

SUMMARY
Isolated methylmalonic acidemia/aciduria (MMA) is a devastating metabolic disorder with poor outcomes despite current medical treatments. Like other mitochondrial enzymopathies, enzyme replacement therapy (ERT) is not available, and although promising, AAV gene therapy can be limited by pre-existing immunity and has been associated with genotoxicity in mice. To develop a new class of therapy for MMA, we generated a 5-methoxyU-modified codon-optimized mRNA encoding human methylmalonyl-CoA mutase (hMUT), the enzyme most frequently mutated in MMA, and encapsulated it into biodegradable lipid nanoparticles (LNPs). Intravenous (i.v.) administration of hMUT mRNA in two different mouse models of MMA resulted in a 75%–85% reduction in plasma methylmalonic acid and was associated with increased hMUT protein expression and activity in liver. Repeat dosing of hMUT mRNA reduced circulating metabolites and dramatically improved survival and weight gain. Additionally, repeat i.v. dosing did not increase markers of liver toxicity or inflammation in heterozygote MMA mice.
In Brief
An et al. find that systemically delivered LNP-encapsulated mRNA results in hepatic protein expression. hMUT mRNA expresses functional mitochondrial MUT enzyme, and MMA mouse models show a metabolic and clinical response after mRNA therapy.
Graphical Abstract
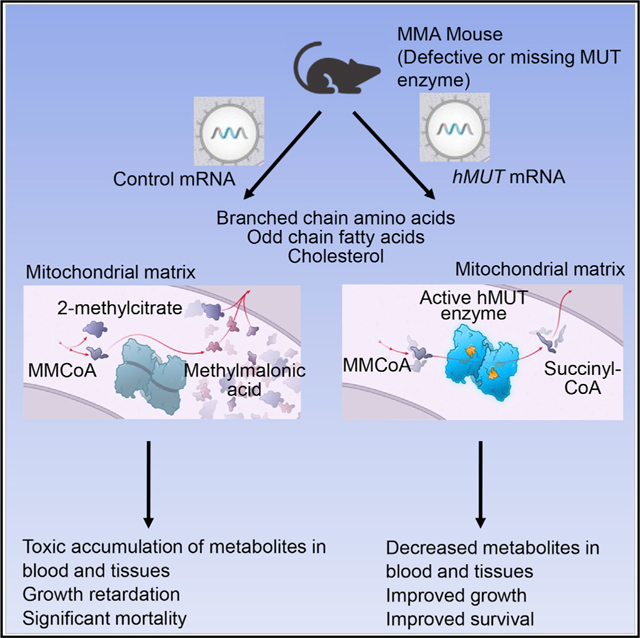
INTRODUCTION
Methylmalonic acidemia/aciduria (MMA) is a devastating metabolic disorder affecting 1 in 50,000 to 100,000 individuals (Chace et al., 2001). MMA is most commonly caused by complete (mut0) or partial (mut−) deficiency of methylmalonyl-CoA mutase (MUT), a vitamin B12-dependent mitochondrial enzyme that mediates the terminal step of valine, isoleucine, and odd chained fatty acid oxidation (Fenton et al., 2001). There are no approved therapies for MMA, and management of the disease is limited to dietary protein restriction, cofactor supplementation, and vigilant medical monitoring (Fraser and Venditti, 2016). For patients with MMA, dietary restriction to reduce precursors of the metabolic pathway is the standard of care and amounts to the prescription of a low protein diet with carnitine supplementation (Baumgartner et al., 2014; Fraser and Venditti, 2016; Manoli et al., 1993). Despite dietary management and vigilant care, the overall outcome for MMA patients remains poor, particularly for individuals with the mut0 enzymatic subtype. Mortality in mut MMA patients remains at ~20% and surviving patients suffer from numerous complications related to disease progression (Cosson et al., 2009; Fraser and Venditti, 2016; Hörster et al., 2007). Even well-managed patients remain at risk for complications associated with MMA, especially intermittent metabolic decompensation and the obligate risk of disease-associated sequelae (de Baulny et al., 2005; Dionisi-Vici et al., 2006; Hörster et al., 2007; Kölker et al., 2015). For these reasons, elective liver and combined liver-kidney transplantation (LKT) have been offered as experimental surgical treatments for severely affected patients (Niemi et al., 2015; van’t Hoff et al., 1998, 1999). Transplanted MMA patients display ameliorated biochemical perturbations and the elimination of acute metabolic decompensations and the associated risks of death and disability, driving the use of elective liver transplantation in younger patients (Hussein et al., 2013; Kamei et al., 2011; Kasahara et al., 2006) and in some cases, treatment with the living-related donor procedure (Sakamoto et al., 2016). While these positive outcomes highlight the effectiveness of increasing hepatic MUT activity to control the major manifestations of MMA, the finite number of liver donors, significant risks associated with surgery (Vernon et al., 2014), and high procedural costs limit the widespread implementation of liver transplantation in all patients with MMA. Clearly, there remain great unmet medical needs in this population.
Efforts to develop gene and cell therapy as an alternative to elective liver transplantation have been examined. The combination of transgenic animal (Manoli et al., 2013) and preclinical gene therapy studies demonstrate the importance of restoring hepatic MUT activity for phenotypic correction of MMA (Carrillo-Carrasco et al., 2010; Wong et al., 2014) and are supported by clinical observations that MMA patients who have received liver transplantation are protected from metabolic instability, the hallmark of the disorder (Niemi et al., 2015; Sakamoto et al., 2016). Like other mitochondrial enzymopathies, enzyme replacement therapy (ERT) is not available, and while promising, AAV gene therapy has variably been associated with hepatic genotoxicity in mice (Chandler et al., 2015, 2017; Donsante et al., 2007). Additionally, immune responses to the vector leads to development of neutralizing antibodies that makes re-dosing less efficient in AAV-mediated gene therapy.
mRNA treatment is an emerging class of therapy, with multiple mRNA-based cancer immunotherapies and vaccines currently in clinical trials (Sahin et al., 2014). A promising application of mRNA therapy is to restore or augment proteins following systemic administration, especially for metabolic enzymes such as MUT. mRNA therapy has a number of unique features that make clinical translation promising for inborn errors of metabolism (IEMs) such as MMA. One advantage of mRNA-based therapy over viral gene delivery is that mRNA does not transit to the nucleus, thereby mitigating insertional mutagenesis risks. mRNA provides transient, half-life-dependent protein expression, while avoiding constitutive gene activation and maintaining dose responsiveness. Systemic mRNA therapy can therefore be envisioned as an alternative to conventional ERT and can restore ‘‘undruggable’’ targets such as transmembrane and intracellular proteins. Recently, preclinical studies have evaluated mRNA-based therapy in various therapeutic areas including hemophilia B (DeRosa et al., 2016; Ramaswamy et al., 2017), sensory nerve disorders (Baba et al., 2015), congenital lung disease (Kormann et al., 2011), cancer (Wang et al., 2013), and liver and lung fibrosis (Schrom et al., 2017).
