

CONSPECTUS:
Redox reactions that take place in enzymes and on the surfaces of heterogeneous catalysts often require active sites that contain multiple metals. By contrast, there are very few homogeneous catalysts with multinuclear active sites, and the field of organometallic chemistry continues to be dominated by the study of single metal systems. Multinuclear catalysts have the potential to display unique properties owing to their ability to cooperatively engage substrates. Furthermore, direct metal-to-metal covalent bonding can give rise to new electronic configurations that dramatically impact substrate binding and reactivity. In order to effectively capitalize on these features, it is necessary to consider strategies to avoid the dissociation of fragile metal–metal bonds in the course of a catalytic cycle. This Account describes one approach to accomplishing this goal using binucleating redox-active ligands.
Graphical Abstract
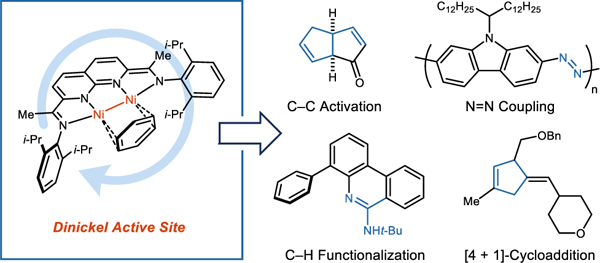
In 2006, Chirik showed that pyridine–diimines (PDI) have sufficiently low-lying π* levels that they can be redox non-innocent in low valent iron complexes. Extending this concept, we investigated a series of dinickel complexes supported by naphthyridine–diimine (NDI) ligands. These complexes can promote a broad range of two-electron redox processes in which the NDI ligand manages electron equivalents, while the metals remain in a Ni(I)–Ni(I) state.
Using (NDI)Ni2 catalysts, we have uncovered cases where having two metals in the active site addresses a problem in catalysis that had not been adequately solved using single-metal systems. For example, mononickel complexes are capable of stoichiometrically dimerizing aryl azides to form azoarenes but do not turn over due to strong product inhibition. By contrast, dinickel complexes are effective catalysts for this reaction and avoid this thermodynamic sink by binding to azoarenes in their higher energy cis form.
Dinickel complexes can also activate strong bonds through the cooperative action of both metals. Norbornadiene has a ring-strain energy that is similar to that of cyclopropane but is not prone to undergoing C–C oxidative addition with monometallic complexes. Using a (NDI)Ni2 complex, norbornadiene undergoes rapid ring opening by oxidative addition of the vinyl and bridgehead carbons. An inspection of the resulting metallacycle reveals that it is stabilized through a network of secondary Ni–π interactions. This reactivity enabled the development of a catalytic carbonylative rearrangement to form fused bicyclic dienones.
These vignettes, and others described in this Account, highlight some of the implications of metal–metal bonding in promoting a challenging step in a catalytic cycle or adjusting the thermodynamic landscape of key intermediates. Given that our studies have focused nearly exclusively on the (NDI)Ni2 system, we anticipate that many more such cases are left to be discovered as other transition metal combinations and ligand classes are explored.
1. Introduction
Cotton’s seminal work on the electronic structure of Re2Cl82– inspired decades of research aimed at understanding the nature of metal-to-metal covalent bonding.5 Unsupported metal–metal bonds can form spontaneously when it is electronically favorable to do so. However, a more reliable synthetic approach is to use binucleating ligands that contain adjacent binding sites with donors suited to the metals and oxidation states of interest. Numerous examples of homobimetallic and heterobimetallic complexes have now been prepared, and key electronic features of metal–metal bonding have been illuminated through a productive interplay of synthetic, spectroscopic, and computational approaches.6
Considering the large number of metal–metal bonded complexes that are now available, it is striking that so few of them have made it into the repertoire of catalysts used by synthetic organic chemists. Many dinuclear complexes have been shown to display catalytic activity. However, the benefits that they offer over existing catalysts, in terms of improved activity or selectivity, often do not justify their added complexity. Cases are fewer still where the involvement of a metal–metal bond in a catalytic cycle enables an entirely new transformation that cannot be carried out with any known mononuclear catalyst.7
Another consideration is that when a dinuclear complex is tested as a catalyst, there is always uncertainty about whether the metal–metal bond is preserved under turnover conditions. For example, a dinuclear [Pd(I)/(II)]2 cycle could be an interesting alternative to canonical mononuclear Pd(0)/(II) cycles for reactions such as cross-coupling. However, in most cases where Pd(I) dimers have been found to promote these reactions, it is thought that they act as pre-catalysts for on-cycle monopalladium species.8 There are only a few studies that provide clear experimental support for a [Pd(I)]29 or a [Pd(III)]210 species being a true catalytic intermediate (Figure 1A). In general, Pd–Pd and other group 10 M–M σ-bonds are only stable in odd-electron oxidation states. Thus, any proposed catalytic process that involves redox changes would inevitably require considering the speciation between dinuclear and mononuclear forms.
Figure 1.

(A) Dinuclear redox reactions often entail the formation or cleavage of a metal–metal bond. (B) Redox active ligands can be used to carry out a two-electron oxidative coupling reactions without perturbing the metal oxidation state. (C) Dinuclear oxidative coupling, oxidative addition, and group transfer reactions at intact Ni(I)–Ni(I) bonds using redox active ligands.
In 2006, Chirik demonstrated that pyridine–diimine (PDI) ligands possess sufficiently low-lying π* levels that they can be redox non-innocent in reduced Fe complexes (Figure 1B).11 This finding led to the development of Fe-catalyzed cycloaddition reactions where Fe remains in a +2 oxidation state, while the ligand traverses 0 and –2 charge states during the key oxidative coupling and reductive elimination steps.12
We wondered whether this concept could prove useful in the design of multinuclear redox processes where a M–M bond would remain intact during the entirety of a catalytic cycle (Figure 1C). In principle, if the responsibility for storing and releasing electrons could be managed by the ligand,13 there should be minimal electronic disruption of the M–M σ-bond during a two-electron reaction. This Account summarizes our exploration of this hypothesis using Ni(I)–Ni(I) bonds supported by redox-active naphthyridine–diimine (NDI) ligands.
2. Synthesis and Electronic Structure of (NDI)Ni2 Complexes
Our initial efforts to design binucleating redox-active ligands focused on the use of 1,8-naphthyridine heterocycles, which have been used in other ligands to generate well-defined dinuclear complexes.14 Upon combining i-PrNDI (1 equiv) and Ni(COD)2 (2 equiv) in C6H6, the dark brown, diamagnetic (i-PrNDI)Ni2(C6H6) complex (1) is formed in quantitative yield.15 The use of an arene solvent is critical for assembling the Ni–Ni bond, and its exclusion during synthesis causes only mononickel complexes to be generated. The weakly bound C6H6 ligand rotates rapidly on the 1H NMR timescale, and it can be readily exchanged with various substrates of interest.
From the bond metrics of the NDI ligand in 1, it is evident that the π-system is reduced by two electrons (Figure 1C). Additionally, the bond length perturbations are symmetrically distributed over the two halves of the ligand, consistent with the redox-active orbital being fully delocalized. A spectroscopic signature of ligand-centered reduction is a prominent band in the NIR region (λmax = 1040 nm, ε = 40,000 cm–1 M–1), which is assigned to a ligand-based π–π* transition. According to DFT models, the HOMO is predominantly ligand-centered but exhibits some mixing with a Ni–Ni π* orbital. Our collective experimental and computational data are consistent with a σ2π4π*4δ4δ*4 description for the Ni–Ni interaction, resulting in a net bond order of one.
The (i-PrNDI)Ni2(C6H6) complex (1) engages in a broad scope of two-electron redox reactions by coupling reactivity at the Ni–Ni bond with electron transfer from the reduced NDI ligand. In the following section, we describe our investigations of stoichiometric oxidative coupling, oxidative addition, and group transfer reactions. Additionally, we highlight cases where the presence of two metals is beneficial for catalysis, either by offering a rate or selectivity advantage over comparable mononuclear catalysts or by enabling new reactivity.
