

Relugolix combination therapy showed sustained improvement of uterine leiomyoma–associated symptoms and patient-reported quality of life through 52 weeks.
OBJECTIVE:
In the LIBERTY 1 and LIBERTY 2 placebo-controlled trials, once-daily relugolix combination therapy reduced menstrual blood loss volume and pain in women with heavy menstrual bleeding associated with uterine leiomyomas and was well tolerated, with preservation of bone mineral density (BMD) through 24 weeks. Here we report the long-term efficacy and safety of relugolix combination therapy treatment for up to 52 weeks.
METHODS:
Women with uterine leiomyoma–associated heavy menstrual bleeding who completed any treatment arm in either the LIBERTY 1 or LIBERTY 2 trial were eligible to enroll in a 28-week long-term extension study. All participants received once-daily relugolix combination therapy (40 mg relugolix, estradiol 1 mg, norethindrone acetate 0.5 mg) in the extension study. The primary efficacy endpoint was the proportion of women who achieved or maintained a menstrual blood loss volume of less than 80 mL and a 50% or greater reduction in menstrual blood loss volume from LIBERTY study baseline to the last 35 days of treatment (defined as responders). Analyses were conducted for all three randomized treatment groups from pivotal studies.
RESULTS:
Overall, 477 women enrolled, 476 were treated, and 363 (76.1%) completed 52 weeks. Among patients treated with relugolix combination therapy through 52 weeks (n=163), sustained improvement in heavy menstrual bleeding was observed in 87.7% (responders). The least squares mean menstrual blood loss volume reduction was 89.9%, with 70.6% of patients achieving amenorrhea. At week 52, 59.0% of patients with anemia at baseline had improvements in hemoglobin concentration of greater than 2 g/dL. Distress due to uterine leiomyoma–associated symptoms measured by the BPD (Bleeding and Pelvic Discomfort) scale score was reduced by 51.3 points. Sustained reductions in uterine and uterine leiomyoma volume were observed. Bone mineral density was preserved through week 52.
CONCLUSION:
Improvements in heavy menstrual bleeding and anemia and reduction of uterine leiomyoma–associated symptom burden were sustained through up to 52 weeks of treatment with relugolix combination therapy in women with uterine leiomyomas. No new safety concerns were identified, and BMD was maintained.
CLINICAL TRIAL REGISTRATION:
FUNDING SOURCE:
Myovant Sciences GmbH.
Uterine leiomyomas or myomas are common, nonmalignant, estrogen- and progesterone-dependent uterine tumors manifesting during a woman's reproductive years.1–3 Relugolix is a once-daily, orally active nonpeptide gonadotropin-releasing hormone receptor antagonist that suppresses secretion of gonadotropins, reducing follicular growth and inhibiting ovulation, thereby decreasing ovarian production of estrogen and progesterone. It is approved in Japan as monotherapy for treatment of uterine leiomyomas (RELUMINA Japanese package insert, ASKA Pharmaceutical Co, Ltd, 2021). However, significant dose-dependent reductions in bone mineral density (BMD) observed during the clinical trials have limited its treatment duration to 6 months.4,5
The combination of relugolix 40 mg with 1 mg estradiol and 0.5 norethindrone acetate, hereafter termed relugolix combination therapy, maintained therapeutic levels of systemic estradiol concentrations while mitigating bone resorption and vasomotor symptoms associated with administration of relugolix alone (Lukes A, Johnson B, Jones L, Berga J, Williams R, Bock B, et al. Pharmacokinetics, pharmacodynamics, and safety of relugolix, a potent oral once-daily gonadotropin-releasing hormone receptor antagonist, as monotherapy and in combination with estradiol/norethindrone acetate add-back therapy [abstract]. Hum Reprod 2017;32[Suppl]:i267–8). In two replicate pivotal phase 3 randomized, double-blind, placebo-controlled studies (LIBERTY 1 and LIBERTY 2), relugolix combination therapy significantly improved uterine leiomyoma–associated symptoms compared with placebo in premenopausal women with uterine leiomyomas over 24 weeks, was well tolerated and preserved BMD.6 Here, we report the long-term efficacy and safety of relugolix combination therapy for up to 52 weeks of treatment. Relugolix combination therapy as once-daily single-tablet therapy has been approved for management of heavy menstrual bleeding associated with uterine leiomyomas in premenopausal women in the United States and for treatment of moderate-to-severe symptoms of uterine leiomyomas in adult women of reproductive age in the European Union (MYFEMBREE [relugolix, estradiol, and norethindrone acetate tablets] prescribing information, Myovant Sciences, 2021; RYEQO [relugolix 40 mg, estradiol 1.0 mg, and norethisterone acetate 0.5 mg], summary of product characteristics, European Medicines Agency, 2021). In addition, relugolix combination therapy has been approved for the treatment of moderate-to-severe pain associated with endometriosis in the United States (MYFEMBREE, prescribing information, 2022).