Given that it is now feasible to systemically deliver lipid nanoparticles (LNPs) that target the liver, we evaluated the potential of mRNA therapy to restore hepatic MUT as a treatment for MMA (Figure 1) using cellular and murine models. Using a bio-degradable lipid nanoparticle (LNP), we encapsulated a codon-optimized hMUT mRNA and systemically delivered the therapeutic to mice via intravenous (i.v.) administration, which resulted in robust hepatic protein expression lasting as long as 7 days. Using two murine models of MMA, we then demonstrated that hMUT mRNA therapy increased hepatic MUT activity and resulted in improved growth and survival, as well as reduced plasma metabolites, thus demonstrating biological efficacy in the treated mutant mice.
Figure 1. Strategy of hMUT mRNA Therapy.

Chemically modified and codon-optimized hMUT mRNA encapsulated in LNP is administrated using an i.v. injection. The LNP delivers the mRNA into hepatocytes where it is translated, undergoes mitochondrial importation and processing, and then forms a homodimer. The MUT enzyme requires the cofactor adenosylcobalamin (AdoCbl) for activity to catalyze the isomerization of L-methylmalonyl-CoA into succinyl-CoA.
RESULTS
hMUT mRNA Restored MUT Activity in Fibroblasts from Patients with MMA after Transfection
MUT is a nuclear-encoded, mitochondria-localized enzyme that catalyzes the reversible isomerization of L-methylmalonyl-CoA to succinyl-CoA. The protein is a homodimer that requires a cofactor, 5′-deoxyadenosylcobalamin (Adocbl), for activity. To test if hMUT mRNA (sequence provided in Table S1) expressed functional hMUT protein in vitro, human fibroblasts from one healthy subject (control) and two mut0 MMA patients were electroporated to introduce hMUT mRNA or control (eGFP) mRNA. All transfected cells showed significantly increased hMUT expression (Figure 2A) and activity (Figure 2B) compared to the eGFP mRNA transfected cells (n = 3 independent experiments). To determine the localization of hMUT expressed by hMUT mRNA, cells were stained with both anti-MUT antibodies and MitoTracker Red. Image analysis demonstrated robust colocalization between hMUT and the mitochondrial marker for all transfected cells, suggesting that hMUT mRNA effectively directs the biosynthesis of active enzyme, with the proper subcellular localization (Figures 2C and S1).
Figure 2. Expression, Activity, and Localization of mRNA-Encoded MUT.

Human fibroblasts (1 × 106) isolated from a healthy subject (control) and 2 mut0 patients were transfected with 1 μg eGFP or hMUT mRNA for 24 hr.
(A) hMUT expression in transfected human fibroblasts was determined by western blotting. Expression levels were quantified and normalized to α-tubulin levels.
(B) MUT activity in transfected human fibroblasts.
(C) Transfected cells were incubated with MitoTracker Red for 30 min to mark mitochondria and then stained with an anti-MUT mouse monoclonal antibody (mAb) to examine cellular localization (scale bars, 10 μm).
Data are presented as mean ± SD. *p < 0.05 compared to corresponding control group of same treatment, #p < 0.05 compared to its own eGFP group. p values obtained from pairwise Tukey’s post hoc test after two-way ANOVA. n = 3 independent experiments.
hMUT mRNA and Carrier LNP Were Rapidly Degraded in Liver after Delivery but Hepatic MUT Protein Persisted for 1 Week in Mice
To characterize the kinetics of hMUT mRNA, the LNP (Benenato et al., 2017), and the encoded hMUT protein in the liver, we performed a pharmacokinetics (PK) study in wild-type CD1 mice (n = 3 mice). After a single i.v. bolus of 0.5 mg/kg hMUT mRNA injected via the tail vein route, the ionizable lipid was fully metabolized in the liver after 6 hr, suggesting that the LNP was highly degradable (Figure 3A). Similar to what was observed with the carrier LNP only, mRNA quantification by branched-chain DNA (bDNA) assay showed that hMUT mRNA was rapidly cleared from the liver (Figure 3B). In situ hybridization showed that the hMUT mRNA distributed primarily to the sinusoidal space at 2 hr and then shifted to hepatocytes by 6 hr, where it remained until 24 hr (Figure 3C). In contrast, hMUT protein expression peaked at 16 hr (Tmax) with a concentration of 85 ng/mg protein, ~3-fold higher than endogenous murine Mut in the liver. hMUT protein expressed 16–48 hr post-injection was higher than endogenous MUT measured in cadaveric human livers (45.1 ± 25.3 ng/mg protein, n = 4 mice). By serially measuring hepatic MUT protein over a period of 7 days in the treated mice, we also determined that the half-life of hMUT expressed after LNP delivery was ~1.2 days (Figure 3D).
Figure 3. Pharmacokinetics of LNP and hMUT mRNA and Protein.

Wild-type mice (CD-1) were administered a single i.v. bolus of 0.5 mg/kg hMUT mRNA encapsulated in a biodegradable LNP and sacrificed at various time points (2, 6, 16 hr, 1, 2, 3, 5, 7 days; n = 3 mice/time point).
(A) Ionizable lipid in liver extracts was quantified by LC-MS/MS.
(B) Hepatic hMUT mRNA following hMUT administration was quantified using a branched-chain DNA (bDNA) assay.
(C) hMUT mRNA distribution was determined by in situ hybridization (ISH) using an hMUT mRNA-specific probe.
(D) Hepatic hMUT protein levels following hMUT mRNA administration. hMUT protein was quantified by LC-MS/MS.
****p < 0.0001, p values obtained from Tukey’s multiple-comparison test after one-way ANOVA.
A Systemic Dose of hMUT mRNA Increased hMUT Protein Expression and Activity and Lowered Plasma Methylmalonic Acid Levels in Mut−/−;TgINS-MCK-Mut Mice
Mut−/−;TgINS-MCK-Mut mice recapitulate the clinical characteristics observed in severe MMA patients where the lethal Mut−/− phenotype is rescued by skeletal muscle expression of a germline transgene (TgINS-MCK-Mut) (Manoli et al., 2011). Mut−/−; TgINS-MCK-Mut mice survive the neonatal period but because they lack hepatic Mut activity, they have 58.5% lower weight compared to heterozygous littermates at 5 weeks and marked elevations of metabolites such as methylmalonic acid (Figure S2). To determine whether systemic administration of hMUT mRNA could restore functional hepatic MUT, Mut−/−; TgINS-MCK-Mut mice received a single i.v. bolus dose of hMUT mRNA (0.5 mg/kg) encapsulated in a LNP (n = 4 mice). Three days post-injection, hepatic hMUT expression was detected (Figure 4A) and was accompanied by increased 1-13C-sodium propionate oxidative capacity to near heterozygote levels (Figure 4B) and a 75% reduction in plasma methylmalonic acid concentrations (Figure 4C). Additionally, we observed acute metabolic correction after hMUT mRNA treatment in these mice, where after a single i.v. injection at 0.5 mg/kg, plasma methylmalonic acid decreased by 48%, 80%, and 90% at 2, 6, and 24 hr, respectively, and remained suppressed over the next 2 days (Figure 4D, n = 4–8 mice). Like plasma methylmalonic acid, methylmalonic acid in various tissues including liver, heart, kidney, skeletal muscle, and brain was acutely reduced 24 hr after a single dose injection and remained suppressed for the next 2 days (Figure 4E).
