, 8.8 TNF-α−/−, and 8.8 Faslgld mice were previously described (18). TNF-α−/− mice were crossed with 8.8 IFN-γ−/− mice to generate homozygous double-knockout mice. Eight- to 12-wk-old female mice were used for all experiments employing Vac infection. Studies of SEAE used both male and female mice with age- and sex-matched 8.8 or WT control mice, except only female 8.8 IFN-γ−/−/TNF-α−/− double-knockout mice were monitored for EAE clinical signs due to the higher incidence of disease in female mice. All mice were bred and maintained in a specific pathogen-free facility at the South Lake Union Campus of the University of Washington. All procedures were approved by the University of Washington Institutional Animal Care and Use Committee.
CD8 T cell enrichment
Single-cell suspensions were prepared from splenocytes from naive C3HeB/FeJ (WT), 8.8, 8.8 IFN-γ−/−, 8.8 Pfp−/−, 8.8 TNF-α−/−, and 8.8 Faslgld mice as previously described (19). CD8 T cell enrichment was performed by incubating cells with biotinylated Abs (all from BioLegend) specific for CD4 (RM4-5), B220 (RA3-6B2), CD11b (M1/70), CD11c (N418), and TER-119 (TER-119) and then removing labeled cells with magnetic streptavidin particles (557812, BD Biosciences) according to the manufacturer’s instructions. The average frequency of CD8 T cells in the enriched populations from mice of all genotypes ranged from 53.6 ± 1.6% to 54.9 ± 1.1%, with the remaining populations consisting mostly of CD45− cells (13–31%), CD4 T cells (0–13%), and B cells (1.8–9%).
Virally induced EAE
Vac-WT and Vac-MBP were obtained and grown as previously described (15). Disease was induced in female intact TCR-transgenic 8.8 mice 8–12 wk of age by i.p. infection with 3–5 × 106 PFU of Vac-WT. Control WT mice also received 3-5 × 106 PFU of Vac-WT. In an alternative disease induction protocol, female WT mice 6–10 wk of age were injected with 1 × 106 CD8 T cells enriched from either WT, 8.8, 8.8 IFN-γ−/−, 8.8 Pfp−/−, 8.8 TNF-α−/−, or 8.8 FasL−/− mice generated as described above and infected with 3–5 × 106 PFU of Vac-MBP on the same day. Mice were monitored for EAE symptoms and weighed daily; they were euthanized when more than >20% of original body weight was lost or a clinical score of ≥5 was reached.
Clinical scoring scale
Classic EAE signs were scored as follows: grade 0.5, compromised tail tone; grade 1, fully paralyzed tail; grade 2, hindlimb weakness; grade 3, one paralyzed hindlimb; grade 4, two paralyzed hindlimbs; grade 5, forelimb weakness; grade 6, moribund. Atypical EAE clinical signs were scored as follows: grade 1, hyperactive; grade 2, slight ataxia, lowered pelvis; grade 3, mild ataxia, mild body lean; grade 4, moderate ataxia, moderate body lean; grade 5, severe ataxia, severe body lean; grade 6, rolling. Grades 5 and 6 were considered severe EAE and mice were euthanized if they reached a clinical score of ≥5.
Flow cytometry
Single-cell suspensions from the brain, spinal cord, spleen, and deep cervical lymph node (dCLN) dissected from perfused mice were prepared as previously described (20). Live and dead cells were discriminated by incubation with an amine-reactive dye (L10119, Invitrogen; P30253, Molecular Probes) for 10 min at 4°C. After washing, cells were incubated with Fc Block (BDB553142, eBioscience) in 5% mouse serum (08642931, MP Biomedicals) for 15 min at 4°C, washed, and stained with Abs for 30 min at 4°C, except for chemokine receptor–specific Abs that were incubated with cells for 1 h at 37°C prior to additional cell staining. T and B cell cytokine production levels were analyzed directly ex vivo by incubating cells only with GolgiPlug (51-2301K7, BD Biosciences) for 4 h at 37°C. Intracellular cytokine staining was performed using the Cytofix/Cytoperm kit (555028, BD Biosciences) according to the manufacturer’s instructions. Cells were gated on single/live/CD45+ or in some cases single/live/MBP/Kk+/CD45+. Neutrophils were identified as CD11b+Ly6G+ cells. T cells were identified as Ly6G−TCRβ+CD4+ or CD8+. B cells were identified as Ly6G−TCRβ−CD19+. Plasma cells were identified as Ly6G−TCRβ−CD11b−CD138+B220− whereas plasmablasts were identified through the same gate as CD138+B220+. Microglia were identified as Ly6G−TCRβ−CD19−CD138−CD11bintCD45intLy6C−. Myeloid cells were identified as Ly6G−TCRβ−CD19−CD138−CD11b+CD45+, with monocytes identified as Ly6C+MHC class II− myeloid cells and monocyte-derived cells (MdCs) were MHC class II+ myeloid cells. T cell phenotypes were characterized as either naive (CD44−CD62L+), central memory (Tcm; CD44+CD62L+), tissue-resident memory (Trm; CD44+CD62L−CD69+CD103+), effector memory (Tem; CD44+CD62L−CD103−KLRG1−), or effector (Teff; CD44+CD62L−CD103−KLRG1+) phenotype. B cell subsets were defined as IgD (IgD+/IgM+/−), IgM (IgD−/IgM+), and class-switched (IgM−IgD−). Data were acquired with an Aurora spectral flow cytometer (Cytek Biosciences) cytometer and analyzed using FlowJo software (Tree Star).
Abs used
Abs specific for CD4 (1:600; 563232), Ly6C (1:600; 561237), I-Ak (1:200; 553536), IFN-γ (1:300; 562019), GM-CSF (1:300; 554406), CD62L (1:600; 560184), KLRG1 (1:300; 564014), CD69 (1:400; 563290), CD103 (1:400; 562771), CD19 (1:600; 552854), CD138 (1:400; 563147), IgM (1:300; 743328), and IgD (1:300; 565988), and isotype controls for GM-CSF (1:300; IgG2a), IL-17 (1:300; IgG1,κ), TNF-α (1:300; IgG1,κ), and IFN-γ (1:300; IgG1,κ) were from BD Biosciences. Abs specific for CD8 (1:600; 67008182), CD38 (1:400; 56-0381-82), GL7 (1:400; 48-5902-82), CD73 (1:300; 25-0731-82), and CD44 (1:1000; 17044182) were from Invitrogen. Abs specific for TCRβ (1:400; 109226), CD45 (1:600; 103138), Ly6G (1:600; 127607), TNF-α (1:300; 506324), IL-17 (1:300; 506912), Thy1.2 (1:200; 105320), TNF-α (1:300; 506333), CD11c (1:300; 117339), CXCR4 (1:200; 146511), and CCR6 (1:200; 129807) were from BioLegend. Abs specific for CD11b (1:1000; 45-0112-82), CD80 (1:300; 12-0801-82), and IL-6 (1:300; 11706182) were from eBioscience. The 12H4 Ab specific for MBP/Kk was generated and validated as previously described (21) and used at a 1:100 dilution.
Histology
Tissues were fixed in 10% neutral-buffered formalin, trimmed, embedded in paraffin, and sectioned for either staining with H&E or immunohistochemistry (IHC) analyses performed by the Histology and Imaging Core at the University of Washington. IHC was performed using a Leica Bond automated immunostainer and Leica reagents and protocols (Leica Microsystems, Buffalo Grove, IL). mAbs and dilutions used for these studies were rat anti-mouse CD4 (1:800; 14-9766-80) and rat anti-mouse CD8 (neural-specific) (1:500; 14-0195-82) from Thermo Fisher Scientific (Waltham MA) and rabbit anti-CD19 (1:1000; ab245235) and rat anti-MBP (1:500, ab7349) from Abcam (Waltham MA). Polyclonal Abs were rabbit anti-Mac2 (galectin-3) (1:25; LS-B5661) from LSBio (Seattle, WA) and rabbit anti-GFAP (1:500; Z033401) from Dako/Agilent Technologies (Santa Clara, CA). Ag retrieval was performed using appropriate dilutions of citrate buffer (CD4, CD8, MBP), proteinase K (GFAP), or EDTA buffer (CD19). All bound Abs were detected using Leica Bond polymer diaminobenzidine. Tissues were examined by a board-certified veterinary pathologist experienced in the evaluation of mouse models of MS who was blinded to group assignments. Tissue injury was evaluated histologically using a semiquantitative grading system. Lesions, principally from H&E-stained sections, were graded for degree of inflammatory cell accumulation and tissue injury on an all-inclusive severity scale of 0 (normal) to 4+ (severe). Based on preliminary studies, histologic evaluation focused on specific regions of the brain including the cerebellum, hippocampus, thalamus, hypothalamus, and pons as well as areas within the immediate vicinity of these sites; however, all regions of the brain were examined and changes noted. Findings that were considered in the overall assessment and score assignments included the intensity and distribution of inflammatory cell accumulation, the presence of necrosis or apoptosis, and the degree of staining with IHC markers.
