

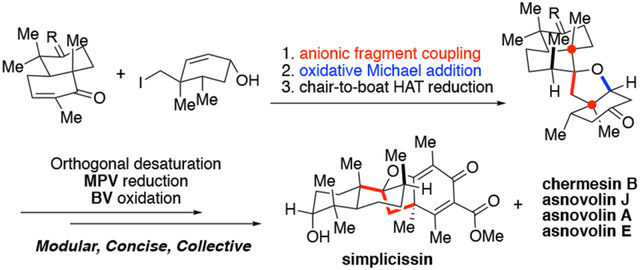
Abstract
DMOA-derived spiromeroterpenoids are a unique natural product family with attractive structures, unconventional stereochemistry, and potent biological activities. Herein, we report the first asymmetric total syntheses of the asnovolins, DMOA-derived spiromeroterpenoids. The spirocyclic skeleton was efficiently assembled through a sterically hindered bis-neopentyl 1,2-addition coupling/oxidative Michael addition sequence. The unusual axial C12-methyl stereochemistry was established via metal hydrogen atom transfer (MHAT) reduction involving a chair-to-boat conformational change. The mechanism of the HAT process was studied through both deuterium labeling and computational studies. Attempted late-stage alkene isomerization of an exocyclic enone proved to be challenging and resulted in hetero-Diels-Alder dimerization which ultimately led to development of an alternative desaturation/coupling sequence. Endgame core modifications including orthogonal desaturation, Sc(III)-promoted regioselective Baeyer-Villiger oxidation, and Meerwein-Ponndorf-Verley reduction enabled collective syntheses of five asnovolin-related natural products. This study demonstrates the utility of anionic fragment coupling to assemble a sterically congested molecular framework and provides a foundation for the synthesis of spiromeroterpenoid congeners with higher oxidation states for subsequent biological studies.
Graphical Abstract
INTRODUCTION
3,5-Dimethylorsellinic acid (DMOA)-derived meroterpenoids are an extensive natural product family with significant structural complexity and diverse biological activity.1 Among DMOA-derived meroterpenoids, the spiromeroterpenoids feature a unique spirocyclic skeleton and varied oxidation states of the terpene and polyketide moieties (Figure 1, A) in comparison to the bridged congeners such as berkeleyone A (7) and the fused derivatives including preandiloid A (9) (Figure 1, B). Fungi are rich sources of hybrid polyketide-terpene natural products. Chronologically, fumigatonin (4) was the first DMOA-derived spiromeroterpenoid isolated in 1984 with a highly complex ring system and high level of oxidation.2 Later, simpler congeners were discovered such as simplicissin (1) which was isolated in 1997 and found to have pollen growth inhibitory activity.3 In 2006, the highly oxidized congener novofumigatonin (5) was isolated from the Aspergillus novofumigatus without indication of biological activity.4 Chermesins A–D5 and asnovolins A–F6 were isolated in 2016. Among these targets, chermesin B (2) exhibited potent antibacterial activity against the pathogen Micrococcus luteus (MIC = 8 μg/mL) and asnovolin E (3) was found to suppress fibronectin expression in normal human neonatal dermal fibroblast cells (Figure 1, A). In 2018, Liu and coworkers isolated fusariumins A (6) and B, the former which exhibits strong antibacterial activity against Staphylococcus aureus and Pseudomonas aeruginosa (MIC = 6.3 μg/mL).7 This report attracted our interest underscoring potential biological activities of highly oxidized family members. Moreover, in 2018, the Abe group reported the complete biosynthetic pathway of novofumigatonin (5); additional biosynthetic intermediates were discovered as natural products which have not been isolated from the fungi previously including asnovolins H, I, J, and the highly oxidized congeners fumigatonoids A, B, and C.8
Figure 1.

DMOA-derived spiromeroterpenoids, related congeners and structural highlights/limitations.
Although the biosyntheses of DMOA-derived meroterpenoids have been studied in detail,9 their chemical syntheses have only appeared recently; all completed syntheses have focused on berkeleyone A (7) and congeners10 (Figure 1, B). Thus far, there have been no reported chemical syntheses of targets in the DMOA-derived spiromeroterpenoid family. The only synthetic study towards simplicissin (1) was described in a Ph.D. dissertation from the Baran group.11 Herein, we report an advancement in the meroterpenoid synthesis field with the chemical synthesis of five DMOA-derived spiromeroterpernoids.
It is noteworthy that, historically, total syntheses of drimane-aromatic type spiromeroterpenoids such as K-76 12(10), stypoldione 13(l1), and corallidictycal D 14(12), stachybotrylactam,15 and related meroterpenoids 16(Figure 1, C) have been studied. In comparison, the presence of the quaternary carbon center at C1’ in DMOA-derived spiromeroterpenoids significantly elevates synthetic challenge (Figure 1, D). Other salient differences include the opposite stereochemistry at the spirocyclic-center, a thermodynamically less stable axial C12 methyl group, and a fully substituted dienone or cyclohexanone subunit. In the synthesis of drimane-aromatic type spiromeroterpenoids, 12-15 the most commonly used strategy to construct the spirocycle employed acid-mediated cyclization of alkenyl phenols which generally affords the equatorial C12 methyl group and the undesired spirocenter stereochemistry due to Ftirstner-Plattner restrictions (Figure 1, E).