Figure 4. Single i.v. Dose of hMUT mRNA Increased Hepatic MUT Expression, Enhanced Propionate Oxidation, and Lowered Plasma and Tissue Methylmalonic Acid in Mut−/−;TgINS-MCK-Mut Mice.

Mut−/−;TgINS-MCK-Mut mice (n = 4) were administered a single i.v. dose (0.5 mg/kg) of hMUT mRNA encapsulated in LNP and sacrificed 3 days later.
(A) Hepatic MUT expression in Mut−/−;TgINS-MCK-Mut mice was quantified using LC-MS/MS.
(B) Propionate oxidation in Mut−/−;TgINS-MCK-Mut mice before and 3 days after hMUT mRNA treatment. The percentage of the administered 1-13C-propionate dose that was oxidized was determined by measuring 13C enrichment in expired CO2 at multiple time points.
(C) Plasma methylmalonic acid concentrations in Mut−/−;TgINS-MCK-Mut mice 3 days after hMUT mRNA treatment analyzed by LC-MS/MS.
(D) Plasma methylmalonic acid concentrations 1 day before, and 2, 6, 24, 48, and 72 hr after a single i.v. injection of hMUT mRNA (0.5 mg/kg) or PBS control in Mut−/−;TgINS-MCK-Mut mice (n = 4–8/group).
(E) Tissue methylmalonic concentrations in liver, heart, skeletal muscle (SM), kidney, and brain in PBS-injected control and hMUT mRNA (0.5 mg/kg)-treated Mut−/−;TgINS-MCK-Mut mice (n = 4–8/group). hMUT mRNA-treated mice were sacrificed at 1 day or 3 days after a single i.v. injection.
Data shown as mean ± SD. *p < 0.05, **p < 0.01. p values obtained from paired t tests to compare post-treatment versus pre-treatment values.
Dose Response and Duration of Action of Plasma MMA in Mut−/−;TgINS-MCK-Mut Mice
To explore the dose response and duration of hMUT mRNA therapy, we administered a single i.v. dose of hMUT mRNA to Mut−/−;TgINS-MCK-Mut mice at three different doses (0.05, 0.2, and 0.5 mg/kg) and bled the mice 4 days before and again 3, 7, 10, and 14 days after injection (n = 3–4 mice). No change of plasma methylmalonic acid was observed when mice were dosed at 0.05 mg/kg (Figure 5A). In contrast, when dosed at 0.2 mg/kg, Mut−/−;TgINS-MCK-Mut mice showed decreased plasma methylmalonic acid compared to basal levels 3, 7, and 10 days after injection, which rebounded 14 days after injection (Figure 5A). A further reduction of plasma methylmalonic acid and slower rebound were observed in mice that were dosed at 0.5 mg/kg (Figure 5A). Similarly, mice treated with 0.2 mg/kg or 0.5 mg/kg hMUT mRNA gained more weight than mice treated with 0.05 mg/kg 2 weeks after a single i.v. injection (Figure S3A). Based on these results, we chose a dosage of 0.2 mg/kg and weekly dosing regimen for a repeat dose study of hMUT mRNA in Mut−/−;TgINS-MCK-Mut mice.
Figure 5. Repeat i.v. Administration of hMUT mRNA Improved Survival and Ameliorated Metabolic Alterations in mut0 MMA Mice.

To explore the dosage and dosing frequency, Mut−/−;TgINS-MCK-Mut mice received a single i.v. injection of hMUT mRNA at 0.05–0.5 mg/kg (n = 6–7). Mice were bled 4 days before and 3, 7, 10, and 14 days after a single i.v. injection.
(A) Plasma methylmalonic acid response and duration of effect across 3 doses (0.05, 0.2, 0.5 mg/kg). Mut−/−;TgINS-MCK-Mut mice received weekly i.v. injections of hMUT mRNA or a control mRNA for 5 doses at 0.2 mg/kg.
(B) Survival curve of untreated, control mRNA and hMUT mRNA-treated Mut−/−;TgINS-MCK-Mut mice. **p < 0.01 from log-rank test.
(C) Plasma methylmalonic acid concentrations 3 days after each dose in Mut−/−;TgINS-MCK-Mut mice. Plasma methylmalonic acid concentrations 6 days after each hMUT mRNA dose were similar to concentrations measured 3 days after each dose (data not presented). Arrows indicate the day of injection. WO = 10 day washout following the last i.v. injection.
(A–C) Data shown as median ± MAD (A) and mean ± SD (B and C). *p < 0.05, p values obtained from Tukey’s multiple-comparison test after one-way ANOVA.
Repetitive Systemic Dosing of hMUT mRNA Ameliorated the Biochemical Phenotype and Resulted in Improved Survival and Growth in Mut−/−;TgINS-MCK-Mut Mice
Although muscular expression of Mut prevents neonatal lethality in Mut−/−;TgINS-MCK-Mut mice, these animals display severe metabolic perturbations, growth retardation, and significant mortality by 6–9 weeks of age, reminiscent of the disease state observed in severely affected humans with mut0 MMA who survive the neonatal period. We therefore examined whether repeat i.v. dosing of hMUT mRNA could improve survival and correct biochemical and growth abnormalities. Mut−/−;TgINS-MCK-Mut mice received weekly i.v. injections of hMUT mRNA or a control mRNA for 5 weeks, both encapsulated in LNP, at a dose of 0.2 mg/kg, followed by a 10-day washout period (n = 6–7 mice). We observed high mortality in Mut−/−;TgINS-MCK-Mut mice that were untreated or received control mRNA injections (Figure 5B). From a cohort of 6 mice, only 1 untreated and 1 control mRNA-treated Mut−/−;TgINS-MCK-Mut mouse survived the first 2 weeks of the repeat-dose study (Figure 5B), and only one control mRNA-treated Mut−/−;TgINS-MCK-Mut mouse survived the entire duration of the 6-week study. In contrast, all Mut−/−; TgINS-MCK-Mut mice that were treated with hMUT mRNA survived the entire duration of the study (Figure 5B). In addition, the treated mice thrived, with 40% more weight gain compared to the sole surviving mouse in the control mRNA group (Figure S3B). Moreover, plasma methylmalonic acid levels in treated mice were significantly decreased, by 62%–89%, from the basal levels in Mut−/−;TgINS-MCK-Mut mice (PRE, Figure 5C), and partially rebounded 10 days after the last 0.2 mg/kg dose (WO, Figure 5C). Consistent with lower plasma methylmalonic acid, decreased plasma 2-methylcitrate (36%–62%) and lower C3/C2 carnitine ratios (51%–73%) were observed in Mut−/−;TgINS-MCK-Mut mice that were treated with hMUT mRNA (Figure S4). In stark contrast, nearly all of the control mice perished with the exception of a single surviving Mut−/−;TgINS-MCK-Mut mouse that manifested no metabolic response (Figure S5) and a decreased weight gain compared to mice that received hMUT mRNA therapy (Figure S3B). Our data demonstrate that systemic hMUT mRNA therapy increased survival and ameliorated both biochemical and growth abnormalities—all cardinal features of the disorder—in a MMA mouse model of the more severe mut0 subtype.