METHODS
Participants enrolled in the LIBERTY 1 or LIBERTY 2 studies, two replicate and independent pivotal studies, were premenopausal women aged 18–50 years who had menstrual blood loss volume of 80 mL or greater per cycle for two cycles or 160 mL or greater during one cycle and a diagnosis of uterine leiomyomas confirmed by transvaginal ultrasonography during the screening period. Patients who completed 24 weeks of treatment in one of the replicate phase 3 randomized, double-blind, placebo-controlled pivotal studies6 and who were not expecting to undergo additional procedures for uterine leiomyomas within the study period were eligible to enroll in the long-term extension study. Patients were to have a negative pregnancy test result and agreed to continue to use acceptable nonhormonal contraceptive methods. Patients with a Z-score for BMD less than −2.0 or with a decrease in BMD of 7% or more at the lumbar spine, total hip, or femoral neck from pivotal study baseline to week 24 were excluded. There was no washout period from investigational product in the pivotal studies, and all patients received their first dose of relugolix combination therapy after being deemed eligible and providing informed consent.
This was a multinational, open-label, single-arm, long-term efficacy and safety extension study (ClinicalTrials.gov: NCT03049735; NCT03103087; NCT03412890). All patients received oral relugolix combination therapy for up to 28 weeks. The study objectives were to evaluate long-term efficacy and safety through up to 52 weeks of treatment with relugolix combination therapy, which included the antecedent 24 weeks of treatment during either pivotal study. Patient visits occurred every 4 weeks. The studies were conducted in accordance with International Conference on Harmonisation guidelines and ethical principles of the Declaration of Helsinki. All patients provided written informed consent. A list of independent ethics committees and institutional review boards is provided in Appendix 1, available online at http://links.lww.com/AOG/C913.
The primary endpoint was the proportion of women who achieved or maintained a menstrual blood loss volume of less than 80 mL and at least a 50% reduction in menstrual blood loss volume from pivotal study baseline to the last 35 days of treatment, as measured by the established alkaline hematin quantification method for menstrual blood volume determination by chemically measuring the blood content of used menstrual products.6 This endpoint also was evaluated for predefined subgroups (ie, geographic region, baseline menstrual blood loss volume, age, race, baseline uterine leiomyoma volume, baseline uterine volume, baseline body mass index [BMI, calculated as weight in kilograms divided by height in meters squared], and maximum baseline numeric rating scale pain score). Considering a high prevalence of uterine leiomyomas in Black and African American women,7 race evaluation was included to ensure that the study’s patient population was reflective of the population of women with leiomyomas in general. Secondary endpoints included change from pivotal study baseline to week 52 in menstrual blood loss volume, proportion of women who achieved or maintained amenorrhea over the last 35 days of treatment, and proportion of women with a hemoglobin concentration of 10.5 g/dL or less at pivotal study baseline who achieved an increase of greater than 2 g/dL at week 52. Change in hemoglobin concentration, UFS-QOL (Uterine Fibroid Symptom and Quality of Life) scale score,8 BPD (Bleeding and Pelvic Discomfort) scale score, HRQOL (Health-Related Quality of Life) subscale scores and total score, uterine volume, and uterine leiomyoma volume were evaluated as secondary endpoints in this study.
Safety evaluations included monitoring of vital signs, physical examinations, adverse events, clinical laboratory parameters, and 12-lead electrocardiograms. Percent change from pivotal study baseline to week 52 in BMD at the lumbar spine (L1–L4), total hip, and femoral neck, as assessed by dual-energy X-ray absorptiometry, were evaluated as part of the safety assessment in this study.
Because this was an extension study, the sample size was determined by the number of patients who completed either the LIBERTY 1 or LIBERTY 2 study and who were eligible and consented to participate in this study. The pivotal study baseline was used as the reference point in the study report for analyzing all change from baseline-related endpoints unless otherwise specified. Safety and efficacy data were analyzed using descriptive statistics for patients who enrolled in the study and who received at least one dose of study drug in the long-term extension period and are described here by randomized treatment group at pivotal study baseline (LIBERTY 1 and LIBERTY 2): 1) relugolix combination therapy: relugolix combination therapy up to 52 weeks; 2) relugolix→relugolix combination therapy: 12 weeks of relugolix 40 mg monotherapy followed by relugolix combination therapy up to 40 weeks; and 3) placebo→relugolix combination therapy: randomized to placebo in the pivotal study and transitioned to relugolix combination therapy for up to 28 weeks (Appendix 2, available online at http://links.lww.com/AOG/C913). No statistical comparisons were performed among treatment groups for this extension study. The methods for analyzing the efficacy endpoints in the long-term extension study, including missing data–handling rules, were similar to those used in the two replicate LIBERTY 1 and LIBERTY 2 studies.6
The severity of all adverse events was evaluated by the investigator based on Common Terminology Criteria for Adverse Events and was coded to preferred term and system organ class using MedDRA version 22.0 or higher. Laboratory values were classified by toxicity grade based on the Common Terminology Criteria for Adverse Events. Additional details regarding the trial design and analysis methods are provided in Appendix 3, available online at http://links.lww.com/AOG/C913.