The biosynthesis of DMOA-derived spiromeroterpenoids8 shares similar early steps to that of other subfamilies: the enzymes NvfB and NvfK catalyze the union of DMOA and farnesyl pyrophosphate (FPP) through dearomatization and epoxidation of the farnesyl terminal double bond, respectively, to produce acid substrate 16. However, 16 undergoes a unique polyene cyclization involving a hydride shift/cyclization cascade mediated by the cyclase enzyme NvfL to forge the unusual stereochemistry of the axial C12 methyl group and the spirocyclic skeleton of asnovolin H (13) (Figure 2). Further oxidative enzyme tailoring and skeletal rearrangement produce highly oxidized members including novofumigatonin (5). Our laboratory’s interest in dearomatization chemistry and biomimetic synthesis led to the successful alkylative dearomatization of the biogenic substrate DMOA17 (Figure 2). However, biomimetic polyene cyclization of the resulting dearomatized methyl ester substrate 17 only produced unnatural tetracyclic meroterpenoid isomers and no spirocyclic products, thereby highlighting the key utility of cyclase enzymes for this process. Even when the biosynthetic precursor 17 was fed to the cyclase NvfL in vitro, asnovolin H also was not produced,18 highlighting the importance of a carboxylic acid moiety for NvfL substrate acceptance. Considering the difficulties in biomimetic approaches to spirocycles, the constraints in traditional spirocyclization, and that the reported synthetic strategies may only be immediately applicable to the berkeleyone A family, we therefore considered an abiotic synthetic strategy to access DMOA-derived spiromeroterpenoids.
Figure 2.

Biosynthesis of spiromeroterpenoids and our group’s previous biomimetic synthesis studies
RESULTS AND DISCUSSION
In our synthetic plan, asnovolin A (19) was designated as an entry target and gateway compound connecting other members with both lower and higher oxidation states (Figure 3). After extensive strategy refinement, the successful conceptual retrosynthetic analysis came from recognition of a bis-neopentyl linkage relationship in all spiromeroterpenoid family members. Accordingly, asnovolin A may be obtained from the spirocyclic precursor 20 through late-stage D ring functionalization. Compound 20 can be obtained from hydroxy-enone 21 by oxa-Michael addition. The highlighted bis-neopentyl linkage of 21 was strategically disconnected to decalin fragment 22 and cyclohexanol neopentyl iodide 23, the latter of which can be viewed as a DMOA surrogate. The syn-dimethyl relationship in 23 clearly suggested application of an asymmetric Diels-Alder cycloaddition between tiglic aldehyde and Rawal’s diene to produce the known adduct 25.19 At the outset of our investigation, the facial selectivity for late-stage stereoselective hydrogenation of 20 to establish the axial C12 methyl group was uncertain. The steric hindrance of the proposed bis-neopentyl 1,2-addition to access 21 was an additional question.
Figure 3.

Retrosynthetic analysis
In the forward synthesis, the known hydroxy enone 2619b was transformed to neopentyl iodide 27 in high yield using a one-pot triflation/SN2 process (Scheme 1). Luche reduction of enone 27 produced allylic alcohol 23 quantitatively as a single diastereomer on gram scale. It should be noted that fragment 23 contains all desired stereochemistry, especially the chiral all-carbon quaternary center, which is established early in the synthesis. Moreover, the allylic alcohol provides a suitable synthon for subsequent oxa-Michael reaction (vide infra). Additionally, decalin fragment 22 is a readily available building block prepared in five steps from (R)-carvone 24 on gram scale.20
Scheme 1.

Synthesis and further reactions of the key spiromeroterpenoid intermediate (30)
With scalable access to both fragments, the decisive anionic fragment coupling21 step was next explored. Bis-neopentyl type 1,2-addition is rare in the literature, but is a powerful transformation for assembling molecular complexity.22 Although decalin 22 and its analogues have been used in 1,2-addition, the nucleophiles in these examples were far less hindered and often used in excess (4–6 equiv.).20b,23 Optimization studies indicated that fast Li-I exchange and a higher temperature for 1,2-addition was crucial for higher product yield. In the optimized protocol,24 Li-I exchange of 23 was executed using n-BuLi (1.0 equiv.) followed by t-BuLi (2.2 equiv.) treatment at −78 °C for 10 min and then r.t. for 15 min for complete consumption of excess t-BuLi and generation of the requisite neopentyl anion nucleophile. Decalin 22 was then added to the neopentyl anion species at r.t. and was reacted for 1.5 h. The coupling product 28 was isolated in 83% yield on a 200 mg scale. The usual longer Li-X exchange time at low temperature (e.g. 1 h, −78 °C) and addition of decalin 22 at lower temperature resulted in significantly lower yield.25 The higher reaction temperature required indicated a much higher energy barrier required for the sterically hindered bis-neopentyl coupling. Diol product 28 was found to be unstable and prone to decompose likely due to intramolecular SN2’ cyclization as the tertiary alcohol lone electron pair is suitably positioned over the π* orbital of allylic alcohol. Accordingly, purified diol 28 was immediately subjected to MnO2 oxidation.26 The expected tandem allylic oxidation/oxa-Michael addition event occurred with good efficiency to afford spirocycle 20 in 67–72% yield. It is noteworthy that other protecting groups (e.g. enol silanes, enolates, and acetals) for enone 27 were not compatible with the key anionic fragment coupling step. Use of an allylic alcohol (i.e. 23) was unique in its ability to enable the key 1,2-addition fragment coupling and consecutive, oxidative oxa-Michael addition. Due to the innate steric shielding of the β-face by the angular C13 methyl group, the 1,2-addition/oxidative Michael addition sequence readily established the correct C9 stereochemistry of 20. This step highlights the strategic value of anionic fragment coupling in Csp3–Csp3 bond formation in complex molecule synthesis
The next key goal was to establish the axial C12 methyl group through stereoselective reduction of the trisubstituted alkene in substrate 20 (Scheme 1). Both DFT models and NOESY experiment indicated spatial proximity of the vinyl methyl and C6’ methine (1.92Å) for substrate 20, but very similar steric environments for both faces of the alkene. We considered that if hydrogenation occurred from the β-face, the resulting equatorial methyl group would have severe steric interactions with the C6’ methine. In initial experiments, hydrogenation of 20 (H2, Pd/C) resulted in full recovery of starting material. Recently, metal hydride atom transfer (MHAT) hydrogenation of alkenes pioneered by Shenvi and coworkers has been widely applied in natural product synthesis to afford thermodynamically stable products.27 Along these lines, production of a thermodynamically less stable axial methyl group through MHAT is not common or obvious. To the best of our knowledge, there is only one literature precedent for this type of process by the Yang group who installed an axial methyl group in their elegant synthesis of waihoensene using HAT reduction to reduce a 1,1-disubstituted, exocyclic olefin.28a In their study, an initially formed tertiary radical abstracted a spatially proximal ketone α-hydrogen. In our system, the initially formed tertiary radical may also abstract the tertiary C6’ methine. However, this reaction should also scramble the C6’methine stereochemistry. Gratifying, HAT reduction of 20 under Baran’s condition (Fe(acac)3, PhSiH3, EtOH)29 afforded compound 29 bearing the axial C12 methyl stereochemistry as a single diastereomer wherein the C6’ stereochemistry was retained. In comparison, the recent synthesis of the aromatic-type spiromeroterpenoid stypodiol30 by Renata and coworkers described production of an equatorial methyl group in a similar HAT reduction, highlighting the decisive role of the C2’ sp3carbon in conformational control. Compared with the cationic hydride shift/cyclization process in nature, this rationally designed HAT process provides an alternative approach to establish the challenging axial methyl stereochemistry.