3. Stoichiometric Reactivity and Catalysis
3.1. Dinuclear Oxidative Coupling
Thermal cycloaddition reactions, such as Diels–Alder reactions, hold high synthetic value in organic chemistry but often require electronically polarized substrates to lower their activation barriers. Low-valent transition metals are capable of coordinating to non-polar π-systems and mediating oxidative coupling reactions to generate metallacycles.16 This process provides the mechanistic basis for metal-catalyzed cycloaddition reactions, which are not constrained by the electronic considerations of their thermal pericyclic counterparts.17 A prototypical example is the Pauson–Khand reaction: a formal [2 + 2 + 1]-cycloaddition of an alkyne, an alkene, and CO mediated by Co2(CO)8.18 The accepted mechanism of the Pauson–Khand reaction is attributed to Magnus, who was seeking to rationalize the diastereoselectivity of an intramolecular variant of the reaction.19 The key step in his proposal was a dinuclear oxidative coupling step to generate a dicobaltacycle. Despite extensive mechanistic investigations of the Pauson–Khand reaction,20 this putative metallacycle has eluded structural characterization. Kinetics studies suggest that it is a fleeting intermediate that undergoes fast and irreversible CO insertion (Figure 2A inset).
Figure 2. Dinuclear Oxidative Coupling Reactions.
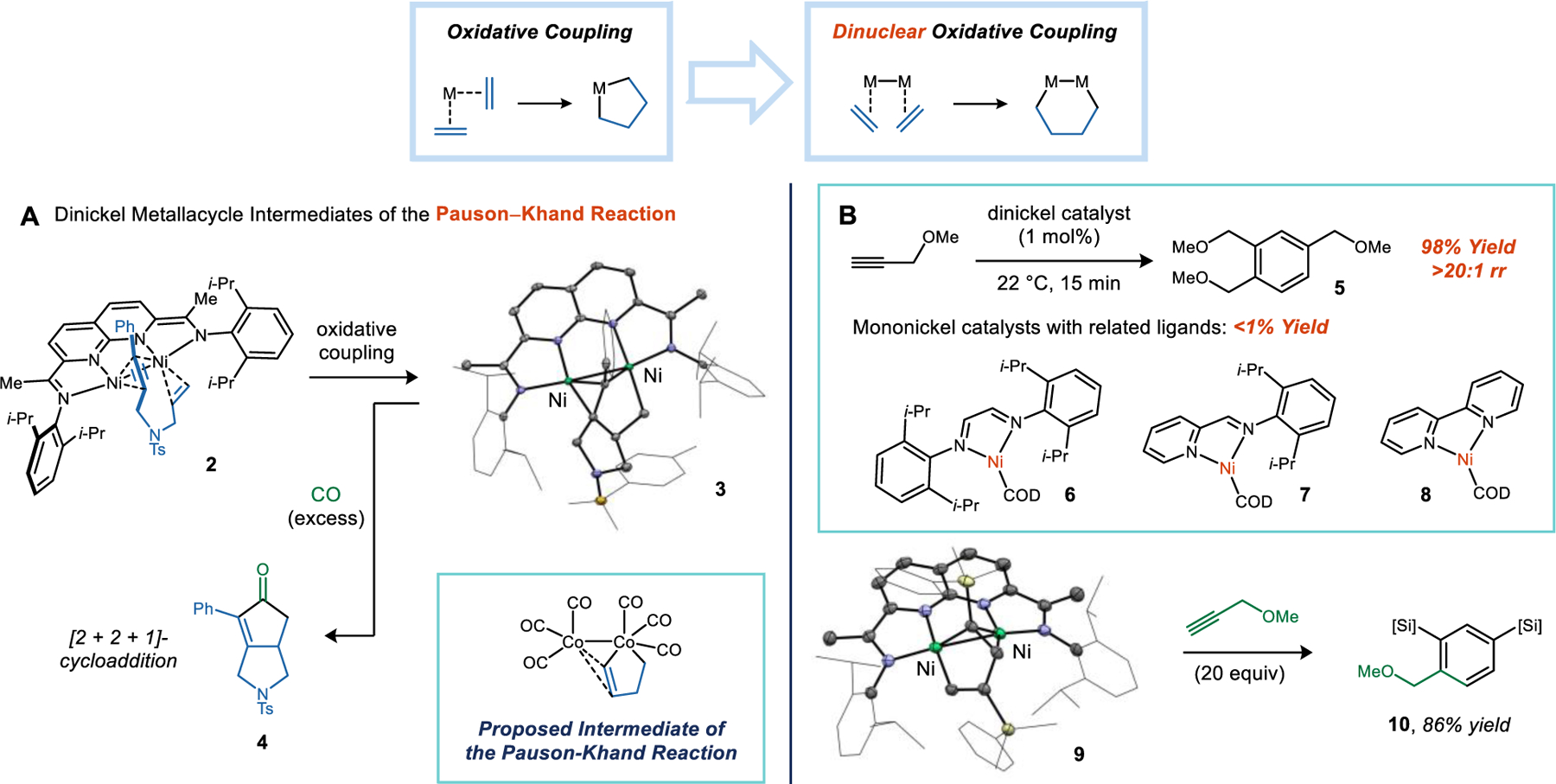
(A) A functional model for the dinuclear intermediates of the Pauson–Khand reaction using a Ni–Ni site as a surrogate for Co2(CO)8. (B) Comparing the activity and selectivity of alkyne cyclooligomerization reactions using dinickel and mononickel catalysts.
The Pauson–Khand reaction, being a rare example of an organometallic process that occurs at a metal–metal bond, captured our interest as we were initially exploring the chemistry of dinuclear Ni(I) (Figure 2A).21 Combining the (i-PrNDI)Ni2(C6H6) complex (1) with a model enyne yielded a transient alkyne adduct (2) that could be observed by 1H NMR spectroscopy. This intermediate decays with clean first-order kinetics (ΔG‡ = 22.7 kcal/mol at 30 °C) to the product of alkyne–alkene oxidative coupling. The solid-state structure of this product (3) maps directly onto Magnus’s proposed Pauson–Khand intermediate and features a five-membered metallacycle with the alkenyl π-system engaged in an η2-interaction with the second Ni. When placed under an atmosphere of CO, complex 3 releases the [2 + 2 + 1]-cycloaddition product (4) in high yield.
The finding that the (NDI)Ni2 platform could mediate kinetically facile oxidative coupling reactions motivated us to consider whether a dinuclear active site could be exploited to achieve an improvement in the rate or selectivity of a catalytic cycloaddition.1 To answer this question, we selected the cyclooligomerization of alkynes as a model reaction, because it was known to be promoted by Ni catalysts but with efficiencies and selectivities that were moderate and highly sensitive to catalyst structure (Figure 2B). Selecting a suitable set of mononickel complexes to serve as a comparison was a non-trivial consideration. Complexes containing bidentate N-donor ligands were an appealing choice because they could be prepared in a low-valent form, featured donors that mirror those of NDI, and were known to perform oxidative coupling reactions.
Previously, tom Dieck showed that Ni complexes of N-donor ligands function as catalysts for the cyclooligomerization of ethyl propiolate, predominantly forming cyclooctatetraene products.22 We confirmed these results in our own studies. Catalysts 6–8 achieve high conversions of ethyl propiolate and form cyclooctatetraene regioisomers, along with a minor fraction of arenes. Interestingly, Ni2 catalyst 1 exhibits a very different product profile. In addition to being more active than any of the mononickel catalysts, 1 yields arenes nearly exclusively with very little cyclooctatetraene formation. We observed the most striking differences when examining more electron-rich alkyl-substituted alkynes. Mononickel catalysts 6–8 exhibit little to no activity for the cyclotrimerization of methyl propargyl ether. By contrast, at 1 mol% loading, Ni2 catalyst 1 affords the 1,2,4-substituted arene (5) in 98% yield within minutes at room temperature.