Functional analyses of 8.8 CD4 and CD8 T cells
Cytokine production
Single-cell suspensions of splenocytes were prepared from 8.8 healthy mice, stained with anti-Thy1.2, and Thy1.2+ T cells were sorted on a FACSAria II (BD Biosciences). Single-cell suspensions of splenocytes were also prepared from β2m−/− and WT mice, labeled with biotinylated anti-Thy1.2 (105304, BioLegend), and Thy1.2+ cells were depleted using magnetic streptavidin particles (557812, BD Biosciences) according to the manufacturer’s instructions. Thy1.2-deficient splenocytes were plated at 6 × 105 cells/well and incubated with 10 μM MBP peptide (MBP79–87, ChinaPeptides) or PBS for 30 min at room temperature (RT). Cells were washed and 3 × 105 8.8 Thy1.2+ T cells were added to each well. Cultures were incubated with GolgiPlug (512301KZ, BD Biosciences) for 4 h at 37°C followed by intracellular staining for TNF-α overnight.
Proliferation
Single-cell suspensions of splenocytes from healthy 8.8 mice were incubated with 5 μM CellTrace Violet dye (C34571, Molecular Probes) for 5 min at RT, washed, and plated at 1 × 107 cells/well. Cells were incubated either with MBP peptide (10 μM) or PBS for 24, 48, and 72 h. Dilution of dye in 8.8 CD4 and CD8 T cells was analyzed with an Aurora spectral flow cytometer (Cytek Biosciences).
ELISA
Assays were conducted in 96-well plates (Corning) coated with 2 μg/ml lyophilized human MBP (M0689-1MG, Sigma-Aldrich) resuspended in PBS and incubated at 4°C overnight. Plates were washed with PBS containing 0.05% Tween 20 (PBST) and incubated with blocking buffer (PBST with 3% powdered milk) for 1 h at RT. Serial dilutions of serum in buffer (PBST with 1% milk) were added to wells in duplicate and incubated for 2 h at RT. Secondary Abs were diluted in buffer as follows: anti-mouse pan-IgG-HRP (A16078, Invitrogen) at 1:1500, and anti-mouse IgM-HRP (1139-05, SouthernBiotech) at 1:3000. Plates were washed with PBST and incubated with secondary Ab for 1 h at RT. Plates were washed and bound secondary Ab was detected with 1× 3,3′,5,5′-tetramethylbenzidine (TMB; 00-4201-56, Invitrogen) and quenched with 1 M HCl. Sample OD was measured at 450–570 nm. Positive controls were serum collected from WT mice immunized with whole lyophilized MBP in CFA. Standard controls were generated using a mouse IgG ELISA kit and mouse IgM ELISA kit (01998, 01999, STEMCELL Technologies). The area under the curve was determined using Prism (GraphPad).
Anti-CD20 treatment
Mice were anesthetized with isoflurane and injected with anti-CD20 Ab (clone 18B12, InvivoGen) at 10 mg/kg in PBS via retro-orbital injection. Control mice were treated with isotype Ab (clone 2B8, InvivoGen) at 10 mg/kg in PBS. Mice were treated on days 1–3 after symptom onset and then every 7 d for 5 wk. Mice were monitored daily for 5 wk by a researcher blinded to treatment groups.
Statistical analysis
Data are presented as mean ± SEM unless otherwise stated in the figure legends. GraphPad Prism (8) was used for all analyses, and p values <0.05 were considered statistically significant.
Results
Virally activated 8.8 T cells require IFN-γ and perforin to cause brain and spinal cord injury
Infection of intact 8.8 TCR-transgenic mice with Vac-WT induced a high incidence of both classic EAE signs associated with spinal cord inflammation and atypical EAE signs associated with brain inflammation (Fig. 1A). To define the mechanisms by which virally activated 8.8 T cells induced disease, we developed an alternative disease induction protocol that was used to generate the data in (Fig. 1B (all other disease induction experiments used intact TCR-transgenic 8.8 mice infected with Vac-WT). This model allowed us to test 8.8 CD8 T cells lacking specific effector functions for their ability to trigger disease. 8.8 CD8 T cells were introduced into the periphery of WT mice, and the recipient mice were infected with Vac-MBP. Vac-MBP was used because the frequency of 8.8 T cells expressing dual TCRs that are activated by Vac-WT is much lower in these recipient mice compared with intact TCR-transgenic mice. Disease susceptibility was compared between recipients of 8.8 CD8 T cells versus 8.8 CD8 T cells deficient in IFN-γ (IFN-γ−/−), perforin (Pfp−/−), TNF-α (TNF-α−/−) or FasL (FasL−/−). Vac-MBP–infected recipients of 8.8 CD8 T cells with no genetic deficiencies exhibited a similar, albeit slightly reduced, incidence of atypical and classic EAE signs compared with Vac-WT–infected intact TCR-transgenic 8.8 mice (Fig. 1B). Atypical symptoms were primarily associated with cerebellar dysfunction, including wide and dragging gait and reduced pelvic elevation (22). Classic EAE was manifested mainly as a limp tail. Recipients of 8.8 CD8 TNF-α−/− and FasL−/− T cells developed EAE with the same incidence, clinical signs, and severity as mice that received 8.8 CD8 T cells (data not shown). In contrast, recipients of 8.8 CD8 IFN-γ−/− or Pfp−/− T cells exhibited no clinical signs (Fig. 1B), indicating that both IFN-γ and perforin, but not TNF-α or FasL, are required for virally activated 8.8 CD8 T cells to induce EAE.
FIGURE 1.

Virally activated 8.8 T cells induce EAE via an IFN-γ– and perforin-mediated mechanism. (A and C–E) Intact 8.8 TCR-transgenic and WT female mice were infected with Vac-WT virus; (B) female WT mice that received either 8.8, 8.8 IFN-γ−/−, or 8.8 Pfp−/− CD8 T cells were infected with Vac-MBP virus. (A) Incidence, day of onset, and mean maximum clinical score corresponding to classic or atypical EAE symptoms are shown for 8.8 mice infected with Vac-WT (n = 23), compiled from five independent experiments). (B) This experiment used the adoptive transfer disease induction protocol in which WT mice were injected with CD8 T cells enriched from either WT mice or 8.8 mice on the indicated genetic backgrounds and then the recipient mice were infected with Vac-MBP. The cumulative incidence of classic (left) and atypical (right) clinical signs seen each day following infection of recipient WT mice that received 8.8 (n = 25), 8.8 IFN-γ−/− (n = 15), or 8.8 Pfp−/− (n = 7) CD8 T cells, compiled from three independent experiments. (C) Absolute numbers of infiltrating immune cells was determined by flow cytometry in the brains of 8.8 (n = 11) and WT (n = 7) mice infected with Vac-WT. Gating strategy is shown in Supplemental Fig. 1A. (D) Percentages of CD8 T cells exhibiting either a naive, Tcm, Trm, Tem, or Teff phenotype in the brains of 8.8 mice (n = 15) or WT mice (n = 8) at 8 d postinfection with Vac-WT were determined by flow cytometry and compiled from four independent experiments. Graphs show mean ± SEM; gating strategy is shown in Supplemental Fig. 1B. (E) The absolute numbers of MBP/Kk+ cells in the brains of 8.8 (n = 11) and WT (n = 7) mice were determined by flow cytometry at 8 d after Vac-WT infection. Graphs show mean ± SEM and are compiled from three independent experiments. Gating strategy is shown in Supplemental Fig. 1C. Statistical significance was determined in (B) with a Fisher’s exact test: *p < 0.05, **p < 0.01, ***p < 0.001, for differences between infected mice that received 8.8 CD8 T cells and mice that received 8.8 IFN-γ−/− cells; ##p < 0.01, for differences between infected mice that received 8.8 CD8 T cells and mice that received 8.8 Pfp−/− cells. Statistical significance in (C)– (E) was determined using a Mann–Whitney U test: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
In EAE induced in intact 8.8 TCR-transgenic mice by infection with Vac-WT virus, T cells were the predominant infiltrating cell type in the brains (Fig. 1C) and spinal cords (data not shown). WT mice infected with Vac-WT virus exhibited a similar composition of infiltrating cells; however, the T cell number was significantly reduced compared with infected 8.8 mice (Fig. 1C), consistent with preferential accumulation in the brain of CNS Ag-specific T cells. CD8 T cells in the brains of both 8.8 and WT mice were primarily Teff and Tem cells (Fig. 1D). The bias in 8.8 mice toward a Tem versus Teff phenotype may reflect more retention of 8.8 compared with WT T cells in the brain. Virally infected intact 8.8 TCR-transgenic mice required euthanasia after 9 d due to weight loss (15), which may account for their low percentage of Trm cells.