In a key step, HAT reduction established the axial C12 methyl and the stereochemistry of the C6’ methine was retained which prompted our interest to explore the reaction mechanism (Scheme 2). Deuterium labeling using PhSiD3 as reductant showed complete labeling of the C7-axial hydrogen as the only labeling site. When the experiment was conducted in EtOD, 47% deuterium labeling was observed at C5’as the only labeling site which indicates the possibility for direct C–H abstraction from the ketone α-hydrogen (C5’-H, BDE = 94 kcal/mol), rather than the C6’-methine (BDE = 96 kcal/mol).31 The moderate labeling content also suggests that the radical intermediate C (cf. Figure 4.) can be either reduced by Fe(II) species to an enolate which may be followed by protonation with EtOD or directly undergo HAT from Fe(III)–H. The stereochemistry of the incorporated deuterium atom was determined by coupling constant analysis.24 When both PhSiD3 and EtOD were used, complete labeling of the C7 axial hydrogen and C5’ ketone α-position were observed. No deuterium labeling at C6’ was observed in all experiments.
Scheme 2.

Deuterium labeling experiments
Figure 4.

DFT calculations for the Curtin-Hammett-driven HAT Reduction at M06-2X/6-31G(d,p)/ PBF (EtOH)
In order to further understand the energetics of the stereoselective HAT reduction process, we performed DFT computations at the M06-2X/6-31G(d,p) /PBF(EtOH) level of theory (298 K, 1 atm) using Jaguar in the Maestro platform32 (Figure 4). Conformational searches indicated that the radical A produced by initial HAT reduction can undergo a chair-to-boat conformational change to B (ΔG = 3.45 kcal/mol). The boat conformer B positions the ketone α-hydrogen proximal to the tertiary radical (2.54 Å) to allow for efficient HAT to occur. Transition state calculations shows that TS1 for ketone α-hydrogen abstraction is 3.7 kcal/mol lower than TS2 for C6’ methine abstraction. Thus, the process appears to kinetically favor the boat HAT pathway. Thermodynamically, the keto radical product C is 2.48 kcal/mol more stable than boat radical conformer B, while radical product D is 3.25 kcal/mol less stable than chair radical conformer A. Overall, keto radical C is 2.28 kcal/mol more stable than tertiary radical D. Thus, we conclude that the HAT reduction process for substrate 20 occurs under a Curtin-Hammett scenario33 and follows the boat 1,6-HAT pathway which explains the complete retention of C6’ stereochemistry.
Having developed an efficient approach to the spirocyclic skeleton of the asnovolins with full stereochemical control in only five steps from 26, the major tasks that remained were to install the vinylogous methyl ester and further elaborate the spirocyclic core. We first designed an Eschenmoser methylenation/isomerization sequence for this non-trivial transformation in terms of step-economy (Scheme 1). DFT calculations showed that the desired endocyclic enone 31 is 18 kcal/mol more stable than the exocyclic enone counterpart 30 due to conjugation with vinylogous oxygen.24 Thus, it is predicted the alkene isomerization should be a facile transformation. Soft enolization of 29 gave complete regiocontrol; the resulting crude TMS enol ether was treated with freshly prepared N,N-dimethylmethyleneiminium iodide (Eschenmoser salt) in one pot. The resulting crude Mannich base was then treated with MeI and NaHCO3 which cleanly afforded exocyclic enone 30 by TLC analysis. However, to our surprise, 30 was found to be reactive and produced a varied and inseparable diastereomeric mixture of dimers in CDCl3 or when stored neat through hetero-Diels Alder (HDA) cycloaddition.34 Despite this undesired reactivity, the exocyclic enone was stable in non-acidic solvents and its clean formation indicated high efficiency if alkene isomerization could be achieved. Shenvi’s and Norton’s radical type Co–H alkene isomerization chemistry36a,b were first evaluated, but they were not effective at r.t., while heating at 60 °C produced the mixture of dimeric hetero-Diels Alder cycloadducts as the major product.24 Extensive screening of other metal-hydride catalysts all proved to be unfruitful.35,36 Only minimal success was found using catalytic RhCl3 in EtOH under reflux conditions (30–33% yield). Even so, the high temperature required resulted in competing dimerization, acetal hydrolysis, and Michael addition of EtOH.
The challenge encountered with the alkene isomerization chemistry promoted the development of an alternative desaturation/cross coupling sequence to install the vinylogous methyl ester moiety. IBX was identified as an optimal oxidant to effect desaturation of 29 in which case the desired enone 32 was produced with complete regiocontrol, in good yield (72%), and without evidence of double desaturation37 (Scheme 3). Addition of 4 Å molecular sieves and Na2HPO4 was crucial to prevent acetal hydrolysis. An iodide handle was installed on the electron-rich vinylogous ester in high yield (84%) using CAN/I2 at 0 °C.38 The methyl group was installed through Suzuki coupling10a in excellent yield (95%) to produce the divergent intermediate 31. Overall, this sequence was more robust and scalable than the elusive alkene isomerization approach.