Using sufficiently hindered terminal alkynes, it is possible to arrest the catalytic process and gain insight into the intermediates of the cyclotrimerization reaction. PhMe2SiCCH (2 equiv) undergoes a head-to-tail oxidative coupling with 1 to yield metallacycle 9. DFT models suggest that the alternative regioisomers are disfavored due to steric interactions with the aryl substituents of the catalyst. The remaining mechanistic question is regarding the addition of the third alkyne equivalent. This step was examined computationally by optimizing stationary points for a concerted [4 + 2]-cycloaddition, a stepwise [2 + 2]-cycloaddition/ring-expansion, and a stepwise migratory insertion/C–C reductive elimination pathway.23 The third mechanism was calculated to be the most energetically favorable, and it accurately reproduced the observed regioselectivity of the reaction.
3.2. Dinuclear Oxidative Addition
Oxidative addition is an elementary organometallic process that can be used to activate a strong, non-polar bond in the course of a catalytic transformation.24 In 1955, Tipper described an oxidative addition of cyclopropane by PtCl2 to generate a high-valent species25 that was later assigned by Chatt as a platinacyclobutane.26 Since this report, activation reactions of cyclopropanes have emerged as useful strategies to generate reactive three-carbon fragments that engage in myriad cycloaddition, isomerization, and rearrangement processes.27 Cyclopropane ring-opening reactions commonly rely on polar activating substituents to enable heterolytic bond cleavage.28 In this context, Louie reported a noteworthy rearrangement of electronically neutral vinylcyclopropanes to cyclopentenes using Ni(0) catalysts bearing strongly σ-donating NHC ligands.29
Activation Reactions of Three-Membered Rings.
When vinylcyclopropane was added to solutions of (i-PrNDI)Ni2(C6H6) (1) in C6D6, the oxidative addition product was not observed.30 However, vinylcyclopropane was catalytically consumed to form cyclopentene (83% yield with 1 mol% 1, 24 h, rt) (Figure 3A). The putative product of cyclopropane C–C oxidative addition does not build up in significant concentrations under turnover conditions, presumably due to the fast rate of C–C reductive elimination. Thus, in order to characterize the metallacyclic intermediate, it was necessary to modify the substrate to slow down reductive elimination. N-Tosyl vinylaziridine undergoes stoichiometric oxidative addition to generate azametallacycle 11, which features an η3-coordinated allyl and an NTs group both occupying bridging positions between the two metals.
Figure 3. Dinuclear Oxidative Addition Reactions.

(A) Catalytic and stoichiometric ring-opening reactions of vinylaziridines and vinylcyclopropanes. (B) Norbornadiene has a similar ring-strain energy to that of cyclopropane but does not readily undergo C–C oxidative addition. (C) Dinuclear oxidative addition of norbornadiene and catalytic carbonylative rearrangements.
Activation Reactions of Bicyclic Compounds.
Norbornadiene (nbd) possesses a similar ring-strain energy to that of vinylcyclopropane32 but is not known to undergo similar C–C oxidative addition reactions (Figure 3B).33 There are likely two factors that contribute to this divergence in reactivity. First, norbornadiene forms stable π-complexes with low-valent transition metals (e.g. PtCl2(ndb)).34 Indeed, norbornadiene and related [2.2.2]-bicyclodienes have been employed as non-reactive supporting ligands in catalytic reactions.35 Second, the alkenes of norbornadiene are sufficiently activated by ring strain such that addition or coupling reactions of the π-bonds occur in preference to cleavage of the σ-bonds. One illustrative example is the reaction between [Ir(cod)Cl]2 and 2 equiv of norbornadiene to yield an Ir(III) metallacyclopentane.36
The (i-PrNDI)Ni2(C6H6) complex (1) undergoes a stoichiometric reaction with norbornadiene to generate the oxidative addition product 12 (Figure 3C).2 The intermediate diene adduct is not detectable even at partial conversions, indicating that the ligand substitution step is slower than C–C activation. DFT models are consistent with this observation. The calculated oxidative addition barrier is only 10.6 kcal/mol and is exothermic by 14.8 kcal/mol. An apparent driving force for this unusual reaction is the ability of the Ni2 active site to form a network of stabilizing Ni–π interactions with the allyl and vinyl fragments. From an electron counting perspective, the ring-opened norbornadienyl fragment in 12 is functioning as an 8 e– donor to the Ni–Ni bond. It is unlikely that a single metal center could geometrically or electronically support such a density of interactions.
Metallacycle 12 reacts with CO to generate a carbonylated product. The carbonylation proceeds with transposition of the allylic system, and the fused bicyclodienone is formed selectively over the alternative bridged isomer. This series of stoichiometric reactions could not be rendered catalytic using CO (g) due to strong inhibition of the catalyst by excess CO. However, turnover could be achieved using Cr(CO)6,37 which slowly releases free CO at high temperatures. Under optimized conditions, several catalytic carbonylative rearrangements were carried out using 7-substituted norbornadienes as substrates.
3.3. Dinuclear Group Transfer
Owing to their valence deficiency, carbenes and nitrenes are highly unstable species. In their free form, they often react indiscriminately with substrates of any structural or functional complexity. Transition metal catalysis has become the primary strategy to control the reactivity of carbenes and nitrenes to achieve high levels of chemo-, regio-, and stereoselectivity. Efforts to characterize the key intermediates of these catalytic processes have largely focused on gaining synthetic access to isolable terminal M=CR2 and M=NR complexes.38 Complexes in which a carbene or nitrene ligand bridge two metals are also known but generally exhibit lower reactivity. For example, Warren found that the reaction between a Cu(I) β-diketiminate complex and an aryl azide generates an equilibrating mixture of dinuclear Cu2(NAr) and mononuclear Cu(NAr) species.39 Kinetics studies indicate that the Cu2(NAr) complex is an off-cycle resting state that must dissociate into a monomer before engaging an alkene.
Carbene and Nitrene Transfer.
The (NDI)Ni2 system provided a suitable platform to study the reactivity of bridging carbenes in the absence of dissociation pathways to form mononuclear M=CR2 species (Figure 4).40 Accordingly, Ph2C=N2 undergoes a ligand exchange reaction with (i-PrNDI)Ni2(C6H6) complex 1 to yield a diazoalkane adduct. The Ni–Ni bond is cleaved in this process (Ni–Ni = 3.135 Å), and the =N2 unit is bound in an end-on fashion to one Ni and is η2-coordinated to the other. Upon gentle heating at 60 °C, N2 is eliminated, and the Ni–Ni bond is reestablished to support a bridging carbene. The carbene is capable of transferring to an isonitrile. t-BuNC first binds terminally to one Ni (complex 14), resulting in cleavage of the Ni–Ni bond. This adduct is metastable and decays unimolecularly in C6H6 at 80 °C to expel a keteneimine product (15) and regenerate 1. This series of stoichiometric reactions can be rendered catalytic by adding the diazoalkane and isonitrile dropwise to solutions of the catalyst.
Figure 4. Dinuclear Group Transfer Reactions.