RESULTS
Of the 770 patients enrolled in LIBERTY, 610 completed the pivotal studies. A total of 477 (78.1%) patients enrolled in the LIBERTY Extension Study between December 5, 2017, and January 24, 2020; 476 were treated, and 363 (76.1%) completed 52 weeks (Appendix 4, available online at http://links.lww.com/AOG/C913; an investigator list is provided in Appendix 5, available online at http://links.lww.com/AOG/C913). Additional information on patient disposition for the LIBERTY 1 and LIBERTY 2 studies has been published previously.6 A similar number of patients in the relugolix combination therapy (11 patients) and placebo groups (eight patients) and a higher number of patients in the relugolix→relugolix combination therapy group (21 patients) were not eligible to participate in the long-term extension due to BMD decrease exclusionary criteria of either percent change 7% or more or Z-score less than −2.0, most of which occurred at the femoral neck (Appendix 6, available online at http://links.lww.com/AOG/C913). Although demographics and baseline characteristics were similar across treatment groups and consistent with those in the pivotal studies (Table 1), there were more participants in the relugolix combination therapy group who were White and had higher menstrual blood loss volume at baseline and slightly lower uterine leiomyoma volume when compared with other groups. Mean cumulative and extension study compliance with relugolix combination therapy was 98.1%. Baseline characteristics of patients completing the extension study were generally similar to those of patients in the pivotal studies (Appendix 7, available online at http://links.lww.com/AOG/C913).
Table 1.
Baseline Characteristics of the Long-term Extension Study Population by LIBERTY 1 and LIBERTY Randomized Treatment Assignment (Data From LIBERTY 1 and LIBERTY 2 Baseline)

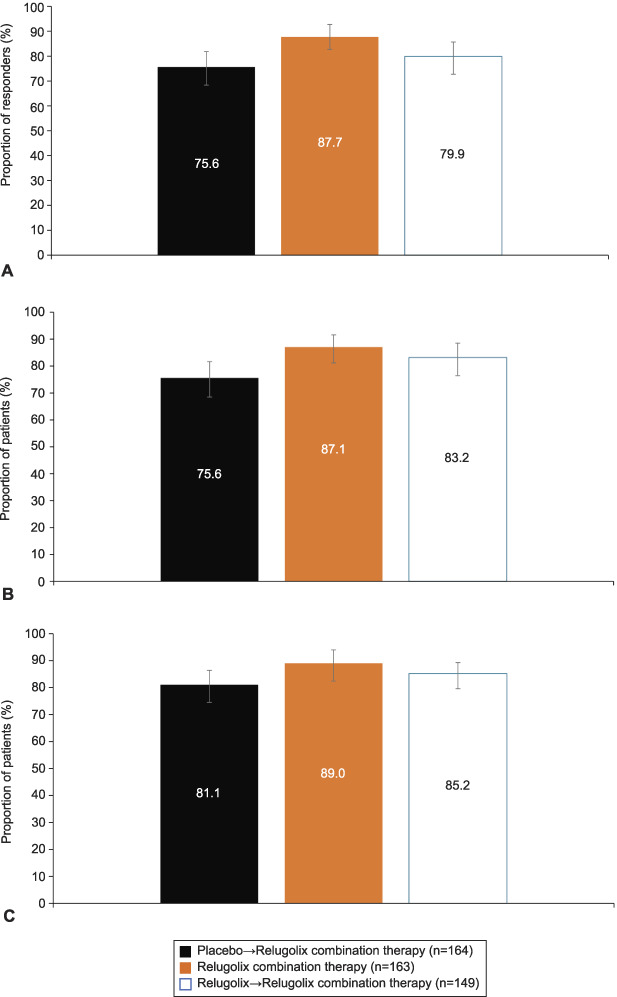
The group receiving relugolix combination therapy continuously for 52 weeks had sustained improvement in heavy menstrual bleeding, with 143 patients (87.7%) meeting the definition for responder (Fig. 1A). The proportion of patients meeting the two criteria (ie, menstrual blood loss volume less than 80 mL or percent change from baseline of at least 50%) of the composite primary endpoint was similar, indicating that no single component drove results for the primary endpoint (Fig. 1B and C). For patients treated with placebo who transitioned to relugolix combination therapy, the proportion of responders observed at week 52 (124 patients, 75.6%) was similar to that observed for relugolix combination therapy at week 24 in the pivotal trials,6 confirming the consistent effect of relugolix combination therapy on improvement in heavy menstrual bleeding. For patients treated with relugolix as monotherapy for 12 weeks in the LIBERTY studies followed by transition to relugolix combination therapy for 40 weeks (12 weeks in LIBERTY+28 weeks in the long-term extension), 119 (79.9%) met the definition for responder. Predefined subgroup analysis demonstrated that the magnitude of the responder rates observed was generally consistent with that observed in the analysis of primary efficacy for the overall population in each treatment group (Appendix 8, available online at http://links.lww.com/AOG/C913).
Fig. 1. A. Primary efficacy analysis results: menstrual blood loss responder rate. Treatment responder: proportion of women who achieved or maintained a menstrual blood loss volume of less than 80 mL and a 50% or greater reduction from pivotal study baseline to the last 35 days of treatment in menstrual blood loss volume. B. Proportion of patients with menstrual blood loss volume less than 80 mL over the last 35 days of treatment. C. Patients with menstrual blood loss reduction of 50% or greater. Error bars represent 95% CIs. Data shown by randomization treatment assignment.
Al-Hendy. Long-term Relugolix in Uterine Leiomyomas. Obstet Gynecol 2022.