Scheme 3.

Collective synthesis of five asnovolin natural products
With advanced intermediate 31 in hand, attempts to access chermesin B (2) with a second IBX desaturation only returned starting material even under more forcing conditions. It was rationalized that the vinylogous oxygen atom may stabilize a radical cation intermediate, thereby suppressing proton elimination.39 After considerable experimentation, we found the benzeneseleninic acid anhydride40 worked well to effect desaturation of 31 to afford chermesin B (2) in 86% yield after one pot acetal hydrolysis. Next, the ester was installed through enolate acylation of 31, which afforded asnovolin J (35) in 77 % yield after acetal hydrolysis. It is of note that KHMDS was more effective than LiHMDS in the enolate acylation. The structure of the ester intermediate 34 was confirmed through single crystal X-ray analysis which also confirmed the axial C12 methyl and C6’ methine stereochemistry established by HAT reduction (cf. Scheme 1). Direct Baeyer-Villiger oxidation of 35 using m-CPBA produced a complex mixture where it appeared that the electron-rich vinylogous ester double bond also underwent epoxidation. Gratifyingly, addition of the rare-earth Lewis acid Sc(OTf)3 to substrate 35 reduced electron density of the enone and increased electrophilicity of the C3 ketone towards Baeyer-Villiger oxidation41 to afford asnovolin A (19) in 76% yield. Treatment asnovolin A (19) with p-TsOH in MeOH produced asnovolin E (3) in 41% yield. Ester intermediate 34 was further desaturated and deprotected to afford the advanced dienone intermediate 36. Final reduction of the C3-ketone to produce simplicissin (1) proved to be very challenging. Bulky reductants including L-Selectride and LiAlH(t-Bu)(i-Bu)2 reduced the dienone moiety preferentially which was followed by reduction of the C3-carbonyl to a diastereomeric mixture (1.5:1 d.r.) favoring the undesired equatorial alcohol. This result indicated that the electron-poor dienone C5’–C6’ double bond was incompatible with reactive metal-hydride reagents. Alternatively, when asnovolin J (35) was subjected to L-Selectride reduction, the equatorial alcohol was again found to be the major product which indicated a non-trivial conformational effect in a complex environment. Other strategies including enone protection via Diels-Alder cycloaddition with CpTMS42 or Ph3P/TMSOTf43 and stereo-inversion of the C3-equatorial hydroxyl group via Mitsunobu reaction all proved to be unsuccessful. Finally, it was found Meerwein-Ponndorf-Verley (MPV) reduction through in-situ generation of the highly reactive and non-aggregated reagent (i-PrO)2AlCl44 was the sole method to produce simplicissin (1) as major product (5:1 d.r.). This highly reactive, yet mild reduction has remarkable chemo- and stereoselectivity and may find applications in other total synthesis endeavors when traditional methods may fail. It is rationalized that intramolecular transfer of the hydride to the C3 ketone through the six-membered transition state TS3 from the β-face is more favored (Figure 5.) where the aluminum chelates to the carbonyl electron lone pair away from the gem-dimethyl and the isopropyl group also steers away from the methyl groups, while the corresponding reduction from the α-face may suffer from severe steric interactions between the isopropyl group and the C1-Hax, C5-H, C7’-Me and the spiro ring system and is thus less favored.
Figure 5.

Proposed transition state of the diastereoselective MPV reduction
CONCLUSION
In summary, we have developed a modular and concise route to construct the spiromeroterpenoid skeleton of the asnovolins and related compounds and have demonstrated its efficiency through the collective synthesis of five spiromeroterpenoids in the asnovolin family in 9–11 steps from known materials (14–16 steps from commercially available (R)-carvone). Key transformations include 1) use of a sterically hindered bis-neopentyl anionic fragment coupling, 2) oxidative oxa-Michael cyclization, 3) stereoselective HAT reduction to install an axial methyl group and 4) late-stage orthogonal ketone desaturation, oxidation, and reduction. Moreover, deuterium labeling and DFT computational studies revealed that the stereoselective MHAT reduction process involves an interesting chair-to-boat conformational change under Curtin-Hammett conditions. The chemistry developed herein should enable access to diverse spiromeroterpenoid analogues for biological studies. The synthesis also provides access to materials for chemoenzymatic and enzyme mechanistic experiments. Access to numerous lower oxidation state spiromeroterpenoids through this approach provides a solid foundation for the synthesis of highly oxidized meroterpenoids such as novofumigatonin using chemical and chemoenzymatic synthesis. Such efforts will be reported from our laboratories in due course.
Supplementary Material
ACKNOWLEDGMENTS
We thank the National Institutes of Health (NIH) (R35 GM 118173) and Prelude Biosciences for financial support. We gratefully acknowledge Prof. Ikuro Abe (University of Tokyo) for providing NMR spectra of asnovolins A and J. Prof. John Snyder and Dr. Paul Ralifo (Boston University) are acknowledged for assistance with the NMR analysis and deuterium labeling experiments. We thank Dr. James McNeely (Boston University) for assistance with computations and Dr. Jeffrey Bacon (Boston University) for assistance with X-ray crystal structure analysis. We thank the National Science Foundation (NSF) for support of NMR (CHE-0619339) and MS (CHE-0443618) facilities at BU and the NIH (S10OD028585) for support of the single crystal XRD system. This paper is dedicated to the memory of Professor David A. Evans for his enormous contribution to organic synthesis and chemistry education.
Footnotes
Supporting Information
The Supporting Information is available free of charge at: Experimental procedures, analytical data, 1H and 13C NMR spectra of all newly synthesized compounds, and X-ray crystallographic analysis of compound 34 and DFT calculation details (PDF)
Accession Codes
CCDC 2171953 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.acuk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystal-lographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax +44 1223 336033.
The authors declare no competing financial interest.