Multiple cell types present MBP/Kk in the CNS of infected 8.8 mice
We employed an Ab that detects the MBP/Kk ligand (21) to identify which cell types present MBP during virally induced EAE. Intact TCR-transgenic 8.8 mice infected with Vac-WT were used to detect only the cells presenting MBP derived from endogenous sources and not cells presenting the ligand as a result of infection with Vac-MBP. Multiple cell types presented MBP/Kk in the brains of 8.8 mice following Vac-WT infection (Fig. 1E), with a significantly higher number of MBP/Kk+ microglia, MdCs, monocytes, T cells, and CD45+/CD11b−/non-T/non-B cells (labeled as Other) found in infected 8.8 versus WT mice. Expression of MBP/Kk on 8.8 T cells is consistent with our observation that 8.8 T cells can strip their ligand from the surface of APCs (14); no MBP/Kk+ T cells were seen in infected WT mice. The few MBP/Kk+ cells seen in infected WT mice belonged to the microglia, B cell, and neutrophil cell subsets (Fig. 1E). Thus, viral infection alone caused little MBP/Kk presentation, but the autoimmune response initiated by virally activated 8.8 T cells resulted in cross-presentation of the MBP ligand by multiple cell types.
EAE induced in 8.8 TCR-transgenic mice by Vac-WT infection targets the CNS parenchyma with minimal demyelination
CNS tissues from eight intact 8.8 TCR-transgenic mice with EAE induced by Vac-WT infection were examined histologically. All brains were minimally to mildly affected (severity of 1–2+). Primary targets were the cerebellum and areas within the immediate vicinity of the hippocampus (Fig. 2A–G). A few mice also displayed minimal or rarely mild lesions in the thalamus, hypothalamus, and pons (Fig. 2H, 2I); no lesions were seen in the meninges, cerebral cortex, or olfactory bulb. Lesions were widely scattered in white and gray parenchymal tissues with CD8, and fewer CD4 T cells accumulated around blood vessels and dispersed in surrounding areas (Fig. 2D, 2E, 2I). No B cells were detected. Lesions occasionally displayed GFAP+ foci reflecting mild astrogliosis (Fig. 2C), but there was no histologic evidence of demyelination (Fig. 2F) and only scant necrotic or apoptotic change. Analyses of the spinal cords revealed that seven of eight mice had minimal to occasionally mild changes with CD8 and CD4 lymphocytes widely scattered throughout the parenchyma without accompanying demyelination or necrosis (data not shown). Overall, the pathological features of lesions associated with parenchymal blood vessels with little meningeal inflammation or demyelination were similar in the brains and spinal cords.
FIGURE 2.
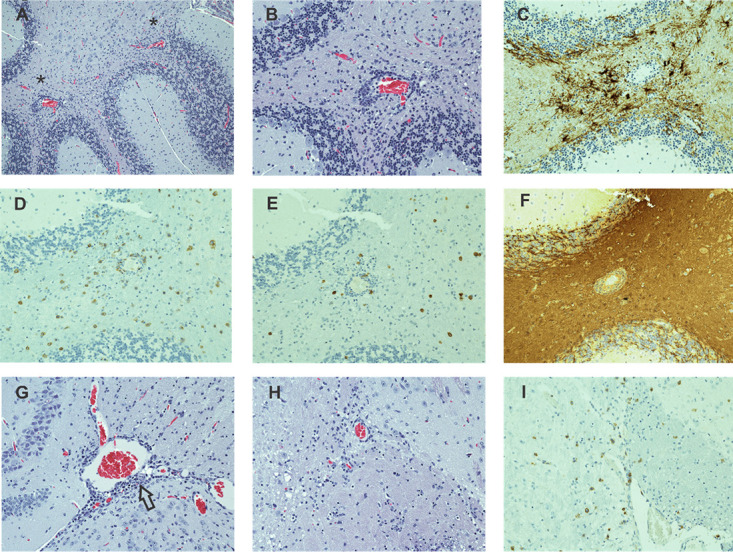
Vac-WT induced EAE affects both the gray and white matter in the brain parenchyma. (A–I) Sections from an intact 8.8 TCR-transgenic mouse with EAE induced by Vac-WT infection are shown from the cerebellum (A–F), hippocampal region (G), and hypothalamus (H, I) (original magnification ×20). (A) H&E analyses of a cerebellar section reveals a predominance of white matter lesions manifested as perivascular accumulations of lymphocytes. (B) Higher power magnification of the region denoted by an asterisk in (A) demonstrates the predominantly perivascular localization of lymphocytes as well as their widely scattered presence in the surrounding parenchyma. (C) IHC staining for GFAP shows increased numbers and prominence of GFAP+ cells associated with inflammatory foci. IHC staining for CD8 (D) and CD4 (E) demonstrates a preponderance of CD8 T cells both in perivascular spaces and widely scattered in the tissue parenchyma, with fewer numbers of intermixed CD4+ cells. (F) IHC staining for MBP in the same region as (D) demonstrates an absence of lesion-associated demyelination. (G) H&E staining of the hippocampal region, another primary site of involvement, reveals histologically mild, mostly perivascular lesions (arrow). (H) H&E staining in the hypothalamic region shows mild lesions with scattered lymphoid cells similar to those seen in the cerebellum. (I) IHC staining for CD8 demonstrates that many of the lymphoid cells in the hypothalamic region are CD8 T cells.
8.8 Mice develop SEAE
SEAE was not seen in our prior studies of 8.8 mice (14); therefore, we were surprised to observe mice develop clinical signs in our breeding colony of 8.8 IFN-γ−/− mice beginning at 16 wk of age. To determine whether development of SEAE required IFN-γ deficiency, we monitored cohorts of 8.8 IFN-γ−/− and 8.8 IFN-γ+/+ mice aged to 33 wk (Fig. 3A). Interestingly, SEAE incidence was significantly biased toward females in 8.8 IFN-γ−/− mice. A lower incidence of SEAE with later onset was observed in 8.8 IFN-γ+/+ mice; the number of 8.8 IFN-γ+/+ mice that developed disease in this study was too small to assess sex differences. The clinical course in both male and female 8.8 IFN-γ−/− mice began with atypical signs of proprioception defects followed by an uneven gait and lowered pelvis, which progressed to ataxia and frequently a body lean. The same clinical signs but with a slower progression developed in IFN-γ+/+ mice. Most mice also exhibited some loss of tail tone but few developed a fully limp tail.
FIGURE 3.