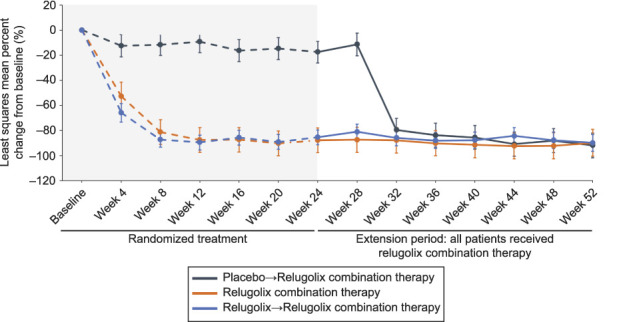
In patients treated with relugolix combination therapy continuously, mean percent reduction in menstrual blood loss volume from pivotal study baseline to week 52 was 89.9% (Fig. 2). Efficacy was observed as early as week 4 after treatment initiation in the pivotal studies, with nearly maximal effect occurring at week 8 and sustained through week 52. Most patients (70.6%) achieved amenorrhea (Appendix 9, available online at http://links.lww.com/AOG/C913). The reductions in menstrual blood loss led to substantial improvements (greater than 2 g/dL) in hemoglobin concentrations at week 52 for more than half of patients (59.0%) with anemia (10.5 g/dL or less) at baseline (Fig. 3A). Least squares mean percent increase in hemoglobin concentration was 28.4% in women treated with relugolix combination therapy up to 52 weeks in the anemia-evaluable population (Fig. 3B).
Fig. 2. Least squares percent change in menstrual blood loss volume from baseline to week 52. Error bars show 95% CIs.
Al-Hendy. Long-term Relugolix in Uterine Leiomyomas. Obstet Gynecol 2022.
Fig. 3. A. Proportion of responders* among patients with anemia at baseline. Anemia-evaluable population: patients with hemoglobin concentration of 10.5 g/dL or less at baseline and with a hemoglobin value at week 24. *Treatment responder (hemoglobin): proportion of women with hemoglobin concentrations of 10.5 g/dL or less at pivotal study baseline who achieved an increase of more than 2 g/dL at week 52. Error bars show 95% CI. B. Least squares mean percent change in hemoglobin concentrations with relugolix combination therapy in women with anemia at baseline. Error bars show 95% CI.

Al-Hendy. Long-term Relugolix in Uterine Leiomyomas. Obstet Gynecol 2022.
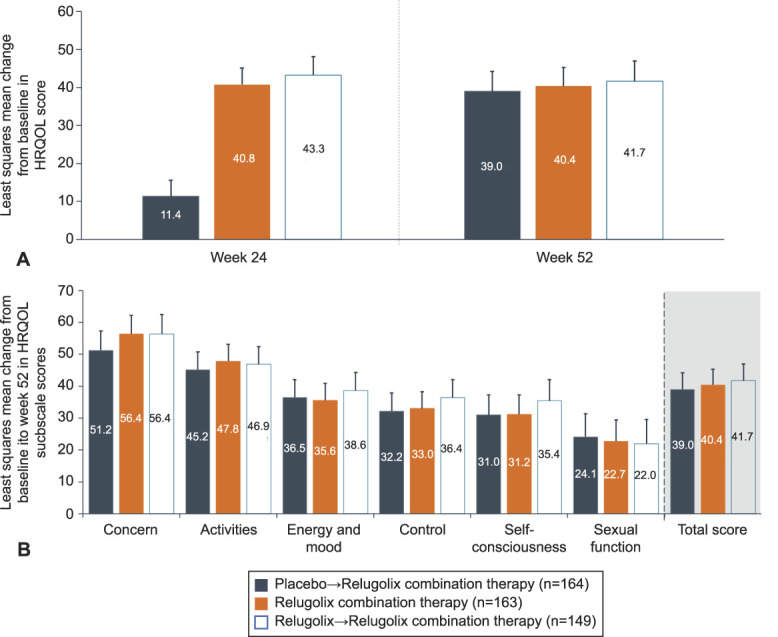
Consistent with the change observed at week 24, the BPD scale score was reduced by 51.3 points (indicating improvement) from baseline to week 52 in the relugolix combination therapy group, reflecting durability of treatment effect on uterine leiomyoma–associated symptoms (Appendix 10, available online at http://links.lww.com/AOG/C913). Relugolix combination therapy resulted in a sustained reduction in the overall burden of uterine leiomyoma symptoms assessed by UFS-QOL scale score (Appendix 11, available online at http://links.lww.com/AOG/C913) and sustained improvement in HRQOL scores over 52 weeks (Fig. 4).
Fig. 4. Least squares mean change in total Health-Related Quality of Life (HRQOL) scores at week 24 and week 52 (A) and least squares mean change in HRQOL subscale scores from baseline to 52 weeks (B). Error bars show upper 95% CI.
Al-Hendy. Long-term Relugolix in Uterine Leiomyomas. Obstet Gynecol 2022.
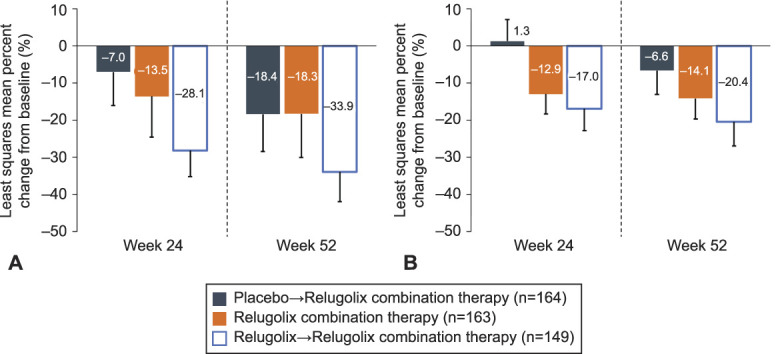
Modest reductions in uterine leiomyomas and uterine volume were also observed with treatment (Fig. 5). In the relugolix combination therapy group, the least squares mean percent change in uterine volume from baseline was −12.9% at week 24 and −14.1% at week 52. Likewise, the least squares mean percent change in uterine leiomyoma volume from baseline was −13.5% at week 24 and −18.3% at week 52 (Fig. 5).