REFERENCES
- (1).(a) For reviews of meroterpenoids, see: Geris R; Simpson TJ Meroterpenoids Produced by Fungi. Nat. Prod. Rep 2009, 26, 1063–1094. [DOI] [PubMed] [Google Scholar]; (b) Jiang M; Wu Z; Liu L; Chen S The Chemistry and Biology of Fungal Meroterpenoids (2009-2019). Org. Biomol. Chem 2021, 19, 1644–1704. [DOI] [PubMed] [Google Scholar]
- (2).Okuyama E; Yamazaki M Fumigatonin, a New Meroterpenoid from Aspergillus Fumigatus. Tetrahedron Lett. 1984, 25 (30), 3233–3234. [Google Scholar]
- (3).Kusano M; Koshino H; Uzawa J; Fujioka S; Kawano T; Kimura Y Simplicissin, a New Pollen Growth Inhibitor Produced by the Fungus, Penicillium Cf. Simplidssimum (Oudemans) Thorn No. 410. Biosci. Biotechnol. Biochem 1997, 61 (12), 2153–2155. [DOI] [PubMed] [Google Scholar]
- (4).Rank C; Phipps RK; Harris P; Fristrup P; Larsen TO; Gotfredsen CH Novofumigatonin, a New Orthoester Meroterpenoid from Aspergillus Novofumigatus. Org. Lett 2008, 10 (3), 401–404. [DOI] [PubMed] [Google Scholar]
- (5).Liu H; Li X-M; Liu Y; Zhang P; Wang J-N; Wang B-G Chermesins A–D: Meroterpenoids with a Drimane-Type Spirosesquiterpene Skeleton from the Marine Algal-Derived Endophytic Fungus Penicillium Chermesinum EN-480. J. Nat. Prod 2016, 79 (4), 806–811. [DOI] [PubMed] [Google Scholar]
- (6).Ishikawa K; Sato F; Itabashi T; Wachi H; Takeda H; Wakana D; Yaguchi T; Kawai KI; Hosoe T Asnovolins A-G, Spiromeroterpenoids Isolated from the Fungus Aspergillus Novofumigatus, and Suppression of Fibronectin Expression by Asnovolin E. J. Nat. Prod 2016, 79 (9), 2167–2174. [DOI] [PubMed] [Google Scholar]
- (7).Yan C; Liu W; Li J; Deng Y; Chen S; Liu H Bioactive Terpenoids from Santalum Album Derived Endophytic Fungus Fusarium Sp. YD-2. RSC Adv. 2018, 8 (27), 14823–14828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Matsuda Y; Bai T; Phippen CBW; Nødvig CS; Kjærbølling I; Vesth TC; Andersen MR; Mortensen UH; Gotfredsen CH; Abe I; Larsen TO Novofumigatonin Biosynthesis Involves a Non-Heme Iron-Dependent Endoperoxide Isomerase for Orthoester Formation. Nat. Commun 2018, 9 (1), 2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).(a) For review of meroterpenoid biosynthesis, see: Matsuda Y; Awakawa T; Mori T; Abe I Unusual Chemistries in Fungal Meroterpenoid Biosynthesis. Current Opinion in Chemical Biology. 2016, 31, 1–7. [DOI] [PubMed] [Google Scholar]; (b) Matsuda Y; Abe I Biosynthesis of Fungal Meroterpenoids. Nat. Prod. Rep. 2016, 33, 26–53. [DOI] [PubMed] [Google Scholar]; (c) Barra L; Abe I Chemistry of Fungal Meroterpenoid Cyclases. Nat. Prod. Rep 2021, 38 (3), 566–585. [DOI] [PubMed] [Google Scholar]
- (10).(a) For chemical synthesis of DMOA-derived meroterpenoids, see: Ting CP; Xu G; Zeng X; Maimone TJ Annulative Methods Enable a Total Synthesis of the Complex Meroterpene Berkeleyone A. J. Am. Chem. Soc 2016, 138, 55. [DOI] [PubMed] [Google Scholar]; (b) Elkin M; Szewczyk SM; Scruse AC; Newhouse TR Total Synthesis of (±)-Berkeleyone A. J. Am. Chem. Soc 2017, 139, 1790–1793. [DOI] [PubMed] [Google Scholar]; (c) Xu G; Elkin M; Tantillo DJ; Newhouse TR; Maimone TJ Traversing Biosynthetic Carbocation Landscapes in the Total Synthesis of Andrastin and Terretonin Meroterpenes. Angew. Chem., Int. Ed 2017, 56(41), 12498–12502. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zhang Y; Ji Y; Franzoni I; Guo C; Jia H; Hong B; Li H Enantioselective Total Synthesis of Berkeleyone A and Preaustinoids. Angew. Chem., Int. Ed 2021, 60 (27), 14869–14874. [DOI] [PubMed] [Google Scholar]
- (11).Lockner JW Ph.D. Dissertation, The Scripps Research Institute, 2011. [Google Scholar]
- (12).(a) K-76 synthesis: Corey EJ; Das J Total Synthesis of the Complement Inhibitor K-76 in Racemic Form. Structural Assignment to “K-76 Monocarboxylic Acid”. J. Am. Chem. Soc 1982, 104 (20), 5551–5553. [Google Scholar]; (b) McMurry JE; Erion MD Stereoselective Total Synthesis of K-76. J. Am. Chem. Soc 1985, 107, 2712–2720. [Google Scholar]
- (13).Falck JR; Chandrasekhar S; Manna S; Chiu C-CS Total Synthesis of the Spiro-Benzoquinonefuran (−)-Stypoldione. J. Am. Chem. Soc 1993, 115, 11606–11607. [Google Scholar]
- (14).(a) Cano MJ; Bouanou H; Tapia R; Alvarez E; Alvarez-Manzaneda R; Chahboun R; Alvarez-Manzaneda E NIS-PPh3: A Selective Reagent for the Spiroannulation of o-Allyl Phenols. Total Synthesis of Corallidictyal D. J. Org. Chem 2013, 78 (18), 9196–9204. [DOI] [PubMed] [Google Scholar]; (b) Markwell-Heys AW; George JH Some Chemical Speculation on the Biosynthesis of Corallidictyals A-D. Org. Biomol. Chem 2016, 14 (24), 5546–5549. [DOI] [PubMed] [Google Scholar]; (c) Dethe DH; Dherange BD; Ali S; Parsutkar MM Enantiospecific Total Syntheses of Meroterpenoids (−)-F1839-I and (−)-Corallidictyals B and D. Org. Biomol. Chem 2017, 15 (1), 65–68. [DOI] [PubMed] [Google Scholar]; (d) Wang JL; Li HJ; Wu YC Divergent Synthesis of Marine Natural Products Siphonodictyal B, Corallidictyals C/D, and Liphagal Based on the Early Presence of an Aldehyde Group Instead of a Late-Stage Introduction. J. Org. Chem 2018, 83 (15), 8716–8723. [DOI] [PubMed] [Google Scholar]