8.8 IFN-γ−/− mice develop SEAE associated with CD4 T cells. (A) Incidence of SEAE observed in cohorts of 8.8 IFN-γ−/− male (n = 32) and female (n = 37) mice, and in 8.8 IFN-γ+/+ male (n = 33) and female (n = 24) mice. (B–E) Data were analyzed by flow cytometry. (B) Absolute numbers of immune cell types found in the brains of 8.8 IFN-γ−/− (n = 13) and 8.8 IFN-γ+/+ (n = 3) mice with SEAE analyzed 2–4 d after disease onset and in age-matched 8.8 healthy mice (n = 11) are shown; gating strategy is shown in Supplemental Fig. 2A. Graphs show mean ± SEM; data are compiled from nine independent experiments. (C) Percentages of CD4 and CD8 T cells found in the brains of 8.8 IFN-γ−/− SEAE mice (n = 10) harvested 2–4 d after disease onset and age-matched 8.8 healthy mice (n = 6) are shown that exhibit either a naive, Tcm, Trm, Tem, or Teff phenotype; gating strategy is shown in Supplemental Fig. 2B. Mean ± SEM is shown; data are pooled from three independent experiments. (D) CD4/CD8 ratio is shown for T cells in the brains of healthy WT (n = 8) and 8.8 (n = 19) mice, as well as 8.8 IFN-γ−/− (n = 27) and 8.8 IFN-γ+/+ (n = 8) SEAE mice (2–4 d after disease onset) and Vac-WT–infected 8.8 (n = 7) and WT (n = 7) mice at 8 d postinfection. Graphs show mean ± SEM; data are compiled from 17 independent experiments. (E) Cytokine production was analyzed directly ex vivo (without in vitro Ag stimulation) by CD4 and CD8 T cells from the brains of 8.8 IFN-γ−/− mice with SEAE at 2–3 d after onset (n = 12) and age-matched 8.8 healthy mice (n = 11). The mean percentages ± SEM of cytokine-producing cells pooled from 12 independent experiments are shown. Statistical significance was determined for (A) using a Fisher’s exact test; *p < 0.05, **p < 0.01, for differences in disease incidence between 8.8 IFN-γ−/− female and male mice; (C and D) a Kruskal–Wallis with Dunn’s posttest (Vac-WT–infected mice in C were compared separately from mice with spontaneous EAE); and (B) and (E) a Mann–Whitney U test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
The infiltrates in the brains of 8.8 IFN-γ−/− and IFN-γ+/+ SEAE mice were very similar but distinct from those seen in the brains of mice with virally induced EAE (Figs. 1C, 3B). In contrast to the absence of B cells in brains of infected 8.8 mice, comparable numbers of B and T cells were seen in the brains of both 8.8 IFN-γ−/− and 8.8 IFN-γ+/+ mice with SEAE (Fig. 3B). Monocytes, MdCs, and neutrophils represented a much smaller percentage of the infiltrate compared with T and B cells in SEAE mice, although their numbers were significantly increased compared with those seen in healthy mice. Both CD4 and CD8 T cells in the brains of SEAE mice were predominantly Tem cells, with more Trm cells observed compared with mice with virally induced EAE (Fig. 3C). The ratio of CD4 to CD8 T cells in the brain was also strikingly different for SEAE mice versus virally induced EAE (Fig. 3D). In healthy 8.8 mice, more CD4 than CD8 T cells populate the periphery; however, only CD8 T cells are present in 8.8 Rag−/− mice (15). This indicates that rearrangement of endogenous TCR chains allows many thymocytes to be selected into the CD4 lineage despite expression of the transgenic 8.8 TCR. Vaccinia infection specifically expanded CD8 T cells in the periphery of both 8.8 and WT mice such that the CD4/CD8 ratio was skewed toward CD8 in both the brain and spleen. In contrast, the CD4/CD8 ratio was similarly skewed toward CD4 in both healthy 8.8 mice and 8.8 IFN-γ+/+ SEAE mice, and the ratio was the same in both brain and spleen. The CD4/CD8 ratio was also skewed toward CD4 in 8.8 IFN-γ−/− with SEAE mice; however, the CD4/CD8 ratio was significantly higher in the brain compared with the spleen in these mice (Fig. 3D). This suggests that CD4 T cells were specifically enriched in the brain compared with the periphery when SEAE developed in the absence of IFN-γ. To confirm that 8.8 CD4 T cells still recognized MBP and that MBP recognition was MHC class I–restricted, we compared the proliferative and functional responses of 8.8 CD4 and CD8 T cells stimulated with MBP. Both 8.8 CD4 and CD8 T cells proliferated to the same extent and with similar kinetics when stimulated with MBP79–87 (Supplemental Fig. 3A). Importantly, 8.8 CD4 and CD8 T cells produced similar amounts of TNF-α when cultured with MBP79–87 presented by WT APCs, but neither subset produced TNF-α when MBP was presented by β2m−/− APCs (Supplemental Fig. 3B). These data indicate that MBP recognition is strictly MHC class I–restricted for both 8.8 CD4 and CD8 T cells, and that expression of endogenously rearranged TCR genes in CD4 T cells does not compromise their response to MBP.
SEAE in 8.8 IFN-γ−/− mice is TNF-α–dependent
To investigate which T cell effector functions contributed to the pathogenesis of SEAE, we analyzed cytokine production by CD4 and CD8 T cells in the brains of sick 8.8 IFN-γ−/− mice directly ex vivo. TNF-α was the predominant cytokine produced by the CD4 T cells, with only a very small percentage of GM-CSF+ and no IL-17+ cells detected (Fig. 3E). A small number (<5%) of CD8 T cells in the brains of these mice also produced TNF-α, which was not significantly different from that seen for CD8 T cells in healthy 8.8 mice (Fig. 3E). No IL-17 or GM-CSF production was observed for CD8 T cells from sick mice. We generated 8.8 IFN-γ−/−TNF-α−/− mice and monitored 15 double-knockout mice for clinical signs until 33 wk of age. None of these mice developed disease. These data indicate that TNF-α is required for SEAE in this model and that CD4 T cells are the predominant producers of this cytokine.
SEAE targets the brain with lesions resembling active lesions in MS
Histological examination of brains and spinal cords from eleven 8.8 IFN-γ−/− SEAE mice revealed consistent development of moderate to severe lesions (grades 3–4+) involving the cerebellum (7/11 affected), the hippocampus (8/11 affected), and closely adjacent areas (Fig. 4). The remainder had mild–moderate changes in these same sites. There was also frequent but generally minimal–moderate involvement of the hypothalamus and less commonly the pons and thalamus. The cerebral cortex and olfactory bulbs were not affected. Focal leptomeningitis was occasionally present especially within deeper areas of the brain and particularly near areas of parenchymal injury (Fig. 4A, 4J). Lesions occurred principally in white matter, although adjacent gray matter could be involved. Lesions were characterized by parenchymal loss, extensive demyelination, and accumulation of lymphoid cells in perivascular sites as well as clustered or scattered in adjacent tissues. There was no evidence of vascular wall injury or thrombosis. In more moderate to severe foci, marked astrogliosis was accompanied white matter injury (Fig. 4L), and striking loss of myelin appeared to extend in a gradient outward from the center of the lesions (Fig. 4G, 4M). Many phagocytes within the demyelinated region contained MBP+ debris (Fig. 4N), and dense accumulations of Mac2+ phagocytes were seen localized within demyelinated regions (Fig. 4H, 4I, 4O). CD4 T cells and B cells were present in moderate to large numbers in perivascular locations (Fig. 4D, 4E, 4P, 4Q) with CD8 T cells rarely detected (data not shown). CD4 T cells were also observed scattered in extraperivascular sites (Fig. 4R), whereas B cells were almost never seen outside of perivascular locations. Tertiary lymphoid structures or meningeal lymphocyte aggregates were not present. The spinal cord from 1 of 11 mice had focal cervical and thoracic lesions similar to those described for the brain (data not shown). The remainder had minimal to mild lesions consisting of widely scattered to focal accumulations of CD4 T cells (6/11) or were normal (4/11). Histologic analyses of liver, lung, and kidney sections from seven randomly selected cases revealed no evidence of systemic disease. Taken together, these analyses revealed a predominance of demyelinated brain lesions with many similarities to active lesions seen in MS brain sections.
FIGURE 4.
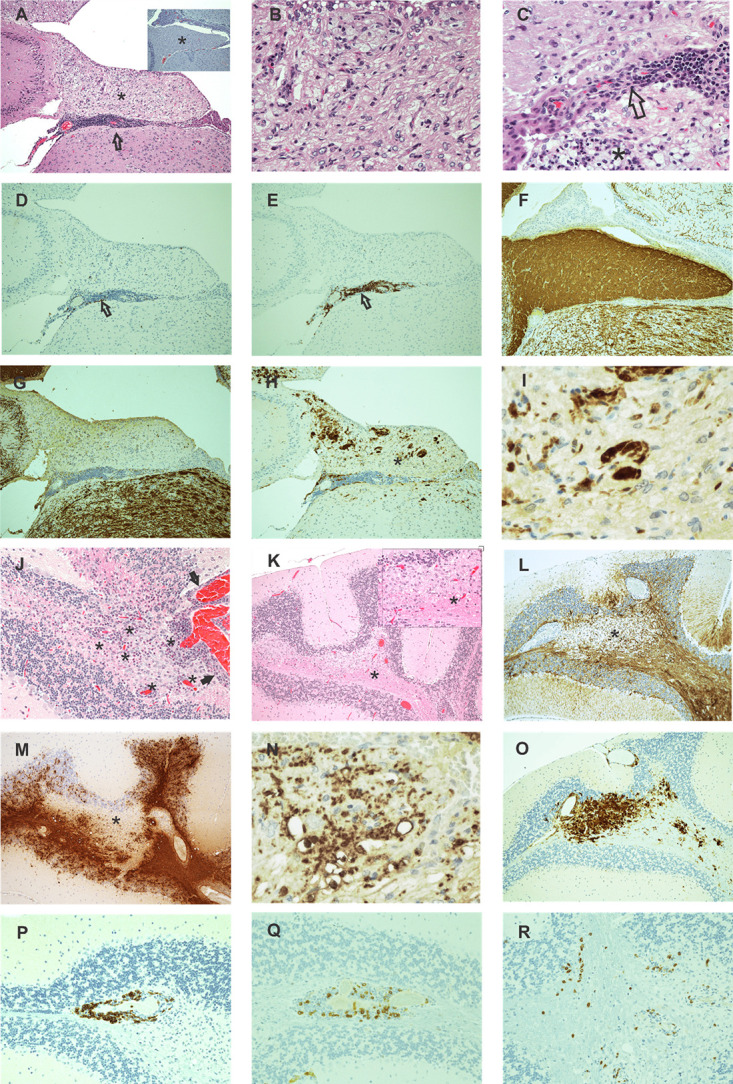
SEAE in 8.8 mice targets white matter in the brain. (A–I) Serial sections of the hippocampal fimbria were analyzed from an 8.8 IFN-γ−/− SEAE mouse. (A) Loss of H&E staining density when compared with normal tissue (inset) is shown. (B) Higher magnification of this same region (asterisk) shows accumulation of foamy macrophages, a few lymphocytes, and loss of normal tissue architecture principally from myelin loss. Lymphocyte accumulations in the hippocampus are prominent in adjacent leptomeningeal tissues (A, arrow) with occasional proximity to ependymal lining structures (C, arrow) or small foci of necrosis and apoptosis (C, asterisk). (D and E) IHC labeling reveals CD4+ (D, arrow) and CD19+ (E, arrow) lymphocytes within lesions. (F and G) IHC MBP staining contrasts uniform brown staining myelin in a normal fimbria (F) with dramatic loss of myelin in an SEAE mouse fimbria (G). (H) IHC MAC2 labeling reveals MAC2+ phagocytic cells within the demyelinated region. (I) Higher magnification of the area marked in (H) (asterisk) shows clusters of MAC2+ cells. (J–R) Cerebellum from another SEAE mouse shows a similar pattern of injury. (J) H&E staining of the cerebellum shows a lesion with numerous lymphocytes, many of which are perivascular (asterisks) along with a few necrotic and apoptotic elements all predominantly in white matter but with limited involvement of adjacent gray matter and leptomeninges (arrows). (K) Lower magnification of an H&E-stained section of a different cerebellar section shows a large pale-staining focus (asterisk) in the medulla. Inset is a higher magnification of the region (asterisk) in which foamy macrophages and other reactive cells predominate. (L) IHC GFAP staining in the same area (deeper section) shown in (K) shows marked astrocytosis surrounding the pale focus. (M) IHC MBP staining shows that the pale focus shown in (K) is devoid of myelin. (N) Higher power magnification of the region in (M) marked with an asterisk reveals MBP+ fragments within phagocytes. (O) IHC MAC2 staining shows accumulation of numerous MAC2+ cells replacing most parenchymal cells within this area. (P) IHC CD19 staining shows B cells tightly localized within the perivascular space while CD4+ cells are present perivascularly (Q) and dispersed in adjacent tissues (R). Original magnification ×10 for all images including the inset in (A) except for the following: original magnification ×40 for (B), (C), and the inset for (K); original magnification ×20 for (J) and (P)–(R); original magnification ×60 for (I) and (N).