Fig. 5. Percent change in uterine leiomyoma volume (A) and uterine volume (B). Error bars show upper and lower 95% CI.
Al-Hendy. Long-term Relugolix in Uterine Leiomyomas. Obstet Gynecol 2022.
For patients who transitioned from placebo to relugolix combination therapy and those treated with monotherapy and then relugolix combination therapy, the least squares mean percent change in menstrual blood loss from pivotal study baseline to week 52 was −91.9% and −89.8%, respectively (Fig. 2). Although 68.5% of patients treated with relugolix→relugolix combination therapy achieved amenorrhea, a lower proportion of women achieved amenorrhea (57.9%) in the placebo→relugolix combination therapy group, which may reflect a shorter time of therapy compared with the longer-term relugolix combination therapy groups. There were improvements in anemia, with 42.1% and 78.9% responders for patients treated with placebo→relugolix combination therapy and relugolix→relugolix combination therapy, respectively.
The least squares mean change from baseline to week 52 in BPD scale score was −48.6 and −51.6 in the placebo→relugolix combination therapy and relugolix→relugolix combination therapy groups, respectively (Appendix 10, http://links.lww.com/AOG/C913). Women treated with placebo in the pivotal studies reported substantial improvements after transitioning from placebo to relugolix combination therapy at week 24, confirming the beneficial effect of treatment on uterine leiomyoma–associated symptoms and HRQOL (Appendix 11, http://links.lww.com/AOG/C913). Similar changes in UFS-QOL scale scores were observed in the relugolix→relugolix combination therapy group as in the relugolix combination therapy group (Fig. 4). Reductions in uterine and uterine leiomyoma volume were modest in patients treated with placebo who transitioned to relugolix combination therapy and were maintained and overall improved from week 24 to week 52 for patients treated with relugolix→relugolix combination therapy (Fig. 5).
In the relugolix combination therapy group, approximately half of the patients (89 patients [54.6%]) had one adverse event during participation in the long-term extension study; cumulatively over the 52-week treatment period, at least one adverse event was reported for 127 patients (77.9%) (Table 2). Few patients were reported to have grade 3 or higher adverse events (7.4% cumulatively, 2.5% during the long-term extension study), serious adverse events (3.7% cumulatively, 0.6% during the long-term extension study), or serious adverse events assessed by the investigator to be related to the study drug (1.2% cumulatively, 0.6% during the long-term extension study). In the long-term extension study, there was a single report of uterine hemorrhage in the continuous relugolix combination therapy group, five events in the relugolix→relugolix combination therapy group (including bone fracture, events of cholecystitis or cholelithiasis, and worsening uterine leiomyoma), and 11 events in the placebo→relugolix combination therapy group (including menorrhagia, appendicitis, cholelithiasis, atrial fibrillation, increased blood pressure, and intervertebral disc protrusion). The disproportionately greater incidence of serious adverse events in the placebo→relugolix combination therapy group during the long-term extension study is mainly driven by serious adverse events across multiple system organ classes that are reflective of general conditions and unlikely to be related to treatment. There was no disproportionate increase in the incidence of either serious or nonserious adverse events in the relugolix combination therapy group through the 52 weeks. The most frequently reported adverse events were headache and hot flush. The overall safety outcomes for the placebo→relugolix combination therapy and relugolix→relugolix combination therapy groups were consistent, with headache and hot flush the most frequently reported adverse events in both groups.
Table 2.
Adverse Events for Study Participants During the Long-term Extension Study and Cumulatively

Assessment of BMD at the lumbar spine showed a minimal decline of −0.37% at week 12 for the relugolix combination therapy group that marked the beginning of a plateau at week 24 of −0.23%, with small changes from the pivotal baseline at week 36 and week 52 of −0.73% and −0.80%, respectively (Fig. 6; Appendix 12, available online at http://links.lww.com/AOG/C913). In the placebo→relugolix combination therapy group, least squares mean percent changes in BMD at the lumbar spine at weeks 12, 24, 36, and 52 were 0.40%, 0.24%, −0.25%, and −0.78%, respectively. Results for the relugolix→relugolix combination therapy group showed a plateau in lumbar spine BMD after initial decline during 12 weeks on the relugolix monotherapy, with least squares mean percent changes from pivotal baseline to week 12, 24, 36, and 52 of −2.27%, −2.18%, −2.11%, and −2.05%, respectively. Similar trends were observed at the total hip (Appendix 9, http://links.lww.com/AOG/C913).
Fig. 6. Percent change in lumbar spine bone mineral density (BMD) (A) and total hip BMD (B) to week 52. Error bars show 95% Cis.