- (15).Kende AS; Deng WP; Zhong M; Guo XC Enantioselective Total Synthesis and Structure Revision of Spirodihydrobenzofuranlactam 1. Total Synthesis of Stachybotrylactam. Org. Lett 2003, 5 (10), 1785–1788. [DOI] [PubMed] [Google Scholar]
- (16).(a) The tetracyclic drimane-aromatic type meroterpenoid family has also been well studied in chemical synthesis, see: Dixon DD; Lockner JW; Zhou Q; Baran PS Scalable, Divergent Synthesis of Meroterpenoids via “Borono-Sclareolide.” J. Am. Chem. Soc 2012, 134 (20), 8432–8435. [DOI] [PubMed] [Google Scholar]; (b) Wildermuth R; Speck K; Haut FL; Mayer P; Karge B; Brönstrup M; Magauer T A Modular Synthesis of Tetracyclic Meroterpenoid Antibiotics. Nat. Commun 2017, 8 (1), 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ma TK; Elliott DC; Reid S; White AJP; Parsons PJ; Barrett AGM Meroterpenoid Synthesis via Sequential Polyketide Aromatization and Cationic Polyene Cyclization: Total Syntheses of (+)-Hongoquercin A and B and Related Meroterpenoids. J. Org. Chem 2018, 83 (21), 13276–13286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Powers Z; Scharf A; Cheng A; Yang F; Himmelbauer M; Mitsuhashi T; Barra L; Taniguchi Y; Kikuchi T; Fujita M; Abe I; Porco JA Biomimetic Synthesis of Meroterpenoids by Dearomatization-Driven Polycyclization. Angew. Chem., Int. Ed 2019, 58 (45), 16141–16146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Mitsuhashi T; Barra L; Powers Z; Kojasoy V; Cheng A; Yang F; Taniguchi Y; Kikuchi T; Fujita M; Tantillo DJ; Porco JA; Abe I Exploiting the Potential of Meroterpenoid Cyclases to Expand the Chemical Space of Fungal Meroterpenoids. Angew. Chem., Int. Ed 2020, 132 (52), 23980–23989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).(a) Huang Y; Iwama T; Rawal VH Highly Enantioselective Diels-Alder Reactions of 1-Amino-3-Siloxy-Dienes Catalyzed by Cr(III)-Salen Complexes. J. Am. Chem. Soc 2000, 122, 7843–7844. [Google Scholar]; (b) You LF; Hsung RP; Bedermann AA; Kurdyumov AV; Tang Y; Buchanan GS; Cole KP An Enantioselective Synthesis of the ABD Tricycle for (−)-Phoinactin A Featuring Rawal’s Asymmetric Diels-Alder Cycloaddition. Adv. Synth. Catal 2008, 350 (18), 2885–2891. [PMC free article] [PubMed] [Google Scholar]
- (20).(a) Odani A; Ishihara K; Ohtawa M; Tomoda H; Omura S; Nagamitsu T Total Synthesis of Pyripyropene A. Tetrahedron. 2011, 67 (42), 8195–8203. [Google Scholar]; (b) Zhong Z; Zhao G; Xu D; Dong B; Song D; Xie X; She X Bioinspired Total Syntheses of Isospongian-Type Diterpenoids (−)-Kravanhins A and C. Chem. - An Asian J 2016, 11 (10), 1542–1547. [DOI] [PubMed] [Google Scholar]
- (21).(a) For an excellent review on fragment coupling strategy in total synthesis, see: Tomanik M; Hsu IT; Herzon SB Fragment Coupling Reactions in Total Synthesis That Form Carbon–Carbon Bonds via Carbanionic or Free Radical Intermediates. Angew. Chem., Int. Ed, 2021, 60, 1116–1150 and references therein. [DOI] [PubMed] [Google Scholar]; (b) This disconnection drew inspiration from Herzon’s elegant synthesis of myrocin G. Economou C; Tomanik M; Herzon SB Synthesis of Myrocin G, the Putative Active Form of the Myrocin Antitumor Antibiotics. J. Am. Chem. Soc 2018, 140 (47), 16058–16061. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) For a recent application of anionic fragment coupling strategy in total synthesis, see: Han A; Tao Y; Reisman SE A 16-Step Synthesis of the Isoryanodane Diterpene (+)-Perseanol. Nature. 2019, 573 (7775), 563–567. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Wong AR; Fastuca NJ; Mak VW; Kerkovius JK; Stevenson SM; Reisman SE Total Syntheses of the C19-Diterpenoid Alkaloids (−)-Talatisamine, (−)-Liljestrandisine, and (−)-Liljestrandinine by a Fragment Coupling Approach. ACS Cent. Sci 2021, 7 (8), 1311–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).(a) There are a few examples of hindered bis-neopentyl carbon-carbon bond constructions through anionic 1,2-addition and 1,4-conjugate addition, see: Zeng M; Murphy SK; Herzon SB Development of a Modular Synthetic Route to (+)-Pleuromutilin, (+)-12-Epi-Mutilins, and Related Structures. J. Am. Chem. Soc 2017, 139 (45), 16377–16388. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Brill ZG; Grover HJ; Maimone TJ Enantioselective Synthesis of an Ophiobolin Sesterterpene via a Programmed Radical Cascade. Science. 2016, 352, 1078–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Okamoto R; Takeda K; Tokuyama H; Ihara M; Toyota M Toward the Total Synthesis of (±)-Andrastin C. J. Org. Chem 2013, 78 (1), 93–103. [DOI] [PubMed] [Google Scholar]
- (24).Please see the SI for complete details.