B cells exhibit an activated/memory, proinflammatory phenotype in SEAE brains
We compared the phenotype and function of B cells isolated from the brains of 8.8 IFN-γ−/− SEAE mice versus healthy 8.8 mice. No differences in the small numbers of plasmablasts and plasma cells were observed between sick and healthy mice (Fig. 3B); however, the B cell numbers in three different subsets defined as IgD (IgD+/IgM+/−), IgM (IgD−/IgM+), and IgM−IgD− (class-switched) were significantly increased in the brains of SEAE mice (Fig. 5A). The frequency of CXCR4+ cells was higher in all three B cell subsets, and the frequency of CCR6+ cells was higher in the IgM and class-switched subsets in SEAE compared with healthy brains (Fig. 5B). CXCR3 was not detected on B cells in the brains of either sick or healthy mice (data not shown). Analyses of expression of CD73 and CD80, markers associated with memory B cells (23), demonstrated that all three B cell subsets in SEAE mice had a higher frequency of CD73+CD80+ cells compared with healthy mice (Fig. 5C). The IgD and IgM subsets in sick mice also had a higher frequency of cells expressing CD80 without coexpressing CD73. The frequency of B cells expressing CD73 without CD80 was very low in both healthy and sick mice. The frequency of CD11c+ B cells was also increased in all subsets in sick compared with healthy mice, especially in the IgM and class-switched subsets (Fig. 5D). A significantly higher frequency of IL-6+ and TNF-α+ B cells was observed directly ex vivo in the brains of 8.8 IFN-γ−/− SEAE mice compared with 8.8 healthy mice (Fig. 5E). No GM-CSF+, granzyme B+, or IL-10+ B cells were detected in the brains of sick or healthy mice (data not shown). These data indicate that many B cells that infiltrated the brain during SEAE exhibit an activated, proinflammatory phenotype.
FIGURE 5.

B cells in the brains of mice with SEAE exhibit an activated, proinflammatory phenotype. (A–E) Mononuclear cells isolated from the brains of 8.8 IFN-γ−/− mice with SEAE (filled bars) or healthy, age-matched 8.8 mice (open bars) were analyzed by flow cytometry; gating strategy is shown in Supplemental Fig. 4. Graphs show mean ± SEM. (A) Absolute numbers of IgD+, IgM+, and IgD−IgM− B cells for 8.8 IFN-γ−/− mice with SEAE (n = 25) and healthy 8.8 mice (n = 15) are shown. (B) The percent of CXCR4+ cells in each B cell subset (top) is shown for 8.8 IFN-γ−/− SEAE (n = 20) and 8.8 healthy mice (n = 10), and the percent of CCR6+ cells in each B cell subset (bottom) is shown for 8.8 IFN-γ−/− SEAE (n = 16) and 8.8 healthy mice (n = 12). (C) The percent in each B cell subset expressing different combinations of CD80 and CD73 is shown for 8.8 IFN-γ−/− SEAE (n = 18) and 8.8 healthy (n = 17) mice. (D) The percent of CD11c+ cells in each B cell subset is shown for 8.8 IFN-γ−/− SEAE (n = 20) and 8.8 healthy mice (n = 13). (E) The percent of cytokine+ B cells for 8.8 IFN-γ−/− SEAE (n = 17) and 8.8 healthy (n = 6) mice was determined directly ex vivo. (A–D) Data are compiled from at least seven independent experiments; (E) data are compiled from five independent experiments. All statistical significance was determined using a Mann–Whitney U test: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
MBP/Kk is presented predominantly by B cells in the brains of SEAE mice
The overall frequency of MBP/Kk+ cells was significantly higher in the brain compared with the spinal cord in 8.8 IFN-γ−/− SEAE mice, with a similar trend seen in 8.8 IFN-γ+/+ SEAE mice (Fig. 6A), consistent with the clinical presentation and histological analyses that indicate the brain is the major target in this model. Unexpectedly, B cells comprised the largest number of MBP/Kk+ cells in the brains of both 8.8 IFN-γ−/− and IFN-γ+/+ SEAE mice, and the absolute number of MBP/Kk+ B cells was significantly higher in these mice compared with 8.8 healthy mice (Fig. 6B). This strongly contrasts with our studies of EAE induced by adoptive transfer of CD4 T cells that identified MdCs and monocytes as the major MBP/Kk+ APCs (18). In support of the notion that the increase in MBP/Kk+ B cells seen in SEAE mice reflects increased Ag presentation to 8.8 T cells, we found that the titers of MBP-specific IgG but not IgM Abs were significantly increased in the serum of SEAE compared with healthy 8.8 mice (Fig. 6C). Interestingly, within the brains of 8.8 IFN-γ−/− SEAE mice, not only was the absolute number but also the frequency of MBP/Kk+ cells significantly increased for B cells compared with MdCs and monocytes (Fig. 6D). A comparison of the MBP/Kk+ frequency between the brain, dCLNs, and spleen for each cell type revealed a greater increase in the frequency of MBP/Kk+ cells in the brain relative to peripheral tissues for B cells compared with other cell types (Fig. 6D). Notably, the frequency of MBP/Kk+ B cells in the brains of 8.8 healthy mice, while much lower than that seen in mice with SEAE, was also significantly increased compared with the dCLNs and spleens (Fig. 6E). Importantly, no difference was seen in the frequency of MBP/Kk+ B cells between brain and peripheral tissues in WT mice, indicating that expression of the MBP-specific TCR in 8.8 mice enhanced B cell cross-presentation of MBP/Kk specifically in the brain. These data demonstrate that B cells are the major APCs that cross-present MBP/Kk within the brains of SEAE mice, and the increased efficiency of B cells cross-presenting MBP/Kk in the brain is likely promoted by interactions with T cells expressing the 8.8 TCR.
FIGURE 6.