A Single Systemic Dose of hMUT mRNA Increased hMUT Protein Expression and Lowered Plasma Methylmalonic Acid in Hypomorphic Mut−/−;TgINS-CBA-G715V Mice
To explore whether hMUT mRNA therapy could also engender a metabolic response in a model of mut− MMA, we further extended our studies using hypomorphic Mut−/−; TgINS-CBA-G715V mice. Mut−/−; TgINS-CBA-G715V mice display 26.4% decreased body weight compared to heterozygous littermates at 5 weeks (Figures S2A and S2B). Moreover, Mut−/−; TgINS-CBA-G715V mice show reduced but not complete loss of Mut activity in the liver and a moderate increase of plasma methylmalonic acid levels (Figures S2C and S2D), similar to patients with mut− MMA who harbor the MUT p.G717V mutation (Senac et al., 2013). Three days after a single i.v. hMUT mRNA administration using a dose of 0.2 mg/kg (n = 4 mice), increased hMUT protein expression (Figure 6A) was detected in the treated mice. Consistent with the results seen with the Mut−/−;TgINS-MCK-Mut mice, Mut−/−; TgINS-CBA-G715V mice showed an 85% reduction in plasma methylmalonic acid levels following a single i.v. administration of hMUT mRNA (Figure 6B). To determine the duration of the biochemical response after a single i.v. injection of hMUT mRNA at an even lower dose (0.1 mg/kg, n = 4 mice), plasma methylmalonic acid was measured at 1 and 2 weeks after dosing, and a >50% reduction in metabolites persisted for 2 weeks (Figure 6C). Finally, we observed acute metabolic correction after hMUT mRNA treatment in these mice similar to what was observed in Mut−/−;TgINS-MCK-Mut mice. After a single i.v. injection at 0.2 mg/kg (n = 7 mice), plasma methylmalonic acid decreased by 30% at 2 hr, reached a nadir at 6 hr, and remained suppressed for the next 3 days (Figure 6D).
Figure 6. i.v. Administration of hMUT mRNA Improved Metabolism for More Than 1 Week in mut− MMA Mice.

(A) Mut−/−;TgINS-CBA-G715V (n = 4) were administered a single i.v. dose (0.2 mg/kg) of hMUT mRNA and sacrificed 3 days later. Hepatic hMUT expression measured by LC-MS/MS in Mut−/−;TgINS-CBA-G715V mice 3 days after a single i.v. hMUT mRNA injection.
(B) Plasma methylmalonic acid concentrations in Mut−/−;TgINS-MCK-Mut mice 1 day before and 3 days after a single i.v. hMUT mRNA injection.
(C) Plasma methylmalonic acid concentrations 1 day before, 1 week after, and 2 weeks after a single i.v. injection of hMUT mRNA (0.1 mg/kg) in Mut−/−;TgINS-CBA-G715V mice (n = 4).
(D) Plasma methylmalonic acid concentrations 1 day before, 2, 6, 48, and 72 hr after a single i.v. injection of hMUT mRNA (0.2 mg/kg) in Mut−/−;TgINS-CBA-G715V mice (n = 7).
(A–D) Data shown as mean ± SD (A–C) and median ± MAD (D). *p < 0.05, **p < 0.01. values obtained from paired t tests to compare post-treatment versus pre-treatment values.
Repeat i.v. Dosing of hMUT mRNA Did Not Increase Markers of Liver Toxicity and Inflammation and Did Not Generate Anti-drug Antibodies against hMUT
To evaluate markers of liver toxicity and inflammation due to repeat hMUT mRNA i.v. administration, Mut+/−;TgINS-CBA-G715V mice were i.v. injected weekly with either PBS or hMUT mRNA for 3 or 5 doses at 0.2 mg/kg (n = 3 mice). There was no difference in liver toxicity markers including ALP, AST, and ALT and inflammatory markers (interleukin [IL]-6, interferon gamma [IFN-γ], transforming growth factor α [TNF-α], and IL-1B) in mice that received 3 or 5 injections compared to mice that received PBS, suggesting that hMUT mRNA did not induce liver toxicity (Figure S6A; Table S2). As immune responses leading to the generation of anti-drug antibodies (ADA) can cause loss of efficacy, we measured anti-hMUT antibody levels in mice that received 3 or 5 injections. There was no significant change in anti-hMUT antibody levels suggesting that hMUT mRNA treatment did not induce an immune response in these mice (Figure S6B).
DISCUSSION
While individually rare, IEMs are relatively common conditions with a collective incidence as high as 1:1,000 (Sanderson et al., 2006). Despite the potential for early diagnosis by newborn screening for a large number of IEMs (American Academy of Pediatrics Newborn Screening Authoring Committee, 2008; Matern et al., 2013), which would enable early therapeutic intervention and improved outcomes, current treatments for these disorders remain limited. The condition under study here, MMA, is typical of the growing number of metabolic diseases that can be detected early, but lack effective treatments. Despite the success of ERT in the treatment of many lysosomal storage diseases, conventional ERT has limitations when applied to IEMs such as MMA because of biophysical and enzymatic constraints surrounding the targeted delivery of enzymes into the mitochondria. Moreover, conventional therapeutic enzymes are manufactured using a variety of human, animal or plant cells, and the biochemical and pharmacological properties of such recombinant enzymes might be different from those made endogenously in humans, such as leader sequence processing, cellular localization signals, and post-translational modifications, all of which could be critical for function.
Delivering mRNA to target cells presents an alternative strategy that could provide an enzyme missing or defective due to mutation(s). In the case of mitochondrial enzyme replacement therapy by mRNA, the resultant proteins would need to be trafficked to the mitochondrial matrix via the endogenous machinery of the cell, increasing enzymatic activity. As we demonstrated in these in vitro and in vivo studies, the delivery of hMUT mRNA results in active, localized, and fully mature MUT.
We developed a potent hMUT mRNA therapy and delivered the drug using an LNP to protect the hMUT mRNA from degradation by RNases in the blood and to improve hepatic uptake. Unlike ERT, in which therapeutic proteins are exposed to immune cells in the blood, LNPs deliver mRNA to hepatocytes, as has been seen in several clinical trials (Barros and Gollob, 2012; Kumar et al., 2014; Maier et al., 2013). The pharmacokinetic data presented here demonstrates that our LNPs are bio-degradable and therefore provide an effective vehicle for mRNA delivery in vivo. Additionally, there was no increase in markers of hepatic toxicity, inflammation or immune response in mice after repeat i.v. dosing.
To establish biochemical and functional efficacy of hMUT mRNA, we first demonstrated cellular activity and then established that it mediated the restoration of hepatic MUT activity in mouse models that recapitulate the mut0 and mut− MMA subtypes. After cell culture studies showed that the MUT enzyme was produced and trafficked to mitochondria in fibroblasts from mut0 patients after mRNA transfection, we performed in vivo pharmacokinetic studies in wild-type mice. The aggregate results suggested that hMUT mRNA therapy could restore enzyme activity in hepatocytes with favorable kinetics for therapeutic applications: hMUT mRNA levels were greatest 2 hr after administration, while hMUT protein expression reached a peak at 16 hr and was sustained for 7 days.