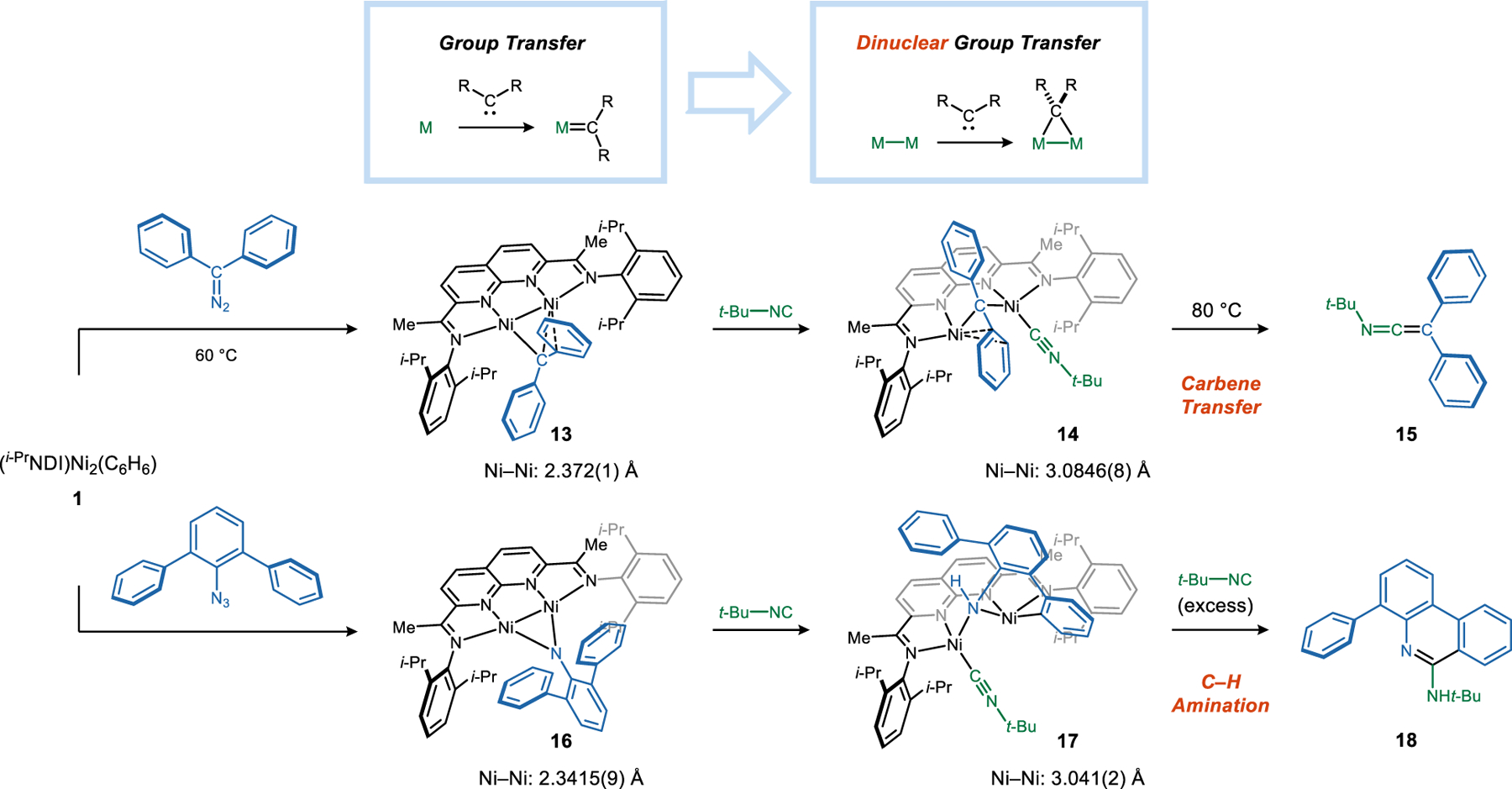
Carbene and nitrene transfer reactions at a flexible dinickel active site.
Aryl azides also react with 1, and N2 elimination is significantly faster than was observed for diazoalkanes (Figure 4).41 Hindered m-terphenylazide reacts with 1 to generate Ni2(μ-NAr) complex 16. The putative azide adduct is not detectable as an intermediate. Interestingly, the addition of t-BuNC to 16 does not result in the expected group transfer to form a carbodiimide. Rather, it triggers a C(aryl)–H activation reaction. The product is a Ni2(aryl)(amide) species (17) that arises from a 1,2-addition of the aryl C–H bond to the Ni2(μ-NAr). This C–H activated intermediate is sufficiently stable that it could be isolated, but additional equivalents of t-BuNC induce a migratory insertion and C–C reductive elimination sequence to form heterocycle 18 in 90% yield. The same product is also generated, along with 1-phenyl-9H-carbazole, if 17 is simply heated at 80 °C. Like the carbene transfer process, the flexibility of the Ni–Ni interaction is critical to the C–H amination. The Ni–Ni distance is short and within bonding range in the Ni2(μ-NR) complex (16) but is capable of expanding to 3.0412(2) Å upon coordination of the isonitrile (17).
When the steric profile of the aryl azide reagent is reduced, the Ni2(μ-NAr) species becomes unstable and is prone to undergoing N=N coupling with a second aryl azide equivalent.3 For example, complex 1 reacts with 2 equiv of 2,6-(i-Pr)2C6H3N3 to generate a cis-azoarene adduct (19). We were motivated by this observation to consider the development of a catalytic N=N coupling reaction (Figure 5). Azoarenes are an important class of dye molecules that have seen a resurgence of interest due to their photoswitching properties.42 Azoarenes have been incorporated into biologically active molecules and molecular machines that use light to trigger conformational changes.43
Figure 5.

Dinickel complexes are efficient catalysts for the dimerization of aryl azides by avoiding inhibition by the azoarene product. Catalytic syntheses of azoarenes and azopolymers.
Catalytic dimerizations of >20 aryl azides were carried out using 1–10 mol% of Ni2 catalyst 1. By using aromatic diazides as substrates, we were also able to develop catalytic polymerization reactions that generate conjugated materials linked through N=N bonds.44 Ongoing studies are aimed at studying the photochemical and electrochemical properties of these polymers.
The rapid and quantitative N=N coupling reaction observed using the (NDI)Ni2 platform was reminiscent of previous findings from Hillhouse that a (P,P)Ni=NAr species reacts with aryl azides to generate azoarene adducts.45 Despite the efficiency of the stoichiometric process, Hillhouse noted in his work that catalytic turnover could not be achieved due to the high binding affinity of the azoarene product relative to the azide starting material. Our mechanistic studies suggest that the ability of the Ni2 catalyst 1 to achieve turnover may be due to the azoarene only binding to the catalyst in its higher energy cis form. Thus, isomerization to the more stable trans-azoarene prevents excessive product inhibition.
Vinylidene Transfer.
There are several classes of carbenes that are difficult to utilize in transition metal catalysis, because their corresponding diazo precursors are either inaccessible or unstable. For example, H2C=C=N2, the precursor to vinylidene, undergoes N2 elimination at –90 °C with a half-life of 0.3 ms.46 In light of this challenge, most catalytic vinylidene transfer reactions use alkynes as vinylidene precursors. The primary drawback of this approach is that only transition metals capable of forming very stable M=C multiple bonds can effect the rearrangement of a metal(alkyne) to a metal(vinylidene), and the reactivity of these metal(vinylidene) species is greatly tempered relative to free vinylidenes.47 Consequently, very few catalytic vinylidene cycloaddition and bond insertion reactions have been developed, particularly those that are enantioselective.
Dinickel catalysts promote methylenecyclopropanation reactions using 1,1-dichloroalkenes as vinylidene precursors and Zn as a stoichiometric reductant (Figure 6A). Various alkyl, aryl, and heteroaryl-substituted vinylidenes are viable substrates. Monosubstituted alkenes are the most reactive class of partners. However, unhindered or moderately activated internal alkenes, such as cyclopentene or norbornene, can also be used. Recently, Cramer developed an asymmetric variant of this transformation using C2-symmetric chiral NDI ligands derived from 2,6-di-(1-arylethyl)anilines.48
Figure 6.