B cells are the predominant cell type cross-presenting MBP in SEAE. (A, B, D, and E) Data obtained by flow cytometric analyses of mononuclear cells are shown as mean ± SEM. (A) The percentages of MBP/Kk+ cells are shown for the brain and spinal cord of 8.8 IFN-γ−/− (n = 21) and 8.8 IFN-γ+/+ (n = 4) mice with SEAE and age-matched 8.8 healthy mice (n = 14). (B) The numbers of MBP/Kk+ cells in the brains of 8.8 IFN-γ−/− (n = 14) and 8.8 IFN-γ+/+ (n = 2) mice with SEAE, and of age-matched 8.8 healthy mice (n = 8) are shown for the indicated cell types; gating strategy is shown in Supplemental Fig. 3C. (C) MBP-binding IgG and IgM Abs were detected in the serum of 8.8 IFN-γ−/− mice with SEAE (n = 25) and 8.8 healthy (n = 6) mice by ELISA; data are plotted as area under the curve (AUC). Data are representative of two independent experiments. (D) The percentages of MBP/Kk+ B cells, MdCs, and monocytes in the brain, deep cervical lymph node (dCLN), and spleen of 8.8 IFN-γ−/− SEAE mice (n = 16) are shown. (E) The percentages of MBP/Kk+ B cells in the brain, dCLN, and spleen of healthy 8.8 (n = 8) and WT mice (n = 7) are shown. Statistical significance was determined using a paired t test (A), Kruskal–Wallis with a Dunn’s posttest (B, E), Mann–Whitney U test (C), and two-way ANOVA with Šidák’s posttest (D). *p < 0.05, **p < 0.01, ****p < 0.0001.
Anti-CD20 treatment halts disease progression in SEAE mice
Anti-CD20 or isotype control Ab was administered weekly to 8.8 IFN-γ−/− SEAE mice beginning 1–3 d after onset of clinical signs, and mice were observed for 35 d following initiation of treatment. B cell depletion in blood was confirmed after 7 d of treatment, and B cells were decreased >81% in the brain and >83% in the spleen compared with isotype-treated mice at the end of the study (data not shown). The severity of clinical signs in mice receiving isotype control Ab steadily worsened over time (Fig. 7A). Importantly, anti-CD20 treatment halted clinical disease progression (Fig. 7A). We monitored some mice after termination of anti-CD20 treatment for an additional 35 d and found that clinical signs again progressed in severity (Fig. 7B). These data demonstrate that B cells play a critical role in promoting progression of disease severity in mice with SEAE.
FIGURE 7.

B cell depletion halts the progression of SEAE in 8.8 IFN-γ−/− mice. (A) 8.8 IFN-γ−/− mice with SEAE were treated with anti-CD20 (n = 18) or isotype (n = 17) Ab 2–3 d after onset of clinical symptoms and then every 7 d for a total of 5 wk. Mice were scored daily by a blinded individual. Statistical significance was determined using a Mann–Whitney U test: ***p < 0.001, ****p < 0.0001. (B) Anti-CD20 treatment of 8.8 IFN-γ−/− mice with SEAE (n = 3) was terminated after 5 wk and clinical signs monitored daily for the following 35 d. Mean clinical scores ± SEM are shown.
Discussion
The 8.8 TCR-transgenic model has allowed investigation of different mechanisms by which MHC class I–restricted myelin-specific T cells can contribute to CNS autoimmunity. Previously, we found that naive 8.8 CD8 T cells recruited to the CNS during CD4 T cell–induced EAE exacerbated brain inflammation via a FasL-dependent but IFN-γ– and perforin-independent mechanism (18). In contrast, we show in the present study that virally activated 8.8 CD8 T cells initiate acute CNS autoimmune disease via an IFN-γ– and perforin-dependent mechanism. These mice exhibited parenchymal lesions in the gray and white matter of the cerebellum and spinal cord with minimal demyelination. This pathology is similar to virally induced CNS disease in GFAP-specific TCR-transgenic mice, although meningeal as well as perivascular inflammation was seen in the GFAP-specific model (16). This difference may reflect the fact that the target Ag for the GFAP-specific TCR-transgenic mice is expressed by astrocytes, which are a component of the glia limitans, rather than by oligodendrocytes. Despite the minimal demyelination seen in 8.8 mice with virally induced EAE, a diverse set of cell types cross-presented endogenous MBP including microglia, MdCs, monocytes, and T cells. Most MBP/Kk presentation by these cell types resulted from the autoimmune response initiated by the 8.8 T cells, as comparable MBP presentation was not seen in infected WT mice. The diversity of MBP/Kk+ APCs seen in infected 8.8 mice differs from CD4 T cell–induced EAE where dendritic cells were the prominent MBP/Kk+ cell type (21). The prominence of MBP/Kk+ microglia in virally induced EAE in 8.8 mice was not seen in CD4 T cell–induced EAE or the spontaneous disease described in the present study, and it may contribute to the greater abundance of gray matter lesions seen in infected 8.8 mice. Similar to other CNS Ag-specific cytotoxic CD8 T cells (16, 17), the inflammatory cascade initiated by 8.8 CD8 T cells does not sustain chronic CNS autoimmunity.
The SEAE observed in 8.8 IFN-γ−/− mice provides a new model of MS with parallels to the human disease not seen in other models. Disease onset in 8.8 IFN-γ−/− female mice begins at ∼16 wk of age, which is later than other SEAE models and better aligned with the 20- to 40-y age range within which most people are diagnosed with MS. SEAE is significantly biased toward females in 8.8 IFN-γ−/− mice, as is the case for MS. IFN-γ deficiency accelerates but is not required for SEAE in 8.8 mice. Clinical signs, the immune cell infiltrate including the prominence of B cells and CD4 T cells, and the number and types of cells presenting MBP/Kk were all similar between 8.8 IFN-γ+/+ and 8.8 IFN-γ−/− mice with SEAE, suggesting a similar pathogenesis. It is not yet clear how IFN-γ deficiency accelerates SEAE; however, multiple groups have shown that IFN-γ production impedes CD4 T cell infiltration of the brain but not spinal cord parenchyma in EAE (24–27). Our observation that CD4 T cells are specifically enriched in the brain compared with the periphery in 8.8 IFN-γ−/− but not IFN-γ+/+ mice supports this notion.
Despite the MHC class I restriction of the 8.8 TCR, CD4 T cells predominate in the brains of 8.8 mice with SEAE and exhibit a much higher frequency of inflammatory cytokine-producing cells compared with CD8 T cells. 8.8 CD4 T cells likely express another TCR in addition to the 8.8 TCR, as CD4 T cells are not generated in 8.8 mice when rearrangement of endogenous TCR chains is prohibited. Expression of the CD4 coreceptor typically reflects positive selection by MHC class II during thymocyte maturation, a developmental pathway that would program 8.8 CD4 T cells to exert different effector functions upon activation compared with the cytotoxic activity of 8.8 CD8 T cells. Disparity in intrinsic effector function between 8.8 CD4 and CD8 T cells is consistent with our earlier observation that adoptive transfer of 8.8 CD8 T cells induced disease similar to virally induced EAE, whereas recipients of 8.8 CD4 T cells remained healthy (15). Taken together, these observations suggest that inefficient allelic exclusion in 8.8 mice allows many 8.8 thymocytes to differentiate into dual TCR-expressing CD4 T cells that lack the potential for cytolytic activity but may recognize both MHC class I– and II–restricted Ags. Initial activation of 8.8 CD4 T cells leading to SEAE could be triggered by interaction with APCs presenting either MBP/Kk or an MHC class II–associated Ag derived from self or an environmental microbe. Generation of dual TCR-expressing T cells in which one TCR is specific for a CNS Ag should be a rare occurrence; however, if the second TCR is specific for a ubiquitous pathogen such as EBV, the likelihood of activating these T cells is increased. This scenario raises the possibility that the association of MHC class II alleles with MS susceptibility may not always be linked to MHC class II–restricted presentation of CNS Ags.
CD4 T cells in the brains of SEAE mice produced TNF-α without IL-17 and IFN-γ and little GM-CSF. This phenotype contrasts with the effector phenotype of pathogenic CD4 T cells elicited by immunization of both C3Heb/Fej and C57BL/6 mice with CNS Ag and adjuvant that generates T cells producing TNF-α but also high levels of IL-17, IFN-γ, and GM-CSF. TNF-α was essential for 8.8 mice to develop SEAE; similarly, neutralizing TNF-α before disease onset in induced EAE significantly attenuated disease (28–30). However, targeting TNF-α therapeutically was not successful, as it signals via two receptors with opposing activities. TNFR1 promotes leukocyte parenchymal migration and triggers oligodendrocyte cell death. These are critical functions as TNFR1-deficient mice are either resistant to EAE (31, 32) or develop attenuated disease (33, 34). TNFR2 exerts a neuroprotective influence and TNFR2-deficient mice develop worse EAE (31, 33). Interestingly, an optimized mAb that antagonizes TNFR1 and ameliorates EAE was recently described (35). This therapeutic strategy might prove beneficial in MS by targeting different types of pathogenic CD4 T cells that share a dependence on TNF-α.