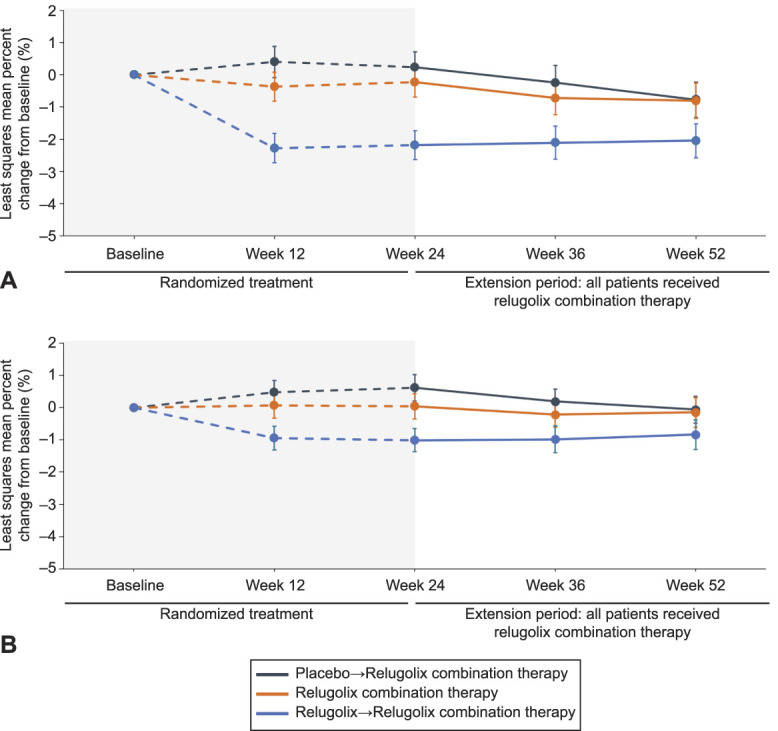
Al-Hendy. Long-term Relugolix in Uterine Leiomyomas. Obstet Gynecol 2022.
There were no pregnancies during the extension study. No clinically important differences were evident in threshold analyses of blood pressure and other laboratory parameters, including liver function tests or lipids.
DISCUSSION
This extension study showed that up to 52 weeks of relugolix combination therapy was well tolerated and resulted in sustained improvement in heavy menstrual bleeding associated with uterine leiomyomas. On average, patients in the relugolix combination therapy group had close to a 90% decrease in menstrual blood loss volume from baseline. Most of the reduction in menstrual blood loss volume was observed by week 4, and the full effect was observed by week 8 and sustained through week 52. Additionally, 70.6% of patients in the relugolix combination therapy group achieved amenorrhea over the last 35 days of treatment. These results indicate that treatment with relugolix combination therapy was associated with a rapid, clinically meaningful, and sustained improvement in bleeding for patients with heavy menstrual bleeding associated with uterine leiomyomas for the duration of 52 weeks of treatment. Treatment effect was independent from race, BMI, and baseline menstrual blood loss volume, as well as uterine and uterine leiomyoma volume.
Results from the placebo→relugolix combination therapy and relugolix→relugolix combination therapy groups were generally supportive of the positive treatment effect observed in the relugolix combination therapy group. Of note, reduction in menstrual blood loss volume in the placebo→relugolix combination therapy group was not observed after first 4 weeks of the extension study due to the necessity of completing menstrual-products collection and, therefore, a later relugolix combination therapy initiation during the menstrual cycle. This may reflect the benefit of starting relugolix combination therapy during menstrual flow.
Long-term treatment with relugolix combination therapy resulted in a sustained reduction in BPD scale score, reflecting improvement in the distress associated with the key uterine leiomyoma symptoms of heavy menstrual bleeding, passing of blood clots, and pelvic pressure and tightness. Additionally, the proportion of responders (reduction in BPD scale scores of 20 points or more) in the relugolix combination therapy group was 68.1% at week 52, consistent with the proportions observed in the pivotal studies. Improvement in HRQOL scores with relugolix combination therapy observed over 24 weeks (Lukes A, Poindexter A, Villarroel V, Li Y, Hunsche E, Stewart E. Relugolix combination improves quality of life in phase 3 studies of symptomatic uterine fibroids [abstract]. Obstet Gynecol 2020;135[Suppl]:7S) was sustained during the 28 weeks of the extension study. This further demonstrates that relugolix combination therapy reduces the distress experienced by patients due to common symptoms of uterine leiomyomas and improves different aspects of quality of life.
Another potentially less obvious aspect of the benefit of relugolix combination therapy involves the effect on uterine and uterine leiomyoma volume (ie, as assessed by imaging changes in response to treatment). A modest but continued reduction in uterine and uterine leiomyoma volume was observed in this study, which is consistent with the estrogen-threshold hypothesis proposing that maintenance of the estradiol concentration between 20 and 50 pg/mL (70 and 180 pmol/L) may stop the progression of leiomyoma growth while minimizing hypoestrogenic adverse effects.9 In addition, the modest reduction in uterine and uterine leiomyoma volume may be beneficial in patients who have bulk symptoms or in the subgroup of patients who plan subsequent surgery.