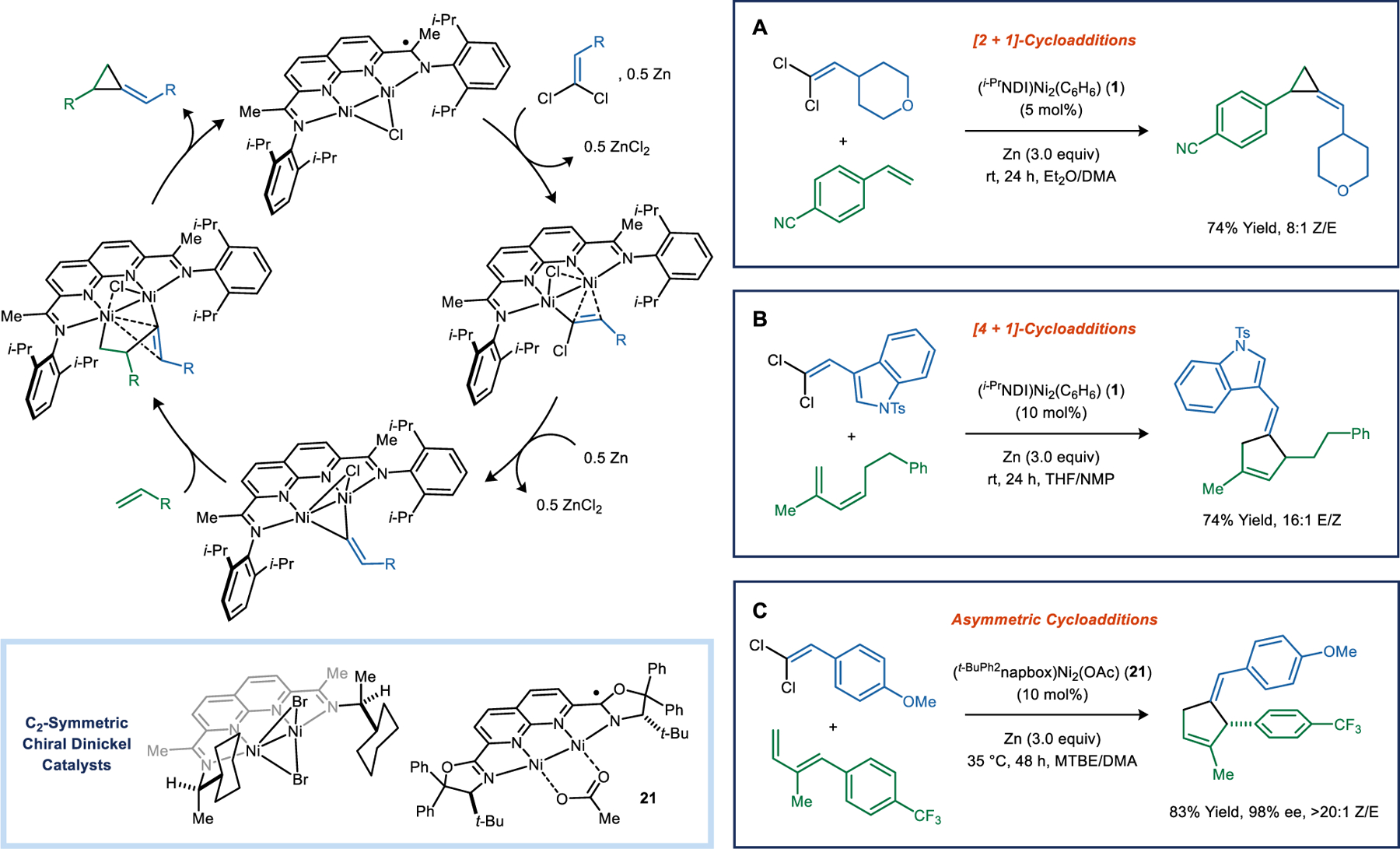
Dinickel catalyzed vinylidene transfer reactions using 1,1-dichloroalkenes. (A) [2 + 1]-cycloaddition and (B) [4 + 1]-cycloaddition reactions. (C) Asymmetric catalysis using chiral naphthyridine–bis(oxazoline) (napbox) ligands.
When a (Z)-alkene is subjected to the standard methylenecyclopropanation conditions, the product is obtained as a cis/trans mixture. This lack of stereospecificity indicates that the two C–C bonds of the cyclopropane are formed in distinct mechanistic steps. This experiment and others led us to propose the general mechanism shown in Figure 6. The 1,1-dichloroalkene is activated by the (i-PrNDI)Ni2Cl complex and, upon reduction with Zn (0.5 equiv), forms a Ni2(C(Cl)=CHR)Cl species. A second reduction triggers isomerization to a bridging vinylidene complex. Addition of the alkene generates a metallacyclic intermediate, and C–C reductive elimination forms the methylenecyclopropane.
Following our development of the methylenecyclopropanation reaction, we wondered whether vinylidene transfers to 1,3-dienes could produce [4 + 1]-cycloadducts.4 Pericyclic [4 + 1]-cycloadditions between 1,3-dienes and free carbenes are disfavored due to closed-shell repulsion of the carbene lone pair and the filled π-orbitals of the diene.49 Instead, these reactions generally produce vinylcyclopropanes. Given that our metal catalyzed vinylidene transfer reactions were likely proceeding by stepwise organometallic mechanisms, they would presumably not be subject to these same orbital constraints.
Under similar conditions to those of the methylenecyclopropanation reaction, a broad scope of 1-substituted, 2-substituted, 1,2-disubstituted, 1,3-disubstituted, and 2,3-disubstituted dienes underwent successful [4 + 1]-cycloaddition (Figure 6B). Notably, competing [2 + 1]-cycloaddition was not observed for any of the dienes that we examined, suggesting that the final C–C reductive elimination strongly favors the formation of five- over three-membered rings. Use of a 2-siloxy-1,3-diene yielded cyclopentenones in a process reminiscent of the Danishefsky diene Diels–Alder reaction.50 Finally, intramolecular [4 + 1]-cycloadditions could be carried out in high yield using substrates in which the 1,1-dichloroalkene and diene were connected by a two or three-atom tether.
Substituted oxazolines are commonly used in chiral ligands for asymmetric catalysis51 but have not been widely incorporated into binucleating ligand frameworks (Figure 6C).52 We found that chiral napthyridine–bis(oxazoline) ligands (napbox) could be prepared and subsequently metallated to form (napbox)Ni2(OAc) complexes.53 Complex 21 is an effective catalyst for enantioselective, intermolecular [4 + 1]-cycloaddition reactions, providing ee values up to 98%.
4. Conclusions and Outlook
The utility of transition metal complexes in catalysis derives from their ability to form and cleave bonds through two-electron redox processes. By combining these elementary steps in different sequences and with different substrate combinations, a broad scope of useful transformations can be developed. Using a redox active ligand, it is also possible to carry out such two-electron oxidations and reductions at metal–metal bonds. In our own work, we have focused on naphthyridine–diimine ligands. However, many other redox-active motifs are currently known. Recently, Tomson described a series of macrocyclic complexes that feature two (PDI)M units connected by alkyl tethers.54 Given our observation that (NDI)Ni2-catalyzed processes can require the reversible formation and cleavage of Ni–Ni bonds, it is interesting to consider the implications of such ligands with added conformational flexibility.
In some of our studies with (NDI)Ni2 complexes, we have observed activity or selectivity properties that diverge significantly from analogous mononickel catalysts. These results suggest that catalyst nuclearity may represent a useful parameter for catalyst optimization that is complementary to ligand effects. Nuclearity can also be used to adjust the thermodynamic landscape of a catalytic cycle to avoid low-energy sinks that slow down or prevent turnover. For example, while mononickel complexes can promote the rapid stoichiometric dimerization of aryl azides to form azoarenes, catalytic turnover is not observed due to product inhibition. (NDI)Ni2 complexes form much less stable azoarene adducts, making the product dissociation step favorable.
Finally, in rare cases, dinuclear complexes can promote transformations that are not known with mononuclear complexes. Such is the case with the catalytic carbonylation of norbornadiene, which relies on an unusual C–C oxidative addition. While it cannot be concluded definitively that mononuclear complexes could not be designed to promote this reaction, none have yet been identified, and it is reasonable to speculate that unique ability of a metal–metal bond to form several stabilizing metal–π interactions would not be readily reproduced with just one metal. Uncovering other such examples is the subject of our ongoing research.