Several different in vivo studies were next performed with various mouse models of MMA to demonstrate efficacy and explore dose response. First, we used MMA mice that were rescued from neonatal lethality by transgene expression, but displayed a high rate of mortality as young adults (Mut−/−;TgINS-MCK-Mut), severe metabolic perturbations, and growth retardation. mRNA therapy restored hepatic MUT expression, normalized propionate oxidation, and lowered circulating metabolites within 72 hr (Figure 4). Moreover, repeat dosing also improved survival, growth, and suppressed metabolites for over 6 days post-therapy, with a partial rebound noted 10 days after withdrawal. The extent of metabolic correction, based on plasma metabolites, appears similar to what was previously documented in Mut−/− mice that received neonatal intrahepatic gene therapy (Chandler and Venditti, 2010). However, because we do not envision hMUT mRNA being delivered via an intrahepatic route (Chandler et al., 2015; Chandler and Venditti, 2008, 2010, 2012), and i.v. injection in neonatal mice can be technically challenging, we did not treat Mut−/− mice. Rather, we extended our murine studies in a hypomorphic model to further explore and define the dose response.
In the next set of studies, a mouse model of mut− MMA, Mut−/−;TgINS-CBA-G715V, was treated and the biochemical response assayed. Interestingly, Mut−/−;TgINS-CBA-G715V mice showed a >50% reduction in plasma methylmalonic acid that persisted for 2 weeks after a single i.v. injection, even with a very low dose of hMUT mRNA (0.1 mg/kg). The longer efficacy observed in Mut−/−;TgINS-CBA-G715Vmice might be explained by a slower turnover of introduced hMUT in the pool of mutant p.G715V Mut, or perhaps by stabilization of the mutant enzyme, which could increase the activity through a chaperone-like mechanism provided by wild-type MUT as a mixed heterodimer (Forny et al., 2014). Additionally, in both Mut−/−;TgINS-MCK-Mut and Mut−/−;TgINS-CBA-G715V mice, hMUT mRNA treatment exhibited rapid lowering of plasma methylmalonic acid, consistent with the observation that hMUT protein expression was detected 2 hr after injection (Figure 3D). Interestingly, in addition to plasma methylmalonic acid, tissue methylmalonic acid was also acutely reduced after hMUT mRNA treatment. The mechanisms of methylmalonic acid reduction in tissues are currently under investigation. Patients with methylmalonic acidemia suffer from acute metabolic decompensations, which require immediate treatment to avoid death and prevent secondary complications such as hyperammonemia. Our data, specifically showing the rapid onset and high hepatic activity achieved after a single i.v. bolus, demonstrate the potential for hMUT mRNA to serve as an emergency treatment for MMA. Taken together, these experiments demonstrate that a single dose of hMUT mRNA, delivered systemically, provides rapid and sustained enzymatic activity for up to 1 week and substantially ameliorates the biochemical abnormalities characteristic of the disorder.
Consistent with previous AAV-mediated gene therapy studies in MMA mice and clinical observations in liver and LKT recipient MMA patients, plasma methylmalonic acid levels were not fully normalized after mRNA therapy, and remained much higher than heterozygous controls (Chandler et al., 2011; Chandler and Venditti, 2010, 2012; Kasahara et al., 2006; Mc Guire et al., 2008; Nagarajan et al., 2005; Sénac et al., 2012; van’t Hoff et al., 1999). One of the reasons for the lack of complete correction is that although the liver is the major organ that produces methylmalonic acid, other tissues such as kidney and skeletal muscle also generate disease-related metabolites (Chandler et al., 2007). Nevertheless, our therapy decreased plasma MMA to levels observed in transplanted patients (Niemi et al., 2015). Moreover, we have observed dramatic improvement in the survival of Mut−/−;TgINS-MCK-Mut mice that received repeat administration of hMUT mRNA therapy as well as significantly increased weight, indicating an improvement in overall health of these mice, and akin to the observations that liver or combined liver-kidney transplant recipients can experience clinical improvement after successful surgery (Niemi et al., 2015).
Although mRNA-based cancer immunotherapies and vaccines have entered clinical development, the use of mRNA therapy as a replacement strategy to treat IEM disorders has not been explored. These studies demonstrate convincing preclinical proof-of-concept efficacy of systemically administered mRNA to target hepatic mitochondrial protein restoration, highlighted here as a new class of genetic medicine for mut MMA. Given the low doses (0.1–0.5 mg/mg) needed for therapeutic effect, rapid onset, sustained efficacy, and ability to redose without hepatic toxicity, inflammation, or immune response, we believe hMUT mRNA therapy presents a versatile and important therapy for MMA, one that could be similarly applied to related IEMs recalcitrant to ERT, both acutely and chronically.
EXPERIMENTAL PROCEDURES
mRNA Production and Formulation
hMUT mRNA was codon optimized using typical approaches in the field (Gustafsson et al., 2004). mRNA was synthesized and formulated with LNP as described previously (Akinc et al., 2008; Leung et al., 2015; Richner et al., 2017). Briefly, mRNA was synthesized in vitro by T7 RNA polymerase-mediated transcription where the UTP was substituted with 5-methoxy UTP, using a linearized DNA template, which incorporates 5′ and 3′ UTRs, including a poly-A tail. A donor methyl group from S-adenosylmethionine (SAM) was added to methylated capped RNA (cap-0), resulting in a cap-1 to increase mRNA translation efficiency. The mRNA was purified and re-suspended in a citrate buffer at the desired concentration. To generate LNP formulations, lipid components (Benenato et al., 2017) were dissolved in ethanol at molar ratios of 50:10:38.5:1.5 (ionizable lipid:DSPC:cholesterol:PEG-lipid). The structure and composition of the LNP was described in Claim 1 in US patent US20170210697 A1 (Benenato et al., 2017). The lipid mixture was combined with a 50 mM citrate buffer (pH 4.0) containing mRNA at a volume ratio of 3:1 (mRNA:lipid) and total flow rate of 14 mL/min using a NanoAssemblr system (Precision NanoSystems, Vancouver, BC). Formulations were dialyzed in 10 kDa membrane dialysis cassettes against phosphate-buffered saline (pH 7.4) for at least 18 hr, followed by concentration using 100 kDa Amicon ultra-centrifugal filters, filtration through a 0.22 μm filter, and storage in pre-sterilized vials at 4°C until use. All formulations were tested for particle size, RNA encapsulation, and endotoxin. The characterization of formulations that were used in the present study is listed in Table S3.
Murine Models of Methylmalonic Acidemia
Animals studies were approved by the Institutional Animal Care and Use Committee of the National Human Genome Research Institute (NHGRI), NIH, or The Institutional Animal Care and Use Committee at Moderna. Both male and female mice were used in the present studies and were evenly distributed in each study group in the same experiment. All mice used in these studies were typically 1.5–3 months of age. Animal details are described in the Supplemental Experimental Procedures.
Quantification of Cationic Lipid by Liquid Chromatography-Tandem Mass Spectrometry
Liver samples were homogenized by Omni probe following addition of 19 eq. (w/v) of water (DF = 20), and protein was precipitated and analyzed against calibration standards prepared in matching blank. Chromatographic separation and quantification was accomplished with a liquid chromatography-tandem mass spectrometry (LC-MS/MS) system. Samples were injected and separated on a Higgins Analytical Clipeus C8-column (Chrom Tech) equilibrated with 35% solvent A containing 5 mM ammonia formate (H2O:MeOH:FA, 50:50:1) and 65% solvent B containing 5 mM ammonia formate in 100:1 MeOH:FA (all from Thermo Fisher Scientific). A triple-quadrupole MS/MS system (Applied Biosystems, API 5500) operated in positive ion mode was used for signal detection.