In this open-label extension study, relugolix combination therapy was generally well tolerated, with no unexpected safety issues identified with the extended administration of relugolix combination therapy for up to 52 weeks. Headache and hot flush were the most frequently reported adverse events, with a higher cumulative reporting rate in women who initially received relugolix monotherapy. Most adverse events were reported within the first 12 weeks of treatment in the pivotal studies. Over the course of the 52-week treatment period, BMD was generally preserved in most women in the relugolix combination therapy group. The findings are consistent with those seen in an observational study of women with uterine leiomyomas (McClung MR, Lukes AS, Venturella R, Santora AC, Zhai D, Wagman RB, et al. Effects of relugolix combination therapy on bone mineral density through 2 years in women with heavy menstrual bleeding associated with uterine fibroids [abstract]. Fertil Steril 2021;116[Suppl]:E13).
Bone mineral density change trajectory in the placebo→relugolix combination therapy group was similar to what has been observed with relugolix combination therapy in the pivotal studies. Minimal decrease in BMD after relugolix combination therapy initiation in the pivotal studies and after transition from placebo likely reflects the new steady state of estradiol concentrations associated with relugolix combination therapy, which are lower than the average value observed over the course of a natural menstrual cycle and consistent with concentrations observed during the early follicular phase.10–12 For women randomized to relugolix→relugolix combination therapy, the initial BMD decline with relugolix monotherapy stabilized after initiation of relugolix combination therapy, though it did not fully recover, which reflected another advantage of initiating treatment with relugolix combination therapy.
Elagolix with add-back therapy is effective in reducing heavy menstrual bleeding in women with uterine leiomyomas,13 and up to 12 months of therapy has shown sustained reduction in menstrual blood loss volume in women with uterine leiomyomas and attenuated, to a certain extent, the hypoestrogenic effects of elagolix alone.14 However, elagolix requires twice-daily dosing due to its short half-life, which may be difficult for long-term adherence with therapy. Although not direct head-to-head studies, the reported rate of vasomotor symptoms and BMD loss at 1 year15 was higher than observed in the LIBERTY Extension Study. Therefore, a therapeutic agent that is well tolerated and provides effective long-term management of the symptoms associated with uterine leiomyomas may help address a significant medical need as well as help support patient demands and choice for uterus-preserving treatment.15
A subset of participants from the LIBERTY pivotal studies enrolled in this study extension, which may have introduced selection bias. Baseline characteristics of the participants were consistent with those in the pivotal studies, with a high disease burden and few participants ineligible due to loss in BMD. There was a lack of a placebo comparator during conduct of the LIBERTY Extension Study, so no cross-group comparisons for efficacy or safety were possible. It would not have been ethical to maintain a placebo group beyond 24 weeks, with the established efficacy of relugolix combination therapy and other available treatments for uterine leiomyomas.
Through 52 weeks of treatment, relugolix combination therapy showed durability of effect in improvement of heavy menstrual bleeding associated with uterine leiomyomas, including rapid and sustained reduction in menstrual blood loss volume, improvement of anemia, increased amenorrhea rate, and sustained improvement in patient-reported outcomes, including distress due to uterine leiomyoma–associated symptoms and HRQOL. No new safety concerns were identified, and BMD was generally preserved. Relugolix combination therapy may represent an option for longer-term treatment in women with heavy menstrual bleeding associated with uterine leiomyomas.
Authors' Data Sharing Statement
Will individual participant data be available (including data dictionaries)? No.
What data in particular will be shared? Not available.
What other documents will be available? Not available.
When will data be available (start and end dates)? Not applicable.
By what access criteria will data be shared (including with whom, for what types of analyses, and by what mechanism)? Not applicable.
Footnotes
Sponsored by Myovant Sciences GmbH. The sponsor designed the study in collaboration with the academic authors. The sponsor had a role in data collection, data analysis, data interpretation and writing of the report. The academic authors worked with the Sponsor to interpret the data. Sponsor authors collaborated with academic authors in the development of the manuscript. The sponsor also funded editorial support from JD Cox, PhD, and Mayville Medical Communications, in compliance with Good Publication Practice 3 ethical guidelines (Battisti et al, Ann Intern Med 2015; 163:461-4).
Financial Disclosure Ayman Al-Hendy has been a Consultant for AbbVie, Bayer, Myovant Sciences, and ObsEva. They have received Research Support from the National Institutes of Health (R01 ES 028615–01, R01HD 087417, R01 HD 094378, R01 HD 094380, R01 HD 10036701, U54 MD 007602) and hold a patent for methods for novel diagnostics and therapeutics for uterine sarcoma (US Pat No. 9,790,562 B2). Andrea Lukes received research support from AbbVie, Astellas, Bayer, Ferring, Merck, Mithra, Mylan, Myovant Sciences, Organon. They have been a consultant for AbbVie and Myovant Sciences and served on the Speaker Bureau for AbbVie. Served on the advisory board for BCD Meetings & Events (December 10, 2021).Roberta Venturella has been a consultant for IBSA Pharmaceuticals and Myovant Sciences Inc. Elizabeth Stewart has been a consultant for AbbVie, Bayer, Myovant Sciences, and ObsEva. She received Research Support from the National Institutes of Health (R01 HD105714) and AHRQ and PCORI (P50 HS023418). She holds a patent for Methods and Compounds for Treatment of Abnormal Uterine Bleeding (US 6440445), which has no commercial activity. She has received royalties from UpToDate and payments for the development of educational content from the Med Learning Group, PER, Massachusetts Medical Society, and Peer View. Laura McKain is a former Myovant Sciences, Inc. employee. She served on the speaker's bureau and recently as consultant. She received payment from Evofem Biosciences (consultant role) and Cooper Surgical (consultant role). She is a Myovant Sciences shareholder. Rachel Wagman and Li are current employees of Myovant Sciences, Inc. Alfred Poindexter and Claudio Villarroel did not report any potential conflicts of interest.