KEY REFERENCES.
Pal, S.; Uyeda, C.* “Evaluating the Effect of Catalyst Nuclearity in Ni-Catalyzed Alkyne Cyclotrimerizations.” J. Am. Chem. Soc. 2015, 137, 8042–8045.1 The activity and selectivity of dinickel and mononickel catalysts are directly compared in alkyne cyclooligomerization reactions. Mechanistic studies provided insight into the structure and reactivity of dinickel metallacycles.
Hartline, D. R.; Zeller, M.; Uyeda, C.* “Catalytic Carbonylative Rearrangement of Norbornadiene via Dinuclear Carbon–Carbon Oxidative Addition” J. Am. Chem. Soc. 2017, 139, 13672–13675.2 Norbornadiene possesses a similar ring-strain energy to that of cyclopropane but is not known to undergo strain-induced C–C oxidative addition. A dinickel complex rapidly activates a C–C σ-bond of norbornadiene, and a catalytic carbonylative rearrangement is described.
Powers, I. G.; Andjaba, J. M.; Luo, X.; Mei, J.; Uyeda, C.* “Catalytic Azoarene Synthesis by a Dinuclear Ni Complex” J. Am. Chem. Soc. 2018, 140, 4110–4118.3 A dinickel catalyst promotes the dimerization of aryl azides to form azoarenes. Mononickel catalysts are not competent as catalysts for this reaction, and the weak binding of azoarenes to the dinickel active site is critical to achieving turnover.
Zhou, Y.-Y.; Uyeda, C.* “Catalytic Reductive [4 + 1]-Cycloadditions of Vinylidenes and Dienes.” Science 2019, 363, 857–862.4 Thermal pericyclic [4 + 1]-cycloadditions suffer from high electronic barriers due to unfavorable filled–filled orbital interactions that develop in the transition state. Dinickel catalysts can promote [4 + 1]-cycloadditions of 1,3-dienes and vinylidenes through a stepwise organometallic pathway.
ACKNOWLEDGMENT
We are grateful to the members of our research group for their contributions to the work described in this Account. We thank Daniel Ess (Brigham Young University) for collaborations on computational modeling studies. Financial support for this program was provided by the NSF (CHE‐1554787, CHE-2101931) and the NIH (R35 GM124791). C.U. is grateful for support from an Alfred P. Sloan Foundation Fellowship, a Camille Dreyfus Teacher–Scholar award, and a Lilly Grantee award.
REFERENCES
- (1).Pal S; Uyeda C “Evaluating the Effect of Catalyst Nuclearity in Ni-Catalyzed Alkyne Cyclotrimerizations” J. Am. Chem. Soc 2015, 137, 8042–8045. [DOI] [PubMed] [Google Scholar]
- (2).Hartline DR; Zeller M; Uyeda C “Catalytic Carbonylative Rearrangement of Norbornadiene via Dinuclear Carbon–Carbon Oxidative Addition” J. Am. Chem. Soc 2017, 139, 13672–13675. [DOI] [PubMed] [Google Scholar]
- (3).Powers IG; Andjaba JM; Luo X; Mei J; Uyeda C “Catalytic Azoarene Synthesis from Aryl Azides Enabled by a Dinuclear Ni Complex” J. Am. Chem. Soc 2018, 140, 4110–4118. [DOI] [PubMed] [Google Scholar]
- (4).Zhou Y-Y; Uyeda C “Catalytic reductive [4 + 1]-cycloadditions of vinylidenes and dienes” Science 2019, 363, 857–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Cotton FA; Curtis NF; Harris CB; Johnson BFG; Lippard SJ; Mague JT; Robinson WR; Wood JS “Mononuclear and Polynuclear Chemistry of Rhenium (III): Its Pronounced Homophilicity” Science 1964, 145, 1305–1307; [DOI] [PubMed] [Google Scholar]; (b) Cotton FA “Discovering and understanding multiple metal-to-metal bonds” Acc. Chem. Res 1978, 11, 225–232. [Google Scholar]
- (6).(a) Liddle ST Molecular metal-metal bonds: compounds, synthesis, properties; John Wiley & Sons, 2015; [Google Scholar]; (b) Berry JF; Lu CC “Metal–Metal Bonds: From Fundamentals to Applications” Inorg. Chem 2017, 56, 7577–7581. [DOI] [PubMed] [Google Scholar]
- (7).(a) Cooper BG; Napoline JW; Thomas CM “Catalytic Applications of Early/Late Heterobimetallic Complexes” Catal. Rev 2012, 54, 1–40; [Google Scholar]; (b) Kornecki KP; Berry JF; Powers DC; Ritter T In Prog. Inorg. Chem; Karlin, K. D., Ed.; John Wiley & Sons, Inc.: Hoboken, New Jersey, 2014; Vol. 58, p 225–302; [Google Scholar]; (c) Buchwalter P; Rosé J; Braunstein P “Multimetallic Catalysis Based on Heterometallic Complexes and Clusters” Chem. Rev 2015, 115, 28–126; [DOI] [PubMed] [Google Scholar]; (d) Pye DR; Mankad NP “Bimetallic catalysis for C–C and C–X coupling reactions” Chem. Sci 2017, 8, 1705–1718; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Powers IG; Uyeda C “Metal–Metal Bonds in Catalysis” ACS Catal 2017, 7, 936–958; [Google Scholar]; (f) Farley CM; Uyeda C “Organic Reactions Enabled by Catalytically Active Metal–Metal Bonds” Trends Chem 2019, 1, 497–509. [Google Scholar]
- (8).(a) Stambuli JP; Kuwano R; Hartwig JF “Unparalleled Rates for the Activation of Aryl Chlorides and Bromides: Coupling with Amines and Boronic Acids in Minutes at Room Temperature” Angew. Chem., Int. Ed 2002, 41, 4746–4748; [DOI] [PubMed] [Google Scholar]; (b) Barder TE “Synthesis, Structural, and Electron Topographical Analyses of a Dialkylbiaryl Phosphine/Arene-Ligated Palladium(I) Dimer: Enhanced Reactivity in Suzuki−Miyaura Coupling Reactions” J. Am. Chem. Soc 2005, 128, 898–904. [DOI] [PubMed] [Google Scholar]