- (25).Bailey WF; Punzalan ER Convenient General Method for the Preparation of Primary Alkyllithiums by Lithium-Iodine Exchange. J. Org. Chem 1990, 55 (19), 5404–5406. [Google Scholar]
- (26).Hu J; Bian M; Ding H Recent Application of Oxa-Michael Reaction in Complex Natural Product Synthesis. Tetrahedron Lett. 2016, 57, 5519–5539. [Google Scholar]
- (27).(a) Iwasaki K; Wan KK; Oppedisano A; Crossley SWM; Shenvi RA Simple, Chemoselective Hydrogenation with Thermodynamic Stereocontrol. J. Am. Chem. Soc 2014, 136 (4), 1300–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Review, see: Green SA; Crossley SWM; Matos JLM; Vásquez-Céspedes S; Shevick SL; Shenvi RA The High Chemofidelity of Metal-Catalyzed Hydrogen Atom Transfer. Acc. Chem. Res 2018. 51. 2628–2640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).(a) Qu Y; Wang Z; Zhang Z; Zhang W; Huang J; Yang Z Asymmetric Total Synthesis of (+)-Waihoensene. J. Am. Chem. Soc 2020, 142 (14), 6511–6515. [DOI] [PubMed] [Google Scholar]; (b) There is related transannular [1,5]-HAT Farney EP; Feng SS; Schä F; Reisman SE Total Synthesis of (+)-Pleuromutilin. J. Am. Chem. Soc 2018, 140 (4), 1267–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Lo JC; Yabe Y; Baran PS A Practical and Catalytic Reductive Olefin Coupling. J. Am. Chem. Soc 2014, 136 (4), 1304–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Li J; Li F; King-Smith E; Renata H Merging Chemoenzymatic and Radical-Based Retrosynthetic Logic for Rapid and Modular Synthesis of Oxidized Meroterpenoids. Nat. Chem 2020, 12 (2), 173–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Luo Y-R Comprehensive Handbook of Chemical Bond Energies; CRC Press: Boca Raton, FL, 2007. [Google Scholar]
- (32).(a) Jaguar, version 9.5, Schrödinger, LLC, New York, NY, 2017. [Google Scholar]; (b) Bochevarov AD; Harder E; Hughes TF; Greenwood JR; Braden DA; Philipp DM; Rinaldo D; Halls MD; Zhang J; Friesner RA Jaguar: A High-Performance Quantum Chemistry Software Program with Strengths in Life and Materials Sciences. Int. J. Quantum Chem 2013. 113 (18). 2110–2142. [Google Scholar]
- (33).Ye T; Zhang FL; Xia HM; Zhou X; Yu ZX; Wang YF Stereoselective Hydrogen Atom Transfer to Acyclic Radicals: A Switch Enabling Diastereodivergent Borylative Radical Cascades. Nat. Commun 2022, 13 (1), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).(a) For hetero-Diels-Alder cycloadditions of exocyclic enones, see: Esmieu WR; Worden SM; Catterick D; Wilson C; Hayes CJ A Formal Synthesis of (−)-Cephalotaxine. Org. Lett 2008. 10 (14). 3045–3048. [DOI] [PubMed] [Google Scholar]; (b) Li C; Yu X; Lei X; Xu J; Shea X-K; Su Y-H; Tian J; Liang J-M; Li S; Liu H-L;; Zhang R-H; A Biomimetic Total Synthesis of (+)-Ainsliadimer A. Org. Lett 2009, 10 (4), 4284–4287. [DOI] [PubMed] [Google Scholar]; (c) Ding C; Wang L; Chen H; Wild C; Ye N; Ding Y; Wang T; White MA; Shen Q; Zhou J Ent-Kaurane-Based Regio- and Stereoselective Inverse Electron Demand Hetero-Diels-Alder Reactions: Synthesis of Dihydropyran-Fused Diterpenoids. Org. Biomol. Chem 2014. 12 (42). 8442–8452. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) D’Souza AM; Paknikar SK; Dev V; Beauchamp PS; Kamat SP Biogenetic-Type Synthesis of (+)-Cymbodiacetal, a Constituent of Cymbopogon Martinii. J. Nat. Prod. 2004, 67 (4), 700–702. [DOI] [PubMed] [Google Scholar]; (e) Hosoyama H; Shigemori H; In Y; Ishida T; Kobayashi J Occurrence of a New Dimeric Compound of 5-Oxotaxinine a through Diels- Alder Cycloaddition. Tetrahedron Lett. 1998, 39 (15). 2159–2162. [Google Scholar]
- (35).(a) A few Ru- and Rh-H conditions for isomerization of exocyclic cyclopentenones have been reported sporadically in literature, see: Disanayaka Bmsara W; Weedon AC A Convenient Synthesis of 2-Methyl-2-Cyclopentene-1-One. Synthesis. 1983, 952. [Google Scholar]; (b) Deng Y; Liang X; Wei K; Yang YR Ir-Catalyzed Asymmetric Total Syntheses of Bisdehydrotuberostemonine D, Putative Bisdehydrotuberostemonine E and Structural Revision of the Latter. J. Am. Chem. Soc 2021, 143 (49), 20622–20627. [DOI] [PubMed] [Google Scholar]; (c) Hirose A; Watanabe A; Ogino K; Nagatomo M; Inoue M Unified Total Syntheses of Rhamnofolane, Tigliane, and Daphnane Diterpenoids. J. Am. Chem. Soc 2021, 143 (31), 12387–12396. [DOI] [PubMed] [Google Scholar]; (d) Zhang J; Liu M; Wu C; Zhao G; Chen P; Zhou L; Xie X; Fang R; Li H; She X Total Synthesis of (−)-Pepluanol B: Conformational Control of the Eight-Membered-Ring System. Angew. Chem., Int. Ed 2020, 59(10), 3966–3970. [DOI] [PubMed] [Google Scholar]