Presented at the American Society of Reproductive Medicine’s Scientific Congress & Expo, held virtually, October 17–21, 2020.
The authors thank the patients who participated in these studies and their families, as well as all the investigators and site staff who made these studies possible. The authors also thank JD Cox, PhD, and Mayville Medical Communications for editorial support, funded by Myovant Sciences, GmbH and in compliance with Good Publication Practice 3 ethical guidelines (Battisti et al, Ann Intern Med 2015; 163:461-4).
Each author has confirmed compliance with the journal's requirements for authorship.
Peer reviews and author correspondence are available at http://links.lww.com/AOG/C914.
REFERENCES
- 1.Stewart EA. Clinical practice. Uterine fibroids. N Engl J Med 2015;372:1646–55. doi: 10.1056/NEJMcp1411029 [DOI] [PubMed] [Google Scholar]
- 2.Stewart EA, Nowak RA. Uterine fibroids: hiding in plain sight. Physiology (Bethesda) 2022;37:16–27. doi: 10.1152/physiol.00013.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stewart EA, Laughlin-Tommaso SK, Catherino WH, Lalitkumar S, Gupta D, Vollenhoven B. Uterine fibroids. Nat Rev Dis Primers 2016;2:16043. doi: 10.1038/nrdp.2016.43 [DOI] [PubMed] [Google Scholar]
- 4.Osuga Y, Enya K, Kudou K, Tanimoto M, Hoshiai H. Oral gonadotropin-releasing hormone antagonist relugolix compared with leuprorelin injections for uterine leiomyomas: a randomized controlled trial. Obstet Gynecol 2019;133:423–33. doi: 10.1097/AOG.0000000000003141 [DOI] [PubMed] [Google Scholar]
- 5.Hoshiai H, Seki Y, Kusumoto T, Kudou K, Tanimoto M. Relugolix for oral treatment of uterine leiomyomas: a dose-finding, randomized, controlled trial. BMC Womens Health 2021;21:375. doi: 10.1186/s12905-021-01475-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Al-Hendy A, Lukes AS, Poindexter AN, III, Venturella R, Villarroel C, Critchley HOD, et al. Treatment of uterine fibroid symptoms with relugolix combination therapy. N Engl J Med 2021;384:630–42. doi: 10.1056/NEJMoa2008283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eltoukhi HM, Modi MN, Weston M, Armstrong AY, Stewart EA. The health disparities of uterine fibroid tumors for African American women: a public health issue. Am J Obstet Gynecol 2014;210:194–9. doi: 10.1016/j.ajog.2013.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spies JB, Coyne K, Guaou Guaou N, Boyle D, Skyrnarz-Murphy K, Gonzalves SM. The UFS-QOL, a new disease-specific symptom and health-related quality of life questionnaire for leiomyomata. Obstet Gynecol 2002;99:290–300. doi: 10.1016/s0029-7844(01)01702-1 [DOI] [PubMed] [Google Scholar]
- 9.Barbieri RL. Hormone treatment of endometriosis: the estrogen threshold hypothesis. Am J Obstet Gynecol 1992;166:740–5. doi: 10.1016/0002-9378(92)91706-g [DOI] [PubMed] [Google Scholar]
- 10.Cramer DW, Barbieri RL, Fraer AR, Harlow BL. Determinants of early follicular phase gonadotrophin and estradiol concentrations in women of late reproductive age. Hum Reprod 2002;17:221–7. doi: 10.1093/humrep/17.1.221 [DOI] [PubMed] [Google Scholar]
- 11.Reed BG, Carr BR. The normal menstrual cycle and the control of ovulation. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dungan K, et al., editors. Endotext. MDText.com, Inc.; 2000. [Google Scholar]
- 12.Stricker R, Eberhart R, Chevailler MC, Quinn FA, Bischof P, Stricker R. Establishment of detailed reference values for luteinizing hormone, follicle stimulating hormone, estradiol, and progesterone during different phases of the menstrual cycle on the Abbott ARCHITECT analyzer. Clin Chem Lab Med 2006;44:883–7. doi: 10.1515/CCLM.2006.160 [DOI] [PubMed] [Google Scholar]
- 13.Schlaff WD, Ackerman RT, Al-Hendy A, Archer DF, Barnhart KT, Bradley LD, et al. Elagolix for heavy menstrual bleeding in women with uterine fibroids. N Engl J Med 2020;382:328–40. doi: 10.1056/NEJMoa1904351 [DOI] [PubMed] [Google Scholar]
- 14.Simon JA, Al-Hendy A, Archer DF, Barnhart KT, Bradley LD, Carr BR, et al. Elagolix treatment for up to 12 months in women with heavy menstrual bleeding and uterine leiomyomas. Obstet Gynecol 2020;135:1313–26. doi: 10.1097/AOG.0000000000003869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Management of symptomatic uterine leiomyomas. ACOG Practice Bulletin No. 228. American College of Obstetricians and Gynecologists. Obstet Gynecol 2021;137:e100–15. doi: 10.1097/AOG.0000000000004403 [DOI] [PubMed] [Google Scholar]