Mitochondrial Isolation, Protein Extraction, and Western Blotting
Freshly isolated mouse livers were homogenized in a buffer containing 10 mM Trizma base-MOPS and 1 mM EGTA-Trizma base and 200 mM sucrose at 4°C using a 15 mL Dounce-type tissue grinder (D9938–1SET, Sigma-Aldrich). Homogenates were centrifuged at 600 × g for 10 min and supernatant was collected and centrifuged at 600 × g for another 10 min. Supernatant was centrifuged at 7,000 × g for 10 min. Pellets were washed, re-suspended in mitochondrial buffer, and re-centrifuged at 7,000 × g for 10 min. Pellets were lysed in lysis buffer containing 0.5% Triton X-100, 2 mM dithiothreitol, and 10 mM HEPES (pH7.4) supplemented with protease inhibitor cocktail (S8830, Sigma-Aldrich). Homogenates were centrifuged at 14,000 × g for 15 min, and supernatants were collected. Protein concentrations were determined by BCA assay. For immunoblotting, lysates (30 μg protein) were separated by SDS-PAGE. Membranes were incubated with MUT (OTI2A8, Origene) and α-tubulin (ab80779, Abcam) antibodies. Membranes were imaged using the LiCOR imaging platform.
MUT Activity Assay
MUT activity was determined as described previously (Ouattara et al., 2013). Briefly, liver homogenates were incubated with AdoCbl (200 μM, C0884, Sigma Aldrich) and racemic mix of methylmalonyl-CoA (1 mM, M1762, Sigma Aldrich) at 37°C for 15 min. MUT enzyme reactions were terminated by the addition of 50 μL 100 g/L TCA with vortexing. Samples were centrifuged at 13,000 × g for 5 min. Chromatographic separation and quantification was accomplished with high pressure liquid chromatography. 20 μL of supernatants were injected and separated on a Poroshell EC-C18 120 HPLC-column equilibrated with 100% Solvent A containing 100 mM acetic acid in 100 mM sodium phosphate buffer, pH 7.0 (all from Sigma Aldrich). Elusion was with a linear methanol gradient: 0–15 min (95% solvent B) and 15–25 min (95%) with a flow rate of 0.5 mL/min.
Plasma Methylmalonic Acid Concentration
Blood was collected through either retro-orbital or submandibular bleeding. Blood samples were immediately centrifuged, and plasma was collected and stored at −80°C. Plasma methylmalonic acid was analyzed and quantified by LC-MS/MS as described previously (Turgeon et al., 2010).
1-13C-Propionate Oxidation
In vivo 1-13C-propionate oxidation was determined as described previously (Chandler and Venditti, 2010). Briefly, mice were injected intraperitoneally with 200 μg of 1-13C-sodium propionate and placed into a respiratory chamber that contained a CO2 probe to allow the direct measurement of CO2 generated. 10 mL of expired air was sampled from the chamber every 5 min from 0–25 min for analysis of 13CO2. The isotope ratio (13C/12C) of the expired gas was determined with a gas isotope ratio mass spectrometer (Metabolic Solutions, Nashua, NH). The percent dose metabolized at each time point was calculated as % dose metabolized = total 13C excreted (mmol/dose (mmol) × 100%).
mRNA In Situ Hybridization
All mRNA in situ hybridization (ISH) assays were automated on the Leica BOND RX platform. Sections were baked and deparaffinized on the instrument, followed by an epitope retrieval for 15 min at 95°C using Leica Epitope Retrieval Buffer 2 and then protease treatment for 15 min at 40°C. Probes were then hybridized using the ACD 1 min hybridization protocol followed by RNAscope amplification using RNAscope 2.5 LS Reagent Kit-BROWN (322100, ACD). The Leica Bond Polymer Refine Detection kit was used as a visualization agent. The following RNAscope probes were used in this study: dihydrodipicolinate reductase (dapB, 312038, ACD), Mm-PPIB (313917, ACD), Hu-MUT custom probe (300038, ACD).
Statistical Analyses
Data are expressed as means ± SD or their nonparametric equivalent (median ± median absolute deviation) for Gaussian and non-Gaussian distributions, respectively. Means were compared by unpaired (two groups), paired t test (to compare pre- and post-treatment values), one-way or two-way ANOVA. Significant ANOVA findings were followed by Tukey’s post hoc test for multiple comparisons. Non-compartmental analysis was used to estimate the t1/2 of mRNA-encoded hMUT in liver. The log rank test was used to compare survival curves between treatment arms. Two-tailed p values <0.05 were considered statistically significant. All statistical analyses were performed using Prism 7 (GraphPad) software.
Supplementary Material
Highlights.
Systemically delivered LNP-encapsulated mRNA results in hepatic protein expression
hMUT mRNA expresses functional mitochondrial MUT enzyme
Two MMA mouse models show a metabolic and clinical response after mRNA therapy
Single and repeat doses are efficacious in mouse models
ACKNOWLEDGMENTS
The authors acknowledge the contribution of Darwin Romero to mouse husbandry at the NIH. J.L.S., R.J.C., and C.P.V. were supported by the Intramural Research Program of the National Human Genome Research Institute at the NIH.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, six figures, and three tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2017.11.081.
DECLARATION OF INTERESTS
D.A., A.F., S.L., X.Z., J.P., M.T., S.H., J.Z., R.R., B.L., R.H., G.B., V.P., S.S., K.E.M., E.S.K., T.S., C.M., C.L., L.T.G., and P.G.V.M. are employees of, and receive salary and stock options from, Moderna Therapeutics. J.L.S., R.J.C., and C.P.V. declare no competing interests.
REFERENCES
- Akinc A, Zumbuehl A, Goldberg M, Leshchiner ES, Busini V, Hossain N, Bacallado SA, Nguyen DN, Fuller J, Alvarez R, et al. (2008). A combinatorial library of lipid-like materials for delivery of RNAi therapeutics. Nat. Biotechnol 26, 561–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Academy of Pediatrics Newborn Screening Authoring Committee (2008). Newborn screening expands: recommendations for pediatricians and medical homes–implications for the system. Pediatrics 121, 192–217. [DOI] [PubMed] [Google Scholar]
- Baba M, Itaka K, Kondo K, Yamasoba T, and Kataoka K. (2015). Treatment of neurological disorders by introducing mRNA in vivo using polyplex nanomicelles. J. Control. Release 201, 41–48. [DOI] [PubMed] [Google Scholar]
- Barros SA, and Gollob JA (2012). Safety profile of RNAi nanomedicines. Adv. Drug Deliv. Rev 64, 1730–1737. [DOI] [PubMed] [Google Scholar]
- Baumgartner MR, Hörster F, Dionisi-Vici C, Haliloglu G, Karall D, Chapman KA, Huemer M, Hochuli M, Assoun M, Ballhausen D, et al. (2014). Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet J. Rare Dis 9, 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benenato KE, Kumarasinghe ES, and Cornebise M. (2017). Compounds and compositions for intracellular delivery of therapeutic agents. US patent US20170210697 A1, filed March 31, 2017, and published July 27, 2017.