- (36).(a) Crossley SWM; Barabé F; Shenvi RA Simple, Chemoselective, Catalytic Olefin Isomerization. J. Am. Chem. Soc 2014, 136 (48), 16788–16791. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Delgado KR; Youmans DD; Diver ST Mild Isomerization of Conjugated Dienes Using Co-Mediated Hydrogen Atom Transfer. Org. Lett 2020, 22 (2), 750–754. [DOI] [PubMed] [Google Scholar]; (c) Hwan JL; Smith CR; RajanBabu TV Facile Pd(II)- and Ni(II)-Catalyzed Isomerization of Terminal Alkenes into 2-Alkenes. J. Org. Chem 2009, 74 (12), 4565–4572. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Sen Ayusman; Lai T-W Mechanism of Palladium(II)-Catalyzed C=C Bond Isomerization in Olefins. J. Org. Chem, 1984, 23, 3257–3258. [Google Scholar]; (e) Ren W; Sun F; Chu J; Shi Y A Pd-Catalyzed Site-Controlled Isomerization of Terminal Olefins. Org. Lett 2020, 22, 1868–1873. [DOI] [PubMed] [Google Scholar]; (f) Wipf P; Waller DL; Reeves JT Transition-Metal-Mediated Cascade Reactions: The Water-Accelerated Carboalumination-Claisen Rearrangement-Carbonyl Addition Reaction. J. Org. Chem 2005, 70 (20), 8096–8102. [DOI] [PubMed] [Google Scholar]; (g) Wakamatsu H; Nishida M; Adachi N; Mori M Isomerization Reaction of Olefin Using RuClH(CO)(PPh3)3. J. Org. Chem 2000, 65 (13), 3966–3970. [DOI] [PubMed] [Google Scholar]; (h) Wüstenberg B; Pfaltz A Homogeneous Hydrogenation of Tri- and Tetrasubstituted Olefins: Comparison of Iridium-Phospinooxazoline [Ir-PHOX] Complexes and Crabtree Catalysts with Hexafluorophosphate (PF6) and Tetrakis[3,5-Bis(Trifluoromethyl) Phenyl]Borate (BArF) as Counterions. Adv Synth. Catal 2008, 350 (1), 174–178. [Google Scholar]; (i) Meng QY; Schirmer TE; Katou K; König B Controllable Isomerization of Alkenes by Dual Visible-Light-Cobalt Catalysis. Angew. Chem., Int. Ed 2019, 58 (17), 5723–5728. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Wakamatsu T; Tanaka M; Mitsuhashi H; Maruno M The Migration of Double Bond under the Neutral Conditions. The Transformation of α-Alkylidene Cyclic Carbonyl Compounds to α,β-Unsaturated Cyclic Carbonyl Compounds. Chem. Lett 1994, 1455–1458. [Google Scholar]; (k) Alsalahi W; Tylus W; Trzeciak AM Green Synthesis of Rhodium Nanoparticles That Are Catalytically Active in Benzene Hydrogenation and 1-Hexene Hydroformylation. ChemCatChem. 2018, 10 (9), 2051–2058. [Google Scholar]
- (37).Nicolaou KC; Montagnon T; Baran PS; Zhong Y-L Iodine (V) Reagents in Organic Synthesis . Part 4 . o-Iodoxybenzoic Acid as a Chemospecific Tool for Single Electron Transfer-Based Oxidation Processes. J. Am. Chem. Soc 2002. 124 (10). 2245–2258. [DOI] [PubMed] [Google Scholar]
- (38).Kuramochi A; Usuda H; Yamatsugu K; Kanai M; Shibasaki M Total Synthesis of (±)-Garsubellin A. J. Am. Chem. Soc 2005, 127 (41), 14200–14201. [DOI] [PubMed] [Google Scholar]
-
(39).According to the SET mechanism proposed in ref. 37, it was rationalized that vinylogous esters may stabilize radical cation intermediate which may inhibit a second IBX desaturation:
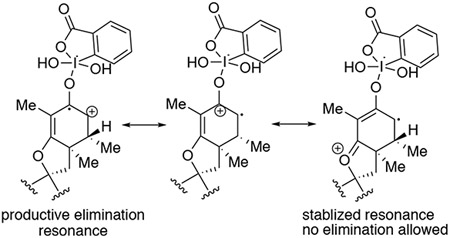
- (40).Barton DHR; Lester DJ; Ley SV Dehydrogenation of Steroidal Ketones Using Benzeneseleninic Anhydride. J. Chem. Soc. Chem. Commun 1978, 3, 130–131. [Google Scholar]
- (41).Kotsuki H; Arimura K; Araki T; Shinohara T Sc(OTf)3 and TfOH-Catalyzed Baeyer-Villiger Oxidation of Carbonyl Compounds with m-Chloroperbenzoic Acid. Synlett. 1999, 4, 462–464. [Google Scholar]
- (42).Herzon SB; Calandra NA; King SM Efficient Entry to the Hasubanan Alkaloids: First Enantioselective Total Syntheses of (−)-Hasubanonine, (−)-Runanine, (−)-Delavayine, and (+)-Periglaucine B. Angew. Chem., Int. Ed 2011, 50 (38), 8863–8866. [DOI] [PubMed] [Google Scholar]
- (43).Yahata K; Minami M; Watanabe K; Fujioka H Selective Transformations of Carbonyl Functions in the Presence of α,β-Unsaturated Ketones: Concise Asymmetric Total Synthesis of Decytospolides A and B. Org. Lett 2014, 16 (14), 3680–3683. [DOI] [PubMed] [Google Scholar]
- (44).Campbell EJ; Zhou H; Nguyen SBT Catalytic Meerwein-Pondorf-Verley Reduction by Simple Aluminum Complexes. Org. Lett 2001, 3 (15), 2391–2393. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.