Remarkably, despite the lack of IL-17, IFN-γ, and GM-CSF produced by T cells in this spontaneous model, lesions in 8.8 SEAE mice were severe and exhibited many similarities to active lesions in MS tissues (3). Lesions were principally found in white matter in the brain parenchyma and were hypercellular with large regions of demyelination extending in a gradient away from perivascular cuffs surrounding central veins. As TNF-α is the dominant cytokine in this model, the gradient of demyelination emanating from the lesion center could reflect diffusion of this toxic cytokine. Foamy macrophages and phagocytes containing engulfed myelin debris were present in the demyelinated areas, which were marked by surrounding areas of astrogliosis. Foci of leptomeningeal inflammation were also present primarily in deeper areas of the brain. Despite the abundance of B cells in the brains of mice with SEAE, no tertiary lymphoid structures or lymphocyte aggregates were observed similar to those seen in the meninges in some EAE models. This may reflect lack of IL-17 production by 8.8 CD4 T cells, as Th17 cells were implicated in the formation of meningeal aggregates in EAE mice (36, 37). The tight perivascular localization of B cells in the brains of 8.8 mice with SEAE may reflect their increased expression of CXCR4, the receptor for CXCL12. CXCL12 is expressed on the basolateral surface of the microvasculature in the CNS, and CXCL12/CXCR4 interactions can retain infiltrating cells within the perivascular space (38). Less than 10% of T cells infiltrating the brains of sick 8.8 mice expressed CXCR4 (data not shown), consistent with their increased migration into the parenchyma.
A striking finding from these studies is that B cells are the predominant APC presenting MBP in the MHC class I pathway. B cells can function as APCs in MHC class II–restricted TCR-transgenic SEAE models; however, they are not considered efficient in cross-presenting exogenous Ag. Nevertheless, the increase in MBP-specific IgG Ab titers in 8.8 mice with SEAE compared with healthy 8.8 mice (Fig. 6C) demonstrates that B cells cross-present MBP to 8.8 T cells during the pathogenesis of SEAE. Two factors account for the role of B cells as the major APCs driving disease in this model. First, B cells comprise a much larger percentage of the infiltrate in the brain compared with MdCs and monocytes. This contrasts with EAE induced in C3Heb/Fej mice by adoptive transfer of MOG-specific CD4 T cells where MdCs and monocytes represent a major portion of the immune infiltrate and B cells are a minor fraction. Second, the frequency of MBP/Kk+ cells in the brain is significantly higher for B cells compared with MdCs and monocytes (Fig. 6D), indicating that B cells are more efficient in cross-presenting MBP specifically in the brain in the setting of SEAE. However, the frequency of MBP/Kk+ B cells was not drastically different between the brains of SEAE 8.8 mice (∼5%, (Fig. 6D) versus EAE conventionally induced in C3Heb/Fej mice (1–3%, data not shown). A much larger difference is seen between the frequency of MBP/Kk+ MdCs and monocytes in the brains of SEAE 8.8 mice (<1%) versus induced EAE (30–40%) (18). The decreased frequency of MBP/Kk+ MdCs and monocytes in the brain in SEAE may reflect the absence of GM-CSF and IFN-γ, which promote myeloid cell maturation and activity. The frequency of MBP/Kk+ B cells in SEAE mice was significantly higher in the brain compared with both dCLNs and spleen. Interestingly, this was not the case for monocytes, and the frequency of MBP/Kk+ MdCs in the brain was only slightly higher compared with the spleen and not different compared with dCLNs (Fig. 6D). Notably, the frequency of MBP/Kk+ B cells in healthy 8.8 mice was also significantly higher in the brain compared with dCLNs and spleen (Fig. 6E), whereas no differences between these tissues were seen for MdCs and monocytes in healthy 8.8 mice (data not shown). The increase in MBP/Kk+ B cells in the brain in healthy mice required expression of the 8.8 TCR, as this enrichment was not seen in WT mice (Fig. 6E). These data suggest that prior to onset of clinical signs, B and T cells may engage in cognate interaction more frequently in the brain where MBP is in greater abundance than in the periphery. Interestingly, the increase in frequency of MBP/Kk+ B cells in the brains of healthy 8.8 mice occurs prior to the differentiation of B cells into an Ag-experienced, proinflammatory phenotype, as expression levels of memory and activation markers, as well as TNF-α and IL-6 production, were all significantly lower in 8.8 healthy compared with SEAE mice (Fig. 5).
The ability of anti-CD20 treatment to halt disease progression in mice after SEAE onset demonstrates that B cells play a critical role in this model. This finding differs from the increased incidence in SEAE seen in GFAP-specific TCR-transgenic mice when crossed onto either a Rag−/− or μMT−/− background (16). However, CD8 rather than CD4 T cells mediate spontaneous disease in the GFAP-specific model, and the target cell type may be astrocytes that are not required to cross-present GFAP. Eliminating B cells in the brains of 8.8 SEAE mice strongly reduced the number of APCs accessible to infiltrating 8.8 T cells. Reducing the levels of IL-6 and TNF-α produced in situ should also be beneficial, as this combination of cytokines inhibits the ability of regulatory T cells to suppress Teff cells (39). The resumption of disease progression in mice following termination of anti-CD20 treatment highlights the key pathogenic role of B cells in perpetuating CNS autoimmunity.
In summary, our studies demonstrate that CD4 T cells that coexpress a myelin-specific, MHC class I–restricted TCR as well as endogenously rearranged TCR chains can spontaneously induce a CNS autoimmune disease that differs dramatically from CNS injury mediated by cytotoxic CD8 T cells expressing the same MHC class I–restricted TCR. The spontaneous disease exhibits several key parallels to MS, including a female bias, early–middle adult age of onset, response to B cell depletion after clinical onset, and lesions with similarities to active lesions in MS. Disease progression was driven by B cells that cross-present the MBP epitope. The CD4 T cells adopt a unique effector phenotype characterized by expression of TNF-α without IL-17 and IFN-γ and only very low levels of GM-CSF. These results suggest that TNF-α alone can induce pathology similar to that seen in MS tissues. Future studies will investigate whether additional TCR specificities on 8.8 CD4 T cells and/or clonal expansion of B cells play a role in triggering spontaneous disease.
Supplementary Material
Acknowledgments
We thank Neal Mausolf and Trevor Mileur for technical assistance and Dr. Catriona Wagner for advice on statistical comparisons.
This article is featured in Top Reads, p.
This work was supported by Division of Intramural Research, National Institute of Allergy and Infectious Diseases Grant R37 AI107494 (to J.M.G.).
Joan M. Goverman is a Distinguished Fellow of AAI.
The online version of this article contains supplemental material.
- dCLN
- deep cervical lymph node
- EAE
- experimental autoimmune encephalomyelitis
- GFAP
- glial fibrillary acidic protein
- IHC
- immunohistochemistry
- β2m
- β2-microglobulin
- MBP
- myelin basic protein
- MdC
- monocyte-derived cell
- MOG
- myelin oligodendrocyte glycoprotein
- MS
- multiple sclerosis
- RT
- room temperature
- SEAE
- spontaneous EAE
- Tcm
- T central memory
- Teff
- T effector
- Tem
- T effector memory
- Trm
- tissue-resident memory
- Vac
- vaccinia virus
- WT
- wild-type
Disclosures
The authors have no financial conflicts of interest.