- (9).(a) Bonney KJ; Proutiere F; Schoenebeck F “Dinuclear Pd(I) complexes-solely precatalysts? Demonstration of direct reactivity of a Pd(I) dimer with an aryl iodide” Chem. Sci 2013, 4, 4434–4439; [Google Scholar]; (b) Aufiero M; Sperger T; Tsang ASK; Schoenebeck F “Highly Efficient C-SeCF3 Coupling of Aryl Iodides Enabled by an Air-Stable Dinuclear PdI Catalyst” Angew. Chem., Int. Ed 2015, 54, 10322–10326; [DOI] [PubMed] [Google Scholar]; (c) Kalvet I; Sperger T; Scattolin T; Magnin G; Schoenebeck F “Palladium(I) Dimer Enabled Extremely Rapid and Chemoselective Alkylation of Aryl Bromides over Triflates and Chlorides in Air” Angew. Chem., Int. Ed 2017, 56, 7078–7082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).(a) Powers DC; Ritter T “Bimetallic Pd(III) complexes in palladium-catalysed carbon–heteroatom bond formation” Nat. Chem 2009, 1, 302–309; [DOI] [PubMed] [Google Scholar]; (b) Powers DC; Geibel MAL; Klein JEMN; Ritter T “Bimetallic Palladium Catalysis: Direct Observation of Pd(III)−Pd(III) Intermediates” J. Am. Chem. Soc 2009, 131, 17050–17051; [DOI] [PubMed] [Google Scholar]; (c) Powers DC; Benitez D; Tkatchouk E; Goddard WA; Ritter T “Bimetallic Reductive Elimination from Dinuclear Pd(III) Complexes” J. Am. Chem. Soc 2010, 132, 14092–14103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Bart SC; Chłopek K; Bill E; Bouwkamp MW; Lobkovsky E; Neese F; Wieghardt K; Chirik PJ “Electronic Structure of Bis(imino)pyridine Iron Dichloride, Monochloride, and Neutral Ligand Complexes: A Combined Structural, Spectroscopic, and Computational Study” J. Am. Chem. Soc 2006, 128, 13901–13912. [DOI] [PubMed] [Google Scholar]
- (12).Bouwkamp MW; Bowman AC; Lobkovsky E; Chirik PJ “Iron-Catalyzed [2π + 2π] Cycloaddition of α,ω-Dienes: The Importance of Redox-Active Supporting Ligands” J. Am. Chem. Soc 2006, 128, 13340–13341. [DOI] [PubMed] [Google Scholar]
- (13).(a) Praneeth VKK; Ringenberg MR; Ward TR “Redox-Active Ligands in Catalysis” Angew. Chem., Int. Ed 2012, 51, 10228–10234; [DOI] [PubMed] [Google Scholar]; (b) Luca OR; Crabtree RH “Redox-active ligands in catalysis” Chem. Soc. Rev 2013, 42, 1440–1459. [DOI] [PubMed] [Google Scholar]
- (14).(a) Gavrilova AL; Bosnich B “Principles of Mononucleating and Binucleating Ligand Design” Chem. Rev 2004, 104, 349–384; [DOI] [PubMed] [Google Scholar]; (b) Bera JK; Sadhukhan N; Majumdar M “1,8-Naphthyridine Revisited: Applications in Dimetal Chemistry” Eur. J. Inorg. Chem 2009, 2009, 4023–4038; [Google Scholar]; (c) Desnoyer AN; Nicolay A; Rios P; Ziegler MS; Tilley TD “Bimetallics in a Nutshell: Complexes Supported by Chelating Naphthyridine-Based Ligands” Acc. Chem. Res 2020, 53, 1944–1956. [DOI] [PubMed] [Google Scholar]
- (15).Zhou Y-Y; Hartline DR; Steiman TJ; Fanwick PE; Uyeda C “Dinuclear Nickel Complexes in Five States of Oxidation Using a Redox-Active Ligand” Inorg. Chem 2014, 53, 11770–11777. [DOI] [PubMed] [Google Scholar]
- (16).(a) Schore NE “Transition metal-mediated cycloaddition reactions of alkynes in organic synthesis” Chem. Rev 1988, 88, 1081–1119; [Google Scholar]; (b) Montgomery J “Nickel-Catalyzed Reductive Cyclizations and Couplings” Angew. Chem., Int. Ed 2004, 43, 3890–3908. [DOI] [PubMed] [Google Scholar]
- (17).Lautens M; Klute W; Tam W “Transition Metal-Mediated Cycloaddition Reactions” Chem. Rev 1996, 96, 49–92. [DOI] [PubMed] [Google Scholar]
- (18).(a) Pauson PL “The Khand reaction : A convenient and general route to a wide range of cyclopentenone derivatives” Tetrahedron 1985, 41, 5855–5860; [Google Scholar]; (b) Schore NE “The Pauson-Khand Cycloaddition Reaction for Synthesis of Cyclopentenones. Org. React 1991, 40, 1–90. [Google Scholar]
- (19).Magnus P; Principe LM “Origins of 1,2- and 1,3-stereoselectivity in dicobaltoctacarbonyl alkene-alkyne cyclizations for the synthesis of substituted bicyclo[3.3.0]octenones” Tetrahedron Lett 1985, 26, 4851–4854. [Google Scholar]
- (20).(a) Gimbert Y; Lesage D; Milet A; Fournier F; Greene AE; Tabet J-C “On Early Events in the Pauson−Khand Reaction” Org. Lett 2003, 5, 4073–4075; [DOI] [PubMed] [Google Scholar]; (b) Banide EV; Müller-Bunz H; Manning AR; Evans P; McGlinchey MJ “X-ray Crystal Structure of an Alkene–Pentacarbonyldicobalt–Alkyne Complex: Isolation of a Stable Magnus-Type Pauson–Khand Reaction Intermediate” Angew. Chem., Int. Ed 2007, 46, 2907–2910; [DOI] [PubMed] [Google Scholar]; (c) Lesage D; Milet A; Memboeuf A; Blu J; Greene AE; Tabet J-C; Gimbert Y “The Pauson–Khand Mechanism Revisited: Origin of CO in the Final Product” Angew. Chem., Int. Ed 2014, 53, 1939–1942. [DOI] [PubMed] [Google Scholar]
- (21).Hartline DR; Zeller M; Uyeda C “Well-Defined Models for the Elusive Dinuclear Intermediates of the Pauson–Khand Reaction” Angew. Chem., Int. Ed 2016, 55, 6084–6087. [DOI] [PubMed] [Google Scholar]
- (22).(a) Diercks R; Stamp L; Dieck HT “Diazadiene als Steuerliganden in der homogenen Katalyse, VII. Katalytische Darstellung und Struktur eines doppelt vicinal substituierten Cyclooctatetraens – 1,4,5,8-Tetrakis[(p-tolyloxy)methyl]-1,3,5,7-cyclooctatetraen” Chem. Ber 1984, 117, 1913–1919; [Google Scholar]; (b) Diercks R; Stamp L; Kopf J; tom Dieck H “Elementary Steps in Catalytic 1-Alkyne Coupling to Substituted Cyclooctatetraenes” Angew. Chem., Int. Ed 1984, 23, 893–894. [Google Scholar]
- (23).Kwon D-H; Proctor M; Mendoza S; Uyeda C; Ess DH “Catalytic Dinuclear Nickel Spin Crossover Mechanism and Selectivity for Alkyne Cyclotrimerization” ACS Catal 2017, 7, 4796–4804. [Google Scholar]
- (24).Labinger JA “Tutorial on Oxidative Addition” Organometallics 2015, 34, 4784–4795. [Google Scholar]
- (25).Tipper CFH “Some reactions of cyclopropane, and a comparison with the lower olefins. Some platinous-cyclopropane complexes. “ J. Chem. Soc 1955, 2045–2046.
- (26).Adams DM; Chatt J; Guy RG; Sheppard N “149. The structure of “cyclopropane platinous chloride”“ J. Chem. Soc 1961, 738–742.