- Carrillo-Carrasco N, Chandler RJ, Chandrasekaran S, and Venditti CP (2010). Liver-directed recombinant adeno-associated viral gene delivery rescues a lethal mouse model of methylmalonic acidemia and provides long-term phenotypic correction. Hum. Gene Ther 21, 1147–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chace DH, DiPerna JC, Kalas TA, Johnson RW, and Naylor EW (2001). Rapid diagnosis of methylmalonic and propionic acidemias: quantitative tandem mass spectrometric analysis of propionylcarnitine in filter-paper blood specimens obtained from newborns. Clin. Chem 47, 2040–2044. [PubMed] [Google Scholar]
- Chandler RJ, and Venditti CP (2008). Adenovirus-mediated gene delivery rescues a neonatal lethal murine model of mut(0) methylmalonic acidemia. Hum. Gene Ther 19, 53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler RJ, and Venditti CP (2010). Long-term rescue of a lethal murine model of methylmalonic acidemia using adeno-associated viral gene therapy. Mol. Ther 18, 11–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler RJ, and Venditti CP (2012). Pre-clinical efficacy and dosing of an AAV8 vector expressing human methylmalonyl-CoA mutase in a murine model of methylmalonic acidemia (MMA). Mol. Genet. Metab 107, 617–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler RJ, Sloan J, Fu H, Tsai M, Stabler S, Allen R, Kaestner KH, Kazazian HH, and Venditti CP (2007). Metabolic phenotype of methylmalonic acidemia in mice and humans: the role of skeletal muscle. BMC Med. Genet 8, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler RJ, Chandrasekaran S, Carrillo-Carrasco N, Senac JS, Hofherr SE, Barry MA, and Venditti CP (2011). Adeno-associated virus serotype 8 gene transfer rescues a neonatal lethal murine model of propionic acidemia. Hum. Gene Ther 22, 477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler RJ, LaFave MC, Varshney GK, Trivedi NS, Carrillo-Carrasco N, Senac JS, Wu W, Hoffmann V, Elkahloun AG, Burgess SM, and Venditti CP (2015). Vector design influences hepatic genotoxicity after adeno-associated virus gene therapy. J. Clin. Invest 125, 870–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler RJ, Sands MS, and Venditti CP (2017). Recombinant adeno-associated viral integration and genotoxicity: insights from animal models. Hum. Gene Ther 28, 314–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosson MA, Benoist JF, Touati G, De chaux M, Royer N, Grandin L, Jais JP, Boddaert N, Barbier V, Desguerre I, et al. (2009). Long-term outcome in methylmalonic aciduria: a series of 30 French patients. Mol. Genet. Metab 97, 172–178. [DOI] [PubMed] [Google Scholar]
- de Baulny HO, Benoist JF, Rigal O, Touati G, Rabier D, and Saudubray JM (2005). Methylmalonic and propionic acidaemias: management and outcome. J. Inherit. Metab. Dis 28, 415–423. [DOI] [PubMed] [Google Scholar]
- DeRosa F, Guild B, Karve S, Smith L, Love K, Dorkin JR, Kauffman KJ, Zhang J, Yahalom B, Anderson DG, and Heartlein MW (2016). Therapeutic efficacy in a hemophilia B model using a biosynthetic mRNA liver depot system. Gene Ther. 23, 699–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dionisi-Vici C, Deodato F, Röschinger W, Rhead W, and Wilcken B. (2006). ‘Classical’ organic acidurias, propionic aciduria, methylmalonic aciduria and isovaleric aciduria: long-term outcome and effects of expanded newborn screening using tandem mass spectrometry. J. Inherit. Metab. Dis 29, 383–389. [DOI] [PubMed] [Google Scholar]
- Donsante A, Miller DG, Li Y, Vogler C, Brunt EM, Russell DW, and Sands MS (2007). AAV vector integration sites in mouse hepatocellular carcinoma. Science 317, 477. [DOI] [PubMed] [Google Scholar]
- Fenton WA, Gravel RA, and Rosenblatt DS (2001). Disorders of propionate and methylmalonate metabolism. In The Metabolic and Molecular Bases of Inherited Disease, Scriver CR, Beaudet AL, Sly WS, and Valle D, eds. (McGraw-Hill; ), pp. 2165–2193. [Google Scholar]
- Forny P, Froese DS, Suormala T, Yue WW, and Baumgartner MR (2014). Functional characterization and categorization of missense mutations that cause methylmalonyl-CoA mutase (MUT) deficiency. Hum. Mutat 35, 1449–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser JL, and Venditti CP (2016). Methylmalonic and propionic acidemias: clinical management update. Curr. Opin. Pediatr 28, 682–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson C, Govindarajan S, and Minshull J. (2004). Codon bias and heterologous protein expression. Trends Biotechnol. 22, 346–353. [DOI] [PubMed] [Google Scholar]
- Hörster F, Baumgartner MR, Viardot C, Suormala T, Burgard P, Fowler B, Hoffmann GF, Garbade SF, Kölker S, and Baumgartner ER (2007). Long-term outcome in methylmalonic acidurias is influenced by the underlying defect (mut0, mut-, cblA, cblB). Pediatr. Res 62, 225–230. [DOI] [PubMed] [Google Scholar]
- Hussein MH, Hashimoto T, Suzuki T, Daoud GA, Goto T, Nakajima Y, Kato T, Hibi M, Tomishige H, Hara F, et al. (2013). Children undergoing liver transplantation for treatment of inherited metabolic diseases are prone to higher oxidative stress, complement activity and transforming growth factor-β1. Ann. Transplant 18, 63–68. [DOI] [PubMed] [Google Scholar]
- Kamei K, Ito S, Shigeta T, Sakamoto S, Fukuda A, Horikawa R, Saito O, Muguruma T, Nakagawa S, Iijima K, and Kasahara M. (2011). Preoperative dialysis for liver transplantation in methylmalonic acidemia. Ther. Apher. Dial 15, 488–492. [DOI] [PubMed] [Google Scholar]
- Kasahara M, Horikawa R, Tagawa M, Uemoto S, Yokoyama S, Shibata Y, Kawano T, Kuroda T, Honna T, Tanaka K, and Saeki M. (2006). Current role of liver transplantation for methylmalonic acidemia: a review of the literature. Pediatr. Transplant 10, 943–947. [DOI] [PubMed] [Google Scholar]
- Kölker S, Valayannopoulos V, Burlina AB, Sykut-Cegielska J, Wijburg FA, Teles EL, Zeman J, Dionisi-Vici C, Barić I, Karall D, et al. (2015). The phenotypic spectrum of organic acidurias and urea cycle disorders. Part 2: the evolving clinical phenotype. J. Inherit. Metab. Dis 38, 1059–1074. [DOI] [PubMed] [Google Scholar]
- Kormann MS, Hasenpusch G, Aneja MK, Nica G, Flemmer AW, Herber-Jonat S, Huppmann M, Mays LE, Illenyi M, Schams A, et al. (2011). Expression of therapeutic proteins after delivery of chemically modified mRNA in mice. Nat. Biotechnol 29, 154–157. [DOI] [PubMed] [Google Scholar]