References
- 1. Bjornevik K., Cortese M., Healy B. C., Kuhle J., Mina M. J., Leng Y., Elledge S. J., Niebuhr D. W., Scher A. I., Munger K. L., Ascherio A.. 2022. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science 375: 296–301. [DOI] [PubMed] [Google Scholar]
- 2. Reich D. S., Lucchinetti C. F., Calabresi P. A.. 2018. Multiple sclerosis. N. Engl. J. Med. 378: 169–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kuhlmann T., Ludwin S., Prat A., Antel J., Brück W., Lassmann H.. 2017. An updated histological classification system for multiple sclerosis lesions. Acta Neuropathol. 133: 13–24. [DOI] [PubMed] [Google Scholar]
- 4. Lucchinetti C., Brück W., Parisi J., Scheithauer B., Rodriguez M., Lassmann H.. 2000. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann. Neurol. 47: 707–717. [DOI] [PubMed] [Google Scholar]
- 5. Wagner C. A., Roqué P. J., Goverman J. M.. 2020. Pathogenic T cell cytokines in multiple sclerosis. J. Exp. Med. 217: e20190460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bettelli E., Baeten D., Jäger A., Sobel R. A., Kuchroo V. K.. 2006. Myelin oligodendrocyte glycoprotein-specific T and B cells cooperate to induce a Devic-like disease in mice. J. Clin. Invest. 116: 2393–2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Krishnamoorthy G., Lassmann H., Wekerle H., Holz A.. 2006. Spontaneous opticospinal encephalomyelitis in a double-transgenic mouse model of autoimmune T cell/B cell cooperation. J. Clin. Invest. 116: 2385–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pöllinger B., Krishnamoorthy G., Berer K., Lassmann H., Bösl M. R., Dunn R., Domingues H. S., Holz A., Kurschus F. C., Wekerle H.. 2009. Spontaneous relapsing-remitting EAE in the SJL/J mouse: MOG-reactive transgenic T cells recruit endogenous MOG-specific B cells. J. Exp. Med. 206: 1303–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fransen N. L., Hsiao C. C., van der Poel M., Engelenburg H. J., Verdaasdonk K., Vincenten M. C. J., Remmerswaal E. B. M., Kuhlmann T., Mason M. R. J., Hamann J., et al. 2020. Tissue-resident memory T cells invade the brain parenchyma in multiple sclerosis white matter lesions. Brain 143: 1714–1730. [DOI] [PubMed] [Google Scholar]
- 10. Sinha S., Boyden A. W., Itani F. R., Crawford M. P., Karandikar N. J.. 2015. CD8+ T-cells as immune regulators of multiple sclerosis. Front. Immunol. 6: 619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Saligrama N., Zhao F., Sikora M. J., Serratelli W. S., Fernandes R. A., Louis D. M., Yao W., Ji X., Idoyaga J., Mahajan V. B., et al. 2019. Opposing T cell responses in experimental autoimmune encephalomyelitis. Nature 572: 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brate A. A., Boyden A. W., Jensen I. J., Badovinac V. P., Karandikar N. J.. 2021. A functionally distinct CXCR3+/IFN-γ+/IL-10+ subset defines disease-suppressive myelin-specific CD8 T cells. J. Immunol. 206: 1151–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cruz F. M., Colbert J. D., Merino E., Kriegsman B. A., Rock K. L.. 2017. The biology and underlying mechanisms of cross-presentation of exogenous antigens on MHC-I molecules. Annu. Rev. Immunol. 35: 149–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Perchellet A., Stromnes I., Pang J. M., Goverman J.. 2004. CD8+ T cells maintain tolerance to myelin basic protein by “epitope theft”. Nat. Immunol. 5: 606–614. [DOI] [PubMed] [Google Scholar]
- 15. Ji Q., Perchellet A., Goverman J. M.. 2010. Viral infection triggers central nervous system autoimmunity via activation of CD8+ T cells expressing dual TCRs. Nat. Immunol. 11: 628–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sasaki K., Bean A., Shah S., Schutten E., Huseby P. G., Peters B., Shen Z. T., Vanguri V., Liggitt D., Huseby E. S.. 2014. Relapsing-remitting central nervous system autoimmunity mediated by GFAP-specific CD8 T cells. J. Immunol. 192: 3029–3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Friese M. A., Jakobsen K. B., Friis L., Etzensperger R., Craner M. J., McMahon R. M., Jensen L. T., Huygelen V., Jones E. Y., Bell J. I., Fugger L.. 2008. Opposing effects of HLA class I molecules in tuning autoreactive CD8+ T cells in multiple sclerosis. Nat. Med. 14: 1227–1235. [DOI] [PubMed] [Google Scholar]
- 18. Wagner C. A., Roqué P. J., Mileur T. R., Liggitt D., Goverman J. M.. 2020. Myelin-specific CD8+ T cells exacerbate brain inflammation in CNS autoimmunity. J. Clin. Invest. 130: 203–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stromnes I. M., Goverman J. M.. 2006. Active induction of experimental allergic encephalomyelitis. Nat. Protoc. 1: 1810–1819. [DOI] [PubMed] [Google Scholar]
- 20. Pierson E. R., Goverman J. M.. 2017. GM-CSF is not essential for experimental autoimmune encephalomyelitis but promotes brain-targeted disease. JCI Insight 2: e92362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ji Q., Castelli L., Goverman J. M.. 2013. MHC class I-restricted myelin epitopes are cross-presented by Tip-DCs that promote determinant spreading to CD8+ T cells. Nat. Immunol. 14: 257–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Guyenet S. J., Furrer S. A., Damian V. M., Baughan T. D., La Spada A. R., Garden G. A.. 2010. A simple composite phenotype scoring system for evaluating mouse models of cerebellar ataxia. J. Vis. Exp. (39):1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cancro M. P., Tomayko M. M.. 2021. Memory B cells and plasma cells: the differentiative continuum of humoral immunity. Immunol. Rev. 303: 72–82. [DOI] [PubMed] [Google Scholar]
- 24. Wensky A. K., Furtado G. C., Marcondes M. C., Chen S., Manfra D., Lira S. A., Zagzag D., Lafaille J. J.. 2005. IFN-γ determines distinct clinical outcomes in autoimmune encephalomyelitis. J. Immunol. 174: 1416–1423. [DOI] [PubMed] [Google Scholar]
- 25. Lees J. R., Golumbek P. T., Sim J., Dorsey D., Russell J. H.. 2008. Regional CNS responses to IFN-γ determine lesion localization patterns during EAE pathogenesis. J. Exp. Med. 205: 2633–2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kroenke M. A., Chensue S. W., Segal B. M.. 2010. EAE mediated by a non-IFN-γ/non-IL-17 pathway. Eur. J. Immunol. 40: 2340–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Simmons S. B., Liggitt D., Goverman J. M.. 2014. Cytokine-regulated neutrophil recruitment is required for brain but not spinal cord inflammation during experimental autoimmune encephalomyelitis. J. Immunol. 193: 555–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ruddle N. H., Bergman C. M., McGrath K. M., Lingenheld E. G., Grunnet M. L., Padula S. J., Clark R. B.. 1990. An antibody to lymphotoxin and tumor necrosis factor prevents transfer of experimental allergic encephalomyelitis. J. Exp. Med. 172: 1193–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Baker D., Butler D., Scallon B. J., O’Neill J. K., Turk J. L., Feldmann M.. 1994. Control of established experimental allergic encephalomyelitis by inhibition of tumor necrosis factor (TNF) activity within the central nervous system using monoclonal antibodies and TNF receptor-immunoglobulin fusion proteins. Eur. J. Immunol. 24: 2040–2048. [DOI] [PubMed] [Google Scholar]
- 30. Selmaj K., Papierz W., Glabiński A., Kohno T.. 1995. Prevention of chronic relapsing experimental autoimmune encephalomyelitis by soluble tumor necrosis factor receptor I. J. Neuroimmunol. 56: 135–141. [DOI] [PubMed] [Google Scholar]
- 31. Suvannavejh G. C., Lee H. O., Padilla J., Dal Canto M. C., Barrett T. A., Miller S. D.. 2000. Divergent roles for p55 and p75 tumor necrosis factor receptors in the pathogenesis of MOG35–55-induced experimental autoimmune encephalomyelitis. Cell. Immunol. 205: 24–33. [DOI] [PubMed] [Google Scholar]
- 32. Kassiotis G., Kollias G.. 2001. Uncoupling the proinflammatory from the immunosuppressive properties of tumor necrosis factor (TNF) at the p55 TNF receptor level: implications for pathogenesis and therapy of autoimmune demyelination. J. Exp. Med. 193: 427–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Eugster H. P., Frei K., Bachmann R., Bluethmann H., Lassmann H., Fontana A.. 1999. Severity of symptoms and demyelination in MOG-induced EAE depends on TNFR1. Eur. J. Immunol. 29: 626–632. [DOI] [PubMed] [Google Scholar]
- 34. Steeland S., Van Ryckeghem S., Van Imschoot G., De Rycke R., Toussaint W., Vanhoutte L., Vanhove C., De Vos F., Vandenbroucke R. E., Libert C.. 2017. TNFR1 inhibition with a Nanobody protects against EAE development in mice. Sci. Rep. 7: 13646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Richter F., Williams S. K., John K., Huber C., Vaslin C., Zanker H., Fairless R., Pichi K., Marhenke S., Vogel A., et al. 2021. The TNFR1 antagonist atrosimab is therapeutic in mouse models of acute and chronic inflammation. Front. Immunol. 12: 705485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Peters A., Pitcher L. A., Sullivan J. M., Mitsdoerffer M., Acton S. E., Franz B., Wucherpfennig K., Turley S., Carroll M. C., Sobel R. A., et al. 2011. Th17 cells induce ectopic lymphoid follicles in central nervous system tissue inflammation. Immunity 35: 986–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pikor N. B., Prat A., Bar-Or A., Gommerman J. L.. 2016. Meningeal tertiary lymphoid tissues and multiple sclerosis: a gathering place for diverse types of immune cells during CNS autoimmunity. Front. Immunol. 6: 657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McCandless E. E., Wang Q., Woerner B. M., Harper J. M., Klein R. S.. 2006. CXCL12 limits inflammation by localizing mononuclear infiltrates to the perivascular space during experimental autoimmune encephalomyelitis. J. Immunol. 177: 8053–8064. [DOI] [PubMed] [Google Scholar]
- 39. Korn T., Reddy J., Gao W., Bettelli E., Awasthi A., Petersen T. R., Bäckström B. T., Sobel R. A., Wucherpfennig K. W., Strom T. B., et al. 2007. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat. Med. 13: 423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.