- (27).Wong HNC; Hon MY; Tse CW; Yip YC; Tanko J; Hudlicky T “Use of cyclopropanes and their derivatives in organic synthesis” Chem. Rev 1989, 89, 165–198. [Google Scholar]
- (28).Reissig H-U; Zimmer R “Donor−Acceptor-Substituted Cyclopropane Derivatives and Their Application in Organic Synthesis” Chem. Rev 2003, 103, 1151–1196. [DOI] [PubMed] [Google Scholar]
- (29).Gang Z; Janis L “Highly Active Nickel Catalysts for the Isomerization of Unactivated Vinyl Cyclopropanes to Cyclopentenes” Angew. Chem., Int. Ed 2004, 43, 2277–2279. [DOI] [PubMed] [Google Scholar]
- (30).Rounds HR; Zeller M; Uyeda C “Dinuclear Pathways for the Activation of Strained Three-Membered Rings” Organometallics 2018, 37, 545–550. [Google Scholar]
- (31).Vogel E “Kleine Kohlenstoff‐Ringe” Angewandte Chemie 1960, 72, 4–26. [Google Scholar]
- (32).Schleyer P. v. R.; Williams JE; Blanchard KR “Evaluation of strain in hydrocarbons. The strain in adamantane and its origin” J. Am. Chem. Soc 1970, 92, 2377–2386. [Google Scholar]
- (33).Schrauzer GN “On Transition Metal-Catalyzed Reactions of Norbornadiene and the Concept of π Complex Multicenter Processes” Adv. Catal 1968, 18, 373–396. [Google Scholar]
- (34).(a) Abel EW; Bennett MA; Wilkinson G “646. Norbornadiene–metal complexes and some related compounds” J. Chem. Soc 1959, 3178–3182;; (b) Defieber C; Grützmacher H; Carreira EM “Chiral Olefins as Steering Ligands in Asymmetric Catalysis” Angew. Chem., Int. Ed 2008, 47, 4482–4502. [DOI] [PubMed] [Google Scholar]
- (35).Hirano M; Komine N; Arata E; Gridneva T; Hatori A; Kaizawa N; Kamakura K; Kuramochi A; Kurita S; Machida S; Okada H; Sawasaki A; Uchino T “Recent advances of achiral and chiral diene ligands in transition-metal catalyses” Tetrahedron Lett 2019, 60, 150924. [Google Scholar]
- (36).Betoré MP; Casado MA; García-Orduña P; Lahoz FJ; Polo V; Oro LA “2,5-Norbornadiene C–C Coupling Reactions Mediated by Iridium Complexes” Eur. J. Inorg. Chem 2016, 2016, 3489–3499. [Google Scholar]
- (37).Mukerjee SL; Nolan SP; Hoff CD; Lopez de la Vega R “Heat of reaction of (norbornadiene)molybdenum tetracarbonyl with monodentate and bidentate ligands. Solution thermochemical study of ligand substitution in the complexes cis-L2Mo(CO)4” Inorg. Chem 1988, 27, 81–85. [Google Scholar]
- (38).(a) Waterman R; Hillhouse GL “Group Transfer from Nickel Imido, Phosphinidene, and Carbene Complexes to Ethylene with Formation of Aziridine, Phosphirane, and Cyclopropane Products” J. Am. Chem. Soc 2003, 125, 13350–13351; [DOI] [PubMed] [Google Scholar]; (b) Shay DT; Yap GPA; Zakharov LN; Rheingold AL; Theopold KH “Intramolecular C-H Activation by an Open-Shell Cobalt(III) Imido Complex” Angew. Chem., Int. Ed 2005, 44, 1508–1510; [DOI] [PubMed] [Google Scholar]; (c) Kogut E; Wiencko HL; Zhang L; Cordeau DE; Warren TH “A Terminal Ni(III)−Imide with Diverse Reactivity Pathways” J. Am. Chem. Soc 2005, 127, 11248–11249; [DOI] [PubMed] [Google Scholar]; (d) King ER; Sazama GT; Betley TA “Co(III) Imidos Exhibiting Spin Crossover and C–H Bond Activation” J. Am. Chem. Soc 2012, 134, 17858–17861; [DOI] [PubMed] [Google Scholar]; (e) Kornecki KP; Briones JF; Boyarskikh V; Fullilove F; Autschbach J; Schrote KE; Lancaster KM; Davies HML; Berry JF “Direct Spectroscopic Characterization of a Transitory Dirhodium Donor-Acceptor Carbene Complex” Science 2013, 342, 351–354. [DOI] [PubMed] [Google Scholar]
- (39).Dai X; Warren TH “Discrete Bridging and Terminal Copper Carbenes in Copper-Catalyzed Cyclopropanation” J. Am. Chem. Soc 2004, 126, 10085–10094. [DOI] [PubMed] [Google Scholar]
- (40).Maity AK; Zeller M; Uyeda C “Carbene Formation and Transfer at a Dinickel Active Site” Organometallics 2018, 37, 2437–2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).(a) Powers IG; Kiattisewee C; Mullane KC; Schelter EJ; Uyeda C “A 1,2-Addition Pathway for C(sp2)−H Activation at a Dinickel Imide” Chem.-Eur. J 2017, 23, 7694–7697; [DOI] [PubMed] [Google Scholar]; (b) Powers IG; Andjaba JM; Zeller M; Uyeda C “Catalytic C(sp2)–H Amination Reactions Using Dinickel Imides” Organometallics 2020, 39, 3794–3801. [Google Scholar]
- (42).Griffiths J “II. Photochemistry of azobenzene and its derivatives” Chem. Soc. Rev 1972, 1, 481–493. [Google Scholar]
- (43).(a) Beharry AA; Woolley GA “Azobenzene photoswitches for biomolecules” Chem. Soc. Rev 2011, 40, 4422–4437; [DOI] [PubMed] [Google Scholar]; (b) Merino E; Ribagorda M “Control over molecular motion using the cis-trans photoisomerization of the azo group” Belstein J. Org. Chem 2012, 8, 1071–1090; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mart RJ; Allemann RK “Azobenzene photocontrol of peptides and proteins” Chem. Commun 2016, 52, 12262–12277. [DOI] [PubMed] [Google Scholar]
- (44).Andjaba JM; Rybak CJ; Wang Z; Ling J; Mei J; Uyeda C “Catalytic Synthesis of Conjugated Azopolymers from Aromatic Diazides” J. Am. Chem. Soc 2021, 143, 3975–3982. [DOI] [PubMed] [Google Scholar]
- (45).Harrold ND; Waterman R; Hillhouse GL; Cundari TR “Group-Transfer Reactions of Nickel−Carbene and −Nitrene Complexes with Organoazides and Nitrous Oxide that Form New C═N, C═O, and N═N Bonds” J. Am. Chem. Soc 2009, 131, 12872–12873. [DOI] [PubMed] [Google Scholar]
- (46).Knorr R “Alkylidenecarbenes, Alkylidenecarbenoids, and Competing Species: Which Is Responsible for Vinylic Nucleophilic Substitution, [1 + 2] Cycloadditions, 1,5-CH Insertions, and the Fritsch−Buttenberg−Wiechell Rearrangement?” Chem. Rev 2004, 104, 3795–3850. [DOI] [PubMed] [Google Scholar]
- (47).(a) Bruneau C; Dixneuf P Metal vinylidenes and allenylidenes in catalysis: from reactivity to applications in synthesis; John Wiley & Sons, 2008; [Google Scholar]; (j) Trost BM; McClory A “Metal Vinylidenes as Catalytic Species in Organic Reactions” Chem.-Asian J 2008, 3, 164–194; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lynam JM “Recent Mechanistic and Synthetic Developments in the Chemistry of Transition-Metal Vinylidene Complexes” Chem.-Eur. J 2010, 16, 8238–8247; [DOI] [PubMed] [Google Scholar]; (d) Roh SW; Choi K; Lee C “Transition Metal Vinylidene- and Allenylidene-Mediated Catalysis in Organic Synthesis” Chem. Rev 2019, 119, 4293–4356. [DOI] [PubMed] [Google Scholar]
- (48).Cramer N; Braconi E “A Chiral Naphthyridine Diimine Ligand Platform Enables Nickel-Catalyzed Asymmetric Alkylidenecyclopropanations” Angew. Chem., Int. Ed, 2020, 59, 16425–16429. [DOI] [PubMed] [Google Scholar]
- (49).Fujimoto H; Hoffmann R “Molecular orbital study of the addition of singlet methylene to butadiene” J. Phys. Chem 1974, 78, 1167–1173. [Google Scholar]
- (50).Danishefsky S; Kitahara T “Useful diene for the Diels-Alder reaction” J. Am. Chem. Soc 1974, 96, 7807–7808. [Google Scholar]
- (51).Yoon TP; Jacobsen EN “Privileged Chiral Catalysts” Science 2003, 299, 1691–1693. [DOI] [PubMed] [Google Scholar]
- (52).Fahrni CJ; Pfaltz A “Synthesis of Chiral C2-Symmetric Binucleating Ligands” Helv. Chim. Acta 1998, 81, 491–506. [Google Scholar]
- (53).Behlen MJ; Uyeda C “C2-Symmetric Dinickel Catalysts for Enantioselective [4 + 1]-Cycloadditions” J. Am. Chem. Soc 2020, 142, 17294–17300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Wang Q; Brooks SH; Liu T; Tomson NC “Tuning metal–metal interactions for cooperative small molecule activation” Chem. Commun 2021, 57, 2839–2853. [DOI] [PMC free article] [PubMed] [Google Scholar]