- Kumar V, Qin J, Jiang Y, Duncan RG, Brigham B, Fishman S, Nair JK, Akinc A, Barros SA, and Kasperkovitz PV (2014). Shielding of Lipid Nanoparticles for siRNA Delivery: Impact on Physicochemical Properties, Cytokine Induction, and Efficacy. Mol. Ther. Nucleic Acids 3, e210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung AK, Tam YY, Chen S, Hafez IM, and Cullis PR (2015). Microfluidic Mixing: A General Method for Encapsulating Macromolecules in Lipid Nanoparticle Systems. J. Phys. Chem. B 119, 8698–8706. [DOI] [PubMed] [Google Scholar]
- Maier MA, Jayaraman M, Matsuda S, Liu J, Barros S, Querbes W, Tam YK, Ansell SM, Kumar V, Qin J, et al. (2013). Biodegradable lipids enabling rapidly eliminated lipid nanoparticles for systemic delivery of RNAi therapeutics. Mol. Ther 21, 1570–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manoli I, Sloan JL, and Venditti CP (1993). Isolated methylmalonic acidemia. In GeneReviews(R), Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Mefford HC, and Smith RJH, et al. , eds. (University of Washington, Seattle: ). [Google Scholar]
- Manoli I, Sysol J, Li L, Chandler R, Senac J, Hoffmann V, Zerfas P, Schnermann J, and Venditti C. (2011). Muscle targeted transgene expression rescues the lethal phenotype of Mut knockout mice. Mol. Genet. Metab 102, 248. [Google Scholar]
- Manoli I, Sysol JR, Li L, Houillier P, Garone C, Wang C, Zerfas PM, Cusmano-Ozog K, Young S, Trivedi NS, et al. (2013). Targeting proximal tubule mitochondrial dysfunction attenuates the renal disease of methylmalonic acidemia. Proc. Natl. Acad. Sci. USA 110, 13552–13557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matern D, Oglesbee D, and Tortorelli S. (2013). Newborn screening for lysosomal storage disorders and other neuronopathic conditions. Dev. Disabil. Res. Rev 17, 247–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mc Guire PJ, Lim-Melia E, Diaz GA, Raymond K, Larkin A, Wasserstein MP, and Sansaricq C. (2008). Combined liver-kidney transplant for the management of methylmalonic aciduria: a case report and review of the literature. Mol. Genet. Metab 93, 22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan S, Enns GM, Millan MT, Winter S, and Sarwal MM (2005). Management of methylmalonic acidaemia by combined liver-kidney transplantation. J. Inherit. Metab. Dis 28, 517–524. [DOI] [PubMed] [Google Scholar]
- Niemi AK, Kim IK, Krueger CE, Cowan TM, Baugh N, Farrell R, Bonham CA, Concepcion W, Esquivel CO, and Enns GM (2015). Treatment of methylmalonic acidemia by liver or combined liver-kidney transplantation. J. Pediatr 166, 1455–1461. [DOI] [PubMed] [Google Scholar]
- Ouattara B, Duplessis M, and Girard CL (2013). Optimization and validation of a reversed-phase high performance liquid chromatography method for the measurement of bovine liver methylmalonyl-coenzyme a mutase activity. BMC Biochem. 14, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswamy S, Tonnu N, Tachikawa K, Limphong P, Vega JB, Karmali PP, Chivukula P, and Verma IM (2017). Systemic delivery of factor IX messenger RNA for protein replacement therapy. Proc. Natl. Acad. Sci. USA 114, E1941–E1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richner JM, Himansu S, Dowd KA, Butler SL, Salazar V, Fox JM, Julander JG, Tang WW, Shresta S, Pierson TC, et al. (2017). Modified mRNA vaccines protect against Zika virus infection. Cell 168, 1114–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin U, Karikó K, and Türeci Ö (2014). mRNA-based therapeutics–developing a new class of drugs. Nat. Rev. Drug Discov 13, 759–780. [DOI] [PubMed] [Google Scholar]
- Sakamoto R, Nakamura K, Kido J, Matsumoto S, Mitsubuchi H, Inomata Y, and Endo F. (2016). Improvement in the prognosis and development of patients with methylmalonic acidemia after living donor liver transplant. Pediatr. Transplant 20, 1081–1086. [DOI] [PubMed] [Google Scholar]
- Sanderson S, Green A, Preece MA, and Burton H. (2006). The incidence of inherited metabolic disorders in the West Midlands, UK. Arch. Dis. Child 91, 896–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrom E, Huber M, Aneja M, Dohmen C, Emrich D, Geiger J, Hasenpusch G, Herrmann-Janson A, Kretzschmann V, Mykhailyk O, et al. (2017). Translation of angiotensin-converting enzyme 2 upon liver- and lung-targeted delivery of optimized chemically modified mRNA. Mol. Ther. Nucleic Acids 7, 350–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sénac JS, Chandler RJ, Sysol JR, Li L, and Venditti CP (2012). Gene therapy in a murine model of methylmalonic acidemia using rAAV9-mediated gene delivery. Gene Ther. 19, 385–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senac JS, Aswani VH, Sysol JR, Manoli I, and Venditti CP (2013). 280. Partial deficiency model of MUT methylmalonic acidemia (MMA) displays diet inducible disease and sensitivity to acetaminophen (APAP). Mol. Ther 21 (Suppl 1), S107. [Google Scholar]
- Turgeon CT, Magera MJ, Cuthbert CD, Loken PR, Gavrilov DK, Tortorelli S, Raymond KM, Oglesbee D, Rinaldo P, and Matern D. (2010). Determination of total homocysteine, methylmalonic acid, and 2-methylcitric acid in dried blood spots by tandem mass spectrometry. Clin. Chem 56, 1686–1695. [DOI] [PubMed] [Google Scholar]
- van’t Hoff WG, Dixon M, Taylor J, Mistry P, Rolles K, Rees L, and Leonard JV (1998). Combined liver-kidney transplantation in methylmalonic acidemia. J. Pediatr 132, 1043–1044. [DOI] [PubMed] [Google Scholar]
- van’t Hoff W, McKiernan PJ, Surtees RA, and Leonard JV (1999). Liver transplantation for methylmalonic acidaemia. Eur. J. Pediatr 158 (Suppl 2), S70–S74. [DOI] [PubMed] [Google Scholar]
- Vernon HJ, Sperati CJ, King JD, Poretti A, Miller NR, Sloan JL, Cameron AM, Myers D, Venditti CP, and Valle D. (2014). A detailed analysis of methylmalonic acid kinetics during hemodialysis and after combined liver/kidney transplantation in a patient with mut (0) methylmalonic acidemia. J. Inherit. Metab. Dis 37, 899–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Su HH, Yang Y, Hu Y, Zhang L, Blancafort P, and Huang L. (2013). Systemic delivery of modified mRNA encoding herpes simplex virus 1 thymidine kinase for targeted cancer gene therapy. Mol. Ther 21, 358–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong ES, McIntyre C, Peters HL, Ranieri E, Anson DS, and Fletcher JM (2014). Correction of methylmalonic aciduria in vivo using a codon-optimized lentiviral vector. Hum. Gene Ther 25, 529–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.